

Abstract
The cleavage and functionalization of recalcitrant carbon─carbon bonds is highly challenging but represents a very powerful tool for value-added transformation of feedstock chemicals. Here, an enzyme-mimic iron single-atom catalyst (SAC) bearing iron (III) nitride (FeN3) motifs was prepared and found to be robust for cleavage and cyanation of carbon–carbon bonds in secondary alcohols and ketones. High nitrile yields are obtained with a wide variety of functional groups. The prepared FeN3-SAC exhibits high enzyme-like activity and is capable of generating a dioxygen-to-superoxide radical at room temperature, while the commonly reported FeN4-SAC bearing FeN4 motifs was inactive. Density functional theory (DFT) calculation reveals that the activation energy of dioxygen activation and the activation energy of the rate-determining step of nitrile formation are lower over FeN3-SAC than FeN4-SAC. In addition, DFT calculation also explains the catalyst’s high selectivity for nitriles.
A practical method for nitrile formation via Fe-catalyzed cleavage and cyanation of C─C bonds in alcohols and ketones.
INTRODUCTION
Nitriles are a ubiquitous class of compounds present in natural products, agrochemicals, and pharmaceuticals and serve as the versatile intermediates in organic synthesis (1). Hence, the development of efficient methodologies toward nitrile synthesis continues to be scientifically interesting and attracts the attention of the synthetic community (2). Classical methods to nitrile synthesis include transition metal–catalyzed cyanation of aryl halides (3), Sandmeyer reaction (4), and direct cyanation of C─H bonds (5, 6). While the methods are successful at accessing arene cyanation under homogeneous conditions, complications associated with stability, separation, and recyclability of catalyst, large-scale production, and metal residues are still a challenge. Moreover, toxic organic or inorganic cyanides and stoichiometric metal oxidants have generally been used, thus posing a notable safety and environment concern (Fig. 1A). Therefore, a sustainable, cyanide-free, and one-step direct construction of aryl nitriles under heterogeneous catalytic conditions remains highly desired, especially in pharmaceutical-oriented organic synthesis (7, 8).
Fig. 1. Representative examples for the cyanation of alcohols and carbonyl compounds.

(A) Traditional cyanation. (B) Representative example for alcohols or carbonyl compounds to nitriles. (C) This work: Oxidative cleavage and cyanation of secondary alcohols or ketones to nitriles.
Alcohols and carbonyl compounds (aldehydes and ketones) are broadly accessible and cheap feedstocks, and, thus, numerous efforts have been undertaken toward the development of versatile methods for the direct conversion of alcohols and carbonyl compounds to nitriles under cyanide-free conditions (9–11). Prominent methods include catalytic aerobic ammoxidation of primary alcohols or aldehydes into nitriles (Fig. 1B) (12). As for the construction of nitriles from secondary alcohols or ketones, the cleavage of C─C bond bearing high bond dissociation energy is inevitably involved (13). To the best of our knowledge, the one-step direct conversion of secondary alcohols or ketones into nitriles is still unexplored with NH3 as the nitrogen source, although a multistep conversion of secondary alcohols or ketones to nitriles via aldehydes or carboxylic acid intermediates has been reported (Fig. 1B) (14, 15).
In recent years, single-atom catalysts (SACs) have emerged as promising heterogeneous systems for various chemical reactions due to their maximal atom utilization, superior stability, and high activity and selectivity (16, 17). In particular, the single metal site of SACs with well-defined atomic structure and electronic coordination environments can effectively mimic the catalytic sites of natural enzymes, which has opened a previously unknown research frontier in the catalysis field (18, 19). It is well known that cytochrome P450 enzymes activate oxygen at heme iron centers to oxidize a broad spectrum of biologically active compounds under mild conditions (20). To mimic the active site of cytochrome P450, we designed two types of SACs bearing atomically dispersed Fe sites with three and four coordinated nitrogen atoms (FeN3-SAC and FeN4-SAC, respectively), which can be selectively fabricated via varying the pyrolysis temperature. The FeN3-SAC shows a high enzyme-like activity and is capable of activating O2 to superoxide radical anion at room temperature, while the commonly reported FeN4-SAC is inactive. Driven by the inherent advantages and exceptional performance for activating O2 of FeN3-SAC and as a continuation of our long-time interest in value-added transformation of chemical feedstocks via cleavage and functionalization of C─C bonds (21, 22), we intended to investigate the utilization of the enzyme-inspired FeN3-SAC for oxidative cleavage and cyanation of secondary alcohols and ketones. Here, we report a general, efficient, and practical method for nitrile formation through FeN3-SAC–catalyzed cleavage and cyanation of C─C bonds in secondary alcohols and ketones, with oxygen as the environmentally benign oxidant and ammonia as a nitrogen source. Density functional theory (DFT) calculation reveals the activation energy for the reduction of O2, and synthesis of nitrile is much lower over FeN3-SAC than FeN4-SAC (Fig. 1C). Therefore, the developed enzyme-mimic FeN3-SAC can serve as a bridge between enzymatic catalysis and heterogeneous catalysis.
RESULTS
Catalyst preparation and characterization
ZIF-8 with the zeolitic imidazolate framework was first prepared by the coordination of Zn2+ with 2-methylimidazole (23). Ferrocene and ZIF-8 were used as the precursors for the preparation of a single Fe atom catalyst (abbreviated as Fe-N3/NC-1100) via the gas diffusion strategy at 1100°C (Fig. 2A). During the pyrolysis process, ferrocene was first sublimed into a gas phase and then diffused into the ZIF-8 framework. At high pyrolysis temperature, ZIF-8 gradually transformed into a nitrogen-doped porous carbon polyhedron with single Fe atoms, while the Zn atoms in ZIF-8 were evaporated off because of the low boiling point of Zn at 908°C. The Fe content in Fe-N3/NC-1100 was determined to be 0.52 weight % (wt %) by inductively coupled plasma atomic emission spectrometry (ICP-AES) (table S1), while the Zn content was below the detection limit. The surface area and pore volume of Fe-N3/NC-1100 were determined to be 811 m2/g and 0.67 cm3/g by Brunauer-Emmett-Teller (BET) measurements (fig. S1 and table S1), respectively. Scanning electron microscope (SEM) and transmission electron microscope (TEM) images of Fe-N3/NC-1100 revealed that the as-prepared catalyst remained the rhombic dodecahedron-like morphology of the parent ZIF-8 (Fig. 2, B and C). No Fe nanoparticles were observed in the TEM and high-resolution TEM (HRTEM) images of Fe-N3/NC-1100 (Fig. 2, D and E). HRTEM image only showed the graphitic carbon layers with a d-spacing of 0.250 nm, corresponding to its (101) lattice plane (Fig. 2E). Powder x-ray diffraction (XRD) patterns of Fe-N3/NC-1100 also gave no diffraction peaks of Fe species but showed two weak peaks at 24.4° and 44.6°, assigning to the (002) and (101) crystal planes of graphitic carbon (fig. S2). Two peaks with the Raman shifts around 1350 cm−1 (called D bond) and 1570 cm−1 (called G bond) were observed in the Raman spectra of Fe-N3/NC-1100 (fig. S3), which were attributed to the defect carbon and graphitic carbon, respectively. The Fe species in Fe-N3/NC-1100 was identified by the aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM). As shown in Fig. 2F, single Fe atoms were identified by isolated bright dots and marked with red circles. X-ray photoelectron spectroscopy (XPS) of Fe 2p can be fitted into two types peaks for Fe2+ and Fe3+, suggesting that Fe atoms were bonded with other atoms with the valence states between +2 and +3 (fig. S4) (24). Five types of nitrogen atoms were fitted in the N 1s XPS spectra (fig. S5): pyridinic N (398.5 eV), Fe-Nx (399.3 eV), pyrrolic N (400.3 eV), graphitic N (401.2 eV), and oxidized N (404.2 eV). The O 1s XPS spectra were deconvoluted into three peaks with the binding energies at 533.6, 532.2, and 530.7 eV (Fig. 2G). The peak at 533.6 eV was assigned to the C─OH and C─O─OH group, and the peak at 532.2 eV was attributed to the C═O groups. The large peak at 530.7 eV was assigned to the absorbed oxygen molecules during the exposure of the Fe-N3/NC-1100 catalyst in the air, suggesting that Fe-N3/NC-1100 had a strong ability to adsorb O2 at room temperature (22, 25, 26).
