

Abstract
The clinical manifestation of coronavirus disease 2019 (COVID‐19) mainly targets the lung as a primary affected organ, which is also a critical site of immune cell activation by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). However, recent reports also suggest the involvement of extrapulmonary tissues in COVID‐19 pathology. The interplay of both innate and adaptive immune responses is key to COVID‐19 management. As a result, a robust innate immune response provides the first line of defense, concomitantly, adaptive immunity neutralizes the infection and builds memory for long‐term protection. However, dysregulated immunity, both innate and adaptive, can skew towards immunopathology both in acute and chronic cases. Here we have summarized some of the recent findings that provide critical insight into the immunopathology caused by SARS‐CoV‐2, in acute and post‐acute cases. Finally, we further discuss some of the immunomodulatory drugs in preclinical and clinical trials for dampening the immunopathology caused by COVID‐19.
Keywords: immnopathology, immune responses, respiratory tract, SARS coronavirus
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the causative agent for coronavirus disease 2019 (COVID‐19), has resulted in the loss of lives, and financial and physical distress worldwide on a large scale. As of September 2022, there have been over 600 million infected people with more than 6 million deaths worldwide. 1 The emergence of new variants of concern as a result of the mutation in the structural and nonstructural proteins (NSP) of SARS‐CoV‐2 is making the vaccine less efficient, creating inevitable hurdles in the vaccination programs. 2
The pathophysiology of COVID‐19 is mainly attributed due to the dysfunction of innate and adaptive immune response by SARS‐CoV‐2. This dysfunctional or uncontrolled innate and/or adaptive immune response leads to delayed viral clearance, inflammation, and tissue damage, which is not only restricted to the lungs but systemically, affecting other organs and leading to multiorgan failure. 3 , 4 One of the hallmarks of COVID‐19 is lymphopenia in the blood, a condition where there is a lower‐than‐normal number of lymphocytes such as T cells, B cells, and innate lymphoid cells. 5 , 6 On the other hand, there is an increased aberrant activation and recruitment of myeloid cells in COVID‐19 that may contribute to immune pathology. 7 , 8 , 9 Furthermore, patients with severe COVID‐19 are characterized by increased circulatory inflammatory cytokines, which are significantly associated with acute lung injury in COVID‐19. 10 , 11 Further, inflammatory cytokines and chemokines are highly expressed in the bronchoalveolar lavage (BAL) fluid as compared to blood in patients with severe COVID‐19, suggesting continuous exposure to viral stimulation in the lung microenvironment resulting in heightened inflammatory status locally. 12 Collectively, all this exacerbated immune response eventually leads to pneumonia with vascular leakage, resulting in respiratory failure due to acute respiratory distress syndrome (ARDS) (Figure 1). 13 In addition, extrapulmonary clinical features have also been reported in several COVID‐19 patients such as cardiovascular disorders, thrombotic events, and kidney and liver injury, suggesting that COVID‐19 is not just limited to lungs but also systemically. Furthermore, the rise in postacute COVID‐19 conditions because of chronic tissue and systemic sequelae has been creating new obstacles in combating the ongoing COVID‐19 pandemic.
FIGURE 1.
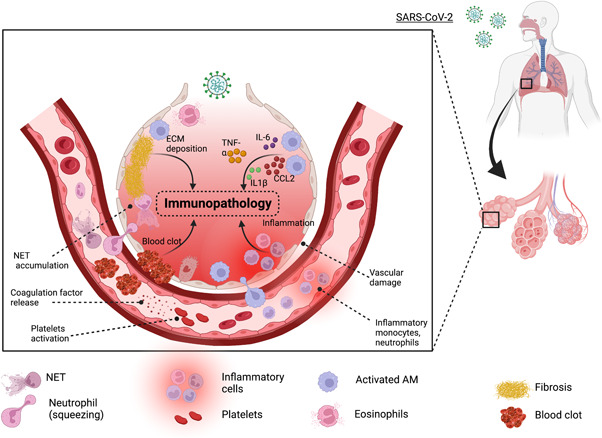
Innate cell‐mediated immunopathology in COVID‐19: Upon viral entry, there is a cascade of events that leads to inflammation, vascular damage, and blot. Tissue‐resident alveolar macrophages (AMs) and interstitial macrophages are among the first responders to SARS‐CoV‐2, which secret inflammatory cytokines including TNF, IL‐6, IL‐1β, and CCL2 that in addition to building up local inflammation but also attract monocyte and neutrophils to the site of infection. Furthermore, IL‐1β favors the expansion of pathological fibroblasts that further contribute to fibrosis. SARS‐CoV‐2 can also stimulate platelets and neutrophils to secrete coagulation factors resulting in the formation of leukocyte–platelet aggregates and NETs, respectively. Lastly, fibroblast proliferation leads to the deposition of extracellular matrix (ECM) and fibrin in alveolar space further complicating the lung alveolar structure. COVID‐19, coronavirus disease 2019; IL‐6, interleukin 6; NET, neutrophil extracellular trap; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TNF, tumor necrosis factor.
In this review, we have summed up some of the recent findings on the innate and adaptive arms of immune response in acute and post‐acute COVID‐19. In addition, we discussed the pathophysiology that arises because of immune dysfunction in COVID‐19 patients, both in acute and chronic sequelae. Finally, we discuss by providing some direct evidence from clinical trials on immunomodulatory drugs that are currently in use for the mitigation of the COVID‐19 pandemic.
2. CLINICAL FEATURES OF ACUTE COVID‐19 IMMUNOPATHOLOGY
Acute COVID‐19 encompasses a multispectrum diseased state with its epicenter majorly in the lungs. Most infected individuals exhibit nonsymptomatic to mild symptoms including fever, coughing, sneezing, running nose, headaches, and fatigue. However, a percentage of individuals may develop severe forms of the diseases, characterized by pulmonary dysfunction and ARDS to systemic organ dysfunction. These patients may often require mechanical ventilation support 14 and typically exhibit an increased risk of mortality. Reports also indicate co‐expression of angiotensin‐converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2) gene at multiple organs, which may suggest the potential for direct viral‐induced pathology to extrapulmonary sites. 15 In addition, excessive systemic inflammation may further contribute to extrapulmonary disease. 16 Cumulatively, these events can be observed as pathological features diagnosed in severe patients, which may progress to multiorgan failure and death.
2.1. Pulmonary clinical features
2.1.1. Acute lung injury and ARDS
Around 6%–10% of SARS‐CoV‐2 infected severe patients experience acute lung injury conditions called ARDS with a high mortality rate. 17 ARDS can be characterized by hypoxemia, ground‐glass opacities, and the presence of bilateral infiltrates in the lungs. 13 , 18 Histological analysis of lungs from COVID‐19 patients identified lung injury as reflected by marked pulmonary inflammation, diffuse alveolar damage, and fibrosis resulting in fatal outcomes. 19 , 20 , 21 Deceased COVID‐19 patient lungs exhibit loss of type II alveolar epithelial cells and show the presence of increased perialveolar lymphocyte cytotoxicity. 22 Furthermore, the accumulation of inflammatory neutrophils and monocytes results in persistent inflammation leading to acute lung injury. 23 The clinical outcome of COVID‐19 is further worsened by endothelial dysfunction, either due to direct infection or systemic inflammation, leveraging the pathological features of COVID‐19. 24 These patients eventually advance to mechanical intubation and ventilator support due to ARDS and may require lung transplantation due to irreversible lung damage. 17 , 25
2.1.2. Fibrosis
Pulmonary fibrosis is characterized by the accumulation of fibroblasts, and excessive deposition of collagen and extracellular matrix, resulting in loss of pulmonary function. 26 The patients that may survive acute illness as a result of ARDS are at high risk of development of pulmonary fibrosis resulting in a high rate of mortality. 27 Intensive fibrosis and collagen deposition have been observed across several COVID‐19 patient autopsy studies. 20 , 21 Lung from COVID‐19 patients with prolonged diseases also showed enhanced pulmonary injury and fibrosis, without the presence of SARS‐CoV‐2 RNA, suggesting sustained tissue damage even after virus clearance. 28
Molecular analyses of the lung tissue from autopsy samples revealed the aberrant activation of interleukin (IL)‐1β‐producing macrophages/monocytes favoring the expansion of pathological fibroblasts that further contribute to fibrosis. 7 Furthermore, fibrosis‐associated genes such as CCL18, LGMN, SPP1, and TGFB1 were enriched in newly recruited CD163+ pulmonary monocyte‐derived macrophages, which also harbor viral transcripts. 29 Moreover, aberrant accumulation of transforming growth factor‐β1 (TGF‐β1) in the lungs as well as type‐III collagen deposition, 30 can further potentiate the risk of terminal pulmonary fibrosis. Of note, patients with pre‐existing idiopathic pulmonary fibrosis (IPF) are at high risk for COVID‐19‐related pathology and clinical outcomes. 31 , 32 Additionally, as the gene signatures from COVID‐19 lungs resemble patients who have IPF, antifibrotic therapy may improve outcomes for COVID‐19 patients with an increased risk of development of fibrosis. 27
2.1.3. Thrombosis
Severe SARS‐CoV‐2 infection is associated with an increased incidence of thrombosis‐associated complications. 33 , 34 , 35 , 36 Pulmonary embolism and deep vein thrombosis are the most prominent thrombosis events that are reported in hospitalized COVID‐19 patients. 37 Currently, there is no clear mechanism for the activation of thrombogenic pathways, although it is believed that a series of complement activation, platelet activation, and/or cytokine storm may trigger thrombotic events in severely infected patients. 37 Microvascular injury and thrombosis have been observed in conjunction with aberrant activation of the alternative and lectin complement pathways. 38 In addition, as platelets express both ACE2 and TMPRSS2, SARS‐CoV‐2 can directly stimulate platelets via the ACE2/mitogen‐activated protein kinase pathway. 39 Upon stimulation, the platelets secrete coagulation factors, resulting in the formation of leukocyte–platelet aggregates. 39 Transcriptomic analysis of platelets from COVID‐19 patients revealed enrichment of pathways including IL‐6, tumor necrosis factor (TNF)‐α, blood coagulation, and hemostasis, suggesting the role of platelet activation in the development of thrombosis. 40 , 41 Postmortem examination of lungs also revealed microvascular thrombi in association with neutrophil extracellular traps (NETs) and platelets, suggesting NET‐triggered thrombosis. 42 As the development of thrombosis has been associated with poor prognosis in hospitalized COVID‐19 patients, early prediction of thrombosis and thromboprophylaxis may improve the clinical outcome. 43
2.2. Extrapulmonary clinical features
COVID‐19 is primarily a respiratory disease, however, increasing evidence suggests that extrapulmonary organs may be subject to direct viral injury or indirect immunopathology caused by SARS‐CoV‐2. 44 , 45 , 46 Organs, such as the brain, heart, kidney, liver, and so forth, are reported to be severely affected as several studies indicate increased risk of neurologic illness, myocardial dysfunction, thrombotic events, kidney injury, and hepatocellular injury following COVID‐19 infection. 45 , 47
The SARS‐CoV‐2 infection has been associated with several cardiovascular disorders including myocardial injury, cardiomyopathy, arrhythmias, and cardiogenic shock. 48 Moreover, individuals with pre‐existing cardiovascular disease exhibit an elevated risk of severe disease and/or death. 49 Some of the studies reported the incidence of acute cardiac injury in COVID‐19 patients. 50 , 51 Patients have also reported neurological and cognitive defects in the aftermath of COVID‐19. Most of the studies showcase the involvement of neurological dysfunction in older patients. 52 However, a case report documents meningitis and seizure in a 24‐year male, 53 posing an alarming threat even to younger individuals. Other neurological symptoms observed in COVID‐19 patients are anxiety, diffusive myalgia, depressive symptoms, headache, and insomnia. 45 In addition, COVID‐19 may result in gastrointestinal complications in some infected patients ranging from nausea, vomiting, and abdominal pain. 45
3. PROTECTIVE INNATE IMMUNE RESPONSES IN ACUTE COVID‐19
As a majority of COVID‐19 infections could be asymptomatic or milder symptomatic, it is suggested that a robust innate immune response may be elicited that is required for viral containment. However, patients with severe disease often had sustained and exacerbated innate responses, which may be induced by sustained viral replication. 54 To completely understand the dynamics of COVID‐19 infection, we need to properly address the recognition of SARS‐CoV‐2 by the innate immune system together with the protective and pathogenic innate response to COVID‐19. Hence, in this section, we will discuss the entry of SARS‐CoV‐2, a protective as well as a pathogenic innate immune response to COVID‐19.
3.1. Recognition of SARS‐CoV‐2
SARS‐CoV‐2 entry to the host cell requires interaction with the ACE2 receptor via viral spike protein. In addition, a host serine protease, TRMPSS2, further facilitates spike protein priming which is important for viral entry. 55 However, to initiate an innate immune response viral genomic single‐stranded RNA and replicative double‐stranded RNA both can be recognized by Toll‐like receptors (TLRs) and RIG‐I‐like receptors (RLRs). In the case of SARS‐CoV‐2, retinoic acid‐inducible gene‐I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA5) can sense viral RNA and drive inflammation in Calu‐3 cells. 56 Conditioned media from these epithelial cells can further lead to propagating inflammation in primary human monocyte‐derived macrophages. 56 However, in primary human epithelial cells, RIG‐I can sense SARS‐CoV‐2 but failed to activate mitochondrial antiviral‐signaling protein‐dependent pathways resulting in reduced interferons (IFNs) and inflammatory cytokines production. 57 Furthermore, TLR2 has been involved in eliciting the proinflammatory immune response in both human and murine macrophages. 58 , 59 A reduction in IL‐6 level was observed in TLR2−/− mice treated with SARS‐CoV‐2 E protein, 58 and TLR2 inhibition in human ACE2 (hACE2) transgenic mice infected with SARS‐CoV‐2 reduces inflammation and mortality. 58 In addition, gene variants in viral sensing such as TLR3 and TLR7 were also observed that are associated with weak IFN response and severity of COVID‐19 in a small number of individuals. 60 , 61 Overall, these observational studies suggest the critical role of the mediators of the innate immune system, which can act differentially following SARS‐CoV‐2 infection. As the current understanding of these mediators is still naïve, we expect more studies are required in this direction.
3.2. The double‐edged sword of IFN responses
Early protection against COVID‐19 can be achieved by balanced and robust innate immune responses. Innate immune cells contribute to providing the first line of defense against viral and bacterial infection. During early infection, IFN response is necessary to limit viral replication. Early IFN levels were reported in COVID‐19 patients, which was further correlated with the lower viral count in BAL fluid and improved outcomes. 62 A study from the SARS‐CoV‐2 infection in macaques also presented that robust IFN response is generated from macrophages and T lymphocyte population during acute infection. This elevated early IFN response eventually serves to clear viremia. 63 Furthermore, transcriptomic analysis of blood and BAL samples from severe COVID‐19 patients revealed diminished IFN‐responsive genes (interferon‐stimulated gene [ISG]) response in BAL fluid as compared to paired blood samples. 12 In addition, downregulation of ISG genes such as MX1, IFITM1, and IFIT2 were reported in critical COVID‐19 patients, and undetected meesenger RNA (mRNA) and protein levels of IFN‐β, and impaired IFN‐α production were observed in the blood of severe patients, 64 suggesting impaired type I IFN responses may promote disease progression. Thus, a robust and early type I IFN response is required to activate a cellular antiviral state and achieve antiviral immunity by stimulating the activation of immune cells such as natural killer (NK) and dendritic cells (DCs). 65 In addition, type I IFN response may promote T and B cell recruitment at the site of infection facilitating viral clearance. 65 SARS‐CoV‐2 infection in cells can block IFN signaling via its proteins such as nsp6, nsp13, and ORF6, which are known to suppress IRF3 phosphorylation and nuclear translocation. 66 Collectively these reports indicate that disease severity is associated with weak IFN response in severe patients. This was further supported by that about 10% of patients with severe COVID‐19 have neutralizing antibodies against type I IFN rendering ineffective IFN response, which may also advocate the protective function of type I IFN. 67 The plasma from these patients was further able to block the protective action of IFN‐ α2 in vitro as evident by enhanced SARS‐CoV‐2 replication in Huh7.5 cells. 67 Type III IFN response shares a similar ISG expression pattern as with type I, only differing in causing lesser inflammation during severe viral infection. 68 Study with influenza infection suggests the protective function of type III IFN in respiratory viral infection, which is also reflected in SARS‐CoV‐2 severity as in mild COVID‐19 patients the levels of type III IFN is higher as compared to severe patients. 69 Nevertheless, more studies are required to delineate the role of type III IFN in the context of COVID‐19.
