

Abstract
Background:
Rearranged during transfection (RET) gene fusions are a validated target in non-small cell lung cancer (NSCLC). RET-selective inhibitors selpercatinib (LOXO-292) and pralsetinib (BLU-667) recently demonstrated favorable antitumor activity and safety profiles in advanced RET fusion-positive NSCLC, and both have received approval by the US Food and Drug Administration for this indication. Insights into mechanisms of resistance to selective RET inhibitors remain limited.
Patients and Methods:
This study was performed at five institutions. Tissue and/or cell-free DNA was obtained from patients with RET fusion-positive NSCLC after treatment with selpercatinib or pralsetinib and assessed by next-generation sequencing (NGS) or MET fluorescence in situ hybridization.
Results:
We analyzed a total of 23 post-treatment tissue and/or plasma biopsies from 18 RET fusion-positive patients who received a RET-selective inhibitor (selpercatinib, n=10; pralsetinib, n=7; pralsetinib followed by selpercatinib, n=1 with biopsy after each inhibitor). Three cases had paired tissue and plasma samples, of which one also had two serial resistant tissue specimens. The median progression-free survival on RET inhibitors was 6.3 months [95% confidence interval (CI), 3.6–10.8 months]. Acquired RET mutations were identified in two cases (10%), both affecting the RET G810 residue in the kinase solvent front. Three resistant cases (15%) harbored acquired MET amplification without concurrent RET resistance mutations, and one specimen had acquired KRAS amplification. No other canonical driver alterations were identified by NGS. Among 16 resistant tumor specimens, none had evidence of squamous or small cell histologic transformation.
Conclusions:
RET solvent front mutations are a recurrent mechanism of RET inhibitor resistance, although they occurred at a relatively low frequency. The majority of resistance to selective RET inhibition may be driven by RET-independent resistance such as acquired MET or KRAS amplification. Next-generation RET inhibitors with potency against RET resistance mutations and combination strategies are needed to effectively overcome resistance in these patients.
Keywords: RET, non-small cell lung cancer, resistance, tyrosine kinase inhibitor, pralsetinib, selpercatinib
INTRODUCTION
The diagnostic and treatment approach to advanced non-small cell lung cancer (NSCLC) continues to be refined, with a growing number of genetic and molecular markers that guide tailored therapy. The oncogenic rearranged during transfection (RET) gene fusions were first identified in NSCLC in 2012.[1–4] Since then, RET fusions have been reported in approximately 1–2% of lung cancer, predominantly associated with a never or light smoking history and adenocarcinoma histology.[5] Importantly, lung cancers harboring RET fusions are sensitive to tyrosine kinase inhibitors (TKIs) with anti-RET activity, and therefore define a distinct molecular subset.[1, 4, 6]
Initial efforts to target RET in lung cancer involved repurposing readily available multikinase inhibitors (MKIs) with potency against RET such as cabozantinib or vandetanib.[7–12] However, these MKIs were limited by modest efficacy and substantial toxicities. In 2017, two novel, potent RET-selective TKIs, selpercatinib (LOXO-292) and pralsetinib (BLU-667), entered clinical testing in patients with advanced RET-altered solid tumors, including RET fusion-positive NSCLC.[13, 14] Both RET TKIs demonstrated favorable tolerability and robust efficacy [including in the central nervous system (CNS)] in patients with RET fusion-positive lung cancer in registrational phase I/II studies, with objective response rates (ORRs) ranging from 55–64% among platinum chemotherapy-pretreated and 66–85% among treatment-naïve patients, respectively.[15, 16] Durable responses were observed regardless of the RET fusion partner or history of prior MKI exposure. On the basis of these data, the US Food and Drug Administration (FDA) recently granted a line-agnostic accelerated approval of selpercatinib and pralsetinib for the treatment of adult patients with metastatic RET fusion-positive NSCLC (with selpercatinib also approved for adult and pediatric patients ≥12 years of age with advanced or metastatic RET-mutant medullary thyroid cancer or RET fusion-positive thyroid cancer who require systemic therapy and are radioactive iodine-refractory).
Despite the encouraging efficacy of selective RET TKIs, experience across the targeted therapy paradigm in NSCLC suggests that the eventual development of acquired resistance will limit the duration of benefit from RET-selective inhibitors. As selpercatinib and pralsetinib are now standard therapies in advanced RET fusion-positive lung cancer and will be more widely used, it is paramount to understand the mechanisms of TKI resistance and inform the development of novel therapeutic strategies that can overcome resistance. In one recent study, Solomon and colleagues reported RET G810R/S/C/V solvent front mutations that mediated acquired resistance to selpercatinib in three RET fusion-positive NSCLC and two RET-mutant medullary thyroid cancer (MTC) cases.[17] The frequency of RET resistance mutations, however, remains undetermined. Furthermore, outside of this study and one case report of a selpercatinib-resistant NSCLC patient harboring MET amplification,[18] insights into mechanisms of resistance to RET-selective TKIs are lacking.
Here, we performed a multi-institutional analysis of repeat tumor or plasma biopsies from a cohort of patients with RET fusion-positive NSCLC after treatment with selpercatinib and pralsetinib, in order to systematically characterize acquired resistance mechanisms to these inhibitors.
