

SUMMARY
We have previously described polyglutamine binding protein 1 (PQBP1) as an adaptor required for the cyclic GMP-AMP synthase (cGAS)-mediated innate response to the human immunodeficiency virus 1 (HIV-1) and other lentiviruses. Cytoplasmic HIV-1 DNA is a transient and low-abundance pathogen-associated molecular pattern (PAMP), and the mechanism for its detection and verification is not fully understood. Here, we show a two-factor authentication strategy by the innate surveillance machinery to selectively respond to the low concentration of HIV-1 DNA, while distinguishing these species from extranuclear DNA molecules. We find that, upon HIV-1 infection, PQBP1 decorates the intact viral capsid, and this serves as a primary verification step for the viral nucleic acid cargo. As reverse transcription and capsid disassembly initiate, cGAS is recruited to the capsid in a PQBP1-dependent manner. This positions cGAS at the site of PAMP generation and sanctions its response to a low-abundance DNA PAMP.
Keywords: PQBP1, cGAS, HIV-1 Capsid, Innate sensing, Two-factor authentication, Uncoating
INTRODUCTION
The cGAS signaling pathway has been established as a critical regulator of the innate immune response to cytoplasmic DNA (Ablasser and Chen, 2019; Chin, 2019; Reinert et al., 2016). Signaling is induced by delivery of exogenous double-stranded DNA > 50 nucleotides long into the cytosol (Gao et al., 2013; Paludan and Bowie, 2013). Upon binding to double-stranded DNA, cGAS synthesizes cyclic GMP-AMP dinucleotide (cGAMP), which serves as a second messenger to induce STING-mediated IRF3/type-I IFN signaling (Gao et al., 2013). cGAS can also sense lentiviral infections but, in contrast to infection by DNA viruses, this requires an adaptor protein, PQBP1 (Yoh et al., 2015). Recently, the non-POU domain-containing octamer-binding protein (NONO) has been implicated in the innate sensing of nuclear HIV-1 DNA (Lahaye et al., 2018).
The HIV-1 capsid is a protein shell, composed of oligomeric capsid proteins (CA), that enfolds other viral proteins and the RNA genome (Rankovic et al., 2017). After virion entry into the cytosol, the capsid undergoes progressive disassembly through a poorly-defined process of uncoating (Campbell and Hope, 2015; Pornillos et al., 2011). The capsid continues to disassemble as the viral reverse transcription complex migrates to the nucleus (Burdick et al., 2020; Hulme et al., 2011; Mamede et al., 2017; Sood et al., 2017). The initiation of capsid disassembly is linked to the early steps of reverse transcription, which starts with first (-) strand cDNA synthesis and is completed with second (+) strand synthesis (Christensen et al., 2020; Hu and Hughes, 2012; Hulme et al., 2011; Mallery et al., 2018; Mamede et al., 2017; Rankovic et al., 2018; Rankovic et al., 2017; Soliman et al., 2017). HIV-1 DNA can serve as a pathogen-associated molecular pattern (PAMP) to activate the cGAS/STING pathway (Cosnefroy et al., 2016; Doitsh et al., 2010; Yoh et al., 2015). Upon completion of reverse transcription, the viral DNA is integrated into the host genome, where it becomes indistinguishable from chromosomal DNA. Only viral RNA nucleic acid species, biochemically indistinguishable from host RNAs, are present during subsequent steps of the viral life cycle. Thus, in contrast to DNA viruses that amplify copies of their DNA genomes upon infection, HIV-1 only produces one DNA molecule per reverse transcription-competent virion (Hu and Hughes, 2012). Its availability for innate sensing is temporally and spatially constrained as the HIV-1 reverse transcription complex transits to the nucleus for integration. The molecular basis of innate sensing of these transient and low-copy lentiviral DNA species, but not low-abundance self-DNAs, including extranuclear DNAs from mitochondrial or nuclear leakage, is not completely understood.
In this report, we find that the innate sensing machinery employs a unique molecular strategy to activate the innate immune response through a two-step authentication process. First, the retrovirus-specific, innate cosensor PQBP1 specifically recognizes intact capsids of incoming HIV-1 viral particles. Subsequently, disassembly of the viral capsid triggers the PQBP1-dependent recruitment of cGAS in a NONO-independent manner, enabling enzymatic activation of the sensor when HIV-1 DNA synthesis is initiated. Thus, PQBP1 recognition of the HIV-1 capsid is the key event that serves to distinguish its cargo from self-nucleic acids, through licensing the recruitment of cGAS at the site of PAMP production. Importantly, this molecular strategy reveals that modular engagement of cofactors to PRRs can enable the innate immune surveillance machinery to respond to an enhanced repertoire of PAMPs, while limiting deleterious responses to host DNA molecules.
RESULTS
PQBP1 colocalizes with viral capsids during early steps of infection.
To better understand the spatial dynamics of HIV-1 innate immune recognition and responses, we relied on super-resolution 3D-SIM microscopy to assess PQBP1 association with incoming virions. Briefly, PMA-differentiated THP-1 cells were infected with HIV-1 viruses labeled with Gag-Integrase (IN)-mRuby3 (Dharan et al., 2017; Hulme et al., 2015; Mamede et al., 2017; Mamede et al., 2021). One hour post-fusion, cells were fixed and stained by immunofluorescence (IF) against PQBP1 protein (green; Figure 1A). Colocalization of PQBP1 with virus particles (red; IN-mRuby3) was assessed by measuring the distance from each labeled virus punctum to the nearest PQBP1 dot centroid (Figure 1B; Figure S1). The distribution of IN-PQBP1 nearest neighbor distances (d) (Figure 1B, blue) exhibited a sharp peak at shorter distances (~0.12 µm), followed by a broad shoulder at distances larger than 0.5 µm, indicating two distinct populations. For comparison, we performed in silico labeling of IN and PQBP1 signals and calculated a randomized d distribution (Figure 1B, magenta) that displayed a single broad peak at 0.8–0.9 µm. This indicates that the observed IN-PQBP1 colocalization peak is probably not due to stochasticity, while the observed broad shoulder (0.5 µm) most likely represents INs that are not colocalizing with PQBP1. To quantify the observed colocalization, we compared the cumulative probability distribution of d across a range of thresholds to establish colocalization frequencies (right, Figure 1B; Figure S1D). At a threshold of 0.4 µm, we find that more than 40% of total INs are colocalizing with PQBP1, compared to less than 9% for the randomized data (right, Figure 1B). Taken together, at thresholds below 1 µm, these data indicate that PQBP1 significantly colocalizes with incoming HIV-1 virions.
Figure 1. PQBP1 colocalizes with capsid of incoming virions during the early steps of infection.
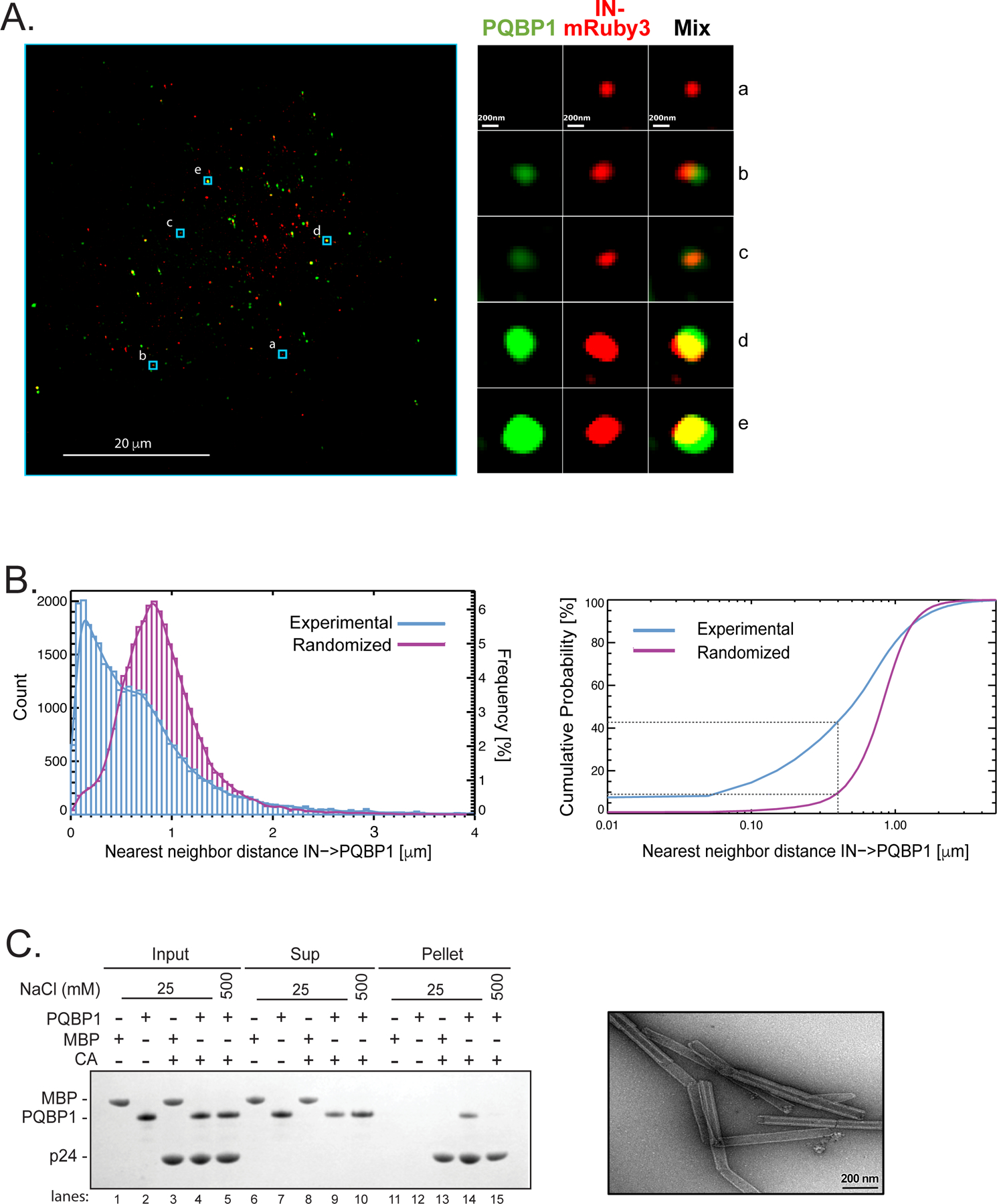
(A) Single Z image of PMA-differentiated THP-1 cells (PMA-THP-1) infected with HIV-1 virions labeled with Gag-IN-mRuby3 (red). Viral fusion was synchronized and, one-hour post-infection, immunostaining of PQBP1 protein (green) was performed. Zoomed images of individual viral particles associating with PQBP1 are shown on the right. Scale bars: 20 μm and 0.2 μm, left and right respectively. (B) Left, distribution of IN-PQBP1 nearest neighbor distances (Count). The distribution of experimental data (blue histogram) is compared with the one generated by in silico randomized dots (magenta histogram). A kernel density estimate of each distribution is overplotted as a solid curve, represented as Frequency, N=32,033 INs. Right, the cumulative probability of nearest neighbor distance (d) measures the percentage of IN dots that have PQBP1 within a defined d. For example, the percentage of INs that have PQBP1 at d < 0.4 µm is a 43% for experimental data and 9% for randomized dots, as highlighted by grey dotted lines. (C) CA tube co-pelleting assay. Insoluble cross-linked CA A14C/E45C tubes were incubated together with PQBP1 or maltose binding protein (MBP) and separated into supernatant (Sup) and pellet fractions, then analyzed by reducing SDS-PAGE. The data are representative of at least three independent experiments. Scale bar, 0.2μm. See also Figure S1.
HIV-1 capsid is composed of ~1500 copies of CA monomers that self-assemble into lattices of hexamers and pentamers to form fullerene-like cones (Perilla and Gronenborn, 2016; Pornillos et al., 2011; Summers et al., 2019). The lattice serves as a binding platform for numerous cellular factors, defining a critical host-pathogen interface after viral entry (Campbell and Hope, 2015; James and Jacques, 2018; Novikova et al., 2019; Summers et al., 2019). Because PQBP1 colocalized with incoming virions, we examined if there was a direct interaction between recombinant PQBP1 and cross-linked CA tubes that recapitulate the hexameric lattices found in intact HIV-1 cores [right, Figure 1C; (Mattei et al., 2016)]. Due to their size (~50 nm in diameter and ~500 nm in length), CA tubes become insoluble upon assembly, which can be exploited to probe binding of cellular factors by cosedimentation [left, Figure 1C; (Summers et al., 2019)]. We find that PQBP1, but not a negative control maltose binding protein (MBP), specifically copelleted with CA tubes (compare lane 13 & 14), and this association could be disrupted by high-salt condition (compare lanes 14 & 15).
