

Abstract
This study assessed the effectiveness of genetic testing in shortening the time to diagnosis of late infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease. Individuals who received epilepsy gene panel testing through Behind the Seizure®, a sponsored genetic testing program (Cohort A), were compared to children outside of the sponsored testing program during the same period (Cohort B). Two cohorts were analyzed: children aged ≥24 to ≤60 months with unprovoked seizure onset at ≥24 months between December 2016 and January 2020 (Cohort 1) and children aged 0 to ≤60 months at time of testing with unprovoked seizure onset at any age between February 2019 and January 2020 (Cohort 2). The diagnostic yield in Cohort 1A (n = 1814) was 8.4% (n = 153). The TPP1 diagnostic yield within Cohort 1A was 2.9‐fold higher compared to Cohort 1B (1.0%, n = 18/1814 vs. .35%, n = 8/2303; p = .0157). The average time from first symptom to CLN2 disease diagnosis was significantly shorter than previously reported (9.8 vs. 22.7 months, p < .001). These findings indicate that facilitated access to early epilepsy gene panel testing helps to increase diagnostic yield for CLN2 disease and shortens the time to diagnosis, enabling earlier intervention.
Keywords: Batten disease, developmental delay, enzyme replacement therapy, genetic epilepsy, genetic testing, neuronal ceroid lipofuscinosis
1. INTRODUCTION
Neuronal ceroid lipofuscinoses (NCLs; Batten disease) are clinically and genetically heterogeneous lysosomal storage disorders representing the most common family of genetic neurodegenerative disorders in children, characterized by seizures, progressive cognitive deterioration, and motor and visual impairment. 1 Late infantile NCL type 2 (CLN2) disease, caused by biallelic pathogenic variants in the TPP1 gene, 2 is estimated to have an incidence of .22–9.0 per 100 000 live births, although this is potentially underestimated. 3 , 4 , 5 , 6 , 7 Affected children present with language delay and seizures between ages 2 and 4 years, and the disease becomes fatal between ages 8 and 12 years. Clinical diagnosis is typically made around age 5 years because of low disease awareness and the nonspecific clinical presentation until neurodegeneration begins. Definitive testing (e.g., TPP1 enzyme testing, TPP1 gene sequencing) is often sought late in the diagnostic odyssey, potentially preventing disease management benefits. 8 , 9 , 10 For example, enzyme replacement therapy (ERT) for TPP1 reduced the rate of loss of ambulation and language skills compared to historical controls in an open‐label, single‐arm study. 11 However, although ERT attenuates disease progression, it does not reverse neurodegeneration that has already occurred; thus, early diagnosis is key.
This report explored the effectiveness of genetic testing to hasten identification of individuals with CLN2 disease.
2. MATERIALS AND METHODS
Unrelated children tested through the Behind the Seizure® (BTS) sponsored testing program between December 5, 2016 and January 6, 2020 were included (WCG Institutional Review Board, 1167406). Relatives tested through the sponsored testing program were excluded. Children aged ≥24 to ≤60 months at the time of testing with unprovoked seizure onset at ≥24 months were eligible. In February 2019, the eligibility criteria expanded to include children aged 0 to ≤60 months at the time of testing with unprovoked seizure onset at any age. Ordering clinicians completed the required eligibility criteria and optional medical history (e.g., language or developmental delays, motor disturbance, family history of epilepsy).
DNA samples extracted from blood or saliva were tested using a multigene epilepsy panel (125–183 genes) and an optional “preliminary‐evidence” panel by next generation sequencing as described previously. 7 TPP1 was analyzed using reference transcript NM_000391.3. Variants were clinically classified using Sherloc, an Invitae variant interpretation system based on guidelines from the American College of Medical Genetics and Genomics and Association for Molecular Pathology. 12 , 13 Variants were categorized as pathogenic (P), likely pathogenic (LP), variant of uncertain significance, likely benign, or benign. A molecular diagnosis (molecular diagnosis/diagnoses [MDx]) was determined based on inheritance patterns (e.g., two P/LP variants in genes associated with autosomal recessive disorders, one P/LP variant in genes associated with autosomal dominant disorders, X‐linked dominant disorders, or X‐linked recessive disorders [male only]). A negative result (No MDx) was defined as the absence of an MDx.
