

Abstract
We present an overview of small molecule glucose-6-phosphate dehydrogenase (G6PD) inhibitors that have potential for use in the treatment of cancer, infectious diseases, and inflammation. Both steroidal and non-steroidal inhibitors have been identified, with steroidal inhibitors lacking target selectivity. The main scaffolds encountered in non-steroidal inhibitors are quinazolinones and benzothiazinones/benzothiazepinones. Three molecules show promise for development as anti-parasitic (25 and 29) and anti-inflammatory (32) agents. Regarding modality of inhibition (MOI), steroidal inhibitors have been shown to be uncompetitive and reversible. Non-steroidal small molecules have exhibited all types of MOI. Strategies to boost the discovery of small molecule G6PD inhibitors include exploration of structure-activity relationships (SARs) for established inhibitors, employment of high-throughput screening (HTS), and fragment-based drug discovery (FBDD) for the identification of new hits. We discuss the challenges and gaps associated with drug discovery efforts of G6PD inhibitors from in silico, in vitro and in cellulo to in vivo studies.
Keywords: small molecules, inhibitors, drug discovery, cancer, infectious diseases, inflammation
Graphical Abstract
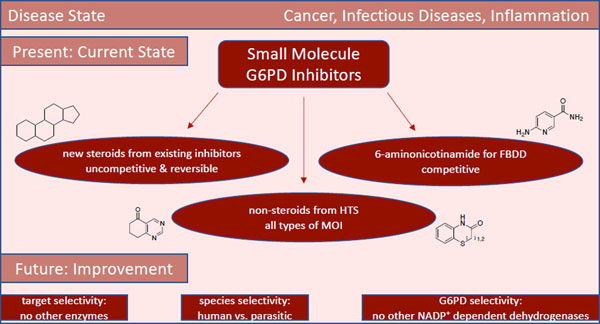
Introduction
Glucose-6-phosphate dehydrogenase (G6PD) catalyzes the conversion of glucose-6-phosphate (G6P) to 6-phospho-glucono-δ-lactone (6PGL), the first and rate-limiting step in the oxidative phase of the pentose phosphate pathway (PPP) (Figure 1). Production of NAPDH in this transformation allows for glutathione disulfide (GSSG) to be reduced to the monomeric thiol, glutathione (GSH). In turn, GSH acts as a radical scavenger and prevents the accumulation of reactive oxygen species (ROS).1–2 The NADPH generated in this process can be used for fatty acid synthesis3–4 whereas ribose-5-phosphate (R-5P), the isomer of ribulose-5-phosphate (Ru-5P) and open form to the respective furanose, is important for nucleotide synthesis.1, 5–7
Figure 1.
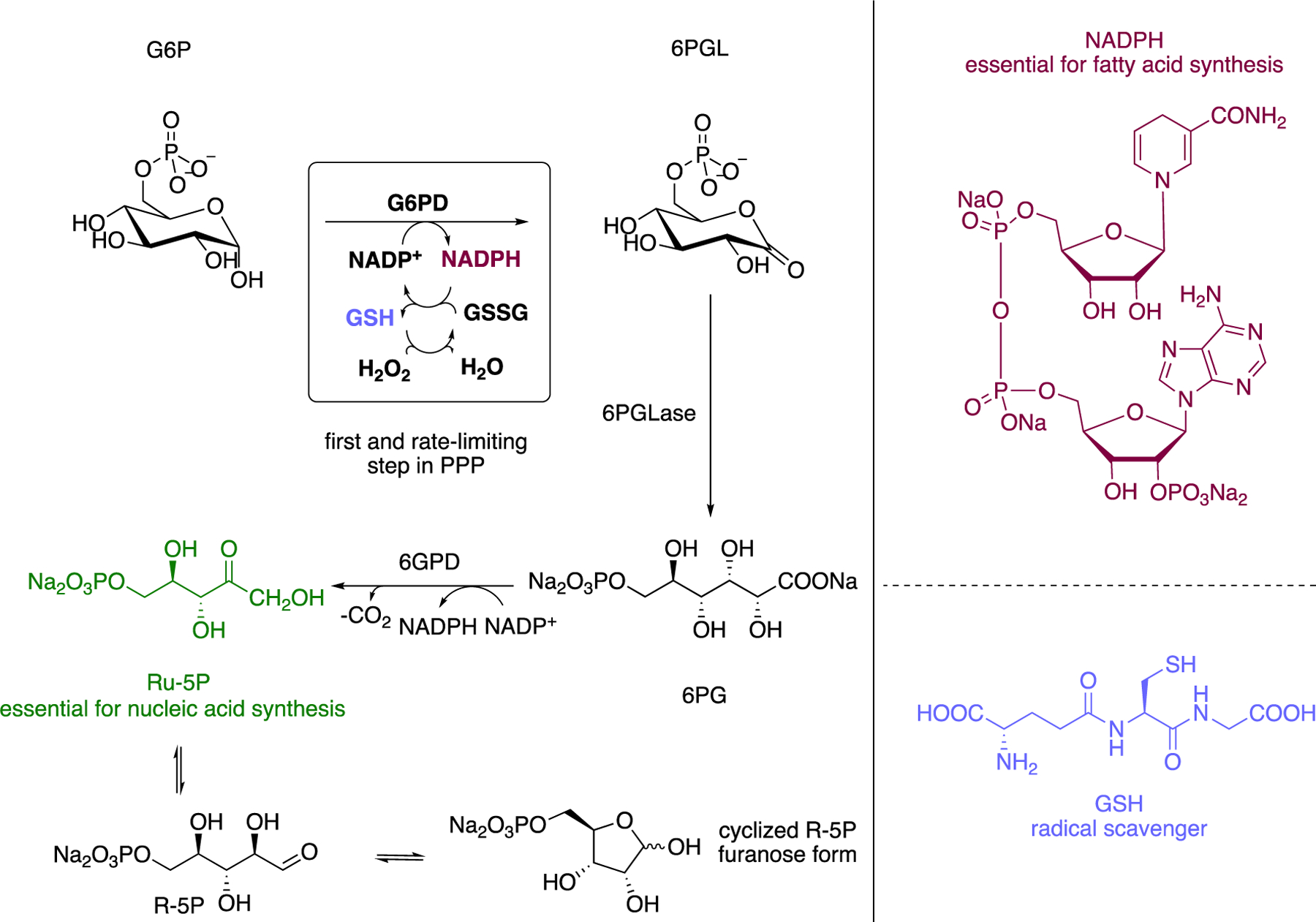
The Pentose Phosphate Pathway (PPP); implication in redox homeostasis and cell proliferation.
Given the vital role of G6PD in nucleic acid synthesis, fatty acid synthesis, and redox homeostasis, G6PD is a compelling target for modulation. Indeed, hypoactive or hyperactive G6PD has been linked to the pathology of many human diseases, including cancer,8–10 hemolytic anemia,11 type II diabetes,12–13 cardiovascular diseases,14–17 and neurogenerative diseases.18–19 In the cases of hemolytic anemia and neurodegenerative disorders, the pathology is caused by reduced G6PD activity and activation of G6PD is of therapeutic interest; an activator of G6PD would mitigate oxidative stress derived from underperforming G6PD. However, in the context of cancer and chronic inflammation, G6PD is hyperactive and requires inhibition. Additionally, the activity of parasitic G6PD, present in parasites that cause trypanosoma or malaria, is critical for parasitic survival and the inhibition of parasitic G6PD has been explored as a treatment against infectious diseases. This Perspective focuses on the discovery and potential development of small molecule inhibitors of G6PD and their use in cancer, infectious diseases, and chronic inflammation.