Type II IFN, IFN‐γ, is secreted by a type I innate lymphoid cells (ILC1s), NK cells, and T‐cells. 70 , 71 Although IFN‐γ also has the antiviral ability but sustained IFN‐γ levels in COVID‐19 patients are associated with mortality. 72 Intriguingly, elevated levels of all IFN such as IFN‐α, IFN‐γ, and IFN‐λ have been reported in severe patients during acute infection; however, only elevated IFN‐λ was correlated with lower viral load. 62 , 73 This report suggests that type I and II IFNs fail to control infection in severe patients and could be associated with pathology if released in an uncontrolled manner. Furthermore, type I and type III IFN have been associated with activation of antiproliferative and cell death pathways in primary murine airway epithelial cells by a respiratory viral infection, 74 suggesting that sustained and/or delayed IFNs could be detrimental in tissue repair. The transcriptome of classical monocyte from severe COVID‐19 patients revealed enrichment of ISG expression. 75 The ISG hence identified in COVID‐19 cases were found to be proinflammatory due to the presence of inflammatory mediators or regulators, 76 advocating detrimental instead of the protective function of IFNs. In this regard, blocking the IFN‐stimulated response with IFN‐alpha and beta receptor subunit 2 antibodies enhanced lung recovery and was observed in humanized mice model of chronic SARS‐CoV‐2 infection. 77 Altogether, these contrasting reports suggest the duality in the IFN response, and hence balanced IFN is required for a protective immune response to COVID‐19. 78 Till this point, it is suggested that IFN protective response is time‐dependent, where early increased levels are beneficial and late can be detrimental. 79 , 80 Nevertheless, this paradoxical nature of IFN signaling is subjected to further clarification.
3.3. Protective cellular innate responses
Alveolar macrophages (AMs) are the tissue‐resident macrophages in the lung and are indispensable for maintaining lung immune homeostasis. AM population was depleted in the BAL fluid of critical COVID‐19 patients, 81 suggesting that AMs are necessary for protection. In a recent preprint study, a monocyte‐derived proliferating Slamf9+ Spp1+ macrophages subset was shown to be resistive to SARS‐CoV‐2 induced cell death and helps us to clear the virus in Syrian hamsters. 82 These macrophages were then differentiated into triggering receptors expressed on myeloid cells 2+ and fructose‐bisphosphatase 1+ macrophages to resolve inflammation and reconstitute AM population, altogether aiding in lung repair. 82 The role of NK cells has not been completely studied in the context of COVID‐19. Although some studies show that the NK population not only decreased but also was in a dysfunctional state in COVID‐19 cases, 83 , 84 , 85 indicating its role in providing protection. In accordance with the latter observations, it was found that NK cells purified from healthy individuals can reduce SARS‐CoV‐2 load in Calu‐3 and Vero E6 cell lines. 86 Relatively abundant NK cells in some COVID‐19 patients were also correlated with the rapid decline of viral load as compared to patients with lower NK levels. 86
Convalescent patients with higher frequencies of ILC subset NK cell‐activating receptor group 2D+ (NKG2D+) ILC2s demonstrated a significant reduction of the hospitalization time, 87 also suggesting the beneficial role of ILCs. Plasmacytoid dendritic cells (pDCs) are capable of IFN‐I production following the viral encounter; however, as pDCs are depleted in peripheral blood mononuclear cells (PBMCs) of COVID‐19 patients, 88 , 89 their protective functions are largely compromised. Altogether these reports point to the fact that even though these cellular innate responses have intrinsic antiviral defense capacity, in COVID‐19 all these responses are either weakened or dysfunctional eventually leading to pathogenic outcomes.
4. PATHOGENIC INNATE RESPONSES IN ACUTE COVID‐19
A balanced and robust innate immune response is critical to encountering COVID‐19. However, an uncontrolled or misfired innate immune response could be detrimental to the host, resulting in acute severe diseases. Here, in this section, we have discussed some of the pathological features of innate immune cells in response to SARS‐CoV‐2 infection.
4.1. Pathogen‐associated molecular patterns and damage‐associated molecular patterns
The innate immune response is elicited by recognition of evolutionarily conserved structures on pathogens known as pathogen‐associated molecular patterns (PAMPs). Damage‐associated molecular patterns (DAMPs) are molecules released by stressed or dead cells. 90 DAMPs and PAMPs are detected by pattern recognition receptors (PRR), such as TLR and RLR, and can initiate inflammation upon binding and may cause tissue damage leading to acute lung injury. 91 Elevated levels of DAMPs and PAMPS have been reported in a recent study comprising a longitudinal evaluation of serum and endotracheal aspirate from severe COVID‐19 patients. 91 Alarmins S100A8 were found to be upregulated by SARS‐CoV‐2 infection in rhesus macaques and in hACE2 transgenic mice. 92 Likewise, high levels of S100A8/9 were reported in the plasma of severe COVID‐19 individuals, which positively correlated with the adversity of the disease. 93 , 94 Another prognosis marker of COVID‐19 severity has been reported is circulating mitochondrial DNA (MT‐DNA), which is a member of a group of mitochondrial DAMPs. 95 In severe or deceased COVID‐19 patients, the levels of MT‐DNA were reportedly high. 95 DAMP molecule IL‐33 levels are high in the serum of COVID‐19 cases and are indicative of disease severity. 87 , 96 IL‐33 has been shown to be secreted by human epithelial cells following SARS‐CoV‐2 infection. 97 However, after disease resolution, induction of IL‐33 in PBMCs of convalescent patients upon T‐cell stimulation suggests persistent secretion of IL‐33 by immune cells. 98 One of the DAMPs, high‐mobility group box 1 protein, levels have been also shown to be upregulated in critically ill patients with COVID‐19 and is related to poor clinical outcomes. 99 , 100
TLR and RLR are among PRRs that can detect nonself RNA. After detecting a viral RNA, RIG‐I and MDA5 trigger the IFN response that is required for viral clearances. However, excessive and prolonged IFN response is determinantal for the host. SARS‐CoV‐2 can be recognized by both RIG‐I and MDA‐5; however, this RNA sensing may differ according to different cell types. 56 , 57 , 101 SARS‐CoV‐2 RNA and proteins such as GU‐rich RNAs, protein E, and viroporin have been shown to activate NLPR3 and hence inflammasome formation. 58 , 102 , 103 NLPR3 activation is a well‐known factor for the proinflammatory event known as pyroptosis. 104 In addition to NOD‐, LRR‐, and pyrin domain‐containing protein 3 (NLRP3) activation, SARS‐CoV‐2 protein E induces enhanced proinflammatory cytokines response in TLR‐2 dependent manner. 58 Hence, these DAMPs and PAMPs could overexaggerate the innate immune system, skewing toward immunopathology instead of disease resolution.
4.2. Neutrophils
Neutrophils are among the first cell types to migrate to the infected sites and encounter pathogens. An increase in the neutrophil count (neutrophilia) in the blood and nasopharyngeal epithelium 105 and BAL fluid 9 of severe patients are among the first findings that suggest the importance of neutrophils in the pathology of SARS‐CoV‐2. Freshly isolated neutrophils showed the presence of inflammasome activation which may play important role in supporting cytokine storm. 106 In a further study, it was shown that neutrophils isolated from COVID‐19 patients have an increased hypoxia‐inducible factor 1 subunit alpha (HIF‐1α) and glycolysis activity. 107 These studies might explain the inflammatory nature of neutrophils in COVID‐19 patients thereby suggesting a pathogenic response of neutrophils in the advent of COVID‐19.
NETs are web‐like structures of DNA containing neutrophil histones and granule‐derived enzymes. 108 The plasma of severe to critical condition patients was found to be enriched in NETs. 108 Recently, it was shown that neutrophils from COVID‐19 patients with ARDS are primed to form NETs as compared to COVID‐19 with non‐ARDS. 109 Furthermore, neutrophils isolated from COVID‐19 patients are more susceptible to release NETs as compared to healthy donors. 108 , 109 In a different study, sera of COVID‐19 patients were demonstrated to have an increased level of myeloperoxidase DNA and citrullinated histone H3, markers for NET. 110 Additionally, serum from these COVID‐19 patients was able to induce NET formation in healthy neutrophils, indicating that both serum and intrinsic factors in neutrophils can govern NET formation. Similarly, a recent preprint study showed that serum from pediatric acute COVID‐19 can trigger the NET formation in healthy neutrophils. 111 Furthermore, this study showed that the spike immune complex generated by the dilution of plasma with spike protein on beads was the major driver for NET formation, suggesting the role of viral spike protein complexes in NET formation. Those NETosing neutrophils have a positive correlation with a novel subset of inflammatory neutrophils in severe and critical COVID‐19 patients. 112
A higher level of NETs was observed in serum, tracheal aspirants, and lung tissues of COVID‐19 patients. 108 , 113 Immunofluorescence and immunohistochemistry studies on lung biopsy tissue from deceased and severe COVID‐19 patients also confirmed the presence of NET. 108 , 114 , 115 The NET formation was further associated with inflammatory interstitial lesions, vascular compartments, and the airways of COVID‐19 injured lungs. 114 , 115 Hence, increased neutrophile‐induced inflammatory NETs are a major cause of pathology in COVID‐19, which is further worsened by delayed tissue repair and thrombosis induced by NETs. 116 These observations suggested that neutrophils not only play a critical role in inducing inflammation in critical COVID‐19 patients but also result in lung damage and interfere with tissue repair through the NET formation.
4.3. Monocytes
Long‐term analysis of monocytes showed that the monocyte number, frequency, and activation markers are deeply influenced in acute and convalescent COVID‐19 patients. 8 The number as well as the absolute count of monocytes increases from 15 to 30 days of infection to 4–5 months postinfection. Similarly, the frequency of monocyte subsets such as classical, intermediate, and nonclassical monocytes, alter with time. 8 Circulating monocyte activation markers, such as soluble CD14, CD163, and C‐reactive protein levels were also found to increase after acute infection, 8 suggesting long‐term activation of monocyte postacute COVID‐19. The SARS‐CoV‐2 infection leads to distinct transcriptomic features in monocytes, 117 which is further regulated by infection kinetics and disease severity. 118 High‐dimensional profiling of human blood and BAL sample from patients with severe COVID‐19 showed upregulation of viral sensing, IFN response genes together with IL‐6, TNF‐α, and IL‐8, which were associated with increased risk of casualties with COVID‐19. 118 , 119 The enhanced inflammatory characteristic in human monocyte is further supported by aerobic glycolysis, which also supports SARS‐CoV‐2 replication in these monocytes. 119 Infected human monocytes, as well as monocytes from severe COVID‐19 patients, observed high expression of HIF‐1α which is stabilized by mitochondria reactive oxygen species production in response to infection. The stabilized HIF‐1α is required to upregulate glycolytic genes during SARS‐CoV‐2 infection and lastly was suggested that targeting HIF‐1α and/or glycolysis may be beneficial for COVID‐19 management. 119
Active NLRP3 inflammasome and elevated levels of caspase‐1 activity in patients in PBMC from COVID‐19 patients have also been reported on the day of hospitalization. The increased caspase‐1 levels dropped significantly thereafter suggesting the role of inflammasome activation in causing acute lung pathology. 120 Recently, it was reported that about 10% of monocyte gets infected by SARS‐CoV‐2 in COVID‐19 patients via Fc‐γ receptors (FcγR)‐mediated uptake of antibody‐coated virus. 121 Additionally, infected monocytes have activated inflammasome, caspase‐1, and gasdermin D (GSDMD) leading to pyroptosis, which further adds up to lung injury. 121 Additionally, monocyte isolated from a healthy individual infected with SARS‐CoV‐2 in vitro also contribute to the fibrotic phenotype, 29 suggesting a role of direct infection of monocytes in promoting fibrosis.
4.4. Macrophages
Myeloid cells population such as interstitial macrophages, monocyte‐derived macrophages, and AMs are among the most enriched immune cells in the lungs of COVID‐19 patients. 122 Using humanized mice model, it was recently demonstrated that SARS‐CoV‐2 can infect and replicate in human macrophages. These infected macrophages have an inflammatory phenotype characterized by inflammasome activation, which also contributes to sustained IFN response. 77 Indeed, these infected macrophages have an inflammatory signature which was evident by enrichment in the expression of several cytokines (IL1A, IL18, and IL27) and chemokines (CXCL10, CCL18, CCL3, CCL7, CCL8, CCL20, and CXCL8). 77 In addition, morphological analysis of the infected macrophages revealed the sign of pyroptosis. Apoptosis‐associated speck‐like protein containing a CARD (ASC), which is a marker for inflammasome activation, was formed in the infected macrophages. Finally, both lactate dehydrogenase (LDH) and GSDMD levels in serum were increased in the infected mice which further suggested the involvement of the pyroptosis pathway. 77 Similarly, clinical data from the COVID‐19 patients also demonstrated enhanced IL‐18, LDH, and GSDMD levels in severe patients. Lung biopsies further revealed activation of ASC more prominently in CD14+‐infected lung macrophages. 121 The activation of pyroptosis‐dependent cell death in macrophages is meant to abort viral replication; however, it also leads to the release of inflammatory mediators that further add up to the immunopathology. 121 These two recent studies have shown conclusive evidence that how infected macrophages can trigger inflammation. Nevertheless, more studies are required to further delineate the underlying mechanism of infected macrophages in the regulation of immunopathology.
AMs are the major sentinels of the lungs and are involved in engulfing inhaled particles and allergens, and aid in tissue repair, which is critical for maintaining lung homeostasis. 123 Following lung insults, the self‐renewal ability of AM is required to repopulate the alveolar space and aid in tissue repair. 123 However, during COVID‐19, AMs can result in an inflammatory response. RNA‐sequencing data from the public dataset reflects that AMs derived from COVID‐19 patients show an increase in inflammatory properties with a concomitant decrease in reparative ability. 123 In COVID‐19 patients, there is a decrease in the AM population in the BAL fluid. 81 The lung is later repopulated by CD11b+ interstitial macrophages, probably to aid lung repair. 124 , 125 As AMs can be readily infected with SARS‐CoV‐2 similar to other coronaviruses, 126 , 127 it is speculated that AMs may be critical for virus propagation. 128 AMs isolated from BAL fluid of severe COVID‐19 patients within 48 h after intubation also showed the presence of SARS‐CoV‐2 viral transcript. 128 These AMs then secret T‐cell chemokines, recruiting more T‐cells in the vicinity resulting in T‐cell‐dependent IFN‐γ secretion, eventually leading to AM inflammatory response. This feedback loop may be functional for long period due to infection of monocyte‐derived macrophages with SARS‐CoV‐2, contributing to lung injury. 128 Furthermore, AMs can be programmed to inflammatory M1 phenotype causing lung damage by SARS‐CoV‐2 infection and facilitating viral replication. 129 , 130 , 131 Furthermore, depletion of AMs by clodronate results in effective virus clearance and lung recovery in the hACE2 transgenic mouse model, 130 suggesting a pathological response of AMs in COVID‐19. However, as these AM are primarily of inflammatory phenotype, the pathological outcome is excepted. Nevertheless, it is still largely unknown how AMs are skewed towards inflammatory phenotype upon direct SARS‐CoV‐2 infection.
4.5. Other innate cell populations
Several other innate cells are depleted in COVID‐19 cases such as DCs, eosinophils, and NK cells. 75 , 132 , 133 Also, among them, the most prominent depletion occurred in DCs, eosinophils, and NK cells and was associated with disease severity. 132 , 133 , 134 COVID‐19‐associated NK cells were found to be in a dysfunctional state with lower antiviral activity. 85 In addition to compromised function, NK cells from COVID‐19 patients also display profibrotic gene expressions such as AREG, DUSP2, ZFP36L2, and TSC22D3, which is similar to that of NK phenotype in lung fibrosis. 84
Likewise, circulatory DCs were diminished in COVID‐19 samples, both in acute and postacute cases. 135 , 136 , 137 pDCs, which are a major contributor to IFN‐α production, were also reduced in COVID‐19 patients, 84 which may answer why there is delayed IFN‐α response in some patients. 64 The DCs isolated from COVID‐19 patients also has a reduced ability to stimulate naïve T‐cells leading to a weak adaptive immune response. 137 Furthermore, an in vitro study showed that despite low expression of the ACE2 receptor, SARS‐CoV‐2 can infect human DCs. Following infection, the infected DCs are unable to mount IFN responses, which are supposedly considered to delay viral clearance and may also contribute to immunopathology. 138 Intriguingly, lung resident DCs are responsible for IFN‐λ production upon viral RNA stimulation via the TLR3 pathway suggesting a pathogenic role of DCs. 139 Furthermore, sustained IFN‐ λ by DCs has been predisposed to lung epithelial damage and secondary bacterial infection. 139
ILCs are among the major innate immune cell population in the lungs and promotes tissue repair after respiratory viral infection. 140 However, its role in the context of SARS‐CoV‐2 infection is poorly studied. ILCs have been reported to be depleted in severe COVID‐19 and were inversely related to inflammation. 141 In addition to depletion, ILC2s, and ILC precursors showed a higher frequency of CD69+ cells, a reflection of an activated state, and dysregulated ILC tissue migration resulting in pathogenic outcomes. 142 Additionally, chemokine receptor expression, CXCR3, and CCR6 were decreased on ILC2s in COVID‐19 individuals. 142 In contrast, convalescent patients that have higher numbers of ILC subset NKG2D+ ILC2s together with elevated serum IL‐13 levels demonstrated a significant reduction in hospitalization length. 87 Overall advocating the protective role of ILCs in SARS‐CoV‐2 infection. However, IL‐13 has been associated with COVID‐19 severity and IL‐13 neutralization by dupilumab in asthmatic patients resulted in lower mortality and hospitalization rate by COVID‐19. 143 Hence, it is still unclear about the role of ILCs in the regulation of COVID‐19 pathogenesis. The role of mast cells has also been studied in COVID‐19‐induced epithelial inflammation and lung injury. The SARS‐CoV‐2 infection triggers mast cell degranulation in lungs in both humanized mice and nonhuman primates, which is further suggested to induce lung injury. 144
5. PROTECTIVE ADAPTIVE IMMUNE RESPONSES IN ACUTE COVID‐19
The adaptive immune response system, including B and T lymphocytes, carries out body defense in humans. Despite they can take days to become established, activated B and T cells have critical roles in controlling and shaping the immune response by providing various immune functions and long‐lasting protection. SARS‐CoV‐2 infection of the respiratory tract induces virus‐specific B and T cells, mediating viral clearance at the infection sites and preventing viral dissemination through antibodies and T cell effector functions. Indeed, many studies have shown that COVID‐19 patients generated neutralizing antibodies and virus‐specific T cells in the peripheral blood and the respiratory tract (Figure 2). 145 , 146 , 147 , 148 , 149 It was also indicated that patients developed SARS‐CoV‐2‐specific CD8+ T and CD4+ T, and B cell memory in the lungs, lung‐associated lymph nodes, and other organs for up to 6 months following natural infection of SARS‐CoV‐2. 150 , 151 Together, these findings suggest the persistence of humoral and cellular immune responses to SARS‐CoV‐2 infection in humans.
