

ABSTRACT
TFEB (transcription factor EB) and TFE3 (transcription factor binding to IGHM enhancer 3) orchestrate the cellular response to a variety of stressors, including nutrient deprivation, oxidative stress and pathogens. Here we describe a novel interaction of TFEB and TFE3 with the FAcilitates Chromatin Transcription (FACT) complex, a heterodimeric histone chaperone consisting of SSRP1 and SUPT16H that mediates nucleosome disassembly and assembly, thus facilitating transcription. Extracellular stimuli, such as nutrient deprivation or oxidative stress, induce nuclear translocation and activation of TFEB and TFE3, which then associate with the FACT complex to regulate stress-induced gene transcription. Depletion of FACT does not affect TFEB activation, stability, or binding to the promoter of target genes. In contrast, reduction of FACT levels by siRNA or treatment with the FACT inhibitor curaxin, severely impairs induction of numerous antioxidant and lysosomal genes, revealing a crucial role of FACT as a regulator of cellular homeostasis. Furthermore, upregulation of antioxidant genes induced by TFEB over-expression is significantly reduced by curaxin, consistent with a role of FACT as a TFEB transcriptional activator. Together, our data show that chromatin remodeling at the promoter of stress-responsive genes by FACT is important for efficient expression of TFEB and TFE3 targets, thus providing a link between environmental changes, chromatin modifications and transcriptional regulation.
Abbreviations: ADNP2, ADNP homeobox 2; ATP6V0D1, ATPase H+ transporting V0 subunit d1; ATP6V1A, ATPase H+ transporting V1 subunit A; ATP6V1C1, ATPase H+ transporting V1 subunit C1; CSNK2/CK2, casein kinase 2; CLCN7, chloride voltage-gated channel 7; CTSD, cathepsin D; CTSZ, cathepsin Z; EBSS, earle’s balanced salt solution; FACT complex, facilitates chromatin transcription complex; FOXO3, forkhead box O3; HEXA, hexosaminidase subunit alpha; HIF1A, hypoxia inducible factor 1 subunit alpha; HMOX1, heme oxygenase 1; LAMP1, lysosomal associated membrane protein 1; MAFF, MAF bZIP transcription factor F; MAFG, MAF bZIP transcription factor G; MCOLN1, mucolipin TRP cation channel 1; MTORC1, mechanistic target of rapamycin kinase complex 1; NaAsO2, sodium arsenite; POLR2, RNA polymerase II; PPARGC1A, PPARG coactivator 1 alpha; PYROXD1, pyridine nucleotide-disulfide oxidoreductase domain 1; RRAGC, Ras related GTP binding C; SEC13, SEC13 homolog, nuclear pore and COPII coat complex component; SLC38A9, solute carrier family 38 member 9; SSRP1, structure specific recognition protein 1; SUPT16H, SPT16 homolog, facilitates chromatin remodeling subunit; TFEB, transcription factor EB; TFE3, transcription factor binding to IGHM enhancer 3; TXNRD1, thioredoxin reductase 1; UVRAG, UV radiation resistance associated; WDR59, WD repeat domain 59.
KEYWORDS: Autophagy, chaperone, FACT, histone, lysosomes, TFEB, TFE3
Introduction
TFEB (transcription factor EB) and TFE3 (transcription factor binding to IGHM enhancer 3) are basic helix-loop-helix leucine zipper transcription factors that are activated in response to stress to modulate expression of multiple genes involved in lysosomal biogenesis, macroautophagy/autophagy, lipid metabolism, and immune response [1].
The main regulatory mechanism for TFEB and TFE3 is the control of their translocation from the cytosol to the nucleus, although changes in stability [2], quaternary structure [3] and nuclear export [4] also modulate their activity. Retention of TFEB and TFE3 in the cytosol is achieved by phosphorylation of specific residues that mediate binding to the chaperone YWHA/14-3-3. Under basal (non-stressed) conditions, TFEB and TFE3 are recruited to the surface of lysosomes through interaction with active Rag GTPases [5]. This brings TFEB and TFE3 in close proximity to the serine/threonine kinase MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1), which phosphorylates the transcription factors on multiple residues [6–9]. MTORC1‐dependent phosphorylation of TFEB on serine 211 (S211) and TFE3 on serine 321 (S321) creates a binding site for YWHA/14-3-3, resulting in sequestration of TFEB and TFE3 in the cytosol [6,7,9]. Other cellular kinases may also contribute to TFEB and TFE3 regulation [10–13]. Under stress conditions, dephosphorylation of TFEB and TFE3, either by inactivation of MTORC1 or activation of specific phosphatases [14,15], causes a rapid translocation of the transcription factors to the nucleus, where they activate multiple transcriptional networks with the goal of eliminating damaged organelles, preserving cellular functions and ultimately, restoring cellular homeostasis.
A growing list of stressors have been reported to induce TFEB and TFE3 activation. This includes nutrient deprivation [6–9,16], inflammation [17], accumulation of unfolded proteins [18], pathogens [19–22], mitochondrial damage [23], oxidative stress [3,15,24], physical exercise [25], increased cytosolic Ca2+ [14] and DNA damage [26,27]. While this indicates an essential role of TFEB and TFE3 in cellular response to stress, it also raises the question of how these transcription factors contribute to stress-specific transcriptional responses. One possibility is that TFEB and TFE3 bind specific transcriptional co-regulators in certain cell types or stress conditions to favor the expression of a particular set of stress response genes. Another poorly characterized aspect of TFEB and TFE3 regulation is the potential presence of specific transcriptional activators or inhibitors that may help modulating the efficiency of the TFEB and TFE3-mediated response.
Here we report the identification of a novel interaction between TFEB/TFE3 and the FAcilitates Chromatin Transcription (FACT) complex, a histone chaperone previously implicated in transcription initiation and elongation [28]. Following stress, TFEB and TFE3 translocate to the nucleus where they bind SSRP1 and SUPT16H, the two subunits that constitute the FACT complex. Depletion of FACT does not affect TFEB stability or its ability to bind to the promoter of target genes. Instead, FACT stimulates expression of antioxidant, autophagic and lysosomal genes through coactivation of TFEB and TFE3, thus providing a link between environmental changes, chromatin modifications and transcriptional regulation of cellular stress response.
Results
Identification of novel TFEB interactors
In this study we sought to identify novel TFEB interactors, with a particular interest in proteins that may function as TFEB transcriptional co-regulators. For this, we performed Rapid Immunoprecipitation Mass spectrometry of Endogenous proteins (RIME). HeLa cells stably expressing TFEB-Flag were subjected to either oxidative stress (incubation with NaAsO2 for 2 h) or starvation (incubation with EBSS for 4 h), two treatments known to induce efficient translocation of TFEB from the cytosol to the nucleus (Figures S1A and S1C). Hela-WT cells treated with NaAsO2 for 2 h were used as a negative control. After treatment, cells were cross-linked with formaldehyde and the isolated nuclei were sonicated to prepare the chromatin for the immunoprecipitation reaction with Flag antibodies. Proteins pulled down by immunoprecipitation were identified by tandem mass spectrometry (LC-MS/MS). As an additional control, we performed a second independent experiment in which ARPE-19 cells were infected with either control (Ad-Null) or TFEB-expressing adenovirus (Ad-TFEB-Flag) for 16 h, followed by treatment with NaAsO2 for 2 h and immunoprecipitation with Flag antibodies. TFEB-Flag interacting proteins were identified by mass spectrometry analysis (Figures S1B and S1C).
From all the potential TFEB interactors detected by MS, we focused on those that met the following criteria: they were identified in the two different cell lines (HeLa and ARPE-19), under two different types of stress conditions (oxidative stress and starvation) and showed at least a 4-fold enrichment when compared with the negative controls (Table 1, Table S1 and Table S2). As expected, the list contained some previously described TFEB interactors, including TFE3, YWHA/14-3-3, and several components of the lysosomal nutrient-sensing complex. We also identified several proteins implicated in nuclear import and protein stability, as well as some ribonucleoproteins. However, we were particularly interested in those interactors involved in transcriptional regulation and we focused our subsequent analysis on two proteins, SSRP1 and SUPT16H, which interact to form the FACT complex, a histone chaperone implicated in transcription initiation and elongation.
Table 1.
Identification of TFEB interactors by RIME and Mass spectrometry analysis.
| Protein class | Gene/Protein |
|---|---|
| Known TFEB interactors | TFE3, Transcription factor E3 |
| YWHAE, 14-3-3 protein epsilon | |
| YWHAB, 14-3-3 protein beta/alpha | |
| YWHAZ, 14-3-3 protein zeta/delta | |
| YWHAQ, 14-3-3 protein theta | |
| YWHAG, 14-3-3 protein gamma | |
| YWHAH, 14-3-3 protein eta | |
| LAMTOR1, Regulator complex protein LAMTOR1 | |
| RRAGA, Ras-related GTP-binding protein A | |
| RPTOR, Regulatory-associated protein of mTOR | |
| ATP6V1A, V-type proton ATPase catalytic subunit A | |
| ATP6V1B2, V-type proton ATPase subunit B brain | |
| isoform | |
| Stress response/protein stability/degradation | HSPH1, Heat shock protein 105 kDa |
| HSPA4L, Heat shock 70 kDa protein 4 L | |
| HSPA4, Heat shock 70 kDa protein 4 | |
| SQSTM1, Sequestosome-1 | |
| Nuclear import | NUP93, Nuclear pore complex protein Nup93 |
| NUP133, Nuclear pore complex protein Nup133 | |
| Histones and ribonucleoproteins | HNRNPM, Heterogeneous nuclear ribonucleoprotein M |
| HNRNPC, Heterogeneous nuclear ribonucleoproteins | |
| C1/C2 | |
| H2AFY, Core histone macro-H2A.1 | |
| H3F3A, Histone H3.3 | |
| Transcriptional regulators | SUPT16H, FACT complex subunit SPT16 |
| SSRP1, FACT complex subunit SSRP1 | |
| XRCC5, X-ray repair cross-complementing protein 5 | |
| XRCC6, X-ray repair cross-complementing protein 6 | |
| NUMA1, Nuclear mitotic apparatus protein 1 | |
| RPA1, Replication protein A 70 kDa DNA-binding | |
| subunit | |
| CBX3, Chromobox protein homolog 3 | |
| SMARCA5, SWI/SNF-related matrix-associated actin- | |
| dependent regulator of chromatin subfamily | |
| A member 5 | |
| TOP2A, DNA topoisomerase 2-alpha |
List of TFEB interactors identified both in HeLa and ARPE-19 cells under oxidative stress and starvation conditions.
