

Abstract
The ability to perform routine structure-guided drug design for selective BACE inhibitors has been limited because of the lack of robust platform for BACE2 expression, purification, and crystallization. To overcome this limitation, we developed a platform that produces 2–3 mg of pure BACE2 protein per liter of E. coli culture, and we used this protein to design macrocyclic compounds that potently and selectively inhibit BACE1 over BACE2. Compound 2 was found to potently inhibit BACE 1 (Ki = 5 nM) with a selectivity of 214-fold over BACE2. The X-ray crystal structures of unbound BACE2 (2.2 Å) and BACE2 bound to compound 3 (3.0 Å and Ki = 7 nM) were determined and compared to the X-ray structures of BACE1 revealing the S1–S3 subsite as a selectivity determinant. This platform should enable a more rapid development of new and selective BACE inhibitors for the treatment of Alzheimer’s disease or type II diabetes.
Keywords: BACE1, BACE2, selective inhibitors, Alzheimer’s disease, type II diabetes, structure-based drug design
Graphical Abstract
Beta-site amyloid precursor protein (APP) cleaving enzymes 1 and 2 (BACE1 and BACE2) are transmembrane aspartic acid proteases. BACE1 was first identified and characterized in 1999 as the major β-secretase. Since then, BACE1 has emerged as a promising therapeutic target for Alzheimer’s disease (AD).1-4 More recently, BACE2 has also been recognized as a potential target for the treatment of type II diabetes (TIID),5 which is a major metabolic disorder characterized by hyperglycemia. Decreased pancreatic β-cell mass and function are the two typical abnormalities observed in TIID. Overexpression of full-length Tmem27, a type I transmembrane protein in the plasma membrane of pancreatic β cells, increases the mass of the β cells in vivo and augments glucose stimulated insulin secretion.6 The shedding process that leads to cleavage in the ectodomain of Tmem27 results in the loss of function of Tmem27 on pancreatic β cells. In 2011, Esterhazy et al. screened numerous protease inhibitors and siRNA and identified BACE2 as the major Tmem27 sheddase.5 Their results implicated that BACE2 controls β-cell maintenance and suggested that it could be a potential therapeutic target for TIID.
More recently, the islet amyloid polypeptide (IAPP, also known as amylin) was identified as another physiological substrate of BACE2.7 The aberrant production or processing of IAPP would trigger the dimerization and rapid production of amyloid fibrils, which is another key feature of TIID.8,9 The accumulation of amyloid fibrils leads to the formation of plaques in the pancreatic islets and results in dysfunction of β cells.10 Therefore, it is suggested that inhibition of BACE2 to reduce the amount of processed IAPP would restore the function β cells and improve insulin secretion.11 BACE2 inhibitors therefore may serve as potential treatment for TIID.
The development of BACE1 inhibitors has been intensively investigated for nearly two decades4. Unfortunately, no FDA approved BACE1 inhibitor drugs have emerged so far. A number of clinical trials on BACE1 inhibitors have failed due in part to severe adverse effects.12,13 The lack of selectivity and off-target inhibition of BACE2 and cathepsin D by these investigational drugs may contribute to the undesired side effects.13 Potential selectivity issues with these drugs are also supported by the considerable side effects observed with BACE1/BACE2 double-knockout mice.14
BACE1 and BACE2 share an overall 52% amino acid sequence identity and 68% similarity. The amino acids within the active sites of BACE1 and BACE2 are even more conserved at greater than 80% identity,15 making the design of selective BACE1 or BACE2 inhibitors exceptionally challenging. Due to the lack of a robust platform for BACE2 purification and crystallization, molecular dynamic simulations had to be utilized as one of the main approaches to understand the selectivity of BACE1 and BACE2 inhibitors.16,17 In the present study, we developed an efficient protein production process for BACE2 that produces highly active enzyme that is amenable to crystallization and X-ray structure determination to aid the structure-based design of selective BACE inhibitors.
Prior to the work presented herein, there were only two published protocols on the production and purification of BACE2 from E. coli inclusion bodies. Ostermann et al.18 designed a construct containing the sequence of proBACE2 from Ala20p to Ala398 where “p” designates the pro domain (Figure S1). They solubilized the inclusion bodies with 8 M guanidinium chloride at pH 8 and refolded the protein at alkaline pH (9.4–10.4). Their refolding buffer contains expensive reagents such as arginine and oxidized/reduced glutathione. In their protocol, a two-step dilution process was applied to achieve refolding of BACE2. The dilution factor for each step is 20-fold, which results in several liters of protein solution at the end of the refolding process, which therefore makes handling inconvenient in the subsequent purification steps. For this method, the pH is dropped to pH 3.2 for autoactivation by release of the pro-domain and then the mature BACE2 protein is purified via hydrophobic interaction and size-exclusion column chromatography.