FIGURE 2.
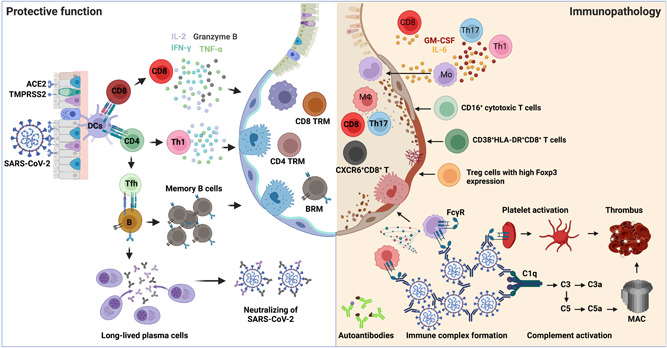
The protective versus pathogenic adaptive immune responses in COVID‐19. Left: When SARS‐CoV‐2 invades the host respiratory tract, the viral antigen can be detected and presented by DCs to either CD4+ T or CD8+ T cells for their activation. Naïve CD4+ T cells mainly differentiate into T helper 1 (Th1) and T follicular helper cells (Tfh). Th1 cells possess antiviral effects by producing higher levels of IFN‐γ, TNF, and IL‐2. Tfh cells provide help to B cells for somatic hypermutations and affinity maturation of germinal center reactions to generate memory B cells and long‐lived antibody‐producing plasma cells. The viral‐specific antibodies secreted by plasma cells play a protective role by neutralizing the virus. Activated CD8+ T cells produce effector cytokines and cytotoxic molecules, including IFN‐γ, TNF, IL‐2, and granzyme B, controlling viral infections. After viral clearance, memory CD4+ T, CD8+ T, and B cells are developed in the circulation and lungs to protect against secondary infections. Right: Excessive T cell responses are associated with severe COVID‐19, including IL‐6‐ and GM‐CSF‐producing Th1 or Th17 cells, CD16+ cytotoxic T cells, CXCR6+ CD8+ T cells, as well as dysregulated Treg cells. On the other hand, the production of autoantibodies, the formation of immune complexes, and complement activation also contribute to the disease progression of COVID‐19. ACE2, angiotensin‐converting enzyme 2; BRM, resident memory B; COVID‐19, coronavirus disease 2019; DC, dendritic cell; FcγR, Fc‐γ receptor; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IFN‐γ, interferon‐γ; IL‐2, interleukin 2; MAC, Membrane attack complex; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TMPRSS2, transmembrane serine protease 2; TNF, tumor necrosis factor; Treg, regulatory T cell; TRM, tissue‐resident memory.
5.1. T cell immunity
Generally, T cells can be divided into two subsets: CD4+ T helper cells and CD8+ cytotoxic T cells, both of which contribute to the protection against respiratory virus infections. Upon activation, naïve CD4+ T cells mainly differentiate into T helper 1 (Th1) and T follicular helper cells (Tfh) during viral infection. Th1 has antiviral properties by triggering cell‐mediated immune responses through activating other immune cells, while Tfh specializes in help to B cells for somatic hypermutations and affinity maturation of germinal center reactions and thus are vital for the generation of high‐affinity neutralizing antibodies, as well as for the development of memory B cells. Activated CD8+ T cells control viral infections by eliminating virus‐infected cells and producing effector cytokines. After viral clearance, memory CD8+ and CD4+ T cells are developed in tissues to protect the host against secondary infections.
SARS‐CoV‐2‐specific T cells are well detected in most donors during acute infection and at the convalescent stage. 152 CD4+ T cells were predominantly exhibiting Th1 phenotype in mild patients, producing higher levels of IFN‐γ, TNF, and IL‐2, and rare Th2‐ and Th17‐related cytokines were detected. 146 , 153 , 154 SARS‐CoV‐2‐specific CD8+ T cells possess high levels of effector molecules, including IFN‐γ, granzyme B, TNF, IL‐2, perforin, and CD107a, which have been associated with a better outcome. 146 , 155 , 156 , 157 One study tracked T cell response and viral burden longitudinally after symptom onset and found patients with the presence of robust early T cell responses were associated with mild disease and rapid viral clearance. 157 Conversely, individuals with very few virus‐specific T cells early on were associated with the persistence of high viral loads and the development of severe COVID‐19. 157 Another study observed a positive association between the presence of SARS‐CoV‐2‐specific CD4+ T and CD8+ T cells and reduced disease severity. 155 Furthermore, a study revealed that SARS‐CoV‐2‐specific CD8+ T cell response was significantly associated with mild disease and high antiviral efficacy. 158 Overall, these studies linked SARS‐CoV‐2‐specific T cell responses to rapid viral clearance and/or better clinical outcomes, suggesting an active role of T cells in the control and clearance of SARS‐CoV‐2. Interestingly, virus‐specific T cells appear to be functionally superior in asymptomatic individuals with a similar frequency of SARS‐CoV‐2‐specific T cells, but higher production of Th1 cytokines IFN‐γ and IL‐2 compared to symptomatic patients. 159
Pre‐existing SARS‐CoV‐2‐specific T cells were also detected in individuals with no history of SARS, COVID‐19, or contact with individuals who had SARS and/or COVID‐19, and these T cells frequently targeted NSP7 and NSP13 of SARS‐CoV‐2 as well as structure nucleocapsid protein, which are highly conserved among different coronavirus. 160 Similarly, a recent study has shown pre‐existing memory T cells that were more frequently directed against replication transcription complex proteins (RTC, including NSP7, NSP12, and NSP13) were enriched and expanded in vivo in seronegative healthcare workers (SN‐HCWs), whereas T cells from mild COVID‐19 individuals preferentially recognized structural proteins. SN‐HCWs with strong RTC‐specific T cells had high induction of IFN‐inducible transcript IFI27 in the blood, a robust early innate signature of SARS‐CoV‐2 infection. 161 These two studies suggest that boosting pre‐existing memory T cells could be a potential target for epitope‐based vaccine design. Additionally, many studies found that SARS‐CoV‐2‐specific memory CD4+ and CD8+ T cell responses were durable over time after infection. 145 , 162 , 163 Wragg et al. 163 reported that SARS‐CoV‐2 infection and/or vaccination‐induced memory CD4+ T cells and circulating T follicular helper are efficiently recalled after antigen re‐exposure, suggesting a long‐term protection capability.
γδ T cells are an innate‐like T cell subset that expresses γδ T‐cell receptor (TCR) and is mainly present in the epithelial layer of mucosa. Upon activation, gd T cells can produce a variety of cytokines, including IFN‐γ, TNF, and IL‐17, as well as the cytotoxic molecules perforin and granzymes, to combat invaders. 164 To date, there is limited information on how γδ T cells are involved in COVID‐19. One study reported that deceased COVID‐19 patients had lower Vγ9Vδ2 T cells, the dominant γδ T‐cell population in adults, compared to surviving patients. 165 In the patients who survived, Vγ9Vδ2 T cell number was comparable to healthy controls, with 26% of cells shifted to an effector (memory) phenotype. 165 Similarly, Carter et al. 166 observed γδ T cell lymphopenia and activation in the acute phase of children with the multisystem inflammatory syndrome and returned to normal by convalescence. Collectively, these studies evidenced that γδ T cells participate in the host immune response against SARS‐CoV‐2 infection. Further investigations are needed to characterize the functional role of γδ T cells in COVID‐19.
5.2. Humoral immunity
Humoral responses are another part of adaptive immunity against viral infection. SARS‐CoV‐2 infection induces robust humoral immune responses and generates potent neutralizing antibodies (nAbs) against the spike (S) protein. 167 , 168 , 169 The receptor‐binding domain of S protein is dominantly targeted by about 90% of nAbs. 170 nAbs prevent the entry of SARS‐CoV‐2 into host cells, primarily by blocking S protein engaging its cognate receptor ACE2. A body of evidence indicates that nAbs are strongly correlated with protection from SARS‐CoV‐2 infection. 169 , 171 , 172 The presence of nAbs induced by a previous infection has also been shown to provide protection against subsequent reinfection. 173 The development of humoral immunity is dependent on the activation of antigen‐specific B cells, which result in the germinal center formation and differentiate into long‐lived plasma cells or memory B cells. 174 nAbs are detectable within 7–14 days postsymptom onsite, peak until 23 days, and maintained for at least 16 months after infection. 175 In addition, S‐specific long‐lived bone marrow plasma cells are still detectable for at least 11 months. 176 SARS‐CoV‐2‐specific memory B cells also persisted for at least 15 months. 162 Memory B cells can be reactivated to elicit an antibody response within a few days upon SARS‐CoV‐2 infection and are likely protective; however, no direct evidence shows the protective role of memory B cells in humans. The mucosal immune system is involved in protection at the sites of infection. As SARS‐CoV‐2 infects the respiratory tract, it could induce robust mucosal immunity. Indeed, studies have demonstrated that COVID‐19 convalescents had significantly higher levels of nAbs against D614G, Delta, and Omicron in the BAL compared to mRNA‐vaccinated individuals. 177
6. PATHOGENIC ADAPTIVE IMMUNE RESPONSES IN ACUTE COVID‐19
6.1. Dysregulated T‐cell responses in COVID‐19
Virus‐specific T‐cell responses are mainly thought to be protective. However, dysregulated T‐cell responses can contribute to disease progression in COVID‐19 patients (Figure 2). In many cohorts of critically ill patients, the numbers of SARS‐CoV‐2‐specific CD4+ T and CD8+ T cells were comparable to or higher than those in mild patients, and such polyfunctional antigen‐specific T cells were predisposed to a cytotoxic phenotype, 159 , 178 , 179 , 180 , 181 which likely play an important role in causing higher disease severity and leading to tissue damage. Consistent with this notion, a recent study revealed that higher frequencies of IFN‐γ‐ and TNF‐α‐producing SARS‐CoV‐2‐specific T cells in the peripheral blood of COVID‐19 patients with postacute syndrome are associated with increased systemic inflammation (plasma IL‐6) and worsen lung function (forced expiratory volume in 1 s). 182 SARS‐CoV‐2‐specific regulatory T cells (Tregs) were also found elevated in fatal COVID‐19 cases, likely associated with the poor SARS‐CoV‐2‐specific T cell responses observed in these patients. 156 Furthermore, activated CD4+ T and CD8+ T cells have been found to infiltrate the lungs of severe COVID‐19 patients and are associated with inflammation, endothelial dysfunction, and fibrosis. 183 , 184
T cell hyperactivation and/or “exhaustion” have been described in COVID‐19. High expression of effector molecules, including GZMH, KLRD1, and SLC9A3R1, by CD8+ T cells in COVID‐19 patients, is linked to improved clinical outcomes. 185 However, excessive T cell activation may be detrimental, as reported by Mathew et al. 179 that hyperactivated CD4+ T and CD8+ T cells are associated with disease severity and poor outcomes. Conversely, upregulation of inhibitory receptor expression on CD8+ T cells including PD‐1, TIM‐3, LAG‐3, TIGIT, CTLA‐4, and NKG2A has been observed during acute infection, reflecting T cell overactivation and dysfunction in acute disease. 179 , 186 , 187 , 188 Nevertheless, these elevated inhibitory receptors may not be exhausted, especially in the early phase, they can represent ongoing activation as evidenced by PD‐1‐expressing SARS‐CoV‐2‐specific CD8+ T cells being functional. 189 Both CD38 and HLA‐DR are well‐known activation markers that are expressed on activated T cells during the acute phase of viral infections in humans, including human immunodeficiency virus, 190 dengue virus, 191 Ebola virus, 192 pandemic H1N1, 193 and H7N9. 194 The increasing number of CD38+HLA‐DR+Ki‐67+ CD4+ T and CD8+ T cells were also found in the acute phase of severe COVID‐19 patients. 155 , 179 , 188 , 195 , 196 These CD38+HLA‐DR+CD8+ T cells express high levels of effector and proinflammatory cytokines, including IFN‐γ and GZMB, contributing to viral control. These studies indicate that early prevalence of an activated CD38+HLA‐DR+CD8+ T cell subset is associated with patient survival, whereas prolonged activated T cells with expression of inhibitory immune checkpoint receptors PD‐1, CTLA‐4, TIM‐3, LAG‐3, and TIGIT may lead to severe and fatal COVID‐19. Yet, it remains unclear whether such T cells are antigen‐specific. Interestingly, bystander‐activated CD38+HLA‐DR+CD8+ T cells were identified in acute hepatitis A patients and chronic hepatitis C patients and are significantly associated with liver injury, 197 , 198 suggesting non‐SARS‐CoV‐2‐specific CD38+HLA‐DR+ CD8+ T and/or CD4+ T cells could play a pathogenic role in fatal COVID‐19 patients. Further studies with larger patient cohorts might provide details on whether such prolonged with functionally exhausted CD38+HLA‐DR+PD‐1+ CD8+ T and CD4+ T cells could predict disease severity and outcome.
Severe COVID‐19 patients have been shown to exhibit elevated BAL and/or serum levels of cytokines, including IL‐6, IL‐2, IL‐1β, IL‐8, IL‐10, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), granulocyte colony‐stimulating factor, IFN‐γ, and TNF‐α, and are associated with disease severity and mortality. 9 , 11 , 105 , 199 , 200 An interesting question is whether acutely activated T cells secrete these cytokines and how they contribute to immunopathogenesis in COVID‐19. Zhou et al. 201 reported that a subset of CD4+ Th1 cells from COVID‐19 patients in both intensive care units (ICU) and non‐ICU express high levels of GM‐CSF, IL‐6, and IFN‐γ compared to healthy controls, while ICU patients with more severe pneumonia had a higher percentage of GM‐CSF+ and IL‐6+ CD4+ T cells. They proposed that these pathogenic CD4+ Th1 cells were rapidly activated to produce GM‐CSF and other cytokines to prime inflammatory monocytes (IL‐6hiCD14+CD16+) entering pulmonary circulation, eventually leading to pulmonary inflammation and injury. 201 The higher proportion of IL‐6‐expressing SARS‐CoV‐2‐specific CD8+ T cells were also detected in the non‐survivor than in the survivor of severe COVID‐19. 156 Similarly, another study found IL‐6 and GM‐CSF were associated with COVID‐19 severity and accompanied by elevated markers of endothelial injury and thrombosis. 202 Of interest, a subset of clonally expanded, GM‐CSF expressing tissue‐resident memory‐like Th17 (TRM17) cells have been identified in the lungs of patients with severe COVID‐19 that persist even after viral clearance. 203 These GM‐CSF expressing TRM17 cells together with IL‐1β‐expressing proinflammatory macrophage and cytotoxic CD8+ T cells forming a pathogenic milieu in the lung could promote inflammatory tissue injury. In general, these studies suggest that pathogenic T cells may contribute to the production of IL‐6 and GM‐CSF in patients with severe COVID‐19.