PQBP1 directly interacts with HIV-1 capsids through its amino-terminus.
To date, at least four binding sites for host factors have been identified on the capsid: (1) the CypA binding loop (residues 85–95, also recognized by Nup358), (2) the FG-binding site located between adjacent CA subunits centered at residue 74 in the hexamer is recognized by CPSF6 and Nup153, (3) the R18 electropositive pore for dNTPs, IP6 and FEZ1 bindings, and (4) the electronegative inter-hexamer junction at the three-fold symmetry of the capsid lattice that recruits the antiviral protein MxB (Bhattacharya et al., 2014; Gamble et al., 1996; Mallery et al., 2018; Price et al., 2014; Smaga et al., 2019). Here, we sought to delineate the binding interface between capsid and PQBP1 using quantitative two-color coincidence detection (TCCD). This technique measures fluorescence intensity fluctuations from cross-linked CA (A204C) self-assembled particles labeled with AF568, and capsid-binding proteins labeled with AF488, diffusing through the confocal volume (Figure 2A, orange and green traces respectively; Figure S2A; (Lau et al., 2021; Lau et al., 2019)). Accumulation of the binding proteins on the capsid results in fluorescence peaks in the binder trace that coincide with the capsid peak (Figure 2A, middle; Figure S2B). Variation in peak amplitudes is due to the size heterogeneity of in vitro-assembled capsid particles. As a control, we assessed the binding of a CPSF6 peptide comprising capsid-binding residues to CA (A204C) particles (Figure 2B; Figure S2B; (Bhattacharya et al., 2014; Lau et al., 2019; Price et al., 2014). Consistent with previously published data, we observed a significant reduction in CPSF6 peptide binding to the N74D capsid mutant, which disables the FG binding site, compared to the strong binding to wild-type and R18G mutant capsids (Mallery et al., 2018). Additionally, CPSF6 peptides were not competed off the capsid by hexacarboxybenzene (HCB), a polyanion that binds to the R18 ring (Figure 2B, Figure S2B; Jacques et al., 2016). In contrast, fluorescein-dATP, which also binds to the R18 ring, was competed off in the presence of HCB and failed to associate with the R18G mutant capsid (Figure 2B, Figure S2B; (Jacques et al., 2016)). Importantly, we observed that the robust PQBP1 binding to CA (A204C) particles was not affected by the N74D mutation, whereas CPSF6 binding was reduced significantly (Figure 2B). Notably, PQBP1 was effectively competed off the capsid by HCB, and showed lack of binding to the R18G mutant of CA (A204C) (Figure 2B, Figure S2B). The R18 residues of capsid pentamers/ hexamers form an electropositive binding site for polyanions, including dNTPs, IP6 and the host protein FEZ1, but do not participate in oligomerization (Mallery et al., 2018; Price et al., 2014) thereby allowing us to confirm the specificity of the PQBP1 association with oligomerized capsid. Complete loss of PQBP1 binding to R18G, but not N74D suggests that PQBP1 overlays the basic pore of the capsid. We further confirmed the PQBP1-pore interaction by demonstrating preferential co-pelleting of PQBP1 with wild-type compared to R18G mutant capsid (Figure S2C). Lastly, we found that the N-terminal domain (residues 1 to 46) of PQBP1, which has a cluster of negatively charged residues, showed greater accumulation on capsid particles than the C-terminal portion of the protein (residues 47–265; Figure 2C; Figure S2B). Taken together, these results suggest that the N-terminal region of PQBP1 and the capsid pore formed by R18 residues make critical contributions to the binding interface.
Figure 2. PQBP1 directly interacts with HIV-1 capsids through its amino-terminus.
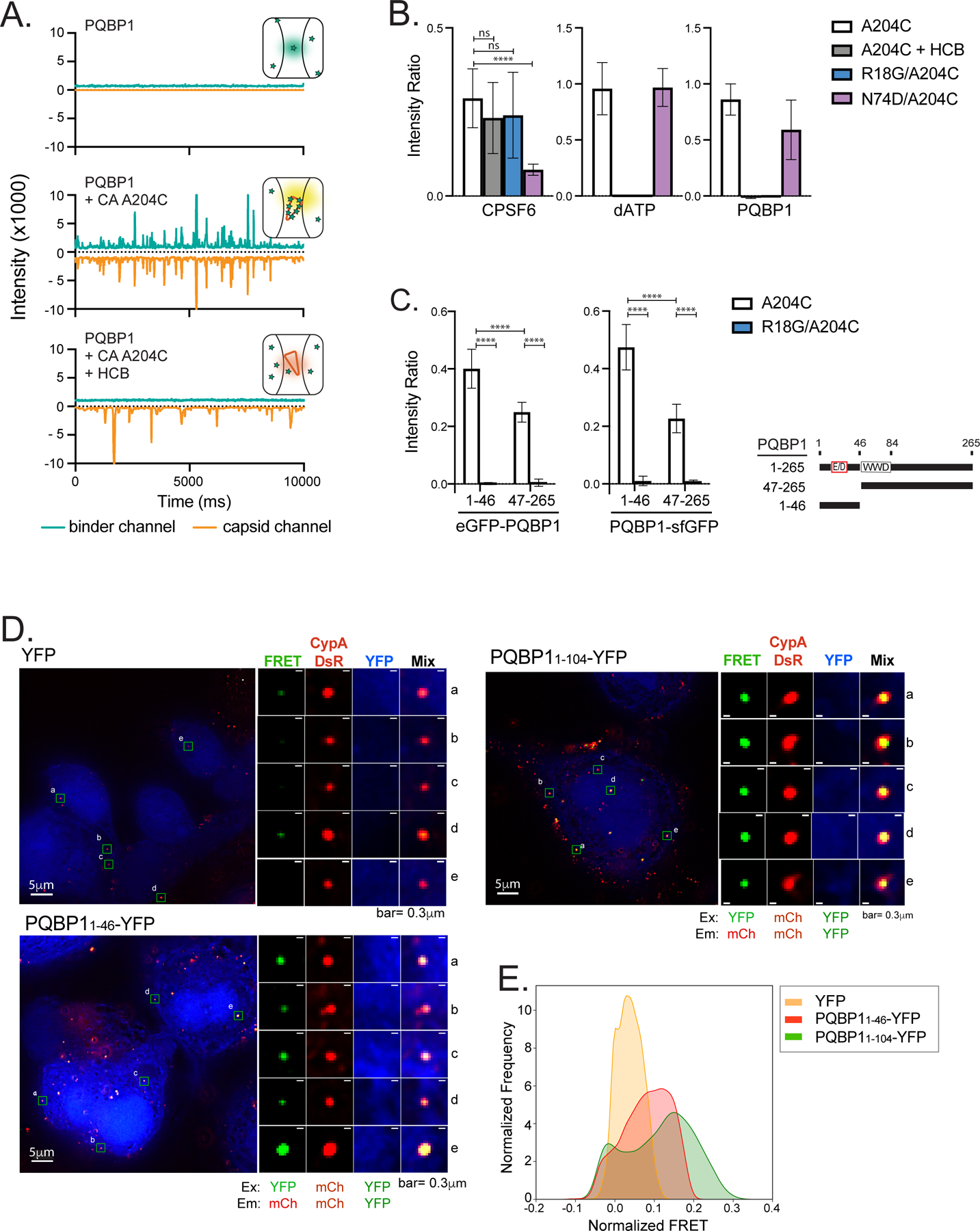
(A) Representative TCCD traces of AF488-PQBP1 (green) and AF568-labeled CA (A204C) particles (orange). The insets show schematics of the species detected by TCCD, with PQBP1 and CA (A204C) particles represented as green stars and orange cones, respectively. Top, featureless PQBP1 trace due to the diffusion of PQBP1 monomers; middle, coincident peaks in both traces due to codiffusion of multiple PQBP1 molecules bound to CA particles; bottom, featureless PQBP1 trace due to non-associated diffusion of PQBP1 monomers and CA particles in the presence of hexacarboxybenzene (HCB). (B) PQBP1 binds to the R18 pore of the capsid. TCCD analysis of AF488-PQBP1, AF488-CPSF6313–327 peptide and fluorescein-dATP to CA (A204C) particles in the absence and presence of HCB, and to R18G or N74D capsids. (C) The N-terminal 46 residues of PQBP1 bind strongly to capsid. TCCD binding analysis of GFP fusions with PQBP11–46 and PQBP147–265 to CA (A204C) particles in the absence and presence of HCB. Data are representative of at least two independent experiments. One-way ANOVA, **** p<0.0001, ns=no significance. (D) and (E) Fluorescence resonance energy transfer (FRET) assay to visualize interactions between PQBP1 and capsid of incoming virions. PMA-THP-1 cells stably expressing either eYFP, PQBP11–46-eYFP or PQBP11–104-eYFP (blue) were infected with HIV-1 packaged with CypA-DsRed (red) for 1.5 hours, followed by PFA fixation, imaging, and FRET analysis. Representative images (D) and distributions of FRET values normalized against an uninfected counterpart (E) were shown (see Star methods for details). FRET excitation and emission wavelengths for YFP and mCherry are as annotated. R0 calculated to be 60.98Å (https://www.fpbase.org/fret/). Data are representative of at least two independent experiments. Scale bars: 5 μm and 0.3 μm, as indicated. See also Figure S2.
To corroborate these in vitro binding studies, the association between the capsid-binding domain of PQBP1 and incoming viral cores in infected cells was assessed by a fluorescence resonance energy transfer (FRET) assay. The strong affinity of CypA-DsRed protein for the HIV-1 capsid enables it to be specifically packaged into virions during budding, and it remains bound to the capsid surface post-fusion (Francis et al., 2016; Francis and Melikyan, 2018; Sood et al., 2017). We reasoned that if a PQBP1-eYFP fusion protein is recruited to the capsid, it should come in close proximity (< 10 nm) of capsid-bound CypA-DsRed, allowing for FRET. THP-1 cells stably expressing eYFP-tagged PQBP1 residues 1–104 or 1–46 showed higher FRET signal compared to eYFP alone when infected by CypA-DsRed-labeled HIV-1 (Figure 2D). Additionally, the FRET signal of each infected sample was normalized for the background signal from a corresponding uninfected sample [(Francis et al., 2016; Francis and Melikyan, 2018; Sood et al., 2017); see Star Method]. The distribution of normalized FRET values of PQBP1-expressing cells was clearly distinct and shifted to higher values than that of cells expressing eYFP alone (Figure 2E). As a control, we showed that the distribution of DsRed signals was similar among infected THP-1 cells subjected to analysis, confirming comparable infection levels (Figure S2D). This observation recapitulates the in vitro PQBP1 association with the capsid. In summary, these data from orthogonal assays support the notion that PQBP1 directly associates with the assembled capsid and this interaction is likely mediated by the N-terminus of PQBP1.
PQBP1 is required for cGAS recruitment to incoming virus particles.
We previously demonstrated that a physical interaction between PQBP1 and cGAS is essential for the innate sensing of HIV-1 infection (Yoh et al., 2015). We hypothesized that cGAS is tethered to incoming capsids via PQBP1; upon loss of capsid integrity, viral DNAs generated by reverse transcription become accessible to cGAS. To address this hypothesis, we performed a super-resolution 3D-SIM IF to examine cGAS recruitment to particles labeled with IN-mRuby3 during the early-stage of infection (Figure 3A; Figure S3A; see Method). Only a fraction of viral particles associated with cGAS one hour post-infection (~8% on average compared to ~60 % for PQBP1) but the majority of these were also associated with PQBP1 (bottom, Figure 3A). Next, we examined the kinetics of PQBP1 and cGAS recruitment to incoming virus particles labeled with a fluid phase marker (HIV-1-iGFP) to monitor the integrity of the capsid. Soon after fusion of the viral and plasma membranes, the HIV-1 core begins to disassemble, allowing the encapsulated GFP to leak out (Chen et al., 2007; Hubner et al., 2007; Hubner et al., 2009; Mamede et al., 2017). This loss of integrity is dependent on reverse transcription. Blocking initiation or first strand transfer delays capsid disassembly (Cosnefroy et al., 2016; Mamede et al., 2017). We used this system to stage the progression of capsid disassembly. MDDCs were infected with HIV-1 particles dual-labeled with iGFP and IN-mRuby3 to allow live-imaging of individual capsids. For our analyses, each intracellular IN-mRuby3 complex was categorized based on iGFP signal decay: (1) unfused (no loss of iGFP), (2) fused but retaining an intact capsid (partial loss of iGFP), and (3) after fusion with loss of capsid integrity (complete loss of iGFP) (Video S1; Figure S3B; iGFP panel, Figure 3B). PQBP1 and cGAS association with each virus particle was then quantified by fixation and immunostaining post-infection the live-imaging, followed by spatial correlation with viral foci (individual panels and quantification graphs in Figure 3B & Figure S3B). We found that PQBP1 associated with post-fusion particles regardless of whether the capsid was intact or open, while cGAS mostly colocalized with capsids that had lost integrity.
