

Abstract
The genus Acinetobacter encompasses a heterogeneous group of bacteria that are ubiquitous in the natural environment due in part to their ability to adapt genetically to novel challenges. Acinetobacter sp. strain ADP1 (also known as strain BD413) is naturally transformable and takes up DNA from any source. Donor DNA can be integrated into the chromosome by recombination provided it possesses sufficient levels of nucleotide sequence identity to the recipient's DNA. In other bacteria, the requirement for sequence identity during recombination is partly due to the actions of the mismatch repair system, a key component of which, MutS, recognizes mismatched bases in heteroduplex DNA and, along with MutL, blocks strand exchange. We have cloned mutS from strain ADP1 and examined its roles in preventing recombination between divergent DNA and in the repair of spontaneous replication errors. Inactivation of mutS resulted in 3- to 17-fold increases in transformation efficiencies with donor sequences that were 8 to 20% divergent relative to the strain ADP1. Strains lacking MutS exhibited increased spontaneous mutation frequencies, and reversion assays demonstrated that MutS preferentially recognized transition mismatches while having little effect on the repair of transversion mismatches. Inactivation of mutS also abolished the marker-specific variations in transforming efficiency seen in mutS+ recipients where transition and frameshift alleles transformed at eightfold lower frequencies than transversions or large deletions. Comparison of the MutS homologs from five individual Acinetobacter strains with those of other gram-negative bacteria revealed that a number of unique indels are conserved among the Acinetobacter amino acid sequences.
The ability to maintain the genetic integrity of bacterial cells is balanced by the need to adapt to rapidly changing environments. The mismatch repair system plays a key role in maintaining this balance by recognizing and correcting mismatched bases that arise in duplex DNA as a result of replication error, DNA damage, and recombination between partially divergent, so-called homeologous, DNA (33, 39). A key component of the mismatch repair system is MutS, which initiates the process by recognizing and binding to mismatched bases in double-stranded DNA (33). The importance of MutS in maintaining the stability of the cellular genome is underscored by its ubiquity in the biological world. MutS homologs have been identified in members of all three biological kingdoms, and with the advent of genome sequence analysis, it has become evident that most organisms encode at least one MutS homolog (9).
The genus Acinetobacter encompasses a diverse group of gram-negative bacteria whose members are found in most aquatic and terrestrial environments (23). The ubiquity of the group can be attributed to its members' ability to adapt genetically to novel environmental challenges. Documented examples of such adaptation include the ability of clinical Acinetobacter isolates to rapidly acquire drug resistance when challenged with antibiotics (48). Such genetic plasticity may also have contributed to the evolution of the diverse nutritional capabilities observed in most Acinetobacter species (1, 23).
Acinetobacter sp. strain ADP1 (also known as strain BD413) (24) possesses a natural transformation system that provides an unusual potential for acquiring foreign DNA. Transformation of strain ADP1 does not require uptake sequences to be present in donor DNA, nor does it have any known requirements for extracellular competence factors. Perhaps most importantly, stationary-phase cells become competent in virtually any growth medium following the addition of a fresh carbon source, and cultures remain competent throughout most of the growth cycle (37). DNA from virtually any source is taken up by strain ADP1 and can be incorporated into its chromosome by recombination provided it possesses sequence identity to the recipient's DNA. Although most of the interspecies transformation experiments done with strain ADP1 have used hybrid donor DNA that possessed sequence identity to the recipient chromosome (13, 27), there is evidence that divergent DNA can also be integrated into the chromosome via homeologous recombination (2, 22).
In this report we describe the cloning and characterization of mutS from Acinetobacter sp. strain ADP1. The Acinetobacter MutS protein recognized mismatches that arose during DNA replication and homeologous recombination, preferentially recognizing transition mismatches and 1-bp frameshifts while having virtually no effect on transversion mismatches or large insertions and deletions. Acinetobacter strains lacking MutS function exhibited increases in spontaneous mutation frequencies and in the frequency of interspecies transformation. We found that the genetic organization of the mutS regions from strain ADP1 and four divergent Acinetobacter strains shares similarities with the mutS regions from members of other bacterial genera. However, comparison of the MutS amino acid sequences from the Acinetobacter strains with those from other gram-negative bacteria clearly showed that the Acinetobacter homologs represent a distinct evolutionary branch within this highly conserved protein family.
MATERIALS AND METHODS
Strains and culture conditions.
Strains and plasmids used in this study are listed in Table 1. Unless otherwise indicated, all Acinetobacter cultures were grown at 30°C in Luria-Bertani broth (LB) (42) or in mineral medium (44) supplemented with 10 mM succinate or 5 mM p-hydroxybenzoate as a sole carbon source. Escherichia coli cultures were grown in LB at 37°C. Liquid cultures were incubated while shaking at 180 rpm. Agar plates were prepared by adding Difco agar (1.8% wt/vol) to liquid media prior to autoclaving. When required, growth media were supplemented with ampicillin, streptomycin, spectinomycin, or rifampin at respective concentrations of 100, 10, 40, and 30 μg/ml. For all transformation and mutation assays, selection plates were incubated for 48 h before counting of colonies, and viable cell counts were performed in parallel by plating dilutions of the cultures on LB plates.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristic(s) | Reference or source |
