

Abstract
Parkinson’s disease is a neurodegenerative disease characterized by the loss of dopaminergic neurons in the substantia nigra with no effective cure available. MicroRNA-124 has been regarded as a promising therapeutic entity for Parkinson’s disease due to its pro-neurogenic and neuroprotective roles. However, its efficient delivery to the brain remains challenging. Here, we used umbilical cord blood mononuclear cell-derived extracellular vesicles as a biological vehicle to deliver microRNA (miR)-124-3p and evaluate its therapeutic effects in a mouse model of Parkinson’s disease. In vitro, miR-124-3p-loaded small extracellular vesicles induced neuronal differentiation in subventricular zone neural stem cell cultures and protected N27 dopaminergic cells against 6-hydroxydopamine-induced toxicity. In vivo, intracerebroventricularly administered small extracellular vesicles were detected in the subventricular zone lining the lateral ventricles and in the striatum and substantia nigra, the brain regions most affected by the disease. Most importantly, although miR-124-3p-loaded small extracellular vesicles did not increase the number of new neurons in the 6-hydroxydopamine-lesioned striatum, the formulation protected dopaminergic neurons in the substantia nigra and striatal fibers, which fully counteracted motor behavior symptoms. Our findings reveal a novel promising therapeutic application of small extracellular vesicles as delivery agents for miR-124-3p in the context of Parkinson’s disease.
Keywords: miR-124, microRNA, small extracellular vesicles, 6-OHDA, dopaminergic degeneration, neurogenesis, neuroprotection, motor behavior, Parkinson’s disease
Graphical abstract
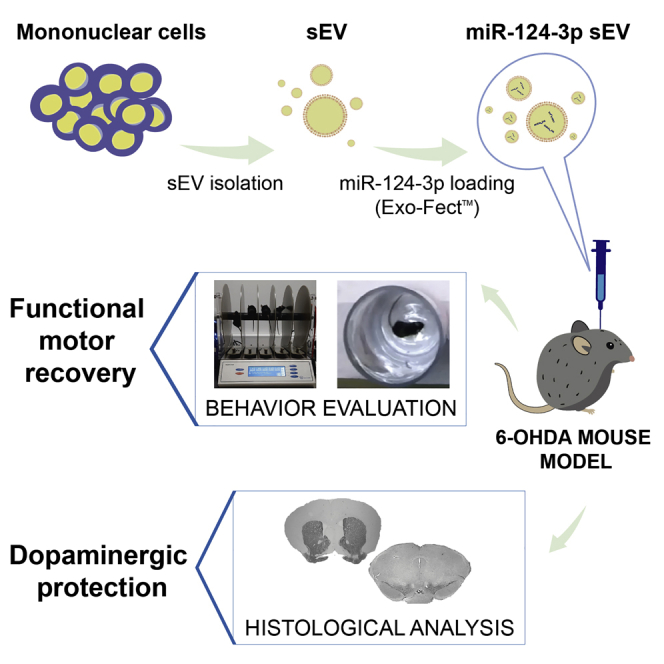
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder and the leading cause of motor disability and death. Esteves et al. report that small extracellular vesicles loaded with microRNA-124-3p protect dopaminergic neurons and counteract motor deficits in a PD mouse model, thus providing a novel, safe, and effective therapy.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease and currently has no effective treatment available. PD is mainly characterized by the progressive degeneration of dopaminergic (DA) neurons in the substantia nigra (SN) of the midbrain, which results in the depletion of dopamine in the striatum. The deficiency of dopamine signaling contributes to motor deficits such as resting tremor, muscle rigidity, and bradykinesia.1 Recent evidence highlight microRNAs (miRNA), small non-coding RNAs that regulate gene expression at the post-transcriptional level, as promising therapeutic targets for neurodegenerative diseases, including PD.2
In particular, the levels of microRNA-124 (miR-124) are decreased in the lesioned brain regions (SN) of PD models and the plasma of patients with PD,3,4 suggesting that it could be a potential biomarker for the diagnosis of PD. At the molecular level, 24% of miR-124-validated target genes are deregulated in PD, stressing the relevance of miR-124 in the regulation of PD.5 In in vivo PD models, miR-124 overexpression counteracted oxidative stress, impaired autophagy, and apoptosis of DA cells, while its knockdown had the opposite effect. These effects were associated with the modulation of AMPK/mTOR, p62/p38, annexinA5 (ANXA5), and Bim signaling pathways.6, 7, 8 Despite this characterization of the cellular and molecular mechanisms that potentially delay/halt disease progression, its therapeutic use is hampered due to its limited and inefficient ability to reach lesioned brain regions. Previously, we have shown that the intracerebroventricular (i.c.v.) delivery of miR-124-loaded polymeric nanoparticles (miR-124 NPs) boosts endogenous neurogenesis in the subventricular zone (SVZ) and increases the number of new neurons in the lesioned striatum, which were associated with the amelioration of PD-related motor deficits.9 However, polymeric delivery systems still have limitations for the delivery of miRNA to DA neurons. In fact, i.c.v.-injected polymeric NPs remain in the SVZ, lining the lateral ventricles, and were not able to migrate to the SN.9 Due to their biocompatibility, safety, and intrinsic ability to target lesioned tissues/cells, small extracellular vesicles (sEVs), also commonly referred to as exosomes, are likely a superior choice, overcoming the limitations observed with viral, liposomal or polymeric drug-delivery systems (e.g., bioaccumulation and toxicity).10, 11, 12, 13 Accordingly, recent strategies based on native or modulated sEVs have been proposed for treating PD.11,14,15 Nevertheless, the therapeutic use of sEVs enriched with miR-124 in PD has not been addressed yet.
Herein, we used sEVs as a biological carrier of the therapeutic entity, miR-124, to boost neuroprotection, neurogenesis, and functional motor recovery in PD preclinical models. In this work, sEVs were isolated from human umbilical cord blood-derived mononuclear cells (hUCB-MNCs) and enriched with miR-124-3p (miR-124-3p sEVs) as a strategy to deliver miR-124-3p to the brain and induce therapeutic effects in PD. sEVs derived from hUCB-MNCs are well characterized in composition, have low immunogenicity, and are easily accessible in cord blood banks.10,16 Several clinical trials are using umbilical cord blood cells to treat neurologic diseases, supporting their safety.16 We found that miR-124-3p sEVs protect DA neurons from 6-hydroxydopamine (6-OHDA)-induced toxicity both in vitro and in vivo, culminating in a full motor function recovery in vivo. This study provides a new and efficient biological sEV-based delivery system for miR-124-3p capable of halting the progression of PD neurodegeneration.
Results
Characterization of hUCB-MNC-derived sEVs
Native sEVs were collected from hUCB-MNCs by differential ultracentrifugation method.17 NP tracking analysis (NTA) showed a canonical size-distribution profile of the native sEVs with an average size of 131 nm (Figure 1A). Native sEVs showed an average zeta potential of -25 mV, while sEVs transfected with the Exo-Fect Exosome Transfection Reagent and a fluorescently labeled miR-124-3p-Cy5 had a zeta potential of -15 mV (Figure 1B). The sEV structure was also characterized by transmission electron microscopy (TEM). Native sEV structure was found to be canonical, with the double-membrane structure clearly observable around cup-shaped particles. The transfection of sEVs with Exo-Fect and miR-124-3p-Cy5 preserved its morphological structure but induced some aggregation (Figure 1C), probably due to the Exo-Fect treatment, as recently reported by us.17 To confirm that samples were enriched in sEVs, we performed western blot analysis for common EV markers and potential contaminants in hUCB-MNC and native sEV samples. Figure 1D shows that sEVs derived from two donors expressed the common EV markers CD9, CD63, and heat shock protein 70 (HSP70), although their expression level seems to be donor dependent. Calnexin, a marker of endoplasmic reticulum and ApoA-1, a component of high-density lipoproteins, were not detected in sEV samples (Figure 1D) but, as expected, were detected in hUCB-MNC samples (Figure 1E). Altogether, these results showed that our samples were enriched in sEVs. Next, the loading efficiency of the sEVs with miR-124-3p-Cy5 was assessed via fluorescence measurement. After the purification step, the sample was separated into a pellet (where sEVs are localized) and a supernatant (where non-bound miR-124-3p-Cy5 is found). Note that only a residual signal was detected in the pellet and supernatant of samples of non-loaded sEVs. When comparing the overall proportion of fluorescence in sEV + miR-124-3p-Cy5 pellet and sEV + miR-124-3p-Cy5 supernatant fractions, we found that the signal intensity present in the sEV pellet is higher than that of the supernatant (Figure 2A). When comparing the overall proportion of fluorescence in each fraction, we found that 74% of the signal was present in the sEV fraction, whereas the remainder was in the supernatant (Figure 2B). Therefore, most of the miR-124-3p was immobilized in sEVs. Then, we quantified miR-124-3p in the loaded sEVs by qRT-PCR (Figure 2C). Relative to native sEVs, there was a fold-change increase in enriched sEV of over 1,000,000 times. Note that the amount of miR-124-3p in the native sEVs is residual (Figures 2C and 2D). We also estimated that the number of miR-124-3p copies in 6.90 × 109 or 1.38 × 1010 sEVs is 6.022 × 106 and 1.2 × 107 copies for native sEVs (not transfected by Exo-Fect) or 1.39 × 1011 and 2.78 × 1011 copies for miR-124-3p sEVs, respectively (Figure S1). Thus, the number of miR-124-3p copies per native sEV is approximately 8.7 × 10-4, i.e., 8.7 copies per 10,000 sEVs, while the number of miR-124-3p copies per miR-124-3p-transfected sEV is approximately 20 copies per sEV. To assess whether the loaded miR-124-3p was exposed or accessible to nucleases after Exo-Fect transfection, sEVs loaded with miR-124-3p were treated with RNase. The RNase treatment did not change miR-124-3p levels in Exo-Fect-modulated miR-124-3p sEVs (Figure 2D). These data suggest that miR-124-3p was either incorporated into the lumen or was entrapped in the membrane of sEVs, therefore inaccessible to nucleases.
Figure 1.

sEV characterization
(A) Size and particle concentration of sEVs collected from human umbilical cord blood-derived mononuclear cells (hUCB-MNCs), evaluated by NTA. (B) Zeta potential analysis of sEVs before and after loading with miR-124-3p (miR-124-3p sEVs). (C) Representative TEM images of native sEVs and sEVs enriched with miR-124-3p. Scale bar: 200 nm. (D) Common native sEV markers (CD63, CD9, and HSP70) and potential contaminants (calnexin and ApoA-1) were further analyzed by western blot, where each lane represents a different donor (n = 2). (E) Non-EV markers were found in hUCB-MNC samples but not in (D) native sEV samples. One donor was used. For all other analyzes, n = 3 biological replicates. Statistical significance was calculated using two-way ANOVA with Sidak’s correction, ∗∗∗∗p < 0.0001. sEV, small extracellular vesicles; NTA, nanoparticle tracking analysis; miR-124-3p sEV, miR-124-enriched small extracellular vesicles; TEM, transmission electron microscopy.
