

Abstract
Black Americans are disproportionately affected by dementia. To expand our understanding of mechanisms of this disparity, we look to Alzheimer's disease (AD) biomarkers. In this review, we summarize current data, comparing the few studies presenting these findings. Further, we contextualize the data using two influential frameworks: the National Institute on Aging–Alzheimer's Association (NIA‐AA) Research Framework and NIA's Health Disparities Research Framework. The NIA‐AA Research Framework provides a biological definition of AD that can be measured in vivo. However, current cut‐points for determining pathological versus non‐pathological status were developed using predominantly White cohorts—a serious limitation. The NIA's Health Disparities Research Framework is used to contextualize findings from studies identifying racial differences in biomarker levels, because studying biomakers in isolation cannot explain or reduce inequities. We offer recommendations to expand study beyond initial reports of racial differences. Specifically, life course experiences associated with racialization and commonly used study enrollment practices may better account for observations than exclusively biological explanations.
Keywords: African American or Black, Alzheimer's disease biomarkers, AT(N) criteria, cerebrospinal fluid and positron emission tomography amyloid, cerebrospinal fluid tau, racial disparities
1. INTRODUCTION—BIOLOGICAL FRAMEWORK FOR ALZHEIMER'S DISEASE
Black Americans experience an increased risk for incident dementia compared to non‐Hispanic White Americans. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 Data, primarily from population‐based samples and meta‐ and systematic analyses, suggest Black adults exhibit a 64% higher rate of progression to Alzheimer's disease (AD) and related dementias (ADRD) compared to non‐Hispanic Whites. 9 , 10 These findings emphasize the urgency to clarify factors contributing to racial inequities—factors that underlie the root causes of the differences.
Recent studies 11 , 12 , 13 , 14 , 15 , 16 , 17 investigating AD biomarkers in Black and White cohorts offer preliminary evidence of how AD neurobiology appears to differ by racial groups. To expand our understanding of these biomarker findings we turn to two influential frameworks: the National Institute on Aging–Alzheimer's Assocation (NIA‐AA) Research Framework 18 and the NIA's Health Disparities Research Framework. 19 By examining biomarker findings from this framework duality, mechanism‐focused explanations for racial differentials in biological determinants of AD dementia emerge as critical targets for future epidemiological and translational research.
The NIA–AA Research Framework represents an effort to describe AD in vivo using biological parameters. Informally known as the AT(N) criteria, the framework defines AD based on the presence of amyloid beta plaques (A), neurofibrillary tangles (T), and neurodegeneration (N). Although the definitive diagnosis of the disease continues to require the presence of neurofibrillary tangles (NFTs) and amyloid plaques upon neuropathological examination at autopsy, 20 the biological framework for AD allows investigators to more precisely define and identify the disease in vivo—a critical step toward addressing this devastating illness.
A noted disadvantage to the AT(N) framework is its reductionistic approach. 21 It may be ill‐suited to fully characterize a complex disease that can co‐occur with other neuropathologies; 22 it is also unknown how the combination of neuropathologies alters presentations of the proteinopathies of tau and amyloid, or contributes to atrophy. Finally, despite the conceptual advantage of a biological definition, its implementation is further complicated given that cut‐points must be determined and used to define pathological versus non‐pathological protein levels. These cut‐points are highly dependent not only on how measurement tools were calibrated and standardized, 23 but, crucially, on the composition of the sample from which data were collected. 24 Given that the field has fallen short in efforts to recruit and retain diverse research cohorts, 25 defining representative standardized cut‐points that can be applied broadly could be impossible for the near future.
2. UNDERSTANDING RACE AND RACIALIZATION WITH THE HEALTH DISPARITIES FRAMEWORK
Race represents a social rather than a biological construct, and questions remain about the importance of racial categories in biomedical research. 26 , 27 Ancestry cannot and should not be used to support or validate self‐identified racial categories, nor serve as a default explanation for observed racial differentials. We propose scientists adopt a multifactorial model of socially‐rooted factors in aging such as the NIA Health Disparities Research Framework 19 when interpreting racial differences in AD biomarkers. Before describing the NIA Health Disparities Research Framework, we review the influence of racialization on health.
2.1. Race versus racialization—experiences altering biology
Leaders of the Human Genome Project acknowledge that genetics characterize only geographic origins of ancestors—not race. 28 Still, given the preponderance of racial health disparities that are observed consistently and persistently across both chronic and infectious disease outcomes, race cannot be ignored in biomedical research. The sociological term “racialized group,” 29 referring to a societally defined group status, may more accurately describe representations of race in medical research. That is, individuals are racialized as Black (i.e., societally defined on the basis of skin color, hair texture, or facial features) and that racialization influences interactions with social institutions including health care and other systems that impact human health. Importantly, even within racialized groups, there is considerable heterogeneity. Racialized groups in the United States differ in their experiences and transgenerational histories of slavery, immigration, genocide, and acculturation, among many other factors. Accordingly, population‐level health outcomes are altered across generations due to exposure to racialization. 30 For example, despite being racialized as Black in the United States, babies of first‐generation African immigrants have birth weights similar to offspring of White US‐born birth mothers. In contrast, the birth weights of babies of second‐ and third‐generation African immigrants are lower than those of children born to White birth mothers, and over time approach the significantly lower birth weights of children born to Black mothers whose families have lived in the United States for generations. 31 Altogether, across disciplines and health outcomes, robust evidence supports the relevance and influence of environmental and societal conditions associated with being racialized as Black in the United States.
A similar phenomenon at the latter end of the life course is observed in seminal work comparing dementia in Black Americans to a West African Black cohort, which found that the Yoruba people of Nigeria evidenced lower prevalence of all‐cause dementia and AD clinical syndrome dementia (not informed by biomarkers) compared to Black Americans. 32 Similarly, incident dementia was higher in Black Americans compared to Black Yorubas. 33 Although apolipoprotein E (APOE) allele counts were similar in the two populations, 32 APOE ε4 carrier status appeared to confer greater risk for AD dementia clinical syndrome in the US population than in Black Africans. 34 Of note, as in many other studies that pre‐date the development of biomarker technologies, it is difficult to know how much of this risk was for AD‐specific versus other causes of dementia. The authors suggested that genetic variation between Africans from Nigeria and Black Americans, as well as differences in diet, may explain geographic differences in prevalence and incidence but do not mention racial disparities in upstream factors like financial resources and access to fresh food that shape dietary “choices” in the United States. 35
In the future, larger samples may allow for more granular and descriptive classifications of race. However, for the purpose of the present review, we sought to be consistent with the extant literature, and cautiously use the racialized group category of Black. In the United States, the group category “Black” captures the variety of experiences of individuals who have ancestral connections to the African continent, including African Americans, Black Caribbeans, and West Africans living in the United States, among others. 36 All of the aforementioned groups are classified as Black/African American according to the US census and other agencies, such as the National Institutes of Health (NIH) and US Office of Management and Budget (OMB). For these discussions, Black race refers to the confluence of African ancestry and experiences engendered by living in the United States. Racialized individuals like other minoritized groups are subject to a number of socioeconomic and health disparities, largely because racial categorizations reflect societal power structures, supporting structural racism. 37
RESEARCH IN CONTEXT
Systematic review: Although there are a limited number of studies examining Alzheimer's disease (AD) biomarkers in non‐White groups, a preliminary body of literature exists. We identified literature using traditional (e.g., PubMed) sources. The few relevant citations are appropriately cited.
Interpretation: We interpreted the extant data using two conceptual frameworks—the National Institute on Aging–Alzheimer's Association (NIA‐AA) Research Framework, and theNIA's Health Disparities Research Framework. This allowed for interpretations founded in the biological disease markers and consideration of life course exposures, including those associated with being racialized as Black.
Future directions: In addition to improving the diversity of research participants, we highlight the importance of blood‐based AD biomarkers as a way to improve inclusion and retention of Black participants. We also note the importance of a more accurate consideration of race in biomedical research. All of these endeavors hold potential to reduce racial disparities in AD incidence and prevalence.
The scope of this review is narrow; however, several factors justify this focus. Although still few in number, AD biomarker studies focused on US‐based Black cohorts are predominant among those examining biomarkers by race. Moreover, Black Americans are among the populations facing the most pronounced health disparities, including disparities in incidence and prevalence of AD clinical syndrome and related dementias (ADRD). Finally, the experiences linked to membership in one racialized or marginalized group—such as Black—may be useful for understanding AD biomarker data in a number of other NIA priority populations, for example, American Indians, Latinos, sexual and gender minorities, and rural communities.
2.2. NIA Health Disparities Research Framework—applied to Alzheimer's disease neuropathology
Toward a fuller understanding of risk and resilience factors, we propose using a multi‐level, life‐course model to interpret data. Such a model permits layered and integrated approaches to understanding disparities in AD dementia and facilitates identification of intervention loci across the life course. It can also enable investigators and institutions to mitigate or eliminate many barriers to participation in ADRD cohorts and clinical trials.
The NIA Health Disparities Research Framework 19 serves as a foundational model for scientists investigating health and aging across diverse populations. The Disparities Framework proposes that environmental, sociocultural, behavioral, and biological factors work in concert to influence aging not only during later decades, but throughout the life span. Specifically, risk and resilience represent the intersection and accumulation of exposures occurring across a spectrum of macro‐ to micro‐level factors. By applying this model, race emerges as a sociocultural rather than biological phenomenon. The NIA model of disparity emphasizes the “downstream” positioning of biological disease, and illustrates how unequal distributions of “upstream” risk exposures can become systematized and cumulative when shaped by a fundamental identity marker like race—or, more accurately, by racism resulting from racialization.
The authors of the Health Disparities Research Framework emphasize that although we ultimately seek to address biological dysfunction, environmental and sociocultural exposures including urban pollutants, food deserts, historical trauma, and discrimination represent contributing factors in the causal pathway. These conditions drive downstream exposures along multiple mechanistic pathways (e.g., poor diet, chronic stress), resulting in biological disorders and disease. In the case of ADRD, resultant biological diseases such as diabetes and high blood pressure are well‐established risk factors for cognitive decline and dementia, but not necessarily AD neuropathology as described by the AT(N) criteria. 18 This reinforces the idea (explained further below) that the effect of health exposures on cognitive impairment and age‐related change may be multi‐factorial and need not act through AD pathology exclusively or at all.