Tregs are a subset of CD4+ T cells that have been critically involved in the regulation of immune responses to maintain immune homeostasis. In humans and mice, during respiratory virus infection or acute lung injury, Tregs could migrate into the inflamed lung to suppress inflammatory responses, ameliorate viral pneumonia and promote lung tissue repair. 204 , 205 , 206 Hence, Tregs are likely protective in COVID‐19 patients with cytokine storm. To date, the changes in Treg cell frequency and cell number in the blood of COVID‐19 patients remain controversial, as many studies have shown decreased proportions of naïve Tregs and a shift towards effector Tregs, especially in those with severe disease, while others observed increased or unchanged Treg frequency. 207 , 208 The limited study reported the increased proportion of Tregs in the BAL fluid of COVID‐19 patients. 209 The expansion of effector Tregs may be attributable to the establishment of a dysfunctional lung immune environment and the pathogenesis of COVID‐19. Interestingly, a study reported that the frequency of Tregs and the expression level of FoxP3 were increased in severe COVID‐19 patients and were correlated with poor outcomes. 210 These Tregs have distinctive transcriptional signatures with high levels of effectors and proinflammatory molecules and share many similarities with tumor‐infiltrating Tregs that are generally associated with poor prognosis, suggesting such Tregs may suppress antiviral T cell responses in the acute phase while promoting inflammatory responses. The authors also noted that IL‐6 and IL‐18 potentially contributed to the upregulation of FoxP3 and the unique transcriptional signatures of these Tregs, respectively. 210 Nevertheless, activated Tregs with high suppressive activity in the early phase of the disease are presumably beneficial for the immune system to avoid tissue damage by activated immune cells. In contrast, the lower number of naïve Tregs in combination with higher active Tregs in severe cases or later stages of the disease may exacerbate the cytokine storm that leads to ARDS.
Chemokine receptors are important in the control of T cell migration to several tissues in disease states or after infections, most notably to the lungs. 211 , 212 , 213 CCR6, CXCR3, and CXCR6 are found to be upregulated in CD4+ and CD8+ T cells in PBMCs and BAL fluid of patients with COVID‐19. 9 , 214 , 215 Early polyfunctional CXCR3+CD8+ T cells infiltration of the lungs have a potential role in disease control. 215 , 216 However, a study reported that CXCR3 and CCR6 are highly expressed in activated CD16+ CD4+ and CD8+ T cells in severe COVID‐19. 196 The SARS‐CoV‐2 infection triggers complement activation, which creates an inflammatory environment that drives the differentiation of CD16+, highly cytotoxic CD4+ and CD8+ T cells. Expression of CXCR3 and CCR6 may facilitate the migration of these activated CD16+ T cells into the lungs, leading to endothelial cell damage and release of chemokines CXCL8 and CCL2. 196 CXCR6 is important for the migration of CD8+ TRM cells to the airways in response to respiratory virus infection. 213 , 217 Recent studies showed PD1+CXCR6+CD8+ T cells were accumulated in patients with nonalcoholic steatohepatitis (NASH) and in the liver of NASH mice and mediated the immune pathology in NASH through “autoaggressive” activation, 218 , 219 suggesting that CXCR6 might play a pathogenic role in T cell homing to inflamed tissues in diseases. Genome‐wide association studies (GWAS) indicated that CXCR6 is associated with COVID‐19 severity. 220 , 221 Bost et al. 222 showed that CXCR6 was only detected in the BAL TRM (resident memory) and TEM (effector memory), suggesting a protective effect of CXCR6+ T cells. Another study demonstrated that circulating CXCR6+CD8+ T cells were significantly reduced in both mild and severe COVID‐19 patients compared to controls, but significantly increased in individuals aged over 65. 223 In aged individuals, those CXCR6+ T cells may drive lung damage, resulting in severe symptoms and poor outcomes. Together, the effector functions of chemokine receptor‐expressing T cells may be beneficial in early antiviral immunity; however, the prolonged activated effect of these T cells may contribute to the persistent respiratory viral symptoms and fibrosis during or after the resolution of acute SARS‐CoV‐2 infection.
6.2. Humoral responses associated with COVID‐19 severity
Severe COVID‐19 distinctly altered the B cell compartment of adaptive immunity. The absence of a germinal center was reported in the spleen and lymph nodes of COVID‐19 patients, probably due to the failure of differentiation of BCL6+ Tfh as well as the aberrant local accumulation of TNF in lymphoid organs. 224 This might partially explain the low levels of somatic hypermutation among B cells seen in some cases of COVID‐19. It also might skew humoral response toward an extrafollicular B cell response. Indeed, one study reported critically ill COVID‐19 patients displayed hallmarks of extrafollicular B cell responses and high nAb titers, similar to those in human systemic lupus erythematosus. Besides, highly prevalent immunoglobulin G (IgG) responses against nonstructural/accessory proteins were observed in COVID‐19 patients and were positively associated with disease severity and worse clinical outcomes. 225 , 226 Taken together, these findings suggest that excessive humoral responses contribute to disease exacerbation.
Antigen‐specific antibodies can form an immune complex with viral particles or viral antigens and induce a hyperinflammatory response via activating FcγRs on myeloid cells. It has been known that human IgG antibodies can worsen pathology by triggering proinflammatory cytokine release. 227 Several studies have revealed aberrant glycosylation, afucosylation, in the Fc tail of anti‐spike (S) IgG in severely ill COVID‐19 patients but not mild patients. 131 , 228 , 229 , 230 , 231 This change increases IgG binding affinity to FcγRs, particularly FcγRIIa and FcγRIIIa. Specifically, the aberrant glycosylation of anti‐S IgG significantly amplified the production of proinflammation cytokines (e.g., IL‐6 and TNF) by AMs or monocytes, resulting in cytokine storms in these patients. 131 Furthermore, the formation of the immune complex between SARS‐CoV‐2 and anti‐S IgG stimulates platelet FcγRIIa and further activates downstream signals to promote platelet activation and thrombus formation. 229 Overall, these studies demonstrate the formation of immune complexes containing aberrant glycosylated IgG bound to activate FcγR could induce excessive inflammatory responses that lead to lung damage in critically ill COVID‐19 patients. More studies are needed to address the detailed mechanisms behind this phenomenon.
Complement activation seems to contribute to the pathophysiology of severe COVID‐19, the deposition of complement components (C1q, C3, C5a, and sC5b‐9) was found in the lung, brain, kidneys, and other organs of severe COVID‐19 patients. 232 , 233 , 234 , 235 , 236 It has been shown that virus‐specific IgG and IgM antibodies could activate the classical pathway, 234 providing evidence that antigen‐antibody immune complex may play a role in complement‐mediated pathogenesis in advanced COVID‐19. However, the role of these antibodies in activating complement and progressing disease has not been fully defined.
6.3. Autoantibody production in COVID‐19
Several studies have described the prevalence of autoantibodies (auto‐Abs) in COVID‐19 patients, particularly those that neutralize type I IFNs, including IFN‐α2 and IFN‐ω, found in about 10% of patients and are associated with critical COVID‐19 pneumonia. 67 , 237 , 238 , 239 , 240 These auto‐Abs were not found in asymptomatic or mild patients and only 0.33% of healthy individuals before the pandemic and in a few patients tested before SARS‐CoV‐2 infection contain detectable auto‐Abs. 67 Notably, one study measured auto‐Abs neutralizing lower, more physiological, the concentration of IFN‐α and/or IFN‐ω (100 pg/ml) in COVID‐19 patients across different disease severity and ages and found auto‐Abs in 6.5% and 13.6% of patients with severe and critical COVID‐19, respectively, and in 18% of deceased patients. 237 Such auto‐Abs were more prevalent in critical patients older than 65 and were greater in men than women. 237 More interestingly, testing a larger cohort of individuals aged 20–100 years from the general population showed a sharp increase of auto‐Abs against IFN‐α and/or IFN‐ω after the age of 70 years. 237 These auto‐Abs might contribute to the higher risk of critical COVID‐19 in the elderly.
Of importance, IFN auto‐Abs were also detected in the upper respiratory tract (nasopharyngeal swabs) and lower respiratory tract (BAL fluid) of COVID‐19 patients and revealed that the IFN auto‐Abs in the nasopharyngeal swabs were linked with poor IFN‐stimulated responses among the nasal epithelial cells in severe COVID‐19 individuals, 241 , 242 , 243 allowing higher or persistent viral replication in the respiratory tract and potentiating excessive respiratory inflammation that could drive severe pneumonia. Indeed, the IFN auto‐Abs were shown to block the antiviral activity of IFN‐α against SARS‐CoV‐2 infection in vitro 67 and in vivo, 238 providing a potential explanation for weaker antiviral immunity in some severe patients in the acute phase. However, if such auto‐Abs are still present in patients with long COVID, particularly in their airways, the potential pathogenic roles of these auto‐Abs need to be investigated.
7. IMMUNOMODULATORY DRUGS FOR ACUTE COVID‐19
In the fight against COVID‐19, currently, antiviral drugs and vaccines are viable options. However, the rise in several variants of concerns has lowered the efficacy of most vaccines and antiviral drugs are usually not effective in severe COVID‐19 patients. A plethora of evidence, both from preclinical and clinical studies, have demonstrated the beneficial effect of immunomodulatory drugs such as corticosteroids, metformin, recombinant IFNs and GM‐CSF, IL‐6, and TNF‐alpha targeting monoclonal antibody (mAb) in treating COVID‐19 (Figure 3). Here, in this section, we have discussed some of the most used immunomodulatory drugs for COVID‐19 and their mode of action.
FIGURE 3.
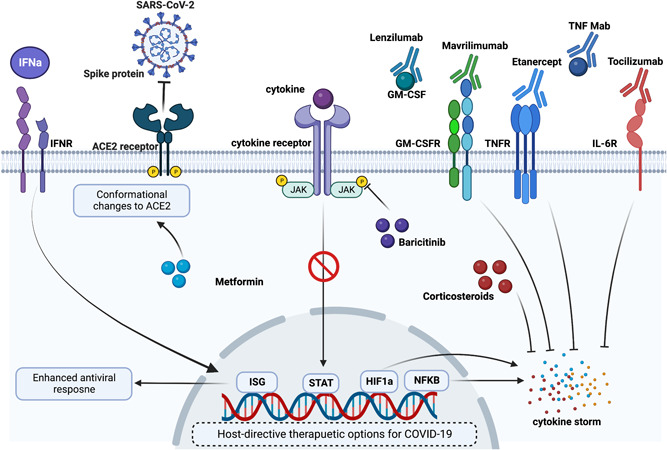
Potential immunomodulatory drugs for acute COVID‐19: Acute COVID‐19 is accompanied by hyperinflammatory responses and hence use of immunomodulatory in several clinical and preclinical settings has shown therapeutic benefits. Immunomodulatory drugs, such as metformin, corticosteroids, and baricitinib have shown reduced inflammation following SARS‐CoV‐2 infection. Additionally, monoclonal antibodies (mAbs) such as lenzilumab, mavrilimumab, etanercept, tocilizumab, and TNF mAb have been studied in various clinical trials for their beneficial role in dampening COVID‐19‐induced inflammation. As early ISG expression is required for effective viral clearance, treatment with recombinant IFNs has also been proposed to mitigate viral load. ACE2, angiotensin‐converting enzyme 2; COVID‐19, coronavirus disease 2019; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; GM‐CSFR, GM‐CSF receptor; HIF‐1a, hypoxia‐inducible factor 1a; IFN, interferon; IFNR, IFN receptor; IL‐6R, interleukin 6R; ISG, interferon‐stimulated gene;NF‐κB, nuclear factor‐κB; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; STAT, signal transducer and activator of transcription; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor.
7.1. Corticosteroids
Methylprednisolone is a frequently recommended corticosteroid to COVID‐19 patients to dampen inflammatory response due to the presence of increased proinflammatory cytokines (IL‐2, TNF‐α, IL‐1β, IFN‐γ, and IL‐6) and chemokines (CCL2 and MIP‐1α). 105 According to some meta‐analysis studies, methylprednisolone treatment has shown reduced mortality in severe patients. 244 , 245 However, some of the clinical findings indicate that the use of methylprednisolone therapy has resulted in delayed viral clearance and prolonged hospitalization, 246 , 247 , 248 further discouraging its use outside the clinical trials.
Dexamethasone is another immunosuppressive corticosteroid that was previously shown to improve mortality in COVID‐19 patients. 249 In severe COVID‐19 patients, dexamethasone administration has been beneficial in improving clinical parameters of lung epithelial and endothelial injury without affecting the viral load. 250 Mechanistically, dexamethasone can suppress IFN‐activated neutrophils and limit neutrophil‐induced immunopathology. 251 Nevertheless, cautious administration of dexamethasone has been recommended, particularly in the early phase of infection, due to its several side effects and possible suppression of antiviral immune responses. 252 In a recently published large multicenter cohort study, severe COVID‐19 patients under dexamethasone treatment are found to develop more risk of ICU‐acquired respiratory tract infection. 253 However, these clinical trials skip the use of antiviral, remdesivir, which further leads to the notion that dexamethasone administration along with antiviral therapy may prove clinically useful. 254
7.2. Metformin
SARS‐CoV‐2 infection is also known to alter the metabolic profile of infected monocytes, 119 which is mediated by its spike protein, 255 leading to HIF‐1α‐dependent enhanced inflammation. 119 , 255 Metformin is an antidiabetic drug that has been suggested as a repurposed drug for COVID‐19 due to its anti‐inflammatory property. 256 In addition to its anti‐inflammatory property, metformin is known to phosphorylate the entry receptor for SARS‐CoV‐2, the ACE2, suggesting its possible role in blocking the entry of SARS‐CoV‐2. 257 Metformin injection in SARS‐CoV‐2 infected hACE2 transgenic mice improved the morbidity and rescued the mice from ARDS. 258 In an in vitro setting, it was also demonstrated that metformin results in the rescue of monocytes from inflammation. 255
However, in clinical trials, metformin showed uncertainty. In a retrospective cohort analysis, the use of metformin was not associated with a reduced risk of mortality in total samples of both men and women from COVID‐19. Of note, in the case of women, there was a reduced risk of mortality, indicating the sex‐dependent effect of metformin. 259 In a recent randomized clinical trial, the effect of early treatment with metformin was assessed for high‐risk patients with early COVID‐19, and metformin treatment failed to improve the primary endpoints including hypoxemia, emergency department visit, hospitalization, or death. 260 Likewise, metformin was not able to provide clinical benefits even given early. 261 These clinical observations failed to indicate any beneficial role of metformin. Nevertheless, in clinical trials involving COVID‐19 patients with type 2 diabetes, there appeared a reduced risk of mortality associated with the metformin treatment. 262 , 263 Altogether, more randomized clinical trials are required to further confirm these claims.
7.3. Baricitinib
Baricitinib is a selective inhibitor of Janus kinase 1 and 2 with known anti‐inflammatory properties. 264 Baricitinib treatment in rhesus monkeys rescued the inflammatory phenotype of macrophages isolated from BAL, in particular, IL‐6 and TNF expression. 265 However, baricitinib was able to suppress SARS‐CoV‐2‐induced pathology of the lung but it did not limit SARS‐CoV‐2 infection in the rhesus monkey. In addition to dampening the inflammatory properties of macrophages, the baricitinib treatment abolished the degranulation of neutrophils and NET formation. 265 In humans, baricitinib administration increased virus‐specific IgG and lowered the serum levels of IL‐6, IL‐1β, and TNF‐α. Furthermore, the treated patients further needed no oxygen support as a result of the improved oxygenation index. 266 Along with antiviral drug remdesivir, baricitinib treatment may help to accelerate the recovery of COVID‐19 patients. 267
7.4. Tocilizumab
Tocilizumab is a mAb that can bind to the membrane‐bound or soluble IL‐6 receptor. 268 Excessive systemic inflammation because of inflammatory cytokines including IL‐6 levels was associated with adverse clinical outcomes in patients hospitalized with COVID‐19. 269 Hence, for achieving therapeutic benefits, the use of several IL‐6 antagonists was studied in several randomized clinical trials. 270 In a randomized clinical trial, tocilizumab was not associated with improved clinical outcomes in severe COVID‐19 patients. 271 However, with oxygen support, the COVID‐19 patients on tocilizumab therapy showed improved mortality. 272 Intriguingly, in a different study tocilizumab treatment at the early inflammatory stage at moderate dosage resulted in improved mortality of severe COVID‐19 patients. 273 , 274 These contradictory reports may prompt clinicians to critically assess the timing and dose of tocilizumab for improved benefits.
