

ABSTRACT
Zaire ebolavirus (EBOV) causes a severe hemorrhagic fever in humans and non-human primates with high morbidity and mortality. EBOV infection is dependent on its structural glycoprotein (GP), but high levels of GP expression also trigger cell rounding, detachment, and downregulation of many surface molecules that is thought to contribute to its high pathogenicity. Thus, EBOV has evolved an RNA editing mechanism to reduce its GP expression and increase its fitness. We now report that the GP expression is also suppressed at the protein level in cells by protein disulfide isomerases (PDIs). Although PDIs promote oxidative protein folding by catalyzing correct disulfide formation in the endoplasmic reticulum (ER), PDIA3/ERp57 adversely triggered the GP misfolding by targeting GP cysteine residues and activated the unfolded protein response (UPR). Abnormally folded GP was targeted by ER-associated protein degradation (ERAD) machinery and, unexpectedly, was degraded via the macroautophagy/autophagy-lysosomal pathway, but not the proteasomal pathway. PDIA3 also decreased the GP expression from other ebolavirus species but increased the GP expression from Marburg virus (MARV), which is consistent with the observation that MARV-GP does not cause cell rounding and detachment, and MARV does not regulate its GP expression via RNA editing during infection. Furthermore, five other PDIs also had a similar inhibitory activity to EBOV-GP. Thus, PDIs negatively regulate ebolavirus glycoprotein expression, which balances the viral life cycle by maximizing their infection but minimizing their cellular effect. We suggest that ebolaviruses hijack the host protein folding and ERAD machinery to increase their fitness via reticulophagy during infection.
Abbreviations: 3-MA: 3-methyladenine; 4-PBA: 4-phenylbutyrate; ACTB: β-actin; ATF: activating transcription factor; ATG: autophagy-related; BafA1: bafilomycin A1; BDBV: Bundibugyo ebolavirus; CALR: calreticulin; CANX: calnexin; CHX: cycloheximide; CMA: chaperone-mediated autophagy; ConA: concanamycin A; CRISPR: clusters of regularly interspaced short palindromic repeats; Cas9: CRISPR-associated protein 9; dsRNA: double-stranded RNA; EBOV: Zaire ebolavirus; EDEM: ER degradation enhancing alpha-mannosidase like protein; EIF2AK3/PERK: eukaryotic translation initiation factor 2 alpha kinase 3; Env: envelope glycoprotein; ER: endoplasmic reticulum; ERAD: ER-associated protein degradation; ERN1/IRE1: endoplasmic reticulum to nucleus signaling 1; GP: glycoprotein; HA: hemagglutinin; HDAC6: histone deacetylase 6; HMM: high-molecular-mass; HIV-1: human immunodeficiency virus type 1; HSPA5/BiP: heat shock protein family A (Hsp70) member 5; IAV: influenza A virus; IP: immunoprecipitation; KIF: kifenesine; Lac: lactacystin; LAMP: lysosomal associated membrane protein; MAN1B1/ERManI: mannosidase alpha class 1B member 1; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MARV: Marburg virus; MLD: mucin-like domain; NHK/SERPINA1: alpha1-antitrypsin variant null (Hong Kong); NTZ: nitazoxanide; PDI: protein disulfide isomerase; RAVV: Ravn virus; RESTV: Reston ebolavirus; SARS-CoV: severe acute respiratory syndrome coronavirus; SBOV: Sudan ebolavirus; sGP: soluble GP; SQSTM1/p62: sequestosome 1; ssGP: small soluble GP; TAFV: Taï Forest ebolavirus; TIZ: tizoxanide; TGN: thapsigargin; TLD: TXN (thioredoxin)-like domain; Ub: ubiquitin; UPR: unfolded protein response; VLP: virus-like particle; VSV: vesicular stomatitis virus; WB: Western blotting; WT: wild-type; XBP1: X-box binding protein 1.
KEYWORDS: Autophagy, ebola, ERAD, ERp57, ER-phagy, EVD, filoviruses, glycoproteins, lysosomes, reticulophagy
Introduction
The virus family Filoviridae includes two genera ebolavirus and marburgvirus that cause severe hemorrhagic fever in humans and nonhuman primates with up to a 90% mortality rate [1]. Ebolaviruses have five major species, including Zaire ebolavirus (EBOV), Sudan ebolavirus (SBOV), Bundibugyo ebolavirus (BDBV), Taï Forest ebolavirus (TAFV), and Reston ebolavirus (RESTV), and EBOV causes the highest mortality rate. Marburgviruses have two species, including Marburg virus (MARV) and Ravn virus (RAVV). Filoviruses are enveloped viruses with a negative-sense single-stranded RNA genome comprising seven genes [2]. Their entry is mediated by the virion associated GP1/GP2 trimer, which is proteolytically processed in the Golgi from the structural GP precursor (GP1,2) produced in the endoplasmic reticulum (ER) from the GP gene [3].
High levels of EBOV-GP1,2 expression triggers cell rounding, detachment, and downregulation of many surface molecules that is thought to contribute to its high pathogenicity [4–9]. The determinants for this cellular effect were mapped to the mucin-like domain (MLD) in GP1 and the transmembrane domain in GP2 [7,9,10]. To increase its fitness, EBOV has evolved an RNA editing mechanism for GP, which reduces the GP1,2 expression [11,12]. Approximately 70% of the total transcripts are not edited, containing a premature stop codon and producing a nonstructural soluble GP (sGP); ~25% of transcripts are edited by a + 1 frameshift and produce GP1,2; the remaining ~5% of transcripts are edited by a + 2 frameshift that produces another nonstructural small soluble GP (ssGP) [13].
Enzyme-mediated disulfide formation, which is catalyzed by protein disulfide isomerases (PDIs), is a key aspect of protein folding in the ER. The PDI family consists of 21 members and most PDIs have an active TXN (thioredoxin)-like domain (TLD) that functions as a thiol-disulfide oxidoreductase and isomerase [14]. PDIs catalyze the formation and breakage of disulfide bonds between cysteine residues within proteins as they fold, ensuring that proteins are in a properly folded state. PDIs contribute to two major folding pathways by interacting with ER chaperones [15]. PDIA1/PDI/P4HB, the first characterized member of this family, interacts with HSPA5/ BiP (heat shock protein family A (Hsp70) member 5), and is a constituent of the general folding pathway. PDIA3/ERp57 (PDI family A member 3) interacts with CANX (calnexin) and CALR (calreticulin), and forms the CANX-CALR cycle that promotes N-glycosylated protein folding [15]. Notably, the ER is easily overloaded due to its inefficient and error-prone protein folding capacity, resulting in accumulation of misfolded proteins that cause ER stress [16]. To regain the ER homeostasis, the unfolded protein response (UPR) is activated, which stalls protein translation to reduce the protein load in the ER and activates gene expression of proteins involved in protein folding. In addition, the ER has also evolved two quality control mechanisms to degrade these abnormal proteins via ER-associated degradation (ERAD) or reticulophagy [17].
EBOV places a heavy burden on the ER protein folding machinery during infection. In EBOV-GP1,2, GP1 has 15 N-glycosylation sites and its MLD has ~80 O-glycosylation sites; GP2 has 2 N-glycosylation sites. In addition, GP1 and GP2 have five or seven cysteine residues. Except for two cysteine residues in GP2 (Cys638, Cys640) that are in the transmembrane domain, all of them are involved in disulfide bond formation. Cys21 in GP1 and Cys577 in GP2 form one intermolecular bridge, and the other eight are cross-linked into four intramolecular bridges, which are all required for viral infection [18]. We now report that PDIs negatively regulate ebolavirus GP1,2 expression via the autophagy-lysosomal pathway, providing a new understanding of mechanisms for regulation of the GP expression.
Results
PDIA3 inhibits EBOV structural GP expression
To understand the regulation of EBOV structural glycoprotein expression, we investigated how EBOV-GP1,2 (hereafter “GP”) interacts with proteins in the ER via affinity tag-purification mass spectrometry (AP-MS). Because genetic deletion of MLD in GP1 greatly increases EBOV entry [18], both the wild-type (WT) EBOV-GP and its MLD-deleted version, GP∆MLD, were tested. FLAG-tagged GP and GP∆MLD were expressed in HEK293T cells, and they were purified by an anti-FLAG affinity column, which were detected at ~130 kDa or ~75 kDa by Western blotting (WB), respectively (Figure 1A). Purified protein complexes were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) as we did recently [19]. A total of 672 and 926 cellular proteins were identified from GP∆MLD- or GP-derived samples (Table S1 & S2). ER-associated proteins were then selected based on the cellular component information obtained from UniProt. Three chaperones (HSPA5/BiP, CANX, CALR), 4 PDIs (PDIA1/PDI/P4HB, PDIA3/ERp57, PDIA4/ERp70, PDIA6/ERp5), 11 ribosomal proteins, and 12 additional proteins were found from both samples (Figure 1B). Identification of CANX and CALR was expected because they bind to EBOV-GP [20].
Figure 1.
