

Abstract
Aims
Inherited or somatic mutations in the MRE11, RAD50 and NBN genes increase the incidence of tumours, including medulloblastoma (MB). On the other hand, MRE11, RAD50 and NBS1 protein components of the MRN complex are often overexpressed and sometimes essential in cancer. In order to solve the apparent conundrum about the oncosuppressive or oncopromoting role of the MRN complex, we explored the functions of NBS1 in an MB‐prone animal model.
Materials and methods
We generated and analysed the monoallelic or biallelic deletion of the Nbn gene in the context of the SmoA1 transgenic mouse, a Sonic Hedgehog (SHH)‐dependent MB‐prone animal model. We used normal and tumour tissues from these animal models, primary granule cell progenitors (GCPs) from genetically modified animals and NBS1‐depleted primary MB cells, to uncover the effects of NBS1 depletion by RNA‐Seq, by biochemical characterisation of the SHH pathway and the DNA damage response (DDR) as well as on the growth and clonogenic properties of GCPs.
Results
We found that monoallelic Nbn deletion increases SmoA1‐dependent MB incidence. In addition to a defective DDR, Nbn +/− GCPs show increased clonogenicity compared to Nbn +/+ GCPs, dependent on an enhanced Notch signalling. In contrast, full Nbn KO impairs MB development both in SmoA1 mice and in an SHH‐driven tumour allograft.
Conclusions
Our study indicates that Nbn is haploinsufficient for SHH‐MB development whereas full Nbn KO is epistatic on SHH‐driven MB development, thus revealing a gene dosage‐dependent effect of Nbn inactivation on SHH‐MB development.
Keywords: haploinsufficiency, MRN complex, Nijmegen breakage syndrome, Notch, Sonic Hedgehog
The dual role of Nbs1 in Sonic Hedgehog (SHH)‐medulloblastoma (MB). In cancer prone SmoA1‐Granule Cell Progenitors (GCPs), Nbn KO causes reduced proliferation and increased cell death, ultimately leading to inhibition of MB development, due to activation of the DDR and impairment of the SHH pathway. On the other hand, Nbn hemizygosis (Nbn HZ) causes DDR activation and Notch‐dependent increase in clonogenic capability, favoring GCP transformation. These results, collectively, indicate a gene dosage‐dependent function of Nbn in SHH‐driven tumorigenesis.
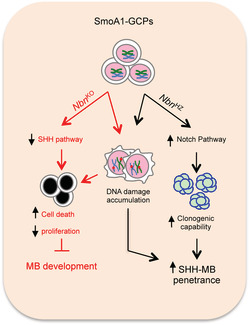
Key points.
Monoallelic Nbn deletion promotes SHH‐MB in the SmoA1 mouse model.
Monoallelic Nbn deletion causes defective DDR in GCPs.
Nbn +/− GCP features enhanced Notch‐dependent clonogenicity.
Full Nbn KO impairs SHH‐driven MB in the SmoA1 model and in tumour allografts.
INTRODUCTION
Medulloblastoma (MB) is one of the most common and frequently lethal childhood malignant brain tumours. Four consensus molecular groups are currently defined by distinct multiomic patterns and clinical features [1]. Groups 1 and 2 are typically linked to genetic aberrations and deregulated gene expression patterns of the homologous Wingless and Int‐1 (WNT) and Sonic Hedgehog (SHH) pathways, respectively. Groups 3‐MB and 4‐MB present more heterogeneous molecular features, sharing relatively frequent alterations of Chromosome 17 and an enrichment for amplification of MYC genes. Based on the more extensive and integrated use of multiomic approaches, histological reports and clinical data, each of these core groups may be further divided into additional subgroups [2, 3, 4].
Mutations in critical regulators of the WNT (e.g., APC) and SHH (e.g., Ptch1, SUFU and Smo) pathways confer increased risk for MB development [5]. In particular, WNT‐MBs develop from progenitor cells of the lower rhombic lip, whereas SHH‐MBs arise from the granule cell progenitors (GCPs) in the external germinal layer of the developing cerebellum. Intriguingly, the DNA damage/repair response (DDR) pathway also plays an important role in MB development. For example, mutations in BRCA2, PALB2 and TP53 confer an increased risk of MB, most often of the SHH type [5]. Studies in animal models complemented these observations by showing that inactivation of Non‐Homologous End‐Joining (NHEJ) and Homologous Recombination (HR) genes, such as Lig4, Xrcc4, Ku80, Xrcc2, Brca2 and Parp1, leads to MB development in p53‐deficient backgrounds [6, 7, 8, 9, 10].
The MRE11/RAD50/NBS1 (MRN) complex is a crucial orchestrator of the DDR [11], disruption of which results in embryonic lethality in mice [12, 13, 14]. In humans, homozygous hypomorphic mutations in MRE11, NBN (coding for the NBS1 protein) and RAD50 genes cause ataxia telangiectasia‐like disorder (ATLD‐OMIM:604391), Nijmegen breakage syndrome (NBS‐OMIM:251260) and NBS‐like disorder (NBSLD‐OMIM:613078), respectively. These are typically associated with genome instability, immunodeficiency and developmental defects of neural tissues [15]. The Nbn‐CNS‐del mouse, which carries full Nbn knockout restricted to the central nervous system (CNS), recapitulates some of the neural phenotypes of the above‐mentioned syndromes such as microcephaly and dramatic cerebellar atrophy associated with ataxia [16, 17], indicating that the MRN complex is essential for cerebellar development and for the expansion of cerebellar GCPs [16, 18].
Notably, the role of the MRN complex in cancer is multifaceted. On one side, MRN‐associated syndromes are characterised by cancer predisposition [15] (http://www.cancerindex.org/Nijmegen_Breakage_Syndrome#section9), highlighting an oncosuppressive function for this complex. This is also supported by mouse models [14, 19, 20]. In particular, monoallelic Nbn knockout significantly increased the occurrence of lymphoma and solid tumours on its own or after γ‐ray exposure, supporting the hypothesis that Nbn is haploinsufficient for cancer suppression [21, 22]. Monoallelic variants of the MRN genes have been reported in many human tumours (reviewed in [23, 24]). With respect to MB, not only has it been sporadically described in NBS patients [25, 26], but heterozygous germline or somatic NBN, RAD50 and MRE11 mutations were discovered in MB patients, implying their potential role as haploinsufficient tumour suppressors for MB development [27, 28, 29, 30].
On the other hand, the MRN complex also exerts oncopromoting functions and confers survival advantages to tumour cells by facilitating resistance to oncogene‐induced RS, as demonstrated for MYCN‐driven neuroblastoma [31] and c‐MYC‐driven B‐cell lymphomas [32]. Increased expression of MRN genes has been observed in MYCN‐driven tumours and other cancer types, including prostate, gastric, rectal and head‐and‐neck cancers [31, 33].
The apparent conundrum generated by the oncosuppressive and oncopromoting functions of the MRN complex has not yet been clearly reconciled. Here, we report that monoallelic or full Nbn deletion in an SHH‐MB‐prone mouse model supports a role of this gene as a haploinsufficient oncosuppressor or an oncopromoting factor, respectively, providing evidence for a gene/dosage‐dependent effect on MB development.
MATERIALS AND METHODS
Mouse models and genotyping
The ND2:SmoA1 mice [34] and the Nbn‐CNS‐del mice [16] were previously described. By crossing ND2:SmoA1 transgenic mice, Nbn F6/F6 mice and nestin‐Cre mice, we obtained ND2:SmoA1/Nbn F6/F6;Cre+ mice (here called SmoA1/Nbn KO) and ND2:SmoA1/Nbn +/F6;Cre+ mice (here called SmoA1/Nbn HZ). The SmoA1 transgene copy number was evaluated via qPCR analysis.
Gli1tm3(Cre/ERT2)Alj [35] (here called Gli1CreER) mice were purchased from Jackson Laboratories and crossed with Nbn F6/F6 mice to obtain Nbn +/F6/Gli1CreER+/− mice. All breeding strategies were designed in order to compare mice with the same genetic background.
Genomic DNA was isolated from tail biopsies. Methods, primers and probe sequences for genotyping are given in Tables S1 and S2.
For mouse allografts, MB neurospheres (500,000 cells per flank) were suspended in an equal volume of medium and Matrigel (BD Biosciences, Heidelberg, Germany) and injected into the posterior flank of female BALB/c nude mice (Nu/Nu) (Charles River Laboratories, Lecco, Italy). When measurable, tumour size was calculated by the formula length × width × 0.5 × (length + width). For survival studies, mice were sacrificed when the tumour size reached 2 cm3. Mice not meeting these criteria earlier were sacrificed 66 days from the beginning of tumour size measurement, at the latest.
GCP cultures, SAG‐dependent neurospheres and MB neurospheres
GCPs were isolated from P5 or P7 mouse cerebella and grown as standard cultures or Smo agonist (SAG)‐dependent cerebellar neurospheres (S‐cNS) as described in [18, 36], respectively. SAG (AG‐CR1‐3506, AdipoGen Life Sciences, San Diego, CA, USA) was added to GCP neurospheres at a 200 nM concentration unless otherwise specified. MB neurospheres were prepared and grown as described [37]. Whenever necessary, GCP and MB neurospheres were pelleted and dissociated by incubation with Accutase (Gibco) to obtain single‐cell suspensions.
For neurosphere‐forming assays, dissociated cells were seeded at 20 cells/well in 96 well plates in the appropriate medium with SAG and with or without mirin (40 μM, Sigma Aldrich), GDC‐0449 (0.5 μM, S1082, Selleckchem, Houston, TX, USA) or DAPT (10 μM, 565,770, Merk Millipore, Darmstadt, Germany). Thirty technical replicates were performed for each experiment. The number of spheres/well was counted after 2 weeks.
In order to induce Nbn monoallelic KO, Nbn +/−/Gli1CreER+/− neurospheres were dissociated and seeded at 1x105 cells/cm2 in poly‐L‐Lysine (40 μg/ml) coated dishes with the adhesion medium (details in Supplementary Information). After three days, subconfluent adhering cells were treated with 1 μM 4‐hydroxytamoxifen (SC‐3542, Santa Cruz Biotechnology) or ethanol for 24 h and analysed 48 h after tamoxifen wash out.