Fig. 2. Preparation and characterization of the Fe-N3/NC-1100 catalyst.

(A) Catalyst preparation. (B to D) STEM images. (E) HRTEM image. (F) The aberration-corrected HAADF-STEM image (single-atom Fe sites are highlighted with red circles). (G) O 1s XPS spectra. (H) X-ray absorption near-edge structure (XANES) spectra at the Fe K-edge. (I) Fourier-transformed extended x-ray absorption fine structure (FT-EXAFS) spectra of the Fe foil, Fe3O4, FeO, and Fe-N3/NC-1100. (J) R space and (K) k space of Fe-N3/NC-1100. a.u., arbitrary units.
X-ray absorption near-edge structure (XANES) and extended x-ray absorption fine structure (EXAFS) spectroscopy at the Fe K-edge are performed to verify the electronic structure and coordination information. The absorption edge energy of Fe-N3/NC-1100 is located between that of FeO and Fe3O4, suggesting that the valence state of Fe-N3/NC-1100 is between +2 (FeO) and +8/3 (Fe3O4) (Fig. 2H). As shown in the Fourier-transformed (FT)–EXAFS spectra (Fig. 2I), Fe─Fe bond at 2.2 Å is not observed by comparison with the spectra of Fe foil, while a primary peak at 1.5 Å is present, which is associated with Fe-N(O) contributions, indicating the formation of the Fe-Nx configuration (27–30). Furthermore, the EXAFS wavelet transform plot exhibited only one intensity maximum at ~3.5 Å−1 in k space that was assigned to Fe-N(O) (fig. S6). A least-squares EXAFS curve fit is performed to achieve the quantitative structural parameters of Fe in Fe-N3/NC-1100 (Fig. 2, J and K, and table S3). The coordination numbers of the single Fe atom were calculated to be 4.8, where the coordination numbers for the nitrogen and oxygen atoms were 2.7 and 2.1, respectively. Therefore, the atomically dispersed Fe center is mainly coordinated with three N atoms and two O atoms.
Because of the instability of Fe─O bond at 1100°C (31) and the presence of adsorbed O2 in the O 1s XPS spectra, the Fe─O bonds should be derived from the axially coordinated O2 molecules. O2 temperature-programmed desorption (TPD) profiles of Fe-N3/NC-1100 also showed a strong desorption peak of O2 at room temperature (Fig. 3A), further confirming that the single Fe atoms had the strong affinity to O2. As far as the coordination of O2 molecules, one model is the only adsorption of one O2 molecule via a side-on coordination mode (fig. S7, A and C), in which the two oxygen atoms in one O2 molecule were simultaneously coordinated to the single Fe atom. The other model is the adsorption of two O2 molecules in the end-on configuration (fig. S7B), where two O2 molecules were coordinated with an Fe-N3 plane from the front and back, respectively. DFT calculation revealed that the optimal structure of Fe-N3/NC-1100 was present in the enclosed Fe-N3 plane with the single Fe atom axially coordinated by one O2 molecule via a side-on coordination mode (Fig. 3C and fig. S7A).
Fig. 3. Characterization and DFT calculation of the as-prepared Fe catalysts.

(A) O2-TPD profiles of Fe-N3/NC-1100 and Fe-N4/NC-800. (B) 5,5-dimethyl-1-pyrroline N-oxide (DMPO) spin-trapping electron paramagnetic resonance (EPR) spectrum of air with Fe catalysts in methanol. The optimized structure of O2 adsorption and activation on (C) Fe-N3/NC-1100 and (D) Fe-N4/NC-800. The projected electron density of (E) O2/Fe-N3/NC-1100 and (F) O2/Fe-N4/NC-800. (G) The charge variation of Fe atom upon O2 adsorption.
Catalyst screening
The catalytic activity of the as-prepared catalysts was first evaluated by the ammoxidation of benzaldehyde with O2, as the aldehydes are the key intermediates of the oxidative transformation of 1,2-diols, ketones, and secondary alcohols into nitriles. To our great pleasure, Fe-N3/NC-1100 demonstrated the super-high catalytic activity at room temperature under atmospheric oxygen pressure (Table 1, entry 1), in consistency with the above statements that Fe-N3/NC-1100–bearing Fe-N3 motifs showed a strong affinity to O2 at room temperature. Fe-N3/NC-1000 was also prepared at the pyrolysis temperature of 1000°C. The Fe content in Fe-N3/NC-1000 was higher than that in Fe-N3/NC-1100 (0.76 wt % versus 0.52 wt %), but 0.54 wt % of Zn was still present in Fe-N3/NC-1000 (table S1). The activity of Zn atoms in Fe-N3/NC-1000 could be excluded, as Zn/NC-1000 showed no activity, which was prepared by the pyrolysis of ZIF-8 at 1000°C (Table 1, entry 3). XRD patterns (fig. S2), Raman spectra (fig. S3), Fe 2p and N 1s XPS spectra (figs. S4 and S5), TEM images (fig. S8), and XANES and EXAFS (fig. S9) of Fe-N3/NC-1000 were almost the same as Fe-N3/NC-1100. Therefore, the Fe species in Fe-N3/NC-1000 should also be present in the single Fe atoms bearing Fe-N3 motifs. Although Fe-N3/NC-1000 had higher Fe weight percentage than Fe-N3/NC-1100, it delivered lower conversion (Table 1, entry 1 versus entry 2), possibly because of its smaller surface area (627 m2/g versus 811 m2/g) (fig. S1 and table S1), because the large specific surface area is more conducive to the adsorption of substrate molecules, and XPS (table S2 and fig. S5) shows that the content of Fe and Fe-Nx on the Fe-N3/NC-1100 surface is higher than content of Fe and Fe-Nx on the Fe-N3/NC-1000 surface. Besides the high activity, the single Fe atom catalysts also demonstrated an excellent selectivity of benzonitrile (>99%) without the formation of benzamide by-product (32). As expected, no target product was formed under nitrogen atmosphere (Table 1, entry 4), which suggested that Fe-N3/NC-1100 could not catalyze the dehydrogenative oxidation of aldimine intermediate (C6H5CH═NH) into benzonitrile. The oxidation of benzaldehyde was not observed in the absence of NH3·H2O (Table 1, entry 5), which suggested that the oxidation of benzaldehyde into benzoic acid was much more difficult than the oxidation of aldimine intermediate (C6H5CH═NH) into benzonitrile, and, thus, the excellent selectivity could be obtained in our catalytic system.
Table 1. Catalyst screening for the ammoxidation of benzaldehyde.*.
