

Abstract
Cardiac stromal cells (CSCs) are the main players in fibrosis. Dysmetabolic conditions (metabolic syndrome—MetS, and type 2 diabetes mellitus—DM2) are strong pathogenetic contributors to cardiac fibrosis. Moreover, modulation of the oxidative state (OxSt) and autophagy is a fundamental function affecting the fibrotic commitment of CSCs, that are adversely modulated in MetS/DM2. We aimed to characterize CSCs from dysmetabolic patients, and to obtain a beneficial phenotypic setback from such fibrotic commitment by modulation of OxSt and autophagy. CSCs were isolated from 38 patients, stratified as MetS, DM2, or controls. Pharmacological modulation of OxSt and autophagy was obtained by treatment with trehalose and NOX4/NOX5 inhibitors (TREiNOX). Flow‐cytometry and real‐time quantitative polymerase chain reaction (RT‐qPCR) analyses showed significantly increased expression of myofibroblasts markers in MetS‐CSCs at baseline (GATA4, ACTA2, THY1/CD90) and after starvation (COL1A1, COL3A1). MetS‐ and DM2‐CSCs displayed a paracrine profile distinct from control cells, as evidenced by screening of 30 secreted cytokines, with a significant reduction in vascular endothelial growth factor (VEGF) and endoglin confirmed by enzyme‐linked immunoassay (ELISA). DM2‐CSCs showed significantly reduced support for endothelial cells in angiogenic assays, and significantly increased H2O2 release and NOX4/5 expression levels. Autophagy impairment after starvation (reduced ATG7 and LC3‐II proteins) was also detectable in DM2‐CSCs. TREiNOX treatment significantly reduced ACTA2, COL1A1, COL3A1, and NOX4 expression in both DM2‐ and MetS‐CSCs, as well as GATA4 and THY1/CD90 in DM2, all versus control cells. Moreover, TREiNOX significantly increased VEGF release by DM2‐CSCs, and VEGF and endoglin release by both MetS‐ and DM2‐CSCs, also recovering the angiogenic support to endothelial cells by DM2‐CSCs. In conclusion, DM2 and MetS worsen microenvironmental conditioning by CSCs. Appropriate modulation of autophagy and OxSt in human CSCs appears to restore these features, mostly in DM2‐CSCs, suggesting a novel strategy against cardiac fibrosis in dysmetabolic patients. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: cardiac stromal cells, cardiac fibroblasts, autophagy, oxidative stress, cardiac fibrosis, metabolic syndrome, type 2 diabetes mellitus, anti‐fibrotic therapy
Introduction
The cardiac stroma is now described as a continuous spectrum of functional phenotypes based on microenvironmental cues [1, 2]. Cardiac homeostasis and architecture are regulated, among others, through the synthesis of extracellular matrix (ECM) and the release of paracrine factors by stromal interstitial cells. Several pathological conditions, such as cardiac mechanical overload, metabolic alterations, and ischemia, are associated with the activation of resident cardiac stromal cells (CSCs), and the consequent enrichment in profibrotic cells (namely myofibroblasts), which contribute to altered ECM deposition, proinflammatory signaling, and overall myocardial remodeling [3, 4]. On the other hand, signals from a pathological cardiac microenvironment can enhance the profibrotic phenotype of stromal cells, including differentiated myofibroblasts and primitive inactivated stromal cells [5]. Reducing proinflammatory and profibrotic signaling in favor of beneficial conditioning is critical to allow effective reparative responses, thus limiting excessive collagen deposition, extensive fibrosis, and cell death [6].
Altered cardiac muscle homeostasis and stress response capacity have been linked to pathological metabolic assets. Specific molecular pathways, including fundamental functions such as the oxidative state (OxSt) and the autophagic machinery [7, 8, 9, 10], are impaired in cardiomyocytes and vascular cells by dysmetabolic conditions. Moreover, type 2 diabetes mellitus (DM2) is known to negatively correlate with the amount and function of resident CSCs with a proreparative potential [11]. Detrimental effects have also been observed in CSCs isolated from patients with hyperglycemia, with a decrease in cell abundance, impaired angiogenic and chemotactic properties, together with a reduced repair capacity observed in vivo [12]. Diabetes, however, is nowadays considered the final step of a chronic pathogenetic process starting with the subclinical alteration of fasting blood levels associated with insulin resistance, obesity, dyslipidemia, and hypertension [13, 14, 15], namely the metabolic syndrome (MetS) [16]. How the above‐mentioned altered phenotypic features may be anticipated in prediabetic conditions, such as MetS, and to what extent they may be reversible is still unknown. Several pathways are known to modulate the behavior and functional properties of CSCs, including hyperglycemia [17, 18, 19, 20, 21, 22, 23, 24, 25]. However, the result of chronic exposure of CSCs in situ to a cardiac microenvironment conditioned by MetS has never been investigated.
Recently, autophagy activation has been shown to favor the cardioprotective and proangiogenic function of murine CSCs under hyperglycemic stress [25]. Indeed, autophagy is a well‐known mechanism for molecules/organelles recycling and stress resistance, known to be significantly affected by dysmetabolic conditions [26, 27]. The OxSt is widely dysregulated in diabetes as well [7, 10]. It has been previously shown that CSCs with an altered phenotype due to detrimental microenvironmental conditioning during cardiac remodeling can modulate their OxSt [28]. Furthermore, reactive oxygen species (ROS) production is functional to fibrosis enhancement in response to hyperglycemia [29].
With these premises, we sought to examine how the complex metabolic imbalance associated with MetS [16, 30] could affect the phenotype, response to stress, and functional properties of human primitive CSCs, compared to cells isolated from DM2 or control nondysmetabolic patients. We also analyzed whether these functional shifts may be reversible, at least in part, by modulation of the fundamental biological functions of OxSt and autophagy, known to be impaired in dysmetabolic conditions.
Materials and methods
Patient enrolment and stratification
This study was approved by the competent Ethics Committee of “Umberto I” General Hospital—Sapienza University of Rome (protocol number 2154/15) and was performed in conformity with the Declaration of Helsinki. All patients provided written informed consent. A total of 38 patients undergoing elective cardiac surgery were included in our analysis, stratified based on the diagnosis of DM2, or impaired glucose tolerance and MetS. Patient stratification for MetS was performed retrospectively according to a modified version of the National Cholesterol Education Program—Adult Treatment Panel III (NCEP‐ATPIII) criteria, presented in Table 1 [31, 32, 33]. Specifically, the body mass index (BMI) was used instead of the abdominal circumference [31], and the presence of dyslipidemia (defined by cholesterol levels and/or treatment for dyslipidemia) were used instead of the triglyceride measurement, as this parameter was missing in the available clinical records. None of the MetS patients were under treatment with hypoglycemic drugs. Patients negative for all dyslipidemia markers and positive for two other criteria were excluded. Basic anthropometric and medical features of donor patients are listed in Tables 2 and 3. Considering the previously described differences in yield and phenotype due to pharmacological treatments [19], in order to exclude any known bias patients were included only if under β‐blocker treatment, since at least 30 days before surgery.
Table 1.
Modified National Cholesterol Education Program—Adult Treatment Panel III (NCEP‐ATPIII) criteria for the diagnosis of metabolic syndrome (MetS) and patient stratification.
| Criterion | Males | Females |
|---|---|---|
| Body mass index | ≥28.8 | ≥26.7 |
| Fasting blood glucose | ≥100 mg dl−1 | ≥100 mg dl−1 |
|
Dyslipidemia, reduced HDL‐C |
HDL‐C ≤40 mg dl−1 Or in treatment with statins Or classified as dyslipidemic by clinician |
HDL‐C ≤50 mg dl−1 Or in treatment with statins Or classified as dyslipidemic by clinician |
| Elevated blood pressure |
Blood pressure ≥130/85 mmHg Or in treatment Or classified as hypertensive by clinician |
Blood pressure ≥130/85 mmHg Or in treatment Or classified as hypertensive by clinician |
HDL‐C, high‐density lipoprotein‐cholesterol.
Table 2.