For Nbn RNA interference, MB neurospheres were dissociated and electroporated with 100 nM of anti‐Nbn Stealth RNAi (set of 3: MSS219515, MSS219516, MSS219517, #1320001, Invitrogen) or negative control Stealth RNAi (#452001, Invitrogen) by the Amaxa Mouse NSC Nucleofector kit (VPG‐1004, Lonza, Basel Switzerland) in a Nuclefector II (Amaxa Biosystems, Gaithersburg, MD, USA).
Immunohistochemistry and immunofluorescence assays
Formalin‐fixed and paraffin‐embedded (FFPE) tissue sections were processed for H&E staining. Images were acquired using a Leica DM1000 microscope.
For EdU incorporation assay, cells were incubated with EdU (10 μM) for 1 h (S‐cNS cultures) or 24 h (GCPs standard cultures) and fixed with 3.7% formaldehyde in PBS. EdU incorporation was evaluated using the Click‐iT™ EdU Cell Proliferation Kit (Thermo Fisher Scientific), according to the manufacturer's instructions. Live/dead cell analysis was performed using the Live/Dead Cell Imaging Kit (R37601, Thermo Fisher Scientific), according to the manufacturer's instructions. Fluorescence images were acquired on a LEICA DM 2500 microscope using the IScapture software (Tucsen Photonics Co., Ltd, Fuzhou, Fujian, China). Phase contrast images were acquired on an EVOS XL CORE microscope (Thermo Fisher Scientific).
RNA and protein extraction, qPCR and Western blot
mRNA and protein extraction were performed as previously described [38]. Quantitative PCR (qPCR) was performed on a ViiA 7 real‐time PCR system (Thermo Fisher Scientific) as previously described [39] or using a custom 384‐Well Microfluidic Card TaqMan Gene Expression Assay (Thermo Fisher Scientific). The list of qPCR assays is given in Supplementary Information. At least three biological replicates were analysed for each experimental condition. All values were normalised on the expression of at least two reference genes. mRNA quantification was quantified through the ΔΔCt method.
Total protein extraction and Western blot protocols were performed as described [38, 40]. Immunoreactive bands were visualised by enhanced chemoluminescence (Advansta Inc., Menlo Park, CA, USA). The list of antibodies is given in Supplementary Table 5.
Sanger sequencing and RNA sequencing
cDNA was amplified by PCR (list of primers in Supplementary Table 6). Sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit and a 3130XL Genetic Analyser (Applied Biosystems).
RNA sequencing in service (Genomix4Life S.r.l., Baronissi [SA] Italy) was performed by Illumina NextSeq and sequenced in paired‐end mode. For each experimental condition, three biological replicates were analysed. The method for quantification of transcripts is given as Supplementary Information. Heatmap of expression values for transcripts (rows) across samples (columns) was generated by using the pheatmap R package. Gene expression values are z‐score normalised across rows and expression levels increase from blue (low) to red (high). Scatter plots are generated in R by using the library plotly on the mean of the expression levels (in log‐scale) across samples.
Public dataset
R2‐Genomics analysis and visualisation platform (http://r2.amc.nl) were used to investigate the expression of NBN, MRE11 and RAD50 genes in MB patients. MB dataset: (SHH)‐Pfister‐73‐MAS5.0, GSE49243; normal cerebellum dataset: Roth‐9‐MAS5.0, GSE3526. Data were downloaded from the website and formatted for publication.
Statistical analysis
Data are presented as mean from at least three independent experiments ± standard deviation (SD) or ± standard error (SEM), as indicated in the figure legends. Statistical analysis was performed by a standard two‐tailed Student's t‐test or ANOVA test, as indicated in the figure legends. Statistical significance of the differences in tumour penetrance between Nbn WT and Nbn HZ mice was computed using the one‐tailed chi‐square test. The survival curve was drawn as Kaplan–Meier Cumulative Proportion Surviving graph, and the corresponding p‐value was calculated with the Gehan‐Breslow‐Wilcoxon test. Software GraphPRISM6 (La Jolla, CA) was used to calculate the median survival. For all tests, a p‐value <0.05 was considered to indicate statistically significant differences.
RESULTS
Generation of SmoA1/Nbn‐CNS‐del mice
To address the role of NBS1 in MB, we used the ND2:SmoA1 mice in which a transgenic mutant Smo receptor activates the SHH pathway in GCPs, inducing SHH‐dependent MB development [34]. By repeatedly interbreeding these mice, MB penetrance in homozygous ND2:SmoA1 mice (the Smo/Smo model) declined significantly compared to the original model (Figure S1A), as also reported by others [41]. This is possibly due to a counterselection of the mice with a stronger phenotype, rendering the model more suitable for investigating the contribution of MRN dysfunction to MB tumorigenesis. These mice were crossed with Nbn‐CNS‐del mice, where Nbn is knocked out specifically in the CNS by nestin‐Cre‐dependent excision of its exon 6 [16]. The phenotypes of both ND2:SmoA1/Nbn +/F6;Cre+ mice (from now on SmoA1/Nbn HZ mice) and ND2:SmoA1/Nbn F6/F6;Cre+ mice (from now on SmoA1/Nbn KO) were studied. Nbn gene deletion and impaired protein expression in SmoA1/Nbn HZ and SmoA1/Nbn KO mice, as well as the expression of the SmoA1 transgene, were confirmed as appropriate (Figure S1B, C, D).
Nbn hemizygosity increases SmoA1‐dependent MB development
Hypomorphic and/or monoallelic MRN mutations and losses occur in human SHH‐MB[25, 26, 27, 28, 29, 30] (Waszak S. and Pfister S, personal communication). Thus, we examined the effect of monoallelic Nbn deletion (Nbn HZ) in the SmoA1 background with reduced MB penetrance, to specifically test whether it may facilitate GCP transformation. SmoA1 mice with wild‐type Nbn (from now on SmoA1/Nbn WT) reached adulthood and as expected, developed clinical signs of MB between 3 and 13 months (Figures 1A,B and S1A). Interestingly, about twice as many SmoA1/Nbn HZ mice, when compared to the SmoA1/Nbn WT ones, showed ataxia, a protruded skull, a tilted head and hunched posture suggesting the presence of a MB, which was further confirmed by macroscopic and histological analyses in all cases (Figure 1A,B). Latency for MB development did not vary significantly between the two genotypes (Figure 1C). Similarly to human MB[27], loss or mutations in the residual Nbn allele did not occur in SmoA1/Nbn HZ mice (data not shown).
FIGURE 1.
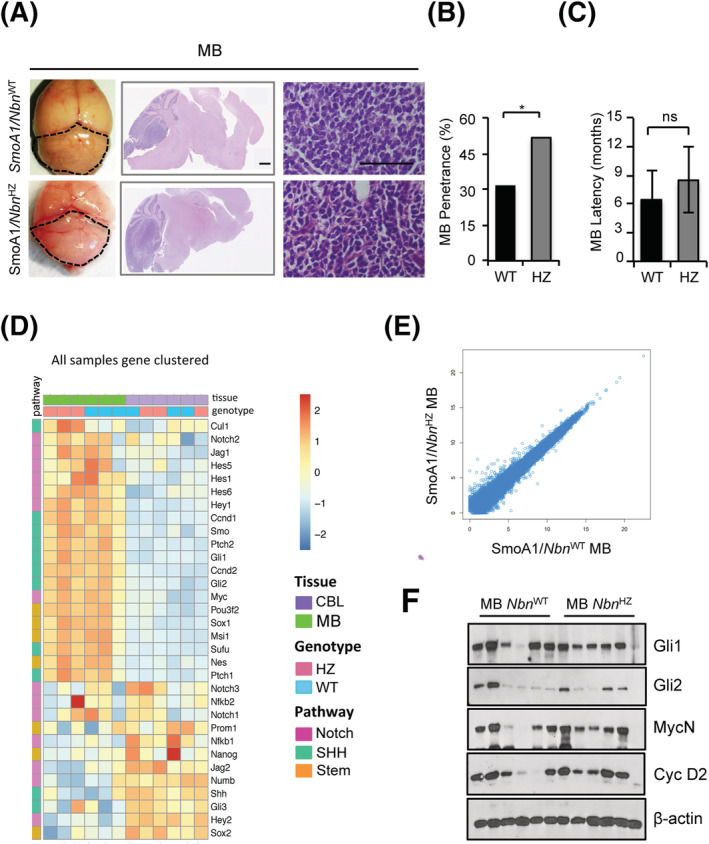
Monoallelic Nbn loss enhances Sonic Hedgehog (SHH)‐dependent medulloblastoma (MB) development. (A) Representative images of macroscopic features (left panels), haematoxylin/eosin‐stained sagittal sections (middle panels, scale bar: 1 mm) and high‐magnification view (right panels; scale bar 100 μM) of the brain/cerebella from adult mice with the indicated genotypes. Dotted lines highlight the size/shape of MB‐carrying cerebella. (B and C) Histograms representing MB penetrance (B) and latency (C) in SmoA1/Nbn WT and SmoA1/Nbn HZ mice. p‐Values were calculated by a one‐tailed chi‐square test (*p < 0.05) or two‐sided Student's t‐test (ns, not significant). (D) Heatmap of the RNA‐Seq data of the expression of the indicated transcripts in MB and healthy adult cerebella (CBL; 15 months) of SmoA1/Nbn WT (WT) and SmoA1/Nbn HZ (HZ) mice. The heatmap colour key denotes the gene expression value increasing from blue (low) to red (high). Gene expression values were clustered according to the column (samples) by exploiting a complete linkage hierarchical clustering algorithm and by using the Euclidean distance as distance metric. Pathway indication for each gene is also reported. (E) Scatter plot of the RNA‐Seq expression data in SmoA1/Nbn WT versus SmoA1/Nbn HZ tumours. Data are reported as the mean of the gene expression levels (in log‐scale) of three independent samples for each genetic background. (F) Western blot (WB) analysis of MB (n = 6) explanted from mice with the indicated genotypes
SmoA1/Nbn HZ and SmoA1/Nbn WT MBs appeared very similar at the histopathological level (Figure 1A). RNA sequencing distinctly showed that all MB samples were different from healthy adult cerebella, showing deregulation of SHH, Notch and stemness pathways (Figures 1D and S2A, B). Even though MBs were correctly clustered for genotype by using a complete linkage hierarchical clustering algorithm (Figure 1D), we found no significant differences between SmoA1/Nbn HZ and SmoA1/Nbn WT MBs (Figure 1E). In particular, analysis of the hedgehog (HH) targets Gli1, MycN and Cyclin D2 proteins confirmed a similar SHH activity in the tumour samples of both backgrounds (Figure 1F).