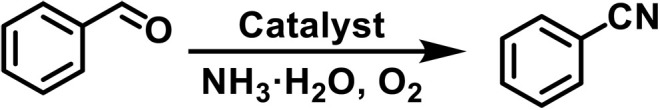
| Entry | Catalyst | T (°C) | Time (hours) | Con. (%) | Sel. (%) |
| 1 | Fe-N3/NC-1100 | 25 | 8 | 58 | >99 |
| 2 | Fe-N3/NC-1000 | 25 | 8 | 49 | >99 |
| 3 | Zn/NC-1000 | 25 | 8 | 0 | − |
| 4† | Fe-N3/NC-1100 | 25 | 8 | 0 | − |
| 5‡ | Fe-N3/NC-1100 | 25 | 8 | 0 | − |
| 6 | Fe-N4/NC-800 | 25 | 8 | 0 | − |
| 7 | Fe-N4/NC-800 | 150 | 8 | 37 | >99 |
| 8 | Ferrocene | 25 | 8 | 0 | − |
| 9 | Iron(II) phthalocyanine | 25 | 8 | 0 | − |
| 10 | FeCl3 | 25 | 8 | 0 | − |
| 11 | CuI | 25 | 8 | 0 | − |
| 12 | Cu(OAc)2 | 25 | 8 | 0 | − |
| 13§ | Se,S,N-CNs-1000 | 80 | 10 | 99 | >99 |
*Reaction conditions: Benzaldehyde (0.50 mmol), catalyst (20 mg), NH3·H2O (26.5 wt %, 200 μl) toluene (1.5 ml), O2 (1 bar), 25°C, and 8 hours.
†The reaction was performed under N2 atmosphere.
‡The reaction was performed in the absence of NH3·H2O.
§ The results were reported from (6), which were performed at 80°C and 10 bars of O2.
To further demonstrate the superior activity of Fe-N3/NC-1100, Fe-N4/NC-800 bearing Fe-N4 motifs was also prepared by the pyrolysis of Fe-coordinated 1,10-phenanthroline complex with the same procedures as reported in the reference (33). Preliminary characterizations (figs. S10 and S11) revealed that the Fe species in Fe-N4/NC-800 were also presented as single Fe atoms with a weight percentage of 1.0, being close to the reported data (33). However, Fe-N4/NC-800 showed no activity at room temperature, and it still delivered much lower conversion at 150°C than that over Fe-N3/NC-1100 at 25°C (Table 1, entry 1 versus entries 6 and 7). The great differences in the activation of O2 between Fe-N3/NC-1100 and Fe-N4/NC-800 were further studied by O2-TPD and electron paramagnetic resonance (EPR) technologies (Fig. 3, A and B) (34). Unlike Fe-N3/NC-1100 with a strong O2 desorption peak at room temperature, a very weak O2 desorption peak around 100°C was only observed for Fe-N4/NC-800 (Fig. 3A). In addition, no reductive oxygen species were detected by EPR technology over Fe-N4/NC-800 at room temperature, while the strong signal of the reductive oxygen species was observed over Fe-N3/NC-1100 (Fig. 3B). These results suggested that Fe-N3/NC-1100 bearing Fe-N3 motifs could more likely be regarded as a mimic of the enzymes such as hemoglobin enzyme and cytochrome P450 (fig. S12) rather than the commonly reported Fe-N4/NC-800–bearing Fe-N4 motifs.
Furthermore, DFT calculation was carried out to illustrate the difference in the catalytic activity between Fe-N4/NC-800 and Fe-N3/NC-1100. As shown in Fig. 3 (C and D), the adsorption energy of O2 over Fe-N3/NC-1100 was −2.467 eV, which was much larger than that over Fe-N4/NC-800 (−1.083 eV), suggesting that the single Fe atom catalyst bearing Fe-N3 motifs demonstrated much stronger affinity to O2 than that bearing Fe-N4 motifs. In addition, the bond length of O─O after the activation by Fe-N3/NC-1100 was larger than that by Fe-N4/NC-800 (1.392 Å versus 1.351 Å). DFT calculation further revealed that the electron transfer from the Fe center to activate O2 was larger over Fe-N3/NC-1100 than that over Fe-N4/NC-800 (0.292 eV versus 0.159 eV; Fig. 3, E to G). Both DFT and characterization data all revealed that Fe-N3/NC-1100 was robust to adsorb and activate O2. After the activation of O2, the oxidative dehydrogenation of the two H atoms (─NH and ─CH) in aldimine intermediate (C6H5CH═NH) generated the benzonitrile product. DFT calculation (Fig. 4) revealed that the oxidative dehydrogenation of the first H atom from N─H in C6H5CH═NH was the rate-determining step. The energy required for this step was 1.086 eV over Fe-N3/NC-1100, while that was high to 1.771 eV over Fe-N4/NC-800. DFT calculation also revealed that the ammoxidation of benzaldehyde over the catalyst with Fe-N3 motifs was much easier than that with Fe-N4 motifs, in good accordance with the experimental data (Table 1). In addition, the superior catalytic performance of Fe-N3/NC-1100 to other homogeneous transition metal catalysts was also compared. At the room temperature, homogenous Fe complex including ferrocene, iron(II) phthalocyanine, and FeCl3 showed no catalytic activity for the ammoxidation of benzaldehyde (Table 1, entries 8 to 10). The reported Cu salts with the ability to activate O2 (35, 36) were also unable to catalyze this transformation at room temperature (Table 1, entries 11 and 12). Moreover, we also compared the as-prepared Fe-N3/NC-1100 catalyst with the state-of-the art Se,S,N-tridoped carbon nanosheets (Se,S,N-CNs-1000) for the same reaction (Table 1, entries 13), which was performed at 80°C and 5 bars of O2 (7). The reaction rate of the Fe-N3/NC-1100 catalyst at room temperature and atmospheric oxygen pressure (1.8 × 10−3 mmol mgcatalyst−1 hour−1) was much higher than that over Se,S,N-CNs-1000 at 80°C and 5 bars of O2 pressure (1.0 × 10−3 mmol mgcatalyst−1 hour−1). In addition, the Fe content (~5 wt %) in Earth for the preparation of the single Fe atom catalyst was much more abundant than Se (trace) for the preparation of Se,S,N-CNs-1000. All these results listed in Table 1 demonstrate the unique advantages of the Fe-N3/NC-1100 room temperature activation of O2 molecules and the followed ammoxidation of aldehyde into nitriles.
Fig. 4. DFT calculation of the Fe-N3/NC-1100 and Fe-N4/NC-800 catalysts.

(A) The energy profile along the benzonitrile formation route [the oxidative dehydrogenation of the two hydrogen atoms (─NH and ─CH) in aldimine intermediate (C6H5CH═NH) generated benzonitrile] on Fe-N3/NC-1100 (blue line) and Fe-N4/NC-800 (red line) catalysts. (B) The main structures of benzonitrile formation on Fe-N3/NC-1100 catalyst.
Oxidative transformation of aldehydes into nitriles at room temperature
Delighted by the excellent performance of Fe-N3/NC-1100 toward the oxidative cyanation of benzaldehyde to benzonitrile, the ammoxidation of different kinds of aldehydes into nitriles were also studied. As shown in Fig. 5, different kinds of aromatic and fused ring aromatic aldehydes (S1 to S19), regardless of the electronic properties of the substituted groups, were successfully transformed into the corresponding benzonitrile derivates with excellent to quantitative yields at room temperature and atmospheric oxygen pressure. Substrates with dialdehyde groups (S8 and S9) were smoothly transformed into the dinitriles with yields in 95 to 97%. No steric hindrance was observed for the substrates with one to three substituted groups due to the introduction of linear nitrile group, which was very important for the synthesis of bioactive molecules bearing multiply substituted groups. For example, 2,6-dichlorobenzonitrile, which severs a key intermediate for the preparation of a series of highly potential pesticides and various special kinds of engineering plastics, has been successfully synthesized in quantitative yield from the ammoxidation of 2,6-dichlorobenzaldehyde (S10). Cyano-substituted heterocycles are highly important compounds with a wide application in medical molecules. In general, cyano-substituted heterocycles are synthesized by cyanation from the corresponding heteroaryl halides (37, 38). To our great pleasure, the Fe-N3/NC-1100 catalyst was effective for the synthesis heterocyclic nitriles along with aryl nitriles bearing heterocyclic backbones from the corresponding aldehydes (S15 to S19). Compared with the state-of-the art methods (7), our method was also effective for the ammoxidation of inert aliphatic aldehydes (S20 to S28), affording the corresponding aliphatic nitriles with high yields at the low reaction temperature of 50°C. Besides the wide substrate scope, the Fe-N3/NC-1100 catalyst also showed the advantages with a good tolerance to many functional groups including halogen, nitro, thio-ether, sulfone, and vinyl groups. For example, substrates S2 and S22 with C═C bonds have been selectively oxidized to give the corresponding nitriles in yields up to 90% without the isomerization, hydrogenation, and hydration of the C═C bonds.
