

Abstract
Neutrophil granulocytes form the first line of host defense against invading pathogens and tissue injury. They are rapidly recruited from the blood to the affected sites, where they deploy an impressive arsenal of effectors to eliminate invading microbes and damaged cells. This capacity is endowed in part by readily mobilizable proteins acquired during granulopoiesis and stored in multiple types of cytosolic granules with each granule type containing a unique cargo. Once released, granule proteins contribute to killing bacteria within the phagosome or the extracellular milieu, but are also capable of inflicting collateral tissue damage. Neutrophil‐driven inflammation underlies many common diseases. Research over the last decade has documented neutrophil heterogeneity and functional versatility far beyond their antimicrobial function. Emerging evidence indicates that neutrophils utilize granule proteins to interact with innate and adaptive immune cells and orchestrate the inflammatory response. Granule proteins have been identified as important modulators of neutrophil trafficking, reverse transendothelial migration, phagocytosis, neutrophil life span, neutrophil extracellular trap formation, efferocytosis, cytokine activity, and autoimmunity. Hence, defining their roles within the inflammatory locus is critical for minimizing damage to the neighboring tissue and return to homeostasis. Here, we provide an overview of recent advances in the regulation of degranulation, granule protein functions, and signaling in modulating neutrophil‐mediated immunity. We also discuss how targeting granule proteins and/or signaling could be harnessed for therapeutic benefits.
Keywords: apoptosis, autoimmunity, degranulation, efferocytosis, inflammation, NET formation, neutrophil trafficking, neutrophils, phagocytosis, resolution of inflammation
Neutrophils respond rapidly to infection and deploy an arsenal of effectors, including proteins stored in cytosolic granules to eliminate invading pathogens. Recent research has documented neutrophil heterogeneity and functional versatility far beyond their antimicrobial function. Here, we review the mechanism of neutrophil degranulation and the emerging roles of granule proteins in orchestrating neutrophil‐mediated immunity. We also discuss how targeting granule proteins and/or signaling could be harnessed for therapeutic benefits.
Abbreviations
- ANCA
antineutrophil cytoplasmic antibody
- CCL5
C‐C motif ligand 5 (aka RANTES)
- CR1
complement receptor 1
- CR3
complement receptor 3 (CD11b/CD18)
- DAG
diacylglycerol
- GPCR
G protein‐coupled receptor
- IP3
inositol‐(1, 4, 5)‐trisphosphate
- JAM‐C
junctional adhesion molecule‐C
- LTB4
leukotriene B4
- MMP‐9
matrix metalloproteinase 9
- MPO
myeloperoxidase
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- PAD‐4
peptidyl arginine deiminase 4
- PAR‐2
protease‐activated receptor 2
- PIP3
phosphatidylinositol‐(3, 4, 5)‐trisphosphate
- PKC
protein kinase C
- PLC
phospholipase C
- SLPI
secretory leukocyte peptidase inhibitor
- SNARE
soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor
- SOC
store‐operated channel
- TEM
transendothelial migration
- TLR9
Toll‐like receptor 9
- TNF
tumor necrosis factor
- VAMP
vesicle‐associated membrane protein
Introduction
Neutrophil granulocytes are the most abundant white blood cells in human circulation and play a central role in host defense against invading pathogens and tissue injury. Typically, neutrophils are rapidly recruited to sites of infection or injury, where they deploy an impressive chemical and enzymatic array of mechanisms, resulting in the elimination of the offending agents and necrotic tissues [1, 2]. A key component of this weaponry is the mobilization and release of preformed cytosolic granules with each granule type containing multiple microbicide proteins and proteases at unique signaling thresholds [3, 4]. Granules can fuse with pathogen‐containing phagosomes to destroy ingested microorganisms. Neutrophil granules can also fuse with the plasma membrane in response to pathogens that cannot be phagocytosed, leading to extracellular release of their content [1, 5]. Release of granule proteins to the extracellular milieu can inflict damage to the surrounding tissues and prolong the inflammatory reaction [6, 7, 8]. Over the past two decades, neutrophil‐driven inflammation has been recognized as a common mechanism underlying many pathological conditions, including atherosclerosis, cardiovascular, respiratory, autoimmune and neurodegenerative diseases, sepsis, and cancer [8, 9]. Neutrophils are also involved in the resolution of inflammation [10]; hence, the balance between their deleterious and beneficial effects will likely be one of the key determinants of the outcome of the inflammatory response.
Research over the last decade has documented the heterogeneity of neutrophils and their functional versatility, extending far beyond their antimicrobial function [11, 12]. Neutrophils interact with innate and adaptive immune cells and orchestrate immune responses [9]. Emerging evidence indicates the involvement of granule proteins, in addition to de novo synthesis of cytokines in this crosstalk [4]. In this paper, we aim to summarize recent advances in our understanding of degranulation, granule protein function, and signaling in neutrophil‐mediated immunity. We also discuss how targeting granule proteins and/or signaling could be harnessed for therapeutic benefits.
Granule formation and composition
Neutrophils contain different granule subsets that are mobilized by stimulation. Based on physical properties and expression markers, at least four different types of secretory compartments have been identified: azurophilic or primary granules [specific markers: myeloperoxidase (MPO) and CD63], specific or secondary granules (markers: lipocalin 2 and CD66b), gelatinase (GG) or tertiary granules (markers: gelatinase B and CD11b), and secretory vesicles (SV) (Fig. 1) [13]. Neutrophils acquire granules and SV sequentially during their maturation in the bone marrow. The ‘targeting by timing’ hypothesis postulates that the synthesis of granule proteins at the time of formation of granule subset would determine granule content [14]. Appropriate timing of mRNA expression during granulopoiesis coincides with granule protein distribution in most proteins used to identify granule subsets [3]. However, the discrepancy in the timing of mRNA expression for a minority of proteins and overlaps in granule content among different subsets (e.g., lysozyme in all three granule types) suggest the involvement of mechanisms in addition to the timing of protein synthesis in the regulation of granule content. Primary granules are the earliest to be formed in promyelocytes (hence their name), stain by the dye azure A (i.e., azurophilic). They contain a large number of antimicrobial proteins, including MPO, serine proteases (elastase, proteinase 3, cathepsin G, and azurocidin), α‐defensins, lysozyme, and Cap57 (bactericidal/permeability‐increasing protein, BPI) [3, 15]. Azurophilic granule proteases are activated by proteolytic processing prior to incorporation into granules, resulting in a highly toxic readily available cargo upon release [16]. Proteomic analysis has identified at least 850 proteins associated with azurophilic granules, of which 135 show the highest relative amounts among granules or are exclusively expressed in this granule subset [3]. Azurophilic granules are heterogeneous in their protein content, which may determine their trafficking to the cell surface or fusion with phagosomes. Plasma membrane‐targeted azurophilic granules express Rab27 and Slp1/JFC1, whereas primary granules targeted to the phagosome lack these proteins [17]. Intriguingly, a subset of primary granules does not contain α‐defensin, while expressing all other granule proteins [18]. The origin and function of this subset remain to be investigated.
Fig. 1.

Main constituents of neutrophil granules and secretory vesicles. The list depicts the most abundant proteins in human resting neutrophils. There are some species differences. For example, while mouse neutrophils contain most of these proteins, azurocidin, α‐defensin 3, and Cap57 are specific for human neutrophils. In contrast, mouse neutrophils harbor abundant expression of neutrophil granular protein, high‐mobility group 1, heat‐shock protein A8, and chitinase‐like protein 3 [4]. In human neutrophils, specific and gelatinase granules exist as a continuum bearing markers of SG (lactoferrin+, gelatinase−), gelatinase granules (lactoferrin−, gelatinase+), and hybrid granules (lactoferrin+, gelatinase+) [3]. A fourth granule type, ficolin 1‐enriched granules have also been described [19].
Specific and GG granules are peroxidase‐negative and are formed throughout myelocyte, metamyelocyte, and band stages. These granules share similar contents and functions, though heterogeneity within these subsets is well recognized. Specific (secondary) granules contain lactoferrin and lipocalin (NGAL), but no GG [matrix metalloproteinase 9 (MMP‐9)], whereas GG (tertiary) granules contain MMP‐9, but lack lactoferrin and lipocalin [18]. Of 1024 proteins associated with specific granules (SG), 111 are maximally expressed in this subset [3]. GG granules express only 30 of 1123 proteins maximally. A hybrid type granule, constituting about two‐thirds of the total peroxidase‐negative granules, contains lactoferrin, lipocalin, and MMP‐9 [18]. GG granules containing the microbe‐binding lectin ficolin 1 have been proposed as a separate granule subset, termed ficolin 1‐rich granules [19]. Besides the rapid exocytosis of these granules in response to the bacterial chemoattractant formyl‐Met‐Leu‐Phe [19, 20], little is known about the function of this granule type. SV are formed by endocytosis during the band and segmented stages [21]. These vesicles (markers: albumin, CD45, Mac‐1/CD11b, and CD13) are enriched in numerous proteins also present in the cell membrane, including phagocytic, chemoattractant and cytokine receptors, adhesion molecules, membrane components of NADPH oxidase, and plasma proteins [3, 22]. Like specific and GG granules, the release of the content of SV is restricted to the plasma membrane [21] and is thought to facilitate neutrophil adherence to the activated vascular endothelial cells (EC), the initial step in neutrophil trafficking into inflamed or injured tissues. Following firm adhesion of neutrophils to the endothelium, chemokine signaling and outside‐in signaling through β2 integrins induce exocytosis of GG and SG [18].
A recent case report shows apical polarization of neutrophil granules in both mature and band neutrophils from patients with severe sepsis [23], which may coincide with their antimicrobial activity. Several granule proteins, such as MPO, lactoferrin, lysozyme, cathepsin G, and α‐defensin, were detected in thin cellular protrusions (termed cytonemes) that are likely specialized for delivering signaling proteins to neighboring cells [24, 25]. Another communication structure, extracellular vesicles also contain granule proteins, including MPO, lactoferrin, cathepsin G, and α‐defensin in a context‐dependent fashion [26]. Little is known of the molecular links between the formation of these structures and neutrophil granules.
Regulation of degranulation
Neutrophil granules can fuse with the nascent phagosome, releasing their cargo into the vacuole to destroy ingested pathogens or with the plasma membrane, resulting in secretion of granule proteins extracellularly (degranulation). The release of granule contents is a tightly regulated receptor‐coupled process that is mediated by distinct signaling events for each granule type, allowing the selective release of granule subsets (Fig. 2) [27]. Ligation of G protein‐coupled receptors (GPCRs), Fcγ receptors, or the β2 integrin Mac‐1(CD11b or CR3) by a wide range of stimuli, such as bacterial formyl‐peptides, chemokines, cytokines, complement fragments, or EC adhesion molecules, triggers a degranulation response [28], which is markedly enhanced by prior exposure of neutrophils to a priming stimulus, such as granulocyte–macrophage colony‐stimulating factor, tumor necrosis factor (TNF), or platelet‐activating factor [29]. Although these receptors activate different sets of downstream signaling molecules, they converge on two signaling pathways, the activation of Rac2 and the induction of a calcium flux [30]. Importantly, the different granule subsets can be selectively released for each granule type requires a different intracellular calcium concentration to trigger its exocytosis [18, 30]. SV have the highest sensitivity to calcium followed in order of decreasing calcium sensitivity by GG, SG, and azurophilic granules.
Fig. 2.