METHODS
Study Population
Patients were identified at five participating institutions: Massachusetts General Hospital (MGH; n=10), Georgetown University (GU; n=2), National Cancer Centre Singapore (NCCS; n=1), University of California Irvine (UCI; n=1), and University of California San Francisco (UCSF; n=4). Patients were eligible if they had advanced or metastatic NSCLC with RET fusion identified by local molecular profiling [e.g., fluorescent in situ hybridization (FISH), DNA-based next-generation sequencing (NGS), or targeted RNA sequencing]. Patients must have received pralsetinib and/or selpercatinib (as any line of systemic therapy) with subsequent resistant tumor or liquid biopsy analyzed by molecular testing. Most of the enrolled patients received pralsetinib or selpercatinib in clinical trials (ClinicalTrials.gov identifier NCT03037385 or NCT03157128), respectively. One patient received selpercatinib through single patient compassionate use access, and one patient received selpercatinib through the expanded access program. The studies were approved by the Institutional Review Board at each participating institution.
Data Collection
Medical records were retrospectively reviewed to extract data on clinical, pathologic, and molecular features. Response to therapy was determined per the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Progression-free survival (PFS) was measured from the time of therapy initiation to clinical/radiographic disease progression or death. Duration of therapy was measured from the time of therapy initiation to therapy discontinuation. Patients continuing on therapy were censored at last follow-up. All data were updated as of June 10, 2020.
Biopsy Genotyping
All patients included in this study underwent tumor or plasma biopsy after treatment with pralsetinib or selpercatinib and genotyping after providing informed consent. Fifteen tissue biopsies were analyzed using one of the following NGS platforms: the previously described MGH SNaPshot DNA-based genotyping panel and a separate RNA-based NGS assay (Solid Fusion Assay) for the detection of fusion transcripts (n=10),[19] FoundationOne (n=1; Foundation Medicine, Inc.), Caris MI profile (n=2; Caris Molecular Intelligence), MSK IMPACT (n=1),[20] or UCSF500 (n=1).[21] One tissue specimen was insufficient for NGS but sufficient for analysis by MET fluorescence in situ hybridization (FISH). Seven liquid biopsies were analyzed using either the commercially available Guardant 360 cell-free DNA (cfDNA) assay (n=5; Guardant Health, Inc.) or the FoundationACT assay (n=2; Foundation Medicine, Inc.).
MET FISH was performed using formalin-fixed paraffin-embedded tumor specimens and the dual-color FISH assay with 07Q001B550 C-MET (7q31) probe (chromosome 7q31 MET locus; Leica Biosystems) and a copy number probe (centromere 7 or CEP7; Abbott-Vysis). Signal quantitation of 50 tumor nuclei was used to generate a MET/CEP7 ratio. A ratio greater than 5.0 or clustered MET signals too numerous to count were considered highly amplified.
A cell line (MGH9009–1) was developed from the lymph node biopsy of case MGH2, as previously described.[22] RET fusion mRNA was PCR-amplified and RET kinase domain was sequenced. Primer sequences were: KIAA1468 F 5′- CGAGGTGTCTCGTATTGCAG -3′, RET R 5′- GCATTATTACAGTCCACCAGCG -3′.
Statistical Analysis
The Kaplan-Meier method was used to estimate PFS and duration of therapy medians and probabilities (Stata version 14.2).
RESULTS
Clinical Characteristics
A total of 18 patients with advanced RET fusion-positive NSCLC were treated with pralsetinib (n=7), selpercatinib (n=10), or pralsetinib followed by selpercatinib (n=1), and underwent post-treatment biopsies between 2017 and 2020 (Table 1). In the cohort, the median age at diagnosis was 56.5 (range, 30–77). All patients had adenocarcinoma and were never or light smokers. The RET fusion partner was known for all patients. The predominant fusion was KIF5B-RET (67%), consistent with the literature.[5] Seven patients (39%) had known brain metastases at the time of starting selpercatinib or pralsetinib.
Table 1.
Baseline characteristics of the RET inhibitor-resistant cohort with RET fusion-positive lung cancer.
| Characteristic | n (%), N = 18 |
|---|---|
| Age at diagnosis, median (range) | 56.5 (30–77) |
| Female | 10 (56) |
| Never or light smoker | 18 (100) |
| Adenocarcinoma | 18 (100) |
| RET fusion | |
| KIF5B-RET | 12 (67) |
| CCDC6-RET | 4 (22) |
| Other | 2 (11) |
| RET inhibitor prior to biopsy | |
| Selpercatinib | 10 (56) |
| Pralsetinib | 7 (39) |
| Pralsetinib, then selpercatinib | 1 (6)* |
| Prior lines of therapy | |
| 0 | 3 (17) |
| 1 | 10 (56) |
| 2 or more | 5 (28) |
| Prior platinum chemotherapy | 13 (72) |
| Prior multikinase inhibitor with anti-RET activity | 4 (22) |
One patient underwent a repeat biopsy at resistance to pralsetinib, then started selpercatinib and had another biopsy at resistance to selpercatinib.
Outcomes on RET Inhibitors and Patterns of Progression
Fifteen patients (83%) had achieved partial response (PR) per RECIST v1.1 on their first RET-selective inhibitor. The remaining three patients had stable disease as the best overall response. The median PFS on the initial RET-selective TKI was 6.3 months [95% confidence interval (CI), 3.6–10.8 months], and the median duration of therapy was 7.2 months (95% CI, 3.7–19.0 months) (Supplemental Figure 1).