|---|---|---|
| Bacterial strains | ||
| Acinetobacter sp. strain ADP1 | Wild-type | 24 |
| Derivatives of strain ADP1 | ||
| ADP1424 | pobR1424 | 26 |
| ADP1451 | pobR1451 | 26 |
| ADP6205 | pcaH9 | 14 |
| ADP6209 | ΔpcaH20 | 14 |
| ADP6314 | pcaH5 | 14 |
| ADP6407 | pcaH12 | 14 |
| ADP6417 | ΔpcaH19 mutS+ | 14 |
| ADP7003 | pcaH+ pcaG+ ΔmutS11::pGEM3Z(+) (Apr) | This study |
| ADP7118 | ΔpcaH20 ΔmutS6::Ω (Str/Spr) | This study |
| ADP7021 | ΔmutS6::Ω (Str/Spr) | This study |
| ADP7037 | ΔpcaH19 ΔmutS6::Ω (Str/Spr) | This study |
| ADP7053 | pcaH5 ΔmutS6::Ω (Str/Spr) | This study |
| ADP7072 | pobR1424 ΔmutS6::Ω (Str/Spr) | This study |
| ADP7073 | pobRI451 ΔmutS6::Ω (Str/Spr) | This study |
| ADP7087 | orf1::Ω (Str/Spr) | This study |
| ADP7088 | ΔfdxA::Ω (Str/Spr) | This study |
| ADP7098 | pcaH12 ΔmutS6::Ω (Str/Spr) | This study |
| ADP7099 | pcaH9 ΔmutS6::Ω (Str/Spr) | This study |
| Wild-type strains | ||
| Acinetobacter sp. strain 93A2 | pcaH+ pcaG+ | 1 |
| Acinetobacter sp. strain AD321 | pcaH+ pcaG+ | This study |
| Acinetobacter sp. strain AD532 | pcaH+ pcaG+ | This study |
| Acinetobacter sp. strain 01B0 | pcaH+ pcaG+ | 1 |
| Acinetobacter sp. strain 48A1 | pcaH+ pcaG+ | 1 |
| A. haemolyticus 40B4 (ATCC 17906) | pcaH+ pcaG+ | 1 |
| A. johnsonii LUH540 | L. Dijkshoorn | |
| Acinetobacter sp. strain AC423D | This study | |
| Pseudomonas putida PRS2000 | pcaH+ pcaG+ | 36 |
| Plasmids | ||
| pGEM 3zf(+) | Cloning vector (Apr) | Promega |
| pHP45Ω | Ω element (Apr) (Str/Spr) | 38 |
| pUI1638 | Ω element (Apr) (Str/Spr) | 10 |
| pZR2 | 2,392-bp fragment containing pcaK′CHG in pUC19 | 14 |
| pZR419 | ΔpobSR::Ω in pUC19 (Str/Spr) (Apr) | Tony DiMarco |
| pZR7000 | 1942-bp PCR fragment of ADP1 mutS inserted in pGEM3Zf(+) (Apr) | This study |
| pZR7006 | pZR7000 with the ΔmutS6::Ω mutation (Kmr) | This study |
| pZR7008 | pZR7000 with the mutS8::Ω mutation (Str/Spr) | This study |
| pZR7009 | 5′ end of ADP1 mutS and 9 kbp of upstream DNA (Apr) | This study |
| pZR7010 | 3′ end of ADP1 mutS and 8 kbp of downstream DNA (Apr) | This study |
| pZR7072 | 1,165-bp PCR fragment, containing orf1, ligated into pGEM3Zf(+) (Apr) | This study |
| pZR7074 | orf1::Ω (Str/Spr) | This study |
| pZR7075 | 1,532-bp PCR product containing a portion of fdxA and downstream DNA, ligated into pGEM3Zf(+) (Apr) | This study |
| pZR7076 | ΔfdxA::Ω (Str/Spr) | This study |
PCR amplification of a mutS fragment.
A segment of mutS from Acinetobacter sp. strain ADP1 was amplified by PCR using degenerate primers MUTSF2 and MUTSR3 (Table 2 and Fig. 1) in a standard PCR reaction (95°C, 45 s; 50°C, 30 s; 72°C, 2 min; 30 cycles) using Pfu polymerase according to the directions of the supplier (Stratagene) and chromosomal DNA as a template.
TABLE 2.
Primers used for PCR amplification of Acinetobacter DNA
| Primer | Nucleotide sequencea | Annealing siteb |
|---|---|---|
| MUTC | 5′-GTCAACTGGGCAGACTTCTAC-3′ | 3′ strand of fdxA |
| MUTF | 5′-CATGGTCGATATTTTGCTGAG-3′ | 5′ strand of fdxA |
| MUTRR5 | 5′-GTTTAACGCCGAGACAAG-3′ | 5′ strand of mutS |
| MUTRR8 | 5′-CCAGATGAGTGCATTGACTGC-3′ | 3′ strand of orf3 |
| MUTSF2 | 5′-AYCGXATGGXGAYTTYTAYGARC-3′ | 5′ strand of mutS |
| MUTSR3 | 5′-GCXGTYTCXGTCATYTCXAC-3′ | 3′ strand of mutS |
Primers MUTSF2 and MUTSR3 are degenerate primers.
Location in the mutS chromosomal region of Acinetobacter sp. strain ADP1.
FIG. 1.

Cloning of mutS and flanking DNA from the chromosome of Acinetobacter sp. strain ADP1. Small arrows indicate the degenerate primers MUTSF2 (F) and MUTSR3 (R) used for PCR amplification of a 1.9-kb segment of mutS. Shaded regions indicate this portion of mutS throughout the figure. Strain ADP7003 was formed by integration of pZR7000 into the chromosome of strain ADP1. pGEM-3Zf(+) does not replicate in strain ADP1, so selection for ampicillin resistance (apr), encoded on the vector, demanded strain ADP7003. Digestion of chromosomal DNA from strain ADP7003 with EcoRI yielded a fragment containing pGEM-3Zf(+) fused to a segment of upstream DNA that included the 5′ end of mutS, and chromosomal DNA extending to the first EcoRI site upstream of the gene. Circularization of the restriction fragments by ligation followed by transformation into E. coli DH5α and selection for Apr resulted in pZR7009. The 3′ end of mutS and downstream DNA were cloned in the same manner except that ADP7003 DNA was digested with BamHI rather than EcoRI, and the resulting plasmid was designated pZR1010.
Cloning and sequencing of the mutS region.
All DNA manipulations were performed according to standard procedures (42). Recombinant plasmids were isolated by transforming E. coli DH5α with the appropriate ligation reaction according to the transformation protocol provided by the supplier (Gibco BRL).
Plasmid pZR7000 was constructed by blunt end ligation of the MUTSF2-MUTSR3 PCR product into the SmaI site of pGEM-3Zf(+). Clones containing the 5′ and 3′ ends of mutS and the genes flanking it were obtained using the vector integration strategy depicted in Fig. 1 (32). The overlapping nucleotide sequences of pZR7000, pZR7009, and pZR7010 made it possible to assemble a contiguous sequence for mutS and flanking DNA from Acinetobacter sp. strain ADP1 (Fig. 2).
FIG. 2.

Genetic organization of the mutS region of
the Acinetobacter sp. strain ADP1 chromosome. The annealing
sites of primers MUTSF2, MUTSR3, and MUTC are indicated as horizontal
arrows. The symbol
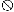 represents
putative transcription terminators downstream from mutS
(5′-ATAAGTAGCCATCGTGCTACTTAT-3′) and downstream from
fdxA (5′-AAAAGATCAGCATTAGCTGATCTTTT-3′).
Horizontal lines indicate inserts in plasmids containing
overlapping portions of mutS.
represents
putative transcription terminators downstream from mutS
(5′-ATAAGTAGCCATCGTGCTACTTAT-3′) and downstream from
fdxA (5′-AAAAGATCAGCATTAGCTGATCTTTT-3′).
Horizontal lines indicate inserts in plasmids containing
overlapping portions of mutS.
Construction of Insertion mutations.
