

Summary
We present draft genome assemblies of Beta patula, a critically endangered wild beet endemic to the Madeira archipelago, and of the closely related Beta vulgaris ssp. maritima (sea beet). Evidence‐based reference gene sets for B. patula and sea beet were generated, consisting of 25 127 and 27 662 genes, respectively. The genomes and gene sets of the two wild beets were compared with their cultivated sister taxon B. vulgaris ssp. vulgaris (sugar beet). Large syntenic regions were identified, and a display tool for automatic genome‐wide synteny image generation was developed. Phylogenetic analysis based on 9861 genes showing 1:1:1 orthology supported the close relationship of B. patula to sea beet and sugar beet. A comparative analysis of the Rz2 locus, responsible for rhizomania resistance, suggested that the sequenced B. patula accession was rhizomania susceptible. Reference karyotypes for the two wild beets were established, and genomic rearrangements were detected. We consider our data as highly valuable and comprehensive resources for wild beet studies, B. patula conservation management, and sugar beet breeding research.
Keywords: genome sequencing, genome assembly, gene annotation, comparative genomics, genome structure and evolution, synteny display, Beta patula, Beta vulgaris ssp. maritima , Beta vulgaris ssp. vulgaris , sugar beet
Significance Statement
Wild beets of the genus Beta are important genetic resources for the improvement of cultivated beet varieties. This work presents genome assemblies and gene sets of Beta vulgaris ssp. maritima (sea beet) and of the critically endangered Beta patula, identifies orthologs and syntenic regions between sugar beet and the two wild beets, and provides valuable genome‐wide molecular resources for wild beet studies and sugar beet breeding.
Introduction
Genetic divergence of wild plant species is commonly exploited to introduce useful properties into domesticated varieties. One of the most important crops is sugar beet as the main source for sugar production, animal feed and sustainable energy in Europe, and competing with sugar cane worldwide. In sugar beet breeding programs, the introduction of wild alleles played and still plays an important role for the acquisition of resistances and for yield improvement (Biancardi et al., 2012; Monteiro et al., 2018).
The Amaranthaceae family (order Caryophyllales) contains many species of nutritional and economic importance, such as spinach, quinoa, amaranth, as well as Beta vulgaris ssp. vulgaris (here referred to as B.v. vulgaris) comprising the cultivars chard, table beet, fodder beet and sugar beet. B.v. vulgaris belongs to the genus Beta, section Beta, and apart from the cultivated forms this section contains the four wild beets B. vulgaris ssp. maritima (sea beet), B. vulgaris ssp. adanensis, Beta macrocarpa and Beta patula (Biancardi et al., 2012). Their habitats are generally coastal regions ranging from the Mediterranean Sea to the Atlantic Ocean in Northern Europe. Beta patula, however, is a rare and critically endangered species endemic to two small uninhabited islets of the Madeira archipelago (Pinheiro de Carvalho et al., 2010). The population is fragmented and consists of less than 3000 individuals spread over 1.5 km2 (Carvalho et al., 2011).
For sugar beet (diploid, 2n = 18 chromosomes), a broad range of resources exists, including sequence‐based genetic and physical maps (Schneider et al., 2007; Dohm et al., 2012; Holtgräwe et al., 2014), expressed sequence tags (ESTs) (Herwig et al., 2002), isogenic bacterial artificial chromosome (BAC) end sequences and fosmid end sequences from the double‐haploid genotype KWS2320 (Lange et al., 2008), as well as a genome assembly of KWS2320 (Dohm et al., 2014). Recently, the five‐generation inbred genotype EL10 was sequenced (Funk et al., 2018).
Studies on wild beets addressed their genetic diversity (Andrello et al., 2017), as well as their phylogeny and geographic distribution (Richards et al., 2014; Andrello et al., 2016; Romeiras et al., 2016; Touzet et al., 2018). These studies used molecular information and generated large‐scale marker sets of several wild beet (sub‐)species. The organization of wild beet chromosomes had been analyzed in fluorescence in situ hybridization (FISH) experiments, showing that wild species of the Beta section have the same number of chromosomes as sugar beet and share many repeat families (Paesold et al., 2012).
However, de novo genome sequencing of wild beets has so far only been described for B. vulgaris ssp. maritima (here referred to as B.v. maritima). Wild species are generally a challenge for genome assembly due to heterozygosity and unavailability of double‐haploid lines. For B.v. maritima, a draft genome sequence and gene set were published along with the identification of the rhizomania resistance gene Rz2 (Capistrano‐Gossmann et al., 2017) using an accession that had been subjected to inbreeding over several generations to reduce heterozygosity. Beta patula was described as a naturally inbreeding species (Pinheiro de Carvalho et al., 2010) and therefore may be expected to show a low level of heterozygosity, qualifying the poorly studied B. patula as an attractive and promising target for de novo genome sequencing.
Here, we present assemblies and gene sets of B. patula and B.v. maritima as well as genome‐wide comparisons between the two wild beets and the domesticated sugar beet B.v. vulgaris. We include reference FISH karyotypes at chromosome‐arm resolution for two additional B.v. maritima accessions and B. patula. We performed a comparative genomic analysis of the Rz2 locus, responsible for rhizomania resistance. For graphical representation of conserved gene order we developed a synteny display tool. These resources are not only important for evaluating the genetic diversity of wild beets and for sugar beet breeding research, but also crucial for monitoring B. patula as a highly endangered species and hence for conservation management.
Results
Beta patula genome assembly and gene set
Using the Illumina platform we generated genomic sequences for B. patula from one paired‐end library (insert size 0.6 kbp) and five mate‐pair libraries (span sizes 2.5–9 kbp). After quality filtering we assembled 336 million paired‐end pairs and 186 million mate‐pairs into 41 354 scaffolds and contigs. The assembly size was 624 Mbp with an N50 size of 271 kbp, and the largest sequence had a length of 1.7 Mbp (Table 1).
Table 1.
Illumina sequencing data after quality filtering as input for assembly (a) and assembly results (b) for Beta patula and B.v. maritima
| (a) | ||||
|---|---|---|---|---|
| Input data | ||||
| Beta patula | Beta vulgaris ssp. maritima | |||
| Insert size (kbp) | Filtered pairs (million) | Insert size (kbp) | Filtered pairs (million) | |
| Paired‐end | 0.6 | 336 | 0.6* | 390 |
| Mate‐pair | 2.5 | 56 | 2.5* | 30 |
| 4 | 17 | 2.5 | 41 | |
| 5 | 58 | 5* | 35 | |
| 6 | 27 | 5 | 42 | |
| 9 | 28 | 10 | 22 | |
| (b) | ||
|---|---|---|
| Assembly results | ||
| Beta patula | Beta vulgaris ssp. maritima | |
| Total size | 624 Mbp | 590 Mbp |
| Sequences ≥ 500 bp | 41 354 | 28 626 |
| N50 size | 271 kbp | 176 kbp |
| N50 number | 654 | 887 |
| N90 size | 20 kbp | 24 kbp |
| N90 number | 2905 | 4177 |
| Avg. seq. length | 15 kbp | 21 kbp |
| Longest sequence | 1.68 Mbp | 2.42 Mbp |
Asterisks refer to data sets generated with previous work (Capistrano‐Gossmann et al., 2017). In (b), only scaffolds and contigs of length 500 bp or larger were considered.
To support gene prediction we generated mRNA‐seq data from B. patula leaves and combined them with existing B.v. vulgaris mRNA‐seq data from five different accessions and different tissues (Dohm et al., 2014; Minoche et al., 2015). Transcript evidence and repeat information were prepared as input for the AUGUSTUS pipeline (Stanke et al., 2004) that was run using Beta‐specific prediction parameters as established previously (Minoche et al., 2015). From the initial number of 29 379 predicted transcripts (27 375 genes), we kept only those that had at least 1% mRNA‐seq evidence resulting in a final set of 25 127 genes (27 119 transcripts) supported by 1–100% evidence (Table 2). The percent evidence refers to the fraction of predicted features (e.g. exon‐intron boundaries) that were supported by mRNA‐seq data. Because most evidence‐supported transcripts included both start and stop codons (92%), we concluded that the gene models as well as the genome assembly were of high quality.
Table 2.
Properties of the Beta patula and Beta vulgaris spp. maritima gene sets
| Beta patula | Beta vulgaris spp. maritima | |||||
|---|---|---|---|---|---|---|
| 100% evid. | 1–99% evid. | 0% evid. | 100% evid. | 1–99% evid. | 0% evid. | |
| Number of transcripts | 15 246 | 11 873 | 2260 | 13 889 | 16 145 | 5804 |
| Number of proteins | 15 237 | 11 823 | 2256 | 13 887 | 16 102 | 5804 |
| Avg. transcript length | 1494.9 bp | 1421.5 bp | 678.7 bp | 1499.8 bp | 1368.5 bp | 690.6 bp |
| Avg. protein length | 355.9 aa | 350.9 aa | 198.9 aa | 359.4 aa | 354.6 aa | 201.2 aa |
| Tr. with start and stop | 92.4% | 91.4% | 48.7% | 90.6% | 79.7% | 46.4% |
| Avg. exon number per tr. | 4.9 | 5.5 | 1.5 | 4.9 | 5.4 | 1.5 |
| Single‐exon transcripts | 29.7% | 16.0% | 70.5% | 29.1% | 16.5% | 71.0% |
| Avg. exon length | 302.6 bp | 257.8 bp | 447.6 bp | 305.5 bp | 253.7 bp | 456.8 bp |
| Avg. intron length | 939.3 bp | 1097.5 bp | 1584.9 bp | 931.8 bp | 1025.1 bp | 1857.2 bp |
Proteins of < 10 aa were removed. The average length of genes that had at least one transcript showing 1–100% mRNA‐seq evidence was 5695.4 bp (B. patula) and 5457.3 bp (B.v. maritima), respectively.
aa, amino acids; avg., average; bp, base pairs; evid., evidence; tr., transcripts.