Demographic and clinical characteristics of the population of biopsy donors. Comparative analysis of demographic and clinical characteristics of patients, according to the diagnosis of metabolic syndrome (MetS) or type 2 diabetes mellitus (DM2).
| Feature* | Control patients, N = 13 | MetS patients, N = 13 | DM2 patients, N = 12 | Significance † |
|---|---|---|---|---|
| Age (years) | 75 (64; 78) | 69 (60; 75) | 65 (59; 74) | 0.313 |
| Gender, male (%) | 9 (69%) | 9 (69%) | 6 (50%) | 0.521 |
| BMI (kg/m2) | 26.6 (22.3; 27.9) | 27.5 (26.8; 28.0) | 27.4 (25.4; 33.8) | 0.493 |
| BSA (m2) | 1.9 (1.7; 2.0) | 1.9 (1.8; 2.0) | 1.8 (1.8; 2.0) | 0.774 |
| Creatinine (mg dl−1) | 1.1 (0.95; 1.2) | 1.1 (0.8; 1.36) | 0.9 (0.86; 1.0) | 0.154 |
| Systolic BP (mmHg) | 120 (110; 127) | 130 (110; 140) | 130 (130; 140) | 0.040 |
| Diastolic BP (mmHg) | 75 (62; 80) | 80 (70; 90) | 80 (70; 90) | 0.504 |
| Heart rate (bpm) | 71 (60; 91) | 70 (58; 75) | 80 (70; 85) | 0.306 |
| Active smoker, N (%) | 5 (42%) | 4 (31%) | 4 (33%) | 0.310 |
| Hypertension, N (%) | 12 (92.3%) | 11 (84.6%) | 12 (100%) | 0.362 |
| Atrial fibrillation, N (%) | 2 (15.4%) | 5 (38.5%) | 0 (0%) | 0.044 |
| Previous myocardial infarction, N (%) | 2 (15%) | 3 (23%) | 4 (33%) | 0.527 |
| Recent (<30 days) myocardial infarction, N (%) | 0 (0%) | 0 (0%) | 1 (8.3%) | 0.329 |
BMI, body mass index; BSA, body surface area; BP, blood pressure; BPM, beats per minute.
Reported as median (1st; 3rd quartile) for continuous variables and count/total (percentage) for categorical variables.
Computed with unpaired Mann–Whitney U‐test for continuous variables and Fisher's exact test for categorical variables.
Table 3.
Surgical characteristics of the study population of biopsy donors. Comparative analysis of surgical characteristics of patients, according to the diagnosis of metabolic syndrome (MetS) or type 2 diabetes mellitus (DM2).
| Feature* | Controls, N = 13 | MetS patients, N = 13 | DM2 patients, N = 12 | Significance † |
|---|---|---|---|---|
| STS mortality risk score | 1.3 (0.8; 2.6) | 1.3 (0.5; 4.2) | 1.6 (1.0; 4.7) | 0.400 |
| ACEF score | 1.3 (1.2; 1.9) | 1.4 (1.2; 2.3) | 1.7 (1.2; 1.8) | 0.950 |
| Euroscore II | 2.3 (1.6; 10) | 2.3 (1.5; 9.8) | 2.9 (1.6; 4.5) | 0.466 |
| Surgical indication | 0.456 | |||
| Coronary bypass surgery, N (%) | 7 (54%) | 8 (61.5%) | 8 (66.7%) | |
| Heart valve surgery, N (%) | 4 (31%) | 4 (31%) | 1 (8.3%) | |
| Coronary bypass + heart valve surgery, N (%) | 1 (7.7%) | 1 (7.7%) | 3 (25%) | |
| Other surgery, N (%) | 1 (7.7%) | 0 (0%) | 0 (0%) | |
| CPB time (min) | 161 (113; 198) | 106 (91; 154) | 158 (119; 196) | 0.140 |
| Cross clamp time (min) | 98 (82; 143) | 84 (66; 123) | 127 (90; 160) | 0.203 |
ACEF, age, creatinine, ejection fraction; CBP, cardiopulmonary bypass.; STS, society of thoracic surgery.
Reported as median (1st; 3rd quartile) for continuous variables and count/total (percentage) for categorical variables.
Computed with unpaired Mann–Whitney U‐test for continuous variables and Fisher's exact test for categorical variables.
Cell cultures and treatments
Surgical biopsies of left atrial appendages were collected and cultured as an explant culture. CSCs were isolated as previously described [19, 34]. In brief, explant outgrowth cells were collected 4 weeks after tissue culture establishment with mild digestion performing sequential washes with Ca2+‐Mg2+ free phosphate‐buffered saline (PBS), 0.48 mm Versene (Thermo Fisher Scientific, Waltham, MA, USA) for 3 min, and 0.05% trypsin–EDTA (Lonza, Basel, Switzerland) for 5 min at room temperature under visual control. Harvests were made weekly up to three times. At each harvest, cells were seeded onto poly‐ D‐lysine (BD‐Biosciences, Heidelberg, Germany)‐coated wells (9,000 cells/cm2). Spheroid cultures, to select primitive inactivated CSCs, were established. The spheroids obtained were collected and expanded on fibronectin (BD‐Biosciences)‐coated surfaces. Since the number of cells obtained from each biopsy was inevitably variable, depending on the biopsy size, and did not allow carrying out all experiments on all CSC lines, samples were randomly allocated in the different analysis performed. To calculate cell yield, the total number of collected EDCs per each biopsy was normalized to the milligrams of tissue plated.
For all treatments, cells were plated at a density of 16,000 cells/cm2. For the starvation treatment, cells were plated in Dulbecco's modified Eagle medium (DMEM) 1 g l−1 glucose (Sigma‐Aldrich, St Louis, MO, USA), 1% L‐glutamine, 1% penicillin–streptomycin stock solution (Sigma‐Aldrich), 20% fetal bovine serum (FBS) at the time of seeding; after 24 h the medium was removed, cells were washed with PBS, and FBS concentration was reduced to 0.1%. For Bafilomycin‐A1 (Sigma‐Aldrich) treatment, the compound was added at a final concentration of 100 nm in the last 2 h of the starvation period. For the autophagy‐enhancing and NADPH‐oxidase (NOX)‐inhibiting treatment, cells were plated in DMEM 1 g l−1 glucose (Sigma‐Aldrich), 1% L‐glutamine, 1% penicillin–streptomycin stock solution, 20% FBS, supplemented with 50 mm trehalose (Sigma‐Aldrich). After 24 h the plates were washed and the medium was replaced with DMEM 1 g l−1 glucose, 1% L‐glutamine, 1% penicillin–streptomycin stock solution, 0.1% FBS, supplemented with 20 μm NOX4 inhibitor (GKT137831, Cayman Chemical, Ann Arbor, MI, USA) and 50 μm NOX5 inhibitor (KN‐93, Cayman Chemical).
Flow cytometry analyses
The percentage of cells expressing CD90 was assessed using flow cytometry on CSC cultures maintained in 20% FBS. CSCs from the first explant harvest and at passage 1 were used for all donors. Semiconfluent cells were harvested using trypsin–EDTA and stained with CD90‐FITC (Dianova, Geneva, Switzerland) or CD90‐PerCP‐Cy 5.5 (Biolegend, San Diego, CA, USA) using 300 ng antibody in 100 μl staining buffer (PBS with 2% FBS) per sample. For apoptosis detection, CSCs were stained with Annexin V/7‐AAD (BD‐Bioscience), according to the manufacturer's guidelines for adherent cells. All acquisitions were performed using a BD FACS‐Aria II equipped with DIVA software (BD‐Biosciences), and data were analyzed with FlowJo software (FlowJo Inc., Ashland, OR, USA).
RNA extraction and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using miRNeasy Micro Kits (Qiagen, Hilden, Germany) and quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific). For gene expression analyses, cDNA was synthesized from 0.5 μg RNA using a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Merelbeke, Belgium). Real‐time qPCR was performed to assess gene expression using Power SYBR Green PCR Master Mix (Life Technologies) and standard thermocycling conditions, according to the manufacturer's protocol. The relative ratio versus reference gene was calculated using the comparative Ct method (2−ΔCt) and fold‐change versus control samples was calculated if needed. The set of genes analyzed, and the sequences of primers, are listed in supplementary material, Table S1. Hypoxanthine Phosphoribosyltransferase 1 (HPRT1) was selected as the reference gene using the Bestkeeper spreadsheet macro (freely available at: www.gene-quantification.de). The PCR data (ΔCt value) were analyzed using R‐studio software (The R Foundation, Boston, MA, USA) and the pheatmap package to generate the heatmap showing Euclidean distance (hclust package embedded in pheatmap package in R).