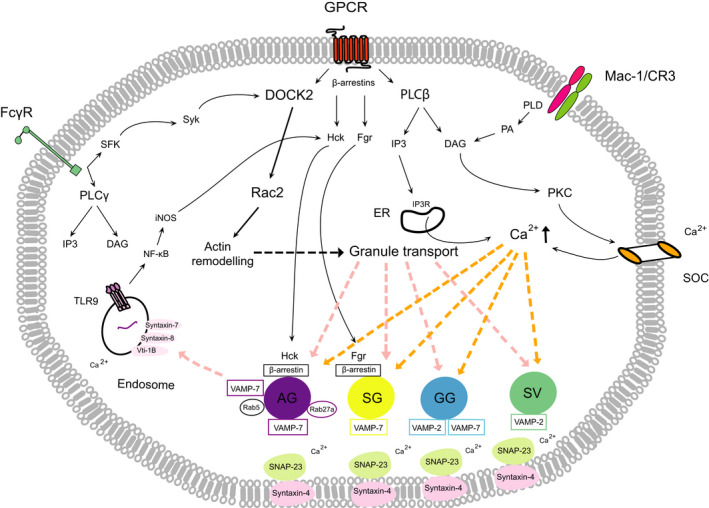
Major signaling pathways regulating neutrophil degranulation. Ligation of GPCRs, Fc receptors, or the phagocytic β2‐integrin Mac‐1/CR3 triggers degranulation through multiple, partially overlapping downstream signaling pathways, converging on activation of Rac2, and increasing intracellular calcium. Rac2 is activated through GPCR‐activated guanine nucleotide exchange factors or the SFK (Src family of tyrosine kinases)‐Syk‐DOCK2 pathway following ligation of Fcγ receptors. Calcium signaling is driven by influx of extracellular calcium via DAG/PKC‐regulated SOCs or calcium release from intracellular stores via the IP3 receptor. Chemokine binding to GPCRs leads to direct binding of β‐arrestins to the receptor, which along with the Src‐family kinases Hck or Fgr translocate to the azurophilic and SG, respectively. Activation of TLR9 in the endosome also triggers degranulation through NF‐κB/iNOS‐mediated activation of Hck. DOCK‐2‐Rac2 signaling induces actin remodeling and granule transport from the cytosol to the plasma membrane. Fusion of granules with the plasma membrane is governed by calcium‐dependent formation of SNARE complexes, consisting of granule VAMPs and cognate SNAPs/syntaxins on the plasma membrane. The Rab GTPase, Rab5, expressed on a subset of azurophilic granules, mediates their fusion with endosomes. AG, azurophilic (primary) granules; IP3R, IP3 receptor; SG, secretory (secondary) granules.
Rac2 is a member of the Rho GTPase subfamily, which plays multiple roles in degranulation. It mediates the remodeling of the actin cytoskeleton required for mobilization of granules from the cytosol to the target membrane and clearing of the cortical actin cytoskeleton that would otherwise block granule docking to the plasma membrane [31, 32, 33, 34]. Genetic deletion of Rac2 in mice leads to a loss of primary granule release without affecting degranulation of specific and GG granules, suggesting a role restricted to primary granules [31, 32, 35]. However, actin reorganization is required for the degranulation of all granule types [36], but it likely depends on granule‐specific signaling mechanisms, such as the Src family of nonreceptor tyrosine kinases. Neutrophils express 3 Src‐family members, Hck, Fgr, and Lyn. Ligation of GPCRs or the phagocytic receptor FcγRIIA (CD64) induces Hck translocation to azurophilic granules and Fgr to SG, albeit some degree of functional overlap between these kinases has also been reported [37, 38, 39]. Hck activates the ELMO‐Dock2 pathway leading to Rac2 activation and degranulation of azurophilic granules [40]. Src‐family kinases also activate p38 MAPK, which through a yet unidentified pathway mediates degranulation of azurophilic and specific/ GG granules [41]. Of note, p38 MAPK can induce actin rearrangements via Hsp27 [42]; hence, it may function in a manner similar to Rac2. Recent results indicate that Hck activation following ligation of Toll‐like receptor 9 (TLR9) with bacterial DNA, a pathogen‐associated molecular pattern or the damage‐associated molecular pattern mitochondrial DNA, leads to the release of primary granule contents, MPO, neutrophil elastase (NE), and proteinase 3 [43]. TLR9 activation induces NF‐κB‐mediated transcription of inducible nitric oxide synthase (iNOS) in human neutrophils [44], which, in turn, could activate Src kinases as observed in macrophages [45]. TLR9 signaling does not activate Lyn or Fgr and does not require neutrophil priming [43].
An increase in intracellular calcium is indispensable for degranulation of all granule subtypes and is required to form the fusion pore between granules and cell or phagosome membranes. Calcium signaling can be induced at multiple points during neutrophil trafficking to inflamed loci and phagocytosis. The most studied receptors driving calcium release are GPCRs. Ligation of GPCRs induces the dissociation of the β/γ heterodimer from the Gα subunit [46]. The ß/γ subunit transduces the majority of the neutrophil signaling through activation of phospholipase C (PLC), which generates inositol‐trisphosphate (IP3) and diacylglycerol (DAG). IP3 releases calcium stored in the endoplasmic reticulum (ER), leading to modest increases in cytosolic calcium [47]. DAG induces protein kinase C (PKC)‐mediated activation of store‐operated channels (SOCs), producing a more robust and prolonged increase in intracellular calcium. Activation of GPCRs also generates phosphatidylinositol‐(3,4,5)‐trisphosphate (PIP3) via protein kinase AKT. Hypoxia through activation of PI3Kγ also evokes PIP3 and AKT signaling and enhances degranulation [48]. A hierarchy of granule exocytosis exists in response to elevated intracellular calcium concentrations with an order of release of SV > GG granules > SG > azurophilic granules [49, 50]. The exocytosis of primary granules depends on ATP and high cytosolic calcium concentration [50]. Activation of neutrophil receptors other than GPCRs also induces calcium fluxes. For instance, FcγRIIA‐mediated phagocytosis of antibody‐opsonized pathogens induces a potent calcium flux through the recruitment of Src‐family kinases, which activates the Syk‐PLC pathway, leading to IP3 and DAG synthesis [51]. Engagement of complement receptor 1 (CR1) and CR3 with C3b‐opsonized pathogens induces phospholipase D (PLD)‐mediated release of phosphatidic acid (PA), which is readily converted into DAG [18].
Rapid mobilization of granules is controlled by effector molecules that include small GTPases and their interacting proteins [52]. The final step of degranulation, docking, and fusion of granule membrane to the target membrane is mediated by the binding of vesicle soluble N‐ethylmaleimide‐sensitive factor attachment protein receptors (SNAREs, also referred to as vesicle‐associated membrane protein or VAMP) to a cognate SNARE (t‐SNARE) expressed at the plasma membrane or phagosome at a ratio of 1 : 3 [53]. Human neutrophils express a range of vSNARES, including VAMP‐2 and VAMP‐7, and t‐SNARES, such as syntaxin A1, 3, 4, 5, 6, 7, 9, 11, and SNAP‐23 [54], though degranulation largely depends on SNAP‐23 and syntaxin 6. VAMPs may impart granule‐specific degranulation. Thus, VAMP‐2 mediates exocytosis of GG granules [55], whereas VAMP‐1 and VAMP‐7 direct release of azurophilic granules [56, 57]. The selective recruitment of VAMP‐7 to azurophilic granules may account for their capability to fuse with both the plasma membrane and phagosomes [56]. VAMP‐7 is thought to interact with phagosome‐resident syntaxin 7, syntaxin 8, and/or Vti‐1B to mediate azurophilic granule–phagosome fusion [18].
The Rab GTPases Rab5 and Rab27a have also been implicated in controlling the degranulation of human neutrophils. Rab5 appears to be selectively associated with azurophilic granules and mediates their fusion with phagosomes, although the underlying mechanism has not been explored in detail [38]. In contrast, Rab27a and its two effectors Munc‐13‐4 and JFC1 are required for extracellular release of azurophilic, SG, and GG granules [18]. JFC1 transports granules to the plasma membrane, where it docks granules to the plasma membrane through bridging Rab27a. Munc13‐4 immobilizes Rab271‐positive granules at the plasma membrane and controls the secretion of azurophilic and tertiary granules, for example MPO release in response to bacterial lipopolysaccharide. Deficiency in Rab27a and Munc13‐4 has been linked to impaired neutrophil functions in some hereditary immune deficiency diseases such as the Griscelli syndrome type 2 and familial hemophagocytic lymphohistiocytosis type 3 [58]. Munc13‐4 was also reported to regulate TLR9 activation directly, upregulate surface expression of Mac‐1 (CD11b), and bind to Rab11, enabling Rab11a‐positive vesicles to dock and presumably fuse with the plasma membrane [59, 60]. At least four additional Rab GTPases have been identified in neutrophils, but their function remains largely unexplored [18, 52].
Biological roles of granule proteins
Much has been written on the mechanisms of neutrophil trafficking into inflamed tissues and the roles of emigrated neutrophils in host defense and inflicting tissue injury [1, 2, 6, 28, 61, 62, 63, 64]. While degranulation at specific times during neutrophil transendothelial migration (TEM) and engagement with invading pathogens equips neutrophils with efficient effectors, the roles of granule proteins are not restricted to antimicrobial and tissue‐damaging actions. In the following sections, we aim to provide a focus on how neutrophils utilize granule proteins to interact with innate and adaptive immune cells and shape innate and adaptive immunity (Fig. 3).
Fig. 3.
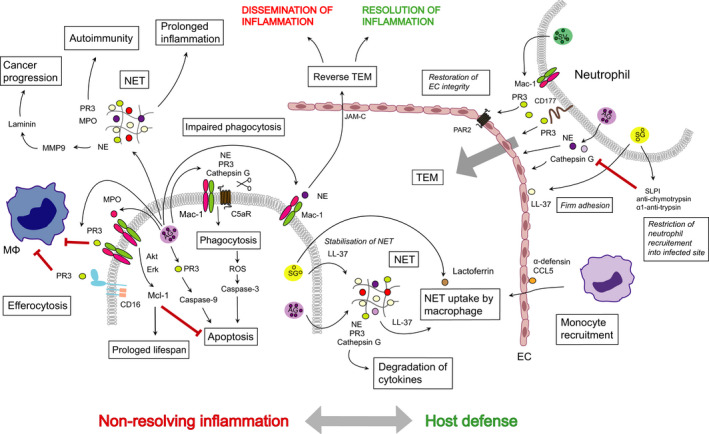
Roles of neutrophil granule proteins in host defense, nonresolving inflammation, autoimmunity, and cancer progression. Release of content of SV and LL‐37 (cathelicidin) bound to ECs allows firm adherence of neutrophils to ECs. PR3, NE, and cathepsin G are required for TEM and extravasation into the inflamed tissue. Neutrophils also secrete protease inhibitors that by cleaving NE and cathepsin G restricts neutrophil recruitment into the infected locus. The α‐defensin‐CCL5 complex immobilized on the EC surface contributes to monocyte recruitment. PR3 through PAR‐2 contributes to restoration of EC integrity following TEM. NE binds to Mac‐1 and may direct neutrophil reverse TEM through interacting with JAM‐C. NE, PR3, and cathepsin G are components of NET. LL‐37 stabilizes NET and together with lactoferrin may promote NET uptake by macrophages. NE, PR3, and cathepsin G can cleave C5aR and by altering the Mac‐1/C5aR ratio impairs phagocytosis, bacterial clearance, and phagocytosis‐induced cell death. MPO binds Mac‐1 and generates survival signals for neutrophils, consistent with prolongation of inflammation. PR3 bound to Mac‐1 or CD16 functions as a ‘don't eat me signal’ for macrophages and attenuates efferocytosis. Extensive NET formation is associated with many pathological conditions. NET‐associated PR3 and MPO are potent autoantigens to trigger autoimmunity. NE can activate MMP‐9, which cleaves laminin, thereby may awaken dormant cancer cells. AG, azurophilic granules; MΦ, macrophage; PR3, proteinase 3.
Neutrophil trafficking into tissues and reverse transendothelial migration
Rapid neutrophil infiltration into microbe‐infected and damaged tissues is a critical component of the innate immune response and a hallmark of acute inflammatory reactions. Neutrophils egress from the blood to tissues is a multistep process involving a sequence of events orchestrated by well‐characterized adhesive and stimulatory pathways, followed by migration through the EC barrier via both paracellular and transcellular paths and chemotactic migration toward the inflammatory locus [61, 62, 63, 64].
