

Abstract
Application of materials capable of energy harvesting to increase the efficiency and environmental adaptability is sometimes reflected in the ability of discovery of some traces in an environment―either experimentally or computationally―to enlarge practical application window. The emergence of computational methods, particularly computer‐aided drug discovery (CADD), provides ample opportunities for the rapid discovery and development of unprecedented drugs. The expensive and time‐consuming process of traditional drug discovery is no longer feasible, for nowadays the identification of potential drug candidates is much easier for therapeutic targets through elaborate in silico approaches, allowing the prediction of the toxicity of drugs, such as drug repositioning (DR) and chemical genomics (chemogenomics). Coronaviruses (CoVs) are cross‐species viruses that are able to spread expeditiously from the into new host species, which in turn cause epidemic diseases. In this sense, this review furnishes an outline of computational strategies and their applications in drug discovery. A special focus is placed on chemogenomics and DR as unique and emerging system‐based disciplines on CoV drug and target discovery to model protein networks against a library of compounds. Furthermore, to demonstrate the special advantages of CADD methods in rapidly finding a drug for this deadly virus, numerous examples of the recent achievements grounded on molecular docking, chemogenomics, and DR are reported, analyzed, and interpreted in detail. It is believed that the outcome of this review assists developers of energy harvesting materials and systems for detection of future unexpected kinds of CoVs or other variants.
Keywords: chemogenomics, computational drug discovery, coronavirus disease, Covid‐19, drug repositioning, polypharmacology
Chemogenomics and DR have become efficient approaches to screening potential therapeutic drugs for COVID‐19.
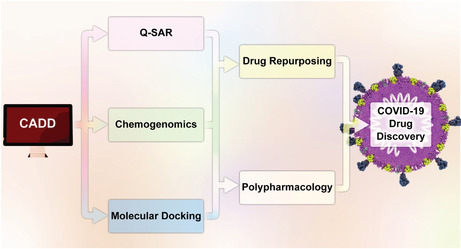
1. INTRODUCTION
Tight and selective interaction between ligands and target proteins is of cardinal importance in drug discovery that various stages of identification and validation of targets and potential drug leads, as well as manifold preclinical and clinical trials, should be carried out until obtaining the final approval from the Food and Drug Administration (FDA) (Abel et al., 2017; Campillos et al., 2008; Olayan et al., 2018). Accurate prediction of drug–target interactions (DTIs) plays a vital role for in silico and modern drug discovery in developing new drug candidates (Olayan et al., 2018). Unfortunately, despite the promising advancements in genomics, proteomics, and systems biology, formidable scientific and regulatory obstacles such as high attrition rates, excessively time‐consuming, and high‐priced procedures have taken a heavy toll on proposing effective biologically active agents, and the success of drugs to pass clinical trials stage is about only 13% (Fotis et al., 2018; Liu et al., 2022; Murray & Rees, 2009). Such attempts enable scientists to design energy harvesting materials and systems for the sake of higher efficiency and enlarging practical window (Massetti et al., 2021; Safaei et al., 2019).
Accordingly, computer‐aided drug discovery (CADD) has revolutionized the history of drug discovery, especially regarding its substantial advantages (Schneider & Fechner, 2005). These favors are (a) giving more information about the beneficial interaction between the drug and target and the availability of three‐dimensional (3D) structure, (b) providing a cost‐efficient way to reduce failures for high‐throughput screening (HTS), (c) producing new ideas for rationally drug design, and (d) leading to rationally anticipate the targeted protein and candidate hits (Bajorath, 2002; Bleicher et al., 2003; Ou‐Yang et al., 2012; Sliwoski et al., 2014). These strategies are currently classified into three major categories: ligand‐based, structure‐based, and chemogenomic approaches (Imam & Gilani, 2017), as summarized in Figure 1.
FIGURE 1.
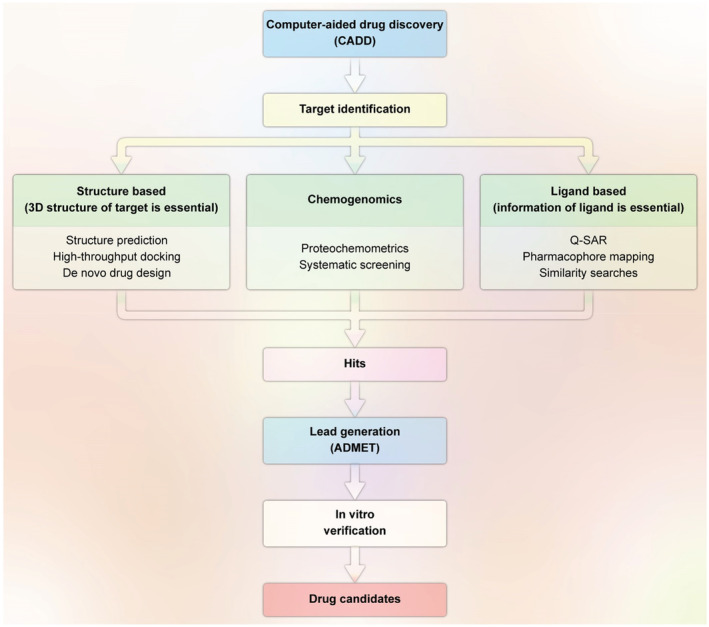
Illustration of the CADD pipeline based on the availability or unavailability of 3D information of the target. The ultimate aim of this process is to obtain a lead compound to be identified as a drug candidate when the desired results from in vitro/in vivo testing are expected (redesigned by the authors of present work with permission from; Imam & Gilani, 2017)
Most importantly, recent studies have illuminated the potential roles of CADD in designing new drug candidates for COVID‐19 treatment. Coronaviruses (CoVs) are cross‐species viruses and can spread expeditiously into new host species, which in turn cause epidemic diseases. The whole members of the CoV family have a similar genome sequence, and the severe acute respiratory syndrome CoV (SARS‐CoV) and Middle East respiratory syndrome CoV (MERS‐CoV) are considered the two prototypical well‐known CoVs (Aghamirza Moghim Aliabadi et al., 2022; Seidi et al., 2021; Yin & Wunderink, 2018). The new CoV, identified as COVID‐19 and caused by SARS‐CoV‐2, has a very similar genome sequence to SARS‐CoV (C. Wang et al., 2020). The lack of effective drugs led to considerable unwholesomeness and death rate of SARS‐CoV‐2, especially in 2020 and 2021, when a novel coronavirus strain brought about a fatal respiratory illness. Since the outgrowth of the SARS‐CoV‐2, more than 528 million people have been infected with up to 6.3 million deaths, as reported by the WHO (https://covid19.who.int/).
Although effective vaccines have amazingly reduced the COVID‐19 cases recently, they will not change the world right away, and importantly, their permanent efficacy, short‐term and long‐term side effects, and long‐term protection against different variants of SARS‐CoV‐2 are still under debate. These issues are accompanied by mass vaccination campaigns (Wen et al., 2022). Furthermore, several studies have reported that vaccinated people can also be infected with novel coronavirus variants (Largent & Miller, 2021). Therefore, the design and introduction of new drugs, either the current drugs or synthesized agents, for eradicating the SARS‐CoV‐2 is of extreme concern. To accelerate this process, repositioning broad‐spectrum antiviral drugs can be a potential strategy concerning the presence of the pharmacokinetic, pharmacodynamic, and toxicity data of these drugs (Serafin et al., 2020).
For instance, the efficacy of some antiviral drugs with previously known treatment of hepatitis, HIV, influenza, and Ebola viral diseases has been investigated. These agents, including adefovir, foscarnet, tenofovir, sofosbuvir, and uprifosbuvir, have phosphoramide and phosphate with various azole rings in their structures. By assessing their effectiveness against COVID‐19, remdesivir was the one that grabbed much attention and is believed to be the most successful drug for the therapy for combating COVID‐19 (Brown et al., 2019; Gholivand et al., 2022). Further potential macromolecular targets of SARS‐CoV‐2 are depicted in Figure 2. Therefore, finding the drugs that can target this enzyme may decrease the possibility of mutation‐mediated drug resistance and show significant antiviral activity (Pachetti et al., 2020).
FIGURE 2.
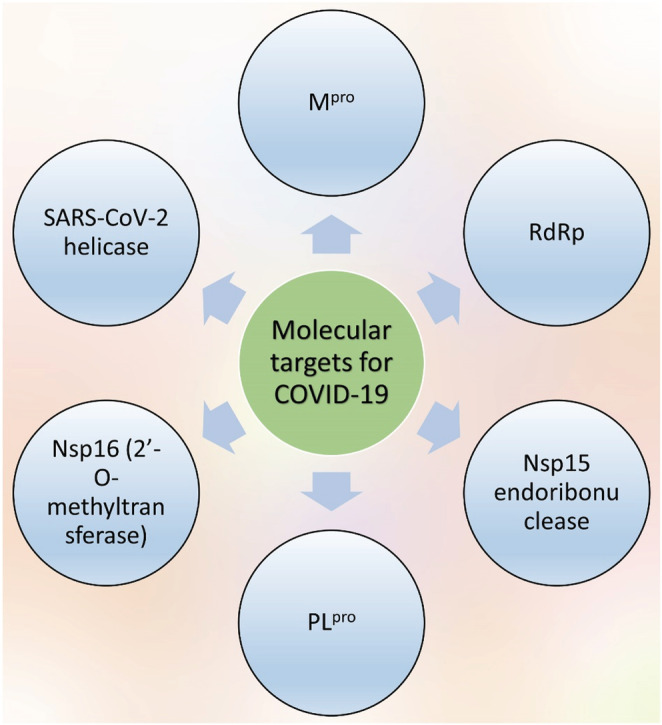
Potential drug targets of SARS‐CoV‐2 . There are different potential drug targets in case of use for SARS‐CoV‐2 including Mpro, RdRp, Nsp15, PLpro, Nsp16, and the helicase forms
Thus, it is likely that CADD might be able to provide us with effective small molecules targeting the main receptors of SARS‐CoV‐2 with fully therapeutical and preventional‐oriented outlooks (Muratov et al., 2021). The more recent update of FDA approval for COVID‐19 drug discovery shows that authorization of remdesivir (inhibits the RNA‐dependent RNA polymerase (RdRp) of the virus), molnupiravir (inhibition of viral reproduction through enhancing the vast mutations in the replication of viral RNA) and paxlovid (3c‐like protease inhibitor) for the treatment of mild‐to‐moderate COVID‐19 (https://www.fda.gov/drugs/emergency‐preparedness‐drugs/coronavirus‐covid‐19‐drugs). These drugs are used for various viral diseases such as hepatitis C, Ebola virus, influenza, etc. (Vangeel et al., 2022). Molnupiravir and remdesivir acted on the RdRp enzyme employed by the virus to increase the transcription and replication capability of the viral RNA genome. While some groups reported the promising results of remdesivir, others showed a lack of efficacy, and therefore, the WHO did not suggest using it (Mahase, 2021).
Our focus in this paper is to explore the role of CADD on COVID‐19; most specifically, chemogenomics and DR, with potential examples of COVID‐19 drug discovery. Unfortunately, with boundless energy and time to discover a successful small molecule by utilizing various databases to inhibit the main protease (Mpro) of SARS‐CoV‐2 via several virtual screening (VS) approaches, there are no effective drugs that have been approved. Only safety protocols such as wearing face masks, face shields, social distancing, and washing hands frequently are the best approaches to minimize the illness's danger and prevent transmission.
Basically, the process of bringing an FDA‐approved medicine to market for COVID‐19 drug discovery is extremely challenging, starting from the initial research of scientists for target identification and lead compound generation. After lead discovery and development of drug‐like candidates and investigation of in vitro and in vivo results, the process will go into three phases, i.e., preclinical and, if successful, into clinical studies, and ultimately a marketed medicine after FDA approval (Schaduangrat et al., 2019). It is where CADD is evolved as a valuable and powerful method to aid the challenging tasks of identifying small bioactive molecules and predicting targets.
2. COMPUTATIONAL DRUG DISCOVERY APPROACHES FOR COVID‐19
In 1981, an influential article appeared in “Fortune magazine” titled “Next Industrial Revolution: Designing Drugs by Computer at Merck”. This article led to a realization of the importance of in silico studies in drug discovery and manifestation of CADD (Sliwoski et al., 2014). Since then, high‐throughput screening (HTS) has been increasingly used in the pharmaceutical sphere and academic institutions to the rapid discovery of hit and lead compounds. This potential and automated approach enables us to screen the entire compound library of either number of chemicals or biological compounds in a target‐based assay or even a cell‐based assay to obtain the desired biological response (Bittker & Ross, 2016; Maghsoudi et al., 2021). It can spring to the mind that the stringent requirement of large resources of targets and ligands in HTS makes it expensive and time‐consuming, and we should overlook the use of the useless ligands having no probability of showing success. But, the CADD technologies are very powerful and able to address this issue easily by employing virtual high‐throughput screening (vHTS) that uses virtual compound libraries and allows experimentalists to focus on ligands which are more likely to have any activity of interest (Leelananda & Lindert, 2016). In the first days of COVID‐19 outbreak, vHTS was employed and developed efficiently to investigate a group of approved drugs to identify the possible anti‐SARS‐CoV‐2 agents at the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH), embracing a broad spectrum of the SARS‐CoV‐2 life cycle, such as viral entry, viral replication, in vitro infectivity, and live virus infectivity (Xu et al., 2021). Many possible drug candidates were selected for targeting various stages of the SARS‐CoV‐2 life cycle. For instance, cepharanthine, an anti‐inflammatory compound, effectively showed ability to rescue the cytopathic effects (CPEs) of SARS‐CoV‐2 (Rogosnitzky et al., 2020), or corilagin, a possible antifibrosis, revealed potential against the angiotensin‐converting enzyme (ACE2) receptor‐binding domain (RBD) (Yang et al., 2021). Therefore, it is urgent to build a high bio‐safety SARS‐CoV‐2 infection model with a high ability for HTS of drugs against SARS‐CoV‐2.