Steroidal G6PD Inhibitors in the Context of Cancer and Infectious Diseases: Optimization of Existing Inhibitors via SAR Exploration
Cancer
The history of G6PD inhibitors as potential therapeutics for cancer treatment starts with dehydroepiandrosterone (DHEA) (1) and epiandrosterone (EA) (2) (Figure 2). Both DHEA (1) and EA (2) are involved in the metabolic pathway of testosterone, with EA having a weaker androgenic activity compared to DHEA. There are three enzymes for which DHEA (1) serves as a substrate in its naturally occurring role: 3-β-hydroxysteroid dehydrogenase 2 (HSD3B2), 17-β-hydroxysteroid dehydrogenase 5 (17BHSD5), and steroid sulfatase (sulfotransferase). Sulfotransferases introduce sulfate groups to hydroxy groups, rendering DHEA and other substrates more water soluble. Given that 1 is involved in testosterone biosynthetic pathways,20–21 it is apparent that lack of target selectivity has raised concerns for its consideration as an anticancer agent.
Figure 2.
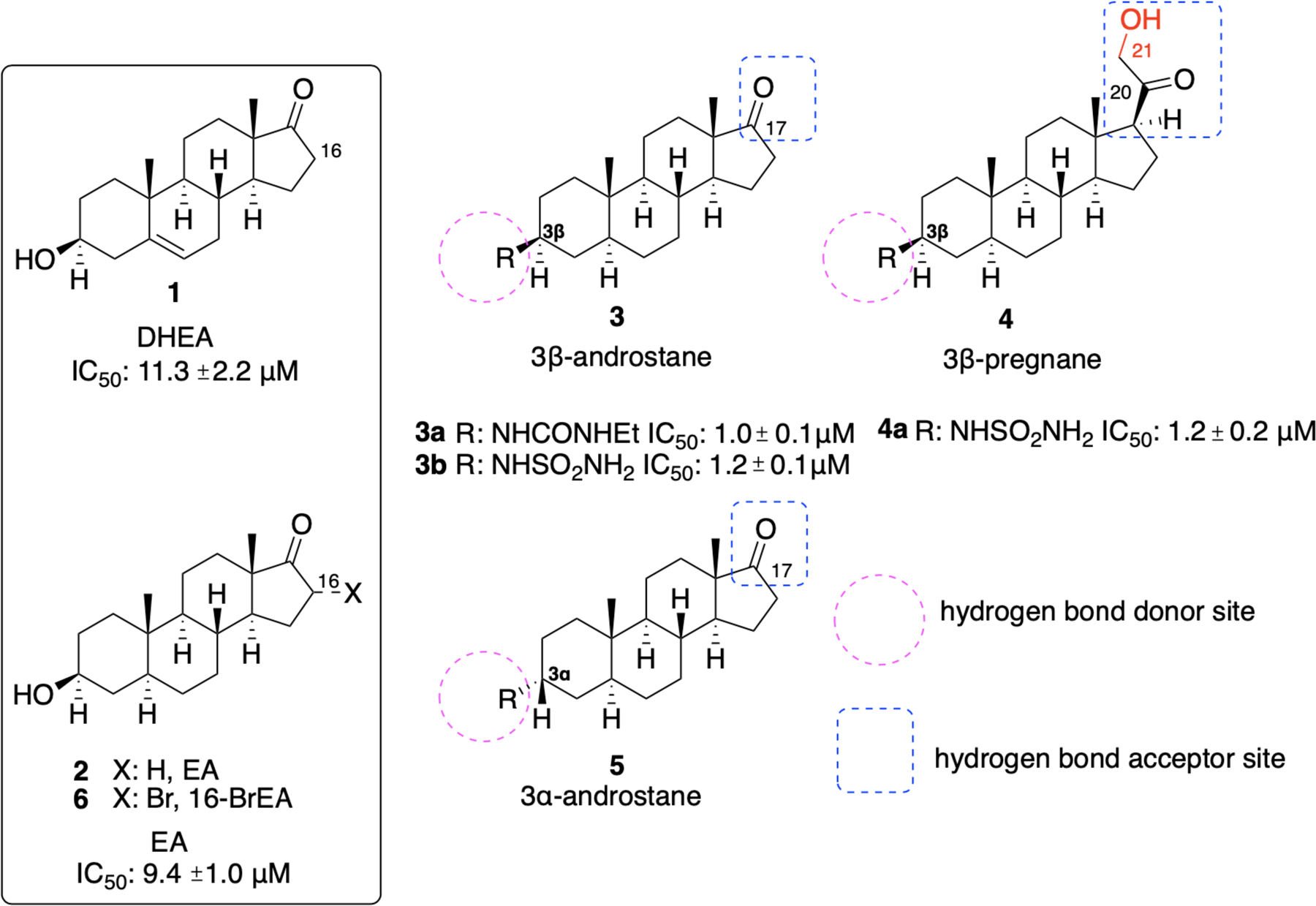
DHEA 1, EA 2, and newer steroidal analogues as inhibitors of hG6PD and TrG6PD.
Marks and Banks were the first to show that 17- and 20-ketosteroids act as inhibitors of mammalian G6PD.22 Over the years, mechanistic investigations have demonstrated that 1 acts as an uncompetitive inhibitor of G6PD with respect to both NADP+ and G6P.23 The KM for both the substrate and NADP+ or the products (lactone and NADPH) across different studies, confirm uncompetitive inhibition (KM,G6P ~ 6KM,NADP+ and KM,lactone ~ 35KM,NADPH).23–25 Additionally, the reported Ki values for the forward and reverse reactions suggest reversible inhibition, implying uncompetitive, yet reversible, inhibition of G6PD by 1 and related steroids.23 This type of inhibition for both NADP+ and G6P indicates that it binds to the ternary enzyme-NADP+-G6P complex.26 Although there have been studies to propose this MOI, since a co-crystal structure of the steroid/G6PD/NADP+ complex is not available, the pharmacophoric interactions which may inform drug design remain unknown. Crystal structures of human (Homo sapiens; tetrameric),27–28 trypanosomal (Trypanosoma cruzi; tetrameric),29 and bacterial (Leuconostoc mesenteroides; dimeric) 30–31 G6PD will assist in analyzing pharmacophoric interactions and will help inform future SAR studies.
In recent years, the groups of Hamilton and Cordeiro have focused on the development of steroidal analogues of 1 and 2 with the goal of improving selectivity for G6PD. Although both groups have focused on the identification of new steroidal analogues, their drug discovery approaches have been different. The Hamilton group has focused on targeting human G6PD (hG6PD) for cancer and the Cordeiro group has targeted trypanosoma (TrG6PD) to selectively inhibit the parasite G6PD as a treatment for trypanosomiasis.