7.5. TNF inhibitor
The concept of blocking TNF as a potential therapy stems from observation clinical studies that show that severe patients have increased TNF in serum and BAL fluid. 9 , 200 , 275 TNF inhibitors that are mostly used in clinical trials are anti‐TNF antibodies (such as infliximab, adalimumab, and golimumab) etanercept (TNF‐R2 Ig‐Fc fusion protein), and certolizumab pegol (monovalent fab fragment of a humanized mAb without Fc region). 276 In a large cohort of more than 6000 COVID‐19 patients, anti‐TNF monotherapy proved to be associated with a lower risk of COVID‐19‐induced pathology. 277 Similarly, a meta‐analysis of 34 studies also advocates the beneficial role of anti‐TNF therapy in lowering the hospitalization rate due to COVID‐19 severity. 278
7.6. IFN treatment
Following SARS‐CoV‐2 infection, there was a reduction in type I and type III IFN response. 279 As robust IFN response is required for antiviral defenses, recombinant IFNs such as IFN‐α, IFN‐β, and IFN‐λ are currently being investigated as a potential therapy in several clinical trials (clinical trial identifier number NCT04276688, NCT04343976, NCT04354259, NCT04388709, and NCT04344600). A recent report involved 446 patients tested for IFN‐α treatment, both during early and late infection. Early treatment with recombinant IFN via aerosol resulted in decreased mortality, whereas late treatment increased mortality. 280 Hence, these studies must proceed with caution due to heterogeneity in IFN response among COVID‐19 samples, 134 and timing of the IFN treatment. 76 , 281
7.7. GM‐CSF mAb
Pathogenic T cells may contribute to the production of GM‐CSF in patients with severe COVID‐19, suggesting GM‐CSF blockade as a therapeutic target in COVID‐19. Human mAbs targeting GM‐CSF, such as otilimab, gimsilumab, lenzilumab, and namilumab, or GM‐CSF receptors, such as mavrilimumab, have been assessed in several clinical trials. 282 A meta‐analysis of GM‐CSF mAbs therapy for COVID‐19 patients was performed with six eligible studies involving 1501 patients. The analysis revealed that the GM‐CSF mAbs therapy was associated with reduced mortality (3.8%–26.9%), a decreased incidence of invasive mechanical ventilation (5.3%–28.7%), and improved ventilation (23.3%–50.0%) in severe COVID‐19 patients. They also found there was no increased incidence of secondary infection in COVID‐19 patients between GM‐CSF mAbs group and control, whereas similar immunomodulatory strategy IL‐6 receptor mAbs therapy showed increased secondary infection. 283 , 284 Given the crucial role of GM‐CSF in AM homeostasis and lung viral clearance, 285 recombinant GM‐CSF administration may be more beneficial in earlier‐stage COVID‐19, whereas GM‐CSF mAbs therapy could be beneficial for more severe COVID‐19 patients. Overall, the safety and efficacy of GM‐CSF blockade in the treatment of COVID‐19 patients are still controversial, and more random clinical trials are required to evaluate these therapeutics in COVID‐19.
8. IMMUNOPATHOLOGY IN LONG COVID
Apart from the acute manifestations of disease during COVID‐19 illness, increasing evidence points to the development of chronic pulmonary and extrapulmonary sequelae termed the postacute sequelae of SARS‐CoV‐2 infection (PASC) or long COVID following the resolution of primary SARS‐CoV‐2 infection. 286 Specifically, PASC is defined by the persistence of disease greater than 28 days following the onset of symptoms, a phenomenon observed in 27%–80% of convalescent individuals. 287 Symptoms range from brain fog, general fatigue, dyspnea, and joint pain to multiorgan impairments (Figure 4). 45 Patients often exhibit diminished lung function and exercise capacity in addition to several radiological anomalies including ground‐glass opacities, atelectasis, and reticulation, with evidence of persistent inflammation and fibrotic‐like changes. 286 , 288 Although the pathophysiology of pulmonary abnormalities has been most widely studied thus far, extrapulmonary manifestations including thrombotic complications, myocardial injury, and neuropsychiatric symptoms have also been frequently observed. 45 , 286 , 287 Despite ongoing efforts, however, PASC the etiology of chronic sequelae following acute COVD‐19 remains poorly understood. Long‐term persistence of SARS‐CoV‐2 viral remnants has been observed in numerous sites including the lungs, brain, kidneys, and the gut suggesting possibly instigating aberrant immune responses and pathology. 289 In support of this notion, longitudinal studies have revealed sustained dysregulation of immune responses in PASC ‐ highly activated myeloid cells, T‐cells, elevated proinflammatory cytokine levels, and a reduction in naïve T‐ and B‐cells. 290 , 291 , 292 , 293 Moreover, sustained reduction of circulating cortisol, an immunosuppressive factor, has been reported in independent PASC cohorts. 294 , 295 Postviral pulmonary sequelae are not unique to SARS‐CoV‐2 and have been reported following several other respiratory viral infections, potentially driven by the immune system as well. 296 For the remainder of this review, we specifically focus on various immune mediators implicated in the development of PASC and highlight potential therapeutic avenues to mitigate chronic disease.
FIGURE 4.
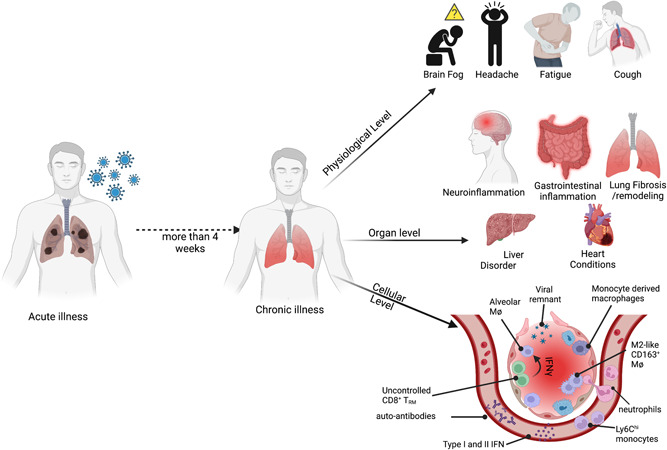
Immunopathology in long COVID: Immunopathology in long COVID is studied at different biological levels. At the physiological levels, individuals recovering from acute SARS‐CoV‐2 infection have been complaining about brain fog, headache, fatigue, cough, and so forth, for a prolonged period. At the organ levels, in infected patients, there have been reports of long‐term neuro‐and gastroinflammation. In some individuals, there have been incidences of liver and heart conditions. However, the cellular insight into this chronic illness remains poorly understood. Some of the recent reports have suggested the presence of viral remnants, prolonged systemic or tissue inflammatory responses, and/or the presence of autoantibodies may contribute to the disease etiology. COVID‐19, coronavirus disease 2019; IFN, interferon; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2.
8.1. Innate cells
The accumulation of a monocyte‐derived CD163+ macrophage pool was observed during severe COVID‐19. 29 The cells exhibited an M2‐like phenotype, which although crucial for the resolution of inflammation and wound repair, may also promote fibrotic changes within the microenvironment. Moreover, these cells were observed in close association with pockets of collagen deposition and exhibited a profibrotic transcriptional phenotype with remarkable similarity to macrophage populations in IPF patients. 29 Notably, the degree of radiological abnormalities correlated with myeloid cell numbers within the BAL fluid. 297 PASC patients also exhibit persistent elevation of chemokines known to recruit monocytes, such as CCL‐2, further suggesting a detrimental role of these cells in long‐term pathology. 293 While rapid induction of IFNs following infection is typically associated with improved viral clearance and outcomes, long‐term studies have identified sustained elevation of type I and type III IFNs up to 8 months postinfection in patients. 290 This chronic activation of IFNs and downstream pathways has been shown to adversely affect epithelial repair following injury. 73 , 139 , 290 However, the exact roles of types I–III IFNs during PASC remain unclear and warrant further investigation. The maintenance of a chronic proinflammatory state is also known to prevent the differentiation of alveolar type II cells to alveolar type I cells during repair, promoting their accumulation and impaired regeneration. 298 In support of this, an accumulation of keratin 8+ transitional cells has been observed in lethal COVID, similar to IPF, potentially driven by monocyte/macrophage‐derived IL‐1β. 298 Chronic elevation of circulating IL‐1β, along with IL‐6 and TNF in PASC patients indicates a self‐sustaining feed‐forward loop, likely contributing to the establishment of a proinflammatory environment. 299 Furthermore, PASC patients with persistent interstitial lung changes maintain an immune signature associated with sustained neutrophilic inflammation, indicating a potential role for neutrophils in driving chronic sequelae. 300 , 301 In addition to pulmonary sequelae, myeloid cells have been found to contribute to cognitive impairments associated with PASC—typically referred to as “brain fog.” Microglia undergo significant perturbations during acute COVID‐19, exhibiting enhanced reactivity which has previously been linked to loss of oligodendrocytes and myelinated axons. 302
8.2. Adaptive cells
During acute infection, AMs were found to recruit CD8+ T‐cells, which in turn secrete IFNγ establishing a positive feedback loop between macrophage activation and T‐cell recruitment. 128 Additional chemokines such as CXCL‐9, CXCL‐10, and CXCL‐11 were also found to remain elevated in PASC patients in the absence of active infection, likely recruiting and maintaining several adaptive immune populations. 297 Further indicating a lack of resolution of inflammation, PASC patients were found to harbor CD8+ TRM cells in the airways at least 90 days following acute disease. 151 , 290 The persistence of CD8+ TRM cells in the BAL fluid was associated with increased epithelial damage, and the CD69+CD103‐ subset, in particular, negatively correlated with lung function in convalescents. 151 , 303 Notably, the cells were enriched for TCR signaling pathway genes suggesting antigen‐mediated stimulation. 291 However, the nature of the antigen – whether residual viral remnants or self‐antigen is unknown and will likely be answered by comprehensive profiling of chronically activated T‐cell subsets following infection. Alternatively, antigen‐independent mechanisms may be at play, as an auto‐aggressive CXCR6+ TRM subset previously described in the liver was also identified in the airways of COVID‐19 convalescents. 291 While the origins of pathological CD8+ TRM subsets are unknown, early COVID‐19 studies identified a deleterious CD8+ T‐cell subset (CXCR4+), which may potentially seed the CD69+CD103‐ TRM population within the lung. 156 , 218 CD4+ TRM cells were also persistently enriched within the airways of PASC patients, potentially orchestrating fibrotic responses and negatively influencing lung repair. 297 In addition to the exuberant activity of the immune system, inhibition of regulatory activities may also contribute to chronic disease. Notch4 expression on TREG cells was found to correlate with disease severity, limiting resolution of inflammation and amphiregulin‐dependent tissue repair. 304
Apart from T‐cell mediated pathology, B‐cells and antibodies induced during acute infection may contribute to the development of chronic sequelae. B‐cell numbers correlated with the incidence of radiological abnormalities and impaired gas exchange. 297 Although direct mechanisms are still under investigation, auto‐Abs have been hypothesized to drive long‐term symptoms, with IFNα‐2 auto‐Abs uniquely correlating with pulmonary sequelae. 238 , 294 However, a recent report profiling auto‐Abs against extracellular and secreted proteins in PASC patients failed to show an association with symptoms. 295 Other immunoglobulin signatures during acute infection have also been found to predict the development of PASC, which may instead reflect the inflammatory milieu responsible for poor control of viral replication. 305 However, further studies are required to elucidate the underlying mechanisms.
8.3. Potential therapy for long COVID‐19
Several studies have identified severity and damage accrued during acute infection to be the strongest predictor of chronic pulmonary sequelae in the context of COVID‐19. 306 , 307 , 308 Furthermore, host factors such as advanced age and comorbidities including metabolic syndrome, cardiovascular disease, immunosuppression, and so forth, have been associated with long‐term adverse outcomes. 309 However, it is unclear if this association is due to the increased risk of severe disease or a direct predisposition toward the development of chronic sequelae. Nevertheless, dysregulated immune responses are a common theme in both scenarios which contribute significantly to postviral lung disease, as exemplified over the course of the COVID‐19 pandemic. 290 , 297 Thus, the aforementioned drugs targeting the immune system as well as vaccines are likely effective in attenuating postviral disease. In addition, cellular and molecular mediators relevant to PASC may also be targeted however further clinical studies are required to determine their efficacy. The exuberant activity of myeloid cells in the aftermath of the acute disease may be dampened by blocking chemokines such as CCL‐2, CXCL‐17, and so forth, and their receptors to prevent continuous recruitments of monocytes and neutrophils. Similar strategies may be utilized to inhibit CXCR3‐mediated recruitment and maintenance of adaptive cells such as CD8+ T‐cells, CD4+ T‐cells, and B‐cells in the lung. Moreover, specific pathologic subsets of CD8+ TRM responsible for impaired pulmonary function and adverse outcomes can be targeted for ablation. Alternatively, cytokines and immunological mediators such as IL‐22 and amphiregulin, known to contribute to epithelial repair may be administered to augment the epithelial repair. Recent studies have also shown potential for the use of IPF drugs in the dampening of fibrotic disease following COVID‐19, however, further investigations are required to characterize their efficacy in PASC. 310
9. CONCLUDING REMARKS
In wake of the current COVID‐19 pandemic, there is an unprecedented growth in the development of vaccines and therapeutics to halt the damage caused by SARS‐CoV‐2. However, to first delineate the course of a successful vaccine or therapeutic design, we must first investigate the pathophysiology caused by SARS‐CoV‐2. By taking advantage of current advanced tools and techniques we are now able to answer any questions that remained unanswered until recently regarding the immunopathology caused by SARS‐CoV‐2. Having a detailed bird's eye view of the pathogenic immune responses in acute and chronic pathophysiology following SARS‐CoV‐2 infection, it is anticipated that critical information may be unearthed, which will be beneficial for designing novel therapeutics.
Apart from vaccines, several immune‐based therapies have shown some success in mitigating the current pandemic. However, due to complexity such as variable immune response to therapies, and heterogeneity in host and virus, there have been lots of challenges in adopting immunomodulatory drugs. Additionally, while acute COVID‐19 is still a threat, complications rising due to PASC are recently making headlines in the scientific community. Nevertheless, these challenges could likely be answered through precision or evidence‐based immune therapies in the future. Some of the highlights of precision therapy are that the therapy is largely influenced by the patient's immune condition, disease course, and use of cointervention apart from drugs. 311 , 312 Therefore, immune profiling and lung function examination before immunotherapy may provide critical insight into a patient's condition. Incorporation of these approaches may help increase the efficacy of current immunomodulatory drugs and could help to curb the current pandemic.
AUTHOR CONTRIBUTIONS
Mohd Arish, Wei Qian, and Harish Narasimhan wrote the original manuscript. Jie Sun edited the manuscript.
CONFLICT OF INTEREST
Jie Sun is a consultant for TeneoFour company.
ACKNOWLEDGMENTS
This study was supported in part by the US National Institutes of Health Grants AI147394, AG069264, AI 112844, and AI 154598 to Jie Sun.
Arish M, Qian W, Narasimhan H, Sun J. COVID‐19 immunopathology: from acute diseases to chronic sequelae. J Med Virol 2022;1‐24. 10.1002/jmv.28122
Mohd Arish, Wei Qian, and Harish Narasimhan contributed equally to this study.
DATA AVAILABILITY STATEMENT
All data reviewed in this manuscript are from published papers.