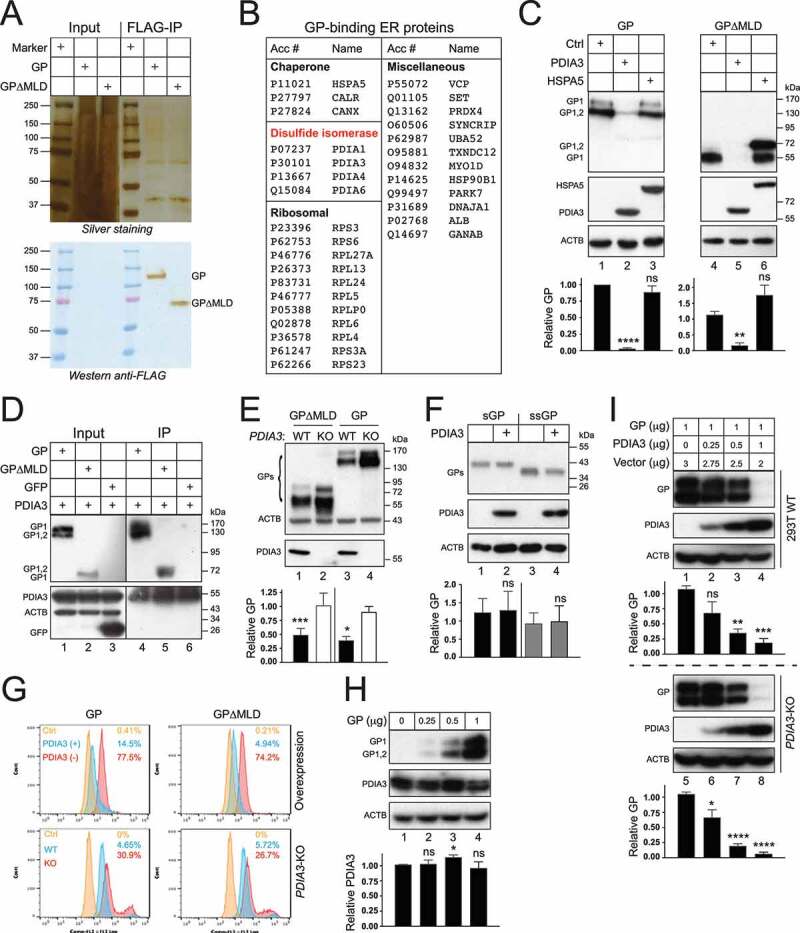
Identification of ER proteins that interact with the EBOV structural GP. (a) FLAG-tagged EBOV-GP and EBOV-GP∆MLD were expressed in HEK293T cells and purified with an anti-FLAG affinity column. Purified proteins were analyzed by SDS-PAGE followed by silver staining (top gel) or WB (bottom gel). (b) Affinity-purified EBOV glycoprotein complexes were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). Identified ER proteins that interact with both GP and GP∆MLD are listed. (c) GP and GP∆MLD with a N-terminal FLAG-tag were expressed with PDIA3/ERp57 or HSPA5/BiP in HEK293T cells, and their expression was determined by WB. (d) Flag-tagged GP, GP∆MLD, and GFP were expressed with PDIA3 and pulled down by anti-PDIA3. Proteins in cell lysate and immunoprecipitation (IP) samples were determined by WB. (e) GP and GP∆MLD were expressed in HEK293T WT and PDIA3-KO cells and their expression was determined by WB. (f) sGP and ssGP with a N-terminal HiBiT-tag were expressed with PDIA3 in HEK293T cells, and their expression was determined by the Nano-Glo® HiBiT Blotting System. (g) HEK293T cells were transfected with 1 μg PDIA3 and 1 μg GP or GP∆MLD expression vectors. Alternatively, HEK293T WT and PDIA3-KO cells were transfected with 1 μg GP or GP∆MLD expression vectors. GP expression on the cell surface was determined by flow cytometry. (h) HEK293T cells were transfected with increasing amounts of a GP expression vector. GP and the endogenous PDIA3 expression were determined by WB. (i) HEK293T WT and PDIA3-KO cells were transfected with 1 μg GP expression vector and increasing amounts of an PDIA3 expression vector. GP and PDIA3 expression were detected by WB. The levels of GP expression in C, E, F, and I, and those of PDIA3 expression in H were further quantified from these Western blots by ImageJ and are presented under each of these blots. Results are shown as relative values, with the value from a control (Ctrl) vector set as 1. Error bars represent the standard error of measurements (SEMs) calculated from three experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (not significant, p > 0.05).
To understand how HSPA5 and PDIA3 affect the GP expression, FLAG-tagged GP and GP∆MLD were expressed with HSPA5 or PDIA3, and their expression was analyzed by WB. Both of their uncleaved, ER form of GP (GP1,2), and cleaved, Golgi form of GP (GP1) were detected (Figure 1C). As we reported previously [20,21], GP1 exhibited a much higher molecular weight than GP1,2 (170 versus 130 kDa) due to the heavy O-glycosylation in the Golgi (lanes 1, 3), and when MLD was deleted, GP1 and GP1,2 were detected at 55 or 72 kDa, respectively (lanes 4, 6). HSPA5 increased the ER form of GP∆MLD expression but did not affect the Golgi form of GP∆MLD or any GP expression (lanes 3, 6). Unexpectedly, PDIA3 strongly decreased both GP and GP∆MLD expression up to 150-fold (lanes 2, 5). The interaction of PDIA3 with GP and GP∆MLD was detected by immunoprecipitation (IP) (Figure 1D). We also tested the endogenous PDIA3 activity after knocking out PDIA3 in HEK293T cells by CRISPR-Cas9. Compared to WT cells, the GP and GP∆MLD expression were both increased in PDIA3-knockout (KO) cells up to 2.5-fold (Figure 1E, lanes 2, 4). PDIA3 had no effect on sGP and ssGP expression (Figure 1F). Both exogenous and endogenous PDIA3 also effectively downregulated GP and GP∆MLD from the cell surface when measured by flow cytometry (Figure 1G). GP did not apparently reduce the endogenous PDIA3 expression (Figure 1H). The PDIA3 inhibitory effect on GP was dose-dependent in both WT and PDIA3-KO cells (Figure 1I). These results demonstrate that PDIA3 inhibits EBOV-GP expression.
Viral assays were conducted to confirm the PDIA3 inhibitory effect on the GP expression. Initially, we used HIV-1 pseudoviruses to measure EBOV entry, which are widely used for studying the GP function [20,21]. Env-deficient HIV-1 luciferase reporter viruses were produced from HEK293T WT, PDIA3-overexpressing, or PDIA3-KO cells after expressing GP or GP∆MLD. An equal amount of pseudoviruses was collected to infect HEK293T cells, and viral entry was determined by measuring intracellular luciferase activity. GP∆MLD-pseudoviruses had much higher entry than GP-pseudoviruses, which is consistent with the observation that MLD is deleterious to the virus entry (Figure 2A, lanes 1, 3, 5, 7). Both exogenous and endogenous PDIA3 reduced the GP- and GP∆MLD-mediated viral entry up to 10-fold (lanes 2, 4, 6, 8). Next, we detected virion-associated GP. EBOV virus-like particles (VLPs) were produced from these three HEK293T cells after expressing GP or GP∆MLD with EBOV matrix protein VP40. Both exogenous and endogenous PDIA3 reduced the GP and GP∆MLD levels in these VLPs up to 4-fold (Figure 2B). Furthermore, we verified the PDIA3 inhibitory effect by using replication and transcription-competent virus-like particles (trVLPs), which model the entire EBOV replication cycle [22]. PDIA3 consistently suppressed EBOV replication approximately 10-fold in three consecutive passages (p0, p1, p2) in HEK293T cells (Figure 2C). These results demonstrate that PDIA3 reduces GP levels in virions, which is consistent with the observation that PDIA3 decreases GP levels in viral producer cells.
Figure 2.
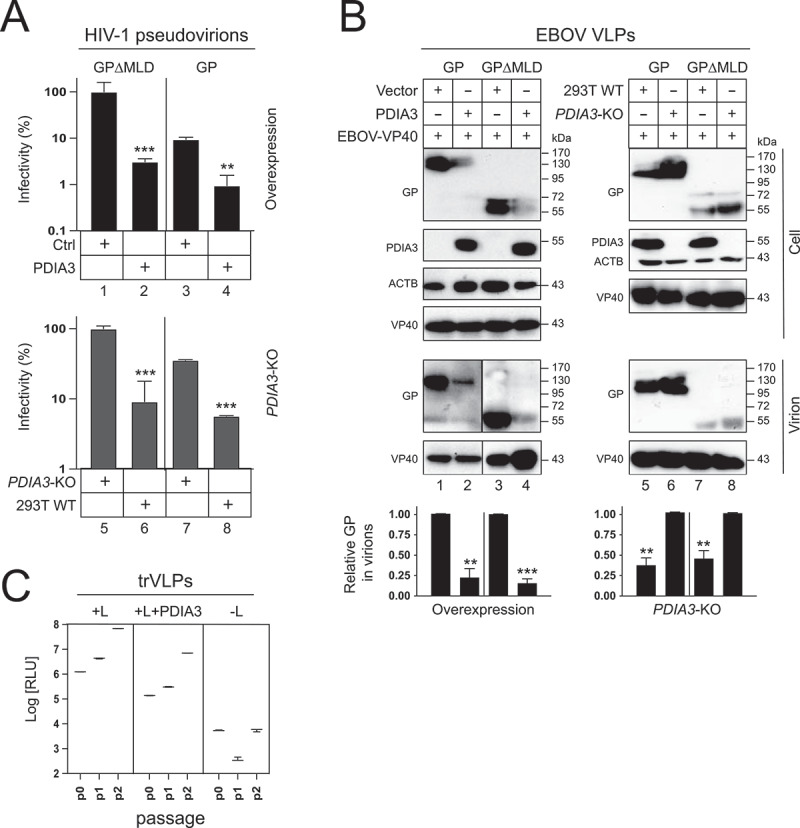
PDIA3 inhibits EBOV entry. (a) HIV-1 firefly luciferase reporter viruses pseudotyped with GP or GP∆MLD were produced from HEK293T WT, PDIA3-overexpressing, or PDIA3-KO cells. After infecting HEK293T cells with an equal number of these different viruses, viral entry was determined by measuring intracellular luciferase activities. Viral entry is shown as relative values, with the entry of GP∆MLD-pseudotyped viruses in the absence of PDIA3 normalized to 100%. (b) EBOV virus-like particles (VLPs) were produced from HEK293T WT, PDIA3-overexpressing, or PDIA3-KO cells after expression of GP and EBOV-VP40. VLPs were purified by ultra-centrifugation and analyzed by WB. Levels of GP in virions were further quantified from these Western blots and are presented as relative values as previously. (c) EBOV replication and transcription-competent virus-like particles (trVLPs) were produced and passaged two times (p0, p1, p2) in HEK293T cells in the presence or absence of PDIA3. EBOV replication was determined by measuring intracellular Renilla luciferase activity. Viral replication was also measured in the absence of EBOV-L, which is a negative control. Results from three independent experiments are presented. Error bars in A and B represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
PDIA3 targets EBOV-GP cysteine residues for inhibition
Tizoxanide (TIZ), the active metabolite of nitazoxanide (NTZ), specifically inhibits PDIA3 disulfide reductase activity [23]. Both TIZ and NTZ antagonized the PDIA3 inhibitory activity on GP expression up to 80% in a dose-dependent manner (Figure 3A). Thus, the PDIA3 inhibitory activity requires its reductase activity.
Figure 3.