Applying the Health Disparities Research Framework to ADRD, racial disparities in rates of all‐cause dementia in the United States arise from multi‐level and multi‐factorial mechanistic origins. Notably, the role of pervasive, long‐standing, and institutionalized racism should be acknowledged as a source of dementia disparities, 38 which contribute to persistently elevated rates of dementia among Black Americans despite current declines in rates of dementia noted in the US White population. 39 , 40
At a practical level, while studies indicate that differences in genetic and cardiovascular risk inadequately account for racial disparities in dementia, 1 , 4 , 41 a growing body of work suggests that life‐course social factors account for persistent late‐life cognitive health disparities in racialized and minoritized communities. For example, historical racial inequalities in education access (e.g., segregation) impact older Black populations and despite formal legal action—specifically the Civil Rights Act of 1964—de facto education disparities persist, in the form of larger student‐to‐teacher ratios, outdated and limited education materials, and less access to high‐quality curriculum, among other factors. A large body of work demonstrates that early‐life disparities in quantity and quality of education partially or completely explain racial disparities in dementia risk. 42 , 43 , 44 , 45 , 46 , 47 Educational access creates opportunity to build cognitive reserve, and is also a powerful socioeconomic resource that subsequently associates with higher‐paying occupations, financial and housing stability, and health insurance coverage. Accordingly, Chen and Zissimopolous 41 have demonstrated that disparity in accumulated wealth also substantially explains disproportionate dementia burden in older Black populations. Moving downstream, racialized social disadvantage associates with a number of environmental exposures across the life course including the experience of acute and chronic stressors, and related physiological processes.
Through a health disparities lens, chronic physiological/psychological stress plausibly influences cognitive outcomes through multiple, synchronous, and likely synergistic pathways. Self‐reported stress associates directly with neurotoxic processes in the brain itself, and when prolonged, chronic stress results in systemic dysregulation and accelerated biological, brain, and cognitive aging. 48 , 49 , 50 Stress may act directly on AD pathology: in animal models, exposure to acute and chronic stressors associates with amyloid accumulation, and neurodegeneration has been observed in animal and human studies. 51 However, in a multi‐site study of chronic post‐traumatic stress from Vietnam‐era US Veterans found that posttraumatic stress disorder was associated with lower cognitive function, but was not associated with amyloid burden, hippocampal volume, or ischemic lesions. 52 Crucially, stressors may contribute dementia risk via downstream behavioral pathways, constraining individual‐ or community‐level resources to engage in and successfully sustain preventive health behaviors, such as smoking cessation 53 and physical activity. 54 And, universal and race‐based stress associates with far‐downstream biomedical risk factors for AD dementia, including hypertension 55 and diabetes, 56 that have provided the only explanatory context explored or hypothesized for a majority of the AD studies to be reviewed here. In recognition of such pathways, Zuelsdorff et al. 57 and others 58 , 59 have investigated stress and cognition in racially diverse cohorts. Those studies examining late‐life stress in Black elders found that disproportionately high stress exposure partially explains racial disparities in cognitive health. 57 , 58
A consideration of the systematized and interactive risk and resilience exposures across multiple levels facilitates an understanding of distinct pathways salient in various populations experiencing health disparities. For instance, substantial evidence suggests that social gradients of health, wherein socioeconomic status is inversely associated with morbidity and mortality, may be attenuated or “flattened” among Black populations for many health outcomes; for example, disparities in pregnancy‐related mortality experienced by Black women in the United States are not mitigated by maternal educational attainment. 60 The most relevant risk and protective exposures for affluent Black Americans likely vary from those impacting low‐income communities, but some factors such as structural and individual‐level racial discrimination play a role across socioeconomic status. In total, the Health Disparities Research Framework allows for consideration of the unique pattern of factors experienced in various groups, and importantly, the unique targets for intervention.
3. AD BIOMARKERS
Increased focus on a biological framework for AD 18 intensifies the need to measure the disease biomarkers in vivo in clinical care settings. Without a biological test for the disease, diagnosis relies solely on the practical—but often imperfect—clinical assessment. Contributing to misdiagnosis, common cognitive assessment practices do not perform equally across races; inherent if unintentional test bias lowers the validity of many screeners and tests for Black patients and participants. 61 , 62 Clinical diagnostic and referral practices can be subject to systemic and interpersonal biases, as clinical 63 and population‐based 64 studies have suggested. Moreover, data 2 , 25 revealing parallel declines with age, but worse average measured cognitive performance in Black individuals compared to White individuals suggests that cognitive tests may over‐pathologize cognitive status in Black populations, and under‐identify cognitive impairment in Whites. In other words, our current cognitive tests and testing environments may not perform equally across races.
Altogether, there are several advantages to applying biomarkers in conjunction with clinical examination. First, biomarkers can assist clinical assessment by increasing the reliability of diagnosis. Second, more reliable diagnosis would help identify those individuals who can benefit the most from participation in clinical trials targeting specific pathologies or symptoms. Third, work is ongoing to refine the prognostic accuracy of biomarkers for pre‐clinical stages of AD, and mild cognitive impairment (MCI), 65 allowing for timely detection of disease. Importantly, biomarker assessment would need to occur in combination with clinical assessment, as is typical for other health conditions including hypercholesterolemia and diabetes.
Biomarkers for AD are evolving. At present, cerebrospinal fluid (CSF) biomarkers for AD‐related molecular changes and positron emission tomography (PET) tracer‐dependent neuroimaging of amyloid and tau provide the most direct measurements of AD pathology. However, rates of biomarker research participation are lower for many marginalized populations including racial and ethnic minoritized communities. In addition to insights on risk and resilience mechanisms, the health disparities lens offers clarity for addressing barriers to participation in and completion of AD biomarker studies. Imaging and lumbar puncture protocols are high burden: they are time‐consuming and may be intimidating to participants. Lumbar puncture can be painful and even when participants have consented and prepared to participate, research teams may not be trained to make minor procedural adaptations and accomodations needed in the presence of obesity and other health conditions more prevalent in socially disadvantaged populations. Further, lumbar puncture is associated with historical trauma for some Black participants given that the procedure was also used in the Tuskegee syphilis study. Table 1 provides a summary of currently available AD biomarker methods. In a later section, we detail findings specific to Black American cohorts for each biomarker methodology.
TABLE 1.
Summary of AD biomarker methods
| Table 1. (A) Neuroimaging AD biomarkers | ||
|---|---|---|
| PET | ||
| Amyloid PET: Quantification of amyloid plaque deposition (A) | ||
| Tracers | Quantification | Interpretation |
|
Amyloid quantified in comparison to a reference brain region, deriving
|
|
| Tau PET: Quantification of neurofibrillary tangles (T) | ||
|---|---|---|
| Tracers | Quantification | Interpretation |
|
Tau quantified in comparison to a reference brain region, deriving
|
|
| FDG‐PET: Quantification of hypometabolism, synaptic dysfunction, metabolic dysfunction, neuronal cell loss (N) | ||
|---|---|---|
| Tracers | Quantification | Interpretation |
|
Cerebral glucose uptake/metabolism
|
|
| MRI | ||
|---|---|---|
| MRI volume: Quantification of atrophy or neurodegeneration (N) | ||
| Technique | Interpretation | |
|
|
|
|
|
|
| MRI Measurement of cerebrovascular dysfunction. Not directly related to AT(N) criteria | |
|---|---|
| Technique | Interpretation |
|
|
|
|
| Table 1 (B) CSF AD biomarkers | ||
|---|---|---|
| CSF Aβ: Quantification of amyloid proteins in CSF (A) | ||
| Proteins | Detection methods | Interpretation |
|
|
|
| CSF tau: Quantification of tau proteins in CSF (T) | ||
|---|---|---|
| Proteins | Detection methods | Interpretation |
|
|
|
| Various other proteins measured in CSF (N) | ||
|---|---|---|
| Proteins | Detection methods | Interpretation |
|
|
|
| Table 1 (C). Plasma AD biomarkers | ||
|---|---|---|
| Plasma Aβ: Quantification of amyloid proteins in plasma (A) | ||
| Proteins | Detection methods | Interpretation |
|
|
|
| Plasma tau: Quantification of tau proteins in plasma (T) | ||
|---|---|---|
| Proteins | Detection methods | Interpretation |
|
|
|
| Various other proteins measured in plasma (N) | ||
|---|---|---|
| Proteins | Detection methods | Interpretation |
|
|
|
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; AT(N) criteria, National Institute on Aging–Alzheimer's Association Research Framework with (A) amyloid beta deposition, (T) tau hyperphosphorylation or tangle formation, and (N) neuronal death; CSF, cerebrospinal fluid; DVR, distribution volume ratio; IP‐MS, immunoprecipitation mass spectrometry; MRI, magnetic resonance imaging; NfL, neurofilament light; PET, positron emission tomography; p‐tau181/217/231, tau phosphorylated at threonine 181/217/231; SUV, standardized uptake value; SUVR, standardized uptake value ratio; t‐tau, total tau; WMH, white matter hyperintensities.
Current methodologies for measuring biomarkers per the AT(N) criteria of the NIA‐AA Research Framework 18 include neuroimaging and fluid biomarkers. Apart from neuropathological evaluation, amyloid imaging with PET, using tracers that bind to fibrillar amyloid beta (Aβ) remains the gold standard for determining AD‐specific pathological status and has been in use for more than 15 years. 66 Amyloid PET tracers are used to determine amyloid positivity and localize regional amyloid accumulation for staging disease severity. In recent years, several compounds have been evaluated for their binding affinity to abnormal aggregates of filamentous tau protein constituting NFTs, including the Food and Drug Administration (FDA)‐approved [18F]‐flortaucipir (Tauvid), which preferentially binds to paired helical filament‐tau containing NFTs. 67 Currently, the availability of tau PET imaging data from diverse populations is very limited.