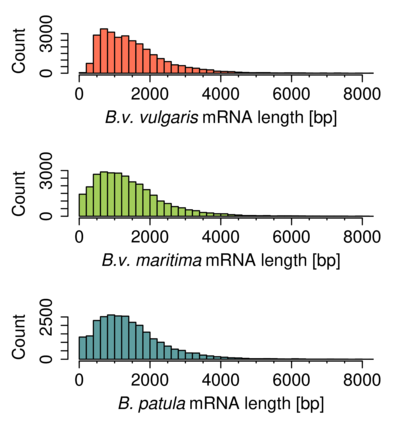
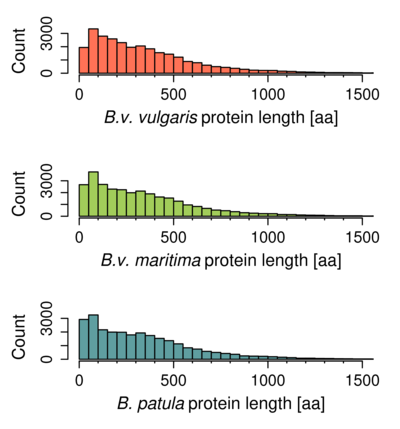
The predicted transcripts of B. patula and B.v. vulgaris (Minoche et al., 2015) showed similar length distribution profiles (Figure S1), as expected for closely related species. Although the genome assembly of B. patula (N50 size of 271 kbp) was much more fragmented than the one of B.v. vulgaris (N50 size of 2 Mbp, version RefBeet‐1.2; Dohm et al., 2014), the assembled contigs and scaffolds mostly contained complete transcripts. This indicates that the gene‐containing fraction of the genome was well represented in the B. patula assembly.
Beta vulgaris ssp. maritima assembly and gene set
A previously reported assembly of B.v. maritima accession WB42 (Capistrano‐Gossmann et al., 2017) had been calculated based on Illumina data from one paired‐end library and two mate‐pair libraries (span sizes 2.5 and 5 kbp) containing 57 361 sequences (largest sequence: 899 kbp) with an N50 size of 59 kbp. To improve the genomic contiguity, we generated Illumina mate‐pair sequences from new libraries of different span sizes (additional 2.5, 5 and 10 kbp libraries) from the same genotype resulting in 105 million newly generated mate‐pairs. Assembling the genome including all data sets, we achieved an N50 size of 176 kbp and an assembly size of 590 Mbp in 28 626 sequences with the largest sequence having a length of 2.42 Mbp (Table 1).
The previous gene prediction had been performed without repeat masking in order to find a specific transposon insertion (Capistrano‐Gossmann et al., 2017). We performed transposon masking in the previously predicted gene set and found that 45% of the predicted coding sequence was masked. We therefore established a reference gene set for B.v. maritima using mRNA‐seq data, repeat information for masking, and Beta‐specific prediction parameters as input for AUGUSTUS. From the 35 838 transcripts predicted based on our B.v. maritima genome assembly, we kept 30 034 transcripts (27 662 genes) that were supported by at least 1% mRNA‐seq evidence (Table 2). A total of 85% among the evidence‐supported transcripts included both start and stop codons.
The length distribution of transcripts and protein sequences was similar to the distributions of B. patula and B.v. vulgaris gene sets (Figure S1).
Validation of assemblies and gene sets
The completeness of assemblies and gene sets was estimated by sequence comparisons to a set of 1440 highly conserved single‐copy plant genes using BUSCO (Simão et al., 2015). We ran BUSCO on our assemblies and gene sets of B. patula and B.v. maritima, and additionally on sugar beet and spinach data (Dohm et al., 2014; Minoche et al., 2015). For each gene set, only one sequence per gene was selected. When using genome assemblies as input we achieved high numbers of matched core plant genes in all assemblies (1309 of 1440 on average, 91%). The numbers were generally higher (94% on average) when running BUSCO on the predicted gene sets (Table S1). The internal gene prediction step of BUSCO during assembly validation may be less accurate than the gene prediction we performed separately. The elevated fraction of matched core plant genes indicates that our clade‐specific adaptions (mRNAseq data, Beta‐specific prediction parameters, Caryophyllales‐specific transposon masking) improved the accuracy of the resulting gene models. Because the gene sets were predicted based on the genome assemblies, we concluded that BUSCO's gene set validation reflected at the same time the genome assembly completeness in gene‐containing regions.