TFEB and TFE3 interact with the FACT complex under stress conditions
To validate the interaction between TFEB and the FACT complex observed in our MS analysis, HeLa cells stably expressing TFEB-Flag were treated with EBSS (starvation), Torin-1 (MTORC1 catalytic inhibitor) or NaAsO2 (oxidative stress) (Figure 1A). TFEB activation by these treatments was confirmed by changes in TFEB electrophoretic mobility and reduced interaction with YWHA/14-3-3. As previously described, EBSS and Torin-1 caused efficient MTORC1 inactivation, as indicated by the reduced phosphorylation of the MTORC1 targets RPS6KB2 and EIF4EBP1. In contrast, 2 h treatment with NaAsO2 resulted in MTORC1 hyperactivation [15]. Importantly, immunoprecipitation of TFEB-Flag pulled down endogenous SSRP1 and SUPT16H under all stress conditions (Figure 1A). The reduced interaction observed following incubation with EBSS for 4 h likely reflects the less efficient dissociation of the Flag-TFEB-YWHA/14-3-3 complex achieved under this experimental condition. These results were further confirmed in ARPE-19 cells infected with either Ad-Null or Ad-TFEB-Flag. As seen in Figure 1B, TFEB co-immunoprecipitated with the FACT complex following treatment with EBSS, Torin-1 or NaAsO2, whereas negligible interaction was observed under control conditions. Furthermore, stress-increased interaction between FACT and recombinant TFE3 was also observed in ARPE-19 cells infected with Ad-TFE3-MYC (Figure S1D).
Figure 1.
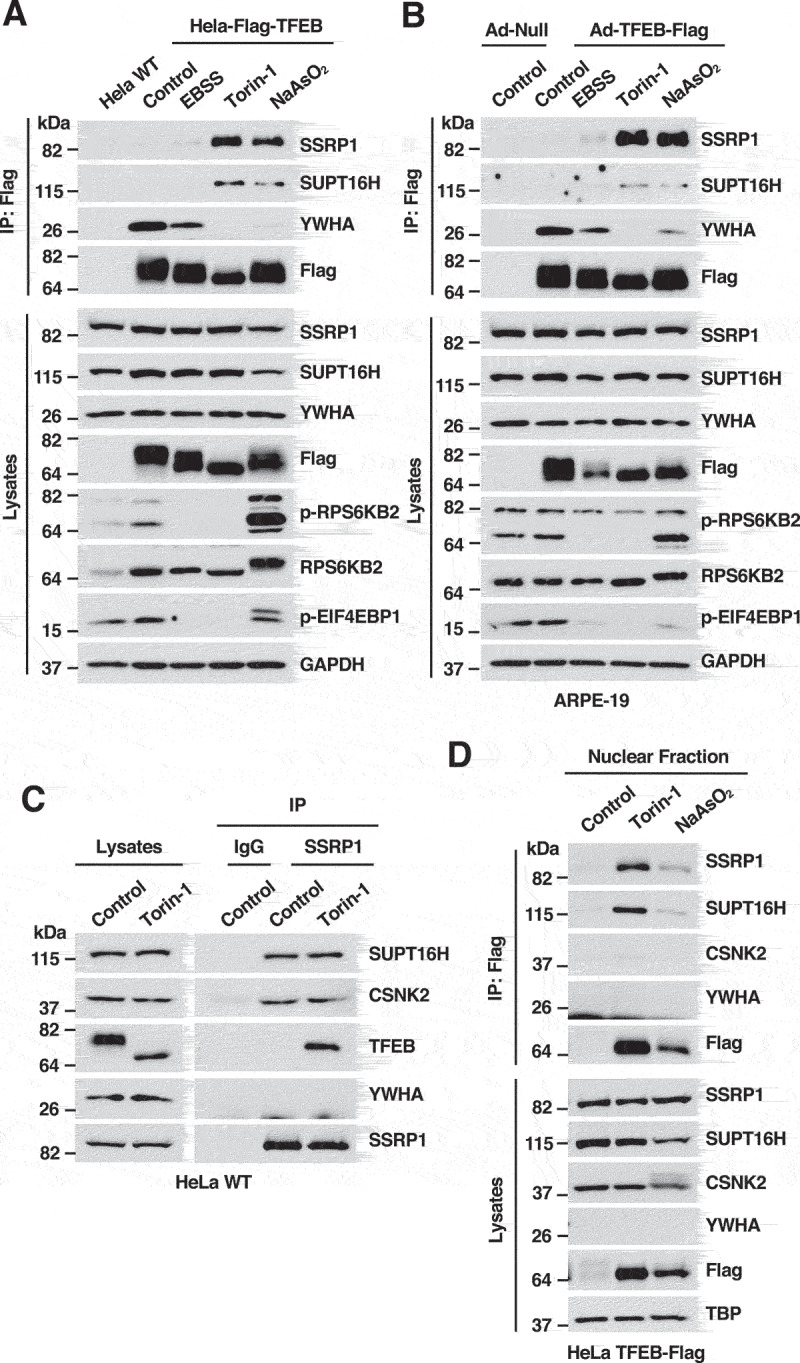
TFEB interacts with the FACT complex under stress conditions. (A) Immunoblot analysis of immunoprecipitated TFEB-Flag from HeLa cells incubated with DMSO (Control), EBSS for 4 h, Torin-1 (250 nM) for 2 h or NaAsO2 (250 μM) for 2 h. (B) Immunoblot analysis of immunoprecipitated TFEB-Flag from ARPE-19 cells infected with either Ad-Null or Ad-TFEB-S211A and incubated with DMSO (Control), EBSS for 4 h, Torin-1 (250 nM) for 2 h or NaAsO2 (250 μM) for 2 h. (C) Immunoblot analysis of immunoprecipitated endogenous SSRP1 from HeLa WT cells treated with DMSO (Control) or Torin-1 (250 nM) for 2 h. (D) Immunoblot analysis of proteins from the nuclear fraction of HeLa-TFEB-Flag cells treated with DMSO (Control), Torin-1 (250 nM) for 2 h or NaAsO2 (250 μM) for 2 h. Immunoblots are representative of at least three independent experiments.
To assess the binding of the FACT complex to endogenous TFEB, we performed reverse immunoprecipitations by pulling down endogenous SSRP1. As expected, SSRP1 and SUPT16H interacted with each other both under control and stress conditions (Figure 1C and Figure S1E). CSNK2/CK2 (casein kinase 2), a kinase known to associate with the SSRP1-SUPT16H heterodimer, also co-immunoprecipitated with the FACT complex in control and stress conditions [29]. In contrast, binding of SSRP1 to endogenous TFEB and TFE3 was only observed under stress, suggesting that the interaction mainly occurs following TFEB and TFE3 activation (Figure 1C and Figure S1E). To confirm this possibility, we performed subcellular fractionation. As seen in Figure 1D, the amount of TFEB in the nucleus was clearly increased by either Torin-1 or NaAsO2 treatment. Interestingly, nuclear TFEB pulled-down SSRP1 and SUPT16H but not CSNK2/CK2. These results indicate that TFEB binds SSRP1 and SUPT16H in the nucleus and suggest the existence of at least two different FACT complex populations, one bound to CSNK2/CK2 and the other bound to TFEB.
The interaction between TFEB and the FACT complex occurs in the nucleus
To further corroborate that TFEB interacts with the FACT complex after translocation to the nucleus, we infected ARPE-19 cells with adenovirus expressing either WT TFEB or the TFEBS211A mutant. We have previously described that mutation of serine 211 to alanine prevents binding of TFEB to YWHA/14-3-3, resulting in its accumulation in the nucleus even under basal (non-stressed) conditions [6]. While the interaction between WT TFEB and the FACT complex was only detected following stress, TFEBS211A bound SSRP1 and SUPT16H both in control and stress conditions (Figure 2A). These results suggest that TFEB presence in the nucleus is both sufficient and necessary for its interaction with the FACT complex.
Figure 2.
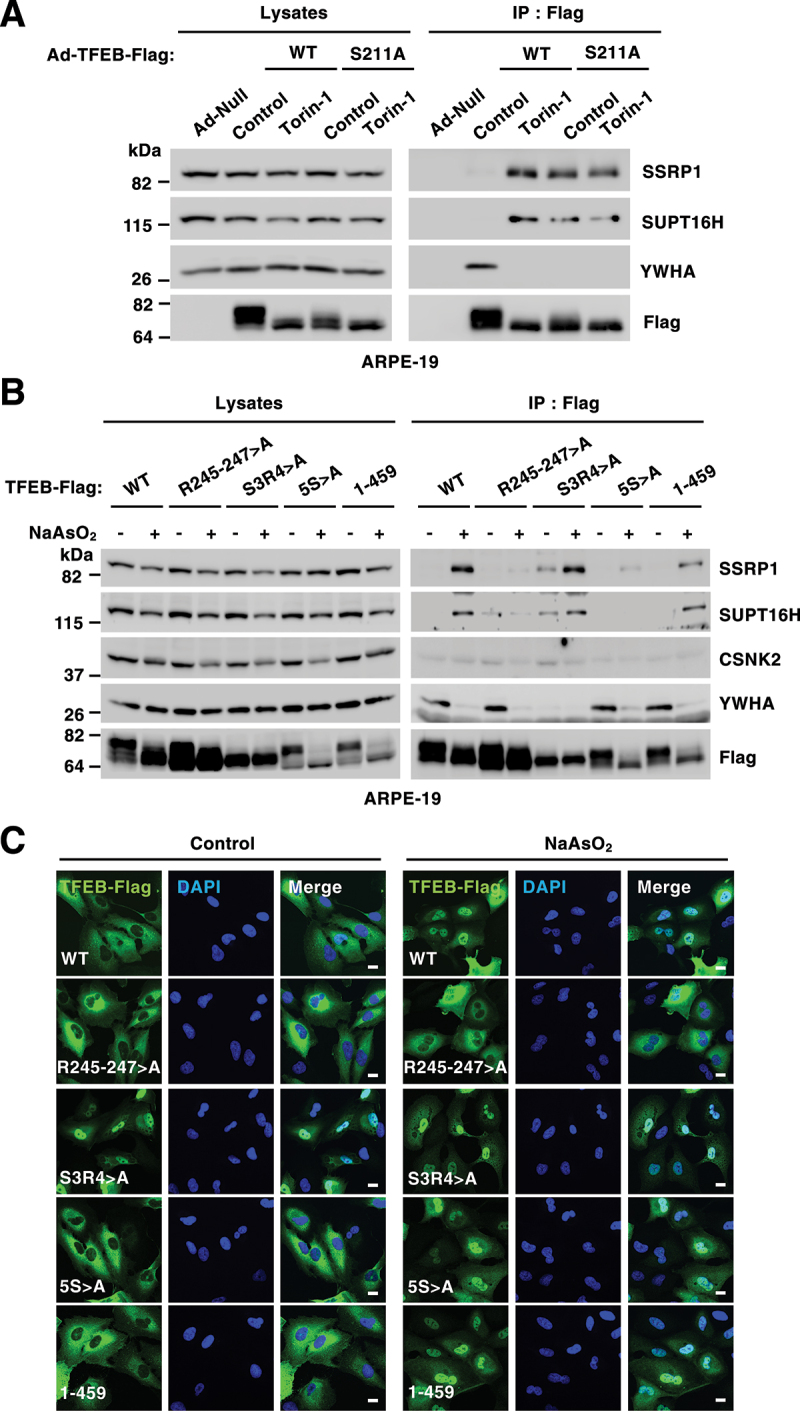
The interaction between TFEB and the FACT complex occurs in the nucleus. (A) Immunoblot analysis of TFEB-Flag immunoprecipitation from ARPE-19cells infected with Ad-Null, Ad-TFEB-WT-Flag or Ad-TFEB-S211A-Flag and treated with DMSO (Control) or Torin-1 (250 nM) for 2 h. (B) Immunoblot analysis of TFEB-Flag immunoprecipitation from ARPE-19 cells expressing TFEB-WT or the indicated TFEB mutants upon treatment with NaAsO2 (250 μM) for 2 h. (C) Immunofluorescence confocal microscopy analysis of ARPE-19 cells showing the intracellular distribution of recombinant TFEB-WT or the indicated TFEB mutants in response to treatment with NaAsO2 (250 μM) for 2 h. Scale bars: 10 μm.