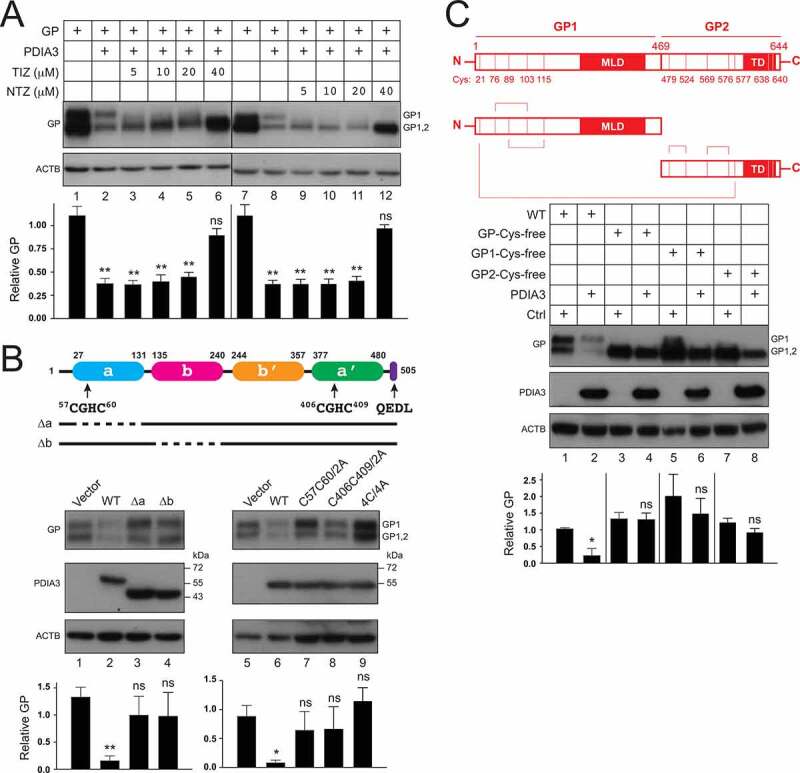
PDIA3 targets cysteine residues in EBOV-GP. (a) HEK293T cells were transfected with PDIA3 and GP expression vectors and treated with increasing concentrations of nitazoxanide (NTZ) or its circulating-metabolite tizoxanide (TIZ). GP expression was analyzed by WB. (b) A schematic diagram of PDIA3 is presented on the top. Two catalytic TLDs, a (blue) and a’ (green) that contain the canonical CGHC active motif, two non-catalytic TLDs, b (purple) and b’ (Orange), and an ER-retention signal (purple) are shown. Indicated PDIA3 mutants were expressed with GP in HEK293T cells and their effect on GP expression was determined by WB. (c) A schematic diagram of EBOV-GP is presented on the top. Ten cysteine residues located within GP1 and GP2 are indicated. Cysteine residues that form one intermolecular and four intramolecular disulfide-bonds are linked by lines. Indicated GP mutants were expressed with PDIA3 in HEK293T cells, and their expression was analyzed by WB. The levels of GP expression in A, B, and C were further quantified and are presented similarly as we did previously. Error bars represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
PDIA3 has two catalytically active TLDs (a, a’) that have a CGHC motif, two inactive TLDs (b, b’), and an ER retention sequence QEDL (Figure 3B, top). The cysteine residues in the catalytic CGHC motif are redox active and mediate active shuffling of disulfide bonds. To map the critical PDIA3 region, we created ∆a and ∆b deletion mutants that do not express the corresponding TLDs, and C57C60/2A, C406C409/2A, and 4C/4A point mutants that disrupt the N-terminal, C-terminal, or both CGHC motifs, respectively. All these PDIA3 mutants were expressed at a similar level as the WT protein (Figure 3B). They all showed a much lower activity than the WT PDIA3 in inhibition of GP expression (Figure 3B). These results not only confirm that the enzymatic activity is required, but also demonstrate that its structural integrity is important for the PDIA3 antiviral activity.
EBOV-GP has five respective cysteine residues in GP1 or GP2 that form disulfide bonds [18]. To determine how they are targeted by PDIA3, we created GP-Cys-free, GP1-Cys-free, and GP2-Cys-free mutants that do not have any cysteines in these proteins. All these GP mutants were expressed at a similar level as the WT protein (Figure 3C). The GP-Cys-free mutant became completely resistant, while the GP1-Cys-free and GP2-Cys-free mutants remained only partially sensitive to the PDIA3 inhibitory activity (Figure 3C). The partial sensitivity of GP1-Cys-free and GP2-Cys-free suggests that Cys residues in both regions are involved in PDIA3 inhibition. These results demonstrate that PDIA3 targets these GP Cys residues or a GP structural feature dependent upon these Cys residues to inhibit GP expression.
Induction of ER stress and involvement of ERAD machinery
To understand how EBOV-GP affects the ER homeostasis, we determined whether ER stress is induced after GP expression. During ER stress, UPR is activated via three ER transmembrane receptors, including the double-stranded RNA (dsRNA)-activated protein kinase EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3), ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1), and ATF6 (activating transcription factor 6). ERN1 activates another transcription factor, XBP1 (X-box binding protein 1), and EIF2AK3 activates the transcription factor ATF4 during UPR. We used luciferase-based reporter assays to detect XBP1, ATF4, and ATF6 activation. As a control, the terminally misfolded human SERPINA1/alpha1-antitrypsin variant null (Hong Kong) (NHK) was used [24]. Because HSPA5 is a master ER stress regulator, its induction was also measured using a similar reporter assay. As expected, NHK upregulated and activated the regulator and all these sensors (Figure 4A). GP also upregulated HSPA5 and activated ATF4 much more strongly than XBP1 and ATF6. Thus, GP expression induces ER stress that is preferentially sensed by ATF4.
Figure 4.
EBOV-GP induces UPR and is targeted by ERAD machinery. (a) HEK293T cells were transfected with a GP or SERPINA1 expression vector and pLightSwitch-BiP, pXBP1u-FLuc, pATF4-UTR-Fluc, or p5× ATF6-GL3. Luciferase activities were measured and are presented as relative values, with the activities in the presence of SERPINA1 normalized to 100%. (b) GP and VSV-G were expressed in HEK293T cells with increasing amounts of SERPINA1 and their expression was analyzed by WB. (c) GP, IAV-HA, and VSV-G were expressed with PDIA3 in HEK293T cells and treated with 0.1 µM thapsigargin (TGN). In addition, GP was expressed with PDIA3 and treated with 1 mM 4-phenylbutyrate (4-PBA). DMSO was used as a vehicle control. Viral protein expression was analyzed by WB. (d) GP and PDIA3 were expressed in HEK293T cells and treated with 20 μM lactacystin (Lac) or 50 μM kifunensine (KIF). DMSO was used as a vehicle control. GP expression was analyzed by WB. (e) GP and VSV-G were expressed with EDEM1, EDEM2, EDEM3, or MAN1B1 in HEK293T cells and their expression was analyzed by WB. GFP was used as a control. EDEM1, EDEM2, EDEM3, MAN1B1, and GFP were detected by an anti-FLAG. (f) GP was expressed with FLAG-tagged GFP, MAN1B1, EDEM1, EDEM2, or EDEM3 in HEK293T cells. Proteins were pulled down by anti-GP and analyzed by WB. (G) GP was expressed with MAN1B1, EDEM1, EDEM2, and their catalytic site-deficient mutants in HEK293T cells, and their expression was analyzed by WB. The levels of GP expression in B, C, D, E, and G were further quantified. Error bars represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
To understand how ER stress affects viral glycoprotein expression, GP was expressed with increasing amounts of NHK. NHK strongly decreased the GP expression in a dose-dependent manner, but it did not affect the vesicular stomatitis virus glycoprotein (VSV-G) expression (Figure 4B). We then tested whether thapsigargin (TGN), which disrupts Ca2+ homeostasis in the ER and induces ER stress, has a similar effect. TGN strongly reduced the GP expression but had little effect on VSV-G or influenza A virus (IAV)-hemagglutinin (HA) expression (Figure 4C, lanes 1, 2). We further tested whether an ER stress suppressor would have an opposite effect by treating cells with 4-phenylbutyrate (4-PBA). 4-PBA increased the GP expression (lanes 3, 4). Collectively, these results demonstrate that ER stress negatively regulates GP expression.
To understand how PDIA3 decreases the GP expression, GP and PDIA3 were expressed and treated with the proteasomal inhibitor lactacystin (Lac) and the class I α-mannosidase inhibitor kifunensine (KIF). The decreased GP expression was effectively rescued by KIF but not lactacystin (Figure 4D), indicating that GP might be specifically targeted by class I α-mannosidases. In the ER, four class I α-mannosidases, including MAN1B1/ERManI (mannosidase alpha class 1B member 1), and EDEM1 (ER degradation enhancing alpha-mannosidase like 1), EDEM2, and EDEM3, are responsible for initiation of ERAD [25]. They cleave α1,2-linked mannose residues in the N-glycan precursor, triggering retro-translocation of misfolded proteins from the ER to the cytoplasm for subsequent degradation in proteasomes. When FLAG-tagged MAN1B1, EDEM1, EDEM2, and EDEM3 were expressed with GP and VSV-G, the expression of GP, but not VSV-G, was decreased by EDEM1, EDEM2, and MAN1B1, whereas EDEM3 did not have such activity (Figure 4E). We then determined how GP interacts with these four ER enzymes by IP using anti-EBOV-GP. This antibody selectively pulled down the ER form of GP (GP1,2) with MAN1B1, EDEM1, and EDEM2, but not EDEM3, which confirms the specific effect of these three enzymes (Figure 4F). To dissect the different roles of MAN1B1, EDEM1, and EDEM2 in inhibition of GP expression, GP was expressed with their mutants bearing an inactive catalytic site [26]. The MAN1B1E330A mutant was inactive, whereas the EDEM1E225A and EDEM2E117A mutants were still active to reduce the GP expression (Figure 4G). Thus, unlike MAN1B1, both EDEM1 and EDEM2 do not depend on their own enzymatic activity to decrease the GP expression. Collectively, these results demonstrate that ERAD initiation machinery is involved in PDIA3 inhibition of the GP expression.
EBOV-GP is targeted to lysosomes via autophagosomes
To understand whether GP is targeted to protein degradation pathways, cells were treated with two proteasome inhibitors lactacystin (Lac) and MG132, or two lysosome inhibitors bafilomycin A1 (BafA1) and concanamycin A (ConA). Again, we found that proteasomal inhibitors did not block the PDIA3 inhibitory activity (Figure 5A, lanes 3, 4). However, it was blocked by two lysosome inhibitors (lanes 5, 6), indicating that GP is targeted to lysosomes for degradation.
Figure 5.
