

SUMMARY
Sterol deficiency triggers SCAP-mediated SREBP activation, whereas hypernutrition together with ER-stress activate SREBP1/2 via caspase-2. Whether these pathways interact and how they are selectively activated by different dietary cues is unknown. Here we reveal regulatory crosstalk between the two pathways that controls the transition from hepatosteatosis to steatohepatitis. Hepatic ER-stress elicited by NASH-inducing diets activates IRE1 and induces expression of the PIDDosome subunits caspase-2, RAIDD and PIDD1, along with INSIG2, an inhibitor of SCAP-dependent SREBP activation. PIDDosome assembly activates caspase-2 and sustains IRE1 activation. PIDDosome ablation or IRE1 inhibition blunt steatohepatitis and diminish INSIG2 expression. Conversely, while inhibiting simple steatosis, SCAP ablation amplifies IRE1 and PIDDosome activation and liver damage in NASH-diet fed animals, effects linked to ER disruption and preventable by IRE1 inhibition. Thus, the PIDDosome and SCAP pathways antagonistically modulate nutrient-induced hepatic ER-stress to control non-linear transition from simple steatosis to hepatitis, a key step in NASH pathogenesis.
Keywords: Steatosis, steatohepatitis, NAFLD, NASH, SREBP, SCAP, Caspase-2, PIDDosome, ER stress, IRE1
eTOC
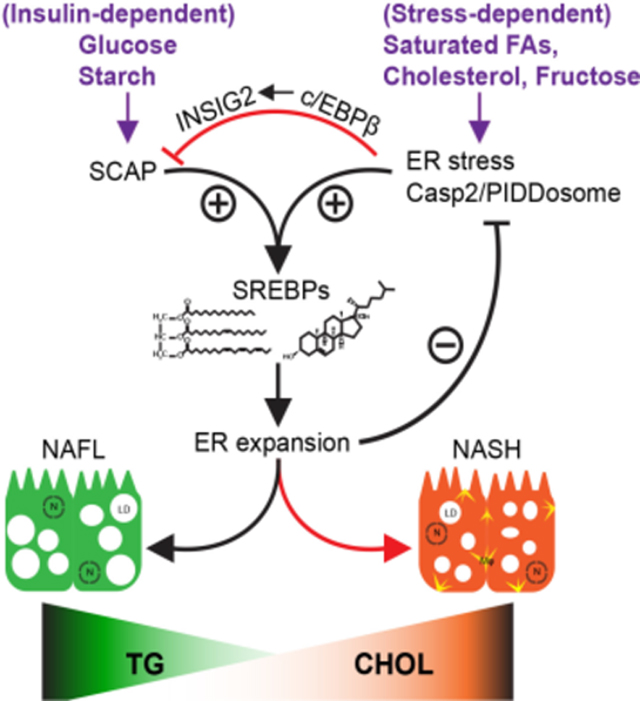
Kim et al. show that while diets rich in glucose lead to insulin dependent SREBP activation via the SCAP pathway, diets rich in the saturated FAs and fructose trigger ER stress and INSIG2 induction, which inhibits SCAP-dependent SREBP activation. Instead, ER stress results in Casp2/PIDDosome-dependent SREBP activation.
Graphical Abstract
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) includes a benign manifestation called NAFL and a more severe form, non-alcoholic steatohepatitis (NASH), which in addition to liver damage, fibrosis, cirrhosis, and cancer, is associated with increased cardiovascular disease risk (Francque et al., 2016; White et al., 2012). Both NAFL, which is often linked to insulin resistance is characterized by simple hepatosteatosis, and NASH, characterized by steatohepatitis (inflammation), liver damage and fibrosis, are strongly affected by alterations in lipid metabolism (Tilg and Moschen, 2010). But what controls the transition from simple steatosis to steatohepatitis remains unclear. Moreover, it is poorly understood why most people with NAFL never progress to NASH, a situation suggesting that the progression from simple steatosis to NASH is non-linear. Moreover, development of liver fibrosis is usually coupled to a decrease in liver fat (Powell et al., 1989; van der Poorten et al., 2013). Sterol response element binding proteins (SREBP) 1 and 2 are the master transcriptional regulators of triglyceride and cholesterol synthesis, respectively (Brown and Goldstein, 1997). By extension, the SREBPs are important for membrane biosynthesis and remodeling (Dobrosotskaya et al., 2002; Rong et al., 2017) and NAFL development (Moon et al., 2017). Both SREBPs, which are embedded in the ER membrane, are activated via a unique proteolytic pathway (Sakai et al., 1996). In sterol depleted cells, SREBP precursors are escorted by the ER resident chaperon SCAP to the Golgi apparatus, where they are sequentially cleaved by site 1 and site 2 proteases (S1P and S2P, respectively). This results in release of the SREBP cytoplasmic portions, which translocate to the nucleus and activate genes coding for lipid and cholesterol biosynthetic enzymes and transporters (Brown and Goldstein, 1997). In addition to sterol binding to SCAP, trafficking of SCAP:SREBP complexes to the Golgi apparatus is inhibited by two other ER embedded proteins: INSIG1 and INSIG2 (Yabe et al., 2002; Yang et al., 2002), whose expression is intricately regulated at the transcriptional and post-translational levels by insulin and other factors (Goldstein et al., 2006; Hwang et al., 2017; Wang et al., 2016; Yabe et al., 2002). Sterol deficiency leads to SCAP-mediated SREBP activation and hepatocyte-specific SCAP ablation prevents simple hepatosteatosis (Moon et al., 2012). But the combination of high fat diet (HFD) and ER-stress or excessive fructose intake trigger paradoxical SREBP activation and upregulate de novo lipogenesis (DNL) as well as liver damage, thereby leading to steatohepatitis (Nakagawa et al., 2014; Softic et al., 2018; Todoric et al., 2020). In ER-stress prone MUP-uPA mice, HFD feeding activates SREBP1 and 2 via a non-canonical, SCAP-independent, caspase-2 (Casp2)-mediated pathway (Kim et al., 2018). Whereas Casp2 mRNA transcription is induced by TNF, its translation is stimulated by the ER-stress responsive ribonuclease and protein kinase IRE1 (Kim et al., 2018), a major sensor of protein misfolding and ER membrane perturbation (Gardner and Walter, 2011). How Casp2 is enzymatically activated is not fully understood, but once activated, Casp2 leads to S1P cleavage and activation (Kim et al., 2018). Unlike the canonical SCAP-dependent pathway, the Casp2-S1P-SREBP pathway is not subject to feedback inhibition by sterols or excessive lipid intake. Moreover, Casp2 activation in HFD-fed MUP-uPA mice is accompanied by hepatosteatosis, liver damage and progression to steatohepatitis. Cleaved and activated Casp2 was detected in livers of people with NASH and alcoholic steatohepatitis (ASH) and found to contribute to ASH in mice (Ma et al., 2020). Although the Casp2 promoter contains SREBP binding sites and is activated on SREBP2 overexpression (Logette et al., 2005), it is unknown whether the SCAP- and Casp2-dependent SREBP activation pathways interact and modulate each other’s activity.
In cells harboring extra centrosomes or stressed by DNA damaging agents, Casp2 undergoes activation as part of a large multi-protein complex, the PIDDosome, which also contains the scaffold protein RAIDD/CRADD and the regulatory subunit PIDD1 (Burigotto et al., 2021; Fava et al., 2017). While PIDD1 expression can be induced in response to p53 activation, PIDDosome-associated active Casp2 stabilizes p53 through proteolytic cleavage of the p53 inhibitor MDM2 (Burigotto et al., 2021; Fava et al., 2017; Oliver et al., 2011). Although PIDDosome formation was originally implicated in the apoptotic death of DNA-damaged and ER-stressed cells, more recent data implicate Casp2 and the PIDDosome in control of cellular differentiation and polyploidization (Sladky and Villunger, 2020). Along this line, the PIDDosome has emerged as an important negative regulator of liver polyploidization during neonatal organogenesis and regeneration (Sladky et al., 2020). However, it is yet to be determined whether the PIDDosome accounts for Casp2 activation and hepatosteatosis in mice fed NASH-inducing diets. Moreover, it is unclear whether Casp2 and the PIDDosome control lipid metabolism in a hepatocyte autonomous manner. The latter is an important question, as whole body Casp2−/− mice do not gain weight on HFD and show elevated energy expenditure relative to wild type (WT) mice (Kim et al., 2018), suggesting extrahepatic sites of Casp2 action.
Here we show that Casp2 acts in hepatocytes to promote fructose-induced hepatosteatosis and that Raidd−/− and Pidd1−/− mice are equally protected from high fructose diet (HFrD)-induced hepatosteatosis and ER-stress. HFrD-induced PIDDosome activation is accompanied by accumulation of INSIG2, an inhibitor of SCAP-dependent SREBP activation. Conversely, SCAP restrains PIDDosome activation, which is strongly potentiated in HFrD-fed hepatocyte specific ScapΔHep knockout mice due to ER disruption and enhanced IRE1 activation, despite absence of hepatosteatosis. While dissociating steatosis from liver damage and fibrosis, these results uncover a previously unknown crosstalk between the two SREBP activation pathways, whose apparent role is adjusting hepatocyte homeostasis and ER health in response to nutritional perturbations.
RESULTS
Hepatocyte-specific Casp2 ablation prevents fructose-induced steatosis
To investigate Casp2’s role in hepatic lipid metabolism in the absence of the MUP-uPA transgene and circumvent the effect of Casp2 ablation on HFD-induced weight gain (Kim et al., 2018), we fed WT BL6 mice with either HFrD or a control cornstarch diet (CSD). In both diets, 70% of the caloric content is provided by carbohydrates, either cornstarch (CSD) alone or 60% fructose plus 10% cornstarch (HFrD). Previously, we found that HFrD feeding stimulates hepatic DNL and steatosis via an inflammation (TNF) dependent mechanism, whose inhibition did not perturb weight gain (Todoric et al., 2020). Likewise, weight gain or food consumption did not significantly differ between CSD- or HFrD-fed mice (Figure S1A). As expected, HFrD caused hepatosteatosis, hepatic cholesterol (Chol) and triglyceride (TG) accumulation (Figures S1B–C) and led to SREBP1/2 activation and increased expression of several of their target genes (Figure 1A). Moreover, HFrD consumption resulted in IRE1 activation (phosphorylation) and increased expression of Casp2 and its PIDDosome partners PIDD1 (which was proteolytically processed) and RAIDD (Figure 1B). HFrD feeding also induced eIF2α phosphorylation and increased BIP/Grp78, Grp94 and TNF mRNAs in liver extracts (Figures S1D–E), suggesting induction of ER-stress and modest inflammation. Although it was reported that acute fructose administration decreases hepatocellular ATP and leads to uric acid production (Abdelmalek et al., 2012), we did not observe a significant difference in ATP concentrations between CSD- and HFrD-fed livers (Figure S1F). HFrD feeding strongly induced INSIG2 and c/EBPβ/LAP proteins and mRNAs (Figure 1C). Previous studies showed that c/EBPβ activates Insig2 gene transcription (Jakobsen et al., 2013).
Figure 1. Hepatocyte-specific Casp2 ablation prevents fructose induced steatosis.