Plasmid pZR7008 was constructed by ligating the Ω-cassette, cut from pHP45Ω as a SmaI fragment, into an EcoRV site located in the middle of the pZR7000 insert. Plasmid pZR7006 was constructed by deleting 637 bp of DNA between the EcoRV and MscI sites in the pZR7000 insert and inserting a 2-kb SmaI fragment containing the Ω element from plasmid pUI1638.
The orf1::Ω mutation was constructed using a 1,165-bp PCR product that was amplified in a standard PCR reaction from the chromosome of strain ADP1 with the primers MUTRR5 and MUTC (Table 2). pGEM-3Zf(+) was digested with SmaI and HincII, and the PCR product was ligated into the vector, creating pZR7072. Plasmid pZR7074 was created by ligating an XbaI fragment, containing the Ω element from pUI1638, into the unique XbaI site located in the center of open reading frame 1 (orf1) within the pZR7072 insert.
The ΔfdxA::Ω mutation was engineered in a 1,532-bp DNA fragment that was PCR amplified from the chromosome of strain ADP1 using the primers MUTRR8 and MUTF (Table 2). The PCR product was digested with SphI, which cut in orf3 near the end of the gene, and the resulting fragment was ligated into pGEM-3Zf(+) that had been digested with HincII and SphI, resulting in pZR7075. Plasmid pZR7076 was then constructed by the following steps. First, the pZR7075 insert was excised as a BamHI-SphI fragment. Second, pZR7072 was digested with PstI and SphI, both of which cleave at sites located in the vector, downstream of fdxA. Third, the Ω element was cut from pUI1638 as a BamHI-PstI fragment. Finally, the three fragments were ligated together in a forced direction ligation, resulting in pZR7076.
Transformation of Acinetobacter strains with engineered mutations.
Engineered mutations were integrated into the chromosomes of Acinetobacter sp. strain ADP1 and its derivatives by transformation as described previously (8).
DNA sequencing.
DNA sequencing was performed as described previously (25), and the PCR primers used in this study were synthesized by the W. M. Keck Foundation Biotechnology Resource Laboratory, Yale University, New Haven, Conn.
Determining mutation frequencies.
Mutation frequencies were determined either by selecting for spontaneous rifampin-resistant mutants or by selecting for reversion of various base substitution mutations in pcaH (14) or pobR (7), genes required for growth with p-hydroxybenzoate. For both types of assays, single colonies were used to inoculate 1-ml LB cultures. Following incubation for 24 h at 30°C, a 0.5-ml sample of each culture was pelleted by centrifugation, resuspended in mineral medium, and plated on p-hydroxybenzoate plates. At least three experiments were performed for each strain examined, and at least seven individual cultures were assayed for each strain per experiment.
Determining marker replacement frequencies during transformation.
Recipients containing pcaH mutations were transformed with a 2,392-bp HindIII-fragment from plasmid pZR2 that contained wild-type Acinetobacter pcaH plus flanking DNA. Liquid overnight cultures of each recipient were diluted 1:10 in fresh succinate media and incubated for an additional 2 h to induce competence. Transformation cultures consisted of 0.4 ml of the competent recipient culture plus 0.1 μg of the donor DNA fragment. Negative control cultures to which no donor DNA was added were also set up for each recipient. After incubation for 1 h, 50 μg of DNAse I was added to each culture and 0.2-ml aliquots were removed and plated on p-hydroxybenzoate plates.
To control for differences in the competence levels of the recipients, a second set of transformation cultures was set up for each strain in parallel to those above. The control cultures were transformed with a 7.1-kb PstI-SacI fragment, excised from plasmid pZR419, which contained the pob genes from strain ADP1 with an Ω element inserted into a pobS-pobR deletion. Since large insertions and deletions are not recognized by MutS (3, 33), determining the transformation frequency of this marker for each of the recipients provided a measure of their relative competence levels. This was used to normalize the marker replacement efficiencies obtained for each strain. Transformations with the pZR419 fragment were performed as described above with one exception: following addition of DNAse I, the cultures were allowed to incubate an additional 1.5 h before plating on LB containing spectinomycin and streptomycin. Very little difference in competence was observed between each of the eight recipients.
Isolation and characterization of mutS-fdxA DNA from divergent Acinetobacter strains.
PCR primers MUTSF2 and MUTC (Table 2 and Fig. 2) were used to amplify DNA extending between the 3′ terminus of mutS and the 5′ terminus of fdxA from different Acinetobacter isolates. PCR reactions were performed under standard conditions (95°C, 45 s; 56°C, 45 s; 72°C, 4 min; 30 cycles) using Taq polymerase (Boehringer) and chromosomal DNA from each strain as a template. The nucleotide sequences of the PCR products were determined as described above.
Sequence analysis.
Multiple sequence alignments were performed using the ClustalW program (18). Phylogenetic trees were constructed from the alignments by the neighbor-joining method (41) with 100 bootstrap trials. National Center for Biotechnology Information (NCBI) database accession numbers for the gram-negative MutS sequences used in the alignments were the following: for Pseudomonas aeruginosa, AAF42850; for E. coli, CAB43497; for Azotobacter vinelandii, AAA16868; for Salmonella typhimurium, A28668; for Vibrio cholerae, AAF93703; and for Haemophilus influenzae, AAC22364. The MutS sequences for Pseudomonas putida, Pseudomonas syringae, and Yersinia pestis were obtained by performing TBLASTX (1) searches of the NCBI unfinished genome database using the P. aeruginosa mutS nucleotide sequence (A220055) as the query sequence.
Determination of interspecies transformation frequencies.
Chromosomal DNA from each donor was purified using standard techniques (42). Transformations were performed as they were for determining marker integration frequencies, except that the overnight recipient culture was diluted 1:25, 0.1 μg of chromosomal DNA was used as donor DNA, and the cultures were incubated for 6 h before adding DNAse I. Two negative controls were performed to account for spontaneous reversion of the recipient marker; in one, 0.1 μg of the recipient's own DNA was added as a donor, and in the other, no donor DNA was added. In each experiment, five replicate transformations were performed per donor.
Nucleotide sequence accession numbers.
The nucleotide sequences presented in this paper were deposited in the NCBI database under the following accession numbers: for Acinetobacter sp. strain ADP1 mutS region, AF400582; Acinetobacter sp. strain 93A2 mutS′-fdxA′, AF400583; Acinetobacter sp. strain AD321 mutS′-fdxA′, AF400584; Acinetobacter sp. strain AC423D mutS′-fdxA′, AF400585; and Acinetobacter johnsonii LUH540 mutS′-fdxA′, AF400586.
RESULTS
Cloning of mutS and its surrounding genes from Acinetobacter sp. strain ADP1.
