

Abstract
Advanced wound scaffolds that integrate active substances to treat chronic wounds have gained significant recent attention. While wound scaffolds and advanced functionalities have previously been incorporated into one medical device, the wirelessly triggered release of active substances has remained the focus of many research endeavors. To combine multiple functions including light-triggered activation, anti-septic, angiogenic, and moisturizing properties, we have developed a 3D printed hydrogel patch encapsulating vascular endothelial growth factor (VEGF) decorated with photoactive and antibacterial tetrapodal zinc oxide (t-ZnO) microparticles. To achieve the smart release of VEGF, t-ZnO was modified by chemical treatment and activated through UV/visible light exposure. This process would also make the surface rough and improve protein adhesion. The elastic modulus and degradation behavior of the composite hydrogels, which must match the wound healing process, were adjusted by changing t-ZnO concentrations. The t-ZnO-laden composite hydrogels can be printed with any desired micropattern to potentially create a modular elution of various growth factors. The VEGF decorated t-ZnO-laden hydrogel patches showed low cytotoxicity and improved angiogenic properties while maintaining antibacterial functions in vitro. In vivo tests showed promising results for the printed wound patches, with less immunogenicity and enhanced wound healing.
Keywords: Zinc oxide tetrapod, 3D Printing, Photoactive, Controlled release, Hydrogel composites, Wound healing
Introduction
Chronic inflammation is a widespread complication of diabetes.[1] Diabetic foot ulcers can lead to amputation in 14–24% of cases[2] and 84% of amputations have been linked to extreme ulcers.[1] Chronic wounds are described as diseased tissues that have failed to heal over a reasonable amount of time, typically within three months.[3] Thus, treatment is important and necessary to avoid the consequences of prolonged inflammation and to reduce the probability of systemic infection. To ensure expeditious healing, a treatment that incorporates multiple factors is ideal. These factors include oxygenation, infection treatment, hydration, and the application of growth- and proliferation-related proteins. To include all of these factors, smart wound scaffold devices have gained considerable interest in the past decade.[4] However, incorporating synergistic therapeutic effects like antibacterial properties and proper angiogenesis into one treatment remains challenging.
To alleviate this, nanoparticle composites can be used as smart delivery platforms due to their unique physical properties, such as high surface area, the ability to respond to various stimuli (i.e. light, temperature, electrical signal, etc.), and the ability to control the release of bioactive molecules like growth factors (GFs) under specific conditions.[5] Their success rate is usually hampered because nanoparticles often penetrate cell membranes by endocytosis and damage the cells through increased reactive oxygen species (ROS) concentration, etc.[6] For example, spherical ZnO nanoparticles have been studied extensively for their antibacterial properties,[6–7] however, due to their size, they are phagocytosed by cells and can be cytotoxic, even at low concentrations (> ~25 μg/mL at around 50% viability).[8] Tetrapod-shaped ZnO particles (t-ZnO) are nanostructured microparticles that exhibit most of the benefits of the nanoscale despite being bigger than normal cells. For example, they retain their antibacterial properties without being endocytosed, which makes them fit for many biomedical and tissue engineering applications.[8a, 9] Due to their unique 3D shape and high aspect ratio, they are accessible from all dimensions, resulting in a large contact area with the surrounding media that can facilitate the efficient loading and release of e.g. vascular endothelial growth factor VEGF. In addition, each arm of t-ZnO is a single-crystal, thus showing unique semiconductor and photoactive properties not inherently found in amorphous ZnO nanoparticles.[10] The surface’s chemical and physical properties can also be altered by chemical treatment. Therefore, by tuning the photoactive function of t-ZnO, we could achieve the controlled release of VEGF by light stimulations while maintaining the antibacterial properties.
To successfully integrate these functional microparticles into the wound area for a long period of time, VEGF decorated t-ZnO particles can then be embedded into a printable hydrogel like gelatin methacryloyl (GelMA), which is chemically functionalized and denatured collagen. Additionally, it is possible to tune GelMA hydrogels in terms of stiffness and elasticity, ideally matching those of the native skin, so that they degrade at a rate that supports their replacement by the regenerating tissue to prevent scar tissue formation.[11] The stiffness of the gels is particularly important, as a mismatch in mechanical properties compared to surrounding tissues can cause stress concentrations at the interface that can result in delamination or hypertrophic scarring. Furthermore, the GelMA hydrogels have microporous structures which allow for the uptake of a large number of biological solutions and the release of various biological factors, while the t-ZnO microparticles exhibit enough contact with the wound area to effectively kill bacteria. Therefore, this t-ZnO-laden printed composite hydrogel forms a synergistic wound healing platform, combining the unique properties of both hydrogels and microparticles such as attractive fluid absorbance properties, tunable mechanical properties, VEGF delivery capabilities and antibacterial properties.[12]
In this work, we show how a combination of materials science and biomedical engineering can drastically reduce fabrication efforts while significantly improving the wound healing aspects of wound scaffolds. A smart wound scaffold strategy based on VEGF-decorated and antibacterial t-ZnO microparticles embedded in GelMA hydrogels is presented. Moreover, chemically-treated t-ZnO microparticles can provide denser protein decoration, which can be released by green light stimulations. These composite hydrogels were printed to provide open and porous 3D constructs that could assist in the release of VEGF and oxygenation penetration while maintaining hydration and having antibacterial and angiogenic functions via VEGF-decorated t-ZnO. The cellular behavior, in vivo biocompatibility, wound healing behaviors, antibacterial functions, and the selective release properties of the wound scaffold and the t-ZnO particles have been investigated and discussed in detail.
Results and Discussions
The working concept for the smart wound scaffold and its fabrication is briefly depicted in Schematic 1. We obtained t-ZnO microparticles with 30–100 μm overall size via flame transport synthesis.[13] The particles’ unique 3D tetrapod shape and high aspect ratio (diameter: ~100 nm - 10 μm, length: ~5 μm −100 μm) makes it difficult for them to penetrate cells while allowing them to retain a large contact area with the surrounding media. Aside from these benefits, t-ZnO also has a strong antibacterial effect. Paired with its semiconductor nature, it can become a beneficial platform for protein and drug adhesion and release. Pristine t-ZnO exhibits a bandgap in the UV range.[14] To improve the release rate of VEGF through natural conditions such as visible light stimulation, which does not require specific devices or tools, it is necessary to tune t-ZnO microparticles to become photoactive and enable them to react to both UV and relatively biosafe visible light ranges, while maintaining their antibacterial properties.
Schematic 1.

Schematic diagram of a fabrication process of the smart wound scaffold and working principle for wound healing process: Preparation of t-ZnO microparticles by H2O2-treatment, subsequent VEGF adsorption, and composite ink with GelMA pre-polymer solution. Light-triggered release of VEGF on the H2O2-treated ZnO microparticles. Improved angiogenesis and antibacterial activity by a VEGF-coated t-ZnO embedded the open porous printing constructs at wound area.
To do this, surface-only chemical treatments like etching t-ZnO with e.g., hydrogen peroxide (H2O2) can introduce defects and lower the bandgap of t-ZnO into the range of green light. This enables the t-ZnO microparticles to interact with blue or green light and can thus allow them to deliver specific proteins depending on the selected responsiveness towards different light sources in the future. The t-ZnO itself can show a large bandgap and inconsistent polarity on its side facets due to a difference in oxygen vacancy concentration. High polarity is required for strong physical interactions with the desired growth factors to ensure homogeneous and thick coating. Converting the surface superficially into t-ZnO2 with H2O2 increases protein adhesion due to increased surface area and higher polarity. Subsequently, proteins like the vascular endothelial growth factor (VEGF) can be used for decoration, to, e.g., increase angiogenesis upon release. The coated t-ZnO can then be mixed into a GelMA pre-polymer solution as a composite ink at varying concentrations, and then 3D printed into patch-like open-pore constructs. Antibacterial activity is additionally ensured by the protruding t-ZnO from the printed constructs.
The treatment of t-ZnO with H2O2 is a viable option to change the surface properties of the t-ZnO, like protein adhesion and light responsiveness, without deteriorating the mechanical integrity of the t-ZnO. The H2O2 treatment can be easily recognized by the color change of t-ZnO from a white color to a yellowish color as shown in Figure 1A. The superficial conversion of ZnO to ZnO2 by H2O2 treatment was investigated by micro-Raman spectroscopy, which is presented in Figure 1B. Untreated t-ZnO and H2O2-treated t-ZnO both show their respective, characteristic peaks at 100 cm−1, 440 cm−1, and 1140 cm-1. Additional peaks at 850 cm−1 and 1550 cm−1, which are related to ZnO2, were observed for the H2O2-treated t-ZnO, indicating a partial conversion from ZnO to ZnO2. The presence of some ZnO peaks in the t-ZnO is likely due to the incomplete conversion of the ZnO to ZnO2, making it possible to maintain the functions of both pristine t-ZnO and H2O2-treated t-ZnO. The duration of the chemical treatment dictates the degree of conversion, so the desired amount of ZnO2 can be tailored to the specific requirements.
Figure 1. Tuned photoactive characterization of chemically-treated t-ZnO.

(A) Photograph images showed color change from white to yellow after H2O2-treatment. (B) Raman spectra of t-ZnO microparticles before and after H2O2-treatment. (C) The pristine t-ZnO powder exhibited smooth surface of individual tetrapod in the SEM image (i) and induced low protein adhesion of IgG AF594 in the fluorescence micrograph (ii). Red and blue fluorescence signals indicated by IgG AF594 and t-ZnO, respectively. (D) The surface of the individual tetrapod of t-ZnO treated with H2O2 has turned to a rough and porous surface as shown in the SEM image (i). A significantly thicker coverage of t-ZnO with IgG AF594 can be observed from the fluorescence micrograph (ii). (E) The working principle of protein-release by UV/green light stimulations. Light induces charge carrier separation in the semiconducting t-ZnO microparticles. The charges travel to the surface and accumulate, resulting in proteins being released from the t-ZnO microparticle surface via electrostatic force of repulsion. (F) SEM image showed VEGF-coated on the individual tetrapod of H2O2-treated t-ZnO. (G) Released VEGF on the H2O2-treated t-ZnO with a low (1 μg/mL) and high concentration of VEGF (5 μg/mL) immobilization upon green light exposure over time. (n = 2). (H) Released the concentration of VEGF from H2O2-treated t-ZnO after green and UV light exposure. (n = 2)
The morphology of the t-ZnO before and after treatment with H2O2 was evaluated by scanning electron microscopy (SEM) as shown in Figures 1 C–i and D–i, respectively. The t-ZnO showed hexagonal arms, each with a length of roughly 30 μm and a thickness of around 2 μm. Prior to the H2O2 treatment, the arms’ surfaces were smooth, and afterwards the surfaces became rough and lumpy, while pristine ZnO remained underneath. These SEM images confirmed the results of the Raman spectroscopy, which showed an incomplete transformation of ZnO to ZnO2. The adhesion of the nanostructured ZnO2 on the surfaces of the ZnO arms was so strong that no ZnO2 nanoparticles were released within the biological solutions.
These nanostructured ZnO2 particles also significantly increased the protein adsorption due to their rough and porous surface morphology. To confirm the protein adhesion, the commonly used secondary antibody IgG Alexa Fluor™ 594 (AF594) was coated on the t-ZnO with and without H2O2 treatment as a model protein to easily confirm protein absorption via fluorescence microscopy. Only a mild and inconsistent coating, indicated by the partially expressed red fluorescence signal, was observed on the pristine t-ZnO, which expressed a blue fluorescence signal (Figure 1C–ii, -iii, and -iv). In contrast, H2O2-treated t-ZnO showed a thick and homogeneous coating that was confirmed by the strong red fluorescence signal expressed on all particles, shown in Figure 1D–ii (high magnification) and -iii (low magnification). Furthermore, no blue fluorescence signal was observed on the H2O2 treated samples, most likely because the protein coverage was thick, preventing the excitation of the UV light from reaching the t-ZnO. Additionally, the bandgap of ZnO2 (3.8 eV, visible light range) is higher than that of ZnO, which is 3.37 eV in the UV light range.[15] The inconsistent coating of protein on pristine t-ZnO could be due to the varying polarity on the particles’ side facets. Oxygen vacancies on the ZnO surface are often attributed to polarity, however, their concentrations may vary laterally and protein adsorption by polar bonding might show large lateral deviations. The mechanism of protein adsorption can be covalent bonding between the amide and these oxygen vacancies. This of course only holds for the first layer of protein. All subsequent layers may be held in place by dipole forces from the polar t-ZnO to the protein. The complicated interactions of semiconductors and liquids are often hard to predict, so the superficial conversion to ZnO2 helps with consistent and thick protein adsorption through rough morphology. From the yellow color of the powder, one can also see an optical activation of pristine ZnO underneath, which is attributed to the narrowing bandgap induced by oxygen vacancies.[16]
Protein release experiments were performed to assess the functionality of the proposed working mechanism of H2O2-treated t-ZnO particles, which were produced based on their tuned photoactive properties as well as their strong protein interactions. The ZnO2 which resulted from H2O2 treatment was much more polar than pristine t-ZnO and could therefore bind indiscriminately yet more efficiently to the polar groups of larger proteins. Additionally, the bandgap after H2O2 treatment was smaller, resulting in photoactivity in the optical spectrum. The proposed mechanism for visible light-controlled protein release is due to the formation of electrons and holes, resulting in the accumulation of charges on the ZnO2 surface as shown in Figure 1E. Later the increased charge density could help to release proteins via electrostatic interaction. The energy of the electron-hole pairs needs to be low enough to prevent photocatalysis, which destroys the adsorbed protein. In this process, the charge carriers react with organic substances in the solution.
For the release of proteins, the binding must be light, yet efficient. A large amount of protein loading via strong bonding to the t-ZnO surface might result in low quantities of protein release upon illumination. To evaluate whether the H2O2 treatment is the best way to efficiently load and release of proteins, different surface treatments that would reduce adhesive forces while maintaining efficient loading were tested for t-ZnO. First, different reagents were tested to determine the most effective coating material for t-ZnO in terms of its capacity to enhance protein adsorption. As shown in Figure S1, the pristine t-ZnO microparticles were coated with bovine serum albumin (BSA), oleic acid, and streptavidin, respectively, and then were further coated with IgG AF594. The t-ZnO particles with various coatings were exposed to UV light for 25 seconds. The BSA-coated and the oleic acid-coated t-ZnO microparticles showed no significant changes in the red fluorescence signal after UV light stimulation. Moreover, protein degradation was observed on the oleic acid-coated t-ZnO microparticles (data not shown). This could be due to the addition of ethanol during the IgG AF594 coating process. In terms of the streptavidin-coated samples, there was no observable coating with IgG AF594. Consequently, all treatments showed low adhesion of the protein and no visible protein release by UV light stimulation.