Hamilton et al.32 classified the newly designed and synthesized analogues into three categories: androstane analogues with 3β-amino-substituents (3), pregnane analogues with 3β-amino-substituents (4) and androstane derivatives with 3α-amino-substituents (5), as summarized in Figure 2. Of the three G6PD inhibitory analogues, the last class of analogues 5 (3α-analogues) showed inferior inhibitory activity compared to 1 and 2, highlighting the importance of retaining the R group above the ring (3β-analogues 3 and 4). Replacement of the 3-OH with other hydrogen donors such as amides, ureas, carbamates, sulfamides and sulfonamides led to analogues with superior activity (Figure 2). From the 3β-androstane analogues, urea 3a and sulfamide 3b demonstrated up to 10-fold higher inhibitory activity compared to 1 and 2, with IC50 values of 1.0 ± 0.1 μM (R = NHCONHEt, urea 3a) and 1.2 ± 0.1 μM (R = NHSO2NH2, sulfamide 3b), respectively.
Of the 3β-pregnane derivatives, the corresponding sulfamide 4a (R = NHSO2NH2) inhibits the enzyme with an IC50 of 1.2 ± 0.2 μM. The presence of a hydroxy group at C21 proved to be essential for this higher potency; in its absence, the IC50 was increased to 2.0 ± 0.4 μM. However, this replacement provided a five-fold more potent analogue compared to the parent 1 and 2. As shown in Figure 2, the groups on the C3 carbon serve as hydrogen bond donors and the groups at C17 (androstane core) or C20 (pregnane core) act as hydrogen bond acceptors. Compounds which demonstrated better activity than the parent 1 and 2 in the enzyme assay (IC50 <9.4 ± 1.0 μΜ) were tested in cellular assays using HEK293T cells. Unfortunately, the pharmacologic profile of the selected compounds was poor and no correlation between the enzymatic and cellular assays could be drawn.
Two main factors may influence the lack of biological activity: aqueous solubility and cell permeability. Two of the most potent compounds with excellent inhibitory enzyme activity, 3β-sulfamides 3b and 4a, were water-soluble; however, they were the least potent in cellular assays. Therefore, poor aqueous solubility does not explain discrepancies between in vitro and in cellulo potency. To determine whether membrane permeability explained reduced compound activity, a Caco-2 permeability assay was employed. The measured permeabilities were good to moderate, suggesting poor permeability does not cause reduced inhibitory activity in cell-based assays. The authors concluded that a combination of factors pertinent to solubility, permeability and inherent assay differences are the cause for the observed discrepancy in inhibitory activities. However, it is possible that since these compounds are structurally similar to 1, they could be sequestered by enzymes recognizing 1 (HSD3B2, 17BHSD5 and sulfotransferase) or be metabolically transformed in cellular assays and, thus, their actual intracellular concentration is insufficient to inhibit G6PD. In vitro studies using human liver microsomes demonstrated good metabolic stability for the compounds, making it unlikely for metabolic transformations catalyzed by P450 enzymes to account for the profile discrepancy observed between the biochemical and assay in cells.
Infectious Diseases
Although research efforts have focused on the development of new steroidal analogues of both 1 and 2 to improve target selectivity between the cognate protein and G6PD in humans, differences in sequence conservation across species have made it an attractive target for the treatment of infectious diseases, by selectively targeting the G6PD enzyme of the pathological microorganisms. Steroidal derivatives of 1 and 2 have been shown to exert anti-infective properties in the parasites Plasmodium falciparum (P. falciparum),33 Cryptosporidium pavnum,34 Schistosoma mansoni,35 Trypasonoma cruzi (T. cruzi),36 Taenia crassiceps37 and Entamoeba histolytica.38 In all cases, the mechanism of action is unknown. In the case of Trypanosoma species, steroidal derivatives have been investigated for development as anti-infective agents.
In the work developed by Cordeiro, 1 and 2 (Figure 2) inhibited TrG6PD,39 acting as uncompetitive inhibitors for both substrates, G6P and NADP+ (in the low micromolar range), providing trypanocidal activity against Trypanosoma brucei (T. brucei). Notably the Ki for TrG6PD was ~6-fold lower than that for hG6PD for both G6P and NADP+, indicating a higher affinity of both steroids 1 and 2 for the parasitic G6PD. Viability assays on cultures of bloodstream forms of T. brucei (427 strain) identified an LD50 for 1 and 2 of 43.8 ± 2 and 24.5 ± 0.7 μM, respectively. Both 1 and 2 were also tested against Leishmania mexicana (L. mexicana) but had no inhibitory effect. These results demonstrate that 1 and 2 may be selective for certain types of trypanosomes. The molecular basis for this difference may shed light on the pharmacophore or its pharmacokinetics in the two trypanosome species.
In a subsequent study, Cordeiro et al. showed that 16-bromoepiandrosterone (16-BrEA, 6) (Figure 2) exerts trypanocidal activity against T. cruzi, the parasitic protozoan causing Chagas disease, via the inhibition of TrG6PD.40 The IC50 value for 6 was 86 ± 8 nM and its LD50 in parasitic cultures (T. cruzi epimastigotes, Y strain) was found to be 12 ± 8 μM, with this 100-fold value difference suggesting that 6 has a broad therapeutic index. For comparison, the authors measured the IC50 values of 1 and 2 for TrG6PD which were found to be 25.0 ± 3.5 and 5.6 ± 1.2 μΜ, respectively. Whereas the authors measured the Ki values for 1 (Ki = 21.5 ± 0.5 μΜ) and 2 (Ki = 4.8 ± 0.3 μΜ), the measurement of the Ki value of 6 would have allowed for a broad comparison across the species T. cruzi and T. brucei, and potency comparison for hG6PD, since this comparison was addressed in the study focusing on T. brucei.39 Notably, the same group has further increased the selectivity for TrG6PD (T. cruzi) by designing and synthesizing derivatives of 6 and achieving high selectivity for the parasitic G6PD.41 In addition to maintaining the bromine at C16, the C3-hydroxy group was either esterified or etherified. This strategy, leading to abolition of hydrogen bond donating features of the C3-hydroxy group, aimed to increase the selectivity of the new compounds for TrG6PD over hG6PD.