In agreement with these results, we also found that mutation of the TFEB nuclear import signal (R245-247>A), which causes TFEB retention in the cytosol under stress conditions [7], prevented the interaction of TFEB with the FACT complex, even though the mutant efficiently dissociated from YWHA/14-3-3 following NaAsO2 treatment (Figures 2B and 2C). Conversely, mutation of serine 3 and arginine 4 to alanine (S3R4>A), a mutation that prevents binding of TFEB to RRAG GTPases causing TFEB nuclear accumulation [5], resulted in TFEB-FACT binding both in control and stress conditions (Figures 2B and 2C). Finally, deletion or amino acid substitution of a serine-rich region in the C terminus of TFEB (462SSRRSSFS469) (mutants 1–459 and 5S>A) did not significantly change the ability of TFEB to bind FACT (Figures 2B and 2C).
It has been shown that the FACT complex distributes between the nucleoli and the nucleoplasm [30]. To better understand where the interaction between TFEB/TFE3 and the FACT complex takes place, we expressed a previously described Flag-tagged SSRP1 construct [31] in ARPE-19 cells. Similar to what we observed with the endogenous protein, recombinant SSRP1 co-immunoprecipitated SUPT16H and CSNK2/CK2 in control and stress conditions, while the binding to endogenous TFEB and TFE3 was only observed following stress (Figure S2A). Immunofluorescence analysis confirmed that SSRP1-Flag localized to both nucleoli and nucleoplasm (Figure S2B). In contrast, active TFE3 was excluded from nucleoli both under Torin-1 and NaAsO2 conditions, even in those cells in which SSRP1 was over-expressed (Figure S2B), suggesting that the interaction between TFEB/TFE3 and the FACT complex occurs primarily in the nucleoplasm.
The FACT complex does not affect stress-induced TFEB nuclear translocation
Next, we investigated whether the FACT complex may affect TFEB activation or stability. For this, we depleted SSRP1 in HeLa cells using siRNAs. Importantly, this also led to a simultaneous reduction of SUPT16H levels, as assessed by immunoblot (Figures 3A and 3B). This interdependence of the two FACT subunits has been observed before [32]. As seen in Figures 3A, 3D, and 3E, depletion of the FACT complex did not affect TFEB or TFE3 activation. In fact, dephosphorylation of TFEB-S211 and TFE3-S321 in response to EBSS, Torin-1 and NaAsO2 was robust and comparable between cells treated with either control or SSRP1 siRNAs (Figure 3A). Similar results were obtained when siRNAs against SUPT16H were used (Figures S3A and S3B). Furthermore, efficient nuclear translocation of both endogenous and recombinant TFEB in response to MTORC1 inactivation and oxidative stress was observed in SSRP1-depleted cells (Figures 3D, 3E S3C, and S3D).
Figure 3.
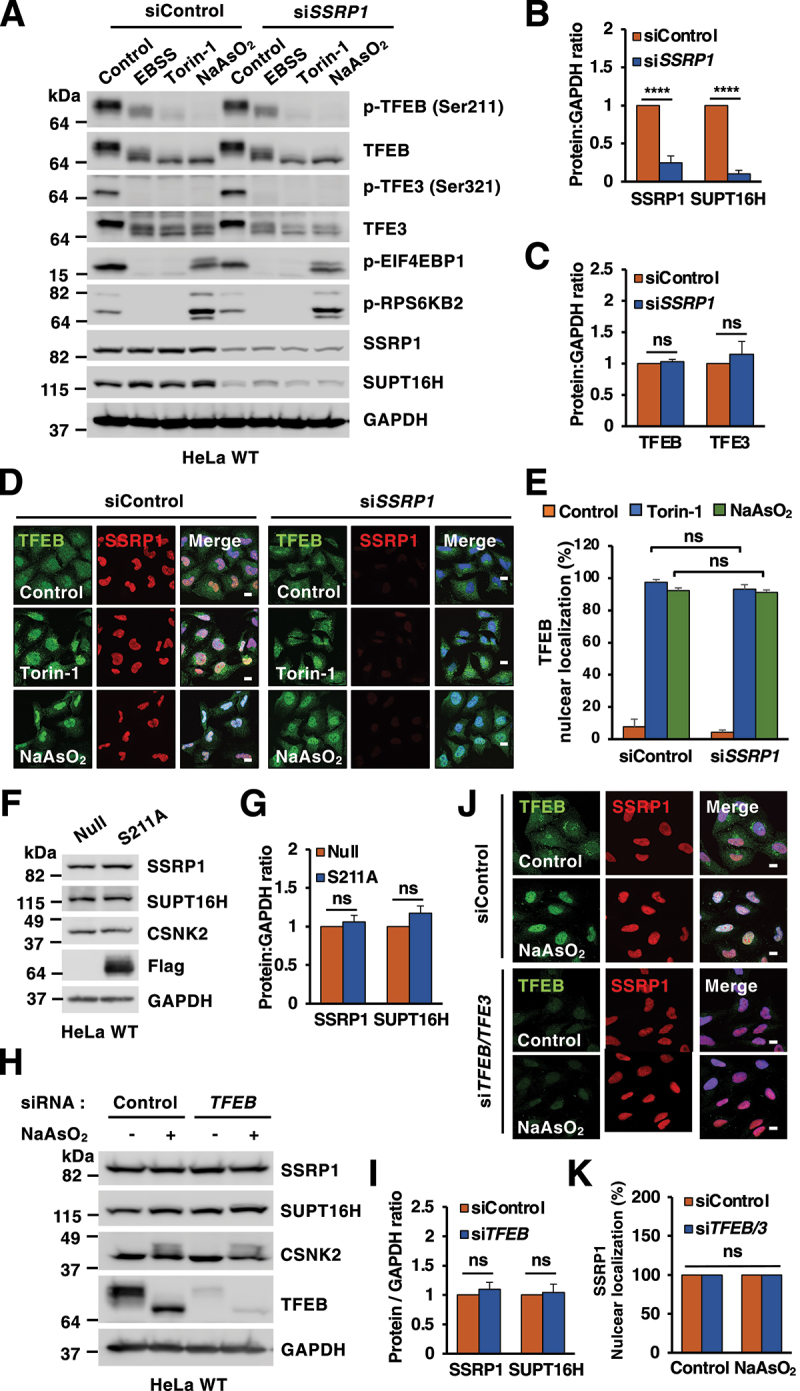
Depletion of the FACT complex does not alter TFEB or TFE3 activation. (A) Immunoblots of protein lysates from HeLa WT cells transfected with the indicated siRNAs upon incubation with DMSO (Control), EBSS for 4 h, Torin-1 (250 nM) for 2 h or NaAsO2 (50 μM) for 6 h. (B and C) Quantification of immunoblots shown in (A). Significance tested with two-way ANOVA with Sidak’s multiple comparisons test (****p < 0.0001) from three independent experiments. (D) Immunofluorescence confocal microscopy analysis of siRNA-transfected HeLa WT cells treated with DMSO (Control), Torin-1 (250 nM) for 2 h or NaAsO2 (50 μM) for 6 h and stained with antibodies against TFEB and SSRP1. Scale bars: 10 μm. (E) Quantification of immunofluorescence images shown in (D). Significance tested with two-way ANOVA with Sidak’s multiple comparisons test from three independent experiments and >100 cells counted per trial. (F) Immunoblot analysis of protein lysates from HeLa WT cells infected with either Ad-Null or Ad-TFEB-S211A-Flag for 40 h. (G) Quantification of immunoblots shown in (F). Significance tested with two-way ANOVA with Sidak’s multiple comparisons testfrom three independent experiments. (H) Immunoblots of protein lysates from HeLa WT cells depleted of TFEB and treated with NaAsO2 (50 μM) for 6 h. (I) Quantification of immunoblots shown in (H). Significance tested with two-way ANOVA with Sidak’s multiple comparisons test from three independent experiments. (J) Immunofluorescence confocal microscopy analysis of HeLa WT cells depleted of TFEB and TFE3 stained with antibodies against endogenous TFEB and SSRP1. Scale bars: 10 μm. (K) Quantification of immunofluorescence images shown in (J). Significance tested with two-way ANOVA with Sidak’s multiple comparisons test from three independent experiments and >100 cells counted per trial.
In addition, depletion of the FACT complex (Figures 3A, 3C, S3A and S3B) or treatment with the FACT complex inhibitor CBL0137 (also known as Curaxin) (Figure S3E) did not change total TFEB or TFE3 levels, suggesting that FACT does not regulate TFEB/TFE3 stability (note that increased phosphorylation of TP53 was used as a control for efficient Curaxin-induced FACT inactivation). Likewise, TFEB over-expression or depletion did not alter SSRP1, SUPT16H and CSNK2/CK2 proteins levels (Figure 3F-I) or SSRP1 intracellular distribution (Figures 3J and 3K).
Depletion of the FACT complex impairs the oxidative stress response
To investigate whether FACT orchestrates transcriptional regulation in response to oxidative stress, we performed RNA-seq analysis in HeLa-TFEB-Flag cells treated with either control or SSRP1 siRNAs after 6 h incubation with NaAsO2. In total we found 8,624 differentially expressed genes, 3,938 upregulated and 4,686 downregulated. While the changes were not very pronounced (with most of the genes showing a shrunker log2 fold change<2), they were highly significant (q-value<0.05) (Figures 4A, 4B and Table S3). Interestingly, close to 70% (500 out of 727) of the genes previously identified as TFEB targets were significantly altered by SSRP1 depletion, suggesting that the FACT complex may function as a TFEB transcriptional regulator (Figure 4C and Table S4). Given that TFEB is a well-established master regulator of lysosomal biogenesis, we first compared expression of lysosomal genes between control and FACT-depleted cells in response to NaAsO2. However, we did not observe a major upregulation of lysosomal genes under these conditions, neither FACT depletion affected the expression of these genes (Figure S4A-C).
Figure 4.
