

Abstract
Alzheimer disease (AD) is a chronic disease characterized by a progressive decline in memory and cognition. AD progression is closely correlated with neuropathologic changes and accumulation of the two main hallmark lesions, senile plaques and neurofibrillary tangles. Nevertheless, deciphering the complex biological aspects of AD requires looking for the neuropathologic changes not only as the cause but also as the collective response to a disease process that is essential to maintaining life during aging but ultimately generates a nonfunctional brain. Chronic conditions, such as AD, represent a new homeostatic balance or disease state, where the organism responds or adapts to maintain life. The pathologic diagnosis of AD still remains the gold standard for precise diagnosis of dementia, commonly in conjunction with cognitive-memory tests and brain image scans. Herein, we present a general overview of the main neuropathologic hallmarks and features of AD and related dementia, revealing the key biological and functional changes as potential drivers of age-dependent brain failure related to AD. The present work reflects some of the main ideas presented during the American Society for Investigative Pathology Rous-Whipple Award Lecture 2021.
Alzheimer disease (AD) is a progressive neurodegenerative disorder characterized by memory loss, diminishing mental functions, and cognitive impairment.1 Around 6.5 million Americans, aged ≥65 years, have a diagnosis of AD and related dementia, and this number is expected to reach 13.8 million by 2060.2 Today, AD is the sixth-leading cause of death in the United States, with 121,499 official death certificates in 2019. AD is by far the most common cause of dementia, comprising 60% to 80% of all cases.
In his original 1907 article, “On an Unusual Illness of the Cerebral Cortex,” Alois Alzheimer described, for the first time, the neuroanatomy of a 51-year–old woman with dementia.3 The article describes an unknown illness characterized by a rapid loss of memory, disorientation, altered behavior, marked difficulties in reading, misspelling, and disorientation, that progressed in severity over 4.5 years of observation. A post-mortem examination showed an evenly atrophic brain and arteriosclerotic changes. Brain tissue revealed striking changes in the neurofibrils, and the presence of thick fibrils in the neurons. He observed that “the change of the fibrils seems to be a parallel process of deposition of a pathological metabolic substance in the neurons whose closer examination is still pending.”3,p.430 Additional interesting comments from Alois Alzheimer included the observation that around one-fourth to one-third of the neurons in the cortex had morphologic changes, and marked neuronal loss in the upper layers of the brain cortex. He concluded that “we are dealing with a special illness” and that “a histological examination will enable us to determine the characteristics of some of these cases.”3,p.430 It is now recognized that elderly patients with AD and related dementia have an abnormal aggregation of misfolded proteins forming neurotoxic bundles within the cerebral cortex.
Neuropathologic Hallmarks of Alzheimer Disease
The hallmark pathologies of AD are the accumulation of amyloid-β (Aβ) peptide into senile plaques (SPs) outside neurons and of twisted strands of hyperphosphorylated tau protein into neurofibrillary tangles (NFTs) inside neurons in the brain.4,5 Figure 1 shows immunocytochemical location of SPs and NFTs in AD brain tissue. The deposition of these protein aggregates is accompanied by complex molecular and cellular responses that lead to synaptic failure, neuroinflammation, and eventually progressive neuronal death.6 Over the last decades, one of the main focuses in AD has been to decipher the mechanisms of formation of SPs and NFTs, with the objective of finding potential avenues to stop or even reverse their formation. This knowledge could be used to reduce the development of AD and for the development of a possible cure or effective treatments of AD and related dementias.
Figure 1.

Neuropathologic hallmarks of Alzheimer disease (AD). A: Immunocytochemistry of AD brain tissue section to locate amyloid-β senile plaques. B: Neurofibrillary tangles of phosphorylated tau. Images courtesy of Sandra Siedlak (Case Western Reserve University, Cleveland, OH). Scale bars = 50 μm (A and B).