Diagnostic yield was calculated based on the proportion of children with any MDx (All MDx) or an MDx in TPP1 (CLN2 disease MDx). Two cohorts tested through the sponsored program were compared to children tested outside of the program. Cohort MDx1A included children aged ≥24 to ≤60 months at the time of testing with unprovoked seizure onset at ≥24 months who were tested through the BTS program at any time during the study period. Cohort 1A was compared to children aged ≥24 to ≤60 months with seizure onset at any age who were tested outside of the program during the same time period (Cohort 1B). Cohort 2A included children aged 0 to ≤60 months at the time of testing with unprovoked seizure onset prior to testing who received testing through the BTS program between February 2019 and January 2020. Cohort 2A was compared to children aged 0 to ≤60 months who were tested outside of the BTS program during the same time period (Cohort 2B). Some children from Cohorts 1A and 1B were also included in Cohorts 2A and 2B due to overlapping criteria. Differences in age were calculated by two‐sample t‐test (R 4.1.1).
Relationships between various clinical symptoms and MDx status were explored in Cohort 1A. Proportions of patients with history of each clinical characteristic were calculated including only data for items where either “yes” or “no” were selected; items that were left blank or where “unknown” was selected were not assumed to be indicative of negative history. T‐tests and Fisher exact tests assessed statistical significance between outcome groups (p ≤ .05 was considered significant). Average time from seizure onset to diagnosis in Cohort 1A was compared to a historical control 8 by calculating T‐score (https://www.socscistatistics.com/tests/studentttest/default.aspx). Statistical comparisons were conducted using a two‐sided p‐value (R 4.1.1).
3. RESULTS
Among 4246 children tested through the BTS program (Table S1, Figure S1), 629 (14.8%) received an MDx in 82 genes. The 10 most common MDx were in PRRT2 (n = 100), SCN1A (n = 91), KCNQ2 (n = 56), UBE3A (n = 25), MECP2 (n = 21), SCN2A (n = 20), STXBP1 (n = 19), TPP1 (n = 18), GABRB3 (n = 17), and TSC2 (n = 17; Table S2). All 18 (.42%) children with an MDx in TPP1 were aged 36–48 months at time of testing. Clinicians reported a high index of suspicion for NCL and/or CLN2 disease for 33 children tested through the sponsored program; eight (24.2%) received an MDx, including CLN2 disease (n = 3; Table S3).
In Cohort 1A (n = 1814), the All MDx rate was 8.4% (n = 153; Figure 1A), with the remaining 91.6% (n = 1661) with No MDx. CLN2 disease MDx was determined in 1.0% (n = 18) of children (Figure 1B), representing 11.8% (18/153) of All MDx. In Cohort 1B (n = 2,303), the All MDx rate was 15.0% (n = 346), with the remaining 85.0% (n = 1957) with No MDx (Figure 1A). Eight (.35%) children with an MDx received a CLN2 disease MDx (Figure 1B). The rate of CLN2 disease MDx in Cohort 1A was 2.9‐fold higher compared to Cohort 1B (1A n = 18/1814 vs. 1B n = 8/2303; p = .0157), likely because of differences in eligibility criteria targeting timing of seizure onset rather than the statistically (but not clinically) significant differences in age at time of testing (43.6 ± 9.8 vs. 41.0 ± 10.4 months, p < .001; Table S1).
FIGURE 1.