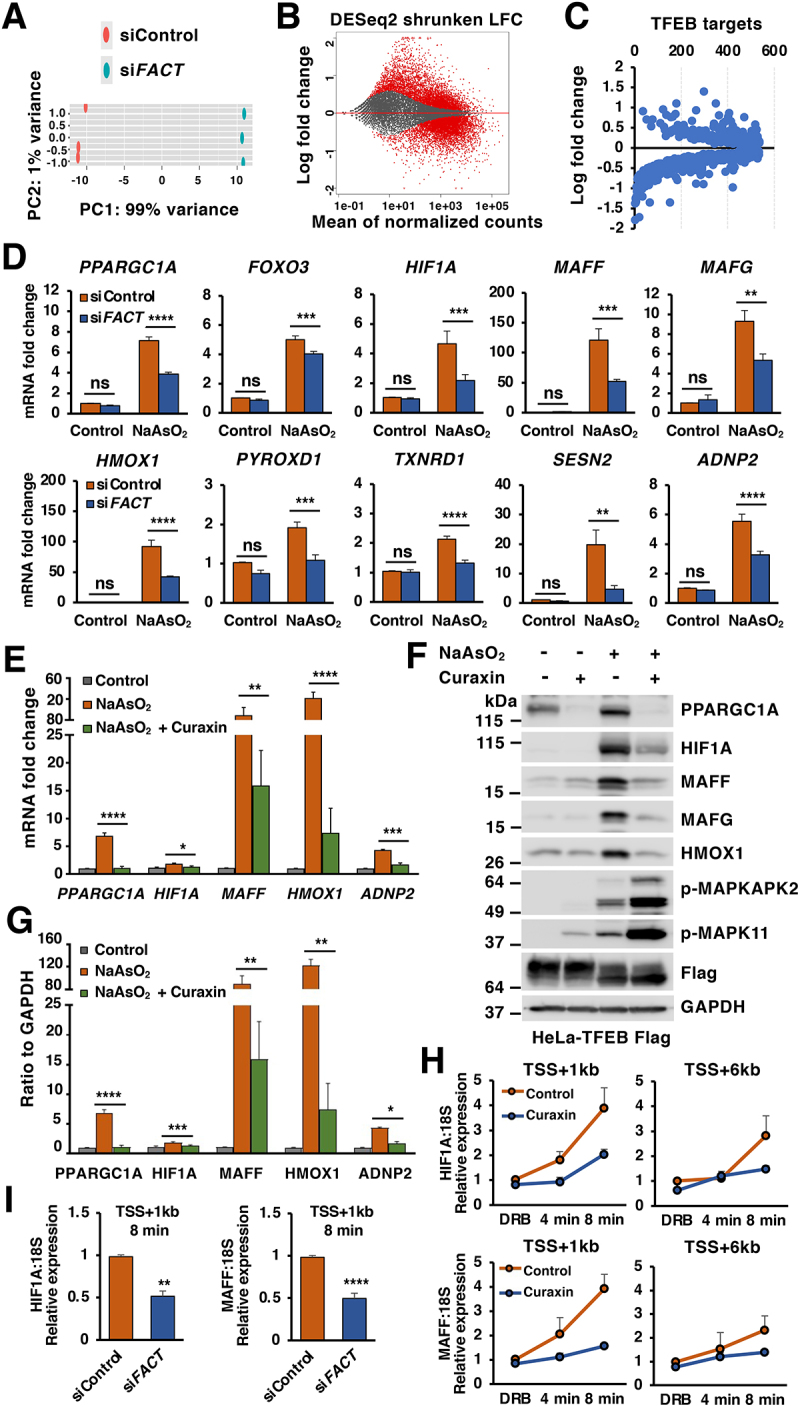
The FACT complex modulates cellular response to oxidative stress. (A) Principal component analysis of genes with q-value < 0.05 reveals distinct clustering of siControl- and siFACT-treated cells upon incubation with NaAsO2 (50 μM) for 6 h. (B) Volcano plot indicating distribution of genes significantly up- and downregulated in control versus FACT complex depleted cells incubated with NaAsO2 (50 μM) for 6 h. Cutoffs indicate genes with q-value < 0.05. (C) Volcano plot indicating distribution of previously identified TFEB target genes in control versus FACT complex depleted HeLa-TFEB-Flag cells as described in (B). (D) Relative quantitative real-time PCR analysis of PPARGC1A, FOXO3, HIF1A, MAFF, MAFG, HMOX1, PYROXD1, TXNRD1, SESN2 and ADNP2 mRNA transcript levels in HeLa-TFEB-Flag cells treated with either control or FACT siRNAs upon incubation with NaAsO2 (50 μM) for 6 h. Significance tested with two-way ANOVA with Sidak’s multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) from three independent experiments. (E) Relative quantitative real-time PCR analysis of PPARGC1A, HIF1A, MAFF, HMOX1 and ADNP2 mRNA transcript levels in HeLa-TFEB-Flag cells treated with DMSO (Control), NaAsO2 (50 μM) or NaAsO2 (50 μM) plus curaxin (1 μM) for 6 h. Data represented as geometric mean ± SD and significance tested with Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) from three independent experiments (F) Immunoblots analysis of protein lysates from HeLa-TFEB-Flag cells treated with NaAsO2 (50 μM), Curaxin (1 μM) alone or NaAsO2 (50 μM) plus curaxin (1 μM) for 6 h. (G) Quantification of immunoblots shown in (F). Data represents mean relative protein levels ± SD with n = 5. Significance tested using Student’s test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (H and I) Transcription elongation rate was analyzed by relative quantitative real-time PCR for HIF1A and MAFF pre-mRNA transcript levels in HeLa-TFEB-Flag cells treated with NaAsO2 (50 μM) plus curaxin (1 μM) for 6 h (H) or siRNA transfected HeLa-TFEB-Flag cells upon NaAsO2 (50 μM) incubation for 6 h (I). Data represents mean relative protein levels ± SD with n = 4. Significance tested using Student’s test (**p < 0.01, ****p < 0.0001).
Among the genes more severely down regulated by FACT depletion in our RNA-seq analysis were many regulators of the oxidative stress response (Table S5). Quantitative-PCR analysis (q-PCR) confirmed that in control cells, treatment with NaAsO2 for 6 h caused an efficient upregulation of multiple genes implicates in cellular response to oxidative stress, including several transcription factors and reactive oxygen species (ROS) scavengers. Interestingly, the induction of antioxidant genes was significantly impaired in FACT-depleted cells (Figure 4D). Similar results were obtained when we used curaxin, a small molecule capable of sequestering FACT, thus preventing its normal function in opening up chromatin structure to allow transcription [33]. Treatment with curaxin strongly inhibited the upregulation of oxidative stress genes as assessed both by q-PCR (Figure 4E) and Western blot (Figures 4F and 4G). Curaxin treatment did not prevent oxidative stress, as indicated by the efficient phosphorylation of MAPK11 and the MAPK11 target MAPKAPK2 (Figure 4F).
It has been reported that the FACT complex promotes nucleosome assembly and disassembly, thus facilitating rapid passage of the POLR2 (RNA polymerase II) and expediting transcription. To further confirm this model, we measured the elongation rates of several antioxidant genes by using 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB), which reversibly blocks transcription in vivo, combined with quantitative reverse transcriptase-PCR (qRT-PCR), as previously described [34]. Because the typical elongation rates are estimated at around a few Kb/min, measurements were performed at 4 and 8 min after DRB removal using primers specific for intronic sequences located 1 Kb (proximal) and 6 Kb (distal) downstream to the transcription start site (TSS). As expected, depletion of the FACT complex or curaxin treatment substantially reduced the transcription elongation rate of oxidative stress response genes, such as HIF1A and MAFF (Figures 4H and 4I). Consequently, the ability to cope with stress is decreased in FACT-depleted cells, as evidenced by the increased accumulation of intracellular ROS (Figure S4D and S4E). Altogether, these findings suggest that the FACT complex plays a role in modulating expression of oxidative stress genes.
The FACT complex promotes TFEB-dependent expression of oxidative stress genes
Next, we asked whether the role of FACT in the expression of oxidative stress genes is mediated by modulation of TFEB transcriptional activity. For this, we performed ChIP-seq analysis on stable HeLa-TFEB-Flag cells treated with NaAsO2 for 6 h and assessed promoter occupancy by TFEB-Flag. Interestingly, we observed binding of TFEB-Flag to the promoters of many of the oxidative stress genes regulated by FACT. The peaks localized in close proximity to the transcription start site and were not detected in HeLa-WT (Figure S5A and Table S6). These data suggest that TFEB mediates expression of multiple genes implicated in cellular response to oxidative stress, consistently with recent studies suggesting a role for TFEB in promoting oxidative stress resistance both in mammals and C elegans [35,36].
To confirm the role of TFEB in oxidative stress, we expressed constitutively active TFEB (TFEBS211A) in HeLa cells and assessed the expression of several potential TFEB-targeted antioxidant genes both by q-PCR and Western blot. As seen in Figure 5A-C, over-expression of TFEBS211A alone (without NaAsO2 treatment) was sufficient to induce a strong mRNA and protein upregulation of critical regulators of the oxidative stress response, including HMOX1, ADNP2, PYROXD1, PPARGC1, HIF1A, MAFF, MAFG, FOXO3, and SRXN1. Furthermore, expression of oxidative stress genes following treatment with NaAsO2 was significantly impaired in double tfeb and tfe3 knockout mouse embryonic fibroblasts (MEFs; Figure S5B). In contrast, depletion of NFE2L2 did not affect the upregulation of antioxidant genes induced by TFEBS211A, suggesting that the two transcription factors function independently of each other (Figure S5C). Altogether, these results confirm that TFEB plays a role in redox homeostasis by regulating expression of key antioxidant genes.
Figure 5.