Western blotting
Protein extracts were made using Laemmli 2× buffer with 5% β‐mercaptoethanol, and stored at −80 °C until analysis. Protein lysates were boiled at 100 °C for 5 min, loaded on a sodium dodecyl sulfate 15% polyacrylamide gel for electrophoresis (SDS‐PAGE), then transferred to PVDF membranes (Sigma‐Aldrich). Membranes were blocked with 3% BSA for 1 h at room temperature, and then incubated with primary antibodies against β‐actin (ACT‐β; Cell Signaling Technology, Danvers, MA, USA), and microtubule‐associated proteins 1A/1B light chain 3B—LC3‐II (MBL International, Woburn, MA, USA) at 4 °C overnight with gentle agitation. Membranes were washed with TBS‐0.01% Tween and incubated with appropriate horseradish peroxidase (HRP)‐conjugated secondary antibodies (Cell Signaling Technology) for 1 h at room temperature with gentle shaking. Chemiluminescent detection was performed after incubation with Clarity western ECL substrate (Bio‐Rad, Hercules, CA, USA), following the manufacturer's instructions using ChemiDoc XRS+ Imager (Bio‐Rad), and analyzed using ImageLab software (Bio‐Rad). Each band density was adjusted for background signal, and LC3‐II band OD was normalized to ACT‐β, as the loading control.
Conditioned medium collection and screening analysis
Cell culture conditioned medium (CM) were collected at the end of the starvation period (24 h following serum concentration decrease to 0.1%) with constant cell number/volume ratios among samples. CMs were centrifuged at 2,000× rcf for 5 min and then stored at −80 °C until analysis. CMs were analyzed using Proteome Profiler Human XL Cytokine Arrays (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. In brief, cell culture supernatants were incubated on blocked array membranes (1 ml of culture medium per membrane) and incubated overnight at 4 °C. Membranes were washed, incubated with biotin‐conjugated antibody at room temperature for 1 h, washed, and incubated for a further 30 min with HRP‐conjugated streptavidin. All incubation steps were performed under agitation on an orbital shaker. Spot detection was performed using the detection buffer provided in the array kit. Images were collected using ChemiDocXRS+ Imager (Bio‐Rad) and densitometric analysis was performed using Image Lab software (Bio‐Rad). Data are presented as optical density (OD) values normalized to assay internal positive control. The log2 OD value was used to generate the heatmap using the R pheatmap package.
Quantification of H2O2 and NO
Assays on conditioned media were performed as previously described [35]. In brief, H2O2 and NO were evaluated using a Colorimetric Detection Kit (Arbor Assays, Ann Arbor, MI, USA) and concentrations expressed in μm. Intraassay and interassay coefficients of variation were 2.1 and 3.7%, respectively. Assays were performed according to the manufacturers' instructions.
Enzyme‐linked immunoassay (ELISA)
The concentrations of specific cytokines were evaluated using ELISA for vascular endothelial growth factor (VEGF), soluble Endoglin, and monocyte chemotactic protein 3—MCP‐3 (RayBiotech, Peachtree Corners, GA, USA). Culture medium and standard solutions (created with a range specific for each tested cytokine) were incubated overnight in antibody precoated wells at 4 °C. At the end of the incubation time each well was washed, incubated with the specific biotinylated antibody, and incubated for 1 h at room temperature with gentle shaking. After removing the antibody solution, each well was washed, the streptavidin solution was added, and incubated for 45 min at room temperature with gentle shaking. Finally, wells were washed, the TMB One‐Step Substrate Reagent was added to each well, and they were incubated for 30 min in the dark. Stop solution was added, and the absorbance at 450 nm was recorded immediately using a Varioskan Lux multimode microplate reader (Thermo Fisher Scientific). Data were analyzed using the Skanit software (Thermo Fisher Scientific).
HUVEC angiogenic assay
The angiogenesis assay was performed as described previously [28, 36]. In brief, human umbilical vein endothelial cells (HUVECs) were cultured for 18 h on Matrigel‐coated 96‐well plates (Growth Factor Reduced Matrigel Matrix Phenol Red Free, BD‐Biosciences) at a density of 2.5 × 104 cells/well in the presence of the CSC CM collected at the end of serum starvation. Nonconditioned starvation medium (DMEM, 1 g l−1 glucose, 0.1% FBS) and endothelial growth media (EGM, Lonza) were used as negative and positive controls, respectively. Since endothelial cells are sensitive to NOX4‐inhibition [37], data with CM collected from TREiNOX treatments were analyzed separately from that of medium from untreated cells, and plain EGM supplemented with TREiNOX was used as the experimental control. Assay quantification was performed using the Angiogenesis Analyzer Plugin of the ImageJ Software (NIH, Bethesda, MD, USA) on randomly captured images with a 4×objective on a Nikon Eclipse TI inverted microscope (Nikon, Tokyo, Japan).
Statistical analyses
For clinical record analyses, variables were reported as median (1st; 3rd quartile) for continuous variables, and count/total (percentage) for categorical variables. Differences were computed with the unpaired Mann–Whitney U test for continuous variables, and Fisher's exact test for categorical variables using SPSS v. 25 (IBM, Armonk, NY, USA). Significance was set at <0.05. For experimental data, results are presented as mean ± SEM, unless specified. For in vitro treatment results, interpatient variability was controlled for by normalizing experimental results versus each patient's internal reference, unless specified, and averaging fold‐change modulations among patients. Normality of data was assessed and significance of differences among groups was determined by one‐way or two‐way analysis of variance (ANOVA), with Fisher LSD post hoc test, as appropriate. P < 0.05 was considered significant.
Results
Resident human CSCs were isolated through primary explant cultures established from human atrial appendage tissue and selected for an inactivated primitive phenotype by spontaneous spheroid growth, as described previously [34, 38]. Donor patients affected by ischemic cardiomyopathy and undergoing elective cardiac surgery were stratified based on the diagnosis of DM2, MetS, or neither of the two, and these latter were considered the control (CTR). DM2 diagnosis was directly mentioned in the clinical records. MetS diagnosis was either directly mentioned in the clinical records, or retrospectively established based on modified NCEP‐ATPIII criteria [31], summarized in Table 1. Multiple anthropometric and clinical parameters of the donors were analyzed for possible correlations with the biological features of isolated CSCs, such as cellular yield of explant cultures, successful spheroid growth, and immunophenotypic profile of the cell culture obtained. The overall descriptive analysis (demographic, clinical, surgical characteristics) of the population enrolled is presented in Tables 2 and 3. We observed few differences between groups. Some could be expected based on the stratification and diagnostic criteria, such as lower blood pressure in controls compared to dysmetabolic patients. Others appeared to be random, as is likely in a small cohort such as this one. Importantly, groups were homogeneous concerning different risk scores and surgical indications (Table 3). No statistically significant correlations could be observed between clinical parameters and CSC explant culture yield based on MetS or DM2 diagnosis, with the overall cell yield from primary cultures being comparable among the three groups (Figure 1A), as well as the efficiency of the spheroid‐forming assay (p = 0.60 by Chi‐squared test) (Figure 1B).
Figure 1.