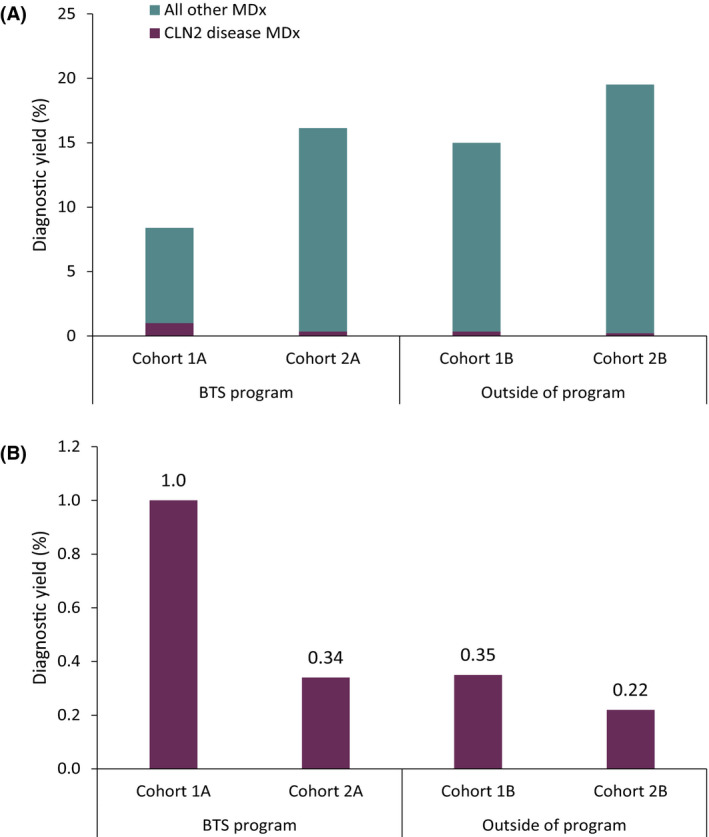
Diagnostic yield of positive molecular diagnoses within and outside of the Behind the Seizure® (BTS) sponsored program. (A) Overall and (B) ceroid lipofuscinosis type 2 (CLN2) disease diagnostic yield among children stratified by age at time of testing and age at seizure onset. Outside of BTS, age at seizure onset was unknown and age at time of testing was used. MDx, molecular diagnosis/diagnoses.
In Cohort 2A (n = 3572), the MDx rate was 16.2% (n = 578), with the remaining 83.8% (n = 2994) receiving No MDx (Figure 1A). Twelve children (.34%) had a CLN2 disease MDx (Figure 1B). In Cohort 2B (n = 1860), the MDx rate was 19.5% (n = 363) and the remaining 80.5% (n = 1497) of children had No MDx (Figure 1A). Four (.22%) children had a CLN2 disease MDx (Figure 1B). The rate of CLN2 disease MDx in Cohort 2A was 1.5‐fold higher compared to Cohort 2B, although age at time of testing was statistically (but not clinically) different (27.5 ± 18.1 vs. 26.0 ± 17.9, p = .00253; Table S1), demonstrating the effectiveness of targeted testing programs in enriching testing cohorts for rare diseases.
Differences in timing of genetic testing and clinical characteristics based on genetic testing results (i.e., No MDx, All MDx, CLN2 disease MDx) were assessed in Cohort 1A. Average time from seizure onset to testing for No MDx, All MDx, and CLN2 disease MDx was 8.4 months, 6.9 months, and 9.8 months, respectively. A significant difference was observed in the mean time from first seizure to genetic testing among the No MDx and All MDx groups (8.4 vs. 6.9 months, p < .05). CLN2 disease was diagnosed significantly earlier in individuals than is typically the case for this disease (average time from first symptom = 9.8 ± 7.4 months vs. 22.7 ± 9.8 months, 8 p < .001). Average age at time of testing was similar for all three groups (Figure S2).
Differences in the clinical presentation were observed; those in the All MDx or CLN2 Disease MDx groups more often had language and/or motor delay, an abnormal electroencephalogram, and an abnormal magnetic resonance imaging compared to the No MDx cohort (p < .05 in all comparisons; Table 1, Figure S3). Developmental delays preceding seizure onset were more prevalent in the All MDx cohort compared to the No MDx group (60.7 vs. 35.0%; p < .001), whereas these differences were observed, but not statistically significant, in the CLN2 Disease MDx group (66.7% vs. 35.0%, p = .074). Family history of epilepsy was not associated with molecular genetic testing outcome (25.5%, All MDx; 36.9%, No MDx; p = .13).