The majority of patients (72%) in this cohort experienced extracranial disease progression. Five patients (28%) had both extracranial and intracranial disease progression.
Summary of Biopsies and Histology
To assess the resistance mechanism to RET inhibitors, tissue biopsies alone were performed in 11 patients (one of whom had a resistant biopsy after pralsetinib and another following selpercatinib), and liquid biopsies alone in four patients. Two patients underwent paired tissue and plasma biopsies. One patient had two serial tumor biopsies of distinct metastatic sites at progression on a RET inhibitor, one of which also had a paired plasma biopsy (summarized in Figure 1 and further delineated in Supplemental Table 1).
Figure 1.
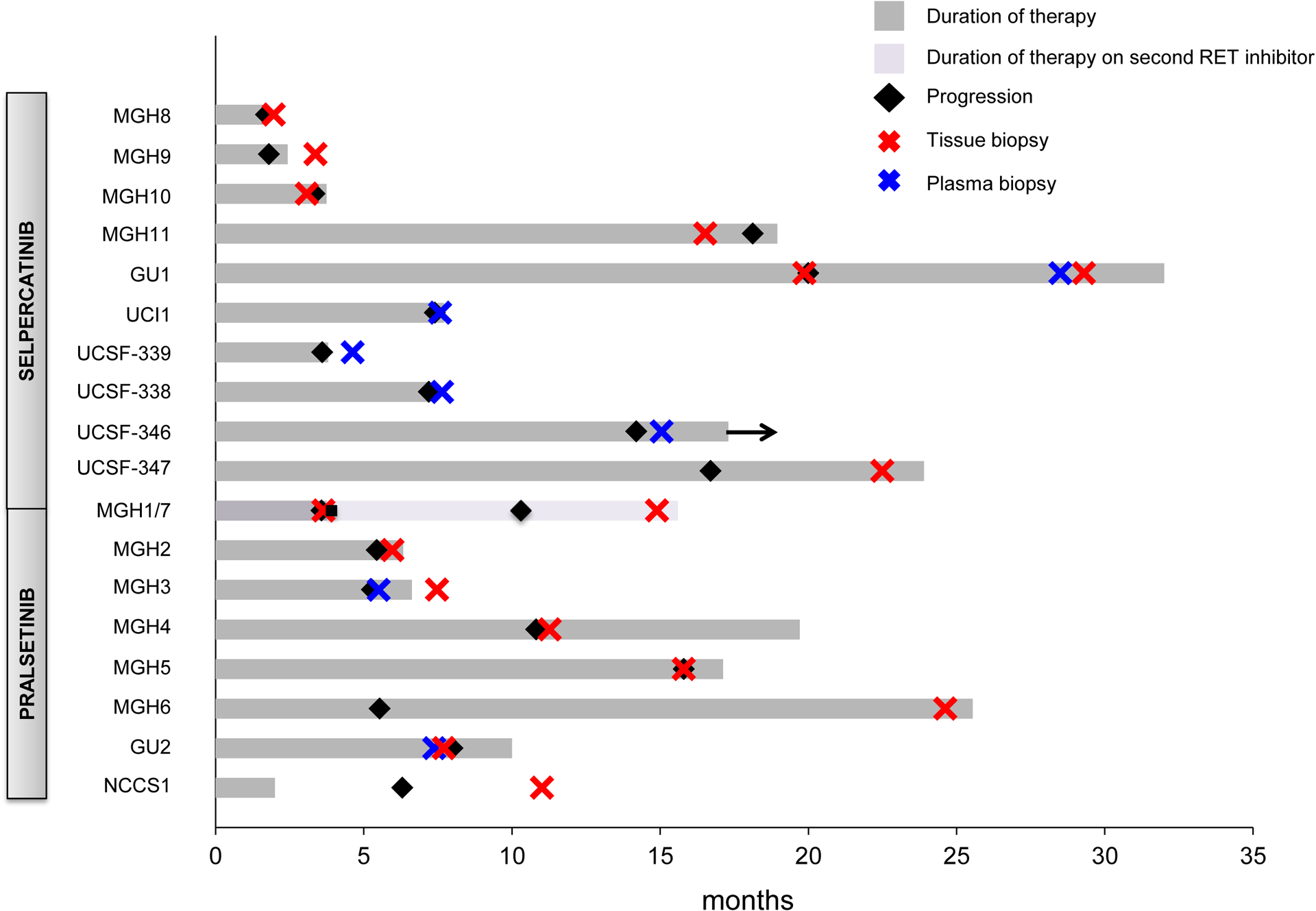
Duration of RET inhibitor treatment and timing of biopsies in the cohort. Arrow indicates ongoing therapy with a RET inhibitor at the time of this analysis. MGH1 and MGH7 biopsies were derived from the same patient (labeled here as MGH1/7), who first had disease progression on pralsetinib and underwent a tissue biopsy, followed by treatment with selpercatinib, again with disease progression and a repeat tissue biopsy.
In total, therefore, 20 distinct selpercatinib- or pralsetinib-resistant cases were analyzed by molecular testing, 3 of which had paired tissue and plasma (Figure 2).
Figure 2.

Summary of gene alterations in RET fusion-positive NSCLC resistant to selective RET inhibition. The heatmap summarizes findings from tissue (top) and cell-free DNA (bottom) analysis after treatment with selpercatinib or pralsetinib. Only those genes included in the MGH SNaPshot assay are shown. TKI, tyrosine kinase inhibitor; SFA, solid fusion assay; mut, mutation; seq, sequencing; CNA, copy number alteration.