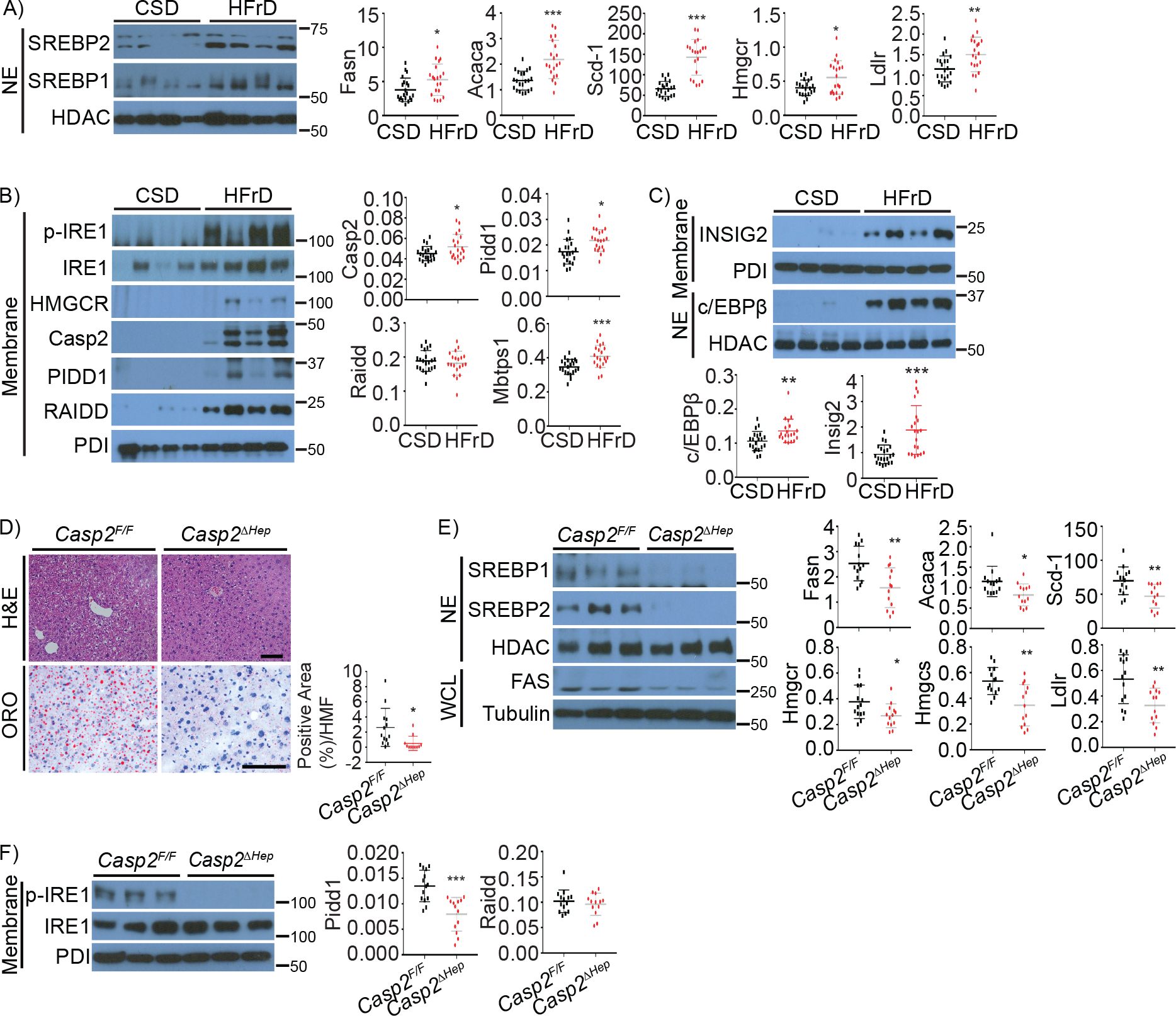
A-C: WT BL6 mice were fed cornstarch (CSD, n=21) or high fructose (HFrD, n=20) diets for 12 weeks and analyzed as indicated.
A. Nuclear SREBP1 and 2 and relative target mRNA amounts.
B. Representative immunoblots (IB) of phosphorylated IRE1 (p-IRE1), HMGCR, and PIDDosome components. The corresponding mRNAs were quantitated by Q-RT-PCR and their relative amounts are shown on the right.
C. Representative IB analysis (top) and mRNA quantification of INSIG2 and c/EBPβ (bottom).
D-F: Casp2 floxed (Casp2F/F, n=14) and liver specific Casp2 knockout (Casp2ΔHep, n=13) mice were fed HFrD for 12 weeks and analyzed.
D. Representative hematoxylin and eosin (H&E) stained formalin fixed paraffin embedded (FFPE) liver sections and oil red O (ORO) stained frozen liver sections. Four high magnification fields (HMF) were taken from each liver and quantified by Image J software. Quantification is on the right.
E. IB of nuclear SREBPs and FAS in whole tissue lysates (left), and relative amounts of lipogenic enzyme mRNAs (right).
F. IRE1 phosphorylation (left) and relative PIDDosome component mRNAs (right).
Results are mean ± SEM. Scale bar, 100 μm. Statistical significance was determined by two-tailed Student’s t test. *p < 0.05, **p < 0.005, ***p < 0.001.
We generated hepatocyte specific Casp2ΔHep knockout mice by crossing Casp2F/F mice with Alb-Cre mice (Figure S1G) and found them to be resistant to HFrD-induced hepatosteatosis (Figure 1D) although their weight gain and food consumption were not altered (Figure S1H). HFrD-fed Casp2ΔHep mice exhibited substantially reduced nuclear SREBP1/2 proteins and lower expression of SREBP target genes and fatty acid synthase (FAS) protein (Figure 1E). Casp2 ablation inhibited IRE1 activation, Pidd1 mRNA induction (Figure 1F), as well as inflammation- and fibrosis-related cytokines and markers (Figure S1I). These results suggest that inhibition of hepatosteatosis prevents fructose-induced ER-stress and that stress-mediated Casp2 activation enhances ER-stress and IRE1 activation.
PIDDosome components are required for fructose-induced hepatosteatosis
Given upregulation of PIDDosome components in HFrD-fed livers and their downregulation on hepatocyte-specific Casp2 ablation, we examined participation of PIDD1 and RAIDD in HFrD-induced hepatosteatosis. We placed WT, Casp2−/−, Pidd1−/− and Raidd−/− mice on CSD and HFrD as above. Ablation of any PIDDosome component prevented fructose-induced hepatosteatosis (Figure 2A), without an effect on HFrD-induced weight gain and food consumption (Figure S2A). Similar results were obtained at two different mouse facilities (UCSD and Medical University of Innsbruck). Consistent with reduced hepatosteatosis, the Casp2, PIDD1 and RAIDD deficiencies also reduced serum and liver Chol and liver TG (Figure 2B) and inhibited SREBP1/2 activation and target gene expression (Figure 2C). Curiously, however, Pidd1 and Raidd, but not Casp2, ablations resulted in a small increase in nuclear SREBP1/2 in CSD-fed mice (Figure 2D), an effect that could be related to reduced INSIG2 expression and the high carbohydrate content of CSD. However, the increase in nuclear SREBP1/2 was too modest to result in elevated target gene expression relative to CSD-fed WT mice. Nonetheless, in WT BL6 mice CSD feeding resulted in modest hepatosteatosis, along with decreased Insig1 mRNA and increased SCAP and SREBP regulated mRNAs, relative to normal chow diet (NCD) (Figures S2B–C). All PIDDosome component ablations prevented IRE1 activation and each of them blocked the HFrD-induced expression of the two other subunits (Figure 2D). These results support the notion that PIDDosome activation enhances HFrD-induced IRE1 activation via a positive autoregulatory loop that may be propagated through hepatocyte ER-stress. Since a diet in which 60% of the calories are supplied by fructose is not representative of human fructose consumption, we placed WT and Casp2−/− mice on Western diet enriched in fat and cholesterol plus water containing 15% fructose and 15% glucose for 10 weeks. Casp2 ablation slightly reduced BW gain in these mice (Figure S2D) and prevented liver damage, TG accumulation, and induction of lipogenic, inflammatory and fibrogenic genes (Figures S2E–H).
Figure 2. PIDDosome components are required for fructose induced hepatosteatosis.
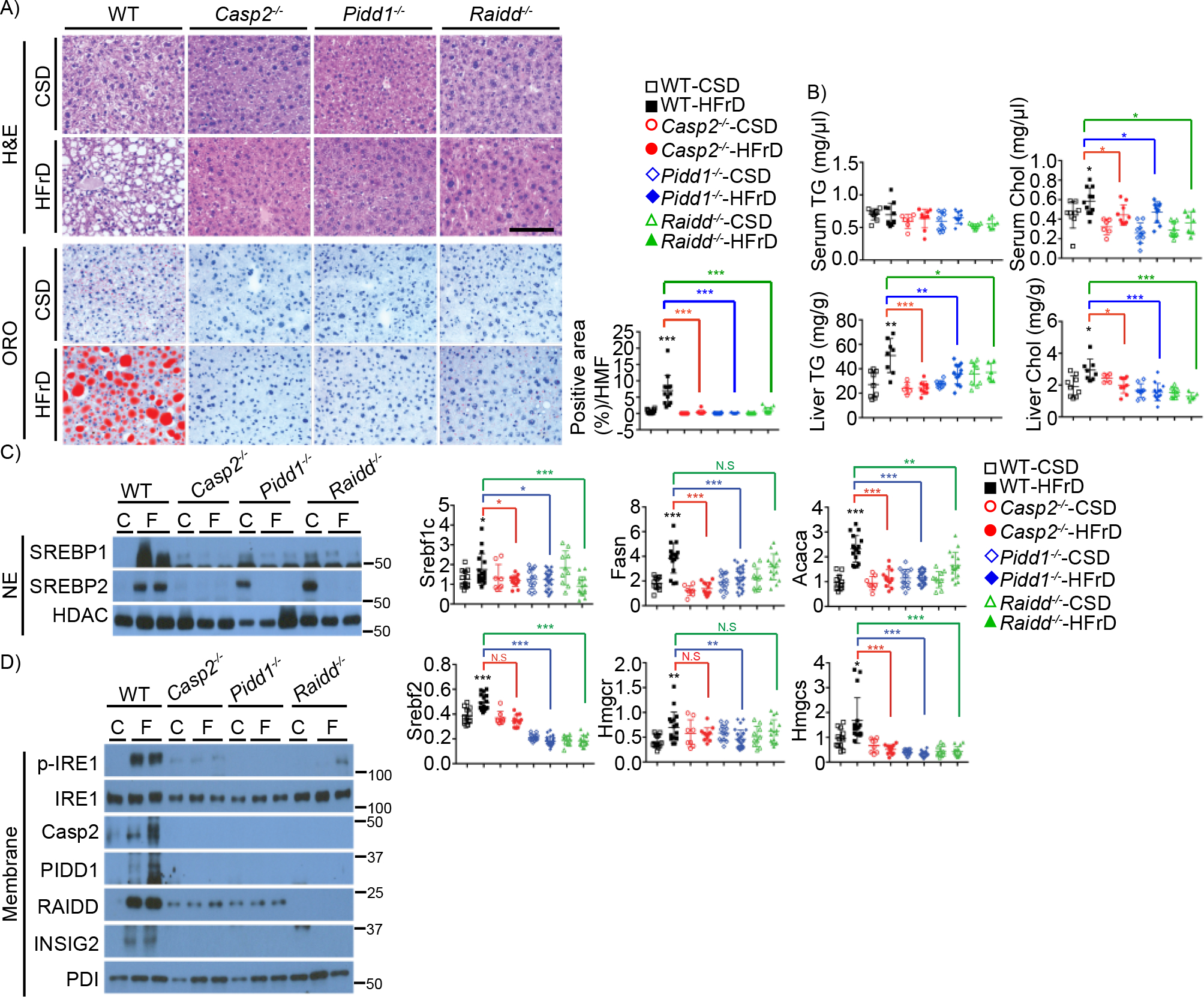
A. WT, Casp2−/−, Pidd1−/−, and Raidd−/− mice were fed CSD or HFrD for 12 weeks and liver sections from indicated mice were H&E and ORO stained. Four HMFs were taken from each section and ORO-positive areas were quantitated and shown on the right.
B. Serum and liver triglyceride (TG) and cholesterol (Chol) from above mice (Serum: WT-CSD: n=9, WT-HFrD: n=11, Casp2−/−-CSD: n=6, Casp2−/−-HFrD: n=10, Pidd1−/−-CSD: n=10, Pidd1−/−-HFrD: n=10, Raidd−/−-CSD: n=9, Raidd−/−-HFrD: n=7; Liver: WT-CSD: n=9, WT-HFrD: n=8, Casp2−/−-CSD: n=5, Casp2−/−-HFrD: n=9, Pidd1−/−-CSD: n=9, Pidd1−/−-HFrD: n=13, Raidd−/−-CSD: n=9, Raidd−/−-HFrD: n=6).
C. Representative IB analysis of nuclear SREBPs (left) and relative lipogenic enzyme mRNAs (right) in livers of indicated mice. (WT-CSD: n=13, WT-HFrD: n=17, Casp2−/−-CSD: n=8, Casp2−/−-HFrD: n=13, Pidd1−/−-CSD: n=14, Pidd1−/−-HFrD: n=27, Raidd−/−-CSD: n=10, Raidd−/−-HFrD: n=16).
D. IB analysis of indicated proteins in the above mouse livers.
Scale bar, 100 μm. Positive areas were quantitated using Image J software. Statistical significance was evaluated by one way ANOVA and Tuckey’s multiple comparison. *p < 0.05, **p < 0.005, ***p < 0.001. C-CSD, F-HFrD.
PIDDosome components control fructose-induced lipid metabolizing genes
To investigate how the PIDDosome affects the hepatic transcriptome, we performed unbiased sequence analysis of RNA extracted from CSD or HFrD fed WT, and HFrD fed Casp2−/−, Pidd1−/−, and Raidd−/− livers. Principal Component Analysis (PCA) showed that the Casp2−/−, Pidd1−/−, and Raidd−/− liver transcriptomes were clearly distinct from the WT liver transcriptome, even though the Pidd1−/− transcriptome was not as closely related to the Casp2−/− and Raidd−/− transcriptomes as they were to each other (Figure 3A). By comparing HFrD-fed WT livers to HFrD-fed Casp2−/−, Pidd1−/−, and Raidd−/− livers, we identified 1,500–2,800 transcripts that were differentially expressed in significant and robust manners (FDR < 0.05, Fold change > |1.5|) and visualized the transcriptomic alterations with heatmaps (Figure S3A; Table S1). Among PIDDosome-dependent transcripts that were upregulated in HFrD fed livers, we found that genes involved in fatty-acid metabolism and cholesterol synthesis were significantly reduced by PIDDosome component ablation (Figure 3B). We analyzed the altered pathways in these livers using EnrichR tools and found that gene sets regulating adipogenesis, fatty acid metabolism, oxidative phosphorylation, mTORC1 pathway and protein secretion were the most significantly downregulated by Casp2, Pidd1 or Raidd gene ablations (Figure 3C). Consistent with increased ER-stress, the UPR pathway was upregulated upon HFrD feeding in the WT liver and down-regulated in the Casp2−/− and Pidd1−/− livers (Figure S3B). Thus, the results of the unbiased RNAseq analysis were in general agreement with those of the targeted analyses depicted above, showing alterations in gene expression that correlate with SREBP and IRE1 activation.