To evaluate protein release by green light stimulation, H2O2-treated t-ZnO microparticles were coated with IgG AF594 before applying UV light for 300 seconds. As a comparison, and to differentiate protein release from protein degradation, hydrophilic and non-photoactive SiO2 particles were coated with IgG AF594 and exposed to UV light (Figure S2). The negative slope despite the photo-inactivity of SiO2 suggests degradation of the material after it had been illuminated. The rate of degradation was around 0.008% per second, which could be explained by photobleaching. Due to the large bandgap of SiO2, it exhibited no semiconductor effects.
Coating efficiency tests were performed on both pristine and H2O2-treated t-ZnO. Human VEGF was coated onto both forms of t-ZnO by incubation in an ice bath for 2 h. The concentration of the protein solution was measured before and after the coating and the adsorption rate of the VEGF was determined as the coating efficiency. For a measured concentration of 1 μg/mL, the maximum concentration of VEGF immobilized on the pristine t-ZnO would be ~9.6 ng/mg of pristine t-ZnO. For H2O2-treated t-ZnO, no remaining VEGF was found in the solution after 2 h of incubation in an ice bath, meaning that the coating efficiency of VEGF on the H2O2-treated t-ZnO was 100% (n=3) and the maximum amount of VEGF would be 10ng/mg of H2O2-treated t-ZnO. The reason for this is the much higher polarity after the etching by H2O2, which can also be observed when putting the dry powder into water. After H2O2-treatment, the t-ZnO powder distributes quickly and efficiently in water, while pristine t-ZnO needs more agitation. The VEGF-coating on the surfaces of H2O2-treated t-ZnO particles was evaluated by SEM. As shown in Figure 1F, the nanostructures of ZnO2 were entirely covered by the VEGF layer and a smooth surface could be seen, but the texture of the surface was completely different from pristine t-ZnO (Figure 1C–i). The formation of these VEGF layers has provided strong evidence for the successful functionalization of t-ZnO with H2O2 treatment, which has generated a hydrophilic ZnO2 nanostructure that allows for uniform protein adhesion.
To confirm the VEGF release, VEGF was coated onto H2O2-treated t-ZnO and was irradiated with strong green light (527 nm at >2000 lx) for a few minutes. Then ELISA assay was performed to monitor the controlled release of VEGF in the supernatant after each irradiation. Here, two t-ZnO groups, one with a high initial concentration during coating and another with a lower initial protein concentration, were compared to optimize the concentration of VEGF loading (Figure 1G). Both low (1 μg/mL) and high (5 μg/mL) concentrations of coating resulted in the same release rate, indicating that the coating was already maximized with the lower coating concentration. Even after 120 seconds with strong green light illumination, there was no decrease in slope, which makes this technique a promising candidate for long term protein release in natural conditions. The concentration of VEGF released per mg t-ZnO was below 1 ng, within the efficient treatment window of VEGF in wound healing on the order of ng/106, which makes this approach suitable for triggering proper angiogenesis without an overdose of VEGF.[17]
When comparing the VEGF release after UV-light and green light stimulations, a strong effect can be observed. In this experiment, the supernatants of the solutions were not removed, and the samples were not washed after each round of illumination. The release from treated microparticles was assessed with an ELISA. The highest concentration of VEGF in the solution was observed after 250 seconds of green light exposure, where the released concentration represented a 600% increase compared to the initial value. When exposed to UV light, only a minor increase in protein concentration could be found. For longer durations of the experiment, while the protein was bound to the t-ZnO surface, the binding may have stabilized the protein and facilitated its integrity. The experiment was carried out at room temperature, and sensitive proteins like VEGF are known to degenerate at higher temperatures after some time. In any case, this set of experiments showed that fine-tuning the semiconductor properties of a ZnO biomaterial is a viable strategy for protein adhesion. VEGF could be successfully coated and released by utilizing green light. The increased coating efficiency and the higher surface area after the H2O2-treatment could provide a reasonable release in the effective range. A significant difference can be observed in the efficiency of protein release between green light illumination and storage under dark conditions, as shown in Figure S3. Although for a thick coating, a certain release solely through the dissolution of the outermost protein layers is also possible, the effect is strongly increased by green light irradiation. This concept could be elaborated on in future works by varying the intensity, duration and wavelength of the light source as well as the treatment of the t-ZnO. Still, this approach is a promising candidate for light-triggered actuators for drug delivery.
To develop a t-ZnO embedded wound scaffold which can protect wounds from outside environmental conditions and infections, a GelMA hydrogel was used to facilitate the wound scaffold due to its attractive fluid absorbance properties, biological properties similar to collagen, structural fidelity via various microfabrication techniques, and easy to tune physical and mechanical properties.[12] The mechanical properties of the wound scaffold should also match those of native skin (Elastic modulus: 100 – 210 kPa)[18] to provide the necessary mechanical cues for accelerated healing of the wound and to minimize scar tissue formation.[19] To resemble the elastic modulus of the native skin, a high concentration GelMA hydrogel with 10% w/v which usually showed > 50 kPa compressive modulus was selected.[12] Furthermore, nanoparticles or microparticles have been used as reinforcing reagents to improve the mechanical properties of hydrogels. Therefore, we studied the effect of t-ZnO microparticles on the physical properties of hydrogels, such as the mechanical and degradation behavior of composite hydrogels with increasing amounts of t-ZnO (from 0 to 5%). Due to the hydrophilic nature of both t-ZnO- and H2O2-treated t-ZnO microparticles, thorough, yet careful vortexing of the t-ZnO particle-laden solution at 37 °C can help with the dispersion of the particles without breaking the particles into a high concentration of GelMA pre-polymer solution. The t-ZnO microparticles were well dispersed in the 10 w/v% GelMA pre-polymer solution without using additional harsh dispersion processes (i.e. using ultrasonicator, strongly acidic or basic solvents, or surfactants) due to hydrophilic nature of t-ZnO microparticles. Also at 37 °C, the increased chain mobility and decreased viscosity of GelMA pre-polymer could help to disperse the t-ZnO particles. To prevent re-agglomeration or precipitation of t-ZnO particles during the crosslinking process, the temperature was rapidly decreased while pouring the t-ZnO-laden GelMA pre-polymer solutions into a mold, resulting in significantly increasing the viscosity of the GelMA pre-polymer solution. After crosslinking, we observed that most of the t-ZnO particles in the GelMA hydrogels were not distributed at equal intervals, but still showed homogeneously distributed t-ZnO particles within the GelMA hydrogels as shown in Figure 2A and Figure S4.
Figure 2. Mechanical properties of t-ZnO-laden hydrogel.
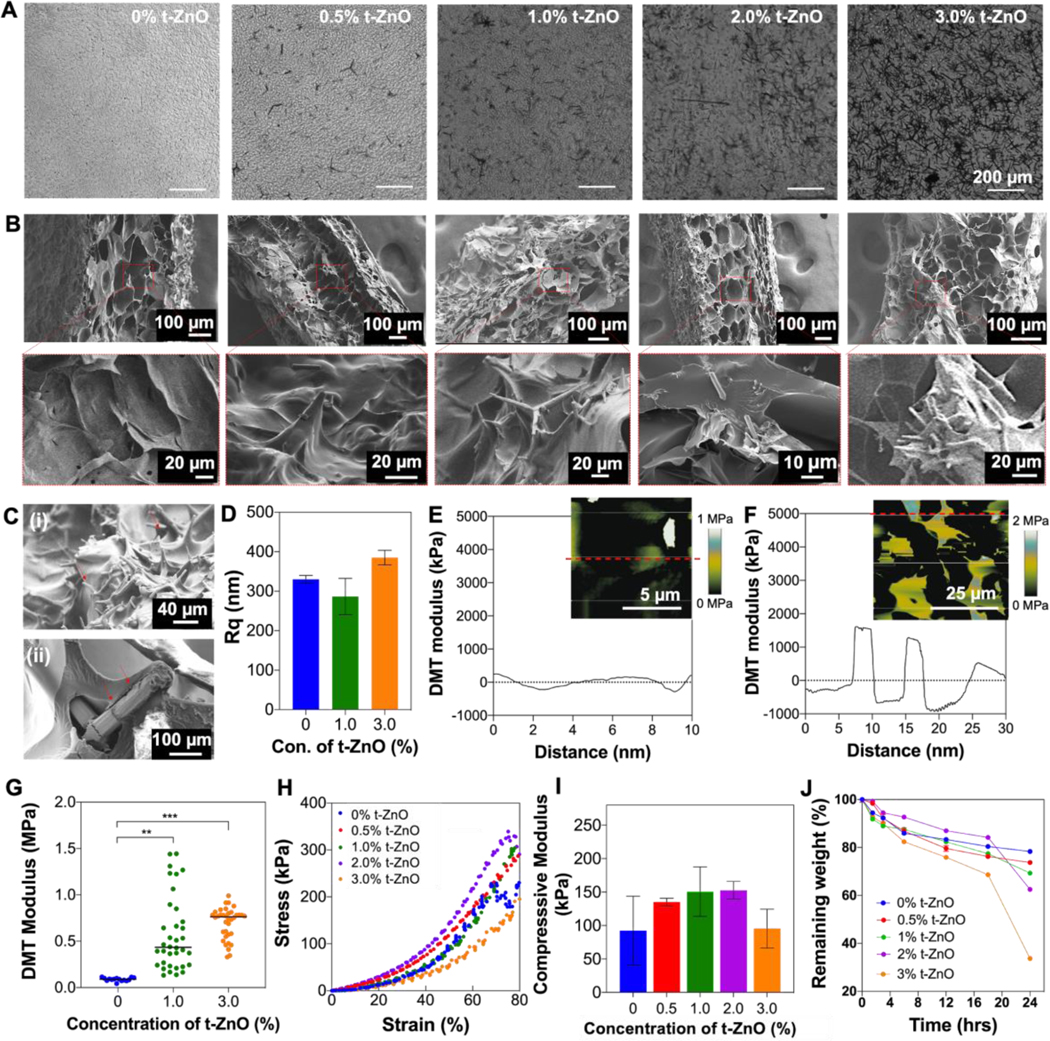
(A) Phase contrast images to evaluate well-dispersed t-ZnO microparticles in GelMA hydrogels. (B) Cross-sectional view of SEM images of t-ZnO-laden GelMA hydrogels with increasing t-ZnO filling (0–3 w/v% t-ZnO) (C) Surface morphology of 2% t-ZnO-laden GelMA hydrogel (i) and tetrapod covered by GelMA gels (ii). (D) Measured root mean square (RMS) roughness (Rq) of t-ZnO-laden GelMA hydrogels with various concentrations of t-ZnO. (n = 3). (E and F) DMT modulus distribution profiles of (E) pristine GelMA and (F) 1 %t-ZnO-GelMA hydrogel along with specified line in inset AFM modulus mapping images. (G) Local DMT modulus of t-ZnO-laden GelMA hydrogels with various concentrations of t-ZnO measured by AFM. (n>15) (H) Stress-strain curves and (I) macroscale compressive modulus of t-ZnO-laden GelMA hydrogels depending on the t-ZnO filling factor at 50 seconds of crosslinking time. (n = 5) (J) Degradation behaviors of t-ZnO-laden GelMA hydrogels depending on the t-ZnO filling factor incubated with collagenase Type II (1 U/mL). (n = 5).
The surface morphology of the smart wound scaffold is of importance since it will be in direct contact with the wound. Hence, SEM analysis was performed to investigate the structure and morphology of the dried GelMA hydrogel. Also, embedded t-ZnO microparticles or extruded tetrapodal arms were clearly observed in the pore walls of composite GelMA hydrogels using an elemental mapping with energy dispersive x-ray spectroscopy (EDX) as shown in Figure S5. The composite hydrogels still maintained the porous nature of a pristine GelMA hydrogel without any significant effect on the morphological change by increasing the concentration of t-ZnO (Figure 2B). Furthermore, the tetrapodal arms of the t-ZnO microparticles stick out on the surfaces of the hydrogels, which has been proven to provide excellent antibacterial properties while supporting cell growth in porous scaffolds.[20] In Figure 2C–i, we observed a large number of tetrapodal arms (indicated by red arrows) on the surfaces of the composite hydrogels. Also, we found that most of the extruded tetrapodal arms, indicated by red arrows in Figure 2C–ii, were coated by the GelMA hydrogel. These extruded tetrapodal arms significantly increased the surface roughness of composite hydrogels, which was measured by topographic mapping using an AFM (Figure 2D). This means that the concentration of t-ZnO can be actively used to determine the number of protruding arms, and t-ZnO microparticles could lead to an increase in the local mechanical properties of the composite hydrogels.
To investigate the microscale local mechanical properties of the composite hydrogels and the impact of the t-ZnO microparticles, a Derjaguin−Muller−Toporov (DMT) modulus was performed using force−deformation plots to reveal the local mechanical properties of different composite hydrogels (Figures 2E and F). With a likeness to the surface roughness of composite hydrogels, a composite hydrogel laden with a high concentration of t-ZnO particles showed the highest level of modulus fluctuation across the surface compared with that of a pristine GelMA hydrogel. Moreover, the local elastic moduli of GelMA hydrogels that incorporated 1.0 % t-ZnO (~ 0.5 MPa) and 3.0 % t-ZnO (~ 0.85 MPa) were higher than that of pristine GelMA (~0.1 MPa), as shown in Figure 2G. The local elastic moduli of the composite hydrogels increased because of the single-crystal structure of ZnO’s tetrapodal arms, which themselves showed high elastic moduli in the ~70 GPa range.[21] Here, we observed a much lower elastic modulus than that of a single crystal of t-ZnO, likely because the extruded tetrapodal arms were covered by the soft GelMA hydrogel around 10 μm in thickness, which was confirmed by Figure 2C–ii. This GelMA was ordered around the arms of the tetrapods and thus became stiffer than the unordered hydrogel in the rest of the composite.