Fig. 5. Substrate scope of the oxidative transformation of aldehydes into nitriles.

Reaction conditions A: aldehydes (0.50 mmol), Fe-N3/NC-1100 (20 mg, 0.93 mmol %), NH3·H2O (26.5 wt %, 200 μl), toluene (1.5 ml), O2 (1 bar), 25°C, and 24 hours. Reaction conditions B: aldehydes (0.50 mmol), Fe-N3/NC-1100 (20 mg, 0.93 mmol %), NH3·H2O (26.5 wt %, 200 μl), toluene (1.5 ml), O2 (1 bar), 50°C, and 24 hours.
Oxidative transformation of 1,2-diols into nitriles
After the success in the ammoxidation of aldehydes into nitriles, the one-pot oxidative transformation of 1,2-diols into nitriles was investigated. The reaction conditions were optimized for the model reaction using R,R-hydrobenzoin as the starting material (tables S4 to S6), which were as follows: toluene as the best solvent, 26.5 wt % of NH3·H2O (400 μl), 25°C, and 1 bar of O2. Benzaldehyde was the intermediate, and the ammoxidation of benzaldehyde into benzonitrile was the rate-determining step (table S6). As shown in Fig. 6, Fe-N3/NC-1100 could successfully catalyze the oxidative transformation of benzylic-benzylic 1,2-diols (S29 to S37) into the corresponding nitriles with high yields (89 to 99%) at room temperature and 1 bar of O2. The steric configuration of the 1,2-diols (S29 and S30) and the electronic properties and position of the substituted groups in 1,2-diols (S31 to S37) showed no significant influence on the catalytic efficiency. In addition, Fe-N3/NC-1100 was also effective for the oxidative transformation of heteroaromatic 1,2-diols (S38 and S39) into the corresponding nitriles. Compared with benzylic-benzylic 1,2-diols, benzylic-aliphatic 1,2-diols and monosubstituted benzylic 1,2-diols (S40 to S46) were much less active, and 120°C was required for the oxidative transformation of them into the corresponding nitriles with good yields. In addition, different kinds of aromatic primary alcohols (S47 to S58) were also successfully transformed into the corresponding nitriles with good yields.
Fig. 6. Substrate scope of the oxidative transformation of diols and primary alcohol into nitriles.

Reaction conditions A: 1,2-diols (0.20 mmol), Fe-N3/NC-1100 (20 mg, 0.93 mmol %), NH3·H2O (26.5 wt %, 400 μl), toluene (1.5 ml), O2 (1 bar), 25°C, and 24 hours. Reaction conditions B: 1,2-diols (0.20 mmol), Fe-N3/NC-1100 (20 mg, 0.93 mmol %), NH3·H2O (26.5 wt %, 400 μl), toluene (1.5 ml), O2 (1 bar), 120°C, and 16 hours. Reaction conditions C: primary alcohols (0.20 mmol), Fe-N3/NC-1100 (20 mg, 0.93 mmol %), NH3·H2O (26.5 wt %, 400 μl), toluene (1.5 ml), O2 (1 bar), 120°C, and 24 hours.
Oxidative transformation of ketones and secondary alcohols into nitriles
Then, the oxidative cleavage of ketones into nitriles was studied, which was more challenging than the use of 1,2-diols as the starting materials, and harsh reaction conditions (150°C and 10 bar of O2) were required to access nitriles from ketones (tables S7 and S8). First, different kinds of (het)aryl-methyl ketones (S59 to S82) were explored for the synthesis of nitriles (Fig. 7). Substrates with electron-withdrawing substituents (S60 to S68) not only demonstrated higher activity than those with electron-donating groups (S69 to S78) but also gave higher selectivity to nitriles. In all cases, benzamides were detected to be the sole by-products. According to the following discussion, the oxidative cleavage of aromatic ketones into nitriles proceeded via benzaldehydes as the intermediates. The benzamide by-products should be formed by the condensation of NH3·H2O with benzoic acids, which were formed by further oxidation of benzaldehydes. Our results indicated that the electron-donating groups in benzaldehydes promoted further oxidation of benzaldehydes into benzoic acids, resulting in the slight decrease in nitrile selectivity. Furthermore, the efficiency was also affected by the position of the substituents. Substrates with the p-substituted groups gave higher selectivity to nitriles than those with o- and m-substituted groups (S69 versus S70 and S71; S74 versus S75). In addition, Fe-N3/NC-1100 was also robust for the oxidative transformation of fused ring substrates (S78 and S79) and heteroaryl-methyl ketones (S80 and S81) into the corresponding nitriles with yields >97%. 1,4-Diacetylbenzene (S82) with two methyl ketone groups was successfully converted into the dinitrile product, showing a great protentional in the synthesis of polymer building blocks. Again, the Fe-N3/NC-1100 catalyst showed a good tolerance to the functional groups in ketones such as halogen, nitro, and nitrile groups. Furthermore, Fe-N3/NC-1100 also demonstrated a high stability during the recycling experiments without the significant loss of its activity (fig. S13).
Fig. 7. Substrate scope of the oxidative transformation of aryl-methyl ketones, aryl-aliphatic ketones, aliphatic carbonyl compounds, and secondary alcohols into nitriles.

Reaction conditions: substrates (0.20 mmol), Fe-N3/NC-1100 catalyst (20 mg, 0.93 mmol %), NH3·H2O (26.5 wt %, 400 μl), toluene (1.5 ml), O2 (10 bar), and 150°C.
Besides aryl-methyl ketones, other kinds of aryl-aliphatic ketones were also investigated. Using standard conditions, aromatic-alkyl ketones (S83 to S88) were also successfully transformed into the corresponding nitriles with high to excellent yields. The carbon chain length in the alkyl fragment showed no significant influence on these transformations with high yields after 10 hours (S83 to S86). However, substrates with the ─CH─ groups at the α-position (S87 and S88) proceeded much more slowly because of the larger steric hindrance. Again, substrate S83 with the electro-withdrawing substituted group gave higher nitrile selectivity than the counterpart (S84) with electro-donating group. In addition, the aryl-aliphatic ketones with α-CH2─ groups connected by ketones and phenol (S90) were also successfully converted into benzonitrile with high to excellent yields. In the abovementioned aryl-aliphatic ketones, the aliphatic fragment was not detected as either aliphatic aldehydes or aliphatic nitriles by gas chromatography–mass spectrometry (GC-MS), but CO2 was detected in the gas phase (fig. S14). These results suggested that the aliphatic fragments were fully oxidized into CO2 via the consecutive C(O)─C bond cleavage. To further study the oxidative process, aryl-aliphatic ketones containing phenyl group in the alkyl fragments were chosen as the substrates. Substrate S91 gave two different kinds of benzonitriles with excellent yields, and substrate S92 gave double yields of benzonitrile. These results indicated that the oxidative transformation of aryl-aliphatic ketones proceeded via the oxidative cleavage of C(O)─C bonds into two kinds of aldehydes. However, the products from aryl-aliphatic ketones with the phenyl group in the alkyl fragments depended on the carbon atoms between the carbonyl group and the phenyl group. Substrate S93 with two carbon atoms to isolate the carbonyl group and the phenyl group gave double yields of benzonitrile. However, substrate S94 with three carbon atoms between the carbonyl group and the phenyl group gave 110% yield of benzonitrile and 74% yield of 3-phenylpropionitrile. Similar results were also observed with phenylacetaldehyde (S95) and 3-phenylpropionaldehyde (S96) as the substrates. The above results indicated that the consecutive cleavage of C(O)─C bonds in aliphatic aldehydes with the terminal phenyl group became much more difficult with much more carbon atoms between the aldehyde group and the phenyl group, and the direct aerobic ammoxidation of the aliphatic aldehydes to generate the aliphatic nitriles became the major reaction route. In addition, Fe-N3/NC-1100 was also effective for the oxidative transformation of aliphatic ketone (S97) and ketene (S98) into benzonitrile with high yields. Besides the wide scope of carbonyl compounds, Fe-N3/NC-1100 was also active for the oxidative transformation of secondary alcohols (S99 to S103) into benzonitriles with excellent yields in one pot, including the first step of oxidation of secondary alcohols into ketones, and followed by oxidative transformation of ketones into nitriles.