Emmons and co-workers19 made an expression construct with the BACE1 pro-segment (TQHGIRLPLRSGLGGAPLG) followed by the BACE2 catalytic sequence. They solubilized the inclusion bodies with 7.5 M urea at pH 10.5–11 and refolded BACE2 by rapid dilution into cold water with a dilution factor of 50–60. In their method, the refolding solution was first stirred at 4 °C for 3 weeks, and then the temperature was raised to 37 °C, and the solution was incubated for another 3 weeks. Finally, to purify BACE2, multiple chromatographic steps were used including an anion-exchange column, a size-exclusion column, and a BACE2-specific affinity column (cross-linked to a BACE2 ligand). The total time for BACE2 purification using this method requires nearly two months.
The current protocols for BACE2 production and purification are either too time-consuming or too costly for the average academic lab to pursue BACE2 research. This is likely the reason why the BACE2 protein is typically purchased from commercial vendors and why the majority of available BACE2 X-ray crystal structures are produced from industry.20,21 We therefore, pursued the development of an efficient and relatively inexpensive method for the production of pure BACE2 from E. coli inclusion bodies in sufficient quantities for kinetic, biochemical, and structural studies on.
Similar to Ostermann et al., we utilized an expression construct that encodes the sequence of proBACE2 (Ala20p-Ala398) see Figure S1. We also introduced a mutation, E269A, to facilitate crystallization,20 as well as a TEV recognition site between the pro and catalytic domains to remove the pro domain (Figure S1B). The final pET28a plasmid encoding the gene of BACE2 catalytic domain was transformed into E. coli. Protein expression was induced by the addition of IPTG to a final concentration of 0.2 mM. The inclusion bodies were collected and solubilized with 8 M urea. Similar to Emmons et al., the BACE2 protein was readily refolded in cold water with a dilution factor of 80. We shortened the refolding time considerably compared to the published protocol.19 The refolding solution was kept at 4 °C for 1 day and then quickly heated up to 25 °C by stirring the 4–5 L refolding solution on a heating plate for 1 h. The temperature-jump approach allows the folding intermediate to rapidly convert to native protein.22 The refolding protein solution was incubated at 25 °C using a tabletop incubator and the activity was monitored daily. The peak activity was observed after 4 days of incubation. The total time for our refolding process is only 4–5 days. Finally, the refolded protein was purified using ammonium sulfate precipitation and only a single chromatographic step, which was a hydrophobic interaction column (HIC). The final purity of BACE2 is over 95% as judged by SDS-PAGE (Figure S2). Using this method, the final yield of active BACE2 protein was 2.5 mg from 1 L culture scale (Table S1). The kinetic parameters of purified BACE2 were determined using the fluorescent peptide substrate derived from the β-secretase cleavage site of the Swedish APP mutation.23,24 The catalytic efficiency (kcat/Km) for BACE2 at pH 4 and at 37 °C is 0.23 μM−1 min−1 (equivalent to 3.8 × 103 M−1 s−1, kcat = 5.7 ± 0.5 min−1, Km= 24.8 ± 4.7 μM).
The BACE2 protein expressed and purified by our method crystallized without any inhibitor bound from a solution containing 0.1 M MES (pH 6), 1.4 M (NH4)2SO4, 6% isopropanol, and 4% tert-butanol. The X-ray structure was determined to a resolution of 2.2 Å using the published BACE2 apoenzyme structure (PDB 3ZKG) as a search model for molecular replacement.20 After several runs of manual building and structure refinement, the final resulting Rwork and Rfree values were 21.9% and 25.7%, respectively (Table S2).
Analysis of this structure in comparison to other X-ray structures of unbound BACE2 (Table S3) reveals different orientations in the flap-loop (83VTVKYTQGSWTG94) and the loop102-110 (Figure 1A). The flap-loop in the BACE2 structure determined here is in a more closed conformation where it resides 2.3 Å closer to the active site compared to a previously published BACE2 structure (PDB 3ZKG, Figure 1B) in its unbound state.20 The orientation of the flap-loop reported here is actually more similar to an inhibitor-bound BACE2 structure (PDB 2EWY, Figure 1B).18 The flap-loop was reported as a flexible region in BACE1 structures as well. In 2012, Xu et al. published a systematic study on the position of the flap-loop in BACE1.25 By comparing all the BACE1 structures in the PDB and performing molecular dynamic simulations, they concluded that the orientation of the flap-loop in BACE1 structures could be affected either by inhibitor binding or by crystal packing. The open conformation is mainly present in unbound BACE1 structures, whereas the closed conformation is observed in inhibitor-bound structures. When inhibitors bind to the BACE1 active site, they can form hydrogen bonds with the residues in the flap-loop, which then produces a closed conformation. The different conformations of the flap-loop observed in BACE1 structures are thought to play a role in regulating the entry of the substrate during catalysis.
Figure 1.