The causes of AD are not completely understood but probably include a combination of many factors, such as aging, genetics, environmental conditions, and lifestyle. Although aging is the most important risk factor for neurodegeneration, it is not a direct cause of AD; only one-third of all people aged ≥85 years may have AD, and many elderly people never develop dementia (https://www.nia.nih.gov/health/what-causes-alzheimers-disease, last accessed June 7, 2022). Findings of SPs and NFTs in normal aging and in patients with traumatic brain injury are common. Although neither fully correlated with cognitive loss, they were seldom questioned as the unique cause of AD.
AD is classified into two types, early-onset familial AD and late-onset or sporadic AD. Remarkably, late-onset or sporadic AD is by far the most common type of AD, whereas only ≤1% of all cases correspond to early-onset familial AD. Genetic factors are implicated in both types. In particular, the apolipoprotein E-ε4 (APOE-ε4) gene on chromosome 19 has showed a higher risk of AD. However, inheriting an APOE-ε4 allele does not always correlate with the development of AD because many carriers do not develop dementia. APOE-ε3 gene is the most common type, with no evidence suggesting it may decrease or increase the risk of AD. In contrast, APOE-ε2 is a rare form of this gene that may provide some protection or lower the risk of AD. Subjects who inherit genes with specific mutations associated with early-onset familial AD usually develop symptoms as early as their late 30s. In particular, genetic studies of AD subjects have identified several mutations in three genes: amyloid-β precursor protein (AβPP) on chromosome 21, presenilin 1 (PSEN1) on chromosome 14, and presenilin 2 (PSEN2) on chromosome 1. Although the function of AβPP is not clear, AβPP has been implicated as a neuronal receptor, in synaptic formation, and on hormone-metabolite regulation. AβPP is the precursor of Aβ40/42 peptide, which is produced by its proteolytic processing by PSEN1, PSEN2, and other enzymes. Mutations in AβPP, PSEN1, and PSEN2 genes are associated with early-onset familial AD and an overproduction of Aβ peptide that forms neurotoxic fibrils and SPs. Nevertheless, the association of these genes with AD has been misinterpreted as directly causal. Causality requires demonstrating that Aβ removal either prevents or reverses the development of AD.7
There are no effective treatments for AD. Current treatments focus solely on the management of symptoms and maintenance of mental function, and have limited efficacy. Over the last years, development of therapeutic compounds against AD has been focused mainly on Aβ. In particular, multiple monoclonal therapeutic antibodies have been developed for the removal of neurotoxic forms of Aβ. However, surprisingly, in over a dozen clinical trials, patients with AD have not benefited significatively, as expected or as dictated by causality.7,8 Cumulative research indicates that Aβ is not the only driving force of AD, but still raises the question of how it is genetically and biologically associated with the development of AD pathology causing progressive neuronal failure. Therapeutic antibodies targeting aggregated tau into NFTs are also being evaluated for removing tau deposits from the brain, but initial studies focused on tau removal have not reversed disease progression. The failure of multiple therapeutic compounds (small drugs, inhibitors, and antibodies) targeting Aβ and tau leads to the question of whether the biological roles of Aβ and tau in AD are still incomplete, wrong, or even completely reversed. There is a striking mismatch between lesions of SPs/NFTs and symptoms of AD; a significant number of subjects show neuropathologic changes with widespread deposition of SPs/NFTs (observed at autopsy) but with relatively normal cognitive conditions and without manifesting dementia.9