Figure 2.

Modulation and loading of sEVs with miR-124-3p by transfection with Exo-Fect
(A) sEVs (non-loaded or loaded with miR-124-3p-Cy5) were purified, and the fluorescence of the pellet (sEVs) and supernatant (leftover probe) were quantified. (B) After the loading protocol, the signal intensity in the sEV fraction (pellet) corresponded to over 74% of the total fluorescence observed in the sample. Loading efficiency in (B) was calculated using the following formula: fluorescence intensity of the pellet/(fluorescence intensity of the pellet + fluorescence intensity of the supernatant). (C) qPCR against miR-124-3p in modified sEVs showed an increase in miR-124-3p of over 1,000,000 times the control. (D) sEVs loaded with Exo-Fect and miR-124-3p were treated with RNase and re-purified, and miR-124-3p levels in the vesicles were quantified by qPCR. RNase treatment does not change miR-124-3p levels in sEVs. For all the experiments, n = 3. For the qPCR analysis, U6 was used as housekeeping gene. sEV, small extracellular vesicles; miR-124-3p sEV, miR-124-enriched small extracellular vesicles.
miR-124-3p sEVs induced neuronal differentiation in vitro
Previously, we have shown that miR-124 loaded into polymeric NPs was able to boost neurogenesis and the migration of new neurons into the 6-OHDA-lesioned striatum.9 Based on these findings, we first evaluated the ability of miR-124-3p sEVs to induce neurogenesis in vitro by using primary SVZ neural stem cell (NSC) cultures. Based on preliminary studies on endothelial cells, we tested two doses, 1.5 × 109 and 3 × 109 particles (part)/mL of native sEVs, miR-124-3p sEVs, or scrambled (SCR) sEVs. First, we found that both sEVs (3 × 109 part/mL: p < 0.05; n = 6) and miR-124-3p sEVs (1.5 × 109 part/mL: p < 0.05; 3 × 109 part/mL: p < 0.01; n = 7) significantly reduced basal cell death (Figures 3A and 3E; mean absolute value in untreated control cultures: 22.4%, standard error of the mean [SEM]: 1.1), as detected by the propidium iodide (PI) assay. The SVZ NSC culture is a mixed primary cell culture containing immature neural stem cells/progenitor cells (NSCs/NPCs) in distinct neuronal and glial lineages stages.9,18,19 In the NSC untreated cultures used in this study (controls), proliferating cells labeled with Ki-67 represent about 16.6% (mean absolute value; Figure S2, SEM: 2.6) of the total number of cells. Among the Ki-67-positive cell pool, immature NSCs/NPCs labeled with Nestin represent about 53.2% (mean absolute value; Figure 3B; SEM: 1.5), and immature neurons labeled with doublecortin (DCX) represent about 34.3% (mean absolute value; Figure 3C; SEM: 3.6). Moreover, control cultures showed about 20.6% NeuN-labeled mature neurons (mean absolute value; Figure 3D; SEM: 1.5) of the total number of cells. The mean absolute values of untreated cultures were then set to 100% in Figures 3B–3D. Next, we evaluated the effects of sEVs on SVZ cell proliferation and neuronal differentiation. Overall, total cell proliferation, analyzed by the labeling against Ki-67, was not affected by sEVs or miR-124-3p sEVs compared with control cultures (Figure S2). Then, we analyzed the effect of sEVs in the proliferation of immature cells by performing co-labeling against Nestin or DCX, a marker for newly born neurons, 48 h after treatments. As shown in Figure 3, miR-124-3p sEVs reduced significantly the proliferation of NSCs/NPCs at both doses (Nestin+/Ki-67+; Figures 3B and 3F; 1.5 × 109 part/mL; n = 6: p < 0.05 and 3 × 109 part/mL; p < 0.001; n = 3), while native sEVs significantly reduced the proliferation of NSCs/NPCs at the dose of 3 × 109 part/mL (Nestin+/Ki-67+; Figure 3B; p < 0.001; n = 3). Regarding the proliferation of newly born neurons, we found no significant effect in SVZ cells treated with miR-124-3p sEVs (DCX+/Ki-67+; Figures 3C and 3G; n = 5) compared with control cultures (n = 6), while native sEVs significantly reduced the proliferation at both doses (DCX+/Ki-67+; Figures 3C and 3G; 1.5 × 109 part/mL: p < 0.01 and 3 × 109 part/mL: p < 0.05; n = 5). No significant effect was observed in the proliferation of NSCs/NPCs or newly born neurons in SVZ cells treated with SCR sEVs (Figures 3B and 3C, respectively; n = 3) compared with control cultures. Then, to evaluate the effect on neuronal differentiation, SVZ cells were cultured in the presence of sEVs for 7 days and processed for immunostaining against NeuN, a neuronal-specific nuclear protein. We have found that miR-124-3p sEVs increased the percentage of NeuN-positive cells compared with control cells (n = 7) (Figures 3D and 3H; 1.5 × 109 part/mL, n = 6, and 3 × 109 part/mL, n = 5: p < 0.0001) or SCR sEV- (Figure 3D; 1.5 × 109 part/mL, n = 3: p < 0.01 and 3 × 109 part/mL, n = 3: p < 0.05) or sEV-treated cells (Figure 3D; 1.5 × 109 part/mL: p < 0.001, n = 3) by about 1.8-fold. This effect in neuronal differentiation was dependent on miR-124-3p because native sEVs and SCR sEVs did not alter the number of NeuN-positive cells (Figure 3D). To exclude the effect of the transfection agent per se, SVZ cultures were also treated with Exo-Fect and miR-124-3p in the absence of sEV. This treatment did not change cell survival (Figure 3A), cell commitment (Figures 3B and 3C), and neuronal differentiation (Figure 3D) compared with the control cultures. Altogether, data suggest that miR-124-3p sEVs induce neuronal differentiation of SVZ NSCs in vitro.
Figure 3.

miR-124-3p sEVs reduce NSCs basal death and immature cell proliferation while increasing neuronal differentiation
Cell survival (A and E), commitment (B, C, F, and G), and neuronal differentiation (D and H) were evaluated in SVZ cells treated with sEVs transfected or not with miR-124-3p or scramble (SCR) using Exo-Fect. Two doses of sEVs were used: 1.5 × 109 (gray symbols) or 3 × 109 particles/mL (black symbols). An additional control was done by incubating cells with Exo-Fect and miR-124-3p in the absence of sEVs (ExoF + miR-124-3p). Representative fluorescence photomicrographs of dead NSCs (E; white arrows), proliferating NSCs stained against Nestin/Ki-67 (F; red arrows), proliferating neuroblasts stained against DCX/Ki-67 (G; white arrows), and (H) mature neurons stained against NeuN in control (CTR) cultures and in 3 × 109 particles/mL of sEVs or miR-124-3p sEV-treated cultures. Nuclei are shown in blue, Ki-67 in green, Nestin in gray, and PI, DCX, and NeuN in red. Scale bar: 20 μm. Cell viability (E) was evaluated by PI incorporation and proliferation of (F) NSCs and (G) neuroblasts was evaluated by colocalization against Ki-67 and Nestin or DCX, respectively, 2 days after treatments. (H) Neuronal differentiation was evaluated by staining against NeuN 7 days after treatments. (B–D) Data are expressed as a percentage of control (mean ± SEM; n = 3–7). ∗∗∗∗p < 0.0001, ∗∗p < 0.01, and ∗p < 0.05 versus control; +p < 0.05 versus 1.5 × 109 particles/mL sEVs; ###p < 0.001, and ##p < 0.01 versus 1.5 × 109 particles/mL miR-124-3p sEVs; and $p < 0.05 versus 3 × 109 particles/mL miR-124-3p sEVs, using unpaired, two-tailed t test. SVZ NSCs, subventricular zone neural stem cells; CTR, control; sEV, non-loaded small extracellular vesicles; miR-124-3p sEV, miR-124-3p-loaded small extracellular vesicles; SCR, scramble miRNA; ExoF, Exo-Fect; PI, propidium iodide; DCX, doublecortin.
miR-124-3p sEVs do not increase the number of newly born neurons found in the 6-OHDA-lesioned striatum in vivo
To evaluate the functional effects of miR-124-3p sEVs in vivo, 20 pmol of miR-124-3p per 2.3 × 1010 particles (10 μg sEVs) were injected into the right lateral ventricles of adult C57BL/6 mice (Figure 4A), as previously described by us.9 In accordance with Figure S1B, the number of miR-124-3p copies in 2.3 × 1010 sEVs of native sEVs or miR-124-3p sEVs (10 μg sEV) is 2 × 107 and 4.6 × 1011 copies, respectively.
Figure 4.

miR-124-3p sEVs do not increase the number of SVZ-derived newly born neurons in the lesioned striatum of 6-OHDA-treated mice.
(A) Design and timeline of the animal experimental procedure. Male C57Bl/6 mice were subjected to unilateral injection of 6-OHDA into the right striatum followed by an i.c.v. injection with miR-124-3p sEVs, SCR sEVs, sEVs, or saline. Then, mice received BrdU injections (every 12 h) during the following 3 days after surgery. Behavioral tests were performed on day -1 and on weeks 1, 2, and 4 after stereotaxic injections. After 4 weeks, mice brains were collected for histological processing. (B) Representative confocal photomicrographs of (B.1) SVZ, striatal and (B.2, B.3) midbrain parenchyma 24 h after i.c.v. injection of non-loaded sEVs labeled with PKH67 (sEV-PKH67; shown in green). White arrows show sEV-PKH67 close to the cell nuclei. TH labeling (red) depicts DA neurons in the SN and nuclei are counterstained with Hoechst (blue). Scale bar: 20 μm. (C) Representative confocal digital images of BrdU (green), NeuN (red), and Hoechst (blue) staining observed in the striatum of saline-treated mice, 6-OHDA-treated mice, or miR-124-3p sEV-treated 6-OHDA mice. Scale bar: 20 μm; white arrows highlight NeuN+/BrdU+ cells. (D) The bar graph depicts the total number of NeuN+BrdU+ cells found in the striatum of mice 4 weeks after treatments. Data are expressed as mean ± SEM, n = 3 mice. ∗∗p < 0.01 and ∗p < 0.05 versus saline-treated mice (control); #p < 0.05 versus miR-124-3p sEV-treated 6-OHDA-lesioned mice group using one-way ANOVA followed by Tukey’s multiple comparison test. 6-OHDA, 6-hydroxydopamine; SCR sEV, scramble-loaded small extracellular vesicles; sEV, non-loaded small extracellular vesicles; miR-124-3p sEV, miR-124-3p-loaded small extracellular vesicles; i.c.v., intracerebroventricular; DA, dopaminergic; BrdU, 5-bromo-2′-deoxyuridine; SN, substantia nigra; TH, tyrosine hydroxylase; SVZ, subventricular zone.