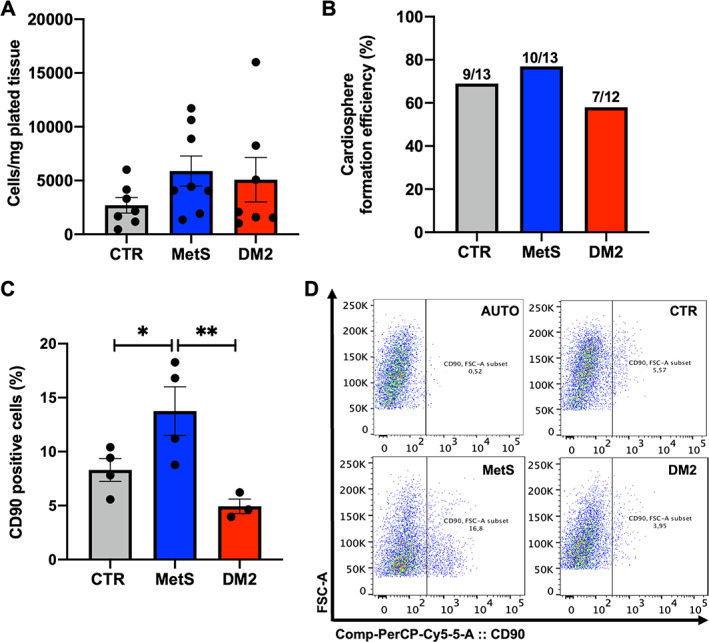
Yield, spheroid growth, and immunophenotype of cardiac stromal cells (CSCs) from different donor groups. (A) CSC yield was calculated as the number of explant‐derived cells per milligram of plated tissue in the explant culture from each biopsy in the different donor groups: control (CTR), metabolic syndrome (MetS), and type 2 diabetes mellitus (DM2). (B) After the spheroid forming selection, which gave similar efficiencies among groups, CSCs at passages 1 or 2 were analyzed by flow cytometry for CD90 positivity. (C,D) The average proportion of CD90+ cells is reported, with representative panels of data analysis, including autofluorescence control (AUTO). *p < 0.05. **p < 0.01. N ≥ 3 for each group.
Concerning CSC surface markers, flow cytometry analysis revealed a statistically significant increase in the proportion of CD90+ cells in MetS‐CSCs, compared with CTR and DM2 (Figure 1C,D), suggesting an enhanced fibrotic profile [39]. Moreover, we analyzed gene transcript levels using RT‐qPCR for a panel of widely used markers related to CSC phenotype and function. Hierarchical clustering analysis for this set of genes evidenced higher similarities in the expression profile of CSCs from CTR and MetS, compared to DM2 (Figure 2A). However, single gene analysis showed that MetS‐CSCs express significantly higher levels of GATA Binding Protein 4 (GATA4), smooth muscle actin (ACTA2), and Thymocyte Antigen 1 (THY1, encoding for CD90) versus both control and DM2 (Figure 2B–D); these are all markers of profibrotic myofibroblast commitment of stromal cells [2]. Interestingly, DM2‐CSCs showed a dramatic downregulation in the expression levels of the cardiac transcription factor NK2 Homeobox 5 (NKX2‐5) (Figure 2E). In contrast, transcripts from embryonic marker POU Class 5 Homeobox 1 (POU5F1), encoding for the octamer‐binding transcription factor 4 (Oct‐4), were significantly upregulated in MetS‐CDCs versus the other groups (Figure 2F).
Figure 2.
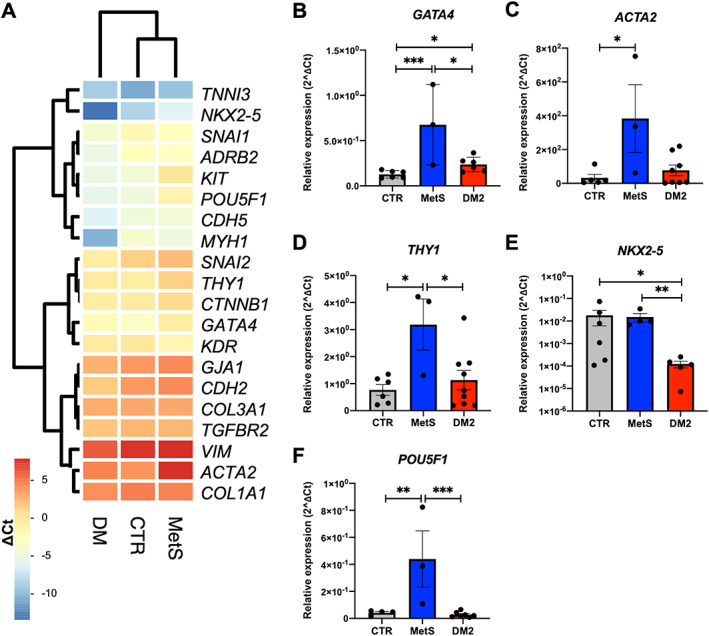
Distinct gene expression profiles of CSCs from different patient groups. (A) A set of genes of interest was analyzed in CSCs from control (CTR), metabolic syndrome (MetS), or type 2 diabetes mellitus (DM2) donors by real‐time qPCR. Hierarchical clustering with Euclidean distance and heatmap analysis were obtained by considering ∆Ct values. Single‐gene analysis of differentially expressed markers evidenced significant modulation for (B) GATA4, (C) ACTA2, (D) THY1, (E) NKX2‐5, and (F) POU5F1. *p < 0.05. **p < 0.01. ***p < 0.001. N ≥ 3 for each group.
In order to study possible additional profibrotic features in MetS‐CSCs revealed by response to stress, CSCs were cultured for 24 h under serum starvation (Figure 3A). This was a sublethal stress, as confirmed by Annexin V/7AAD labeling and flow cytometry analysis, with no detectable differences in the percentage of late apoptotic cells among the groups (Figure 3B,C). Gene expression analysis after starvation evidenced a significant upregulation of collagen type I alpha 1 chain (COL1A1) and collagen type III alpha 1 chain (COL3A1) in MetS‐CSCs versus control (Figure 3D,E), without significant modulation of the ECM resorption marker Matrix Metallopeptidase 1 (MMP1) (Figure 3F), suggesting a commitment of CSCs toward matrix deposition in response to stress.
Figure 3.
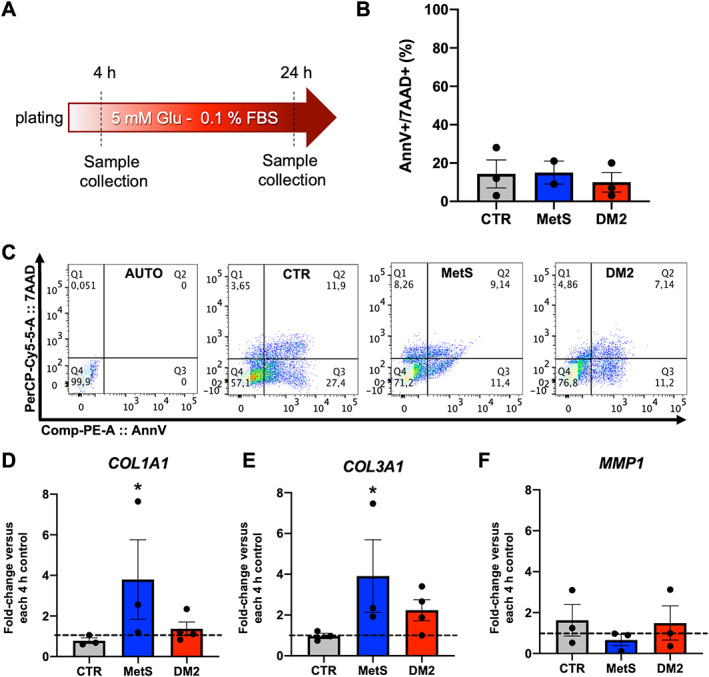
Viability and gene expression of CSCs after starvation. (A) A protocol for serum starvation was optimized, as depicted in the panel. (B) Cell viability was evaluated by Annexin V/7AAD labeling in flow cytometry, and similar percentages of late apoptotic cells (annV+/7AAD+) were detected among groups, as also shown (C) by representative panels of data analysis, including autofluorescence control (AUTO). Gene expression analysis after starvation revealed that CSCs from metabolic syndrome (MetS) patients significantly modulated markers of increased extracellular matrix (ECM) deposition, such as (D) collagen I—COL1A1 and (E) collagen III—COL3A1 versus each 4‐h control, without modulation of (F) the ECM resorption enzyme matrix‐metallo‐protease 1—MMP1. DM2: type 2 diabetes mellitus. *p < 0.05 versus each 4‐h control. N ≥ 3 for each group.