Fluorodeoxyglucose (FDG) PET and magnetic resonance imaging (MRI) have been widely used to evaluate abnormal glucose utilization and neurodegeneration in AD. 68 , 69 , 70 Additionally, MRI can provide an estimate of the extent of cerebrovascular dysfunction or injury. Although vascular injury is not included within the AT(N) framework, determining vascular contributions to cognitive impairment is still an important focus in the study of ADRD 71 (see Table 1). 72 , 73 , 74
CSF Aβ42 and the CSF Aβ42/Aβ40 ratio are established biomarkers for the A criterion in the AT(N) framework. Recent studies indicate that the Aβ42/Aβ40 ratio is concordant with amyloid PET positivity and that the two biomarkers can be used more or less interchangeably. Originally described as markers of neurodegeneration and tangle pathology markers, CSF total tau (t‐tau) and phosphorylated tau (p‐tau) correlate highly 75 and are remarkably AD‐specific 76 ; that is, their increase in CSF is a reaction to Aβ exposure. 77 Regarding neurodegeneration, CSF neurofilament light (NfL) might be a better direct marker; it correlates with imaging evidence of neurodegeneration, and tracks age‐related brain changes. 78
4. AD BIOMARKER COMPARISONS BETWEEN BLACK AND NON‐HISPANIC WHITE SAMPLES
In this section, we summarize the handful of studies examining AD biomarker data in US Black cohorts. In all but one case, 17 Black participants’ ethnicity was not reported. Table 2 provides a summary of study characteristics and findings. In Table 3 and sections below, we organize our discussion of extant literature by the AT(N) criteria and by cohort type.
TABLE 2.
Summary of published research, comparing AD biomarkers in Black and non‐Hispanic White adults
| Authors | Study design | Participants | AD biomarker(s) | Findings | Conclusions |
|---|---|---|---|---|---|
| Gottesman et al. (2016) a | Subset of participants recruited from population‐based study. Cross‐sectional analysis. |
Diagnostic categories included: MCI and HC |
[18]Florbetapir PET; global cortical amyloid from 9 ROIs provided dichotomous outcome of Amyloid positive v. negative (SUVR ≤ 1.2); WMH measured with MRI |
|
Presence of vascular risk factors in late life does not account for racial differences in cortical amyloid. Authors considered whether sociocultural factors could account for observed differences, but dismissed as unlikely. Proposed differences in metabolomic or genetic factors may contribute to differences between. |
| Howell et al. (2017) | Convenience sample recruited from ADC, clinic and community outreach events. Cross‐sectional analysis. |
|
CSF levels of Aβ42, Aβ40, Aβ42/Aβ40, t‐tau, p‐tau181, t‐tau/Aβ42, p‐tau181/Aβ42 and NfL; and WMH and HV measured with MRI |
|
Considered several possible reasons for racial differences in CSF AD biomarkers. Misdiagnosis (White being HC) and differences in level of neurodegeneration were considered, but dismissed because amyloid levels were similar between racial groups. Also examined whether vascular disease or comorbid neuro‐pathologies may explain observed differences, finding no support for either theory. Concluded difference may be due to potentiated effects of WMH on cognition observed for Black individuals. |
| Garrett et al. (2019) | Case‐control, convenience sample of participants enrolled in B‐SHARP, recruited from an ADC. Cross‐sectional analysis. |
Diagnostic categories included: HC and MCI |
CSF levels of Aβ42, Aβ40, t‐tau, P‐tau181, t‐tau/Aβ42, and P‐tau181/Aβ42; and HV measured with MRI; and ROC analyses with AUC |
|
Differences in CSF biomarkers for AD were not accounted for by differences in cognitive performance, hippocampal neurodegeneration or vascular disease risk factors. Proposed possible contributions from other neuropathologies, i.e., mixed pathology. Also hypothesized that groups exhibited differences in cognitive reserve, with Whites having greater reserve than Black participants. Based on finding of ROC analyses, authors recommended caution when using cut‐off scores to characterize AD biomarker status across races. |
| Morris et al. (2019) | Convenience sample recruited from ADC. Cross‐sectional analysis. |
Diagnostic categories included: HC, MCI, and AD dementia |
HV measured with MRI (N = 1032 [13.9% Black]); partial volume corrected PET PIB SUVR (N = 569 [12.9% Black]); CSF levels of Aβ42, Aβ40, t‐tau, p‐tau181 (N = 903 [9.6% Black]) |
|
CSF tau level differences suggesting decreased neuropathology in Black participants compared to Whites were hypothesized to be related to a differential effect of the APOE ε4 carrier status, such that the gene shows a closer association with AD in Whites than Black participants. In contrast, in those with a family history of AD, HV were smaller in Black than White participants. Based on the similarities between racial groups in CSF amyloid levels and PET amyloid deposition, authors propose that the disease presents with an “identical AD biosignature” across the two groups. Rather, race was hypothesized to modify risk and expression of the disease. |
| Meeker et al. (2020) | Convenience sample recruited from ADC. Cross‐sectional analysis. |
Diagnostic categories included: HC |
PET amyloid imaging: [11C] (PiB) or [18F]‐Florbetapir (AV45); PET tau imaging: [18F]‐Flortaucipir (AV1451)38 with SUVRs calculated for the 80 to 100‐minute post‐injection window. Brain volume atrophy in AD signature regions with structural MRI and resting state functional connectivity BOLD imaging. |
|
Greater cerebral volume loss in Black participants compared to whites reflect both direct and indirect contributors. Racial differences comprised lower education levels and higher polygenic risk for AD among Black participants. |
| Kumar et al. (2020) | Convenience sample recruited from ADC. Cross‐sectional analysis. |
Diagnostic categories included: HC with at least one biological parent with AD |
CSF levels of t‐tau, p‐tau, Aβ42; vascular ultrasound of PWV, FMD, EndoPAT; |
|
Lower levels of tau in Black participants than Whites suggests that AD neuropathology begins during middle age. White participants displayed higher scores on all cognitive tests than Black individuals, suggesting that cognitive tests may have implicit cultural biases favoring Whites. As race modified relationship between tau and executive function such that small changes in tau resulted in worse cognition among Black participants compared to Whites. neuropathology of tau deposition may differ in Black participants. Existing cut‐off values for CSF biomarkers may be inappropriate for Black patients. |
| Deters et al. (2021) | Convenience sample recruited from A4 study. Cross‐sectional analysis. |
Diagnostic categories included: HC |
PET amyloid imaging; raw continuous amyloid SUVR; participants were classified into amyloid groups using a cut‐point of ≥1.17. |
|
Less amyloid among Black participants suggests other risk factors may contribute to AD dementia among Black participants, such as vascular risk factors, TDP43 pathology, other age‐related pathologies, and other social determinants of health, e.g., increased lifespan exposure to stress. Representing an important gap in the literature, it is unknown whether PET measures of AD pathology equally predict future progression to dementia in Black participants compared to Whites. Baseline neuropsychological tests may not accurately capture true cognitive ability in Black participants, instead reflecting biases and inadequate norms. The authors emphasized that race is a social construct predominantly used in the United States and current data may not adequately its effect on risk of AD and amyloid. |
Abbreviations: A4, Anti‐Amyloid Treatment in Asymptomatic Alzheimer's Disease; Aβ, amyloid beta; AD, Alzheimer's disease; ADC, Alzheimer's Disease Center; ADI, Area Disadvantage Index; APOE ε4, ε4 allele of the apolipoprotein E gene; AUC, area under the curve; BMI, body mass index; BOLD, blood oxygen level dependent; B‐SHARP, Brain Stress Hypertension and Aging Research Program; CDR, Clinical Dementia Rating; CSF, cerebrospinal fluid; DM, diabetes; EndoPAT, pulsatile arterial tonometry; FMD, flow‐mediated vasodilation; HbA1c, hemoglobin A1c test; HC, healthy control; HTN, hypertension; HV, hippocampal volume; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; MoCA, Montreal Cognitive Assessment; MRI, magnetic resonance imaging; NfL, neurofilament light chain; OR, odds ratio; PET, positron emission tomography; PiB, Pittsburgh compound B; SES, socioeconomic status; p‐tau181, tau phosphorylated at threonine 181; PWV, pulse wave velocity; ROI, regions of interest; ROC, receiver operating curves; SD, standard deviation; SUVR, standardized uptake value ratio; TDP43, TAR DNA‐binding protein 43; t‐tau, total tau; WMH, white matter hyperintensities.
Gottesman et al. (2017) also included Black participants. However, rather than contrasting AD biomarker in Black and White participants, study compared relationship between vascular risk in midlife and amyloid deposition, examining whether the relationship differed by race.
Notes: Overall, AD biomarkers measured in these studies behaved as expected, for example, increasing age and clinical symptoms, and APOE ε4 carrier status were associated with elevated PET amyloid levels, decreased CSF Aβ42, increased CSF T‐tau, P‐tau181, and neurodegeneration
TABLE 3.