The high degree of sequence conservation shared among members of the MutS protein family facilitated the design of PCR primers for amplification of a 1.9-kb portion of mutS from Acinetobacter sp. strain ADP1. Nucleotide sequencing revealed that the mutS ORF is 2,646 bp in length and is predicted to encode a 97-kDa product that possesses approximately 50% amino acid sequence identity to the MutS homolog from E. coli (43). Sequence analysis of about 3 kb of DNA on both sides of mutS disclosed six additional ORFs (Fig. 2). Of particular interest was a 324-bp ORF located downstream of mutS that was designated fdxA because its predicted product displayed more than 75% amino acid sequence identity with a family of closely related 7Fe ferredoxins (21). fdxA homologs have also been reported directly downstream of mutS in the chromosomes of A. vinelandii (30), P. aeruginosa (35), and P. putida (28), and a similar gene arrangement is also evident in the unfinished genome sequence of P. syringae (see Materials and Methods).
Unlike the mutS-fdxA regions of A. vinelandii and the three Pseudomonas species, where fdxA is located immediately downstream of mutS, in strain ADP1 the two genes are separated by a 450-bp ORF, designated orf1 in Fig. 2. The predicted product of orf1 shares its closest similarity with AppA, a redox regulator involved in photosystem formation in Rhodobacter sphaeroides (15). However, functional similarity of the orf1 product and AppA cannot be inferred, since the amino acid sequence similarity is confined to the N-terminal portions of the proteins, a region containing a novel flavin-binding domain in AppA (15). Of the four remaining ORFs, only orf3 shared significant sequence similarity with known proteins. The predicted product of orf3 is a member of the o-methyltransferase protein family, and its function in strain ADP1 is unknown.
The effect of mutS region mutations on spontaneous mutation frequencies in Acinetobacter.
Defects in mutS have been shown to increase spontaneous mutation frequencies in bacteria (5, 46). To determine if this generalization holds true for Acinetobacter, we examined the effect of a knockout mutation in mutS on the frequency of rifampin resistance (Rifr) mutations in strain ADP1. To explore the possibility that the genetically linked genes, orf1 and fdxA, might also contribute to mutation repair, the effects of blocks in these genes were also determined. As shown in Table 3, inactivation of mutS increased the frequency of Rifr mutations 54-fold over that for the wild-type strain. No significant change in mutation frequency was observed in strains defective in orf1 or fdxA (Table 3). Thus, it appears that these genes are not directly involved in mismatch repair under the conditions examined here.
TABLE 3.
Frequency of Rifr mutation for Acinetobacter strains containing mutations in the mutS region
| Strain | Mutation | No. of Rifr mutants/viable cella |
|---|---|---|
| ADP1 | None | 4.6 × 10−9 |
| ADP7021 | mutS | 250.0 × 10−9 |
| ADP7087 | orf1 | 6.6 × 10−9 |
| ADP7088 | fdxA | 2.8 × 10−9 |
Mean of at least 21 replicates, with standard deviation of <0.2 of the mean.
Previous reports demonstrated that transition mutations were more frequent than transversions in mutS-deficient bacteria (3). As shown in Table 4, similar results were obtained with Acinetobacter, where mutS inactivation increased reversion frequencies for strains containing the transition mutations pcaH5, pcaH12, and pobR1451 more than 100-fold to frequencies between 2.2 × 10−8 and 5.5 × 10−8. Inactivation of mutS caused no more than a twofold increase in the frequencies of transversions required for reversion of pobR1424 and pcaH9.
TABLE 4.
Reversion frequencies for mutations in mutS+ and mutS mutant strains
| Mutation | Mutation type | No.
of revertants/viable
cella
|
mutS mutant/ mutS+b | |
|---|---|---|---|---|
| mutS+ | mutS mutant | |||
| pcaH5 | Transition (C→T) | 2.3 × 10−10 | 300.0 × 10−10 | 130.0 |
| pcaH12 | Transition (C→T) | 1.9 × 10−10 | 260.0 × 10−10 | 137.0 |
| pobR1451 | Transition (T→C) | 4.8 × 10−10 | 550.0 × 10−10 | 115.0 |
| pobR1424 | Transversion (T→A) | 1.9 × 10−10 | 2.3 × 10−10 | 1.2 |
| pcaH9 | Transversion (G→T) | 1.5 × 10−10 | 3.1 × 10−10 | 2.1 |
Mean of at least 20 replicates with standard deviation of <0.2 of the mean.
Ratio of revertants in mutS mutant strains to those in mutS+ strains.
Effect of mutS mutations on variations in marker replacement frequencies during transformation.
In other naturally transformable bacteria, single mismatches arising during heteroduplex formation are recognized with varying efficiencies by the mismatch repair system, resulting in variable integration efficiencies for the donor fragments (3). We examined whether mismatch repair had a similar effect during transformation of Acinetobacter. Recipient strains that contained either a transition, a transversion, a 1-bp frameshift, or a 128-bp deletion in pcaH were transformed with a DNA fragment that contained the wild-type pcaH allele. As shown in Table 5, in mutS+ recipients, transformation of the transversion and the 128-bp deletion alleles occurred at five- to eightfold higher frequencies than transformation of either the transition or 1-bp frameshift. As evidence that the differences in transformation frequencies were due to the activity of the mismatch repair system, strains in which mutS had been inactivated exhibited similar frequencies for all four recipient alleles (Table 5). The transformation frequencies for the transversion (pcaH9) and large deletion (ΔpcaH19) were unaffected by MutS.
TABLE 5.
Marker replacement frequencies for mutS+ and mutS mutant Acinetobacter recipients
| Recipient mutation | No. of transformants/viable recipient
cella
|
|
|---|---|---|
| mutS+ | mutS mutant | |
| pcaH12 (C→T transition) | 4.6 × 10−4 (±1.3 × 10−4) | 40.0 × 10−4 (±1.3 × 10−3) |
| ΔpcaH20 (1-bp frameshift) | 4.4 × 10−4 (±7.1 × 10−3) | 40.0 × 10−4 (±1.0 × 10−3) |
| pcaH9 (G→T transversion) | 36.0 × 10−4 (±1.1 × 10−3) | 42.0 × 10−4 (±1.1 × 10−3) |
| ΔpcaH19 (128-bp deletion) | 24.0 × 10−4 (±4.3 × 10−4) | 26.0 × 10−4 (±9.2 × 10−4) |
Mean (±standard deviation) of at least 10 replicates.
Inactivation of mutS increases the frequency of interspecies transformation in Acinetobacter.
Experiments with other bacteria have demonstrated that mutations in mutS reduced the requirement for sequence identity between donor and recipient alleles during interspecies recombination following conjugation (39), transduction (39, 50), and transformation (19). To determine whether mutS played a similar role in controlling the frequency of interspecies transformation in Acinetobacter, recipients containing the pcaH9 mutation in either a mutS+ or mutS mutant background were transformed with chromosomal DNA from divergent Acinetobacter strains.