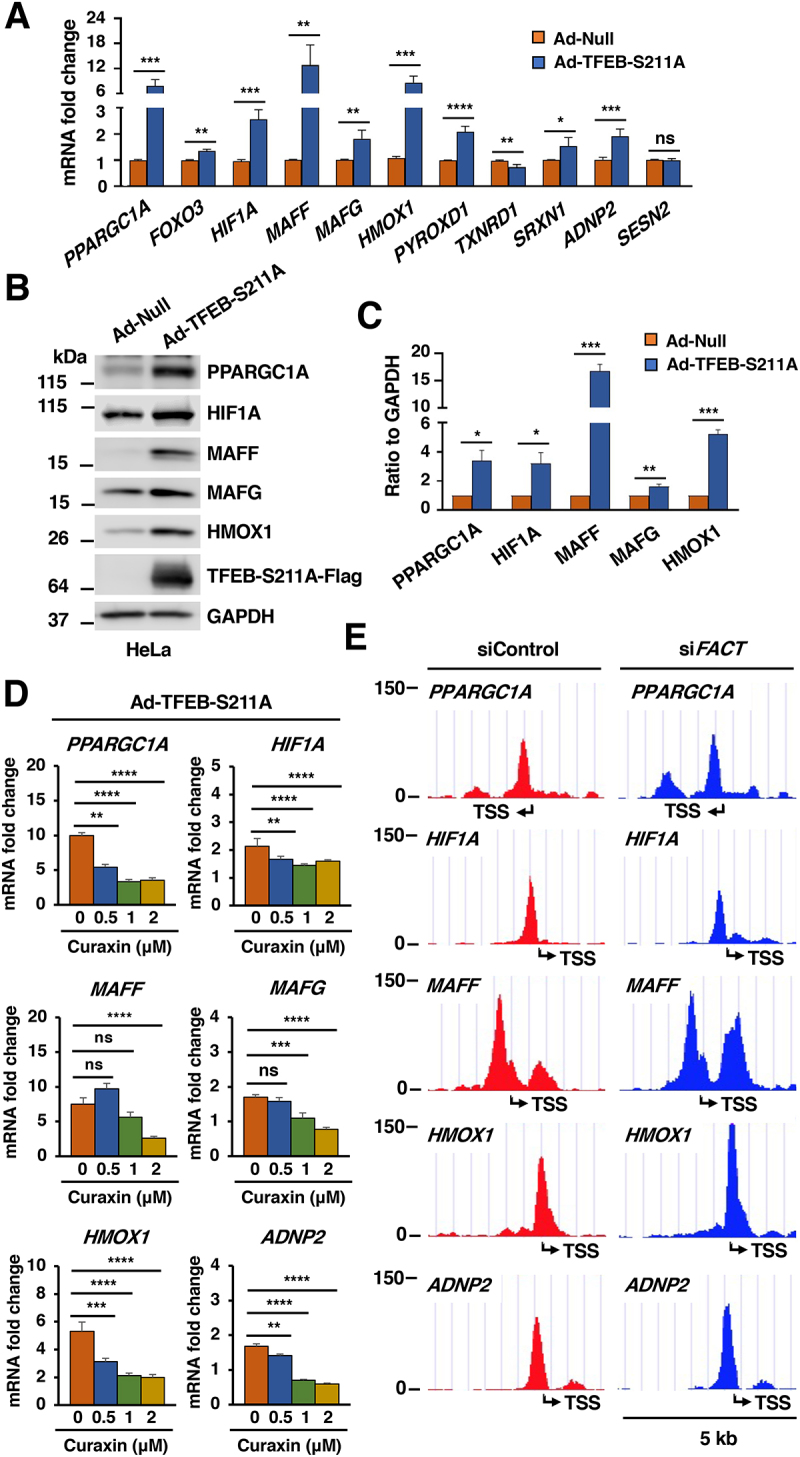
The FACT complex promotes TFEB-dependent expression of oxidative stress genes. (A) Relative quantitative real-time PCR analysis of PPARGC1A, FOXO3, HIF1A, MAFF, MAFG, HMOX1, PYROXD1, TXNRD1, SRXN1, ADNP2 and SESN2 mRNA transcript levels in HeLa WT cells infected with adenovirus expressing TFEB-S211A-Flag compared to Null for 40 h. Data represented as geometric mean ± SD and significance tested with Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) from at least three independent experiments. (B) Immunoblot analysis of protein lysates from HeLa WT cells infected with either Null adenovirus or adenovirus expressing TFEB-S211A-Flag for 40 h. (C) Quantification of immunoblots shown in (B). Data represents mean relative protein levels ± SD with n = 3. Significance tested using Student’s test (*p < 0.05, **p < 0.01, ***p < 0.001). (D) Relative quantitative real-time PCR analysis of PPARGC1A, HIF1A, MAFF, MAFG, HMOX1 and ADNP2 mRNA transcript levels in HeLa WT cells infected with adenovirus expressing TFEB-S211A-Flag upon incubation with curaxin for 4 h at different doses. Significance tested with two-way ANOVA with Sidak’s multiple comparisons test (**p < 0.01, ***p < 0.001, ****p < 0.0001) from three independent experiments. (E) Schematic representations of the TFEB-Flag binding region in the promoter of PPARGC1A, HIF1A, MAFF, HMOX1 and ADNP2 in HeLa-TFEB-Flag cells transfected with either control or FACT siRNAs treated with NaAsO2 (50 μM) for 6 h. The transcription start site is indicated as TSS.
To further assess whether the TFEB-mediated expression of antioxidant genes is modulated by FACT, we expressed TFEBS211A either alone or in the presence of the FACT inhibitor curaxin. Interestingly, curaxin significantly inhibited the TFEB-mediated upregulation of oxidative genes, indicating that the FACT complex was required for efficient TFEB activity (Figure 5D). Furthermore, ChIP-seq analysis showed that depletion of FACT did not noticeably affect TFEB’s ability to bind to the promoter of oxidative stress genes (Figure 5E), thus suggesting that FACT likely functions as a TFEB transcriptional activator.
The FACT complex modulates TFEB transcriptional activity under starvation conditions
We next assessed whether the FACT complex functions as a general TFEB/TFE3 transcriptional activation or it may contribute to stress-specific transcriptional responses. It is well established that TFEB and TFE3 induce expression of multiple lysosomal and autophagic genes in response to nutrient deprivation (Figure S6A) [9,16,37]. Treatment of HeLa-TFEB-Flag cells with EBSS for 16 h resulted in robust accumulation of TFEB-Flag in the nucleus and increased binding to endogenous SSRP1 and SUPT16H (Figures 6A and 6B). Prolonged starvation caused a significant increase in the expression of multiple lysosomal TFEB targets, including several subunits of the v-ATPase (ATP6V1C1, ATP6V0D1), lysosomal hydrolases (CTSD, HEXA), components of the nutrient sensing machinery (RRAGC, WDR59, NPRL3, DEPDC5, SEC13), and lysosomal transmembrane proteins (LAMP1, MCOLN1, CLCN7, SLC38A9, NPC2). Importantly, the upregulation of most of these genes was significantly impaired upon depletion of the FACT complex (Figure 6C). Furthermore, the transcriptional upregulation of numerous autophagic (ATG4, ATG12, ATG13, ATG16L1, ATG101, BCL2, GABARAPL1, SNCA, UVRAG, and WIPI1) and antioxidant (HMOX1, PPARGC1A, ADNP2, HIF1A, MAFF, and MAFG) TFEB/TFE3 targets was also strongly reduced in FACT-depleted cells (Figure S6B and 6D). In contrast, the expression of several ribosomal genes was not affected by FACT depletion (Figure S6C), suggesting that the FACT complex does not function as a generic transcriptional activator but specifically regulates expression of TFEB and TFE3 targets under a variety of stress conditions. Finally, we measured the transcription elongation rates of LAMP1 and MCOLN1 in starved cells and, as expected, found a significant reduction upon FACT depletion (Figure S6D).
Figure 6.
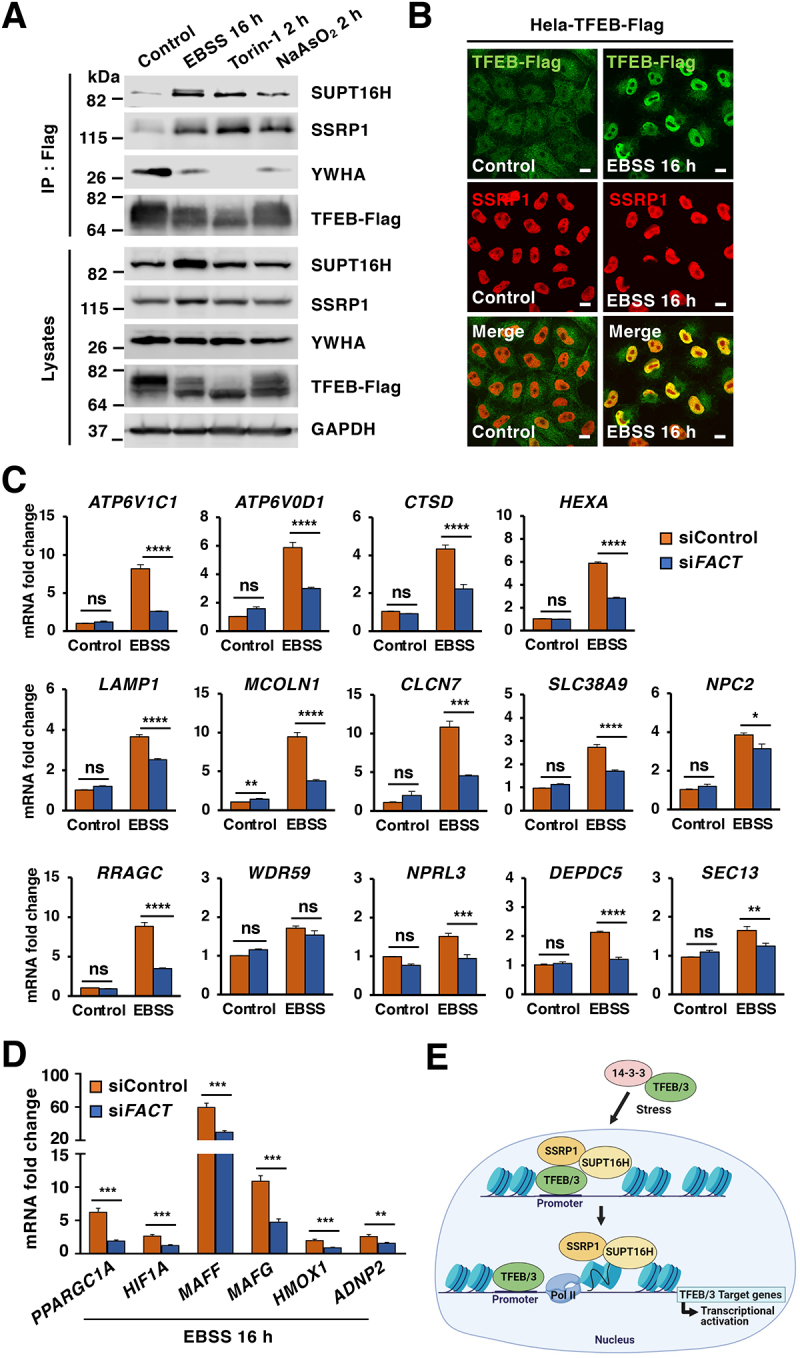
The FACT complex modulates TFEB transcriptional activity under starvation conditions. (A) Immunoblot analysis of immunoprecipitated TFEB from HeLa-TFEB-Flag cells incubated with DMSO (Control), EBSS for 16 h, Torin-1 (250 nM) for 2 h or NaAsO2 (250 μM) for 2 h. (B) Immunofluorescence images of HeLa-TFEB-Flag cells treated with EBSS for 16 h and stained with antibodies against TFEB and SSRP1. Scale bars: 10 μm. (C) Relative quantitative real-time PCR analysis of ATP6V1C1, ATP6V0D1, CTSD, HEXA, LAMP1, MCOLN1, CLCN7, SLC38A9, NPC2, RRAGC, WDR59, NPRL3, DEPDC5, and SEC13 in control and FACT-depleted HeLa-TFEB-Flag cells upon incubation with EBSS for 16 h. Significance tested with two-way ANOVA with Sidak’s multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) from three independent experiments. (D) Relative quantitative real-time PCR analysis of PPARGC1A, HIF1A, MAFF, HMOX1, and ADNP2 mRNA transcript levels in control and FACT-depleted cells incubated with EBSS for 16 h compared to control condition. Data represented as geometric mean ± SD and significance tested with Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001) from three independent experiments (E) Model depicting the proposed pathway of TFEB/TFE3-FACT complex activated by stress.