First, we performed a qualitative evaluation of the biodistribution of sEVs 24 h after the injection of hUCB-MNC-derived sEVs labeled with PKH67, a fluorescent membrane lipophilic dye commonly used to label sEVs.17,20 PKH67-labeled sEVs were detected lining the SVZ (Figure 4B.1) of both lateral ventricles and in the striatal parenchyma at the vicinity of the intraventricular injection. Importantly, sEVs were also detected in midbrain sections (Figure 4B.2), mainly in the aqueduct but also in the SN co-localizing with tyrosine hydroxylase (TH)-positive DA neurons (Figure 4B.3), the most susceptible neuronal population in PD. Then, we evaluated the ability of miR-124-3p sEVs to trigger the migration of new neurons likely derived from SVZ into the 6-OHDA-lesioned striatum, the most likely regenerative response in PD.9 Consistent with our previous study,9 the striatal administration of 6-OHDA increased the number of newly born neurons (NeuN+/BrdU+) found in the lesioned striatum compared with saline-treated mice (Figures 4C and 4D; mean absolute value in saline-treated mice: 9.37%, SEM: 1.2; p < 0.05, n = 3). A similar increase was observed in 6-OHDA-lesioned mice administered with SCR sEVs (p < 0.01, n = 3). However, the i.c.v. administration of miR-124-3p sEVs did not alter the number of newly born neurons in the lesioned striatum compared with saline-treated mice (n = 3) (Figures 4C and 4D). These results suggest that our formulation does not increase the number of newly born neurons in the lesioned striatum of 6-OHDA-treated mice.
miR-124-3p sEVs promote neuroprotection against 6-OHDA-induced toxicity in PD models
Before proceeding to the in vivo studies, we first evaluated the putative neuroprotective effect of miR-124-3p sEVs in an in vitro model of PD using the Cell Counting Kit-8 (CCK-8) assay. The viability of N27 DA cells decreased about 30% after exposure to 50 μM 6-OHDA for 24 h, while 3 × 109 part/mL miR-124-3p sEVs were able to protect cells from 6-OHDA-induced toxicity (Figure S3; p < 0.05, n = 3). Native sEVs (1.5 × 109 part/mL: n = 3; 3 × 109 part/mL: n = 2) did not promote neuroprotection, suggesting that the protective effect was due to miR-124-3p and not to the sEV itself. Moreover, none of the sEV formulations per se were toxic to DA cells (Figure S3). Next, we validated these results using the 6-OHDA-induced PD model in vivo. In accordance with in vitro studies, miR-124-3p sEV treatment fostered significant neuroprotection of DA neurons, as detected by the number of TH-positive neurons compared with the 6-OHDA-lesioned group (Figures 5A and 5B; p < 0.001, n = 4). Also, the 6-OHDA injection caused a significant decrease in the intensity and percentage of area occupied by TH-positive fibers in the striatum compared with saline-treated animals (set to 100%, n = 4; Figures 5A and 5C; p < 0.0001), while the exposure to miR-124-3p sEVs significantly reduced the loss of TH staining in striatal fibers (Figures 5A and 5C; intensity and percentage of area: p < 0.01, n = 4). As expected, the treatment with SCR sEVs did not protect striatal TH-positive fibers and TH-positive neurons in SN from 6-OHDA-induced toxicity, suggesting that miR-124-3p is responsible for the neuroprotection of the nigrostriatal pathway in lesioned animals. Altogether, these results suggest that the administration of miR-124-3p sEVs effectively protected DA neurons against the toxicity induced by 6-OHDA.
Figure 5.

miR-124-3p sEVs counteract dopaminergic degeneration induced by 6-OHDA
(A) Representative photomicrographs of SN and striatal sections immunostained for TH. Scale bars: 200 and 500 μm, respectively.
(B and C) Quantitative analysis of (B) TH+ cells in the SN and (C) intensity and area occupied by TH+ fibers 4 weeks after stereotaxic injections (black symbols: intensity; gray symbols: percentage of area). Data are expressed as mean ± SEM, n = 4 mice. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001 versus saline-treated mice (control; C, set to 100%); ###p < 0.001 and ##p < 0.01 versus 6-OHDA-lesioned mice group; $$$p < 0.001 and $p < 0.05 versus miR-124-3p sEV + 6-OHDA-lesioned mice group using one-way ANOVA followed by Tukey’s multiple comparison test. 6-OHDA, 6-hydroxydopamine; SCR sEV, scramble-loaded small extracellular vesicles; sEV, non-loaded small extracellular vesicles; miR-124-3p sEV, miR-124-3p-loaded small extracellular vesicles; TH, tyrosine hydroxylase; SN, substantia nigra; ST, striatum; PD, Parkinson’s disease.
miR-124-3p sEVs counteracted PD-related motor deficits in the 6-OHDA mouse model
To unveil if our formulation counteracts the motor symptoms of 6-OHDA-lesioned mice, we performed the rotarod and apomorphine-induced rotation behavioral tests (Figure 4A). These tests, widely used in PD models, evaluate motor function, balance, and grip strength (rotarod) and nigrostriatal dopamine depletion (apomorphine-induced rotation test). In the rotarod test, the latency time to fall off from the rod was reduced in 6-OHDA-lesioned mice (n = 5) compared with saline-treated mice (n = 4) (about 70% of saline) (Figure 6A; p < 0.0001). Notably, the treatment with miR-124-3p sEVs significantly increased the latency time to fall off from the rod by about 70% compared with 6-OHDA-lesioned animals (p < 0.0001, n = 5), resulting in no difference with saline-treated mice (Figure 6A). These results indicate that miR-124-3p sEVs fully counteracted 6-OHDA-induced motor performance and coordination deficits. We further evaluated motor behavior by the apomorphine-induced rotation test. Apomorphine is a dopamine receptor agonist that can uncover asymmetrical dopamine loss (occurring with the presently used unilateral 6-OHDA administration) through its induction of contralateral rotations. As expected, saline-treated mice exhibited a net rotation near zero, while 6-OHDA-lesioned mice showed a significant increase (p < 0.0001, n = 4 for saline and n = 5 for the 6-OHDA group) in the contralateral net rotations that was reverted by the miR-124-3p sEVs treatment (Figure 6B; week 2: p < 0.05; week 4: p < 0.01, n = 4). The treatment of lesioned animals with SCR sEVs (n = 5) or sEVs (n = 3) showed significant differences compared with miR-124-3p sEVs treatment; however, they did not reveal any differences when compared with 6-OHDA-injured animals in both rotarod (Figure 6A) and apomorphine (Figure 6B) tests. Overall, the behavior data attest to the therapeutic efficacy of miR-124-3p sEVs in counteracting motor deficits in the PD mouse model.
Figure 6.

miR-124-3p sEVs ameliorate motor symptoms in a mouse model of Parkinson’s disease.
(A and B) Behavioral data. (A) Latency remaining on the rotarod was tested in the first 2 weeks post 6-OHDA lesion. Data shown represent the four-trial average time on the rotarod. Data are expressed as mean of latency to fall from rod ± SEM; saline: n = 4; 6-OHDA and miR-124-3p sEV + 6-OHDA: n = 5; SCR sEV + 6-OHDA: n = 6 and sEV + 6-OHDA: n = 3. (B) Number of contralateral rotations induced by apomorphine were measured for 45 min, 2 and 4 weeks after 6-OHDA lesion. Data are expressed as mean of contralateral net turns ± SEM; saline and miR-124-3p sEV + 6-OHDA: n = 4; 6-OHDA and SCR sEV + 6-OHDA: n = 5; and sEV + 6-OHDA: n = 3. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, and ∗∗p < 0.01 versus saline-treated mice (control); ####p < 0.0001, ###p < 0.001, ##p < 0.01, and #p < 0.05 versus 6-OHDA-lesioned mice group; $$$$p < 0.0001, $$$p < 0.001, $$p < 0.01, and $p < 0.05 versus miR-124-3p sEV + 6-OHDA-lesioned mice group using one-way ANOVA followed by Tukey’s multiple comparison test. 6-OHDA, 6-hydroxydopamine; SCR sEV, scramble-loaded small extracellular vesicles; sEV, non-loaded small extracellular vesicles; miR-124-3p sEV, miR-124-3p-loaded small extracellular vesicles.