RET Solvent Front Mutations
The gene alterations detected in the resistant biopsies are summarized in Figure 2. A RET resistance mutation was detected in two cases (10%), both affecting the G810 residue in the RET solvent front. In the first patient with CCDC6-RET fusion, a RET G810S mutation was detected at progression on selpercatinib (case MGH7, previously published).[17] This patient had previously received multiple multikinase inhibitors (e.g., ponatinib, alectinib, vandetanib) as well as pralsetinib, and had a post-pralsetinib/pre-selpercatinib biopsy (MGH1) which did not reveal any RET resistance mutations (Supplemental Table 2). Thus, RET G810S was most likely acquired on selpercatinib.
A second patient (GU1) with CCDC6-RET fusion-positive adenocarcinoma was initially treated with chemoradiation followed by durvalumab for stage 3 disease, with a biopsy at that time demonstrating the CCDC6-RET fusion but no evidence of RET mutations. This patient subsequently received multiple lines of immunotherapy, chemotherapy, and a multikinase inhibitor (RXDX-105), before enrolling in the clinical trial of selpercatinib. A soft tissue biopsy at progression on selpercatinib obtained after approximately 20 months on therapy did not reveal RET resistance mutations (GU1-T1 in Figure 2). She received radiation and continued therapy, but had further disease progression. A repeat biopsy of a progressing liver metastasis approximately 9 months later and paired cfDNA both revealed an acquired RET G810C mutation (GU1-T2 in Figure 2).
One case (MGH11) had a RET G597V mutation, which lies outside the RET kinase domain and is of unknown functional significance. Of note, this RET 597V mutation was also detected in the patient’s treatment-naïve plasma sample, and the patient went on to achieve a PR on RET-selective inhibitor with duration of response lasting 16.9 months. Therefore, this RET mutation was presumed not to be a driver of resistance. In addition, a RET V804 gatekeeper mutation was not detected in this series of post-treatment biopsies.
RET-Independent Resistance
Given the infrequency of on-target molecular mechanisms of resistance, we next investigated potential target-independent mechanisms of resistance. Among a total of 16 selpercatinib- or pralsetinib-resistant tissue biopsies, none had evidence of transformation to small cell or squamous cell histology.
Nineteen of the 20 distinct resistant cases were analyzed by broad NGS-based testing, with the one remaining case analyzed by MET FISH only due to insufficient tumor tissue for NGS. MET amplification is a recurrent bypass signaling pathway across oncogenic drivers, such as in NSCLC with EGFR mutations or ALK fusions.[23–26] We identified MET amplification in three post-RET TKI cases (15%), none of which harbored a concomitant RET resistance mutation (Supplemental Table 2). Two selpercatinib-resistant cases with KIF5B-RET fusions (GU2, PFS of 8 months; UCI1, PFS of 7.4 months and previously published[18]) were assessed by cfDNA sequencing and found to have MET amplification (plasma copy numbers of 2.7 by Guardant360 and ~17 by FoundationACT, respectively). For both cases, pre-selpercatinib cfDNA analyses did not demonstrate evidence of pre-existing baseline MET amplification. Of note, GU2 had a paired selpercatinib-resistant liver tumor biopsy that was also found to harbor MET amplification by NGS testing.
Another patient (MGH2) had received pralsetinib after prior chemotherapy, achieving RECIST PR. He had disease progression after 5.3 months, and a biopsy was performed of the resistant retroperitoneal lymph node. Tissue proved insufficient for NGS analysis. Sanger sequencing of the cDNA extracted from the corresponding patient-derived cell line did not reveal RET resistance mutations. Given the finding of MET amplification in other specimens, we pursued MET FISH testing, which demonstrated a high-level focal amplification of MET with MET/CEP7 ratio of greater than 25:1 (Figure 3). NGS and MET FISH analysis of the treatment-naïve tumor from this patient did not detect evidence of MET amplification.
Figure 3.