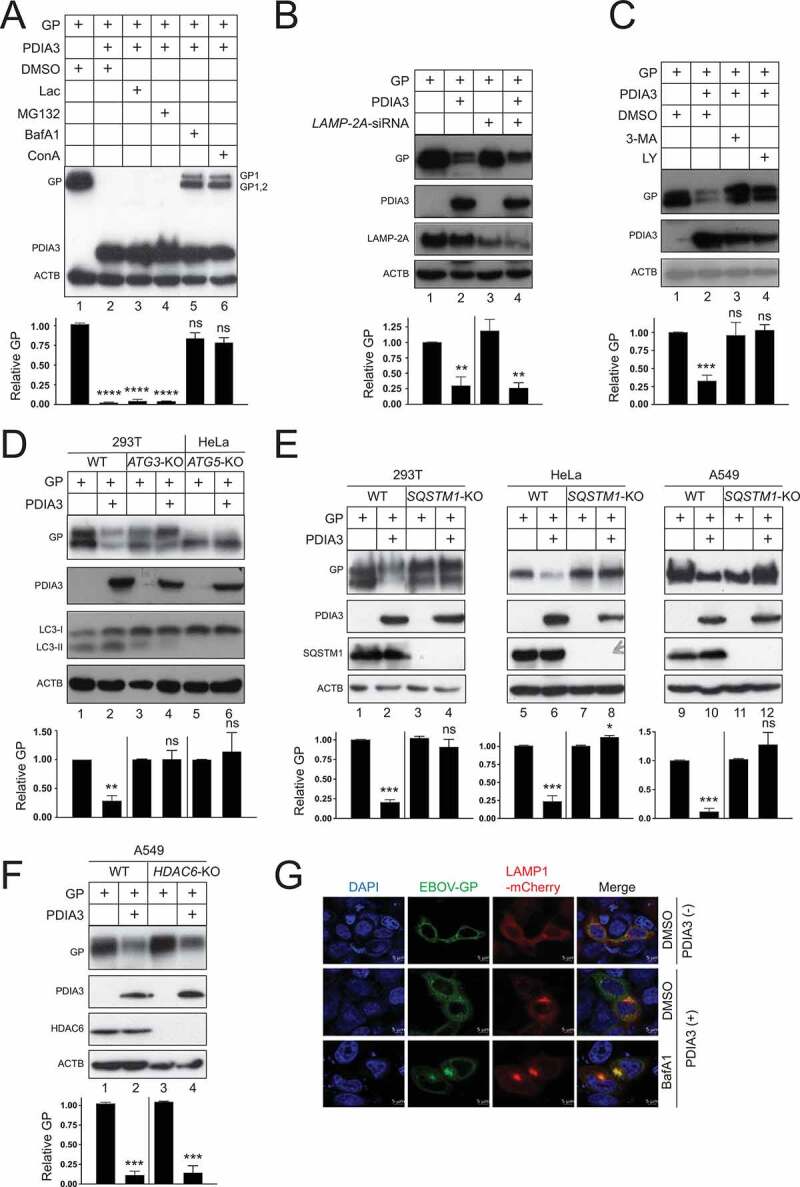
EBOV-GP is targeted to lysosomes via macroautophagy/autophagy. (a) GP and PDIA3 were expressed in HEK293T cells and treated with 20 μM lactacystin (Lac), 20 μM MG132, 100 nM bafilomycin A1 (BafA1), and 20 nM concanamycin A (ConA). DMSO was used as a vehicle control. GP expression was analyzed by WB. (b) GP and PDIA3 were expressed in HEK293T cells in the presence of LAMP-2A-siRNA and their expression was analyzed by WB. (c) GP and PDIA3 were expressed in HEK293T cells and treated with 10 mM 3-methyladenine (3-MA) or 20 μM LY294002 (LY). Their expression was analyzed by WB. (d) GP and PDIA3 were expressed in indicated WT, ATG3-KO, and ATG5-KO cells and their expression was analyzed by WB. (e) GP and PDIA3 were expressed in indicated WT and SQSTM1-KO cells and their expression was analyzed by WB. (f) GP and PDIA3 were expressed in A549 WT and HDAC6-KO cells and protein expression was analyzed by WB. (g) GP was expressed with LAMP1-dsRed in the presence or absence of a PDIA3 expression vector in HeLa cells. Cells with ectopic PDIA3 were treated with DMSO or 100 nM BafA1. The colocalization of GP with LAMP1 was determined by confocal microscopy after staining GP with its specific antibody. The levels of GP expression in A, B, C, D, E, and F were further quantified. Error bars represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
Cytoplasmic materials are targeted to lysosomes via three types of autophagy, namely macroautophagy/autophagy, chaperone-mediated autophagy (CMA), and microautophagy [27]. CMA requires LAMP2A (lysosomal associated membrane protein 2A) as a receptor. When LAMP2A was silenced using small interfering RNAs (siRNAs), PDIA3 could still reduce the GP expression (Figure 5B). Macroautophagy/autophagy is mediated by autophagosomes. 3-methyladenine (3-MA) and LY 294002 (LY) block autophagosome synthesis by inhibiting the phosphatidylinositol 3-kinase (PtdIns3K). Both 3-MA and LY blocked the PDIA3 activity (Figure 5C). Thus, GP is likely targeted by macroautophagy/autophagy.
Next, we used a series of KO cell lines to confirm the involvement of autophagosomes. Autophagosome formation is dependent on a number of ATG (autophagy related) proteins, which induce lipidation of the soluble MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-I to become LC3-II on phagophore membranes [28]. When ATG3 or ATG5 was knocked out in HEK293T or HeLa cells, both LC3-II synthesis and the PDIA3 inhibitory activity were blocked (Figure 5D). We then knocked out an autophagy receptor SQSTM1/p62 (sequestosome 1) in HEK293T, HeLa, and A549 cells, and another receptor, histone deacetylase 6 (HDAC6), in A549 cells. The PDIA3 inhibitory activity was blocked in all three SQSTM1-KO cell lines (Figure 5E), but not in the HDAC6-KO cell line (Figure 5F). Thus, GP is recruited to autophagosomes in a SQSTM1-dependent, but HDAC6-independent manner.
We then tested whether GP is delivered into lysosomes. When GP was expressed alone, it did not colocalize with the lysosome marker LAMP1 (Figure 5G). However, when GP was expressed with PDIA3, the GP-LAMP1 colocalization was detected, and importantly, the colocalization signal was increased by treatment with BafA1. Thus, GP is recruited into lysosomes for degradation in the presence of PDIA3.
EBOV-GP is ubiquitinated and degraded in the presence of PDIA3
Most cytosolic proteins are degraded by lysine (K)48- or K63-mediated polyubiquitination [29]. We attempted to rescue the GP expression by expressing three ubiquitin (Ub) mutants with one or both sites mutated to arginine (R), including UbK48R, UbK63R, and UbK48,K63R. When His-tagged WTUb and these three mutants were expressed with GP in HEK293T WT, PDIA3-overexpressing, and PDIA3-KO cells, none of them rescued the decreased GP expression by PDIA3 (Figures 6A, B). Notably, when proteins were probed with anti-His, a high-molecular-mass (HMM) protein (>170 kDa) was detected only in the presence of GP and these Ub proteins, indicating that GP was likely polyubiquitinated (Figure 6A, B, lanes 3–10). We then tested another Ub mutant with all seven lysine residues mutated to arginine (Ub[7 K/R]). When HA-tagged WT Ub and Ub[7 K/R] were expressed with GP and PDIA3, unlike WT Ub, this Ub[7 K/R] mutant completely rescued decrease of the GP expression by PDIA3 (Figure 6C). In addition, the specific HMM protein was also detected (lanes 3, 4, 7, 8). These results demonstrate that polyubiquitination is required for reduction of the GP expression by PDIA3, which should not be solely dependent on the K48 or K63 site in ubiquitin. To confirm that GP is indeed polyubiquitinated, GP with a FLAG-tag was expressed with His-tagged UbK48,K63R in the presence or absence of PDIA3 and pulled down by anti-FLAG. The HMM protein was detected again in pulldown samples by anti-His, which was significantly increased by PDIA3 (Figure 6D, lanes 3, 4). These results demonstrate that GP is polyubiquitinated, which is enhanced by PDIA3.
Figure 6.
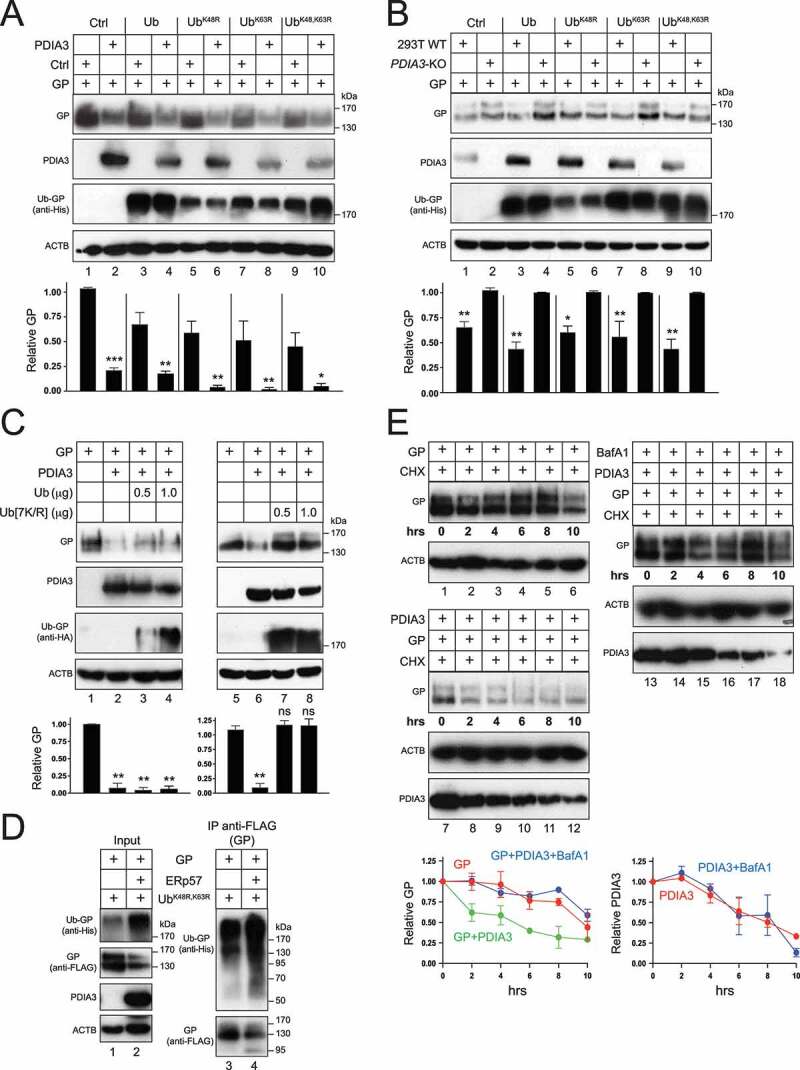
EBOV-GP is ubiquitinated and degraded in the presence of PDIA3. (a) GP was expressed with WT Ub or indicated Ub mutants in HEK293T cells in the presence or absence of PDIA3. Protein expression was analyzed by WB. (b) GP was expressed with WT Ub or indicated Ub mutants in HEK293T WT and PDIA3-KO cells. Protein expression was analyzed by WB. (c) GP was expressed with increasing amounts of WT Ub or Ub[7 K/R] in the presence or absence of PDIA3 in HEK293T cells. Protein expression was analyzed by WB. (d) GP was expressed with UbK48,63R in the presence or absence of PDIA3 in HEK293T cells. GP was pulled down by anti-FLAG and protein expression in cell lysate (Input) and IP samples was analyzed by WB. (e) GP was expressed in the presence or absence of PDIA3 in HEK293T cells. Cells were treated with 50 μM cycloheximide (CHX) in the presence or absence of 100 nM BafA1. Cells were collected at indicated time points and protein expression was analyzed by WB. The levels of GP in A, B, C, D, and E were further quantified. Error bars represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
We then tested how PDIA3 affects the GP stability after treatment with a protein synthesis inhibitor, cycloheximide (CHX) that inhibits translation. GP alone was quite stable, with a half-life around 10 h (Figure 6E, lanes 1–6, and bottom panel for quantification). However, PDIA3 strongly reduced its half-life to 2–4 h (lanes 7–12, and bottom panel). Consistently, BafA1 restored GP stability that was decreased by PDIA3, and it did not affect the PDIA3 stability (lanes 13–18, and bottom panel). Collectively, these results demonstrate that PDIA3 promotes GP polyubiquitination, which results in decrease of GP at steady-state and GP degradation in lysosomes.