Our results indicate that there is not an absolute barrier to interspecies transformation in strain ADP1. All but the most divergent donor DNA yielded pcaH+ transformants even when the recipient contained a functional mutS gene (Table 6). However, the transformation frequencies for most of the donors were 103- to 106-fold lower than the frequencies obtained using isogenic donor DNA. Only Acinetobacter sp. strain 93A2, which is 1 to 2% divergent relative to strain ADP1 at the nucleotide level, transformed the pcaH9 mutation as efficiently as strain ADP1.
TABLE 6.
Interspecies transformation frequencies for mutS+ and mutS mutant Acinetobacter recipients
| Donor | % pcaH sequence identityc | No. of transformants
per viable recipient
cellab
|
mutS mutant/ mutS+d | |
|---|---|---|---|---|
| mutS+ | mutS mutant | |||
| Acinetobacter sp. strain ADP1 | 100 | 2.9 × 10−2 | 1.9 × 10−2 | 0.65 |
| Acinetobacter sp. strain 93A2 | 99 | 2.9 × 10−2 | 2.4 × 10−2 | 0.83 |
| Acinetobacter sp. strain AD321 | 88 | 2.9 × 10−5 | 9.9 × 10−5 | 3.4 |
| Acinetobacter sp. strain AD532 | 81 | 6.5 × 10−7 | 76.0 × 10−7 | 11.7 |
| Acinetobacter sp. strain 48A1 | 81 | 9.3 × 10−8 | 110.0 × 10−8 | 11.8 |
| A. haemolyticus strain 40B4 | 80 | 7.9 × 10−8 | 69.0 × 10−8 | 8.7 |
| Acinetobacter sp. strain 01B0 | 80 | 1.6 × 10−8 | 28.0 × 10−8 | 17.5 |
| P. putida PRS2000 | 55 | <1.0 × 10−9 | <1.0 × 10−9 | |
Recipients (ADP6205 and ADP7099) contained the pcaH9 mutation, and transformants were selected on p-hydroxybenzoate plates.
Mean frequencies of three experiments, each of which consisted of at least five replicates of each donor/recipient transformation pair. Standard deviations were ≤0.2 of the mean.
pcaH sequence identity of each donor relative to that of strain ADP1 is based on partial sequence data. At least 80% of the total pcaHG sequence was determined for each donor (Young and Ornston, unpublished results).
Ratio of transformants in mutS mutant recipients to those in mutS+ recipients.
Although MutS did not completely block interspecies exchange in Acinetobacter, its inactivation did significantly increase the transformation frequencies for most of the divergent donors (Table 6). The effect of mutS inactivation ranged from 3-fold for strain AD321, whose pcaH gene is approximately 12% divergent from that of strain ADP1, to 17-fold for strain 01B0, whose pcaH gene is about 20% divergent. Inactivation of mutS had no effect on transformation with DNA that was identical (strain ADP1) or nearly identical (strain 93A2) to the recipient. Likewise, MutS did not affect transformation with extremely divergent DNA such as that from P. putida, which failed to yield transformants in either the mutS+ or mutS mutant recipients.
PCR amplification combined with restriction analysis and DNA sequencing was used to examine representative transformants and confirm that the recombinant phenotype was due to replacement of the recipient allele by the divergent donor DNA and not spontaneous reversion (results not shown). These analyses indicated that in most of the transformants, donor DNA had replaced not only the pcaH allele but various amounts of flanking DNA as well. In some cases as much as 7 to 8 kb of the recipient's DNA had been replaced.
Close genetic linkage of mutS and fdxA in Acinetobacter strains and other gram-negative bacteria.
Our observation that mutS and fdxA are closely linked in Acinetobacter sp. strain ADP1 and in members of the genera Pseudomonas and Azotobacter made it of interest to determine if such linkage was conserved among other Acinetobacter strains. It was also of interest to determine whether the interposition of orf1 between the two genes, a trait that distinguishes the mutS-fdxA region of strain ADP1 from those of A. vinelandii and the Pseudomonas species, was conserved as well. The PCR primers MUTC and MUTSF2 were used in an attempt to amplify DNA extending from the 5′ end of mutS to the 5′ end of fdxA in 10 Acinetobacter strains (Fig. 2). Eight of the 10 strains examined yielded distinct PCR products that were either 3.6, 3.2, or 2.7 kb in size (data not shown). Sequence analysis of the products from four strains revealed considerable variability. Acinetobacter sp. strain 93A2, similar to strain ADP1 in that it is competent for natural transformation (Young and Ornston, unpublished result), possesses 98% nucleotide sequence identity to strain ADP1 throughout the 3.6-kb amplified region which includes a homolog of orf1. The 3.2-kb amplified segment from strain AD321 includes a noncoding, 655-bp intergenic region between mutS and fdxA. In the 2.7-kb amplified region from strains AC423D and LUH540, fdxA is about 200 bp downstream from mutS, similar to the genetic organization and intergenic spacing observed in the Pseudomonas species and A. vinelandii.
Conserved and divergent sequences within MutS.
Alignment of the Acinetobacter sp. strain ADP1 MutS amino acid sequence with the MutS sequences of nine different gram-negative bacteria (see Materials and Methods) revealed substantial sequence conservation (≥43% amino acid similarity), particularly among amino acids comprising the mismatch-binding domain located near the N terminus and those in the ATP-binding and helix-turn-helix domains near the C terminus (Fig. 3) (29, 34). Despite the overall sequence conservation, MutS from strain ADP1 did contain notable signs of divergence. The length of the region spanning from the mismatch binding domain (amino acids 2 to 115 in the E. coli MutS sequence [29]) to the helix-turn-helix domain (E. coli amino acids 766 to 800) varies by no more than four amino acids in nine other gram-negative MutS homologs, while in the strain ADP1 homolog, six indels were present that resulted in its being 13 to 17 amino acids longer than the other homologs over the same interval.
FIG. 3.

Conserved and divergent amino acid sequences in Acinetobacter MutS. Horizontal arrows indicate degenerate primers originally used to amplify a portion of mutS from the chromosome of strain ADP1. Boxes surround amino acid residues conserved in the Acinetobacter MutS and in the E. coli MutS for which the crystal structure has been determined (29). A vertical arrow indicates a conserved phenylalanine residue that has been shown to be required for mismatch binding in other MutS homologs. Numbers in parentheses indicate positions in the E. coli MutS sequence corresponding to the starts of indels that distinguish the primary sequences of the two MutS proteins.