The assembly sequence of B.v. vulgaris (version RefBeet‐1.2), assembled from medium‐long reads and long‐range paired sequences (BAC ends and fosmid ends), had a much higher N50 size than the short‐read assemblies of B. patula, B.v. maritima and spinach. However, the estimated completeness of the predicted gene sets was comparable among all four species (Table S1).
There were 47 plant genes from the BUSCO data set that were not matched in any of the Beta or spinach gene sets. Their functional assignments were extracted from OrthoDB (Kriventseva et al., 2019; Table S2). However, manually inspected tBLASTn searches against the B.v. vulgaris genome (RefBeet‐1.2) of these missed genes revealed that 39 out of 47 genes matched annotated exons at considerable E‐values (largest E‐value 6e‐04). This group of genes may have evolved differently in Caryophyllales compared with the species that were used to establish the BUSCO database and its default homology cutoffs. Another explanation would be that these genes were inaccurately predicted, for example due to repeat vicinity as some OrthoDB annotations suggest (Table S2). We conclude that the average of 94% completeness as determined by BUSCO was the lower limit, and the actual completeness of the four gene sets may be higher.
Genome size estimation
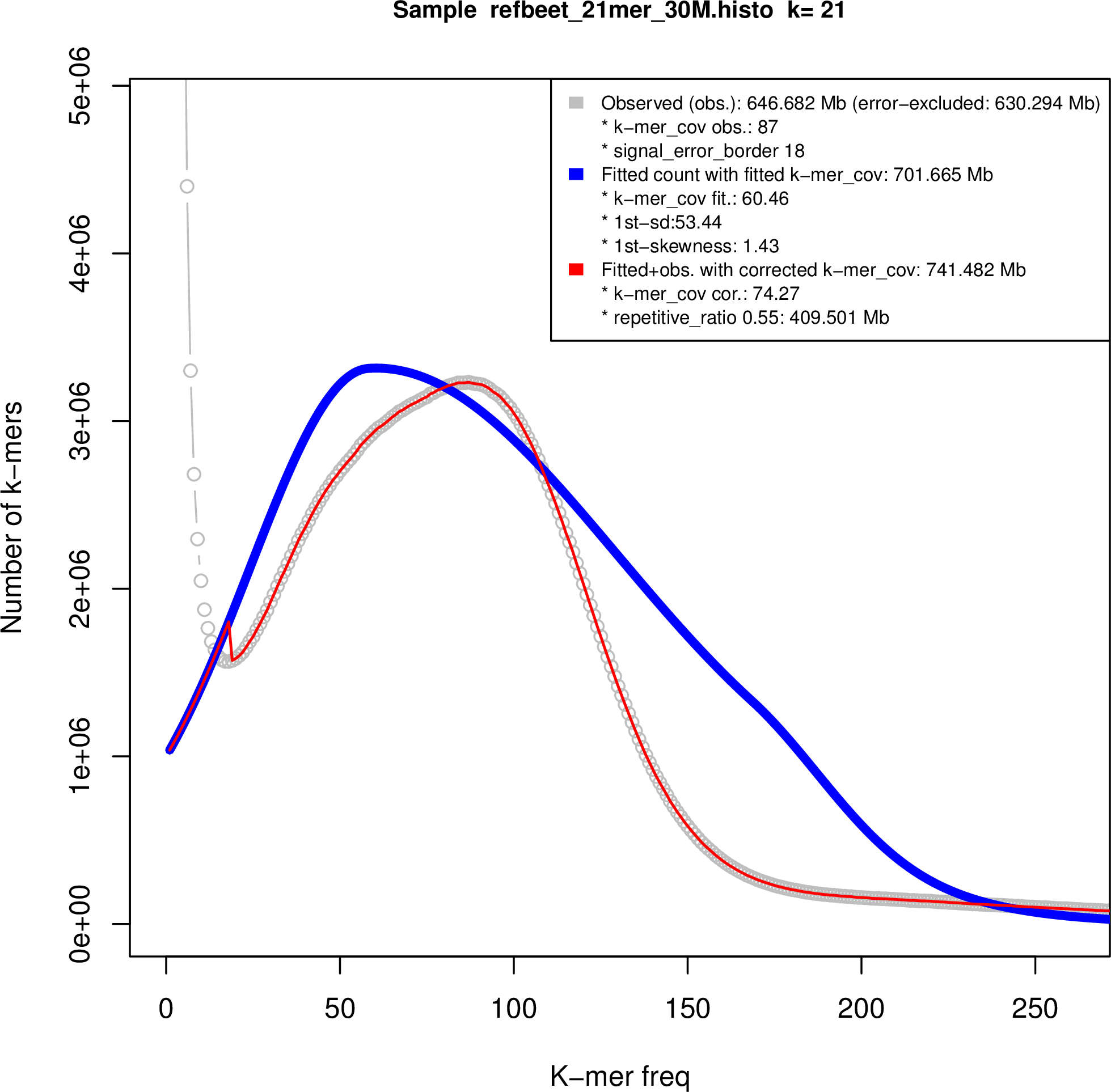
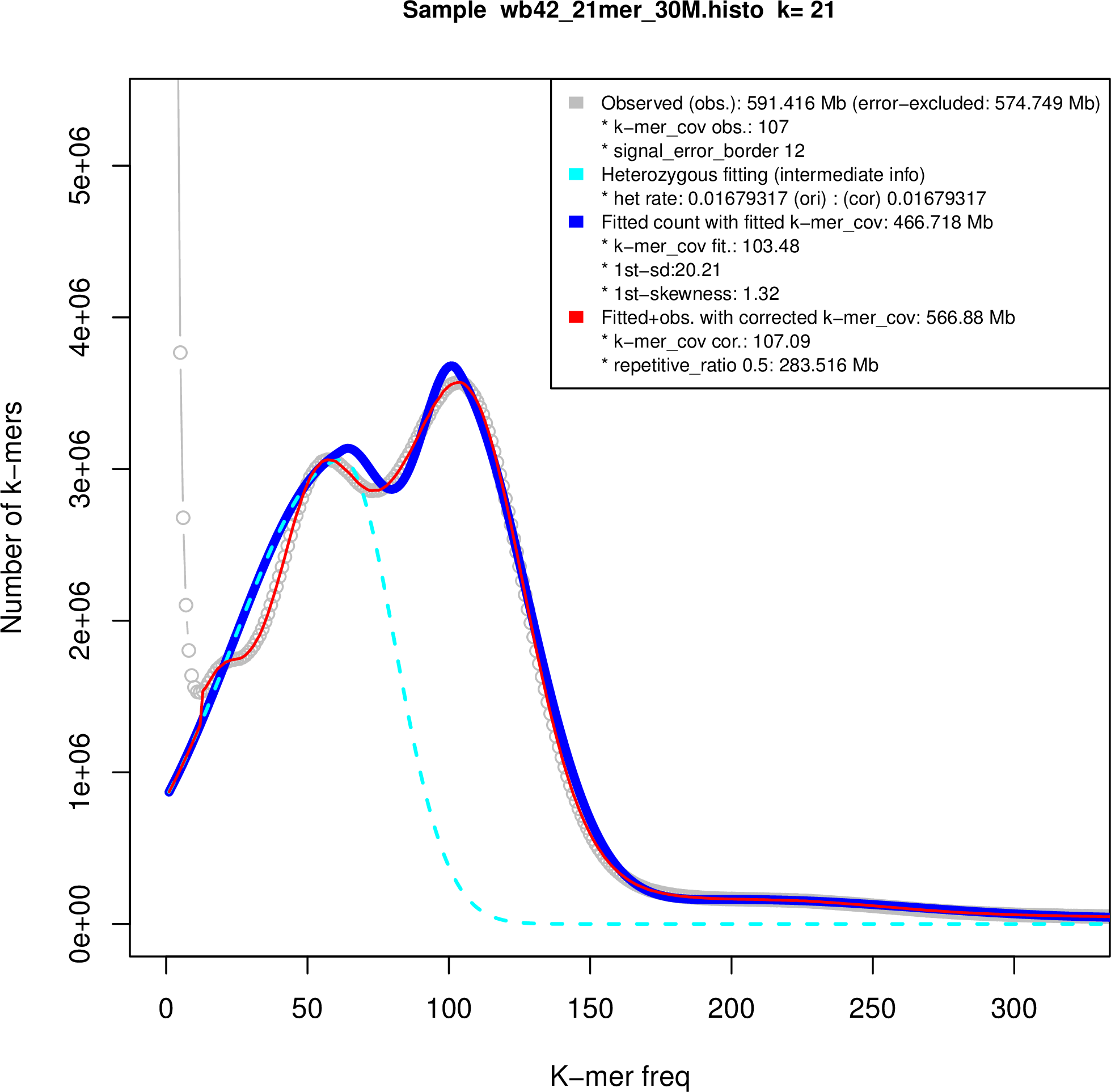
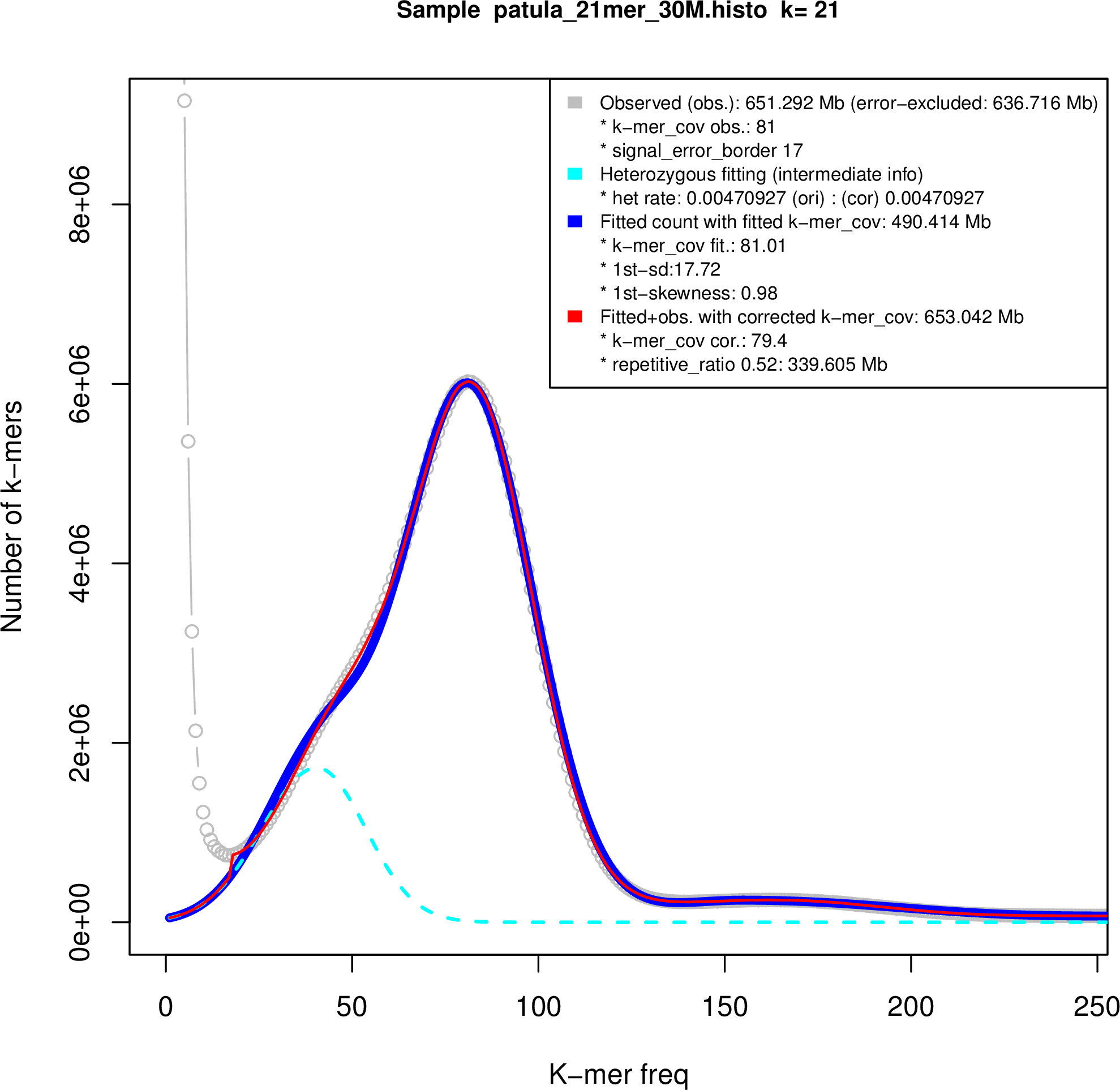
We used the sequencing reads to estimate the genome sizes of B.v. vulgaris, B.v. maritima and B. patula based on k‐mer frequencies. Subsequences of length k = 21 derived from quality‐filtered paired‐end reads as used for the wild beet assemblies (this work) and for a sugar beet assembly named RefBv (Dohm et al., 2014) were analyzed. The k‐mer frequency distributions were refined by fitting of skew normal distributions as implemented in findGSE (Sun et al., 2018), which also takes heterozygosity into account (Figure S2). According to this method, the wild beet genomes had a smaller size than sugar beet. The sugar beet genome size had been estimated previously in a range of 731 Mbp (Dohm et al., 2014) to 758 Mbp (Arumuganathan and Earle, 1991). The result of findGSE indicated genome sizes of 741 Mbp for sugar beet, 567 Mbp for B.v. maritima and 653 Mbp for B. patula, respectively.
The heterozygosity fitting of the k‐mer frequency distributions exhibited two peaks in the wild beet data plots as expected for heterozygous genomes. The homozygous fraction was more pronounced in B. patula, indicating that the naturally inbreeding B. patula had a lower level of heterozygosity than the targeted inbred B.v. maritima genotype WB42. The estimation was 0.47% heterozygosity for B. patula and 1.68% heterozygosity for WB42.