The increased local elastic modulus, which correlated with increases in the concentration of t-ZnO, might have a significant effect on the macroscale mechanical strength of composite hydrogels. To ensure the mechanical integrity of the constructs for different concentrations of t-ZnO microparticles, compression tests were performed (Figure 2H and I). Although fluctuations were not significantly high, the highest compressive modulus was found with 1% and 2% t-ZnO-laden GelMA hydrogel. Lower compressive moduli were found for both the pristine GelMA hydrogel and the highest concentrations of 3% t-ZnO-laden composite hydrogels (Figure 2I and Figure S6). Due to the relatively low concentration and the softness of the hydrogel, the incorporation of the t-ZnO particles does not significantly impact the mechanical properties. However, t-ZnO is a wide bandgap semiconductor, which can absorb UV-light during curing. The absorbed light is therefore not available for crosslinking reactions at very high filling factors. Induced electron-hole-pairs of the t-ZnO can significantly absorb radicals which are generated by the photoinitiator under UV exposure during the photocrosslinking process. These two competing effects may be the reason why the highest measured compressive modulus was at 2% t-ZnO (Figure S6). In any case, the constructs showed an appropriate mechanical stiffness, which enabled handling without problems.
For wound healing and protein release, the degradation of the hydrogels over time also plays a crucial role. To assess the interaction between t-ZnO and the hydrogels in terms of degradation, bulk hydrogel samples with varying concentrations of t-ZnO were subdued to a forced degradation test by incubating them at 37 °C in a collagenase type II dissolved in PBS for 24 hours (Figure 2J). As depicted in Figure 2J, the resulting measurements from the degradation test confirmed that a correlation exists between the hydrogels’ structural integrity and the percentage of t-ZnO within the constructs. With increasing concentrations of t-ZnO, the GelMA polypeptide chains have less available space to form a more solid arrangement, hence, compromising the structural integrity and favoring exposition to enzymatic degradation. Considering their wound healing applications, constructs with 1% t-ZnO were stable enough to withstand the effects of collagenase degradation, making them feasible to function efficiently for prolonged treatment times.
The high-concentration GelMA hydrogels (> 5 w/v%) have dense structures, which, in high concentrations might induce slow degradation behavior in vitro and in vivo and slow down the release of biomolecules or drugs. To solve these issues, microfabricated hydrogels with open porous microstructures, which have large surface areas compared with bulk hydrogels, could help to adjust the degradation rate and achieve the desired release behaviors. They can also help to flow oxygen to a wound site through open porous structures, which could improve wound healing.[22] To do this, a 3D printing technique was developed to fabricate reproducible open porous 3D structures using composite hydrogel inks, such as t-ZnO embedded GelMA hydrogels. The printing process is shown schematically in Figure 3A. As depicted in the schematic, the protein-coated, H2O2-treated, t-ZnO suspended composite inks were successfully printed with good shape fidelity on the print bed, which was kept at 4 °C to maintain 3D structures before crosslinking the hydrogels by UV exposure (Video 1). To improve the printing ability, a gelatin was added to the t-ZnO-laden GelMA pre-polymer solutions. Controlling the temperature of the composite ink was necessary because GelMA-gelatin pre-polymer solutions exhibit strong changes in viscosity with varying temperatures.[23] This is due to the tangling and untangling of individual GelMA-gelatin strands below ~20 °C.[24] We characterized the rheological properties of the inks to confirm the effect of incorporation of t-ZnO and H2O2 treated t-ZnO on the thermal responsive GelMA-gelatin pre-polymer solutions (Figure S7A). The hydrophilic nature of both t-ZnO and H2O2 treated t-ZnO may decrease physical interactions within GelMA-gelatin chains, resulting in minimal changes to the sol-gel transition of the GelMA-gelatin pre-polymer solution. Also, the viscosity of the GelMA-gelatin pre-polymer solution after adding both t-ZnO and H2O2 treated t-ZnO particles at 30 °C was minimally increased (Figure S7B). Therefore, all inks still exhibited lower viscosity values and were easily printable. To prevent the fractures from occurring in the tetrapods, a relatively large nozzle size (Inner diameter: 0.4 mm) was used due to the complex shape and high aspect ratio of the tetrapods (some particles are up to 0.1 mm in diameter). These measures resulted in a successful printing as depicted in Figures 3B and 3C. We observed a homogeneous and fine dispersion of the t-ZnO particles with minimal breaking in the printed microfibers, which is important to secure a sufficient contact area for t-ZnO in the hydrogel. In Figure 3B, we also observed the protruding arms (arrows) from the printed microfibers, which may also help to prevent separations between the scaffold and the injured area via physical interlocking.[25]
Figure 3. 3D printing of t-ZnO-laden GelMA hydrogel constructs.

(A) Schematic diagram of 3D printing of t-ZnO-laden composite ink on the printing bed at 4 °C and crosslinked by UV light exposure. Phase contrast images showed (B) printed microfilaments of 10 % t-ZnO-laden GelMA hydrogels, individual tetrapods of t-ZnO that protruded out from the filaments, and (C) well-dispersed t-ZnO in the filaments. (D) Different shapes of VEGF-coated t-ZnO-laden printed hydrogel. (E) Photograph of the highly dense printed construct (3 layers) under UV and confocal micrograph of the 2% t-ZnO microparticles embedded in the printed construct. (F) Photograph showing free-standing printed construct with excellent mechanical stability to touch and grasp. (G and H) Photographs of the 3D printed constructs with grid-like (G) and layered (H) arrangements using two colored t-ZnO-laden composite inks with multi-nozzle printer in grid-like arrangement for synergetic effect of two inks. (I) SEM image of printed construct that showed microporous morphology and embedded t-ZnO microparticles.
After optimizing the printing parameters, patch-like printed constructs with various shapes were successfully obtained after crosslinking by UV exposure using VEGF-coated 1% H2O2-treated t-ZnO-laden GelMA (Figure 3D) and 2% t-ZnO-laden GelMA (Figure 3E, no VEGF coating) inks. During the crosslinking process, the UV light could not trigger VEGF release from the printed constructs. The UV light might initially be strongly absorbed by the GelMA matrix which has methacrylic groups and photoinitiator. Furthermore, we observed that UV light (100 mW/cm2) was ineffective in triggering VEGF release, as shown in Figure 1H. A fluorescence microscope image of the hydrogels was also taken, showing the blue photoluminescence of the t-ZnO inside the gels when irradiated with UV-light. The printed constructs with various shapes could withstand the touch, could be picked up, and were generally easy to handle (Figure 3F). Patient-specific therapy might need to rely on a mixture of multiple materials for optimal treatment. Multi-material constructs were therefore fabricated using fluorescent dyes to demonstrate the feasibility of construction. Two printed constructs are shown in Figures 3G and 3H, possessing a cross-like and a layered pattern of pink-colored ink with spaces filled with green colored ink, respectively.
In an SEM image, we observed well distributed t-ZnO microparticles within the printed constructs with microporous structures (Figure 3I). Exposure to UV light triggered the crosslinking reaction of the GelMA, and the amount of methacrylic groups and the intensity and duration of the exposure determined the degree of crosslinking. For the treatment of sensitive wounds, and to have a sufficient water uptake, a soft and mildly crosslinked hydrogel is beneficial. Additionally, the UV light should not trigger the premature release of protein. For these two reasons, the UV exposure time should be kept as short as possible. To evaluate the swelling behavior of the hydrogels and the influence of t-ZnO, incubation experiments in PBS at 37 °C were carried out for the 3D-printed constructs for 1 week. Representative images of the constructs that were crosslinked for 10 to 100 seconds can be seen in Figure S8. The constructs did not show significant degradation upon incubation, and the hydrogels only swelled in the solution after 6 days. No significant impact of the added t-ZnO could be observed.
After optimizing the printing parameters, VEGF-coated t-ZnO-laden GelMA hydrogels were characterized to study the feasibility of cell functionality, attachment, and proliferation on the surfaces of composite hydrogels (Figure 4 and Figure S9). Wound healing is contingent upon numerous factors, such as the interactions of different cell lineages with specific functions.[26] Human umbilical vein endothelial cells (HUVEC), a representative cell type involved in the key roles of angiogenesis and wound healing,[20] were cultured and seeded on top of hydrogels. C2C12 myoblasts, which have been previously used to measure the cytotoxicity of t-ZnO particles, were also used to assess the cell compatibility of the t-ZnO microparticles inside the constructs.[27]
Figure 4. In vitro characterization of VEGF-coated t-ZnO GelMA hydrogels.
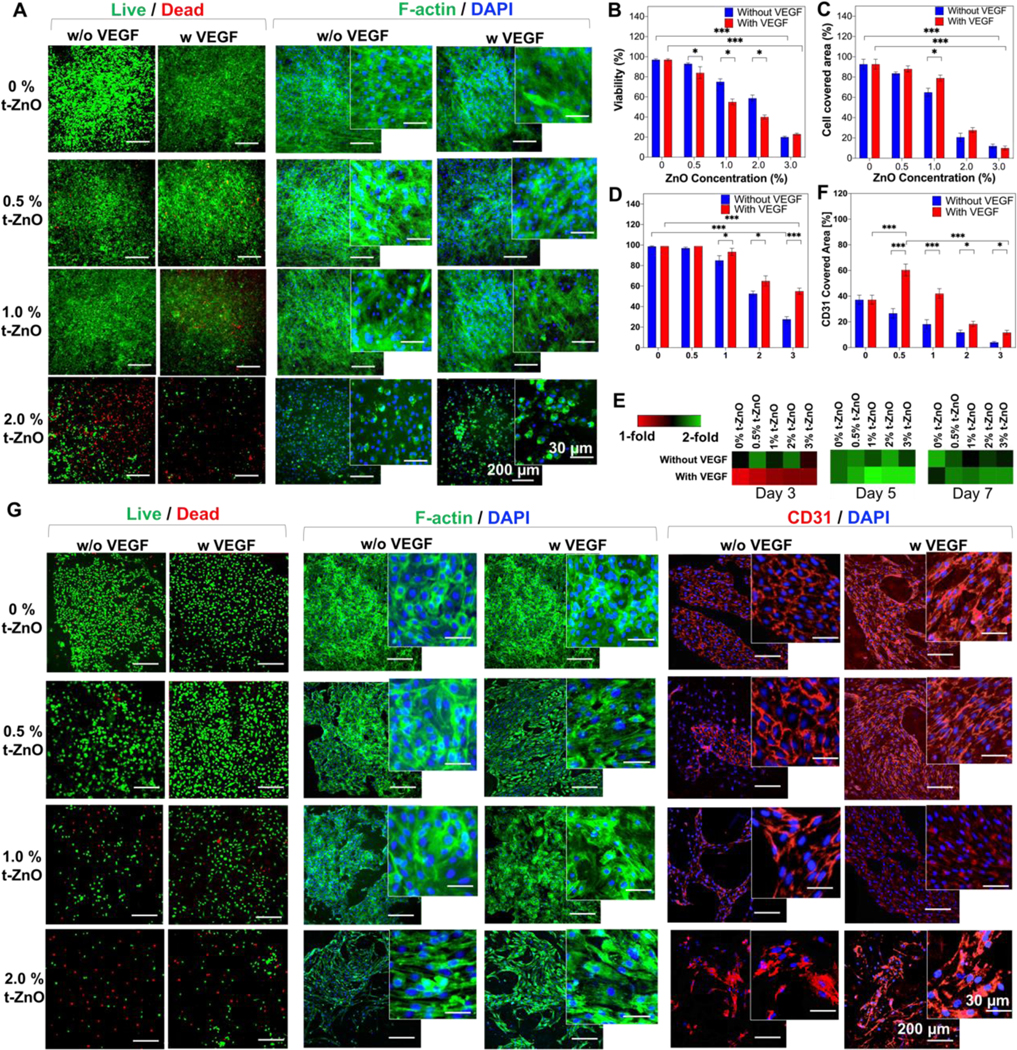
(A) Live/Dead and F-Actin/DAPI staining on C2C12 behaviors cultured on the t-ZnO-laden GelMA hydrogels with and without VEGF coating at various concentrations of t-ZnO. (Β) Quantified viability of cultured C2C12 cells on the constructs with and without VEGF coating and different concentrations of t-ZnO. (n = 3, 2 random images/sample, *p < 0.05, ***p < 0.001). (C) Quantified cell covered area that was calculated from F-Actin/DAPI staining images of cultured C2C12 cells on the t-ZnO-laden GelMA hydrogels obtained on day 3 of culture. (n = 3, 2 random images/sample, *p < 0.05, ***p < 0.001). (D) Quantified viability of cultured HUVECs on the t-ZnO-laden GelMA hydrogels with and without VEGF coating at various concentrations of t-ZnO on day 1 of culture. (E) PrestoBlue™ assay of HUVECs seeded on different concentrations of t-ZnO and t-ZnO VEGF-coated hydrogels over 7 days of culture. (n = 3). (F) Higher CD31 covered area observed on VEGF-coated t-ZnO-laden GelMA hydrogels which were calculated from CD31/DAPI staining images obtained on day 7 of culture. (n = 5, *p < 0.05, ***p < 0.001). (G) Representative fluorescence microscope images of live/dead, F-Actin/DAPI, and CD31/DAPI staining for cultured HUVECs on t-ZnO-laden GelMA hydrogels with and without VEGF coating at various concentrations of t-ZnO on day 7 of culture.