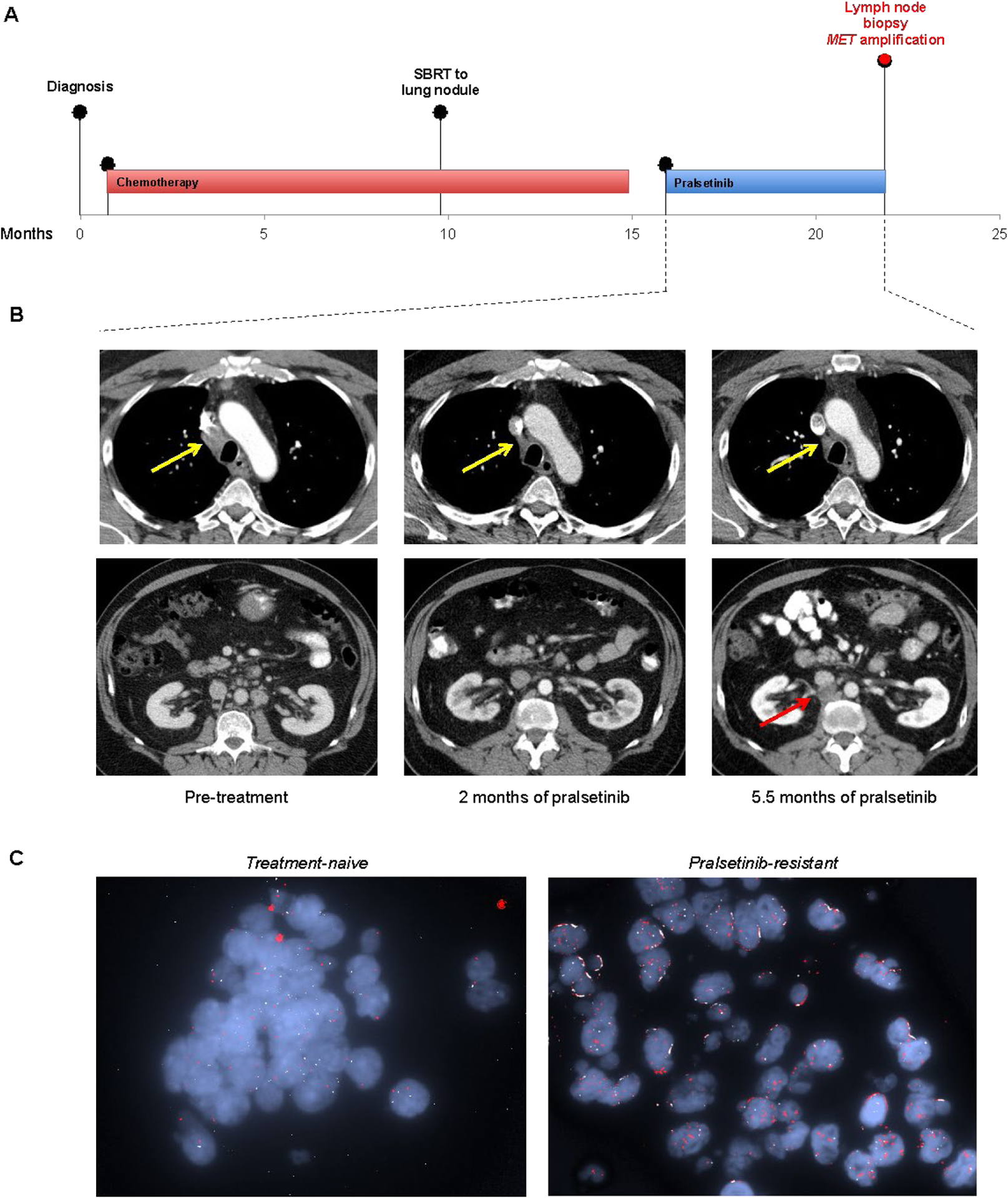
Emergence of high-level focal MET amplification after selective RET inhibition. (A) Treatment and biopsy timeline of MGH2 who had RET fusion-positive NSCLC with disease progression on pralsetinib. (B) Representative axial computed tomography images at baseline, 2 months, and 5.5 months after starting pralsetinib. Yellow arrows indicate interval response in the mediastinal lymph node. Red arrow indicates progression of a retroperitoneal lymph node on therapy, which was biopsied. (C) Fluorescence in situ hybridization images capture the de novo increase in MET copy number at resistance.
We identified a MET single nucleotide variant in two cases: MET M822I (NCCS1) and MET S108L (UCSF-339) (Figure 2). These mutations reside outside the MET kinase domain and are of unclear functional significance.
KRAS amplification is another genetic event which has been reported as a driver of resistance to targeted therapies in multiple contexts, including EGFR-directed therapies in colorectal cancer, and MET TKIs or ALK TKIs in NSCLC with MET exon 14 skipping or ALK fusions, respectively.[27–29] We detected KRAS amplification in a post-selpercatinib bone biopsy from a patient with KIF5B-RET fusion-positive NSCLC who had PR on selpercatinib and subsequently had disease progression after 16.7 months (UCSF-347, Figure 2; Supplemental Table 2). This resistant specimen was also found to have FGFR2 amplification, CCNE1 amplification, LRP1B deletion, and variants in TP53 and KMT2A. By comparison, a treatment-naïve lymph node specimen from this patient harbored CCNE1 amplification, LRP1B deletion, and TP53 variant, but no evidence of KRAS or FGFR2 amplification.
We did not identify acquired oncogenic mutations in other canonical drivers previously implicated in targeted therapy resistance, such as EGFR or ERBB2 (Figure 2, Supplemental Table 3). One pralsetinib-resistant case (MGH6 in Figure 2) had a PIK3CA H1047R mutation detected in post-treatment lung biopsy, which had not been detected in the TKI-naïve bone biopsy. BRAF N236S and ROS1 D2213E variants, both of unknown functional significance, were noted in one case each (UCSF-346 and NCCS1, respectively, in Figure 2). De novo oncogenic fusions involving ALK, ROS1, NTRK1-3, BRAF, NRG1, or MET genes were not detected.
DISCUSSION
In this multi-institutional study, we examined a total of 23 tumor and liquid biopsies derived from advanced RET fusion-positive NSCLC patients who were treated with RET-selective inhibitors pralsetinib and selpercatinib. To our knowledge, this is the largest study to date to examine mechanisms of resistance to RET-selective inhibitors. We identified RET solvent front mutations and MET amplification as recurrent mechanisms of resistance, and additionally identified KRAS amplification in one resistant case.