Altogether, our results suggest that translocation of TFEB and TFE3 to the nucleus upon stress conditions, such as starvation or oxidative stress, allows the interaction of these transcription factors with the FACT complex, thus facilitating TFEB and TFE3 transcriptional response (Figure 6E). Therefore, FACT functions as a TFEB and TFE3 transcriptional activator to help maintaining cellular homeostasis under stress.
Discussion
Gene expression requires recruitment of transcription factors as well as POLR2 to specific promoters. However, the presence of tightly wrapped nucleosomes represents a major obstacle for POLR2. Chromatin remodeling, which is mediated by histone-modifying enzymes, ATP-dependent chromatin remodelers and histone chaperones, is therefore critical for efficient transcription.
The FACT complex possesses intrinsic histone chaperone function and plays an important role regulating nucleosome disassembly and reassembly [38]. Nucleosomes consist of 146 bp of DNA wrapped around a histone octamer (one H3–H4 tetramer and two H2A–H2B dimers). When POLR2 approaches, the SSRP1 subunit of the FACT complex binds the H3-H4 tetramer, while the SUPT16H subunit removes one of the H2A-H2B dimers, resulting in the formation of a transient histone hexasome that facilitates the passage of the polymerase [39,40]. In some cases, the FACT complex can also promote nucleosome assembly by reducing the displacement of the dimers, thus repressing transcription initiation. It has been suggested that the interaction with specific transcription factors may favor FACT disassembling activity or reduce its reassembling function, allowing local regulation of gene expression. The role of FACT as a chromatin remodeler extends beyond the regulation of transcriptional initiation and elongation, as it has also been shown to participate in DNA replication and repair [41].
The requirement of the FACT complex for efficient transcription is highly variable between cell types, with cancer cells being highly dependent and showing high levels of SSRP1 and SUPT16H [42,43], whereas some differentiated cells grow normally in its absence. This suggests that FACT is not an essential component of the transcription machinery but may be needed when cells experience high transcriptional demand. Furthermore, FACT seems to regulate the expression of only a small fraction of genes in some cancer cells [32], further suggesting that it may function as a selective transcriptional regulator.
In this study we identified a novel interaction between FACT and the transcription factors TFEB and TFE3. Following activation by different stress conditions, such as starvation, oxidative stress, or Torin1-mediated MTORC1 inactivation, TFEB and TFE3 translocate from the cytosol to the nucleus, where they associate with the SSRP1-SUPT16H heterodimer. Previous studies have reported a role of FACT in regulating the stability of several transcription factors, including TP53, NFE2L2, and MYC/c-Myc [42,44]. However, neither TFEB/TFE3 protein levels, nor their translocation from the cytosol to the nucleus in response to stress, were altered in FACT-depleted cells. Furthermore, our ChIP-seq analysis revealed that FACT is not required for TFEB recruitment to the promoter of its target genes. In contrast, FACT depletion or inactivation by Curaxin caused a marked reduction in the expression of multiple TFEB target genes, suggesting that FACT exerts transcriptional co-activator function on antioxidant, autophagic, and lysosomal genes.
Our findings provide new insight into the mechanism of TFEB transcriptional regulation. Previous studies reported enhanced expression of TFEB-regulated autophagy genes by the histone arginine methyltransferase CARM1 (coactivator associated arginine methyltransferase 1) [45]. Here we suggest that TFEB facilitate nucleosome eviction by activating FACT disassembly activity or suppressing its repressor function, thus enabling rapid transcriptional elongation of target genes. It is, therefore, becoming clear that epigenetic regulation and chromatin remodeling are essential for maintaining homeostasis and viability in response to stress.
Several studies have reported that the global efficiency of transcription is not dependent on FACT, suggesting that the relevance of the complex may depend on the rate of transcription in specific cell types or environmental conditions. Consistently, FACT contributes to the expression of several inducible genes in yeast and mammals [46–52]. Our data further establish a role of FACT in facilitating expression of specific genes in response to stress through regulation of TFEB and TFE3.
It is also important to keep in mind that the FACT complex is frequently upregulated in human cancers and might be a potential target for cancer therapeutics [53]. For example, FACT was shown to accelerate expression of antioxidant genes in a model of hepatocellular carcinoma, thus increasing cancer cell survival. Targeting the FACT complex with Curaxin increased the vulnerability of cancer cells to oxidative stress and efficiently suppressed their growth [52]. Considering that aberrant TFEB and TFE3 activity has been reported in a variety of tumors, including renal cell carcinoma, alveolar soft part sarcoma, and pancreatic ductal adenocarcinoma [54], it is tempting to hypothesize that Curaxin might represent an interesting strategy for the treatment of these type of cancers.
In summary, our study underlines the importance of chromatin remodeling for a sustained and efficient stress response, and sheds new light on the epigenetic regulation of lysosomal biogenesis and redox homeostasis.
Material and methods
Cell line cultures and treatments
HeLa cells (CCL-2, ATCC) and HeLa cells stably expressing TFEB-Flag (CF7) were grown in DMEM, high glucose, GlutaMAX, sodium pyruvate (Gibco, 10569044) supplemented with 10% fetal bovine serum (Invitrogen, 21041–025), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, 2114). HeLa (CF7) cells were a kind gift of Dr. Andrea Ballabio (Baylor College of Medicine, Houston, TX, USA and Telethon Institute of Genetics and Medicine, Napoli, Italy). ARPE-19 cells (ATCC, CRL-2302) were grown in DMEM/F-12, GlutaMAX, sodium pyruvate (Gibco, 10565018) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Control and tfeb tfe3 KO MEFs were generated by transducing wild-type MEF cells with lentiviruses containing control or Tfeb and Tfe3 CRISPR-Cas9 guide RNA-targeting sequences and grown in DMEM, high glucose, GlutaMAX, sodium pyruvate supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin, as described previously [18], Adenovirus expressing Null, TFEB-S211A-Flag and TFE3-WT-MYC were prepared, amplified and purified by Welgen, Inc. as previously described [6,9]. For drug treatment experiments, cells were incubated for the indicated periods of time at 37°C in medium containing the following reagents: DMSO (Invitrogen, D12345), Torin-1 (TOCRIS, 4247/10), sodium arsenite solution (Sigma-Aldrich, 1062,771000), curaxin (Cayman, 19110), 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (DRB, Sigma-Aldrich, d1916). For starvation experiments, cells were washed three times in PBS (Gibco, 70011069) and incubated for 4–16 h at 37°C in Earle’s balanced salt solution (Gibco, 24010043).
Antibodies
The following antibodies were used in this study: anti-TFEB (Bethyl Laboratories, A303-673A), anti-TFEB (Cell Signaling Technology, 4240), anti-phospho S211 TFEB (YenZym Antibodies) [15], anti-TFE3 (Sigma HPA023881), anti-phospho S321 TFE3 (YenZym Antibodies) [18], anti-Flag (clone M2; Sigma-Aldrich, F1804), anti-LAMP1 from the Developmental Studies Hybridoma Bank deposited by August, J.T. (DSHB, 1D4B), anti-SSRP1 (clone 10D1; BioLegend, 609702), anti-PPARGC1A/PGC1a (Novus Biologicals, NBP1-04676), anti-MAFF (Proteintech, 12771–1AP), anti-MAFG (Genetex, GTX Inc., 114541), anti-GAPDH (Santa Cruz Biotechnology, sc-365062), anti-MYC/cMyc (Santa Cruz Biotechnology, sc-40), anti-SUPT16H (Cell Signaling Technology, 12191), anti-SSRP1 (Cell Signaling Technology, 13421), anti-phospho-RPS6KB/p70 S6 Kinase (Cell Signaling Technology, 9205), anti-RPS6KB/p70 S6 Kinase (Cell Signaling Technology, 2708), anti-phospho-EIF4EBP1/4E-BP1 (Cell Signaling Technology, 2855), anti-EIF4EBP1/4E-BP1 (Cell Signaling Technology, 9644), anti-YWHA/14-3-3 (Cell Signaling Technology, 8312), anti-CSNK2/CK2a (Cell Signaling Technology, 2656), anti- phospho-TP53/p53 (Ser392; Cell Signaling Technology, 9281), anti-HIF1A (Cell Signaling Technology, 36169), anti-HMOX1 (Cell Signaling Technology, 43966), anti-phospho-MAPK11/p38 (Cell Signaling Technology, 4511), anti-phospho-MAPKAPK2 (Cell Signaling Technology, 3041), anti-WDR59 (Cell Signaling Technology, 53385), anti-RRAGC/RagC (Cell Signaling Technology, 5466), anti-RHEB (Cell Signaling Technology, 13879), anti-LAMTOR1 (Cell Signaling Technology, 8975), anti-FLCN (Cell Signaling Technology, 3697), anti-TSC2 (Cell Signaling Technology, 4308), anti-UVRAG (Cell Signaling Technology, 13115), anti-KEAP1 (Cell Signaling Technology, 8047), anti-NFE2L2 (Cell Signaling Technology, 12721), HRP-conjugated anti-mouse (Cell Signaling Technology, 7076), HRP-conjugated anti-rabbit IgG (Cell Signaling Technology, 7074), Alexa Fluor 568-conjugated goat anti-mouse IgG (Invitrogen, A21090), Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, A-11008) Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen, A-11001).
Recombinant DNA plasmid
ARPE-19 cells were transfected using Cell Line Nucleofector Kit V (Lonza, VCA-1003) according to the manufacturer’s instructions. TFEB-Flag expression vectors were generated by cloning the full-length encoding sequence of human TFEB into p3× FLAG-cytomegalovirus with a triple FLAG tag fused to the C termini of TFEB [5]. Amino acid substitutions in TFEB were made using the QuikChange Lightning site-directed mutagenesis kit (Stratagene, 200522/200521) as previously described [5,6]. SSRP1-Flag expression vector was a kind gift from Dr. Vincenzo Costanzo (FIRC Institute of Molecular Oncology, Milan, Italy).
RNA interference (RNAi)
For siRNA knockdown, cells were transfected using Lipofectamine RNAiMAX transfection reagent (Invitrogen, 13778075) with ON-TARGETplus non-targeting pool siRNA duplexes or ON-TARGETplus smart pool siRNA duplexes targeted against SSRP1, SUPT16H, TFEB, TFE3 or NFE2L2 genes (Dharmacon-Thermo Scientific, L-011783-00-0005, L-009517-00-0005, L-009798-00-0005, L-009363-00-0005 and L-003755-00-0005 respectively). Transfected cells were analyzed 72 h after transfection.