2.1. Ligand‐based drug design for COVID‐19
This strategy just relies on ligand information—one of the broadly known and widely used ligand‐based approaches in QSAR modeling. The advantages accruing from the QSAR methods in drug discovery and development are noteworthy. This computational technique attempts to construct mathematical models of compounds' physical and chemical properties associated with their chemical structure. QSAR studies are extensively employed to predict pharmacokinetic properties such as ADME and toxicity by building a training set of measured properties of known compounds and making a mathematical model of the encoded chemical structure, and also is useful in sensory applications as well (Alam & Khan, 2019; Farshchi et al., 2021; Kholafazad‐Kordasht et al., 2021; Nosrati et al., 2022; Seidi et al., 2021).
Generating molecular descriptors from compound structures is also significant in assessing compounds' different physical, chemical, and topological features. After computing molecular descriptors, machine learning techniques play indispensable roles in the quality of QSAR predictions (Popelier & Smith, 2006; Rad & Maghsoudi, 2016; Sliwoski et al., 2014). Using machine learning and QSAR, Muratov et al. identified 32 drugs and their 73 selected binary combinations for testing against SARS‐CoV‐2. They found that the combinations of four of these drugs, including nitazoxanide, remdesivir, amodiaquine, and umifenovir, could synergize against SARS‐CoV‐2. Interestingly, the combination of nitazoxanide and remdesivir showed a great synergistic interaction, and the combination of remdesivir and hydroxychloroquine exhibited a powerful antagonist role against SARS‐CoV‐2 (Bobrowski et al., 2021).
The two proteins pp1a and pp1ab are quickly translated after entry into the host cells and further separated by two viral proteases, 3 C‐like proteases (3CLpro) and papain‐like protease (PLpro) (Amin et al., 2021). The latter is a critical virus enzyme responsible for producing a functional replicase complex and stimulating viral spread (Shin et al., 2020). Amin et al. used chemical‐informatics approaches to build a Monte Carlo optimization based on (a) QSAR data of various PLpro inhibitors, (b) VS to choose the best in‐house molecules among 67 with the high likelihood of targeting against PLpro, and (c) eventually verification of these inhibitors by using receptor–ligand interaction investigation. Out of 67 compounds, 56 were anticipated as active from a QSAR model. Afterward, these 56 similar compounds were screened through SwissADME, and eventually, 13 compounds were approved for the ADME criteria. Furthermore, molecular docking has proved the potentiality of these candidates against SARS‐CoV‐2 PLpro (Figure 3; Amin et al., 2021).
FIGURE 3.
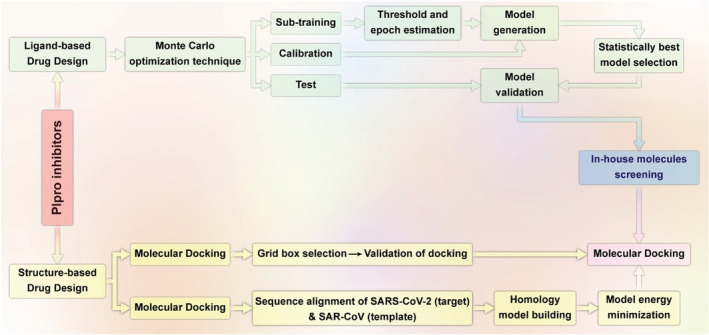
Illustration of ligand‐ and structure‐based drug design on SARS‐CoV‐2 PLpro (redesigned by the authors of present work with permission from Amin et al. (2021))
In an interesting work, Tropsha et al. developed open‐source data QSAR models of 42 inhibitors from DrugBank by employing QSAR models, docking, and similarity searching and utilized these models for virtual screening of all drugs in the DrugBank database. The 42 virtual hits were finalized for availability and cost using in‐house ZINC Express software. The National Center for Advancing Translational Sciences (NCATS) analyzed 11 of total 42 hits which were potential compounds, and three of them, cenicriviroc (AC50 of 8.9 μM), proglumetacin (tested twice independently, with AC50 of 8.9 μM and 12.5 μM), and sufugolix (AC50 12.6 μM). While molecular docking was unsuccessful to accurately distinguish between experimentally active and inactive compounds, QSAR was able to identify three inhibitors of SARS‐CoV‐2 replication through Mpro inhibition. Overall, these results show that QSAR models proposed using SARS‐CoV‐2 Mpro data can be useful to introduce compounds active against SARS‐CoV‐2 (Alves et al., 2021).
2.2. Structure‐based drug design and molecular docking
This strategy relies more on target information. The structure‐based drug design, also called target‐based drug design, rests on the assumption that the bioactivity of a ligand comes from the physical interaction of the ligand with the corresponding target protein. The prerequisite of this approach is the 3D structure of the biological target, obtained from crystal structures, NMR data, or molecular modeling. Molecular docking is an established structure‐based approach frequently used to predict the binding affinity and analyze the interactive mode between a ligand and a target/receptor. In other words, docking enables us to identify novel drug candidates by predicting ligand–target interactions at a molecular level (Pinzi & Rastelli, 2019; Sargazi et al., 2021).
There are several references in which the significant role and applications of docking in drug design and development have been discussed (De Vivo & Cavalli, 2017; Keretsu et al., 2020; Kitchen et al., 2004; Meng et al., 2011). Furthermore, in the current spread of SARS‐CoV‐2, molecular docking is important in screening potential drugs (Keretsu et al., 2020; Lokhande et al., 2020; Marinho et al., 2020; Yu et al., 2020). The recent work of Khelfaoui et al. (2020) could be a good example of using molecular docking combined with MD to find promising candidate drugs against COVID‐19. They selected a library of 18 drugs that three of which include ramipril, delapril, and lisinopril, bound to angiotensin‐converting enzyme 2 (ACE2) receptor and [SARS‐CoV‐2/ACE2] complex well. ACE2 is one of the receptors in the human body with which the S proteins from coronaviruses have a great binding affinity and serve as the entry point into cells for SARS‐CoV‐2. In fact, due to the long time required to discover specific antiviral agents or a vaccine, the best strategy would be to repurpose existing drugs to treat COVID‐19, which will be discussed in the DR section.
Using VS and molecular docking, Abdellatiif et al. identified some promising antimicrobial and chemotherapeutic agents having coumarin and pyridine derivatives in their structures. In order to identify the most potential drug–target interactions, a molecular operating environment (MOE) program was carried out against the human ACE2, SARS‐CoV‐main peptidase, serine protease hepsin, human coronavirus papain‐like proteases, and SARS‐Coronavirus NSP12. As shown in Figure 4, among compounds a, b, c, d, and e with binding score energy values of −6.08, −6.48, −6.78, −7.08, and −6.63, respectively, compound d had the binding score energy against the aforementioned targets. Thus, compound d needs to be additionally evaluated in clinical trials as a candidate for potential inhibitor of the SARS‐CoV‐2 (Abdellatiif et al., 2021).
FIGURE 4.
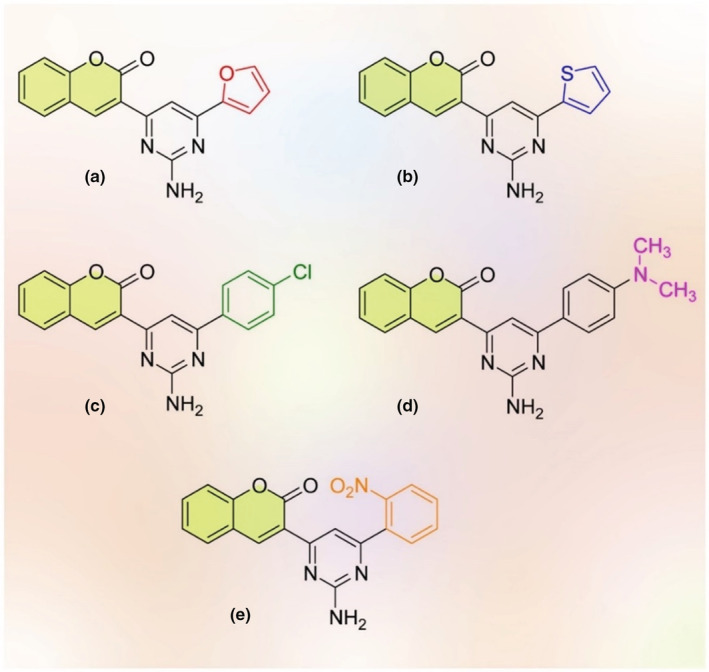
The structures of docked molecules docked by MOE having coumarin (yellow rings) compounds (redesigned by the authors of present work with permission from Abdellatiif et al. (2021))
Hosseini et al. evaluated a virtual screening using docking of 1615 FDA‐approved drugs to identify novel potential inhibitors for protease protein of COVID‐19. They showed that among all investigated FDA‐approved drugs, simeprevir, a Hepatitis C virus (HCV) NS3/4A protease inhibitor, manifested high affinity to protease binding pocket and placed effectively into the binding pocket more beneficially than the lopinavir‐ritonavir. This study suggested this compound as a potential inhibitor of COVID‐19 (Hosseini & Amanlou, 2020). In another docking study, Motiwale et al. worked to identify potential inhibitors against Mpro of SARS‐CoV‐2 from previously reported SARS‐3CL protease inhibitors where among 61 previously reported potential inhibitors, 4‐benzoic acid and 4‐(4‐methoxyphenyl)‐6‐oxo‐2‐[(2‐phenylethyl)sulfanyl]‐1,6‐dihydropyrimidine‐5‐carbonitrile revealed a minimum and maximum binding energy, respectively (Motiwale et al., 2022).
Table 1 presents a selection of recent studies that researched the docking strategy of different pharmacological classes as treatments of SARS‐Co and SARS‐CoV‐2. Although many studies are carried out and many drugs are being tested to open a new road for effective treatment of COVID‐19, there are no fully beneficial antiviral drugs against the disease. Many of these studies are now in clinical trials to assess their safety and efficacy against infected patients.
TABLE 1.
Recent studies used the docking strategy of different pharmacological classes against COVID‐19
| Biomolecule | Target proteins | Therapeutic assessment | Refs. |
|---|---|---|---|
| Curcumin | Spike glycoproteins, nucleocapsid phosphoprotein, membrane glycoprotein & nsp10, RNA‐dependent RNA polymerase |
Promising binding affinity against nucleocapsid & nsp10 High antiviral activity |
Suravajhala et al. (2020) |
| Ivermectin | 3CLpro and S protein | Disrupting viral replication and attachment. | Low et al. (2022) |
| Sofosbuvir, Ribavirin, Galidesivir, Remdesivir, Favipiravir, Cefuroxime, Tenofovir, Hydroxychloroquine, and IDX‐184 | RdRp |
Tightly binds to RdRp active site Setrobuvir, YAK, and IDX‐184 showed higher affinity to RdRp |
Elfiky (2021) |
| Iron oxide nanoparticles (Fe2O3 and Fe3O4) | Spike protein receptor‐binding domain (S1‐RBD) | Effective interaction with the S1‐RBD | Abo‐Zeid et al. (2020) |
| Teicoplanin | Cathepsin L |
Blocking the Cathepsin L Preventing virus entrance into the cytoplasm |
Vimberg (2021) |
| Viomycin | 3CLpro |
High ‐CDocker energy High H‐bonds with Mpro Placed well in the binding pocket |
Mahanta et al. (2021) |
| Jensenone | Mpro/chymotrypsin‐like protease (3CLpro) | A strong complex formed between Mpro/Jensenone | Sharma and Kaur (2020) |
| Leupeptin, hemisulphate, pepstatin A, nelfinavir, lypression, birinapant, and octreotide | Mpro |
Have significant MM‐GBSA score Forming stable interactions with hot‐spot residues |
Mittal et al. (2021) |
| Terpenoid (T3) from marine sponge Cacospongia mycofijiensis | Mpro | Remarkable SARS CoV‐2 Mpro inhibitory activity | Sabe et al. (2021) |
| Amodiaquine & Ribavirin | Mpro | Great affinity with a high‐lying HOMO, electrophilicity index, basicity, & dipole moment. | Hagar et al. (2020) |
| Eucalyptol (1,8 cineole) | Mpro/3CLpro | Strong complex formed between Mpro/eucalyptol | Sharma (2020) |
| Dithymoquinone (DTQ) | ACE2 | High affinity and stability at SARS‐CoV‐2:ACE2 | Ahmad et al. (2020) |
| Zanamivir, Indinavir, Saquinavir, & Remdesivir | Spike glycoprotein & the 3CL protease | Potential 3CLpro proteinase inhibitors | Hall Jr and Ji (2020) |
| Nigellidine, & α‐ Hederin | 3CLpro/Mpro | Strong inhibitor effect on 3CLpro/Mpro | Bouchentouf and Missoum (2020) |
2.3. Chemogenomic approaches for COVID‐19 drug discovery
Chemical genetics, chemical biology, and chemogenomics (chemical genomics) are some cutting‐edge applied fields in target and drug discovery (Rognan, 2007; Urán Landaburu et al., 2020). Among the presented strategies, chemogenomics is an emerging discipline that principally investigates all molecules that can interact with any biological target. In humans, the number of the gene is about 26,000, whereas the amount of various proteins is approximately more than 1 million (Andersson et al., 2011). However, due to the extensive number of available molecules and biological targets, chemogenomics is limited to screening congeneric compound libraries against a selected target family. This alternative proposes an unprecedented chance to seek drug‐like compounds with a noteworthy target or significant subtype (Jones & Bunnage, 2017; Kubinyi, 2006).