Figure 3. PIDDosome components control fructose induced lipid metabolizing genes.
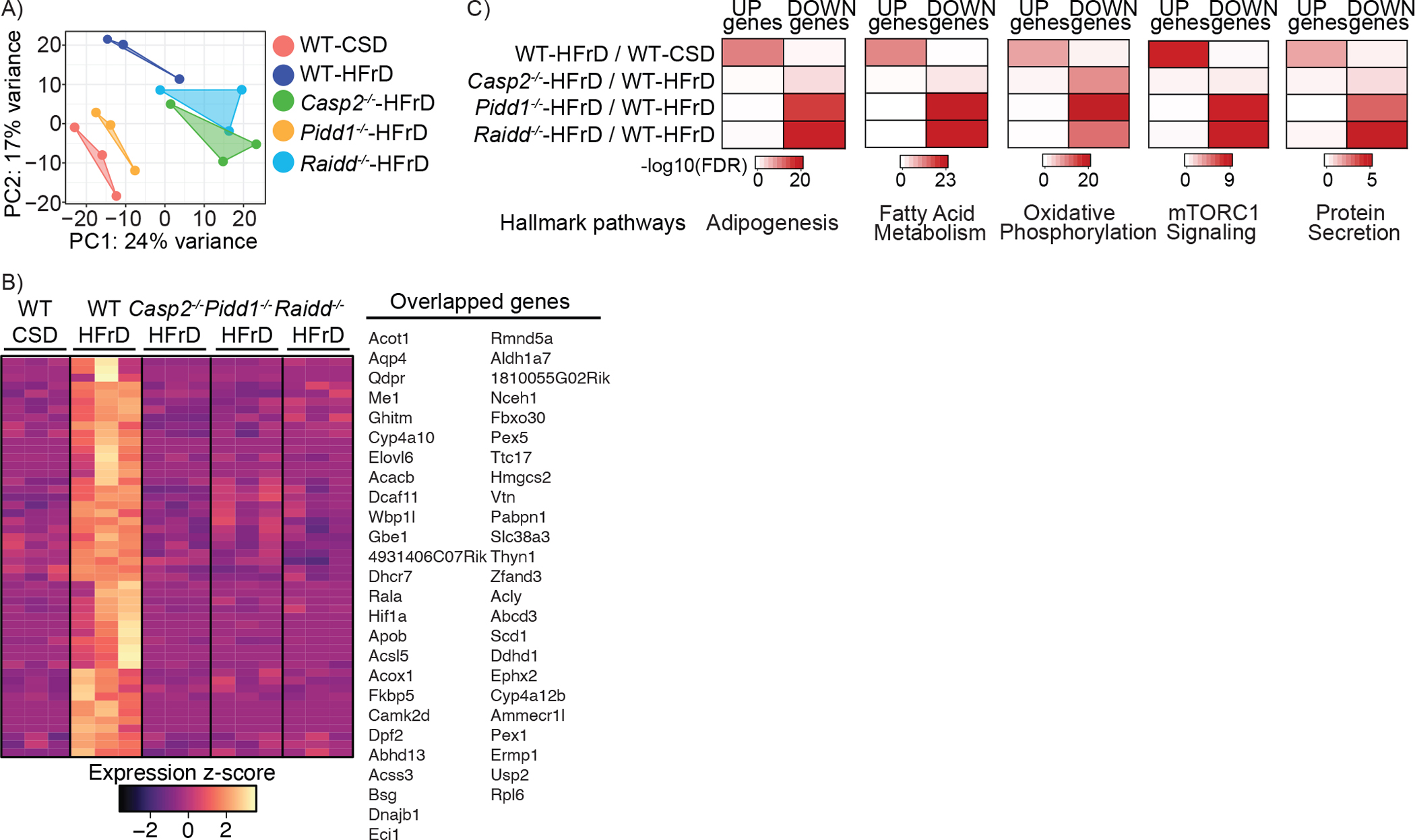
WT, Casp2−/−, Pidd1−/−, and Raidd−/− mice were fed CSD or HFrD for 12 weeks. Total liver RNA was extracted and subjected to whole genome RNA-seq analysis.
A. Principal Component Analysis (PCA) plot showing 5 groups with 3 biological replicates each. The PCA captured the variation of the samples among 5 groups, showing PC1 and PC2 scores. Each group is shown with geom_polygon function of ggplot2.
B. Venn diagram showing overlapping genes from 4 comparative analyses. Selected overlapping genes are listed on the right.
C. Heatmap showing significantly altered genes among the 5 groups. Overlapping pathways among 4 comparisons (n=50) were visualized in z-score scale.
PIDD1 and RAIDD are needed for Casp2 activation
Co-immunoprecipitation experiments confirmed that co-transfection of Casp2, PIDD1 and RAIDD into HEK293 cells resulted in PIDDosome formation (Figures S4A–B). Previously, we showed that co-transfection of Casp2, S1P and SREBP2 expression vectors into HEK293 cells results in S1P proteolytic cleavage at a Casp2 consensus site and SCAP-independent SREBP2 activation that is refractory to feedback inhibition (Kim et al., 2018). To determine whether this response depends on endogenous PIDD1 and RAIDD, we ablated PIDD1 and RAIDD by CRISPR-Cas9 gene editing. As opposed to HEK293 cells expressing all PIDDosome components, no SREBP2 activation took place in PIDD1Δ or RAIDDΔ cells co-transfected with Casp2, S1P and SREBP2 expression vectors (Figure 4A). Restoration of PIDD1 expression in PIDD1Δ cells restored S1P cleavage and SREBP2 activation (Figures 4B and S4C). Similar results were obtained after restoration of RAIDD expression in RAIDDΔ cells (Figures 4C and S4D). PIDD1 binds RAIDD through its death domain (DD) (Tinel et al., 2007). We generated DD deficient PIDD1 (PIDD1ΔDD) (Figure S4E), which failed to restore SREBP2 activation in PIDD1Δ cells (Figure S4F). As previously observed (Kim et al., 2018), addition of 25-OH-cholesterol (25-HC) to HEK293 cells transfected with the SREBP2 reporter, SCAP and S1P inhibited SREBP2 activation but had no effect on its SCAP-independent activation by the PIDDosome (Figure 4D).
Figure 4. PIDD1 and RAIDD are needed for Casp2 activation.
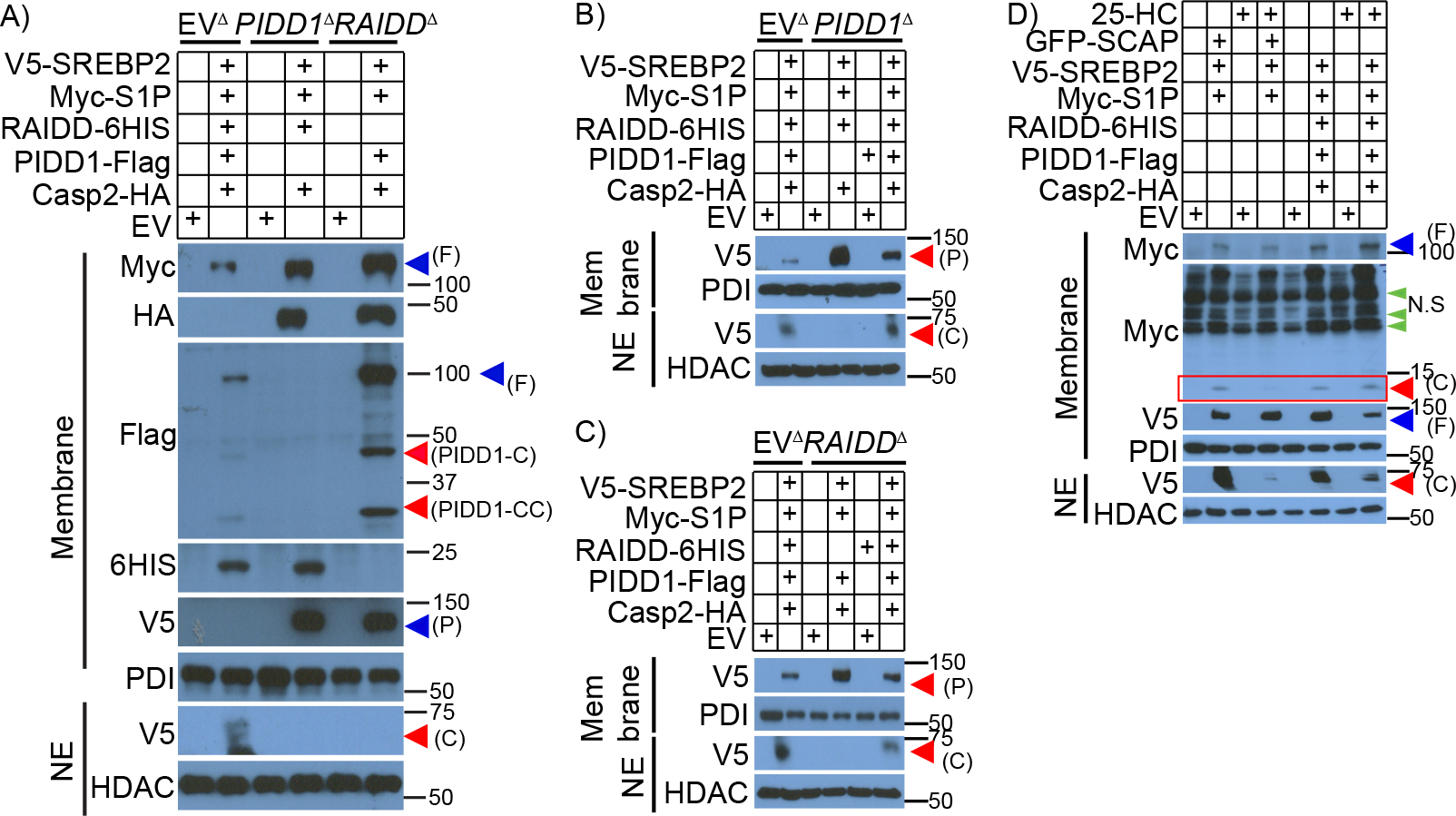
A-C. Parental (EVΔ), PIDD1- (PIDD1Δ) or RAIDD-ablated (RAIDDΔ) HEK293 cells were transfected with plasmids encoding epitope-tagged PIDDosome components, S1P, and SREBP2 as indicated and cultured for 16 hrs. Membrane and nuclear fractions were isolated and the indicated proteins were IB analyzed.
D. HEK293 cells were transfected with the indicated plasmids and supplemented with 25-hydroxycholesterol (25-HC, 1 μg/ml) for 16 hrs. Indicated proteins were IB analyzed. Cleaved S1P is shown in the red box.
Each experiment was repeated at least 3 times and representative results are shown. F-Full length, C-Cleaved, P-precursor, N.S-non-specific, NE-nuclear extract.
IRE1 inhibition prevents fructose induced PIDDosome and SREBP activation.
Treatment of HFD-fed MUP-uPA mice with the IRE1 ribonuclease inhibitor MKC3946 blocked Casp2 induction and prevented SREBP activation and NASH development (Kim et al., 2018). To determine whether IRE1 also plays a role in fructose-induced hepatosteatosis, PIDDosome induction and SREBP activation, we treated HFrD-fed WT mice with MKC3946 every two days for 4 weeks. MKC3946 treatment inhibited hepatosteatosis and fructose-induced expression of PIDDosome subunits (Figures 5A–B). MKC3946 also inhibited SREBP1/2 activation and target gene expression (Figure 5C), and reduced liver TG and Chol in HFrD-fed mice (Figure 5D) without an effect on BW, food intake, and serum insulin (Figures S5A–B). c/EBPβ and INSIG2 induction (Figures 5E and S5C) were also inhibited by MKC3946, which reduced expression of inflammatory and fibrogenic markers (Figure S5D). These results confirm the importance of IRE1 for fructose induced PIDDosome activation, hepatosteatosis, and c/EBPβ and INSIG2 induction.
Figure 5. IRE1 inhibition prevents PIDDosome and SREBP activation.