Immunoprecipitation and immunoblotting
Cells were washed with cold PBS and lysed in lysis buffer containing 150 mM NaCl, 20 mM HEPES, pH 7.4, 5 mM EDTA, 10% glycerol, 1% Triton X-100 (Sigma-Aldrich, X100) with phosphatase inhibitor (Roche, 4906837001) and protease inhibitor (Roche, 11836170001). Whole cell lysates were homogenized and incubated on ice for 30 min and then centrifuged at 22,000 x g for 15 min at 4°C. For immunoprecipitation, the soluble fractions were incubated with 1 μg of antibody overnight at 4°C. The Antibody-protein complexes were incubated with protein G-Sepharose beads (GE Healthcare, GE17-0618-01) for 2 h at 4°C and collected, washed three times with lysis buffer and then proteins were mixed with NuPage 4X loading buffer (Life Technologies, NP0007). Samples were analyzed by SDS-PAGE 4–20% gradient gels (Invitrogen, XP04202BOX) and transferred to nitrocellulose membranes. Membranes were immunoblotted with the indicated antibodies. Horseradish peroxidase-conjugated anti-mouse, anti-rabbit IgG, or anti-rat IgG were used at a dilution of 1:5,000. Immunoblots were developed with Radiance Plus Chemiluminescent Substrate (Azure Biosystems, 10147–298) and exposed using a GE Healthcare Life Sciences Amersham Imager 600. Immunoblots were quantitated with densitometric analysis using ImageJ (NIH) and normalized to GAPDH as a loading control.
Subcellular fractionation
Cells were lysed with cytosolic extraction buffer (10 mM HEPES, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol) containing protease and phosphatase inhibitors and kept on ice for 10 min. After a brief vortexing, cells were centrifugated at 800 x g for 10 min at 4°C, and then supernatants were collected for the cytosolic fraction. Pellets were washed with cytosol extraction buffer and centrifugated at 800 x g for 10 min at 4°C and lysed with nuclear extraction buffer (5 mM HEPES, pH 7.4, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol) containing protease inhibitors. After incubation on ice for 30 min, the nuclear fraction was obtained by centrifugation at 22,000 x g for 30 min at 4°C.
Immunofluorescence confocal microscopy
Cells grown on glass coverslips were washed three times with PBS and fixed with 4% formaldehyde (Electron Microscopy Sciences, 15710) diluted in PBS for 15 min at room temperature. After fixation, cells were washed three times with PBS and permeabilized for 10 min in 0.2% Triton X-100 in PBS. Cells were then incubated with primary antibodies for 1 h at room temperature in IF buffer (PBS containing 10% fetal bovine serum and 0.1% [w:v] saponin (Sigma-Aldrich, S-4521). Cells were washed three times with PBS and incubated with secondary antibodies for 30 min at room temperature followed by an additional three times wash in PBS. Coverslips were mounted with ProLong Diamond Antifade Mountant reagent with DAPI (Invitrogen, P36966). Images were acquired with an LSM 510 Meta confocal microscope (Zeiss, Oberkochen, Germany) with 63x numerical aperture 1.4 oil immersion objective with a Zeiss AxioCam camera.
Rapid Immunoprecipitation Mass spectrometry of Endogenous proteins (RIME)
HeLa or HeLa-TFEB-Flag cells (6 x 107) were treated with NaAsO2 (250 µM; Sigma-Aldrich, 1062771000) for 2 h or incubated with EBSS for 4 h. Cells were then crosslinked by adding to the existing media 1/10 volume of Formaldehyde Solution (11% methanol free formaldehyde, 0.1 M NaCl, 1 mM EDTA, 50 mM HEPES, pH 7.9) for 8 min at room temperature. Crosslinking was then quenched by adding 1/20 volume of 2.5 M glycine for 5 min at room temperature. Crosslinked cells were then harvested and centrifuged at 800 x g for 10 min at 4°C. Cell pellets were resuspended and washed two times with cold PBS containing 0.5% Igepal CA-630 (Sigma-Aldrich, I8896). Cell pellets were resuspended and washed one more time with cold PBS containing 0.5% Igepal CA-630 and 1 mM PMSF. Washed cell pellets were snap-frozen on dry ice and processed for RIME [55,56] by Active Motif, Inc. The detailed RIME protocol is provided in Supplementary Materials.
Mass spectrometry
ARPE-19 cells infected with either adenovirus Null or adenovirus expressing TFEB-Flag were treated with NaAsO2 (250 µM) for 2 h at 37°C. Cell lysates were subjected to immunoprecipitation using an antibody against Flag (Sigma-Aldrich, F1804) as described in the “Immunoprecipitation and immunoblotting” section. Immunoprecipitated proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue R-250. Protein bands were excised and sequentially reduced with tris (2-carboxyethyl) phosphine hydrochloride and alkylated with chloroacetamide. Proteins were then digested with trypsin or chymotrypsin (Promega, V1061). The resulting peptide mixtures were analyzed with an Orbitrap Fusion Lumos equipped with a Dionex Ultimate 3000 nanoLC system (Thermo Fisher). Peptide IDs were assigned with Mascot V2.5 (Matrix Science) and manually validated using Scaffold 5 software (Proteome Software). All peptides were filtered out at 1% false discovery rate (FDR) and their relative abundances were compared based on the areas under curve (AUC) of their corresponding chromatographic peaks.
RNA-seq sample processing
HeLa-TFEB-Flag cells (1 x 106) were treated with NaAsO2 (50 µM) for 6 h and transferred to a 15 ml conical tube and then centrifuged at 800 x g for 5 min at 4°C. Cell pellets were resuspended with cold PBS and centrifuged again at 800 x g for 5 min. Washed cell pellets were snap-frozen on dry ice and then processed for RNA-seq assay by Active Motif, Inc. Total RNA was isolated from cells using the Qiagen RNeasy Mini Kit (Qiagen, 74104). For each sample, 0.5 ng of total RNA was then used in Illumina’s TruSeq Stranded mRNA Library kit (Cat# 20020594). Libraries were sequenced on Illumina NextSeq 500 as paired-end 42-nt reads. Sequence reads were analyzed with the STAR alignment – DESeq2 software pipeline.
RNA-Seq analysis
Read Mapping: The paired-end 42 bp sequencing reads (PE42) generated by Illumina sequencing (using NextSeq 500) were mapped to the genome using the STAR algorithm with default settings. Alignment information for each read was stored in the BAM format.
Fragment Assignment: The number of fragments overlapping predefined genomic features of interest (e.g. genes) were counted. Only read pairs that have both ends aligned were counted. Read pairs that have their two ends mapping to different chromosomes or mapping to same chromosome but on different strands are discarded. The gene annotations were obtained from Subread package. These annotations were originally from NCBI RefSeq database and then adapted by merging overlapping exons from the same gene to form a set of disjoint exons for each gene. Genes with the same Entrez gene identifiers were also merged into one gene.
Differential Analysis: After obtaining the gene table containing the fragment counts of genes, differential analyses were performed to identify statistically significant differential genes using DESeq2. The following lists the pre-processing steps before differential calling:
Data Normalization: DESeq2 expects un-normalized count matrix of sequencing fragments. The DESeq2 model internally corrects for library size using their median-of-ratios method. The gene table obtained from “Fragment Assignment” was used as input to perform the DESeq2ʹs differential test.
Filtering before multiple testing adjustment: After a differential test has been applied to each gene except the ones with zero counts, the p-value of each gene was calculated and adjusted to control the number of false positives among all discoveries at a proper level.
Differential Calling: Differential genes were detected by DESeq2 at 0.1 (or 10%) FDR (i.e. adjusted p-value).
Chip-seq
Sample preparation: HeLa-TFEB-Flag cells (1 x 107) were treated with NaAsO2 (50 µM) for 6 h and then crosslinked with 1/10 volume of freshly prepared Formaldehyde Solution (described in the “RIME” section) to the existing media of cells and agitated for 15 min at room temperature. Fixation was stopped by adding 1/20 volume Glycine to the existing media. Cells were washed by transferring to a 15 ml conical tube and centrifuged at 800 x g for 10 min at 4°C. Supernatants were removed and cells were resuspended with cold PBS containing 0.5% Igepal CA-630. Cell pellets were resuspended and washed one more time with cold PBS containing 0.5% Igepal CA-630 and 1 mM PMSF. Cells were centrifuged again to pellet and snap-frozen on dry ice and then processed for Chip-seq assay by Active Motif, Inc.
Chromatin Immunoprecipitation: Chromatin was isolated by adding lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris-HCl, pH 8.1 containing protease inhibitors), followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp with Active Motif’s EpiShear probe sonicator (EpiShear, 53051). Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by SPRI beads clean up (Beckman Coulter) and quantification by Clariostar (BMG Labtech). Extrapolation to the original chromatin volume allowed determination of the total chromatin yield. An aliquot of chromatin (30 ug) was precleared with protein G agarose beads (Invitrogen, 20398). Genomic DNA regions of interest were isolated using 5 ug of monoclonal antibody against Flag. Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation.
ChIP Sequencing (Illumina): Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. Steps were performed on an automated system (Apollo 342, Wafergen Biosystems/Takara). After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina’s NextSeq 500 (75 nt reads, single end). Reads were aligned to the human genome using the BWA algorithm (default settings). Duplicate reads were removed, and only uniquely mapped reads (mapping quality ≥ 25) were used for further analysis. Alignments were extended in silico at their 3’-ends to a length of 200 bp, which is the average genomic fragment length in the size-selected library and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peak locations were determined using the MACS algorithm (v2.1.0) with a cutoff of p-value = 1e-7. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations, and gene annotations.
Software: bcl2fastq2 (v2.20) was used for processing of Illumina base-call data and demultiplexing, Samtools (v0.1.19) was used for processing of BAM files, BEDtools (v2.25.0) was used for processing of BED files and wigToBigWig (v4) was used for the generation of bigWIG files.
Intracellular ROS analysis
siRNA transfected HeLa-TFEB-Flag cells were washed, trypsinized and collected. Cells were diluted with H2DCFDA (20 µM; Invitrogen, C400) for 30 min and then incubated with NaAsO2 (50 µM) for 6 h. After incubation, cells were washed with PBS and flow cytometry was performed by using BD LSR Fortessa.
Relative quantitative real-time PCR
RNA was isolated from samples with the PureLink RNA Mini Kit (Invitrogen, 12183018A) and reverse transcribed using SuperScript III First-Strand Synthesis SuperMix kit (Invitrogen, 11752050). Relative quantitative real-time PCR reactions were performed at least triplicate with SYBR Green PCR Master Mix (Applied Biosystems, A25742) using a QuantStudio 12 K Flex Real-Time PCR system (Applied Biosystems, Life Technologies). Expression levels were displayed relative to control conditions and normalized using Actb (mouse genes) or ACTB (human genes) using the ΔΔCT method. Human primers for PPARGC1A, HIF1A, MAFF, MAFG, HMOX1, ADNP2, FOXO3, PYROXD1, TXNRD1, LAMP1, MCOLN1, ATP6V1C1, RRAGC, WIPI1, RPS8, RPS28, RPL24, SSRP1, SUPT16H, 18SRNA, ACTIN and mouse primers for Adnp2, Maff, Atp6v0d1, Atp6v1c1, Ctsd, Hexa, Clcn7, Mcoln1, Lamp1, Actin were purchased from Qiagen (QuantiTect primer assays). Additional primers used in this study are provided in Table S7.