Chemogenomics is different from the traditional pharmaceutical methods due to linking the widespread software library directly to the methodology of modern genomics. In order to achieve this purpose, chemogenomics, along with a contribution of several chemical disciplines, including synthetic chemistry, combinatorial chemistry, cheminformatics, bioinformatics, and genomics, screen synthetic compound libraries for their impact on biological targets (Bredel & Jacoby, 2004; Quinlan & Brennan, 2021). The ultimate goal of chemogenomics is the rapid screening as well as the rational development of target‐specific chemical ligands to decrease the cost and enhance the drug discovery period, especially in the case of COVID‐19 drug investigation (Jones & Bunnage, 2017).
Basically, the main role of chemogenomics is unifying the discovery of ligands and targets by taking advantage of active compounds as probes to identify proteome functions. Similar to genetics, and if the operation of investigation arises from the target to phenotype, or vice‐versa, there are two concepts: (a) forward and (b) reverse. In ‘forward chemogenomics’, the ligand is discovered through their phenotypic condition that could affect the entire biological system instead of the base of their mechanism of suppression of a certain desired protein. In other words, the molecular basis of the phenotype interest is unknown and phenotypic screening is carried out in cells of organisms having a single‐cell or multicellular utilizing a group of ligands. (Figure S1).
On the other hand, in “reverse chemogenomics,” the desired sequence of genes is cloned and explicit at the first step as target proteins; afterward, they are preselected in a high‐throughput screening (HTS) ‘target‐based’ way by a panel of compounds. Here, the assays are normally classified as (i) organismal assays, (i) cell‐free, and (ii) cell‐based organismal assays.
Cell‐free assays are generally straightforward but precise and extremely automated. They are comprehensive binding trials in which manifold compounds are examined concurrently for their binding affinity toward a variety of particular goals. Here, DTIs are clearly recognized in the absence of confounding variables. However, in cell‐based and organismal assays, chosen compounds are delivered right to cells or organisms in vitro to select hits within a relevant cellular context. However, the hits need further mechanistic characterization due to simultaneously interacting with multiple targets.
The former approach is usually employed in reverse chemogenomics, whether target information is at hand, while organismal assays and cell‐based are mainly employed in forwarding chemogenomics to test a wide variety of compound effects on whole biological systems (Figure S1) (Bredel & Jacoby, 2004).
In forward chemogenomics, cells or organisms are distributed in multi‐well cell culture plates for incubation. Subsequently to the incubation step, an aliquot from donor plates is taken to the recipient and mock plates (mock treatment), in which the ligand–target binding assay is performed. The endpoint of most cell‐based HTS assays is a spectrophotometric measurement in a spectrophotometer equipped with suitable software that calculates the final data, including SAR analysis and in silico QC. Then, active compounds that obtain the intended changes in the phenotype are chosen to elucidate their targets at the molecular level. This strategy could be performed using affinity chromatography, microarray, phage display, or transcriptional or proteomic profiling.
In reverse chemogenomics, the main focus is to elucidate the protein families and the gene. In this regard, the sequencing of the target gene demonstrates a specific level of homology that is transfected in a host cell. These family targets are sequentially gathered, purified, and subjected to trial design. Finally, similar to forwarding chemogenomics, the measurement data on the spectrophotometer are directed to a suitable data analysis system.
2.3.1. Target fishing Chemogenomics approaches for COVID‐19 drug discovery
The biological target is extracted from the existent information in the biologically annotated chemical database in the target fishing approach (Huang et al., 2021). However this procedure needs an abundant effort from a time, cost, and resource perspective to identify all plausible chemical target associations; thus, computational target prediction techniques are strongly being pursued with the contribution of advanced data‐mining algorithms (Anwar et al., 2021). This current mode of action or the computational target approach is classified into four classes: data‐mining, pharmacophore searching (analogy searching), analysis of bioactivity spectra, and molecular docking. In the past, these methods were applied to investigate new compounds of known targets, but recently, they have been applied to predict novel targets for the known compounds via similar approaches (Campbell & Marchant, 2018; Ekins et al., 2007).
Target‐based (or receptor‐based chemogenomics) refers to the discovered biologically active compounds with phenotypically readout information. Nevertheless, no existing target data; thus, by applying the chemogenomics strategy, the hypotheses can be generated of the target by comparing the new biological compounds to the same known target (Fourches et al., 2015; Weston et al., 2019). For example, the target receptors or proteins that are necessary for replication and survival of SARS‐CoV‐2 are viral proteases involving papain‐like protease (PLpro), the 3‐chymotrypsin‐like protease (3CLpro), and Mpro, RNA‐dependent RNA polymerase (RdRp) complex, the spike (S) receptor binding glycoprotein, the nucleocapsid (N) protein, nonstructural protein (NSP), and the membrane (M) protein including transmembrane (TM) as well as small envelope E protein (Cavasotto et al., 2021).
As an example of this part, it can be referred to the work of Mishra and his coworkers that reported an inclusive computational approach for the identification of drug molecules that are multi‐targeted struggling with the SARS‐CoV‐2 proteins. These drugs are essentially included in virus replication inside the host body, the viral–host interaction, disease development, and transmission of Sars‐CoV‐2 infection. They screened 72 potential antiviral drugs approved by the FDA against the S glycoprotein, hACE2, 3CLpro, Cathepsin L, N protein, RdRp, and NSP6 proteins.
Furthermore, they investigated the molecular interaction by applying free energy landscape, simulation of Molecular Dynamics, and binding free energy estimation using MM‐PBSA. Their finding showed favorable results for seven drugs, among which catechin (flavan‐3‐ol) can effectively bind to 3CLpro, Cathepsin L, RBD of S protein, NSP‐6, and Nucleocapsid protein to be used as a potential multi‐targeted agent in combating SARS‐CoV‐2. However, due to the limited timeframe of combating this pandemic, it is not realistic to be assured of finding a definitive therapy for combating the pandemic. Hence, vigorous attempts are still continuing for the DR as a clinical treatment strategy to fight COVID‐19 (Mishra et al., 2020). In another research, Omotuyi et al. investigated molecular docking protocols for fast screening of the FDA database for high‐affinity interaction of the resolved 3D structure of SARS‐CoV‐2 receptor‐binding domain (RBD) in complex with its receptor hACE‐2 interface.
Ubrogepant, as one of the preferable candidate drugs, was studied utilizing the atomistic molecular dynamics simulation method. They reported that UBR breaks the interaction at the RBD/ACE‐2 interface residues of SARS‐CoV‐2 RBD/ACE‐2 complex that might result in protein function losses with direct inference on the oligomerization in RBD and function loss in ACE‐2. Therefore, leading to a binding and cellular receptor recognition unattainable. Therefore, UBR exhibits a novel therapeutic candidate in combating SARS‐CoV‐2 since it binds to proportionally high affinity with free RBD, ACE‐2 receptor, and SARS‐CoV‐2 RBD/ACE‐2 complex based on only binding affinity calculations (Omotuyi et al., 2020). The other studies in this area are demonstrated in Table 2.
TABLE 2.
Drug repurposing studies and computational DR for coronavirus disease (COVID‐19), drug discovery, and various diseases
| Drug(s)/name | Screening technique | Target protein | No. of drugs | Data Bank | Refs. |
|---|---|---|---|---|---|
| Carfilzomib, Eravacycline, Valrubicin, Lopinavir, and Elbasvir | Docking | Mpro | 2201 | DrugBank | Wang (2020) |
| Paritaprevir, Simeprevir, Ledipasvir, Glycyrrhizic acid, TMC‐310911 | Docking | 3CLpro, PLpro, cleavage site, HR1 and RBD | 2471 |
Protein data bank Drugbank |
Mahdian et al. (2020) |
| Binifibrate, bamifylline, rilmazafon, afatinib, ezetimibe, macimorelin, and acetate | E‐pharmacophore based virtual screening, structure based virtual screening | 3CLpro | 4600 | SuperDRUG2 database | Arun et al. (2021) |
| Sovaprevir, Danoprevir, Samatasvir, Ritonavir, Rebamipide, Zabofloxacin, Pralatrexate, … | Docking | Mpro, PLpro, and the S‐protein | 11,552 |
Protein data bank Drugbank, ChEMBL DrugCentral database of approved drugs, Selleck Chemicals library |
Cavasotto and Di Filippo (2021) |
| Melatonin, Mercaptopurine, Sirolimus | Network‐based | Membrane (M), spike (S), nucleocapsid (N) proteins envelope (E), and Replicase complex (ORF1ab) nucleocapsid proteins | 2000 | DrugBank Database (TTD), PharmGKB, ChEMBL, BindingDB89, and IUPHAR/BPS Guide Pharmacology | Zhou et al. (2020) |
| Penciclovir, Ribavirin, and Zanamivir | Docking | Spike protein, isolate spike protein RBD, NSP 10, NSP 16, main protease, and RdRp polymerase | 48 | FDA and SUS databanks | Grahl et al. (2021) |
| Drug(s)/Name | Computation | Target protein | No. of drugs | Data Bank | Refs. |
|---|---|---|---|---|---|
|
Paromomycin Phensuximide Magnesium ascorbate Tizanidine Cefotiam Voriconazole, Tobramycin Kanamycin, |
Docking | Mpro | 1490 |
UniProt Drugbank |
Sencanski et al. (2020) |
|
Lopinavir, Asunaprevir, Indinavir, Ritonavir, Remdesivir, Methisazone Paritaprevir |
Docking |
Protease inhibitor Inhibits viral DNA polymerase Protease inhibitor HIV protease inhibitor Viral RNA polymerase Inhibits mRNA and protein synthesis Inhibitor of the nonstructural protein 3‐4A (NS3/4A) serine protease |
61 |
PubChem database Protein Data Bank (http:// www.rscb.org) |
Shah et al. (2020) |
|
Talampicillin, Lurasidone, Rubitecan and Loprazolam Talampicillin, Lurasidone, ZINC000015988935, ZINC000103558522 (drug‐like molecules) |
Docking | Mpro and TMPRSS2 enzymes |
4500 30,000 (drug‐like molecules) |
ChEMBL, DrugBank and Selleckchem ZINC15 database |
Elmezayen et al. (2020) |
| Darunavir, Nelfinavir and Saquinavir |
(MOE) Docking Molecular Operating Environment |
Mpro | >2000 | drugbank.ca | Farag et al. (2020) |
|
Remdesivir, Saquinavir and Darunavir Small molecules: flavone and coumarin derivatives |
(MOE) Docking Molecular dynamics simulation |
3CLpro | 16 |
in‐house database RCSB Protein Data Bank |
Khan et al. (2020) |
| Ritonavir |
Docking Molecular dynamics simulation |
Mpro | 18 | Drug Bank | Ibrahim et al. (2021) |
| Paritaprevir, Semeprevir, Grazoprevir Velpatasvir |
Docking Supervised machine learning |
Spike protein, nucleocapsid protein, and 2′‐o‐ribose methyltransferase | 1577 | ZINC database | Kadioglu et al. (2020) |
2.3.2. Drug repurposing application of Chemogenomics for COVID‐19 drug discovery
A primary example of the beneficial application of chemogenomics is DR, which accelerates drug discovery by identifying novel clinical targets for the available approved drugs. This method possesses the distinctive merit of rapid clinical development without bearing the long‐lasting and expensive procedures included in drug discovery. Therefore, according to the advantages of these proposed drug discovery methods, many COVID research orientations have been focused on DR (Galindez et al., 2021; R. Liu et al., 2021).
Computational DR applies available drugs for new or existing diseases that do not have an absolute cure, while the available drugs were not originally designed for the novel disease treatment. Furthermore, utilizing the enormous volumes of available omics information in digital form in conjunction with in silico screening intrigued the researcher to use this appropriate ground to significantly increase the speed of shortlisting promising candidates in reply to spreading diseases like SARS‐CoV‐2. To be exemplified, Lucchetta, M. and M. Pellegrini reported a methodology for preclinical computational DR called DrugMerge, and it is based on combining several drug rankings gained via a set of Disease Active Subnetwork construction algorithms.