Overall, these data are consistent with the hypothesis that full Nbn gene dosage protects from MB development in SHH‐MB‐prone mice.
DDR markers do not differ in Nbn HZ and Nbn WT SmoA1‐driven MBs, but can be induced by acute Nbn monoallelic deletion in GCP neurospheres
Nbn hypomorphic mutations in humans and Nbn hemizygosity in mice are associated with genetic instability and increased cancer susceptibility 22, 42, 43], suggesting that Nbn might be haploinsufficient in protecting against cancer possibly via impairment of the DDR. However, we failed to detect significant differences in H2AX phosphorylation between SmoA1/Nbn HZ and SmoA1/Nbn WT MBs (Figure S3A) and we did not detect relevant differences in the expression of DDR genes among MBs from the two genotypes (Figure S3A‐F). Rather, we noticed a general upregulation of DNA repair pathways in MB samples compared to healthy cerebella (Figure S3B‐F). Moreover, we found no significant variation in the expression of NBS1 in SmoA1/Nbn HZ versus SmoA1/Nbn WT MBs (Figure S4A and D), overall confirming that full‐blown MB of SmoA1/Nbn HZ and SmoA1/Nbn WT animals do not differ significantly.
In contrast, we observed a significantly lower NBS1 expression in P7 and P14 cerebella from SmoA1/Nbn HZ compared to SmoA1/Nbn WT mice (Figure S4B, C and D), indicating that Nbn hemizygosis results in decreased NBS1 expression in developing cerebella and potentially suggesting that an increased compensatory expression by the single Nbn allele restores its protein levels in Nbn HZ MBs (Figure S4E).
To address the consequences of hemizygous Nbn KO and reduced NBS1 expression in the early steps of GCP development, we established primary SAG‐dependent GCP neurospheres (S‐cNS) [36] from P7 Nbn F6/+;Gli1CreER+/− mice. In this cellular context, the SHH pathway is constitutively active due to SAG stimulation [36], and Cre‐mediated Nbn monoallelic KO can be induced via 4‐hydroxytamoxifen (4‐OHT) administration in vitro (Figures 2A,B and S5A). Acute Nbn monoallelic KO reduced NBS1 expression in 4‐OHT‐treated S‐cNS and induced γH2AX, p53S15P and total p53 accumulation (Figure 2B), indicating a DDR activation. Therefore, the higher levels of cleaved PARP (Figure 2B) and induced cell death by 4‐OHT (Figure 2C) are likely due to an excessive accumulation of unrepaired DNA damage. Consistently, the occurrence of cell death was also confirmed in adherent S‐cNS (Figure 2D). Finally, we did not observe similar results in 4‐OHT‐treated S‐cNS from Nbn F6/+;Gli1CreER−/−mice (Figure S5B), excluding that DDR activation was directly due to 4‐OHT.
FIGURE 2.
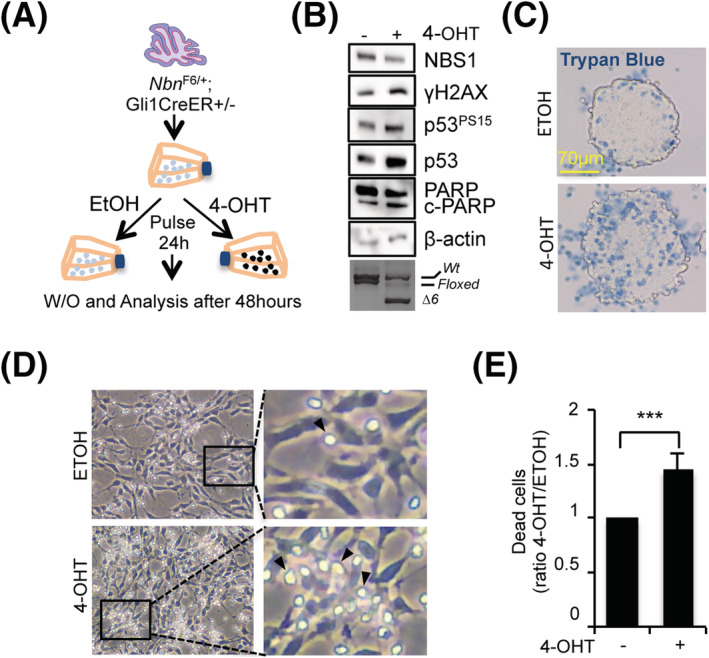
Acute monoallelic Nbn loss induces DNA damage response (DDR) activation in Smo agonist (SAG)‐dependent cerebellar neurospheres (S‐cNS). (A) Schematic protocol depicting the generation and manipulation of Nbn F6/+;Gli1CreER+/− S‐cNS. (B) Western blot (WB) analysis of the Nbn F6/+;Gli1CreER+/− S‐cNS treated with ethanol (ETOH) or 4‐hydroxytamoxifen (4‐OHT) for 24 h and collected 48 h after the washout. Blots were probed with the indicated antibodies, and β‐actin was used as loading control. c‐PARP, cleaved PARP. Cre‐induced Nbn gene cleavage identified by PCR is provided in the lowest panel, as an internal control; the size of wild type and floxed alleles are indicated as well as the Δ6 deleted allele. Data are representative of four replicates. (C) Representative images of Nbn F6/+;Gli1CreER+/− S‐cNS treated with ETOH or 4‐OHT for 24 h and collected 48 h after the washout. Cells were suspended in a Trypan blue solution to mark dead cells. (D) Representative phase‐contrast images of Nbn F6/+;Gli1CreER+/− adherent S‐cNS treated with ETOH or 4‐OHT for 24 h and photographed 48 h after washout. Arrows highlight dead cells (original magnification: 40×). (E) Quantification of cell death in cells treated as in D. Data are representative of three replicates.
Thus, Nbn hemizygosis leads to DDR activation in SHH‐driven GCPs.
Nbn hemizygosis associates with higher clonogenic capability in GCPs
Despite the biochemical activation of a typical DDR, we did not observe reduction either in EdU incorporation nor reduction of the HH targets Gli1 and MycN in surviving cells (Figure 3A,B,C). To our surprise, we found that S‐cNS with 4‐OHT‐induced Nbn hemizygosis was reproducibly able to re‐form more neurospheres compared to the control‐treated cells (Figure 3D), suggesting that Nbn hemizygosis may increase clonogenic ability in GCPs. By contrast, we did not observe similar results in 4‐OHT‐treated S‐cNS from Nbn F6/+;Gli1CreER−/− mice (Figure S5C), excluding that the clonogenic increase was directly due to 4‐OHT. Similar data were also obtained in the SmoA1‐dependent tumorigenic context (Figure 3E), where the differences between SmoA1/Nbn HZ and SmoA1/Nbn WT cultures were exacerbated by decreasing the concentration of SAG (Figure 3E), implicating that additional SHH‐independent activity may also contribute to clonogenesis in SmoA1/Nbn HZ S‐cNS compared to SmoA1/Nbn WT ones.
FIGURE 3.
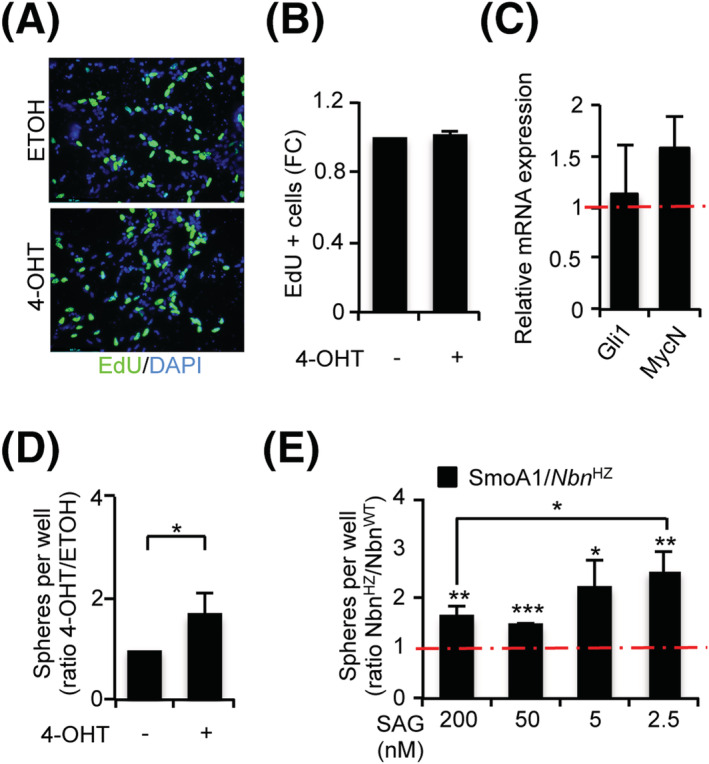
Monoallelic Nbn loss enhances clonogenicity in primary granule cell progenitors (GCPs). (A, B) Representative images of EdU staining (A) and graph (B) indicating the percentage of EdU‐positive cells in Nbn F6/+;Gli1CreER+/− GCPs treated as in Figure 2C. Data are reported as mean (±SD). (C) qPCR analysis of the indicated transcripts in Nbn F6/+;Gli1CreER+/− S‐cNS treated with ethanol (ETOH) or 4‐hydroxytamoxifen (4‐OHT) for 24 h and collected 48 h after washout. mRNA expression levels were normalised on the mean expression of two reference genes (β2‐microglobulin and Hprt), and relative mRNA quantification was expressed as fold change. Data are reported as mean (±SD). p‐Values were calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, ***p < 0.001). (D) Neurosphere formation assay from P7 Nbn F6/+;Gli1CreER+/− GCPs. Neurospheres were treated with ETOH or 4‐OHT for 24 h, dissociated, and plated for the analysis. Data are expressed as the ratio of 4‐OHT‐ versus ETOH‐treated samples and reported as mean (±SD). The p‐value was calculated by two‐sided Student's t‐test (*p < 0.05). (E) Neurosphere formation assay from P7 SmoA1/Nbn WT and SmoA1/Nbn HZ GCPs. Neurospheres were dissociated and plated for analysis in the presence of the indicated concentrations of SAG. Data are expressed as the ratio of SmoA1/Nbn HZ versus SmoA1/Nbn WT spheres for each concentration. p‐Values were calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, ***p < 0.001).