Plausible reaction mechanism
First, the active sites for the oxidative transformation of 1,2-diols, ketones, and secondary alcohols into nitriles were explored by control experiments (tables S9 and 10). The NC-1100 catalyst exhibits no catalytic activity, indicating that the presence of Fe is critical. Compared with Fe-N3/NC-1100, the catalytic activity of the Fe-N3/NC-1100-H+ catalyst after acid etching did not decrease, but addition of KSCN will greatly decrease the catalytic activity of the Fe-N3/NC-1100-H+ catalyst, indicating that the active site for the oxidative transformation of 1,2-diols, ketones, and secondary alcohols into nitriles is the Fe-N3 site; because SCN− can form stable chelate complexes with Fe cations, KSCN would poison the Fe-N3 site (33). Then, the reactive oxygen species were explored by radical inhibition experiments (table S11). The reaction almost stopped with a radical scavenger of 2,6-di-tert-butyl-4-methylphenol, revealing that the reaction was a radical reaction process. The reaction stopped in the presence of the superoxide (·O2−) scavenger of 1,4-benzoquinone, while no significant influence was observed with singlet oxygen (1O2) scavenger of furfuryl alcohol and hydroxyl radical (·OH) scavenger of isopropyl alcohol. There results confirmed that ·O2− was the reactive oxygen species, which was further confirmed by EPR technology (Fig. 3B). Second, the plausible reaction pathways were unified for the oxidative transformation of 1,2-diols, secondary alcohols, and ketones into nitriles over Fe-N3/NC-1100, using 1-phenyl-ethanol, 1-phenyl-1,2-ethanediol, and acetophenone as the representative substrates. The direct oxidation of 1-phenyl-ethanol without NH3·H2O at 150°C gave low amounts of acetophenone and benzaldehyde within 2 hours (fig. S15), where benzoic acid from the oxidation of benzaldehyde and its ester were observed after 8 hours (fig. S15). These results suggested that the oxidative transformation of 1-phenyl-ethanol into benzonitrile proceeded via the oxidation of 1-phenyl-ethanol into acetophenone, followed by the oxidative cleavage C─C bond of acetophenone into benzaldehyde (21) and the final step of the ammoxidation of benzaldehyde into benzonitrile. To further identify the potential intermediates of the oxidative cleavage C─C bond of acetophenone into benzaldehyde, the oxidative transformation of acetophenone was performed with trace amount of NH3·H2O. In this case, 2,2-dihydroxy-acetophenone, phenylglyoxylic acid, benzaldehyde, benzoic acid, benzamide, and benzonitrile were identified by GC-MS spectra (fig. S16), in which 2,2-dihydroxy-acetophenone (7; Fig. 8), phenylglyoxylic acid (8; Fig. 8), and benzaldehyde (9; Fig. 8) were the intermediates.
Fig. 8. Proposed mechanism of the oxidative cleavage C─C bonds of diols, secondary alcohols, and ketones into nitriles over the Fe-N3/NC-1100 catalyst.

According to the above results, the plausible reaction mechanism is proposed (Fig. 8). O2 first adsorbs and then axially coordinates to the single Fe atom in the Fe-N3/NC-1100 catalyst via a side-on coordination mode (Fig. 3C). Because of the flexible valence state of Fe2+ and Fe3+, the electron transfer from Fe2+ to O2 generates the reductive oxygen species (·O2−/Fe3+-N3), and electron transfer from Fe atom to O2 was also confirmed by DFT calculation (Fig. 3, E and G). As far as the oxidation of 1-phenyl-ethanol (1) into acetophenone (2), the H+ from ─OH is transferred to ·O2−/Fe2+-N3 to generate ·O─O─H/Fe3+-N3 species and then captures one H− from C─H bond to generate acetophenone. Meanwhile, ·O─O─H/Fe3+-N3 species combines with H− to release H2O2, which was detected using an ultraviolet-visible spectrophotometer (fig. S17) (39), and the Fe3+-N3 complex is transformed to the initial Fe2+-N3 complex state to start the catalytic cycle or subsequent oxidation reactions.
The oxidative cleavage C─C bond of acetophenone begins with the oxidation of C─H in acetophenone to give the 2-hydroxy-1-phenylethanone intermediate (5), but it was not detected by GC-MS, suggesting that it was the active intermediate. As far as the oxidation of C─H in acetophenone, ·O2−/Fe3+-N3 captures the ·H from C─H in acetophenone to generate H─O─O−─/Fe3+-N3 species and C6H5COC·(H2) intermediate (3). Then, the H─O─O−─/Fe3+-N3 species decomposes into H─O─O· radical and Fe2+-N3 species. The combination of C6H5COC·(H2) with H─O─O· generates the C6H5COC(O-O-H)(H2) intermediate (4). As shown in Fig. 8, the intermediate (4) can undergo two pathways to give 2-hydroxy-1-phenylethanone (5) or phenylglyoxal (6) via the cleavage of O─O bond in the ─C─O─O─H group (40). However, phenylglyoxal (6) was not observed by GC-MS, but the hydration product of 2,2-dihydroxy-acetophenon (7) was detected (fig. S16) possibly because of the much higher stability of 2,2-dihydroxy-acetophenon (7). Phenylglyoxal (6) can also be produced from the oxidation of 2-hydroxy-1-phenylethanone (5). 2,2-Dihydroxy-acetophenon can be produced by the second oxidation of C─H bond in 2-hydroxy-1-phenylethanone (5) into C─OH. The further oxidation of phenylglyoxal (6) or 2,2-dihydroxy-acetophenon (7) generates the important phenylglyoxylic acid intermediate (8) as detected by GC-MS (fig. S16) (41). Then, the C─C bond cleavage of phenylglyoxylic acid (8) produces benzaldehyde (9) and simultaneously released CO2 (42). Last, the ammoxidation of benzaldehyde produced benzonitrile (11). As far as the use of 1-phenyl-1,2-ethanediol (15) as the starting materials, the oxidation of 1-phenyl-1,2-ethanediol into 2-hydroxy-1-phenylethanone (5) should be the first step, as the hydroxyl group near the phenyl group was more active than the other hydroxyl group. Then, other steps were the same as described for the oxidative transformation of acetophenone. Some of the abovementioned intermediates were subjected to the standard conditions, and they also gave excellent yield of benzonitrile (fig. S18), which further confirmed that the proposed mechanism was plausible.