DrugMerge utilizes differential transcriptomic data from drug perturbation assays and cell lines/tissues of patients affected by the illnesses on the subject of a considerable gene co‐expression network. Application of DrugMerge to COVID‐19 shows that DrugMerge can mimic human expert judgment (Lucchetta & Pellegrini, 2021). Rima Hajjo and Alexander Tropsha developed a systems biology workflow for drug and vaccine repurposing. They reported that the identification of Bacillus Calmette–Guérin (BCG) and small molecules of BGC mimics involving antiviral drugs (raltegravir, emetine, and lopinavir as high confidence hits) are capable of boosting immunity and preventing emerging new viruses. Their results approved that BGC impacted the generation and maturation of naïve T cells, resulting in increasing long‐lasting trained intrinsic immune replies to combat novel viruses, including SARS‐CoV‐2 in vitro as a protective measurement from the lethal consequences (Onozuka et al., 2022). Due to the lengthy development cycle of novel drugs, DR turns into an efficient approach to screening drugs of potential therapeutic use for SARS‐CoV‐2. Che et al., in another attempt, screened potential therapeutic drugs for COVID‐19 based on a knowledge‐graph‐based DR method. In their experiment, five drugs predicted by the models exhibited efficiency in clinical treatment.
The experimental outputs prove that the model can predict drug–disease interaction effectively for normal diseases and SARS‐CoV‐2 (Lv et al., 2021). Therefore, DR presents a constant source of novel knowledge in virus biology and molecules with previously undefined antiviral properties, which can be applied as molecular tools in unknown molecular mechanisms of virus replication and pathogenesis (Mercorelli et al., 2018). Further investigation of SARS‐CoV‐2 in related research is summarized in Table 2.
2.3.3. Predicting the bio‐profile of drugs via Chemogenomics for COVID‐19 drug discovery
A fundamental role of chemogenomics is predicting the activity profile of the compounds for a group of targets that can be implemented by using ligand‐based or target‐based approaches. The ligand‐based philosophy is underlying that molecules sharing enough similarity to available biologically defined ligands have an increased probability of sharing the same biological profile. Therefore, the efforts are devoted to organizing chemical libraries with biological information, including in vitro affinity, targets, and toxicology (Harris & Stevens, 2006). As a result, designing chemical libraries is of great interest for a certain individual target or target family, which is attempted via utilizing machine‐learning methods.
Herein, concentration is on protein–ligand interaction via applying physics‐based protein‐ligand docking on machine learning. Computational chemogenomics or quantitative multiple structure–activity relationships (QMSPR) modeling consists of the compound–protein interaction space modeling of large datasets at once, usually for drug discoveries, where the available techniques chiefly incorporate escalating numbers of bioactive samples or concentrate on individual subfamilies of proteins and ligands (Cheng et al., 2012; Xue et al., 2018). However, the 3D information of protein targets is needed most of the time. Thus, the application of precise target data in chemogenomics is confined to the explicitly known X‐ray structures or structures that homology models can evaluate. As a result, many QMSAR is centered on ligand‐based studies or similarity principles (Gawriljuk et al., 2021).
In this context, compounds of phenotype‐based preselection possess merits in discovering target‐based drugs due to unclimbable and missing perceptions of the mechanism of action of the drugs. Therefore, a handful of methods can carry out new chemical compound screening which is phenotype‐based. In this regard, Thai‐Hoang Pham et al. extended a sound learning framework for a high‐throughput mechanism‐driven neural network‐based method for phenotype compound screening. They applied this framework for COVID‐19 DR. Their proposed novel data escalating technique extracts beneficial information from untrustful assays in the dataset of L1000. Furthermore, they showed the value of DeepCE by applying it to the DR of COVID‐19 and produced novel lead compounds that represented clinical evidence. Therefore, DeepCE can provide a potentially strong framework for rapid predictive modeling via using noisy omics data and screening new chemicals to modulate an organized response to illnesses (Wang et al., 2021).
A computational approach for increasing the rate of development of drugs and comprehension of the mechanism of action of small molecules is the identification of drug–target interaction (DTIs). Lee and his coworkers represented an AI‐DTI, a new method for predicting activation and inhibition of DTIs, by combining the mol2vec and genetically perturbed transcriptomes. Their finding showed that this methodology was a success in discovering about half of the DTIs for drugs that are employed for treating COVID‐19 (Lee et al., 2021). A myriad of compounds represent potential drug candidates, but merely a few of them will be formulated, approved, and registered as a drug for individual health issues which investigation for that compound will be a long, intricate, and costly process, while sometimes there is an urgent need for developing a drug like COVID‐19 pandemic (Muratov et al., 2021). Table S1 demonstrates chemogenomics related to COVID‐19 research.
To conclude, chemogenomics is an emerging interdisciplinary field at the interface of chemistry, biology, and informatics which decreases the cost and time of drug discovery dramatically via three main approaches, including target fishing, predicting the bio‐active profile of drugs, and DR. Herein, of much current importance is chemogenomics, is in COVID‐19 drug discoveries, which this review paper highlighted specifically.
3. DRUG REPOSITIONING
Drug repositioning, also known as drug repurposing, drug redirecting, drug reprofiling, and therapeutic switching, is defined as identifying new indications and therapeutic targets for existing drug compounds (Lotfi Shahreza et al., 2017). DR has received significant attention caused of its crucial role in finding and developing new therapies for complex, orphan, and chronic diseases with unknown or less effective treatments (Shameer et al., 2015). Since developing a new drug in the traditional drug discovery process is time‐consuming and takes approximately between 10 to 15 years to enter the market, repurposing existing drug compounds is capable of overcoming these shortcomings, and this type of research has been growing drastically (Figure 5) (Kumar et al., 2019). Therefore, DR plays a versatile role in developing new drugs because it can reduce the costs of the whole process and shorten the development pathways of approved or investigational drugs (Pushpakom et al., 2019). DR works with two main approaches. The first is that a drug can bind to various targets simultaneously, which is good for searching novel target sites of action for the known biological agents, and the second is that targets related to a disease are often associated with different biological processes of pathogenesis, which is important for choosing of a new indication for the known target (Badary, 2021; Kamel et al., 2022). These are indispensable in proposing new drug candidates for unprecedented diseases like COVID‐19.
FIGURE 5.
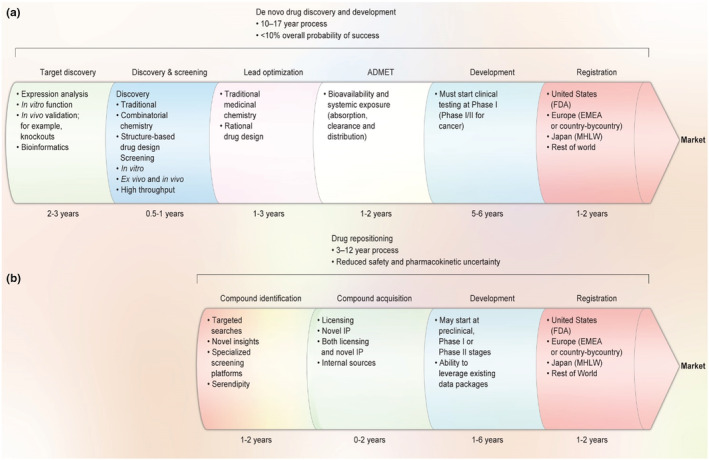
A comparison of traditional de novo drug discovery and development (a) versus DR (b); (redesigned by the authors of present work with permission from Ashburn and Thor (2004))
It has emerged as an alternative way of accelerating the process of developing new treatments against COVID‐19 (Yu et al., 2021). Based on data brought from PubMed, the number of works of literature on DR has been growing dramatically, and this number has increased since the outbreak of SARS‐CoV‐2 (Galindez et al., 2021). It should be noted that polypharmacolgy and DR are inextricably interrelated with each other (Cheng, 2019). DR has become an influential tactic from both academia and pharmaceutical industries as an alternative strategy to traditional drug discovery that is related to when new therapeutic applications are pinpointed for existing drugs (Karakida et al., 2019; Turanli et al., 2018).
Compared to traditional de novo drug discovery and development, this approach provides a new and fast way for drug development and reduces risk, particularly for drugs that cannot pass phase I clinical trials (Figure 5; Ashburn & Thor, 2004). It is able to reprocess the information on known drugs such as pharmacokinetics and safety for humans and manufacturing pathways, resulting in reduced time and expenses in traditional de novo drug discovery (Sawada et al., 2015). There are a wide variety of examples of repositioned drugs. Sildenafil is considered the most prominent one, beginning with cardiovascular treatment followed by accidentally treating erectile dysfunction clinical trial and later in low dosages, treating pulmonary hypertension (Ghofrani et al., 2006; Prasad et al., 2000). Furthermore, as repurposed drugs pass the preliminary clinical trials (phase I or safety level), the time needed for drugs to reach the market can be reduced significantly. Hence, scientists can identify the unintended secondary targets through CADD.
The history of discovering the effect of an established drug on new indications dates back to World War II, since considerable improvement had been observed for cutaneous rashes and arthritis in soldiers who were under treatment with an antimalarial medicine, Chloroquine (Schlitzer, 2007). Chloroquine is a well‐known antimalarial drug that was synthesized in 1934 and later on demonstrated successful various therapeutic effects, including anti‐inflammatory/immunomodulating, anti‐infective, antithrombotic, and metabolic. Furthermore, antitumoral properties make it available for the treatment of a number of cancers (Plantone & Koudriavtseva, 2018). In 1942, Marcel Jonbon used sulfonamide antibiotics for typhoid treatment, but he faced hypoglycemia as an unexpected side effect in some patients; therefore, sulfonamides presented the role of an anti‐diabetic drug (Quianzon & Cheikh, 2012). Aspirin is one of the most famous DR examples with an original indication as an analgesic drug that for the first time repositioned as an antiplatelet aggregation drug in 1980 and a second indication still is commonly used for cardiovascular problems (Jourdan et al., 2020). Furthermore, several types of research have been conducted on the anticancer effect of aspirin such as colorectal cancer (Drew et al., 2016), lung, and breast cancer (Li, Hu, et al., 2020). The notable potential and extensive applications of DR in novel therapies are entirely apparent and accordingly, the number of acceptable research studies which have been done or are under processing are growing every day. There are various classifications for DR methods which typically can be categorized into computational approaches and experimental approaches (Park, 2019). Experimental approaches refer to testing drugs in evaluations based on available comprehensive clinical compound databases in which the possible indications of drugs can be discovered by target‐based or cell‐based analysis (Luo et al., 2021). There are some hurdles for experimental identifications because they are mostly expensive and time‐consuming, requiring a set of marketed drugs, technical equipment, and also screen tests. In contrast to experimental methods, computational drug repositioning methods which are mainly data‐driven utilizes databases and bioinformatics tools to find out interactions between drugs, targets, and diseases (Pillaiyar et al., 2020). Analyzing a large type of data (like chemical structures, gene expression, genotype or proteomic data, and electronic health records (EHRs)) provides the possibility of understanding novel and unexpected functional gene interactions and unclear drug response or mechanisms of disease (Pushpakom et al., 2019).
3.1. Drug repositioning for COVID‐19 drug discovery
Since there is no specific and registered therapy to cure the infection of this virus, finding effective therapeutics by applying existing drugs is a pressing exigency and established strategy (Galindez et al., 2021). Concerning the time‐consuming regulatory process of the newly approved drugs, the DR strategy strongly utilizes the identification of new treatment methods by presenting FDA‐approved drugs. In addition, many repurposed drugs with acceptable results are accomplished by computational methods (Wang & Guan, 2021).
There are myriad reasons why we need to go through DR for different diseases like COVID‐19 if we intend to accelerate the process of developing a new drug. These reasons can be (i) availability of marketed drugs and their excipients and APIs for formulation and distribution, (ii) the likelihood of taking advantage of a synergistic effect of various drugs with each other, (iii) cheap and less time‐consuming process for drugs to be marketed and (IV) emergence of new therapeutic pathways for former drugs and new categories of medicine (Chung et al., 2021). Therefore, repositioned drugs have very important impacts on COVID‐19 drug discovery. Therefore, different categories of drugs are being repositioned in in vivo and in vitro studies to treat SARS‐CoV‐2 (Wu et al., 2021). These potential agents are namely Ebola virus (remdesivir), antimalarial (chloroquine), anthelmintic and antiprotozoal drugs (niclosamide), diarrhea (nitazoxanide), antiviral (lopinavir and ritonavir), antineoplastic (carfilzomib and bortezomib), and antibacterial (azithromycin) (Figure 6).
FIGURE 6.