(A) Overlay of BACE2 structures of 3ZKG (marine) and 6UJ0 (magenta, structure determined in this study). The boxed regions and associated insets show the enlargement of the overlay of flap-loop (top) and mobile loop102-110 (bottom). (B) Overlay of BACE2 flap-loop from different crystal forms. The BACE2 protein structure is shown in gray surface. The catalytic residues are shown in sticks and colored based on the atom types. The flap loops from different crystal forms are shown as ribbons. Structures for unbound BACE2 are shown for 6UJ0 (magenta, determined in this study) and 3ZKG (marine), as well as for inhibitor bound BACE2 (orange, PDB 2EWY). The double arrow indicates the observed flexibility of the flap-loop.
A previous study reported that the flap-loop in BACE1 appears to adopt different orientations when crystallized at different pH values.26 It was observed that the flap-loop tends to reside closer to the active site adopting an inactive conformation at high pH (pH 6–7). In contrast, an open conformation is adopted when crystallizing at low pH (pH 4–5), indicating that the pH of the buffer used for crystallization may affect the orientation of the flap-loop. In our current studies, the BACE2 crystals were grown from MES buffer at pH 6. Similar to what has been reported for BACE1, we observe a closed conformation of the flap-loop in our structure of BACE2. However, it is not clear if the closed conformation observed here is simply due to the effect of pH since there are no BACE2 structures determined at various pH values for comparison as has been done for BACE1.
In order to investigate the factors that could affect the orientation of the flap-loop in BACE2, we conducted a systematic comparison of all of the BACE2 structures reported in the PDB, including wild-type and the BACE2-E269A mutant, and the results are summarized in Table S3. The E269A surface mutation was first reported by Banner et al.20 to significantly improve the resolution of BACE2 crystal structures. Among 15 reported structures, nearly two-thirds of them were determined in the presence of a crystal-aide binding protein, Xaperone, Fab, or Fynomer. We compared and analyzed the unbound and bound structures as well as structures determined in different space groups. However, the inhibitor-induced movement or crystal packing could not simply explain the flexibility of the flap-loop observed in the BACE2 structures. The open conformation was observed in both unbound and inhibitor-bound structures of BACE2 crystallizing in the same space group (3ZKG vs 3ZKI and 3ZKQ vs 3ZKS, Table S3). Different conformations were also observed in the same space group (3ZKM vs 3ZKQ). More BACE2 structures will be required to make any definitive conclusions.
Residues within the flap-loop involved in the ligand binding are highly conserved between BACE1 and BACE2, except for Pro70 in BACE1, which is a Lys86 in BACE2. The substitution of a proline with a lysine in the BACE2 flap-loop might affect the flexibility of the flap-loop as well as its interactions with the ligand, thereby making this region a potentially good target for designing inhibitor selectivity.21
Other than the flap-loop, a significant shift was also observed in the 102TIPKGFNTS110 (loop102-110) loop (Figure 1A). However, the movement of loop102-110 could simply be interpreted as a result of crystal packing effects since this loop is located on the surface and close to the dimer in the next unit cell (Figure S3). Besides the difference observed in the flap-loop and loop102-110, strong, residual (Fo – Fc) electron density surrounding residues 278SQLACW283 was identified in our unbound structure. This region has been reported as a binding hot spot for different crystal-aide binding proteins.18 The currently unidentified molecule that fits in the residual electron density could act as a crystal-aide and play a role in stabilizing the crystal packing (Figure S4A). We modeled the residual electron density in this region with a hepta-alanine peptide into the A-chain and a penta-alanine peptide into the B-chain, which resulted in a good overall fit (Figure S4B,C). Residual positive density was observed in the difference map (Fo – Fc map), which is likely to be specific side chain density (Figure S4B,C). The extra density may arise from a peptide in the BACE2 pro-domain, which is produced through the autocleavage process and was not removed during the purification process. However, we were not able to confirm the identity of the extra density.
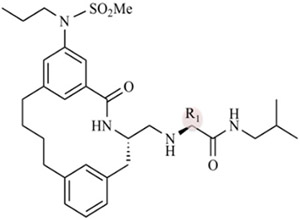
Macrocyclic BACE1 inhibitors have been designed to improve potency and drug-like properties.27-29 Three potent macrocyclic compounds including one that has been characterized in a murine model27 have been evaluated in our previous work on developing a drug discovery platform for BACE14. In the present study, we determined the Ki values of the three designed macrocyclic compounds against BACE2 as a parallel study to our BACE1 work. The degree of selectivity of each compound was calculated using our previously reported Ki values for BACE1 (Table 1).30 These three compounds were designed to have the same macrocyclic scaffold but different substitutions at the P1′ moiety (R1 position in Table 1). All three compounds turn out to be selective BACE1 inhibitors. Among these three compounds, compound 2 contains a 2-propanol group at the P1′ position and exhibits a 214-fold selectivity for BACE1 over BACE2. Compound 2 is also the most potent inhibitor against BACE1 (Ki = 5 nM), but it is the weakest inhibitor against BACE2 (Ki = 1114 nM) among the three compounds. Upon substitution of a larger pentane or pentanol group at the P1′ position, we observe increases in the potency of compounds 1 and 3 for BACE2 with slight decreases in potencies toward BACE1.