Study of the product selectivity by DFT calculation
Besides the super-high activity of Fe-N3/NC-1100, the other great advantage of our developed method is the high selectivity to nitriles. As depicted in Fig. 8, the competitive side reaction is the overoxidation of benzaldehyde into benzoic acid (13), which further undergoes the condensation with NH3 to generate the benzamide by-product (14) (41). DFT calculation was further used to probe the two competitive reaction routes (Fig. 9). As far as the oxidation of benzaldehyde into benzoic acid, the first step is the addition of one water to benzaldehyde to generate the geminal diol intermediate (12) (41), which underwent the consecutive release of two H atoms (─OH and ─CH) with the assistance of ·O2− to give benzoic acid. On the similar way, the oxidative dehydrogenation of the two hydrogen atoms (─NH and ─CH) in aldimine intermediate (C6H5CH═NH) (10) generated benzonitrile. DFT calculation revealed that the active energy for the oxidation of benzaldehyde into benzoic acid was much higher than that for the oxidation of aldimine intermediate (C6H5CH═NH) to benzonitrile (2.212 eV versus 1.086 eV). DFT calculation revealed that the oxidation of benzaldehyde into benzoic acid was much more difficult than the oxidation of benzaldehyde into benzonitrile; thus, the side reaction could be successfully inhibited over Fe-N3/NC-1100. The DFT results were consistent with our experimental data (Table 1), which had been discussed above.
Fig. 9. DFT calculation of benzonitrile formation and benzoic acid formation.

(A) Comparison of the energy profiles of benzonitrile route [blue line; the oxidative dehydrogenation of the two hydrogen atoms (─NH and ─CH) in aldimine intermediate (C6H5CH═NH) generated benzonitrile (Fig. 8, 10 and 11)] and benzoic acid route [red line; the oxidative dehydrogenation of the two hydrogen atoms (─OH and ─CH) in geminal diol intermediate generated benzoic acid (Fig. 8, 12 and 13)]. (B) The main structures of benzoic acid formation on Fe-N3/NC-1100 catalyst.
Practical applications in the synthesis of a single product from mixed substrates and the bioactive molecules
Last, the practical application of our developed method was studied. One of the most important applications in the chemical industry was to get the single product from a crude mixture of aromatic ketones derived from fossil fuels and biomass. For example, a mixture of aromatic-alkyl ketones was successfully converted into a single product of benzonitrile with a high yield of 82% (fig. S19A). Second, the developed catalytic system can also be applied for the synthesis of bioactive molecules. Bioactive molecules such as tocopherol (substrate S104) were worked well over the developed method, affording the corresponding nitriles with isolated yields of 66% at 150°C after 36 hours (fig. S19B).
Practical applications in the large-scale preparation of catalyst and the products
The practical application of a developed catalytic method requires the facile preparation of the catalyst and the products in large scale. The procedure of the catalyst preparation is very simple, and the single Fe atom catalyst can be prepared in a large scale of 5.12 g (table S12). The catalytic activity of Fe-N3/NC-1100 remained stable with different scales from 160 mg to 5.12 g by the test of several representative substrates including aldehydes and ketones (table S13).
With the facile and large-scale preparation of the catalyst in a large scale, the large-scale synthesis of the nitrile products was also performed. As shown in table S14, some representative substrates including 2-naphthaldehyde, 2,6-dichlorobenzaldehyde, mesitaldehyde, 1,4-phthalaldehyde, 6-nitrobenzo[d][1,3]dioxole-5-carbaldehyde, and 4-chlorobenzaldehyde were also performed with the loading of 2 g. To our great pleasure, the corresponding products of 2-naphthonitrile, 2,6-dichlorobenzonitrile, mesitonitrile, 1,4-dicyanobenzene, 6-nitrobenzo[d][1,3]dioxole-5-carbonitrile, and 4-chlorobenzonitrile were obtained in the isolated yields from 83 to 95%. Furthermore, the catalytic activity did not decrease after five large-scale cycle experiments (table S15), and the structure of Fe-N3/NC-1100 after five large-scale cycle experiments also did not change (figs. S20 and S21), indicating that Fe-N3/NC-1100 exhibits high stability.
DISCUSSION
In conclusion, we have developed a method for the aerobic oxidative cleavage of 1,2-diols, secondary alcohols, and ketones into nitriles over the FeN3-SAC catalyst bearing FeN3 motifs, easily prepared from the simple pyrolysis method. Compared with the FeN4-SAC bearing Fe-N4 motifs, FeN3-SAC was robust toward the activation of O2 to generate the superoxide species (·O2−) at room temperature. FeN3-SAC successfully promoted the oxidative transformation of a broad range of the substrates including 1,2-diols, secondary alcohols, and ketones into nitriles with yields in 72 to 99% via C─C bond cleavage strategy. DFT calculation reveals that the activation energy of O2 activation and the activation energy of the rate-determining step of nitrile formation are lower over FeN3-SAC than FeN4-SAC. The side reaction of the overoxidation of aldehydes into carboxylic acids was successfully inhibited because of its much higher activation energy over FeN3-SAC. The absence of precious metals, toxic solvents, and reagents and the use of O2 and NH3 as the oxidant and the nitrogen source with H2O as the only by-product make this catalyst more appealing for the synthesis of nitriles by the oxidative cleavage of 1,2-diols, secondary alcohols, and ketones than any previously reported systems.
MATERIALS AND METHODS
Materials
All of the chemicals including the starting materials for the catalyst preparation and the chemical reactions were purchased from Aladdin Ltd. (Shanghai, China). All the solvents were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). All chemicals used in this study were analytical grade and used without further purification.
Preparation of Fe-N3/NC-1100 and Fe-N3/NC-1100-H+
Ferrocene (7 mg) and ZIF-8 (500 mg) were placed on the front and rear ends of a corundum boat with a cover, respectively. The corundum boat was placed in the center of a tube furnace, and then the tube furnace was heated from room temperature to 1100°C with a ramp rate of 5°C/min under the flow of nitrogen and then held at 1100°C for 2 hours. After cooling to room temperature under nitrogen atmosphere, the nitrogen-coordinated single Fe atom catalyst was obtained with a mass of 160 mg. According the characterization, the as-prepared catalyst represents the single Fe atom catalyst, in which each Fe atom is coordinated with three nitrogen atoms. Thus, the as-prepared catalyst is labeled as Fe-N3/NC-1100. Then, the obtained Fe-N3/NC-1100 was stirred with 50 ml of 2 M HNO3 solution at 60°C for 24 hours. The recovered solid was washed with water until the filtrate became neutral and then dried at 60°C for 12 hours. The resultant sample is labeled as Fe-N3/NC-1100-H+.
Preparation of Zn/NC-1000 and NC-1100
ZIF-8 (500 mg) was placed on a corundum boat with a cover. The corundum boat was placed in the center of a tube furnace, and then the tube furnace was heated from room temperature to 1000°C with a ramp rate of 5°C/min under the flow of nitrogen and then held at 1000°C for 2 hours. After cooling to room temperature under nitrogen atmosphere, the Zn/NC-1000 was obtained with mass of 150 mg. The preparation of NC-1100 is the same as that of Zn/NC-1000 except that the calcination temperature is changed to 1100°C.
Preparation of Fe-N4/NC-800
A mixture of Fe(OAc)2 (0.5 mmol, 106 mg) and 1,10-phenanthroline monohydrate (1.5 mmol, 330 mg) was added to 50 ml of ethanol and sonicated for 10 min, followed by addition of 3.16 g of MgO-nano and sonicated for another 10 min. Then, the mixture was stirred under reflux at 60°C overnight. After ethanol was removed by rotary evaporation, the remaining solid was dried at 60°C for 12 hours and then transferred to a quartz boat in the oven and fluxed with N2 for 30 min. The oven was then heated to 800°C in nitrogen atmosphere at a ramp of 2°C/min and was held at 800°C for 2 hours. The obtained black solid was stirred with 100 ml of 1 M HNO3 solution at room temperature for 2 hours, and this leaching procedure was repeated three times to remove the MgO support. The recovered solid was washed with water until the filtrate became neutral and then dried at 60°C for 12 hours. The resultant sample is labeled as Fe-N4/NC-800.