Neutrophil activation results in secretion of NE, proteinase 3, and cathepsin G from the azurophilic granules, leading to degradation of extracellular matrix and neutrophil transmigration [1, 63]. In vivo, the activity of these proteases is inhibited by protease inhibitors, such as leukocyte protease inhibitor (SLPI), antichymotrypsin, and α1‐antitrypsin [4], thereby restricting protease activity to the infected or injured locus. Proteinase 3 binds to the neutrophil surface through an association with NB1 (CD177), a heterophilic binding partner for platelet–EC adhesion molecule 1 [65, 66]. This interaction protects proteinase 3 from proteolytic inactivation. Thus, NB1 may direct proteinase 3 to EC junctions where transmigration occurs [67] and facilitates neutrophil diapedesis through degradation of junctional proteins or the extracellular matrix [66] or by proteolysis of endothelial cadherin [68]. Acting through the endothelial protease‐activated receptor 2 (PAR‐2), proteinase 3 counters the actions of PAR‐1 agonists on the endothelium and contributes to restoring junctional integrity and barrier function following neutrophil TEM [69]. Proteinase 3 was also reported to maintain calcium transients in EC independent of its proteolytic activity [69]. This action may contribute to protecting ECs from permeability changes, though the underlying molecular mechanism requires future investigations.
Studies in zebrafish embryos [70] and mouse cremaster circulation [71] reported the ability of neutrophils to exhibit motility away from inflamed tissues back to the vessel lumen, known as reverse TEM [71, 72]. Neutrophil reverse TEM is most prevalent in tissues subjected to ischemia–reperfusion injury and is driven by a leukotriene B4 (LTB4)‐ NE axis [72] in a paracellular manner [73]. Under ischemia‐reperfusion, locally generated LTB4, through the LTB4 receptor BLT1, induces elastase release from neutrophils [72]. NE binds to Mac‐1 [74], which is also a ligand for junctional adhesion molecule‐C (JAM‐C), and then cleaves JAM‐C [72]. Reversely transmigrated neutrophils display a phenotype (ICAM‐1high, CXCR1low) distinct from tissue‐resident (ICAM‐1low, CXCR1low) or circulating neutrophils (ICAM‐1low, CXCR1high) and have increased capacity to produce superoxide [71, 75]. The pathophysiological relevance of neutrophils undergoing reverse TEM remains unclear. Reverse TEM might be a protective response as it may dampen the inflammatory response through facilitating the removal of neutrophils from the inflamed site [70, 76]. Alternatively, retrograde neutrophil migration might disseminate local inflammation and lead to distant organ injury [71, 72].
Regulation of neutrophil life span, apoptosis, and efferocytosis
Neutrophils are typically short‐lived with a half‐life in the range of 1–8 h in the circulation [77, 78]; however, some reports estimated that their life span can extend to 5.4 days [79], especially once activated. Blood neutrophils die via apoptosis [80]. At sites of inflammation, neutrophil life span is extended through delaying intrinsic apoptosis by prosurvival cues from neutrophil adhesion to ECs, inflammatory mediators, and microbial constituents [4, 12]. Upon their release, granule proteins act in an autocrine/paracrine fashion to modulate neutrophil function, life span, and mode of cell death. Neutrophil subsets may undergo apoptosis, NETosis [81], or necroptosis [82]. Cell death is an important mechanism to remove neutrophils from inflamed tissues for assuring the timely resolution of inflammation. Pathways that lead to apoptosis have been extensively studied, and the execution of this intrinsic death program depends on activation of the caspase cascade [83, 84]. However, in mouse neutrophils, activation of caspase 3 may occur independently of the canonical caspase 8 or caspase 9‐triggered pathway. Instead, proteinase 3 was detected in the cytosol of aging neutrophils, where it cleaves procaspase 3 at a site upstream of the canonical caspase 9 cleavage site [85]. Consistently, pharmacological inhibition or knockdown of proteinase 3 delays neutrophil death in vitro and in a murine model of peritonitis [85].
MPO can regulate neutrophil function through its nonenzymatic actions. MPO binds to Mac‐1 on human neutrophils [86]. Ligation of Mac‐1 evokes release of additional MPO and leads to upregulation of Mac‐1 expression [86, 87]. MPO also activates the MAPK/ERK and PI3K/Akt pathways, resulting in the preservation of the expression of the anti‐apoptotic protein Mcl‐1, a key regulator of neutrophil life span [88], and delay of intrinsic apoptosis [87]. Thus, activation of a MPO‐centered feed‐forward loop would amplify neutrophil responses and perpetuate the inflammatory response [87]. Consistently, genetic deletion of MPO in mice attenuates local neutrophil accumulation and distant organ damage after renal ischemia–reperfusion [89] and reduces Escherichia coli septicemia‐induced lung injury and mortality [90]. Administration of exogenous MPO prolongs neutrophil‐mediated acute lung injury in mice [87]. Decomposition of MPO, which consists of two identical protomers, into the monomers results in a partial loss in prolonging neutrophil life span and inducing degranulation [91], suggesting a mechanism that may control MPO‐mediated neutrophil activation. Such mechanism may be operational in vivo as suggested by the detection of monomeric MPO in the serum of patients with acute inflammation [91]. However, the mechanism of MPO decomposition remains to be investigated.
Proteinase 3 is constitutively expressed on the surface of a subset of human neutrophils [92, 93] and is externalized through association with phospholipid scramblase 1 in neutrophils undergoing apoptosis [94]. Membrane‐bound proteinase 3 decreases macrophage phagocytosis, indicating that proteinase 3 may function as a ‘don't eat me’ signal, thereby delaying neutrophil clearance [94].
Prompt removal of apoptotic cells, including neutrophils by tissue‐resident phagocytes (such as macrophages and dendritic cells), is central for returning to homeostasis. The molecular mechanisms that underpin the recognition and clearance of apoptotic cells are increasingly better defined [95]. Recent data imply the involvement of granule proteins in this process. During the resolution of experimental peritonitis, macrophages process and cleave lactoferrin initially released by neutrophils [96]. A 17 kDa lactoferrin fragment was found to enhance efferocytosis of apoptotic neutrophils and to stimulate secretion of the proresolution cytokine IL‐10 from macrophages, thereby facilitating the resolution of peritonitis in mice [96]. Apoptotic human cells of diverse lineage can also secrete lactoferrin, which selectively inhibits migration and activation of neutrophils but not mononuclear phagocytes [97], suggesting a regulatory role for lactoferrin in dampening the inflammatory response.
Neutrophil apoptosis and subsequent efferocytosis are important mechanisms of removing neutrophils from inflamed sites. Clinical studies documented extended neutrophil life span through delayed apoptosis in a wide range of pathologies, including acute coronary artery disease [98], severe asthma [99], acute respiratory distress syndrome [100], and sepsis [101] and its association with disease severity and poor prognosis. Consistent with the clinical observations, in experimental models, delaying neutrophil apoptosis, for example with administration of exogenous MPO, can adversely affect the outcome of the inflammatory response [87, 102].
Phagocytosis and phagocytosis‐induced cell death
Phagocytosis of opsonized bacteria or necrotic cells is initiated by lateral clustering of Mac‐1 (complement receptor 3 or CR3) [51] and governed by a delicate balance between Mac‐1 and the complement C5a receptor (C5aR or CD88) [103, 104]. C5aR is highly expressed in cells of myeloid origin [105]. Diminished expression of Mac‐1 [106] or pharmacological blockade or genetic deletion of C5aR leads to defective phagocytosis and intracellular killing of bacteria by both human and mouse neutrophils [104]. Neutrophil serine proteases have been shown to proteolytically reduce the expression of several molecules involved in immunity, including CXCR1 (IL‐8 receptor), CD16, CR1, and C5aR. NE, proteinase 3, and cathepsin G can also cleave C5aR [43, 107], though their involvement appears to be context‐dependent. For example, C5a reduces C5aR expression through inducing NE release [107], whereas TLR9 activation‐induced reduction of C5aR and defective phagocytosis are mediated by the release of NE and proteinase 3 [43]. Bronchoalveolar lavage fluid from patients with cystic fibrosis was found to contain NE and cathepsin G at concentrations sufficient to cleave C5aR [107]. Typically, phagocytosis of opsonized bacteria accelerates neutrophil apoptosis through ROS‐dependent activation of caspase 8, which counters the survival signals generated by ligation of Mac‐1 [12, 108, 109]. Consistently, activation of TLR9 with either bacterial or mitochondrial DNA impaired bacterial phagocytosis and delayed apoptosis in both human and mouse neutrophils, and prolonged E. coli‐evoked lung injury in mice [43].
Reduced C5aR expression is a common finding ex vivo in neutrophils from patients with sepsis [110, 111] and in experimental models of sepsis [112, 113]. C5a‐induced neutrophil serine protease‐mediated cleavage of C5aR may explain why high levels of activated C5a in the serum are associated with neutrophil unresponsiveness to C5a in sepsis [103]. TLR9‐evoked cleavage of C5aR may explain defects in neutrophil function to clear bacteria under pathological conditions that are associated with release of bacterial or mitochondrial DNA, as observed in sepsis [114], acute respiratory distress syndrome [115], inflammation, and tissue injury [116, 117].
Neutrophil extracellular trap formation
Neutrophils target invading pathogens through several processes, including phagocytosis, degranulation, and formation of neutrophil extracellular traps (NETs) [1, 118]. NETs capture and kill an extensive range of microbes [119], degrade cytokines and chemokines [120], and immobilize activated platelets [121], microparticles [122], and coagulation factors [123]. NETs are a meshwork of chromatin decorated with granule proteins, including MPO, NE, azurocidin, proteinase 3, and LL‐37 (cathelicidin), and absorb penetrating 3 [124] and complement [125]. Recent reviews detail NADPH oxidase 2‐dependent and NADPH oxidase 2‐independent pathways leading to NET formation as well as differences in its composition [126, 127, 128]. Quantitative proteomics of NET proteins produced by neutrophils from healthy subjects and patients with rheumatoid arthritis or systemic lupus erythematosus indicates that the nature of the stimulant rather than neutrophil physiology may determine NET protein profiles irrespective of the disease background [129].
Neutrophil extracellular traps may be formed by the release of a DNA scaffold consisting of mitochondrial DNA that binds granule proteins without affecting cell viability [130]. This process depends on glycolytic ATP production for cytoskeletal rearrangements that are essential for releasing both mitochondrial DNA and granule proteins. By contrast, the process commonly designated as NETosis is a form of programmed necrotic cell death, involving the release of nuclear DNA [131]. This process involves ROS generation through PKC‐dependent activation of NADPH oxidase, followed by MPO‐dependent release and migration of elastase from azurophilic granules to the nucleus [131], where it cleaves histones to elicit chromatin decondensation [132]. While the molecular mechanisms eliciting the release of nuclear or mitochondrial DNA appear to differ [130], the pathological relevance of these processes to innate immunity remains to be investigated. Of note, phagocytosis of bulky phosphatidylserine‐exposing particles, such as apoptotic bodies or platelets, renders neutrophils unable to release NET [121, 133]. Hence, impaired phagocytosis by MPO, proteinase 3, or NE may represent another mechanism to govern NET formation.
Complexes of extracellular DNA and LL‐37 can form stable structures that protect NET against degradation by bacterial nucleases and activate TLR9 in monocytes and dendritic cells [134, 135]. NETs can trigger thrombus formation, which prevents the dissemination of microorganisms [123, 136]. Neutrophil serine proteases may function as a reciprocal link between coagulation and innate immunity [123]. However, the role of serine proteases may be context‐dependent. For instance, NE is not required to form NETs in a mouse model of deep vein thrombosis [137]. NETs are eventually degraded by DNase 1 released predominantly from dendritic cells [138, 139] or following engulfment by macrophages through the cytosolic exonuclease TREX1 (DNase III) [139, 140]. LL‐37 is required for the uptake of NET by macrophages [139].