Figure 3. PQBP1 is required for cGAS recruitment to incoming virus particles.
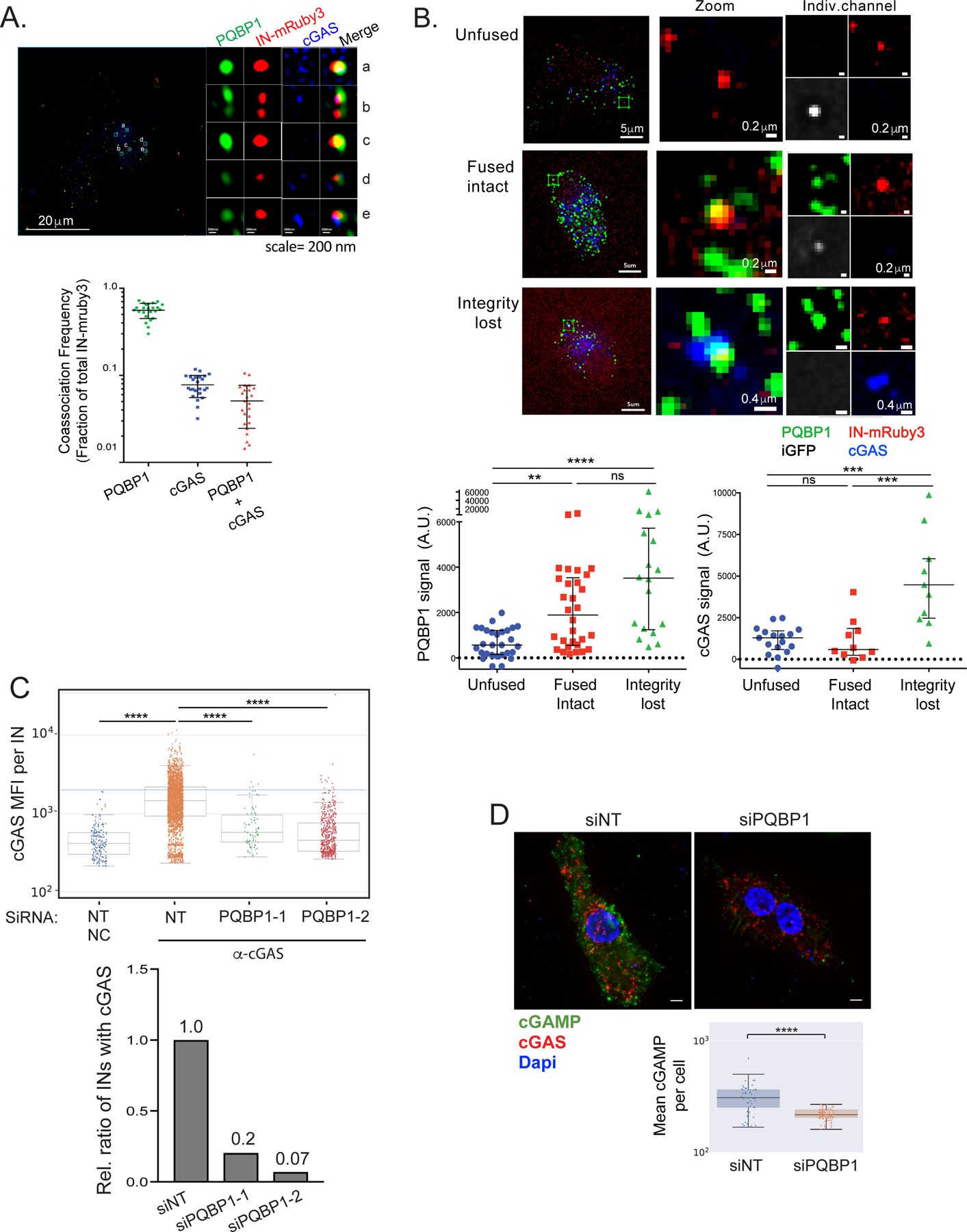
(A) cGAS and PQBP1 association with incoming viral particles. Single Z slice images of PMA-THP-1 cells infected with HIV-1 labeled with Gag-IN-mRuby3 (red) in the presence of VLP-Vpx coinfection for 1 hr, followed by antibody detection of PQBP1 protein (green) and cGAS (blue). Detailed, cropped images of individual viral complexes associating with PQBP1 and cGAS at different levels and distributions are shown on the right. Scale bars: 20 μm (left) and 0.2 μm for panels a to e. Fractions of total IN-mRuby3 foci showing association with PQBP1, cGAS and PQBP1 plus cGAS are quantified at the bottom. (B) PQBP1 recruitment to viral particles precedes cGAS recruitment. MDDCs were infected with HIV-1 labeled with iGFP fluid phase marker and IN-mRuby3 at low MOI and subjected to time-lapse imaging for an hour, followed by fixed IF for PQBP1 and cGAS. Signal intensity of PQBP1/cGAS for each viral particle is quantified and plotted according to the structural state of capsids, assessed by iGFP signal status (see Star method, bottom graphs). Scale bars: 5 μm (left column panels), 0.2 μm and 0.4 μm (middle and right column panels as indicated). Kruskal Wallis H; KW (p<0.001), followed by Dunn’s multiple comparisons *** p<0.001, ** p<0.01. Data represent the sum of 2 to 3 independent experiments. (C) PQBP1 is required for cGAS recruitment to viral particles. Top, THP-1 PMA-treated cells with either a non-targeting siRNA (NT) or siRNAs targeting PQBP1 (PQBP1–1 and -2), were infected with HIV-1 (Gag-IN-mRuby3) for 2.5 hours, followed by fixed imaging for cGAS. NC denotes the negative control where cells were only stained with secondary antibodies. Mean fluorescence intensity (MFI) of cGAS signal per viral particle (IN) is quantified. Median and error bars (-/+ 1.5*IQR) are shown. Box indicates the interquartile range (IQR). Dunn’s column comparison **** p<0.0001. Bottom, relative ratio of viral particles (INs) where co-associating cGAS MFI signals above the maximum value of NC sample are plotted from the data shown in top graph. All the HIV-1 infections of PMA-THP-1 cells were performed in the presence of VLP-Vpx coinfection. (D) Knockdown of PQBP1 results in decreased cGAMP production. PMA-THP-1, transfected with non-targeting siRNAs (NT) or siRNA targeting PQBP1, were challenged with HIV-1 for 2.5 hours and stained for cGAMP (green), cGAS (red), and DAPI (blue). Mean cGAMP signals per cell were graphed (bottom). Scale bar, 10 μm. Median and Error bars (-/+ 1.5*IQR) are shown. Box indicates the interquartile range (IQR). Mann-Whitney **** p<0.0001. Data are representative of three independent infections. All the data presented, unless otherwise stated, are representatives of at least two independent experiments. See also Figure S3.
The temporal disparity between PQBP1 and cGAS recruitment to the capsid suggests a mechanism whereby PQBP1 binding to the capsid is a prerequisite for cGAS recruitment. However, cGAS recruitment additionally requires loss of capsid integrity associated with reverse transcription. Because genetic ablation of PQBP1 is lethal (Iwasaki and Thomsen, 2014; Tamura et al., 2013; Yoh et al., 2015), we transiently depleted PQBP1 from THP-1 cells and evaluated the frequency of cGAS association with virus particles (Figure 3C). Depletion was confirmed by decreases in both PQBP1 mRNA and protein levels (Figure S3C, top). Importantly, down-regulation of PQBP1 resulted in a marked decrease in cGAS association per virus particle (Figure 3C), reflected by a marked drop in the fraction of virus particles targeted by cGAS (Figure 3D). We did not observe a significant overall reduction in cGAS protein levels (Figure S3C bottom). Consistent with a model where cGAS is specifically recruited to PQBP1-decorated virions, we observed a significantly higher density of cGAS foci in the vicinity of virions compared to their random distribution in the cytoplasm (data not shown). Lastly, we addressed the contribution of the PQBP1/cGAS/capsid complex to cGAS activation by assessing levels of its enzymatic product, cGAMP (Hall et al., 2017). THP-1 cells, either infected with HIV-1 or transfected with HT-DNA for 3 hours, revealed cGAMP staining comparable to levels observed in cells where cGAMP is transfected directly (Figure S3D). Consistent with a robust induction of cGAMP in the challenged cells, we also observed nuclear localization of IRF3, a hallmark of cGAS/IRF3 pathway activation (IRF3 panel, Figure S3D). Accordingly, a selective depletion of PQBP1 resulted in a marked decrease in cGAMP levels in response to HIV-1 challenge, with a concomitant reduction in IFN-β mRNA (Figure S3F). These results further confirm that cGAS activation depends on PQBP1 (Figures 3D and S3E).
Previously, Lahaye et. al. showed that NONO associates with nuclear HIV-1 capsids and is needed for innate immune responses to infection in MDDCs (Lahaye et al., 2018). Consistent with these results, we observed that HIV-1-induced cGAS signaling is NONO-dependent in MDDCs (Figures 4A and S4A). However, NONO depletion in THP-1 cells infected either with HIV-1 (in the presence of Vpx to induce SAMHD1 degradation) or HIV-2, did not significantly downregulate cGAS signaling (Figures 4B), which is otherwise intact in these cells (Collins et al., 2015; Gao et al., 2013; Sumner et al., 2020; Sun et al., 2013; Wiser et al., 2020; Yoh et al., 2015). To determine if NONO recognizes incoming viral particles in MDDCs, we first analyzed the distribution of nearest neighbor distances between IN-mRuby3 and NONO. This association displayed a single broad peak at ~0.9 µm (blue lines, Top), similar to a randomized distribution generated in silico (magenta, Top, Figure 4C), and unlike the distinctive peak at ≤0.5 µm that we observe with PQBP1 (blue lines, Bottom). At the 0.5 µm threshold, only ~8 % of labeled capsids associate with NONO (similar to the level of randomized control) whereas ~23% associate with PQBP1 (Bottom left, Figure 4C). These data indicate that, unlike PQBP1, NONO does not significantly colocalize with incoming virus particles in MDDCs.
Figure 4. NONO is not required for the PQBP1-dependent cGAS sensing in the early stages of infection.