Cell viability was assessed on day 1 of culture for both cell types, and the results showed a decrease in cell viability with higher t-ZnO concentrations. Previous studies regarding the cytotoxic effects of t-ZnO and regular ZnO nanoparticles on different cell types, including the C2C12 cell lineage, have shown similar behaviors. For the nanoparticles, this reduction in viability was already observed at concentrations as low as 40 μg/mL, which is 25 times lower than the concentration of t-ZnO used in this study.[8b, 28] The primary mechanisms for cytotoxicity from ZnO particles have been linked to the induction of Ca+2 influxes, reduced mitochondrial membrane potential, inactivation of key enzymatic processes, loss of membrane integrity and increased levels of reactive oxygen species (ROS).[28b] Since the first influences cannot occur, because endocytosis of ZnO nanoparticles is the trigger to these effects, only increased levels of ROS play a role. Therefore, the presence of elevated ROS concentrations in the t-ZnO hydrogel samples compared to controls was probed, and these findings may also be a contributing factor for cell toxicity (Figure S10). A significant increase in the concentration of ROS was found when t-ZnO was incorporated into the cells, which may also be the reason for their antibacterial properties. In regards to ZnO nanoparticles, further studies have demonstrated that cytotoxicity was drastically increased by the cellular uptake of the nanoparticles, and Zn2+ ions were released into the solution due to their high surface area and the possibility of ZnO nanoparticles penetrating through the cells’ membranes.[8b] In our approach, the particles were much bigger and could not penetrate the cells. This can be observed in cell culture experiments shown in Figure 4A. The release of reactive oxygen species (ROS) in low concentrations offers a therapeutic window for the proliferation of cells and antibacterial properties.[29]
Differences in cell viability between t-ZnO and t-ZnO-VEGF-coated hydrogels were present in both cell types following different trends. In C2C12, 0.5 to 2% of groups with uncoated t-ZnO had higher cell viabilities compared with VEGF-coated groups (Figure 4B). There may be some beneficial interaction between t-ZnO and the C2C12 proliferation, or the presence of VEGF could put stress onto this cell type. On the other hand, for HUVECs, viability was significantly increased for 1 to 3% t-ZnO VEGF-coated constructs (Figure 4D). The metabolic activities of C2C12 and HUVECs were also different from each other. Whereas C2C12 showed a dramatic reduction in metabolic activity after 5 days of culture on both groups, increased metabolic activity was always maintained within the t-ZnO-VEGF-coated hydrogel groups for HUVECs after 3 days of culture. Although VEGF has been associated with promoting the cell growth of different cell types, this peptide has a special tropism for endothelial cells as a mitogen and chemotactic agent.[20, 30] These findings can explain the differences seen which favor cell growth on VEGF-coated samples. HUVECs on VEGF-coated groups showed slow proliferation at first (until day 3 of culture) but exhibited a sustained increase in proliferation rate thereafter (Figure 4E). This behavior can be explained since VEGF has been shown to experience loss of activity in vitro under cellular conditions with a half-life of 40 to 96 min at 37 °C due to adaptive cellular mechanisms, uptake, clearance, and receptor expression changes.[31] In contrast, the HUVECs cultured on uncoated t-ZnO groups showed minimally decreased proliferation rates until day 7, especially at high concentrations of t-ZnO. This result exhibited similar behavior to the viability result shown in Figure 4D. Therefore, the presented VEGF here maintained and improved the endothelial cells’ viability and proliferation.
To evaluate cell adhesion and cell growth, area immunostainings with F-actin and CD31 were performed on days 3 and 7 of culture, respectively. The F-actin covered area showed a decremental tendency with higher concentrations of t-ZnO, while better cell morphology and elongation were found at lower concentrations of t-ZnO (Figure 4C). It has been observed that direct contact between tetrapods and cells can increase toxicity, in comparison with indirect contact with zinc ions from transwell models.[8b] That means that it is feasible that the reduction in the cell covered area was caused by direct cell damage accumulated from contact with t-ZnO. Interestingly, the cell covered area from F-actin staining was higher at 0.5 to 2% on non-VEGF-coated samples compared with coated ones, following the same tendency as that which occurred with metabolic activity. The local concentration of VEGF on the protruding tetrapodal arms may be significantly higher than the requirement for healthy cell activity. This is supported by the C2C12 cells, which did not show significant differences in the cell covered area between VEGF-coated and non-coated groups.
CD31 is a transmembrane protein whose primary functions are promoting endothelial cell migration, angiogenesis, and stabilization of endothelial cell-cell junctions; it also plays an important role as a marker of endothelial cell survival and cytoprotection.[32] These functions are indirectly associated with endothelial integrity and are needed for successful wound healing. HUVECs showed a higher CD31 covered area for 0.5 to 3% VEGF-coated t-ZnO samples compared to non-coated ones (Figures 4F and 4G). The CD31 covered area was also significantly higher in 0.5% t-ZnO VEGF-coated hydrogels compared to controls and other groups. These results show that the presented VEGF here plays a substantial role in endothelial cell proliferation, adhesion, and integrity. VEGF-coated t-ZnO hydrogels at concentrations ranging from 0.5% to 1% seem to sustain an adequate microenvironment for endothelial cell growth, a key cell lineage involved in the wound healing process.
Although bacteria colonies are usually found on normal skin, the loss of integrity in the epidermis may impair mechanisms associated with controlling bacterial growth and could facilitate wound infections. Bacterial invasion leads to increased metabolic strain and a proinflammatory state. This results in tissue injury and chronic wound formation.[33] Therefore, the implementation of antibacterial qualities is crucial to the proper healing of chronic wounds. The principles of antibacterial activity of the t-ZnO are shown schematically in Figure 5A. The formation of ROS species by the t-ZnO can provide a good therapeutic window, where both antibacterial activity, angiogenesis and thus wound healing are high. Additionally, direct bacterial contact may damage the bacteria directly. As shown in Figures 2B and 2D, the number of penetrating arms of t-ZnO increased by increasing the concentration of the microparticles, which may lead to varying degrees of antibacterial activity. The antibacterial effects of both t-ZnO-laden hydrogels and t-ZnO powder were evaluated by measuring the growth of different bacterial strains. Both S. aureus and P. aeruginosa are common pathogens associated with infections in chronic wounds.[34]
Figure 5. Antibacterial function of t-ZnO-laden GelMA hydrogels compared with t-ZnO powder.

(A) The schematic diagram of bacterium and t-ZnO-interactions via oxidative stress, DNA damage and lipid peroxidation, which all damage the bacteria. (B and C) Photographs of cultured samples on the LB-agar after incubation for 12 hours. For both S. aureus and P. aeruginosa, no zone of inhibition was detected. (D) The number of colony forming units of S. aureus and P. aeruginosa cultured on t-ZnO-laden GelMA composite hydrogels. (n = 3, **p < 0.001). (E) The number of colony forming units after exposition to t-ZnO powders with and without H2O2-treatment. A significant decrease in bacterial activity can be seen for all experiments containing t-ZnO after 24 hours. S. aureus is completely inactive, while the amount of colony forming units for P. aeruginosa is significantly decreased after 24 hours. (n = 3, ****p < 0.0001). (F) Inhibition zone tests for 2% H2O2-treated t-ZnO hydrogels. Inhibition zones were found for both S. aureus and P. aeruginosa, for the latter only a reduction in bacteria population density for the former complete inactivation can be observed (n=9).
First, the mechanism of the antibacterial testing was investigated by performing an inhibition zone test. We expected the release of low concentrations of Zn2+ ions due to the low surface area of the t-ZnO microparticles in comparison to traditionally used nanoparticles. When too many Zn2+ ions are released from t-ZnO particles, they may become cytotoxic as well, and if this is the only mechanism for antibacterial behavior, it may be difficult to find a therapeutic window. To test this, the pristine, printed GelMA hydrogel, and t-ZnO-laden composite hydrogels were placed into bacteria-seeded agar plates (Figures 5B, 5C and 5F). If Zn2+ ions were released and killed the bacteria, an inhibition zone would form. In Figure 5B and 5C, the t-ZnO-laden hydrogels are depicted after 12 h of treatment. No inhibition zone can be observed. Inhibition zones could be observed for the 2% t-ZnO-laden hydrogels only after 72 h of treatment on the agar plates. This is a strong indication that a Zn2+ ion release is present but takes a long build up time. In Figure 5F the inhibition zones for S. aureus and P. aeruginosa can be observed, which are much more pronounced for S. aureus than for P. aeruginosa. Seeding agar from these inhibition zones on a fresh plate showed that the P. aeruginosa bacteria were still active, while S. aureus became inactive. It is well known that P aeruginosa is much more resistant toward metal ions than S. aureus.[35] It is therefore likely that the formation of these inhibition zones after 72 h is linked to Zn2+ ions released from the hydrogel.
A different result was obtained for antibacterial tests in liquid media where even short term exposure led to an effective reduction in bacterial activity. The printed composite hydrogels with and without H2O2 treatment as well as an additional protein coating with BSA were observed to have antibacterial properties even at low concentrations of 0.5 % t-ZnO. All hydrogels limited the bacterial growth of S. aureus compared to the pristine GelMA hydrogel as control (Figures 5D). A similar reduction in bacterial growth was also found for P. aeruginosa, except for 2% H2O2-treated t-ZnO-laden GelMA hydrogels, which only were able to reduce the colony formation of the pathogen while bacterial growth continued to be observable.
Additionally, the antibacterial activity of t-ZnO powder was also evaluated over a short time (Figure 5E). 1% H2O2-treated t-ZnO and 1% pristine t-ZnO powders were shown to eliminate the bacterial growth of S. aureus after 24 hours of culture, and significant statistical differences were found on both powders in comparison to the control. A different behavior was found with P. aeruginosa, where no significant differences were found for bacterial growth with 1% H2O2-treated t-ZnO and 1% pristine t-ZnO powders compared to the control. Previous studies have demonstrated antibacterial activity from ZnO nanoparticles for Salmonella, Listeria monocytogenes, Escherichia coli, S. aureus, and other gram-positive and negative bacteria.[36] Different mechanisms of t-ZnO antibacterial activity have been proposed, including direct bacterial cell wall damage, the release of ROS and resultant DNA damage, oxidative stress, and lipid peroxidation (in agreement with the working principle depicted in Figure 5A).[36] Due to the quick impact that the t-ZnO-laden composites and the pure powder have on the bacteria’s activity, a release of Zn2+ ions is unlikely in these experiments. This is in agreement with the in vitro results, where an increased amount of ROS was found (cf. Figure S10). Moreover, the exposure to of HUVEC and C2C12 cells to the t-ZnO-laden hydrogels did not have the same dramatic impact as for the bacteria. In this regard, previous studies have found ZnO nanoparticles to release large amounts of Zn2+ ions due to their large surface-to-volume ratios, leading to apoptosis in, e.g., mouse macrophage Ana-1. While for the same ZnO nanoparticles a certain quantity of ROS was also found, it was only believed to be harmful to the cells when the particles entered the cells.[37] The t-ZnO particles are multiple orders of magnitude larger than ZnO nanoparticles, and therefore have a much lower surface-to-volume ratios. For this reason, we believe that the main antibacterial mechanism stems from ROS generation by nanoparticles. That means that it is possible to use the antibacterial activity of t-ZnO for prompt treatment before bacterial colonization is chronically established. These results show that t-ZnO-laden hydrogels have potential antibacterial properties and can have a great impact on the success of wound healing.
A subcutaneous in vivo study was performed to evaluate the biological stability of the produced hydrogels in terms of biodegradability and biocompatibility with neighboring tissues (Figure 6A). After subcutaneous transplantation, H&E staining was conducted to identify the reaction between the hydrogel and its surrounding tissues. Encapsulated fibrous tissue, which could be a problem during tissue remodeling, was not observed nearby for all transplanted hydrogels due to their good biocompatibility (Figure 6B). Especially, materials with t-ZnO were observed to have an excellent connection with neighboring tissues (Figure 6B). Most of the hydrogels remained 4 weeks after the transplant. 1% VEGF-coated H2O2/t-ZnO showed the greatest tendency for biodegradability (Figure 6C). The differences in biodegradability in the hydrogels are believed to be due to the different components in the hydrogels. VEGF upregulates in an inflammatory environment and induces inflammatory cells such as macrophages to contribute to wound healing.[38] Such macrophages can play a role in accelerating the biodegradation of hydrogels.[39] It has also been reported from earlier research that t-ZnO induced macrophages secreted enzymes, causing materials to be bio-degraded faster. In Figure 6C, 1% t-ZnO-VEGF showed faster biodegradation than 2% t-ZnO-VEGF. This result could be due to macrophage recruitment being most active in 1% t-ZnO-VEGF, causing rapid degradation.
Figure 6. Evaluation of biocompatibility and angiogenesis of VEGF-coated t-ZnO-laden GelMA hydrogels using in vivo subcutaneous tests.
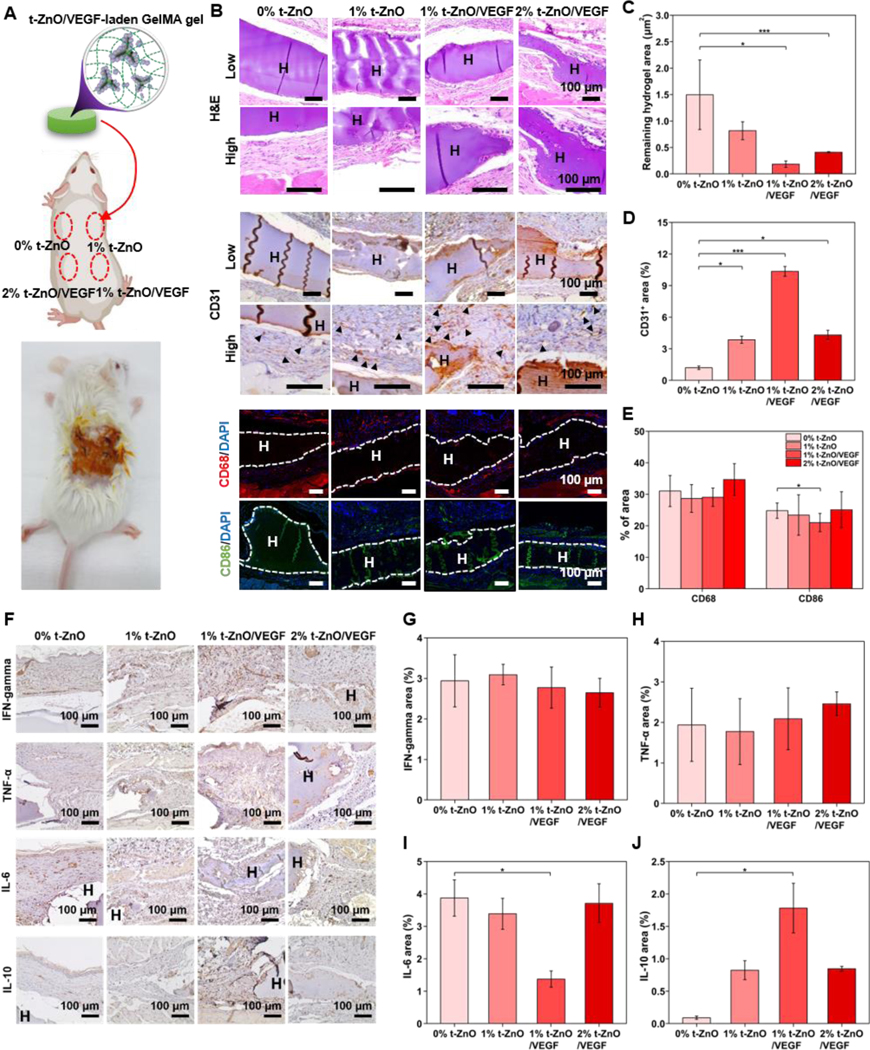
(A) Representative photos of mice subcutaneous surgeries and a scheme showing the inserted locations of our samples. (B, F) H&E, Angiogenesis marker (CD31), Monocyte marker (CD68) and M1 macrophage (CD86), Macrophage activation (IFN-γ), Role in the initiation and perpetuation of inflammation (TNF-α), Pro-inflammation marker (IL-6) and anti-inflammation marker (IL-10) stained histological sections of mice skins 28 days after treatment in different groups. H&E staining of the skin tissue collected at 28 days. The characteristics of the wound healing process with samples are highlighted. H, hydrogel, the dotted lines indicate the interaction between the hydrogel and the skin tissue. Immunohistochemistry of CD31 confirms the ability to induce angiogenesis. H, hydrogel, and arrows indicate new blood vessels. CD68/CD86 expressed in monocytes/macrophages was confirmed by immunofluorescence. Representative photographs of 0% t-ZnO, 1% t-ZnO, 1% t-ZnO-VEGF and 2% t-ZnO-VEGF after immunofluorescence labeling with CD68 in red, DAPI in blue and CD86 in Green. (C) Statistical analysis of hydrogel degradation at 28 days after surgery; the hydrogel density is the area of hydrogel in a field in the upright microscope with a magnification of 40×, (n = 3). (D) Statistical analysis of vessel density at 28 days after surgery; the vessel density is the area of vessels in a field in the upright microscope with a magnification of 400×, (n = 3). (E) Statistical analysis of monocyte and macrophage intensity at 28 days after surgery; the CD68 and CD86 intensities are the luminescence of monocytes and macrophages in a field in the upright confocal with magnification of 10x, n = 3. (F) IFN-γ/ TNF-α/ IL-6/IL-10 were confirmed by Immunohistochemistry. (G, H, I, J) Statistical analysis of macrophages activation density at 28 days after surgery; the macrophages activation is the area of skin tissue around the hydrogel in a field the upright microscope with a magnification of 400×, (n = 3).*p ≤ 0.05, **p ≤ 0.005 and ***p ≤ 0.001, mean ± SD, (n = 3).