REFERENCES
- 1. COVID‐19 dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Accessed September 7, 2022. https://coronavirus.jhu.edu/map.html
- 2. Harvey WT, Carabelli AM, Jackson B, et al. SARS‐CoV‐2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lopes‐Pacheco M, Silva PL, Cruz FF, et al. Pathogenesis of multiple organ injury in COVID‐19 and potential therapeutic strategies. Front Physiol. 2021;12:593223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang L, Liu S, Liu J, et al. COVID‐19: immunopathogenesis and immunotherapeutics. Signal Transduct Target Ther. 2020;5(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang W, Berube J, McNamara M, et al. Lymphocyte subset counts in COVID‐19 patients: a meta‐analysis. Cytometry A. 2020;97(8):772‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lagunas‐Rangel FA. Neutrophil‐to‐lymphocyte ratio and lymphocyte‐to‐C‐reactive protein ratio in patients with severe coronavirus disease 2019 (COVID‐19): a meta‐analysis. J Med Virol. 2020;92(10):1733‐1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Melms JC, Biermann J, Huang H, et al. A molecular single‐cell lung atlas of lethal COVID‐19. Nature. 2021;595(7865):114‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajamanickam A, Kumar NP, Pandiarajan AN, et al. Dynamic alterations in monocyte numbers, subset frequencies and activation markers in acute and convalescent COVID‐19 individuals. Sci Rep. 2021;11(1):20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liao M, Liu Y, Yuan J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26(6):842‐844. [DOI] [PubMed] [Google Scholar]
- 10. Chen LD, Zhang ZY, Wei XJ, et al. Association between cytokine profiles and lung injury in COVID‐19 pneumonia. Respir Res. 2020;21(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Costela‐Ruiz VJ, Illescas‐Montes R, Puerta‐Puerta JM, Ruiz C, Melguizo‐Rodríguez L. SARS‐CoV‐2 infection: the role of cytokines in COVID‐19 disease. Cytokine Growth Factor Rev. 2020;54:62‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu G, Qi F, Li H, et al. The differential immune responses to COVID‐19 in peripheral and lung revealed by single‐cell RNA sequencing. Cell Discov. 2020;6:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Batah SS, Fabro AT. Pulmonary pathology of ARDS in COVID‐19: a pathological review for clinicians. Respir Med. 2021;176:106239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dong M, Zhang J, Ma X, et al. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID‐19. Biomed Pharmacother. 2020;131:110678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ning Q, Wu D, Wang X, et al. The mechanism underlying extrapulmonary complications of the coronavirus disease 2019 and its therapeutic implication. Signal Transduct Target Ther. 2022;7(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kurihara C, Manerikar A, Querrey M, et al. Clinical characteristics and outcomes of patients with COVID‐19‐associated acute respiratory distress syndrome who underwent lung transplant. JAMA. 2022;327(7):652‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Ma X. Acute respiratory failure in COVID‐19: is it “typical” ARDS? Crit Care. 2020;24(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schaller T, Hirschbühl K, Burkhardt K, et al. Postmortem examination of patients with COVID‐19. JAMA. 2020;323(24):2518‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Q, Shi Y, Cai J, et al. Pathological changes in the lungs and lymphatic organs of 12 COVID‐19 autopsy cases. Natl Sci Rev. 2020;7(12):1868‐1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valdebenito S, Bessis S, Annane D, et al. COVID‐19 lung pathogenesis in SARS‐CoV‐2 autopsy cases. Front Immunol. 2021;12:735922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chait M, Yilmaz MM, Shakil S, et al. Immune and epithelial determinants of age‐related risk and alveolar injury in fatal COVID‐19. JCI Insight. 2022;7:e157608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loo J, Spittle DA, Newnham M. COVID‐19, immunothrombosis and venous thromboembolism: biological mechanisms. Thorax. 2021;76(4):412‐420. [DOI] [PubMed] [Google Scholar]
- 25. Zhang B, Zhou X, Qiu Y, et al. Clinical characteristics of 82 cases of death from COVID‐19. PLoS One. 2020;15(7):e0235458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sime PJ, O'Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99(3):308‐319. [DOI] [PubMed] [Google Scholar]
- 27. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID‐19: the potential role for antifibrotic therapy. Lancet Respir Med. 2020;8(8):807‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bharat A, Querrey M, Markov NS, et al. Lung transplantation for patients with severe COVID‐19. Sci Transl Med. 2020;12(574):eabe4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wendisch D, Dietrich O, Mari T, et al. SARS‐CoV‐2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. 2021;184(26):6243‐6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaz de Paula CB, Nagashima S, Liberalesso V, et al. COVID‐19: immunohistochemical analysis of TGF‐β signaling pathways in pulmonary fibrosis. Int J Mol Sci. 2021;23(1):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Naqvi SF, Lakhani DA, Sohail AH, et al. Patients with idiopathic pulmonary fibrosis have poor clinical outcomes with COVID‐19 disease: a propensity matched multicentre research network analysis. BMJ Open Respir Res. 2021;8(1):e000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Esposito AJ, Menon AA, Ghosh AJ, et al. Increased odds of death for patients with interstitial lung disease and COVID‐19: a case‐control study. Am J Respir Crit Care Med. 2020;202(12):1710‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. El‐Qutob D, Alvarez‐Arroyo L, Barreda I, et al. High incidence of pulmonary thromboembolism in hospitalized SARS‐CoV‐2 infected patients despite thrombo‐prophylaxis. Heart Lung. 2022;53:77‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N Engl J Med. 2020;383(2):120‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iba T, Levy JH, Levi M, Connors JM, Thachil J. Coagulopathy of coronavirus disease 2019. Crit Care Med. 2020;48(9):1358‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID‐19: a prospective cohort study. Ann Intern Med. 2020;173(4):268‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanff TC, Mohareb AM, Giri J, Cohen JB, Chirinos JA. Thrombosis in COVID‐19. Am J Hematol. 2020;95(12):1578‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID‐19 infection: a report of five cases. Transl Res. 2020;220:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang S, Liu Y, Wang X, et al. SARS‐CoV‐2 binds platelet ACE2 to enhance thrombosis in COVID‐19. J Hematol Oncol. 2020;13(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaur S, Singh A, Kaur J, et al. Upregulation of cytokine signalling in platelets increases risk of thrombophilia in severe COVID‐19 patients. Blood Cells Mol Dis. 2022;94:102653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ji W, Chen L, Yang W, et al. Transcriptional landscape of circulating platelets from patients with COVID‐19 reveals key subnetworks and regulators underlying SARS‐CoV‐2 infection: implications for immunothrombosis. Cell Biosci. 2022;12(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID‐19 acute respiratory distress syndrome. Blood. 2020;136(10):1169‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang L, Feng X, Zhang D, et al. Deep vein thrombosis in hospitalized patients with COVID‐19 in Wuhan, China: prevalence, risk factors, and outcome. Circulation. 2020;142(2):114‐128. [DOI] [PubMed] [Google Scholar]
- 44. Behzad S, Aghaghazvini L, Radmard AR, Gholamrezanezhad A. Extrapulmonary manifestations of COVID‐19: radiologic and clinical overview. Clin Imaging. 2020;66:35‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gupta A, Madhavan MV, Sehgal K, et al. Extrapulmonary manifestations of COVID‐19. Nat Med. 2020;26(7):1017‐1032. [DOI] [PubMed] [Google Scholar]
- 46. Finsterer J, Scorza FA, Scorza CA, Fiorini AC. Extrapulmonary onset manifestations of COVID‐19. Clinics. 2021;76:e2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elrobaa IH, New KJ. COVID‐19: pulmonary and extra pulmonary manifestations. Front Public Health. 2021;9:711616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Clerkin KJ, Fried JA, Raikhelkar J, et al. COVID‐19 and cardiovascular disease. Circulation. 2020;141(20):1648‐1655. [DOI] [PubMed] [Google Scholar]
- 49. Driggin E, Madhavan MV, Bikdeli B, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID‐19 pandemic. J Am Coll Cardiol. 2020;75(18):2352‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Doyen D, Moceri P, Ducreux D, Dellamonica J. Myocarditis in a patient with COVID‐19: a cause of raised troponin and ECG changes. Lancet. 2020;395(10235):1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hingorani KS, Bhadola S, Cervantes‐Arslanian AM. COVID‐19 and the brain. Trends Cardiovasc Med. 2022;32:323‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moriguchi T, Harii N, Goto J, et al. A first case of meningitis/encephalitis associated with SARS‐Coronavirus‐2. Int J Infect Dis. 2020;94:55‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Diamond MS, Kanneganti TD. Innate immunity: the first line of defense against SARS‐CoV‐2. Nat Immunol. 2022;23(2):165‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thorne LG, Reuschl AK, Zuliani‐Alvarez L, et al. SARS‐CoV‐2 sensing by RIG‐I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021;40(15):e107826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamada T, Sato S, Sotoyama Y, et al. RIG‐I triggers a signaling‐abortive anti‐SARS‐CoV‐2 defense in human lung cells. Nat Immunol. 2021;22(7):820‐828. [DOI] [PubMed] [Google Scholar]
- 58. Zheng M, Karki R, Williams EP, et al. TLR2 senses the SARS‐CoV‐2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Potapov I, Kanneganti TD, Del Sol A. Fostering experimental and computational synergy to modulate hyperinflammation. Trends Immunol. 2022;43(1):4‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Asano T, Boisson B, Onodi F, et al. X‐linked recessive TLR7 deficiency in ~1% of men under 60 years old with life‐threatening COVID‐19. Sci Immunol. 2021;6(62):eabl4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science. 2020;370(6515):eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Galani IE, Rovina N, Lampropoulou V, et al. Untuned antiviral immunity in COVID‐19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol. 2021;22(1):32‐40. [DOI] [PubMed] [Google Scholar]
- 63. Singh DK, Aladyeva E, Das S, et al. Myeloid cell interferon responses correlate with clearance of SARS‐CoV‐2. Nat Commun. 2022;13(1):679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369(6504):718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sodeifian F, Nikfarjam M, Kian N, Mohamed K, Rezaei N. The role of type I interferon in the treatment of COVID‐19. J Med Virol. 2022;94(1):63‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xia H, Cao Z, Xie X, et al. Evasion of type I interferon by SARS‐CoV‐2. Cell Rep. 2020;33(1):108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science. 2020;370(6515):eabd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Galani IE, Triantafyllia V, Eleminiadou EE, et al. Interferon‐λ mediates non‐redundant Front‐Line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46(5):875‐890. [DOI] [PubMed] [Google Scholar]
- 69. Fukuda Y, Homma T, Inoue H, et al. Downregulation of type III interferons in patients with severe COVID‐19. J Med Virol. 2021;93(7):4559‐4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9:2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Akamatsu MA, de Castro JT, Takano CY, Ho PL. Off balance: interferons in COVID‐19 lung infections. EBioMedicine. 2021;73:103642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gadotti AC, de Castro Deus M, Telles JP, et al. IFN‐γ is an independent risk factor associated with mortality in patients with moderate and severe COVID‐19 infection. Virus Res. 2020;289:198171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature. 2020;584(7821):463‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Major J, Crotta S, Llorian M, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee JS, Park S, Jeong HW, et al. Immunophenotyping of COVID‐19 and influenza highlights the role of type I interferons in development of severe COVID‐19. Sci Immunol. 2020;5(49):eabd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID‐19 patients. Cell Host Microbe. 2020;27(6):883‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sefik E, Qu R, Junqueira C, et al. Inflammasome activation in infected macrophages drives COVID‐19 pathology. Nature. 2022;606(7914):585‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. King C, Sprent J. Dual nature of type I interferons in SARS‐CoV‐2‐Induced inflammation. Trends Immunol. 2021;42(4):312‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Park A, Iwasaki A. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID‐19. Cell Host Microbe. 2020;27(6):870‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type I interferon and inflammatory monocyte‐macrophage responses cause lethal pneumonia in SARS‐CoV‐infected mice. Cell Host Microbe. 2016;19(2):181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wauters E, Van Mol P, Garg AD, et al. Discriminating mild from critical COVID‐19 by innate and adaptive immune single‐cell profiling of bronchoalveolar lavages. Cell Res. 2021;31(3):272‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cong B, Dong X, Yang Z, et al. Spatiotemporal landscape of SARS‐CoV‐2 pulmonary infection reveals Slamf9 + Spp1 + macrophages promoting viral clearance and inflammation resolution. Biorxiv . 2022.
- 83. Jiang Y, Wei X, Guan J, et al. COVID‐19 pneumonia: CD8+ T and NK cells are decreased in number but compensatory increased in cytotoxic potential. Clin Immunol. 2020;218:108516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Krämer B, Knoll R, Bonaguro L, et al. Early IFN‐α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID‐19. Immunity. 2021;54(11):2650‐2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bi J. NK cell dysfunction in patients with COVID‐19. Cell Mol Immunol. 2022;19(2):127‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Witkowski M, Tizian C, Ferreira‐Gomes M, et al. Untimely TGFβ responses in COVID‐19 limit antiviral functions of NK cells. Nature. 2021;600(7888):295‐301. [DOI] [PubMed] [Google Scholar]
- 87. Gomez‐Cadena A, Spehner L, Kroemer M, et al. Severe COVID‐19 patients exhibit an ILC2 NKG2D. Cell Mol Immunol. 2021;18(2):484‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Saichi M, Ladjemi MZ, Korniotis S, et al. Single‐cell RNA sequencing of blood antigen‐presenting cells in severe COVID‐19 reveals multi‐process defects in antiviral immunity. Nat Cell Biol. 2021;23(5):538‐551. [DOI] [PubMed] [Google Scholar]
- 89. Arunachalam PS, Wimmers F, Mok CKP, et al. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369(6508):1210‐1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roh JS, Sohn DH. Damage‐Associated molecular patterns in inflammatory diseases. Immune Netw. 2018;18(4):e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Naqvi I, Giroux N, Olson L, et al. DAMPs/PAMPs induce monocytic TLR activation and tolerance in COVID‐19 patients; nucleic acid binding scavengers can counteract such TLR agonists. Biomaterials. 2022;283:121393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Guo Q, Zhao Y, Li J, et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID‐19. Cell Host Microbe. 2021;29(2):222‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shi H, Zuo Y, Yalavarthi S, et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID‐19. J Leukoc Biol. 2021;109(1):67‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Silvin A, Chapuis N, Dunsmore G, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID‐19. Cell. 2020;182(6):1401‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Scozzi D, Cano M, Ma L, et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID‐19. JCI Insight. 2021;6(4):e143299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Burke H, Freeman A, Cellura DC, et al. Inflammatory phenotyping predicts clinical outcome in COVID‐19. Respir Res. 2020;21(1):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liang Y, Ge Y, Sun J. IL‐33 in COVID‐19: friend or foe? Cell Mol Immunol. 2021;18(6):1602‐1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Stanczak MA, Sanin DE, Apostolova P, et al. IL‐33 expression in response to SARS‐CoV‐2 correlates with seropositivity in COVID‐19 convalescent individuals. Nat Commun. 2021;12(1):2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chen L, Long X, Xu Q, et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID‐19 patients. Cell Mol Immunol. 2020;17(9):992‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sivakorn C, Dechsanga J, Jamjumrus L, et al. High mobility group box 1 and interleukin 6 at intensive care unit admission as biomarkers in critically ill COVID‐19 patients. Am J Trop Med Hyg. 2021;105(1):73‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yin X, Riva L, Pu Y, et al. MDA5 governs the innate immune response to SARS‐CoV‐2 in lung epithelial cells. Cell Rep. 2021;34(2):108628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Campbell GR, To RK, Hanna J, Spector SA. SARS‐CoV‐2, SARS‐CoV‐1, and HIV‐1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non‐classical pathway. iScience. 2021;24(4):102295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Xu H, Akinyemi IA, Chitre SA, et al. SARS‐CoV‐2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology. 2022;568:13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aymonnier K, Ng J, Fredenburgh LE, et al. Inflammasome activation in neutrophils of patients with severe COVID‐19. Blood Adv. 2022;6(7):2001‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Borella R, De Biasi S, Paolini A, et al. Metabolic reprograming shapes neutrophil functions in severe COVID‐19. Eur J Immunol. 2021;52:484‐502. [DOI] [PubMed] [Google Scholar]
- 108. Veras FP, Pontelli MC, Silva CM, et al. SARS‐CoV‐2‐triggered neutrophil extracellular traps mediate COVID‐19 pathology. J Exp Med. 2020;217(12):e20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Panda R, Castanheira FV, Schlechte JM, et al. A functionally distinct neutrophil landscape in severe COVID‐19 reveals opportunities for adjunctive therapies. JCI Insight. 2022;7:e152291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID‐19. JCI Insight. 2020;5(11):e138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Boribong BP, LaSalle TJ, Bartsch YC, et al. Neutrophil profiles of pediatric COVID‐19 and multisystem inflammatory syndrome in children. bioRxiv . 2021. [DOI] [PMC free article] [PubMed]
- 112. de Kay JT, Emery IF, Rud J, et al. DEspR high neutrophils are associated with critical illness in COVID‐19. Sci Rep. 2021;11(1):22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor‐enriched neutrophil extracellular traps are key drivers in COVID‐19 immunothrombosis. J Clin Invest. 2020;130(11):6151‐6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Radermecker C, Detrembleur N, Guiot J, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID‐19. J Exp Med. 2020;217(12):e20201012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Obermayer A, Jakob LM, Haslbauer JD, Matter MS, Tzankov A, Stoiber W. Neutrophil extracellular traps in fatal COVID‐19‐associated lung injury. Dis Markers. 2021;2021:5566826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134‐147. [DOI] [PubMed] [Google Scholar]