Summary of published research, comparing AD biomarkers in US‐based Black and non‐Hispanic White cohorts in relation to the AT(N) criteria, that is, NIA‐AA Research Framework
| Biomarker | Study by | Indicator of | Summary of racial comparison | Relation to AT(N) criteria |
|---|---|---|---|---|
| [18]Florbetapir PET and PET PiB SUVR |
Gottesman et al. (2016) Morris et al. (2019) Meeker et al. (2020) Deters et al. (2021) |
Higher levels amyloid deposition in brain tissue |
|
Believed to reflect A; i.e., Aβ deposition |
| CSF levels of Aβ42 and Aβ40 |
Howell et al. (2017) Garrett et al. (2019) Morris et al. (2019) Kumar et al. (2020) |
Lower levels suggest deposition of amyloid in tissue, i.e., toxic forms of proteins are not cleared into CSF, thus levels are lower. |
|
Believed to reflect A; i.e., Aβ deposition |
| PET tau | Meeker et al. (2020) | Higher levels tracer signal indicate more Tau deposition in brain tissue |
|
Believed to reflect T |
| CSF levels of t‐tau and p‐tau181 |
Howell et al. (2017) Garrett et al. (2019) Morris et al. (2019) Kumar et al. (2020) |
Higher levels are associated with cell death caused by Aβ pathology, causing the release of tau protein into the CSF. |
|
Believed to reflect T and possibly N; i.e., tau hyperphosphorylation or tangle formation, and neuronal death |
| CSF levels of NfL | Howell et al. (2017) | Higher levels suggest neuronal injury caused by multiple factors, including AD associated neurodegeneration, cerebrovascular disease and neuroinflammation. |
|
Believed to reflect N; i.e, neuronal death |
| Cerebral volume, e.g., HV measured with MRI |
Garrett et al. (2019) Morris et al. (2019) Meeker et al. (2020) |
Lower HV indicates neurodegeneration in brain region effected early in AD neuropathological time course. |
|
Believed to reflect N; i.e., neuronal death |
| Cerebrovascular disease, ischemic burden, WMH |
Gottesman et al. (2016) Howell et al. (2017) Morris et al. (2019) Meeker et al. (2020) |
Marker of cerebral small vessel disease |
|
Believed to reflect N; i.e., neuronal death |
Abbreviations: AA, Alzheimer's Association; Aβ, amyloid beta; AD, Alzheimer's disease; APOE ε4, ε4 allele of the apolipoprotein E gene; ATN criteria, National Institute on Aging–Alzheimer's Association Research Framework with (A) amyloid beta deposition, (T) tau hyperphosphorylation or tangle formation, and (N) neuronal death; CSF, cerebrospinal fluid; HC, healthy control; HV, hippocampal volume; MCI, mild cognitive impairment; NfL, neurofilament light chain; NIA, National Institute on Aging; PET, positron emission tomography; PiB, Pittsburgh compound B; p‐tau181, tau phosphorylated at threonine 181; SUVR, standardized uptake value ratio; T‐tau, total tau; WMH, white matter hyperintensities.
Of note, efforts to compare data from Black participants to White participants are problematic as others have highlighted. 79 The comparisons described here should not be interpreted as implying that White participants are a “gold standard” or reference group. Rather, we highlight these comparisons between Black and White populations to emphasize (1) the problems with applying cut‐points derived from predominantly White samples, and (2) how environmental, sociocultural, and behavioral factors associated with race in the United States may contribute to AD pathology.
4.1. Amyloid biomarkers
4.1.1. Amyloid PET biomarker from subset of a population‐based cohort
Contrasting two analyses from an ancillary study linked to the population‐based Atherosclerosis Risk in Communities (ARIC) study highlights how adopting a life course perspective may alter the interpretation of racial differences in amyloid PET findings. 14 , 80 Initial findings from the ARIC amyloid PET study indicated that Black race was associated with a greater than 2‐fold increased odds of exhibiting a positive amyloid status, with Black participants demonstrating elevated odds of amyloid deposition in the cingulate gyrus; the precuneus; and the orbitofrontal, prefrontal, and superior frontal cortices. 14 While concurrent vascular risk factors were examined, the authors did not find that they accounted for associations between Black race and amyloid positivity. The paper concluded that genetic and/or metabolic factors may account for racial differences in amyloid status. However, limitations of unmeasured confounding remained, and social forces that are likely to drive hypothesized or reported findings were not discussed. Subsequent analysis of the data 80 could be considered an early and preliminary adoption of a life course perspective. Specifically, the investigators examined the relationship between midlife vascular risk factors (ages 45–64) in Black and White participants, and elevated amyloid status approximately 20 years later. Having two or more vascular risk factors at midlife was associated with risk of elevated amyloid deposition later in life, but no race–by–risk factor interactions were observed, indicating that Black and White adults demonstrated a similar relationship between midlife vascular risk and elevated risk for AD pathology. Simply stated, the authors concluded that Black individuals may have more vascular risk factors at midlife, and in turn that these disparities in vascular risk factors may drive disparities in ADRD later in life. Below we discuss further opportunities to embed the findings in a multi‐level, mechanistic framework.
4.1.2. Amyloid PET biomarker convenience samples
Three studies, two from the same center, provided amyloid PET comparisons from convenience samples of Black and White individuals, revealing disparate results. Morris et al. 12 examined amyloid PET measured with partial volume‐corrected mean Pittsburgh compound B (PiB) standardized uptake value ratios (SUVRs) in older adults in three diagnostic categories: cognitively healthy, MCI, or AD. Amyloid burden increased with age as expected and equally for Black and non‐Hispanic White participants. Meeker et al. 15 included only cognitively healthy adults from the same Alzheimer's Disease Center (ADC) as Morris et al., expanded amyloid imaging methods to include both PiB and [18F]‐florbetapir measurements, and obtained consistent results with Morris et al.
In contrast, Deters et al. 17 found that non‐Hispanic Black participants demonstrated lower cerebral amyloid deposition compared to non‐Hispanic Whites. Using [18F]‐florbetapir imaging data from the Anti‐Amyloid Treatment in Asymptomatic Alzheimer's Disease (A4) study, which included cognitively healthy adults being screened for a clinical study, the investigators further examined whether African ancestry estimated with genome‐wide single nucleotide polymorphisms (SNP) array data or APOE ε4 status influenced the association between race and amyloid status. Black race and African ancestry were associated with decreased amyloid deposition, and an interaction effect of APOE ε4 carrier status was found, such that non‐Hispanic Black participants who were APOE ε4 positive evidenced lower amyloid deposition than non‐Hispanic White individuals who were APOE ε4 positive. Deters et al. 17 note that social determinants including neighborhood disadvantage and stress are likely to shape brain health and that these data will be crucial to collect and explore in future studies of dementia risk in Black populations.
4.1.3. CSF amyloid biomarkers from ADC cohorts
Several studies enrolling from ADCs have examined CSF amyloid in Black and White participants. 11 , 12 , 13 , 16 In general, most studies found minimal racial differences in the various Aβ‐analyte levels, and none found between‐group differences in CSF Aβ42. Variations in findings related to which analyte was examined—Aβ38, Aβ40, or Aβ42—or to which diagnostic category participants belonged: cognitively healthy, MCI, or AD dementia. Howell et al. 13 found that CSF Aβ40 was lower in Black than White participants but only for cognitively healthy individuals. Also, with cognitively healthy adults, Kumar et al. 16 found a difference only in CSF Aβ38 levels with Black participants having lower levels of the analyte. Garrett et al. 11 found isolated differences by race; specifically, Black participants with MCI demonstrated higher Aβ40 levels than White participants with MCI. Overall, given the small sample sizes and paucity of studies, more replication is needed before conclusions about differences by race can be made.
4.1.4. Summary of amyloid comparisons
Table 3 summarizes findings from studies examining racial differences in amyloid biomarkers. Briefly, amyloid burden as indexed by CSF Aβ42 appears to be similar in Black and White participants. Importantly, all CSF amyloid data were derived from ADC samples. In contrast there was greater variability in comparisons of amyloid imaging. It is possible that the site of acquisition could influence findings. For example, amyloid imaging from ADCs suggests similar levels of deposition between Black and White participants. In contrast, the ARIC‐PET study 14 and an examination of A4 screening amyloid PET data 17 reported racial differences in amyloid deposition, but in divergent directions (i.e., higher and lower cortical amyloid in Black individuals compared to White individuals).
4.2. Tau biomarkers
Among studies examining in vivo tau pathology, all but one relied on CSF levels of tau isoforms, t‐tau and p‐tau181. 11 , 12 , 13 , 16 Meeker al. 15 used PET tau imaging using [18F]‐flortaucipir. Across all CSF studies, tau isoform levels were lower in Black compared to White participants. Small variations in subgroups were observed. For example, Howell et al. 13 observed these differences in cognitively healthy participants, whereas Garrett et al. 11 noted these differences only in participants with MCI. Morris et al. 12 found the differences in both cognitively healthy and impaired participants.
None of the above studies found that CSF tau levels were associated with other pertinent comorbidities, including cerebrovascular disease, cardiovascular risk factors, or white matter hyperintensities (WMH). Most concluded like Howell et al. 13 that cerebrovascular disease, estimated with MRI measures of ischemic lesion burden, did not account for the differences in CSF t‐tau or p‐tau181. Rather, the authors hypothesized that possible differences in how APOE ε4 carrier status may interact with tau production/dysregulation might account for the racial differences in CSF tau levels, supported by other data suggesting that the APOE ε4 genotype has a diminished association with AD in Black participants. 81
In contrast to CSF measurement of tau, a tau imaging study found no racial differences in tau deposition. 15 However, few studies have been conducted in diverse groups.
4.2.1. Summary of tau comparisons
Among the studies published to date, there is general agreement that CSF tau protein levels differ between Black and White participants, with the caveat that all data were obtained from ADCs (see Table 3). In all studies examining CSF p‐tau181 and t‐tau, values were lower in Black than White individuals, suggesting lower levels of tau pathology or lower amyloid‐induced tau pathology. In contrast a tau imaging study suggested no difference in deposition between Black and White participants. 15 The role of social factors rooted in racialization have in generating in these differences remains unclear.
4.3. Neurodegeneration biomarkers
A large number of studies have examined racial differences in various indicators of neurodegeneration. We focus here on those examinations of neurodegeneration in conjunction with amyloid and tau biomarkers (see Table 3). Several studies already highlighted included markers of neurodegeneration in combination with other AD biomarkers.
In some cases, 11 , 12 , 15 atrophy was compared by race, with most findings suggesting less global and/or regional atrophy in White participants than in Black participants. However, Garrett et al. 11 found the reverse was true among individuals with MCI, observing that models correcting for covariates reduced or eliminated volume differences.
Of those studies examining WMH volume or cerebrovascular disease burden, 12 , 13 , 14 , 15 none found differences between Black and White adults despite the greater prevalence of cardiovascular risk factors in Black participants. On the other hand, Howell et al. 13 found that race modified the relationship between WMH and cognition with Black participants showing more cognitive impairment than Whites with every unit increase in WMH.
Only Howell et al. 13 measured CSF NfL, a marker of neurodegeneration. Among cognitively unimpaired participants alone, Whites exhibited higher NfL levels, that is, more neurodegeneration than cognitively normal African Americans.
4.3.1. Summary of neurodegeneration comparisons
Findings related to neurodegeneration were variable (see Table 3), likely dependent on how neurodegeneration was estimated. All studies found Black participants to have lower cerebral volume measurements, but differences were not universally apparent after models were corrected for differences in sociodemographic factors. In a single study, CSF NfL values suggested less neurodegeneration in cognitively healthy Black participants than White participants, but values were similar across races when cognitively impaired groups were compared.