Neutrophil extracellular traps formation plays an important role in host defense and limits the extent of infected areas [136, 141]. However, aberrant NET formation is increasingly being recognized to participate in a range of human pathologies, including atherosclerosis, thrombosis [142], and sepsis‐associated or COVID‐19‐associated acute respiratory distress syndrome [143, 144, 145]. Excessive NET formation and/or impaired NET degradation have also been implicated as a trigger of autoimmune diseases [119].
Autoimmunity
Many granule proteins (e.g., MPO, proteinase 3) are recognized autoantigens in autoimmunity [119, 146]. Since these molecules are externalized through NETosis together with other well‐known autoantigens, such as double‐stranded DNA and histones, aberrant NET formation, or degradation has been implicated in the initiation of systemic autoimmune responses in susceptible individuals [119]. Some granule proteins are target antigens in antineutrophil cytoplasmic antibody (ANCA)‐associated vasculitides. MPO is the main target antigen in microscopic polyangiitis and Churg–Strauss syndrome, whereas Wegener's granulomatosis is predominantly associated with ANCA directed against proteinase 3 [147]. MPO might trigger autoimmunity during uncontrolled inflammation [148], though it is unclear whether this response would also involve NET formation. The functions of proteinase 3 depend on its localization [92, 93]. Membrane‐bound proteinase 3 likely contributes to neutrophil activation in co‐operation with its binding partners, CD16, Mac‐1, and CD177 [93, 149], and inhibits macrophage phagocytosis [94]. Proteinase 3 on apoptotic neutrophils instructs plasmocytoid dendritic cell‐driven generation of Th9/Th2 cells, disrupting immune silencing [150]. Antiproteinase 3 ANCA can be triggered by cPR3(105‐201), a complimentary protein translated from proteinase 3 antisense DNA [151]. Neutrophils from vasculitis patients exhibit an altered gene expression profile (e.g., re‐expression of mRNA for several azurophilic granule proteins), and newly synthesized proteinase 3 partner proteins that are not expressed in neutrophils from healthy subjects might associate with proteinase 3 and function as autoantigens [93]. An alternative possibility is that NET‐associated proteinase 3 or proteinase 3 released during cell death other than apoptosis [152] could act as sources of modified autoantigens.
The antimicrobial protein LL‐37 (cathelicidin) was identified as an autoantigen in psoriasis [153]. Recent studies with the cathelicidin protein CRAMP (a truncated form of the mouse homolog of human CAP18) have linked this protein to atherosclerosis as a potential self‐antigen in ApoE‐deficient mice [154]. Immunization with CRAMP resulted in differential outcomes depending on the dose used; at a lower dose reducing atherosclerosis, whereas at a higher dose exacerbating the disease. While clinical studies indicate an association between psoriasis and cardiovascular disease, the pathological relevance of LL‐37 to human atherosclerosis remains to be investigated.
Regulation of cytokine/chemokine activity
Human neutrophils produce cytokines belonging to different families, including pro‐ and anti‐inflammatory cytokines, chemokines, colony‐stimulating factors (e.g., G‐CSF), angiogenic, and fibrinogenic factors [4, 155]. A peculiar characteristic of neutrophils is their ability to regulate cytokine activity extracellularly through the release of serine proteases. Neutrophil serine proteases can either negatively or positively modulate the activity of cytokines/chemokines produced by neutrophils or other cells. For example, elastase, cathepsin G, and proteinase 3 bound to aggregated NETs degrade cytokines and chemokines and protect from antiproteases, thereby facilitating the resolution of inflammation [156]. These serine proteases, either individually or collectively, can inactivate mature IL‐1β and IL‐33, while they process immature IL‐1α, IL‐33, and IL‐36 to yield mature active cytokines [157]. Other studies showed proteinase 3 to cleave IL‐32 synthesized by natural killer cells, T lymphocytes, and epithelial cells, yielding a more potent form than the parent molecule to stimulate macrophage differentiation [158]. Upregulation of cathepsin G expression in neutrophils led to increased formation of active IL‐1β, which, in turn, promoted tumor progression in a murine model of lung cancer [159]. NET released during inflammation can also awaken dormant cancer cells. Thus, NET‐bound elastase and MMP‐9 were shown to sequentially cleave laminin, uncovering an epitope that triggered the proliferation of dormant tumor cells in a mouse model of lung injury [160]. These findings suggest another intriguing link between chronic inflammation and cancer.
Neutrophil granule proteins can also synergize with chemokines. For example, MMP‐9 (gelatinase B) cleaves the chemoattractant cytokine CXCL8 (interleukin‐8) at the N terminus to yield a truncated form that displays increased binding affinity to its cognate receptor and more potent biological activities than the parent chemokine [161, 162]. The truncated CXCL8 enhances MMP‐9 release and promotes neutrophil chemotaxis, forming a feed‐forward circuit to amplify the inflammatory response [163]. Cathepsin G or cathelicidin immobilized on the surface of ECs promotes firm adhesion of rolling monocytes in the vasculature [164, 165] as reported in mouse models of atherosclerosis [166]. Another granule protein α‐defensin (also known as HNP1) forms a heterodimer with platelet‐derived C‐C motif ligand 5 (aka RANTES) (CCL5), which enhances monocyte adherence in the mouse microvasculature [167].
Granule constituents and neutrophil phenotypic heterogeneity
Consideration of neutrophil functional versatility and novel paradigms in neutrophil biology (as discussed above and reviewed in [4, 11, 12]) provide clues to characterizing neutrophil phenotypic heterogeneity. Recent reviews detailed distinct neutrophils subsets, characterized by expression of various surface markers, such as NB1 (CD177), olfactomedin 4, IL‐17+, CD63+, or variable T‐cell receptor‐like immune receptors [11, 12]. Neutrophil populations with different density properties have also been identified (reviewed in Ref. [168, 169]). The spectrum of neutrophil density may partially link to their stage of maturation with high‐density and low‐density populations representing mature and immature neutrophils, respectively [169]. Low‐density neutrophils (also known as granulocytic myeloid‐derived suppressor cells) consist of a heterogeneous population, which may either indicate in vivo activation/degranulation of mature neutrophils or represent a distinct lineage of cells (i.e., immature neutrophils released from the bone marrow) [169]. Expression of certain granule markers on the surface of neutrophils, such as CD63 (azurophilic granules), CD66b (SG), or CD11b (GG granules), has been proposed as indicators of cell activation [169]. Alternatively, the activated phenotype might be acquired within inflamed tissues, thus indicating neutrophils that underwent reverse TEM [71, 75]. Low‐density neutrophils have been detected in pregnancy [170] and in patients with sepsis [171], diabetes [172], cancer [173, 174], or autoimmune diseases [168]. Intriguingly, some low‐density neutrophil subsets share the ability to suppress immune response, whereas systemic lupus erythematosus‐associated low‐density neutrophils display pro‐inflammatory properties and are highly susceptible to form NET [168]. The phenotypical and functional divergence of low‐density neutrophil subsets requires further investigations.
Potential therapeutic modulation of granule proteins
The role of neutrophils in the initiation and progression of pathological processes underlying tissue damage makes these cells attractive therapeutic targets. However, global reduction of neutrophil numbers or functional responses limits the usefulness of these therapies because of compromised host defense to bacterial infections. Considering the importance of granule proteins in mediating neutrophil responses, an attractive alternative approach may be targeting the release of these proteins and/or their actions without impairing the ability of neutrophils to contain the microbial invasion. Accumulating data indicate the feasibility of this approach.
Inhibition of granule trafficking and docking
Azurophilic granules contain the most toxic protein cargo and their exocytosis is selectively regulated by Rab27a through interaction with the effector Slp1/JFC1 [18]. Structural modeling and high‐throughput screening analysis led to identifying small molecule neutrophil‐specific exocytosis inhibitors, termed Nexinhibs, which selectively interrupt the Rab27a‐JFC1 interaction and release of the azurophilic granule cargo without interfering with phagocytosis [57, 175]. Nexinhib 20‐treated mice were reported to exhibit decreases in plasma levels of neutrophil granule proteins [176] and reduced neutrophil accumulation in the kidney and liver in a mouse model of LPS‐induced systemic inflammation [175], similar to the phenotype observed in Rab27a‐deficient mice [177]. Peptide aptamers derived from SNARE domains that compete for binding between intact SNARE proteins have also been developed [57]. SNARE mimicking peptides were fused with the cell‐penetrating peptide HIV TAT to facilitate uptake by neutrophils [57]. Fusion proteins containing the N‐terminal SNAP‐23 SNARE domain (TAT‐SNAP‐23) efficiently inhibited formyl‐Met‐Leu‐Phe‐stimulated degranulation of SG, GG granules, and SV, but not azurophilic granule exocytosis, whereas fusion proteins containing syntaxin 4 SNARE domain (TAT‐STX‐4) inhibited degranulation of all four granule subsets in vitro [57]. In preclinical models, TAT‐SNAP‐23 reduced neutrophil‐mediated acute lung injury induced by pulmonary immune complex deposition [178] or sepsis [179]. Since SNARES expression is not restricted to neutrophils, SNARE inhibitors lack selectivity for neutrophils [57]. Further studies are needed to define the cell types other than neutrophils affected by SNARE inhibitors in vivo.
Modulation of NET formation or degradation
Neutrophil extracellular traps formation may contribute to the pathophysiology of many diseases; hence, modulating this process opens potential avenues for therapy. Preclinical studies showed that ROS scavengers, such as N‐acetyl cysteine [180], and MPO inhibitors [180, 181] could reduce NET release both in vitro and in vivo. Similar to MPO inhibitors, TLR4 [182] or peptidyl arginine deiminase (PAD) inhibitors [183, 184, 185] reduced NET formation and tissue damage in murine models of arthritis, atherosclerosis, and lupus. Studies in PAD4‐knockout mice [186, 187], however, suggest that bacterial infections may shift the balance of the protective and deleterious effects of NETs in host defense. Another potential approach is to facilitate the degradation of already formed NETs with DNase 1 and enhance their clearance. Indeed, treatment with exogenous DNase 1 reduced tissue damage and mortality in tumor‐bearing mice [188], lupus‐prone mice [189], acid inspiration‐induced lung injury [190], and transplantation‐associated lung injury in mice [191]. Of note, the synthetic DNase 1 analog dornase‐α is currently being tested in a phase III clinical trial in patients with severe trauma‐associated respiratory failure [192].
Blocking the actions of granule proteins
Disrupting granule protein‐mediated crosstalk and signaling circuits is another promising therapeutic avenue. Of particular interest is the members of the superfamily of specialized pro‐resolving lipid mediators (SPMs), including lipoxins, resolvins, protectins, and maresins [193, 194, 195]. SPMs are formed within the inflammatory environment during the resolution phase of acute inflammation [193, 195] and resolution of clots in venous thrombosis [196]. SPMs are typically generated from arachidonic acid and polyunsaturated fatty acids by transcellular biosynthesis in inflammatory exudates and by neutrophils and macrophages [195]. Aspirin and statins trigger the biosynthesis of R‐epimeric forms of SPMs [193, 197]. In addition to transcellular mechanisms, macrophages can also produce various SPMs when interacting with apoptotic neutrophils [198], pro‐resolving microparticles [199], or high‐density lipoprotein [200]. While each mediator carries defining biological functions with specific receptors and cell and organ‐specific properties, their primary cellular targets are myeloid cells [201]. This intricate signaling network is mapped onto the searchable Atlas of Inflammation Resolution [202]. SPMs attenuate neutrophil accumulation in inflamed tissues partly by preventing upregulation of Mac‐1 [193, 194, 195, 202]. Resolvin D1 may limit neutrophil trafficking into tissues through countering the exocytosis of SV [203]. Aspirin triggered 15‐epi‐lipoxin A4, acting through the lipoxin A4/formyl peptide receptor 2, disrupts the MPO‐based self‐amplifying loop by attenuating MPO‐induced Mac‐1 upregulation and degranulation and redirects neutrophil to apoptosis [204]. 15‐epi‐lipoxin A4 and 17‐epi‐resolvin D1 counter TLR9 activation‐triggered release of NE and proteinase 3 and restore the balance between Mac‐1 and C5aR expression to enhance phagocytosis of bacteria and phagocytosis‐induced apoptosis in human neutrophils [43]. The therapeutic potential of these lipids is illustrated by the observations that treatment with these lipids limits neutrophil accumulation, accelerates bacterial clearance, and enhances neutrophil apoptosis and efferocytosis, resulting in accelerated resolution of inflammation in mouse models of MPO [204] or E. coli‐induced acute lung injury [43]. Select SPMs, such as resolvin D4, limit NETosis in the formation of clots in murine models of deep vein thrombosis [196]. Other SPMs, such as resolvin E1, which signals through the LTB4 receptor BLT1 [205], and resolvin D5, which acts through GPR32 [206], facilitate phagocytosis of bacteria by naïve neutrophils via distinct receptors and molecular mechanisms. Whether these SPMs could also modulate the actions of granule proteins remain to be investigated.