We compared the paracrine profile of CSCs from the three donor groups to verify potential differences in their capacity to condition the microenvironment. Cell culture‐conditioned media (obtained by plating the same cell number‐to‐volume ratio for each sample) were analyzed by protein arrays, and log2 OD of each dot (corresponding to one cytokine) was calculated. Hierarchical clustering analysis evidenced a difference of the paracrine profile among groups, and closer clustering between dysmetabolic cells (DM2 and MetS) compared to control cells (Figure 4A). Several cytokines were secreted at lower levels from DM2 and MetS‐CSCs compared to control (<0.5‐fold in at least one dysmetabolic group); in particular, a downregulated cluster was evidenced that includes VEGF, soluble endoglin, and monocyte chemotactic protein 3 (MCP‐3). Since these factors were released at high levels in control cells and reached the highest relative modulation in CSCs from dysmetabolic patients, they were more accurately quantified by ELISA. Results consistently showed significantly lower concentrations of VEGF (Figure 4B) and MCP‐3 (Figure 4C) in conditioned media of both DM2 and MetS‐CSCs versus control, and significantly reduced concentration of soluble endoglin from DM2‐CSCs versus CTR (Figure 4D). A consistent trend was observed in MetS‐CSC‐conditioned media as well. Given the biological roles described for VEGF and soluble endoglin, the proangiogenic properties of CSC‐conditioned media were tested in a functional assay with human umbilical vein endothelial cells (HUVECs) plated on Matrigel to assess their proangiogenic capacity. The results showed lower loop formation capacity associated with the paracrine effects of dysmetabolic donors of CSCs, with a statistically significant reduction in loop number when HUVECs were cultured in conditioned media from DM2‐CSCs versus control CSCs (Figure 4E,F). These data suggest overall a detrimental microenvironmental conditioning ability of MetS‐ and DM2‐CSCs, both deficient in angiogenic support, although to a different extent.
Figure 4.
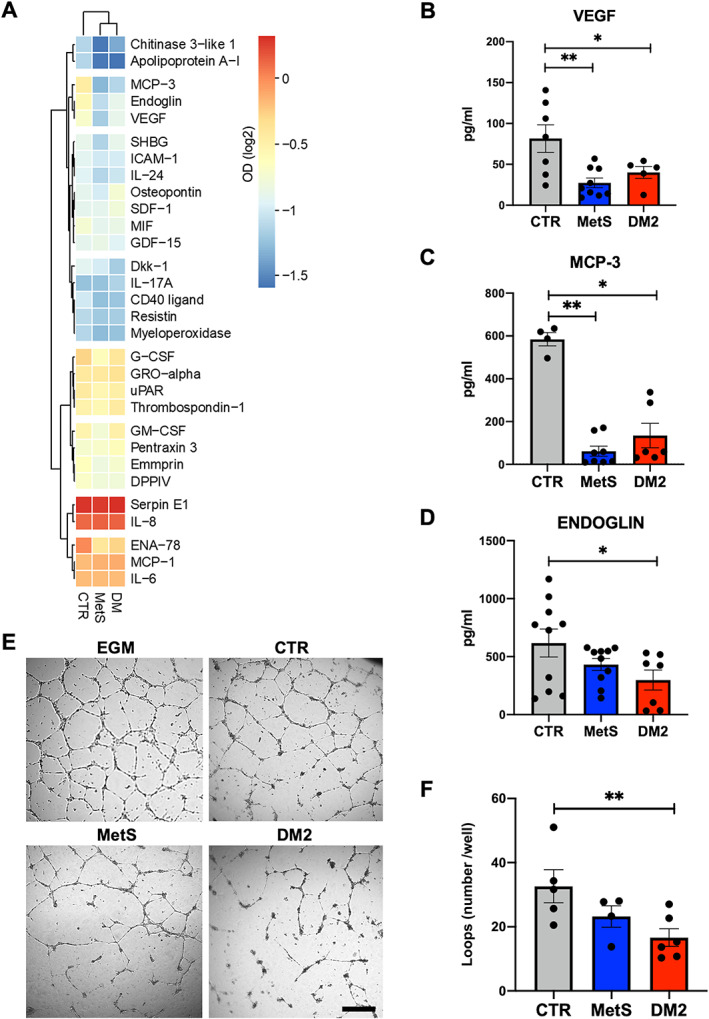
Differential paracrine and proangiogenic features of CSCs. (A) Heatmap of the normalized optical density (OD) values obtained from protein array‐based screening of conditioned media from diabetic (DM2), metabolic syndrome (MetS), or control (CTR) CSCs, plotted with hierarchical clustering of Euclidean distance. Single ELISA assays for (B) VEGF, (C) MCP‐3, and (D) soluble endoglin on the different conditioned media are also reported. (E) Representative microscopy images (including the experimental control with endothelial growth media‐EGM) and (F) loop number quantification are shown for the tube‐forming assay with human umbilical vein endothelial cells (HUVECs) exposed to CTR‐, MetS‐, or DM2‐CSC‐conditioned media. *p < 0.05. **p < 0.01. Scale bar for all panels, 200 μm.
We also analyzed the OxSt in cell cultures after starvation: DM2‐CSCs released a significantly higher amount of H2O2 in conditioned media, compared to control cells (supplementary material, Figure S1A). Also, MetS‐CSCs released a higher amount of H2O2 compared to controls, albeit to a lower extent than cells from DM2 donors. Consistent with this, DM2‐CSCs displayed significantly higher gene expression of the NADPH‐oxidase (NOX) isoforms NOX4 and NOX5 versus both control and Met‐CSCs at baseline (supplementary material, Figure S1B,C). This appears fully consistent with the overall oxidative derangement typical of diabetes [7, 10].
Autophagy has been shown to limit the profibrotic polarization of CSCs under both starvation and hyperglycemic stress [25]. Thus, we assessed the differential ability of human CSCs to activate autophagy after starvation. In line with previous results, protein levels of the autophagy initiator autophagy‐related protein 7 (ATG7, supplementary material, Figure S1D,E) and the autophagosome marker microtubule‐associated protein 1A/1B‐light chain 3 (LC3‐II, supplementary material, Figure S1F,G) were dramatically lower in DM2‐CSCs compared to MetS and control CSCs, suggesting a reduced capacity of human stromal cells from diabetic hearts to increase, or even maintain, autophagy in response to stress. The dynamics of autophagy after starvation in the CSC cell type have been already thoroughly investigated [25], thus here we confirmed that the lower LC3‐II protein level was not caused by lysosome degradation. In fact, the concomitant 2h treatment with 100 nm of the lysosomal inhibitor bafilomycin a1 (BAF) showed LC3‐II protein accumulation in all starved CSCs (supplementary material, Figure S2A).
In order to test whether the observed differences in basic biological functions (i.e. autophagy and OxSt) may cause the fibrotic polarization of CSCs in dysmetabolic patients, stromal cell cultures were treated with a cocktail of molecules to simultaneously boost autophagy and inhibit NOX activity. The autophagy activator trehalose (already validated in this cell type [25]), and specific NOX4 and NOX5 inhibitors (GKT137831 and KN‐93, respectively) were used (TREiNOX treatment) before subjecting the cells to serum starvation for 24 h. The specific effect of TREiNOX on autophagy and OxSt was confirmed by increased LC3‐II protein levels (supplementary material, Figure S2B), and reduced H2O2 release into the culture media by treated cells (supplementary material, Figure S2C). Gene expression analysis (Figure 5) showed that multiple markers of fibroblast activation and myofibroblast differentiation (GATA4, ACTA2, THY1, COL1A1, COL3A1) were significantly downregulated (Figure 5A–E, respectively) in CSCs from dysmetabolic patients by TREiNOX treatment (except for GATA4 and THY1 in MetS‐CSC), together with NOX4 (Figure 5F), which is the most highly expressed NOX isoform in these cells. With regard to the paracrine functions of the cells, TREiNOX significantly increased VEGF release in culture media by DM2‐CSCs (Figure 6A), as well as endoglin release in culture media by both MetS and DM2‐CSCs, versus untreated controls (Figure 6B). On the other hand, MCP‐3 release was significantly decreased by TREiNOX only in the culture media of DM2‐CSCs (Figure 6C). Consistent with the transcript and paracrine changes, TREiNOX treatment was able to significantly recover the proangiogenic ability of DM2‐CSC conditioned media. As assessed by the tube‐forming assay, HUVECs treated with the DM2‐CSC+TREiNOX conditioned media grew similar numbers of loops versus CTR‐CSC+TREiNOX medium, while the loop number remained significantly reduced in HUVECs treated with MetS‐CSC+TREiNOX conditioned media, compared to CTR‐CSC+TREiNOX (Figure 6D,E).
Figure 5.