Notch and stemness pathways have been previously involved in GCP expansion [44, 45]. Interestingly, most of the representative transcripts for these pathways were slightly more expressed in SmoA1/Nbn HZ than SmoA1/Nbn WT S‐cNS (Figure 4A). For Hes1 and Hes5 transcripts, such differences reached statistical significance, suggesting a causal role of the Notch pathway in promoting clonogenesis in SmoA1/Nbn HZ. Of interest, although the SHH inhibitor GDC‐0049 inhibited clonogenesis and Gli1 expression in both SmoA1/Nbn HZ and SmoA1/Nbn WT S‐cNS (Figure 4B, S6A), confirming they are both strictly dependent on the SHH pathway, the Notch inhibitor DAPT impaired clonogenesis only in the SmoA1/Nbn HZ cultures (Figure 4B). Because DAPT inhibited Notch pathway in both S‐cNS cultures, as indicated by a similar decrease in Hes5 expression (Figure 4C), the data suggest that the Notch pathway is essential for clonogenesis only in the SmoA1/Nbn HZ S‐cNS. Importantly, DAPT did not alter Gli1 expression indicating that the HH pathway remained unaffected (Figure 4C). Similar results were also reproduced in Nbn F6/+;Gli1CreER+/− S‐cNS with a Nbn knockout acutely induced by 4‐OHT treatment (Figure S6B).
FIGURE 4.
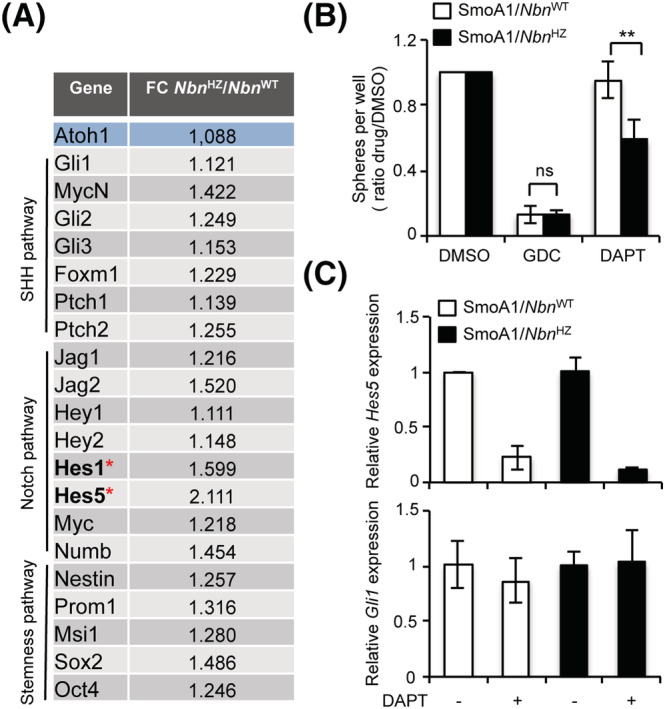
The enhanced clonogenicity of SmoA1/Nbn HZ granule cell progenitors (GCPs) depends on Notch signalling. (A) Microfluidic card qPCR analysis of the indicated transcripts in SmoA1/Nbn WT and SmoA1/Nbn HZ GCP neurospheres. mRNA expression levels were normalised on the mean expression of four reference genes (Pgk1, Hprt, Gusb, and Tfrc), and relative mRNA quantification was expressed as fold change. Data are reported as mean (±SD) of five replicates. p‐Values were calculated by two‐sided Student's t‐test. Statistically significant differences (p < 0.05) were observed for Hes1 and Hes5 (marked with a red asterisk). (B) Neurosphere formation assay from P7 SmoA1/Nbn WT and SmoA1/Nbn HZ GCPs. Neurospheres were dissociated and plated for the analysis in the presence of DMSO, DAPT or GDC‐0449. Data are expressed as the ratio of drug‐ versus DMSO‐treated samples. p‐Values were calculated by two‐sided Student's t‐test (**p < 0.01; ns, not significant). (C) qPCR analysis of the indicated transcripts in GCP neurospheres treated as in B
Collectively, these data indicate that Nbn hemizygosis increases clonogenic capability, at least partially through Notch signalling, in preneoplastic GCPs.
Nbn is essential for SHH‐driven MB development
Twelve out of 38 SmoA1/Nbn WT mice developed clinically evident MBs (Figure 5A,B). This was preceded by the occurrence of hyperplastic lesions in the EGL of fully developed cerebella (Figure 5B, box g), as described [34]. Coherently, enhanced SHH activity was revealed by the increased expression of its target genes Gli1, MycN and CycD2 and enlargement of the EGL during postnatal cerebellar development (Figure 5C,D). In sharp contrast, biallelic Nbn deletion resulted in complete suppression of SmoA1‐dependent EGL hyperplasia and MB development (Figure 5A,B). Rather, SmoA1/Nbn KO mice presented precocious signs of ataxia, growth retardation and a short lifespan, dying around weaning, substantially identical to the SmoWT/Nbn KO mice (Figure S7A‐C). Moreover, biallelic Nbn deletion resulted in impaired postnatal development of the cerebellum, which maintained an embryo‐like morphology with no fissures or folia formation, impaired expansion of the EGL, and persistently disorganised granule and Purkinje cell layers, all of which occurred equally in both WT and SmoA1 backgrounds (Figure 5C). This was accompanied by reduced cerebellar expression of SHH targets and of the GCP marker Zic1, in WT and SmoA1 backgrounds correspondingly (Figure 5D and S7D).
FIGURE 5.
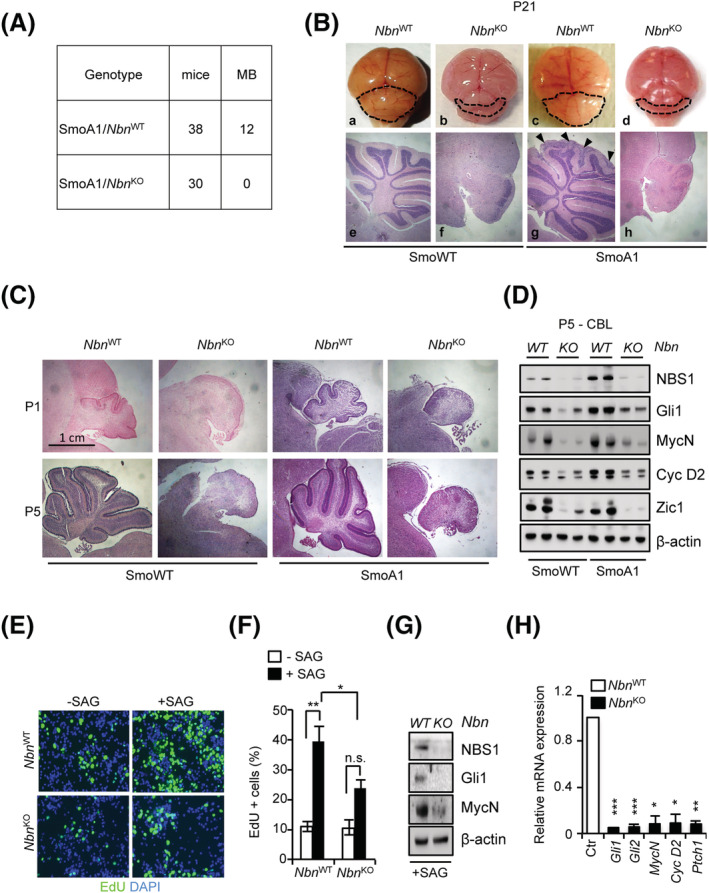
Nbn KO abolishes SmoA1‐dependent medulloblastoma (MB) and cerebellar development. (A) Summary of the animals developing MB in SmoA1/Nbn WT and SmoA1/Nbn KO mice. (B) Representative images of the macroscopic features (upper panel) and haematoxylin/eosin‐stained sagittal sections (lower panel) of the brain/cerebellum from P21 mice with the indicated genotypes. Dotted lines highlight the size/shape of the cerebella. Arrows in box g indicate regions of EGL hyperplasia. Original magnification (4×). (C) Representative images of haematoxylin/eosin‐stained sagittal sections of the P1/P5 cerebella explanted from mice with the indicated genotypes. Original magnification (4×). (D) Western blot (WB) analysis of protein extracts from P5 cerebella (n = 2). Blots were probed with the indicated antibodies, and β‐actin was used as a loading control. Data are representative of at least three replicates. (E, F) Representative images of EdU staining (E) and percentage of EdU‐positive cells (F) in primary GCPs were obtained from SmoWT/Nbn WT and SmoWT/Nbn KO cerebella, treated with or without SAG for 48 h. Data are reported as mean (±SD). p‐Values were calculated by ordinary one‐way ANOVA test. (G) WB analysis of primary granule cell progenitor (GCP) cultures generated from SmoWT/Nbn WT and SmoWT/Nbn KO cerebella. Data are representative of at least three replicates. (H) qPCR analysis of the indicated transcripts in primary GCP cultures. mRNA expression levels were normalised on two reference genes (β2‐microglobulin and Hprt), and relative mRNA quantification was expressed as fold change (FC). Data are reported as mean (±SD). p‐Values were calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant).
Importantly, we observed reduced SAG‐dependent proliferation and induction of SHH targets in primary GCPs isolated from P5 cerebella from Nbn KO compared to Nbn WT backgrounds (Figure 5E‐H). Consistently, we also observed impaired SHH‐dependent clonogenic growth of S‐cNS cultures from Nbn KO mice (Figure S7E).
Collectively, these results indicate that full Nbn KO impairs GCP postnatal expansion, preventing SmoA1‐driven hyperplasia and MB development. Because the phenotype of the SmoA1 model is driven by SHH activity, these results suggest that reduced proliferation and cell death driven by Nbn KO are epistatic on the SmoA1‐driven constitutive activation of the SHH pathway.