TABLE 1.
Clinical characteristics of children aged ≥24 to ≤60 months with seizure onset after 24 months of age tested through the Behind the Seizure® program (Cohort 1A)
| Clinical characteristic, n (%) | No MDx, n = 1661 | All MDx, n = 152 | CLN2 MDx, n = 17 | p, No MDx vs. | ||||
|---|---|---|---|---|---|---|---|---|
| Yes | No | Yes | No | Yes | No | All MDx | CLN2 MDx | |
| Language delay | 688 (46.5) | 792 (53.5) | 96 (66.7) | 48 (33.3) | 12 (80.0) | 3 (20.0) | <.001 | .002 |
| Motor delay | 383 (27.4) | 1015 (72.6) | 74 (54.4) | 62 (45.6) | 11 (84.6) | 2 (15.4) | <.001 | <.001 |
| Language OR motor delay | 756 (51.4) | 716 (48.6) | 104 (71.7) | 41 (28.3) | 14 (93.3) | 1 (6.7) | <.001 | .001 |
| Language AND motor delay | 315 (22.4) | 1091 (77.6) | 66 (48.9) | 69 (51.1) | 9 (69.2) | 4 (30.8) | <.001 | <.001 |
| Developmental delay precedes seizure onset | 403 (35.0) | 750 (65.0) | 74 (60.7) | 48 (39.3) | 6 (66.7) | 3 (33.3) | <.001 | .07 |
| Abnormal EEG | 707 (51.8) | 658 (48.2) | 87 (65.9) | 45 (34.1) | 13 (81.3) | 3 (18.7) | .002 | .02 |
| Abnormal MRI | 178 (15.0) | 998 (85.0) | 29 (24.6) | 89 (75.4) | 6 (60.0) | 4 (40.0) | .01 | .002 |
| Family history of epilepsy | 215 (36.9) | 368 (63.1) | 13 (25.5) | 38 (74.5) | 2 (40.0) | 3 (60.0) | .13 | 1.00 |
Total number of patients in each group includes only patients with available medical history data for at least one clinical characteristic. Percentages are based on the number of patients for whom the ordering clinician indicated the presence (“Yes”) or absence (“No”) of each clinical characteristic, which may not total the overall n indicated in the top row. MDx = definitive molecular diagnosis determined based on genetic testing results, defined as two P/LP variants in genes associated with autosomal recessive disorders, one P/LP variant in genes associated with autosomal dominant disorders, X‐linked dominant disorders, or X‐linked recessive disorders (male only); No MDx = negative result defined as the absence of any reportable results, including variant of uncertain significance and single P/LP variants in a gene associated with autosomal recessive disorders; All MDx = children with any positive MDx; CLN2 MDx = children with biallelic TPP1 P/LP variants. See Figure S2 for a graphic representation of these data.
Abbreviations: CLN2, ceroid lipofuscinosis type 2; EEG, electroencephalogram; LP, likely pathogenic; MRI, magnetic resonance imaging; P, pathogenic.
4. DISCUSSION
Although up to 40% of early life epilepsies have been reported to have genetic causes, 14 genetic testing is underutilized, and there are limited professional consensus guidelines advocating such testing early in the diagnostic workup in routine clinical practice. However, a recent CLN2 disease management guideline does recommend genetic testing to confirm biochemical findings, 15 and multigene testing has been recommended in cases of unexplained seizures for early diagnosis of CLN2 disease. 5 The offering of an expanded genetic testing panel allowed the BTS program to provide an MDx to children affected with both CLN2 disease and other genetic epilepsies even when that program had targeted a clinical presentation consistent with CLN2 disease.