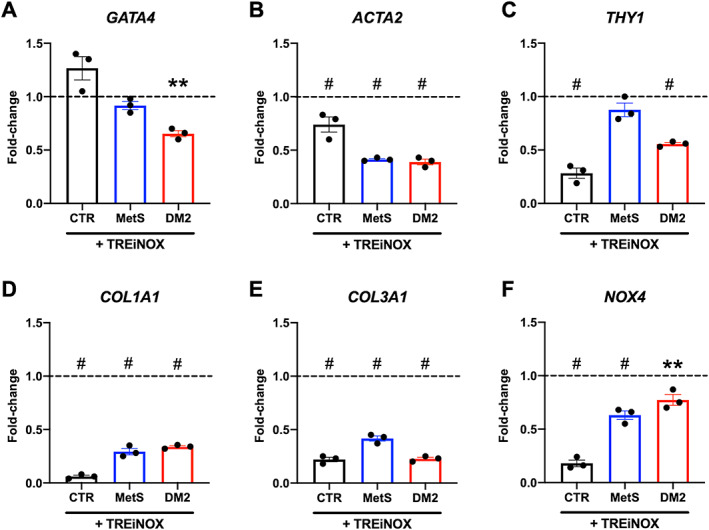
Gene expression analysis after trehalose and NOX‐inhibitors treatment of CSCs. Real‐time PCR was used to assess gene expression levels of different markers of interest for fibroblast activation and oxidative stress, including (A) GATA4, (B) ACTA2, (C) THY1, (D) COL1A1, (E) COL3A1, and (F) NOX4, in CSCs after starvation with concomitant treatment with the autophagy inducer trehalose, in combination with NOX4/NOX5 inhibitors (TREiNOX). Data are plotted as fold‐change versus untreated samples, and the dotted lines represent the level of untreated controls. **p < 0.01, and # p < 0.0001 versus each untreated control. N = 3 for each group.
Figure 6.
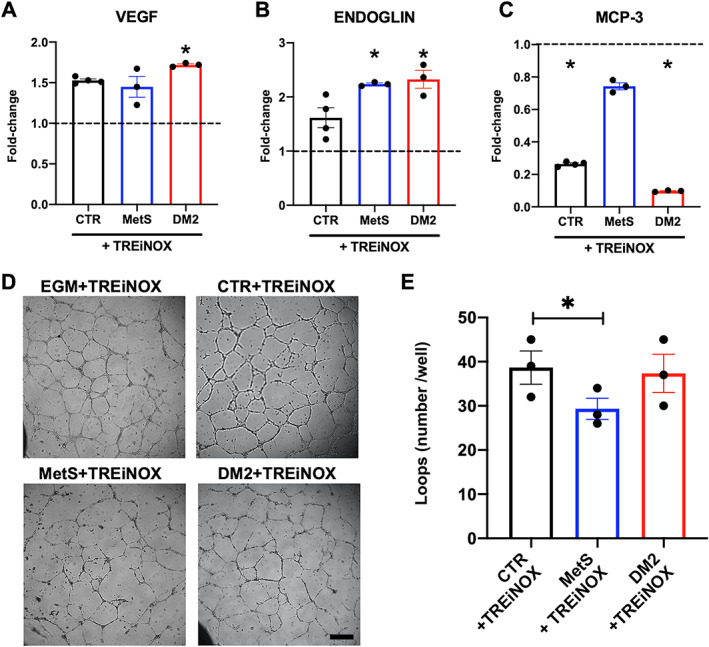
Paracrine and angiogenic profile of CSCs after trehalose and NOX inhibitors treatment. Conditioned media from CSCs isolated from the different donor groups, treated or not with the autophagy activator trehalose in combination with NOX4/NOX5 inhibitors (TREiNOX treatment) were analyzed by ELISA to quantify the differential release of (A) VEGF, (B) soluble endoglin, and (C) MCP‐3, plotted as fold‐change versus untreated controls (n = 3). Dotted lines represent the level of untreated controls. *p < 0.05 versus each untreated control. (D) Representative microscopy images (including the experimental control with endothelial growth media‐EGM supplemented with TREiNOX) and (E) loop number quantification are shown from the tube‐forming assay with human umbilical vein endothelial cells (HUVECs) exposed to conditioned media from CTR‐, MetS‐, or DM2‐CSCs treated with TREiNOX (*p < 0.05). N = 3 for each group. Scale bar for all panels, 200 μm.
Discussion
Several reports have demonstrated that type 2 diabetes impairs repair mechanisms in multiple tissues and cell types, including stromal/mesenchymal populations, particularly by affecting their commitment and paracrine features [40, 41, 42]. The mechanisms driving a phenotypic shift in the cardiac stroma in dysmetabolic conditions, however, and whether it can be overturned, still need thorough elucidation. Primitive CSCs have been described as a nonactivated pool contributing to homeostasis, that is, significantly triggered after injury toward a proliferating state, and then toward distinct functional populations, including myofibroblasts and other cellular pools with different profiles (including protective and prorepair subsets) [2, 3, 4]. The action of CSCs has a relevant role for angiogenesis support in the heart, particularly after injury, and diabetes is known to impair the angiogenetic process through multiple mechanisms that act directly on endothelial cells in various organs [43]. Very little evidence has been collected so far on the proangiogenic profile of CSCs in the presence of dysmetabolic conditions, despite angiogenesis being one of the main mechanisms by which these cells support cardiac repair after injury [44, 45].
Here we showed that different stages of dysmetabolic conditions, from MetS to DM2, in patients suffering from cardiovascular diseases, are associated with specific phenotypes of human resident primitive stromal cells. In fact, CSCs isolated from DM2 patients (undergoing elective pharmacological treatments) seem to be similar to cells from control patients for features such as transcriptional and phenotypic markers. For other characteristics, such as their paracrine, oxidative, and autophagic states, DM2‐CSCs display an altered profile instead, in line with well‐known aspects of the diabetic condition [8, 9]. Concerning the expression level of the transcription factor NKX2‐5, its dramatic drop detected in DM2‐CSCs, compared to control and MetS cells, is fully consistent with data from the literature from animal models of diabetic heart [46, 47], thus supporting the consistency of the phenotypic features of DM2‐CSCs. Interestingly, CSCs from MetS patients displayed an overall divergent phenotype from control donors at the transcriptional, phenotypic, paracrine, and oxidative level, albeit to a lower extent in the comparison versus DM2‐CSCs for this latter. This appears notable when considering that cells isolated from MetS patients derive from a less‐advanced dysmetabolic microenvironment. Furthermore, MetS‐CSCs displayed significant upregulation of profibrotic markers (GATA4, ACTA2, THY1/CD90) and of the pluripotency gene POU5F1/Oct‐4. This latter could be interpreted as a compensatory response, since Oct‐4 activation in adult tissues has been associated with increased plasticity and repair capacity of vascular smooth muscle cells [48]. Overall, these differences appear most likely due to the lack of any specific pharmacological regimen in prediabetic MetS patients, compared to diagnosed DM2 patients, thus suggesting another previously ignored positive effect of the clinical management in diabetic patients. Nevertheless, DM2 and MetS CSCs displayed highly overlapping features concerning their paracrine profile.
Hyperglycemia has been shown to enhance fibrosis in rats through enhanced differentiation of cardiac fibroblasts, in terms of collagen expression, cell migration, and proliferation [49]. Primitive CSCs from diabetic mice display an altered metabolic and phenotypic profile [12, 50]. Moreover, increased levels of advanced glycation end‐products (AGEs) and proinflammatory cytokines, collateral to dysmetabolic conditions, can autonomously increase ECM synthesis and remodeling. This leads to a chronic microenvironmental drive toward myocardial fibrosis and stiffening, that may not be overridden by simple changes in glucose availability [51]. In fact, despite metabolic reconditioning in low glucose culture has been shown to normalize CSC metabolic profile in a diabetic mouse model, the decrease in glucose availability in culture could not fully recover a proreparative phenotype [52], consistent with the concept of “metabolic memory.” Genetic intervention on the key metabolic enzyme glyoxalase‐1 has yielded functional recovery of cardioprotective features in CSCs isolated from diabetic mice [18]. Once cells have been chronically primed in situ by a dysmetabolic microenvironment, however, it is most likely difficult to intervene through extrinsic reconditioning, while other altered pathways may become decisive in controlling CSC phenotype.