Characterization
The BET surface area, pore volume, pore size distribution, and average pore diameter of catalysts were characterized by nitrogen adsorption/desorption analysis at 77 K on a Quantachrome Quadrasorb SI instrument. Before the measurements, the samples were degassed at 150°C for 4 hours. TEM images were acquired on a Hitachi S-4800 SEM operated at 15 kV. The sample was first dispersed in ethanol and dropped onto copper grids for observation. Raman spectra were obtained in a Thermo Fisher Scientific DXR2 xi spectrometer with an excitation wavelength at 532 nm. Powder XRD studies were conducted on a Bruker advanced D8 diffractometer with a Cu Kα source at 40 kV and 20 mA. The morphology and sizes of the samples were studied by HRTEM (JEM-2100 from JEOL). XPS measurements were performed on a Thermo Fisher Scientific ESCALAB Xi+ system equipped with a hemispherical analyzer and using a monochromatic Al Kα radiation source. The iron content in the catalyst was determined by ICP-AES on an Agilent 720ES instrument. XANES and EXAFS data reduction and analysis were processed by the Athena software. The O2-TPD measurements were performed using a MicroActive for AutoChem II 2920 Version with a flowing 5% O2/He stream (50 ml min−1) at −50°C. EPR measurements were performed at room temperature on a Bruker Benchtop EMX Nano EPR spectrometer.
General procedures
In a typical procedure, the substrate (0.20 mmol), Fe-N3/NC-1100 [20 mg, 0.93 millimole % (mmol %)], and toluene (1.5 ml) were charged into a 4-ml high-pressure stainless steel reactor (Xi’an Taikang Biotechnology Co., ltd, China). The air in the autoclave was removed by the flush of oxygen gas, and then the autoclave was equipped with the oxygen balloon or charged with 10 bar of O2. The reaction was performed at room temperature or the setting temperature with a magnetic stirring at 800 RPM. After the reaction, ethyl benzene was added into the reaction mixture as the internal standard, and the mixture was analyzed by GC. The products were confirmed by GC-MS (Thermo Trace 1300 GC-ISQ) and nuclear magnetic resonance (NMR) technologies.
To isolate these products, the reaction mixture was first concentrated under reduced pressure and then purified by silica gel chromatography eluting using hexane and dichloromethane (hexane/ethyl acetate = 4/1) as the eluent. The structures of the purified products were characterized by 1H and 13C NMR.
Analytic methods
Products analysis was performed on an Agilent 7890A GC instrument with a cross-linked capillary HP-5 column (30 m by 0.32 mm by 0.4 mm), which was equipped with a flame ionization detector. N2 was used as the carrier gas with a flow rate of 40 ml min−1. Standard analysis conditions were described as follows: injector temperature of 300°C, detector temperature of 300°C, and column temperature program from 50°C (hold for 1.5 min) to 300°C (hold for 3 min) at a heating rate of 10°C min−1. The content of each compound was determined on the basis of the internal standard. The structures of products were identified by NMR technology (Bruker TCI IIITM 400 MHz).
Acknowledgments
Funding: We thank the National Natural Science Foundation of China (21922513). This work was supported by the National Natural Science Foundation of China (21922513).
Author contributions: J.Q., X.Li., and Z.Z. proposed the project, designed the experiments, and wrote the manuscript. J.Q. and X.Li. performed the whole experiments except the O2-TPD and the reactions on bioactive molecules. B.H. carried out the DFT calculation. H.L. performed the O2-TPD and the reactions on bioactive molecules. Y.W., X. Lu, J.N., and C.X. performed the analysis of experimental data. W.D. contributes the discussion of the results. Z.Z. supervised the whole project. All authors discussed the results and commented on the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Supplementary Materials
This PDF file includes:
Figs. S1 to S22
Tables S1 to S15
Supplementary NMR data
Supplementary mass spectrometry and NMR spectrometry
REFERENCES AND NOTES
- 1.Nakao Y., Metal-mediated C–CN bond activation in organic synthesis. Chem. Rev. 121, 327–344 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Chandrashekhar V. G., Senthamarai T., Kadam R. G., Malina O., Kašlík J., Zbořil R., Gawande M. B., Jagadeesh R. V., Beller M., Silica-supported Fe/Fe-O nanoparticles for the catalytic hydrogenation of nitriles to amines in the presence of aluminium additives. Nat. Catal. 5, 20–29 (2022). [Google Scholar]
- 3.Kim J., Kim H. J., Chang S., Synthesis of aromatic nitriles using nonmetallic cyano-group sources. Angew. Chem. Int. Ed. 51, 11948–11959 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Akhtar R., Zahoor A. F., Rasool N., Ahmad M., Ali K. G., Recent trends in the chemistry of Sandmeyer reaction: A review. Mol. Divers. 26, 1837–1873 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu D.-G., Gensch T., Azambuja F. D., Vásquez-Céspedes S., Glorius F., Co(III)-catalyzed C-H activation/formal SN-type reactions: Selective and efficient cyanation, halogenation, and allylation. J. Am. Chem. Soc. 136, 17722–17725 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Wang T., Jiao N., Direct approaches to nitriles via highly efficient nitrogenation strategy through C-H or C-C bond cleavage. Acc. Chem. Res. 47, 1137–1145 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Hua M., Song J., Huang X., Liu H., Fan H., Wang W., He Z., Liu Z., Han B., Highly efficient oxidative cyanation of aldehydes to nitriles over Se,S,N-tri-doped hierarchically porous carbon nanosheets. Angew. Chem. Int. Ed. 60, 21479–21485 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Dongbang S., Ellman J. A., Synthesis of nitrile bearing acyclic quaternary centers through Co(III)-catalyzed sequential C-H bond addition to dienes and N-cyanosuccinimide. Angew. Chem. Int. Ed. 60, 2135–2139 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han P. F., Tang C., Sarina S., Waclawik E. R., Du A. J., Bottle S. E., Fang Y. F., Huang Y. P., Li K., Zhu H.-Y., Wavelength-specific product desorption as a key to raising nitrile yield of primary alcohol ammoxidation over illuminated Pd nanoparticles. ACS Catal. 12, 2280–2289 (2022). [Google Scholar]
- 10.Murugesan K., Donabauer K., Kçnig B., Visible-light-promoted metal-free synthesis of (hetero)aromatic nitriles from C(sp3)-H bonds**. Angew. Chem. Int. Ed. 60, 2439–2445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senthamarai T., Chandrashekhar V. G., Rockstroh N., Rabeah J., Bartling S., Jagadeesh R. V., Beller M., A “universal” catalyst for aerobic oxidations to synthesize (hetero)aromatic aldehydes, ketones, esters, acids, nitriles, and amides. Chem 8, 508–531 (2022). [Google Scholar]