Nbn knockdown impairs SHH‐MB in vitro and in vivo
Although apparently counterintuitive, the idea that NBS1 is necessary for MB development is consistent with the previous data indicating that the MRN complex is required to control MYCN‐dependent RS and DNA damage in primary GCPs and in MYCN‐dependent tumours [18, 31, 46]. Consistent with this, we found higher MRN protein levels in SmoA1 and Ptch1 KO MBs compared to adult (P21) WT cerebella (Figure 6A). Moreover, human SHH‐MBs had a significantly higher expression of MRN complex mRNA compared to normal cerebellar tissues (Figure S8), supporting the hypothesis that MRN functions might be essential in SHH‐MBs. To directly test this, we interfered Nbn gene expression in Ptch KO primary MB neurospheres. As expected, Nbn depletion induced DDR activation, indicated by the increase of p53 and KAP1 phosphoepitopes (Figure 6B). Notably, it also resulted in reduced expression of the HH targets Gli1 and Gli2 (Figure 6B). Similar data were obtained by treating MB neurospheres with the MRE11 inhibitor mirin (Figure 6C and S9A), which also led to cell proliferation arrest and cell death (Figure S9B, C). To test the dependency of SHH‐MB on MRN integrity in vivo, we allografted control and Nbn‐depleted primary MB neurospheres on the flank of nude mice (n = 5/condition) and monitored tumour growth. Remarkably, Nbn depletion slowed down tumour growth in vivo, allowing an increase in mice survival (Figure 6D,E).
FIGURE 6.
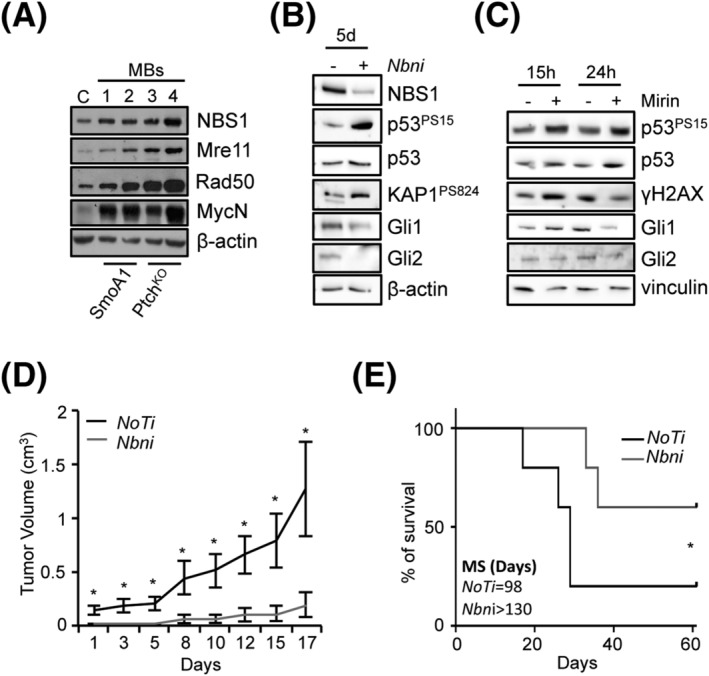
Nbn depletion impairs Sonic Hedgehog (SHH)‐medulloblastoma (MB) growth in vitro and in vivo. (A) Western blot (WB) analysis of healthy wild‐type (WT) cerebella at P21 (C) and MBs explanted from mice with the indicated genotypes. Blots were probed with the indicated antibodies, and β‐actin was used as a loading control. Data are representative of at least three replicates. (B) WB analysis of MB‐neurospheres subjected to Nbn RNAi (Nbni) or no target RNAi (NoTi) for 5 days. (C) WB analysis of MB‐neurospheres treated with mirin or vehicle. Data are representative of three replicates. (D) evaluation of tumour growth in MB‐neurosphere allografts; data are expressed as mean volume ± SEM (n = 5 per group). Day 1 represents the first day in which tumour mass reached a measurable size. p‐Values were calculated by two‐sided Student's t‐test (*p < 0.05). (E) Kaplan–Meier survival curve of MB‐neurosphere allografts. Mice were sacrificed when tumours reached a volume of ≥2 cm3 or alternatively at Day 66. MS, median survival. p‐Values were calculated by a Gehan–Breslow–Wilcoxon test (*p < 0.05).
These data indicate that Nbn is required for the growth of SHH‐MBs in vitro and in vivo and that its depletion affects the SHH pathway.
DISCUSSION
The MRN complex may exert both oncosuppressive and oncopromoting functions; however, as of today, this has not yet been clearly reconciled. Here, we report on a gene dosage‐dependent effect of Nbn KO on SHH‐MB development. In particular, we found that Nbn is haploinsufficient for SHH‐MB, for DDR and for the clonogenic growth of cerebellar GCPs, both of which emerge as relevant biological mechanisms involved in the development of SHH‐driven MB (Figure 7). In sharp contrast, full Nbn loss prevents both SHH‐MB and cerebellar development in a SmoA1‐dependent model (Figure 7).
FIGURE 7.
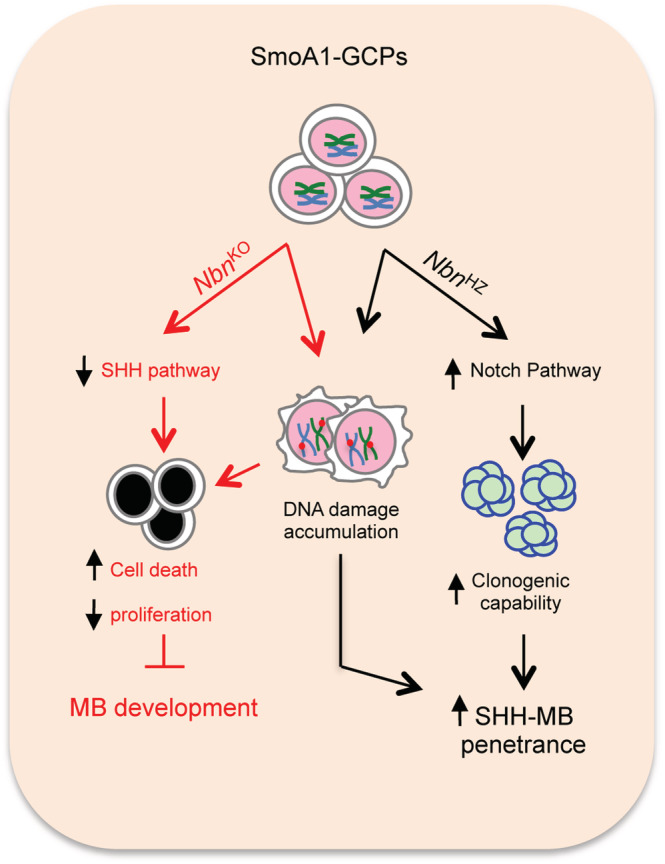
The Dual Role Of Nbs1 in Sonic Hedgehog (SHH)‐medulloblastoma (MB). In cancer prone SmoA1‐Granule Cell Progenitors (GCPs), Nbn KO causes reduced proliferation and increased cell death, ultimately leading to inhibition of MB development due to activation of the DNA damage response (DDR) and impairment of the SHH pathway. Whether these two events are linked or fully independent is currently not known. On the other hand, Nbn hemizygosis (Nbn HZ) causes DDR activation and a Notch‐dependent increase in clonogenic capability, favouring GCP transformation. These results, collectively, indicate a gene dosage‐dependent function of Nbn in SHH‐driven tumorigenesis.
The occurrence of MB in NBS patients and in heterozygous carriers and the presence of somatic NBN mutations in sporadic MB patients suggest that NBN might be haploinsufficient for MB development [27, 28, 29]. This has now been confirmed by our SmoA1/Nbn HZ model, where monoallelic Nbn KO significantly increases the likelihood of developing SHH‐MB in the absence of LOH. Rather, the reduced levels of NBS1 detectable in preneoplastic SmoA1/Nbn HZ cerebella appear to be compensated by an increased expression by the remaining allele in full‐blown MBs, which is in line with a widespread transcriptional activation of DNA repair and cell cycle genes in MB compared to healthy cerebella.
Full‐blown MBs appear morphologically identical in SmoA1/Nbn HZ and SmoA1/Nbn WT backgrounds and fail to show dramatic gene expression differences, perhaps suggesting that the mechanism/s enhancing cancer proneness in the former genotype operates at early stages of transformation. The increased level of chromosomal aberrations observed in Nbn HZ fibroblasts and tumour cells [22] and in NBS1 and RAD50 hypomorphic mutation carriers[42, 43, 47] suggests that the defective DDR in these individuals is the outcome of Nbn haploinsufficiency and the origin of genetic instability and cancer proneness. By showing that acute Nbn monoallelic KO induces H2AX and p53 phosphorylation and apoptosis in primary S‐cNS, we now add direct evidence that Nbn is haploinsufficient for the DDR‐associated replication‐born DNA damage in proliferating GCPs, the cells of origin of SHH‐MB. Much to our surprise, however, this did not lead to a reduction in EdU incorporation nor to the silencing of the SHH pathway in these cultures. Rather, Nbn HZ S‐cNS showed enhanced clonogenicity compared to Nbn WT cultures. This was further confirmed in SmoA1/Nbn HZ S‐cNS, where we also revealed upregulation of the Notch pathway compared to SmoA1/Nbn WT cultures. Consistently, the enhanced clonogenicity of SmoA1/Nbn HZ S‐cNS could be reduced by the Notch inhibitor DAPT. A role for the Notch pathway in SHH‐MB had been previously established by Hallahan et al, reporting increased expression of Notch targets in primary MBs and impaired growth/survival of SHH‐MB cells upon inhibition of Notch signalling [48]. Interestingly, Notch signalling appears to be mostly active in GCPs with more proliferative and immature properties, suggesting its expression is involved in regulating the proportion between GCPs and postmitotic GCs [49]. Our data, indicating the role of Nbn HZ on the enhanced and Notch‐dependent clonogenicity of GCPs, are also in line with the upregulation of the Notch pathway in lymphoblastoid cell lines from NBS heterozygous carriers as reported by Cheung et al. [50]. The emerging connection between NBS1 and the Notch pathway is further sustained by the observation that Nbn deletion elevates the expression of Notch targets via regulation of NICD‐RBPJ‐mediated transcriptional activity in a DDR‐independent manner [51]. Thus, both the Notch‐dependent enhanced clonogenicity and impaired DNA repair may be responsible for genetic instability and tumour proneness in Nbn HZ GCPs.