Here we have shown that intervention on fundamental biological functions, such as autophagy enhancement and oxidative enzyme inhibition, exerts a positive effect on the deranged phenotype of human stromal cells from dysmetabolic patients. In fact, it reduced markers of profibrotic commitment and restored a beneficial paracrine profile, particularly in CSCs from DM2 patients where derangements in OxSt and autophagy appear to reach the most advanced stage. The OxSt has been shown to induce greater deposition of collagen I by CSCs in diabetic conditions directly through a ROS‐dependent signal transduction pathway [29]. Moreover, autophagy enhancement has been recently shown to preserve a less fibrotic and inflammatory phenotype in murine CSCs under metabolic stress [25]. Therefore, the integrated data presented here on the simultaneous opposite modulation of autophagy and OxSt show overall consistency with previous reports, while providing novel insights on the relationship among autophagy, OxSt, and human CSC phenotype.
CSCs act through paracrine effects on surrounding cells [45, 53, 54]; therefore, modulation of their secretome represents a key feature to monitor their phenotype and function. Soluble endoglin plays important antifibrotic effects on the cardiac stroma by inhibiting the signaling from the master fibrotic cytokine TGF‐β1, and collagen I deposition [55]. Moreover, it mediates proangiogenic mechanisms in cardiac repair, counteracting cardiac remodeling after ischemia [56, 57]. Interestingly, the paracrine profile of CSCs from dysmetabolic patients displayed significantly reduced levels of secreted endoglin, which were consistently increased by TREiNOX treatment in both MetS and DM2‐CSCs. This observation suggested that an antifibrotic and proangiogenic phenotype could be restored through autophagy enhancement and decreased oxidative stress. Moreover, CSCs from dysmetabolic patients released significantly lower levels of MCP‐3. Previous reports on human primitive CSCs have shown no differences in the release of this cytokine by cells isolated from failing versus nonfailing hearts [58]. Reduced MCP‐3 release, however, has been reported from CSCs depleted of a CD90+ profibrotic fraction [39], consistent with the results reported here. This cytokine has proinflammatory effects, and it has been studied also as a homing factor to recruit cardiovascular reparative cells into the injured myocardium [59, 60]; therefore, such differential expression at basal conditions could be interpreted in a bivalent compensatory way. However, TREiNOX treatment reduced MCP‐3 release by CTR cells, as well as by DM2‐CSCs, thus suggesting a relative reduction in proinflammatory signaling, with an opposite trend compared with VEGF and endoglin.
In line with the profiles of secreted cytokines, functional evaluation of DM2‐CSC‐conditioned media showed significantly reduced proangiogenic support to HUVECs, with a consistent biological trend observed with MetS‐CSC‐conditioned media. TREiNOX treatment consistently recovered the proangiogenic paracrine capacity of DM2‐CSCs to the level of CTR‐CSCs, in line with our previous report about trehalose treatment in murine CSCs [25]. Nonetheless, it appeared not to be sufficient to recover the proangiogenic function of MetS‐CSCs in our conditions.
The main limitation of our study concerns the ex vivo design. Regrettably, we do not have access to any clinical data allowing us to associate the phenotypic changes observed in vitro with parameters of myocardial fibrosis or angiogenesis in situ. Future studies will be needed to investigate this in more detail.
In conclusion, chronic dysmetabolic conditions in patients affected by ischemic cardiomyopathy significantly influence the biological and functional features of CSCs. Moreover, MetS and DM2 appear to diminish the positive conditioning on the microenvironment by CSCs. This phenomenon seems to be reversible, at least in part, and mostly in CSCs from DM2 donors, by modulating two key mechanisms active in CSCs, that is, autophagy and the OxSt. These results suggest novel potential strategies to interfere with cardiac fibrotic mechanisms in the myocardium of dysmetabolic patients.
Author contributions statement
IC, FP, SS and GF contributed to the conception and design of the study. MP, FM, GBZ and GF provided study material. FP, VP, AB, EC, CN, CC, EF, FA, AS, RC and IC performed data collection, analysis, and interpretation. IC, FP and VP drafted the article. VP, AB, EC, CN, FA, MP, GBZ, EDF, RC and SS contributed with critical article, revising for important intellectual content. IC, FP and GF gave final approval of the article submitted.
Supporting information
Figure S1. Oxidative state and autophagy modulation in CSCs after starvation
Figure S2. Validation of autophagy activation and TREiNOX treatment in CSCs
Table S1. Sequences of the primers used for RT‐qPCR
Acknowledgements
The authors thank the funders of this work that was supported by: grant # GR‐2013‐02355401 from the Italian Health Ministry to SS and IC; grant # RG11916B85CDBF76 from Sapienza University to IC; grant # AR120172B8B543B3 from Sapienza University to VP. FP is supported by Grant # A0375‐2020‐36621 from Regione Lazio (POR‐FESR 2014‐2021).
Conflict of interest statement: GBZ has consulted for Cardionovum, Crannmedical, Innovheart, Meditrial, Opsens Medical, Replycare and Terumo on topics not related to this research project. No other conflicts of interest were declared.
Data availability statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.
References
- 1. Litviňuková M, Talavera‐López C, Maatz H, et al. Cells of the adult human heart. Nature 2020; 588: 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Picchio V, Bordin A, Floris E, et al. The dynamic facets of the cardiac stroma: from classical markers to omics and translational perspectives. Am J Transl Res 2022; 14: 1172–1187. [PMC free article] [PubMed] [Google Scholar]
- 3. Farbehi N, Patrick R, Dorison A, et al. Single‐cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019; 8: e43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forte E, Skelly DA, Chen M, et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep 2020; 30: 3149–3163.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schirone L, Forte M, Palmerio S, et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev 2017; 2017: 3920195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chimenti I, Sattler S, Del Monte‐Nieto G, et al. Editorial: fibrosis and inflammation in tissue pathophysiology. Front Physiol 2022; 12: 830683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frati G, Schirone L, Chimenti I, et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res 2017; 113: 378–388. [DOI] [PubMed] [Google Scholar]
- 8. Peña‐Oyarzun D, Bravo‐Sagua R, Diaz‐Vega A, et al. Autophagy and oxidative stress in noncommunicable diseases: a matter of the inflammatory state? Free Radic Biol Med 2018; 124: 61–78. [DOI] [PubMed] [Google Scholar]
- 9. Sarparanta J, García‐Macia M, Singh R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr Diabetes Rev 2017; 13: 352–369. [DOI] [PubMed] [Google Scholar]
- 10. Luc K, Schramm‐Luc A, Guzik TJ, et al. Oxidative stress and inflammatory markers in prediabetes and diabetes. J Physiol Pharmacol 2019; 70: 809–824. [DOI] [PubMed] [Google Scholar]
- 11. Hu S, Yan G, He W, et al. The influence of disease and age on human cardiac stem cells. Ann Clin Biochem 2014; 51: 582–590. [DOI] [PubMed] [Google Scholar]
- 12. Molgat AS, Tilokee EL, Rafatian G, et al. Hyperglycemia inhibits cardiac stem cell‐mediated cardiac repair and angiogenic capacity. Circulation 2014; 130: S70–S76. [DOI] [PubMed] [Google Scholar]
- 13. Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech 2009; 2: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Badimon L, Bugiardini R, Cenko E, et al. Position paper of the European Society of Cardiology‐working group of coronary pathophysiology and microcirculation: obesity and heart disease. Eur Heart J 2017; 38: 1951–1958. [DOI] [PubMed] [Google Scholar]
- 15. Cosentino F, Grant PJ, Aboyans V, et al. 2019 ESC Guidelines on diabetes, pre‐diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J 2020; 41: 255–323. [DOI] [PubMed] [Google Scholar]
- 16. Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120: 1640–1645. [DOI] [PubMed] [Google Scholar]
- 17. Pagano F, Angelini F, Siciliano C, et al. Beta2‐adrenergic signaling affects the phenotype of human cardiac progenitor cells through EMT modulation. Pharmacol Res 2018; 127: 41–48. [DOI] [PubMed] [Google Scholar]