Indels represent insertions and deletions that punctuate evolutionary divergence of orthologous proteins (17), and so identification of potential indels in the MutS protein from strain ADP1 warranted determining how conserved the polymorphisms were among other Acinetobacter homologs. As shown in Fig. 4, all six indels set representatives of the genus Acinetobacter apart from other gram-negative bacteria. Unlike the other indels, indel 1 also separates the Acinetobacter strains ADP1, 93A2, and AD321 from strains AC423D and LUH540. The groupings that are based upon the distribution of MutS indels match those found in the phylogenetic tree that is based upon MutS amino acid sequence alignments (Fig. 5). This tree is essentially congruent to one generated from an alignment of the 16S rDNA sequences from these strains (Young and Ornston, unpublished result).
FIG. 4.

Indels that distinguish Acinetobacter MutS homologs from those of other gram-negative bacteria. Portions of a multiple sequence alignment depicting Acinetobacter MutS indels relative to other gram-negative homologs. Dashes indicate gaps in the aligned sequences. Numerals indicate positions corresponding to amino acids in the E. coli MutS primary sequence (29) that are located immediately prior to the start of the indel. Bold type indicates amino acids that are identical to those in Acinetobacter sp. strain ADP1.
FIG. 5.

Phylogenetic tree based on alignments of MutS from Acinetobacter with those from nine other gram-negative bacteria. The tree was generated as described in Materials and Methods. Bootstrap values are indicated on tree branches.
DISCUSSION
Influence of MutS on the frequency of natural transformation in Acinetobacter sp. strain ADP1.
The efficiency with which MutS binds different mismatches in vivo affects not only the spectrum and frequency of spontaneous mutations in the cell but also the frequency in which mutant alleles are integrated into the chromosome by recombination. Variations in marker transformation frequencies were first reported for Streptococcus pneumoniae (16), whose natural transformation system is similar to that of strain ADP1 in many respects (37). As shown in Table 5, the Acinetobacter mismatch repair system influenced transformation frequencies in a marker-specific manner as well. The effect of the Acinetobacter MutS protein on marker integration frequencies and its specificity towards transition and frameshift mismatches generally parallels the effect reported for HexA, the MutS homolog in S. pneumoniae (3, 12). The specificity of MutS was also apparent in the results of spontaneous mutation assays that were used to examine its role in postreplication repair (Table 4).
Although the single-transversion mismatch formed during recombination between the pcaH9 allele and wild-type DNA was not efficiently recognized by MutS (Table 5), MutS strongly influenced transformation frequencies when pcaH9 was transformed with heterospecific donor DNA (Table 6), which results in the formation of heteroduplexes containing multiple mismatches. In a wild-type mutS background, the frequency of recombinant formation with divergent Acinetobacter donors varied more than 106-fold (Table 6). The lowest frequencies were observed with donor DNA possessing only 80% nucleotide sequence identity to the recipient, and these frequencies were increased 3- to 17-fold by inactivation of MutS (Table 6).
We have shown that mismatch repair in strain ADP1 is capable of reducing the integration efficiencies of donor fragments containing single transition mismatches (Table 5), yet chromosomal DNA from strain 93A2 transformed the mutS+ recipient with approximately the same efficiency as isogenic DNA (Table 6), even though its pcaH allele contains multiple transition substitutions in the vicinity of the target mutation. One possible explanation for this result is that the mismatch repair system of strain ADP1 may be susceptible to saturation by high numbers of mismatches, similar to that shown in S. pneumoniae (19) and in E. coli (31). Saturation results in a transient mutator phenotype as functional MutS homodimers are titrated from the system by binding to numerous mismatches simultaneously. Chromosomal DNA from strain 93A2 could saturate mismatch repair during attempts to undergo homeologous recombination at other sites on the chromosome in addition to the target locus. In S. pneumoniae, transformation with highly divergent DNA resulted in saturation even during formation of a single heteroduplex at the target locus (19). Further study is required to determine how much of an effect saturation of mismatch repair has on interspecies transformation frequencies in Acinetobacter.
The mutator phenotype of MutS-deficient strains.
Inactivation of Acinetobacter mutS increased the frequency of Rifr mutations 54-fold over that for wild-type cells (Table 3). Though a significant value, it is less than the 100- to 1,000-fold effects reported for corresponding mutants of E. coli (4) and P. putida (28) and more closely resembles the effects reported for mismatch repair-deficient strains of A. vinelandii (64-fold) (30) and S. pneumoniae (4- to 30-fold) (46, 47). A previous comparison of data reported for the mismatch repair systems of S. pneumoniae and E. coli indicated that they reduced the frequency of spontaneous mutations at single sites by about 100- and 1,000-fold respectively (3). However, it was pointed out that despite the reduced effect in S. pneumoniae, the overall mutation rates for the two bacteria were still within the same range (10−8 to 10−10 mutations cell−1 generation−1), suggesting that the initial fidelity of DNA synthesis may be greater in S. pneumoniae (3).
Further investigation is required to determine conclusively whether mismatch repair has a reduced role in maintaining the fidelity of DNA replication in naturally transformable bacteria like S. pneumoniae and Acinetobacter. Such a situation is not inconceivable, given that attempts to undergo homeologous recombination can result in a transient mutator phenotype due to saturation of the mismatch repair system. Naturally transformable bacteria that are capable of taking up large amounts of heterologous DNA may have evolved mechanisms to increase the fidelity of DNA replication to compensate for frequent mismatch repair saturation. This would allow them to take advantage of the potential for rapid adaptation via interspecies genetic exchange without increasing the frequency of less beneficial spontaneous replication errors.
Chromosomal organization and mutS sequence divergence in Acinetobacter and other gram-negative bacteria.
The observation that fdxA homologs are located downstream of mutS in members of the genera Azotobacter and Pseudomonas and in five Acinetobacter strains suggested that conservation of this genetic organization might have been selected due to a functional relationship between these genes. However, inactivation of fdxA did not result in increased spontaneous mutation frequencies for either Acinetobacter (Table 3) or A. vinelandii (30), indicating that the genes does not contribute to mutation avoidance when the cells are grown in nutrient broth. Interestingly, the A. vinelandii fdxA gene product, AvFdI, was recently shown to be a redox sensor involved in gene regulation under oxidative stress conditions (40). Since MutS is believed to participate in the repair of oxidative DNA damage (45, 51), it will be of interest to see whether inactivation of fdxA affects mutation frequencies, or mutS expression, when cells are exposed to oxidative stress.
The observation that several indels are conserved among the Acinetobacter MutS (Fig. 4) protein sequences raises questions concerning why these polymorphisms have been preserved. In natural populations of E. coli, mutS undergoes frequent horizontal transfers as an adaptive mechanism in which mutation and recombination rates are modulated through recurrent losses and reacquisition of mutS (6). Reacquisition is often the result of homeologous recombination in which the defective mutS allele is replaced by a functional allele from a divergent strain. Frequent horizontal transfer of mutS between Acinetobacter strains could serve to homogenize its nucleotide sequence and result in conservation of the MutS indels. However, the relatively high levels of sequence divergence exhibited by the Acinetobacter MutS sequences, including many amino acid substitutions that are located immediately adjacent to indels (Fig. 4), suggests that this is not the case, since these residues would also be expected to be conserved due to frequent exchange. Frequent horizontal transfer of mutS may occur between closely related Acinetobacter strains; however, even if such exchange occurs it is unlikely that it would result in the conservation of polymorphisms between members of separate genomic species.