5. CONTEXTUALIZING FINDINGS—EXPLANATIONS AND IMPLICATIONS
There is a scarcity of studies examining AD biomarkers in Black cohorts. These seven studies represent, to our knowledge, the bulk of all imaging and CSF biomarker publications at this time that analyze and discuss Black race as a key participant characteristic. Still, the papers reviewed here included cohorts and outcomes too disparate to warrant attempting meta‐analytic summaries. This important limitation highlights the pressing need for diverse cohorts of research participants. Available data, largely from older ADC cohorts, represent a heterogeneity of cognitive statuses and describe preliminary racial differences in only one of the three AT(N) criteria. Specifically, CSF t‐tau and p‐tau levels (T) levels were generally lower in Black individuals compared to Whites. It might be hypothesized that more extensive cerebrovascular disease among Black adults could result in greater cognitive impairment even in the context of less AD‐related tau neuropathology, or that racial inequities in access to resources like education result in inequities in cognitive reserve. Notably, no study found that the differences in tau levels were explained by measured coexisting vascular or cerebrovascular disease. Similarly, the available results so far on NfL, a marker highly sensitive to axonal degeneration induced by cerebrovascular disease, 78 show no race‐associated differences.
Critically, additional studies are needed with cohorts that are representative of the racially heterogeneous population of the United States in addition to studies that consider multiple pathologies, to better understand the potential impacts of racialization in the United States. In the following sections we start to contextualize findings from studies identifying racial differences and similarities in biomarker levels. Specifically, we apply the NIA Health Disparities Reseacrch Framework to findings and consider the influence of commonly used study enrollment practices.
5.1. Applying the NIA Health Disparities Research Framework to AT(N) findings in a racialized group
In several of the reviewed papers, the studies’ authors proposed genetic differences could account for racial differences in biomarker status. A growing body of research is exploring genetic risk and resilience associated with African ancestry in Black populations historically excluded from biomedical research. 17 In general, these studies could also help elucidate how environmental and societal factors may converge with genetic factors to influence health disparities. However, as noted above, current evidence for social determinants of racial disparities in cognitive health suggests that race remains an imprecise concept, more accurately defined as a societal construct, rather than a biological or genetic characteristic. 82 , 83 As such, it is most certainly social factors that underpin observed differences. Of the reviewed articles, only one explicitly described the social construction of race and the systematized risk exposures that may foment racial brain health disparities. 17 Thus, research on brain health disparities would benefit from using a framework that can take a multifactorial approach. In this section, we propose that applying the NIA Health Disparities Research Framework 19 to AT(N) findings would open new lines of inquiry and offer new targets for intervention and prevention.
Figure 1 offers an example of how commonly studied downstream ADRD risk factors can be contextualized with the Health Disparities Research Framework. 19 Doing so expands interpretation of factors contributing to the AT(N) pathology and broadens opportunities for translational research and intervention. Distal factors like structural racism plausibly link to proximal factors like metabolic diseases, inflammation, and mood disorders, which initial reports suggest associate with the AT(N) signal, 84 , 85 , 86 , 87 via risk exposures including poorer health care and constraints on physical activity. In the case of Black Americans, stress related to racialization and structural racism may be a life‐long or chronic exposure, resulting in “weathering”—the process of premature aging hypothesized to play a major role in many racial health disparities. As noted earlier, stress associations with neurotoxicity, metabolic dysfunction, and health behaviors suggest multiple mechanistic targets for modifying both AT(N) pathologies and cognitive reserve. 55 , 56 , 57 Howell et al. 13 proposed a related idea, suggesting that “non‐AD copathology” (p. 5) may cause greater cognitive decline in Black individuals than Whites. As such, Black participants could potentially be identified as impaired—with either MCI or dementia—at a younger age than White participants and with less advanced tau pathology.
FIGURE 1.
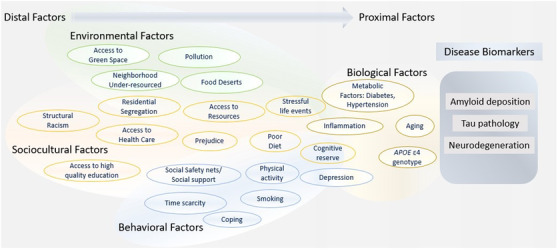
Hypothetical model integrating the National Institute on Aging (NIA) Health Disparities Research Framework with the amyloid/tau/neurodegeneration (AT[N]) criteria from the NIA–Alzheimer's Assocation Research Framework. Evidence supporting individual examples association with Alzheimer's disease biomarkers is hypothetical in some instances. Examples are generally categorized as environmental, sociocultural, behavioral, or biological factors. However, we acknowledge that many examples can belong in multiple categories
Analyses like those from the ARIC‐PET 80 substudy offered further insights on life‐course risk—another important domain in the Health Disparities Research Framework. Specifically, excess vascular risk at midlife but not late life was associated with elevations in cortical amyloid for both Black and White participants. The authors concluded that the greater prevalence of AD clinical syndrome in Black individuals could result from higher rates of midlife vascular risks in Black compared to White adults. A deep examination of factors contributing to the higher prevalence of mid‐life metabolic disease would provide footholds for prevention strategies—specifically, strategies that account for and address both the social determinants of vascular health for Black populations as well as population‐salient barriers to managing vascular conditions like hypertension and diabetes in middle age.
In summary, we should not stop at identifying what individual‐level behaviors or isolated risk factors are associated with ADRD in Black populations. The Health Disparities Research Framework 19 could be used to guide both data collection and inquiry into what policies would reduce dementia disparities. Recent publications call for more robust assessment of social and environmental factors associated with ADRD health disparities, 88 as well as a public health approach to ADRD risk reduction across the life span through health promotion, improved access to care, and other policy‐ and system‐level changes. 89 In total, examining ADRD risk through the lens of the Health Disparities Research Framework can not only clarify mechanisms, but also identify targets for intervention. In Figure 1 policy changes would target several distal factors, personalized medicine approaches could improve outcomes for proximal factors, and public health interventions for those factors in between.
5.2. Influence of enrollment practices
Another explanation that takes research context into account is that differences noted in this review may be partially explained by selection or ascertainment biases within study cohorts. Available data were largely from highly selected cohorts. Moreover, studies often use divergent enrollment practices when recruiting minoritized versus non‐minoritized racial groups. We recently reported 90 findings using data collected at US ADCs, revealing that cognitively healthy Black participants showed no greater hazard for incident MCI or dementia than White participants and that Black participants with MCI evidenced a lower hazard for incident dementia than similar White participants. Notably, White participants were more likely to have been referred to an ADC by a medical provider and to report a family history of AD than Black participants.
It may be that Black participants enrolled at ADCs are healthier and better‐resourced than non‐participating Black peers, that is, exhibit a healthy participant bias, which could potentially explain biomarker results in which Black participants have lower levels of tau pathology compared to White participants. It is equally likely that White participants from ADCs represent a highly selected and unique sample, given that they report high levels of reserve‐building resources like educational attainment—and they are also more likely to have been recruited by a clinician, in many cases after presenting with subjective memory complaints and/or a family history of dementia. Specifically, both AD pathology and cognitive reserve may be over‐represented in affluent White ADC samples, as was suggested by our analyses. 90 Taken together, it should not be surprising that Black and White participants recruited in different settings would present with differences in underlying AD risks and biomarker profiles. It follows that interpretations of patterns of cognitive status, AD biomarkers, and exposures would be flawed due to differences in enrollment patterns—differences secondary to disparate barriers to research participation.
As such, caution is warranted when considering data from these and similar cohort‐derived comparisons, especially when studies involve intensive and invasive procedures such as lumbar puncture collection of CSF. Examples include neuropathological studies 91 as well as our own neuroimaging analyses, 92 , 93 wherein differences in underlying disease mechanism are suggested from racial comparisons of ADC participants. Indeed, we suggest that across many volunteer cognitive aging cohorts, the White cohort more than the Black cohort is atypical and divergent from the general population, that is, at higher biological risk for AD than the general population despite the disproportionate prevalence of high educational attainment and other protective factors.
It is possible that fully inclusive population‐based studies may find more similarity than dissimilarity across race and ethnicity in AD biomarkers. Further, it may be the case that a greater burden of cumulative ADRD risk factors—including elevated midlife vascular risk factors but also many seldom‐measured, upstream social determinants—explains elevations in ADRD incidence and prevalence in Black Americans, a notion consistent with the NIA Health Disparities Research Framework. 19 However, as we have repeatedly emphasized, more research in this area, and in larger and more representative samples, is greatly needed.
6. THE PROMISE OF PLASMA BIOMARKERS AS A MEANS TO IMPROVE DIVERSITY IN AD RESEARCH
Over the past decade, with refinement of collection and processing procedures as well as analytics, blood‐based or plasma AD biomarkers are emerging as reliable and valid alternatives to biomarkers obtained with more invasive collection procedures, for example, lumbar puncture and radioactive tracers (see O'Bryant et al. 94 and Blennow & Zetterberg 95 for reviews). Blood‐based biomarkers could be described as a “holy grail” of biomarker AD research. A simple, inexpensive, non‐invasive test for AD can accelerate observational epidemiological, genetic, and clinical trial research; diagnostics in the clinic; and, when specific AD treatments are developed, allow appropriate identification of patients who would benefit most from AD‐targeted treatment. Crucially, advances in blood‐based biomarkers also present an opportunity to move data collection out of the laboratory and into the community, offering a practical, patient‐oriented strategy for broad population assessment. Likewise, blood‐based biomarkers could also offer a means to mitigate significant barriers to participation in research, 96 and especially barriers unique to AD biomarker research. 97 However, data on these blood biomarkers have been largely collected in non‐Hispanic White populations; this is a knowledge gap that now can and must be addressed.