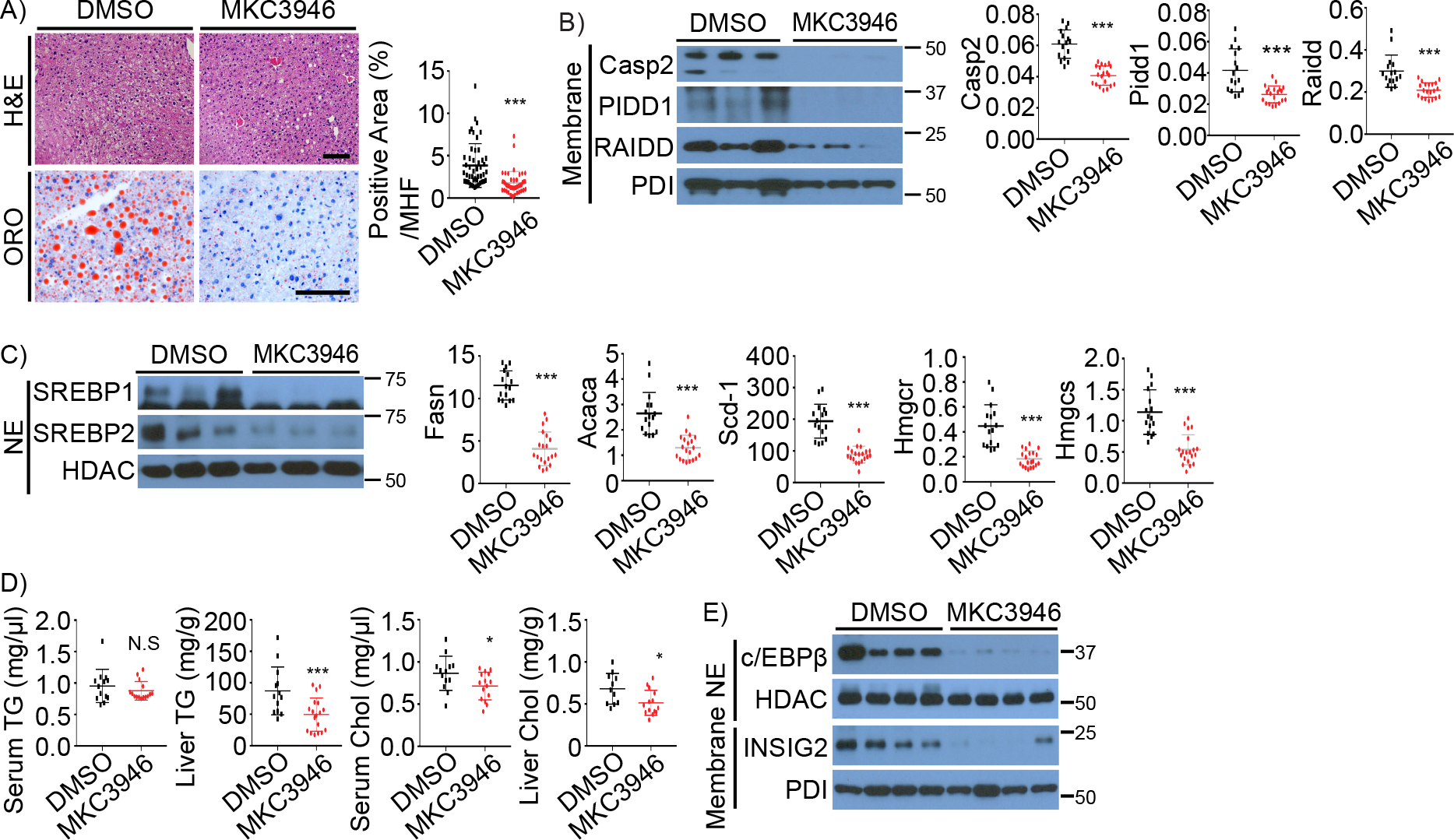
WT mice were fed HFrD for 8 weeks, followed by DMSO (n=16) or MKC3946 (3 mg/kg, n=20) treatments every two days for 4 weeks.
A. H&E and ORO staining of liver sections. Quantification is shown on the right.
B, C. The indicated proteins in liver lysates of above mice were IB analyzed (left). PIDDosome components (B) and relative lipogenic enzyme mRNAs (C) were quantitated and the results shown on the right.
D. Serum and liver TG and Chol in above mice.
E. IB of c/EBPβ and INSIG2 in liver lysates of above mice.
Results are mean ± SEM. Scale bar, 100 μm. Statistical significance was performed by two-tailed Student’s t test. *p < 0.05, **p < 0.005, ***p < 0.001. N.E-nuclear extract.
The PIDDosome regulatory subunit, PIDD1, is p53-inducible (Lin et al., 2000). We therefore examined the effect of hepatocyte-specific p53 ablation on fructose-induced steatosis. However, HFrD feeding did not induce p53 and p53 ablation did not prevent fructose-induced hepatosteatosis (Figure S5E).
Scap ablation potentiates fructose-induced ER stress and PIDDosome activation
To further exclude SCAP involvement in stress and Casp2 mediated SREBP activation, we crossed MUP-uPA mice with ScapF/F and ScapΔHep mice and fed the compound mutants with HFD as before (Kim et al., 2018). SCAP deficiency in MUP-uPA mice did not prevent HFD-induced hepatic inflammation and damage and further enhanced liver fibrosis, despite a modest decrease in hepatosteatosis (Figure S6A). Enhanced fibrosis most likely reflects increased liver damage. Scap ablation in the MUP-uPA liver also augmented IRE1 activation and Casp2 and PIDD1 expression (Figures S6B–C). Scap ablation, which resulted in disappearance of the precursor forms of SREBP1/2 (Figure 6A), resulted in almost complete absence of hepatosteatosis in WT mice given HFD plus 30% fructose water (HFHFD) or HFD (Figures S6D–F), consistent with a previous report by Horton and co-workers (Moon et al., 2012). HFrD-fed ScapΔHep mice were also showed a decrease in circulating and liver TG and circulating, but not liver, Chol, as well as greatly reduced ACC1 and FAS expression (Figures S6G–H). Rather surprisingly, ScapΔHep mice given either HFrD or HFHFD exhibited clear signs of liver damage with areas of cytoplasmic clearing in the periportal region, anisonucleosis, apoptosis, and fibrosis (Figures 6B, S6E, Table S2). Consistent with these results, HFrD or HFHFD feeding of ScapΔHep mice markedly increased liver IRE1 activation/phosphorylation, Casp2, PIDD1, S1P(Mbtps1) mRNA expression and protein cleavage and eIF2α phosphorylation (Figures 6C–D, S6I–K). Correlating with HFrD-induced liver damage and eIF2α phosphorylation, RNA-seq analysis revealed that the mTORC1 pathway was significantly suppressed along with the cholesterol metabolic pathway, but inflammatory response, ROS, and apoptosis were the top upregulated pathways in the HFrD-fed ScapΔHep liver (Figure 6E), implying extensive cell damage which was probably augmented by p-eIF2α related inhibition of protein synthesis.
Figure 6. Scap ablation potentiates fructose-induced ER stress and PIDDosome activation.
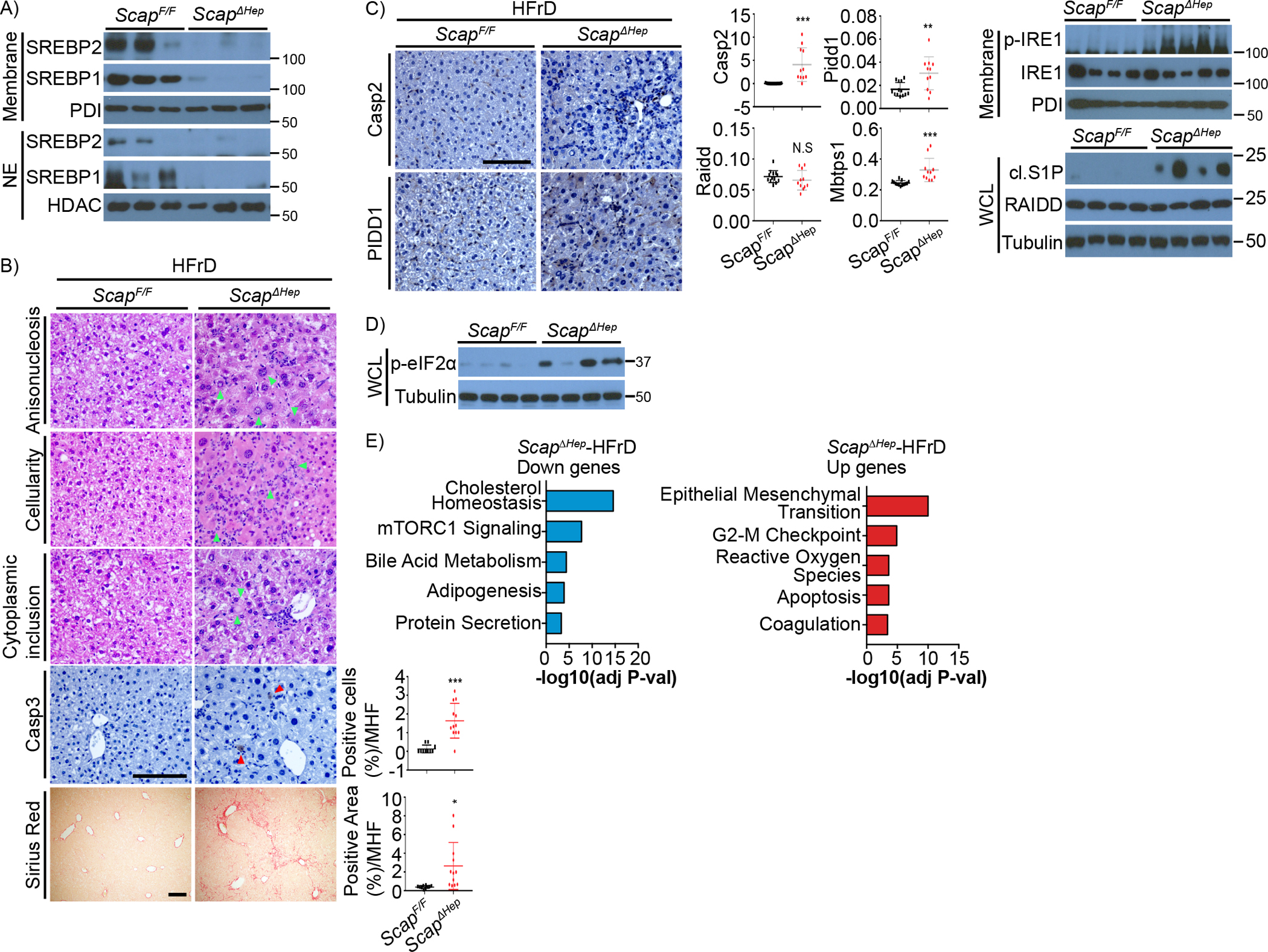
ScapF/F (n=13) and ScapΔhep (n=11) mice were fed HFrD for 12 weeks.
A. IB analysis of precursor and cleaved SREBP1/2 in above livers.
B. Liver sections from above mice were stained with H&E, Sirius red, and cleaved Casp3 antibody. Anisonucleosis, portal cellularity, cytoplasmic inclusion, indicated by light green arrowheads, and Casp3-positive hepatocytes, indicated by red arrowheads, and fibrosis were visualized in above livers. Quantification is shown on the right.
C. IHC analysis of Casp2 and PIDD1 in livers of above mice and relative mRNA expression of PIDDosome components and S1P (MBTPS1) are shown on the right. IB analysis of phospho-IRE1 and cleaved (cl.) S1P is at the bottom.
D. eIF2α phosphorylation in livers of above mice.
E. Hallmark pathways from Enrich analysis using up- or down-regulated genes in HFrD-ScapΔhep compared to HFrD-ScapF/F livers with 3 biological replicates each.
Results are mean ± SEM. Scale bar, 100 μm. Statistical significance was performed by two-tailed Student’s t test. *p < 0.05, **p < 0.005, ***p < 0.001.
To investigate the role of IRE1 activation in HFrD-induced damage to the ScapΔHep liver, we treated the mice with MKC3946, which reversed much of the observed damage, including hepatocyte death and anisonucleosis (Figure 7A). MKC3946 also inhibited HFrD-induced IRE1 and eIF2α phosphorylation, as well as Casp2 and PIDD1 expression (Figure 7B). Interestingly, IRE1 inhibition restored SREBP1c expression and activation (Figure 7C), although SREBP2 and its targets remained suppressed (Figure S7A). IRE1 inhibition also resulted in normalization of the rough ER (RER), which was distended and disrupted in the HFrD-fed SCAP-deficient liver (Figure 7D).
Figure 7. IRE1 inhibition alleviates ER stress and damage in the HFrD-fed ScapΔhep liver.