In sharp contrast, complete Nbn deletion fully prevents SmoA1‐dependent MB development due to an impairment in GCP proliferation and massive cell death, which are likely to overshadow other effects (i.e., on Notch signalling). Genetic or pharmacological inhibition of the MRN complex also impairs the growth of MB spheres in vitro and MB allografts in vivo, overall implying that MRN complex function/s are essential for SHH‐MBs. This is likely due to its role in preventing the deleterious accumulation of replication‐born DNA damage [52] exacerbated by MYCN‐induced replication stress in primary GCPs [18], which would result in a DDR and p53‐dependent cell cycle inhibition and/or cell death [16, 18, 52]. Consistently, p53KO significantly rescues cerebellar defects in Nbn KO mice [16]. Of interest, the SmoA1/Nbn KO phenotype is essentially identical to the CNS‐Nbn KO, suggesting that Nbn deletion is epistatic on constitutive SHH pathway activation because it not only impairs SmoA1‐dependent GCPs expansion in vivo but also inhibits SHH pathway and GCPs proliferation in a cell autonomous context in vitro. Due to the essential role of the SHH pathway for cerebellar development, our observations raise the hypothesis that the Nbn KO‐dependent cerebellar defects and SHH‐MB prevention might also be linked to inhibition of the SHH pathway in addition to the well‐established link to DDR and p53 activation. It is worth recalling that p53 negatively controls Gli1 expression, its nuclear localization and activity, thus counteracting SHH signalling [45, 53, 54]. Moreover, CNS‐restricted deletion of diverse DNA repair genes systematically leads to SHH‐MBs in p53−/− mice [6, 7, 8, 9], which might also be interpreted as the result of a specific function of p53 in shielding against SHH‐MB via repression of the SHH pathway. Notwithstanding these hypotheses, the molecular mechanism/s leading to the suppression of the SHH pathway in Nbn KO mice deserve further characterisation.
In conclusion, our work indicates that Nbn is haploinsufficient for SHH‐MB due to both DDR impairment and increased clonogenicity linked to the deregulation of the Notch pathway. Finally, biallelic Nbn KO appears epistatic on constitutive SHH pathway activation in GCPs, the molecular mechanisms of which needs to be further elucidated.
CONFLICTS OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ETHICS STATEMENT
Animals were housed in cages with numbers and conditions according to the Italian ministry of Health and directive 2010/637EU guidelines. Animal experiments were approved by local ethics authorities (ministry autorization no. 379/2016‐PR).
AUTHOR CONTRIBUTIONS
MP, FF and SDG performed the majority of the experiments, acquired and analysed the results, and wrote the manuscript. GG (corresponding author) conceived and supervised the project and wrote the manuscript. VNdR, MM and SC performed subcutaneous xenograft experiment and reviewed the manuscript. VLM and FBu assisted with all experiments and reviewed the manuscript. FBe performed Sanger sequencing and reviewed the manuscript. PP performed statistical and bioinformatic analyses and reviewed the manuscript. ACor performed histological analysis and reviewed the manuscript. ACop, EDS, GC, LDM and ZQW critically helped in project design, data analysis and manuscript writing.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12837.
Supporting information
Table S1. Primers sequences for PCR genotyping.
Table S2. Primers and probes sequences for qPCR genotyping.
TableS 3. List of qPCR assays used for mRNA analysis.
Table S4. List of CARD Assays used for mRNA analysis.
Figure S1. Generation of the SmoA1/Nbn KO and SmoA1/Nbn HZ mice models.
A, Kaplan‐Meier survival curve of SmoA1 homozygous mice after repeated inter crossing. B, C PCR analyses (B) showing Nbn gene deletion and Western Blot (C) showing NBS1 protein down‐regulation in P1 cerebella. Wt: Wild type allele; Floxed: allele modified by flanking exon 6 with two loxP sites; Δ6: cleaved allele. β‐actin was used as loading control. WT: Wild type; HZ: mono‐allelic KO; KO: bi‐allelic KO. D, Western Blot analysis confirming the expression of SmoA1 transgene in the new mouse models by the Myc‐tag detection in P1 cerebella. Cerebella from SmoA1 and SmoWT mice were used as positive and negative controls for SmoA1 expression, respectively.
Figure S2. Global gene expression analysis of MBs and healthy cerebella in SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
A, Scatter plot of the RNA‐Seq expression data in SmoA1/Nbn WT MB versus healthy cerebella. Data are reported as the mean of the gene expression levels (in log‐scale) of three independent experiments. B, Scatter plot of the RNASeq expression data in SmoA1/Nbn HZ MB versus healthy cerebella. Data are reported as the mean of the gene expression levels (in log‐scale) of three independent experiments.
Figure S3. Analysis of the DDR pathway in MBs originated from SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
A, WB analysis of MB (n=5) explanted from mice with the indicated genotypes. Blots were probed with the indicated antibody and β‐actin was used as loading control. B, C, D, E, F, heatmaps from RNA‐seq of the indicated transcripts in MBs and healthy adult cerebella (CBL; 15 months) of SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
Figure S4. Analysis of the NBS1 expression in pre‐tumor and MBs originated from SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
A, WB analysis of MBs (n=5) explanted from symptomatic mice with the indicated genotypes at around 7 months. Blot was probed with a Nbn antibody and β‐actin was used as loading control B, C, WB analysis of developing cerebella (n=6) explanted from mice with the indicated genotypes at P7 (A) and P14 (B). Blots were probed with a Nbn antibody and β‐actin was used as loading control. *P7 N°4 sample was loaded in all three blots to allow a proper comparison of NBS1 expression levels in the different blots. D, Box Blot representing the median Nbn protein expression in the indicated samples. Densitometry was performed from WB in A, B and C using ImageJ software (version 2.0.0‐rc‐69/1.52n). p value was calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, n.s. not significant).
Figure S5. Generation and characterization of the Nbn F6/+ ;Gli1CreER +/− cell model.
A, Schematic representation of the constructs used for the generation of the Nbn F6/+;Gli1CreER+/− mice. B, WB analysis of the Nbn F6/+;Gli1CreER−/− cells treated with ETOH or 4‐OHT for 24 hours and collected after 48 and 72 hours from the wash out. Blots were probed with the indicate antibodies and vinculin was used as loading control. C, Neurosphere formation assay from P7 Nbn F6/+;Gli1CreER−/− GCPs. Cultures were treated with ETOH or 4‐OHT for 24 hours, dissociated and plated for the analysis. Data are expressed as ratio of 4‐OHT versus ETOH treated samples and reported as mean (±SD).
Figure S6. Involvement of the Notch pathway in the enhanced clonogenicity of the SmoA1/NbnHZ S‐cNS.
A, Q‐PCR analysis of Gli1 mRNA expression in GCP neurospheres treated with GDC‐0449 for 24 hours. B, Neurosphere formation assay from P7 Nbn F6/+; Gli1CreER+/− GCPs. Neurospheres were treated with ETOH or 4‐OHT for 24 hours and then dissociated and plated for the analysis, in the presence of DMSO, DAPT or GDC‐0449. Data are expressed as ratio of 4‐OHT versus ETOH treated samples and reported as mean (±SD) of three independent experiments. P value was calculated by two‐sided Student's t‐test (*p < 0.05).
Figure S7. Additional features of SmoA1/Nbn KO and Smo/Nbn KO mice.
A, Representative pictures of the mice with the indicated genotypes at P21. B, Relative weight analysis of mice with the indicated genotypes at P14 and P21. Data are reported as mean (±SE) of at least 6 independent biological replicates. The values for each condition are represented as FC with respect to the corresponding weights at P7. P value was calculated by two‐sided Student's t‐test (*p < 0.05, ***p < 0.001, ns: not significant). C, Survival analysis of the Nbn KO mice in the SmoWT and SmoA1 backgrounds. Data are reported as mean (±SE) of at least 4 animals. D, qPCR analysis of the indicated transcripts in cerebella explanted from P5 mice with the indicated genotype. mRNA expression levels were normalized on the mean of expression of two reference genes (β2‐microglobulin, Hprt) and relative mRNA quantification was expressed as FC. Data are reported as mean (±SD). P values were calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, ***p < 0.001). E, Neurosphere formation assay from P5 Nbn WT and Nbn KO cerebellar explants grown with SAG. Data are expressed as ratio of Nbn KO versus Nbn WT samples and reported as mean (±SD). (**p < 0.01).
Figure S8. Expression of MRN complex genes in human samples. Box plots representing MRE11, RAD50 and NBN mRNA expression in healthy cerebella and SHH‐MBs (Roth‐9‐MAS5.0 and SHH‐Pfister ‐73‐MAS5.0 datasets, respectively) obtained from the R2‐Genomics platform.
Figure S9. Mirin impairs both SHH pathway and survival in MB‐neurospheres.
A, qPCR analysis of the indicated transcripts in MB‐neurospheres treated with mirin or DMSO for 15 hours. mRNA expression levels were normalized on the mean of expression of two reference genes (β2‐microglobulin, Hprt) and relative mRNA quantification was expressed as FC. Data are reported as mean (±SD). P values were calculated by two‐sided Student's t‐test (**p < 0.01, ***p < 0.001). B, Representative images of EdU staining in MB‐neurospheres treated as in A. C, Representative images of alive (green) or dead (red) MB‐neurospheres treated with mirin or DMSO for 72 hours.
ACKNOWLEDGEMENTS
Financial Support: This work was supported by: Italian Ministry of Education, Universities and Research – Dipartimenti di Eccellenza – L. 232/2016; Associazione Italiana per la Ricerca sul Cancro (AIRC) grants IG17734 and IG24329, Fondazione Telethon ‐ Italy (Grant no. GGP20135), Istituto Pasteur‐Fondazione Cenci Bolognetti and fondi Ricerca Ateneo La Sapienza to G. Giannini; AIRC grant IG17575, AFM‐Telethon grant #21025, and Istituto Pasteur‐Fondazione Cenci Bolognetti to G. Canettieri; DFG grants (WA2627/1‐1, WA2627/5‐1) Germany, grants from the Leibniz Association (SAW2014, SAW2015), a grant from the German‐Israel Foundation (GIF) (I‐1307‐418.13/2015) to Z.‐Q. Wang.