In order to closely observe the biological characteristics of the hydrogels in vivo, immunostaining was performed. The phenomenon for angiogenesis (vascularization) was observed through CD31. CD68 and CD86, markers to confirm monocytes and macrophages, were used to analyze the immune response and inflammatory reaction, respectively. In Figure 6B, the expression of CD31 in all groups was confirmed. 1% t-ZnO-VEGF especially showed a higher level of CD31 expression compared to other groups. The CD31 expression of the dermis per designated area (0.01 mm2) was quantified in Figure 6D. The expression of CD31 in 0% t-ZnO was lower than the other groups. Interestingly, 1% t-ZnO-VEGF showed a higher value of 10.35% ± 0.46%, compared to 2% t-ZnO-VEGF showing 4.31% ± 0.44%, indicating that angiogenesis had further proceeded in 1% t-ZnO-VEGF.
The CD68 expression in immunofluorescence confirms that monocytes differentiate into macrophages and cause an immune response. The tendency of a monocyte to be located within or induced to the tissue was confirmed through the immunofluorescence results of CD68. A monocyte, a type of leukocyte in the blood, can differentiate into either a macrophage or a dendritic cell that immunizes. Monocytes also produce inflammatory cytokines.[41] The cytokine expression acts as proof that the damaged tissues are healing. The M1 macrophage, which performs pro-inflammatory, bactericidal, and phagocytic functions, was identified through CD86 staining. The CD68 and CD86 expressions were noticeable in the tissues of the hydrogels with t-ZnO (Figure 6B). In Figure 6E, CD68 was not significant for all samples, but CD86, a macrophage marker, was significant between 0% t-ZnO and 1% t-ZnO-VEGF. The 1% t-ZnO-VEGF also showed a low intensity. This shows that 1% t-ZnO-VEGF was more degraded than the other groups and that macrophages had a low expression level in the neighboring tissues.
Macrophages were classified according to their functionality, pro-inflammatory macrophages were named M1 and tissue-repairing ones were named M2. M1 and M2 macrophages are activated by pro-inflammatory cytokines (TNF-α, IL-6) and anti-inflammatory cytokines (IL-4, IL-10 or IL-13), respectively.[40] Interferon-γ (IFN-γ) as the primary activator of macrophages can either promote or inhibit immune or inflammatory responses through improving macrophage activation, mediating antiviral and antibacterial immunity, enhancing antigen presentation, etc.[41] Therefore, IFN-γ expression was investigated for all samples (Figure 6G), and there was no significant difference in IFN-γ expression levels within the samples, as well as CD68 (Figure 6E and Figure 6G). Tumor necrosis factor-α (TNF-α) can be induced by tissue injury and infection. TNF-α and lipopolysaccharides (LPS) regulate inflammatory cytokine production and macrophage function.[42] As presented in Figure 6H, all samples showed TNF-α expression due to the wound creation and foreign body response imposed by the implanted hydrogels.
However, the hydrogels with 1% t-ZnO-VEGF revealed the lowest interleukin-6 (IL-6) expression compared to the other samples. IL-6 is a pro-inflammatory cytokine and is released in response to tissue damage, chronic inflammatory diseases, or the presence of TNF-α.[43] It is well known that the suppression of IL-6 expression is effective for the treatment of infections and the prevention of inflammatory diseases. Indeed, Figure 6I indicates that 1% t-ZnO-VEGF can most appropriately function for inflammation reduction or prevention. The result shown in Figure 6I is similar to the M1-specific marker CD86 expression in Figure 6E and can be confirmed through the expression of IL-10. IL-10 secreted from M2 macrophages is a cytokine critical to anti-inflammatory effects, which inhibit pro-inflammatory mediators and promote wound healing.[44] According to the results shown in Figure 6J, the expression of IL-10 was significantly increased in the tissues of the hydrogels with t-ZnO, and the highest level was observed in 1% t-ZnO-VEGF samples. These significantly reduced IL-6 and increased IL-10 expressions might have resulted in an enhanced expression of CD31 as an angiogenesis marker (Figure 6D). These results verified that the hydrogel with 1% t-ZnO-VEGF was a better candidate for wound healing due to its biocompatibility and inflammation reduction.
The effects of the produced hydrogel scaffold on wound healing were assessed by non-infected wound model through changes in the wound size and histological analysis of the wound site. Two sections of a mouse’s skin were incised (0.9 cm x 0.9 cm) to compare the left, non-treated, and the right, applied with either 0% t-ZnO or 1% t-ZnO-VEGF (Figures 7A and 7B). The healing of the wound was visually monitored for 2 weeks following surgery (Figure 7C). In the case of no treatment (control), wound contraction, scab formation, and a slow healing process were observed. This is because the exposure of the wound without the dressing could cause excessive moisture loss in the epidermis, leading to skin damage.[45] On the other hand, 0% t-ZnO and 1% t-ZnO-VEGF showed fast wound healing processes without inflammatory phenomena and scab formation, compared to the non-treated tissue. Especially, 1% t-ZnO-VEGF revealed an excellent wound healing process without contraction. The wound healing rate of each group was quantified by the healing time in relation to the area of the wound (Figure 7D). The wound healing rate was effectively facilitated by the scaffold containing t-ZnO and VEGF. The size of the wound treated with 1% t-ZnO-VEGF decreased to 24.90% ± 4.21% after 1 week and further decreased to 3.48% ± 6.03% after 2 weeks.
Figure 7. Evaluation of wound healing of 3D printed VEGF-coated t-ZnO-laden GelMA hydrogel patches.
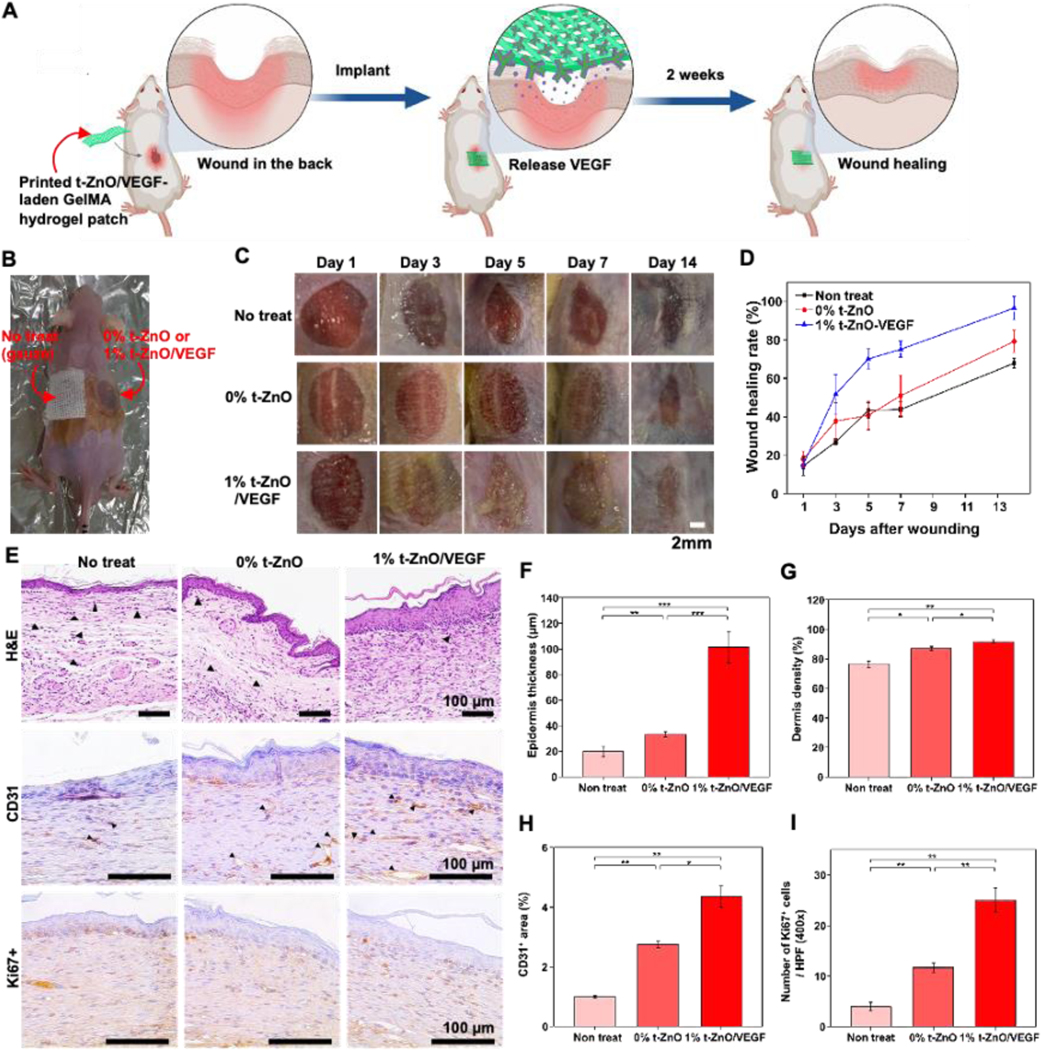
(A) Schematic illustration for wound healing process. (B) Representative photos of mice wound healing surgeries and a scheme showing inserted locations of our samples. (C) Representative photos of the skin wounds treated with no treatment, 0% t-ZnO and 1% t-ZnO-VEGF. (D) The wound healing rate of 9 mm punch biopsies was measured on days 1, 3, 5, 7 and 14. Wound healing of surgery mice represent the baseline, 1% t-ZnO-VEGF significantly improved the rate of wound healing compared with that of 0% t-ZnO and non-treated. (E) Histomorphological evaluation of regenerated skin wounds treated with non-treated control, 0% t-ZnO and 1% t-ZnO-VEGF in excision wound model. Images of H&E staining of sections at 14 days. The characteristics of the wound healing process with time are highlighted. H, hydrogel and arrows indicate neutrophils. (F) Quantification of epidermis thickness and (G) dermis density in respective groups. (H) CD31 in wound sections of control and hydrogels-treated mice at 14 days after surgery. Quantification of CD31 area. *p ≤ 0.05, **p ≤ 0.005 and ***p ≤ 0.001, mean ± SD, (n = 3). (I) Representative images of Immunohistochemistry for Ki67. Quantification of cell proliferation. *p ≤ 0.05, **p ≤ 0.005 and ***p ≤ 0.001, mean ± SD, (n = 3).
Immunohistological staining was performed to investigate the interaction between the wound and scaffold materials in-depth. The wound healing progress can be determined in terms of re-epithelization and collagen deposition of the wounded tissue. In the early stages of wound healing, re-epithelization, contraction of connective tissue, collagen formation in the wound, and angiogenesis are needed to start the skin regeneration. In the later stages, cell proliferation, matrix remodeling, and granulation tissue formation by re-epithelization are required to complete wound healing.[46] H&E staining showed the representative histological sections of the wound healing of each group after 2 weeks (Figure 7E). Although a new layer of skin tissue was generated in all groups, the non-treated case showed an incomplete wound recovery, creating a thin epidermis and lots of keratin. The wound covered with 1% t-ZnO-VEGF formed a thick epidermis and had relatively little keratin compared to the non-treated wound. The measured epidermal thickness of the 1% t-ZnO-VEGF-treated wound, 101.2 um ± 12.3 um, can be confirmed to be more than twice the thickness of the non-treated wound, 19.7 um ± 4.1 μm (Figure 7F).