Comparison of gene sets and synteny analysis
The gene sets of B. patula and B.v. maritima and the current reference gene set of B.v. vulgaris (BeetSet‐2; Minoche et al., 2015) were compared with each other in order to find sequence‐based relationships. Only one transcript per gene was used in its translated form, i.e. 26 385 peptide sequences for B.v. vulgaris, 27 617 for B.v. maritima, and 25 068 for B. patula (sequences shorter than 10 amino acids were removed). The input sequences were clustered into gene families, referred to as ‘orthogroups’, by applying OrthoFinder (Emms and Kelly, 2015). The initial orthogroups (Appendix S1, Figures S3 and S4) were refined by taking conserved gene order into account. The Beta species studied are very closely related, with B.v. vulgaris and B.v. maritima being two subspecies of B. vulgaris, and B. patula being a member of the same section and genus. Thus, we expected the structure and organization of the three genomes to reflect this relationship by widespread similarity in their gene order. In order to confirm this, we explored the relative order of the B.v. vulgaris genes and their orthologs of B. patula and B.v. maritima. We transferred the orthogroup clustering into 1:1 orthologous gene connections between B.v. vulgaris and each of the two other species. For the two sets of orthologous gene pairs (one set for B.v. vulgaris and B.v. maritima, one for B.v. vulgaris and B. patula), we sorted the genes by their genomic positions within scaffolds. Sparsely connected scaffolds were removed. The remaining scaffolds showed large blocks of genes in conserved order (Figure 1). Between B.v. vulgaris and B.v. maritima, we found 274 B.v. vulgaris scaffolds and 1097 B.v. maritima scaffolds that showed syntenic regions including a total of 13 759 B.v. vulgaris genes and 13 648 B.v. maritima genes. Continuous syntenic blocks comprised up to 73 genes (the largest in scaffold Bvchr9.sca026); or 92 genes (in Bvchr7.sca021) if ignoring single missing genes between neighboring syntenic blocks. A similar number of scaffolds showing synteny between B.v. vulgaris and B. patula was found (286 B.v. vulgaris scaffolds and 953 B. patula scaffolds involved), and a higher total number of connected genes (15 297 B.v. vulgaris genes, 15 021 B. patula genes). The largest syntenic block of B.v. vulgaris and B. patula contained 48 genes (in Bvchr8.sca018); the three largest blocks after ignoring single missing genes showed 106 (in Bvchr9.sca026), 85 (in Bvchr5.sca023) and 77 genes (in Bvchr6.sca002). We graphically represented the syntenic regions along each reference scaffold (see Experimental procedures) and provide the genome‐wide collection as pictures together with text files containing positions and gene names at http://bvseq.boku.ac.at/Genome/Download/Bmar/Synteny (B.v. maritima) and http://bvseq.boku.ac.at/Genome/Download/Bpat/Synteny (B. patula), respectively. Refinement of the orthogroups based on syntenic relations (after filtering sparse scaffold connections) resulted in 15 540 orthogroups containing 17 189 B.v. vulgaris genes, 13 648 B.v. maritima genes and 15 021 B. patula genes (Table S3). There were 10 911 orthogroups containing genes from all three species, and 9861 orthogroups containing exactly one gene from each species.
Figure 1.
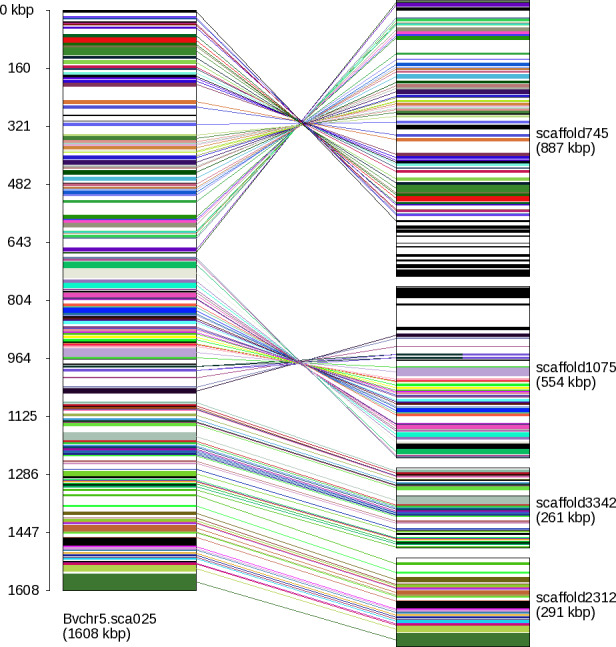
Gene connections based on orthology relations in one Beta vulgaris scaffold (Bvchr5.sca025) and four Beta patula scaffolds (scaffold745, scaffold1075, scaffold3342, scaffold2312).
In these scaffolds, 144 B. vulgaris genes were linked to 144 B. patula genes in total, all showing perfect colinearity. The two B. patula scaffolds on top have reversed orientation indicated by the crossed lines. The upper part of the reversed B. patula scaffold745 matches the adjacent B. vulgaris scaffold Bvchr5.sca024. The lower part of the reversed B. patula scaffold1075 shows sparse matches to three different chromosomes (ignored after filtering) potentially indicating a misassembly in scaffold1075.
Phylogenetic relationship
Identifying orthologous genes is a crucial step in phylogeny reconstruction (Delsuc et al., 2005). We performed alignments for the 9861 genes showing 1:1:1 orthology in each species and inferred a phylogenetic tree based on the concatenated alignments of all genes. In current taxonomies B. patula is considered a species, whereas B.v. maritima and B.v. vulgaris are classified as subspecies (Biancardi et al., 2012). The topology of our phylogeny placed B.v. vulgaris and B.v. maritima closer together than to B. patula as expected, considering that the crop B.v. vulgaris was derived from B.v. maritima by breeding activities (Figure 2). All distances were very small, raising the question if B. patula actually has the status of a species or rather a subspecies. In a previous phylogenetic study based on nuclear and chloroplast DNA markers, the relationship of B.v. maritima, B.v. vulgaris and B. patula remained unresolved (Romeiras et al., 2016). However, morphology and geographic distribution support the status of B. patula as a species (Pinheiro de Carvalho et al., 2010).
Figure 2.

Phylogenetic tree of Beta vulgaris, Beta vulgaris spp. maritima and Beta patula based on sequence similarity of 9861 orthologous genes. Distances in Newick format: [(B.v. maritima:0.00606, B.v. vulgaris:0.00606):0.00047, B. patula:0.00653].
Wild beet karyotype analysis
We performed a molecular cytogenetic chromosome analysis, and compared the karyotypes of sugar beet and wild beets. To establish wild beet reference karyotypes, we used a set of chromosome‐specific terminal BACs developed in B.v. vulgaris (Paesold et al., 2012) as probes for double target FISH experiments in B.v. maritima and B. patula accessions. The sugar beet BAC probes consistently produced FISH signals in terminal positions on two homoeologous metaphase chromosomes in each of the wild beets enabling pairing and numbering of the 2n = 18 chromosomes (Figure 3). The overall signal pattern was similar in sugar beet and wild beets; however, both B.v. maritima accessions showed chromosomal rearrangements. The simultaneous hybridization with probe pZR18S (specific for the 18S‐5.8S‐25S rRNA genes) and a cytogenetic marker for B.v. vulgaris chromosome 1 pXV2 (specific for the 5S rRNA genes) revealed polymorphisms in the ribosomal gene arrays. While the B.v. maritima accessions BETA 1101 had an additional minor site on chromosome 9, the rearrangement in accession BETA 2322 was considerably larger. Here, a translocation resulting from breakage in the 18S‐5.8S‐25S rRNA gene array of one chromosome 1 copy and incorporation into a single chromosome nine was detected. In B. patula no chromosomal rearrangements affecting the terminal regions of BAC hybridization were observed.
Figure 3.
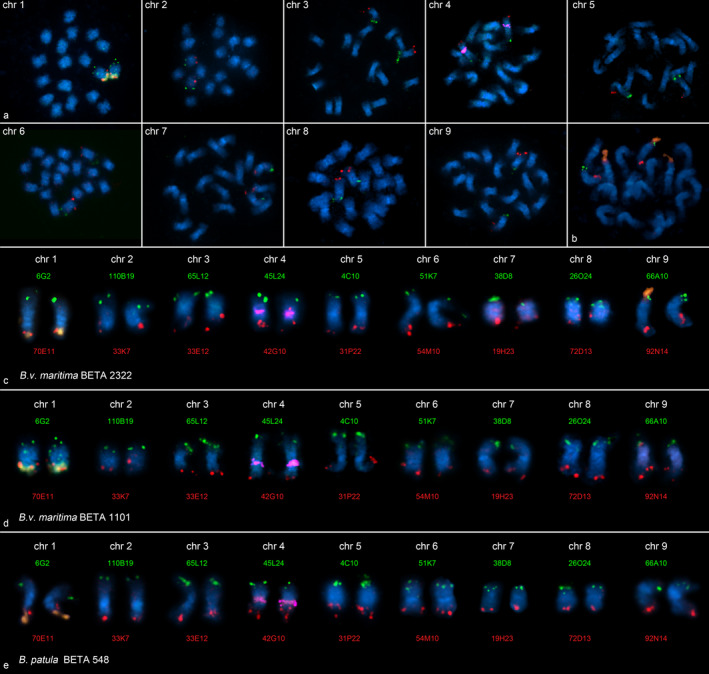
Reference fluorescence in situ hybridization (FISH) karyotypes of wild beets.
Terminal, arm‐specific BAC markers representing the most distal genetic markers of the nine genetic linkage groups of sugar beet were hybridized to mitotic metaphases of Beta vulgaris spp. maritima accessions BETA 1101 (originating from The Netherlands). In each panel, one pair of chromosomes was detected (a). Multi‐color FISH of differently labeled probes enabled the identification of each arm of mitotic metaphase chromosomes in B.v. maritima accessions BETA 1101 (c) and BETA 2322 (originating from Portugal) (d) as well as the Beta patula accession BETA 548 (e). Cytogenetic markers for chromosomes 1 (18S‐5.8S‐25S rRNA genes, orange in a–e) and 4 (5S rRNA genes, magenta in c–e) were hybridized simultaneously. A translocation consisting of a large part of the 18S‐5.8S‐25S rRNA gene array from chromosome 1 (orange signal) to a single chromosome 9 (orange signal) was detected in both B.v. maritima accessions BETA 1101 (top left in a, d) and BETA 2322 (b, c). The rDNA translocation in B.v. maritima BETA 2322 was larger. The chromosomes were counterstained with DAPI (blue) and arranged according to the sugar beet kayotype.