F. Belardinilli is the recipient of a fellowship from the Pezcoller Foundation. Open Access Funding provided by Universita degli Studi di Roma La Sapienza within the CRUI‐CARE Agreement.
Petroni M, Fabretti F, Di Giulio S, et al. A gene dosage‐dependent effect unveils NBS1 as both a haploinsufficient tumour suppressor and an essential gene for SHH‐medulloblastoma. Neuropathol Appl Neurobiol. 2022;48(6):e12837. doi: 10.1111/nan.12837
Marialaura Petroni, Francesca Fabretti and Stefano Di Giulio contributed equally to this work.
Funding information AFM‐Telethon; Associazione Italiana per la Ricerca sul Cancro (AIRC); DFG grants, Germany; Fondazione Telethon ‐ Italy; German‐Israel Foundation (GIF); Fondi Ricerca Ateneo La Sapienza; Italian Ministry of Education, Universities and Research ‐ Dipartimenti di Eccellenza ‐ L. 232/2016; The Leibniz Association; Istituto Pasteur‐Fondazione Cenci Bolognetti
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Northcott PA, Jones DTW, Kool M, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12(12):818‐834. doi: 10.1038/nrc3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31(6):737‐754.e6. doi: 10.1016/j.ccell.2017.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole‐genome landscape of Medulloblastoma subtypes. Nature. 2017;547(7663):311‐317. doi: 10.1038/nature22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood Medulloblastoma: A cohort study. Lancet Oncol. 2017;18(7):958‐971. doi: 10.1016/S1470-2045(17)30243-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waszak SM, Northcott PA, Buchhalter I, et al. Spectrum and prevalence of genetic predisposition in Medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018;19(6):785‐798. doi: 10.1016/S1470-2045(18)30242-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yan CT, Kaushal D, Murphy M, et al. XRCC4 suppresses Medulloblastomas with recurrent translocations in P53‐deficient mice. Proc Natl Acad Sci. 2006;103(19):7378‐7383. doi: 10.1073/pnas.0601938103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frappart P‐O, Lee Y, Lamont J, McKinnon PJ. BRCA2 is required for neurogenesis and suppression of Medulloblastoma. EMBO J. 2007;26(11):2732‐2742. doi: 10.1038/sj.emboj.7601703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holcomb VB, Vogel H, Marple T, Kornegay RW, Hasty P. Ku80 and P53 suppress Medulloblastoma that Arise independent of Rag‐1‐induced DSBs. Oncogene. 2006;25(54):7159‐7165. doi: 10.1038/sj.onc.1209704 [DOI] [PubMed] [Google Scholar]
- 9. Frappart P‐O, Lee Y, Russell HR, et al. Recurrent genomic alterations characterize Medulloblastoma arising from DNA double‐Strand break repair deficiency. PNAS. 2009;106(6):1880‐1885. doi: 10.1073/pnas.0806882106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tong W‐M, Ohgaki H, Huang H, Granier C, Kleihues P, Wang Z‐Q. Null mutation of DNA Strand break‐binding molecule poly (ADP‐ribose) Polymerase Causes Medulloblastomas in P53−/− Mice. Am J Pathol. 2003;162(1):10‐352. doi: 10.1016/S0002-9440(10)63825-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Syed A, Tainer JA. The MRE11–RAD50–NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. 2018;87(1):263‐294. doi: 10.1146/annurev-biochem-062917-012415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11(2):105‐109. doi: 10.1016/S0960-9822(01)00019-7 [DOI] [PubMed] [Google Scholar]
- 13. Buis J, Wu Y, Deng Y, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135(1):85‐96. doi: 10.1016/j.cell.2008.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo G, Yao MS, Bender CF, et al. Disruption of MRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci. 1999;96(13):7376‐7381. doi: 10.1073/pnas.96.13.7376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor AMR, Rothblum‐Oviatt C, Ellis NA, et al. Chromosome Instability Syndromes. Nat Rev Dis Primers. 2019;5(1):64. doi: 10.1038/s41572-019-0113-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frappart P‐O, Tong W‐M, Demuth I, et al. An essential function for NBS1 in the prevention of Ataxia and cerebellar defects. Nat Med. 2005;11(5):538‐544. doi: 10.1038/nm1228 [DOI] [PubMed] [Google Scholar]
- 17. Li R, Yang Y‐G, Gao Y, Wang Z‐Q, Tong W‐M. A distinct response to endogenous DNA damage in the development of Nbs1‐deficient cortical neurons. Cell Res. 2012;22(5):859‐872. doi: 10.1038/cr.2012.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petroni M, Sardina F, Heil C, et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2016;23(2):197‐206. doi: 10.1038/cdd.2015.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta GP, Vanness K, Barlas A, Manova‐Todorova KO, Wen YH, Petrini JHJ. The Mre11 complex suppresses oncogene‐driven breast tumorigenesis and metastasis. Mol Cell. 2013;52(3):353‐365. doi: 10.1016/j.molcel.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JH, Penson AV, Taylor BS, Petrini JHJ. Nbn−Mre11 interaction is required for tumor suppression and genomic integrity. Proc Natl Acad Sci U S A. 2019;116(30):15178‐15183. doi: 10.1073/pnas.1905305116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang J, Bronson RT, Xu Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J. 2002;21(6):1447‐1455. doi: 10.1093/emboj/21.6.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dumon‐Jones V, Frappart P‐O, Tong W‐M, et al. Nbn Heterozygosity Renders Mice Susceptible to Tumor Formation and Ionizing Radiation‐Induced Tumorigenesis. Vol. 8. [PubMed] [Google Scholar]
- 23. Rahman S, Canny MD, Buschmann TA, Latham MP. A survey of reported disease‐related mutations in the MRE11‐RAD50‐NBS1 complex. Cell. 2020;9(7):1678. doi: 10.3390/cells9071678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bian L, Meng Y, Zhang M, Li D. MRE11‐RAD50‐NBS1 complex alterations and DNA damage response: implications for Cancer treatment. Mol Cancer. 2019;18(1):169. doi: 10.1186/s12943-019-1100-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bakhshi S, Cerosaletti KM, Concannon P, et al. Medulloblastoma with adverse reaction to radiation therapy in Nijmegen breakage syndrome. J Pediatr Hematol Oncol. 2003;25(3):248‐251. doi: 10.1097/00043426-200303000-00013 [DOI] [PubMed] [Google Scholar]
- 26. Wegner R‐D, German JJ, Chrzanowska KH, Digweed M, Stumm M. Chromosomal Instability Syndromes Other than Ataxia‐Telangiectasia. In: Ochs HD, Smith CIE, Puck JM, eds. Primary Immunodeficiency Diseases. Oxford University Press; 2013:632‐661. doi: 10.1093/med/9780195389838.003.0048. [DOI] [Google Scholar]
- 27. Ciara E, Piekutowska‐Abramczuk D, Popowska E, et al. Heterozygous germ‐line mutations in the NBN gene predispose to Medulloblastoma in pediatric patients. Acta Neuropathol. 2010;119(3):325‐334. doi: 10.1007/s00401-009-0608-y [DOI] [PubMed] [Google Scholar]
- 28. Trubicka J, Żemojtel T, Hecht J, et al. The germline variants in DNA repair genes in pediatric Medulloblastoma: A challenge for current therapeutic strategies. BMC Cancer. 2017;17(1):239. doi: 10.1186/s12885-017-3211-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang J, Grotzer MA, Watanabe T, et al. Mutations in the Nijmegen breakage syndrome gene in Medulloblastomas. Clin Cancer Res. 2008;14(13):4053‐4058. doi: 10.1158/1078-0432.CCR-08-0098 [DOI] [PubMed] [Google Scholar]
- 30. Viana‐Pereira M, Almeida I, Sousa S, et al. Analysis of microsatellite Instability in Medulloblastoma. Neuro Oncol. 2009;11(5):458‐467. doi: 10.1215/15228517-2008-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petroni M, Sardina F, Infante P, et al. MRE11 inhibition highlights a replication stress‐dependent vulnerability of MYCN‐driven tumors. Cell Death Dis. 2018;9(9):895. doi: 10.1038/s41419-018-0924-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spehalski E, Capper KM, Smith CJ, et al. MRE11 promotes tumorigenesis by facilitating resistance to oncogene‐induced replication stress. Cancer Res. 2017;77(19):5327‐5338. doi: 10.1158/0008-5472.CAN-17-1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altan B, Altan B, Yokobori T, et al. High expression of MRE11–RAD50–NBS1 is associated with poor prognosis and Chemoresistance in gastric Cancer. Anticancer Res. 2016;36(10):5237‐5247. doi: 10.21873/anticanres.11094 [DOI] [PubMed] [Google Scholar]
- 34. Hatton BA, Villavicencio EH, Tsuchiya KD, et al. The Smo/Smo model: hedgehog‐induced Medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68(6):1768‐1776. doi: 10.1158/0008-5472.CAN-07-5092 [DOI] [PubMed] [Google Scholar]
- 35. Ahn S, Joyner AL. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell. 2004;118(4):505‐516. doi: 10.1016/j.cell.2004.07.023 [DOI] [PubMed] [Google Scholar]
- 36. Petroni M, Sahùn Roncero M, Ramponi V, et al. SMO‐M2 mutation does not support cell‐autonomous hedgehog activity in cerebellar granule cell precursors. Sci Rep. 2019;9(1):19623. doi: 10.1038/s41598-019-56057-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway–Dependent Tumors. Cancer Res. 2015;75(17):3623‐3635. doi: 10.1158/0008-5472.CAN-14-2999-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Di Giulio S, Colicchia V, Pastorino F, et al. A combination of PARP and CHK1 inhibitors efficiently antagonizes MYCN‐driven tumors. Oncogene. 2021;40(43):6143‐6152. doi: 10.1038/s41388-021-02003-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petroni M, Veschi V, Prodosmo A, et al. MYCN sensitizes human neuroblastoma to apoptosis by HIPK2 activation through a DNA damage response. Mol Cancer Res. 2011;9(1):67‐77. doi: 10.1158/1541-7786.MCR-10-0227 [DOI] [PubMed] [Google Scholar]