Due to the ability of NE to degrade extracellular matrix proteins, a critical event in the development of chronic inflammatory diseases, such as pulmonary emphysema, adult respiratory distress syndrome, cystic fibrosis, chronic obstructive pulmonary disease, and rheumatoid arthritis [57, 207], extensive research efforts have been directed to develop inhibitors. Recombinant endogenous elastase inhibitors, including recombinant α1‐proteinase inhibitor and secretory leukocyte inhibitor, became available [208], and some (e.g., sivelestat) were evaluated in clinical trials [209]. However, the toxicity and off‐target effects of synthetic inhibitors considerably limit their clinical use. Several natural compounds (for example, flavonoids, tannins, and cinnamic acid derivatives) were found to exert direct inhibitory activity on NE in vitro [210], though little information is available on their in vivo actions. Of note, by degrading various pro‐inflammatory cytokines, including IL‐1, TNF, and IL‐6 [211] and mediating neutrophil reverse TEM [72], NE may play a role in dampening the inflammatory reactions. In addition to NE, preclinical data imply a role for proteinase 3 in the development of chronic obstructive pulmonary disease [212] and possibly other chronic diseases characterized by tissue destruction. However, validating proteinase 3 as a relevant therapeutic target in patients requires additional investigation and development of selective inhibitors.
Concluding remarks
Proteins expressed and stored in neutrophils during granulopoiesis represent a readily mobilizable pool of molecules that mediate neutrophil effector functions. In addition to microbicidal and tissue‐damaging actions, neutrophil granule proteins are increasingly being recognized as mediators of neutrophil orchestration of innate and adaptive immunity.
Consideration of novel paradigms in functions of granule proteins provides a different vantage point of the neutrophils’ roles in host defense, causing local tissue damage and systemic complications, in particular under chronic inflammatory conditions. However, whether neutrophil subsets (identified by phenotypic markers) exhibit differences in granule protein content and secretion remains to be defined. A better understanding of the roles of neutrophil granule proteins is essential to further the development of neutrophil granule‐specific therapies. Results from preclinical models indicate that this can be accomplished by selective targeting of granule exocytosis or interference with signaling from granule constituents released to the extracellular environment with small molecule inhibitors, recombinant protein inhibitors, and SPMs, such as lipoxins and resolvins, which can dampen neutrophil‐mediated inflammation without interfering with antimicrobial defense. Clinical trials with synthetic NE inhibitors yielded disappointing results due to toxicity. While large‐scale clinical trials with selective inhibitors of other granule proteins seem distant, results from an ongoing trial targeting granule protein functions (i.e., promoting degradation of NETs) hold promise as a disease‐modifying intervention. Further studies are needed to investigate whether therapeutic interventions aimed to counter the actions of individual granule proteins could limit the deleterious actions of neutrophils in inflamed tissues, perhaps in a partial tissue‐specific manner, and whether this will facilitate the resolution of inflammation underlying many chronic diseases.
Conflict of interest
The authors declare no conflicts of interest.
Author contributions
AO, MS, and JGF wrote the review.
Acknowledgements
This study was supported by grants from the Canadian Institutes of Health Research (MOP‐97742 and MOP‐102619) and the Natural Science and Engineering Research Council of Canada (RGPIN‐2017‐04980) (to JGF).
References
- 1. Nauseef WM & Borregaard N (2014) Neutrophils at work. Nat Immunol 15, 602–611. [DOI] [PubMed] [Google Scholar]
- 2. Gordon S (2016) Phagocytosis: an immunobiologic process. Immunity 44, 463–475. [DOI] [PubMed] [Google Scholar]
- 3. Rørvig S, Østergaard O, Heegaard NHH & Borregaard N (2013) Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol 94, 711–721. [DOI] [PubMed] [Google Scholar]
- 4. Cassatella MA, Östberg NK, Tamassia N & Soehnlein O (2019) Biological roles of neutrophil‐derived granule proteins and cytokines. Trends Immunol 40, 648–664. [DOI] [PubMed] [Google Scholar]
- 5. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y & Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- 6. Nathan C & Ding A (2010) Nonresolving inflammation. Cell 140, 871–882. [DOI] [PubMed] [Google Scholar]
- 7. Klebanoff SJ (2005) Myeloperoxidase: friend or foe. J Leukoc Biol 77, 598–625. [DOI] [PubMed] [Google Scholar]
- 8. Liew PX & Kubes P (2019) The neutrophil’s role during health and disease. Physiol Rev 99, 1223–1248. [DOI] [PubMed] [Google Scholar]
- 9. Mantovani A, Cassatella MA, Costantini C & Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11, 519–531. [DOI] [PubMed] [Google Scholar]
- 10. Jones HR, Robb CT, Perretti M & Rossi AG (2016) The role of neutrophils in inflammation resolution. Semin Immunol 28, 137–145. [DOI] [PubMed] [Google Scholar]
- 11. Silvestre‐Roig C, Hidalgo A & Soehnlein O (2016) Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127, 2173–2181. [DOI] [PubMed] [Google Scholar]
- 12. Filep JG & Ariel A (2020) Neutrophil heterogeneity and fate in inflamed tissues: implications for the resolution of inflammation. Am J Physiol Cell Physiol 319, C510–C532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Molinedo F (2019) Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol 40, 228–242. [DOI] [PubMed] [Google Scholar]
- 14. Mora‐Jensen H, Jendholm J, Fossum A, Porse B, Borregaard N & Theilgaard‐Mönch K (2011) Technical advance: immunophenotypical characterization of human neutrophil differentiation. J Leukoc Biol 90, 629–634. [DOI] [PubMed] [Google Scholar]
- 15. Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA & McLeish KR (2005) Proteomic analysis of human neutrophil granules. Mol Cell Proteomics 4, 1503–1521. [DOI] [PubMed] [Google Scholar]
- 16. Kettritz R (2016) Neutral serine proteases of neutrophils. Immunol Rev 273, 232–248. [DOI] [PubMed] [Google Scholar]
- 17. Munafó DB, Johnson JL, Ellis BA, Rutschmann S, Beutler B & Catz SD (2007) Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem J 402, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yin C & Heit B (2018) Armed for destruction: formation, function and trafficking of neutrophil granules. Cell Tissue Res 371, 455–471. [DOI] [PubMed] [Google Scholar]
- 19. Rørvig S, Honore C, Larsson LI, Ohlsson S, Pedersen CC, Jacobsen LC, Cowland JB, Garred P & Borregaard N (2009) Ficolin‐1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J Leukoc Biol 86, 1439–1449. [DOI] [PubMed] [Google Scholar]
- 20. Clemmensen SN, Udby L & Borregaard N (2014) Subcellular fractionation of human neutrophils and analysis of subcellular markers. Methods Mol Biol 1124, 53–76. [DOI] [PubMed] [Google Scholar]
- 21. Uriarte SM, Powell DW, Luerman GC, Merchant ML, Cummins TD, Jog NR, Ward RA & McLeish KR (2008) Comparison of proteins expressed on secretory vesicle membranes and plasma membranes of human neutrophils. J Immunol 180, 5575–5581. [DOI] [PubMed] [Google Scholar]
- 22. Jethwaney D, Islam MR, Leidal KG, de Bernabe DB, Campbell KP, Nauseef WM & Gibson BW (2007) Proteomic analysis of plasma membrane and secretory vesicles from human neutrophils. Proteome Sci 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mantripragada KC & Quesenberry PJ (2018) Polarization of neutrophil granules ‐ A characteristic of inflammatory states. Blood Cells Mol Dis 69, 74. [DOI] [PubMed] [Google Scholar]
- 24. Galkina SI, Fedorova NV, Serebryakova MV, Romanova JM, Golyshev SA, Stadnichuk VI, Baratova LA, Sud'ina GF & Klein T (2012) Proteomic analysis identified human neutrophil membrane tubulovesicular extensions (cytonemes, membrane tethers) as bactericide trafficking. Biochim Biophys Acta 1820, 1705–1714. [DOI] [PubMed] [Google Scholar]
- 25. Galkina SI, Fedorova NV, Serebryakova MV, Arifulin EA, Stadnichuk VI, Gaponova TV, Baratova LA & Sud'ina GF (2015) Inhibition of the GTPase dynamin or actin depolymerisation initiates outward plasma membrane tabulation/vesiculation (cytoneme formation) in neutrophils. Biol Cell 107, 144–158. [DOI] [PubMed] [Google Scholar]
- 26. Dalli J, Montero‐Melendez T, Norling LV, Yin X, Hinds C, Haskard D, Mayr M & Perretti M (2013) Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol Cell Proteomics 12, 2205–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lacy P & Eitzen G (2008) Control of granule exocytosis in neutrophils. Front Biosci 13, 5559. [DOI] [PubMed] [Google Scholar]
- 28. Mayadas TN, Cullere X & Lowell CA (2014) The multifaceted functions of neutrophils. Ann Rev Pathol 9, 181–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miralda I, Uriarte SM & McLeish KR (2017) Multiple phenotypic changes define neutrophil priming. Front Cell Infect Microbiol 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Freeman SA & Grinstein S (2014) Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev 262, 193–215. [DOI] [PubMed] [Google Scholar]
- 31. Abdel‐Latif D, Steward M, Macdonald DL, Francis GA, Dinauer MC & Lacy P (2004) Rac2 is critical for neutrophil primary granule exocytosis. Blood 104, 832–839. [DOI] [PubMed] [Google Scholar]
- 32. Abdel‐Latif D, Steward M & Lacy P (2005) Neutrophil primary granule release and maximal superoxide generation depend on Rac2 in a common signaling pathway. Can J Physiol Pharmacol 83, 69–75. [DOI] [PubMed] [Google Scholar]
- 33. Sun CX, Magalhães MAO & Glogauer M (2007) Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J Cell Biol 179, 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitchell T, Lo A, Logan MR, Lacy P & Eitzen G (2008) Primary granule exocytosis inhuman neutrophils is regulated by Rac‐dependent actin remodeling. Am J Physiol Cell Physiol 295, C1354–C1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eitzen G, Lo AN, Mitchell T, Kim JD, Chao DV & Lacy P (2011) Proteomic analysis of secretagogue‐stimulated neutrophils implicates a role for actin and actin‐interacting proteins in Rac2‐mediated granule exocytosis. Proteome Sci 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bengtsson T, Dahlgren C, Stendahl O & Andersson T (1991) Actin assembly and regulation of neutrophil function: effects of cytochalasin B and tetracaine on chemotactic peptide‐induced O2‐ production and degranulation. J Leukoc Biol 49, 236–244. [DOI] [PubMed] [Google Scholar]
- 37. Mócsai A, Ligeti E, Lowell CA & Berton G (1999) Adhesion‐dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol 162, 1120–1126. [PubMed] [Google Scholar]
- 38. Perskvist N, Roberg K, Kulyté A & Stendahl O (2002) Rab5a GTPase regulates fusion between pathogen‐containing phagosomes and cytoplasmic organelles in human neutrophils. J Cell Sci 115, 1321–1330. [DOI] [PubMed] [Google Scholar]
- 39. Kovács M, Németh T, Jakus Z, Sitaru C, Simon E, Futosi K, Botz B, Helyes Z, Lowell CA & Mócsai A (2014) The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med 211, 1993–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sai J, Raman D, Liu Y, Wikswo J & Richmond A (2008) Parallel phosphatidylinositol 3‐kinase(PI3K)‐dependent and Src‐dependent pathways lead to CXCL8‐mediated Rac2 activation and chemotaxis. J Biol Chem 283, 26538–26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mócsai A, Jakus Z, Vántus T, Berton G, Lowell CA & Ligeti E (2000) Kinase pathways in chemoattractant‐induced degranulation of neutrophils: the role of p38 mitogen‐activated protein kinase activated by Src family kinases. J Immunol 164, 4321–4331. [DOI] [PubMed] [Google Scholar]