- 117. Jiang Y, Rosborough BR, Chen J, et al. Single cell RNA sequencing identifies an early monocyte gene signature in acute respiratory distress syndrome. JCI Insight. 2020;5(13):e135678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cillo AR, Somasundaram A, Shan F, et al. Critically ill COVID‐19 patients exhibit peripheral immune profiles predictive of mortality and reflective of SARS‐CoV‐2 viral burden in the lung. Cell Rep Med. 2021;2:100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Codo AC, Davanzo GG, Monteiro LB, et al. Elevated glucose levels favor SARS‐CoV‐2 infection and monocyte response through a HIF‐1α/glycolysis‐dependent axis. Cell Metab. 2020;32(3):437‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rodrigues TS, de Sá KSG, Ishimoto AY, et al. Inflammasomes are activated in response to SARS‐CoV‐2 infection and are associated with COVID‐19 severity in patients. J Exp Med. 2021;218(3):e20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Junqueira C, Crespo Â, Ranjbar S, et al. FcγR‐mediated SARS‐CoV‐2 infection of monocytes activates inflammation. Nature. 2022;606:576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Paludan SR, Mogensen TH. Innate immunological pathways in COVID‐19 pathogenesis. Sci Immunol. 2022;7(67):eabm5505. [DOI] [PubMed] [Google Scholar]
- 123. Zhu B, Wu Y, Huang S, et al. Uncoupling of macrophage inflammation from self‐renewal modulates host recovery from respiratory viral infection. Immunity. 2021;54(6):1200‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rendeiro AF, Ravichandran H, Bram Y, et al. The spatial landscape of lung pathology during COVID‐19 progression. Nature. 2021;593(7860):564‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fahlberg MD, Blair RV, Doyle‐Meyers LA, et al. Cellular events of acute, resolving or progressive COVID‐19 in SARS‐CoV‐2 infected non‐human primates. Nat Commun. 2020;11(1):6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhou J, Chu H, Li C, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209(9):1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yip MS, Leung NH, Cheung CY, et al. Antibody‐dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. 2014;11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Grant RA, Morales‐Nebreda L, Markov NS, et al. Circuits between infected macrophages and T cells in SARS‐CoV‐2 pneumonia. Nature. 2021;590(7847):635‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wang S, Yao X, Ma S, et al. A single‐cell transcriptomic landscape of the lungs of patients with COVID‐19. Nat Cell Biol. 2021;23(12):1314‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lv J, Wang Z, Qu Y, et al. Distinct uptake, amplification, and release of SARS‐CoV‐2 by M1 and M2 alveolar macrophages. Cell Discov. 2021;7(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hoepel W, Chen HJ, Geyer CE, et al. High titers and low fucosylation of early human anti‐SARS‐CoV‐2 IgG promote inflammation by alveolar macrophages. Sci Transl Med. 2021;13(596):eabf8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Jesenak M, Brndiarova M, Urbancikova I, et al. Immune parameters and COVID‐19 infection—associations with clinical severity and disease prognosis. Front Cell Infect Microbiol. 2020;10:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wilk AJ, Rustagi A, Zhao NQ, et al. A single‐cell atlas of the peripheral immune response in patients with severe COVID‐19. Nat Med. 2020;26(7):1070‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pérez‐Gómez A, Vitallé J, Gasca‐Capote C, et al. Dendritic cell deficiencies persist seven months after SARS‐CoV‐2 infection. Cell Mol Immunol. 2021;18(9):2128‐2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhou R, To KK, Wong YC, et al. Acute SARS‐CoV‐2 infection impairs dendritic cell and T cell responses. Immunity. 2020;53(4):864‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Winheim E, Rinke L, Lutz K, et al. Impaired function and delayed regeneration of dendritic cells in COVID‐19. PLoS Pathog. 2021;17(10):e1009742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yang D, Chu H, Hou Y, et al. Attenuated interferon and proinflammatory response in SARS‐CoV‐2‐infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5):734‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Broggi A, Ghosh S, Sposito B, et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369(6504):706‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cells promote lung‐tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Silverstein NJ, Wang Y, Manickas‐Hill Z, et al. Innate lymphoid cells and COVID‐19 severity in SARS‐CoV‐2 infection. eLife. 2022;11:e74681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. García M, Kokkinou E, Carrasco García A, et al. Innate lymphoid cell composition associates with COVID‐19 disease severity. Clin Transl Immunology. 2020;9(12):e1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Donlan AN, Sutherland TE, Marie C, et al. IL‐13 is a driver of COVID‐19 severity. JCI Insight. 2021;6(15):e150107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wu ML, Liu FL, Sun J, et al. SARS‐CoV‐2‐triggered mast cell rapid degranulation induces alveolar epithelial inflammation and lung injury. Signal Transduct Target Ther. 2021;6(1):428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dan JM, Mateus J, Kato Y, et al. Immunological memory to SARS‐CoV‐2 assessed for up to 8 months after infection. Science. 2021;371(6529):eabf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181(7):1489‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gaebler C, Wang Z, Lorenzi JCC, et al. Evolution of antibody immunity to SARS‐CoV‐2. Nature. 2021;591(7851):639‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Rodda LB, Netland J, Shehata L, et al. Functional SARS‐CoV‐2‐Specific immune memory persists after mild COVID‐19. Cell. 2021;184(1):169‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lu Z, Laing ED, Pena DaMata J, et al. Durability of SARS‐CoV‐2‐specific T‐cell responses at 12 months postinfection. J Infect Dis. 2021;224(12):2010‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Poon MML, Rybkina K, Kato Y, et al. SARS‐CoV‐2 infection generates tissue‐localized immunological memory in humans. Sci Immunol. 2021;6(65):eabl9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Cheon IS, Li C, Son YM, et al. Immune signatures underlying post‐acute COVID‐19 lung sequelae. Sci Immunol. 2021;6(65):eabk1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sette A, Crotty S. Adaptive immunity to SARS‐CoV‐2 and COVID‐19. Cell. 2021;184(4):861‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Neidleman J, Luo X, Frouard J, et al. SARS‐CoV‐2‐specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Rep Med. 2020;1(6):100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sekine T, Perez‐Potti A, Rivera‐Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID‐19. Cell. 2020;183(1):158‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rydyznski Moderbacher C, Ramirez SI, Dan JM, et al. Antigen‐specific adaptive immunity to SARS‐CoV‐2 in acute COVID‐19 and associations with age and disease severity. Cell. 2020;183(4):996‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Neidleman J, Luo X, George AF, et al. Distinctive features of SARS‐CoV‐2‐specific T cells predict recovery from severe COVID‐19. Cell Rep. 2021;36(3):109414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Tan AT, Linster M, Tan CW, et al. Early induction of functional SARS‐CoV‐2‐specific T cells associates with rapid viral clearance and mild disease in COVID‐19 patients. Cell Rep. 2021;34(6):108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Peng Y, Felce SL, Dong D, et al. An immunodominant NP105‐113‐B*07:02 cytotoxic T cell response controls viral replication and is associated with less severe COVID‐19 disease. Nat Immunol. 2022;23(1):50‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Le Bert N, Clapham HE, Tan AT, et al. Highly functional virus‐specific cellular immune response in asymptomatic SARS‐CoV‐2 infection. J Exp Med. 2021;218(5):e20202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Le Bert N, Tan AT, Kunasegaran K, et al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature. 2020;584(7821):457‐462. [DOI] [PubMed] [Google Scholar]
- 161. Swadling L, Diniz MO, Schmidt NM, et al. Pre‐existing polymerase‐specific T cells expand in abortive seronegative SARS‐CoV‐2. Nature. 2022;601(7891):110‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Marcotte H, Piralla A, Zuo F, et al. Immunity to SARS‐CoV‐2 up to 15 months after infection. iScience. 2022;25(2):103743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Wragg KM, Lee WS, Koutsakos M, et al. Establishment and recall of SARS‐CoV‐2 spike epitope‐specific CD4(+) T cell memory. Nat Immunol. 2022;23:768‐780. [DOI] [PubMed] [Google Scholar]
- 164. Yazdanifar M, Mashkour N, Bertaina A. Making a case for using gammadelta T cells against SARS‐CoV‐2. Crit Rev Microbiol. 2020;46(6):689‐702. [DOI] [PubMed] [Google Scholar]
- 165. Rijkers G, Vervenne T, van der Pol P. More bricks in the wall against SARS‐CoV‐2 infection: involvement of gamma9delta2 T cells. Cell Mol Immunol. 2020;17(7):771‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Carter MJ, Fish M, Jennings A, et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS‐CoV‐2 infection. Nat Med. 2020;26(11):1701‐1707. [DOI] [PubMed] [Google Scholar]
- 167. Brouwer PJM, Caniels TG, van der Straten K, et al. Potent neutralizing antibodies from COVID‐19 patients define multiple targets of vulnerability. Science. 2020;369(6504):643‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Suthar MS, Zimmerman MG, Kauffman RC, et al. Rapid generation of neutralizing antibody responses in COVID‐19 patients. Cell Rep Med. 2020;1(3):100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Rogers TF, Zhao F, Huang D, et al. Isolation of potent SARS‐CoV‐2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369(6506):956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Piccoli L, Park YJ, Tortorici MA, et al. Mapping neutralizing and immunodominant sites on the SARS‐CoV‐2 spike receptor‐binding domain by structure‐guided high‐resolution serology. Cell. 2020;183(4):1024‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Garcia‐Beltran WF, Lam EC, Astudillo MG, et al. COVID‐19‐neutralizing antibodies predict disease severity and survival. Cell. 2021;184(2):476‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Khoury DS, Cromer D, Reynaldi A, et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS‐CoV‐2 infection. Nat Med. 2021;27(7):1205‐1211. [DOI] [PubMed] [Google Scholar]
- 173. Addetia A, Crawford KHD, Dingens A, et al. Neutralizing antibodies correlate with protection from SARS‐CoV‐2 in humans during a fishery vessel outbreak with a high attack rate. J Clin Microbiol. 2020;58(11):e02107‐e02120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Laidlaw BJ, Ellebedy AH. The germinal centre B cell response to SARS‐CoV‐2. Nat Rev Immunol. 2022;22(1):7‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Seow J, Graham C, Merrick B, et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS‐CoV‐2 infection in humans. Nat Microbiol. 2020;5(12):1598‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Turner JS, Kim W, Kalaidina E, et al. SARS‐CoV‐2 infection induces long‐lived bone marrow plasma cells in humans. Nature. 2021;595(7867):421‐425. [DOI] [PubMed] [Google Scholar]
- 177. Tang J, Zeng C, Cox TM, et al. Mucosal immunity against SARS‐CoV‐2 variants of concern including Omicron following vaccination. Sci Immunol . 2022;eadd4853. [DOI] [PMC free article] [PubMed]
- 178. Thieme CJ, Anft M, Paniskaki K, et al. Robust T cell response toward spike, membrane, and nucleocapsid SARS‐CoV‐2 proteins is not associated with recovery in critical COVID‐19 patients. Cell Rep Med. 2020;1(6):100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Meckiff BJ, Ramirez‐Suastegui C. Fajardo V, et al. imbalance of regulatory and cytotoxic SARS‐CoV‐2‐reactive CD4(+) T cells in COVID‐19. Cell. 2020;183(5):1340‐1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Arcanjo A, Pinto KG, Logullo J, et al. Critically ill COVID‐19 patients exhibit hyperactive cytokine responses associated with effector exhausted senescent T cells in acute infection. J Infect Dis . 2021. [DOI] [PMC free article] [PubMed]
- 182. Littlefield KM, Watson RO, Schneider JM, et al. SARS‐CoV‐2‐specific T cells associate with inflammation and reduced lung function in pulmonary post‐acute sequalae of SARS‐CoV‐2. PLoS Pathog. 2022;18(5):e1010359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chioh FW, Fong SW, Young BE, et al. Convalescent COVID‐19 patients are susceptible to endothelial dysfunction due to persistent immune activation. eLife. 2021;10:e64909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Kaneko N, Boucau J, Kuo HH, et al. Expansion of cytotoxic CD4+ T cells in the lungs in severe COVID‐19. medRxiv . 2021. doi:10.1101/2021.03.23.21253885 [DOI] [PMC free article] [PubMed]
- 185. Su Y, Chen D, Yuan D, et al. Multi‐Omics resolves a sharp disease‐state shift between mild and moderate COVID‐19. Cell. 2020;183(6):1479‐1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Zheng HY, Zhang M, Yang CX, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID‐19 patients. Cell Mol Immunol. 2020;17(5):541‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Song JW, Zhang C, Fan X, et al. Immunological and inflammatory profiles in mild and severe cases of COVID‐19. Nat Commun. 2020;11(1):3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Rha MS, Jeong HW, Ko JH, et al. PD‐1‐Expressing SARS‐CoV‐2‐Specific CD8(+) T cells are not exhausted, but functional in patients with COVID‐19. Immunity. 2021;54(1):44‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Ndhlovu ZM, Kamya P, Mewalal N, et al. Magnitude and kinetics of CD8+ T cell activation during hyperacute HIV infection impact viral set point. Immunity. 2015;43(3):591‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chandele A, Sewatanon J, Gunisetty S, et al. Characterization of human CD8 T cell responses in dengue virus‐infected patients from India. J Virol. 2016;90(24):11259‐11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. McElroy AK, Akondy RS, Davis CW, et al. Human Ebola virus infection results in substantial immune activation. Proc Natl Acad Sci U S A. 2015;112(15):4719‐4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Fox A, Le NM, Horby P, et al. Severe pandemic H1N1 2009 infection is associated with transient NK and T deficiency and aberrant CD8 responses. PLoS One. 2012;7(2):e31535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Wang Z, Zhu L, Nguyen THO, et al. Clonally diverse CD38(+)HLA‐DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat Commun. 2018;9(1):824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Stephenson E, Reynolds G, Botting RA, et al. Single‐cell multi‐omics analysis of the immune response in COVID‐19. Nat Med. 2021;27(5):904‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Georg P, Astaburuaga‐García R, Bonaguro L, et al. Complement activation induces excessive T cell cytotoxicity in severe COVID‐19. Cell. 2022;185(3):493‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kim J, Chang DY, Lee HW, et al. Innate‐like cytotoxic function of Bystander‐activated CD8(+) T cells is associated with liver injury in acute hepatitis A. Immunity. 2018;48(1):161‐173. [DOI] [PubMed] [Google Scholar]
- 198. Huang CH, Fan JH, Jeng WJ, et al. Innate‐like bystander‐activated CD38(+) HLA‐DR(+) CD8(+) T cells play a pathogenic role in patients with chronic hepatitis C. Hepatology. 2022;76:803‐818. [DOI] [PubMed] [Google Scholar]
- 199. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Del Valle DM, Kim‐Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26(10):1636‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zhou Y, Fu B, Zheng X, et al. Pathogenic T‐cells and inflammatory monocytes incite inflammatory storms in severe COVID‐19 patients. Natl Sci Rev. 2020;7(6):998‐1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Thwaites RS, Sanchez Sevilla Uruchurtu A, Siggins MK, et al. Inflammatory profiles across the spectrum of disease reveal a distinct role for GM‐CSF in severe COVID‐19. Sci Immunol. 2021;6(57):eabg9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Zhao Y, Kilian C, Turner JE, et al. Clonal expansion and activation of tissue‐resident memory‐like Th17 cells expressing GM‐CSF in the lungs of severe COVID‐19 patients. Sci Immunol. 2021;6(56):eabf6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. 2020;38:541‐566. [DOI] [PubMed] [Google Scholar]
- 205. Veiga‐Parga T, Sehrawat S, Rouse BT. Role of regulatory T cells during virus infection. Immunol Rev. 2013;255(1):182‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. D'alessio FR, Tsushima K, Aggarwal NR, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119(10):2898‐2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Wang H, Wang Z, Cao W, Wu Q, Yuan Y, Zhang X. Regulatory T cells in COVID‐19. Aging Dis. 2021;12(7):1545‐1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Rahimzadeh M, Naderi N. Toward an understanding of regulatory T cells in COVID‐19: a systematic review. J Med Virol. 2021;93(7):4167‐4181. [DOI] [PubMed] [Google Scholar]
- 209. Ronit A, Berg RMG, Bay JT, et al. Compartmental immunophenotyping in COVID‐19 ARDS: a case series. J Allergy Clin Immunol. 2021;147(1):81‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Galván‐Peña S, Leon J, Chowdhary K, et al. Profound Treg perturbations correlate with COVID‐19 severity. Proc Natl Acad Sci U S A. 2021;118(37):e2111315118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Slutter B, Pewe LL, Kaech SM, Harty JT. Lung airway‐surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity. 2013;39(5):939‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Lim K, Hyun YM, Lambert‐Emo K, et al. Neutrophil trails guide influenza‐specific CD8(+) T cells in the airways. Science. 2015;349(6252):aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Wein AN, McMaster SR, Takamura S, et al. CXCR6 regulates localization of tissue‐resident memory CD8 T cells to the airways. J Exp Med. 2019;216(12):2748‐2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. De Biasi S, Meschiari M, Gibellini L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID‐19 pneumonia. Nat Commun. 2020;11(1):3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Saris A, Reijnders TDY, Reijm M, et al. Enrichment of CCR6(+) CD8(+) T cells and CCL20 in the lungs of mechanically ventilated patients with COVID‐19. Eur J Immunol. 2021;51(6):1535‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Adam L, Rosenbaum P, Quentric P, et al. CD8+PD‐L1+CXCR3+ polyfunctional T cell abundances are associated with survival in critical SARS‐CoV‐2‐infected patients. JCI Insight. 2021;6(18):e151571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Takamura S, Kato S, Motozono C, et al. Interstitial‐resident memory CD8(+) T cells sustain frontline epithelial memory in the lung. J Exp Med. 2019;216(12):2736‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Dudek M, Pfister D, Donakonda S, et al. Auto‐aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592(7854):444‐449. [DOI] [PubMed] [Google Scholar]
- 219. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti‐tumour surveillance in immunotherapy‐treated HCC. Nature. 2021;592(7854):450‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Dai Y, Wang J, Jeong HH, Chen W, Jia P, Zhao Z. Association of CXCR6 with COVID‐19 severity: delineating the host genetic factors in transcriptomic regulation. Hum Genet. 2021;140(9):1313‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Kasela S, Daniloski Z, Bollepalli S, et al. Integrative approach identifies SLC6A20 and CXCR6 as putative causal genes for the COVID‐19 GWAS signal in the 3p21.31 locus. Genome Biol. 2021;22(1):242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Bost P, De Sanctis F, Canè S, et al. Deciphering the state of immune silence in fatal COVID‐19 patients. Nat Commun. 2021;12(1):1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Payne D, Dalal S, Leach R, et al. The CXCR6/CXCL16 axis links inflamm‐aging to disease severity in COVID‐19 patients. bioRxiv . 2021;01.25.428125:.