The necessity for C3-hydrogen bond donating groups was projected as an essential feature for inhibitory activity toward hG6PD by Hamilton et al. Non-brominated derivatives were also synthesized for some analogues. Although these non-brominated derivatives were not cytotoxic to the host, they were not potent inhibitors of T. cruzi. This further demonstrates the necessity for bromine at C16 for optimal potency against the parasite. In Figure 3, we show C3 esterified and etherified derivatives of type 7 and 8, respectively. These compounds were tested in rat H9C2 rat cardiomyocytes infected with epimastigotes (Y strain) of T. cruzi. Compounds of type 7 and 8 that at a concentration of 20 μΜ reduced the cell viability to 25% or less were subjected to a dose-response assay starting at lower concentrations. The purpose was to measure efficacy against the parasite and toxicity against the host cell. In addition, the selectivity index (SI) (EC50-host/EC50-T.cruzi) was measured to assess selectivity between the TrG6PD and hG6PD. Carboxylic esters 7a and 7b were shown to be the least potent (EC50 values 8.5 ± 1.5 and 16.7 ± 4.2 μΜ, respectively); however, 7b had the broader therapeutic window between the parasite and the host (EC50 value for the host for 7b: 54 ± 3.3 μΜ). On the other hand, sulfonate 7c and ether derivatives 8a-c were more potent (range of EC50 values: ~2–4 μΜ) against T. cruzi but insufficient selectivity was achieved between the parasite and the host. A reason for the discrepancy in potency between the carboxylate esters and the ether derivatives could also be that the esters are cleaved by esterases in the cytosol, therefore, a greater dose is required to achieve the same pharmacological profile compared to the ether derivatives. Although compounds 7b and 8c had nearly the same SI, the EC50 for 8c was as low as 8.5 ± 1.9 μM for the host, rendering it cytotoxic. Future development for preclinical studies would need to focus on increasing the SI through additional rounds of SAR optimization and evaluation of the amine congeners of 8b and 8c for optimal dissolution in the stomach; amines, in contrast to amides, can be protonated and that contributes to their better solubility. Although optimal dissolution depends on protonation of the molecule, the determinant for absorption is the availability of the uncharged molecule, that is, the free base. Unless the internalization mechanism involves organic anion or cation transporters, only the free base is permeable through cell membranes via diffusion. Other non-brominated analogues of 2 with non-hydrolyzable groups at C3 but which possess substituents with hydrogen bond donating or accepting groups were in general weak inhibitors of TrG6PD.
Figure 3.
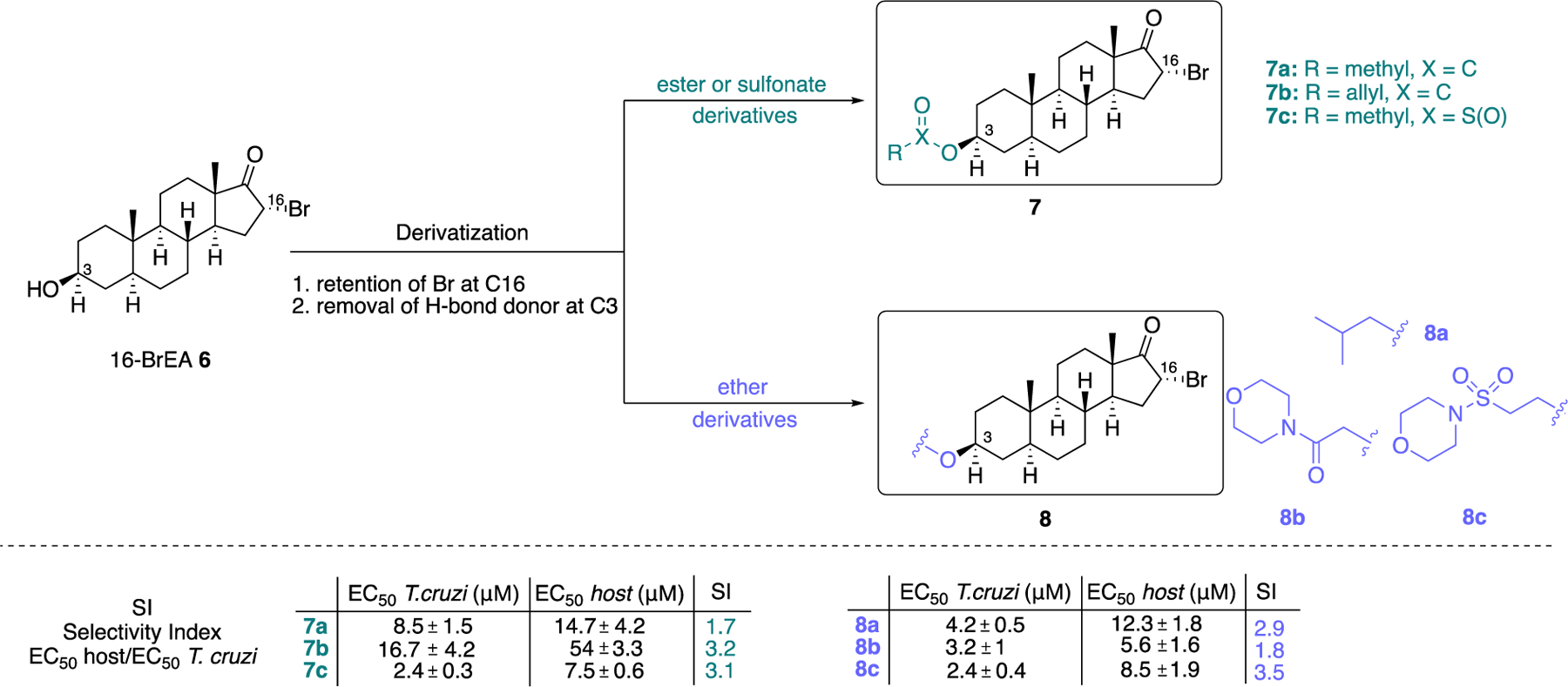
Derivatives of 6 as selective (7a-7b, selective for TrG6PD over hG6PD) and potent (7c, 8a-c) steroidal inhibitors.
An advantage associated with all steroidal inhibitors, encountered both in the context of cancer and infectious diseases, is that they lack functional groups which would serve as Michael acceptors. Thus, their promiscuity as substrates for Michael donors, such as thiolates from cysteines, is attenuated. Despite 1 and 2 exhibiting lower Ki values for TrG6PD (for T. brucei) compared to hG6PD and 6 exhibiting a lower IC50 value compared to 1 and 2 for T. cruzi, the lack of selectivity for parasitic G6PD can cause complications in the mammalian host. These complications are due to the androgenic effects of steroids.42–43
Non-Steroidal G6PD Inhibitors from HTS in the Context of Infectious Diseases, Cancer, and Inflammation
Infectious Diseases
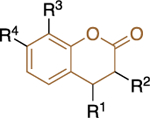
The lack of species and target selectivity led Cordeiro et al. to pursue non-steroidal G6PD inhibitors that are selective toward the protozoic G6PD enzyme by conducting high throughput screening.44 Hits identified in the primary screen and their analogues were subjected to follow-up studies; compounds that inhibited TrG6PD between 40% and 100% at 20 μΜ were subjected to dose-response for IC50 determination. Those compounds with IC50 values of 0.48<IC50<32 μM were grouped into two chemical categories: thienopyrimidines 9 and quinazolinones 10 (Figure 4).
Figure 4.