Table 1.
Ki of Macrocyclic Compounds against BACE1 and BACE2
| ||||
|---|---|---|---|---|
| compd | R1 |
Ki against BACE1 (nM) |
Ki against BACE2 (nM) |
BACE1 selectivity |
| 1 | pentane | 21 ± 3 | 576 ± 38 | 28 |
| 2 | 2-propanol | 5 ± 3 | 1114 ± 40 | 214 |
| 3 | 2-pentanol | 7 ± 5 | 99 ± 29 | 15 |
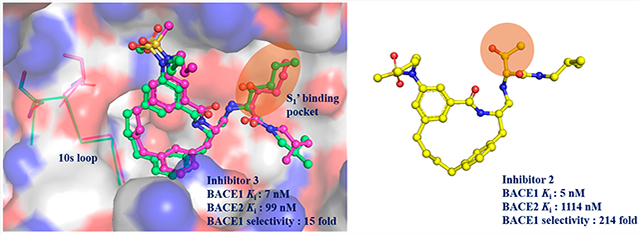
Since inhibitor 3 most potently inhibited BACE2, we determined the X-ray structure of the complex to 3 Å resolution (Table S2). The electron density for inhibitor 3 was well-defined for accurately modeling inhibitor 3 into the active site (Figure S5A). Inhibitor 3 binds in the same orientation as it does in the BACE1 active site (PDB: 6NV7, Figure S5B). The network of hydogen bonds between inhibitor 3 and the active sites of BACE1 and BACE2 are nearly identical (Figure 2A,B). However, the P1–P3 linkage in inhibitor 3 makes significant van der Waals contacts with residues in the 10s loop (Gly11, Gln12, and Gly13) in the BACE1 active site but not in the active site of BACE2 (Figure 2A,B).
Figure 2.

Ligand interaction plot of inhibitor 3 with (A) BACE1 and (B) BACE2. The plots are adapted from the output of LIGPLOT. The original plots are shown in Figure S6. Inhibitor 3 is shown in green (BACE1) and magenta (BACE2) line structures, and the non-carbon atoms are colored according to the atom types. The polar contacts are indicated by the dash lines with distances shown in angstroms, and the interacting residues are represented as black line. The residues involved in the hydrophobic interactions are labeled and shown as red hashes. Residues involved in BACE1 specific hydrophobic interactions are highlighted with pink circles. (C) Overlay of inhibitor 3 in the active site of BACE1 (green) and BACE2 (magenta). Inhibitor 3 is shown in ball and stick and colored according to the atom types. The 10s loop is labeled and shown as lines. Ser10 (Ser26 in BACE2) and Thr232 (Thr245 in BACE2) are shown in stick and colored based on atom types. The distance from the hydroxyl side chain of Thr232 (Thr245 in BACE2) to the carbonyl oxygen of Ser10 (Ser26 in BACE2) is shown in angstroms. The location of the residues involved in BACE1 specific interactions are highlighted within the pink circle (GQG in 10s loop). See also panel A and Figure S7. (D) Open conformation of 10s loop (shown as a ribbon representation) in unbound BACE1 (PDB: 3TPJ, colored in yellow) and inhibitor 3 bound BACE1 (PDB: 6NV9, colored in green). Ser10, Thr232, inhibitor 3, and an ordered water molecule are shown in ball and stick. The distance between the ordered water and the hydroxyl side chain of Thr232 is shown in angstroms.
Based on our X-ray structural models, we suspect that the aforementioned interactions between inhibitor 3 and BACE1 are absent in BACE2, which would result in different orientations of the 10s loop. The 10s loop adopts different conformations in the active sites of BACE1 and BACE2 when bound to inhibitor 3 (Figure 2C). In the BACE2–inhibitor 3 complex, the distance between the hydroxyl side chain of Thr245 and the carbonyl oxygen of Ser26 is 3 Å, which is within hydrogen-bonding distance. The hydrogen bond between these two residues maintains the 10s loop in a closed conformation.31 However, this hydrogen bond is replaced by an interaction between Thr232 and an ordered water molecule in BACE, which precludes the contact of Thr232 with Ser10 and thus results in the open conformation of the 10s loop in the BACE1 active site (Figure 2D). The ordered water molecule is observed in unbound BACE1 structures (Figure 2D), and its role may be to help maintain the open conformation of the 10s loop. It has been shown that the S3 substituent plays a role in determining the orientation of 10s loop by breaking the hydrogen bond between the ordered water and Thr232 in BACE1.31 The different conformations of the10s loop might affect the binding properties of inhibitor 3 in the S1–S3 pocket. The P1–P3 linkage in inhibitor 3 binds deeper in the S1–S3 pocket, and is capable of making more favorable van der Waals interactions with residues Gly11 to Gly13 in the 10s loop in the active site of BACE1 (Figure S7), which likely explains the better inhibitory potency and selectivity of the macrocyclic scaffold for BACE1.