Solvent front mutations in the target kinase are known to confer on-target resistance in other fusion oncogene-driven lung cancers. For example, ALK G1202R and ROS1 G2032R are refractory solvent front mutations that cause resistance to a number of available TKIs in ALK or ROS1 fusion-positive NSCLC, respectively.[30, 31] Earlier this year, Solomon and colleagues reported RET G810 solvent front mutations as a mechanism of resistance to selpercatinib in five patients with RET fusion-positive NSCLC and RET-mutant MTC, predicted to hinder drug binding based on structural modeling.[17] However, the frequency of these RET mutations remained unknown. Here, we detected the RET G810C and G810S mutations in two cases (10%), supporting solvent front mutations as a recurrent mechanism of resistance to RET inhibitors and underscoring the importance of developing next-generation RET TKIs with potency against these mutations.
Overall, RET resistance mutations were detected at a low frequency in this cohort, and other, non-solvent front RET mutations including gatekeeper mutations affecting the V804 residue (known to confer resistance to MKIs such as vandetanib[32]) did not emerge in our series. The relatively low prevalence and narrow spectrum of RET mutations may reflect the high anti-RET potency of selpercatinib and pralsetinib, although our findings will require validation in larger cohorts. Interestingly, despite the potency of pralsetinib and selpercatinib against the gatekeeper RET V804 mutations based on preclinical studies,[13, 14] the study by Solomon et al. identified RET V804 and G810 mutations in trans in two selpercatinib-resistant cases and in cis in a minority of reads in one selpercatinib-resistant case.[17] Further studies are needed to elucidate whether the gatekeeper mutations can confer resistance to selpercatinib and/or pralsetinib despite the preclinical evidence, and whether the spectra of RET resistance mutations (and non-RET resistance alterations) differ between selpercatinib and pralsetinib.
Importantly, our findings indicate that the majority of cases progressing on RET-selective inhibitors are likely driven by off-target, RET-independent mechanisms of resistance. Indeed, the preponderance of resistant cases without RET resistance mutations is striking when compared to EGFR-mutant or ALK fusion-positive NSCLC, where approximately 50–60% of resistance to next-generation TKIs is driven by target-independent mechanisms.[25, 30] This observation highlights the importance of identifying putative potentially targetable RET-independent resistance drivers, with the ultimate goal of designing new treatment approaches.
We identified MET gene amplification as a recurring RET-independent resistance mechanism in RET fusion-positive lung cancer, observed in 15% of cases in this cohort. MET amplification is an established mechanism of resistance to EGFR inhibitors in EGFR-mutant NSCLC and has been identified in up to 20% of EGFR TKI-resistant biopsies.[23–25] Notably, a combination of EGFR and MET inhibitors, such as osimertinib plus savolitinib or osimertinib plus capmatinib, is able to effectively overcome this MET-driven resistance in clinic.[33, 34] Similarly, MET amplification can mediate resistance to next-generation ALK inhibitors in ALK fusion-positive lung cancer.[26] Within the framework of this collective knowledge, our findings support MET amplification as a recurring, clinically relevant driver of resistance across multiple distinct subsets of oncogene-driven lung cancer.
Furthermore, our findings naturally raise the question of whether combined RET and MET inhibition could represent a viable therapeutic strategy to target resistance in a subset of patients progressing on selpercatinib or pralsetinib. Certainly, studies evaluating combinations of a RET-selective inhibitor with a MET inhibitor will be required in order to explore this possibility. Multikinase inhibitors with activity against both MET and RET (e.g., cabozantinib) may represent an alternative and perhaps more readily accessible option, though likely less desirable in terms of potency and tolerability. The identification of potentially targetable resistance gene alterations, such as MET amplification or RET solvent front mutations in this study, implies that repeat biopsies will have clinical value in patients progressing on RET inhibitors.
Finally, it is worth noting that over a quarter of patients in our cohort had both intracranial and extracranial disease progression, despite the known favorable CNS activity of selpercatinib and pralsetinib.[15, 30, 35] This observation serves to emphasize that CNS penetration and efficacy should be an integral feature of next-generation RET inhibitors. If successfully developed, next-generation RET TKIs could enable a sequential treatment paradigm in RET fusion-positive disease, reminiscent of that seen in ALK or ROS1 fusion-positive lung cancer.
This study had several important limitations. First, although this is the largest study to date to analyze a series of selective RET TKI-resistant biopsies, the cohort remains small in size, and the possibility of ascertainment bias cannot be excluded. Second, various NGS platforms including cfDNA assays were used to detect gene alterations, with no standardized definition for calling gene amplification (such as MET amplification). This was a limitation inherent to the retrospective analysis of real-world, clinical genotyping results. Second, it is plausible that the spectrum and relative frequencies of detected resistance alterations may vary with longer follow-up, particularly if certain alterations are associated with an earlier versus more delayed onset of resistance. It should be noted that the median PFS and duration of therapy in this cohort were 6.3 months and 7.2 months, respectively, which are shorter than has been reported from the phase I/II trials of selpercatinib and pralsetinib.[15, 16] Thus, this cohort may have been biased towards early progressors, and further, larger studies are needed with additional resistant biopsies and functional studies. Our analysis was also limited to genetic alterations detected through different assays and did not assess for non-genetic mechanisms of resistance that may additionally have a role in RET fusion-positive lung cancer. While histologic transformation—such as from adenocarcinoma to squamous cell or small cell histology as identified in resistant EGFR-mutant or ALK fusion-positive lung cancer[24, 25, 36–39]—was not observed in our series, we speculate that this was likely due to the relatively low frequency of such events and a small number of cases analyzed herein. Despite these limitations, our study offers important early insights into the relative prevalence and spectrum of mechanisms of resistance to RET-selective inhibitors.