PDIA3 specificity to ebolaviruses
We determined whether glycoproteins from other filoviruses and other enveloped viruses are targeted by PDIA3. First, we tested MARV-GP. We found that MARV-GP did not inhibit cell proliferation as EBOV-GP when tested in HEK293T cells (Figure 7A), which is consistent with that MARV-GP does not induce cellular detachment as reported previously [30]. The EBOV-GP inhibitory activity was counteracted by PDIA3 (Figure 7A), which is consistent with that PDIA3 targets EBOV-GP for degradation. Increasing amounts of MARV-GP with a HiBiT-tag were then expressed with a fixed amount of PDIA3, and MARV-GP expression was analyzed (Figure 7B) and quantified (Figure 7C) by measuring the levels of HiBiT expression. Strikingly, PDIA3 increased the MARV-GP expression up to 10-fold in these measurements. When MARV-GP expression was compared in HEK293T WT and PDIA3-KO cells, it was also increased up to 3-fold by the endogenous PDIA3 (Figure 7D). Thus, in contrast to EBOV-GP, PDIA3 increases the MARV-GP expression. Second, we tested GPs from the other ebolaviruses, including BDBV, RESTV, SBOV, and TAFV. When these GPs were expressed with either ectopic or endogenous PDIA3 (Figure 7E), all of them showed reduced expression up to 10-fold by PDIA3. Third, we tested glycoproteins from four other enveloped viruses, including IAV-HA, severe acute respiratory syndrome coronavirus (SARS-CoV) spike (S), VSV-G, and HIV-1 envelope glycoproteins (Env). Neither ectopic nor endogenous PDIA3 affected their expression (Figure 7F), except that exogenic PDIA3 slightly increased the IAV-HA expression (lane 3, 4). Finally, we used HIV-1 pseudoviruses to further confirm these results. PDIA3 increased the MARV-GP mediated virus entry more than 2-fold but had poor effect on the VSV-G and HIV-1 Env mediated virus entry (Figure 7G). Collectively, these results demonstrate that the PDIA3 inhibitory activity is highly specific to ebolaviruses.
Figure 7.
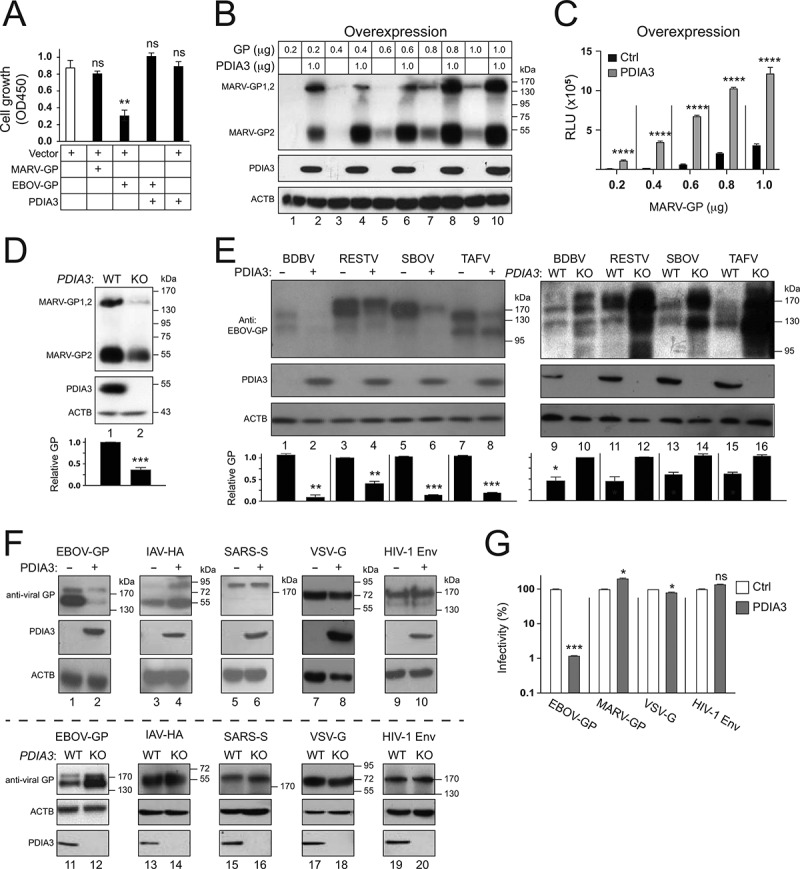
Specificity of the PDIA3 inhibitory activity on viral glycoprotein expression. (a) HEK293T cells were transfected with a control (Ctrl) vector, or vectors expressing MARV-GP, EBOV-GP, and/or PDIA3. After 48 h, cells were incubated with WST-8 and viable cells were counted by a microplate reader with a 450 nm filter. Results are presented as optical density (OD450). (b) HEK293T cells were transfected with increasing amounts of MARV-GP with a HiBiT-tag and 1.0 μg PDIA3 expression vectors. The GP expression was determined by Nano-Glo® HiBiT Blotting System. (c) MARV-GP expression in B was further quantified by Nano-Glo® HiBiT Lytic Reagent and are presented relative luminescence units (RLU). (d) MARV-GP was expressed in HEK293T WT and PDIA3-KO cells, and its expression was determined by the HiBiT Blotting system. (e) GPs from indicated ebolaviruses were expressed with PDIA3 in HEK293T cells. Alternatively, they were expressed in HEK293T WT and PDIA3-KO cells. GP expression was determined by WB using anti-EBOV-GP. (f) Indicated viral glycoproteins were expressed with PDIA3 in HEK293T cells. Alternatively, they were expressed in HEK293T WT and PDIA3-KO cells. Their expression was analyzed by WB using their specific antibodies. (g) HIV-1 firefly luciferase reporter viruses with authentic HIV-1 Env or pseudotyped with EBOV-GP, MARV-GP, or VSV-G were produced from HEK293T cells in the presence of ectopic PDIA3. The entry of viruses with HIV-1 Env was determined in TZM-bI cells, and that of pseudoviruses was determined in HEK293T cells. Viral entry is shown as relative values, with the value in the absence of PDIA3 normalized to 100%. The levels of GP in D and E were further quantified. Error bars in A, C, D, and G represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
Broadness of the PDIA3 activity
We next addressed whether the PDIA3 activity is cell type specific. EBOV initially replicates in macrophages and dendritic cells, and later in hepatocytes and endothelial cells [1]. We already confirmed that PDIA3 has a similar activity in HEK293T, A549, and HeLa cells (Figure 5). Three other cell types were further tested, including human hepatoblastoma cell line Hep G2, human monocytic THP1 cell-derived macrophages (THP1-MΦ), and mouse primary macrophages (MΦ). PDIA3 inhibited the GP expression up to 10-fold in all these cell types (Figure 8A). Thus, the PDIA3 inhibitory activity is not cell type specific.
Figure 8.
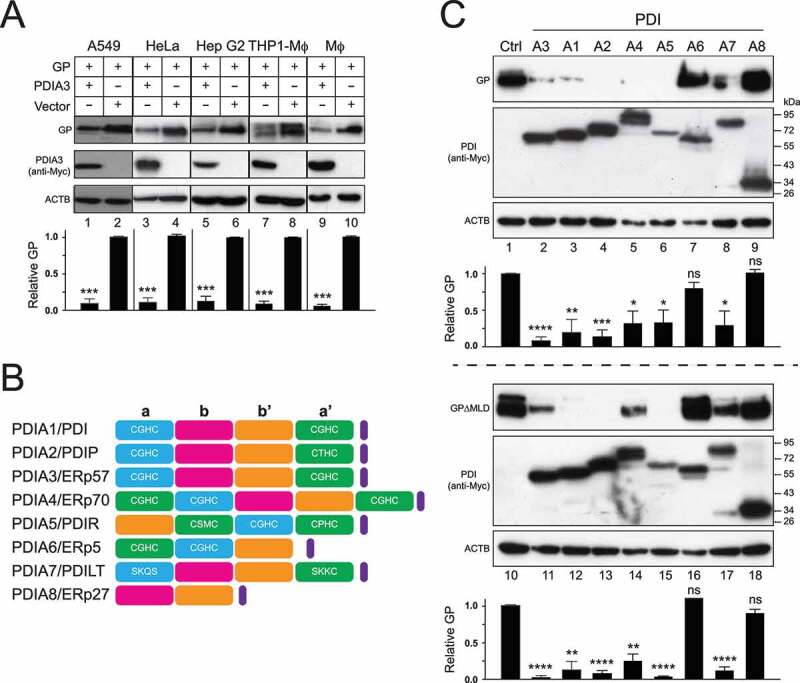
Broadness of the PDIA3 inhibitory activity. (a) GP and PDIA3 were expressed in A549 cells, HeLa cells, Hep G2 cells, THP1-derived macrophages (THP1-Mϕ), and primary mouse macrophages (Mϕ), and their expression was detected by WB. (b) Schematic diagrams of eight PDI family members are shown. TLDs (a, a’, b, and b’) are shown in blue, green, purple, or Orange, respectively. An ER retention sequence is shown in purple. The canonical CGHC or its similar motifs in TRX-like domains are indicated. (c) GP and GP∆MLD were expressed with indicated eight PDI family members in HEK293T cells and their expression was analyzed by WB. The levels of GP in A and C were further quantified. Error bars in A and C represent SEMs calculated from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (p > 0.05).
Lastly, we determined whether any other PDI family members have a similar activity. Another seven PDI members were expressed with GP and GP∆MLD, and their effects on GP expression were compared to PDIA3 (Figure 8B). Notably, five of them, PDIA1/PDI, PDIA2/PDIP, PDIA4/ERp70, PDIA5/PDIR, and PDIA7/PDILT, had a similar activity as PDIA3/ERp57 that decreased both GP and GP∆MLD expression, whereas PDIA6/ERp5 and PDIA8/ERp27 did not show any activity (Figure 8C). Thus, the PDIA3 inhibitory activity against EBOV-GP is conserved and shared by at least five other PDI members. In addition, like PDIA3, all these five active members PDIA1, PDIA2, PDIA4, PDIA5, and PDIA7 have two active TLDs, whereas PDIA6 and PDIA8 do not. Thus, the inhibitory activity may require two active TLDs, which is consistent with our previous mapping results (Figure 3B).
Discussion
Unlike EBOV-GP, MARV-GP does not induce cell rounding and detachment [30]. In addition, the MARV-GP expression is not regulated at RNA levels, resulting in protein expression from a single open reading frame [3]. Now, we show that EBOV-GP expression is also strongly inhibited at the protein levels in the ER by PDIs. In contrast, PDIA3 strongly increases the MARV-GP expression. The activity of PDIs to decrease EBOV-GP expression rather than promote its expression likely represents an evolutionary adaptation of EBOV to limit its cellular effect in hosts. Presumably, this novel mechanism for suppressing EBOV-GP expression increases the viral fitness by enhancing survival of infected cells. Importantly, our work suggests that further enhancement of PDIA3 suppression of the EBOV-GP expression might block the assembly of infectious virions and limit viral infection. More understanding of the mechanism of PDIA3 suppression of the EBOV-GP expression will hopefully give direction to this new strategy for control of Ebola virus disease (EVD).