Roles of Oxidative Stress in Alzheimer Disease
As discussed previously, the etiology and pathogenesis of AD are not completely described, but oxidative stress is a key component. Oxidative stress refers to a state of cellular imbalance between the production of reactive oxygen species and reactive nitrogen species with the antioxidant defense systems.10 Proteins, lipids, and genetic material may experience oxidative damage during oxidative stress, altering their functions-structure and promoting mutations. Oxidative damage in the AD brain is higher than that in the brains of healthy elderly individuals. This damage includes elevated levels of intracellular reactive oxygen species/reactive nitrogen species, protein carbonyls, and 3-nitrotyrosine, increased lipid peroxidation, significant DNA damage as 8-hydroxy-deoxyguanosine produced by reactive oxygen species/reactive nitrogen species, and 8-hydroxyguanine in RNA.10
Almost three decades ago, we performed pioneering studies of oxidative damage in AD, particularly to understand the chemical and biological properties of SPs and NFTs. Oxidative modification of neuronal proteins through nitration was identified in AD brain tissue, and within NFTs (Figure 2, A and B).11 Conversely, the levels of nitrotyrosine in age-matched controls were undetectable through immunocytochemistry in the cerebral cortex. Furthermore, oxidative damage in AD is also present in lipid membranes, as demonstrated by immunocytochemical detection of lipid peroxidation (4-hydroxynonenal) in brain tissue from AD patients (Figure 2, C and D).12 Surprisingly, the lipid peroxidation colocated with NFT lesions but not with SPs. The oxidative damage in AD is extended to the genetic material, as demonstrated by the presence of 8-hydroxy-deoxyguanosine and 8-hydroxyguanine in neurons within the hippocampus, subiculum, entorhinal cortex, as well as frontal, temporal, and occipital neocortex (Figure 2, E and F).13 The subcellular localization of the oxidative damage primarily in the cytoplasm allowed us to hypothesize that mitochondrial components may be the source of free radicals promoting oxidative stress in AD. The signs of oxidative damage are more prominent early in the disease and reduce with disease progression; this relationship is more significant in APOE-ε4 carriers.14 These studies indicate that the damage of oxidative stress conditions is widely extended in AD to all biomolecules, including lipids, proteins, sugars, and nucleic acids in brain cells. Moreover, the oxidative damage to each type of biomolecule specifically increased in vulnerable populations of neurons during AD. The range of oxidative damage types in AD suggested the involvement of Fenton reactions as the source of free radicals that promote oxidative damage, particularly focusing on abnormal levels of oxidation-reduction (redox)–active metal ions, such as copper and iron, which are the source of reactive oxygen species and other redox-generated free radicals.15 Figure 3, A and B, shows the histochemical detection of iron in AD, the deposition of iron collocated mainly with SPs and NFTs.
Figure 2.
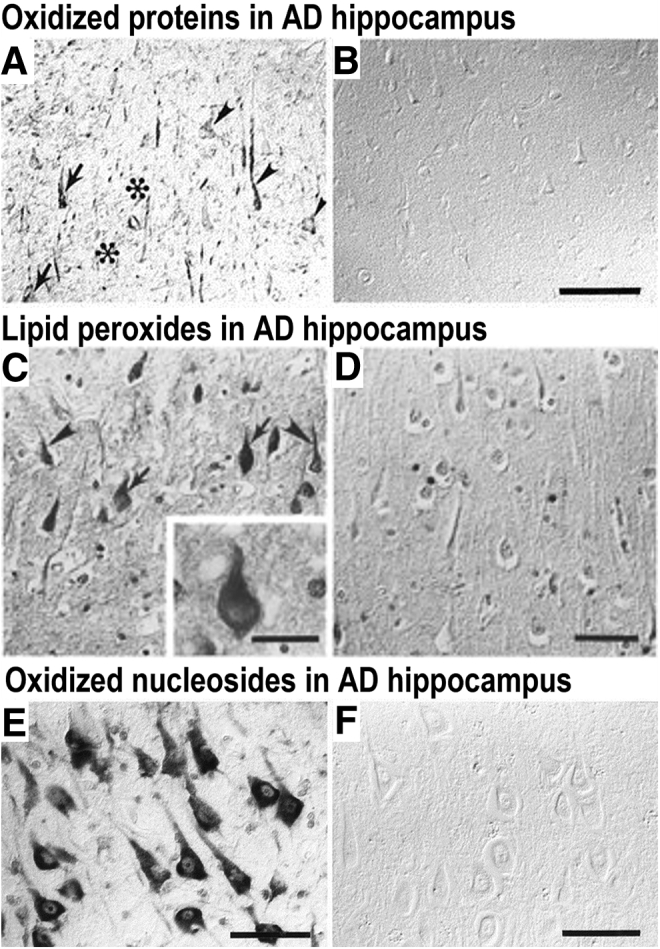