- 42. Jog NR, Jala VR, Ward PA, Rane MJ, Haribabu B & McLeish KR (2007) Heat shock protein 27 regulates neutrophil chemotaxis and exocytosis through two independent mechanisms. J Immunol 178, 2421–2428. [DOI] [PubMed] [Google Scholar]
- 43. Sekheri M, El Kebir D, Edner N & Filep JG (2020) 15‐Epi‐LXA4 and 17‐epi‐RvD1 restore TLR9‐mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc Natl Acad Sci USA 117, 7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. József L, Khreiss T, El Kebir D & Filep JG (2006) Activation of TLR‐9 induces IL‐8 secretion through peroxynitrite signaling in human neutrophils. J Immunol 176, 1195–1202. [DOI] [PubMed] [Google Scholar]
- 45. Maa MC, Chang MY, Li J, Li YY, Hsieh MY, Yang CJ, Chen YJ, Li Y, Chen HC, Cheng WE et al. (2011) The iNOS/Src/FAK axis is critical in Toll‐like receptor‐mediated cell motility in macrophages. Biochim Biophys Acta 1813, 136–147. [DOI] [PubMed] [Google Scholar]
- 46. Ghosh E, Kumari P, Jaiman D & Shukla AK (2015) Methodological advances: the unsung heroes of the GPCR structural revolution. Nat Rev Mol Cell Biol 16, 69–81. [DOI] [PubMed] [Google Scholar]
- 47. Hamada K, Miyatake H, Terauchi A & Mikoshiba K (2017) IP3‐mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X‐ray crystallography. Proc Natl Acad Sci USA 114, 4661–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lodge KM, Cowburn AS, Li W & Condliffe AM (2020) The impact of hypoxia on neutrophil degranulation and consequences for the host. Int J Mol Sci 21, 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sengeløv H, Kjeldsen L & Borregaard N (1993) Control of exocytosis in early neutrophil activation. J Immunol 150, 1535–1543. [PubMed] [Google Scholar]
- 50. Theander S, Lew DP & Nüsse O (2002) Granule‐specific ATP requirements for Ca2+‐induced exocytosis in human neutrophils. Evidence for substantial ATP‐independent release. J Cell Sci 115, 2975–2983. [DOI] [PubMed] [Google Scholar]
- 51. Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, Hinz B, van der Merwe PA, Das R & Grinstein S (2016) Integrins form an expanding diffusion barrier that coordinates phagocytosis. Cell 164, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramadass M & Catz SD (2016) Molecular mechanisms regulating secretory organelles and endosomes in neutrophils and their implications for inflammation. Immunol Rev 273, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu C, Ahmed M, Melia TJ, Söllner TJ, Mayer T & Rothman JE (2003) Fusion of cells by flipped SNAREs. Science 300, 1745–1749. [DOI] [PubMed] [Google Scholar]
- 54. Martín‐Martín B, Nabokina SM, Lazo PA & Mollinedo F (1999) Coexpression of several human syntaxin genes in neutrophils and differentiating HL‐60 cells: variant isoforms and detection of syntaxin 1. J Leukoc Biol 65, 397–406. [DOI] [PubMed] [Google Scholar]
- 55. Mollinedo F, Martín‐Martín B, Calafat J, Nabokina SM & Lazo PA (2003) Role of vesicle‐associated membrane protein‐2, through Q‐soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor/R‐soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor interaction, in the exocytosis of specific and tertiary granules. J Immunol 170, 1034–1042. [DOI] [PubMed] [Google Scholar]
- 56. Mollinedo F, Calafat J, Janssen H, Martin‐Martin B, Canchado J, Nabokina SM & Gajate C (2006) Combinatorial SNARE complexes modulate the secretion of cytoplasmic granules in human neutrophils. J Immunol 177, 2831–2841. [DOI] [PubMed] [Google Scholar]
- 57. Catz SD & McLeish KR (2020) Therapeutic targeting of neutrophil exocytosis. J Leukoc Biol 107, 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Monfregola J, Johnson JL, Meijler MM, Napolitano G & Catz SD (2012) MUNC13‐4 protein regulates the oxidative response and is essential for phagosomal maturation and bacterial killing in neutrophils. J Biol Chem 287, 44603–44618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson JL, He J, Ramadass M, Pestonjamasp K, Kiosses WB, Zhang J & Catz SD (2016) Munc13‐4 is a Rab11‐binding protein that regulates Rab11‐positive vesicle trafficking and docking at the plasma membrane. J Biol Chem 291, 3423–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. He J, Johnson JL, Monfregola J, Ramadass M, Pestonjamasp K, Napolitano G, Zhang J & Catz SD (2016) Munc13‐4 interacts with syntaxin seven and regulates late endosomal maturation, endosomal signaling, and TLR9‐initiated cellular responses. Mol Biol Cell 27, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ley K, Laudanna C, Cybulsky MI & Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7, 678–689. [DOI] [PubMed] [Google Scholar]
- 62. Nourshargh S, Hordijk PL & Sixt M (2010) Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol 11, 366–378. [DOI] [PubMed] [Google Scholar]
- 63. Kolaczkowska E & Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13, 159–175. [DOI] [PubMed] [Google Scholar]
- 64. Nourshargh S & Alon R (2014) Leukocyte migration into inflamed tissues. Immunity 41, 694–707. [DOI] [PubMed] [Google Scholar]
- 65. Sachs UJ, Andrei‐Selmer CL, Maniar A, Weiss T, Paddock C, Orlova VV, Choi EY, Newman PJ, Preissner KT, Chavakis T et al. (2007) The neutrophil‐specific antigen CD177 is a counter‐receptor for platelet‐endothelial cell adhesion molecule‐1 (CD31). J Biol Chem 282, 23603–23612. [DOI] [PubMed] [Google Scholar]
- 66. Kuckleburg CJ, Tilkens SB, Santoso S & Newman PJ (2012) Proteinase 3 contributes to transendothelial migration of NB1‐positive neutrophils. J Immunol 188, 2419–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sun J, Paddock C, Shubert J, Zhang HB, Amin K, Newman PJ & Albelda SM (2000) Contribution of the extracellular rand cytoplasmic domains of platelet‐endothelial cell adhesion molecule‐1 (PECAM‐1/CD31) in regulating cell‐cell localization. J Cell Sci 113, 1459–1469. [DOI] [PubMed] [Google Scholar]
- 68. Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T & Gulino‐Debrac D (2013) Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem 278, 14002–14012. [DOI] [PubMed] [Google Scholar]
- 69. Kuckleburg CJ & Newman PJ (2013) Neutrophil proteinase 3 (PR3) acts on protease‐activated receptor‐2 (PAR‐2) to enhance vascular endothelial cell barrier function. Arterioscler Thromb Vasc Biol 33, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT & Huttenlocher A (2006) Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol 80, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 71. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE et al. (2011) The junctional adhesion molecule JAM‐C regulates polarized transendothelial migration of neutrophils in vivo . Nat Immunol 12, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA & Nourshargh S (2015) Leukotriene B4‐neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo . Immunity 42, 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Filippi MD (2019) Neutrophil transendothelial migration: updates and new perspectives. Blood 133, 2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cai TQ & Wright SD (1996) Human leukocyte elastase is an endogenous ligand for the integrin CR3 (CD11b/CD18, Mac‐1, alpha M beta 2) and modulates polymorphonuclear leukocyte adhesion. J Exp Med 184, 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, Stone PC, Salmon M, Matharu NM, Vohra RK et al. (2006) Identification of a phenotypically and functionally distinct population of long‐lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol 79, 303–311. [DOI] [PubMed] [Google Scholar]
- 76. Nourshargh S, Renshaw SA & Imhof BA (2016) Reverse migration of neutrophils: where, when, how and why? Trends Immunol 37, 273–286. [DOI] [PubMed] [Google Scholar]
- 77. Lahoz‐Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, Niederalt C, Asquith B & Macallan D (2016) Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half‐lives. Blood 127, 3431–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tak T, Tesselaar K, Pillay J, Borghans JA & Koenderman L (2013) What’s your age again? Determination of human neutrophil half‐lives revisited. J Leukoc Biol 94, 595–601. [DOI] [PubMed] [Google Scholar]
- 79. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, Tesselaar K & Koenderman L (2010) In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116, 625–627. [DOI] [PubMed] [Google Scholar]
- 80. Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM & Haslett C (1989) Macrophage phagocytosis of aging neutrophils in inflammation: programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest 83, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V & Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pasparakis M & Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517, 311–320. [DOI] [PubMed] [Google Scholar]
- 83. Luo HR & Loison F (2008) Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 83, 288–295. [DOI] [PubMed] [Google Scholar]
- 84. Riedl SJ & Salvesen GS (2007) The apoptosome: signaling platform of cell death. Nat Rev Mol Cell Biol 8, 405–413. [DOI] [PubMed] [Google Scholar]
- 85. Loison F, Zhu H, Karatepe K, Kasorn A, Liu P, Ye K, Zhou J, Cao S, Gong H, Jenne DE et al. (2014) Proteinase 3‐dependent caspase‐3 cleavage modulates neutrophil death and inflammation. J Clin Invest 124, 4445–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brümmer J, Rudolph V, Münzel T, Heitzer T et al. (2005) Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA 102, 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. El Kebir D, József L, Pan W & Filep JG (2008) Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res 103, 352–359. [DOI] [PubMed] [Google Scholar]
- 88. Dzhagalov I, St. John A & He Y‐W (2007) The antiapoptotic protein Mcl‐1 is essential for the survival of neutrophils but not macrophages. Blood 109, 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Matthijsen RA, Huugen D, Hoebers NT, de Vries B, Peutz‐Kootstra CJ, Aratani Y, Daha MR, Tervaert JW, Buurman WA & Heeringa P (2007) Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am J Pathol 171, 1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Brovkovych V, Gao XP, Ong E, Brovkovych S, Brennan ML, Su X, Hazen SL, Malik AB & Skidgel RA (2008) Augmented inducible nitric oxide synthase expression and increased NO production reduce sepsis‐induced lung injury and mortality in myeloperoxidase‐null mice. Am J Physiol Lung Cell Mol Physiol 295, L96–L103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gorudko IV, Grigorieva DV, Sokolov AV, Shamova EV, Kostevich VA, Kudryavtsev IV, Syromiatnikova ED, Vasilyev VB, Cherenkevich SN & Panasenko OM (2018) Neutrophil activation in response to monomeric myeloperoxidase. Biochem Cell Biol 96, 592–601. [DOI] [PubMed] [Google Scholar]
- 92. Witko‐Sarsat V, Cramer EM, Hieblot C, Guichard J, Nusbaum P, Lopez S, Lesavre P & Halbwachs‐Mecarelli L (1999) Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood 94, 2487–2496. [PubMed] [Google Scholar]
- 93. Witko‐Sarsat V, Reuter N & Mouthon L (2010) Interaction of proteinase 3 with its associated partners: implications in the pathogenesis of Wegener's granulomatosis. Curr Opin Rheumatol 22, 1–7. [DOI] [PubMed] [Google Scholar]
- 94. Kantari C, Pederzoli‐Ribeil M, Amir‐Moazami O, Gausson‐Dorey V, Moura IC, Lecomte MC, Benhamou M & Witko‐Sarsat V (2007) Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood 110, 4086–4095. [DOI] [PubMed] [Google Scholar]
- 95. Pon IKH, Lucas CD, Rossi AG & Ravichandran KS (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lutaty A, Soboh S, Schif‐Zuck S, Zeituni‐Timor O, Rostoker R, Podolska MJ, Schauer C, Herrmann M, Muñoz LE & Ariel A (2018) A 17‐kDa fragment of lactoferrin associates with the termination of inflammation and peptides within promote resolution. Front Immunol 9, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, Rossi AG & Gregory CD (2009) Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest 119, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Garlichs CD, Eskafi S, Cicha I, Schmeisser A, Walzog B, Raaz D, Stumpf C, Yilmaz A, Bremer J, Ludwig J et al. (2004) Delay of neutrophil apoptosis in acute coronary syndromes. J Leukoc Biol 75, 828–835. [DOI] [PubMed] [Google Scholar]