The S1′ pocket in the BACE1 active site is formed by residues Tyr198, Lys224, and Ile226, and loop Thr329–Val332 (Figure 3A). These residues are conserved in the BACE2 active site except for three loop residues, Thr329, Gly330, and Thr331 (Figure 3A). This loop adopts different conformations in the active sites of BACE1 and BACE2 likely due to their sequence differences (Figure 3A). The orientation of this loop leads to a more compact S1′ pocket in the active site of BACE1 compare to BACE2 (Figure 3B). When the P1′ pentane group (inhibitor-3) is substituted with a smaller functional moiety (inhibitor-2), it still retains its potency against BACE1 but exhibits a 10-fold reduction in potency against BACE2 (Table 1). A possible explanation could be that the smaller propane moiety might still maintain sufficient van der Waals interactions in the more compact S1′ pocket in the BACE1 active site but not in the expanded S1′ pocket in BACE2.
Figure 3.

(A) Overlay of inhibitor 3 in the active site of BACE1 (green) and BACE2 (magenta). The set of amino acid residues surrounding the S1′ binding pocket are shown as stick structures. Loop Thr329–Val332 in BACE1 and Asn341–Val344 are shown in ribbon. (B) Closer view of the S1′ binding pocket in the active site of BACE1 (top) and BACE2 (bottom). BACE1 and BACE2 are shown in surface presentation and colored in gray. Flap-loop is removed for clarity. Inhibitor 3 is shown in sphere presentation and colored according to the atom types. The S1′ binding pocket in the active site of BACE1 and BACE2 is highlighted with orange circles.
In conclusion, we developed an efficient protein production and purification protocol for BACE2 that is convenient and scaleable. The BACE2 protein purified by this method is highly suitable for enzyme kinetics and X-ray structural studies aimed at performing structure-based drug design for the development of selective BACE inhibitors. Three BACE1 selective macrocyclic compounds were evaluated using this platform and are reported. We determined the X-ray structures of BACE1 and BACE2 bound with the same compound, and to the best of our knowledge, this is the first example of X-ray structures of a BACE inhibitor bound to both enzymes. The structural information provided molecular insights into the BACE1 selectivity of the macrocyclic scaffold. Based on the structural comparison, we determined that the P1–P3 linkage in the macrocyclic scaffold plays a role in the selectivity.
Future directions on optimization of this scaffold to improve the selectivity toward BACE1 include varying the length or bonding properties of the P1–P3 scaffold through the introduction of different functional groups. The removal of the P2-sulfoamide group would improve BACE2 selectivity based on our previous work on selective peptidomimetic BACE2 inhibitors.32,33 Nevertheless, a comprehensive structure–activity relationship study would better guide us on the optimization of this scaffold in the future for possible new treatments for Alzheimer’s diseases and type 2 diabetes.
MATERIAL AND METHODS
Construct Design.
The expression plasmid containing BACE2 pro and catalytic domain (Ala21p to Ala398, the residue numbering used here is shown in Figure S1A) was synthesized for optimal expression in E. coli (BioBasicInc.). The gene was cloned into a pET28a expression vector with an N-terminal His8 tag. In addition, a TEV cleavage recognition sequence had been engineered between the pro and catalytic domain for the N-terminal truncation to create a mature BACE2.The E269A surface mutation was introduced to facilitate crystallization, which was reported previously by Banner et al.20 The scheme of BACE2 construct is shown in Figure S2B. The expression construct was verified by DNA sequencing at Purdue University Genomics Core Facility.
Expression and Refolding of BACE2 Catalytic Domain.
Protein expression, inclusion body isolation, and solubilization are as described in the previous publication.30 To achieve refolding, the solubilized protein was diluted 80-fold into cold water and stirred at 4 °C for 1 day. Next day, the refolding solution was heated from 4 to 25 °C using a heating plate and then further incubated at 25 °C for another 4 days. The refolded protein sample was concentrated using a stirred ultrafiltration cell (Amicon) for the following purification steps.
Hydrophobic Interacting Chromatography.
After refolding, (NH4)2SO4 was added into the concentrated protein solution to a final concentration of 0.6 M to precipitate out high molecular weight aggregates. The precipitated aggregates were removed by centrifugation at 26727g for 20 min. The supernatant from the (NH4)2SO4 precipitation step was then loaded onto a 30 mL phenyl sepharose hydrophobic interacting column (Amersham Biosciences) equilibrated in buffer A (20 mM Tris-HCl, pH 8, 0.15 M NaCl, 0.6 M (NH4)2SO4). The column was then washed with 1 column volume of buffer A until the signals at UV280 were stable. Protein was eluted with a linear gradient to 100% buffer B (20 mM Tris-HCl, pH 8, 0.15 M NaCl). Fractions were collected and pooled based on the SDS-PAGE and the activity assay described in the previous publication.30 The purified BACE2 protein was concentrated and stored at 4 °C until further use.
Crystallization and Determination of X-ray Structure of BACE2 Protein.