A CLN2 disease MDx was returned to 1.0% of children in Cohort 1A, representing 11.8% of all MDx in Cohort 1A (n = 18/153), and was 2.9‐fold higher compared to Cohort 1B, demonstrating how the program has helped identify affected individuals for a rare condition with a nonspecific phenotype by increasing awareness among clinicians and reducing barriers for patients to pursue testing. The overall MDx rate was lower in Cohort 1A (8.4%) compared to all other cohorts (1B, 15.0%; 2A, 16.2%; 2B, 19.5%). Although the diagnostic yield is expected to be lower for the older cohort, the variance is likely due not only to differences in age at testing but also to age at seizure onset per eligibility criteria (≥24 months), highlighting the benefits of expanding the eligibility criteria of the sponsored program.
Furthermore, the average time from the first symptom to CLN2 disease MDx in the sponsored program was significantly shorter (by approximately 1 year) compared to the natural history published average (9.8 vs. 22.7 months 8 ), suggesting that the program encourages earlier testing. Early MDx has significant clinical impact in children with epilepsy by enabling earlier initiation of treatment that may attenuate clinical deterioration (e.g., US Food and Drug Administration‐approved treatments for CLN2 disease 11 ) or by avoiding therapies that are contraindicated (e.g., valproic acid for POLG, 16 sodium channel blockers for loss of function MDx in SCN1A 17 ), as well as helping patients and family with early disease and genetic counseling for reproductive decision‐making. 8 , 17 The BTS testing program has demonstrated that the association of language delay and motor impairments are more common in younger children with a recognizable monogenic etiology for epilepsy, including CLN2 disease, as compared to children without a single‐gene disorder. Individuals in the latter group possibly suffer from genetic generalized epilepsies. Although this study was limited by the change in eligibility criteria and limited clinical information from children tested outside of the program, the data presented here support the calls for clinical practice guidelines to consider the utilization of genetic testing as a first‐tier test for individuals with epilepsy early in the diagnostic journey, or with neurodegenerative diseases presenting with epilepsy, such as CLN2 disease. 18 Ways to improve clinical decision‐making and outcomes through better awareness of the possibility of a definitive MDx of CLN2 disease is yet another area with great potential for future study.
CONFLICTS OF INTEREST
F.L.‐P. was an employee of BioMarin Pharmaceutical during the conduct of this study and is currently an employee of Taysha Gene Therapies and shareholder of BioMarin Pharmaceutical and Taysha Gene Therapies. T.Y.P., J.C.‐P., and E.I. are employees and shareholders of BioMarin Pharmaceutical, which funded the study. R.T., D.A.M., B.J., A.M., S.L.B., and S.A. are employees of and shareholders in Invitae and provided the genetic analyses. B.J. is also a shareholder in BioMarin Pharmaceutical. R.S. has served on advisory boards and the speakers bureau for BioMarin and has received honoraria and travel expenses from BioMarin, which funded the study. S.K. has no conflicts to report. E.C.W. has received honoraria from BioMarin, which funded the study. J.J.M. has received honoraria from BioMarin, which funded the study. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
AUTHOR CONTRIBUTIONS
F.L.‐P., R.T., J.C.‐P., E.I., D.A.M., and B.J. were involved in the conception and design of the study and in the acquisition and analysis of data, as well as critical review and revision of the manuscript. T.Y.P., R.S., S.K., E.C.W., and J.J.M. were involved in the acquisition and analysis of data and critically reviewed and revised the manuscript. S.L.B. was involved in analysis of the data and drafted a significant portion of the manuscript and figures. S.A. was involved in the conception and design of the study, in the acquisition and analysis of data, and in drafting a significant portion of the manuscript. A.M. was involved in project administration, resources, as well as critical review and revision of the manuscript.
Supporting information
Tables S1‐S3
Fig S1
Fig S2
Fig S3
ACKNOWLEDGMENTS
We would like to thank Darlene Riethmaier and Nila Patil of Invitae for their contributions to the conception and design of the study and critical review of the manuscript. We would also like to thank Mitch Bailey of BioMarin Pharmaceutical for input into interpretation of the data and critical review of the manuscript and Nicole Miller for input into interpretation of the data. This study was funded by BioMarin Pharmaceutical.