Oxidative stress in Alzheimer disease (AD). A: Immunolocalization of oxidized proteins (nitrotyrosine) in AD hippocampus [arrows indicate neurons with neurofibrillary tangles (NFTs), arrowheads indicate neurons without NFTs, and asterisks indicate location of senile plaques]. B: Control case (non-AD). C: Lipid peroxidation (4-hydroxy-2-nonenal–pyrrole) immunostaining in AD hippocampus (arrows indicate location of neurons with NFTs, and arrowheads indicate location of neurons without NFTs). Inset: A high-magnification view of a neuron containing an NFT and lipid peroxidation. D: Control (non-AD). E: Immunolocalization of oxidized nucleosides (anti–8-hydroxy-deoxyguanosine and anti–8-hydroxyguanine) in AD hippocampus. F: Control (non-AD). Reproduced with permission from J Neurosci, 1997, 17:2653–2657 (A and B)11; J Neurochem, 1997, 68:2092–2097 (C and D)12; and J Neurosci, 1999, 19:1959–1964 (E and F).13 Scale bars: 100 μm (B); 50 μm (C–F).
Figure 3.

Histochemical location of oxidation-reduction–active iron in Alzheimer disease (AD). A: Tissue section from AD brain (arrowheads indicate the presence of neurofibrillary tangles, and arrows indicate amyloid senile plaques). B: Control (non-AD) brain. C: X-ray spectromicroscopy (STXM) image showing the overall plaque morphology. D: Composite STXM image showing plaque morphology (blue), Cu2+ (green), Cu+/Cu0 (red), and iron (gray) content. E: Iron oxidation state difference map of the region highlighted in D. Strongly absorbing oxidized iron (Fe3+) is shown as light contrast, and chemically reduced iron (Fe2+ and/or Fe0) is shown as dark contrast. F: High-resolution (HR) composite image. G: Copper oxidation state difference map of particles G1–G6 of the region highlighted in D. In the oxidation state difference map, oxidized copper (Cu2+) is shown as light contrast, and chemically reduced copper (Cu+ and/or Cu0) is shown as dark contrast. Reproduced with permission from Proc Natl Acad Sci U S A, 1997, 19:9866 (A and B)15; and Sci Adv, 2021, 7:eabf6707 (C–G).16 Scale bars: 200 μm (B); 5 μm (C and D); 500 nm (E–G).
The oxidative stress conditions correlate with a transition from normal aging to the onset of cognitive impairment, and eventually on to prodromal AD.17 This occurs primarily by affecting the hippocampus and temporal cortex with oxidative damage, and is mostly restricted to the neuronal components in the cytoplasm, not SPs and NFTs. In fact, affected neurons with NFTs have reduced oxidative damage, and Aβ levels negatively correlate with oxidative damage in AD and Down syndrome.14,18 Overall, these observations suggest that the overexpression and aggregation of Aβ and tau could be part of complex neuronal molecular responses that are responsive to oxidative stress conditions in the aging brain. Experiments to understand the response of the brain to oxidative stress indicated, first, that NFTs are not formed by the aggregation of pure tau protein but also are intimately associated with other proteins, such as heme oxygenase-1, an enzyme that converts heme to biliverdin/bilirubin—transforming an oxidant to an antioxidant.19 Second, the levels of tau phosphorylation are correlated with heme oxygenase-1 expression in neuronal cells, and tau is regulated through signal transduction pathways that are modulated by oxidative stress to play a role in the cytoprotection of vulnerable neurons.20 Third, Aβ peptide binds different redox-active metals, including Cu(II), Fe(II/III), and Zn(II), to stabilize them from redox cycling reactions in the AD brain.21,22 The interactions between Aβ peptide and redox-active metal ions promotes the oxidation of Met residues of Aβ, and favors their aggregation into SPs.