In summary, we demonstrated that RET resistance mutations, though recurrent, are identified in a low frequency of RET fusion-positive NSCLC after progression on selpercatinib or pralsetinib. The majority of resistance appears to be driven by RET-independent mechanisms, such as MET amplification or KRAS amplification detected in our series. Moving forward, it will be important to continue to assess and validate mechanisms of resistance in larger cohorts of RET-altered solid tumors. Our findings should help inform the development of next-generation RET inhibitors and other treatment approaches such as combination strategies, with the goal of overcoming resistance and improving outcomes in patients with RET fusion-positive lung cancer.
Supplementary Material
HIGHLIGHTS.
Resistance is a major challenge in RET fusion-positive lung cancer treated with RET tyrosine kinase inhibitors (TKIs).
RET mutations involving the solvent front residue G810 are a recurrent yet infrequent mechanism of resistance to RET TKIs.
The majority of resistance to selective RET inhibition is driven by RET-independent resistance, such as MET amplification.
RET TKIs with potency against RET solvent front mutations and combination strategies are needed to overcome resistance.
Funding Sources:
This work was supported by Be a Piece of the Solution and by the Targeting a Cure for Lung Cancer Research Fund at MGH. LVS is partially supported by funding from R01-CA137008.
Disclosures:
JJL has served as a compensated consultant or received honorarium from Chugai Pharma, Boehringer-Ingelheim, Pfizer, C4 Therapeutics, Nuvalent, Turning Point Therapeutics, Blueprint Medicines, and Genentech; received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Roche/Genentech, Pfizer, and Novartis; received CME funding from OncLive, MedStar Health, and Northwell Health; and received travel support from Pfizer.
SVL served as a compensated consultant or on the advisory board for AstraZeneca, Blueprint, Boehringer-Ingelheim, Bristol-Myers Squibb, Celgene, G1 Therapeutics, Genentech/Roche, Guardant Health, Inivata, Janssen, Jazz, Lilly, Merck/MSD, PharmaMar, Pfizer, Regeneron, and Takeda; and received institutional research funding from Alkermes, AstraZeneca, Blueprint, Bristol-Myers Squibb, Corvus, Genentech, Lilly, Merck, Pfizer, Rain Therapeutics, RAPT, Spectrum, and Turning Point Therapeutics.
CEM received honorarium from Novartis and Guardant Health; served on the advisory board for Genentech; and received research funding from Novartis and Revolution Medicines.
ACT has served as a compensated consultant or received honorarium from ThermoFisher.
IDJ has received honoraria from Foundation Medicine, consulting fees from Boehringer Ingelheim and AstraZeneca; research support from Array, Genentech, Novartis, Pfizer, and Guardant Health; and travel support from Array and Pfizer.
LJW has received personal fees from Bayer, Blueprint Medicines, Cue Biopharma, Exelexis, Genentech, and Rakuten Medical; received personal fees and non-financial support from Eisai, Lilly, Loxo Oncology, and Merck; and received institutional research funding from Loxo Oncology.
ANH has received research support from Amgen, Pfizer, Novartis, Blueprint Medicines, Eli Lilly, Roche/Genentech, and Relay Therapeutics.
MMK has served as a compensated consultant for H3 Biomedicine and AstraZeneca; received institutional research support from Novartis.
SIO has received personal fees from Pfizer, Merck, Takeda, AstraZeneca, Roche/Genentech, Daiichi Sankyo, Blueprint Medicines, and Janssen JNJ; and has stock ownership in Turning Point Therapeutics.
DTSW has served as a compensated consultant or received honorarium from Amgen, Boehringer-Ingelheim, Pfizer, C4 Therapeutics, Takeda, Bristol-Myers Squibb, MSD, Bayer, and Novartis; received institutional research funds from AstraZeneca, Pfizer, and Amgen; and received travel support from Pfizer and MSD.
JFG has served as a compensated consultant or received honoraria from Bristol-Myers Squibb, Genentech, Ariad/Takeda, Loxo/Lilly, Blueprint, Oncorus, Regeneron, Gilead, AstraZeneca, Pfizer, Incyte, Novartis, Merck, Agios, Amgen, and Array; research support from Novartis, Genentech/Roche, and Ariad/Takeda; institutional research support from Bristol-Myers Squibb, Tesaro, Moderna, Blueprint, Jounce, Array Biopharma, Merck, Adaptimmune, Novartis, and Alexo; and has an immediate family member who is an employee of Ironwood Pharmaceuticals. All remaining authors have declared no conflicts of interest.