- 18. Villanueva M, Michie C, Parent S, et al. Glyoxalase 1 prevents chronic hyperglycemia induced heart‐explant derived cell dysfunction. Theranostics 2019; 9: 5720–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chimenti I, Pagano F, Cavarretta E, et al. B‐blockers treatment of cardiac surgery patients enhances isolation and improves phenotype of cardiosphere‐derived cells. Sci Rep 2016; 6: 36774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mauretti A, Spaans S, Bax NAM, et al. Cardiac progenitor cells and the interplay with their microenvironment. Stem Cells Int 2017; 2017: 7471582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Angelini F, Pagano F, Bordin A, et al. Getting old through the blood: circulating molecules in aging and senescence of cardiovascular regenerative cells. Front Cardiovasc Med 2017; 4: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Secco I, Barile L, Torrini C, et al. Notch pathway activation enhances cardiosphere in vitro expansion. J Cell Mol Med 2018; 22: 5583–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pagano F, Angelini F, Castaldo C, et al. Normal versus pathological cardiac fibroblast‐derived extracellular matrix differentially modulates Cardiosphere‐derived cell paracrine properties and commitment. Stem Cells Int 2017; 2017: 7396462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pesce M, Messina E, Chimenti I, et al. Cardiac mechanoperception: a life‐long story from early beats to aging and failure. Stem Cells Dev 2017; 26: 77–90. [DOI] [PubMed] [Google Scholar]
- 25. Chimenti I, Picchio V, Pagano F, et al. The impact of autophagy modulation on phenotype and survival of cardiac stromal cells under metabolic stress. Cell Death Discov 2022; 8: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sciarretta S, Boppana VS, Umapathi M, et al. Boosting autophagy in the diabetic heart: a translational perspective. Cardiovasc Diagn Ther 2015; 5: 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Menikdiwela KR, Ramalingam L, Rasha F, et al. Autophagy in metabolic syndrome: breaking the wheel by targeting the renin‐angiotensin system. Cell Death Dis 2020; 11: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Belviso I, Angelini F, Di Meglio F, et al. The microenvironment of decellularized extracellular matrix from heart failure myocardium alters the balance between angiogenic and fibrotic signals from stromal primitive cells. Int J Mol Sci 2020; 21: 7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fiaschi T, Magherini F, Gamberi T, et al. Hyperglycemia and angiotensin II cooperate to enhance collagen I deposition by cardiac fibroblasts through a ROS‐STAT3‐dependent mechanism. Biochim Biophys Acta 2014; 1843: 2603–2610. [DOI] [PubMed] [Google Scholar]
- 30. American Diabetes Association . 6. Glycemic targets: Standards of Medical Care in Diabetes‐2021. Diabetes Care 2021; 44: S73–S84. [DOI] [PubMed] [Google Scholar]
- 31. Gierach M, Gierach J, Ewertowska M, et al. Correlation between body mass index and waist circumference in patients with metabolic syndrome. ISRN Endocrinol 2014; 2014: 514589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alberti KG, Zimmet P, Shaw J, et al. The metabolic syndrome–a new worldwide definition. Lancet 2005; 366: 1059–1062. [DOI] [PubMed] [Google Scholar]
- 33. Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005; 112: 2735–2752. [DOI] [PubMed] [Google Scholar]
- 34. Chimenti I, Gaetani R, Forte E, et al. Serum and supplement optimization for EU GMP‐compliance in cardiospheres cell culture. J Cell Mol Med 2014; 18: 624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pagano F, Nocella C, Sciarretta S, et al. Cytoprotective and antioxidant effects of steen solution on human lung spheroids and human endothelial cells. Am J Transplant 2017; 17: 1885–1894. [DOI] [PubMed] [Google Scholar]
- 36. Frati G, Forte M, di Nonno F, et al. Inhibition of miR‐155 attenuates detrimental vascular effects of tobacco cigarette smoking. J Am Heart Assoc 2020; 9: e017000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hakami NY, Ranjan AK, Hardikar AA, et al. Role of NADPH Oxidase‐4 in human endothelial progenitor cells. Front Physiol 2017; 8: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. White AJ, Smith RR, Matsushita S, et al. Intrinsic cardiac origin of human cardiosphere‐derived cells. Eur Heart J 2013; 34: 68–75. [DOI] [PubMed] [Google Scholar]
- 39. Cheng K, Ibrahim A, Hensley MT, et al. Relative roles of CD90 and c‐kit to the regenerative efficacy of cardiosphere‐derived cells in humans and in a mouse model of myocardial infarction. J Am Heart Assoc 2014; 3: e001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fadini GP, Albiero M, Vigili de Kreutzenberg S, et al. Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care 2013; 36: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y, Li Z, Liu T, et al. Impaired cardioprotective function of transplantation of mesenchymal stem cells from patients with diabetes mellitus to rats with experimentally induced myocardial infarction. Cardiovasc Diabetol 2013; 12: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan J, Tie G, Wang S, et al. Type 2 diabetes restricts multipotency of mesenchymal stem cells and impairs their capacity to augment postischemic neovascularization in db/db mice. J Am Heart Assoc 2012; 1: e002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fadini GP, Albiero M, Bonora BM, et al. Angiogenic abnormalities in diabetes mellitus: mechanistic and clinical aspects. J Clin Endocrinol Metab 2019; 104: 5431–5444. [DOI] [PubMed] [Google Scholar]
- 44. Finan A, Richard S. Stimulating endogenous cardiac repair. Front Cell Dev Biol 2015; 3: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pagano F, Picchio V, Angelini F, et al. The biological mechanisms of action of cardiac progenitor cell therapy. Curr Cardiol Rep 2018; 20: 84. [DOI] [PubMed] [Google Scholar]
- 46. Sun Y, Wang Q, Fang Y, et al. Activation of the Nkx2.5‐Calr‐p53 signaling pathway by hyperglycemia induces cardiac remodeling and dysfunction in adult zebrafish. Dis Model Mech 2017; 10: 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim HS, Woo JS, Joo HJ, et al. Cardiac transcription factor Nkx2.5 is downregulated under excessive O‐GlcNAcylation condition. PLoS One 2012; 7: e38053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cherepanova OA, Gomez D, Shankman LS, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med 2016; 22: 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shamhart PE, Luther DJ, Adapala RK, et al. Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can J Physiol Pharmacol 2014; 92: 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salabei JK, Lorkiewicz PK, Mehra P, et al. Type 2 diabetes dysregulates glucose metabolism in cardiac progenitor cells. J Biol Chem 2016; 291: 13634–13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hutchinson KR, Lord CK, West TA, et al. Cardiac fibroblast‐dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS One 2013; 8: e72080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mehra P, Guo Y, Nong Y, et al. Cardiac mesenchymal cells from diabetic mice are ineffective for cell therapy‐mediated myocardial repair. Basic Res Cardiol 2018; 113: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Plikus MV, Wang X, Sinha S, et al. Fibroblasts: origins, definitions, and functions in health and disease. Cell 2021; 184: 3852–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Siciliano C, Chimenti I, Ibrahim M, et al. Cardiosphere conditioned media influence the plasticity of human mediastinal adipose tissue‐derived mesenchymal stem cells. Cell Transplant 2015; 24: 2307–2322. [DOI] [PubMed] [Google Scholar]
- 55. Kapur NK, Wilson S, Yunis AA, et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 2012; 125: 2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Redgrave RE, Tual‐Chalot S, Davison BJ, et al. Cardiosphere‐derived cells require endoglin for paracrine‐mediated angiogenesis. Stem Cell Reports 2017; 8: 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tseliou E, Reich H, de Couto G, et al. Cardiospheres reverse adverse remodeling in chronic rat myocardial infarction: roles of soluble endoglin and Tgf‐β signaling. Basic Res Cardiol 2014; 109: 443. [DOI] [PubMed] [Google Scholar]
- 58. Cheng K, Malliaras K, Smith RR, et al. Human cardiosphere‐derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. JACC Heart Fail 2014; 2: 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schenk S, Mal N, Finan A, et al. Monocyte chemotactic protein‐3 is a myocardial mesenchymal stem cell homing factor. Stem Cells 2007; 25: 245–251. [DOI] [PubMed] [Google Scholar]
- 60. Ullah M, Liu DD, Thakor AS. Mesenchymal stromal cell homing: mechanisms and strategies for improvement. iScience 2019; 15: 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Oxidative state and autophagy modulation in CSCs after starvation
Figure S2. Validation of autophagy activation and TREiNOX treatment in CSCs
Table S1. Sequences of the primers used for RT‐qPCR
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.