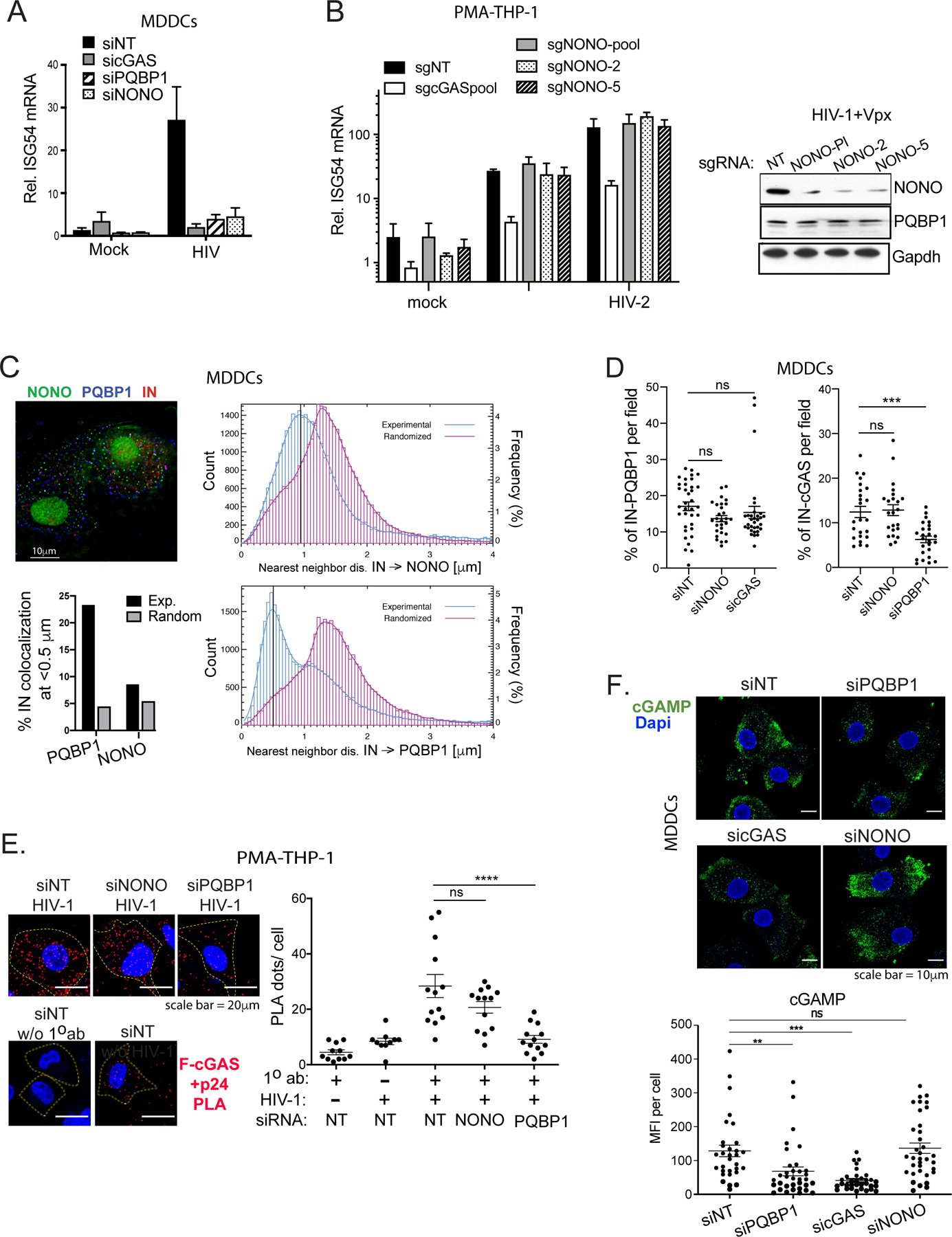
(A) NONO is required for HIV-1-induced ISG54 expression in MDDCs. Cells targeted by the indicated siRNAs were either mock-treated or infected with HIV-1 luciferase virus, followed by ISG54 mRNA quantification at 16 hrs post-infection. (B) NONO is not required for ISG54 induction during HIV-1 infection in PMA-differentiated THP-1 cells. Left, Cells were subjected to CRISPR gene editing with indicated sgRNAs (see Star method) and assessed for ISG54 mRNA inductions at 16 hours post-infection with either mock, HIV-1 in the presence of VLP-Vpx or HIV-2. Right, levels of NONO and PQBP1 proteins in cells targeted with indicated sgRNAs were shown accordingly. (C) Left, representative image of MDDCs infected with HIV-1 virus (IN-mRuby3) and stained for PQBP1 (blue) and NONO (green). Scale bar, 10 mm. Right, distribution of IN-NONO (top) or IN-PQBP1 (bottom) nearest neighbor distances (d). The distribution of experimental data (blue histograms) is compared with distribution generated from in silico randomized dots (magenta histograms). A kernel density estimate of each distribution is overplotted as a solid curve, represented as Frequency, N=27,932 INs. The percentage of INs that have either PQBP1 or NONO at d < 0.5 µm as well as the values for the randomized controls are graphed bottom left. (D) NONO depletion does not impair cGAS recruitment to incoming capsids. MDDCs treated with indicated siRNAs were infected with HIV-1 viruses (Gag-IN-mRuby3/GFP) for 2.5 hours, followed by immunostaining for cGAS and imaging. Fractions of total IN foci overlap with cGAS signal are graphed. Mean and SEM are shown. One-way ANOVA, *** p<0.001. The results are based on four independent experiments. (E) NONO is not required for cGAS recruitment to capsid in PMA-THP-1 cells. Efficiency of Flag-cGAS and viral p24 interactions in PMA-THP-1 cells targeted by indicated siRNAs was determined by a proximal ligation assay (PLA). The cells were infected with HIV-1 virus in the presence of VLP-Vpx for 2 hrs followed by paraformaldehyde fixation and PLA. Representative images (left) and quantification (right) of PLA dots (red) per cell are shown. Dapi (blue) and cell boundary (dotted line) are shown. Scale bar, 20 μm. Mean and SEM are shown. One-way ANOVA, **** p<0.0001. ns denotes no significance. (F) cGAMP production upon HIV-1 infection is not impaired by depletion of NONO. MDDCs, transfected with indicated siRNAs, were challenged with HIV-1 for 2.5 hours, then stained for cGAMP (green) and DAPI (blue). Scale bar, 10 μm. Mean cGAMP signals per cell were graphed (bottom). Mean and SEM are shown. One-way ANOVA, ** p<0.01, *** p<0.001, ns denotes no significance. All the data, unless noted otherwise, are representative of at least two independent experiments. See also Figure S4.
Next, we examined whether NONO is required for cGAS recruitment to incoming virus particles. Analogous to Figure 3A, the puncta overlap between IN-mRuby3 (virus) and cGAS signal was assessed in infected MDDCs to determined % of total capsids positive for cGAS. We observed that depletion of NONO had no noticeable impact on either PQBP1 or cGAS coassociation with viral capsids (Figure 4D; Figure S4B). Similarly, we found that NONO depletion did not affect cGAS association with the viral capsid in THP-1 cells. Specifically, a proximal ligation assay between stably expressed Flag-cGAS and CA in infected THP-1 cells confirms that NONO does not participate in cGAS recruitment to incoming capsids (Figure 4E; Figure S4C). Lastly, we confirmed that loss of NONO did not affect cGAMP production in HIV-1 infected MDDCs (Figure 4F; Figure S5C). Taken together, these data suggest that, in certain cellular contexts, NONO is dispensable for the innate response to HIV-1. Moreover, these results also indicate that NONO does not participate in the recruitment of cGAS to incoming HIV-1 capsids and its subsequent enzymatic activation.
PQBP1 interaction with the capsid licenses cGAS sensing of HIV-1 infection.
We next asked whether the HIV-1 capsid serves as a platform for facilitating the PQBP1-cGAS interactions. IF-based analysis revealed that HIV-1 infection selectively enhanced PQBP1 and cGAS colocalization whereas a direct activation of cGAS by herring-testis (HT) DNA did not (Figure 5A). Briefly, colocalization analysis was determined by three-dimensional detection of spots, followed by image segmentation. We utilized a threshold distance of 0.4 μm to delineate true molecular interactions. We observed a seven-fold increase in the frequency of PQBP1-cGAS colocalization in HIV-1 infected cells, while no enhancement in association was observed in cells transfected with HT-DNA, an established cGAS ligand (right, Figure 5A). Complementing the IF result, we also demonstrated that PQBP1 coprecipitated more efficiently with cGAS after HIV-1 infection but not after HT-DNA treatment compared to the no treatment control in MDDCs and PMA-treated THP-1 cells (Figure 5B).
Figure 5. HIV-1 infection promotes the formation of a PQBP1/cGAS innate sensing complex.
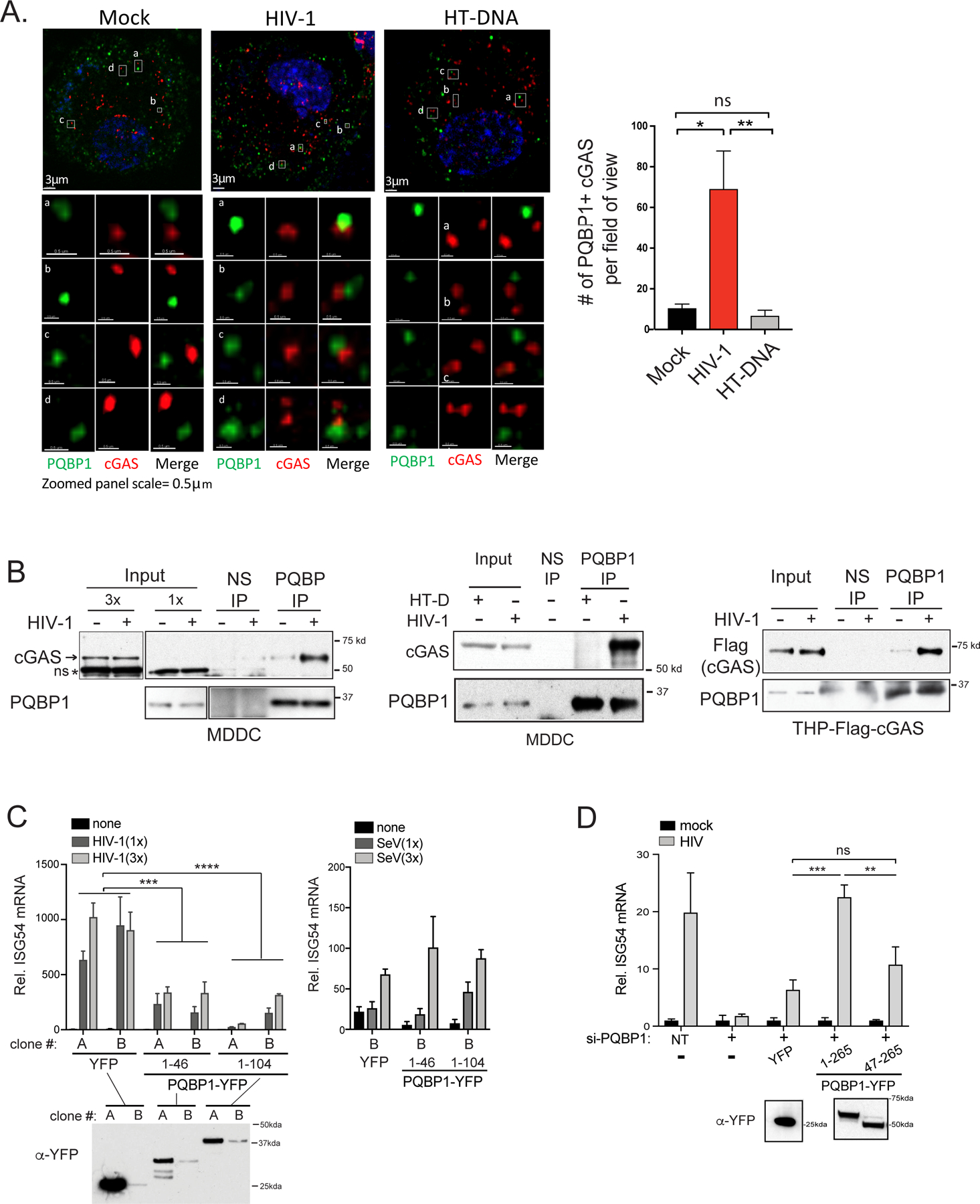
HIV-1 infection enhances PQBP1 and cGAS interactions. (A) THP-1 cells treated with PMA were fixed 2.5 hours post HIV-1 infection (Gag-IN-mRuby) or HT DNA transfection, followed by immunostaining of PQBP1 and cGAS for confocal microscopy. Z-section images (left) and the quantification of coassociating foci (right) are shown. A threshold for colocalization is set at a distance of 0.4 µm. Averages and SEM are shown. One-way ANOVA, ** p<0.01, * p<0.05, ns denotes no significance. Scale bars: 3μm (top) and 0.5 μm (bottom). (B) HIV-1 infection (+) enhances coimmunoprecipitation of cGAS or Flag-cGAS with PQBP-IP in MDDCs or PMA-THP-1, respectively. Cells were infected with HIV-1 luciferase virus in the presence of VLP-Vpx and subjected to co-IPs at 3 hrs post-infection. HT-DNA was delivered to cells instead of viral infection, where indicated. Endogenous cGAS or Flag-cGAS co-precipitating with PQBP1 was assayed. 1x and 3 x denote the relative amounts of inputs loaded on a gel. Normal IgG (NS) or antibody against PQBP1 were used as indicated. ns denotes non-specific bands. Cropped images are enclosed by boxed frames. See Figure S5A for original uncropped images. (C) The capsid interaction domain of PQBP1 is a dominant inhibitor of cGAS-mediated innate sensing of HIV-1 infection. Two independent PMA-THP-1 cell clones (A and B clones) stably expressing either YFP, PQBP11–46-YFP or PQBP11–104-YFP were infected with HIV-1 luciferase virus in the presence of VLP-Vpx or Sendai virus as indicated. ISG54 mRNAs were measured 16 hours post infection by qRT-PCR. (D) The capsid interaction domain of PQBP1 is needed for the innate response against HIV-1 infection. THP-1 cells treated with PMA, either un-transduced or stably expressing the indicated PQBP1-YFP proteins, were treated with siRNA against endogenous PQBP1 (+), followed by HIV-1 infection and ISG54 mRNA detection as in (C). NT denotes non-targeting siRNA. ISG54 mRNA levels were expressed as fold-induction over their uninfected counterparts. Expression level of either YFP or PQBP-YFPs in the cells used in (C) and (D) were determined by anti-GFP/YFP western blots. Equal numbers of cells were analyzed. T-test (unpaired; two-tailed) *p<0.05, **p<0.01, ***p<0.001. All data, unless noted otherwise, are representative of at least two independent experiments. See also Figure S5.