Recent studies demonstrate that SPs from AD contain metallic elements within them, including copper and iron in multiple valence states (Cu+, Cu2+, Fe2+, and Fe3+), as observed with synchrotron-based X-ray spectromicroscopy (Figure 3, C–G).16 The advanced imaging and quantitative spectroscopic techniques were used to directly demonstrate the involvement of redox-active metal ions in redox cycling metabolism. Furthermore, advanced X-ray spectromicroscopy imaging revealed the presence of nanometer size metallic aggregates of copper (Cuᵒ) and iron (Feᵒ) within purified amyloid plaque cores, and in correlation with deposition of iron in AD pathology. Remarkably, SPs from advanced AD also contain iron aggregates formed by redox-active species, Fe3+, Fe2+, and metallic iron (Feᵒ), and forming nanostructured aggregates.23,24 Some of these metallic species should not be stable to air, demonstrating that Aβ peptide has unique properties responsible for its antioxidant activity through binding of redox-active ions and promoting their aggregation within SPs.
Overall, not only do SPs and NFTs play a role in oxidative stress responses in brain cells, but also several signal transduction pathways are induced during the development of neurodegenerative processes of AD, such as mitogen-activated protein kinase/extracellular receptor kinase,25 neuroinflammation, and cell cycle reentry (https://www.alzforum.org/news/research-news/targeting-glial-cells-towards-selective-cns-specific-antiinflammatory-drugs, posted June 2, 2002, last accessed June 7, 2022). What may be the most fundamental: in AD, dysfunctional glucose metabolism by induction of the pentose phosphate pathway and increased neuronal reduction may produce the cellular antioxidant compound glutathione.26
Mitochondrial Dysfunction in the Pathogenesis of Alzheimer Disease
Another neuropathologic feature related to the neurodegenerative processes of AD is mitochondrial dysfunction. Specifically, this includes mitochondrial structural and functional integrity alterations, mitochondrial biogenesis and dynamics, axonal transport, mitophagy, and mitochondrial proteostasis.27 These biological changes raise the question of where and how they occur in vulnerable cell populations in the AD brain. Mitochondrial responses during AD were investigated because mitochondria contain abundant levels of metalloproteins and are the main source of free radicals and highly oxidant compounds, such as superoxide (O2–). Just as most oxidative damage is dependent on redox-active ions Cu/Fe, the mitochondria oxidative metabolism is almost completely dependent on these metals as cofactors and acceptors. Mitochondria from AD brain samples were examined with probes to mitochondrial DNA, enzyme proteins, and enzyme-prosthetic groups, such as lipoic acid. The same population of neurons displaying a high degree of oxidative damage also contain abundant mitochondrial debris, mostly located within autophagosomes.28 These subcellular structures also contain ferroxidase-ferroreductase, which change iron redox state to catalyze both redox silencing and oxidative damage.15
Close examination of an aging brain series revealed those older than 40 years showed similar, albeit reduced, mitochondrial autophagy to that observed in patients with AD. These observations suggest a new molecular mechanism related to AD pathogenesis, one where the activation of mitochondria autophagy is at the center of neuronal damage. Figure 4 illustrates the neuronal responses to oxidative stress and their impact on mitochondrial dysfunction. Mitochondrial transport, fusion-fission, and turnover studies all have consistently shown morphologic and functional abnormalities related to the progression of AD. Activation of mitochondrial autophagy poses several challenges for affected neurons. Specifically, the alterations in metal ion transport/homeostasis are key components, because redox-active Cu/Fe ions that are liberated during the process of mitochondria turnover can cause massive oxidative damage to all compartments of neurons and even the entire brain if not properly counterbalanced by the antioxidant systems. Based on the cumulative evidence, we hypothesize that Aβ and tau may play a neuroprotectant role by binding free metal ions to reduce their reactivity. This molecular function of Aβ and tau is essential during neurodegeneration, and mutations in AβPP may lead to improper functionality in neuronal cells and, consequently, lead to early-onset AD.