- 99. Uddin M, Nong G, Ward J, Seumois G, Prince LR, Wilson SJ, Cornelius V, Dent G & Djukanovic R (2010) Prosurvival activity for airway neutrophils in severe asthma. Thorax 65, 684–689. [DOI] [PubMed] [Google Scholar]
- 100. Matute‐Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD & Martin TR (1997) Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med 156, 1969–1977. [DOI] [PubMed] [Google Scholar]
- 101. Keel M, Ungethum U, Steckholzer U, Niederer E, Hartung T, Trentz O & Ertel W (1997) Interleukin‐10 counterregulates proinflammatory cytokine‐induced inhibition of neutrophil apoptosis during severe sepsis. Blood 90, 3356–3363. [PubMed] [Google Scholar]
- 102. Jonsson H, Allen P & Peng SL (2005) Inflammatory arthritis requires Foxo3a to prevent Fas ligand‐induced neutrophil apoptosis. Nat Med 11, 666–671. [DOI] [PubMed] [Google Scholar]
- 103. Guo RF & Ward PA (2005) Role of C5a in inflammatory responses. Annu Rev Immunol 23, 821–852. [DOI] [PubMed] [Google Scholar]
- 104. Mollnes TE, Brekke OL, Fung M, Fure H, Christiansen D, Bergseth G, Videm V, Lappegård KT, Köhl J & Lambris JD (2002) Essential role of the C5a receptor in E coli‐induced oxidative burst and phagocytosis revealed by a novel lepirudin‐based human whole blood model of inflammation. Blood 100, 1869–1877. [PubMed] [Google Scholar]
- 105. Monk PN, Scola AM, Madala P & Fairlie DP (2007) Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol 152, 429–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Berger M, Sorensen RU, Tosi MF, Dearborn DG & Döring G (1989) Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas‐infected lung in cystic fibrosis. J Clin Invest 84, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. van den Berg CW, Tambourgi DV, Clark HW, Hoong SJ, Spiller OB & McGreal EP (2014) Mechanism of neutrophil dysfunction: neutrophil serine proteases cleave and inactivate the C5a receptor. J Immunol 192, 1787–1795. [DOI] [PubMed] [Google Scholar]
- 108. Mayadas TN & Cullere X (2005) Neutrophil β2 integrins: modulators of life or death decision. Trends Immunol 26, 388–395. [DOI] [PubMed] [Google Scholar]
- 109. Filep JG & El Kebir D (2009) Neutrophil apoptosis: a target for enhancing the resolution of inflammation. J Cell Biochem 108, 1039–1046. [DOI] [PubMed] [Google Scholar]
- 110. Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, Mackenzie SJ, Reid H, Davidson DJ, Haslett C et al. (2009) C5a mediates peripheral blood neutrophil dysfunction in critically ill patients. Am J Respir Crit Care Med 180, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Unnewehr H, Rittirsch D, Sarma JV, Zetoune F, Flierl MA, Perl M, Denk S, Weiss M, Schneider ME, Monk PN et al. (2013) Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. J Immunol 190, 4215–4225. [DOI] [PubMed] [Google Scholar]
- 112. Huber‐Lang MS, Riedeman NC, Sarma JV, Younkin EM, McGuire SR, Laudes IJ, Lu KT, Guo RF, Neff TA, Padgaonkar VA et al. (2002) Protection of innate immunity by C5aR antagonist in septic mice. FASEB J 16, 1567–1574. [DOI] [PubMed] [Google Scholar]
- 113. Guo RF, Riedemann NC, Bernacki KD, Sarma VJ, Laudes IJ, Reuben JS, Younkin EM, Neff TA, Paulauskis JD, Zetoune FS et al. (2003) Neutrophil C5a receptor and the outcome in a rat model of sepsis. FASEB J 17, 1889–1891. [DOI] [PubMed] [Google Scholar]
- 114. Ratanarat R, Cazzavillan S, Ricci Z, Rassu M, Segala C, de Cal M, Cruz D, Corradi V, Manfro S, Roessler E et al. (2007) Usefulness of a molecular strategy for the detection of bacterial DNA in patients with severe sepsis undergoing continuous renal replacement therapy. Blood Purif 25, 106–111. [DOI] [PubMed] [Google Scholar]
- 115. Schwartz DA, Quinn TJ, Thorne PS, Sayeed S, Yi AK & Krieg AM (1997) CpG motifs in bacterial DNA cause inflammation in the lower respiratory tract. J Clin Invest 100, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K & Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. West AP & Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bardoel BW, Kenny EF, Sollberger G & Zychlinsky A (2014) The balancing act of neutrophils. Cell Host Microbe 15, 526–536. [DOI] [PubMed] [Google Scholar]
- 119. Gupta S & Kaplan MJ (2016) The role of neutrophil NETosis in autoimmune and renal diseases. Nat Rev Nephrol 12, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, Lell M, Manger B, Rech J, Naschberger E et al. (2014) Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med 20, 511–517. [DOI] [PubMed] [Google Scholar]
- 121. Manfredi AA, Ramirez GA, Rovere‐Querini P & Maugeri N (2018) The neutrophil’s choice: phagocytose vs make neutrophil extracellular traps. Front Immunol 9, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rother N, Pieterse E, Lubbers J, Hilbrands L & van der Vlag J (2017) Acetylated histones in apoptotic microparticles drive the formation of neutrophil extracellular traps in active lupus nephritis. Front Immunol 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB et al. (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16, 887–896. [DOI] [PubMed] [Google Scholar]
- 124. Erreni M, Manfredi AA, Garlanda C, Mantovani A & Rovere‐Querini P (2017) The long pentraxin PTX3: a prototypical sensor of tissue injury and a regulator of homeostasis. Immunol Rev 280, 112–125. [DOI] [PubMed] [Google Scholar]
- 125. Yuen J, Pluthero FG, Douda DN, Riedl M, Cherry A, Ulanova M, Kahr WH, Palaniyar N & Licht C (2016) NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non‐alternative pathways. Front Immunol 7, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sorensen OE & Borregaard N (2016) Neutrophil extracellular traps – the dark side of neutrophils. J Clin Invest 126, 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jorch SK & Kubes P (2017) An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23, 279–287. [DOI] [PubMed] [Google Scholar]
- 128. Papayannopoulos V (2017) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18, 134–147. [DOI] [PubMed] [Google Scholar]
- 129. Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ & Wright HL (2019) Caught in a Trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front Immunol 10, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C & Simon HU (2019) Untangling “NETosis” from NETs. Eur J Immunol 49, 221–227. [DOI] [PubMed] [Google Scholar]
- 131. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A & Papayannopoulos V (2014) A myeloperoxidase‐containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep 8, 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tessarz P & Kouzarides T (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15, 703–708. [DOI] [PubMed] [Google Scholar]
- 133. Maugeri N, Rovere‐Querini P, Evangelista V, Covino C, Capobianco A, Bertilaccio MT, Piccoli A, Totani L, Cianflone D, Maseri A et al. (2009) Neutrophils phagocytose activated platelets in vivo: a phosphatidylserine, P‐selectin, and β2 integrin‐dependent cell clearance program. Blood 113, 5254–5265. [DOI] [PubMed] [Google Scholar]
- 134. Garcia‐Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL et al. (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3, 73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V et al. (2011) Neutrophils activate plasmocytoid dendritic cells by releasing self‐DNA peptide complexes in systemic lupus erythematosus. Sci Transl Med 3, 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH & Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA 107, 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Martinod K, Witsch T, Farley K, Gallant M, Remold‐O’Donnell E & Wagner DD (2016) Neutrophil elastase‐deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost 14, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll Re & Zychlinsky A (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 107, 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lazzaretto B & Fadeel B (2019) Intra‐ and extracellular degradation of neutrophil extracellular traps by macrophages and dendritic cells. J Immunol 203, 2276–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Farrera C & Fadeel B (2013) Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 191, 2647–2656. [DOI] [PubMed] [Google Scholar]
- 141. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD et al. (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13, 463–469. [DOI] [PubMed] [Google Scholar]
- 142. Döring Y, Libby P & Soehnlein O (2020) Neutrophil extracellular traps participate in cardiovascular diseases. Recent experimental and clinical insights. Circ Res 126, 1228–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lefrançais E, Mallavia B, Zhuo H, Calfee CS & Looney MR (2018) Maladaptive role of neutrophil extracellular traps in pathogen‐induced lung injury. JCI Insight 3, e98178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, Borczuk A, Cools‐Lartigue J, Crawford JM, Daßler‐Plenker J, Guerci P, Huynh C, Knight JS et al. (2020) Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med 217, e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M et al. (2020) Neutrophil extracellular traps in COVID‐19. JCI Insight 5, e138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Jennette JC, Xiao H & Falk RJ (2006) Pathogenesis of vascular inflammation by antineutrophil cytoplasmic antibodies. J Am Soc Nephrol 17, 1235–1242. [DOI] [PubMed] [Google Scholar]
- 147. Morgan MD, Harper L, Williams J & Savage C (2006) Antineutrophil cytoplasm‐associated glomerulonephritis. J Am Soc Nephrol 17, 1224–1234. [DOI] [PubMed] [Google Scholar]
- 148. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ & Jennette JC (2002) Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 110, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. David A, Kacher Y, Specks U & Aviram I (2003) Interaction of proteinase 3 with CD11b/CD18 (beta2 integrin) on the cell membrane of human neutrophils. J Leukoc Biol 74, 551–557. [DOI] [PubMed] [Google Scholar]
- 150. Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, Terrier B, Kerstein A, Tamassia N, Satyanarayanan SK et al. (2015) Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Invest 125, 4107–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Pendergraft WF 3rd, Preston GA, Shah RR, Tropsha A, Carter CW Jr, Jennette JC & Falk RJ (2004) Autoimmunity is triggered by cPR‐3(105–201), a protein complementary to human autoantigen proteinase‐3. Nat Med 10, 72–79. [DOI] [PubMed] [Google Scholar]
- 152. Tait SW, Ichim G & Green DR (2014) Die another way‐ non‐apoptotic mechanisms of cell death. J Cell Sci 127, 2135–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Morrizane S & Gallo RL (2012) Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol 39, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Mihailovic PM, Lio WM, Yano J, Zhao X, Zhou J, Chyu KY, Shah PK, Cercek B & Dimayuga PC (2017) The cathelicidin protein CRAMP is a potential atherosclerosis self‐antigen in ApoE(‐/‐) mice. PLoS One 12, e0187432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC & Catz SD (2018) Neutrophils: new insights and open questions. Sci Immunol. 3, eaat4579. [DOI] [PubMed] [Google Scholar]
- 156. Hahn J, Schauer C, Czegley C, Kling L, Petru L, Schmid B, Weidner D, Reinwald C, Biermann MHC, Blunder S et al. (2019) Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J 33, 1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, Lavelle EC & Martin SJ (2018) Extracellular neutrophil proteases are efficient regulators of IL‐1, IL‐33, and IL‐36 cytokine activity but poor effectors of microbial killing. Cell Rep 22, 2937–2950. [DOI] [PubMed] [Google Scholar]