REFERENCES
- 1.Kohno T, Ichikawa H, Totoki Y et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med 2012; 18: 375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeuchi K, Soda M, Togashi Y et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012; 18: 378–381. [DOI] [PubMed] [Google Scholar]
- 3.Ju YS, Lee WC, Shin JY et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res 2012; 22: 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipson D, Capelletti M, Yelensky R et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 2012; 18: 382–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gautschi O, Milia J, Filleron T et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J Clin Oncol 2017; 35: 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol 2018; 15: 151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drilon A, Rekhtman N, Arcila M et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016; 17: 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SH, Lee JK, Ahn MJ et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: a phase II clinical trial. Ann Oncol 2017; 28: 292–297. [DOI] [PubMed] [Google Scholar]
- 9.Yoh K, Seto T, Satouchi M et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med 2017; 5: 42–50. [DOI] [PubMed] [Google Scholar]
- 10.Hida T, Velcheti V, Reckamp KL et al. A phase 2 study of lenvatinib in patients with RET fusion-positive lung adenocarcinoma. Lung Cancer 2019; 138: 124–130. [DOI] [PubMed] [Google Scholar]
- 11.Drilon A, Fu S, Patel MR et al. A Phase I/Ib Trial of the VEGFR-Sparing Multikinase RET Inhibitor RXDX-105. Cancer Discov 2019; 9: 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gainor JF, Gadgeel S, Ou S-HI et al. A Phase II Study of the Multikinase Inhibitor Ponatinib in Patients With Advanced, RET-Rearranged NSCLC. JTO Clinical and Research Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subbiah V, Velcheti V, Tuch BB et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 2018; 29: 1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subbiah V, Gainor JF, Rahal R et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov 2018; 8: 836–849. [DOI] [PubMed] [Google Scholar]
- 15.Drilon A, Oxnard G, Wirth L et al. PL02.08 Registrational Results of LIBRETTO-001: A Phase 1/2 Trial of LOXO-292 in Patients with RET Fusion-Positive Lung Cancers. Journal of Thoracic Oncology 2019; 14: S6–S7. [Google Scholar]
- 16.Gainor JF, Curigliano G, Kim D-W et al. Registrational dataset from the phase I/II ARROW trial of pralsetinib (BLU-667) in patients (pts) with advanced RET fusion+ non-small cell lung cancer (NSCLC). Journal of Clinical Oncology 2020; 38: 9515–9515. [Google Scholar]
- 17.Solomon BJ, Tan L, Lin JJ et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J Thorac Oncol 2020; 15: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu VW, Madison R, Schrock AB, Ignatius Ou S-H. Emergence of High Level of MET Amplification as Off-Target Resistance to Selpercatinib Treatment in KIF5B-RET NSCLC. Journal of Thoracic Oncology 2020; 15: e124–e127. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Z, Liebers M, Zhelyazkova B et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014; 20: 1479–1484. [DOI] [PubMed] [Google Scholar]
- 20.Cheng DT, Mitchell TN, Zehir A et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015; 17: 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afshar AR, Damato BE, Stewart JM et al. Next-Generation Sequencing of Uveal Melanoma for Detection of Genetic Alterations Predicting Metastasis. Transl Vis Sci Technol 2019; 8: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crystal AS, Shaw AT, Sequist LV et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014; 346: 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 24.Piotrowska Z, Isozaki H, Lennerz JK et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov 2018; 8: 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoenfeld AJ, Chan JM, Kubota D et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin Cancer Res 2020; 26: 2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dagogo-Jack I, Yoda S, Lennerz JK et al. MET Alterations Are a Recurring and Actionable Resistance Mechanism in ALK-Positive Lung Cancer. Clin Cancer Res 2020; 26: 2535–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valtorta E, Misale S, Sartore-Bianchi A et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer 2013; 133: 1259–1265. [DOI] [PubMed] [Google Scholar]
- 28.Bahcall M, Awad MM, Sholl LM et al. Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 2018; 24: 5963–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hrustanovic G, Olivas V, Pazarentzos E et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med 2015; 21: 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gainor JF, Dardaei L, Yoda S et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 2016; 6: 1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Awad MM, Katayama R, McTigue M et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med 2013; 368: 2395–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dagogo-Jack I, Stevens SE, Lin JJ et al. Emergence of a RET V804M Gatekeeper Mutation During Treatment With Vandetanib in RET-Rearranged NSCLC. J Thorac Oncol 2018; 13: e226–e227. [DOI] [PubMed] [Google Scholar]
- 33.Sequist LV, Han JY, Ahn MJ et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol 2020; 21: 373–386. [DOI] [PubMed] [Google Scholar]
- 34.Gautschi O, Menon R, Bertrand M et al. Capmatinib and Osimertinib Combination Therapy for EGFR-Mutant Lung Adenocarcinoma. J Thorac Oncol 2020; 15: e13–e15. [DOI] [PubMed] [Google Scholar]
- 35.Subbiah V, Gainor JF, Oxnard GR et al. Intracranial activity of selpercatinib (LOXO-292) in RET fusion-positive non-small cell lung cancer (NSCLC) patients on the LIBRETTO-001 trial. Journal of Clinical Oncology 2020; 38: 9516–9516. [Google Scholar]
- 36.Sequist LV, Waltman BA, Dias-Santagata D et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011; 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyamoto S, Ikushima S, Ono R et al. Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib. Jpn J Clin Oncol 2016; 46: 170–173. [DOI] [PubMed] [Google Scholar]
- 38.Fujita S, Masago K, Katakami N, Yatabe Y. Transformation to SCLC after Treatment with the ALK Inhibitor Alectinib. J Thorac Oncol 2016; 11: e67–72. [DOI] [PubMed] [Google Scholar]
- 39.Takegawa N, Hayashi H, Iizuka N et al. Transformation of ALK rearrangement-positive adenocarcinoma to small-cell lung cancer in association with acquired resistance to alectinib. Ann Oncol 2016; 27: 953–955. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.