To set up the crystallization screening trays, the pooled BACE2 protein from hydrophobic interacting column fractions was first buffer exchanged into buffer B and then concentrated to 5 mg/mL. A final concentration of 500 μM potent inhibitor was mixed with the concentrated protein solution if needed. The mixture of the protein and inhibitor was incubated for at least 1 h at 4 °C in order to form the BACE2–inhibitor complex. To set up the screening trays, 300 nL of BACE2 protein or BACE2–inhibitor complex was then mixed with 300 nL of reservoir in a sitting drop using the crystallization robot (Mosquito crystal, TTP labtech). Eleven different crystallization trays were prepared with commercial screening suites (MCSG crystallization suite 1–4 from Anatrace, PEGs I, PEGs II, PACT, pH clear I, pH clear II, cations, anions crystallization suites from Qiagene). The trays were stored in a 20 °C cabinet and monitored regularly. Needle cluster crystals were observed 8 weeks after setting up the tray from a condition containing 0.1 M MES (pH 6) and 1.26 M (NH4)2SO4. Different attempts have been made to optimize this condition, including additive screening (HRT2–138, Hampton research). Eventually, a single needle apo crystal was obtained in a condition containing 0.1 M MES (pH 6), 1.4 M (NH4)2SO4, 6% isopropanol, and 4% tertbutanol. The crystal was fresh frozen into liquid nitrogen supplemented with 25% glycerol as cryo-protectant. The diffraction data was collected at beamline 31-ID-D at the Lilly Research Laboratories Collaborative Access Team (LRL-CAT), at the Advanced Photon Source (APS), at Argonne National Laboratories. The structure was solved with molecular replacement using the published BACE2 apo structure (PDB 3ZKG) as a model.20
Steady-State Kinetic Assays.
To determine Km and kcat values, 100 nM of BACE2 enzyme and various substrate concentrations (0 μM, 0.78 μM, 1.56 μM, 3.12 μM, 6.25 μM, 12.5 μM, 25 μM, and 50 μM) of a fluorogenic 8-mer peptide substrate (Mca-S-E–V-N-L-D-A-E–F-K-Dnp) were mixed in the reaction buffer containing 0.1 M sodium acetate (pH 4). All reactions were initiated by the addition of the peptide substrate. The initial slope of each reaction (RFU/min) was measured and converted to specific activity (μM·min−1·mg−1) using the experimentally derived fluorescence extinction coefficient of the peptide substrate. The BACE2 enzyme concentration was quantified using BioRad Bradford protein assay. The specific activity values were plotted as a function of substrate concentration, and the data were then fit to the Michaelis–Menten equation using nonlinear regression and the program Graphpad Prism 6.
BACE1 Enzyme Purification and BACE 1 and 2 Enzyme Kinetics Studies.
The human BACE1 protein was purified using our recently described method.30 The BACE1 and BACE2 enzyme kinetic assays and data analysis were performed using our recently described approach.30 Compound 2a32 is used as a standard compound in the titration assay for determining the active BACE2 concentration. For curve fitting, the Ki value was constrained to 14 nM, the substrate concentration was constrained to 1000 nM, and the Km value was constrained to 24810 nM.
Supplementary Material
ACKNOWLEDGMENTS
We thank the LRL-CAT beamline staff for their help in acquiring X-ray data. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding
Partial financial support by the National Institutes of Health (AG 18933) to A.K.G. and 5U54AG065181 to A.D.M. is gratefully acknowledged. Partial financial support from the Purdue EVPRP was also provided to A.D.M. Crystallization and DNA sequencing were partially supported by the Purdue Center for Cancer Research Macromolecular Crystallography and DNA Sequencing Shared Resources, which are partially supported by NIH grant P30 CA023168.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00629.
Amino acid sequence of BACE1 and the expression construct scheme, SDS-PAGE analysis throughout the purification of BACE2, crystal packing of unbound BACE2, extra density observed in unbound BACE2 structure, electron density map of inhibitor 3 in the active site, ligand interaction plot of inhibitor 3 with BACE1 and BACE2, inhibitor 3 in the active site of BACE1 and BACE2, BACE2 purification from 2 L E. coli culture, data collection and refinement statistics, and comparison of BACE2 structures crystallized under different conditions and space group (PDF)
Accession Codes
PDB codes: unbound BACE2 structure 6UJ0; BACE2–inhibitor 3 complex 6UJ1.
The authors declare no competing financial interest.
Contributor Information
Yu-Chen Yen, Department of Biological Sciences, Purdue University, West Lafayette, Indiana 47907, United States.
Annalissa M. Kammeyer, Department of Biological Sciences, Purdue University, West Lafayette, Indiana 47907, United States
Jagannadharao Tirlangi, Department of Chemistry, Purdue University, West Lafayette, Indiana 47907, United States.
Arun K. Ghosh, Department of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, Indiana 47907, United States.
Andrew D. Mesecar, Department of Biological Sciences, Department of Chemistry, and Department of Biochemistry, Purdue University, West Lafayette, Indiana 47907, United States.