A systematic truncation analysis of PQBP1 confirmed that the cGAS and capsid interaction domains are distinct (Figure S5A). Briefly, either full-length or truncated PQBP1-YFP proteins were over-expressed with MBP-tagged cGAS proteins in 293T cells and subjected to coimmunoprecipitation assays (coIPs). Regions encompassing the capsid-associating N-terminal domain of PQBP1, aa 1–46 and aa 1–104, failed to coprecipitate with MBP-cGAS. In contrast, PQBP1 (aa 47–265) lacking the capsid-binding domain coprecipitated with MBP-cGAS as efficiently as the full-length protein (aa 1–265) (Figure S5A). Previously, we showed that a point mutation within the WW domain (aa 47–86) of PQBP1 impaired binding to cGAS. However, the N-terminal fragment containing the WW domain (aa 1–104) required additional C-terminal regions of PQBP1 to bind to cGAS (compare aa 1–104 with aa 1–211 in Figure S5; (Yoh et al., 2015)). These findings suggest that cGAS and capsid bind two distinct surfaces on PQBP1. We asked whether expression of PQBP1 mutants comprising either the capsid- or cGAS- binding domains could exert dominant negative effects on cGAS sensing of HIV-1 infection. Ectopic expression of residues 1–46 or 1–104 of PQBP1 inhibited ISG54 mRNA induction by HIV-1 infection when compared to cells expressing YFP alone (left, Figure 5C). This effect was specific to HIV-1 because Sendai virus infection was not inhibited (right, Figure 5C). Reciprocally, THP-1 cells expressing PQBP1 (aa 47–265) (Figure S6A) did not induce ISG54 mRNA in response to HIV-1 challenge (Figure 5D).
DISCUSSION
Our findings show that the assembled HIV-1 capsid acts as a preliminary PAMP that authenticates incoming viral nucleic acid cargo. Specifically, we find that PQBP1 recognizes the positively charged arginine pore on the multimerized CA lattice, but not monomeric CA (data not shown). The conservation of the charged pore among lentiviral capsids (Mallery et al., 2019) supports our previous observation that PQBP1 is a lentivirus-specific cofactor for cGAS sensing of viral DNA. The intact HIV-1 capsid cone has an estimated ~250 CA hexamers plus twelve pentamers that provide as many PQBP1 docking sites (Perilla and Gronenborn, 2016; Pornillos et al., 2011). Our in vitro binding assays indicate that multiple PQBP1 proteins bind to each assembled CA lattice (compare AF488-PQBP1 signal peaks in the absence and presence of CA-A204C, Figure 2B). cGAS is positioned at the site of viral DNA synthesis by virtue of its interactions with capsid-bound PQBP1. Its recruitment licenses a second step of innate immune sensing, which is cGAMP production and activation of down-stream STING/IRF3 signaling. This two-step recognition of pathogen protein and DNA probably ensures both sensitivity and specificity such that IFN pathways are not activated by independent recognition of low-abundance self PAMPs. This results in a powerful yet specific inflammatory response.
The finding that cGAS is recruited to the PQBP1-capsid platform only after capsid disassembly is initiated (Figure 3B) suggests that additional molecular signals are required for the formation of a functional PRR complex. These may include oligomerization of and conformational changes in PQBP1, as well as reverse-transcription or cytosolic exposure of transcribed DNA due to loss of core integrity (Christensen et al., 2020; Mamede et al., 2017; Manel et al., 2010; Sumner et al., 2020). Insights into the potential contributions of the reverse transcription complex, PQBP1 and cGAS come from various studies. Inhibition of reverse transcription by nevirapine blocks loss of capsid structural integrity and innate immune responses (Felts et al., 2011; Ilina et al., 2012; Mamede et al., 2017; Wang et al., 2021; Yoh et al., 2015). Blocking of first strand transfer by reverse transcriptase with an RNase H inhibitor (Ilina et al., 2012; Julias et al., 2002) does not compromise the innate immune response (data not shown), suggesting that the production of strong stop HIV-1 DNA is sufficient to activate cGAS. The coupling of reverse transcription and capsid disassembly is reinforced by the identification of a capsid mutant that displays both accelerated enzyme kinetics and capsid disassembly (Sultana et al., 2019). Lastly, cryo-electron tomography of in vitro-assembled reverse transcription complexes reveals strand-like loops, believed to be nascent DNA extruding from disassembling capsids (Christensen et al., 2020). Collectively, these findings underscore the crosstalk between reverse transcription, capsid disassembly and cGAS-mediated innate sensing.
Lahaye et. al. reported that cGAS sensing of HIV can occur in the nucleus through NONO binding of disassembled capsids (Lahaye et al., 2018). In contrast, we present evidence that intact, incoming HIV-1capsids interact with PQBP1 then cGAS in the cytoplasm to initiate the innate immune response, independently of NONO. These observations raise the possibility that cytoplasmic and nuclear sensing of HIV may be distinct processes, regulated by different capsid configurations and different cofactors. Collectively, our data reveal a unique, early, two-factor authentication strategy that activates the innate immune response to transient and low-abundance retroviral DNA species. The modular association of PQBP1 with cGAS triggers a robust response to viral DNA while circumventing activation by extranuclear, host-derived DNAs. This molecular strategy represents an expedient mechanism that expands the versatility of germline-encoded sensors, allowing them to mount effective immune responses to a range of invading pathogens with unique features that label them as PAMPs.
Limitation of the study.
Cyclophilin A (CypA) is a well-characterized capsid binding protein with a KD of ~ 10 μM as measured by total internal reflection fluorescence (TIRF) microscopy (Lau et al., 2019). Although we observe that PQBP1 and CypA bind to the capsid with comparable strength in TCCD analysis (data not shown), additional studies are warranted to determine the precise affinity of PQBP1 for the HIV-1 capsid. Secondly, further investigation is needed to understand the precise mechanism by which structural changes in the capsid and the progression of viral DNA synthesis result in the recruitment and activation of cGAS. Lastly, our observations raise the possibility that cytoplasmic and nuclear sensing of HIV-1 may be distinct processes that depend on different capsid configurations and different cofactors. Examination of a large number of different viral capsids, including HIV-1 mutants, naturally occurring variants or other lentiviruses, for their potential to be sensed by PQBP1/cGAS and further characterization on NONO's involvement in this process will certainly deepen our understanding of innate sensing of HIV-1 infection.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to Sumit K. Chanda (schanda@scripps.edu).
Materials Availability
All reagents generated in this study will be made available on request, but we may require a completed Materials Transfer Agreement if potential for commercial application.
Data and Code Availability
Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication DOIs are listed int the key resources table. Software may be used and modified for any non-commercial use under the GPL license terms.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-IRF3 (D9J5Q) | Cell Signaling Technology | Cat# 109495; |
| Rabbit polyclonal anti-PQBP1 | Bethyl Laboratory | Cat# A302–802A |
| Mouse monoclonal anti-PQBP1 | Santa Cruz Biotechnology | Cat# sc-376039; RRID:AB_10989350 |
| Mouse monoclonal anti-PQBP1 (1A11) | Sigma Aldrich | Cat# SAB1402496; RRID:AB_10640643 |
| Rabbit polyclonal anti-cGAS | Novus | Cat# NBP1–8676 |
| Mouse monoclonal anti-cGAS (D-9) | Santa Cruz Biotechnology | Cat# sc-515777; RRID:AB_2734736 |
| Mouse monoclonal anti-cGAS (5G10) | Caymanchem | Cat# 23853 |
| Rabbit monoclonal anti-cGAS (D1D3G) | Cell Signaling Technology | Cat# 49705 |
| Mouse monoclonal anti-GFP (GF28R) | Thermo Scientific | MA5–15256 |
| Rabbit polyclonal anti-GFP | Takarabio | Cat# 632592 |
| Mouse monoclonal anti-Flag | Sigma Aldrich | Cat# F1804; RRID:AB_262044 |
| Human monoclonal anti-HIV-1 p24 (71–31) | AIDS Reagent | Cat# ARP-530 |
| Mouse monoclonal anti-cGAMP | Pfizer (Hall et al., 2017) | PF-07043030 |
| Mouse monoclonal anti-MBP magnetic beads | New England Biolabs | Cat# 28037S |
| Rabbit monoclonal anti-beta-actin (13E5) | Cell Signaling Technology | Cat# 49705 |
| Normal rabbit/mouse IgG | Santa Cruz Biotechnology | Cat# sc-2027; RRID:AB_737197 |
| Bacterial and virus strains | ||
| Human lentiORF cDNA clone of PQBP1 | Open Biosystems (Yoh et. al., 2015) | N/A |
| pNL43 env- luc. | Manel et. al., 2010; Yoh et. al., 2015 | N/A |
| SIV3+ for production of VLP-VPX | D. R. Litman (Manel et. al., 2010; Yoh et. al., 2015) | N/A |
| Gag-IN-mRuby3 | Mamede et al., 2017 | N/A |
| HIV-Gag-iGFP | Mamede et al., 2017 | N/A |
| CypA-dsRed | Francis et al., 2016; Francis and Melikyan, 2018. | N/A |
| Cantell strain of Sendai virus | Mount Sinai Department of Microbiology Virus Collection | N/A |
| Biological samples | ||
| De-identified healthy donor blood | San Diego Blood bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 2’ 3’ -cGAMP | Invivogen | Cat# tirl-nacga23–02 |
| Herring testis DNA | Sigma Aldrich | Cat# D6898 |
| Lipofectamine 2000 | Life Technology | Cat# 11668030 |
| Stemfect RNA transfection kit | ReproCell | Cat# 00–0069 |
| Critical commercial assays | ||
| Duolink in situ detection reagent | Sigma | Cat# DUO92008 |
| Deposited data | ||
| None | N/A | N/A |
| Experimental models: Cell lines | ||
| Dual-KO-cGAS-THP-1 | Invivogen | Cat# thpd-kocgas |
| Dual-KO-cGAS-Flag-cGAS-THP-1 | This paper | |
| THP1-Dual Cells | Invivogen | Cat# thpd-nfis |
| THP-1 | ATCC | Cat# TIB-202 |
| 293T | ATCC | Cat# CRL-3216 |
| Experimental models: Organisms/strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| siNT (5’-AATCGATCATAGGACGAACGC-3’) | This paper | N/A |
| siPQBP1–1 (5’- AAGCTCAGAAGCAGTAATGCA-3’) | This paper | N/A |
| siPQBP1–2 (5’-AAAGCCATGACAAGTCGGACA-3’) | This paper | N/A |
| Non-targeting guide crRNA guide sequence | Dharmacon | U-007502 |
| MB21D1(cGAS) crRNA guides: 1–5 | Dharmacon | CM-015607-(1 to 5), see Guide RNA sequences under Star Method |
| NONO (4841) targeting guides: 2 and 5 | Dharmacon | CM-007756-(02 and 05), see Guide RNA sequences under Star Method |
| Recombinant DNA | ||
| pEASIL-Flag-cGAS | This paper | N/A |
| pEASIL | M. Malim | N/A |
| pCDFDuet-PQBP1 | This paper | N/A |
| pCDNA 3.1-MBP-cGAS | This paper | N/A |
| Software and algorithms | ||
| Imaris | Bitplane | RRID:SCR_007370 |
|
Python Programming Language
|
Python Software Foundation | RRID:SCR_008394 |
| Custom Scripts for imaging analysis | this article, PMID:33154161, | 10.5281/zenodo.6551125 |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary cells and cell-lines
This study was approved by the National Institutes of Health (NIH) through our Institute Biosafety Committee (IBC). Primary monocyte-derived dendritic cells (MDDCs) were prepared from fresh, healthy donor blood from the San Diego Blood Bank as described previously (Yoh et al., 2015). The human monocyte-like THP-1 cell line, grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), was differentiated by treatment with 20–40 ng/ml PMA (Phorbol myristate acetate) for 2 days. HEK293T cells were grown in DMEM high glucose supplemented with 10% FBS. Both THP-1 and HEK293T cells were purchased from ATCC (Cat# TIB-202 and CRL-3216). THP-1 Dual and THP-1-Dual KO-cGAS cells were purchased from Invivogen (Cat# thpd-kocGAS and Cat# thpd-nfis) and maintained in RPMI 1640+10% FBS. All cell culture media were further supplemented with 2 mM Glu, 100 IU/mL penicillin/streptomycin.
Authentication:
The listed cell-lines were not further authenticated after purchase from ATCC and Invivogen.