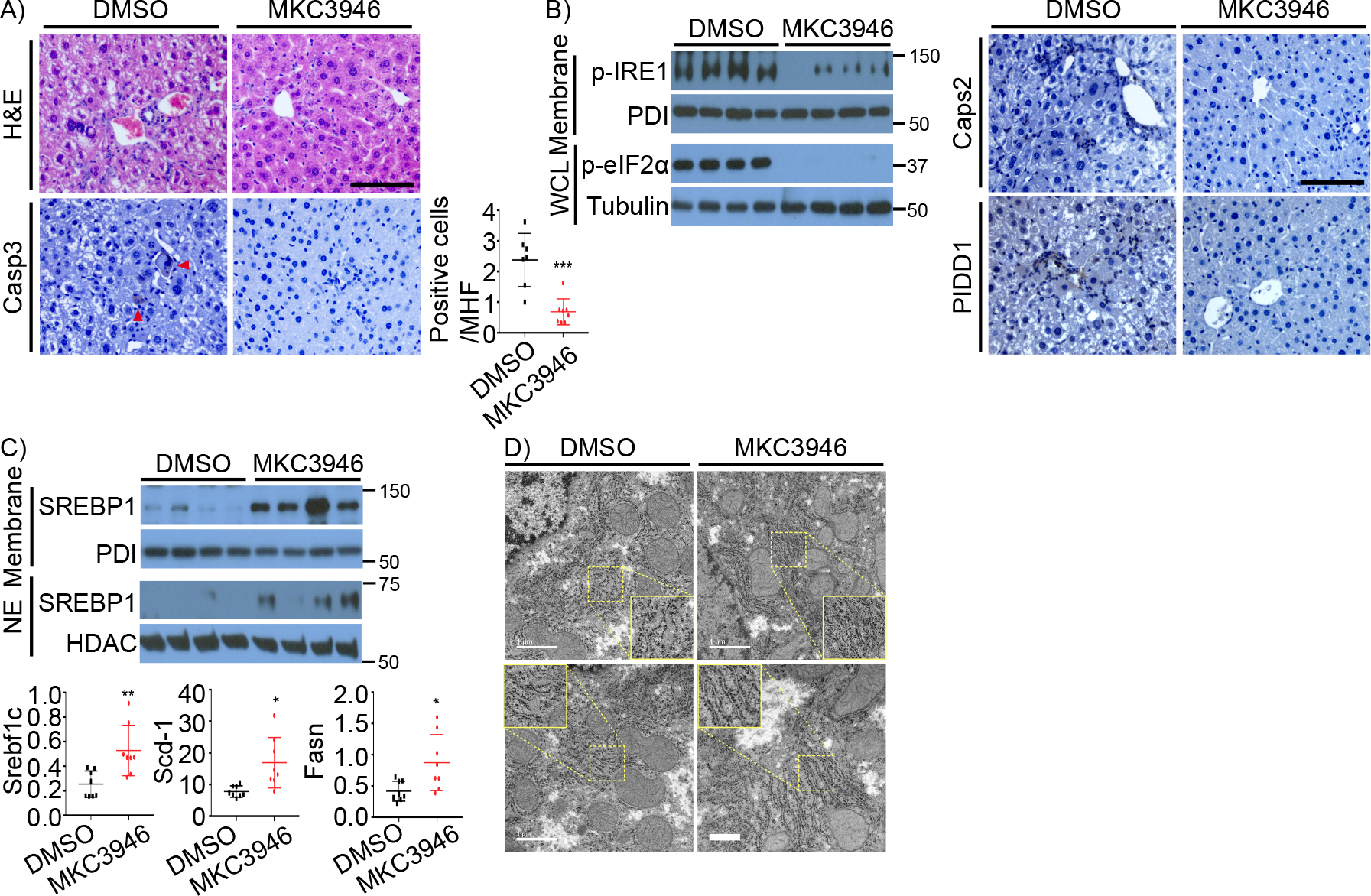
ScapΔhep mice were fed HFrD for 8 weeks, followed by DMSO (n=8) or MKC3946 (3 mg/kg, n=8) treatment every two days for 4 weeks.
A. H&E and Casp3 staining of FFPE liver sections. Quantification of cleaved Casp3-positive hepatocytes is on the right.
B. B of ER stress markers (left) and Casp2 and PIDD IHC (right) of above livers.
C. IB analysis of SREBP1 (left) and mRNA of lipogenic enzymes (right) in above livers.
D. EM images of above livers. Disrupted and well-organized RER are highlighted and enlarged in the dotted yellow boxes.
Results are mean ± SEM. Scale bar in H&E image, 100 μm. Scale bar in EM images, 1000 nm. Statistical significance was performed by two-tailed Student’s t test. *p < 0.05, **p < 0.005, ***p < 0.001.
DISCUSSION
Why certain high energy diets, for instance glucose-rich diets, only cause benign steatosis, whereas fructose (Febbraio and Karin, 2021) or cholesterol (Van Rooyen et al., 2011) rich diets induce NASH, is not entirely clear. Our results illustrate the existence of a previously unrecognized regulatory system that maintains liver homeostasis, ER membrane integrity and controls the transition from simple steatosis to hepatitis and fibrosis by adjusting fatty acid and cholesterol metabolism according to dietary cues. This system is composed of two mutually exclusive SREBP activation pathways: the canonical SCAP-dependent activation pathway (Goldstein et al., 2006) and the more recently identified stress-activated Casp2 pathway (Kim et al., 2018). The SCAP-dependent pathway is turned on in response to sterol deficiency, caused by insufficient dietary uptake. In sterol deplete cells, SCAP binds SREBP1/2 in the ER and chaperons them to the Golgi apparatus (Brown et al., 2018) through an interaction with COPII on the surface of coated vesicles (Lee et al., 2020). Cholesterol binding to SCAP disrupts the interaction with COPII and at high cholesterol concentrations that exceed 4–5% of total cellular lipids, SCAP undergoes a further conformational change that enhances binding to INSIG proteins, which anchor the SREBP:SCAP complex in the ER to prevent SREBP cleavage by S1P and S2P in the Golgi apparatus (Goldstein et al., 2006). The major function of this pathway under non-stressed conditions is to maintain sterol homeostasis and prevent excessive accumulation of cholesterol in biological membranes, which can cause membrane malfunction due to decreased fluidity (Cooper, 1978). The feedback-regulated SCAP-activated pathway also prevents accumulation of triglycerides and toxic cholesterol metabolites (Afonso et al., 2018). Paradoxically, however, SREBP1c, which is predominantly expressed in hepatocytes, is also activated by insulin, which increases its transcription and decreases INSIG2 expression, contributing to insulin-induced lipogenesis (Matsuzaka and Shimano, 2013). Pathological cholesterol buildup in the ER due to excessive dietary intake or alcohol abuse triggers ER-stress and abnormal SREBP activation, instead of inhibition (Mandl et al., 2013). The mechanism by which ER-stress downstream to cholesterol overload causes SREBP activation cannot be SCAP mediated, because SCAP-dependent SREBP activation is inhibited by sterols. Moreover, activation of ATF6, which like SREBP1/2 depends on S1P and S2P mediated cleavage, is SCAP independent (Ye et al., 2000). Indeed, ER-stress driven SREBP activation proceeds via the Casp2 pathway, which is activated by TNF and IRE1 (Kim et al., 2018). IRE1 is an ER resident protein kinase and a nuclease that undergoes self-activation in response to membrane perturbations (Promlek et al., 2011). Here we show that in addition to inducing Casp2 and PIDDosome expression and activation, IRE1 leads to induction of INSIG2, an inhibitor of SCAP-dependent SREBP activation (Yabe et al., 2002). INSIG2 induction correlates with activation of c/EBPβ, previously shown to induce Insig2 transcription (Jakobsen et al., 2013). Thus, while activating the Casp2 pathway, IRE1 inhibits the SCAP pathway, which promotes a switch from simple steatosis to liver damage, hepatitis and fibrosis.
Chronic ER-stress, PIDDosome induction and Casp2 activation are linked to NASH and ASH development in mouse models and were observed in human liver specimens (Kim et al., 2018; Ma et al., 2020). Probably, the most detrimental outcome of Casp2-mediated SREBP activation is accumulation of free cholesterol, which differentiates simple hepatosteatosis from NASH (Arguello et al., 2015; Puri et al., 2007) and greatly potentiates TNF-induced hepatocyte death (Marí et al., 2006). Hepatocyte death is directly related to liver fibrosis, a hallmark of NASH. On the other hand, NAFL, in which no liver damage or inflammation are observed, is often linked to type 2 diabetes (T2D) (Targher et al., 2021). Likewise, in ob/ob mice T2D only leads to simple steatosis, which depends on SCAP-mediated SREBP activation, rather than NASH (Moon et al., 2012). SCAP-dependent SREBP1 activation may protect against cholesterol toxicity by stimulating DNL and increasing production of fatty acids that combine with cholesterol to form non-toxic cholesterol esters that are sequestered within lipid droplets.
IRE1, PIDDosome and Casp2 activation
Elevated Casp2 expression in HFD-fed MUP-uPA mice depends on IRE1 (Kim et al., 2018), whose ribonuclease activity enhances Casp2 mRNA translation through cleavage of micro RNAs (Upton et al., 2012), including miR-34a (Li et al., 2019). Here we show that IRE1 activity is also needed for Casp2 and PIDDosome induction in HFrD-fed WT mice. However, how increased Casp2 protein expression results in its activation was heretofore unknown. The results shown above indicate that Casp2-mediated S1P cleavage and SREBP2 activation in HEK293 cells require PIDDosome assembly, which triggers Casp2 auto-activation via induced proximity (Sladky and Villunger, 2020). PIDDosome assembly is triggered in response to p53-mediated PIDD1 induction (Sladky and Villunger, 2020), but p53 does not account for PIDDosome induction in HFrD-fed mice. Sustained PIDDosome expression under diet-induced ER-stress depends on its assembly, as ablation of any PIDDosome subunit suppresses expression of the other two subunits. Moreover, PIDDosome induction sustains IRE1 activation, perhaps by increasing cholesterol synthesis and cholesterol-induced ER membrane perturbations.
SCAP as an inhibitor of diet induced ER-stress
As previously reported (Moon et al., 2012), hepatocyte specific Scap ablation protects WT mice from HFD-induced hepatosteatosis. However, in HFD-fed MUP-uPA mice, SCAP depletion did not prevent NASH development and actually enhanced liver fibrosis despite inhibition of hepatosteatosis. These results show a disconnect between hepatosteatosis and NASH, with the latter being more dependent on liver damage than on TG accumulation. Indeed, it has been observed that as NASH livers become more fibrotic their lipid content declines (Powell et al., 1989; van der Poorten et al., 2013). Even more dramatic increases in hepatocyte apoptosis, liver damage and fibrosis were seen in HFrD- or HFHFD-fed ScapΔHep mice. Although excessive fructose intake results in intestinal and hepatic ER-stress (Febbraio and Karin, 2021; Todoric et al., 2020), in HFrD-fed WT mice hepatic ER-stress is modest and does not result in liver damage and fibrosis. Moreover, the amount of fructose is substantially lower in HFHFD, which still leads to liver damage in ScapΔHep mice. A remarkable feature of ScapΔHep mice is the absence of precursor and nuclear SREBP1/2, previously reported by Horton and colleagues (Moon et al., 2012). The absence of SREBP precursors was accompanied by enhanced IRE1 activation and eIF2α phosphorylation in both HFrD- and HFHFD-fed ScapΔHep mice, as well as HFD-fed ScapΔHep/MUP-uPA mice. We postulate that the absence of SCAP results in SREBP deficiency and potentiation of ER-stress by causing the misfolding of SREBP1/2 and other SCAP clients. Misfolded membrane proteins are often eliminated by the ER-associated degradation (ERAD) machinery, whose expression is induced by IRE1 (Hampton, 2003; Hetz et al., 2020). Indeed, the yeast SREBP homolog Sre1 is degraded via ERAD in the absence of the SCAP homolog Scp1 (Hughes et al., 2009). Moreover, IRE1 inhibition with MKC3946 restored SREBP1 expression and activation and RER membrane integrity. Further and unbiased evidence for fructose-induced ER stress and liver damage in ScapΔHep mice is provided by the RNA-seq analysis, which showed marked downregulation of genes related to cholesterol homeostasis and mTORC1 signaling and upregulation of the inflammatory response, ROS signaling and apoptosis.
Given its important role in SREBP activation, SCAP has been considered as a target for obesity treatment. One agent used to inhibit SCAP function is fatostatin, which interferes with SCAP dissociation from INSIGs and reduces lipogenesis and steatosis in obese mice (Kamisuki et al., 2009). Although fatostatin is a general inhibitor of ER-to-Golgi transport (Shao et al., 2016) and not a specific inhibitor of SCAP, it may prevent early NAFL driven by TG accumulation. Nonetheless, our results suggest that fatostatin and other SCAP inhibitors may aggravate liver damage and fibrosis in NASH linked to fructose and cholesterol intake. We therefore postulate that for advanced NASH, which is thought to be driven by ER-stress (Nakagawa et al., 2014; Tilg and Moschen, 2010), IRE1 inhibitors present a more viable therapeutic approach. Indeed, MKC3946 blocked NASH in HFD-fed MUP-uPA mice (Kim et al., 2018) and reversed fructose-induced liver damage and RER disruption in ScapΔHep mice. Our results suggest that dietary factors that contribute to the switch from benign steatosis to steatohepatitis, liver damage and fibrosis need to be considered when designing pharmacological treatments to NASH, whose development had suffered numerous setbacks.