REFERENCES
- (1).Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson- Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, and John V (1999) Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402 (6761), 537–40. [DOI] [PubMed] [Google Scholar]
- (2).Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, and Citron M (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286 (5440), 735–41. [DOI] [PubMed] [Google Scholar]
- (3).Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashler JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, and Gurney ME (1999) Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402 (6761), 533–7. [DOI] [PubMed] [Google Scholar]
- (4).Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, and Vassar R (2001) Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci 4 (3), 231–2. [DOI] [PubMed] [Google Scholar]
- (5).Esterházy D, Stützer I, Wang H, Rechsteiner MP, Beauchamp J, Döbeli H, Hilpert H, Matile H, Prummer M, Schmidt A, Lieske N, Boehm B, Marselli L, Bosco D, Kerr-Conte J, Aebersold R, Spinas GA, Moch H, Migliorini C, and Stoffel M (2011) Bace2 is a β cell-enriched protease that regulates pancreatic β cell function and mass. Cell Metab. 14 (3), 365–77. [DOI] [PubMed] [Google Scholar]
- (6).Akpinar P, Kuwajima S, Krützfeldt J, and Stoffel M (2005) Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab. 2 (6), 385–97. [DOI] [PubMed] [Google Scholar]
- (7).Rulifson IC, Cao P, Miao L, Kopecky D, Huang L, White RD, Samayoa K, Gardner J, Wu X, Chen K, Tsuruda T, Homann O, Baribault H, Yamane H, Carlson T, Wiltzius J, and Li Y (2016) Identification of Human Islet Amyloid Polypeptide as a BACE2 Substrate. PLoS One 11 (2), No. e0147254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, and Reid KB (1987) Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. U. S. A 84 (23), 8628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dupuis NF, Wu C, Shea JE, and Bowers MT (2011) The amyloid formation mechanism in human IAPP: dimers have β-strand monomer-monomer interfaces. J. Am. Chem. Soc 133 (19), 7240–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ehrlich JC, and Ratner IM (1961) Amyloidosis of the islets of Langerhans. A restudy of islet hyalin in diabetic and non-diabetic individuals. Am. J. Pathol 38, 49–59. [PMC free article] [PubMed] [Google Scholar]
- (11).Alcarraz-Vizán G, Casini P, Cadavez L, Visa M, Montane J, Servitja JM, and Novials A (2015) Inhibition of BACE2 counteracts hIAPP-induced insulin secretory defects in pancreatic β-cells. FASEB J. 29 (1), 95–104. [DOI] [PubMed] [Google Scholar]
- (12).Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, Tintelnot-Blomley M, Laue G, Beltz K, Vogg B, Schmid P, Frieauff W, Shimshek DR, Staufenbiel M, and Jacobson LH (2015) A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegener 10, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shimshek DR, Jacobson LH, Kolly C, Zamurovic N, Balavenkatraman KK, Morawiec L, Kreutzer R, Schelle J, Jucker M, Bertschi B, Theil D, Heier A, Bigot K, Beltz K, Machauer R, Brzak I, Perrot L, and Neumann U (2016) Pharmacological BACE1 and BACE2 inhibition induces hair depigmentation by inhibiting PMEL17 processing in mice. Sci. Rep 6, 21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D’Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, and De Strooper B (2005) Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J. Biol. Chem 280 (35), 30797–806. [DOI] [PubMed] [Google Scholar]
- (15).Mirsafian H, Mat Ripen A, Merican AF, and Bin Mohamad S (2014) Amino acid sequence and structural comparison of BACE1 and BACE2 using evolutionary trace method. Sci. World J 2014, 482463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen J, Wang J, Yin B, Pang L, Wang W, and Zhu W (2019) Molecular Mechanism of Binding Selectivity of Inhibitors toward BACE1 and BACE2 Revealed by Multiple Short Molecular Dynamics Simulations and Free-Energy Predictions. ACS Chem. Neurosci 10 (10), 4303–4318. [DOI] [PubMed] [Google Scholar]
- (17).Chen J, Yin B, Wang W, and Sun H (2020) Effects of Disulfide Bonds on Binding of Inhibitors to β-Amyloid Cleaving Enzyme 1 Decoded by Multiple Replica Accelerated Molecular Dynamics Simulations. ACS Chem. Neurosci 11 (12), 1811–1826. [DOI] [PubMed] [Google Scholar]
- (18).Ostermann N, Eder J, Eidhoff U, Zink F, Hassiepen U, Worpenberg S, Maibaum J, Simic O, Hommel U, and Gerhartz B (2006) Crystal structure of human BACE2 in complex with a hydroxyethylamine transition-state inhibitor. J. Mol. Biol 355 (2), 249–61. [DOI] [PubMed] [Google Scholar]