METHOD DETAILS
Reagents
50 – 100 ng of cGAMP (Invivogen) and 2 ng to 10 ng of Herring testis (HT) DNA (Sigma) were transfected into 2.5 x 104 PMA-differentiated THP-1 using Lipofectamine 2000 (Life Technologies). The following antibodies were used: IRF3 (Cell Signaling D9J5Q), PQBP1 [Bethyl Laboratory A302–802A; Santa Cruz Biotechnology sc-376039; Sigma Aldrich (1A11)], cGAS [Novus NBP1–8676; Santa Cruz Biotechnology (D-9); Cayman (5G10); Cell signaling (D1D3G)], normal rabbit/mouse IgG (Santa Cruz Biotechnology SC-2027), β-actin (Cell Signaling Technology 49705), GFP (Thermo Scientific MA5–15256: Clontech 632592), FLAG (Sigma Aldrich F1804), p24 (AIDS Reagent 71–31). Anti-cGAMP [PF-07043030 Pfizer (Hall et al., 2017)], Anti-MBP magnetic beads (NEB).
Plasmids, siRNAs and qRT-PCR
Human LentiORF cDNA clones of PQBP1 (Open Biosystems), both wild-type and mutant constructs, in-frame fused with eYFP proteins, were generated by a standard Gibson cloning approach, and packaged into lentiviruses according to the manufacturer’s protocol. The silent mutations were introduced to PQBP1-eYFP constructs to render the proteins resistant to siPQBP1 RNAs as described previously (Yoh et al., 2015). Flag-cGAS is cloned into pEASIL doxycycline inducible lentivirus vector (a generous gift from M. Malim). MBP-cGAS and 6xHIS-PQBP1 constructs were cloned into pCDNA 3.1 and pCDFDuet vectors respectively. THP-1 cells, either wildtype or Dual-KO-cGAS (Invivogen) were infected with each lentivirus to generate cells with stable expression of the protein of interest.
siRNAs were introduced into PMA-THP-1 cells using Stemfect RNA transfection kits. Typically, 5 pmol of siRNAs and 0.17 µl of Stemfect are used per 2.5 x 104 cells. Forty-eight hours after siRNA transfection, cells were infected with either HIV-1 in the presence of VLP-VPX (Manel et al., 2010; Yoh et al., 2015) or 2.5 HAU/ml of Sendai virus for 16 hours, followed by RNA isolation and qRT-PCR analysis. Alternatively, for IF analysis, cells were fixed 2–3 hours post-infection. The following siRNAs were used: siNT (5’-AATCGATCATAGGACGAACGC-3’); siPQBP1–1 (5’- AAGCTCAGAAGCAGTAATGCA-3’); siPQBP1–2 (5’-AAAGCCATGACAAGTCGGACA-3’). qPCR primers were described previously (Yoh et al., 2015).
Viral infection
MDDCs or PMA-differentiated THP-1 cells were infected with VSV-G pseudotyped HIV-1 virus and VLP-Vpx, as described previously (Manel et al., 2010; Yoh et al., 2015) unless otherwise specified. In general, 2–10 ng of p24 or 0.1 to 0.5 RT units of HIV-1 were used per 25K cells in 100 μl of media and harvested between 1.5 to 16 hours post infection, as specified. All HIV-1 were generated by transient transfection of provirus plasmids in 293T in 10cm dishes at 50% confluency and harvested 48 hours post-transfection, followed by DNase-treatment. Viral particles were concentrated through 25% sucrose TNE by centrifugation for 16 hours at 3000 x g at 4oC. VSV-G-pseudotyped HIV-1 firefly luciferase (luc) was prepared by transfecting 10 µg of NL4–3 R+ E-firefly-luc plasmid, provided by Dr. Nathaniel Landau and 1.5 μg of pCMV-VSV-G with 40 µl of PEI (pH 4.5, 1 µg/ml) or 30 µl of lipofectamine 2000. HIV-Gag-IN-mRuby3 viruses were produced by transfecting 5 μg of pNL43 env-, 2 μg of Gag-IN-mRuby3 and 4 μg of pCMV-VSV-G. HIV-iGFP and Gag-IN-mRuby3 viruses were produced by transfection of 5 μg of HIV-Gag-iGFP, 3 μg of pGag-IN-mRuby3, and 4 μg of pCMV–VSV-G (Mamede et al., 2017; Sultana et al., 2019). CypA-dsRed packaged HIV-1 virus were produced by transfection of 5 μg of NL43dEnv-, 3 μg of pCMV-VSV-G and 4 μg of CypA-dsRed plasmids (Francis et al., 2016; Francis and Melikyan, 2018).
The Mount Sinai Department of Microbiology Virus Collection provided the Cantell strain of Sendai virus, grown for 2 days in 10-day old embryonated chicken eggs, and tittered using turkey RBC HA assays (Lampire Biological Laboratories).
CRISPR-Cas9 RNP Production and THP-1 Electroporation
Detailed protocols for RNP production have been previously published (Hultquist et al., 2019). All crRNA guide sequences used in this study were derived from the Dharmacon pre-designed Edit-R library for gene knock-out, including the non-targeting guide (U-007502), cGAS-targeting guides (equimolar pool of CM-015607–01 through CM-015607–05), and NONO-targeting guides (CM-007756–02 and CM-007756–05).
| Gene | Guide Number | Sequence | Catalog Number |
|---|---|---|---|
| MB21D1 (cGAS) | 1 | TTGAATGCGCAGGCCTTCTT | CM-015607–01 |
| MB21D1 (cGAS) | 2 | CTGGGTACATACGTGAAAGA | CM-015607–02 |
| MB21D1 (cGAS) | 3 | GAACTTTCCCGCCTTAGGCA | CM-015607–03 |
| MB21D1 (cGAS) | 4 | CCGCGATGATATCTCCACGG | CM-015607–04 |
| MB21D1 (cGAS) | 5 | GCATCCCTCCGTACGAGAAT | CM-015607–05 |
| NONO (4841) | 2 | GTTTATGCCCATAGCACCTA | CM-007756–02 |
| NONO (4841) | 5 | ATGGGAGATATACCGCATCA | CM-007756–05 |
Guide RNA Sequences
Immunofluorescence, image acquisition and analysis
PMA-THP-1 cells were seeded at a density of 2.5 x 104 cells/ well in a 96 well glass plate for 48 hours followed by infection with HIV-1 with or without VLP-Vpx, as specified, or transfection of either HT DNA or cGAMP for 1.5 hrs to 3 hrs. Cells were then fixed with 4% w/v paraformaldehyde in PBS, permeabilized using a standard protocol and subjected to immunostaining. Cells were imaged by fluorescence wide-field deconvolution or confocal microscopy using DV-Elite, GE-Ultra, OMX-SR, Nikon TIe-2, Zeiss LSMv880. Images obtained in GE microscopes were deconvolved with the SoftWorx package using standard vendor software definitions and camera biases. Images obtained with the Nikon TIe-2 microscope were deconvolved with FlowDec using the Lucy-Richardson algorithm with the support of pims relying on python 3.x with PSFs generated in FIJI/ImageJ. Super resolution Images were obtained in 3D-SIM mode in a GE OMX-SR microscope equipped with 4 laser lines, 60x/1.42 NA oil immersion lens (Olympus), and 3 independent cameras. Images were calculated/reconstructed and channel registered with the vendor’s software (SoftWorx) with 0.001 Wiener constant values.
Fluorescence resonance energy transfer assay
Viral infections with dsRed-CypA labeled particles were imaged in cells expressing eYFP or PQBP1-eYFP truncations. Images were acquired as a Z-stack with a GE-Deltavision Ultra microscope equipped with a PCO edge CMOS camera and an oil immersion 60x/1.42 NA Olympus lens with 1.42 NA, and deconvolved using the vendors software (SoftWorkx). Excitation was done with an SSI-LED light source, and the emitted wavelength collection was cleaned up with appropriate excitation/emission filters: for FRET (YFP/mCherry); YFP (YFP/YFP); dsRed (mCherry/mCherry). Analysis was performed by identifying the coordinates of viral particles in the deconvolved images and measuring the intensities from the raw images in each corresponding channel in the regions containing viral particles. Cross-talk was controlled by measuring intensities of dsRed-CypA labeled particles in cells in the absence and presence of eYFP or PQBP1-eYFPs constructs. The cross-talk control also included measurement of signals form the cells without viral particles. The values for the normalized FRET of viral particle intensities were calculated using the following formula and methods as in (Jiang and Sorkin, 2002; Sood et al., 2017; Zal and Gascoigne, 2004):
Viral capsid integrity assay - Correlative Live and fixed immunofluorescent Imaging Analysis
Delta Vision wide-field microscopes (DV-ELITE or OMX-SR, GE Life Sciences) equipped with an electron multiplying charge-coupled device (EMCCD) camera and a CMOS cameras (PCO edge) with solid state illumination (SSI-LED) light path were used to acquire time-lapsed fluorescent snapshots of HIV-iGFP/IN-mRuby3 viruses infecting THP-1 or MDDCs plated in Delta T culture dishes (Bioptechs) or μ-Slide I Luer (Ibidi) that were coated with fibronectin (SIGMA) for overnight adhesion of MDDCs (but not THP-1), according to the supplier’s instructions. Cells were kept in a 37°C heated chamber, 5% CO2 and20% oxygen, throughout the imaging process. Cells were incubated with RPMI without phenol red with 10% FBS, L-glutamine, and MEM-NEAA. All infections were done with Polybrene at a concentration of 5 μg/ml. Z-stacking spacing was set to 0.5 µm, with a total of 12-μm z-axis imaging for the fluorescence snapshots, and a single Z reference image was taken in bright field for cell edge identification. Nominal magnification was 60x/1.42NA lens for all experiments. Cells were washed once with PBS, promptly after the last time point of imaging, and fixed with 3.7% formaldehyde in piperazine-N,N′-bis (PIPES) buffer for 5 min, followed by three PBS washes, as in (Mamede et al., 2017). Culture dishes were then permeabilized with blocking media made of TX-10 donkey serum and stained with antibodies probing for PQBP1 (Sigma) and cGAS (Novus) followed by anti-mouse-AF647 and anti-Rb-dylight405 (Jackson Immunoresearch) secondary antibodies, respectively. Using the same microscope, or synchronized trays between DV-ELITE and OMX-SR, the previously time-lapse imaged cells and fields of view were found and imaged after staining. The viral particles were identified by their IN-mRuby3 signal present in the same area as the last time-lapse frame, when still present in this second phase. ROIs were set in ImageJ/Fiji based on the IN-mRuby3 puncta and the mean intensities for all channels in such ROI were quantified and plotted. The second verification of capsid integrity loss was performed in the fixed imaging stage by the disappearance of iGFP signal.
Z-stacks were deconvolved and z-projected using SoftWorx (GE Life Sciences) before each individual IN-mRuby3 particle were tracked over time was performed using FIJI/ImageJ (NIH). Mean intensities of HIV-iGFP were automatically measured in the same x-y coordinates where the IN particle was identified by the tracking algorithm. Centered particle video recordings were automatically generated as in the references (Mamede et al., 2017; Sultana et al., 2019) using Matplotlib libraries, with the data analyzed and exported from Fiji/ImageJ.
Proximal Ligation Assay (PLA)
PMA-THP-1 cells, 3 hours post-infection, were fixed in 4% PFA in PBS for 20 min, permeabilized with 0.2% Triton X-100 in PBS, and subjected to the PLA assay according to the manufacturer’s protocol for the Duolink in situ detection reagents red kit (DUO92008, Sigma-Aldrich). Antibody incubations were used at 1;400 dilution: anti-rabbit HIV1 p24 (Abcam-ab32352) and mouse anti-FLAG® M2 antibody (Sigma). PLA dots were detected using Nikon A1R HD confocal with 60X objective. Analysis was done using ImageJ.
Image Analysis
Identification of viral particles from infected THP-1 cells was done with python with skimage, scipy, pims, and trackpy tools (v0.4.2 - http://soft-matter.github.io/trackpy/doi.org/10.5281/zenodo.3492186). The original scripts were deposited in doi.org/10.5281/zenodo.6551125. In short, cells were subjected to a binary threshold so that no extracellular signals (viruses or cell debris) would confound the analysis. Viral particles were detected using thresholding and watershed segmentation methods or by using trackpy. For virus analysis, the masks of the pixels that are positive for viral particles were then quantified for each independent channel (PQBP1, cGAS, iGFP, IN label, etc).
Co-immunoprecipitation
MDDCs or PMA-THP-1 cells expressing Flag-cGAS proteins were infected with VSV-G pseudo-typed HIV-1 luciferase virus at MOI of 1 in the absence and presence of VLP-Vpx co-infection, respectively. Three hours post-infection, cells were lysed and subjected to immunoprecipitation against endogenous PQBP1 proteins, followed by probing the IPs for endogenous cGAS or Flag-cGAS presence. HEK293T lysates expressing eYFP or eYFP fused either to wild type or mutant PQBP1 and MBP-cGAS were subjected to immunoprecipitation against MBP tags, followed by probing for PQBP1-YFP proteins. Typically, 300 μg of protein lysates were incubated with anti-MBP-magnetic beads (NEB) in 100 mM KCl, 12.5mM MgCl2, 0.5% Triton-X, 20 mM HEPES, 0.2 mM EDTA, 10% glycerol, 0.2 μM PMSF, and protease inhibitors. The beads were washed three times with 20 mM HEPES, 0.2 mM EDTA, 300 mM KCl, 0.5% Triton-X, 10% glycerol and 0.2 μM PMSF, followed by a conventional western blot analysis.