Highly precise blood Aβ biomarkers, listed in Table 3, were recently reported to have high concordance with both amyloid PET scans and CSF Aβ42/Aβ40 ratio, 98 the gold standard biomarkers for amyloid plaques. These recent developments were enabled by highly specific and precise methods to measure Aβ in plasma by mass spectrometry, 99 but there are also promising results from immunoassays with diagnostic accuracies for Aβ pathology of greater than 80% and receiver operating characteristics (ROC) area under the curve (AUC) of > 0.85. 100 In individuals who are amyloid PET‐negative but Aβ‐positive by blood test, there is a 15‐fold greater risk of converting to amyloid PET positivity over time compared to those with a negative blood test result. 98 These results seem to suggest that plasma Aβ may be an earlier detector of Aβ plaque deposition than amyloid PET.
Recently developed methods have made it possible to measure p‐tau181 concentration in blood. Tau PET, CSF p‐tau181, and plasma p‐tau181 are highly correlated when an ultra‐sensitive assay for the latter biomarker is used. 101 Very similar results were generated using an immunoassay with electrochemiluminescence detection. 102 , 103 Interestingly, high plasma p‐tau181 concentration has been found in tau PET‐negative individuals who have evidence of brain amyloidosis by amyloid PET. 101 The validity of plasma p‐tau181 tests are further supported by evidence that this biomarker predicts subsequent AD dementia in cognitively unimpaired individuals and patients with MCI, 102 shows a significant increase in pre‐symptomatic familial AD (FAD) mutation carriers more than a decade before estimated symptom onset, 104 and accurately (AUC 0.97) predicts AD post mortem pathology. 105 Importantly, the increase in plasma p‐tau181 has consistently been found to be specific for Aβ plaque pathology in AD in that levels are normal in other neurodegenerative disorders. 105 Recent data suggest that p‐tau217 might be an even better blood biomarker for AD, 106 potentially by being more central nervous system–specific and/or more related to AD‐specific tau phosphorylation compared to p‐tau181, which might also reflect physiological plasticity processes. 107
Finally, among the fluid biomarkers, NfL is the blood biomarker that has been the easiest to develop into a blood test. Virtually all CSF NfL findings described above have been replicated in blood with sensitive assays, corroborating that it can be used as a general blood test for neuronal injury/neurodegeneration. 108
Taken together, the impact of blood‐based measures of AD pathology would be profound in the clinic, as well as in epidemiological and clinical studies. The blood‐based biomarkers could democratize our currently imperfect (biased) clinical diagnostics and give information on the A (plasma Aβ42/Aβ40), T (plasma p‐tau, with the potential caveat that it may be a predictive rather than a direct tau pathology marker), and N (plasma NfL) criteria of the AT(N) framework. Further, blood‐based biomarkers could facilitate access to future anti‐amyloid treatments, and relevant for the current paper, greater representation in research studies and clinical trials.
7. CONCLUSIONS
In this paper we propose applying the NIA Health Disparities Research Framework within the NIA–AA Research Framework. In particular, we believe considering the effects of racialization using the Health Disparities Research Framework will shed light on those factors driving AT(N) changes described in the NIA–AA Research Framework.
For example, we can improve our methods and current data collection practices. Self‐reported indicators of specific ethnicity should be collected routinely, and participants must be given the opportunity to describe a multiracial background. Data on social determinants of health can improve AD research rigor and reach across areas: it can illuminate both sources of selection bias for biomarker study recruitment, and mechanisms of brain aging. Collection should be broadened to include variables more nuanced than years of education; for example, quality of education, experiences of discrimination, neighborhood characteristics, acute and chronic stress, and social network and availability of support, among other life course factors. When assessing cognitive function, inclusive normative data should be used to avoid over‐pathologizing participants from minoritized communities. In the meantime, our analytic approaches should move beyond Black and White racial comparisons to recenter brain health and AD prevention within Black populations as an achievable goal. 79 Adequately powered samples allowing for stratified analyses, for instance, could clarify population‐salient risk distributions and effect sizes. Similarly, biomarkers should be interpreted using inclusive cut‐points. Additionally, the results of the current review suggest numerous opportunities to improve diversity in ADRD research to more accurately reflect the general population. Recruitment efforts should seek to include more population‐representative participants, not just those with socioeconomic advantages that enable participation in high‐burden research. For example, population‐based biomarker studies with representative samples would help address concerns about recruitment and ascertainment biases. Finally, given the historical trauma and practical burden associated with CSF collection for many Black participants, examining amyloid and tau could be accomplished with plasma measurements rather than CSF.
We close with an optimism for a future of improved inclusion of diverse populations in ADRD research, such that we more accurately represent the general population. AD biomarkers are increasingly more accessible. Blood‐based biomarkers could help overcome barriers to research participation and facilitate sample collection in community‐based settings and away from academic medical centers. The methods would also make longitudinal sample collection and life course studies easier, offering opportunities to more rigorously explore timing, including sensitive periods as well as temporal order of exposures and outcomes. We could use community‐based participatory research methods and population‐based enrollment practices to partner with groups typically under‐represented in ADRD research. 109 An integrated approach framed within a multi‐level life course model can give rise to innovative research designs, relevant data contextualization, and comprehensive interventions. Such an approach holds promise to accelerate discoveries serving to promote health equity. Inarguably, there is much work to be done.
CONFLICTS OF INTEREST
CEG receives funding from the NIA‐NIH (R01 AG054059, RF1 AG027161, R01AG070883, RF1AG057784, P30 AG062715, AARF‐18‐562958). As a member of the Alzheimer's Association Scientific Program Committee, CEG receives support for travel to AAIC. CEG is a member of Executive Board for the Alzheimer's and Dementia Alliance of Wisconsin. MZ receives funding from NIA‐NIH (R03 AG063304, P30AG062715) and Alzheimer's Association (AARF‐18‐562958). MZ is Vice Chair of ISTAART Diversity & Disparities PIA. DCG is a board member of Society for Research in Psychopathology (SRP) and Schizophrenia International Research Society (SIRS). Additionally, DCG serves as Vice President of NAMI‐Dane County Board, Deputy Editor of Psychiatry Research (honorarium paid), and advisory board member of Black Leaders for Brain Health. AJHK receives funding from the National Institutes of Health National Institute on Aging and National Institute on Minority Health and Health Disparities and from the US Department of Veterans Affairs. AJHK has received speaking honoraria at a variety of US academic universities, as well as travel costs paid for by university academic entities for grand rounds presentations, visiting professorships, or external advisory committee work. AJL receives funding from ADRD Prevention Messaging to Increase Cessation Attempts in Older Adult Smokers (NIH‐NIA K23AG067929) and Wisconsin ADRC Developmental Project Award (NIH‐NIA P30AG534255). AJL is a Treatment Network–Educational subcommittee Co‐Chair for the Society for Research in Nicotine & Tobacco (SRNT) annual meetings. TTJ receives funding from NIA minority supplement: grant to the Institution. NHL has no conflicts of interest. MFW receives funding from the University of Wisconsin ICTR Pilot Funding Program. MFW was supported in part by fellowship funding from the US Department of Veterans Affairs. This content does not represent the views of the U.S. Department of Veterans Affairs or the United States Government. MFW has received financial support for the Vanderbilt University/Middle Tennessee Geriatrics Conference Keynote address, as an employee of Veteran Affairs, and as a trainee of the University of Wisconsin. MFW is involved in the Veterans Affairs Dementia Friendly Hospital Committee of the W.S. Middleton Memorial Veterans Hospital. FK has no conflicts of interest. AG has no conflicts of interest. SCJ receives funding from the NIH and Cerveau technologies to institution. SCJ serves on an advisory board for Roche Diagnostics. BBB receives funding from NIA‐NIH (R01AG070973, R01AG070883, R01AG062285, R01AG062285, R01AG059312, RF1AG057784, R01AG037639). BBB participated as DSMB chair for R21AG056882 Network‐Level Mechanisms for Preclinical Alzheimer's Disease Development, Levetiracetam (Keppra) Clinical Trial. BBB has received precursors and imaging agents from Avid Radiopharmaceuticals. HZ has served on scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx; has given lectures in symposia sponsored by Fujirebio, Alzecure, and Biogen; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). HZ serves as chair of the Alzheimer's Association Biofluid Biomarker‐Based Professional Interest Area. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL.
ACKNOWLEDGMENTS
This work was supported by the National Institute on Aging–National Institutes of Health (NIA‐NIH; R01 AG054059, PI Carey E. Gleason, PhD; RF1 AG027161, PI Sterling C. Johnson, PhD; R01AG070883, PI Amy J.H. Kind, MD, PhD, Co‐PI Barbara Bendlin, PhD; RF1AG057784, PI Amy J.H. Kind, MD, PhD, Co‐PI Barbara Bendlin, PhD; and the Wisconsin ADRC P30 AG062715, PI Sanjay Asthana, MD); the Alzheimer's Association (AARF‐18‐562958, PI Megan Zuelsdorff, PhD); and by use of resources and facilities at the William S. Middleton Memorial Veterans Hospital, Madison, WI.
This is GRECC manuscript number: 002‐2021
Gleason CE, Zuelsdorff M, Gooding DC, et al. Alzheimer's disease biomarkers in Black and non‐hispanic White cohorts: A contextualized review of the evidence. Alzheimer's Dement. 2022;18:1545–1564. 10.1002/alz.12511
Barbara B. Bendlin and Henrik Zetterberg are co‐senior authors.