Limitations of Study
This study demonstrates that SCAP-mediated SREBP activation protects the mouse liver from ER stress induced damage caused by hepatocyte-specific uPA overexpression or consumption of fructose-rich diets, including HFrD and HFD supplemented with a 30% fructose-water drink. Although a convenient model for induction of ER stress, uPA overexpression is not physiological and the two diets used herein contain higher amounts of fructose than most humans ever consume. Nonetheless, this study provides a cautionary note regarding the therapeutic use of SCAP inhibitors, showing that the outcome of SCAP ablation is diet dependent and suggesting that such inhibitors should not be used in patients with advanced NASH, whose liver is ER stressed. Moreover, enhanced liver damage caused by SCAP ablation was also seen in another NASH model, the PtenΔHep mouse, which spontaneously develops NASH due to excessive AKT activation (Kawamura et al., 2022). Liver damage in PtenΔHep/ScapΔHep mice was also due to ER stress and was prevented by restoration of SREBP function.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ju Youn Kim (juk005@health.ucsd.edu).
Materials availability
All reagents generated in this study are available upon reasonable request.
Data and code availability
The sequence data was deposited at NCBI under Bioproject: PRJNA775619.
Original data for creating all graphs in the paper are provided in Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from Dr. Ju Youn Kim upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
ANIMALS
All mouse lines were either in a pure C57BL/6 genetic background or crossed into it for at least nine generations. All mice were maintained in filter-topped cages on autoclaved food and water, and experiments were performed in accordance with UCSD Institutional Animal Care and Use Committee and NIH guidelines. Animal Protocol, S00218 was approved by the UCSD Institutional Animal Care and Use Committee. Animal experiments performed at the Medical University of Innsbruck were approved by the Austrian Federal Ministry of Education, Science and Research (Tierversuchsgesetz, 2012, BGBl I Nr. 114/2012, GZ 66.011/0099/V/3b/2019). Generation and genotyping of Casp2−/−, Pidd1−/−, and Raidd−/− mice were described (Berube et al., 2005; Manzl et al., 2009; O’Reilly et al., 2002). The mice were housed at UCSD or the Medical University of Innsbruck under SPF conditions with a 12 hrs light/dark cycle. Male littermates were randomly assigned to experimental groups and were fed with normal chow diet (NCD), high fat diet (HFD), cornstarch diet (CSD), high fructose diet (HFrD), HFD plus 30% fructose-water, or western diet (WD) plus 15% fructose+glucose water from 8 weeks of age and continued for 12 weeks. Body weight and food consumption were monitored biweekly throughout the entire feeding period. Mice were fasted for 3 hrs before sacrifice and livers and other organs were excised and weighted.
Generation of Casp2 floxed mice
Targeting vector construction and knock-out strategy was performed by genOway (Lyon, France). Genomic region of interest containing the murine Casp2 locus was isolated by PCR from C57BL/6 ES cell genomic DNA. PCR fragments were subcloned into the pCR4-TOPO vector (Invitrogen, Carlsbad, California). The resulting sequenced clones (containing intron 4 to intron 6) were used to construct the targeting vector. Briefly, a 1-kb region comprising exon 6 and Casp2S-specific exon was flanked by a Neo cassette (FRT site-PGK promoter-Neo cDNA-FRT site-LoxP site) and a distal loxP site to have access to the constitutive and conditional knock-out lines by deleting Casp2 exon 6 and Casp2S specific exon. Deletion of exons 6 and Casp2S-specific exon leads to the splicing of exon 5 to exon 7, leading in turn to a frame shift and a premature stop codon in exon 7. The linearized targeting vector was transfected into C57BL/6 ES cells (genOway, Lyon, France) through electroporation (260Volt, 500μF) of 108 ES cells in the presence of 100μg of linearized plasmid. Positive selection was started 48 hrs after electroporation by addition of 200μg/ml of G418 (150μg/ml of active component, Life Technologies, Inc.). 1042 resistant clones were isolated and amplified in 96-well plates. Duplicates of 96-well plates were made. The set of plates containing ES cell clones amplified on gelatin were genotyped by both PCR and Southern blot analysis. For PCR analysis, one primer pair was designed to amplify sequences spanning the 3’ homology region. This primer pair was designed to specifically amplify the targeted locus:
sense, 5’- ATGAGAGTAGGTGTGATCAGAGGTTCTCACAG-3’;
antisense (Neo cassette) 5’ -ATGCTCCAGACTGCCTTGGGAAAAG -3’.
The targeted locus was confirmed by Southern blot analysis using internal and external probes on both 3’ and 5’ ends. 21 clones were identified as correctly targeted at the Casp2 locus. Clones were microinjected into albino C57BL/6J-Tyrc-2J/J blastocysts and gave rise to male chimeras with a significant ES cell contribution (as determined by a black coat color). Mice were bred to C57BL/6 mice expressing Flp recombinase to remove the Neo cassette (Casp2lox mice). The following PCR genotyping primers were used to characterize Casp2lox mice:
sense, 5’- GCCTCCTTTCTTTACCATTCTTGGAGC-3’;
antisense, 5’-AACTCAAGATGCTCCACACACACTTGC-3’.
The WT allele gave rise to a 607-bp product and floxed allele gave rise to a 724-bp product. Validation was achieved by Southern blot analysis of AvrII–digested DNA using a 3’ external probe. The WT allele gave rise to a 7.6-kb signal while the Casp2lox allele gave rise to a 4.8-kb signal. Thereafter, heterozygous animals were interbred to generate the homozygous floxed mouse line.
Cell Lines
HEK293 cells originated from a female fetus were used for the generation of PIDD1, or RAIDD knock-out cells (293ΔPIDD1 or 293ΔRAIDD, respectively) using the LentiCRISPRv2 system (Sanjana et al., 2014). Two oligonucleotides containing guide sequences against PIDD1 and RAIDD were synthesized and annealed to the lentiCRISPRv2 vector. Insertion of oligos into the vector was verified by sequencing. Guide sequences were delivered into HEK293 cells through viral transduction and 293ΔPIDD1 or 293ΔRAIDD were selected by incubation of DMEM/F12 medium supplemented with puromycin (10 μg/ml) at 37°C in the presence of 5% CO2.
The following primers were used for deletion of PIDD1 or RAIDD in HEK293 cells:
PIDD1-F: 5’-CAC CGG CTT CAG AGG ATT CGG ACG C-3’;
PIDD1-R: 5’-AAA CGC GTC CGA ATC CTC TGA AGC C-3’;
RAIDD-F: 5’-CAC CGA GTA CTC CGC TCA CTT CGC C-3’;
RAIDD-R: 5’-AAA CGG CGA AGT GAG CGG AGT ACT C-3’;
METHOD DETAILS
Reagents
PNGase F (#P0704), Endo H (#P0702), Nhe1 (#R3131S), BamH1 (#R3136S), EcoR1 (R3101S), and Not1 (#R3189S) were from New England BioLabs (Ipswich, MA). 25-hydroxycholesterol (HY-113134) was from MedChemExpress (Monmouth Junction, NJ). Puromycin (#ant-pr-1) was from InvivoGen (San Diego, CA). Lipofectamine 3000 Transfection Reagent (# L3000015) and OmniMax™ 2 T1R Chemically competent cells (C854003) were from Thermo Fisher Scientific (Pittsburgh, PA). MKC3946 (#532758) was from EMD Millipore (Burlington, MA). Antibodies used for immunoblotting (IB) and IHC (when indicated) were: anti-Casp2 for IB analysis (#ALX-804-356, Enzo Life Sciences. Inc., Farmingdale, NY), anti-Casp2 for IHC (#Ab182657, Abcam, Cambridge, UK), anti-PIDD1 for IB (#AG-20B-0038-C100, AdipoGen Life Science, San Diego, CA), anti-PIDD1 for IHC (#Ab231315, Abcam), anti-RAIDD (#10401-1-AP, Proteintech, Rosemont, IL), anti-SREBP1 (#Ab3259, Abcam), anti-SREBP2 (#Ab30682, Abcam), anti-S1P for detection of N-terminus (#sc-271916, Santa Cruz Biotechnologies, Inc., Dallas TX), anti-INSIG2 (#PA5-41707, Invitrogen), anti-phospho-IRE1 (#NB100-2323, Novos Biologicals, LLC., Centennial, CO), anti-IRE1 (#9102, Cell Signaling Technologies, Danvers, MA), anti-c/EBPβ (#3087, Cell Signaling Technologies), anti-ACC (#3662, Cell Signaling Technologies), anti-FAS (#3180, Cell Signaling Technologies), PDI (#7150, Cell Signaling Technologies), anti-HA (#1867431, Roche, Basel, Switzerland), anti-Myc (#Ab32, Abcam), anti-Flag (#F7425, Sigma-Aldrich, St. Louis, MO), anti-V5 (#3180, Cell Signaling Technologies) and anti-tubulin (#T5168, Sigma-Aldrich). Triglyceride (#MAK266) and Cholesterol (#MAK043) Quantification kits were purchased from Sigma-Aldrich.
Expression vectors
Mouse RAIDD (CRADD) cDNA (MR202001) was from OriGene Technologies, Inc. (Rockville, MD) and C-terminal 6His tagged mouse RAIDD (pCDH-mRAIDD-6HIS) was generated by PCR amplification and subsequently cloned between the Nhe1 and BamH1 sites of pCDH-CMV-MCS-EF1-Puro (#CD500-CD700, System Bioscience). C-terminal Flag tagged mouse PIDD1 cDNA was amplified by PCR reaction and subcloned between the Nhe1 and BamH1 sites of pCDH-CMV-MCS-EF1-Puro to generate pCDH-mPIDD1-Flag. The death domain (DD)-deleted PIDD1 (PIDD1ΔDD) was generated from parental pCDH-mPIDD1-Flag. Briefly, PIDD1 cDNA fragments from Met1 to Leu792 and from Glu879 to Stop codon were PCR amplified. PCR products were purified by QIAquick Gel Extraction kit (#28706X4, QIAGEN, Inc., Valentia, CA) and used for subsequent PCR reaction to generate DD-deleted mouse PIDD1. The PCR product was inserted between the Nhe1 and BamH1 sites of pCDH-CMV-MCS-EF1-Puro. N-terminal V5 tagged SREBP2 was produced by replacement of Flag with V5 by PCR amplification. Platinum pfx DNA polymerase (#11708-021, Thermo Fisher Scientific) was used for cDNA amplification. The cDNA sequence was verified (Eton Bioscience, Inc., San Diego, CA) and the cDNA was purified by QIAGEN Plasmid Plus Maxi kit (#12963, QIAGEN, Inc.). HA tagged mouse Casp2 and Myc tagged S1P cDNA were described (Kim et al., 2018).
The following primers were used for generation of the above plasmids.
Nhe1-Kozak-mCRADD-F:
5’-AGATCTGCTAGCACCATGGCAATGGAAGCCAGAGACAAGCA-3’;
mCRADD-6HIS-STOP-BamH1-R:
5’-GATGAGGGATCCTCAGTGGTGGTGGTGGTGGTGGTCCAGCATGTGCTGGAGCAGGGA-3’
Nhe1-Kozak-mPIDD1-F:
5’-GCGTTTGCTAGCACCATGGCAATGGCTGCAGTGTTGGA-3’;
mPIDD1-Flag-STOP-BamH1-R:
5’-TCTAGAGGATCCCTACTTGTCGTCATCGTCTTTGTAGTCGCTGC-3’
mPIDD1-DD-2634-F:
5’-AGAAACTGGATTCCTGGAGCTTGGCCGCCACAAGTACCAGGACAG-3’;
mPIDD1-DD-2379-R:
5’-CTTGTGGCGGCCAAGCTCCAGGAATCCAGTTTCTGCATCACCCAGGT-3’
EcoR1-V5-mSREBP2-F:
5’-TTCACCGAATTCATGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACG-3’;
mSREBP2-STOP-NOT1-R: 5’-GCGGCGGCTCAGGAGGCAGCGATGGCAGTGCCA-3’
Transfections and Subcellular fractionation
Cells were plated at a density of 2 × 106 per plate. The next day, 1 μg of indicated cDNAs were transfected using lipofectamine 3000 (Thermo Fisher Scientific, MA) according to the manufacturer’s instructions. After 4 hrs the cells were incubated in DMEM/F12 supplemented with 10% FBS and 1% penicillin streptomycin. The next day, nuclear, membrane and cytosolic fractions were prepared as described (DeBose-Boyd et al., 1999). Briefly, cells were incubated in hypotonic buffer A (10 mM HEPES-KOH, pH 7.6, 10 mM KCl, 1.5 mM MgCl2,1 mM sodium EDTA, 1 mM sodium EGTA, 250 mM sucrose, protease, and phosphatase cocktail) for 1 hr and homogenized in Dounce homogenizer 13 times, followed by passing through a needle 15 times. Cell lysates were centrifuged at 890 g at 4 °C for 10 mins to collect nuclei. Pellets were resuspended in lysis buffer (150 mM Tris-Hcl, pH 7.4, 10% sodium-deoxycholate, 100 mM NaCl, 100 mM EDTA, 200 mM NaF, 100 mM Na3VO4, and a mixture of protease and phosphatase inhibitors) to collect nuclear extracts. The supernatants from the original 890 g spin were centrifuged at 13,000 g for 20 mins and supernatants from the 13,000 g spin were subjected to another spin at 60,000 g for 1.5 hr at 4°C in a Beckman centrifuge (TLA 100.4 rotor) to collect membranes and cytosolic fractions. Pellets were subjected to sonication and removal of carbohydrate moieties by PNGase F or Endo H. Prepared nuclear and membrane extracts were subjected to IB analysis.