Recombinant protein production
C-terminal MBP-6xHis tag was fused to CA (A14C/E45C) using a SARS main protease cleavage site linker in pET11a as previously described (Summers et al., 2019). 6xHis-PQBP1 was cloned into pCDFDuet using BamHI/HindIII restriction sites. MBP, used as a negative control in co-pelleting, was obtained from cleaved CA(A14C/E45C) during purification, before tube assembly. Protein was over expressed in BL21(DE3) cells grown to an OD of 0.6–0.8, and induced with 0.5 mM IPTG at 25 °C for CA(A14C/E45C)-MBP-6xHis or 18˚C for 6xHis-PQBP1 for 18 hours. Cells were lysed by microfluidization in 50 mM Tris, pH 8.0, 500 mM NaCl, 5% v/v glycerol, 0.1 mM TCEP with a protease inhibitor tablet (Roche). CA(A14C/E45C)-MBP-6xHis was purified as described previously (Summers et al., 2019). 6xHis-PQBP1 clarified lysate was purified by Ni-NTA (Qiagen) affinity and size exclusion chromatography (GE). 6xHis-PQBP1 was concentrated to 5–10 mg/mL in 50 mM Tris, pH 8.0, 50 mM NaCl, 0.1 mM TCEP and flash frozen in liquid nitrogen until use. CA tubes were assembled as described previously (Pornillos et al., 2009) from the purified, MBP-6xHis cleaved CA(A14C/E45C) and stored at 4˚C in 50 mM Tris, pH 8.0, until use.
Co-pelleting assays were performed by incubating a final concentration of 10 µM PQBP1 or MBP with 100 µM assembled CA tubes in the presence of 25 or 500 mM NaCl on ice. The reaction buffer contained the noted amount of NaCl with 50 mM Tris, pH 8.0, 25 mM NaCl, 0.1% v/v NP-40. Samples were centrifuged at 14,500 g for 20 min at 4°C,and supernatant was separated from the pellet. The pellet was dissolved in an equal volume of buffer and samples were analyzed via SDS-PAGE on NuPage™ gel (Invitrogen) and developed with SimplyBlue™ stain (Invitrogen). Gels were imaged directly with a CCD camera.
Covalent labelling of Recombinant PQBP1, CypA, and CPSF6313–327
Recombinant PQBP1 protein in PBS (1X) and 0.1 mM TCEP was incubated with AlexaFluor488-C5-malamide dye (Thermo Fischer Scientific) in a 1:1.2 molar ratio for 15 min at room temperature. Unreacted dye was removed by size exclusion chromatography using a Superdex 200 Increase 5/150 GL column (Cytiva) equilibrated in 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl flowing at 0.2 mL/min on a HPLC (Shimadzu). Fractions were collected manually and fractions containing labelled AF488-PQBP1 conjugate were identified by SDS PAGE and imaging with the Alexa Fluor 488 filter on the ChemiDoc MP imaging system (BIORAD). Proteins were stored at -80˚C in 10% v/v glycerol and dialyzed into 20 mM Tris-HCl (pH 8.0) and 75 mM NaCl prior to TCCD measurements. Purification and labelling of CypA and CPSF6313–327 peptide with Alexa Fluor dyes were previously described (Lau et al., 2019; Peng et al., 2019).
Cell-Free Expression and Purification of GFP-tagged PQBP1 fragments
The coding sequences for PQBP11–46 and PQBP147–265 were amplified by PCR and cloned into Gateway™ vectors containing either an N-terminal 8xHis-eGFP or a C-terminal sfGFP-8xHis tag using Gibson assembly (NEB). For each construct, purified plasmid DNA was added to 100 µL Leishmania tarentolae cell-free expression mix to 60 nM DNA and expressed for 2.5 hrs at 28 ˚C.(Lau et al., 2019) The expressed PQBP1 fragments were bound to Ni-NTA beads (BIORAD) for 30 min on ice and washed with 300 µL of 20 mM Tris-HCl (pH 8.0) and 100 mM NaCl before bound protein was eluted with 40 µL of the same buffer containing 0.5 M imidazole. Purified PQBP1 constructs were then dialyzed into 20 mM Tris-HCl (pH 8.0) and 75 mM NaCl for 1 hr and were immediately used for TCCD measurements.
In vitro CA lattice assembly
Recombinant HIV-1 CA with and without additional mutations (R18G, N74D) and CA K158C-AF568 were purified and labeled as described previously (Lau et al., 2019). CA lattices were assembled in vitro at 80 µM CA (1:99 ratio of CAK158C-AF568:CAA204C) in Tris-HCl (pH 8.0) and 1 M NaCl. The assembly solution was incubated at 37°C for 15 min followed by 4°C overnight. Assembled CA lattices were dialyzed into 20 mM Tris-HCl (pH 8.0) containing 75 mM NaCl.
Two-color coincidence detection (TCCD) spectroscopy
The TCCD fluorescence fluctuation spectroscopy approach has been described previously (Lau et al., 2021). Binding reactions were performed in 20 mM Tris-HCl (pH 8.0) and 75 mM NaCl containing 8 µM of CA (monomeric equivalent) that were assembled under high salt conditions with binders as follows: 100 nM GFP-tagged PQBP1, 20 nM PQBP1-AF488, 50 nM CypA-AF488, 10 nM CPSF6313–327-AF488, and 10 nM of fluorescein-12-dATP (PerkinElmer, NEL465001EA). Where applicable, hexacarboxybenzene (HCB) was added to a final concentration of 10 µM. At least 100 s of data were collected for each condition in 10 s acquisitions at 1000 Hz binning on an inverted microscope equipped with 488 nm (2.6 mW), 561 nm (0.5 mW) lasers and a water immersion 40x/1.2 NA objective (Zeiss). The emitted fluorescence from each fluorophore was separated into two channels using a dichroic mirror (565 nm) and filtered through a 525/50 nm band pass filter (AF488/GFP signal) and 590 nm long pass filter (AF568 signal), respectively, prior to focusing onto separate single photon avalanche diodes (Micro Photon Devices). The coincidence intensity ratios were calculated using a custom in-house software (TRISTAN, https://github.com/lilbutsa/Tristan/tree/master/v0_2/Matlab_Programs) as the slope of a curve obtained using weighted linear correlation between the binder fluorescence intensity and capsid signal intensities from each 10 s acquisition (Lau et al., 2021). Coincidence ratios in different conditions were compared using ordinary one-way ANOVA using GraphPad Prism (v8.4).
PQBP1 and IN distance distribution analysis
The distances, d, from each IN to the nearest PQBP1 spot was used as an indicator of their association. The probability distribution of nearest neighbor distances, P(d), shows a strong peak around d=0.125 µm, and a broader and weaker peak around d=0.7 µm (blue; left, Figure S1D). These two peaks likely represent two different aspects of protein localization. To distinguish between stochastic and biologically relevant colocalization, we manipulated the positions of IN and PQBP1 in silico. First, we randomized the labels, shuffling IN and PQBP1 spots together while keeping the density and the IN/PQBP1 ratio constant. After label randomization, P(d) presents as a single broad peak around d=0.75 µm (purple; left, Figure S1D). Next, we added a random uniform jitter J to PQBP1 positions and calculated P(d). We find that any added jitter weakens the narrow peak at d=0.125 µm in favor of the peak at d=0.75 µm. At J=1 µm the jittered curve (yellow; left, Figure S1D) overlays the randomized curve. One way to quantify colocalization is to calculate the percentage ϕ of IN spots that have a PQBP1 within a chosen threshold, δ(right, Figure S1D). Formally, ϕ is defined by . A reasonable value for δ accounts for optical aberrations, which can be wavelength dependent. While the two fluorophores may be in close physical proximity, their image on a microscope could be shifted and deformed. The safest way to proceed is then to choose a reasonable value for δ and calculate the colocalization using that threshold. Finally, the resistance to small variations in δ, known as sensitivity analysis, can be used to support the original choice of parameter. We plot ϕ as a function of jitter J and note that for reasonable values of δ ( 0.1 µm < δ < 0.75 µm ), the dependence of ϕ on J follows a similar behavior, implying that δ is robust against small changes. The biphasic decrease in ϕ again indicates the presence of two aspects of protein colocalization. We note that the step in ϕ happens when J~δ. This does not occur when the labels are randomly shuffled, indicating that once the jitter exceeds the nearest neighbor distance, colocalization is destroyed in real data, but not in randomized shuffling of acquired data. Collectively, the distance analysis confirmed a robust co-localization between PQBP1 protein and incoming virions (above 40% at δ, = 0.5 µm) that is not due to a random distribution of cellular PQBP1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses used GraphPad Prism 8 software, with a minimum of three biologically independent infection for significance. Statistical details of experiments (statistical tests used, value and definition of n and definition of center and dispersion and precision measures) can be found in the figure legends. All experiments were reproduced at least two times, unless otherwise indicated in legends.
Supplementary Material
Video S1. Time-lapse imaging to monitor structural integrity of an incoming viral particle with iGFP fluid phase marker (iGFP+IN-mRuby3) in MDDCs. See Figures 3B and S3B for detail.
ACKNOWLEDGEMENTS
The authors would like to thank Zeli Zhang for reagent preparations; SBP Flow Cytometry core for technical assistance; Tanya Dragic for manuscript editing. Research reported in this publication was supported by NIAID of the National Institutes of Health under award numbers (R01 AI127302–01A1, R01AI162260, P50AI150481, P50AI150464, P50 AI150476, P30 AI117943, R01 AI150455, R01 AI165236, R01AI150998, R01 AI105184–02 Supplement, K22AI140963 and AI150464) and cFAR Development Grant and CHRP Basic Bio Pilot BB19-SBMR-0, Gilead Sciences Research Scholars Program in HIV and by the German Research Foundation (DFG; SPP1923 Project KO4573/1–2). 90%/$ 500,000 of the total project costs were financed with Federal funding. 10%/$ 55,000 of the total costs were financed with non-Federal funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DECLARATION OF INTERESTS
The A.G.-S. laboratory has received research support from Pfizer, Senhwa Biosciences, Kenall Manufacturing, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N-fold LLC, Model Medicines, Atea Pharma, Applied Biological Laboratories and Merck, outside of the reported work. A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories, Pharmamar, Paratus, CureLab Oncology, CureLab Veterinary, Synairgen and Pfizer, outside of the reported work. A.G.-S. has been an invited speaker in meeting events organized by Sequirus, Janssen and Astrazeneca outside of the reported work. A.G.-S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work.
The SKC laboratory has received research support from Eli Lilly & Co., Boehringer Ingelheim, Merck & Co., Cidara Therapeutics, outside of the reported work. SKC has consulting agreements for the following companies involving cash and/or stock: Pagoda Genomics, Cidara Therapeutics, and Samsara Biocapital, outside of the reported work. SKC is inventor on patents and patent applications on the use of antivirals, adjuvants, and immunotherapies for the treatment and prevention of virus infections and cancer, owned by Sanford Burnham Prebys Medical Discovery Institute and Scripps Research, outside of the reported work.