Discussion
To the best of our knowledge, this is the first study using hUCB-MNC-derived sEVs as a biological vehicle to deliver miR-124 in the context of PD. miRNAs are increasingly considered impactful molecular agents for the treatment of this neurodegenerative disease. miR-124 is particularly attractive as it is downregulated in the SN of PD models and plasma of patients with PD, and enhancing its expression boosts neuroprotection and neurogenesis in PD models.3,4,6,7,9,21,22 We have previously found that miR-124-loaded NPs induce SVZ neurogenesis and counteract motor deficits in a 6-OHDA PD model in vivo.9 miR-124 regulates several stages of neuronal differentiation, from early neuronal commitment to maturation by targeting jagged canonical Notch ligand 1 (JAG1),23,24 enhancer of zeste homolog 2 (EZH2),25 signal transducer and activator of transcription 3 (STAT3),26 and SRY-box transcription factor 9 (SOX9).23 Moreover, miR-124 enhances axonogenesis and neurite maturation by regulating cytoskeleton proteins and ras homology growth-related (RhoG) pathway, respectively.27,28 In our mixed SVZ cell culture, containing distinct cell populations at different stages of development, miR-124 may regulate various steps during neuronal lineage differentiation and maturation. Interestingly, overexpression of miR-124 (among other factors) can also be involved in transforming human fibroblasts into functional mature neurons.29 Some reports also suggest a synergistic role for miR-9 and miR-124 on the specification and survival of DA neurons in mice.30 Additionally, several other studies have also reported the neuroprotective role of miR-124 in PD. miR-124-3p was reported to attenuate DA degeneration found in in vitro and in vivo PD models by targeting several signaling pathways, including the ANXA5/ERK pathway,8 STAT3,31 AMPK/mTOR pathway,7 the calpain 1/p25/cdk5 signaling,3 EDN2,32 and Bim.33
Altogether, these evidence highlights the relevance of increasing miR-124 levels as a novel therapeutic strategy for PD. However, miRNAs are easily degraded by nucleases, and their delivery presents poor uptake efficiency due to their hydrophilic nature and negative charge.34 To overcome these challenges, we previously developed polymeric NPs able to release miR-124.9 Knowing the limitations of the previously developed NP-based delivery carriers, such as degradation, bioaccumulation, retention in the basal lamina, and toxicity, we developed a new and biological sEV-based carrier to deliver miR-124 in the brain. sEVs have attracted considerable attention as drug-delivery vehicles for treating several brain diseases. It has been previously reported that the administration of sEVs loaded with anti-inflammatory or antioxidant drugs (curcumin and catalase, respectively) significantly decreased brain inflammation35 and increased neuronal survival in a 6-OHDA PD mouse model.11 Also, blood-derived sEVs loaded with dopamine were shown to reach the striatum and SN, thus promoting the amelioration of the disease phenotype in the same model.14
In this study, native sEVs were obtained from hUCB-MNCs and then loaded with miR-124-3p or SCR using the Exo-Fect Exosome Transfection Reagent.17 The native sEVs obtained had an average size of 131 nm and an average zeta potential of around -25 mV, while miR-124-loaded sEVs had a zeta potential of -15 mV. This shift in zeta potential is likely due to the adsorption of some Exo-Fect molecules to the membrane of sEVs. These results agree with our recent study showing that Exo-Fect interferes with sEV membrane structure, albeit not promoting the disruption of sEV membrane integrity,17 as well as other studies showing that Exo-Fect was an efficient strategy to load sEVs with small nucleic acids.36 Because residual non-absorbed Exo-Fect onto sEVs may have the ability to transfect cells in a similar way as it does to EVs, we have treated SVZ cell cultures with Exo-Fect and miR-124-3p in the absence of sEVs. Our results showed that in contrast to Exo-Fect-modulated miR-124-3p sEVs, a mixture of Exo-Fect and miR-124-3p per se did not alter cell survival, cell commitment/proliferation, and neuronal differentiation compared with the control cultures. Therefore, our data indicate that the miR-124-3p was delivered to cells via sEVs. Using fluorescent-labeled miRNA, we found that 74% of the signal was present in the sEV fraction, suggesting that most miR-124-3p-Cy5 was immobilized in sEVs. Other studies used a similar quantification strategy, showing that upon transfection of sEVs with Exo-Fect, around 80% of the fluorescent signal remained in the sEV fraction of the reaction.17,37 Altogether, these data confirm that Exo-Fect treatment efficiently transfect sEVs with miR-124-3p. However, the exact location of the miR-124-3p in the sEV remains unknown. Importantly, Exo-Fect-modulated miR-124-3p sEVs and those treated with RNase had no change in miR-124-3p levels, showing that the miR-124-3p was inaccessible to nucleases. Further tests are needed to disclose whether miR-124-3p is entrapped in the sEV membrane or its lumen.
Then, we evaluated the effect of native sEVs or sEVs loaded with miR-124-3p or SCR in SVZ neurogenesis. We found that native sEVs are able to counteract basal cell death and reduce the proliferation of NSCs/NPCs and neuroblasts in SVZ cell cultures. sEV cargo includes several bioactive molecules, namely proteins, mRNA, miRNA, and lipids, among others. Some evidence suggests that the biological effects of sEVs are mainly dependent on the intravesicular miRNA signature. We have previously characterized the cargo of the sEVs used in this study in terms of miRNA by RNA sequencing (RNA-seq).10 Importantly, miR-124 was not found in the heatmap of miRNA in these sEVs. Some of the enriched miRNAs were reported to decrease proliferation in cancer cells, such as miR-let-7a.38 Interestingly, Ohno and colleagues showed that intravenously injected GE11-targeted exosomes containing miR-let-7a inhibit tumor growth in mice.39 Other miRNAs enriched in the sEVs were reported to increase cell survival, such as miR-223 and miR-21.40,41 These putative miRNA feedback loops may be responsible for the bioactivity of sEVs found in our study. In addition, these miRNAs may have selectivity toward a specific cell phenotype. The SVZ cell culture is a mixed primary cell culture containing immature NSCs/NPCs, cells in distinct stages of the neuronal, oligodendroglial, and astrocytic lineages. Therefore, differences in the dynamics on other cell populations, not studied in this work, could justify the proliferation data that we obtained. In the particular case of miR-124-3p sEVs, we found a reduction of the population of proliferative immature cells (Nestin+/Ki-67+) and an increase in the number of mature neurons (NeuN) without any significant effect on neuroblast proliferation (DCX+/Ki-67+). One possibility is that some NSCs/NPCs undergo direct neuronal differentiation without proliferating, as reported by others.42 This process is accompanied by a decrease in the NSCs pool. The loaded miR-124-3p may cooperate with intrinsic sEV miRNA cargo, fostering neuronal differentiation.43 For example, it was shown that miR-21-5p and miR-124-3p delivered by NPs may cooperate to induced a pro-osteogenic differentiation effect, which might impact the regeneration of osteoporotic bone.44 We hypothesize that some feedback loops may also occur when loading sEVs with miR-124-3p, boosting neuronal differentiation. Moreover, we cannot exclude the involvement of other components in sEVs in their biological activities (e.g., growth factor-associated proteins, mRNA, lipids, etc.). Better knowledge about the cargo of these vesicles may help design more efficient bioengineering drug-delivery platforms using sEVs as a biological vehicle of therapeutic molecules. In terms of a gene or RNA delivery, an innovative approach would be to empty sEVs of their cargo before loading them with the therapeutic agent of interest. These innovative drug-delivery approaches should be explored in future studies. The in vivo data regarding neurogenesis disagrees with the in vitro data and our previous data using miR-124-loaded NPs.9 While in our previous report, we performed the overall analysis of neurogenesis in the SVZ, olfactory bulb, and striatum, in the current work, our analysis focused on pathologically relevant brain regions only, namely the striatum. Therefore, we cannot exclude that miR-124-3p sEVs may also impact olfactory bulb neurogenesis in vivo. On the other side, we also showed that miR-124-loaded NPs increase SVZ neurogenesis in vitro but not in the in vivo model of ischemic stroke.45 The net effect in SVZ neurogenesis in vivo may depend on the pathology, experimental conditions, and the type of drug-delivery system used.
Regarding biodistribution in vivo, we found previously that polymeric NPs were detected only in the SVZ, meaning they were easily trapped inside the basal lamina, thus limiting their diffusion across the brain parenchyma.46 sEVs were found in the SVZ, striatum, and SN, particularly DA neurons. Due to their biological nature, sEVs have the exceptional ability to interact with recipient cell proteins having a specific cell tropism that can target them to disease tissues, which contrasts to a relatively lower penetration efficiency of polymeric NPs. For example, the intranasal administration of mesenchymal stem cell-derived sEVs loaded with glucose-coated gold NPs led to a higher brain accumulation/distribution than free gold NPs.47 Furthermore, the mesenchymal stem cell-derived sEVs accumulated preferentially in injured brain regions, including the striatum of 6-OHDA PD rat model, and the large majority of the sEVs were found in neurons.12 Interestingly, Haney and collaborators developed macrophage-derived exosomes loaded with catalase, a potent antioxidant, to treat PD.11 These exosomes showed superior intraneuronal accumulation (PC12 cells) compared with the considerably lower uptake of polymeric PLGA NPs or liposomes. Ultimately, intranasal delivery of catalase-loaded exosomes exerted neuroprotective effects in in vivo 6-OHDA model of PD. Other studies suggest that membrane vesicles fused more effectively with the plasma membrane of cancer cells than polymeric NPs and enabled the delivery of hydrophobic photosensitizers in spheroids and in vivo tumors more efficiently, thereby enhancing the therapeutic efficacy.48 This superior uptake of sEVs occurs mainly due to adhesion proteins, tetraspanins, and integrins, which are absent in synthetic NPs. Moreover, sEVs may comprise additional advantages over synthetic nanocarriers and cell-mediated drug delivery, avoiding the rapid clearance by phagocytosis and engulfment by lysosomes and toxicity.49 sEVs can also cross the blood-brain barrier and reach their targets. In fact, sEVs loaded with catalase, dopamine, or GAPDH small interfering RNA (siRNA) were detected in PD mouse brain after intravenous administration.11,14,50 Altogether, these evidence suggest that sEVs have a superior ability than polymeric NPs to enhance the delivery of incorporated drugs to target lesioned brain regions/cells, ultimately increasing therapeutic drug efficacy. Future research should better investigate in vivo trafficking and biodistribution of sEVs and their impact on pathological lesioned brain regions/cells for efficient and specific drug delivery.
Importantly, our study shows that miR-124-3p sEVs counteract DA degeneration and PD-related motor deficits in the 6-OHDA mouse model of PD. Some have demonstrated the bioactivity of sEVs themselves in the context of neurodegenerative diseases, including PD. For example, Chen and collaborators showed that sEVs isolated from human umbilical cord mesenchymal stem cells promoted the proliferation of 6-OHDA-intoxicated SH-SY5Y cells while inhibiting apoptosis. In vivo, these sEVs reduced DA neuronal loss in the SN while increasing dopamine levels in the striatum, alleviating the apomorphine-induced rotational behavior in a 6-OHDA rat model of PD.51 In our study, native sEVs and SCR sEVs did not protect against 6-OHDA-induced lesion both in vitro and in vivo. Thus, the therapeutic neuroprotective effect promoted by miR-124-3p sEVs in the in vivo PD model is likely mediated specifically by the miR-124-3p.
In our study, striatal injection of 6-OHDA caused ∼80% cell death in SN and a strong DA denervation in the striatum. However, the 6-OHDA model does not fully mimic all the pathophysiologic mechanisms in PD. The toxin 6-OHDA induces mitochondrial impairment, proteasomal and lysosomal dysfunction, and neuroinflammation. Before moving on to clinical trials, it is imperative to test the therapeutic effect of miR-124-3p sEVs in other relevant preclinical models that address distinct but complementary aspects of PD pathophysiology. Toxin-induced models (e.g., 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP], paraquat, and rotenone) also induce oxidative stress and DA neurodegeneration but cause a severe and fast DA death, which does not resemble the disease’s evolution in humans and fails to induce alpha-synuclein accumulation and aggregation. Alternatively, genetic models targeting PD-related genes, such as SNCA, LRRK2, and PINK1, could mimic other pathologic aspects of the disease.52 While neurotoxins can be used to evaluate neuroprotective mechanisms, genetic models may be used to study mechanisms associated with alpha-synuclein aggregation and prodromal symptoms. Moreover, to approach the clinic translation, sEVs should be administered ideally after the lesion and delivered using a non-invasive approach, such as the intranasal route.11,12,47
Conclusions
Herein, we reported for the first time the therapeutic potential of miR-124-3p-enriched sEVs for PD treatment. The delivery of miR-124-3p by sEVs induced neuronal differentiation of SVZ NSCs under physiological conditions in vitro. In vivo, we showed that i.c.v. injection of miR-124-3p sEVs protected the nigrostriatal pathway against 6-OHDA-induced neurodegeneration and significantly improved motor performance of lesioned mice. Our findings support the use of miR-124-3p sEVs as a new and efficient therapeutic approach to halt PD pathogenesis progression and open new perspectives for the treatment of other neurodegenerative diseases.