RNA analysis
Liver RNAs were extracted using TRIzol Reagent (#15596026, Invitrogen) according to the manufacturer’s instructions. cDNAs were synthesized from total RNAs using SuperScript VILO cDNA Synthesis Kit (#11754250, Thermo Fisher Scientific) according to the supplier’s instructions. The cDNAs were quantified by real-time PCR analysis using SYBR Green Master mix (Bio-Rad, CA). The following primers were used:
Casp2-F: 5’-CTA CAT GAC CAG ACC GCA CA-3’; Casp2-R:5’-GTG CCA CTA CGC AGG AGT G-3’,
Pidd1-F: 5’-TTG TTC TGC ACA GCA ACC TC-3’; Pidd1-R: 5’-GGG ATA TGT CTG GGG GAC TT-3’
Raidd-F: 5’-TTT TCT CAG CAG GTG GCT TT-3’; Raidd-R: 5’-TTA AAG GCC AGC CAC AAA GT-3’
Bip-F: 5’-ACT TGG GGA CCA CCT ATT CCT -3’; Bip-R: 5’-ATC GCC AAT CAG ACG CTC C -3’
Insig1-F: 5’-CAC GAC CAC GTC TGG AAC TAT -3’; Insig1-R: 5’-TGA GAA GAG CAC TAG GCT CCG -3’
Insig2-F: 5’-GGA GTC ACC TCG GCC TAA AAA -3’; Insig2-R: 5’-CAA GTT CAA CAC TAA TGC CAG GA -3’
Scap-F: 5’-ATT TGC TCA CCG TGG AGA TGT T -3’; Scap-R: 5’- GAA GTC ATC CAG GCC ACT AAT G-3’
Mean Cq values were measured from triplicate PCR analyses for each sample and normalized to the amount of Cyclophilin A transcripts
Primer sequences corresponding to lipogenic proteins including mouse SCAP, SREBP1c, SREBP2, FASN, SCD-1, HMGCR, HMGCS, LDLR, and others were described (Kim et al., 2018; Yang et al., 2001).
Immunohistochemistry
Livers were fixed in 4% paraformaldehyde for 24 hrs and embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) to evaluate gross morphology, or Sirius red to determine fibrosis. The antibodies listed above were used for IHC as described (Kim et al., 2018). Frozen liver sections were prepared by embedding in Tissue-Tek OCT compound (Sakura Finetek), sectioned, and stained with Oil red O (ORO) to visualize TG accumulation. Four high magnification (HMF) fields were blindly taken on a Zeiss Axio Imager, A.2 (Oberkochen, Germany) and positive areas were quantified with Image J software.
Triglyceride and cholesterol analysis
Liver TG were extracted by homogenization in a 5% NP40 solution followed by heating in an 80°C water bath. Samples were then centrifuged and the supernatant containing TG was saved. Liver cholesterol was extracted by homogenization in a 7:11:0.1 mix of chloroform:isopropanol:NP40. The lower organic phase was taken and air dried in the hood, and then redissolved according to the Cholesterol Quantification kit (Sigma Alderich, MO) instructions. Plasma and liver TG and cholesterol were determined using Triglyceride Colorimetric Assay kit (#MAK266, Sigma Aldrich, MO) and Cholesterol Quantification kit (MAK043, Sigma Aldrich, MO) according to manufacturer’s instruction.
ATP measurement
The kit used was the ATP Assay Kit by abcam (ab83355). Liver ATP was extracted from 0.02g of sample tissue homogenized in 200uL of assay buffer according to the manufacturer’s instructions. The assay was performed according to the manufacturer’s instructions. A set of 6 standards and 20uL of each sample were run in duplicate on a clear-bottom 96 well plate and brought up to a final volume of 50uL using the provided assay buffer. Background controls were run as well. After adding the reaction mix, the plate incubated at room temperature at 30min protected from light, before being read at OD 570nm.
Insulin measurement
The kit used was the Mouse INSULIN ELISA Kit by invitrogen (cat# EMINS). 55uL of mouse serum was diluted 4-fold with the provided Assay Diluent C and assay was performed according to the manufacturer’s instructions. A set of 8 standards and 100uL of each sample were run in duplicate on the provided 96 well plate. The plate was covered and incubated for 2.5 hours at room temperature with gentle shaking; afterwards the plate was washed 4 times with the provided wash buffer. Biotin conjugate was added to the plate and allowed to incubate for 1 hour at room temperature with gentle shaking, and then the washing step was repeated. Streptavidin-HRP solution (provided) was added to the plate and allowed to incubate for 45 minutes at room temperature with gentle shaking, and the washing step was repeated. TMB substrate (provided) was added to each well and allowed to incubate for 30 minutes at room temperature protected from the light and with gentle shaking. Finally, stop solution (provided) was added to each well, and the plate was read at OD 450nm.
RNA-Seq
Sequencing libraries were prepared from 100–500 ng total RNA using the TruSeq RNA Sample Preparation Kit v2 (Illumina) according to the manufacturer’s protocol. Briefly, mRNA was purified, fragmented, and used for first-, then second-strand cDNA synthesis followed by adenylation of 3’ ends. For adapter addition, samples were ligated to unique adapters and subjected to PCR amplification. RNA quality was confirmed using the 2100 BioAnalyzer (Agilent) and samples were pooled for sequencing. For sequencing, RNA-seq libraries were prepared and sequenced on an Illumina NovaSeq using barcoded multiplexing with a single-end type. Read quantification was accomplished using Salmon v1.1.0 (Patro et al., 2017) using an option for single-end. Then quantified sf files were loaded into the R environment. For differentially expressed transcripts, we used DESeq2 (Love et al., 2014). To import outcome, DESeqDataSetFromTximport function of the tximport package was performed using the option as follows: type=‘salmon’, txOut=TRUE. After establishment of DESeq2 data set with estimate SizeFactors, we normalized the counts using the ‘counts’ function with the option, normalized=TRUE. To identify significant transcripts/genes, we used the cut-off as follows: False Discovery Rate (FDR) < 0.05 and fold change > |1.5|. PCA plots were generated with vst and plotPCA commands. The R function, heatmap.2, was utilized to generate from overlapped transcripts with viridis color settings. For pathway analysis, transcript names were converted into gene symbols, then significant gene sets (up- or down-regulated) were used with the EnrichR tool. Hallmark Pathway 2020 outputs were visualized in heatmap plots.
Electron Microscopy
Mice were anesthetized with ketamine-xylazine and livers were immediately fixed with fixative (SC buffer: 2%paraformaldehyde, 2.5% of glutaraldehyde in 0.15 M Sodium Cacodylate, pH. 7.4). Trimmed liver pieces 1 mm3 in size were incubated in 1% osmium in 0.15 M sodium cacodylate for 1–2 hrs on ice. After washing with 0.15M SC buffer, the liver pieces were incubated in 2% of uranyl acetate. After dehydration in ethanol and drying in acetone, the tissues were imbedded in Durcupan. Ultrathin sections (60 nm) were cut on Leica microtome with Diamond knife and followed by post staining with uranyl acetate and lead. Images were captured on JEOL 1400 plus TEM at 80KV with Gatan 4kx4k camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are represented as mean ± SEM as indicated. Differences in mean values were analyzed by Student t-test or one-way ANOVA (for more than 2 groups). Quantification and statistical analysis of each experiment can be found in the figure legend. P value < 0.05 was considered as significant (N.S: p ≥ 0.05, *: p < 0.05, **: p < 0.005, ***: p < 0.001). Statistical analyses were performed using GraphPad Prism 7 software (San Diego, CA).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-Casp2 | Enzo Life Science | Cat# ALX-804-356; RRID: AB_2275199 |
| Rabbit polyclonal anti-Casp2 | Abcam | Cat# Ab182657; RRID: |
| Mouse monoclonal anti-PIDD1 | Adipogen | Cat# AG-20B-0038-C100; RRID: |
| Rabbit polyclonal anti-PIDD1 | Abcam | Cat# Ab231315; RRID: |
| Rabbit polyclonal anti-RAIDD | Proteintech | Cat# 10401-1-AP; RRID:AB_2085477 |
| Mouse monoclonal anti-SREBP1 | Abcam | Cat# ab3259; RRID: AB_303650 |
| Rabbit polyclonal anti-SREBP2 | Abcam | Cat# ab30682; RRID:AB_779079 |
| Mouse monoclonal anti-S1P | Santa Cruz Technologies | Cat# sc271916; RRID: AB_10610623 |
| Rabbit polyclonal anti-INSIG2 | ThermoFisher Scientific | Cat# PA5-41707; RRID:AB_2576306 |
| Rabbit polyclonal anti-p-IRE1 | Novos Biologicals, LLC. | Cat# NB100-2323; RRID:AB_10145203 |
| Rabbit monoclonal anti-IRE1 | Cell Signaling Technology | Cat# 3294; RRID:AB_10830732 |
| Rabbit polyclonal anti-c/EBPbeta/LAP | Cell Signaling Technology | Cat# 3087; RRID:AB_2260365 |
| Rabbit polyclonal anti-ERK | Cell Signaling Technology | Cat#9 102; RRID: AB_330744 |
| Rabbit polyclonal anti-ACC | Cell Signaling Technology | Cat# 3662; RRID: AB_2219400 |
| Rabbit monoclonal anti-FAS | Cell Signaling Technology | Cat# 3180; RRID: AB_2100796 |
| Rabbit polyclonal anti-Hmgcr | Santa Cruz Technologies | Cat#s c33827; RRID:AB_2118193 |
| Rabbit monoclonal anti-PDI | Cell Signaling Technology | Cat# 3501; RRID: AB_2156433 |
| Rat monoclonal anti-HA | Roche | Cat# 1867431; RRID:AB_390918 |
| Rabbit polyclonal anti-Flag | Sigma Aldrich | Cat# F7425; RRID: AB_439687 |
| Mouse monoclonal anti-Tubulin | Sigma Aldrich | Cat# T5168; RRID: AB_477579 |
| Mouse monoclonal anti-Myc tag | Abcam | Cat# Ab32; RRID:AB_303599 |
| Rabbit monoclonal anti-V5 | Cell Signaling Technology | Cat# 13202; RRID:AB_2687461 |
| Bacterial and virus strains | ||
| OmniMax™ 2 T1R Chemically competent cells | ThermoFisher Scientific | C854003 |
| Chemicals, peptides, and recombinant proteins | ||
| 25-Hydroxycholesterol | MedChemExpress | Cat# HY-113134 |
| Puromycin | InvivoGen | Cat# Ant-pr-1 |
| Lipofectamine 3000 Transfection Reagent | ThermoFisher Scientific | Cat# L3000015 |
| MKC3946 | EMD Millipore | Cat#532758 |
| TRIzol Reagent | Invitrogen | Cat# 15596026 |
| PNGaseF | New England BioLabs | Cat#P0704 |
| EndoH | New England BioLabs | Cat#P0702 |
| Nhe1 | New England BioLabs | Cat#R3131S |
| BamH1 | New England BioLabs | Cat#R3136S |
| Not1 | New England BioLabs | Cat#R3189S |
| EcoR1 | New England BioLabs | Cat#R3101S |
| Critical commercial assays | ||
| Triglyceride Quantification Kit | Sigma Aldrich | Cat# MAK266 |
| Cholesterol Quantification Kit | Sigma Aldrich | Cat# MAK043 |
| Mouse INSULIN ELIZA Kit | Invitrogen | Cat# EMINS |
| ATP Assay Kit | Abcam | Cat# Ab83355 |
| Super Script VILO cDNA Synthesis Kit Scientific | ThermoFisher Scientific | Cat# 11754050 |
| Deposited data | ||
| RNAseq | This paper | PRJNA775619 |
| Data S1_Source Data | This paper | |
| Experimental models: Cell lines | ||
| HEK293 cells | ATCC | CRL-1573 |
| PIDD1-ablated HEK293 cells | This paper | N/A |
| RAIDD-ablated HEK293 cells | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Casp2 flox | This paper | N/A |
| Mouse: Casp2−/− | O’Reilly et al., 2002 | N/A |
| Mouse: PIDD1−/− | Manzl et al., 2009 | N/A |
| Mouse: RAIDD−/− | Berube et al., 2005 | N/A |
| Mouse: SCAP flox | Moon et al., 2011 | N/A |
| Oligonucleotides | ||
| Guide sequence against human PIDD1, see experimental model and subject details | This paper | N/A |
| Guide sequence against human CRADD, see experimental model and subject details | This paper | N/A |
| Primers for generation of PIDD1-Flag, PIDD1ΔDD-Flag, RAIDD-6HIS, and V5-SREBP2, see experimental model and subject details | This paper | N/A |
| Primers for analysis of mRNA analyses, see experimental model and subject details | This paper | N/A |
| Recombinant DNA | ||
| pCDH-CMV-MCS-EF1-Puro | System Bioscience | Cat#CD500-CD700 |
| plentiCRISPRv2 vector | Sanjana et al., 2014 | N/A |
| pCMV6-Myc-DDK-Cradd | OriGene Technologies, Inc. | Cat#MR202001 |
| pCDH-CMV-MCS-EF1-Puro-mCasp2-HA | Kim J.Y et al., Cell 2018 | N/A |
| pCDH-CMV-MCS-EF1-Puro-mPIDD1-Flag | This Paper | N/A |
| pCDH-CMV-MCS-EF1-Puro-mRAIDD-6HIS | This Paper | N/A |
| pCDH-CMV-MCS-EF1-Puro-Myc-S1P | Kim J.Y et al., Cell 2018 | N/A |
| pCDH-CMV-MCS-EF1-Puro-V5-mSREBP2 | This Paper | N/A |
| pEGFP-SCAP | Nohturfft et al., 2000 (PMID: 10975522) | N/A |
| Software and algorithms | ||
| Illustrator | Adobe | http://www.adobe.com |
| Photoshop | Adobe | http://www.adobe.com |
| Image J | Open Source/National Institute of Health | http://imagej.nih.gov/ij/ |
| GraphPad Prizm 7.0 software | GraphPad Software. Inc. | http://www.graphpad.com/scientificsoftware/prism/ |
| Other | ||
| Diet: Normal chow diet | LabDiet | Rodent Diet 5053 |
| Diet: Cornstarch diet | Research Diets, Inc. | D12450K |
| Diet: Fructose diet | Research Diets, Inc. | D02022704K |
| Diet: High fat diet | Bio-Serv | S3282 |
| Diet: Western Diet | ENVIGO | TD88137 |
| Diet: D-(−)-Fructose | Sigma Aldrich | F3510 |
| Diet: D-(+)-Glucose | Sigma Aldrich | G5767 |
Highlights.