REFERENCES
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, et al. (2014). Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 111, 18625–18630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick RC, Li C, Munshi M, Rawson JMO, Nagashima K, Hu WS, and Pathak VK (2020). HIV-1 uncoats in the nucleus near sites of integration. Proc Natl Acad Sci U S A 117, 5486–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, and Hope TJ (2015). HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 13, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Hubner W, Spinelli MA, and Chen BK (2007). Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J Virol 81, 12582–12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AC (2019). Neuroinflammation and the cGAS-STING pathway. J Neurophysiol 121, 1087–1091. [DOI] [PubMed] [Google Scholar]
- Christensen DE, Ganser-Pornillos BK, Johnson JS, Pornillos O, and Sundquist WI (2020). Reconstitution and visualization of HIV-1 capsid-dependent replication and integration in vitro. Science 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. (2015). Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 17, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosnefroy O, Murray PJ, and Bishop KN (2016). HIV-1 capsid uncoating initiates after the first strand transfer of reverse transcription. Retrovirology 13, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharan A, Opp S, Abdel-Rahim O, Keceli SK, Imam S, Diaz-Griffero F, and Campbell EM (2017). Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc Natl Acad Sci U S A 114, E10707–E10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, and Greene WC (2010). Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts AK, Labarge K, Bauman JD, Patel DV, Himmel DM, Arnold E, Parniak MA, and Levy RM (2011). Identification of alternative binding sites for inhibitors of HIV-1 ribonuclease H through comparative analysis of virtual enrichment studies. J Chem Inf Model 51, 1986–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AC, Marin M, Shi J, Aiken C, and Melikyan GB (2016). Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLoS Pathog 12, e1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AC, and Melikyan GB (2018). Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 23, 536–548 e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, and Hill CP (1996). Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, and Chen ZJ (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, Aulabaugh A, Aivazian D, Castro D, Chen M, et al. (2017). Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One 12, e0184843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WS, and Hughes SH (2012). HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner W, Chen P, Del Portillo A, Liu Y, Gordon RE, and Chen BK (2007). Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol 81, 12596–12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FY, Li XD, Asmuth DM, Huser T, and Chen BK (2009). Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323, 1743–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme AE, Kelley Z, Okocha EA, and Hope TJ (2015). Identification of capsid mutations that alter the rate of HIV-1 uncoating in infected cells. J Virol 89, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme AE, Perez O, and Hope TJ (2011). Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci U S A 108, 9975–9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Hiatt J, Schumann K, McGregor MJ, Roth TL, Haas P, Doudna JA, Marson A, and Krogan NJ (2019). CRISPR-Cas9 genome engineering of primary CD4(+) T cells for the interrogation of HIV-host factor interactions. Nat Protoc 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilina T, Labarge K, Sarafianos SG, Ishima R, and Parniak MA (2012). Inhibitors of HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Biology (Basel) 1, 521–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki Y, and Thomsen GH (2014). The splicing factor PQBP1 regulates mesodermal and neural development through FGF signaling. Development 141, 3740–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques DA, McEwan WA, Hilditch L, Price AJ, Towers GJ, and James LC (2016). HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 536, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James LC, and Jacques DA (2018). The Human Immunodeficiency Virus Capsid Is More Than Just a Genome Package. Annu Rev Virol 5, 209–225. [DOI] [PubMed] [Google Scholar]
- Jiang X, and Sorkin A (2002). Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol Biol Cell 13, 1522–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julias JG, McWilliams MJ, Sarafianos SG, Arnold E, and Hughes SH (2002). Mutations in the RNase H domain of HIV-1 reverse transcriptase affect the initiation of DNA synthesis and the specificity of RNase H cleavage in vivo. Proc Natl Acad Sci U S A 99, 9515–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJ, et al. (2018). NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 175, 488–501 e422. [DOI] [PubMed] [Google Scholar]
- Lau D, Walsh JC, Dickson CF, Tuckwell A, Stear JH, Hunter DJB, Bhumkar A, Shah V, Turville SG, Sierecki E, et al. (2021). Rapid HIV-1 Capsid Interaction Screening Using Fluorescence Fluctuation Spectroscopy. Anal Chem [DOI] [PubMed] [Google Scholar]
- Lau D, Walsh JC, Peng W, Shah VB, Turville S, Jacques DA, and Bocking T (2019). Fluorescence Biosensor for Real-Time Interaction Dynamics of Host Proteins with HIV-1 Capsid Tubes. ACS Appl Mater Interfaces 11, 34586–34594. [DOI] [PubMed] [Google Scholar]
- Mallery DL, Faysal KMR, Kleinpeter A, Wilson MSC, Vaysburd M, Fletcher AJ, Novikova M, Bocking T, Freed EO, Saiardi A, et al. (2019). Cellular IP6 Levels Limit HIV Production while Viruses that Cannot Efficiently Package IP6 Are Attenuated for Infection and Replication. Cell Rep 29, 3983–3996 e3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, Marquez CL, McEwan WA, Dickson CF, Jacques DA, Anandapadamanaban M, Bichel K, Towers GJ, Saiardi A, Bocking T, et al. (2018). IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamede JI, Cianci GC, Anderson MR, and Hope TJ (2017). Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc Natl Acad Sci U S A 114, E7169–E7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamede JI, Griffin J, Gambut S, and Hope TJ (2021). A New Generation of Functional Tagged Proteins for HIV Fluorescence Imaging. Viruses 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, and Littman DR (2010). A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467, 214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei S, Glass B, Hagen WJ, Krausslich HG, and Briggs JA (2016). The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 354, 1434–1437. [DOI] [PubMed] [Google Scholar]
- Novikova M, Zhang Y, Freed EO, and Peng K (2019). Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol Sin 34, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, and Bowie AG (2013). Immune sensing of DNA. Immunity 38, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Shi J, Marquez CL, Lau D, Walsh J, Faysal KMR, Byeon CH, Byeon IL, Aiken C, and Bocking T (2019). Functional analysis of the secondary HIV-1 capsid binding site in the host protein cyclophilin A. Retrovirology 16, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perilla JR, and Gronenborn AM (2016). Molecular Architecture of the Retroviral Capsid. Trends Biochem Sci 41, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, and Yeager M (2009). X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, and Yeager M (2011). Atomic-level modelling of the HIV capsid. Nature 469, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, and James LC (2014). Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10, e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic S, Ramalho R, Aiken C, and Rousso I (2018). PF74 Reinforces the HIV-1 Capsid To Impair Reverse Transcription-Induced Uncoating. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic S, Varadarajan J, Ramalho R, Aiken C, and Rousso I (2017). Reverse Transcription Mechanically Initiates HIV-1 Capsid Disassembly. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, Mogensen TH, Meyer M, Vaegter C, Nyengaard JR, et al. (2016). Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun 7, 13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaga SS, Xu C, Summers BJ, Digianantonio KM, Perilla JR, and Xiong Y (2019). MxB Restricts HIV-1 by Targeting the Tri-hexamer Interface of the Viral Capsid. Structure 27, 1234–1245 e1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman M, Srikrishna G, and Balagopal A (2017). Mechanisms of HIV-1 Control. Curr HIV/AIDS Rep 14, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood C, Francis AC, Desai TM, and Melikyan GB (2017). An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J Biol Chem 292, 20196–20207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana T, Mamede JI, Saito A, Ode H, Nohata K, Cohen R, Nakayama EE, Iwatani Y, Yamashita M, Hope TJ, et al. (2019). Multiple Pathways To Avoid Beta Interferon Sensitivity of HIV-1 by Mutations in Capsid. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers BJ, Digianantonio KM, Smaga SS, Huang PT, Zhou K, Gerber EE, Wang W, and Xiong Y (2019). Modular HIV-1 Capsid Assemblies Reveal Diverse Host-Capsid Recognition Mechanisms. Cell Host Microbe 26, 203–216 e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner RP, Harrison L, Touizer E, Peacock TP, Spencer M, Zuliani-Alvarez L, and Towers GJ (2020). Disrupting HIV-1 capsid formation causes cGAS sensing of viral DNA. EMBO J 39, e103958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Sone M, Nakamura Y, Shimamura T, Imoto S, Miyano S, and Okazawa H (2013). A restricted level of PQBP1 is needed for the best longevity of Drosophila. Neurobiol Aging 34, 356 e311–320. [DOI] [PubMed] [Google Scholar]
- Wang L, Casey MC, Vernekar SKV, Sahani RL, Kirby KA, Du H, Zhang H, Tedbury PR, Xie J, Sarafianos SG, et al. (2021). Novel PF74-like small molecules targeting the HIV-1 capsid protein: Balance of potency and metabolic stability. Acta Pharm Sin B 11, 810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiser C, Kim B, Vincent J, and Ascano M (2020). Small molecule inhibition of human cGAS reduces total cGAMP output and cytokine expression in cells. Sci Rep 10, 7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, De Jesus PD, Ruan C, de Castro E, Ruiz PA, et al. (2015). PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell 161, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zal T, and Gascoigne NR (2004). Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J 86, 3923–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Time-lapse imaging to monitor structural integrity of an incoming viral particle with iGFP fluid phase marker (iGFP+IN-mRuby3) in MDDCs. See Figures 3B and S3B for detail.
Data Availability Statement
Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication DOIs are listed int the key resources table. Software may be used and modified for any non-commercial use under the GPL license terms.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-IRF3 (D9J5Q) | Cell Signaling Technology | Cat# 109495; |
| Rabbit polyclonal anti-PQBP1 | Bethyl Laboratory | Cat# A302–802A |
| Mouse monoclonal anti-PQBP1 | Santa Cruz Biotechnology | Cat# sc-376039; RRID:AB_10989350 |
| Mouse monoclonal anti-PQBP1 (1A11) | Sigma Aldrich | Cat# SAB1402496; RRID:AB_10640643 |
| Rabbit polyclonal anti-cGAS | Novus | Cat# NBP1–8676 |
| Mouse monoclonal anti-cGAS (D-9) | Santa Cruz Biotechnology | Cat# sc-515777; RRID:AB_2734736 |
| Mouse monoclonal anti-cGAS (5G10) | Caymanchem | Cat# 23853 |
| Rabbit monoclonal anti-cGAS (D1D3G) | Cell Signaling Technology | Cat# 49705 |
| Mouse monoclonal anti-GFP (GF28R) | Thermo Scientific | MA5–15256 |
| Rabbit polyclonal anti-GFP | Takarabio | Cat# 632592 |
| Mouse monoclonal anti-Flag | Sigma Aldrich | Cat# F1804; RRID:AB_262044 |
| Human monoclonal anti-HIV-1 p24 (71–31) | AIDS Reagent | Cat# ARP-530 |
| Mouse monoclonal anti-cGAMP | Pfizer (Hall et al., 2017) | PF-07043030 |
| Mouse monoclonal anti-MBP magnetic beads | New England Biolabs | Cat# 28037S |
| Rabbit monoclonal anti-beta-actin (13E5) | Cell Signaling Technology | Cat# 49705 |
| Normal rabbit/mouse IgG | Santa Cruz Biotechnology | Cat# sc-2027; RRID:AB_737197 |
| Bacterial and virus strains | ||
| Human lentiORF cDNA clone of PQBP1 | Open Biosystems (Yoh et. al., 2015) | N/A |
| pNL43 env- luc. | Manel et. al., 2010; Yoh et. al., 2015 | N/A |
| SIV3+ for production of VLP-VPX | D. R. Litman (Manel et. al., 2010; Yoh et. al., 2015) | N/A |
| Gag-IN-mRuby3 | Mamede et al., 2017 | N/A |
| HIV-Gag-iGFP | Mamede et al., 2017 | N/A |
| CypA-dsRed | Francis et al., 2016; Francis and Melikyan, 2018. | N/A |
| Cantell strain of Sendai virus | Mount Sinai Department of Microbiology Virus Collection | N/A |
| Biological samples | ||
| De-identified healthy donor blood | San Diego Blood bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 2’ 3’ -cGAMP | Invivogen | Cat# tirl-nacga23–02 |
| Herring testis DNA | Sigma Aldrich | Cat# D6898 |
| Lipofectamine 2000 | Life Technology | Cat# 11668030 |
| Stemfect RNA transfection kit | ReproCell | Cat# 00–0069 |
| Critical commercial assays | ||
| Duolink in situ detection reagent | Sigma | Cat# DUO92008 |
| Deposited data | ||
| None | N/A | N/A |
| Experimental models: Cell lines | ||
| Dual-KO-cGAS-THP-1 | Invivogen | Cat# thpd-kocgas |
| Dual-KO-cGAS-Flag-cGAS-THP-1 | This paper | |
| THP1-Dual Cells | Invivogen | Cat# thpd-nfis |
| THP-1 | ATCC | Cat# TIB-202 |
| 293T | ATCC | Cat# CRL-3216 |
| Experimental models: Organisms/strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| siNT (5’-AATCGATCATAGGACGAACGC-3’) | This paper | N/A |
| siPQBP1–1 (5’- AAGCTCAGAAGCAGTAATGCA-3’) | This paper | N/A |
| siPQBP1–2 (5’-AAAGCCATGACAAGTCGGACA-3’) | This paper | N/A |
| Non-targeting guide crRNA guide sequence | Dharmacon | U-007502 |
| MB21D1(cGAS) crRNA guides: 1–5 | Dharmacon | CM-015607-(1 to 5), see Guide RNA sequences under Star Method |
| NONO (4841) targeting guides: 2 and 5 | Dharmacon | CM-007756-(02 and 05), see Guide RNA sequences under Star Method |
| Recombinant DNA | ||
| pEASIL-Flag-cGAS | This paper | N/A |
| pEASIL | M. Malim | N/A |
| pCDFDuet-PQBP1 | This paper | N/A |
| pCDNA 3.1-MBP-cGAS | This paper | N/A |
| Software and algorithms | ||
| Imaris | Bitplane | RRID:SCR_007370 |
|
Python Programming Language
|
Python Software Foundation | RRID:SCR_008394 |
| Custom Scripts for imaging analysis | this article, PMID:33154161, | 10.5281/zenodo.6551125 |
| Other | ||