Chromosome assignment and scaffold ordering
Although the two wild beets and sugar beet showed the same number of chromosomes, there may be rearrangements that are different from rDNA translocations and were not detectable by hybridization of terminal BAC markers. Structural differences become obvious during synteny analysis in cases where a scaffold of a wild beet is connected via syntenic regions to two different sugar beet chromosomes or where distant regions of a sugar beet chromosome are linked together within one syntenic wild beet scaffold. The reason for inconsistencies may also be misassemblies or wrongly assigned sugar beet scaffolds. However, inconsistent connections that accumulate in certain chromosomes may indicate structural changes. We analyzed all wild beet scaffolds that have syntenic relations via at least five genes to more than one sugar beet scaffold of RefBeet‐1.2. There were 54 B. patula scaffolds and 40 B.v. maritima scaffolds that bridged two or more sugar beet scaffolds, of which 14 connections were confirmed by both B. patula and B.v. maritima. In one case, both wild beets confirmed a suspected misassembly in RefBeet, in three cases so far unplaced RefBeet scaffold were linked to chromosomally assigned scaffolds, and 10 cases were in line with the established order of RefBeet or linked so far unplaced but chromosomally assigned RefBeet scaffolds to scaffolds of the same chromosome. Among the remaining linking wild beet scaffolds, we found a number of cross‐chromosome connections that concentrated in chromosome 3 (B. patula, seven cases, and B.v. maritima, one case) and chromosome 9 (B. patula, two cases, and B.v. maritima, two cases; Figure 4). Because misassemblies should rather appear randomly throughout the genome, we assume that these cases may represent genomic rearrangements (Table S4). Chromosomal rearrangements that distinguish cultivars and their wild progenitors were also described in wheat (Ma et al., 2014). Additional data, for example long sequencing reads or large‐distance mate‐pairs, may resolve structural differences in more detail. Despite the described uncertainties, we ordered and chromosomally assigned a total of 920 B. patula scaffolds (335.0 Mbp) and 1060 B.v. maritima scaffolds (275.4 Mbp), respectively, based on their syntenic relations to sugar beet (Table S5).
Figure 4.
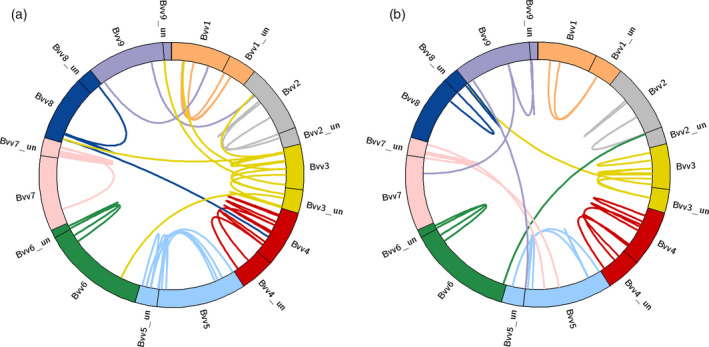
Circular representation of the nine sugar beet reference chromosomes (RefBeet‐1.2).
Connecting lines indicate scaffolds from Beta patula (a) or Beta vulgaris spp. maritima (b), resepectively, that bridge two RefBeet scaffolds based on sequence homology of genes. While most B. patula and B.v. maritima scaffolds linked scaffolds of the same chromosome together (many links between the „un” = unassigned portion and chromosomally assigned scaffolds), there are some cross‐chromosome connections that either indicate misassemblies or structural variation.
Genomic comparison
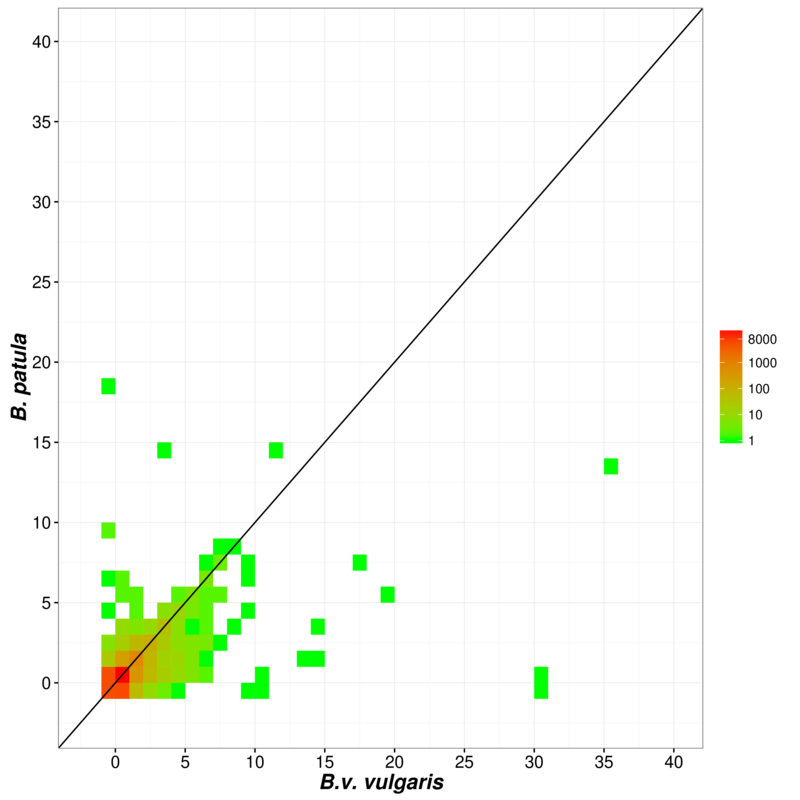
The close relationship of sugar beet and the two wild beets B.v. maritima and B. patula was reflected by the sequence similarity of their genomes. We aligned sequences larger than 1 kbp of the wild beet assemblies against the repeat‐masked RefBeet‐1.2 assembly and counted only non‐overlapping unique matches. A total of 289 Mbp was covered in RefBeet‐1.2 by B.v. maritima, and 259 Mbp were covered by B. patula; of these RefBeet‐1.2 regions 223 Mbp were matched by both wild beets. The average sequence identity of the matching regions was 94.80% for B.v. maritima and 94.97% for B. patula. Increasing the minimum sequence identity to 98%, we found 72 Mbp matching sequence between B.v. vulgaris and B.v. maritima and 57 Mbp between B.v. vulgaris and B. patula (Table S6). Highly conserved regions shared among the three genomes comprised 27 Mbp. The longest continuous shared region (18.7 kbp) was located in chromosome five of sugar beet.
Rhizomania resistance
A well‐described genomic region is the Rz2 locus linked to rhizomania resistance in B.v. maritima (Barzen et al., 1995; Grimmer et al., 2007; Capistrano‐Gossmann et al., 2017; Funk et al., 2018). It has been reported that in sugar beet the corresponding gene was disrupted by a transposon of 8 kbp length resulting in rhizomania susceptibility. Rhizomania leads to dramatic yield loss (Biancardi and Tamada, 2016). We studied the Rz2 region in our B.v. maritima assembly as well as in B. patula by comparing the rhizomania resistance gene from the previously reported gene set (Capistrano‐Gossmann et al., 2017) with the genome assemblies and gene annotations generated in this work. The question was if B. patula showed a genomic structure like B.v. maritima suggesting rhizomania resistance, or rather like B.v. vulgaris suggesting rhizomania susceptibility.
We located the genetic markers for the Rz2 region in our assemblies to identify the corresponding scaffolds and to verify that these KASP (Kompetitive Allele‐Specific PCR) markers were detectable in their correct order. The order of the five markers closest to the Rz2 gene determined by genetic mapping was CAU_4088, CAU_3881, CAU_3880, CAU_4188, CAU_3882 (Capistrano‐Gossmann et al., 2017). The correct marker order was perfectly matched in our assembled B.v. maritima scaffold106 (Figure 5). In B. patula and sugar beet (RefBeet‐1.2), four of the five markers matched in the correct order as well, namely in scaffold35 (B. patula) and Bvchr3_un.sca001 (RefBeet‐1.2), respectively, but we did not find matches for CAU_3880. The expected location of the Rz2 gene was flanked by genes Bv3_066660_tfftt and Bv3_066670_zxwn in the RefBeet annotation, and two genes were predicted between them, i.e. Bv3_066661z821 („z821”) and Bv3_066661z822 („z822”), both lacking mRNA evidence.
Figure 5.
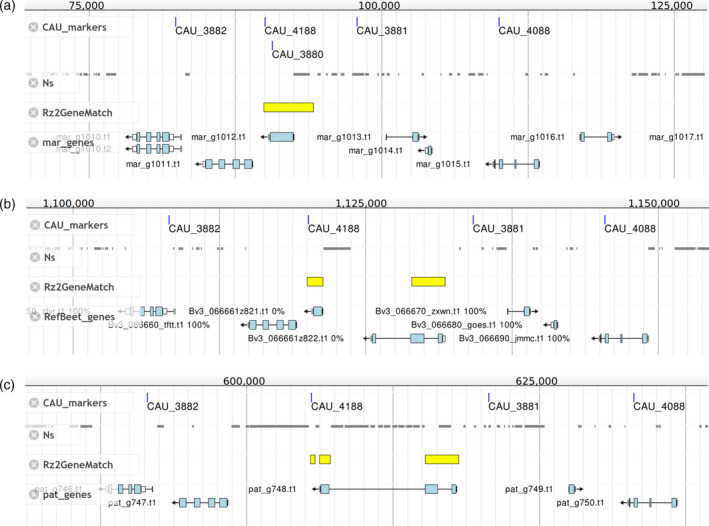
Comparative view of the Rz2 locus in Beta vulagrais spp. maritima (mar_scaffold106) (a), B. vulgaris (Bvchr3_un.sca001 of RefBeet‐1.2) (b), and Beta patula (pat_scaffold35) (c). The four tracks are: „CAU_markers” showing the matching positions of genetic markers from a previous study (Capistrano‐Gossmann et al., 2017), “Ns” showing regions that were masked before gene prediction or assembly gaps, „Rz2GeneMatch” showing the matching positions of Rz2 gene BmChr3_scaffold3841.g9573 from a previous B.v. maritima gene set, “pat_genes, RefBeet_genes, mar_genes” showing the current gene annotation of the respective assembly. Gene names are indicated lefthand to the genes. The Rz2 gene match (yellow) is interrupted in B. vulgaris (matching the genes z821 and z822) and B. patula (matching the gene pat_g748), whereas in B.v. maritima there is a contiguous match spanning the predicted gene mar_g1012.