Leal‐Pardinas F, Truty R, McKnight DA, Johnson B, Morales A, Bristow SL, et al. Value of genetic testing for pediatric epilepsy: Driving earlier diagnosis of ceroid lipofuscinosis type 2 Batten disease. Epilepsia. 2022;63:e68–e73. 10.1111/epi.17269
REFERENCES
- 1. Johnson TB, Cain JT, White KA, Ramirez‐Montealegre D, Pearce DA, Weimer JM. Therapeutic landscape for Batten disease: current treatments and future prospects. Nat Rev Neurol. 2019;15(3):161–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gardner E, Bailey M, Schulz A, Aristorena M, Miller N, Mole SE. Mutation update: review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease. Hum Mutat. 2019;40(11):1924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mole S, Williams R, Goebel H. The neuronal ceroid lipofuscinoses (Batten disease). Oxford, UK: Oxford University Press; 2011. [Google Scholar]
- 4. Claussen M, Heim P, Knispel J, Goebel HH, Kohlschütter A. Incidence of neuronal ceroid‐lipofuscinoses in West Germany: variation of a method for studying autosomal recessive disorders. Am J Med Genet. 1992;42(4):536–8. [DOI] [PubMed] [Google Scholar]
- 5. Fietz M, AlSayed M, Burke D, Cohen‐Pfeffer J, Cooper JD, Dvořáková L, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119(1–2):160–7. [DOI] [PubMed] [Google Scholar]
- 6. Martin JA, Hamilton BE, Ventura SJ, Osterman MJK, Wilson EC, Mathews TJ. Births: final data for 2010. Natl Vital Stat Rep. 2012;61(1):1–72. [PubMed] [Google Scholar]
- 7. Truty R, Patil N, Sankar R, Sullivan J, Millichap J, Carvill G, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open. 2019;4(3):397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, Van de Graaf B, et al. Disease characteristics and progression in patients with late‐infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. 2018;2(8):582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steinfeld R, Heim P, von Gregory H, Meyer K, Ullrich K, Goebel HH, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112(4):347–54. [DOI] [PubMed] [Google Scholar]
- 10. Worgall S, Kekatpure MV, Heier L, Ballon D, Dyke JP, Shungu D, et al. Neurological deterioration in late infantile neuronal ceroid lipofuscinosis. Neurology. 2007;69(6):521–35. [DOI] [PubMed] [Google Scholar]
- 11. Schulz A, Ajayi T, Specchio N, de Los RE, Gissen P, Ballon D, et al. Study of intraventricular cerliponase alfa for CLN2 disease. N Engl J Med. 2018;378(20):1898–907. [DOI] [PubMed] [Google Scholar]
- 12. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho Y‐Y, et al. Sherloc: a comprehensive refinement of the ACMG‐AMP variant classification criteria. Genet Med. 2017;19(10):1105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berg AT, Coryell J, Saneto RP, Grinspan ZM, Alexander JJ, Kekis M, et al. Early‐life epilepsies and the emerging role of genetic testing. JAMA Pediatr. 2017;171(9):863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mole SE, Schulz A, Badoe E, Berkovic SF, de Los Reyes EC, Dulz S, et al. Guidelines on the diagnosis, clinical assessments, treatment and management for CLN2 disease patients. Orphanet J Rare Dis. 2021;16(1):185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saneto RP, Lee I‐C, Koenig MK, Bao X, Weng S‐W, Naviaux RK, et al. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure. 2010;19(3):140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Specchio N, Pietrafusa N, Trivisano M. Changing times for CLN2 disease: the era of enzyme replacement therapy. Ther Clin Risk Manag. 2020;16:213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berkovic SF. Genetics of epilepsy in clinical practice. Epilepsy Curr. 2015;15(4):192–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1‐S3
Fig S1
Fig S2
Fig S3