- 12.Sun K. K., Shan H. B., Neumann H., Lu G.-P., Beller M., Efficient iron single-atom catalysts for selective ammoxidation of alcohols to nitriles. Nat. Commun. 13, 1848 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu B., Jiang Q., Zhao A., Jia J., Liu Q., Luo W., Guo C., Copper-catalyzed aerobic conversion of the C=O bond of ketones to a C≡N bond using ammonium salts as the nitrogen source. Chem. Commun. 51, 11264–11267 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Escande V., Lam C. H., Coish P., Anastas P. T., Heterogeneous sodium-manganese oxide catalyzed aerobic oxidative cleavage of 1,2-diols. Angew. Chem. Int. Ed. 56, 9561–9565 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Huang X. Q., Li X. Y., Zou M. C., Song S., Tang C. H., Yuan Y. Z., Jiao N., From ketones to esters by a Cu-catalyzed highly selective C(CO)-C(alkyl) bond cleavage: Aerobic oxidation and oxygenation with air. J. Am. Chem. Soc. 136, 14858–14865 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Singh B., Gawande M. B., Kute A. D., Varma R. S., Fornasiero P., McNeice P., Jagadeesh R. V., Beller M., Zboril R., Single-atom (iron-based) catalysts: Synthesis and applications. Chem. Rev. 121, 13620–13697 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Kaiser S. K., Chen Z. P., Akl D. F., Mitchell S., Pérez-Ramírez J., Single-atom catalysts across the periodic table. Chem. Rev. 120, 11703–11809 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Deng Q., Li X., Gao R. J., Wang J., Zeng Z. L., Zou J. J., Deng S. G., Tsang S. C. E., Hydrogen-catalyzed acid transformation for the hydration of alkenes and epoxy alkanes over Co-N frustrated lewis pair surfaces. J. Am. Chem. Soc. 143, 21294–21301 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Vijay S., Ju W., Brückner S., Tsang S. C., Strasser P., Chan K. R., Unified mechanistic understanding of CO2 reduction to CO on transition metal and single atom catalysts. Nat. Catal. 4, 1024–1031 (2021). [Google Scholar]
- 20.Nett R. S., Lau W., Sattely E. S., Discovery and engineering of colchicine alkaloid biosynthesis. Nature 584, 148–153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo H. H., Wang L. Y., Shang S. S., Li G. S., Lv Y., Gao S., Dai W., Cobalt nanoparticles-catalyzed widely applicable successive C-C bond cleavage in alcohols to access esters. Angew. Chem. Int. Ed. 59, 19268–19274 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Xie C., Lin L. F., Huang L., Wang Z. X., Jiang Z. W., Zhang Z. H., Han B. X., Zn-Nx sites on N-doped carbon for aerobic oxidative cleavage and esterification of C(CO)-C bonds. Nat. Commun. 12, 4823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T. T., Sang X. H., Zheng W. Z., Yang B., Yao S. Y., Lei C. J., Li Z. J., He Q. G., Lu J. G., Lei L. C., Dai L. M., Hou Y., Gas diffusion strategy for inserting atomic iron sites into graphitized carbon supports for unusually high-efficient CO2 electroreduction and high-performance Zn–CO2 batteries. Adv. Mater. 32, 2002430 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Gan G. Q., Li X. Y., Fan S. Y., Yin Z. F., Wang L., Chen G. H., Ultrathin Fe–N –C single-atom catalysts with bifunctional active site for simultaneous production of ethylene and aromatic chlorides. Nano Energy 80, 105532 (2021). [Google Scholar]
- 25.Datsyuk V., Kalyva M., Papagelis K., Parthenios J., Tasis D., Siokou A., Kallitsis I., Galiotis C., Chemical oxidation of multiwalled carbon nanotubes. Carbon 46, 833–840 (2008). [Google Scholar]
- 26.Shang S. S., Chen P. P., Wang L. Y., Lv Y., Li W.-X., Gao S., Metal-free nitrogen- and boron-codoped mesoporous carbons for primary amides synthesis from primary alcohols via direct oxidative dehydrogenation. ACS Catal. 8, 9936–9944 (2018). [Google Scholar]
- 27.Zhang Z. P., Sun J. T., Wang F., Dai L. M., Efficient oxygen reduction reaction (ORR) catalysts based on single iron atoms dispersed on a hierarchically structured porous carbon framework. Angew. Chem. Int. Ed. 57, 9038–9043 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Zhang Z. W., Jia G. R., Zheng L. R., Zhao J. X., Cui X. Q., Elucidating the mechanism of the structure-dependent enzymatic activity of Fe-N/C oxidase mimics. Chem. Commun. 55, 5271–5274 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Ma W. J., Mao J. J., Yang X. T., Pan C., Chen W. X., Wang M., Yu P., Mao L. Q., Li Y. D., A single-atom Fe–N4 catalytic site mimicking bifunctional antioxidative enzymes for oxidative stress cytoprotection. Chem. Commun. 55, 159–162 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Sun X. H., Tuo Y. X., Ye C. L., Chen C., Lu Q., Li G. N., Jiang P., Chen S. H., Zhu P., Ma M., Zhang J., Bitter J. H., Wang D. S., Li Y. D., Phosphorus induced electron localization of single iron sites for boosted CO2 electroreduction reaction. Angew. Chem. Int. Ed. 60, 23614–23618 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Pan Y., Chen Y. J., Wu K. L., Chen Z., Liu S. J., Cao X., Cheong W.-C., Meng T., Luo J., Zheng L. R., Liu C. G., Wang D. S., Peng Q., Li J., Chen C., Regulating the coordination structure of single atom Fe-NxCy catalytic sites for benzene oxidation. Nat. Commun. 10, 4290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul B., Maji M., Kundu S., Atom-economical and tandem conversion of nitriles to N-methylated amides using methanol and water. ACS Catal. 9, 10469–10476 (2019). [Google Scholar]
- 33.Liu W. G., Zhang L. L., Liu X., Liu X. Y., Yang X. F., Miao S., Wang W. T., Wang A. Q., Zhang T., Discriminating catalytically active FeNx species of atomically dispersed Fe-N-C catalyst for selective oxidation of the C-H bond. J. Am. Chem. Soc. 139, 10790–10798 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Zhao X., Wang F. L., Kong X. P., Dual-metal hetero-single-atoms with different coordination for efficient synergistic catalysis. J. Am. Chem. Soc. 143, 16068–16077 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Zhang L., Bi X. H., Guan X. X., Li X. Q., Liu Q., Barry B.-D., Liao P. Q., Chemoselective oxidative C(CO)-C(methyl) bond cleavage of methyl ketones to aldehydes catalyzed by CuI with molecular oxygen. Angew. Chem. Int. Ed. 52, 11303–11307 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Tang C. H., Jiao N., Copper-catalyzed aerobic oxidative C-C bond cleavage for C-N bond formation: From ketones to amides. Angew. Chem. Int. Ed. 53, 6528–6532 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Coughlin M. M., Kelly C. K., Lin S., MacArthur A. H. R., Cyanation of aryl chlorides using a microwave-assisted, copper-catalyzed concurrent tandem catalysis methodology. Organometallics 32, 3537–3543 (2013). [Google Scholar]
- 38.Mondal B., Acharyya K., Howlader P., Mukherjee P. S., Molecular cage impregnated palladium nanoparticles: Efficient, additive-free heterogeneous catalysts for cyanation of aryl halides. J. Am. Chem. Soc. 138, 1709–1716 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Su F. Z., Mathew S. C., Lipner G., Fu X. Z., Antonietti M., Blechert S., Wang X. C., Mpg-C3N4-catalyzed selective oxidation of alcohols using O2 and visible light. J. Am. Chem. Soc. 132, 16299–16301 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Zhang P. F., Lu H. F., Zhou Y., Mesoporous MnCeOx solid solutions for low temperature and selective oxidation of hydrocarbons. Nat. Commun. 6, 8446 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarbajna A., Dutta I., Daw P., Dinda S., Rahaman S. M. W., Sarkar A., Bera J. K., Catalytic conversion of alcohols to carboxylic acid salts and hydrogen with alkaline water. ACS Catal. 7, 2786–2790 (2017). [Google Scholar]
- 42.Bhattacharya S., Rahaman R., Chatterjee S., Paine T., Aliphatic C-C bond cleavage in α-hydroxy ketones by a dioxygen-derived nucleophilic iron-oxygen oxidant. Chem. A Eur. J. 23, 3815–3818 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figs. S1 to S22
Tables S1 to S15
Supplementary NMR data
Supplementary mass spectrometry and NMR spectrometry