We compared the sequence of the previously predicted single‐exon Rz2 gene „BmChr3_scaffold3841.g9573” (Capistrano‐Gossmann et al., 2017) with the corresponding scaffolds of our assemblies. In our B.v. maritima assembly, the Rz2 gene sequence had one continuous match over its full length at 100% sequence identity covering the location of marker CAU_3880 and the predicted gene mar_g1012, whereas in sugar beet the match was split into two parts separated by 7.6 kbp and covering z821 and z822. In B. patula the match was split up into three matches, of which two were close to each other and the third one was separated by 8.1 kbp, all three covering the predicted gene pat_g748. These matches suggest that z821 and z822 in sugar beet should be considered as one gene corresponding to pat_g748 in B. patula and mar_g1012 in B.v. maritima. Because the B. patula gene showed a large disruption similar to sugar beet and different from B.v. maritima, we would expect the sequenced B. patula accession to be rhizomania susceptible.
Discussion
We generated sequencing data for two wild beet taxa of the Beta genus, section Beta, i.e. for B. patula and B.v. maritima, and obtained a variety of valuable genomic resources: Genome assemblies and gene sets of high quality, orthology relationships including sugar beet genes, genome‐wide synteny display as well as chromosomal assignments and ordering. Cytogenetic analysis established FISH reference karyotypes for three accessions of the two wild beets.
Compared with the previously described B.v. maritima assembly (Capistrano‐Gossmann et al., 2017), we achieved a three times larger N50 size demonstrating that additional mate‐pair data, in particular larger span‐sizes, have substantially improved the assembly. For gene prediction we applied lineage‐specific transposon masking and obtained a similar number of genes compared with the well‐established sugar beet gene set. The assembly of B. patula showed an even larger N50 size than the B.v. maritima assembly, reflecting its low level of heterozygosity and allowing gene predictions of high quality and completeness. The wild beet genes were clustered into orthogroups together with sugar beet genes. Because the gene set of sugar beet had been functionally annotated (Dohm et al., 2014), the orthology relations provide direct access to the presumed functions of the corresponding genes in B. patula and B.v. maritima. Species‐specific genes may be candidates to search for differing phenotypic or physiological traits of the wild beet species. While disease resistances were already widely exploited (Frese et al., 2000), there might be abiotic stress tolerance of interest to sugar beet breeders: B. patula has adapted to a warm and dry habitat, growing on rocky soil of low soil moisture and high salinity (Pinheiro de Carvalho et al., 2010).
Orthologous genes were not only inferred from sequence homology but also confirmed based on their genomic location. After filtering out sparsely connected scaffolds, the remaining orthologs revealed large syntenic regions in the three beet genomes. Further systematic searches for missing genes within syntenic scaffolds may discover diverged orthologs to complete such regions. We developed a software tool to graphically display the scaffolds and their connecting genes, automatically arranged even for very large scaffolds with hundreds of genes.
The 1:1:1 orthologous genes were used for phylogeny reconstruction supporting the results of genomic analyses and genome‐wide synteny and showing that there was only little genomic divergence between B. patula and B.v. martima. Additional data from other members of the Beta section, for example B. macrocarpa and B. vulgaris ssp. adanensis, may support a potential reclassification of B. patula as a subspecies of B. vulgaris. The close relationship of B.v. maritima and B. patula and their geographic distribution suggests that B. patula is a derivative of B.v. maritima solely due to acquisition of self‐fertility, which is known to occur in the species (Biancardi et al., 2012). The emergence of B. patula may be an example that inbreeding in isolated populations contributes to speciation.
Genomic and cytogenetic studies on B. patula as a wild relative of sugar beet are an important step to explore and maintain the genepool for sugar beet breeding. Knowledge about the genetic diversity is important for conservational efforts for this highly endangered species in a very specialized and limited habitat.
Experimental Procedures
Plant material
The wild beet B. vulgaris ssp. maritima accession WB42 originated from Denmark and was partly selfed at KWS SE, Einbeck, Germany, as described (Capistrano‐Gossmann et al., 2017). Seeds of B. patula accession BETA 548 and B. vulgaris ssp. maritima accessions BETA 1011 and BETA 2322 were obtained from the Genebank of the Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany. DNA was extracted from leaves using the NucleoSpin Plant kit (Macherey‐Nagel, Düren, Germany). All plants were grown in a greenhouse under long‐day conditions.
Sequencing, assembly and gene prediction
For B. patula, we generated one paired‐end library with insert size 600 bp and five mate‐pair libraries using the Illumina Nextera technology (Illumina, San Diego, CA, USA) of span sizes 2.5, 4, 5, 6 and 9 kbp from a single plant. The data were sequenced on an Illumina HiSeq2000 instrument with 2 × 100 nt (paired‐ends) and 2 × 50 nt (mate‐pairs), respectively (Table 1). After quality filtering (Minoche et al., 2011), the data were assembled using SOAPdenovo‐V1.05 (Luo et al., 2012) with k = 49, and gaps were closed using SOAP GapCloser release 2011 with parameters ‐l 100 ‐p 31. For AUGUSTUS gene prediction (Stanke et al., 2004), the assembly was repeat‐masked by RepeatMasker (www.repeatmasker.org) using a comprehensive catalog of transposable elements derived from B.v. vulgaris and spinach (Dohm et al., 2014; Minoche et al., 2015) as repeat library and with parameters ‐norna ‐nolow ‐no is ‐gff. AUGUSTUS applies ab initio gene prediction methods in combination with user‐defined evidence to generate accurate gene models. Transcript evidence was provided by about 1 billion Illumina mRNA‐seq reads from five B.v. vulgaris accessions and different tissues (Dohm et al., 2014; Minoche et al., 2015). Additionally, we used 17.8 million single‐end mRNA‐seq reads (Illumina HiSeq2000, 50nt) generated from B. patula leaves (quality filtered according to Minoche et al., 2011). AUGUSTUS pipeline v2.7 was applied on the repeat‐masked assemblies with Beta‐specific prediction parameters as established in Minoche et al. (2015). The gene model was set to ‘complete’, no in‐frame stop codons were allowed and the prediction for untranslated regions (UTRs) was switched on. For the final gene sets we kept only those transcripts that had at least 1% of their features supported by mRNA‐seq evidence.
The previous B.v. maritima assembly was calculated based on one Illumina paired‐end library (600 bp insert size) and two mate‐pair libraries (2.5 and 5 kbp span size). We generated additional mate‐pair data on the Illumina HiSeq2000 instrument from new mate‐pair libraries (Illumina Nextera technology) of span sizes 2.5, 5 and 10 kbp (Table 1) from the same DNA sample, and assembled the previous data and the new data together using SOAPdenovo‐V1.05 (k = 49) for assembly and SOAP GapCloser release 2011 (parameters ‐l 100 ‐p 31) for gap closing. Gene prediction was performed as described above, including B.v. vulgaris mRNA‐seq data as evidence and transposon masking.
Genome size estimation
k‐mer frequency distributions were calculated using jellyfish v2.2.3 (Marçais and Kingsford, 2011) applied on quality‐filtered sequencing read data sets (2 × 100 nt, Illumina paired‐end sequencing). Raw k‐mer counts were obtained by running jellyfish count (parameters ‐C ‐m 21 ‐t 20 ‐s ‐20G) and used as input for jellyfish histo (‐h 30000000). Skew normal distributions were fit to the jellyfish results by findGSE (Sun et al., 2018) with default parameters and size k = 21.
Validation of assemblies and gene sets
BUSCO v3.0.2 (Simão et al., 2015) was run on assemblies and gene sets with default parameters and the database embryophyta_odb9 (downloaded from https://busco.ezlab.org/) comprising 1440 highly conserved plant genes. Only one protein sequence per gene was selected (same as for the gene set comparisons, see below). The sequences of missing BUSCO genes were compared with RefBeet‐1.2 (Dohm et al., 2014) using tBLASTn (Gertz et al., 2006) at http://bvseq.boku.ac.at/blast/ (expect 1e‐3, word size 3, scoring matrix BLOSUM62). Matching regions were manually inspected for overlaps with coding sequences of BeetSet‐2 (Minoche et al., 2015) using Gbrowse (Stein et al., 2002). Functional annotations were obtained from OrthoDB (Kriventseva et al., 2019).
Gene set comparisons
The comparisons and orthogroup clustering were run using OrthoFinder v1.1.4 (Emms and Kelly, 2015) with default parameters applied on the translated sequences of the gene sets (minimum length 10 amino acids), each gene represented by only one peptide. The primary transcript as assigned by AUGUSTUS was selected, i.e. the transcript showing the highest fraction of features being supported by mRNA‐seq evidence. The heatmaps were generated using the ggplot2 package (Wickham, 2009) in R v3.1.2 (R Core Team, 2015).
Phylogenetic analysis
Each triple of the 9861 1:1:1 orthologs (peptide sequences) was separately aligned with MUSCLE v3.8.31 (Edgar, 2004), subsequently all alignments were concatenated. Gap sites were removed with trimAl v1.2rev59 (Capella‐Gutiérrez et al., 2009), including conversion to phylip format. We used phylip v3.696 (Felsenstein, 2005) for phylogeny reconstruction, i.e. distance calculation with „protdist” (default parameters), tree construction with „neighbor” (UPGMA tree) and display with „drawgram” (default parameters).