Although none of the Acinetobacter indels are located in highly conserved regions of the MutS, there still may be a functional basis for their selection. It has been noted that indels are most likely to occur in loop regions near the surface of a protein's three-dimensional structure (11). There is also evidence that MutS functions as part of multimeric protein complexes, interacting with other mismatch repair enzymes such as MutL and potentially interacting with proteins involved in DNA replication, recombination, and other repair pathways (20). Therefore, although none of the Acinetobacter MutS indels are located in regions that have been identified as essential for postreplication repair (49), they may be important for other MutS functions in Acinetobacter by facilitating interactions with other proteins.
We have demonstrated that MutS from Acinetobacter shares functional similarity with homologs in other bacteria while displaying sequence polymorphisms that appear to set it apart from other gram-negative strains. Continued genetic and biochemical studies of mismatch repair systems in organisms such as Acinetobacter allow comparison to be made with paradigmatic systems, such as those of S. pneumoniae and E. coli. These comparisons will help determine whether individual traits, such as the ability to undergo natural transformation, have affected the evolution of MutS within a particular group of bacteria.
ACKNOWLEDGMENTS
This research was supported by grants DAAG55-98-1-0232 from the Army Research Office and MCB-9603980 from the National Science Foundation.
We thank David Lee for his help with the initial transformation experiments and Jason Medeiros for his work in isolating and characterizing Acinetobacter natural isolates.
Footnotes
Publication 29 from the Biological Transformations Center in the Yale Institute for Biospherics Studies.
REFERENCES
- 1.Baumann P, Doudoroff M, Stanier R Y. A study of the Moraxella group. II. Oxidative-negative species (genus Acinetobacter) J Bacteriol. 1968;95:1520–1541. doi: 10.1128/jb.95.5.1520-1541.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks K, Sodeman T. Clinical studies on a transformation test for identification of Acinetobacter (Mima and Herellea) Appl Microbiol. 1974;27:1023–1026. doi: 10.1128/am.27.6.1023-1026.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Claverys J P, Lacks S A. Heteroduplex deoxyribonucleic acid base mismatch repair in bacteria. Microbiol Rev. 1986;50:133–165. doi: 10.1128/mr.50.2.133-165.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox E C. Bacterial mutator genes and the control of spontaneous mutation. Annu Rev Genet. 1976;10:135–156. doi: 10.1146/annurev.ge.10.120176.001031. [DOI] [PubMed] [Google Scholar]
- 5.Cox E C, Degnen G E, Scheppe M L. Mutator gene studies in Escherichia coli: the mutSgene. Genetics. 1972;72:551–567. doi: 10.1093/genetics/72.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denamur E, Lecointre G, Darlu P, Tenaillon O, Acquaviva C, Sayada C, Sunjevaric I, Rothstein R, Elion J, Taddei F, Radman M, Matic I. Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell. 2000;103:711–721. doi: 10.1016/s0092-8674(00)00175-6. [DOI] [PubMed] [Google Scholar]
- 7.DiMarco A A, Averhoff B, Ornston L N. Identification of the transcriptional activator pobR and characterization of its role in the expression of pobA, the structural gene for p-hydroxybenzoate hydroxylase in Acinetobacter calcoaceticus. J Bacteriol. 1993;175:4499–4506. doi: 10.1128/jb.175.14.4499-4506.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doten R C, Ngai K L, Mitchell D J, Ornston L N. Cloning and genetic organization of the pca gene cluster from Acinetobacter calcoaceticus. J Bacteriol. 1987;169:3168–3174. doi: 10.1128/jb.169.7.3168-3174.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisen J A. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 1998;26:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eraso J M, Kaplan S. prrA, a putative response regulator involved in oxygen regulation of photosynthesis gene expression in Rhodobacter sphaeroides. J Bacteriol. 1994;176:32–43. doi: 10.1128/jb.176.1.32-43.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freimuth P I, Taylor J W, Kaiser E T. Introduction of guest peptides into Escherichia colialkaline phosphatase. Excision and purification of a dynorphin analogue from an active chimeric protein. J Biol Chem. 1990;265:896–901. [PubMed] [Google Scholar]
- 12.Gasc A M, Sicard A M, Claverys J P. Repair of single- and multiple-substitution mismatches during recombination in Streptococcus pneumoniae. Genetics. 1989;121:29–36. doi: 10.1093/genetics/121.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebhard F, Smalla K. Transformation of Acinetobactersp. strain BD413 by transgenic sugar beet DNA. Appl Environ Microbiol. 1998;64:1550–1554. doi: 10.1128/aem.64.4.1550-1554.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerischer U, Ornston L N. Spontaneous mutations in pcaH and -G, structural genes for protocatechuate 3,4-dioxygenase in Acinetobacter calcoaceticus. J Bacteriol. 1995;177:1336–1347. doi: 10.1128/jb.177.5.1336-1347.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomelsky M, Kaplan S. AppA, a redox regulator of photosystem formation in Rhodobacter sphaeroides2.4.1, is a flavoprotein. Identification of a novel fad binding domain. J Biol Chem. 1998;273:35319–35325. doi: 10.1074/jbc.273.52.35319. [DOI] [PubMed] [Google Scholar]
- 16.Green D M. A host-specific variation affecting relative frequency of transformation of two markers in pneumococcus. J Exp Cell Res. 1959;16:466–480. doi: 10.1016/0014-4827(59)90312-x. [DOI] [PubMed] [Google Scholar]
- 17.Gupta R S. Protein phylogenies and signature sequences: a reappraisal of evolutionary relationships among archaebacteria, eubacteria, and eukaryotes. Microbiol Mol Biol Rev. 1998;62:1435–1491. doi: 10.1128/mmbr.62.4.1435-1491.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higgins D G, Thompson J D, Gibson T J. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- 19.Humbert O, Prudhomme M, Hakenbeck R, Dowson C G, Claverys J P. Homeologous recombination and mismatch repair during transformation in Streptococcus pneumoniae: saturation of the Hex mismatch repair system. Proc Natl Acad Sci USA. 1995;92:9052–9056. doi: 10.1073/pnas.92.20.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh P. Molecular mechanisms of DNA mismatch repair. Mutat Res. 2001;486:71–87. doi: 10.1016/s0921-8777(01)00088-x. [DOI] [PubMed] [Google Scholar]