Materials and methods
sEV isolation
sEVs were isolated from conditioned medium of hUCB-derived MNC culture. All hUCB samples were obtained upon signed informed consent, in compliance with Portuguese legislation. The collection was approved by the ethics committee of Centro Hospitalar e Universitário de Coimbra, Portugal (HUC-01-11, approved on March 3, 2011). The samples were stored and transported to the laboratory in sterile bags with anticoagulant solution (citrate-phosphate-dextrose) and processed within 48 h after collection. Briefly, MNCs were isolated by density gradient separation (Lymphoprep - STEMCELL Technologies SARL, Grenoble, France). To obtain MNC-derived sEVs, hUCB MNCs were cultured in X-VIVO 15 serum-free cell culture medium (Lonza Group, Basel, Switzerland) supplemented with Flt-3 (100 ng/mL, PeproTech) and stem cell factor (100 ng/mL, PeproTech) under hypoxia (0.5% O2) conditions for 18 h. Conditioned medium was collected and centrifuged at 300 × g, for 10 min, at 4°C to remove cells (pellet). Supernatant was collected for a new tube and centrifuged at 2,000 × g, for 20 min, at 4°C to deplete cellular debris. Then, sEVs were purified by differential centrifugation as described previously.17 Briefly, samples were ultracentrifuged twice at 10,000 × g, for 30 min, at 4°C, and the pellet was discarded. The supernatant was ultracentrifuged at 100,000 × g, for 2 h, at 4°C, to pellet sEVs. Finally, the pellet obtained was washed with cold PBS, ultracentrifuged again at 100,000 × g, for 2 h, at 4°C, resuspended in 150 μL of cold PBS, and stored at -80°C. Ultracentrifugation steps were performed using Optima XPN 100K ultracentrifuge (Beckman Coulter, Brea, CA, USA) with a swinging bucket rotor SW 32 Ti and 28.7 mL polyallomer conical tubes (Beckman Coulter). We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK: EV210107).53
sEV characterization by NTA
Analysis of sEV size distribution and concentration was performed through NTA by NanoSight NS300 (Malvern Instruments, Malvern, UK). The system used an O-Ring Top Plate, and the sample was injected manually at an approximate flow of 1 mL every 20 s. sEVs were diluted in PBS until they reached a concentration between 15 and 45 particles/frame. Five 30 s measurements were done for each sample with the camera level set at 16. All the videos were processed with NTA 3.2 analytical software, using the software threshold between 2 and 4 depending on the quality of the videos.
sEV characterization by protein quantification
The total protein content of sEVs was measured by Micro BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Briefly, sEV samples were diluted 22 times in 2% (v/v) sodium dodecyl sulfate (SDS) to disrupt the sEV membrane, and subsequently, 50 μL of the previous mix was pipetted, in duplicate, into 96-well cell culture plates. Reaction solution provided in the kit was added and incubated for 2 h at 37°C. Next, the plates were equilibrated at room temperature (RT) for 15 min, and the absorbance was read at 562 nm in the microplate reader Synergy H1 (Biotek, Winooski, VT, USA). Total protein concentration was determined using a linear standard curve established with bovine serum albumin (BSA).
sEV characterization by zeta potential
NanoBrook ZetaPALS Potential Analyzer (Brookhaven Instruments, Holtsville, NY, USA) was used for sEV surface-charge measurement. Briefly, 5 μL purified sEVs were diluted in 1,500 μL of biological grade ultrapure water (Thermo Fisher Scientific, Portsmouth, NH, USA) and filtered twice through a 0.2 μM filter. sEVs were then placed in a disposable polystyrene cuvette, and the electrode was immersed within the cuvette. Each sample was measured five times (using Smoluchowski module) at RT.
Western blot analysis
Western blot analysis was performed to detect EV markers and contaminants in EV samples, as previously described.17 Briefly, up to 15 μL of concentrated EV preparations in PBS (0.5 to 4μg) were mixed with 5 μL 4× Laemmli buffer (0.25 M Tris base, 8% SDS, 40% glycerol, 200 mg bromophenol blue, 10% 2-mercaptoethanol) and boiled at 96°C for 10 min. For the analysis of tetraspanins, Laemmli buffer was prepared without reducing agents. Samples were loaded in 30 μL wells of Any kD Mini-PROTEAN TGX Stain-Free Protein Gel (Bio-Rad) and gel electrophoresis was performed in 1× Tris/Glycine/SDS buffer prepared from a commercial 10× concentrated stock (10× Tris/Glycine/SDS Electrophoresis Buffer; Bio-Rad) at a constant voltage of 120 V for 75 min. Afterward, gels were placed in blotting buffer (25 mM Tris, 192 mM glycine, 20% methanol in water) for 10 min to equilibrate. Then, the gel was stacked on top of a nitrocellulose membrane (GE Healthcare), and both were assembled within a transfer system. The transfer was performed in wet conditions at 200 mA for 90 min. Then, the membrane was blocked in a 1:1 PBS-Tween 20 (PBS-T; 0.2% (v/v)) with Intercept Blocking Buffer (Li-cor) solution for 1 h at RT. Afterward, membranes were incubated overnight at 4°C with the appropriate primary antibodies and according to the manufacturer recommendation (antibody details below). Then, membranes were incubated for 1 h at RT with secondary antibodies. Membranes were viewed in the Odyssey CLx system (Li-cor) at the 700 and 800 nm wavelengths. Antibodies used in this study were as follows: CD63 (BD Pharmingen, Franklin Lakes, NJ, USA), ApoA-1 (Santa Cruz Biotechnology), Calnexin (Santa Cruz Biotechnology), HSP70 (Enzo Life Sciences), CD9 (BD Pharmingen), and IRDye 800CW Goat anti-Mouse IgG Secondary Antibody (Li-cor). To detect non-EV markers in hUCB-MNC samples, MNCs were lysed in 2× RIPA buffer for 30 min in ice. The cellular extracts were centrifuged at 16,000 RPM for 10 min at 4°C, and the supernatant containing the proteins was stored at -80°C. The protein (4 mg) was boiled in 1× Laemmli buffer for 5 min and then loaded in a 12% SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes (Amersham Hybond 0.45 PVDF). The membranes were blocked with 5% BSA in TBS-T for 1 h. Then, the membranes were incubated with the monoclonal antibodies against calnexin (Abcam) or ApoA-1 (Affinity Biosciences) overnight at 4°C. The next day, the membranes were washed (3 times for 5 min in TBS-T) and then incubated with polyclonal goat anti-mouse immunoglobulins/HRP (Cell Signaling) for 1 h at RT. Proteins were visualized using the enhanced chemiluminescence detection system (ECL, Advansta).
sEV characterization by TEM
TEM analyses of sEVs were performed as previously described.17 Briefly, samples were diluted 1:1 in 4% (v/v) paraformaldehyde (PFA) and placed on Formvar-carbon-coated grids (TAAB Technologies) for 20 min at RT. After washing 4 times with PBS, grids were placed on a drop of 1% (v/v) glutaraldehyde for 5 min, followed by 5 washes with distilled water, for 1 min each. The grids were incubated in the dark with uranyl-oxalate solution (pH = 7) for 5 min and then placed on ice in contact with a solution of methyl cellulose (9:1) for 10 min. sEV imaging was obtained using a Tecnai G2 Spirit BioTWIN electron microscope (FEI) at 80 kV.
Exo-Fect loading of sEVs
sEVs were loaded with miR-124-3p or SCR (GE Healthcare Dharmacon, Lafayette, CO, USA) by Exo-Fect Exosome Transfection Reagent (10 μL; System Biosciences, Palo Alto, CA, USA) for 10 min at 37°C. Samples were purified using ExoQuick, according to the manufacturer’s instructions. Briefly, samples were incubated with ExoQuick reagent in 1:5 (v/v) for 30 min on ice and centrifuged at 13,000 × g for 3 min, the supernatant was discarded, and the pellets were resuspended in PBS. For in vitro studies, final samples contained 20 pmol (0.27 μg) of miR-124-3p or SCR per 3.5 × 1010–7.5 × 1010 particles (80–90 μg sEVs), whereas for in vivo studies, the final samples contained 20 pmol (0.27 μg) of miR-124-3p or SCR per 2.3 × 1010 part (10 μg sEVs) in a total volume of 5 μL. Twenty pmol miR-124-3p was incubated with Exo-Fect in the absence of sEVs and was used as control. sEV, SCR sEV, and miR-124-3p sEV formulations were eventually stored at -80°C for the next experiments. The emission spectra of all samples, excited at λex = 5 nm, was measured from λem = 5 nm until λem = 700 nm in a microplate reader Synergy H1 (Biotek), and the highest point for each sample was considered to calculate the loading efficiency of each method. The loading efficiency on each condition, including the control without sEVs, was calculated using the following formula: fluorescence intensity of the pellet/(fluorescence intensity of the pellet + fluorescence intensity of the supernatant). For each condition and each type of sEV, the fluorescence value of the respective control was subtracted from the measured value, and this number was expressed, in percentage, as the loading efficiency.
qPCR analysis
To evaluate miR-124-3p expression in sEVs, total RNA was extracted using the RNeasy Micro Kit (Qiagen) as per the manufacturer's instructions and quantified using the Qubit 2.0 system (Thermo Fisher Scientific). cDNA was synthesized for each sample from the amount of RNA extracted from 40 μg (9.2 × 1010 particles) of sEVs using the Mir-X miRNA First-Strand Synthesis Kit (Takara), from which the amount corresponding to roughly 3 μg (6.9 × 109 particles) of EVs was used per qPCR reaction. Finally, qPCR was performed on the CFX Connect Real-Time System (Bio-Rad) using the NZYSpeedy qPCR Green Master Mix (2×) (Nzytech). The reverse primer was the universal 3′ mRQ primer (Takara). The forward primer sequence for miR-124-3p was 5′-TAAGGCACGCGGTGAATGCC-3′, and for RNU6 (RNA, U6 small nuclear) amplification, the forward primer 5′-TCGGCAGCACATATACTAA-3′ and the reverse primer 5′-GAATTTGCGTGTCATCCT-3′ were used. U6 RNA served as housekeeping control, and the native EV samples (non-transfected) as a sample control. To estimate the number of copies of miR-124-3p in sEVs, serial dilutions were prepared from miR-124-3p standards ranging from 2 × 10-6 to 20 pmol, being then correlated with respective Cq values. The resultant value, obtained in pmol, was converted to copy number by multiplication through Avogadro’s constant (6.022 × 1023 mol-1).