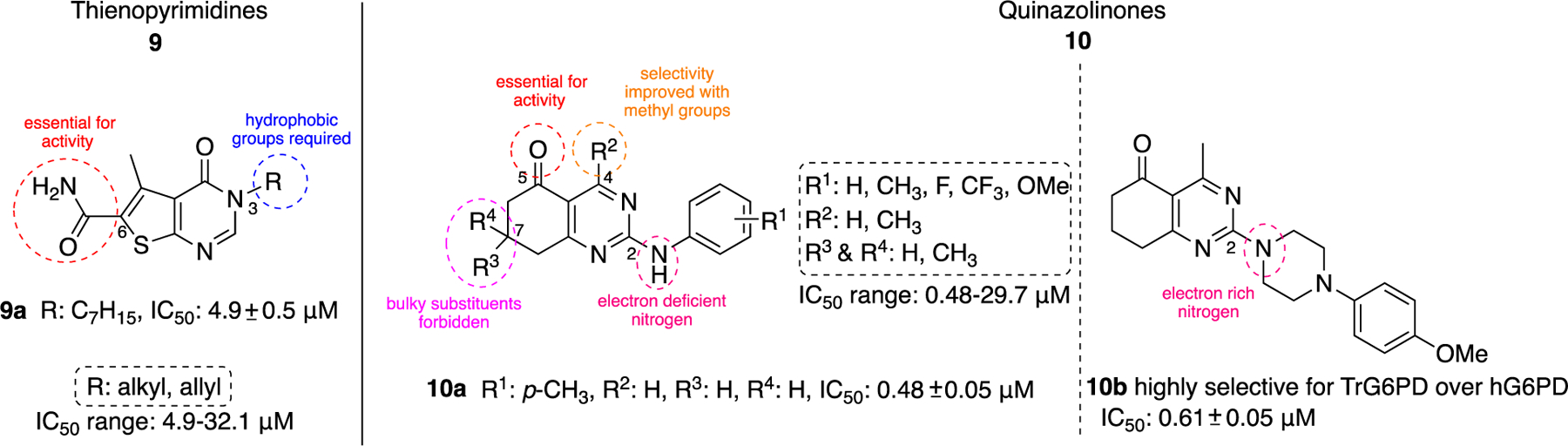
Thienopyrimidine- and quinazoline-containing compounds of type 9 and 10: potent against recombinant TrG6PD; IC50 values and ranges come from measurements against recombinant TrG6PD.
SAR studies with the thienopyrimidines 9 revealed that the amido group at the C6 position of the core was required for the expression of inhibitory activity and long chain hydrophobic aliphatic substituents at the N-3 position are required in order to observe inhibitory activity in the low micromolar range. Compound 9a was found to be the most potent inhibitor with an IC50 of 4.9 ± 0.5 μM. The replacement of the long chains with shorter or more hydrophilic chains reduced the potency.
Regarding the second class, quinazolinones 10, it was shown that the carbonyl at C5 was essential for inhibitory activity. The presence of the methyl group at C4 increased selectivity toward TrG6PD and it should be noted that the most potent compound in this series, quinazolinone 10a, (IC50: 0.48 ± 0.05 μM) lacked the methyl group (R2 = H instead of CH3), highlighting that potency does not guarantee selectivity. The presence of bulky substituents R3/R4 at C7 of the nucleus proved detrimental for inhibitory activity, resulting in a dramatic increase in the IC50 values. Finally, the SAR analysis for C2 of the quinazolinone ring implied that the amine attached at that position can either be an aniline (a weakly basic, electron deficient, nitrogen atom) or a piperazine (a more basic, electron rich nitrogen atom). Notably, the piperazine linker between the quinazolinone moiety and the aromatic ring in 10b delivered a highly selective compound for TrG6PD over hG6PD (IC50: 0.61 ± 0.05 vs. >80 μΜ, respectively).
The most potent compounds from each category are uncompetitive, reversible inhibitors of TrG6PD, as was the case for all steroidal TrG6PD inhibitors (Figure 4). In addition, these compounds compete with 2 for binding to the target enzyme, suggesting that non-steroidal and steroidal compounds are binding to the same region in TrG6PD.45–46
The authors used these compounds in tests with epimastigote forms of T. cruzi, Y strain, and classified the compounds in accordance with the percentage of remaining viable cells. Although thienopyrimidine 9a was the most active against recombinant TrG6PD, it had no effect on cell viability. The most active compounds in the viability assay stemmed from the quinazolinone class, with 10a being the most active in the in vitro biochemical assay and exhibiting comparable activity to benznidazole (8.5 ± 1.5 μΜ for 10a, 10 ± 0.8 μΜ for benznidazole) against epimastigotes T.cruzi. Other quinazolinones bearing a methyl group at C4 (R2 = CH3) and a para-fluoro (R1 = F) or a meta-methyl (R1 = CH3) substituent on the aniline exhibited even lower EC50 values than benznidazole in the viability assay, providing an anchor for the development of potential trypanocidal agents. Thienopyrimidines 9 have a pyrimidinone moiety whereas quinazolinones 10 have a pyrimidine heterocycle as part of their scaffold, suggesting that the pyrimidine nitrogens in both cases may play a role in the pharmacophoric interactions with the target. However, the presence of the primary amide group in thienopyrimidines confers significant polarity to the molecules. This indicates that the thienopyrimidine series requires further optimization, most likely via the introduction of lipophilic substituents on the amido nitrogen to achieve translatable activity from in vitro to in vivo assays.
Although co-crystal structures for human or trypanosomal G6PD with an inhibitor bound do not exist, the crystal structures of both, including substrates and co-factors, allow for comparison between the two and can guide drug discovery efforts (Figure 5). Both hG6PD (PDB ID 2BH9) and TrG6PD (PDB ID 5AQ1) have a catalytic site for the substrate (G6P) and co-factor (NADP+). However, only the mammalian hG6PD has an additional NADP+-binding site, known as the structural or allosteric NADP+-binding site, which determines long term protein stability and structural integrity of the enzyme.
Figure 5.