In the observation of the dermis, non-treated wounds were not filled by generated tissues due to incomplete dermis formation and the concentrated distribution of inflammatory cells such as neutrophils around the wound. Meanwhile, the wound under 1% t-ZnO-VEGF showed the densest dermal layer with 95.3% ± 0.5% per designated area (0.3 mm2). The dermis, which is composed of dense connective tissue, plays an important role in thermoregulation and cutaneous nerve signal sensing because it has a rich layer of blood, lymphatic vessels, and numerous nerve endings. Therefore, the regeneration of a dense dermis can be expected to greatly recover skin functions (Figure 7G).[47]
Angiogenesis is an important process of wound repair because it allows blood vessels to provide nutrients and oxygen to the tissue and it promotes granulation tissue formation.[24] In Figure 7E, the angiogenesis was most active in the wound covered by the scaffold with 1% t-ZnO-VEGF. The results showed that the CD31 expression was significantly different between the 1% t-ZnO-VEGF and the others (0% t-ZnO and control). VEGF is known as a very important growth factor that enables angiogenesis for tissue regeneration.[38a, 48] It has been reported that chronic wounds caused by cell migration and vascularization deficiencies can be cured by VEGF treatment.[22, 49] Zinc ions are also well known for having effective antibacterial production and angiogenesis acceleration. Therefore, it was expected for the components in the scaffold to contribute to angiogenesis. From the experiment results, it was proven that, although the existence of t-ZnO induces angiogenesis, the existence of VEGF dramatically promotes angiogenesis (Figure 7H).
New cell growth in the wound site was assessed by Ki67 staining (Figure 7E). The existence and proliferation of essential cells such as keratinocytes, fibroblasts, and endothelial cells are very important in re-epithelialization for wound remodeling.[50] Figure 7I shows that Ki67 appeared to have the highest activity from 1% t-ZnO-VEGF. Consequently, it was confirmed through the in vivo study that the scaffold with 1% t-ZnO-VEGF enhanced and anti-inflammatory reaction, angiogenesis, and cell proliferation in the wound site, allowing for the fastest wound healing.
Conclusion
In summary, we have demonstrated a method that uses both materials science and biomedical engineering for the treatment of chronic wounds. Printed GelMA hydrogels containing tetrapodal ZnO microparticles could effectively decrease bacteria concentrations while promoting the re-growth of C2C12 and HUVECs in vitro and wound healing in vivo. The open porous structures of the hydrogels and their high-water content enabled the diffusion of adsorbed, angiogenic proteins while maintaining mechanical integrity. The printability of the gels prior to cross-linking showed excellent results, with the option of multi-material printing for enhanced wound healing therapy with multiple growth-factors, drugs, or other proteins. An additional treatment of t-ZnO microparticles with H2O2 led to the formation of a highly protein-adsorbent surface while maintaining their biomedical qualities. The treatment also allowed for the controlled release of VEGF with light in the optical range over the duration of 120 seconds. The VEGF immobilized on the t-ZnO created adequate microenvironments for improving endothelial cell growth while maintaining antibacterial activity that can have a great impact on the success of wound healing. Through in vivo studies, we confirmed that the printed scaffold with t-ZnO-VEGF showed adequate biological stability and enhanced anti-inflammatory reaction, angiogenesis, and cell proliferation in the wound site, resulting in achieving the fastest wound healing. All of these factors combined make this approach an interesting candidate for future smart wound dressing platform.
Materials and methods
Materials
Zinc oxide tetrapods (t-ZnO) were obtained by employing the newly developed flame transport synthesis by Mishra et al.[13] T-ZnO is pure and single-crystalline. The following reagents were purchased from Sigma Aldrich, USA: gelatin from porcine skin, methacrylic anhydride, hydrogen peroxide solution (30 wt. % in H2O), trypsin solution 10X, Triton™ X-100, bovine serum albumin (BSA); Thermo Fisher Scientific, USA: fetal bovine serum (FBS), 100X penicillin-streptomycin, VEGF recombinant human protein, PrestoBlue™, calcein AM, ethidium homodimer-1 (EthD-1), Alexa Fluor™ 488 Phalloidin, 4’,6-diamidino-2-phenylindole (DAPI), CD31 (PECAM-1) monoclonal antibody, goat anti-mouse IgG2a cross-adsorbed secondary antibody Alexa Fluor™ 594; Lonza Biosciences, USA: Dulbecco’s modified eagle medium (DMEM), endothelial cell basal medium-2 (EBM-2), endothelial cell growth medium-2 (EGM-2) SingleQuots™ kit; Electron Microscopy Sciences, USA: paraformaldehyde aqueous solution 16% (w/v).
Methods
GelMA synthesis:
The photo-crosslinkable gelatin methacryloyl (GelMA) hydrogel was used as a base for a biocompatible scaffold. The synthesis method involved the reaction between gelatin and methacrylic anhydride (MA). 10 g of porcine skin gelatin was mixed with 100 mL sterile phosphate-buffered saline (PBS) (Thermo Fisher Scientific, USA) and dissolved at 50 °C. Once the gelatin melted, 1.25 mL of MA was added, and the emulsion was left in rotation (240rpm) for two hours. 100 mL of sterile PBS was added (giving a total volume of 200 mL) and left in heating rotation (10 min at 50 °C). GelMA was transferred to dialysis membranes (Spectro/Por molecularporous membrane tubing, MWCO 12–14,000, Fisher Scientific) and left in distilled water for dialysis (40 °C for 5 days at 500 rpm.) Toxic unreacted MA was removed by changing the distilled water twice a day. Once dialysis was finalized, 200 mL of ultrapure water was added to GelMA and heated at 40 °C. The liquid was filtered, in sterile conditions, using the vacuum Express Plus (0.22 μm) Millipore filtration cup with a coffee filter. The sterilized polymer was transferred into 50 mL Falcons and stored at 80 °C. GelMA was freeze-dried for 3 days at a maximum pressure of 0.13 mbar before use.
H2O2-treatment for improving protein adhesion:
4 g of t-ZnO was added to 100 mL of a 30% w/v solution of hydrogen peroxide solution. The suspension was heated to around 70 °C and left until a noticeable gas evolution occurred. The liquid phase was extracted by filtering and the remaining t-ZnO was washed multiple times with deionized water and then dried at 80 °C in an oven. The dry t-ZnO was then filled into a container, covered with aluminum foil for light protection, and stored under room temperature conditions.
Micro-Raman spectroscopy for investigation of chemical treatment:
Both pristine and H2O2 treated t-ZnO were investigated by micro-Raman (WITec, Germany) at room temperature using a WITec system in a backscattering configuration. A Nd:YAG laser with a wavelength of λex= 532 nm and a power was below 4 mW was used for excitation. Each spectrum was taken using 10 accumulations at an integration time of 0.5 sec.
H2O2–treated t-ZnO coating with VEGF:
A 10% w/v solution of H2O2-treated t-ZnO in 10 μg/mL VEGF was prepared, protected from light with aluminum foil and incubated on ice for 2 hours, shaking every 30 min. The solution was then centrifuged for 10 min at 5,000 rpm and the supernatant was discarded. The remaining pellet was then washed with 1 mL of PBS buffer (centrifugation and washing steps were repeated 3 times). After the last supernatant was removed, the VEGF-coated H2O2/t-ZnO was resuspended in the PBS buffer at a 10% w/v concentration. The solution was stored at −80 °C until further use. Both high and low concentrations of VEGF were coated onto H2O2-treated t-ZnO in the following way: The low concentration coating was done using 1 mL of 10 μg/mL VEGF/PBS solution with 50 mg of t-ZnO. The high concentration coating was done by mixing 400 μL of a 5 μg/mL solution with 50 mg of t-ZnO. The solutions were kept in an Eppendorf container wrapped with aluminum foil at 37 °C for 2 hours. The solutions were shaken every 30 minutes. After coating, the t-ZnO was washed several times and the supernatant was kept for measurement with the ELISA kit to confirm the complete removal of superfluous VEGF. Subsequently, the 50 mg t-ZnO was transferred into 1 mL of PBS and the protein release test was performed.
Protein coating experiments:
Optical protein release tests were performed by coating both pristine t-ZnO, H2O2-treated t-ZnO, and other types of treated t-ZnO with IgG Alexa Fluor™ 594. For these tests, 1 mg of the respective t-ZnO were coated in 100 μL of a 10 μg/mL solution of the IgG Alexa Fluor™ 594. The solution was made by diluting the IgG Alexa Fluor™ 594 obtained from Thermo Fisher to a 10 μg/mL solution with PBS. The coating was performed at room temperature in an Eppendorf container wrapped with aluminum foil for light protection. After coating, the t-ZnO was washed at least 3 times with PBS to remove superfluous antibodies. The 1 mg of t-ZnO was then given into 1 mL of PBS for protein release experiments. SiO2 microparticles with a mean particle size of 20 μm were obtained from Sigma Aldrich and also coated in the same fashion as the t-ZnO.
Oleic acid was coated onto the t-ZnO by adding 1 mg of oleic acid into 1 mL of 97% ethanol. Then 1 mg of t-ZnO was added and the solution was kept for 2 hours. The coated t-ZnO was washed several times with ethanol and then dried at 80 °C for 2 hours. The t-ZnO was then hydrophobic and a small amount of ethanol was added into 1 mL of PBS to break surface tension and to put the t-ZnO inside. Streptavidin and BSA were coated on the t-ZnO in the same manner as the IgG Alexa Fluor™ 594. 10 μg/mL solutions were made in PBS and then coated with 1 mg of t-ZnO in a 1 mL solution.
VEGF coating efficiency tests (n=3) were carried out with both pristine and H2O2-treated t-ZnO. For this, 50 mg of t-ZnO were incubated for 2 h in 0.5 mL of a 1 μg/mL human VEGF solution in an ice bath. Before and after coating, the concentrations were measured with a human VEGF ELISA kit.
Optical protein release experiments:
For protein release, a fluorescence microscope was used with the red filter to check for the 594 nm signal of the IgG Alexa Fluor™ 594 fluorescence. A small lightproof box was laser-cut from black acrylic, to allow for irradiation with a UV-light source while performing the protein release in situ. The t-ZnO/PBS dispersion was placed into a confocal dish that was fitted into the acrylic box and the particles were investigated from the bottom, while irradiated from the top. Fluorescence microscopy was performed during in situ UV irradiation. Images were taken at varying intervals, maintaining set parameters for exposure time and gain throughout the experiment for comparability of the image series. For green-light treatment, a 1 W, 547 nm LED was mounted into the acrylic box with a mean light density of 25,000 lx. During green light illumination, the red fluorescence was much brighter, so those images were not considered during measurement. For the release of VEGF measured with the ELISA kit, the same light density of 25,000 lx with the same green LED was used. The supernatant was taken from the illuminated samples and kept for ELISA 5, 20, 60, and 120 seconds. After each illumination step, the whole solution was discarded, and fresh PBS added.
Preparation of the t-ZnO hydrogels:
The t-ZnO powder was treated with 30% hydrogen peroxide as previously mentioned, combined with 10 μg/mL VEGF and PBS to yield a 100 mg/mL VEGF-coated H2O2/t-ZnO stock solution. By diluting this solution and mixing it with previously made GelMA, gelatin, and photoinitiator, five bioinks containing VEGF-coated H2O2/t-ZnO were prepared, along with five more with uncoated t-ZnO. A ‘spacer’ was created by building two 300 μm-height towers of stacked cover glasses, with a 5 mm distance between each tower. To make the t-ZnO hydrogels, a droplet of the bioink was placed in the 5 mm gap in the spacer and pressed against a previously TMSPMA-coated-and-cut glass slide, creating a round-shaped hydrogel with a 300 μm thickness and a 5 mm diameter.
Mechanical properties of the t-ZnO hydrogels:
For mechanical testing, constructs containing varying amounts of H2O2-treated t-ZnO were printed with a height of roughly 1 mm and 100% filling factor and subsequently crosslinked. Round biopsy punches with a diameter of 8 mm were used to take multiple samples from each printed construct. The samples were stored in humid conditions at 4 °C for hydration. Compression tests were performed using the ADMET MTEST QUATTRO universal test machine (ADMET, Inc., USA). Compression and stress were computed using the difference in height and the area of the constructs, respectively. The compressive moduli were calculated using a linear least-square fit for each compression curve. The mean and the standard deviation were calculated by using the values obtained from multiple samples.
Degradation test:
1cm diameter samples with 600μm thickness with different concentrations of t-ZnO were prepared by using the previously described protocol.[48] Hydrogels were incubated for up to 24 hours in a solution of 1U/mL of collagenase type II (Thermo Fisher Scientific, USA) in an incubator at 37°C. At the end of each time-lapse, the supernatant was removed, and samples were washed with 1mL of PBS and then frozen overnight. After this procedure, the samples underwent lyophilization for 24 hours, and weight measurements were performed.
Scanning electron microscopy (SEM):
1 cm diameter samples with 600 μm thickness with different concentrations of t-ZnO and VEGF coated t-ZnO were prepared by using the previously described protocol.[51] Hydrogel samples were stored in 1.5 mL centrifuge tubes and frozen during 24 hours at −20°C. After this procedure, the samples underwent lyophilization for 24 hours, and several transversal cuts were performed in different sections of the hydrogels. Scanning electron microscopy images were obtained using an FSEM Supra55VP (Zeiss, Germany).
AFM experiments:
All experiments were performed in wet conditions using in-fluid imaging set up of the AFM (Dimension Icon, Bruker, USA). ScanAsyst-Fluid+ probes having a sharpened tip with frequency 150 kHz and spring constant 0.7 N/m, ideal for high-resolution imaging and edge detection in fluid was used. Imaging was performed at a scan rate of 1 Hz with a sampling rate of 512 samples/line. Using offline AFM analysis software, images were first flattened, followed by whole image roughness and section analysis for height and DMT modulus to quantitate surface stiffness and roughness of the samples.
Ink preparation:
The bioinks used for the experiments were composed of 10% w/v of GelMA, 0.5% w/v of photoinitiator (Igracure 2959, Sigma Aldrich), 2% w/v gelatin (Type A, 300 bloom from porcine skin, Sigma Aldrich) and 0.1–10% VEGF-coated H2O2–treated t-ZnO dissolved in PBS buffer (Thermo Fisher Scientific, USA) at 37 °C. The bioink was deposited into 3 mL cartridges covered with aluminum foil to prevent premature crosslinking and was then homogenized and stored at 4 °C.
Rheological characterization:
The rheological properties of the inks were investigated by a DHR rheometer (TA Instruments, USA). A 40 mm diameter 2° cone geometry and ~ 1mL of GelMA-gelatin based inks with various concentrations of t-ZnO microparticles were used for all tests. For loss and storage modulus, temperature sweeps were performed between 4 and 37 °C; all samples were allowed to equilibrate for at least 30 seconds at 37 °C prior to measurement. The viscosity of each ink was measured at 30 °C. The results were recorded and analyzed using the TRIOS software (TA Instruments). Residual friction was compensated for prior to each measurement by calibration of the measuring system inertia (of the upper geometry). The gap was then recalibrated, and the samples were loaded and allowed to equilibrate for 30 min or until the normal force had decayed. Frequency sweeps were performed at the start and end of the measurements and suggested that sample drying was not a significant issue.