- 224. Kaneko N, Kuo HH, Boucau J, et al. Loss of Bcl‐6‐expressing T follicular helper cells and germinal centers in COVID‐19. Cell. 2020;183(1):143‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Li Y, Xu Z, Lei Q, et al. Antibody landscape against SARS‐CoV‐2 reveals significant differences between non‐structural/accessory and structural proteins. Cell Rep. 2021;36(2):109391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Lei Q, Yu CZ, Li Y, et al. Anti‐SARS‐CoV‐2 IgG responses are powerful predicting signatures for the outcome of COVID‐19 patients. J Adv Res. 2022;36:133‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody‐dependent enhancement. Nat Rev Immunol. 2020;20(10):633‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Larsen MD, de Graaf EL, Sonneveld ME, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID‐19 severity. Science. 2021;371(6532):eabc8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Bye AP, Hoepel W, Mitchell JL, et al. Aberrant glycosylation of anti‐SARS‐CoV‐2 spike IgG is a prothrombotic stimulus for platelets. Blood. 2021;138(16):1481‐1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Chakraborty S, Gonzalez J, Edwards K, et al. Proinflammatory IgG Fc structures in patients with severe COVID‐19. Nat Immunol. 2021;22(1):67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Chakraborty S, Gonzalez JC, Sievers BL, et al. Early non‐neutralizing, afucosylated antibody responses are associated with COVID‐19 severity. Sci Transl Med. 2022;14(635):eabm7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Noris M, Benigni A, Remuzzi G. The case of complement activation in COVID‐19 multiorgan impact. Kidney Int. 2020;98(2):314‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement‐induced coagulopathy in COVID‐19. Nat Rev Nephrol. 2021;17(1):46‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Holter JC, Pischke SE, de Boer E, et al. Systemic complement activation is associated with respiratory failure in COVID‐19 hospitalized patients. Proc Natl Acad Sci U S A. 2020;117(40):25018‐25025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Carvelli J, Demaria O, Vély F, et al. Association of COVID‐19 inflammation with activation of the C5a‐C5aR1 axis. Nature. 2020;588(7836):146‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Lee MH, Perl DP, Steiner J, et al. Neurovascular injury with complement activation and inflammation in COVID‐19. Brain. 2022;145:2555‐2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Bastard P, Gervais A, Le Voyer T, et al. Autoantibodies neutralizing type I IFNs are present in ∼4% of uninfected individuals over 70 years old and account for ∼20% of COVID‐19 deaths. Sci Immunol. 2021;6(62):eabl4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Wang EY, Mao T, Klein J, et al. Diverse functional autoantibodies in patients with COVID‐19. Nature. 2021;595(7866):283‐288. [DOI] [PubMed] [Google Scholar]
- 239. van der Wijst MGP, Vazquez SE, Hartoularos GC, et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID‐19. Sci Transl Med. 2021;13(612):eabh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Koning R, Bastard P, Casanova JL, Brouwer MC, van de Beek D, Amsterdam U.M.C. COVID‐19 Biobank Investigators . Autoantibodies against type I interferons are associated with multi‐organ failure in COVID‐19 patients. Intensive Care Med. 2021;47(6):704‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Ziegler CGK, Miao VN, Owings AH, et al. Impaired local intrinsic immunity to SARS‐CoV‐2 infection in severe COVID‐19. Cell. 2021;184(18):4713‐4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Lopez J, Mommert M, Mouton W, et al. Early nasal type I IFN immunity against SARS‐CoV‐2 is compromised in patients with autoantibodies against type I IFNs. J Exp Med. 2021;218(10):e20211211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. de Prost N, Bastard P, Arrestier R, et al. Plasma exchange to rescue patients with autoantibodies against type I interferons and Life‐Threatening COVID‐19 pneumonia. J Clin Immunol. 2021;41(3):536‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Cano EJ, Fuentes XF, Campioli CC, et al, Edison J Cano 1, Xavier Fonseca Fuentes 2, Cristina Corsini Campioli . Impact of corticosteroids in coronavirus disease 2019 outcomes: systematic review and meta‐analysis. Chest. 2021;159(3):1019‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. van Paassen J, Vos JS, Hoekstra EM, Neumann KMI, Boot PC, Arbous SM. Corticosteroid use in COVID‐19 patients: a systematic review and meta‐analysis on clinical outcomes. Crit Care. 2020;24(1):696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Ma Y, Zeng H, Zhan Z, et al. Corticosteroid use in the treatment of COVID‐19: a multicenter retrospective study in Hunan, China. Front Pharmacol. 2020;11:1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019‐nCoV lung injury. Lancet. 2020;395(10223):473‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Wang J, Yang W, Chen P, et al. The proportion and effect of corticosteroid therapy in patients with COVID‐19 infection: a systematic review and meta‐analysis. PLoS One. 2021;16(4):e0249481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. RECOVERY Collaborative G, Horby P, Lim WS, et al. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med. 2021;384(8):693‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Kim WY, Kweon OJ, Cha MJ, Baek MS, Choi SH. Dexamethasone may improve severe COVID‐19 via ameliorating endothelial injury and inflammation: a preliminary pilot study. PLoS One. 2021;16(7):e0254167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Sinha S, Rosin NL, Arora R, et al. Dexamethasone modulates immature neutrophils and interferon programming in severe COVID‐19. Nat Med. 2021;28:201‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Salem MA. A response to the recommendations for using dexamethasone for the treatment of COVID‐19: the dark side of dexamethasone. J Pharm Pract. 2021;34(2):179‐180. [DOI] [PubMed] [Google Scholar]
- 253. Reyes LF, Rodriguez A, Bastidas A, et al. Dexamethasone as risk‐factor for ICU‐acquired respiratory tract infections in severe COVID‐19. J Crit Care. 2022;69:154014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Matthay MA, Thompson BT. Dexamethasone in hospitalised patients with COVID‐19: addressing uncertainties. Lancet Respir Med. 2020;8(12):1170‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Cory TJ, Emmons RS, Yarbro JR, Davis KL, Pence BD. Metformin suppresses monocyte immunometabolic activation by SARS‐CoV‐2 spike protein subunit 1. Front Immunol. 2021;12:733921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Samuel SM, Varghese E, Büsselberg D. Therapeutic potential of metformin in COVID‐19: reasoning for its protective role. Trends Microbiol. 2021;29(10):894‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Sharma S, Ray A, Sadasivam B. Metformin in COVID‐19: a possible role beyond diabetes. Diabetes Res Clin Pract. 2020;164:108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Xian H, Liu Y, Rundberg Nilsson A, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity. 2021;54(7):1463‐1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Bramante CT, Ingraham NE, Murray TA, et al. Metformin and risk of mortality in patients hospitalised with COVID‐19: a retrospective cohort analysis. Lancet Healthy Longev. 2021;2(1):e34‐e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Bramante CT, Huling JD, Tignanelli CJ, et al. Randomized trial of metformin, ivermectin, and fluvoxamine for Covid‐19. N Engl J Med. 2022;387(7):599‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Reis G, Dos Santos Moreira Silva EA, Medeiros Silva DC, et al. Effect of early treatment with metformin on risk of emergency care and hospitalization among patients with COVID‐19: the TOGETHER randomized platform clinical trial. Lancet Reg Health Am. 2022;6:100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Lalau JD, Al‐Salameh A, Hadjadj S, et al. Metformin use is associated with a reduced risk of mortality in patients with diabetes hospitalised for COVID‐19. Diabetes Metab. 2021;47(5):101216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Crouse A, Grimes T, Li P, Might M, Ovalle F, Shalev A. Metformin use is associated with reduced mortality in a diverse population with covid‐19 and diabetes. Front Endocrinol. 2020;11:600439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Keystone EC, Taylor PC, Drescher E, et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann Rheum Dis. 2015;74(2):333‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Hoang TN, Pino M, Boddapati AK, et al. Baricitinib treatment resolves lower‐airway macrophage inflammation and neutrophil recruitment in SARS‐CoV‐2‐infected rhesus macaques. Cell. 2021;184(2):460‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Bronte V, Ugel S, Tinazzi E, et al. Baricitinib restrains the immune dysregulation in patients with severe COVID‐19. J Clin Invest. 2020;130(12):6409‐6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus remdesivir for hospitalized adults with Covid‐19. N Engl J Med. 2021;384(9):795‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. McElvaney OJ, Curley GF, Rose‐John S, McElvaney NG. Interleukin‐6: obstacles to targeting a complex cytokine in critical illness. Lancet Respir Med. 2021;9(6):643‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Jones SA, Hunter CA. Is IL‐6 a key cytokine target for therapy in COVID‐19? Nat Rev Immunol. 2021;21(6):337‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Angriman F, Ferreyro BL, Burry L, et al. Interleukin‐6 receptor blockade in patients with COVID‐19: placing clinical trials into context. Lancet Respir Med. 2021;9(6):655‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Rosas IO, Bräu N, Waters M, et al. Tocilizumab in hospitalized patients with severe Covid‐19 pneumonia. N Engl J Med. 2021;384(16):1503‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Albuquerque AM, Tramujas L, Sewanan LR, Williams DR, Brophy JM. Mortality rates among hospitalized patients with COVID‐19 infection treated with tocilizumab and corticosteroids: a Bayesian reanalysis of a previous meta‐analysis. JAMA Netw Open. 2022;5(2):e220548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Durán‐Méndez A, Aguilar‐Arroyo AD, Vivanco‐Gómez E, et al. Tocilizumab reduces COVID‐19 mortality and pathology in a dose and timing‐dependent fashion: a multi‐centric study. Sci Rep. 2021;11(1):19728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Li P, Lu Z, Li Q, et al. Administration timing and efficacy of tocilizumab in patients with COVID‐19 and elevated IL‐6. Front Mol Biosci. 2021;8:651662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Zaid Y, Doré É, Dubuc I, et al. Chemokines and eicosanoids fuel the hyperinflammation within the lungs of patients with severe COVID‐19. J Allergy Clin Immunol. 2021;148(2):368‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Porter C, Armstrong‐Fisher S, Kopotsha T, et al. Certolizumab pegol does not bind the neonatal Fc receptor (FcRn): consequences for FcRn‐mediated in vitro transcytosis and ex vivo human placental transfer. J Reprod Immunol. 2016;116:7‐12. [DOI] [PubMed] [Google Scholar]
- 277. Izadi Z, Brenner EJ, Mahil SK, et al. Association between tumor necrosis factor inhibitors and the risk of hospitalization or death among patients with immune‐mediated inflammatory disease and COVID‐19. JAMA Netw Open. 2021;4(10):e2129639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Kokkotis G, Kitsou K, Xynogalas I, et al. Systematic review with meta‐analysis: COVID‐19 outcomes in patients receiving anti‐TNF treatments. Aliment Pharmacol Ther. 2022;55(2):154‐167. [DOI] [PubMed] [Google Scholar]
- 279. Blanco‐Melo D, Nilsson‐Payant BE, Liu WC, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181(5):1036‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Wang N, Zhan Y, Zhu L, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID‐19 patients. Cell Host Microbe. 2020;28(3):455‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Wong LR, Perlman S. Immune dysregulation and immunopathology induced by SARS‐CoV‐2 and related coronaviruses—are we our own worst enemy? Nat Rev Immunol. 2022;22(1):47‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Lang FM, Lee KM, Teijaro JR, Becher B, Hamilton JA. GM‐CSF‐based treatments in COVID‐19: reconciling opposing therapeutic approaches. Nat Rev Immunol. 2020;20(8):507‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Kimmig LM, Wu D, Gold M, et al. IL‐6 inhibition in critically ill COVID‐19 patients is associated with increased secondary infections. Front Med. 2020;7:583897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Aziz M, Haghbin H, Abu Sitta E, et al. Efficacy of tocilizumab in COVID‐19: a systematic review and meta‐analysis. J Med Virol. 2021;93(3):1620‐1630. [DOI] [PubMed] [Google Scholar]
- 285. Hamilton JA. GM‐CSF in inflammation. J Exp Med. 2020;217(1):e20190945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Nalbandian A, Sehgal K, Gupta A, et al. Post‐acute COVID‐19 syndrome. Nature Med. 2021;27(4):601‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Lopez‐Leon S, Wegman‐Ostrosky T, Perelman C, et al. More than 50 long‐term effects of COVID‐19: a systematic review and meta‐analysis. Sci Rep. 2021;11(1):16144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Han X, Fan Y, Alwalid O, et al. Six‐month follow‐up chest CT findings after severe COVID‐19 pneumonia. Radiology. 2021;299(1):E177‐E186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Daniel AM, Treece KS. Law enforcement pathways to mental health: secondary traumatic stress, social support, and social pressure. Nature Portfolio. 2022;37:132‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Phetsouphanh C, Darley DR, Wilson DB, et al. Immunological dysfunction persists for 8 months following initial mild‐to‐moderate SARS‐CoV‐2 infection. Nature Immunol. 2022;23:210‐216. [DOI] [PubMed] [Google Scholar]
- 291. Cheon IS, Li C, Son YM, et al. Immune signatures underlying post‐acute COVID‐19 lung sequelae. Science Immunology. 1741;6(65):eabk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Ryan FJ, Hope CM, Masavuli MG, et al. Long‐term perturbation of the peripheral immune system months after SARS‐CoV‐2 infection. BMC Med. 2022;20(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Talla A, Vasaikar SV, Lemos MP, et al. Longitudinal immune dynamics of mild COVID‐19 define signatures of recovery and persistence. bioRxiv . 2021.doi:10.1101/2021.05.26.442666
- 294. Su Y, Yuan D, Chen DG, et al. Multiple early factors anticipate post‐acute COVID‐19 sequelae. Cell. 2022;185(5):881‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Klein J, Wood J, Jaycox J, et al. Distinguishing features of long COVID identified through immune profiling. medRxiv . 2022. 10.1101/2022.08.09.22278592 [DOI] [PMC free article] [PubMed]
- 296. Narasimhan H, Wu Y, Goplen Nick P, Sun J. Immune determinants of chronic sequelae after respiratory viral infection. Science Immunology. 2022;7(73):eabm7996. [DOI] [PubMed] [Google Scholar]
- 297. Vijayakumar B, Boustani K, Ogger PP, et al. Immuno‐proteomic profiling reveals aberrant immune cell regulation in the airways of individuals with ongoing post‐COVID‐19 respiratory disease. Immunity. 2022;55(3):542‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Choi J, Jang YJ, Dabrowska C, et al. Release of notch activity coordinated by IL‐1β signalling confers differentiation plasticity of airway progenitors via Fosl2 during alveolar regeneration. Nature Cell Biol. 2021;23(9):953‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Schultheiß C, Willscher E, Paschold L, et al. The IL‐1β, IL‐6, and TNF cytokine triad is associated with post‐acute sequelae of COVID‐19. Cell Rep Med . 2022;3(6):100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. George P, Reed A, Desai S, et al. A persistent neutrophil‐associated immune signature characterises post‐COVID19 pulmonary sequelae. Res Sq . 2022. 10.21203/rs.3.rs-1293175/v1 [DOI] [PubMed]
- 301. Chun HJ, Coutavas E, Pine AB, et al. Immunofibrotic drivers of impaired lung function in postacute sequelae of SARS‐CoV‐2 infection. JCI Insight. 2021;6(14):e148476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Fernández‐Castañeda A, Lu P, Geraghty AC, et al. Mild respiratory COVID can cause multi‐lineage neural cell and myelin dysregulation. Cell. 2022;185(14):2452‐2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Vijayakumar B, Boustani K, Ogger PP, et al. Immuno‐proteomic profiling reveals aberrant immune cell regulation in the airways of individuals with ongoing post‐COVD‐19 respiratory disease. Immunity, 55:542‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Harb H, Benamar M, Lai PS, et al. Notch4 signaling limits regulatory T‐cell‐mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity. 2021;54(6):1186‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Cervia C, Zurbuchen Y, Taeschler P, et al. Immunoglobulin signature predicts risk of post‐acute COVID‐19 syndrome. Nat Commun. 2022;13(1):446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Al‐Aly Z, Xie Y, Bowe B. High‐dimensional characterization of post‐acute sequelae of COVID‐19. Nature. 2021;594(7862):259‐264. [DOI] [PubMed] [Google Scholar]
- 307. Huang C, Huang L, Wang Y, et al. 6‐month consequences of COVID‐19 in patients discharged from hospital: a cohort study. Lancet. 2021;397(10270):220‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Shaw B, Daskareh M, Gholamrezanezhad A. The lingering manifestations of COVID‐19 during and after convalescence: update on long‐term pulmonary consequences of coronavirus disease 2019 (COVID‐19). Radiol Med. 2021;126(1):40‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Xie Y, Bowe B, Al‐Aly Z. Burdens of post‐acute sequelae of COVID‐19 by severity of acute infection, demographics and health status. Nat Commun. 2021;12(1):6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Dinnon KH, Leist SR, Okuda K, et al. A model of persistent post SARS‐CoV‐2 induced lung disease for target identification and testing of therapeutic strategies. bioRxiv . 2022.doi:10.1101/2022.02.15.480515
- 311. Arish M, Naz F. Personalized therapy: can it tame the COVID‐19 monster? Per Med. 2021;18(6):583‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. van de Veerdonk FL, Giamarellos‐Bourboulis E, Pickkers P, et al. A guide to immunotherapy for COVID‐19. Nat Med. 2022;28(1):39‐50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data reviewed in this manuscript are from published papers.