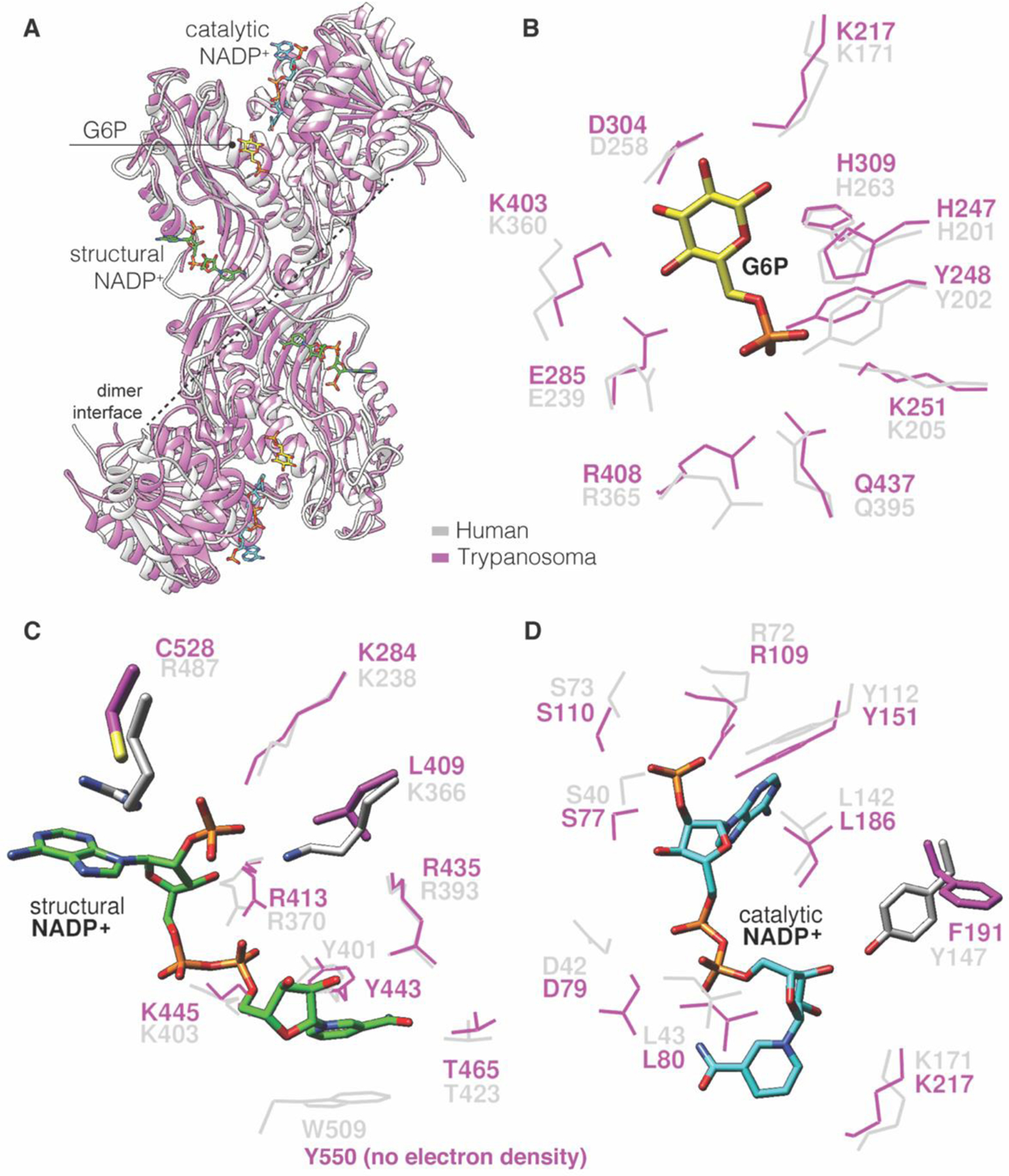
A. Overlay hG6PD (PDB ID: 2BH9) and TrG6PD (PDB ID: 5AQ1), grey for hG6PD and purple for TrG6PD; B. Overlay of the catalytic G6P site hG6PD-TrG6PD; C. Overlay of the structural NADP+ in hG6PD with respective region in TrG6PD lacking the structural NADP+. D. Overlay of the catalytic NADP+ site hG6PD-TrG6PD.
By overlaying hG6PD and TrG6PD crystal structures, we show the point-by-point amino acid comparison for each G6PD species, with emphasis placed on ligand-binding sites. There are three key differences between the two that can be exploited for selectivity of one over the other. Two are found at the structural NADP+-binding site. At this site, Arg487 in hG6PD is replaced by Cys528 in TrG6PD. An antimalarial compound that covalently interacts with the TrG6PD cysteine may allow for selectivity between the human and Trypanosoma G6PD. In addition, Lys366 in hG6PD is Leu409 in TrG6PD, thus in the latter case, the interactions between an envisioned inhibitor and that residue would be limited to hydrophobic. Thus, Cys528 and Leu409 found in Trypanosoma can be exploited as targets for cross-linking with small molecules or hydrophobic interactions, respectively.
The third difference between the two structures is at the catalytic NADP+-binding site; Tyr147 in hG6PD is Phe191 in TrG6PD. Although both residues are aromatic, their orientation is almost perpendicular to each other. A plausible explanation is that the phenolic OH from Tyr147 acts as a H-bond donor toward the OH of the ribose (H-bond acceptor) attached to the pyridinium ring of the co-factor (NADP+). This structural difference has the potential to be considered for the design of competitive NADP+ inhibitors selective for each species. Regarding the catalytic G6P site, it is nearly identical between each species and selective competitive inhibitors for G6P is less likely.
Another parasite for which non-steroidal G6PD inhibitors have been developed is P. falciparum, the parasite that causes malaria. Different from the hG6PD, P. falciparum contains a bifunctional chimeric enzyme, composed of both G6PD and 6-phosphoglucono-δ-lactonase (6PGLase), (PfGluPho), which together catalyze the first and second step of the PPP (Figure 1).47–48 The amino acid sequence for 6PGLases is well conserved across species;49 however, the G6PD sequence has a 62 residue insertion at the N-terminus relative to hG6PD, and this insertion is conserved in other Plasmodium species.47–50 This conserved sequence, present only in the parasite, can potentially be used for the discovery of selective antimalarial drugs. However, the C-terminus of PfGluPho is homologous to the other G6PDs, which may present a challenge for drug discovery.
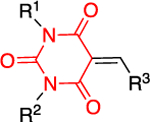
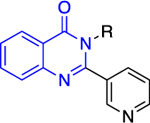
The Bode group used a library screen to identify new antimalarial agents that inhibit PfGluPho.51 Four major classes of commercially available compounds were identified based on their core: pyrimidinones 11, quinazolinones 12, chromenones 13, and sulfonamides 14 (Table 1). Pyrimidinetriones 11a-c (pyrimidinedione 11d) and quinazolinones 12a-b showed appreciable inhibitory activity toward recombinant PfGluPho [IC50 range: (4.5 ± 1.6)-(19.9 ± 7.2) μM]. In in vitro P. falciparum culture assays (3D7 strain), chromenone 13a and sulfonamides 14a-b suppressed the viability of the parasite with IC50 values ranging from (0.97 ± 0.15)-(5.3 ± 2) μM. The same compounds also inhibited recombinant PfGluPho, with 13a being at least a 10-fold more potent inhibitor compared to 14a-b in the biochemical assay (Table 1).
Table 1.
Structural classification of PfGluPho inhibitors identified by the Bode group; Numbers in parenthesis (as taken from the original paper).
| Compounds | Class 1 pyrimidinones 11 |
Class 2 quinazolinones 12 |
Class 3 chromenones 13 |
Class 4 sulfonamides 14 |
PfGluPho IC50 (μΜ) recomb. |
PfGluPho IC50 (μΜ) cult. |
|---|---|---|---|---|---|---|
|
|
|
|
|||
| 11a (CB64) |
|
19.9 ± 7.2 | - | |||
| 11b (CB62) |
|
11.6 ± 1.0 | - | |||
| 11c (CB61) |
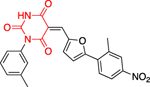
|
5.1 ± 3.2 | - | |||
| 11d (CB22) |
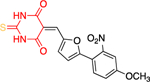
|
4.6 ± 2.4 | - | |||
| 12a (CB104) |
|
7.6 ± 4.0 | - | |||
| 12b (CB70) |
|
4.5 ± 1.6 | - | |||
|
| ||||||
| 13a (CB103) |
|
1.5 ± 0.4 | 0.97 ± 0.15 | |||
|
| ||||||
| 14a (CB83) |
|
22.0 ± 8.0 | 5.3 ± 2 | |||
| 14b (CB90) |
|
>30 | 3.9 ± 0.14 | |||
To assess time-dependence and reversibility, the authors measured different IC50 values following the protocol of: 1 h pre-incubation and 1 h post-dilution (1:1), 1 h preincubation and 0 h post-dilution (1:0), and 0 h pre-incubation and 0 h post-dilution (0:0). The last experimental arrangement corresponds to the control experiment. If there was a decrease in the IC50 value after pre-incubation (1:0) compared to the control (0:0), the inhibition occurred in a time-dependent manner. If there was a decrease in the IC50 value after pre-incubation and post-dilution (1:1) compared to the control (0:0), the inhibition was irreversible. If the IC50 value remained the same after pre-incubation and post-dilution, the inhibition was reversible.