Treatment of sEVs with RNase
sEVs (2 × 1010 total particles) were loaded with miR-124-Cy5 (10 pmol) through Exo-Fect. Samples where then purified via ExoQuick as described above. Subsequently, purified sEV pellets were exposed to 2 μg/mL RNase (Sigma- Aldrich), in a final volume of 150 μL, for 30 min at RT and re-purified via ExoQuick. Finally, qPCR was performed on the RNase treated and non-treated samples.
sEV labeling with PKH67
sEVs were labeled with PKH67 (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer's instructions. Briefly, 20 μg sEVs were diluted in the kit buffer (diluent C) 1:1, and then PKH67 in diluent C (1:75) was mixed with the diluted sample. Subsequently, samples were incubated at RT for 3 min, following purification by ultracentrifugation.
Primary SVZ cell cultures and experimental treatments
One- to three-day-old C57BL/6J mice were used to obtain SVZ cell cultures as previously described by us.9 Briefly, SVZ fragments were dissected from 450-μm-thick coronal brain sections and digested in 0.025% trypsin and 0.265 mM EDTA (all from Life Technologies), followed by mechanical dissociation. The cell suspension was diluted in serum-free medium (SFM) composed of Dulbecco’s modified Eagle medium ((DMEM)/F12 + GlutaMAX-l)) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 1% B27 supplement, 10 ng/mL epidermal growth factor, and 5 ng/mL basic fibroblast growth factor 2 (all from Life Technologies) and plated onto uncoated petri dishes (Corning Life Sciences, Corning, NY, USA). Five- to six-day-old neurospheres were then seeded onto 0.1 mg/mL poly-D-lysine (PDL; Sigma-Aldrich)-coated glass coverslips in 24-well plates in SFM devoid of growth factors. SVZ cells were allowed to form a cell monolayer for 2 days and then were treated with 1.5 (3 μg/mL sEVs) or 3 × 109 part/mL (6 μg/mL sEVs) of sEVs loaded or not with miR-124-3p or SCR or Exo-Fect plus miR-124-3p (the same volume as the miR-124-3p sEV condition was added). Two or 7 days after the treatments, cell death and commitment or neuronal differentiation were evaluated by immunocytochemistry, respectively.
Propidium iodide incorporation
Propidium iodide (PI; 5 μg/mL; Sigma-Aldrich) was used to quantify the number of necrotic and late apoptotic cells. PI was added to SVZ cells 10 min before the end of the 48 h treatments at 37°C. Thereafter, cells were fixed using 10% formalin solution, for 15 min, at RT and then rinsed with PBS. Cell nuclei were stained with Hoechst-33342 (1:500; Life Technologies) for 5 min and then mounted in Fluoroshield Mounting Medium (Abcam). Five random microscopic fields were acquired per replicate using an Axio Imager microscope (from Carl Zeiss) under a magnification of 40×.
Immunocytochemistry
SVZ cells were fixed with 10% formalin, washed with PBS, and then incubated in PBS containing 0.3% Triton X-100 and 3% BSA (cytoplasmatic staining) or 6% BSA (nuclear staining) for 30 min or 1 h at RT, respectively. Cells were subsequently incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-Ki-67 (1:50; Abcam); goat polyclonal anti-DCX (1:200; Santa Cruz Biotechnology); mouse monoclonal anti-Nestin (1:100; Abcam); and mouse monoclonal anti-NeuN (1:100; Merck Millipore, Darmstadt, Germany), all prepared in 0.3% BSA and 0.1% Triton X-100. On the next day, after washing with PBS, cells were incubated for 1 h with the corresponding secondary antibody followed by Hoechst-33342 nuclear staining and then mounted in Fluoroshield mounting medium (Abcam). Secondary antibodies used were Alexa Fluor 488 donkey anti-rabbit, Alexa Fluor 546 donkey anti-goat, and Alexa Fluor 594 or 647 donkey anti-mouse (all 1:200; Life Technologies), all prepared in PBS. Photomicrographs were taken using LSM 710 confocal microscope (Carl Zeiss). Analysis of immunocytochemistry experiments was performed at the border of seeded neurospheres, where cells formed a pseudo-monolayer. The experiments were performed in three independent SVZ cultures from C57BL/6 pups, and for each experimental condition, at least 2 coverslips were analyzed per culture. Percentage of PI+, Nestin+/Ki-67+, DCX+/Ki-67+, and NeuN+ cells were calculated from cell counts in five independent microscopic fields (an average of 150 cells per field) from each coverslip with a 40× magnification.
N27 cell line and experimental treatments
Immortalized N27 cell line, derived from rat mesencephalon, were grown in RPMI 1640 medium containing 2 g/L sodium bicarbonate and supplemented with 10% fetal bovine serum (FBS; Millipore), 100 IU/L penicillin, and 10 g/mL streptomycin in humidified 95% air and 5% CO2, at 37°C. For experiments, the cells were seeded at a density of 1 × 104 cells/well in 96-well culture plates. After 24 h, cells were fed with fresh medium and treated with 50 μM 6-OHDA (Sigma-Aldrich) and/or 1.5 (3 μg/mL) or 3 × 109 part/mL (6 μg/mL) of miR-124-3p sEVs or sEVs for 24 h.
Cell viability assay in N27 cells
Viability of N27 cells was evaluated by using CCK-8 (Dojindo Laboratories, Kumamoto, Japan) following the manufacturer’s instructions with modifications. Briefly, 24 h after the treatments, 5 μL CCK-8 solution was added to each well. Cells were incubated in humidified 95% air and 5% CO2, for 3 h at 37°C. The absorbance was measured at 450 nm using a microplate reader XMark Microplate Spectrophotometer (Bio-Rad).
Animals
Young adult (2- to 3-month-old) male C57BL/6 were used for this study. The experimental procedures were performed following protocols approved by the Directorate-General for Food and Veterinary (DGAV), the Portuguese National Authority for Animal Health (21/01/2019; reference number 0421/000/000/2019), and by the Directive 2010/63/EU of the European Parliament and the Council on the protection of animals used for scientific purposes. All animals were kept in appropriate cages in the same room under temperature-controlled conditions with a fixed 12 h light/dark cycle with free access to food and water. All efforts were made to reduce the number of animals used and to minimize their suffering.
Stereotaxic injections and BrdU administration
Mice were anesthetized with an intraperitoneal (i.p.) injection of ketamine (90 mg/kg of mouse weight) and xylazine (10 mg/kg of mouse weight) and placed in a stereotaxic apparatus (51900 Stoelting, Dublin, Ireland). The skull was exposed, and the scales were defined after setting the zero at the bregma point. Mice were then injected in the right lateral ventricle with 10 μg miR-124-3p sEVs (2.3 × 1010 particles loaded with 20 pmol of miR-124-3p/mouse), SCR sEVs (2.3 × 1010 particles loaded with 20 pmol of SCR/mouse), non-loaded sEVs (2.3 × 1010 particles/mouse), or sterile 0.1 M PBS (saline) (anteroposterior [AP]: -0.5 mm; mediolateral [ML]: -0.7 mm; dorsoventral [DV]: -2.9 mm from bregma) through a 10 μL Hamilton syringe at a speed of 0.5 μL/min in a total volume of 5 μL.9 To find out the effect of the miR sEV formulation in a mouse model of PD, mice were subjected to another stereotaxic injection to deliver 10 μg 6-OHDA (dissolved in 0.02% of ascorbic acid) into the right striatum (AP: -0.6 mm; ML: -2.0 mm, DV: -3.0 mm from bregma) using a 10 μL Hamilton syringe at a speed of 0.2 μL/min, in a total volume of 2 μL.9 The needle was left in place for an additional 5 min before being slowly withdrawn. After the injections, mice were kept warm (37°C) until they fully recovered from the surgery. Four experimental groups were tested: (1) “saline mice” injected with saline both in the right striatum and in the lateral ventricle; (2) “6-OHDA” mice injected with 6-OHDA in the right striatum and saline in the lateral ventricle; (3) “sEV + 6-OHDA” mice injected with 6-OHDA in the right striatum and sEVs in the lateral ventricle; (4) “miR-124-3p sEV + 6-OHDA” mice injected with 6-OHDA in the right striatum and miR-124-3p sEVs in the lateral ventricle; and (5) “SCR sEV + 6-OHDA” mice injected with 6-OHDA in the right striatum and SCR sEVs in the lateral ventricle. To label dividing cells, BrdU dissolved in a sterile solution of 0.9% NaCl was administered i.p. (50 mg/kg of animal weight) for 3 days (every 12 h) after the stereotaxic procedure. Then, animals were maintained in appropriate cages for 4 weeks until they were euthanized. To evaluate the distribution of the sEVs in the brain, mice were injected i.c.v. with PKH67-labeled sEVs (2.3 × 1010 particles), as described above, and 24 h after stereotaxic injections, mice were euthanized, and the brain removed for posterior analysis.
Behavioral studies
All behavioral tests were performed during the light cycle. Mice were transferred to the experimental room at least 20 min before the test to let them acclimatize to the environment.
Rotarod
Motor coordination, balance and grip strength function were analyzed using a mice rotarod apparatus (Ugo Basile, Gemonio, Italy). The rotarod test was performed at weeks 1 and 2 after stereotaxic injections under an accelerating protocol, starting at 4 RPM and reaching 40 RPM over 5 min, and the latency to fall was recorded. The trial stopped when the mouse fell (activating a switch that automatically stopped the timer) or when 5 min had been completed. Each animal was given four independent trials with a 30 min inter-trial period to reduce stress and fatigue. All mice were pretrained on the rotarod to reach a stable performance. The training consisted of one session per day during 2 consecutive days under a fixed accelerating protocol. Each session included 4 separate test trials, each lasting 5 min. Mice were trained at 12 RPM on day 1 and 22 RPM on day 2. The animals were allowed to rest 30 min between each trial. Each full passive rotation performed by mice onto the rod was considered a fall.
Apomorphine-induced rotation test
Apomorphine-induced rotation was measured at weeks 2 and 4 after stereotaxic injections. Mice received a subcutaneous injection of freshly prepared apomorphine hydrochloride (Sigma-Aldrich) (0.5 mg/kg), a dopamine D1/D2 receptor agonist, dissolved in 1% ascorbic acid, and 0.9% NaCl. They were placed in individual round glass bowls and were allowed to adapt to their environment for 5 min before the test. After apomorphine administration, full body ipsilateral and contralateral turns were recorded using a digital camera for 45 min. Subsequently, each 360° rotation of the body axes was manually counted as a rotation. Values were expressed as a mean of net contralateral turns (equal to the number of contralateral turns minus ipsilateral turns).