REFERENCES
- 1. Mayeda ER, Glymour MM, Quesenberry CP, Whitmer RA. Inequalities in dementia incidence between six racial and ethnic groups over 14 years. Alzheimer's. Dement. 2016; 12: 216‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weuve J, Barnes LL, Mendes de Leon CF, et al. Cognitive aging in black and White Americans: cognition, cognitive decline, and incidence of Alzheimer's disease dementia. Epidemiology. 2018; 29: 151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hebert LE, Bienias JL, Aggarwal NT, et al. Change in risk of Alzheimer's disease over time. Neurology. 2010; 75: 786‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tang MX, Cross P, Andrews H, et al. Incidence of AD in African‐Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology. 2001; 56: 49‐56. [DOI] [PubMed] [Google Scholar]
- 5. Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer's disease incidence: a prospective cohort study. Arch Neurol. 2002; 59: 1737‐1746. [DOI] [PubMed] [Google Scholar]
- 6. Plassman BL, Langa KM, McCammon RJ, et al. Incidence of dementia and cognitive impairment, not dementia in the United States. Ann Neurol. 2011; 70: 418‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perkins P, Annegers JF, Doody RS, Cooke N, Aday L, Vernon SW. Incidence and prevalence of dementia in a multiethnic cohort of municipal retirees. Neurology. 1997; 49: 44‐50. [DOI] [PubMed] [Google Scholar]
- 8. Rajan KB, Weuve J, Barnes LL, Wilson RS, Evans DA. Prevalence and incidence of clinically diagnosed Alzheimer's disease dementia from 1994 to 2012 in a population study. Alzheimer's Dement. 2019; 15: 1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steenland K, Goldstein FC, Levey A, Wharton W. A meta‐analysis of Alzheimer's disease incidence and prevalence comparing African‐Americans and Caucasians. J Alzheimer's Dis. 2016; 50: 71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mehta KM, Yeo GW. Systematic review of dementia prevalence and incidence in United States race/ethnic populations. Alzheimer's. Dement. 2017; 13: 72‐83. [DOI] [PubMed] [Google Scholar]
- 11. Garrett SL, McDaniel D, Obideen M, et al. Racial disparity in cerebrospinal fluid amyloid and tau biomarkers and associated cutoffs for mild cognitive impairment. JAMA Netw Open. 2019; 2: e1917363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris JC, Schindler SE, McCue LM, et al. Assessment of racial disparities in biomarkers for Alzheimer's disease. JAMA Neurol. 2019; 76: 264‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Howell JC, Watts KD, Parker MW, et al. Race modifies the relationship between cognition and Alzheimer's disease cerebrospinal fluid biomarkers. Alzheimers Res Ther. 2017; 9: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gottesman RF, Schneider AL, Zhou Y, et al. The ARIC‐PET amyloid imaging study: brain amyloid differences by age, race, sex, and APOE. Neurology. 2016; 87: 473‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meeker KL, Wisch JK, Hudson D, et al. Socioeconomic status mediates racial differences seen using the AT(N) framework. Ann Neurol. 2021; 89: 254‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar VV, Huang H, Zhao L, et al. Baseline results: the association between cardiovascular risk and preclinical Alzheimer's disease pathology (ASCEND) study. J Alzheimers Dis. 2020; 75: 109‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deters KD, Napolioni V, Sperling RA, et al. Amyloid PET imaging in self‐identified non‐Hispanic Black participants of the anti‐amyloid in asymptomatic Alzheimer's disease (A4) Study. Neurology. 2021; 96: e1491‐e500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018; 14: 535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hill CV, Perez‐Stable EJ, Anderson NA, Bernard MA. The National Institute on aging health disparities research framework. Ethn Dis. 2015; 25: 245‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Montine KS, Montine TJ. Anatomic and clinical pathology of cognitive impairment and dementia. J Alzheimers Dis. 2013; 33 Suppl 1: S181‐S184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Glymour MM, Brickman AM, Kivimaki M, et al. Will biomarker‐based diagnosis of Alzheimer's disease maximize scientific progress? Evaluating proposed diagnostic criteria. Eur J Epidemiol. 2018; 33: 607‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zuliani G, Trentini A, Rosta V, et al. Increased blood BACE1 activity as a potential common pathogenic factor of vascular dementia and late onset Alzheimer's disease. Sci Rep. 2020; 10: 14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mattsson N, Zegers I, Andreasson U, et al. Reference measurement procedures for Alzheimer's disease cerebrospinal fluid biomarkers: definitions and approaches with focus on amyloid beta42. Biomark Med. 2012; 6: 409‐417. [DOI] [PubMed] [Google Scholar]
- 24. Manly JJ, Comment on do Alzheimer's biomarkers vary by race? Do Alzheimer's Biomarkers Vary by Race?, 2020. ed. AlzForum2019.
- 25. Barnes LL, Bennett DA. Alzheimer's disease in African Americans: risk factors and challenges for the future. Health Aff (Millwood). 2014; 33: 580‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caulfield T, Fullerton SM, Ali‐Khan SE, et al. Race and ancestry in biomedical research: exploring the challenges. Genome Med. 2009; 1: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ali‐Khan SE, Krakowski T, Tahir R, Daar AS. The use of race, ethnicity and ancestry in human genetic research. Hugo J. 2011; 5: 47‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collins FS. What we do and don't know about ‘race’, ‘ethnicity’, genetics and health at the dawn of the genome era. Nat Genet. 2004; 36: S13‐S15. [DOI] [PubMed] [Google Scholar]
- 29. Omi M, Winant H. Racial Formation in the United States. 3rd ed. New York: Routledge/Taylor & Francis Group; 2015. [Google Scholar]
- 30. Kalewold KH. Race and medicine in light of the new mechanistic philosophy of science. Biology & Philosophy. 2020; 35: 1‐22. [Google Scholar]
- 31. Collins JW Jr, Wu SY, David RJ. Differing intergenerational birth weights among the descendants of US‐born and foreign‐born Whites and African Americans in Illinois. Am J Epidemiol. 2002; 155: 210‐216. [DOI] [PubMed] [Google Scholar]
- 32. Hendrie HC, Osuntokun BO, Hall KS, et al. Prevalence of Alzheimer's disease and dementia in two communities: Nigerian Africans and African Americans. Am J Psychiatry. 1995; 152: 1485‐1492. [DOI] [PubMed] [Google Scholar]
- 33. Hendrie HC, Ogunniyi A, Hall KS, et al. Incidence of dementia and Alzheimer's disease in 2 communities: yoruba residing in Ibadan, Nigeria, and African Americans residing in Indianapolis, Indiana. JAMA. 2001; 285: 739‐747. [DOI] [PubMed] [Google Scholar]
- 34. Hendrie HC, Murrell J, Baiyewu O, et al. APOE ɛ4 and the risk for Alzheimer's disease and cognitive decline in African Americans and Yoruba. Int Psychogeriatr. 2014; 26: 977‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hendrie HC, Hall KS, Ogunniyi A, Gao S. Alzheimer's disease, genes, and environment: the value of international studies. Can J Psychiatry. 2004; 49: 92‐99. [DOI] [PubMed] [Google Scholar]
- 36. Simms M, Say African American or Black, but first acknowledge the persistence of structural racism. Urban Wire: Race and Ethnicity: Urban Institute; 2018.
- 37. Clemons AM. New Blacks: language, DNA, and the construction of the African American/Dominican boundary of difference. Genealogy. 2021; 5: 1. [Google Scholar]
- 38. Babulal GM, Quiroz YT, Albensi BC, et al. Perspectives on ethnic and racial disparities in Alzheimer's disease and related dementias: update and areas of immediate need. Alzheimers Dement. 2019; 15: 292‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hall KS, Gao S, Baiyewu O, et al. Prevalence rates for dementia and Alzheimer's disease in African Americans: 1992 versus 2001. Alzheimers Dement. 2009; 5: 227‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stephan BCM, Birdi R, Tang EYH, et al. Secular trends in dementia prevalence and incidence worldwide: a systematic review. J Alzheimers Dis. 2018; 66: 653‐680. [DOI] [PubMed] [Google Scholar]
- 41. Chen C, Zissimopoulos JM. Racial and ethnic differences in trends in dementia prevalence and risk factors in the United States. Alzheimers Dement (N Y). 2018; 4: 510‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fillenbaum GG, Heyman A, Huber MS, et al. The prevalence and 3‐year incidence of dementia in older Black and White community residents. J Clin Epidemiol. 1998; 51: 587‐595. [DOI] [PubMed] [Google Scholar]
- 43. Katz MJ, Lipton RB, Hall CB, et al. Age‐specific and sex‐specific prevalence and incidence of mild cognitive impairment, dementia, and Alzheimer's dementia in blacks and whites: a report from the Einstein Aging Study. Alzheimer's. Dis Assoc Disord. 2012; 26: 335‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sisco S, Gross AL, Shih RA, et al. The role of early‐life educational quality and literacy in explaining racial disparities in cognition in late life. J Gerontol B Psychol Sci Soc Sci. 2015; 70: 557‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manly JJ, Jacobs DM, Touradji P, Small SA, Stern Y. Reading level attenuates differences in neuropsychological test performance between African American and White elders. J Int Neuropsychol Soc. 2002; 8: 341‐348. [DOI] [PubMed] [Google Scholar]
- 46. Fitzpatrick AL, Kuller LH, Ives DG, et al. Incidence and prevalence of dementia in the cardiovascular health study. J Am Geriatr Soc. 2004; 52: 195‐204. [DOI] [PubMed] [Google Scholar]
- 47. Yaffe K, Falvey C, Harris TB, et al. Effect of socioeconomic disparities on incidence of dementia among biracial older adults: prospective study. BMJ. 2013; 347: f7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zsoldos E, Filippini N, Mahmood A, et al. Allostatic load as a predictor of grey matter volume and white matter integrity in old age. The Whitehall II MRI study Sci Rep. 2018; 8: 6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karlamangla AS, Miller‐Martinez D, Lachman ME, Tun PA, Koretz BK, Seeman TE. Biological correlates of adult cognition: midlife in the United States (MIDUS). Neurobiol Aging. 2014; 35: 387‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seeman TE, McEwen BS, Rowe JW, Singer BH. Allostatic load as a marker of cumulative biological risk: macArthur studies of successful aging. Proc Natl Acad Sci U S A. 2001; 98: 4770‐4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Justice NJ. The relationship between stress and Alzheimer's disease. Neurobiol Stress. 2018; 8: 127‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weiner MW, Harvey D, Hayes J, et al. Effects of traumatic brain injury and posttraumatic stress disorder on development of Alzheimer's disease in Vietnam Veterans using the Alzheimer's Disease neuroimaging initiative: preliminary report. Alzheimer's Dement (N Y). 2017; 3: 177‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bello MS, Liautaud MM, De La Cerda JT, et al. Association of frequency of perceived exposure to discrimination with tobacco withdrawal symptoms and smoking lapse behavior in African Americans. Addiction. 2021; 116: 914‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lutz RS, Lochbaum MR, Lanning B, Stinson LG, Brewer R. Cross‐lagged relationships among leisure‐time exercise and perceived stress in blue‐collar workers. J Sport Exerc Psychol. 2007; 29: 687‐705. [DOI] [PubMed] [Google Scholar]