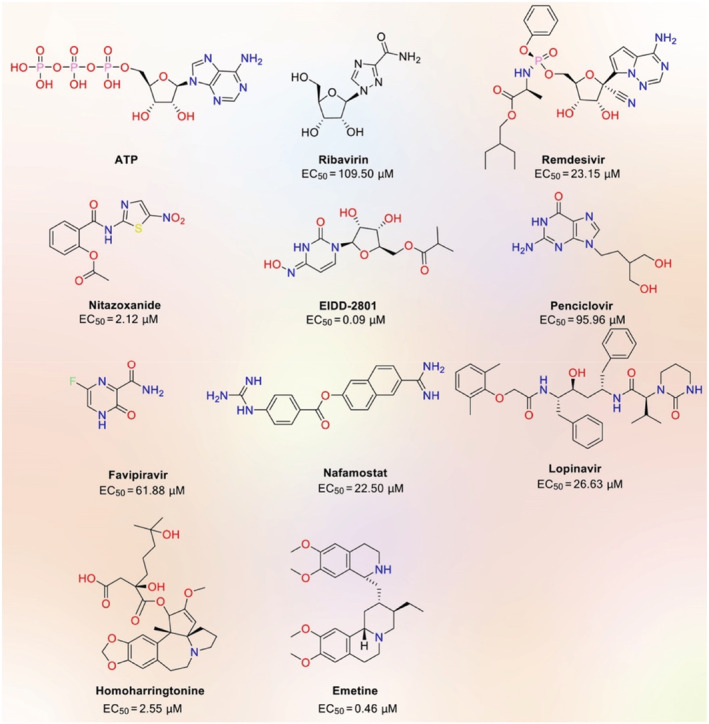
Potential drug candidates against COVID‐19. EC50: Effective concentration (redesigned by the authors of present work with permission from Wu et al. (2021))
In this regard, the WHO supported repositioning remdesivir, lopinavir/ritonavir, and chloroquine/hydroxychloroquine as a potential for COVID‐19, although most might be only supportive symptomatic (Won & Lee, 2020). Nevertheless, they will likely be approved for COVID‐19 therapy if they pass clinical trials and are published in literature and guidelines from leading health organizations (Nain et al., 2021). In order to do this, many groups have been attempting enthusiastically to discover the first approved drug for COVID‐19 therapy (Erlanson, 2020; Low et al., 2020; Muratov et al., 2021). As a result, we identified several clinical trials (Table S2), in which 18 studies were at clinical phases 2, 3, or 4 (https://clinicaltrials.gov/ct2/who_table).
Luo et al. investigated the free energy perturbation‐absolute binding free energy (FEP‐ABFE) method and bioassay validation to reposition 25 potential therapeutic agents to inhibit Mpro. By using the viral proteinase crystal structure, Mpro (PDB ID: 6 LU7), up to 2500 FDA‐approved drugs were first screened via Glide's molecular docking program. They found that Cys145‐His41/Ser144‐His163 could be the nucleophilic agent and acid responsible for hydrolysis of the substrate proteins, and Gly143 and Gln166 can form a hydrogen bond with the “CO‐NH‐Cα‐CO‐NH‐Cα” structure of the backbone of the substrate protein. Of 25 possible agents, 15 showed strong inhibitory properties against Mpro. Among these agents, additionally, dipyridamole passed its first‐round clinical trials with considerable potencies (Li, Hu, et al., 2020; Li, Li, et al., 2020).
The network‐based methods have also given satisfying results in investigating the DTIs and CoV–host interactions. Zho et al. introduced 16 possible anti‐HCoV drugs such as melatonin, mercaptopurine, toremifene, mesalazine, and sirolimus from more than 2000 FDA‐approved drugs by applying a combination of network‐based approaches and systems pharmacology‐based network medicine platform with the ability to quantify the interactions between the drug targets and virus–host interactome in the human protein–protein interactions network. (Zhou et al., 2020). The docking screening method was applied for a set of 2201 approved drugs to find inhibitory activities at SARS‐CoV‐2 main protease, and based on that, carfilzomib, eravacycline, valrubicin, lopinavir, and elbasvir have nominated as an inhibitor for SARS‐CoV‐2 main protease (Wang, 2020). Table 2 includes recent studies in terms of computational DR for coronavirus drug discovery and various diseases, respectively.
Various transcriptomic datasets in relation to SARS‐CoV‐2 were reported since the COVID‐19 outbreak. Basically, the idea is that for a specific disease signature composing of a class of up and downregulated genes, if there is available drug candidate, where those same classes of genes are alternatively downregulated and up‐regulated, respectively, this drug might be potential for that disease (Hoffmann et al., 2020; Le et al., 2021). Le et al. investigated existing computational drug repositioning with remarkably reversed differential gene expression in comparison with manifold input signatures for SARS‐CoV‐2 effects on human cells. They tried to predict the potential drugs toward differentially expressed gene sets from both cell line and organoid disease models and human samples. They found 102 compounds, and among them, 25 were selected in at least two of the signatures, and some of them have been already analyzed in clinical trials. They eventually tested 16 of their shortlisted candidates in live SARS‐CoV‐2 antiviral assays. If a drug could notably (FDR < 0.05) reverse the disease signature, then the drug could be a potential inhibitor for the SARS‐CoV‐2. In the end, they found that 16 of their high predicted hits showed antiviral assay in live SARS‐CoV‐2. Four potential inhibitors were tested for inhibition of SARS‐CoV‐2 in a human lung cell line, Calu‐3, infected with SARS‐CoV‐2. Three drugs revealed in vitro antiviral efficacy—bacampicillin, clofazimine, and haloperidol with no toxicity effects (Le et al., 2021). Ge et al. used an integrative drug repositioning analysis through machine learning to integrate and mine large‐scale knowledge graph, literature together with transcriptome data to identify the potential compounds against SARS‐CoV‐2. They chose to work on CVL218, a poly‐ADP‐ribose polymerase 1 (PARP1) inhibitor. Their in vitro data showed an efficient inhibitory effect against SARS‐CoV‐2 replication without a clear cytopathic effect. They further revealed that CVL218 can effectively bind to the nucleocapsid (N) protein of SARS‐CoV‐2 and could suppress the LPS‐induced production of many inflammatory cytokine, which are highly related to the prevention of immunopathology induced by SARS‐CoV‐2 infection (Ge et al., 2021). Another PARP1 inhibitor, PJ‐34 was previously studied for acute inflammatory including inflammatory diseases of the lung. It was observed that PJ‐34 can interact with the N‐terminal domain of the coronavirus nucleocapsid (N) protein and decrease the RNA binding ability to inhibit virus replication (Ge et al., 2021).
Beclabuvir is an antiviral drug for the treatment of hepatitis C virus (HCV) infection studied for inhibitory activity against SARS‐CoV‐2. It was reported that beclabuvir could be a potential compound for the treatment of SARS‐CoV‐2 through virtual screening and molecular docking in order to bind to RdRp of the SARS‐CoV‐2 (Talluri, 2021). Raltegravir belongs to a class of HIV integrase inhibitors used in the treatment of HIV‐1 infection. It obstructs the insertion of HIV‐1 DNA into the host cell genome. Raltegravir was selected as a highly effective drug against Mpro and has been repurposed for the potential treatment of COVID‐19 in many studies (Nabi et al., 2021).
4. CONCLUSION AND FUTURE PROSPECTS
There has been considerable progress in the practical development and the rational application of computational drug discovery, giving rise to a paradigm shift in both industry and academics. CADD is a powerful approach for proposing new drug candidates, particularly when used in tandem with current chemogenomic methods. Even though CADD takes advantage of manifold dilemmas and approximations, this knowledge‐driven approach plays a significant role in the drug discovery process because of its ability to fast‐track drug discovery. Since CADD has provided several achievements, there would be a promising future to aid drug discovery of many more medicines in the future. Chemogenomics is a superior interdisciplinary field at the interface of chemistry, biology, and informatics, which dramatically decreases the cost and time of drug discovery.
This technique allows sequential genetics that is further interpreted into potential biological targets. Over the last years, there has been great attention to DR in pharmaceutical and biomedical companies, highlighting the paramount importance of repurposing. This interest has led to victorious launches in new, highly economical, and appealing indications compared to traditional drug discovery with high‐priced drug development. Over the last 3 years, the COVID‐19 pandemic has killed many people globally since there are no effective drugs against it. In this sense, we need to highlight that DR attempts the search for effective drugs in an urgent situation such as the COVID‐19. Utilizing DR is strongly recommended to continue investigating the repositioning of existing drugs against different diseases. However, how effectively CADD, including DR and chemogenomics, will produce feasible treatments across different disease areas remains to be seen. Therefore, it should not be surprising that success is limited at times.
Several basic computational problems still need to be resolved. To ameliorate drug design and development dilemmas, rapid developments in chemical and structural biology, bioinformatics, clinical experiments, and computational technology are essential. For example, high‐throughput screening (HTS) has been increasingly used in the pharmaceutical sphere and academic institutions to discover hit and lead compounds rapidly. This potential automated approach lets us screen the entire compound library of the number of chemicals or biological compounds in a target‐based assay or even a cell‐based assay to obtain the desired biological response. In order to use HTS against SARS‐CoV‐2, it is important to take advantage of a similar homology of virus in genomics, specifically, chemogenomics, as a versatile way of drug screening model that provides better bio‐safety.
Hitherto, CADD is the only way for screening drugs against COVID‐19 and is based on a single viral protein or small‐scale drug screening of formerly FDA antiviral drugs utilizing wild‐type SARS‐CoV‐2. Nevertheless, this approach is not well‐suited to HTS when focusing on the bio‐security requirements of SARS‐CoV‐2. Moreover, developing new drugs takes too much time without having a precise crystal structure of the viral target (Liu et al., 2020).
Thus, the best way to rapidly find a drug for the treatment of COVID‐19 would be the use of CADD methods in order to discover lead compounds for clinical use; for instance, we can build a program that takes advantage of structure‐based drug design, HTS, and virtual drug screening to identify new leads targeting the Mpro. More recently, a library of 10,000 compounds involving approved drugs, drug candidates in clinical trials, and biologically active compounds to inhibit Mpro was examined. As a result, six compounds were found with a great IC50 against Mpro and strong antiviral activity in cell‐based assays. Moreover, using CADD, the crystal structure of Mpro in complex with Michael acceptor inhibitor known as N3 can be determined (Jin et al., 2020).
It is also possible to identify drug leads against SARS‐CoV‐2 by targeting the 3‐chymotrypsin‐like cysteine protease (3CLpro) enzyme performing a vital role in the life cycle of CoVs, including MERS‐CoV and SARS‐CoV (Ul Qamar et al., 2020). Scientists employed vHTS tools to predict the 3D structure of the SARS‐CoV‐2 3CLpro enzyme by constructing a 3D homology model and screening it against 32,297 potential antiviral phytochemicals/traditional Chinese medicinal compounds of a medicinal plant library. This effort led to the discovery of nine hits that need further optimization to be identified as anti‐SARS‐CoV‐2 leads. In addition, an interesting investigation based on an HTS assay employing a compound library of 2000 approved drugs and pharmacologically active compounds has also been carried out to find broad‐spectrum inhibitors against the replication of CoVs (Shen et al., 2019). From their HTS data, 56 hits were screened, and 36 compounds were validated in vitro using genetically engineered human CoV OC43 (HCoV‐OC43). Consequently, seven effective compounds were selected as broad‐spectrum inhibitors in which lycorine protected BALB/c mice against lethal human coronavirus OC43 (HCoV‐OC43), and emetine blocked MERS‐CoV entry following pseudo‐virus entry assays. These studies prove the efficacy of screening strategies in the rapid discovery of drug leads, resulting in new drugs for infectious diseases having no effective drugs or available vaccine.
FUNDING INFORMATION
No funds were received for this article.
Supporting information
Appendix S1
ACKNOWLEDGEMENT
Open access publishing facilitated by Macquarie University, as part of the Wiley ‐ Macquarie University agreement via the Council of Australian University Librarians. Open access publishing facilitated by Macquarie University, as part of the Wiley ‐ Macquarie University agreement via the Council of Australian University Librarians.