Figure 4.

Possible mechanisms for oxidative stress in Alzheimer disease. Mitochondria autophagy is responsible for oxidation-reduction (Redox) metal release, leading to cytoplasmic Redox centers (Fenton chemistry) and primary oxidant release (O2−). Aβ, amyloid-β; NFT, neurofibrillary tangle.
Mitochondria are at the center of aerobic energy production, producing >95% of all cellular ATP, also controlling programmed cell death processes, and a master neuronal signaling component, calcium (Ca2+). Calcium controls microtubule assembly. While these microtubules are diminished in the brian during normal aging, they are decreased by up to 90% in neurons affected by AD.29 Proteolysis is also controlled by calcium, as is synaptic vesicle fusion. Loss of mitochondrial functions seems to be at the center of oxidative stress responses, promoting autophagy, altered redox-active biometal dynamics, and activation of neuronal survival mechanisms in neurodegeneration. During aging, diminished mitochondrial functions are met with critical compensations by Aβ/tau/heme oxigenase-1/autophagy/survival responses that are all essential to maintaining brain function. As aging progresses, the compensation mechanisms deployed in the brain grow and may ultimately lead to pathologic state. With continued aging, some of the compensation mechanisms could fail to maintain normal brain function and may redirect cellular metabolism to survival rather than function. This choice is evolutionarily selected as a response to temporary brain stress insults rather than the ever-present effects of normal aging. This interpretation is consistent with the lack of correlation between neuronal loss and cognition. Stated simply, in AD, the neurons are present but nonfunctional.30 Although these affected neurons might correspond to senescent cells, all neurons of the same population display pre-apoptotic changes without death, and their therapeutic removal will only advance disease progression. Moreover, mutations in AβPP lead to the improper molecular processing of Aβ rather than primary causation. Finally, and most significantly, the removal of Aβ, tau, or even redox-active metals does not benefit AD patients by reversal of the primary driver because each is an important neuroprotective response, explaining why many therapies focused on Aβ removal have had no benefit in cognition, memory, or brain function.
Mitochondria are highly responsive to changes in cell homeostasis, cellular stress, inflammation responses, environment, nutrition, and exercise status, all of which play major factors in the potential development of AD. All these factors are directly or indirectly linked to the dysfunctions in energy metabolism as key drivers to the development of AD. Potential therapies maintaining mitochondrial functions delay AD, as observed with some anti-diabetes drugs (metformin and insulin, among others). Similarly, promoting more efficient autophagy or restoring calcium levels could lower neuronal oxidative stress. Focusing on the complex biology of AD through pathology will continue to reveal potential or alternative therapeutic insights that will benefit AD patients and their families.
Conclusions
Alzheimer disease is a complex chronic neurologic disease with no effective treatments. The molecular and structural changes observed in AD brain have helped to establish the key mechanisms involved in the progression of the disease, and to differentiate them from normal aging processes. The main neuropathologic features are synaptic failure, neuronal loss, and mainly the widespread presence of amyloid plaque cores and neurofibrillary tangles, but these abnormal protein aggregates are more complex than previously thought. Over the last years, with the advancement of novel quantitative and advanced imaging techniques, the ultrastructural and chemical signatures of SPs and NFTs have revealed the presence of inorganic aggregates formed by biometals, such as Fe, Cu, Zn, and Ca. Furthermore, additional pathologic features not frequently analyzed include alterations in mitochondria, lipids, and inflammation responses, and altered neuronal phenotype. Remarkably, clinical decisions are often based on histopathologic/microscopic results, such as in cancer, but in the case of AD this is not possible, and the confirmation of neuropathology is performed postmortem. While AD studies still face significant challenges, effective therapeutic approaches and sensitive/effective molecular diagnostic tools are expected to be available soon.