Tissue preparation
Four weeks post-injections, mice were deeply anesthetized through i.p. injection of ketamine (90 mg/kg of mouse weight) and xylazine (10 mg/kg of mouse weight) and euthanized by transcardial perfusion with saline followed by 4% PFA. Following perfusion, brains were removed, post-fixed overnight with 4% PFA, and cryoprotected in 30% sucrose solution. Thereafter, brains were snap frozen in liquid nitrogen and stored at -80°C until further use. Brains were cut into 40-μm-thick coronal sections on a cryostat (Leica CM 3050S, Leica Microsystems, Wetzlar, Germany). The sections corresponding to the striatum and SN of each animal were collected sequentially in 6 compartments of 24-well plates containing cryopreservation solution and stored at -20°C until processing for immunohistostaining.
Immunohistochemistry
Free-floating coronal brain sections were rinsed in PBS-T and then incubated with 10% FBS and 0.1% Triton X-100 in PBS for at least 1 h at RT. Endogenous peroxidase activity was inhibited by incubation with 3% hydrogen peroxide (H2O2) in water for 10 min at RT, and the sections were then washed with PBS-T. Sections were incubated with primary antibody mouse anti-TH (1:2,500; BD Biosciences) diluted in PBS containing 5% FBS overnight at 4°C. After several rinses with PBS-T, sections were incubated with a biotinylated goat anti-mouse secondary antibody (dilution 1:200; Vector Laboratories, Burlingame, CA, USA) diluted in PBS containing 1% FBS for 1 h at RT. The avidin-biotin peroxidase complex reagent (Vectastain ABC KIT, Vector Laboratories, Burlingame, CA, USA) was then added for at least 30 min at RT. The reaction product was visualized using DAB chromogen/HRP substrate-buffer (Dako) until color develops (5–10 min). The reaction was stopped by adding TBS. Sections were mounted on slides, dried, and dehydrated in graded ethanol dilutions, cleared in xylene, and cover slipped using a permanent mounting medium (Entellan, Merck, Branchburg, NJ, USA). Quantitative analysis of DA neurons in SN was carried out by serial section analysis of the total number of TH-positive neurons throughout the rostro-caudal axis. The region corresponding to the ipsilateral SN pars compacta was carefully delineated, and the total number of TH-positive neurons in the full extent of this structure was counted per section. The total number of TH-positive neurons for each representative mesencephalic section was quantified in four coronal sections per mouse from -2.80 to -3.88 mm relative to bregma, under the magnification of 10× at the Axio Imager A1 microscope (Carl Zeiss, Heidenheim, Germany). Quantitative analysis of the intensity and area occupied by TH-positive fibers staining in striatum was carried out in 4 coronal sections of ipsilateral striatum, from 1.34 to 0.38 mm relative to bregma of each mouse, selected throughout the rostro-caudal axis, under the magnification of 5× at the Axio Imager A1 microscope.
For BrdU staining, tissue sections were rinsed in PBS and incubated with 2 M HCl for 25 min at 37°C to induce DNA denaturation and exposure of BrdU. Tissue sections were then incubated in blocking solution 2% of horse serum (Life Technologies) and 0.3% Triton X-100 in PBS for 2 h at RT, followed by a 48 h incubation at 4°C with the following primary antibodies: rat monoclonal anti-BrdU (1:500, AbD Serotec, Raleigh, NC, USA) and mouse monoclonal anti-NeuN (1:1000, Merck Millipore). Thereafter, sections were incubated for 2 h at RT with Hoechst-33342 (1:10,000) and the respective secondary antibodies: Alexa Fluor 488 donkey anti-rat and Alexa Fluor 594 donkey anti-mouse (all 1:1000; from Life Technologies). Then, a simplified version of this protocol was used for sEV distribution/location studies. Briefly, 40-μm-thick coronal sections were incubated in a blocking solution for 2 h at RT and then incubated for 48 h at 4°C with the mouse anti-TH (1:1,000, BD Biosciences). Thereafter, sections were incubated for 2 h at RT with Hoechst (1:10,000) and the secondary antibody Alexa Fluor 594 donkey anti-mouse (1:1,000, Abcam). Finally, sections were mounted in Fluoroshield mounting medium (Abcam). Photomicrographs were obtained using LSM 710 confocal microscope. Quantification of NeuN/BrdU-double positive cell number was performed in the striatum of 3 animals, as described previously by us.9 Two different z axis positions (40× magnification) from 9 fields from 4 slices spaced by 240 μm each were counted per animal.
Statistical analysis
The software used for cell counting and quantification of TH staining in the striatum was FIJI ImageJ (NIH Image, Bethesda, MD, USA). Data were expressed as mean ± SEM of at least three independent experiments performed in triplicate (in vitro experiments) or at least three different animals (in vivo experiments). The percentages of the number of PI+ cells (Figure 3A), NeuN+ cells (Figure 3D), and Ki-67+ cells (Figure S2) were calculated relative to the number of total cells; the percentages of Nestin+ cells (Figure 3B) and of DCX+ cells (Figure 3C) were calculated relative to the total number Ki-67+ cells. The controls (untreated cultures) were set to 100% in all analysis except for the evaluation of cell viability (PI; Figure 3A). Statistical analysis was performed using GraphPad Prism 7 software (GraphPad, San Diego, CA, USA) by using one- or two-way ANOVA followed by Tukey’s or Sidak’s test, respectively, or by unpaired, two-tailed t test. Values of p < 0.05 were considered significant.
Acknowledgments
This work was partially supported by the Portuguese “Fundação para a Ciência e a Tecnologia (FCT)” through the ICON project (Interdisciplinary Challenges On Neurodegeneration; CENTRO-01-0145-FEDER-000013), projects 007630 UID/QUI/00313/2019 and POCI-01-0145-FEDER-029919, co-funded by COMPETE2020-UE and CENTRO-01-0145-FEDER-000014 through “Programa Operacional Regional do Centro” CENTRO2020 and under doctoral grant SFRH/BD/121822/2016; by the Interreg program, specifically the project “2IQBIONEURO: Impulso de una red de I + i en química biológica para diagnóstico y tratamiento de enfermedades neurológicas” (Ref: 1654) and NEUROATLANTIC entitled “An Atlantic innovation platform on diagnosis and treatment of neurological diseases and aging” (Ref: EAPA_791/2018). The authors also acknowledge the support of CICS-UBI and the PPBI-Portuguese Platform of BioImaging (POCI-01-0145-FEDER-022122).
Author contributions
M.E. designed and performed the experiments, collected, assembled, and analyzed the data, and wrote the manuscript. R.A., C.S.-A., P.A.T.M., and M.B. performed the experiments and analyzed the data. H.F., A.C.C., C.S., R.F., L.F., and L.B. gave technical and scientific support to the experiments. A.C.C., R.F., L.F., and L.B. provided resources needed for the completion of the work. R.F., L.F., and L.B. edited the manuscript. L.B. designed the study, supervised the work, analyzed the data, and wrote the manuscript. All authors gave final approval of the manuscript.
Declaration of interests
The authors declare no conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.06.003.
Supplemental information
References
- 1.Dauer W., Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 2.Saraiva C., Esteves M., Bernardino L. MicroRNA: basic concepts and implications for regeneration and repair of neurodegenerative diseases. Biochem. Pharmacol. 2017;141:118–131. doi: 10.1016/j.bcp.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Kanagaraj N., Beiping H., Dheen S.T., Tay S.S.W. Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience. 2014;272:167–179. doi: 10.1016/j.neuroscience.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 4.Li N., Pan X., Zhang J., Ma A., Yang S., Ma J., Xie A. Plasma levels of miR-137 and miR-124 are associated with Parkinson’s disease but not with Parkinson’s disease with depression. Neurol. Sci. 2017;38:761–767. doi: 10.1007/s10072-017-2841-9. [DOI] [PubMed] [Google Scholar]
- 5.Sonntag K.-C. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 2010;1338:48–57. doi: 10.1016/j.brainres.2010.03.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao L., Zhu Z., Wu J., Zhang Y., Zhang H., Sun X., Qian C., Wang B., Xie L., Zhang S., et al. MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease. FASEB J. 2019;33:8648–8665. doi: 10.1096/fj.201900363R. [DOI] [PubMed] [Google Scholar]
- 7.Gong X., Wang H., Ye Y., Shu Y., Deng Y., He X., Lu G., Zhang S. miR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson’s disease. Am. J. Transl. Res. 2016;8:2127–2137. [PMC free article] [PubMed] [Google Scholar]
- 8.Dong R.F., Zhang B., Tai L.W., Liu H.M., Shi F.K., Liu N.N. The neuroprotective role of MiR-124-3p in a 6-hydroxydopamine-induced cell model of Parkinson’s disease via the regulation of ANAX5. J. Cell. Biochem. 2018;119:269–277. doi: 10.1002/jcb.26170. [DOI] [PubMed] [Google Scholar]
- 9.Saraiva C., Paiva J., Santos T., Ferreira L., Bernardino L. MicroRNA-124 loaded nanoparticles enhance brain repair in Parkinson’s disease. J. Control. Release. 2016;235:291–305. doi: 10.1016/j.jconrel.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Henriques-Antunes H., Cardoso R.M.S., Zonari A., Correia J., Leal E.C., Jiménez-Balsa A., Lino M.M., Barradas A., Kostic I., Gomes C., et al. The kinetics of small extracellular vesicle delivery impacts skin tissue regeneration. ACS Nano. 2019;13:8694–8707. doi: 10.1021/acsnano.9b00376. [DOI] [PubMed] [Google Scholar]
- 11.Haney M.J., Klyachko N.L., Zhao Y., Gupta R., Plotnikova E.G., He Z., Patel T., Piroyan A., Sokolsky M., Kabanov A.V., et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release. 2015;207:18–30. doi: 10.1016/j.jconrel.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perets N., Betzer O., Shapira R., Brenstein S., Angel A., Sadan T., Ashery U., Popovtzer R., Offen D. Golden exosomes selectively target brain pathologies in neurodegenerative and neurodevelopmental disorders. Nano Lett. 2019;19:3422–3431. doi: 10.1021/acs.nanolett.8b04148. [DOI] [PubMed] [Google Scholar]
- 13.Guo S., Perets N., Betzer O., Ben-Shaul S., Sheinin A., Michaelevski I., Popovtzer R., Offen D., Levenberg S. Intranasal delivery of mesenchymal stem cell derived exosomes loaded with phosphatase and tensin homolog siRNA repairs complete spinal cord injury. ACS Nano. 2019;13:10015–10028. doi: 10.1021/acsnano.9b01892. [DOI] [PubMed] [Google Scholar]
- 14.Qu M., Lin Q., Huang L., Fu Y., Wang L., He S., Fu Y., Yang S., Zhang Z., Zhang L., et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release. 2018;287:156–166. doi: 10.1016/j.jconrel.2018.08.035. [DOI] [PubMed] [Google Scholar]