Maghsoudi, S. , Taghavi Shahraki, B. , Rameh, F. , Nazarabi, M. , Fatahi, Y. , Akhavan, O. , Rabiee, M. , Mostafavi, E. , Lima, E. C. , Saeb, M. R. , & Rabiee, N. (2022). A review on computer‐aided chemogenomics and drug repositioning for rational COVID‐19 drug discovery. Chemical Biology & Drug Design, 00, 1–23. 10.1111/cbdd.14136
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- Abdellatiif, M. H. , Ali, A. , Ali, A. , & Hussien, M. A. (2021). Computational studies by molecular docking of some antiviral drugs with COVID‐19 receptors are an approach to medication for COVID‐19. Open Chemistry, 19(1), 245–264. [Google Scholar]
- Abel, R. , Wang, L. , Harder, E. D. , Berne, B. , & Friesner, R. A. (2017). Advancing drug discovery through enhanced free energy calculations. Accounts of Chemical Research, 50(7), 1625–1632. [DOI] [PubMed] [Google Scholar]
- Abo‐Zeid, Y. , Ismail, N. S. , McLean, G. R. , & Hamdy, N. M. (2020). A molecular docking study repurposes FDA approved iron oxide nanoparticles to treat and control COVID‐19 infection. European Journal of Pharmaceutical Sciences, 153, 105465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliabadi, H. A. M. , Eivazzadeh‐Keihan, R. , Parikhani, A. B. , Mehraban, S. F. , Maleki, A. , Fereshteh, S. , Bazaz, M. , Zolriasatein, A. , Bozorgnia, B. , Rahmati, S. , Saberi, F. , Najafabadi, Z. Y. , Damough, S. , Mohseni, S. , Salehzadeh, H. , Khakyzadeh, V. , Madanchi, H. , Kardar, G. A. , Zarrintaj, P. , … Mozafar, M. (2022). COVID‐19: A systematic review and update on prevention, diagnosis, and treatment. MedComm, 3(1), e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, S. , Abbasi, H. W. , Shahid, S. , Gul, S. , & Abbasi, S. W. (2020). Molecular docking, simulation and MM‐PBSA studies of nigella sativa compounds: A computational quest to identify potential natural antiviral for COVID‐19 treatment. Journal of Biomolecular Structure and Dynamics, 39(12). 4255–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam, S. , & Khan, F. (2019). 3D‐QSAR, docking, ADME/tox studies on flavone analogs reveal anticancer activity through Tankyrase inhibition. Scientific Reports, 9(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves, V. M. , Bobrowski, T. , Melo‐Filho, C. C. , Korn, D. , Auerbach, S. , Schmitt, C. , Muratov, E. N. , & Tropsha, A. (2021). QSAR modeling of SARS‐CoV Mpro inhibitors identifies Sufugolix, Cenicriviroc, proglumetacin, and other drugs as candidates for repurposing against SARS‐CoV‐2. Molecular Informatics, 40(1), 2000113. [DOI] [PubMed] [Google Scholar]
- Amin, S. A. , Ghosh, K. , Gayen, S. , & Jha, T. (2021). Chemical‐informatics approach to COVID‐19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in‐house molecules as papain‐like protease (PLpro) inhibitors. Journal of Biomolecular Structure and Dynamics, 39(13), 4764–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, C. R. , Gustafsson, M. G. , & Strömbergsson, H. (2011). Quantitative chemogenomics: Machine‐learning models of protein‐ligand interaction. Current Topics in Medicinal Chemistry, 11(15), 1978–1993. 10.2174/156802611796391249 [DOI] [PubMed] [Google Scholar]
- Anwar, T. , Kumar, P. , & Khan, A. U. (2021). Modern tools and techniques in computer‐aided drug design. In Molecular docking for computer‐aided drug design (pp. 1–30). Elsevier. [Google Scholar]
- Arun, K. , Sharanya, C. , Abhithaj, J. , Francis, D. , & Sadasivan, C. (2021). Drug repurposing against SARS‐CoV‐2 using E‐pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. Journal of Biomolecular Structure and Dynamics, 39(13), 4647–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn, T. T. , & Thor, K. B. (2004). Drug repositioning: Identifying and developing new uses for existing drugs. Nature Reviews Drug Discovery, 3(8), 673–683. [DOI] [PubMed] [Google Scholar]
- Badary, O. A. (2021). Pharmacogenomics and COVID‐19: Clinical implications of human genome interactions with repurposed drugs. The Pharmacogenomics Journal, 21(3), 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorath, J. (2002). Integration of virtual and high‐throughput screening. Nature Reviews. Drug Discovery, 1(11), 882–894. 10.1038/nrd941 [DOI] [PubMed] [Google Scholar]
- Bittker, J. A. , & Ross, N. T. (2016). High throughput screening methods: Evolution and refinement. Royal Society of Chemistry. [Google Scholar]
- Bleicher, K. H. , Bohm, H. J. , Muller, K. , & Alanine, A. I. (2003). Hit and lead generation: Beyond high‐throughput screening. Nature Reviews. Drug Discovery, 2(5), 369–378. 10.1038/nrd1086 [DOI] [PubMed] [Google Scholar]
- Bobrowski, T. , Chen, L. , Eastman, R. T. , Itkin, Z. , Shinn, P. , Chen, C. Z. , Guo, H. , Zheng, W. , Michael, S. , Simeonov, A. , Hall, M. D. , Zakharov, A. V. , & Muratov, E. N. (2021). Synergistic and antagonistic drug combinations against SARS‐CoV‐2. Molecular Therapy, 29(2), 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchentouf, S. , & Missoum, N. (2020). Identification of Compounds from Nigella Sativa as New Potential Inhibitors of 2019 Novel Coronasvirus (Covid‐19): Molecular Docking Study.
- Bredel, M. , & Jacoby, E. (2004). Chemogenomics: An emerging strategy for rapid target and drug discovery. Nature Reviews Genetics, 5(4), 262–275. [DOI] [PubMed] [Google Scholar]
- Brown, A. J. , Won, J. J. , Graham, R. L. , Dinnon, K. H., III , Sims, A. C. , Feng, J. Y. , Cihlar, T. , Denison, M. R. , Baric, R. S. , & Sheahan, T. P. (2019). Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Research, 169, 104541. 10.1016/j.antiviral.2019.104541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, E. J. , & Marchant, N. J. (2018). The use of chemogenetics in behavioural neuroscience: Receptor variants, targeting approaches and caveats. British Journal of Pharmacology, 175(7), 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillos, M. , Kuhn, M. , Gavin, A.‐C. , Jensen, L. J. , & Bork, P. (2008). Drug target identification using side‐effect similarity. Science, 321(5886), 263–266. [DOI] [PubMed] [Google Scholar]
- Cavasotto, C. N. , & Di Filippo, J. I. (2021). In silico drug repurposing for COVID‐19: Targeting SARS‐CoV‐2 proteins through docking and consensus ranking. Molecular Informatics, 40(1), 2000115. [DOI] [PubMed] [Google Scholar]
- Cavasotto, C. N. , Lamas, M. S. , & Maggini, J. (2021). Functional and druggability analysis of the SARS‐CoV‐2 proteome. European Journal of Pharmacology, 890, 173705. 10.1016/j.ejphar.2020.173705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. (2019). In silico oncology drug repositioning and polypharmacology. Cancer Bioinformatics, 243–261. [DOI] [PubMed] [Google Scholar]
- Cheng, F. , Zhou, Y. , Li, J. , Li, W. , Liu, G. , & Tang, Y. (2012). Prediction of chemical–protein interactions: Multitarget‐QSAR versus computational chemogenomic methods. Molecular BioSystems, 8(9), 2373–2384. [DOI] [PubMed] [Google Scholar]
- Chung, J. Y. , Thone, M. N. , & Kwon, Y. J. (2021). COVID‐19 vaccines: The status and perspectives in delivery points of view. Advanced Drug Delivery Reviews, 170, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vivo, M. , & Cavalli, A. (2017). Recent advances in dynamic docking for drug discovery. Wiley Interdisciplinary Reviews: Computational Molecular Science, 7(6), e1320. [Google Scholar]
- Drew, D. A. , Cao, Y. , & Chan, A. T. (2016). Aspirin and colorectal cancer: The promise of precision chemoprevention. Nature Reviews Cancer, 16(3), 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins, S. , Mestres, J. , & Testa, B. (2007). In silico pharmacology for drug discovery: Applications to targets and beyond. British Journal of Pharmacology, 152(1), 21–37. 10.1038/sj.bjp.0707306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2021). SARS‐CoV‐2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 39(9), 3204–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen, A. D. , Al‐Obaidi, A. , Şahin, A. T. , & Yelekçi, K. (2020). Drug repurposing for coronavirus (COVID‐19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. Journal of Biomolecular Structure and Dynamics, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson, D. A. (2020). Many small steps towards a COVID‐19 drug. Nature Communications, 11(1), 5048. 10.1038/s41467-020-18710-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farag, A. , Wang, P. , Ahmed, M. , & Sadek, H. (2020). Identification of FDA approved drugs targeting COVID‐19 virus by structure‐based drug repositioning. ChemRxiv. [Google Scholar]
- Farshchi, F. , Saadati, A. , Kholafazad‐Kordasht, H. , Seidi, F. , & Hasanzadeh, M. (2021). Trifluralin recognition using touch‐based fingertip: Application of wearable glove‐based sensor toward environmental pollution and human health control. Journal of Molecular Recognition, 34(11), e2927. [DOI] [PubMed] [Google Scholar]
- Fotis, C. , Antoranz, A. , Hatziavramidis, D. , Sakellaropoulos, T. , & Alexopoulos, L. G. (2018). Network‐based technologies for early drug discovery. Drug Discovery Today, 23(3), 626–635. 10.1016/j.drudis.2017.12.001 [DOI] [PubMed] [Google Scholar]
- Fourches, D. , Muratov, E. , & Tropsha, A. (2015). Curation of chemogenomics data. Nature Chemical Biology, 11(8), 535. 10.1038/nchembio.1881 [DOI] [PubMed] [Google Scholar]
- Galindez, G. , Matschinske, J. , Rose, T. D. , Sadegh, S. , Salgado‐Albarrán, M. , Späth, J. , Baumbach, J. , & Pauling, J. K. (2021). Lessons from the COVID‐19 pandemic for advancing computational drug repurposing strategies. Nature Computational Science, 1(1), 33–41. [DOI] [PubMed] [Google Scholar]
- Gawriljuk, V. O. , Zin, P. P. K. , Puhl, A. C. , Zorn, K. M. , Foil, D. H. , Lane, T. R. , Hurst, B. , Tavella, T. A. , Costa, F. T. M. , Lakshmanane, P. , Bernatchez, J. , Godoy, A. S. , Oliva, G. , Siqueira‐Neto, J. L. , Madrid, P. B. , & Ekins, S. (2021). Machine learning models identify inhibitors of SARS‐CoV‐2. Journal of Chemical Information and Modeling, 61(9), 4224–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, Y. , Tian, T. , Huang, S. , Wan, F. , Li, J. , Li, S. , Wang, X. , Yang, H. , Hong, L. , Wu, N. , Yuan, E. , Luo, Y. , Cheng, L. , Hu, C. , Lei, Y. , Shu, H. , Feng, X. , Jiang, Z. , Wu, Y. , … Zeng, J. (2021). An integrative drug repositioning framework discovered a potential therapeutic agent targeting COVID‐19. Signal Transduction and Targeted Therapy, 6(1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghofrani, H. A. , Osterloh, I. H. , & Grimminger, F. (2006). Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nature Reviews. Drug Discovery, 5(8), 689–702. 10.1038/nrd2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholivand, K. , Mohammadpanah, F. , Pooyan, M. , & Roohzadeh, R. (2022). Evaluating anti‐coronavirus activity of some phosphoramides and their influencing inhibitory factors using molecular docking, DFT, QSAR, and NCI‐RDG studies. Journal of Molecular Structure, 1248, 131481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahl, M. V. C. , Alcará, A. M. , Perin, A. P. A. , Moro, C. F. , Pinto, É. S. M. , Feltes, B. C. , Ghilardi, I. M. , Rodrigues, F. V. F. , Dorn, M. , da Costa, J. C. , de Souza, O. N. , & Ligabue‐Braun, R. (2021). Evaluation of drug repositioning by molecular docking of pharmaceutical resources available in the Brazilian healthcare system against SARS‐CoV‐2. Informatics in Medicine Unlocked, 23, 100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar, M. , Ahmed, H. A. , Aljohani, G. , & Alhaddad, O. A. (2020). Investigation of some antiviral N‐heterocycles as COVID 19 drug: Molecular docking and DFT calculations. International Journal of Molecular Sciences, 21(11), 3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, D. C., Jr. , & Ji, H.‐F. (2020). A search for medications to treat COVID‐19 via in silico molecular docking models of the SARS‐CoV‐2 spike glycoprotein and 3CL protease. Travel Medicine and Infectious Disease, 35, 101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, C. J. , & Stevens, A. P. (2006). Chemogenomics: Structuring the drug discovery process to gene families. Drug Discovery Today, 11(19–20), 880–888. [DOI] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , Schroeder, S. , Krüger, N. , Herrler, T. , Erichsen, S. , Schiergens, T. S. , Herrler, G. , Wu, N.‐H. , Nitsche, A. , Müller, M. A. , Drosten, C. , & Pöhlmann, S. (2020). SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181(2), 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini, F. S. , & Amanlou, M. (2020). Anti‐HCV and anti‐malaria agent, potential candidates to repurpose for coronavirus infection: Virtual screening, molecular docking, and molecular dynamics simulation study. Life Sciences, 258, 118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , Huang, X. , Ma, M. , Hu, C. , Seidi, F. , Yin, S. , & Xiao, H. (2021). Recent advances on the bacterial cellulose‐derived carbon aerogels. Journal of Materials Chemistry C, 9(3), 818–828. [Google Scholar]