Compound 13a had an IC50 value of (1.1 ± 0.4)-(1.5 ± 0.4) μM regardless of whether it was subject to pre-incubation or post-dilution conditions. Similarly, for 14a and 14b, the IC50 values were >30 μM regardless of whether or not the compound was pre-incubated with the PfGluPho enzyme. After post-dilution, the IC50 for 14a decreased to 22 ± 8 μM but remained unchanged for 14b. Regarding translatability, the most potent compounds identified in the biochemical assay did not suppress the viability of the Plasmodium. This may reflect chemical or enzymatic instability of the compound; the duration of the biochemical assay was 20–30 min whereas the in vitro assay with Plasmodium culture (3D7 strain) was 72 h.
All of these PfGluPho inhibitors exhibit mixed kinetics for inhibition of G6P and are non-competitive or competitive with NADP+. Reversibility of inhibition was observed for 11c and 11d since the IC50 values were recovered after post-dilution (Table 1). Compounds 12a and 12b exhibited mixed-type of inhibition of G6P and were non-competitive with NADP+. In addition, similar Ki values after post-dilution were obtained and these compounds were found to be irreversible inhibitors. Notably, 12a and 12b lack functionalities with which they would covalently bind to the target.
As was the case for trypanosomiasis, the above compounds were tested for selectivity using the recombinant hG6PD; only 13a was more potent toward PfGluPho (Table 1), regardless of pre- and post-dilution times (IC50 values: 1.1 ± 0.4 or 1.5 ± 0.4 μM for PfGluPho and 2.2 ± 0.7 or 2.6 ± 0.7 μM for hG6PD). Moreover, when both human cells and the parasite species were present in the culture, compound 13a demonstrated an enhanced inhibitory activity on parasitic growth, with IC50 values in the low μΜ range (IC50 = 0.97 ± 0.15 μM) (Table 1). In contrast, compounds 14a and 14b inhibited recombinant hG6PD in a time-dependent manner; the IC50 value fell from >30 μM to 0.4 ± 0.0 and 0.6 ± 0.0 μM, respectively, after pre-incubation. In addition, these values remained in the same range after post-dilution, suggesting irreversibility of inhibition. Although these latter compounds targeted the G6PD element of Plasmodium PfGluPho, species selectivity was not observed.
Of the four categories of compounds 11–14 presented in Table 1, classes 12 and 13 are less promiscuous and, therefore, present as the preferred vehicles to consider for “hit to lead” optimization. The promiscuity of compounds 11 lies in their functionality which presents Michael acceptors that are activated by two carbonyl moieties (vide infra). The scaffold of pyrimidine-2,4,6(1H,3H,5H)-trione is present in barbiturates and this structural feature may cause off-target effects; barbiturates elicit their sedative effect by acting on GABAA receptors in the mammalian central nervous system. However, what renders barbiturates active is the appropriate substitution at the carbon between the two carbonyl functionalities.52 In the projected structures 11a-c (Figure 6), that carbon is substituted with an alkylidene moiety and, therefore, the resulting acrylamide moiety renders the molecules as potential Michael acceptors. Upon exposure to GSH or cysteine, compounds of type 15 can be obtained, suggesting the potential to react with cysteines present in G6PD. GSH cellular concentrations range from 0.1 to 10 mM,53–54 a range that ensures the presence of sufficient GSH even if minor portions of it are depleted by Michael acceptors. Regarding class 14, the presence of the p-aminophenol skeleton poses the risk for oxidation to the corresponding sulfonated parabenzoquinone imines 16. These benzoquinone imines 16 are potent Michael acceptors and can be captured by thiolates to deliver products 17. This is the pathway observed for paracetamol metabolism and rationalizes the administration of N-acetylcysteine as an antidote in case of paracetamol poisoning. N-acetylcysteine can be converted into cysteine in the blood. Thus, it can provide a cysteine source for GSH biosynthesis or the liberated cysteine directly cross-couple with N-acetyl-p-benzoquinone imine which is the metabolite of paracetamol.55
Figure 6.
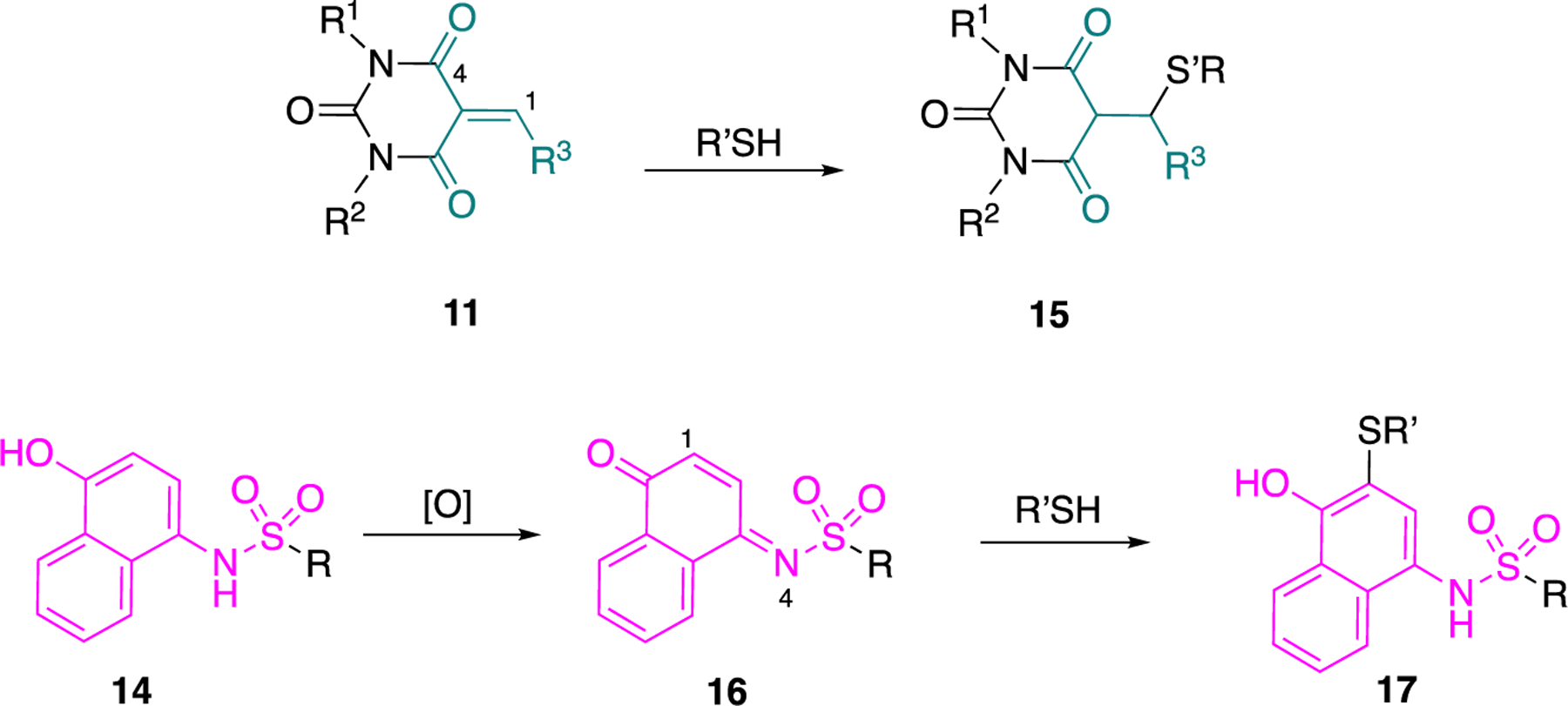
Compounds of type 11 and 14 as promiscuous substrates for Michael addition.