- 15.Cooper J.M., Wiklander P.B.O., Nordin J.Z., Al-Shawi R., Wood M.J., Vithlani M., Schapira A.H.V., Simons J.P., El-Andaloussi S., Alvarez-Erviti L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014;29:1476–1485. doi: 10.1002/mds.25978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steel H.C., Alessandrini M., Mellet J., Dessels C., Oloyo A.K., Pepper M.S. In: Stem Cell Processing. Stem Cells in Clinical Applications. Van Pham P., editor. Springer; 2016. Cord blood stem cell banking; pp. 163–180. [Google Scholar]
- 17.de Abreu R.C., Ramos C.V., Becher C., Lino M., Jesus C., da Costa Martins P.A., Martins P.A.T., Moreno M.J., Fernandes H., Ferreira L. Exogenous loading of miRNAs into small extracellular vesicles. J. Extracell. Vesicles. 2021;10:e12111. doi: 10.1002/jev2.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agasse F., Bernardino L., Silva B., Ferreira R., Grade S., Malva J.O. Response to histamine allows the functional identification of neuronal progenitors, neurons, astrocytes, and immature cells in subventricular zone cell cultures. Rejuvenation Res. 2008;11:187–200. doi: 10.1089/rej.2007.0600. [DOI] [PubMed] [Google Scholar]
- 19.Agasse F., Bernardino L., Kristiansen H., Christiansen S.H., Ferreira R., Silva B., Grade S., Woldbye D.P.D., Malva J.O. Neuropeptide Y promotes neurogenesis in murine subventricular zone. Stem Cell. 2008;26:1636–1645. doi: 10.1634/stemcells.2008-0056. [DOI] [PubMed] [Google Scholar]
- 20.Maas S.L.N., De Vrij J., Van Der Vlist E.J., Geragousian B., Van Bloois L., Mastrobattista E., Schiffelers R.M., Wauben M.H.M., Broekman M.L.D., Nolte-’T Hoen E.N.M. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. J. Control. Release. 2015;200:87–96. doi: 10.1016/j.jconrel.2014.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao L., Ye Y., Mao H., Lu F., He X., Lu G., Zhang S. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson’s disease. J. Neuroinflammation. 2018;15:13. doi: 10.1186/s12974-018-1053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song Y., Li Z., He T., Qu M., Jiang L., Li W., Shi X., Pan J., Zhang L., Wang Y., et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics. 2019;9:2910–2923. doi: 10.7150/thno.30879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng L.-C., Pastrana E., Tavazoie M., Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 2009;12:399–408. doi: 10.1038/nn.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X.S., Chopp M., Zhang R.L., Tao T., Wang X.L., Kassis H., Hozeska-Solgot A., Zhang L., Chen C., Zhang Z.G. MicroRNA profiling in subventricular zone after stroke: MiR-124a regulates proliferation of neural progenitor cells through notch signaling pathway. PLoS One. 2011;6:1–11. doi: 10.1371/journal.pone.0023461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neo W.H., Yap K., Lee S.H., Looi L.S., Khandelia P., Neo S.X., Makeyev E.V., Su I.H. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning Ezh2 expression. J. Biol. Chem. 2014;289:20788–20801. doi: 10.1074/jbc.M113.525493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krichevsky A.M., Sonntag K.-C., Isacson O., Kosik K.S. Specific MicroRNAs modulate embryonic stem cell–derived neurogenesis. Stem Cell. 2006;24:857–864. doi: 10.1634/stemcells.2005-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu X., Meng S., Liu S., Jia C., Fang Y., Li S., Fu C., Song Q., Lin L., Wang X. MiR-124 represses ROCK1 expression to promote neurite elongation through activation of the PI3K/Akt signal pathway. J. Mol. Neurosci. 2014;52:156–165. doi: 10.1007/s12031-013-0190-6. [DOI] [PubMed] [Google Scholar]
- 28.Franke K., Otto W., Johannes S., Baumgart J., Nitsch R., Schumacher S. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling. EMBO J. 2012;31:2908–2921. doi: 10.1038/emboj.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo A.S., Sun A.X., Li L., Shcheglovitov A., Portmann T., Li Y., Lee-Messer C., Dolmetsch R.E., Tsien R.W., Crabtree G.R. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 2011;476:228–231. doi: 10.1038/nature10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang T., Liu Y., Huang M., Zhao X., Cheng L. Wnt1-cre-mediated conditional loss of dicer results in malformation of the midbrain and cerebellum and failure of neural crest and dopaminergic differentiation in mice. J. Mol. Cell Biol. 2010;2:152–163. doi: 10.1093/jmcb/mjq008. [DOI] [PubMed] [Google Scholar]
- 31.Geng L., Liu W., Chen Y. miR-124-3p attenuates MPP+-induced neuronal injury by targeting STAT3 in SH-SY5Y cells. Exp. Biol. Med. 2017;242:1757–1764. doi: 10.1177/1535370217734492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J., Wang W., Zhai H. MicroRNA-124 enhances dopamine receptor expression and neuronal proliferation in mouse models of Parkinson’s disease via the hedgehog signaling pathway by targeting EDN2. Neuroimmunomodulation. 2019;26:174–187. doi: 10.1159/000501339. [DOI] [PubMed] [Google Scholar]
- 33.Wang H., Ye Y., Zhu Z., Mo L., Lin C., Wang Q., Wang H., Gong X., He X., Lu G., et al. MiR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson’s disease by targeting to Bim. Brain Pathol. 2016;26:167–176. doi: 10.1111/bpa.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y., Zhao H., Tan Z., Zhang C., Fu X. Bottleneck limitations for microRNA-based therapeutics from bench to the bedside. Pharmazie. 2015;70:147–154. [PubMed] [Google Scholar]
- 35.Zhuang X., Xiang X., Grizzle W., Sun D., Zhang S., Axtell R.C., Ju S., Mu J., Zhang L., Steinman L., et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011;19:1769–1779. doi: 10.1038/mt.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng Z., Li Y., Pan Y., Lan X., Song F., Sun J., Zhou K., Liu X., Ren X., Wang F., et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018;9:1–14. doi: 10.1038/s41467-018-07810-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pi F., Binzel D.W., Lee T.J., Li Z., Sun M., Rychahou P., Li H., Haque F., Wang S., Croce C.M., et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018;13:82–89. doi: 10.1038/s41565-017-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo C., Zhang J., Zhang Y., Zhang X., Chen Y., Fan W. Low expression of miR-let-7a promotes cell growth and invasion through the regulation of c-Myc in oral squamous cell carcinoma. Cell Cycle. 2020;19:1983–1993. doi: 10.1080/15384101.2020.1786633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T., et al. Systemically injected exosomes targeted to EGFR deliver antitumor MicroRNA to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang B., Luo Q., Han Y., Huang D., Tang Q., Wu L. MiR-223/PAX6 Axis regulates glioblastoma stem cell proliferation and the chemo resistance to TMZ via regulating PI3K/Akt pathway. J. Cell. Biochem. 2017;118:3452–3461. doi: 10.1002/jcb.26003. [DOI] [PubMed] [Google Scholar]
- 41.Gao X., Xiong Y., Li Q., Han M., Shan D., Yang G., Zhang S., Xin D., Zhao R., Wang Z., et al. Extracellular vesicle-mediated transfer of miR-21-5p from mesenchymal stromal cells to neurons alleviates early brain injury to improve cognitive function via the PTEN/Akt pathway after subarachnoid hemorrhage. Cell Death Dis. 2020;11:363. doi: 10.1038/s41419-020-2530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barbosa J.S., Sanchez-Gonzalez R., Di Giaimo R., Baumgart E.V., Theis F.J., Gotz M., Ninkovic J. Live imaging of adult neural stem cell behavior in the intact and injured zebrafish brain. Science. 2015;348:789–793. doi: 10.1126/science.aaa2729. [DOI] [PubMed] [Google Scholar]
- 43.Lu Y.-L., Liu Y., McCoy M.J., Yoo A.S. MiR-124 synergism with ELAVL3 enhances target gene expression to promote neuronal maturity. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2015454118. e2015454118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marycz K., Smieszek A., Marcinkowska K., Sikora M., Turlej E., Sobierajska P., Patej A., Bienko A., Wiglusz R.J. Nanohydroxyapatite (nHAp) doped with iron oxide nanoparticles (IO), miR-21 and miR-124 under magnetic field conditions modulates osteoblast viability, reduces inflammation and inhibits the growth of osteoclast – a novel concept for osteoporosis treatment: Part 1. Int. J. Nanomed. 2021;16:3429–3456. doi: 10.2147/IJN.S303412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saraiva C., Talhada D., Rai A., Ferreira R., Ferreira L., Bernardino L., Ruscher K. MicroRNA-124-loaded nanoparticles increase survival and neuronal differentiation of neural stem cells in vitro but do not contribute to stroke outcome in vivo. PLoS One. 2018;13:e0193609. doi: 10.1371/journal.pone.0193609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arends F., Lieleg O. In: Composition and Function of the Extracellular Matrix in the Human Body. Travascio F., editor. InTechOpen; 2016. Biophysical properties of the basal lamina: a highly selective extracellular matrix. [Google Scholar]
- 47.Betzer O., Perets N., Angel A., Motiei M., Sadan T., Yadid G., Offen D., Popovtzer R. In vivo neuroimaging of exosomes using gold nanoparticles. ACS Nano. 2017;11:10883–10893. doi: 10.1021/acsnano.7b04495. [DOI] [PubMed] [Google Scholar]
- 48.Lee J., Kim J., Jeong M., Lee H., Goh U., Kim H., Kim B., Park J.-H. Liposome-based engineering of cells to package hydrophobic compounds in membrane vesicles for tumor penetration. Nano Lett. 2015;15:2938–2944. doi: 10.1021/nl5047494. [DOI] [PubMed] [Google Scholar]
- 49.Conlan R.S., Pisano S., Oliveira M.I., Ferrari M., Mendes Pinto I. Exosomes as reconfigurable therapeutic systems. Trends Mol. Med. 2017;23:636–650. doi: 10.1016/j.molmed.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 51.Chen H., Liang F., Gu P., Xu B., Xu H., Wang W., Hou J. Exosomes derived from mesenchymal stem cells repair a Parkinson ’ s disease model by inducing autophagy. Cell Death Dis. 2020;11:288. doi: 10.1038/s41419-020-2473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cenci M.A., Björklund A. Animal models for preclinical Parkinson’s research: an update and critical appraisal. Prog. Brain Res. 2020;252:27–59. doi: 10.1016/bs.pbr.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 53.Van Deun J., Mestdagh P., Agostinis P., Akay Ö., Anand S., Anckaert J., Martinez Z.A., Baetens T., Beghein E., Bertier L., et al. EV-TRACK: transparent reporting and centralizing knowledge in EV research. Nat. Methods. 2017;14:228–232. doi: 10.1038/nmeth.4185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.