- Ibrahim, M. A. , Abdelrahman, A. H. , Allemailem, K. S. , Almatroudi, A. , Moustafa, M. F. , & Hegazy, M.‐E. F. (2021). In silico evaluation of prospective anti‐COVID‐19 drug candidates as potential SARS‐CoV‐2 main protease inhibitors. The Protein Journal, 40(3), 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam, S. S. , & Gilani, S. J. (2017). Computer aided drug design: A novel loom to drug discovery. Organic & Medicinal Chemistry International Journal, 1(3), 113–118. [Google Scholar]
- Jin, Z. , Xiaoyu, D. , Xu, Y. , Deng, Y. , Liu, M. , Zhao, Y. , Zhang, B. , Li, X. , Zhang, L. , Peng, C. , Duan, Y. , Yu, J. , Lin, W. , Yang, K. , Liu, F. , Jiang, R. , Yang, X. , You, T. , Liu, X. , … Yang, H. (2020). Structure of M pro from SARS‐CoV‐2 and discovery of its inhibitors. Nature, 582(7811), 289–293. [DOI] [PubMed] [Google Scholar]
- Jones, L. H. , & Bunnage, M. E. (2017). Applications of chemogenomic library screening in drug discovery. Nature Reviews. Drug Discovery, 16(4), 285–296. 10.1038/nrd.2016.244 [DOI] [PubMed] [Google Scholar]
- Jourdan, J.‐P. , Bureau, R. , Rochais, C. , & Dallemagne, P. (2020). Drug repositioning: A brief overview. Journal of Pharmacy and Pharmacology, 72(9), 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadioglu, O. , Saeed, M. , Greten, H. J. , & Efferth, T. (2021). Identification of novel compounds against three targets of SARS CoV‐2 coronavirus by combined virtual screening and supervised machine learning. Computers in biology and medicine, 133, 104359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel, A. M. , Monem, M. S. , Sharaf, N. A. , Magdy, N. , & Farid, S. F. (2022). Efficacy and safety of azithromycin in Covid‐19 patients: A systematic review and meta‐analysis of randomized clinical trials. Reviews in Medical Virology, 32(1), e2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakida, T. , Onuma, K. , Saito, M. M. , Yamamoto, R. , Chiba, T. , Chiba, R. , Hidaka, Y. , Fujii‐Abe, K. , Kawahara, H. , & Yamakoshi, Y. (2019). Potential for drug repositioning of midazolam for dentin regeneration. International Journal of Molecular Sciences, 20(3), 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keretsu, S. , Bhujbal, S. P. , & Cho, S. J. (2020). Rational approach toward COVID‐19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Scientific Reports, 10(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. A. , Zia, K. , Ashraf, S. , Uddin, R. , & Ul‐Haq, Z. (2021). Identification of chymotrypsin‐like protease inhibitors of SARS‐CoV‐2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics, 39(7), 2607–2616. [DOI] [PubMed] [Google Scholar]
- Khelfaoui, H. , Harkati, D. , & Saleh, B. A. (2021). Molecular docking, molecular dynamics simulations and reactivity, studies on approved drugs library targeting ACE2 and SARS‐CoV‐2 binding with ACE2. Journal of Biomolecular Structure and Dynamics, 39(18), 7246–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholafazad‐Kordasht, H. , Hasanzadeh, M. , & Seidi, F. (2021). Smartphone based immunosensors as next generation of healthcare tools: Technical and analytical overview towards improvement of personalized medicine. TrAC Trends in Analytical Chemistry, 145, 116455. [Google Scholar]
- Kitchen, D. B. , Decornez, H. , Furr, J. R. , & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery, 3(11), 935–949. [DOI] [PubMed] [Google Scholar]
- Kubinyi, H. (2006). Chemogenomics in drug discovery. Chemical Genomics, 1–19. [DOI] [PubMed] [Google Scholar]
- Kumar, R. , Harilal, S. , Gupta, S. V. , Jose, J. , Uddin, M. S. , Shah, M. A. , & Mathew, B. (2019). Exploring the new horizons of drug repurposing: A vital tool for turning hard work into smart work. European Journal of Medicinal Chemistry, 182, 111602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largent, E. A. , & Miller, F. G. (2021). Problems with paying people to be vaccinated against COVID‐19. JAMA, 325(6), 534–535. [DOI] [PubMed] [Google Scholar]
- Le, B. L. , Andreoletti, G. , Oskotsky, T. , Vallejo‐Gracia, A. , Rosales, R. , Yu, K. , Kosti, I. , Leon, K. E. , Bunis, D. G. , Christine, L. , Renuka Kumar, G. , White, K. M. , García‐Sastre, A. , Ott, M. , & Sirota, M. (2021). Transcriptomics‐based drug repositioning pipeline identifies therapeutic candidates for COVID‐19. Scientific Reports, 11(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W.‐Y. , Lee, C.‐Y. , & Kim, C.‐E. (2021). Predicting activatory and inhibitory drug–target interactions based on mol2vec and genetically perturbed transcriptomes. bioRxiv. 10.1101/2021.03.18.436088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelananda, S. P. , & Lindert, S. (2016). Computational methods in drug discovery. Beilstein Journal of Organic Chemistry, 12(1), 2694–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Hu, M. , Wang, T. , Chen, H. , & Xu, L. (2020). Repositioning aspirin to treat lung and breast cancers and overcome acquired resistance to targeted therapy. Frontiers in Oncology, 9, 1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Li, X. , Huang, Y.‐Y. , Wu, Y. , Zhou, L. , Liu, R. , Wu, D. , Zhang, L. , Liu, H. , Xu, X. , Zhang, Y. , Cui, J. , Wang, X. , & Luo, H.‐B. (2020). FEP‐based screening prompts drug repositioning against COVID‐19. BioRxiv. [Google Scholar]
- Liu, J. , Li, K. , Lin, C. , Shao, J. , Yang, S. , Zhang, W. , Zhou, G. , de Vries, A. A. F. , & Zhiyi, Y. (2021). A high‐throughput drug screening strategy against Conroviruses. International Journal of Infectious Diseases, 103, 303–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, R. , Wei, L. , & Zhang, P. (2021). A deep learning framework for drug repurposing via emulating clinical trials on real‐world patient data. Nature Machine Intelligence, 3(1), 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Li, R. , Liang, F. , Deng, C. , Seidi, F. , & Xiao, H. (2022). Fluorescent paper‐based analytical devices for ultra‐sensitive dual‐type RNA detections and accurate gastric cancer screening. Biosensors and Bioelectronics, 197, 113781. [DOI] [PubMed] [Google Scholar]
- Lokhande, K. B. , Doiphode, S. , Vyas, R. , & Swamy, K. V. (2021). Molecular docking and simulation studies on SARS‐CoV‐2 Mpro reveals mitoxantrone, leucovorin, Birinapant, and Dynasore as potent drugs against COVID‐19. Journal of Biomolecular Structure and Dynamics, 39(18), 7294–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi Shahreza, M. , Ghadiri, N. , Mousavi, S. R. , Varshosaz, J. , & Green, J. R. (2017). A review of network‐based approaches to drug repositioning. Briefings in Bioinformatics, 19(5), 878–892. 10.1093/bib/bbx017 [DOI] [PubMed] [Google Scholar]
- Low, Z. Y. , Farouk, I. A. , & Lal, S. K. (2020). Drug repositioning: New approaches and future prospects for life‐debilitating diseases and the COVID‐19 pandemic outbreak. Viruses, 12(9), 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low, Z. Y. , Yip, A. J. W. , & Lal, S. K. (2022). Repositioning ivermectin for Covid‐19 treatment: Molecular mechanisms of action against SARS‐CoV‐2 replication. Biochimica et Biophysica Acta (BBA)‐Molecular Basis of Disease, 1868(2), 166294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchetta, M. , & Pellegrini, M. (2021). Drug repositioning by merging active subnetworks validated in cancer and COVID‐19. Scientific Reports, 11(1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, H. , Li, M. , Yang, M. , Wu, F.‐X. , Li, Y. , & Wang, J. (2021). Biomedical data and computational models for drug repositioning: A comprehensive review. Briefings in Bioinformatics, 22(2), 1604–1619. [DOI] [PubMed] [Google Scholar]
- Lv, H. , Shi, L. , Berkenpas, J. W. , Dao, F.‐Y. , Zulfiqar, H. , Ding, H. , Zhang, Y. , Yang, L. , & Cao, R. (2021). Application of artificial intelligence and machine learning for COVID‐19 drug discovery and vaccine design. Briefings in Bioinformatics, 22(6), bbab320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoudi, S. , Rabiee, N. , Ahmadi, S. , Rabiee, M. , Bagherzadeh, M. , & Karimi, M. (2021). An overview of microfluidic devices. Biomedical Applications of Microfluidic Devices, 1–22. [Google Scholar]
- Mahanta, S. , Chowdhury, P. , Gogoi, N. , Goswami, N. , Borah, D. , Kumar, R. , Chetia, D. , Borah, P. , Buragohain, A. K. , & Gogoi, B. (2021). Potential anti‐viral activity of approved repurposed drug against main protease of SARS‐CoV‐2: An in silico based approach. Journal of Biomolecular Structure and Dynamics, 39(10), 3802–3811. [DOI] [PubMed] [Google Scholar]
- Mahase, E. (2021). Covid‐19: Molnupiravir reduces risk of hospital admission or death by 50% in patients at risk, MSD reports. British Medical Journal Publishing Group. [DOI] [PubMed] [Google Scholar]
- Mahdian, S. , Ebrahim‐Habibi, A. , & Zarrabi, M. (2020). Drug repurposing using computational methods to identify therapeutic options for COVID‐19. Journal of Diabetes & Metabolic Disorders, 19, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinho, E. M. , de Andrade Neto, J. B. , Silva, J. , da Silva, C. R. , Cavalcanti, B. C. , Marinho, E. S. , & Júnior, H. V. N. (2020). Virtual screening based on molecular docking of possible inhibitors of Covid‐19 main protease. Microbial Pathogenesis, 148, 104365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massetti, M. , Jiao, F. , Ferguson, A. J. , Zhao, D. , Wijeratne, K. , Würger, A. , Blackburn, J. L. , Crispin, X. , & Fabiano, S. (2021). Unconventional thermoelectric materials for energy harvesting and sensing applications. Chemical Reviews, 121(20), 12465–12547. [DOI] [PubMed] [Google Scholar]
- Meng, X.‐Y. , Zhang, H.‐X. , Mezei, M. , & Cui, M. (2011). Molecular docking: A powerful approach for structure‐based drug discovery. Current Computer‐Aided Drug Design, 7(2), 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercorelli, B. , Palù, G. , & Loregian, A. (2018). Drug repurposing for viral infectious diseases: How far are we? Trends in Microbiology, 26(10), 865–876. 10.1016/j.tim.2018.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, C. B. , Pandey, P. , Sharma, R. D. , Mongre, R. K. , Lynn, A. M. , Prasad, R. , Prasad, R. , Jeon, R. , & Prakash, A. (2020). Discovery of Natural Phenol Catechin as a Multitargeted Agent Against SARS‐CoV‐2 For the Plausible Therapy of COVID‐19. [DOI] [PMC free article] [PubMed]
- Mittal, L. , Kumari, A. , Srivastava, M. , Singh, M. , & Asthana, S. (2021). Identification of potential molecules against COVID‐19 main protease through structure‐guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 39(10), 3662–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiwale, M. , Yadav, N. S. , Kumar, S. , Kushwaha, T. , Choudhir, G. , Sharma, S. , & Singour, P. K. (2022). Finding potent inhibitors for COVID‐19 main protease (Mpro): An in silico approach using SARS‐CoV‐3CL protease inhibitors for combating CORONA. Journal of Biomolecular Structure and Dynamics, 40(4), 1534–1545. [DOI] [PubMed] [Google Scholar]
- Muratov, E. N. , Amaro, R. , Andrade, C. H. , Brown, N. , Ekins, S. , Fourches, D. , Isayev, O. , Kozakov, D. , Medina‐Franco, J. L. , Merz, K. M. , Oprea, T. I. , Poroikov, G. S. , Todd, M. H. , Varnek, A. , Winkler, D. A. , Zakharov, A. V. , Cherkasov, A. , & Tropsha, A. (2021). A critical overview of computational approaches employed for COVID‐19 drug discovery. Chemical Society Reviews, 50, 9121–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, C. W. , & Rees, D. C. (2009). The rise of fragment‐based drug discovery. Nature Chemistry, 1(3), 187–192. 10.1038/nchem.217 [DOI] [PubMed] [Google Scholar]
- Nabi, F. , Ahmad, O. , Khan, Y. A. , Nabi, A. , Md Amiruddin, H. , Abul Qais, F. , Masroor, A. , Hisamuddin, M. , Uversky, V. N. , & Khan, R. H. (2021). Computational studies on phylogeny and drug designing using molecular simulations for COVID‐19. Journal of Biomolecular Structure and Dynamics, 1–10. [DOI] [PubMed] [Google Scholar]
- Nain, Z. , Rana, H. K. , Liò, P. , Islam, S. M. S. , Summers, M. A. , & Moni, M. A. (2021). Pathogenetic profiling of COVID‐19 and SARS‐like viruses. Briefings in Bioinformatics, 22(2), 1175–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosrati, H. , Seidi, F. , Hosseinmirzaei, A. , Mousazadeh, N. , Mohammadi, A. , Ghaffarlou, M. , Danafar, H. , Conde, J. , & Sharafi, A. (2022). Prodrug polymeric nanoconjugates encapsulating gold nanoparticles for enhanced X‐ray radiation therapy in breast cancer. Advanced Healthcare Materials, 11(3), 2102321. [DOI] [PubMed] [Google Scholar]
- Olayan, R. S. , Ashoor, H. , & Bajic, V. B. (2018). DDR: Efficient computational method to predict drug–target interactions using graph mining and machine learning approaches. Bioinformatics, 34(7), 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omotuyi, O. , Nash, O. , Ajiboye, B. , Metibemu, D. , Oyinloye, B. , & Ojo, A. (2020). The disruption of SARS‐CoV‐2 RBD/ACE‐2 complex by Ubrogepant Is mediated by interface hydration.