DRB-transcription elongation assay
HeLa-TFEB-Flag cells incubated with curaxin (1 µM) together with NaAsO2 (50 µM) for 6 h or siRNA transfected HeLa-TFEB-Flag cells incubated with EBSS for 16 h or NaAsO2 (50 µM) for 6 h were treated with DRB for 3 h and total RNA were immediately extracted 0 min, 4 min, and 8 min after DRB removal. RNA was isolated from samples with the miRNeasy kit (Qiagen, 217004) and reverse transcribed using M-MLV reverse transcriptase (Invitrogen, 28025013) and random hexamers (Invitrogen, N8080127). Relative quantitative real-time PCR reactions were performed at least triplicate with SYBR Green PCR Master Mix (Applied Biosystems, A25742) using a QuantStudio 12 K Flex Real-Time PCR system (Applied Biosystems, Life Technologies). Pre-mRNA levels were displayed relative to control conditions and normalized using RNA18S (QuantiTect primer assays) using the ΔΔCT method. Intronic primers for HIF1A, MAFF, MCOLN1 and LAMP1 used in this study are provided in Table S7.
Statistical analysis
Obtained data were processed in Excel (Microsoft Corporation) and Prism (GraphPad Software) to generate bar charts and perform statistical analyses. Student’s t‐test or two‐way ANOVA and pairwise post‐tests were run for each dependent variable, as specified in each figure legend. All data are presented as mean ± SD. P ≤ 0.05 was considered statistically significant (*) and P ≤ 0.001 extremely significant (***). P > 0.05 was considered not significant (ns).
Supplementary Material
Funding Statement
This work was supported by the NHLBI Division of Intramural Research [ZIA HL006151-10].
Ackowledgments
We thank Professor Vincenzo Costanzo (FIRC Institute of Molecular Oncology, Milan, Italy) for providing the SSRP1-Flag plasmid. We also thank Dr. Yong Chen and Dr. Marjan Gucek, NHLBI Proteomics Core Facility, for help to perform and interpret the mass spectrometry analysis. This project was supported by the Intramural Research Program of the NIH, National Heart, Lung, and Blood Institute (NHLBI).
Data availability
The RNA-sequencing and ChIP-sequencing data from this publication have been deposited in the NCBI GEO database and together assigned the identifier GSE180324 (RNA-sequencing: GSE180323; ChIP-sequencing: GSE180322).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Raben N, Puertollano R.. TFEB and TFE3: linking Lysosomes to Cellular Adaptation to Stress. Annu Rev Cell Dev Biol. 2016. Oct 6;32(1):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sha Y, Rao L, Settembre C, et al. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017. Sep 1;36(17):2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Martina JA, Guerrero-Gomez D, Gomez-Orte E, et al. A conserved cysteine-based redox mechanism sustains TFEB/HLH-30 activity under persistent stress. EMBO J. 2021. Feb 1;40(3):e105793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Napolitano G, Esposito A, Choi H, et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018. Aug 17;9(1):3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013. Feb 18;200(4):475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012. Jun;8(6):903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012. Jun 12;5(228):ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012. Mar 7;31(5):1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014. Jan 21;7(309):ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li Y, Xu M, Ding X, et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol. 2016. Oct;18(10):1065–1077. [DOI] [PubMed] [Google Scholar]
- [11].Palmieri M, Pal R, Nelvagal HR, et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 2017. Feb 6;8(1):14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Paquette M, El-Houjeiri L, CZ L, et al. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy. 2021;18:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018. Jun 1;37(11). DOI: 10.15252/embj.201798804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015. Mar;17(3):288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martina JA, Puertollano R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J Biol Chem. 2018. Aug 10;293(32):12525–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011. Jun 17;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pastore N, Brady OA, Diab HI, et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy. 2016. Aug 2;12(8):1240–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martina JA, Diab HI, Brady OA, et al. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016. Mar 1;35(5):479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gray MA, Choy CH, Dayam RM, et al. Phagocytosis Enhances Lysosomal and Bactericidal Properties by Activating the Transcription Factor TFEB. Curr Biol. 2016. Aug 8;26(15):1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Visvikis O, Ihuegbu N, Labed SA, et al. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity. 2014. Jun 19;40(6):896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Campbell GR, Rawat P, Bruckman RS, et al. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015. Jun;11(6):e1005018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].El-Houjeiri L, Possik E, Vijayaraghavan T, et al. The Transcription Factors TFEB and TFE3 Link the FLCN-AMPK Signaling Axis to Innate Immune Response and Pathogen Resistance. Cell Rep. 2019. Mar 26;26(13):3613–3628 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nezich CL, Wang C, Fogel AI, et al. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015. Aug 3;210(3):435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang X, Yu L, Xu H. Lysosome calcium in ROS regulation of autophagy. Autophagy. 2016. Oct 2;12(10):1954–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mansueto G, Armani A, Viscomi C, et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017. Jan 10;25(1):182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Brady OA, Jeong E, Martina JA, et al. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife. 2018. Dec 6;7. DOI: 10.7554/eLife.40856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Slade L, Biswas D, Ihionu F, et al. A lysosome independent role for TFEB in activating DNA repair and inhibiting apoptosis in breast cancer cells. Biochem J. 2020. Jan 17;477(1):137–160. [DOI] [PubMed] [Google Scholar]
- [28].Belotserkovskaya R, Oh S, Bondarenko VA, et al. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003. Aug 22;301(5636):1090–1093. [DOI] [PubMed] [Google Scholar]
- [29].Keller DM, Zeng X, Wang Y, et al. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell. 2001. Feb;7(2):283–292. [DOI] [PubMed] [Google Scholar]
- [30].Dejmek J, Iglehart JD, Lazaro JB. DNA-dependent protein kinase (DNA-PK)-dependent cisplatin-induced loss of nucleolar facilitator of chromatin transcription (FACT) and regulation of cisplatin sensitivity by DNA-PK and FACT. Mol Cancer Res. 2009. Apr;7(4):581–591. [DOI] [PubMed] [Google Scholar]
- [31].Falbo L, Raspelli E, Romeo F, et al. SSRP1-mediated histone H1 eviction promotes replication origin assembly and accelerated development. Nat Commun. 2020. Mar 12;11(1):1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Garcia H, Miecznikowski JC, Safina A, et al. Facilitates chromatin transcription complex is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell Rep. 2013. Jul 11;4(1):159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jin MZ, Xia BR, Xu Y, et al. Curaxin CBL0137 Exerts Anticancer Activity via Diverse Mechanisms. Front Oncol. 2018;8:598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009. Nov;16(11):1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li D, Shao R, Wang N, et al. Sulforaphane Activates a lysosome-dependent transcriptional program to mitigate oxidative stress. Autophagy. 2021. Apr;17(4):872–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin XX, Sen I, Janssens GE, et al. DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat Commun. 2018. Oct 23;9(1):4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013. Jun;15(6):647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen P, Dong L, Hu M, et al. Functions of FACT in Breaking the Nucleosome and Maintaining Its Integrity at the Single-Nucleosome Level. Mol Cell. 2018. Jul 19;71(2):284–293 e4. [DOI] [PubMed] [Google Scholar]
- [39].Stuwe T, Hothorn M, Lejeune E, et al. The FACT Spt16 “peptidase” domain is a histone H3-H4 binding module. Proc Natl Acad Sci U S A. 2008. Jul 1;105(26):8884–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tsunaka Y, Fujiwara Y, Oyama T, et al. Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev. 2016. Mar 15;30(6):673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gurova K, Chang HW, Valieva ME, et al. Structure and function of the histone chaperone FACT - Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech. 2018. Jul 25;1861(9):892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gasparian AV, Burkhart CA, Purmal AA, et al. Curaxins: anticancer Compounds That Simultaneously Suppress NF-κB and Activate p53 by Targeting FACT. Sci Transl Med. 2011. Aug 10;3(95):95ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Koman IE, Commane M, Paszkiewicz G, et al. Targeting FACT Complex Suppresses Mammary Tumorigenesis in Her2/neu Transgenic Mice. Cancer Prev Res (Phila). 2012. Aug;5(8):1025–1035. [DOI] [PubMed] [Google Scholar]
- [44].Carter DR, Murray J, Cheung BB, et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci Transl Med. 2015. Nov 4;7(312):312ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shin HJ, Kim H, Oh S, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016. Jun 23;534(7608):553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].O’Donnell AF, Brewster NK, Kurniawan J, et al. Domain organization of the yeast histone chaperone FACT: the conserved N-terminal domain of FACT subunit Spt16 mediates recovery from replication stress. Nucleic Acids Res. 2004;32(19):5894–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rowley A, Singer RA, Johnston GC. CDC68, a yeast gene that affects regulation of cell proliferation and transcription, encodes a protein with a highly acidic carboxyl terminus. Mol Cell Biol. 1991. Nov;11(11):5718–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schwabish MA, Struhl K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2004. Dec;24(23):10111–10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shakya A, Callister C, Goren A, et al. Pluripotency transcription factor Oct4 mediates stepwise nucleosome demethylation and depletion. Mol Cell Biol. 2015. Mar;35(6):1014–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ransom M, Williams SK, Dechassa ML, et al. FACT and the proteasome promote promoter chromatin disassembly and transcriptional initiation. J Biol Chem. 2009. Aug 28;284(35):23461–23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yu Y, Yarrington RM, Stillman DJ. FACT and Ash1 promote long-range and bidirectional nucleosome eviction at the HO promoter. Nucleic Acids Res. 2020. Nov 4;48(19):10877–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shen J, Chen M, Lee D, et al. Histone chaperone FACT complex mediates oxidative stress response to promote liver cancer progression. Gut. 2020. Feb;69(2):329–342. [DOI] [PubMed] [Google Scholar]
- [53].Attwood K, Fleyshman D, Prendergast L, et al. Prognostic value of histone chaperone FACT subunits expression in breast cancer. Breast Cancer (Dove Med Press). 2017;9:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Perera RM, Di Malta C, Ballabio A. MiT/TFE Family of Transcription Factors, Lysosomes, and Cancer. Annu Rev Cancer Biol. 2019. Mar;3(1):203–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mohammed H, D’Santos C, Serandour AA, et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013. Feb 21;3(2):342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mohammed H, Taylor C, Brown GD, et al. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc. 2016. Feb;11(2):316–326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing and ChIP-sequencing data from this publication have been deposited in the NCBI GEO database and together assigned the identifier GSE180324 (RNA-sequencing: GSE180323; ChIP-sequencing: GSE180322).