PIDDosome is activated in hepatocytes by fructose intake.
PIDDosome accounts for fructose dependent SREBP activation and hepatosteatosis.
SCAP induced SREBP activation expands ER, preventing ER stress and PIDDosome activation.
SCAP ablation enhances diet-induced ER stress, PIDDosome activation, and liver damage.
ACKNOWLEDGEMENTS
We thank M. K. laboratory members for helpful discussions and Cell signaling technology, and Santa Cruz Technology for antibodies and other reagents. Research was supported by grants to M.K. (R01DK120714, R01CA234128 and R01CA211794), M.K. and R.M.E. (P42ES010337), R.M.E. (P01HL147835, R01DK057978, R01DK120480), J.Y.K. (Prevent Cancer Foundation), E.J (WT094823 and Fondation iXCore-iXlife-iXBlue pour la Recherche), A.V. (ERC-AdG POLICE (787171), a Ph.D fellowship of the Austrian Academy of Sciences to F.E., and CCSG Grant (P30CA23100) to Tissue Technology Shared Resource.
Footnotes
DECLARATION OF INTERESTS
M.K. is the founder and scientific advisory board member of Elgia Therapeutics, and receives research support from Merck, Jenssen, and Gossamer. J.Y.K. and M.K. are authors/inventors of patent titled Method for inhibition Nonalcoholic Steatohepatitis, Nonalcoholic Fatty Liver disease, And/Or De Novo Lipogenesis (2017-152-2) (Patent is pending approval).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdelmalek MF, Lazo M, Horska A, Bonekamp S, Lipkin EW, Balasubramanyam A, Bantle JP, Johnson RJ, Diehl AM, Clark JM, et al. (2012). Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 56, 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonso MS, Machado RM, Lavrador MS, Quintao ECR, Moore KJ, and Lottenberg AM (2018). Molecular Pathways Underlying Cholesterol Homeostasis. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello G, Balboa E, Arrese M, and Zanlungo S (2015). Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta 1852, 1765–1778. [DOI] [PubMed] [Google Scholar]
- Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R, Yeh WC, Mak TW, and Benchimol S (2005). Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc Natl Acad Sci U S A 102, 14314–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. [DOI] [PubMed] [Google Scholar]
- Brown MS, Radhakrishnan A, and Goldstein JL (2018). Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu Rev Biochem 87, 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A, et al. (2021). Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. Embo j 40, e104844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RA (1978). Influence of increased membrane cholesterol on membrane fluidity and cell function in human red blood cells. J Supramol Struct 8, 413–430. [DOI] [PubMed] [Google Scholar]
- DeBose-Boyd RA, Brown MS, Li WP, Nohturfft A, Goldstein JL, and Espenshade PJ (1999). Transport-dependent proteolysis of SREBP: relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell 99, 703–712. [DOI] [PubMed] [Google Scholar]
- Dobrosotskaya IY, Seegmiller AC, Brown MS, Goldstein JL, and Rawson RB (2002). Regulation of SREBP processing and membrane lipid production by phospholipids in Drosophila. Science 296, 879–883. [DOI] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al. (2017). The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio MA, and Karin M (2021). “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francque SM, van der Graaff D, and Kwanten WJ (2016). Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J Hepatol 65, 425–443. [DOI] [PubMed] [Google Scholar]
- Gardner BM, and Walter P (2011). Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, and Brown MS (2006). Protein sensors for membrane sterols. Cell 124, 35–46. [DOI] [PubMed] [Google Scholar]
- Hampton RY (2003). IRE1: a role in UPREgulation of ER degradation. Dev Cell 4, 144–146. [DOI] [PubMed] [Google Scholar]
- Hetz C, Zhang K, and Kaufman RJ (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes BT, Nwosu CC, and Espenshade PJ (2009). Degradation of sterol regulatory element-binding protein precursor requires the endoplasmic reticulum-associated degradation components Ubc7 and Hrd1 in fission yeast. J Biol Chem 284, 20512–20521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Nguyen AD, Jo Y, Engelking LJ, Brugarolas J, and DeBose-Boyd RA (2017). Hypoxia-inducible factor 1α activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J Biol Chem 292, 9382–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen JS, Waage J, Rapin N, Bisgaard HC, Larsen FS, and Porse BT (2013). Temporal mapping of CEBPA and CEBPB binding during liver regeneration reveals dynamic occupancy and specific regulatory codes for homeostatic and cell cycle gene batteries. Genome Res 23, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, et al. (2009). A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol 16, 882–892. [DOI] [PubMed] [Google Scholar]
- Kawamura S, Matsushita Y, Kurosaki S, Tange M, Fujiwara N, Hayata Y, Hayakawa Y, Suzuki N, Hata M, Tsuboi M, et al. (2022). Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. J Clin Invest 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, and Karin M (2018). ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 175, 133–145.e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Lee JH, and Im SS (2020). The cellular function of SCAP in metabolic signaling. Exp Mol Med 52, 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hong Y, He H, Jiang G, You W, Liang X, Fu Q, Han S, Lian Q, and Zhang Y (2019). FGF21 Mediates Mesenchymal Stem Cell Senescence via Regulation of Mitochondrial Dynamics. Oxid Med Cell Longev 2019, 4915149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Ma W, and Benchimol S (2000). Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet 26, 122–127. [DOI] [PubMed] [Google Scholar]
- Logette E, Le Jossic-Corcos C, Masson D, Solier S, Sequeira-Legrand A, Dugail I, Lemaire-Ewing S, Desoche L, Solary E, and Corcos L (2005). Caspase-2, a novel lipid sensor under the control of sterol regulatory element binding protein 2. Mol Cell Biol 25, 9621–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY, Alexandrov LB, Koyama Y, Nishio T, Benner C, et al. (2020). IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol 72, 946–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl J, Mészáros T, Bánhegyi G, and Csala M (2013). Minireview: endoplasmic reticulum stress: control in protein, lipid, and signal homeostasis. Mol Endocrinol 27, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F, Logette E, Tschopp J, and Villunger A (2009). Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol 185, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, and García-Ruiz C (2006). Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab 4, 185–198. [DOI] [PubMed] [Google Scholar]
- Matsuzaka T, and Shimano H (2013). Insulin-dependent and -independent regulation of sterol regulatory element-binding protein-1c. J Diabetes Investig 4, 411–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JH, Kim KM, Oh TJ, Choi SH, Lim S, Park YJ, Park DJ, and Jang HC (2017). The Effect of TSH Suppression on Vertebral Trabecular Bone Scores in Patients With Differentiated Thyroid Carcinoma. J Clin Endocrinol Metab 102, 78–85. [DOI] [PubMed] [Google Scholar]
- Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Brown MS, Goldstein JL, and Horton JD (2012). The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab 15, 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, et al. (2002). Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ 9, 832–841. [DOI] [PubMed] [Google Scholar]
- Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, and Jacks T (2011). Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell 43, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell EE, Cooksley WGE, Hanson R, Searle J, Halliday JW, and Powell LW (1989). The Natural History of Nonalcoholic Steatohepatitis: A Follow-up Study of Forty-two Patients for Up to 21 Years. Hepatology, 74–80. [DOI] [PubMed] [Google Scholar]
- Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, and Kimata Y (2011). Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell 22, 3520–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, and Sanyal AJ (2007). A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46, 1081–1090. [DOI] [PubMed] [Google Scholar]
- Rong X, Wang B, Palladino EN, de Aguiar Vallim TQ, Ford DA, and Tontonoz P (2017). ER phospholipid composition modulates lipogenesis during feeding and in obesity. J Clin Invest 127, 3640–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, and Goldstein JL (1996). Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Machamer CE, and Espenshade PJ (2016). Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J Lipid Res 57, 1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Soratroi C, Heppke J, Eichin F, Rocamora-Reverte L, Szabo TG, Bongiovanni L, Westendorp B, Moreno E, et al. (2020). E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev Cell 52, 335–349.e337. [DOI] [PubMed] [Google Scholar]
- Sladky VC, and Villunger A (2020). Uncovering the PIDDosome and caspase-2 as regulators of organogenesis and cellular differentiation. Cell Death Differ 27, 2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, Willoughby J, Harbison C, Fitzgerald K, Ilkayeva O, et al. (2018). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 128, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targher G, Corey KE, Byrne CD, and Roden M (2021). The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nat Rev Gastroenterol Hepatol 18, 599–612. [DOI] [PubMed] [Google Scholar]
- Tilg H, and Moschen AR (2010). Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52, 1836–1846. [DOI] [PubMed] [Google Scholar]
- Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, and Tschopp J (2007). Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. Embo j 26, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todoric J, Di Caro G, Reibe S, Henstridge DC, Green CR, Vrbanac A, Ceteci F, Conche C, McNulty R, Shalapour S, et al. (2020). Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat Metab 2, 1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, et al. (2012). IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poorten D, Samer CF, Ramezani-Moghadam M, Coulter S, Kacevska M, Schrijnders D, Wu LE, McLeod D, Bugianesi E, Komuta M, et al. (2013). Hepatic fat loss in advanced nonalcoholic steatohepatitis: are alterations in serum adiponectin the cause? Hepatology 57, 2180–2188. [DOI] [PubMed] [Google Scholar]
- Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, Lee SP, Teoh NC, and Farrell GC (2011). Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141, 1393–1403, 1403.e1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhao M, Sud N, Christian P, Shen J, Song Y, Pashaj A, Zhang K, Carr T, and Su Q (2016). Glucagon regulates hepatic lipid metabolism via cAMP and Insig-2 signaling: implication for the pathogenesis of hypertriglyceridemia and hepatic steatosis. Sci Rep 6, 32246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DL, Kanwal F, and El-Serag HB (2012). Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 10, 1342–1359.e1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe D, Brown MS, and Goldstein JL (2002). Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A 99, 12753–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Goldstein JL, Hammer RE, Moon YA, Brown MS, and Horton JD (2001). Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci U S A 98, 13607–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, and Brown MS (2002). Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500. [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, and Goldstein JL (2000). ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Protease that Process SREBPs. Molecular Cell 6, 1355–1364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data was deposited at NCBI under Bioproject: PRJNA775619.
Original data for creating all graphs in the paper are provided in Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from Dr. Ju Youn Kim upon request.