- Onozuka, D. , Tanoue, Y. , Nomura, S. , Kawashima, T. , Yoneoka, D. , Eguchi, A. , Ng, C. F. S. , Matsuura, K. , Shi, S. , Makiyama, K. , Uryu, S. , Kawamura, Y. , Takayanagi, S. , Gilmour, S. , Hayashi, T. I. , Miyata, H. , Sera, F. , Sunagawa, T. , Takahashi, T. , … Hashizume, M. (2022). Reduced mortality during the COVID‐19 outbreak in Japan, 2020: A two‐stage interrupted time‐series design. International Journal of Epidemiology, 51(1), 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou‐Yang, S. S. , Lu, J. Y. , Kong, X. Q. , Liang, Z. J. , Luo, C. , & Jiang, H. (2012). Computational drug discovery. Acta Pharmacologica Sinica, 33(9), 1131–1140. 10.1038/aps.2012.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachetti, M. , Marini, B. , Benedetti, F. , Giudici, F. , Mauro, E. , Storici, P. , Masciovecchio, C. , Angeletti, S. , Ciccozzi, M. , Gallo, R. C. , Zella, D. , & Ippodrino, R. (2020). Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. Journal of Translational Medicine, 18(1), 179. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, K. (2019). A review of computational drug repurposing. Translational and Clinical Pharmacology, 27(2), 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar, T. , Meenakshisundaram, S. , Manickam, M. , & Sankaranarayanan, M. (2020). A medicinal chemistry perspective of drug repositioning: Recent advances and challenges in drug discovery. European Journal of Medicinal Chemistry, 195, 112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzi, L. , & Rastelli, G. (2019). Molecular docking: Shifting paradigms in drug discovery. International Journal of Molecular Sciences, 20(18), 4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantone, D. , & Koudriavtseva, T. (2018). Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: A mini‐review. Clinical Drug Investigation, 38(8), 653–671. [DOI] [PubMed] [Google Scholar]
- Popelier, P. , & Smith, P. (2006). QSAR models based on quantum topological molecular similarity. European Journal of Medicinal Chemistry, 41(7), 862–873. [DOI] [PubMed] [Google Scholar]
- Prasad, S. , Wilkinson, J. , & Gatzoulis, M. A. (2000). Sildenafil in primary pulmonary hypertension. The New England Journal of Medicine, 343(18), 1342. 10.1056/NEJM200011023431814 [DOI] [PubMed] [Google Scholar]
- Pushpakom, S. , Iorio, F. , Eyers, P. A. , Jane Escott, K. , Hopper, S. , Wells, A. , Doig, A. , Guilliams, T. , Latimer, J. , McNamee, C. , Norris, A. , Sanseau, P. , Cavalla, D. , & Pirmohamed, M. (2019). Drug repurposing: Progress, challenges and recommendations. Nature Reviews Drug Discovery, 18(1), 41–58. [DOI] [PubMed] [Google Scholar]
- Quianzon, C. C. , & Cheikh, I. E. (2012). History of current non‐insulin medications for diabetes mellitus. Journal of Community Hospital Internal Medicine Perspectives, 2(3), 19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, R. B. , & Brennan, P. E. (2021). Chemogenomics for drug discovery: Clinical molecules from open access chemical probes. RSC Chemical Biology, 2(3), 759–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rad, M. S. , & Maghsoudi, S. (2016). Two‐step three‐component process for one‐pot synthesis of 8‐alkylmercaptocaffeine derivatives. RSC Advances, 6(74), 70335–70342. [Google Scholar]
- Rognan, D. (2007). Chemogenomic approaches to rational drug design. British Journal of Pharmacology, 152(1), 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogosnitzky, M. , Okediji, P. , & Koman, I. (2020). Cepharanthine: A review of the antiviral potential of a Japanese‐approved alopecia drug in COVID‐19. Pharmacological Reports, 72(6), 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabe, V. T. , Ntombela, T. , Jhamba, L. A. , Maguire, G. E. , Govender, T. , Naicker, T. , & Kruger, H. G. (2021). Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. European Journal of Medicinal Chemistry, 224, 113705. [DOI] [PubMed] [Google Scholar]
- Safaei, M. , Sodano, H. A. , & Anton, S. R. (2019). A review of energy harvesting using piezoelectric materials: State‐of‐the‐art a decade later (2008–2018). Smart Materials and Structures, 28(11), 113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargazi, S. , Shahraki, S. , Shahraki, O. , Zargari, F. , Sheervalilou, R. , Maghsoudi, S. , Rad, M. N. S. , & Saravani, R. (2021). 8‐Alkylmercaptocaffeine derivatives: Antioxidant, molecular docking, and in‐vitro cytotoxicity studies. Bioorganic Chemistry, 111, 104900. 10.1016/j.bioorg.2021.104900 [DOI] [PubMed] [Google Scholar]
- Sawada, R. , Iwata, H. , Mizutani, S. , & Yamanishi, Y. (2015). Target‐based drug repositioning using large‐scale chemical–protein interactome data. Journal of Chemical Information and Modeling, 55(12), 2717–2730. [DOI] [PubMed] [Google Scholar]
- Schaduangrat, N. , Anuwongcharoen, N. , Phanus‐umporn, C. , Sriwanichpoom, N. , Wikberg, J. E. , & Nantasenamat, C. (2019). Proteochemometric modeling for drug repositioning. In In Silico drug design (pp. 281–302). Elsevier. [Google Scholar]
- Schlitzer, M. (2007). Malaria chemotherapeutics part I: History of antimalarial drug development, currently used therapeutics, and drugs in clinical development. ChemMedChem: Chemistry Enabling Drug Discovery, 2(7), 944–986. [DOI] [PubMed] [Google Scholar]
- Schneider, G. , & Fechner, U. (2005). Computer‐based de novo design of drug‐like molecules. Nature Reviews. Drug Discovery, 4(8), 649–663. 10.1038/nrd1799 [DOI] [PubMed] [Google Scholar]
- Seidi, F. , Deng, C. , Zhong, Y. , Liu, Y. , Huang, Y. , Li, C. , & Xiao, H. (2021). Functionalized masks: Powerful materials against COVID‐19 and future pandemics. Small, 17(42), 2102453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sencanski, M. , Perovic, V. , Pajovic, S. B. , Adzic, M. , Paessler, S. , & Glisic, S. (2020). Drug repurposing for candidate SARS‐CoV‐2 main protease inhibitors by a novel in silico method. Molecules, 25(17), 3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafin, M. B. , Bottega, A. , Foletto, V. S. , da Rosa, T. F. , Hörner, A. , & Hörner, R. (2020). Drug repositioning is an alternative for the treatment of coronavirus COVID‐19. International Journal of Antimicrobial Agents, 55(6), 105969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, B. , Modi, P. , & Sagar, S. R. (2020). In silico studies on therapeutic agents for COVID‐19: Drug repurposing approach. Life Sciences, 117652, 117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shameer, K. , Readhead, B. , & Dudley, T. (2015). Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Current Topics in Medicinal Chemistry, 15(1), 5–20. [DOI] [PubMed] [Google Scholar]
- Sharma, A. D. (2020). Eucalyptol (1, 8 cineole) from eucalyptus essential oil a potential inhibitor of COVID 19 corona virus infection by molecular docking studies.
- Sharma, A. D. , & Kaur, I. (2020). Molecular docking studies on Jensenone from eucalyptus essential oil as a potential inhibitor of COVID 19 corona virus infection. arXiv preprint arXiv, 10.48550/arXiv.2004.00217 [DOI] [Google Scholar]
- Liang, S. , Niu, J. , Wang, C. , Huang, B. , Wang, W. , Zhu, N. , Deng, Y. , Wang, H. , Ye, F. , Cen, S. , & Tan, W. (2019). High‐throughput screening and identification of potent broad‐spectrum inhibitors of coronaviruses. Journal of Virology, 93(12), e00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, D. , Mukherjee, R. , Grewe, D. , Bojkova, D. , Baek, K. , Bhattacharya, A. , Schulz, L. , Widera, M. , Mehdipour, A. R. , Tascher, G. , Geurink, P. P. , Wilhelm, A. , van der Heden van Noort, G. J. , Ovaa, H. , Müller, S. , Knobeloch, K.‐P. , Rajalingam, K. , Schulman, B. A. , Cinatl, J. , … Dikic, I. (2020). Papain‐like protease regulates SARS‐CoV‐2 viral spread and innate immunity. Nature, 587(7835), 657–662. 10.1038/s41586-020-2601-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwoski, G. , Kothiwale, S. , Meiler, J. , & Lowe, E. W. (2014). Computational methods in drug discovery. Pharmacological Reviews, 66(1), 334–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suravajhala, R. , Parashar, A. , Malik, B. , Nagaraj, V. A. , Padmanaban, G. , Kishor, P.B. K. , Polavarapu, R. , & Suravajhala, P. (2020). Comparative docking studies on curcumin with COVID‐19 proteins. [DOI] [PMC free article] [PubMed]
- Talluri, S. (2021). Molecular docking and virtual screening based prediction of drugs for COVID‐19. Combinatorial Chemistry & High Throughput Screening, 24(5), 716–728. [DOI] [PubMed] [Google Scholar]
- Turanli, B. , Grøtli, M. , Boren, J. , Nielsen, J. , Uhlen, M. , Arga, K. Y. , & Mardinoglu, A. (2018). Drug repositioning for effective prostate cancer treatment. Frontiers in Physiology, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar, M. T. , Alqahtani, S. M. , Alamri, M. A. , & Chen, L.‐L. (2020). Structural basis of SARS‐CoV‐2 3CLpro and anti‐COVID‐19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis, 10(4), 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urán Landaburu, L. , Berenstein, A. J. , Videla, S. , Maru, P. , Shanmugam, D. , Chernomoretz, A. , & Agüero, F. (2020). TDR targets 6: Driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Research, 48(D1), D992–D1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangeel, L. , Chiu, W. , De Jonghe, S. , Maes, P. , Slechten, B. , Raymenants, J. , André, E. , Leyssen, P. , Neyts, J. , & Jochmans, D. (2022). Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS‐CoV‐2 omicron and other variants of concern. Antiviral Research, 198, 105252. 10.1016/j.antiviral.2022.105252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimberg, V. (2021). Teicoplanin—A new use for an old drug in the COVID‐19 era? Pharmaceuticals, 14(12), 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Horby, P. W. , Hayden, F. G. , & Gao, G. F. (2020). A novel coronavirus outbreak of global health concern. The Lancet, 395(10223), 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. (2020). Fast identification of possible drug treatment of coronavirus disease‐19 (COVID‐19) through computational drug repurposing study. Journal of Chemical Information and Modeling., 60, 3277–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , & Guan, Y. (2021). COVID‐19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Medicinal Research Reviews, 41(1), 5–28. 10.1002/med.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , He, Y. , Huang, J. , & Yang, X. (2021). Integrative web‐based analysis of omics data for study of drugs against SARS‐CoV‐2. Scientific Reports, 11(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, W. , Chen, C. , Tang, J. , Wang, C. , Zhou, M. , Cheng, Y. , Zhou, X. , Wu, Q. , Zhang, X. , Feng, Z. , Wang, M. , & Mao, Q. (2022). Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID‐19: A meta‐analysis. Annals of Medicine, 54(1), 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston, M. , Kaserer, T. , Wu, A. , Mouravlev, A. , Carpenter, J. C. , Snowball, A. , Knauss, S. , von Schimmelmann, M. , During, M. J. , Lignani, G. , Schorge, S. , Young, D. , Kullmann, D. M. , & Lieb, A. (2019). Olanzapine: A potent agonist at the hM4D (Gi) DREADD amenable to clinical translation of chemogenetics. Science Advances, 5(4), eaaw1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won, J.‐H. , & Lee, H. (2020). The current status of drug repositioning and vaccine developments for the COVID‐19 pandemic. International Journal of Molecular Sciences, 21(24), 9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Li, Z. , Zhao, Y. S. , Huang, Y. Y. , Jiang, M. Y. , & Luo, H. B. (2021). Therapeutic targets and potential agents for the treatment of COVID‐19. Medicinal Research Reviews, 41(3), 1775–1797. [DOI] [PubMed] [Google Scholar]
- Xu, T. , Zheng, W. , & Huang, R. (2021). High‐throughput screening assays for SARS‐CoV‐2 drug development: Current status and future directions. Drug Discovery Today, 26(10), 2439–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Y. , Feng, Z.‐w. , Li, X.‐y. , Hu, Z.‐h. , Xu, Q. , Wang, Z. , Cheng, J.‐h. , Shi, H.‐t. , Wang, Q.‐b. , Wu, H.‐y. , Xie, X.‐Q. , & Lv, Q.‐z. (2018). The efficacy and safety of cilostazol as an alternative to aspirin in Chinese patients with aspirin intolerance after coronary stent implantation: A combined clinical study and computational system pharmacology analysis. Acta Pharmacologica Sinica, 39(2), 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanga, L. J. , Chena, R. H. , Hamdoun, S. , Coghi, P. , Ng, J. P. L. , Zhang, D. W. , Guo, X. , Xia, C. , Law, B. Y. K. , & Wong, V. K. W. (2021). Corilagin prevents SARS‐CoV‐2 infection by targeting RBD‐ACE2 binding. Phytomedicine, 87, 153591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y. , & Wunderink, R. G. (2018). MERS, SARS and other coronaviruses as causes of pneumonia. Respirology, 23(2), 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J.‐L. , Dai, Q.‐Q. , & Li, G.‐B. (2021). Deep learning in target prediction and drug repositioning: Recent advances and challenges. Drug Discovery Today, 27(7), 1796–1814. [DOI] [PubMed] [Google Scholar]
- Yu, R. , Chen, L. , Lan, R. , Shen, R. , & Li, P. (2020). Computational screening of antagonists against the SARS‐CoV‐2 (COVID‐19) coronavirus by molecular docking. International Journal of Antimicrobial Agents, 56(2), 106012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Hou, Y. , Shen, J. , Huang, Y. , Martin, W. , & Cheng, F. (2020). Network‐based drug repurposing for novel coronavirus 2019‐nCoV/SARS‐CoV‐2. Cell Discovery, 6(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.