- 55. Spruill TM, Butler MJ, Thomas SJ, et al. Association between high perceived stress over time and incident hypertension in Black adults: findings from the Jackson Heart Study. J Am Heart Assoc. 2019; 8: e012139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Whitaker KM, Everson‐Rose SA, Pankow JS, et al. Experiences of discrimination and incident type 2 diabetes mellitus: the multi‐ethnic study of atherosclerosis (MESA). Am J Epidemiol. 2017; 186: 445‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zuelsdorff M, Okonkwo OC, Norton D, et al. Stressful life events and racial disparities in cognition among middle‐aged and older adults. J Alzheimers Dis. 2020; 73: 671‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zahodne LB, Morris EP, Sharifian N, Zaheed AB, Kraal AZ, Sol K. Everyday discrimination and subsequent cognitive abilities across five domains. Neuropsychology. 2020; 34: 783‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barnes LL, Wilson RS, Everson‐Rose SA, Hayward MD, Evans DA, Mendes de Leon CF. Effects of early‐life adversity on cognitive decline in older African Americans and Whites. Neurology. 2012; 79: 2321‐2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mehta PK, Kieltyka L, Bachhuber MA, et al. Racial inequities in preventable pregnancy‐related deaths in Louisiana, 2011‐2016. Obstet Gynecol. 2020; 135: 276‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Werry AE, Daniel M, Bergstrom B. Group differences in normal neuropsychological test performance for older non‐Hispanic White and Black/African American adults. Neuropsychology. 2019; 33: 1089‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rossetti HC, Lacritz LH, Hynan LS, Cullum CM, Van Wright A, Weiner MF. Montreal cognitive assessment performance among community‐dwelling African Americans. Arch Clin Neuropsychol. 2017; 32: 238‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kalkonde YV, Pinto‐Patarroyo GP, Goldman T, et al. Ethnic disparities in the treatment of dementia in veterans. Dement Geriatr Cogn Disord. 2009; 28: 145‐152. [DOI] [PubMed] [Google Scholar]
- 64. Gianattasio KZ, Prather C, Glymour MM, Ciarleglio A, Power MC. Racial disparities and temporal trends in dementia misdiagnosis risk in the United States. Alzheimers Dement (N Y). 2019; 5: 891‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Maurik IS, Vos SJ, Bos I, et al. Biomarker‐based prognosis for people with mild cognitive impairment (ABIDE): a modelling study. Lancet Neurol. 2019; 18: 1034‐1044. [DOI] [PubMed] [Google Scholar]
- 66. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol. 2004; 55: 306‐319. [DOI] [PubMed] [Google Scholar]
- 67. Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F‐18]‐AV‐1451 (T807) on postmortem brain tissue. Ann Neurol. 2015; 78: 787‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. de Leon MJ, Ferris SH, George AE, et al. Computed tomography and positron emission transaxial tomography evaluations of normal aging and Alzheimer's disease. J Cereb Blood Flow Metab. 1983; 3: 391‐394. [DOI] [PubMed] [Google Scholar]
- 69. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer's disease: a longitudinal study. JAMA Neurol. 2019; 76: 915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scheltens P, Leys D, Barkhof F, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer's disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry. 1992; 55: 967‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gladman JT, Corriveau RA, Debette S, et al. Vascular contributions to cognitive impairment and dementia: research consortia that focus on etiology and treatable targets to lessen the burden of dementia worldwide. Alzheimers Dement (N Y). 2019; 5: 789‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wardlaw JM, Valdes Hernandez MC, Munoz‐Maniega S. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc. 2015; 4: 001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rivera‐Rivera LA, Turski P, Johnson KM, et al. 4D flow MRI for intracranial hemodynamics assessment in Alzheimer's disease. J Cereb Blood Flow Metab. 2016; 36: 1718‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Banerjee G, Wilson D, Jager HR, Werring DJ. Novel imaging techniques in cerebral small vessel diseases and vascular cognitive impairment. Biochim Biophys Acta. 2016; 1862: 926‐938. [DOI] [PubMed] [Google Scholar]
- 75. Van Hulle CA, Jonaitis E, Betthauser TJ. An examination of a novel multipanel of CSF biomarkers in the Alzheimer's disease clinical and pathological continuum. Alzheimers Dement. 2021; 17: 431‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Scholl M, Maass A, Mattsson N, et al. Biomarkers for tau pathology. Mol Cell Neurosci. 2019; 97: 18‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sato C, Barthelemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018; 98: 861‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bridel C, van Wieringen WN, Zetterberg H, and the NFLG et al, . Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019; 17: 1035‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Whitfield KE, Allaire JC, Belue R, Edwards CL. Are comparisons the answer to understanding behavioral aspects of aging in racial and ethnic groups?. J Gerontol B Psychol Sci Soc Sci. 2008; 63: P301‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gottesman RF, Schneider AL, Zhou Y, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA. 2017; 317: 1443‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer's disease. A meta‐analysis. APOE and Alzheimer's Disease meta analysis consortium. JAMA. 1997; 278: 1349‐1356. [PubMed] [Google Scholar]
- 82. Burchard EG, Ziv E, Coyle N, et al. The importance of race and ethnic background in biomedical research and clinical practice. N Engl J Med. 2003; 348: 1170‐1175. [DOI] [PubMed] [Google Scholar]
- 83. Kaplan JB, Bennett T. Use of race and ethnicity in biomedical publication. JAMA. 2003; 289: 2709‐2716. [DOI] [PubMed] [Google Scholar]
- 84. Willette AA, Pappas C, Hoth N, et al. Inflammation, negative affect, and amyloid burden in Alzheimer's disease: insights from the kynurenine pathway. Brain Behav Immun. 2021; 95: 216‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Willette AA, Johnson SC, Birdsill AC, et al. Insulin resistance predicts brain amyloid deposition in late middle‐aged adults. Alzheimer's Dement. 2015; 11: 504‐510.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Willette AA, Xu G, Johnson SC, et al. Insulin resistance, brain atrophy, and cognitive performance in late middle‐aged adults. Diabetes Care. 2013; 36: 443‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hoscheidt SM, Starks EJ, Oh JM, et al. Insulin resistance is associated with increased levels of cerebrospinal fluid biomarkers of Alzheimer's disease and reduced memory function in at‐risk healthy middle‐aged adults. J Alzheimer's dis. 2016; 52: 1373‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wilkins CH, Schindler SE, Morris JC. Addressing health disparities among minority populations: why clinical trial recruitment is not enough. JAMA Neurology. 2020; 77: 1063‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Olivari BS, French ME, McGuire LC. The public health road map to respond to the growing dementia crisis. Innov Aging. 2020; 4: igz043‐igz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gleason CE, Norton D, Zuelsdorff M, et al. Association between enrollment factors and incident cognitive impairment in blacks and whites: data from the Alzheimer's disease center. Alzheimer's Dement. 2019; 15: 1533‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Barnes LL, Leurgans S, Aggarwal NT, et al. Mixed pathology is more likely in black than white decedents with Alzheimer's dementia. Neurology. 2015; 85: 528‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Clark LR, Zuelsdorff M, Norton D, et al. Association of cardiovascular risk factors with cerebral perfusion in Whites and African Americans. J Alzheimers Dis. 2020; 75: 649‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Clark LR, Norton D, Berman SE, et al. Association of cardiovascular and Alzheimer's disease risk factors with intracranial arterial blood flow in Whites and African Americans. J Alzheimers Dis. 2019; 72: 919‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. O'Bryant SE, Mielke MM, Rissman RA, et al. Blood‐based biomarkers in Alzheimer's disease: current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017; 13: 45‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Blennow K, Zetterberg H. Biomarkers for Alzheimer's disease: current status and prospects for the future. J Intern Med. 2018; 284: 643‐663. [DOI] [PubMed] [Google Scholar]
- 96. George S, Duran N, Norris K. A systematic review of barriers and facilitators to minority research participation among African Americans, Latinos, Asian Americans, and Pacific Islanders. Am J Public Health. 2014; 104: e16‐e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Howell JC, Parker MW, Watts KD, Kollhoff A, Tsvetkova DZ, Hu WT. Research lumbar punctures among African Americans and Caucasians: perception predicts experience. Front Aging Neurosci. 2016; 8: 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma beta‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019; 93: e1647‐e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zetterberg H, Bendlin BB. Biomarkers for Alzheimer's disease‐preparing for a new era of disease‐modifying therapies. Mol Psychiatry. 2021; 26: 296‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lifke V, Kollmorgen G, Manuilova E, et al. Elecsys((R)) total‐tau and phospho‐tau (181P) CSF assays: analytical performance of the novel, fully automated immunoassays for quantification of tau proteins in human cerebrospinal fluid. Clin Biochem. 2019; 72: 30‐38. 81]Kaplan JB, Bennett T. Use of race and ethnicity in biomedical publication. JAMA 2003;289:2709‐16. [DOI] [PubMed] [Google Scholar]
- 101. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020; 19: 422‐433. [DOI] [PubMed] [Google Scholar]
- 102. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's. dementia Nat Med. 2020; 26: 379‐386. [DOI] [PubMed] [Google Scholar]
- 103. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020; 26: 387‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. O'Connor A, Karikari TK, Poole T, et al. Plasma phospho‐tau181 in presymptomatic and symptomatic familial Alzheimer's disease: a longitudinal cohort study. Mol Psychiatry. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lantero Rodriguez J, Karikari TK, Suarez‐Calvet M, et al. Plasma p‐tau181 accurately predicts Alzheimer's disease pathology at least 8 years prior to post‐mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020; 140: 267‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho‐tau217 for Alzheimer's disease vs other neurodegenerative disorders. JAMA. 2020; 324: 772‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Barthelemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated‐tau isoforms track CNS change in Alzheimer's disease. J Exp Med. 2020; 217: e20200861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019; 90: 870‐881. [DOI] [PubMed] [Google Scholar]
- 109. NIH guidelines on the inclusion of women and minorities as subjects in clinical research In: Health NIo, editor. 59 FR 11146‐11151. Bethesda, MD, 1993. [Google Scholar]