Chromosome preparation and fluorescent in situ hybridization
Mitotic chromosomes were prepared from the leaf meristem of young plants. After incubation for 3–5 h in 2 mm 8‐hydroxyquinoline, leaves were fixed in methanol:acetic acid (3:1). Fixed plant material was macerated in an enzyme mixture consisting of 0.3% (w/v) cytohelicase (Sigma‐Aldrich, St. Louis, MO, USA), 1.8% (w/v) cellulase from Aspergillus niger (Sigma‐Aldrich), 0.2% (w/v) cellulase Onozuka‐R10 (Serva, Heidelberg, Germany), and 20% (v/v) pectinase from A. niger (Sigma‐Aldrich) in citrate buffer (4 mm citric acid, 6 mm sodium citrate, pH 4.5). The resulting nuclei suspension was washed and concentrated in methanol:acetic acid (3:1) by centrifugation followed by spreading of the nuclei suspension onto pre‐cleaned slides.
The probes were labeled by nick‐translation in the presence of biotin‐11‐dUTP or digoxigenin11‐dUTP. FISH was performed as described previously (Schmidt et al., 1994); 30 μl of the hybridization mixture containing 50% formamide in 2 × saline sodium citrate (SSC; stringency 77%), 7.5–10% dextran sulfate, 0.2% sodium dodecyl sulfate, 1 μg blocking Co t‐100 DNA and up to 400 ng of the labeled FISH probes were applied per slide.
Cross‐hybridization of repetitive DNA was blocked by competitive in situ suppression with B.v. vulgaris Co t‐100 DNA prepared from B.v. vulgaris genomic DNA isolated by the standard cetyltrimethylammonium bromide (CTAB) method (Saghai‐Maroof et al., 1984). The DNA was fragmented by sonication, denatured by boiling and reannealed (18 h, 49 min) as described (Zwick et al., 1997). After application of the hybridization mixture, the slides were covered with plastic cover slips, denatured at 80°C, cooled down stepwise to 37°C in an in situ Omnislide Thermal Cycler and hybridized overnight at 37°C in a humid chamber. Stringent post‐hybridization washes were performed at 42°C in 20% formamide/0.1× SSC (stringency 85%). Biotin‐labeled probes were detected by streptavidin coupled to the fluorochromes DY‐415, DY‐495 or DY‐590 (Dyomics, www.dyomics.com). Digoxigenin‐labeled probes were detected with antidigoxigenin coupled to fluorescein isothiocyanate (FITC; Roche, www.roche-applied-science.com). DAPI (4′,6′‐diamidino‐2‐phenylindole) in citifluor AF1 (Chem Lab, www.citifluor.co.uk) antifade solution was used to counterstain chromosomes.
Microscopy and image acquisition
Slides were examined using a Zeiss Axioplan2 fluorescence microscope (Zeiss, www.zeiss.de) equipped with a DAPI filter (Zeiss 01) and the following hard coated mFISH filter sets: F36‐710 (emission at 467 nm), F36‐720 (emission at 521 nm), F36‐730 (emission at 574 nm), F36‐740 (emission at 599 nm) and F36‐760 (emission at 672 nm, AHF, www.ahf.de). Images were acquired with the Applied Spectral Imaging v3.3 software coupled with the high‐resolution CCD camera BV300‐20A (ASI, www.spectral-imaging.com). The contrast of grayscale digital captures was optimized using functions affecting the whole image equally, and individual channels were pseudocolored to visualize the sites of probe hybridization. The images were arranged using Adobe Photoshop v7.0 software (Adobe Systems, www.adobe.com).
Synteny analysis and display
Gene positions were extracted from the predicted genes in AUGUSTUS gff output files. Genes were ordered by their position within each scaffold. Only those scaffolds of B. patula or B.v. maritima, respectively, were considered that had at least five different genes showing orthology relations to B.v. vulgaris genes. The script to display the scaffolds and connections between genes was written in Perl (Data S1). The script can handle a two‐column table of gene names where each line contains one orthologous gene pair from two species. Genes that do not have orthologs can either be provided in a separate file or can be included in the two‐column file with a dash in one of the columns. Gene names need to indicate the scaffold name and genomic positions so that genes can be sorted and drawn in their genomic order. The script processes each scaffold of species 1 as reference and places the corresponding scaffold(s) of species 2 next to it (non‐overlapping, automatically arranged). Colors are assigned randomly to each gene pair, scaffold names and sizes are added as labels. Orthology relations are shown as connecting lines between genes of the two input species. The input table can contain multiple scaffolds, for each reference scaffold one image file is generated. In this way, the display tool allows genome‐wide synteny visualization for all input scaffolds in one run. Detailed information on formats and usage are stated in the header section of the script.
Chromosome assignment
Scaffolds of the wild beets B.v. maritima or B. patula, respectively, that showed syntenic relations via at least five genes with sugar beet were sorted by the corresponding chromosomal order of sugar beet scaffolds (RefBeet‐1.2). In most cases a sugar beet scaffold spanned several wild beet scaffolds as visualized in the genome‐wide display using Gene_connect.pl (see above). In some cases a wild beet scaffold bridged two sugar beet scaffolds. Such linking wild beet scaffolds were selected and displayed with circos v0.69‐3 (Krzywinski et al., 2009). The assignment and ordering in Tables S4 and S5 were based on the order of connected genes within RefBeet‐1.2 scaffolds and were manually inspected.
Genomic comparison
Sequences larger than 1 kbp were selected from the B.v. maritima and B. patula assemblies using bioawk (https://github.com/lh3/bioawk) and aligned against RefBeet‐1.2 using nucmer from the mummer suite v3.1 (Kurtz et al., 2004) with default settings and ‐g 2000. On the nucmer results delta‐filter was applied with parameters ‐r ‐q ‐o 0 and show‐coords with parameters ‐rldcHT. The resulting tables were parsed using awk (https://github.com/onetrueawk/awk) and BEDtools v2.27.1 intersect (Quinlan and Hall, 2010).
Analysis of the Rz2 region
The sequences of the genetic markers CAU_4088, CAU_3881, CAU_3880, CAU_3882, CAU_4089 spanning the Rz2 region were extracted from table S6 of Capistrano‐Gossmann et al. (2017), CAU_4188 primer sequences were taken from the section Experimental procedures. The sequences were matched against the assemblies of B.v. vulgaris (RefBeet‐1.2), B. patula (this work), B.v. maritima (this work) and the previous B.v. maritima assembly (Capistrano‐Gossmann et al., 2017) using BLAT v35 (Kent, 2002) with default settings. The mRNA sequence of the Rz2 gene „BmChr3_scaffold3841.g9573” from the previous gene prediction (Capistrano‐Gossmann et al., 2017) was matched against scaffold106 of our B.v. maritima assembly, against scaffold35 of our B. patula assembly, and against scaffold Bvchr3_un.sca001 of RefBeet‐1.2 using BLASTn (Altschul et al., 1990; Nucleotide‐Nucleotide BLAST 2.2.30+). The output was converted to gff3 format using awk and loaded into jbrowse v1.12.1 (Buels et al., 2016).
Conflict of interest
The authors declare no conflict of interest.
Data statement
Raw sequencing data are available in the NCBI Short Read Archive (SRA) under BioProject number PRJNA527013 with BioSample numbers SAMN11125680 (B. patula) and SAMN11125681 (B.v. maritima), respectively. Assemblies are available at http://bvseq.boku.ac.at/Genome/Download/ and in the NCBI Genomes database under accession numbers VASJ00000000 (B. patula) and VASK00000000 (B.v. maritima). Gene sets and synteny pictures are available at http://bvseq.boku.ac.at/Genome/Download/.
Supporting information
Figure S1. Length distribution of predicted genes.
{kind=link}
{kind=link}
Figure S2. Genome size estimation.
{kind=link}
{kind=link}
{kind=link}
Figure S3. Number of initial orthogroups.
{kind=link}
Figure S4. Number of genes in initial orthogroups.
{kind=link}
{kind=link}
Appendix S1. Initial orthogroups description.
Table S1. BUSCO validation.
Table S2. Functional assignment of missing BUSCO genes.
Table S3. Refined orthogroups.
Table S4. Syntenic wild beet scaffolds connecting two or more sugar beet scaffolds.
Table S5. Order and orientation of syntenic wild beet scaffolds.
Table S6. Highly conserved genomic regions.
Data S1. Gene_connect.pl: Synteny display Perl script.
Acknowledgements
Sequencing was performed at the Genomics Unit of the Centre for Genomic Regulation (CRG), Barcelona, Spain. Calculations were in part performed at the Vienna Scientific Cluster (VSC).