- 40. Giannini G, Kim CJ, Marcotullio LD, et al. Expression of the HMGI(Y) gene products in human neuroblastic tumours correlates with differentiation status. Br J Cancer. 2000;83(11):1503‐1509. doi: 10.1054/bjoc.2000.1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanghez V, Chen M, Li S, et al. Efficacy of asparaginase erwinia chrysanthemi with and without temozolomide against glioma cells and intracranial mouse medulloblastoma. Anticancer Res. 2018;38(5):2627‐2634. doi: 10.21873/anticanres.12504 [DOI] [PubMed] [Google Scholar]
- 42. Neubauer S, Arutyunyan R, Stumm M, et al. Radiosensitivity of ataxia telangiectasia and Nijmegen breakage syndrome homozygotes and heterozygotes as determined by three‐color FISH chromosome painting. Radiat Res. 2002;157(3):312‐321. doi: 10.1667/0033-7587(2002)157[0312:ROATAN]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 43. Tanzarella C, Antoccia A, Spadoni E, et al. Chromosome instability and Nibrin protein variants in NBS heterozygotes. Eur J Hum Genet. 2003;11(4):297‐303. doi: 10.1038/sj.ejhg.5200962 [DOI] [PubMed] [Google Scholar]
- 44. Solecki DJ, Liu X, Tomoda T, Fang Y, Hatten ME. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31(4):557‐568. doi: 10.1016/S0896-6273(01)00395-6 [DOI] [PubMed] [Google Scholar]
- 45. Stecca B, Ruiz i Altaba, A. A GLI1‐P53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009;28(6):663‐676. doi: 10.1038/emboj.2009.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petroni M, Giannini G. A MYCN‐MRN complex axis controls replication stress for the safe expansion of neuroprogenitor cells. Mol Cell Oncol. 2016;3(2):e1079673. doi: 10.1080/23723556.2015.1079673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heikkinen K. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis. 2005;27(8):1593‐1599. doi: 10.1093/carcin/bgi360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that Notch signaling is critical for the growth and survival of sonic hedgehog‐induced medulloblastomas. Cancer Res. 2004;64(21):7794‐7800. doi: 10.1158/0008-5472.CAN-04-1813 [DOI] [PubMed] [Google Scholar]
- 49. Adachi T, Miyashita S, Yamashita M, et al. Notch signaling between cerebellar granule cell progenitors. eNeuro. 2021;8(3):ENEURO.0468‐20.2021. doi: 10.1523/ENEURO.0468-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheung VG. Heterozygous carriers of Nijmegen breakage syndrome have a distinct gene expression phenotype. Genome Res. 2006;16(8):973‐979. doi: 10.1101/gr.5320706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou Z‐W, Kirtay M, Schneble N, et al. NBS1 interacts with notch signaling in neuronal homeostasis. Nucleic Acids Res. 2020;48(19):10924‐10939. doi: 10.1093/nar/gkaa716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bruhn C, Zhou Z‐W, Ai H, Wang Z‐Q. The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep. 2014;6(1):182‐195. doi: 10.1016/j.celrep.2013.12.018 [DOI] [PubMed] [Google Scholar]
- 53. Mazzà D, Infante P, Colicchia V, et al. PCAF ubiquitin ligase activity inhibits hedgehog/Gli1 signaling in P53‐dependent response to genotoxic stress. Cell Death Differ. 2013;20(12):1688‐1697. doi: 10.1038/cdd.2013.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoon JW, Lamm M, Iannaccone S, et al. P53 modulates the activity of the GLI1 oncogene through interactions with the shared coactivator TAF9. DNA Repair. 2015;34:9‐17. doi: 10.1016/j.dnarep.2015.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers sequences for PCR genotyping.
Table S2. Primers and probes sequences for qPCR genotyping.
TableS 3. List of qPCR assays used for mRNA analysis.
Table S4. List of CARD Assays used for mRNA analysis.
Figure S1. Generation of the SmoA1/Nbn KO and SmoA1/Nbn HZ mice models.
A, Kaplan‐Meier survival curve of SmoA1 homozygous mice after repeated inter crossing. B, C PCR analyses (B) showing Nbn gene deletion and Western Blot (C) showing NBS1 protein down‐regulation in P1 cerebella. Wt: Wild type allele; Floxed: allele modified by flanking exon 6 with two loxP sites; Δ6: cleaved allele. β‐actin was used as loading control. WT: Wild type; HZ: mono‐allelic KO; KO: bi‐allelic KO. D, Western Blot analysis confirming the expression of SmoA1 transgene in the new mouse models by the Myc‐tag detection in P1 cerebella. Cerebella from SmoA1 and SmoWT mice were used as positive and negative controls for SmoA1 expression, respectively.
Figure S2. Global gene expression analysis of MBs and healthy cerebella in SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
A, Scatter plot of the RNA‐Seq expression data in SmoA1/Nbn WT MB versus healthy cerebella. Data are reported as the mean of the gene expression levels (in log‐scale) of three independent experiments. B, Scatter plot of the RNASeq expression data in SmoA1/Nbn HZ MB versus healthy cerebella. Data are reported as the mean of the gene expression levels (in log‐scale) of three independent experiments.
Figure S3. Analysis of the DDR pathway in MBs originated from SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
A, WB analysis of MB (n=5) explanted from mice with the indicated genotypes. Blots were probed with the indicated antibody and β‐actin was used as loading control. B, C, D, E, F, heatmaps from RNA‐seq of the indicated transcripts in MBs and healthy adult cerebella (CBL; 15 months) of SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
Figure S4. Analysis of the NBS1 expression in pre‐tumor and MBs originated from SmoA1/Nbn WT and SmoA1/Nbn HZ mice.
A, WB analysis of MBs (n=5) explanted from symptomatic mice with the indicated genotypes at around 7 months. Blot was probed with a Nbn antibody and β‐actin was used as loading control B, C, WB analysis of developing cerebella (n=6) explanted from mice with the indicated genotypes at P7 (A) and P14 (B). Blots were probed with a Nbn antibody and β‐actin was used as loading control. *P7 N°4 sample was loaded in all three blots to allow a proper comparison of NBS1 expression levels in the different blots. D, Box Blot representing the median Nbn protein expression in the indicated samples. Densitometry was performed from WB in A, B and C using ImageJ software (version 2.0.0‐rc‐69/1.52n). p value was calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, n.s. not significant).
Figure S5. Generation and characterization of the Nbn F6/+ ;Gli1CreER +/− cell model.
A, Schematic representation of the constructs used for the generation of the Nbn F6/+;Gli1CreER+/− mice. B, WB analysis of the Nbn F6/+;Gli1CreER−/− cells treated with ETOH or 4‐OHT for 24 hours and collected after 48 and 72 hours from the wash out. Blots were probed with the indicate antibodies and vinculin was used as loading control. C, Neurosphere formation assay from P7 Nbn F6/+;Gli1CreER−/− GCPs. Cultures were treated with ETOH or 4‐OHT for 24 hours, dissociated and plated for the analysis. Data are expressed as ratio of 4‐OHT versus ETOH treated samples and reported as mean (±SD).
Figure S6. Involvement of the Notch pathway in the enhanced clonogenicity of the SmoA1/NbnHZ S‐cNS.
A, Q‐PCR analysis of Gli1 mRNA expression in GCP neurospheres treated with GDC‐0449 for 24 hours. B, Neurosphere formation assay from P7 Nbn F6/+; Gli1CreER+/− GCPs. Neurospheres were treated with ETOH or 4‐OHT for 24 hours and then dissociated and plated for the analysis, in the presence of DMSO, DAPT or GDC‐0449. Data are expressed as ratio of 4‐OHT versus ETOH treated samples and reported as mean (±SD) of three independent experiments. P value was calculated by two‐sided Student's t‐test (*p < 0.05).
Figure S7. Additional features of SmoA1/Nbn KO and Smo/Nbn KO mice.
A, Representative pictures of the mice with the indicated genotypes at P21. B, Relative weight analysis of mice with the indicated genotypes at P14 and P21. Data are reported as mean (±SE) of at least 6 independent biological replicates. The values for each condition are represented as FC with respect to the corresponding weights at P7. P value was calculated by two‐sided Student's t‐test (*p < 0.05, ***p < 0.001, ns: not significant). C, Survival analysis of the Nbn KO mice in the SmoWT and SmoA1 backgrounds. Data are reported as mean (±SE) of at least 4 animals. D, qPCR analysis of the indicated transcripts in cerebella explanted from P5 mice with the indicated genotype. mRNA expression levels were normalized on the mean of expression of two reference genes (β2‐microglobulin, Hprt) and relative mRNA quantification was expressed as FC. Data are reported as mean (±SD). P values were calculated by two‐sided Student's t‐test (*p < 0.05, **p < 0.01, ***p < 0.001). E, Neurosphere formation assay from P5 Nbn WT and Nbn KO cerebellar explants grown with SAG. Data are expressed as ratio of Nbn KO versus Nbn WT samples and reported as mean (±SD). (**p < 0.01).
Figure S8. Expression of MRN complex genes in human samples. Box plots representing MRE11, RAD50 and NBN mRNA expression in healthy cerebella and SHH‐MBs (Roth‐9‐MAS5.0 and SHH‐Pfister ‐73‐MAS5.0 datasets, respectively) obtained from the R2‐Genomics platform.
Figure S9. Mirin impairs both SHH pathway and survival in MB‐neurospheres.
A, qPCR analysis of the indicated transcripts in MB‐neurospheres treated with mirin or DMSO for 15 hours. mRNA expression levels were normalized on the mean of expression of two reference genes (β2‐microglobulin, Hprt) and relative mRNA quantification was expressed as FC. Data are reported as mean (±SD). P values were calculated by two‐sided Student's t‐test (**p < 0.01, ***p < 0.001). B, Representative images of EdU staining in MB‐neurospheres treated as in A. C, Representative images of alive (green) or dead (red) MB‐neurospheres treated with mirin or DMSO for 72 hours.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.