- 21.Iismaa S E, Vazquez A E, Jensen G M, Stephens P J, Butt J N, Armstrong F A, Burgess B K. Site-directed mutagenesis of Azotobacter vinelandiiferredoxin I. Changes in [4Fe-4S] cluster reduction potential and reactivity. J Biol Chem. 1991;266:21563–21571. [PubMed] [Google Scholar]
- 22.Juni E. Interspecies transformation of Acinetobacter: genetic evidence for a ubiquitous genus. J Bacteriol. 1972;112:917–931. doi: 10.1128/jb.112.2.917-931.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juni E. Genetics and physiology of Acinetobacter. Annu Rev Microbiol. 1978;32:349–371. doi: 10.1146/annurev.mi.32.100178.002025. [DOI] [PubMed] [Google Scholar]
- 24.Juni E, Janik A. Transformation of Acinetobacter calco aceticus (Bacterium anitratum) J Bacteriol. 1969;98:281–288. doi: 10.1128/jb.98.1.281-288.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kok R G, D'Argenio D A, Ornston L N. Combining localized PCR mutagenesis and natural transformation in direct genetic analysis of a transcriptional regulator gene, pobR. J Bacteriol. 1997;179:4270–4276. doi: 10.1128/jb.179.13.4270-4276.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kok R G, D'Argenio D A, Ornston L N. Mutation analysis of PobR and PcaU, closely related transcriptional activators in Acinetobacter. J Bacteriol. 1998;180:5058–5069. doi: 10.1128/jb.180.19.5058-5069.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kok R G, Young D M, Ornston L N. Phenotypic expression of PCR-generated random mutations in a Pseudomonas putida gene after its introduction into an Acinetobacterchromosome by natural transformation. Appl Environ Microbiol. 1999;65:1675–1680. doi: 10.1128/aem.65.4.1675-1680.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurusu Y, Narita T, Suzuki M, Watanabe T. Genetic analysis of an incomplete mutS gene from Pseudomonas putida. J Bacteriol. 2000;182:5278–5279. doi: 10.1128/jb.182.18.5278-5279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamers M H, Perrakis A, Enzlin J H, Winterwerp H H, de Wind N, Sixma T K. The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 30.Le O, Shen B, Iismaa S E, Burgess B K. Azotobacter vinelandii mutS: nucleotide sequence and mutant analysis. J Bacteriol. 1993;175:7707–7710. doi: 10.1128/jb.175.23.7707-7710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maas W K, Wang C, Lima T, Hach A, Lim D. Multicopy single-stranded DNA of Escherichia colienhances mutation and recombination frequencies by titrating MutS protein. Mol Microbiol. 1996;19:505–509. doi: 10.1046/j.1365-2958.1996.392921.x. [DOI] [PubMed] [Google Scholar]
- 32.Mejean V, Claverys J P, Vasseghi H, Sicard A M. Rapid cloning of specific DNA fragments of Streptococcus pneumoniaeby vector integration into the chromosome followed by endonucleolytic excision. Gene. 1981;15:289–293. doi: 10.1016/0378-1119(81)90139-6. [DOI] [PubMed] [Google Scholar]
- 33.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 34.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 35.Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosain cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 36.Ornston L N, Stanier R Y. The conversion of catechol and protocatechuate to beta-ketoadipate by Pseudomonas putida. J Biol Chem. 1966;241:3776–3786. [PubMed] [Google Scholar]
- 37.Palmen R, Hellingwerf K J. Uptake and processing of DNA by Acinetobacter calcoaceticus—a review. Gene. 1997;192:179–190. doi: 10.1016/s0378-1119(97)00042-5. [DOI] [PubMed] [Google Scholar]
- 38.Prentki P, Krisch H M. In vitro insertional mutagenesis with a selectable DNA fragment. Gene. 1984;29:303–313. doi: 10.1016/0378-1119(84)90059-3. [DOI] [PubMed] [Google Scholar]
- 39.Rayssiguier C, Thaler D S, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimuriumis disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- 40.Regnstrom K, Sauge-Merle S, Chen K, Burgess B K. In Azotobacter vinelandii, the E1 subunit of the pyruvate dehydrogenase complex binds fprpromoter region DNA and ferredoxin I. Proc Natl Acad Sci USA. 1999;96:12389–12393. doi: 10.1073/pnas.96.22.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd edition. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 43.Schlensog V, Bock A. The Escherichia coli fdv gene probably encodes mutS and is located at minute 58.8 adjacent to the hyc-hypgene cluster. J Bacteriol. 1991;173:7414–7415. doi: 10.1128/jb.173.23.7414-7415.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shanley M S, Neidle E L, Parales R E, Ornston L N. Cloning and expression of Acinetobacter calcoaceticus catBCDE genes in Pseudomonas putida and Escherichia coli. J Bacteriol. 1986;165:557–563. doi: 10.1128/jb.165.2.557-563.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terato H, Masaoka A, Kobayashi M, Fukushima S, Ohyama Y, Yoshida M, Ide H. Enzymatic repair of 5-formyluracil. II. Mismatch formation between 5-formyluracil and guanine during DNA replication and its recognition by two proteins involved in base excision repair (AlkA) and mismatch repair (MutS) J Biol Chem. 1999;274:25144–25150. doi: 10.1074/jbc.274.35.25144. [DOI] [PubMed] [Google Scholar]
- 46.Tiraby J G, Fox M S. Marker discrimination in transformation and mutation of pneumococcus. Proc Natl Acad Sci USA. 1973;70:3541–3545. doi: 10.1073/pnas.70.12.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiraby G, Sicard M A. Integration efficiencies of spontaneous mutant alleles of amiAlocus in pneumococcal transformation. J Bacteriol. 1973;116:1130–1135. doi: 10.1128/jb.116.3.1130-1135.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Towner K J. Clinical importance and antibiotic resistance of Acinetobacterspp. J Med Microbiol. 1997;46:721–746. doi: 10.1099/00222615-46-9-721. [DOI] [PubMed] [Google Scholar]
- 49.Wu T H, Marinus M G. Dominant negative mutator mutations in the mutS gene of Escherichia coli. J Bacteriol. 1994;176:5393–5400. doi: 10.1128/jb.176.17.5393-5400.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zahrt T C, Mora G C, Maloy S. Inactivation of mismatch repair overcomes the barrier to transduction between Salmonella typhimurium and Salmonella typhi. J Bacteriol. 1994;176:1527–1529. doi: 10.1128/jb.176.5.1527-1529.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao J, Winkler M E. Reduction of GC → TA transversion mutation by overexpression of MutS in Escherichia coliK-12. J Bacteriol. 2000;182:5025–5028. doi: 10.1128/jb.182.17.5025-5028.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]