References
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andrello, M. , Henry, K. , Devaux, P. , Desprez, B. and Manel, S. (2016) Taxonomic, spatial and adaptive genetic variation of Beta section Beta. Theor. Appl. Genet. 129, 257–271. [DOI] [PubMed] [Google Scholar]
- Andrello, M. , Henry, K. , Devaux, P. , Verdelet, D. , Desprez, B. and Manel, S. (2017) Insights into the genetic relationships among plants of Beta section Beta using SNP markers. Theor. Appl. Genet. 130, 1857–1866. [DOI] [PubMed] [Google Scholar]
- Arumuganathan, K. and Earle, E.D. (1991) Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218. [Google Scholar]
- Barzen, E. , Mechelke, W. , Ritter, E. , Schulte‐Kappert, E. and Salamini, F. (1995) An extended map of the sugar beet genome containing RFLP and RAPD loci. Theor. Appl. Genet. 90, 189–193. [DOI] [PubMed] [Google Scholar]
- Biancardi, E. and Tamada, T. (2016) Rhizomania. Cham, Switzerland: Springer. [Google Scholar]
- Biancardi, E. , Panella, L.W. and Lewellen, R.T. (2012) Beta maritima: the Origin of Beets. New York: Springer. [Google Scholar]
- Buels, R. , Yao, E. , Diesh, C.M. et al . (2016) JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez, J.M. and Gabaldón, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capistrano‐Gossmann, G.G. , Ries, D. , Holtgräwe, D. et al . (2017) Crop wild relative populations of Beta vulgaris allow direct mapping of agronomically important genes. Nat. Commun. 8, 15 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, M. , Frese, L. , Duarte, M.C. , Magos Brehm, J. , Tavares, M. , Santos Guerra, A. and Draper, D. (2011) Beta patula. The IUCN Red List of Threatened Species. Available at: https://www.iucnredlist.org/species/162088/5532483 [Accessed February 21, 2019].
- Core Team, R. (2015) R: a language and environment for statistical computing. https://www.R-project.org Accessed February 15, 2019. [Google Scholar]
- Delsuc, F. , Brinkmann, H. and Philippe, H. (2005) Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 6, 361–375. [DOI] [PubMed] [Google Scholar]
- Dohm, J.C. , Lange, C. , Holtgräwe, D. , Sörensen, T.R. , Borchardt, D. , Schulz, B. , Lehrach, H. , Weisshaar, B. and Himmelbauer, H. (2012) Palaeohexaploid ancestry for Caryophyllales inferred from extensive gene‐based physical and genetic mapping of the sugar beet genome (Beta vulgaris). Plant J. 70, 528–540. [DOI] [PubMed] [Google Scholar]
- Dohm, J.C. , Minoche, A.E. , Holtgräwe, D. et al . (2014) The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 505, 546–549. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D.M. and Kelly, S. (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J. (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome. Seattle: Sciences, University of Washington. Available at: http://evolution.genetics.washington.edu/phylip.html [Accessed February 15, 2019]. [Google Scholar]
- Frese, L. , Desprez, B. and Ziegler, D. (2000) Potential of genetic resources and breeding strategies for base‐broadening in Beta. In Broadening the genetic base of crop production. ( Cooper, H.D. , Spillane, C. and Hodgkin, T. , eds). FAO: IBPRGI Jointly with CABI Publishing, Rome, Italy, pp. 295–309. [Google Scholar]
- Funk, A. , Galewski, P. and McGrath, J.M. (2018) Nucleotide‐binding resistance gene signatures in sugar beet, insights from a new reference genome. Plant J. 95, 659–671. [DOI] [PubMed] [Google Scholar]
- Gertz, E.M. , Yu, Y.‐K. , Agarwala, R. , Schäffer, A.A. and Altschul, S.F. (2006) Composition‐based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmer, M.K. , Trybush, S. , Hanley, S. , Francis, S.A. , Karp, A. and Asher, M.J.C. (2007) An anchored linkage map for sugar beet based on AFLP, SNP and RAPD markers and QTL mapping of a new source of resistance to Beet necrotic yellow vein virus. Theor. Appl. Genet. 114, 1151–1160. [DOI] [PubMed] [Google Scholar]
- Herwig, R. , Schulz, B. , Weisshaar, B. et al . (2002) Construction of a “unigene” cDNA clone set by oligonucleotide fingerprinting allows access to 25 000 potential sugar beet genes. Plant J. 32, 845–857. [DOI] [PubMed] [Google Scholar]
- Holtgräwe, D. , Sörensen, T.R. , Viehöver, P. , Schneider, J. , Schulz, B. , Borchardt, D. , Kraft, T. , Himmelbauer, H. and Weisshaar, B. (2014) Reliable in silico identification of sequence polymorphisms and their application for extending the genetic map of sugar beet (Beta vulgaris). PLoS ONE 9, e110113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. (2002) BLAT–the BLAST‐like alignment tool. Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva, E.V. , Kuznetsov, D. , Tegenfeldt, F. , Manni, M. , Dias, R. , Simão, F.A. and Zdobnov, E.M. (2019) OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S.J. and Marra, M.A. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, S. , Phillippy, A. , Delcher, A.L. , Smoot, M. , Shumway, M. , Antonescu, C. and Salzberg, S.L. (2004) Versatile and open software for comparing large genomes. Genome Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, C. , Holtgräwe, D. , Schulz, B. , Weisshaar, B. and Himmelbauer, H. (2008) Construction and characterization of a sugar beet (Beta vulgaris) fosmid library. Genome 51, 948–951. [DOI] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. et al . (2012) SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. Gigascience 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. , Stiller, J. , Wei, Y. , Zheng, Y.‐L. , Devos, K.M. , Doležel, J. and Liu, C. (2014) Extensive pericentric rearrangements in the bread wheat (Triticum aestivum L.) genotype “Chinese Spring” revealed from chromosome shotgun sequence data. Genome Biol. Evol. 6, 3039–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais, G. and Kingsford, C. (2011) A fast, lock‐free approach for efficient parallel counting of occurrences of k‐mers. Bioinformatics 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoche, A.E. , Dohm, J.C. and Himmelbauer, H. (2011) Evaluation of genomic high‐throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 12, R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoche, A.E. , Dohm, J.C. , Schneider, J. , Holtgräwe, D. , Viehöver, P. , Montfort, M. , Sörensen, T.R. , Weisshaar, B. and Himmelbauer, H. (2015) Exploiting single‐molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 16, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro, F. , Frese, L. , Castro, S. , Duarte, M.C. , Paulo, O.S. , Loureiro, J. and Romeiras, M.M. (2018) Genetic and genomic tools to asssist sugar beet improvement: the value of the crop wild relatives. Front Plant Sci. 9, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paesold, S. , Borchardt, D. , Schmidt, T. and Dechyeva, D. (2012) A sugar beet (Beta vulgaris L.) reference FISH karyotype for chromosome and chromosome‐arm identification, integration of genetic linkage groups and analysis of major repeat family distribution. Plant J. 72, 600–611. [DOI] [PubMed] [Google Scholar]
- Pinheiro de Carvalho, M. , Nóbrega, H. , Frese, L. , Freitas, G. , Abreu, U. , Costa, G. and Fontinha, S. (2010) Distribution and abundance of Beta patula Aiton and other crop wild relatives of cultivated beets on Madeira. Journal für Kulturpflanzen, 62(10), 357–366. [Google Scholar]
- Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, C.M. , Reeves, P.A. , Fenwick, A.L. and Panella, L. (2014) Genetic structure and gene flow in Beta vulgaris subspecies maritima along the Atlantic coast of France. Genet. Resour. Crop Evol. 61, 651–662. [Google Scholar]
- Romeiras, M.M. , Vieira, A. , Silva, D.N. , Moura, M. , Santos‐Guerra, A. , Batista, D. , Duarte, M.C. and Paulo, O.S. (2016) Evolutionary and biogeographic insights on the Macaronesian Beta‐Patellifolia species (Amaranthaceae) from a time‐scaled molecular phylogeny. PLoS ONE 11, e0152456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghai‐Maroof, M.A. , Soliman, K.M. , Jorgensen, R.A. and Allard, R.W. (1984) Ribosomal DNA spacer‐length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl Acad. Sci. USA 81, 8014–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, T. , Schwarzacher, T. and Heslop‐Harrison, J.S. (1994) Physical mapping of rRNA genes by fluorescent in‐situ hybridization and structural analysis of 5S rRNA genes and intergenic spacer sequences in sugar beet (Beta vulgaris). Theor. Appl. Genet. 88, 629–636. [DOI] [PubMed] [Google Scholar]
- Schneider, K. , Kulosa, D. , Soerensen, T.R. et al . (2007) Analysis of DNA polymorphisms in sugar beet (Beta vulgaris L.) and development of an SNP‐based map of expressed genes. Theor. Appl. Genet. 115, 601–615. [DOI] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Stanke, M. , Steinkamp, R. , Waack, S. and Morgenstern, B. (2004) AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L.D. , Mungall, C. , Shu, S. et al . (2002) The generic genome browser: a building block for a model organism system database. Genome Res. 12, 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H. , Ding, J. , Piednoël, M. and Schneeberger, K. (2018) findGSE: estimating genome size variation within human and Arabidopsis using k‐mer frequencies. Bioinformatics 34, 550–557. [DOI] [PubMed] [Google Scholar]
- Touzet, P. , Villain, S. , Buret, L. , Martin, H. , Holl, A.‐C. , Poux, C. and Cuguen, J. (2018) Chloroplastic and nuclear diversity of wild beets at a large geographical scale: insights into the evolutionary history of the Beta section. Ecol. Evol. 8, 2890–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2009) ggplot2 ‐ Elegant Graphics for Data Analysis. New York: Springer. [Google Scholar]
- Zwick, M.S. , Hanson, R.E. , Islam‐Faridi, M.N. , Stelly, D.M. , Wing, R.A. , Price, H.J. and McKnight, T.D. (1997) A rapid procedure for the isolation of C0t‐1 DNA from plants. Genome 40, 138–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Length distribution of predicted genes.
Figure S2. Genome size estimation.
Figure S3. Number of initial orthogroups.
Figure S4. Number of genes in initial orthogroups.
Appendix S1. Initial orthogroups description.
Table S1. BUSCO validation.
Table S2. Functional assignment of missing BUSCO genes.
Table S3. Refined orthogroups.
Table S4. Syntenic wild beet scaffolds connecting two or more sugar beet scaffolds.
Table S5. Order and orientation of syntenic wild beet scaffolds.
Table S6. Highly conserved genomic regions.
Data S1. Gene_connect.pl: Synteny display Perl script.