3D-printing process:
Rectangular constructs of 20 mm x 20 mm and 0.4 mm (2 layers) were printed at 4 °C with a printing velocity of 0.1 mm/sec and an extrusion pressure of 400 kPa using the BIOMAKER by SUNP BIOTECH (China). The nozzles were standard tapered nozzles with orifices of 0.4 mm. Printed constructs were exposed to UV light at 100 mW/cm2 for 60 seconds.
3D-printing of multi-material constructs:
The ink was prepared as before, containing 2% w/v t-ZnO, and 0.5% w/v of green and pink fluorescence dye was added to two batches of ink. The colored inks were then loaded into the 3 mL cartridges and printed in custom paths. The print settings were not altered compared to the uncolored inks. One construct was a cross-like pattern of pink ink filled with green ink. Another pattern alternated stripe-wise between green and pink ink.
Cell culture:
To culture the needed cells, vials of C2C12 and HUVECs were purchased from Lonza Bioscience, along with the corresponding Dulbecco’s modified eagle medium (DMEM) and Endothelial Cell Basal Medium-2 (EBM-2). For the latter, the endothelial cell growth medium-2 (EGM-2) SingleQuots™ Kit was also acquired to enrich the basal media as it contains supplements and growth factors needed for HUVEC culture. The C2C12 cells were cultured using DMEM with 10% fetal bovine serum and 1% penicillin-streptomycin diluted at 1X from the 100X solution. For both cell lines, only the amount of required basal and complete media for the desired number of T-75 (Thermo Fisher Scientific, USA) cell flasks was first aliquoted in 50 mL Falcon® (Corning, USA) tubes; media tubes and liquid nitrogen-frozen cell aliquots containing ~0.7×106 cells were slowly warmed up to approximately 37 °C using a water bath. Once the cells were thawed, the volume of the aliquot ~1 mL was quickly transferred to a 15 mL Falcon® tube with 9 mL of the corresponding basal media to neutralize the freezing media containing 10% DMSO (Thermo Fisher Scientific, USA), and the solution was centrifuged at 1,000 rpm for 5 min. Following centrifugation, the supernatant was carefully removed, the cell pellet was resuspended in 15 mL of the proper complete media previously warmed and transferred to a T-75 flask for incubation at 37 °C. A cell density of 0.1×106 was required in order to proceed with the cell characterization assays.
Cell-laden viability, proliferation, and attachment assays:
Cell viability and proliferation of the cells seeded on hydrogels were assessed using live/dead assay (Thermo Fisher Scientific, USA) and PrestoBlue™ kit (Thermo Fisher Scientific, USA). PrestoBlue™ assays were performed using the manufacturer’s reagent protocol and measured using a plate reader (SpectraMax® i3, Molecular Devices, USA) by measuring absorbance at 570 nm with reference to 600 nm. The results were normalized by day 1 of culture. For live and dead assay, samples were stained with Calcein AM and Ethidium Homodimer-1 (EthD-1) for live and dead cells respectively. In order to evaluate cell adhesion and cell covered area, fixation of hydrogels was done on days 3 and 7 with 4% paraformaldehyde solution (Electron Microscopy Sciences, USA) for 20 min and then permeabilized using 0.1% Triton X (Sigma-Aldrich) in 1% BSA (Invitrogen). Alexa Fluor™ 488 Phalloidin (1:40, Invitrogen) and CD31 (PECAM-1, 1:50 Invitrogen) antibodies were added to hydrogels for staining. The hydrogels were incubated with the primary antibody overnight at 4°C. Secondary antibody goat anti-mouse Alexa Fluor™ 594 (Invitrogen) was used to image the CD31 staining and was added to the hydrogels followed by incubation at 4 °C for 4 hours. DAPI (4’, 6-diamino-2-phenylindole, 1:300, Sigma Aldrich) was added 25 min prior to imaging with the inverted fluorescence microscope. Images of all assays including phase contrast images were taken by an inverted optical microscope (Nikon Eclipse TE 2000U, Japan) and processed and analyzed by using ImageJ and Fiji software.
Antibacterial Testing:
For the antibacterial tests we used two typical wound pathogens, Staphylococcus aureus (ATCC 25923) and Pseudomonas aeruginosa (ATCC27853). To test the pure t-ZnO, 100 μL of an overnight culture of bacteria was incubated with 5 mL LB medium at 37°C. After two hours, the cell count was estimated by measuring the optical density at 600 nm. 2×105 bacteria/mL in 0.85% NaCl, 1% LB-medium were incubated with 1 mg/mL tetrapods for 1 hour or 24 hours at 37°C on a rotary mixer. After the incubation period, the number of surviving bacteria was determined by plating serial dilutions on LB-agar. For testing of the hydrogels containing t-ZnO, 100 μL of an overnight culture of bacteria was incubated with 5 mL LB medium at 37°C. After two hours, the cell count was estimated by measuring the optical density at 600 nm. A solution of 0.85% NaCl, 1% LB-medium and 6×103 bacteria/50 μL was prepared. On a quartered hydrogel, 30 μL of the prepared solution was added. The hydrogel was incubated in a Petri dish at 37 °C with moistened filter papers. After 24 hours, the hydrogels were transferred into 5 mL 1% LB medium with 0.85% NaCl and vigorously vortexed for 30 seconds. The number of surviving bacteria was determined by plating serial dilutions on LB-agar.
For the long-term incubation zone tests five to ten colonies of P. aeruginosa or S. aureus were resuspended in 500 μl liquid culture medium. From this suspension, 300 μl was plated out onto a Müller-Hinton-Agar plate and allowed to dry for 5 minutes. Three hydrogels containing 2% of H2O2-treated t-ZnO and each with a diameter of approx. 5 mm were placed on the agar and the plate was incubated for 72 h at 37 °C.
In vivo investigation of t-ZnO/GelMA:
The animal care and experimental protocols were approved by the Animal Care and Use Committee of Dankook University, Cheonan Campus, South Korea (DKU 17–011).[52] The wound healing and subcutaneous model animal experiments were conducted using 6-week-old male SKH-1 hairless mice and BALB/C mice, each weighing 20~25 g and purchased from Orient Bio Inc. located in Seongnam, Korea. The breeding timespan of the mice was set to 12 hours, day and night, in constant humidity and with temperatures of RH 55% ± 5% and 25 °C ± 2 °C, respectively. Standard feed and water were freely fed, and all experimental animal care and use committees are at Dankook University, Republic of Korea all experimental animal care and use.
In the case of the subcutaneous model experiment, 4% avertin was used to anesthetize the BALB/C mouse. After resecting the back of the mouse 1 cm into 4 sections, the samples, GelMA hydrogel without t-ZnO (0% t-ZnO), GelMA with 1% coated t-ZnO (1% t-ZnO), GelMA with 1% VEGF-coated t-ZnO (1% t-ZnO-VEGF), and GelMA with 2% VEGF-coated t-ZnO (2% t-ZnO-VEGF) with dimensions 3 mm x 3 mm, were inserted into the subcutaneous tissue and the incisions were sutured using 6–0 black silk (AILEE, Korea). Histological analysis was conducted after 4 weeks.[53]
In the case of the wound healing experiment to study the efficacy of VEGF-laden t-ZnO scaffolds, a non-infected wound mouse model was used. 4% avertin was used to anesthetize the SKH-1 hairless mouse. Light illumination was not used explicitly during these experiments. Two circle defects with the diameter 9 mm were then created symmetrically on the bilateral sides of the backs of mice. These defects were covered with samples non-treated, GelMA hydrogel without t-ZnO (0% t-ZnO) and GelMA with 1% VEGF-coated t-ZnO (1% t-ZnO-VEGF). Defects covered with samples were sealed with an adhesive bandage in order to prevent wound exposures or detachment of the wound scaffold. The wound healing process was monitored by an optical microscope and a confocal microscope for up to two weeks.[54]
Histological analysis:
The experimental mice of the subcutaneous model and wound healing model were sacrificed after 28 days and 14 days, respectively. Based on the location of the wound, the tissues with samples were cut into dimensions of 1 cm x 1 cm, then fixated through 4% PFA (Paraformaldehyde) solution. The fixed tissues were formed into paraffin blocks and were sliced into a thickness of 5 um using the Leica rotary microtome (Leica, Germany). The sliced sections were dyed using hematoxylin and eosin (H&E) and observed through the CKX53 microscope (Olympus, Tokyo, Co).[55]
In this subcutaneous in vivo study, the biocompatibility between the implanted material and the surrounding tissue, and the biodegradation of the materials measured by the remaining area of the material were evaluated through the H&E-stained histological tissue sections (n=3).
The wound healing process with re-epithelialization was observed for 14 days through a digital camera. The area of the healing from the wound compared to the initial wound was measured through an image program (Image J) (n=4), and its healing rate was calculated through the equation as follows:
Ao and At indicate the area of the initial wound and the wound area measured in its corresponding date, respectively.[56] In the H&E staining, the thickness of the recovered epidermis (n=5) and the density of the dermal layer (n=3) from the area of the dermal cells in the designated area (0.3 mm2) of the dermal layer were measured.
The immunohistochemistry staining utilized for observing the vessel structures was used to remove the paraffin from the sliced section using xylene. In order to eliminate the endogenous peroxidase activity, the sliced sections were incubated in methanol containing 3% H2O2 for 30 min. Then samples were heated for 20 min in the sodium citrate buffer and were treated as blocking (3% BSA in PBS) for 1 hour at room temperature.[57]
In order to stain CD31 (Abcam, USA, ab28364), Ki67 (Abcam, USA, ab16667) IFN-γ (Bioss, USA, BS-0480R) TNF-α (Elabscience, USA, E-AB-63550), IL-6 (Elabscience, USA, E-AB-65677), and IL-10 (Bioss, USA, BS-6761R), the primary antibodies, were left overnight at 4 °C. After the antibody treatment, the samples were further treated with 3% H2O2 solution, biotinylated secondary antibody, avidinhorseradish peroxidase, and diaminobenzidine substrate. Finally, hematoxylin solution was stained at room temperature. The stained tissues were visualized through a CKX53 microscope (Olympus, Tokyo, Co). Angiogenesis and cell proliferation levels in the designated area (0.01 mm2) were quantifiably analyzed from the CD31, IFN-γ, TNF-α, IL-6, and IL-10-dyed areas and the number of Ki67-dyed cells, respectively (n=3). The immunofluorescence investigations were conducted through CD68 (Abcam, USA, ab125212), and CD86 (Santacruz, USA, sc-28347) immunostaining. The sliced sections, of which the paraffin removal and blocking were conducted, were prepared. The prepared sliced sections (specimen) were treated with primary antibodies at 4 °C overnight and reacted in the secondary antibody Alexa Fluor™ 555 (Invitrogen, USA) and DAPI (Thermo Fisher Scientific, Germany). After staining, all slices were observed with an LSM700 confocal microscope (Zeiss, Germany).[58] All quantitative results were acquired from at least 3 specimens for analysis. Statistical analysis was performed between the groups using t-test, and ANOVA was performed for comparison between the groups. The value is expressed as ± SD on average. p < 0.05 difference is considered to be statistically significant.[59]
Statistical Analysis:
The average and standard deviation were calculated in Prism8 software by GraphPad. To analyze the results of protein release tests (Figure 1G, H) student’s t test was employed to study the presence of significant differences among groups. Mechanical properties of t-ZnO laden hydrogels (Figure 2D, G, I, J), antibacterial tests (Figure 5D, E), in vivo subcutaneous tests (Figure 6C, D, E, G, H, I, J) and wound healing properties of printed constructs were analyzed by using one way analysis of variance (ANOVA) with Tukey’s multiple comparison method. The results of the PrestoBlue™ assay (Figure 4E), cell viability (Figure 4B, D), cell covered area (Figure 4C), and CD31 (Figure 4F) covered area were analyzed by using a two-way analysis of variance method (ANOVA) to compare different treatment groups. Error bars represented mean ± standard deviation of measurement in each group. Statistical significance in all graphs was indicated by (ns) (p > 0.05), *(p < 0.05) and ***(p < 0.001).
Supplementary Material
Acknowledgements
The authors declare no conflict of interests in this work. All statements made herein are the sole responsibility of the authors. This paper was funded by the National Institutes of Health (R01AR074234 and R21EB026824), the Brigham Research Institute Stepping Strong Innovator Award, and Qatar University grants NPR9-144-3021 funded by Qatar National Research Foundation, a part of Qatar Foundation. This research was also supported by the National Research Foundation of Korea (NRF) Grant funded by the Ministry of Science and ICT for Bio-inspired Innovation Technology Development Project (NRF-2018M3C1B7021994) and by the Research Training Group “Materials for Brain” (GRK 2154) received from the German Research Foundation (DFG). M.A. Hussain and S.R. Shin extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (325).”
Contributor Information
Leonard Siebert, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.; Functional Nanomaterials, Institute for Materials Science, Faculty of Engineering, Kiel University, Kaiserstr. 2, D-24143, Kiel, Germany.
Eder Luna-Cerón, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA..
Luis Enrique García-Rivera, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA..
Junsung Oh, Department of Nano-biomedical Science & BK21 PLUS NBM Global Research Center for Regenerative Medicine, Dankook University, Cheonan 31116, Republic of Korea..
JunHwee Jang, Department of Nano-biomedical Science & BK21 PLUS NBM Global Research Center for Regenerative Medicine, Dankook University, Cheonan 31116, Republic of Korea..
Diego A. Rosas-Gómez, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.
Mitzi D. Pérez-Gómez, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.
Dr Gregor Maschkowitz, Institute for Infection Medicine, Kiel University and University Medical Center Schleswig-Holstein, Brunswiker Str. 4, D-24105 Kiel, Germany..
Helmut Fickenscher, Institute for Infection Medicine, Kiel University and University Medical Center Schleswig-Holstein, Brunswiker Str. 4, D-24105 Kiel, Germany.
Daniela Oceguera-Cuevas, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA..
Carmen G. Holguín-León, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA.
Dr. Batzaya Byambaa, 3D BioLabs, LLC, 700 Main St, Cambridge, MA 02138
Mohammad A. Hussain, Department of Electrical and Computer Engineering, King Abdulaziz University, Jeddah 21569, Saudi Arabia.