Acknowledgments
We thank the Semmes Foundation, the Lowe Foundation, the Kleberg Foundation, the Alzheimer's Association, the NIH National Institute on Aging, and the UTSA Brain Health Consortium.
Footnotes
Supported by the Semmes Foundation, the Lowe Foundation, the Kleberg Foundation, the Alzheimer's Association (AARFD-17-529742), and the NIH/National Institute on Aging (R01AG066749).
Disclosures: None declared.
The Rous-Whipple Award is given by the American Society for Investigative Pathology (ASIP) to a senior pathologist with a distinguished career in experimental pathology research and continued productivity at the time of the award. George Perry, Ph.D., recipient of the 2021 ASIP Rous-Whipple Award, delivered a lecture entitled “Pathology in Alzheimer Disease: A Protective Response?” on April 27, 2021, at the 2021 ASIP Annual Meeting at Experimental Biology (held virtually).
References
- 1.Knopman D.S., Amieva H., Petersen R.C., Chételat G., Holtzman D.M., Hyman B.T., Nixon R.A., Jones D.T. Alzheimer disease. Nat Rev Dis Primers. 2021;7:33. doi: 10.1038/s41572-021-00269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer’s Association 2022 Alzheimer's disease facts and figures. Alzheimers Dement. 2022;18:700–789. doi: 10.1002/alz.12638. [DOI] [PubMed] [Google Scholar]
- 3.Stelzmann R.A., Norman Schnitzlein H., Reed Murtagh F. An English translation of Alzheimer's 1907 paper, “über eine eigenartige erkankung der hirnrinde.”. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 4.Castellani R.J., Plascencia-Villa G., Perry G. In: Pathogenesis of Alzheimer’s Disease: Handbook of Neurotoxicity. Kostrzewa R.M., editor. Springer Nature International Publishing; Cham, Switzerland: 2021. pp. 1–20. [Google Scholar]
- 5.Castellani R., Perry G. Molecular pathology of Alzheimer's disease. Colloquium Ser Neurobiol Alzheimers Dis. 2013;1:1–91. [Google Scholar]
- 6.Perry G., Zhu X., Smith M.A. Do neurons have a choice in death? Am J Pathol. 2001;158:1–2. doi: 10.1016/S0002-9440(10)63936-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearl J. ed 2. Cambridge University Press; Cambridge, UK: 2009. Causality: Models, Reasoning and Inference. [Google Scholar]
- 8.Ackley S.F., Zimmerman S.C., Brenowitz W.D., Tchetgen Tchetgen E.J., Gold A.L., Manly J.J., Mayeda E.R., Filshtein T.J., Power M.C., Elahi F.M., Brickman A.M., Glymour M.M. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ. 2021;372:n156. doi: 10.1136/bmj.n156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gómez-Isla T., Frosch M.P. Lesions without symptoms: understanding resilience to Alzheimer disease neuropathological changes. Nat Rev Neurol. 2022;18:323–332. doi: 10.1038/s41582-022-00642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butterfield D.A., Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20:148–160. doi: 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith M.A., Richey Harris P.L., Sayre L.M., Beckman J.S., Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sayre L.M., Zelasko D.A., Harris P.L., Perry G., Salomon R.G., Smith M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 13.Nunomura A., Perry G., Pappolla M.A., Wade R., Hirai K., Chiba S., Smith M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nunomura A., Perry G., Aliev G., Hirai K., Takeda A., Balraj E.K., Jones P.K., Ghanbari H., Wataya T., Shimohama S., Chiba S., Atwood C.S., Petersen R.B., Smith M.A. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 15.Smith M.A., Harris P.L., Sayre L.M., Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett J., Lermyte F., Brooks J., Tjendana-Tjhin V., Plascencia-Villa G., Hands-Portman I., Donnelly J.M., Billimoria K., Perry G., Zhu X., Sadler P.J., O'Connor P.B., Collingwood J.F., Telling N.D. Biogenic metallic elements in the human brain? Sci Adv. 2021;7 doi: 10.1126/sciadv.abf6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nunomura A., Tamaoki T., Motohashi N., Nakamura M., McKeel D.W., Jr., Tabaton M., Lee H.G., Smith M.A., Perry G., Zhu X. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol. 2012;71:233–241. doi: 10.1097/NEN.0b013e318248e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nunomura A., Perry G., Pappolla M.A., Friedland R.P., Hirai K., Chiba S., Smith M.A. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 19.Smith M.A., Kutty R.K., Richey P.L., Yan S.D., Stern D., Chader G.J., Wiggert B., Petersen R.B., Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol. 1994;145:42–47. [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda A., Perry G., Abraham N.G., Dwyer B.E., Kutty R.K., Laitinen J.T., Petersen R.B., Smith M.A. Overexpression of heme oxygenase in neuronal cells, the possible interaction with tau. J Biol Chem. 2000;275:5395–5399. doi: 10.1074/jbc.275.8.5395. [DOI] [PubMed] [Google Scholar]
- 21.Dong J., Atwood C.S., Anderson V.E., Siedlak S.L., Smith M.A., Perry G., Carey P.R. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: raman microscopic evidence. Biochemistry. 2003;42:2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura M., Shishido N., Nunomura A., Smith M.A., Perry G., Hayashi Y., Nakayama K., Hayashi T. Three histidine residues of amyloid-beta peptide control the redox activity of copper and iron. Biochemistry. 2007;46:12737–12743. doi: 10.1021/bi701079z. [DOI] [PubMed] [Google Scholar]
- 23.Everett J., Collingwood J.F., Tjendana-Tjhin V., Brooks J., Lermyte F., Plascencia-Villa G., Hands-Portman I., Dobson J., Perry G., Telling N.D. Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer's disease subjects. Nanoscale. 2018;10:11782–11796. doi: 10.1039/c7nr06794a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plascencia-Villa G., Ponce A., Collingwood J.F., Arellano-Jiménez M.J., Zhu X., Rogers J.T., Betancourt I., José-Yacamán M., Perry G. High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer's disease. Sci Rep. 2016;6:24873. doi: 10.1038/srep24873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry G., Roder H., Nunomura A., Takeda A., Friedlich A.L., Zhu X., Raina A.K., Holbrook N., Siedlak S.L., Harris P.L., Smith M.A. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- 26.Russell R.L., Siedlak S.L., Raina A.K., Bautista J.M., Smith M.A., Perry G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch Biochem Biophys. 1999;370:236–239. doi: 10.1006/abbi.1999.1404. [DOI] [PubMed] [Google Scholar]
- 27.Wang W., Zhao F., Ma X., Perry G., Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol Neurodegener. 2020;15:30. doi: 10.1186/s13024-020-00376-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirai K., Aliev G., Nunomura A., Fujioka H., Russell R.L., Atwood C.S., Johnson A.B., Kress Y., Vinters H.V., Tabaton M., Shimohama S., Cash A.D., Siedlak S.L., Harris P.L., Jones P.K., Petersen R.B., Perry G., Smith M.A. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cash A.D., Aliev G., Siedlak S.L., Nunomura A., Fujioka H., Zhu X., Raina A.K., Vinters H.V., Tabaton M., Johnson A.B., Paula-Barbosa M., Avíla J., Jones P.K., Castellani R.J., Smith M.A., Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perry G., Nunomura A., Smith M.A. A suicide note from Alzheimer disease neurons? Nat Med. 1998;4:897–898. doi: 10.1038/nm0898-897. [DOI] [PubMed] [Google Scholar]