We found that ebolavirus GPs are very vulnerable to PDIA3, even at physiological levels that more generally promote oxidative protein folding [31]. Thus, PDIA3 also plays a negative role in glycoprotein expression, suggesting that this ER protein folding machinery is a double-edged sword that can either re-shape or degrade its substrate via manipulating disulfide bond formation (Figure 9). We found that EBOV Cys-free GP and nonstructural GPs are all resistant to PDIA3. These results suggest not only that these disulfide bridges are critical for EBOV-GP folding and function, but also that their formation is tightly regulated. We speculate that higher levels of PDIA3 expression may cause Cys mismatches, which triggers EBOV-GP misfolding and recognition by the ER quality control pathways. In addition, five other PDI members, PDIA1, PDIA2, PDIA4, PDIA5, and PDIA7, which are not involved in the CANX-CALR cycle, have a similar inhibitory activity toward EBOV-GP. These results demonstrate that these PDIs can act independently of CANX and CALR, and function as a negative regulatory network that inhibits EBOV-GP.
Figure 9.
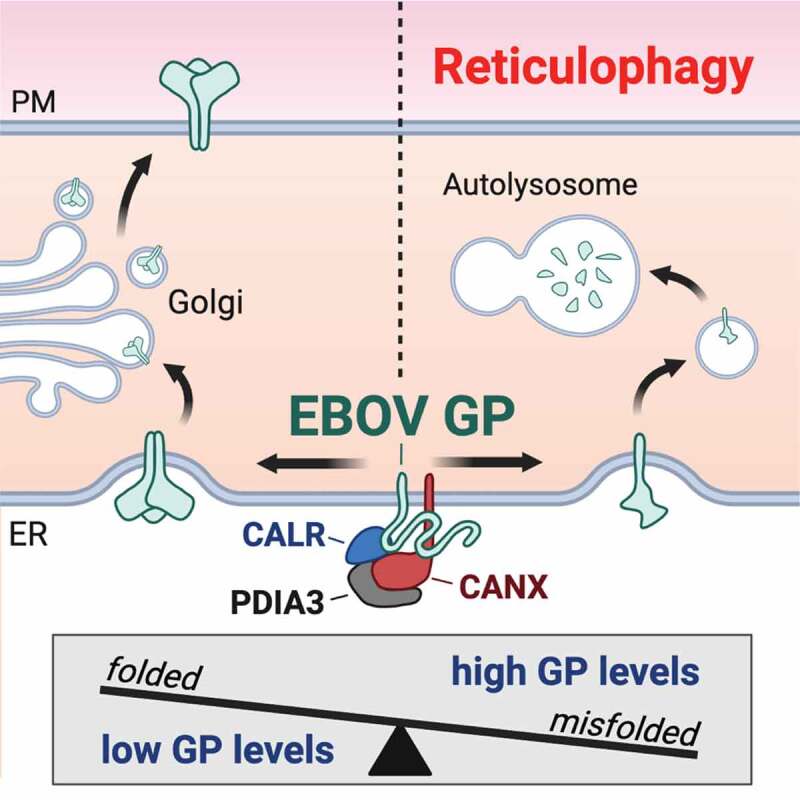
A model of how protein-folding machinery regulates EBOV-GP expression in the ER. PDIA3 interacts with CANX and CALR, which comprises the CANX-CALR cycle in the ER that promotes N-glycosylated protein folding. However, this protein-folding machinery acts as a double-edged sword during EBOV infection. It can increase the GP expression by promoting its folding for viral infection. Alternatively, it also decreases the GP expression by degradation via reticulophagy for cell survival. Thus, PDIA3 increases the viral fitness by tightly controlling the levels of GP expression during ebolavirus infection.
Please remove the thin color line on the left side of ER in this figure.
There are two major quality control mechanisms in the ER. ERAD specifically targets and degrades misfolded proteins via the ubiquitin proteasome system, whereas reticulophagy targets parts of the ER and degrades protein aggregates via the autophagy-lysosomal system [17]. We found that among four ER-resident class I α-mannosidases, MAN1B1, EDEM1, and EDEM2 could accelerate GP degradation, indicating that some parts of ERAD in the ER are likely involved in this degradation process. As observed in our previous studies on IAV-HA proteins, these three mannosidases seem to play different roles in this process [26]. When their catalytic sites are mutated, only MAN1B1 becomes inactive, whereas EDEM1 and EDEM2 are still active. We also reported that HIV-1 Env is sensitive to MAN1B1, but not the three EDEM proteins when these mannosidases are expressed ectopically [32]. Thus, EBOV-GP and IAV-HA demand much more EDEM1 and EDEM2 than does HIV-1 Env during recognition of these viral proteins by the ERAD machinery, which could be due to a difference in their glycosylation. Nonetheless, GP degradation was not solely dependent on the K48- and K63-linked ubiquitination that normally triggers proteasomal degradation and was not blocked by proteasomal inhibitors. Thus, EBOV-GP is not degraded via classical ERAD.
Our results strongly suggest that EBOV-GP is degraded via reticulophagy (Figure 9). We found that unlike proteasomal inhibitors, lysosomal inhibitors blocked the GP degradation. GP degradation was also blocked by inhibiting autophagosome formation, either by two specific inhibitors 3-MA and LY or by knocking out ATG3 and ATG5. We also detected GP localization in the lysosomes in the presence of PDIA3. Thus, GP degradation is clearly dependent on the autolysosomes. We observed that PDIA3 promoted the GP polyubiquitination and that GP is degraded in a SQSTM1-dependent and HDAC6-indepenent manner. Although both SQSTM1 and HDAC6 bind to ubiquitinated substrates and recruit them into autophagosomes, they have different preferences: SQSTM1 prefers misfolded proteins, whereas HDAC6 prefers protein aggregates [33]. These results suggest that PDIs likely trigger ebolavirus GP misfolding, but not aggregation, which may explain why these GPs are recognized by class I α-mannosidases that are essential to ERAD initiation. Collectively, we propose that PDIs promotes ebolavirus GP degradation via reticulophagy in an ERAD-dependent manner, which reduces its cellular effect and increases the fitness of EBOV in hosts.
Materials and methods
Antibodies and inhibitors
Commercial reagents include: mouse monoclonal anti-PDIA3/ERp57 and anti-MYC and bafilomycin A1 (Santa Cruz Biotechnology, sc-23,886, sc-40, sc-201,550); rabbit polyclonal anti-EBOV-GP, anti-VP40, anti-IAV HA, and anti-HDAC6 (Sino Biological, 40,442-T48, 40,446-T48, 11,048-RP02, 100,768-T08); rabbit polyclonal anti-SARS CoV-S (Novus Biologicals, NB100-56,047); mouse monoclonal anti-VSV-G, anti-FLAG, and anti-ACTB/β-actin, and rabbit polyclonal anti-SQSTM1/p62 (Sigma-Aldrich, SAB4200695, F3165, A5441, P0067); horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and anti-rabbit IgG (Jackson ImmunoResearch Laboratories, 115–035-003, 111–035-003); kifunensine, MG132, cycloheximide (CHX), lactacystin, 3-methyladenine, concanamycin A, and nitazoxanide (NTZ) (Sigma-Aldrich, K1140, C2211, L6785, M9281, C9705, N0290); tizoxanide (TIZ; MedChemExpress, HY-12687); LY294002 (Promega, V1201). Mouse monoclonal anti-HIV-1 gp120 was from NIH HIV Reagent Program.
Cell lines
Human embryonic kidney (HEK) 293 cell line transformed with SV40 large T antigen (293 T), human cervical carcinoma cell line HeLa, African green monkey kidney epithelial cell line Vero E6, human lung carcinoma cell line A549, and human hepatoblastoma cell line Hep G2 were purchased (ATCC, CRL-3216, CRM-CCL-2, CRL-1586, CRM-CCL-185). HEK293T PDIA3-KO cells [34] and HEK293T ATG3-KO cells [35] were reported previously. HeLa ATG5-KO and SQSTM1/p62-KO cells were provided by Honglin Jia (Harbin Veterinary Research Institute, China). TZM-bI cells were from NIH HIV Reagent Program. All these cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific, 11965092) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (pen-strep; Thermo Fisher Scientific, 10378016), and cultivated at 37°C in humidified atmosphere in a 5% CO2 incubator.
To differentiate THP1 into macrophages (THP1-MΦ), human monocytic THP1 cells (ATCC, TIB-202) were grown in RPMI 1640 medium (Thermo Fisher Scientific, A4192301) supplemented with 10% FBS and 1% pen-strep in 6-well plates (1.5 × 106 cells per well) and treated with 100 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, P8139) for 2 days. Primary peritoneal macrophages (MΦ) were isolated from mouse peritoneum and cultured in RPMI 1640 supplemented with 10% FBS and 1% pen-strep.
CRISPR-Cas9
HEK293T SQSTM1/p62-KO, A549 SQSTM1/p62-KO, and A549 HDAC6-KO cells were generated through CRISPR-Cas9. To create KO vectors, small guide RNA (sgRNA) encoding DNA oligos were designed and directly cloned into the pSpCas9 (BB)-2A-GFP vector after digestion with BbsI. One pair of oligos was designed for SQSTM1/p62-KO: 5’-CACCGCGAGGGAAAGGGCTTGCAC-3’/5’-AAACGTGCAAGCCCTTTCCCTCGC-3’. Three pairs of oligos were designed for HDAC6-KO: 5’-CACCGAACAGAAACACCGCATCCGG-3’/5’-AAACCCGGATGCGGTGTTTCTGTTC-3’, 5’-CACCGCTAGCAGTGGGGAAACGAG-3’/5’-AAACCTCGTTTCCCCACTGCTAGC-3’, and 5’-CACCGATTTACCCTGCTCGTAGCGG-3’/5’-AAACCCGCTACGAGCAGGGTAGGTC-3’. Cells were transfected with one SQSTM1/p62-KO vector or three HDAC6-KO vectors. After 48 h of transfection, GFP-expressing cells were collected by fluorescence-activated cell sorting (FACS) and cloned by limiting dilution. KO clones were screened by WB.