- 158. Netea MG, Lewis EC, Azam T, Joosten LA, Jaekal J, Bae SY, Dinarello CA & Kim SH (2008) Interleukin‐32 induces the differentiation of monocytes into macrophage‐like cells. Proc Natl Acad Sci USA 105, 3515–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. McLoed AG, Sherrill TP, Cheng DS, Han W, Saxon JA, Gleaves LA, Wu P, Polosukhin VV, Karin M, Yull FE et al. (2016) Neutrophil‐derived IL‐1β impairs the efficacy of NF‐κB inhibitors against lung cancer. Cell Rep 16, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Albrengues J, Shields M, Ng D, Park CG, Ambrico A, Poindexter M, Upadhyay P, Uyeminami D, Pommier A, Küttner V et al. (2018) Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, eaao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Van den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J & Opdenakker G (2003) Gelatinase B/MMP‐9 and neutrophil collagenase /MMP‐8 process the chemokines human GCP‐2/CXCL6, ENA‐78/CXCL5 and mouse GCP‐2/LIX and modulate their physiological activities. Eur J Biochem 270, 3739–3749. [DOI] [PubMed] [Google Scholar]
- 162. Proost P, Struyf S, Van Damme J, Fiten P, Ugarte‐Berzal E & Opdenakker G (2017) Chemokine isoforms and processing in inflammation and immunity. J Autoimmun 85, 45–57. [DOI] [PubMed] [Google Scholar]
- 163. Van Lint P & Libert C (2007) Chemokine and cytokine processing by matrix metalloproteinases and its effects on leukocyte migration and inflammation. J Leukoc Biol 82, 1375–1381. [DOI] [PubMed] [Google Scholar]
- 164. Soehnlein O, Zernecke A, Eriksson EE, Rothfuchs AG, Pham CT, Herwald H, Bidzhekov K, Rottenberg ME, Weber C & Lindbom L (2008) Neutrophil secretion products pave the way for inflammatory monocytes. Blood 112, 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wantha S, Alard J‐E, Megens RTA, van der Does A, Döring Y, Drechsler M, Pham CT, Wang MW, Wang JM, Gallo RL et al. (2013) Neutrophil‐derived cathelicidin promotes adhesion of classical monocytes. Circ Res 112, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ortega‐Gomez A, Salvermoser M, Rossaint J, Pick R, Brauner J, Lemnitzer P, Tilgner J, de Jong RJ, Megens RTA, Jamasbi J et al. (2016) Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation 134, 1176–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Alard J‐E, Ortega‐Gomez A, Wichapong K, Bongiovanni D, Horckmans M, Megens RTA, Leoni G, Ferraro B, Rossaint J, Paulin N et al. (2015) Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med 7, 317ra196. [DOI] [PubMed] [Google Scholar]
- 168. Carmona‐Rivera C & Kaplan MJ (2013) Low‐density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol 35, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Hassani M, Hellebrekers P, Chen N, van Aaalst C, Bongers S, Hietbrink F, Koenderman L & Vrisekoop N (2020) On the origin of low‐density neutrophils. J Leukoc Biol 107, 809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Köstlin N, Kugel H, Spring B, Leiber A, Marmé A, Henes M, Rieber N, Hartl D, Poets CF & Gille C (2014) Granulocytic myeloid derived suppressor cells expand in human pregnancy and modulate T‐cell responses. Eur J Immunol 44, 2582–2591. [DOI] [PubMed] [Google Scholar]
- 171. Janols H, Bergenfelz C, Allaoui R, Larsson AM, Rydén L, Björnsson S, Janciauskiene S, Wullt M, Bredberg A & Leandersson K (2014) A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram‐positive cases. J Leukoc Biol 96, 685–693. [DOI] [PubMed] [Google Scholar]
- 172. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR & Wagner DD (2015) Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 21, 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov RV et al. (2015) Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep 10, 562–573. [DOI] [PubMed] [Google Scholar]
- 174. Moses K & Brandau S (2016) Human neutrophils: their role in cancer and relation to myeloid‐derived suppressor cells. Semin Immunol 28, 187–196. [DOI] [PubMed] [Google Scholar]
- 175. Johnson JL, Ramadass M, He J, Brown SJ, Zhang J, Abgaryan L, Biris N, Gavathiotis E, Rosen H & Catz SD (2016) Identification of neutrophil exocytosis inhibitors (nexinhibs), small molecule inhibitors of neutrophil exocytosis and inflammation: druggability of the small GTPase Rab27a. J Biol Chem 291, 25965–25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Wang H, Ishizaki R, Xu J, Kasai K, Kobayashi E, Gomi H & Izumi T (2013) The Rab27a effector exophilin 7 promotes fusion of secretory granules that have not been docked to the plasma membrane. Mol Biol Cell 24, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Johnson JL, Hong H, Monfregola J & Catz SD (2011) Increased survival and reduced neutrophil infiltration of the liver in Rab27a‐ but not Munc13‐4‐deficient mice in lipopolysaccharide‐induced systemic inflammation. Infect Immun 79, 3607–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Uriarte SM, Rane MJ, Merchant ML, Jin S, Lentsch AB, Ward RA & McLeish KR (2015) Inhibition of neutrophil exocytosis ameliorates acute lung injury in rats. Shock 39, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Bai J, Tang L, Lomas‐Neira J, Chen Y, McLeish KR, Uriarte SM, Chung C‐S & Ayala A (2015) TAT‐SNAP‐23 treatment inhibits the priming of neutrophil functions contributing to shock and/or sepsis‐induced extra‐pulmonary acute lung injury. Innate Immun 21, 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Patel S, Kumar S, Jyoti A, Srinag BS, Keshari RS, Saluja R, Verma A, Mitra K, Barthwal MK, Krishnamurthy H et al. (2010) Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide 22, 226–234. [DOI] [PubMed] [Google Scholar]
- 181. Zheng W, Warner R, Ruggeri R, Su C, Cortes C, Skoura A, Ward J, Ahn K, Kalgutkar A, Sun D et al. (2015) PF‐1355, a mechanism‐based myeloperoxidase inhibitor, prevents immune complex vasculitis and anti‐glomerular‐basement membrane glomerulonephritis. J Pharmacol Exp Ther 353, 288–298. [DOI] [PubMed] [Google Scholar]
- 182. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez‐Álvarez C, Hernández‐Ramírez D, Bockenstedt PL, Liaw PC, Cabral AR et al. (2015) Antiphospholipid antibodies promote the release of neutrophil extracellular traps: a new mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol 67, 2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, Luo Y, Levitt B, Glogowska M, Chandra P et al. (2011) N‐α‐benzoyl‐N5‐(2‐chloro‐1‐iminoethyl)‐L‐ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen‐induced arthritis. J Immunol 186, 4396–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR & Kaplan MJ (2012) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 123, 2981–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT et al. (2014) Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 114, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M, Wang Y & Wagner DD (2015) PAD4‐deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 125, 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N & Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A & Olsson AK (2015) Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor‐bearing animals. Cancer Res 75, 2653–2662. [DOI] [PubMed] [Google Scholar]
- 189. Macanovic M, Sinicropi D, Shak S, Baughman S, Thiru S & Lachmann PJ (1996) The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice: studies with recombinant murine DNase and with dexamethasone. Clin Exp Immunol 106, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S, Dai M, Li Y, Li Q, Mao Z et al. (2018) Neutrophil extracellular traps contribute to the pathogenesis of acid‐aspiration‐induced ALI/ARDS. Oncotarget 9, 1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Sayah DM, Mallavia B, Liu F, Ortiz‐Muñoz G, Caudrillier A, Der Hovanessian A, Ross DJ, Lynch JP 3rd, Saggar R, Ardehali A et al. (2015) Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med 191, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Pottecher J. Inhaled Dornase Alpha to Reduce Respiratory Failure After Severe Trauma (Online). https://clinicaltrials.gov/ct2/show/NCT03368092 [10 December 2020].
- 193. Serhan CN (2014) Pro‐resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Serhan CN (2017) Treating inflammation and infection in the 21st century: new hints for decoding resolution mediators and mechanisms. FASEB J 31, 1273–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Serhan CN & Levy BD (2018) Resolvins in inflammation: emergence of the pro‐resolving superfamily of mediators. J Clin Invest 128, 2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Cherpokova D, Jouvene CC, Libreros S, De Roo EP, Chu L, de la Rosa X, Norris PC, Wagner DD & Serhan CN (2019) Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood 134, 1458–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Dalli J, Chiang N & Serhan CN (2015) Elucidation of novel 13‐series of resolvins that increase with atorvastatin and clear infections. Nat Med 21, 1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Dalli J & Serhan CN (2012) Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro‐resolving mediators. Blood 120, e60–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Norling LV, Spite M, Yang R, Flower RJ, Perretti M & Serhan CN (2011) Cutting edge: humanized nano‐proresolving medicines mimic inflammation‐resolution and enhance wound healing. J Immunol 186, 5543–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Tsuda S, Shinohara M, Oshita T, Nagao M, Tanaka N, Mori T, Hara T, Irino Y, Toh R, Ishida T et al. (2017) Novel mechanism of regulation of the 5‐lipoxygenase/leukotriene B4 pathway by high‐density lipoprotein in macrophages. Sci Rep 7, 12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Chiang N & Serhan CN (2017) Structural elucidation and physiologic functions of specialized pro‐resolving mediators and their receptors. Mol Aspects Med 58, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Serhan CN, Gupta SK, Perretti M, Godson C, Brennan E, Li Y, Soehnlein O, Shimizu T, Werz O, Chiurchiù V et al. (2020) The Atlas of Inflammation Resolution (AIR). Mol Aspects Med 74, 100894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Norling LV, Dalli J, Flower RJ, Serhan CN & Perretti M (2012) Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: receptor‐dependent actions. Arterioscler Thromb Vasc Biol 32, 1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN & Filep JG (2009) 15‐epi‐lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med 180, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. El Kebir D, Gjorstrup P & Filep JG (2012) Resolvin E1 promotes phagocytosis‐induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci USA 109, 14983–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA & Serhan CN (2012) Infection regulates pro‐resolving mediators that lower antibiotic requirements. Nature 484, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Korkmaz B, Moreau T & Gauthier F (2008) Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 90, 227–242. [DOI] [PubMed] [Google Scholar]
- 208. Tremblay GM, Jenelle MF & Bourbonnais Y (2003) Anti‐inflammatory activity of neutrophil elastase inhibitors. Curr Opin Investig Drugs 4, 556–565. [PubMed] [Google Scholar]
- 209. Guay C, Laviolette M & Tremblay GM (2006) Targeting serine proteases in asthma. Curr Top Med Chem 6, 393–402. [DOI] [PubMed] [Google Scholar]
- 210. Siedle B, Hrenn A & Merfort I (2007) Natural compounds as inhibitors of human neutrophil elastase. Planta Med 73, 401–420. [DOI] [PubMed] [Google Scholar]
- 211. Fitch PM, Roghaninan A, Howie SE & Sellenave JM (2006) Human neutrophil elastase inhibitors in innate and adaptive immunity. Biochem Soc Trans 34, 279–282. [DOI] [PubMed] [Google Scholar]
- 212. Crisford H, Sapey E & Stockley RA (2018) Proteinase 3; a potential target in chronic obstructive pulmonary disease and other chronic inflammatory diseases. Resp Res 19, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]