- (19).Emmons TL, Mildner AM, Lull JM, Leone JW, Fischer HD, Heinrikson RL, and Tomasselli AG (2009) Large scale refolding and purification of the catalytic domain of human BACE-2 produced in E. coli. Protein Pept. Lett 16 (2), 121–31. [DOI] [PubMed] [Google Scholar]
- (20).Banner DW, Gsell B, Benz J, Bertschinger J, Burger D, Brack S, Cuppuleri S, Debulpaep M, Gast A, Grabulovski D, Hennig M, Hilpert H, Huber W, Kuglstatter A, Kusznir E, Laeremans T, Matile H, Miscenic C, Rufer AC, Schlatter D, Steyaert J, Stihle M, Thoma R, Weber M, and Ruf A (2013) Mapping the conformational space accessible to BACE2 using surface mutants and cocrystals with Fab fragments, Fynomers and Xaperones. Acta Crystallogr., Sect. D: Biol. Crystallogr 69 (6), 1124–37. [DOI] [PubMed] [Google Scholar]
- (21).Fujimoto K, Matsuoka E, Asada N, Tadano G, Yamamoto T, Nakahara K, Fuchino K, Ito H, Kanegawa N, Moechars D, Gijsen HJM, and Kusakabe KI (2019) Structure-Based Design of Selective β-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitors: Targeting the Flap to Gain Selectivity over BACE2. J. Med. Chem 62 (10), 5080–5095. [DOI] [PubMed] [Google Scholar]
- (22).Xie Y, and Wetlaufer DB (1996) Control of aggregation in protein refolding: the temperature-leap tactic. Protein Sci. 5 (3), 517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, and Selkoe DJ (1995) The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med 1 (12), 1291–6. [DOI] [PubMed] [Google Scholar]
- (24).Ermolieff J, Loy JA, Koelsch G, and Tang J (2000) Proteolytic activation of recombinant pro-memapsin 2 (Pro-beta-secretase) studied with new fluorogenic substrates. Biochemistry 39 (51), 16263. [DOI] [PubMed] [Google Scholar]
- (25).Xu Y, Li MJ, Greenblatt H, Chen W, Paz A, Dym O, Peleg Y, Chen T, Shen X, He J, Jiang H, Silman I, and Sussman JL (2012) Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr., Sect. D: Biol. Crystallogr 68 (1), 13–25. [DOI] [PubMed] [Google Scholar]
- (26).Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, and Nukina N (2008) Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol. Cell. Biol 28 (11), 3663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Stachel SJ, Coburn CA, Sankaranarayanan S, Price EA, Pietrak BL, Huang Q, Lineberger J, Espeseth AS, Jin L, Ellis J, Holloway MK, Munshi S, Allison T, Hazuda D, Simon AJ, Graham SL, and Vacca JP (2006) Macrocyclic inhibitors of beta-secretase: functional activity in an animal model. J. Med. Chem 49 (21), 6147–50. [DOI] [PubMed] [Google Scholar]
- (28).Ghosh AK, Devasamudram T, Hong L, DeZutter C, Xu X, Weerasena V, Koelsch G, Bilcer G, and Tang J (2005) Structure-based design of cycloamide-urethane-derived novel inhibitors of human brain memapsin 2 (beta-secretase). Bioorg. Med. Chem. Lett 15 (1), 15–20. [DOI] [PubMed] [Google Scholar]
- (29).Machauer R, Veenstra S, Rondeau JM, Tintelnot-Blomley M, Betschart C, Neumann U, and Paganetti P (2009) Structure-based design and synthesis of macrocyclic peptidomimetic beta-secretase (BACE-1) inhibitors. Bioorg. Med. Chem. Lett 19 (5), 1361–5. [DOI] [PubMed] [Google Scholar]
- (30).Yen YC, Kammeyer A, Jensen KC, Tirlangi J, Ghosh AK, and Mesecar AD (2019) Development of an efficient enzyme production and structure-based discovery platform for BACE1 inhibitors. Biochemistry 58 (44), 4424–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McGaughey GB, Colussi D, Graham SL, Lai MT, Munshi SK, Nantermet PG, Pietrak B, Rajapakse HA, Selnick HG, Stauffer SR, and Holloway MK (2007) Beta-secretase (BACE-1) inhibitors: accounting for 10s loop flexibility using rigid active sites. Bioorg. Med. Chem. Lett 17 (4), 1117–21. [DOI] [PubMed] [Google Scholar]
- (32).Ghosh AK, Brindisi M, Yen YC, Lendy EK, Kovela S, Cardenas EL, Reddy BS, Rao KV, Downs D, Huang X, Tang J, and Mesecar AD (2019) Highly Selective and Potent Human β-Secretase 2 (BACE2) Inhibitors against Type 2 Diabetes: Design, Synthesis, X-ray Structure and Structure-Activity Relationship Studies. ChemMedChem 14 (5), 545–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ghosh AK, Reddy BS, Yen YC, Cardenas E, Rao KV, Downs D, Huang X, Tang J, and Mesecar AD (2016) Design of Potent and Highly Selective Inhibitors for Human β-Secretase 2 (Memapsin 1), a Target for Type 2 Diabetes. Chem. Sci 7, 3117–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.