Plasmids
pcDNA3.1-EBOV-GP, pcDNA3.1-EBOV-GPΔMLD, pCMV3-EBOV-VP40, and pNL-Luc-ΔEnv expression vectors were described previously [20]. The EBOV-GP was from the Zaire ebolavirus isolate (Ebolavirus/H.sapiens-wt/SLE/2014/Makona-J0038, GenBank Acc# KP759618). CMV-driven vectors expressing human MAN1B1/ERManI, EDEM1, EDEM2, EDEM3 and their catalytic mutants were described previously [26]. EBOV trVLP vectors including p4cis-vRNA-RLuc, pCAGGS-L, pCAGGS-NP, pCAGGS-VP30, pCAGGS-VP35, pCAGGS-Tim1, and pCAGGS-T7 were provided by Heinz Feldmann (NIH). The vector expressing SARS-CoV S expression vector was provided by Shan-Lu Liu (Ohio University); the vector expressing misfolded human SERPINA1/α-1-antitrypsin null Hong Kong (NHK) was provided by Richard N. Sifers (Baylor University); the vector expressing VSV-G was provided by Tannishtha Reya through Addgene (14888); the HSPA5/BiP promoter-luciferase reporter construct pLightSwitch-BiP was purchased from Switchgear Genomics (S719178); the XBP1-activation reporter construct pXBP1u-FLuc was obtained from Yi-Ling Lin (Academia Sinica, Taipei); the ATF4-expression reporter construct pATF4-UTR-Fluc was obtained from Dong-Yan Jin (The University of Hong Kong); the ATF6-activation reporter construct p5xATF6-GL3 was obtained from Ron Prywes through Addgene (11976). pSpCas9 (BB)-2A-GFP (PX458) was obtained from Feng Zhang through Addgene (48138). pcDNA3.1-FLAG-BiP was provided by Sansanee Noisakran (National Center for Genetic Engineering and Biotechnology, Thailand).
sGP and ssGP that contain a N-terminal HiBiT-tag (VSGWRLFKKIS) immediately after the signal peptide were expressed from pcDNA3.1. Lake Victoria Marburgvirus (GenBank Acc# DQ217792) GP that contains a C-terminal 3x(GGGGS)-linker followed by a HiBiT-tag was expressed from pCAGGS. To express Ebolavirus glycoproteins from other species, the GP cDNAs from BDBV (Bundibugyo virus/H. sapiens-tc/UGA/2007/Bundibugyo-200706291, Acc# KU182911), RETV (Reston strain Reston08-E, Acc# FJ621585), SEBOV (Sudan virus/H.sapiens-tc/UGA/2000/Gulu-808892, Acc# NC_006432), and TAFV (Ebola virus/H.sapiens-tc/Cote d’Ivoire/1994/Tai Forest-R4371, Acc# MH121167) were synthesized by GenScript and cloned into pCDNA3.1. To express PDIA3, its cDNAs containing either a 3’-MYC or 3’-HA tag were amplified from pCMV6-PDIA3 (Origene, RC205940) and subcloned into the pCAGGS vector after EcoRI/BglII digestion. The EBOV GP-Cys-free, GP1-Cys-free, and GP2-Cys-free mutants, and the PDIA3 C57C60/2A, C406C409/2A and combined 4C/4A mutants were generated by introducing Cys-to-Ala substitutions using the QuikChange site-directed mutagenesis kit (OMEGA BIO-TEK, D6492-01). The PDIA3 deletion mutants were created by homologous recombination after EcoRI digestion (Vazyme, C113-01). The pCMV-Myc vectors expressing human PDIA2/PDIP, PDIA4/ERp70, PDIA5/PDIR, PDIA6/ERp5, PDIA7/PDILT and PDIA8/ERp27 were purchased (FENGHUISHENGWU, ZL-001, 002, 003, 004, 005, 006). To express PDIA1/PDI, its cDNA was prepared from 293 T by RT-PCR (Toyobo, FSK-101) and cloned into pCMV-Myc via SacI/NotI digestion. All vectors were sequenced by Sanger DNA sequencing to confirm their inserts. Detailed experimental procedures and primer sequences for construction of these vectors are available upon request. Plasmids were prepared using Maxiprep kits (TIANGEN Biotech, DP117).
Transfection
293 T cells were transfected using linear 25-kDa polyethyleneimine (PEI; Polysciences, 23966–2). Briefly, to transfect cells in a 10-cm culture dish with 70% confluency, 9 μg plasmid DNAs were mixed with 27 μg PEI (stock solution at 1 mg/ml) and diluted into 500 μl Opti-MEM (Thermo Fisher Scientific, 31985062). After a 15-min incubation at room temperature, DNA-PEI complexes were added into the cell culture. After culturing cells for 4 to 6 h, cells were washed with PBS and fresh media was added for culturing another 42 h before samples were harvested. HeLa and A549 cells were transfected using Lipofectamine 3000 according to the manufacturer’s protocol (Thermo Fisher Scientific, L3000).
Preparation of EBOV virus-like particles
EBOV VLPs were generated as previously [20]. Briefly, 293 T cells were transfected with EBOV-GP and EBOV-VP40 expression vectors in the presence or absence of ectopic PDIA3 expression. After 48 h of transfection, the culture supernatants were collected, clarified by low-speed centrifugation, and passed through a 0.45-μm syringe filter to remove any cell debris. To analyze the levels of EBOV-GP incorporation, supernatants containing EBOV VLPs were centrifuged at 120,000 x g at 4°C for 2 h using a Beckman SW-32Ti rotor.
Preparation of HIV-1 pseudoviruses for entry analysis
Lentiviral pseudo-virions expressing EBOV-GP were generated as we did previously [20]. Briefly, 293 T cells were transfected with pNL-Luc-ΔEnv and a GP-expression vector in the presence or absence of ectopic PDIA3 expression. After 48 h of transfection, pseudo-virion-containing culture supernatants were collected, clarified by low-speed centrifugation, and passed through a 0.45-μm syringe filter to remove any cell debris. After quantifying by HIV-1 p24 Gag ELISA, an equal number of pseudo-virions were used to infect 293 T and Vero E6 cells. After 48 h, luciferase activities were determined using Bright-Glo Luciferase Assay kit (Promega, N1130).
Production of EBOV trVLPs and infection
Experiments were performed as reported previously [22]. Briefly, to produce p0 trVLPs, HEK293T cells were seeded in 6-well plates at 4 × 105 per well in 2 mL medium and cultured for 24 h. EBOV trVLP system plasmids were diluted from stocks into 125 μL Opti-MEM per well that included 125 ng pCAGGS-NP, 125 ng pCAGGS-VP35, 75 ng pCAGGS-VP30, 1,000 ng pCAGGS-L, 250 ng p4cis-vRNA-Rluc, and 250 ng pCAGGS-T7. The plasmids were combined with 7.5 μL TransIT®-LT1 Transfection Reagent (Mirus Bio, 2305). After incubation at room temperature for 15 min to promote complex formation, the transfection mixes were gently added into cell culture. After 24 h of transfection, the media was exchanged for 4 mL growth media with 5% FBS, and further incubated for 72 h. All supernatants from p0 producer cells were collected followed by centrifugation at 3,000 x g for 5 min and stored at −80°C until use. To generate p1 target cells, HEK293T cells were transfected similarly with EBOV trVLP system plasmids but had p4cis-vRNA-Rluc and pCAGGS-T7 replaced with 250 ng pCAGGS-Tim1. Transfected p1 cells were infected with p0 trVLPs, and after 72 h, p1 trVLPs were collected and stored. P2 target cells were prepared similarly and infected with p1 trVLPs. Cells from p0, p1, and p2 were lysed to quantify virus production by Renilla-Glo® Luciferase Assay System (Promega, E2820).
Analysis of UPR
HSPA5/BiP activation was measured by expressing BiP-RLuc reporter construct and measuring the intracellular Renilla luciferase activities. XBP1, ATF4, and ATF6 activation were measured by expressing XBP1-Fluc, pATF4-UTR-Fluc or p5× ATF6-GL3 reporter construct and measuring the intracellular Firefly luciferase activities. Briefly, 293 T cells were cultured in 6-well plates and transfected with reporter vectors. Luciferase activities were analyzed by Dual-Luciferase® Reporter Assay System (Promega, E1910).
Western blotting
Cells were lysed in ice-cold RIPA lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P-40 [Sigma-Aldrich, R0278]) supplemented with protease inhibitor cocktail (Sigma-Aldrich, P8340). Approximately 0.1 mL buffer was used for a total of 2 × 106 cells. Cell lysates was centrifuged at 12,000 x g at 4°C for 10 min. Total protein from virion or cell extracts was boiled in SDS-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (Solarbio Life Sciences, P1015) and resolved by SDS-PAGE. Separated proteins were transferred onto PVDF membranes and membranes were blocked with 5% nonfat milk powder in TBST (Tris-buffered saline [20 mM Tris, pH 7.4, 150 mM NaCl] containing 0.1% Tween 20; Solarbio Life Sciences, T8220) for 1 h at room temperature. Membranes were then probed by primary antibody followed by HRP-conjugated secondary antibodies. Chemiluminescence signals were then measured by incubating the membrane with SuperSignal substrate (Thermo Fisher Scientific, 34,580).
Quantification of Western blots
Gray analyses were performed with ImageJ software (https://imagej.nih.gov/ij/). Brightness and contrast were adjusted equally for all images within a panel. Levels of target proteins such as EBOV-GP or PDIA3 were normalized to those of ACTB, and results were obtained from three independent experiments. Results are shown as relative values, with the value from a control (Ctrl) vector set as 1.
HiBiT blotting
Proteins were similarly separated by SDS-PAGE and transferred to a PVDF membrane. After that, proteins with HiBiT-tag were detected by Nano-Glo® HiBiT Blotting System (Promega, N2410). Briefly, the membrane was incubated with LgBiT/buffer solution (Promega, N2421) for 2 h at room temperature with gentle rocking and then at 4°C overnight. The membrane was incubated with 20 μL of substrate for 5 min at room temperature and placed between transparent plastic sheets for imaging.
HiBiT lytic detection
Viral glycoproteins with a HiBiT-tag were expressed in HEK293T cells in 6-well plates. Luciferase activities were analyzed by Nano-Glo® HiBiT Lytic Reagent (Promega, N3040).
Immunoprecipitation
After transfection of HEK293T cells cultured in a 10-cm dish, cells were lysed in 0.9 mL RIPA lysis buffer for 30 min on ice. After removal of nuclei via low-speed centrifugation and collecting 100 μL as input, the remaining 800 μL lysate was incubated with a specific antibody followed by addition of protein G-Sepharose beads (Thermo Fisher Scientific, 22,852) and rotated at 4°C overnight. For target proteins expressing FLAG- and HA-tag, anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220) or anti-HA-agarose (Sigma-Aldrich, A2095) were used for overnight rotation. After being washed 3 times with 1 mL pre-cooled RIPA lysis buffer, proteins were removed from beads after boiling in 40 μL RIPA lysis buffer plus 20 μL sample loading buffer (4×) and analyzed by WB.
Flow cytometry
After transfection of HEK293T cells with EBOV-GP and PDIA3 expression vectors, cells were fixed with 4% paraformaldehyde and blocked with 10% FBS in PBS for at least 6 h at 4°C. Cells were then incubated with an anti-GP monoclonal antibody at 4°C overnight. After washing, cells were incubated with Alexa Fluor 488-conjugated goat anti-rabbit antibody (Thermo Fisher Scientific, A32731) for 45 min. GP expressions on the cell surface were determined by flow cytometry.