Eduardo Enciso-Martinez, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA..
Minsung Cho, AltrixBio inc., Cambridge, MA 02139, USA..
Dr. Yuhan Lee, Department of Anesthesiology, preoperative and pain medicine, Center for Nanomedicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Dr. Nebras Sobahi, Department of Electrical and Computer Engineering, King Abdulaziz University, Jeddah 21569, Saudi Arabia.
Anwarul Hasan, Department of Mechanical and Industrial Engineering, College of Engineering, Qatar University, Doha, Qatar. Biomedical Research Centre (BRC), Qatar University, Doha, Qatar.
Dennis P. Orgill, Division of Plastic Surgery, Department of Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Yogendra K. Mishra, Mads Clausen Institute, NanoSYD, University of Southern Denmark, Alsion 2, 6400, Sønderborg, Denmark. Functional Nanomaterials, Institute for Materials Science, Faculty of Engineering, Kiel University, Kaiserstr. 2, D-24143, Kiel, Germany.
Rainer Adelung, Functional Nanomaterials, Institute for Materials Science, Faculty of Engineering, Kiel University, Kaiserstr. 2, D-24143, Kiel, Germany.
Eunjung Lee, Department of Nano-biomedical Science & BK21 PLUS NBM Global Research Center for Regenerative Medicine, Dankook University, Cheonan 31116, Republic of Korea..
Su Ryon Shin, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA..
References
- [1].Pecoraro RE, Reiber GE, Burgess EM, Diabetes Care 1990, 13, 513. [DOI] [PubMed] [Google Scholar]
- [2].James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, Costerton JW, Stewart PS, Wound Repair Regen 2008, 16, 37. [DOI] [PubMed] [Google Scholar]
- [3].a) Augustine R, Zahid AA, Hasan A, Wang M, Webster TJ, Int J Nanomedicine 2019, 14, 8573; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zahid AA, Ahmed R, Raza ur Rehman S, Augustine R, Tariq M, Hasan A, International Journal of Biological Macromolecules 2019, 136, 901; [DOI] [PubMed] [Google Scholar]; c) Augustine R, Rehman SRU, Ahmed R, Zahid AA, Sharifi M, Falahati M, Hasan A, International Journal of Biological Macromolecules 2020, 156, 153. [DOI] [PubMed] [Google Scholar]
- [4].a) Kassal P, Kim J, Kumar R, de Araujo WR, Steinberg IM, Steinberg MD, Wang J, Electrochemistry Communications 2015, 56, 6; [Google Scholar]; b) Black S, in Smart Textiles for Medicine and Healthcare, DOI: 10.1533/9781845692933.1.3 (Ed: Van Langenhove L), Woodhead Publishing; 2007, p. 3; [DOI] [Google Scholar]; c) Tomić SL, Mićić MM, Dobić SN, Filipović JM, Suljovrujić EH, Radiation Physics and Chemistry 2010, 79, 643. [Google Scholar]
- [5].Haidar ZS, Hamdy RC, Tabrizian M, Biomaterials 2008, 29, 1207. [DOI] [PubMed] [Google Scholar]
- [6].Sirelkhatim A, Mahmud S, Seeni A, Kaus NHM, Ann LC, Bakhori SKM, Hasan H, Mohamad D, Nanomicro Lett 2015, 7, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ahmed R, Tariq M, Ali I, Asghar R, Noorunnisa Khanam P, Augustine R, Hasan A, Int J Biol Macromol 2018, 120, 385. [DOI] [PubMed] [Google Scholar]
- [8].a) Mishra YK, Adelung R, Materials Today 2018, 21, 631; [Google Scholar]; b) Papavlassopoulos H, Mishra YK, Kaps S, Paulowicz I, Abdelaziz R, Elbahri M, Maser E, Adelung R, Röhl C, PLOS ONE 2014, 9, e84983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nasajpour A, Ansari S, Rinoldi C, Rad AS, Aghaloo T, Shin SR, Mishra YK, Adelung R, Swieszkowski W, Annabi N, Khademhosseini A, Moshaverinia A, Tamayol A, Advanced Functional Materials 2018, 28, 1703437. [Google Scholar]
- [10].Dejneka A, Aulika I, Makarova MV, Hubicka Z, Churpita A, Chvostova D, Jastrabik L, Trepakov VA, Journal of The Electrochemical Society 2010, 157, G67. [Google Scholar]
- [11].a) Ghobril C, Grinstaff MW, Chemical Society Reviews 2015, 44, 1820; [DOI] [PubMed] [Google Scholar]; b) Rehman SRU, Augustine R, Zahid AA, Ahmed R, Tariq M, Hasan A, in Int J Nanomedicine, Vol. 14, 2019, 9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mishra YK, Kaps S, Schuchardt A, Paulowicz I, Jin X, Gedamu D, Freitag S, Claus M, Wille S, Kovalev A, Gorb SN, Adelung R, Particle & Particle Systems Characterization 2013, 30, 775. [Google Scholar]
- [14].Srikant V, Clarke DR, Journal of Applied Physics 1998, 83, 5447. [Google Scholar]
- [15].Companion AL, Journal of Physics and Chemistry of Solids 1962, 23, 1685. [Google Scholar]
- [16].Wang J, Wang Z, Huang B, Ma Y, Liu Y, Qin X, Zhang X, Dai Y, ACS Applied Materials & Interfaces 2012, 4, 4024. [DOI] [PubMed] [Google Scholar]
- [17].Ozawa CR, Banfi A, Glazer NL, Thurston G, Springer ML, Kraft PE, McDonald DM, Blau HM, J Clin Invest 2004, 113, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Akhmanova M, Osidak E, Domogatsky S, Rodin S, Domogatskaya A, Stem Cells International 2015, 2015, 167025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brandl F, Sommer F, Goepferich A, Biomaterials 2007, 28, 134. [DOI] [PubMed] [Google Scholar]
- [20].Bao P, Kodra A, Tomic-Canic M, Golinko MS, Ehrlich HP, Brem H, J Surg Res 2009, 153, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim H, Jung US, Kim SI, Yoon D, Cheong H, Lee CW, Lee SW, Current Applied Physics 2014, 14, 166. [Google Scholar]
- [22].Dreifke MB, Jayasuriya AA, Jayasuriya AC, Mater Sci Eng C Mater Biol Appl 2015, 48, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kirsch M, Birnstein L, Pepelanova I, Handke W, Rach J, Seltsam A, Scheper T, Lavrentieva A, Bioengineering (Basel) 2019, 6, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jungst T, Smolan W, Schacht K, Scheibel T, Groll J, Chem Rev 2016, 116, 1496. [DOI] [PubMed] [Google Scholar]
- [25].Jin X, Strueben J, Heepe L, Kovalev A, Mishra YK, Adelung R, Gorb SN, Staubitz A, Advanced Materials 2012, 24, 5676. [DOI] [PubMed] [Google Scholar]
- [26].Velnar T, Gradisnik L, Med Arch 2018, 72, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Pandurangan M, Kim DH, Saudi J Biol Sci 2015, 22, 679; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pandurangan M, Veerappan M, Kim DH, Appl Biochem Biotechnol 2015, 175, 1270. [DOI] [PubMed] [Google Scholar]
- [28].a) Heng BC, Zhao X, Xiong S, Ng KW, Boey FY-C, Loo JS-C, Food Chem Toxicol 2010, 48, 1762; [DOI] [PubMed] [Google Scholar]; b) Pandurangan M, Kim DH, Journal of Nanoparticle Research 2015, 17, 158; [Google Scholar]; c) Vandebriel RJ, De Jong WH, Nanotechnol Sci Appl 2012, 5, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Memar MY, Ghotaslou R, Samiei M, Adibkia K, Infect Drug Resist 2018, 11, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Ferrara N, Gerber HP, LeCouter J, Nat Med 2003, 9, 669; [DOI] [PubMed] [Google Scholar]; b) Rosenstein JM, Krum JM, Ruhrberg C, Organogenesis 2010, 6, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vempati P, Popel AS, Mac Gabhann F, Cytokine Growth Factor Rev 2014, 25, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lertkiatmongkol P, Liao D, Mei H, Hu Y, Newman PJ, Curr Opin Hematol 2016, 23, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Warriner R, Burrell R, Adv Skin Wound Care 2005, 18 Suppl 1, 2. [DOI] [PubMed] [Google Scholar]
- [34].Tom IM, Ibrahim MM, Umoru AM, Umar JB, Bukar MA, Haruna AB, Aliyu A, Open Access Library Journal 2019, 6, 1. [Google Scholar]
- [35].Chudobova D, Dostalova S, Ruttkay-Nedecky B, Guran R, Rodrigo MAM, Tmejova K, Krizkova S, Zitka O, Adam V, Kizek R, Microbiological Research 2015, 170, 147. [DOI] [PubMed] [Google Scholar]
- [36].Xie Y, He Y, Irwin PL, Jin T, Shi X, Appl Environ Microbiol 2011, 77, 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Song W, Zhang J, Guo J, Zhang J, Ding F, Li L, Sun Z, Toxicol Lett 2010, 199, 389. [DOI] [PubMed] [Google Scholar]
- [38].a) Li H, Chang J, Acta Biomater 2013, 9, 6981; [DOI] [PubMed] [Google Scholar]; b) Bhang SH, Jang WS, Han J, Yoon J-K, La W-G, Lee E, Kim YS, Shin J-Y, Lee T-J, Baik HK, Kim B-S, Advanced Functional Materials 2017, 27, 1603497. [Google Scholar]
- [39].a) Gao H, Dai W, Zhao L, Min J, Wang F, Journal of Immunology Research 2018, 2018, 6872621; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang S, Ermann J, Succi MD, Zhou A, Hamilton MJ, Cao B, Korzenik JR, Glickman JN, Vemula PK, Glimcher LH, Traverso G, Langer R, Karp JM, Sci Transl Med 2015, 7, 300ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang N, Liang H, Zen K, Front Immunol 2014, 5, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kang K, Bachu M, Park SH, Kang K, Bae S, Park-Min K-H, Ivashkiv LB, Nat Commun 2019, 10, 3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Parameswaran N, Patial S, Crit Rev Eukaryot Gene Expr 2010, 20, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].a) Tanaka T, Narazaki M, Kishimoto T, Cold Spring Harb Perspect Biol 2014, 6, a016295; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gabay C, Arthritis Res Ther 2006, 8 Suppl 2, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].King A, Balaji S, Le LD, Crombleholme TM, Keswani SG, Adv Wound Care (New Rochelle) 2014, 3, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhao X, Lang Q, Yildirimer L, Lin ZY, Cui W, Annabi N, Ng KW, Dokmeci MR, Ghaemmaghami AM, Khademhosseini A, Adv Healthc Mater 2016, 5, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].a) Deng J, Tang Y, Zhang Q, Wang C, Liao M, Ji P, Song J, Luo G, Chen L, Ran X, Wei Z, Zheng L, Dang R, Liu X, Zhang H, Zhang YS, Zhang X, Tan H, Advanced Functional Materials 2019, 29, 1809110; [Google Scholar]; b) Yao X, Zhu G, Zhu P, Ma J, Chen W, Liu Z, Kong T, Advanced Functional Materials 2020, 30, 1909389. [Google Scholar]
- [47].a) Ludatscher RM, Virchows Arch B Cell Pathol 1978, 27, 347; [DOI] [PubMed] [Google Scholar]; b) Takeo M, Lee W, Ito M, Cold Spring Harb Perspect Med 2015, 5, a023267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Tokatlian T, Cam C, Segura T, Adv Healthc Mater 2015, 4, 1084; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ziche M, Morbidelli L, J Neurooncol 2000, 50, 139. [DOI] [PubMed] [Google Scholar]
- [49].Zeng Q, Han Y, Li H, Chang J, Journal of Materials Chemistry B 2015, 3, 8856. [DOI] [PubMed] [Google Scholar]
- [50].Gurtner GC, Werner S, Barrandon Y, Longaker MT, Nature 2008, 453, 314. [DOI] [PubMed] [Google Scholar]
- [51].Mehrotra S, de Melo BAG, Hirano M, Keung W, Li RA, Mandal BB, Shin SR, Advanced Functional Materials 2020, 30, 2070079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].a) Zhang C, Zhang L, Wu W, Gao F, Li R-Q, Song W, Zhuang Z-N, Liu C-J, Zhang X-Z, Adv Mater 2019, 31, e1901179; [DOI] [PubMed] [Google Scholar]; b) Kim T-H, Eltohamy M, Kim M, Perez RA, Kim J-H, Yun Y-R, Jang J-H, Lee E-J, Knowles JC, Kim H-W, Acta Biomaterialia 2014, 10, 2612. [DOI] [PubMed] [Google Scholar]
- [53].Yan LP, Silva-Correia J, Ribeiro VP, Miranda-Gonçalves V, Correia C, da Silva Morais A, Sousa RA, Reis RM, Oliveira AL, Oliveira JM, Reis RL, Sci Rep 2016, 6, 31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].a) Zhang S, Ou Q, Xin P, Yuan Q, Wang Y, Wu J, Biomaterials Science 2019, 7, 4230; [DOI] [PubMed] [Google Scholar]; b) Xu M, Khan A, Wang T, Song Q, Han C, Wang Q, Gao L, Huang X, Li P, Huang W, ACS Applied Bio Materials 2019, 2, 3329. [DOI] [PubMed] [Google Scholar]
- [55].Oh JS, Lee EJ, Mater Sci Eng C Mater Biol Appl 2019, 103, 109815. [DOI] [PubMed] [Google Scholar]
- [56].Chen W-Y, Chang H-Y, Lu J-K, Huang Y-C, Harroun SG, Tseng Y-T, Li Y-J, Huang C-C, Chang H-T, Advanced Functional Materials 2015, 25, 7189. [Google Scholar]
- [57].Yu H, Peng J, Xu Y, Chang J, Li H, ACS Applied Materials & Interfaces 2016, 8, 703. [DOI] [PubMed] [Google Scholar]
- [58].Dos Santos BP, Garbay B, Fenelon M, Rosselin M, Garanger E, Lecommandoux S, Oliveira H, Amédée J, Acta Biomater 2019, 99, 154. [DOI] [PubMed] [Google Scholar]
- [59].Castleberry SA, Almquist BD, Li W, Reis T, Chow J, Mayner S, Hammond PT, Adv Mater 2016, 28, 1809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.