Confocal microscopy
Approximately 1.5 × 105 ~ 2.0 × 105 HeLa cells were seeded on glass bottom cell culture dish (NEST Biotechnology, 801001) and transfected with various vectors using Lipofectamine 3000. After 24 h, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 (Solarbio Life Sciences, T8200), and then blocked with 5% bovine serum albumin (BSA; APPLYGEN, P1622) solution. Cells were then incubated with rabbit polyclonal anti-Zaire EBOV-GP (Sino Biological, 40442-T48) at 1:1,000 dilution in PBS with 5% BSA for 2 h. After being washed 5 times with PBS, cells were stained with Alexa Fluor 488-conjugated goat anti-rabbit IgG at 1:700 dilution for 1 h. Cells were washed again with PBS 5 times and then incubated with DAPI for 3 min for nuclear staining. After further washing with PBS, cells were observed under a confocal microscope (LSM880, Zeiss, White Plains, New York). At least 100 random cells per slide were analyzed, and the most representative images from each slide were selected for presentation.
Cell growth assay
Effects of EBOV-GP and MARV-GP on cell proliferation were compared by Cell Counting Kit-8 (CCK-8) by following the manufacture’s instruction (APExBIO, K1018). Briefly, HEK293T cells were cultured in 24-well plates and transfected with 1 μg/well viral GP expression vectors. After 48 h, 30 uL CCK-8 solution was added to each well of the plate and cells were further cultured for 4 h by avoiding lights. Cell viability was determined by measuring optical density (OD) at 450 nm using a microplate reader.
Mass spectrometry
To purify Ebola glycoproteins, HEK293T cells were cultured in twenty 10-cm cell culture dishes at 2 × 106 cells in 10 ml medium per dish. Cells were transfected with 9 μg pcDNA3.1-EBOV-GP, or pcDNA3.1-EBOV-GPΔMLD in 27 μg PEI and cultured for 48 h. After collecting cells and washing with PBS, cells were incubated with a lysis buffer [50 mM Tris, pH 7.5, 150 mM NaCl, 5% glycerol, 1% n-dodecyl β-D-maltoside (DDM; Sigma-Aldrich, D4641)], and EDTA-free protease inhibitor cocktail (Roche, 11836170001)] at 4°C for 2 h. Total cell lysate was centrifuged at 17,000 x g using a F13-14x50cy rotor in a SORVALL RC6+ centrifuge at 4°C for 30 min. Supernatant was collected and incubated with anti-FLAG M2 affinity gel.
Proteins associated with anti-FLAG M2 affinity gels or with control beads were washed three times with PBS, and treated with 20 µL denaturing buffer (8 M urea, 100 mM NH4HCO3, pH 8.0) at 37°C for 30 min. Proteins were then reduced with addition of 2 µL, 5 mM dithiothreitol (DTT) for 20 m at 37°C. After alkylation with addition of 5 µL, 5 mM iodoacetamide (IAA) for 10 min at room temperature in the dark, the sample was diluted 4 times with addition of 60 µL of 100 mM NH4HCO3 to reduce the concentration of urea. After digestion with 0.2 µg of trypsin at 37°C for overnight, 1 µL formic acid (FA) was added to quench the digestion. The sample tube was centrifuged at 3,000 x g for 30s and supernatant was taken out for desalting using Zip-tip (Millipore, ZTC18S096). After lyophilization, protein digest was resuspended in 10 µL buffer A (2% Acetonitrile [ACN], 0.1% FA), and 1 µL sample was loaded for nanoRPLC-MS/MS analysis.
An EASY nanoLC-1200 system (Thermo Fisher Scientific) equipped with a C18 RPLC column (75 µm i.d. x 50 cm, C18, 2 µm, 100 Å; Thermo Fisher Scientific, 164,942) was connected to a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) for nanoRPLC-MS/MS analysis. A 100-min LC gradient was used for peptide separation and a Top10 data dependent acquisition (DDA) method was used. The full MS resolution was 60,000 and the maximum injection time was 50 ms. The AGC target was 3e6. The MS/MS resolution was 30,000 and the maximum injection time was 50 ms. The AGC target was set 1e5 for MS/MS. The isolation window was set 2.0 m/z.
For the database search, Proteome Discoverer 2.2 (Thermo Fisher Scientific) with SEQUEST HT searching engine was used. UniProt human proteome database (UP000005640) was used. The mass tolerance for precursor ion and fragment ion was set 20 ppm and 0.05 Da, respectively, for the database searching. Oxidation on Methionine, acetylation on Protein N-terminal, and deamination on Asparagine or Glutamine were set as variable modifications. Carbamidomethylation on cysteine was set as the fixed modification. The peptides were filtered with a 1% false discovery rate.
The cellular component information of the identified proteins was obtained from the UniProt (https://www.uniprot.org/) through the “Retrieve/ID mapping” tool.
Statistical analysis
Statistical tests were performed using GraphPad Prism 8. Variance was estimated by calculating the standard error of measurements (SEMs) and represented by error bars. Significance of differences between samples was assessed using two-way ANOVA with Bonferroni posttest. All experiments were performed independently at least three times, with representative experiment being shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (not significant, p > 0.05).
Supplementary Material
Acknowledgments
We thank Jeffrey MacKeigan for critical reading and comments on this manuscript. We thank Shan-Lu Liu, Feng Zhang, Honglin Jia, Heinz Feldmann, Richard N. Sifers, Yi-Ling Lin, Dong-Yan Jin, Tannishtha Reya, Sansanee Noisakran, and Ron Prywes, as well as the NIH HIV Reagent Program for providing various reagents. BW is supported by grants from National Natural Science Foundation of China (31873013) and Natural Science Foundation of Heilongjiang Province (QC2017028).
Funding Statement
This work was supported by the National Natural Science Foundation of China [31873013].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Feldmann H, Sprecher A, Geisbert TW.. Ebola. N Engl J Med. 2020;382(19):1832–1842. [DOI] [PubMed] [Google Scholar]
- [2].Hoenen T, Groseth A, Feldmann H. Therapeutic strategies to target the Ebola virus life cycle. Nat Rev Microbiol. 2019;17(10):593–606. [DOI] [PubMed] [Google Scholar]
- [3].Geisbert TW. Marburg and ebola hemorrhagic fevers (filoviruses). Mandell, Douglas, and Bennett’s principles and practice of infectious diseases. 2015. 1995–9.e1. [Google Scholar]
- [4].Francica JR, Matukonis MK, Bates P. Requirements for cell rounding and surface protein down-regulation by Ebola virus glycoprotein. Virology. 2009;383(2):237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Francica JR, Varela-Rohena A, Medvec A, et al. Steric shielding of surface epitopes and impaired immune recognition induced by the ebola virus glycoprotein. PLoS Pathog. 2010;6(9):e1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Simmons G, Wool-Lewis RJ, Baribaud F, et al. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J Virol. 2002;76(5):2518–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sullivan NJ, Geisbert TW, Geisbert JB, et al. Immune protection of nonhuman primates against ebola virus with single low-dose adenovirus vectors encoding modified GPs. PLoS Med. 2006;3(6):e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Takada A, Watanabe S, Ito H, et al. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology. 2000;278(1):20–26. [DOI] [PubMed] [Google Scholar]
- [9].Yang ZY, Duckers HJ, Sullivan NJ, et al. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat Med. 2000;6(8):886–889. [DOI] [PubMed] [Google Scholar]
- [10].Hacke M, Bjorkholm P, Hellwig A, et al. Inhibition of Ebola virus glycoprotein-mediated cytotoxicity by targeting its transmembrane domain and cholesterol. Nat Commun. 2015;6(1):7688. [DOI] [PubMed] [Google Scholar]
- [11].Alazard-Dany N, Volchkova V, Reynard O, et al. Ebola virus glycoprotein GP is not cytotoxic when expressed constitutively at a moderate level. J Gen Virol. 2006;87(5):1247–1257. [DOI] [PubMed] [Google Scholar]
- [12].Volchkov VE, Volchkova VA, Muhlberger E, et al. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science. 2001;291(5510):1965–1969. [DOI] [PubMed] [Google Scholar]
- [13].Mehedi M, Falzarano D, Seebach J, et al. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J Virol. 2011;85(11):5406–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Galligan JJ, Petersen DR. The human protein disulfide isomerase gene family. Hum Genomics. 2012;6(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kozlov G, Gehring K. Calnexin cycle - structural features of the ER chaperone system. FEBS J. 2020;287(20):4322–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. [DOI] [PubMed] [Google Scholar]
- [17].Molinari M. ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev Cell. 2021;56(7):949–966. [DOI] [PubMed] [Google Scholar]
- [18].Jeffers SA, Sanders DA, Sanchez A. Covalent modifications of the ebola virus glycoprotein. J Virol. 2002;76(24):12463–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chai Q, Li S, Collins MK, et al. HIV-1 Nef interacts with the cyclin K/CDK13 complex to antagonize SERINC5 for optimal viral infectivity. Cell Rep. 2021;36(6):109514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang B, Wang Y, Frabutt DA, et al. Mechanistic understanding of N-glycosylation in Ebola virus glycoprotein maturation and function. J Biol Chem. 2017;292(14):5860–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yu C, Li S, Zhang X, et al. MARCH8 inhibits Ebola virus glycoprotein, human immunodeficiency virus type 1 envelope glycoprotein, and avian influenza virus H5N1 hemagglutinin maturation. mBio. 2020;11(5):e01882–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hoenen T, Watt A, Mora A, et al. Modeling the lifecycle of Ebola virus under biosafety level 2 conditions with virus-like particles containing tetracistronic minigenomes. J Vis Exp. 2014;91:52381. DOI: 10.3791/52381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Piacentini S, La Frazia S, Riccio A, et al. Nitazoxanide inhibits paramyxovirus replication by targeting the Fusion protein folding: role of glycoprotein-specific thiol oxidoreductase ERp57. Sci Rep. 2018;8(1):10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu Y, Swulius MT, Moremen KW, et al. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci U S A. 2003;100(14):8229–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012;37(10):404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Frabutt DA, Wang B, Riaz S, et al. Innate sensing of influenza A virus hemagglutinin glycoproteins by the host endoplasmic reticulum (ER) stress pathway triggers a potent antiviral response via ER-associated protein degradation. J Virol. 2018;92(1):e01690–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mizushima N, Yoshimori T, Ohsumi Y. The role of ATG proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132. [DOI] [PubMed] [Google Scholar]
- [29].Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73(18):3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chan SY, Ma MC, Goldsmith MA. Differential induction of cellular detachment by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J Gen Virol. 2000;81(9):2155–2159. [DOI] [PubMed] [Google Scholar]
- [31].Hudson DA, Gannon SA, Thorpe C. Oxidative protein folding: from thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic Biol Med. 2015;80:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhou T, Frabutt DA, Moremen KW, et al. ERManI (endoplasmic reticulum class I alpha-mannosidase) is required for HIV-1 envelope glycoprotein degradation via endoplasmic reticulum-associated protein degradation pathway. J Biol Chem. 2015;290(36):22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hu D, Wang B, Leng Y, et al. Genetic knockout of erp57 gene in host cells inhibits influenza virus replication. Chin J Preventive Vet Med. 2016;38. 429–433. [Google Scholar]
- [35].Liu X, Wang B, Yao X, et al. The role of autophagy-related gene 3 in the assembly process of Ebola virus. Chin J Virol. 2019;35:888–894. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.