In a study focusing on the chemical genetics of P. falciparum, 18 (ChemDiv: C276–1187) and 19 (ChemDiv: D052–0147) (Figure 7a) were shown to selectively target the 6PGLase part in PhGluPho.56 Compound 18 is a quinolinone (shown in blue, Figure 7a), which is structurally similar to the quinazolinone chemotype 10 (Figure 4, Table 1). However, it was later shown that 18 inhibited both PfGluPho and hG6PD, as did the above compounds.57 Regarding its chemical reactivity, 18 is an enaminone and, thus, its properties as a Michael acceptor are attenuated.
Figure 7.
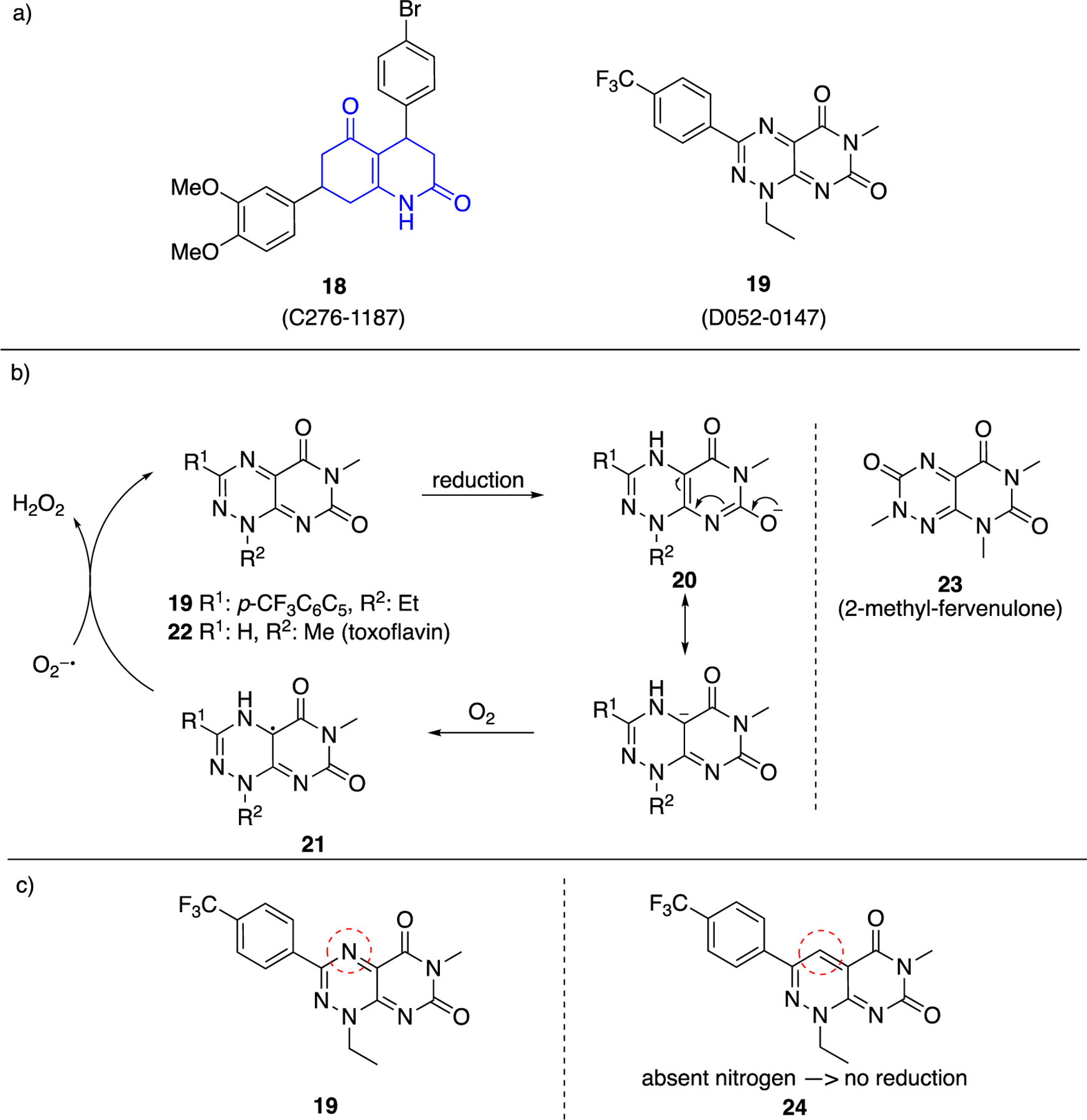
a. Commercially available 18 (C276–1187) and 19 (D052–0147) shown to inhibit P.falciparum; b. Pyrimidine triazinediones 19, 22, 23 lead to radical intermediates 21 and H2O2 release; c. Strategy of replacing triazine with pyridazine to prevent first reduction step shown in 7b.
In contrast to compounds 11, 14 (once oxidized to 16, Figure 6) and 18, which are prone to undergoing two-electron transfer reactions with intracellular components, 19 is a structural alert for a single electron transfer reaction.58 Concerns surrounding this structural alert problem may explain the absence of follow up studies with 19. Specifically, after reduction intermediate 20 transfers a single electron to molecular oxygen to form superoxide ion (O2−●) and the radical intermediate 21 (Figure 7b). Intermediate 21 is oxidized back to 19 with concomitant release of hydrogen peroxide (H2O2). This redox cycling activity is characteristic of pyrimidinetriazinediones (PTDs); other examples include toxoflavin 22 and 2-methyl-fervenulone 23. A necessity for these reactions to occur in vitro is the presence of strong reducing agents in the buffers, such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine.59 Assays to evaluate such reactivity include measurement of released hydrogen peroxide in vitro and ATP depletion in cells. Assays based on the fluorescent signal of resorufin have been employed for the segregation of well-performing caspase-8 and cathepsin L inhibitors from false-positive hits.60
A strategy to circumvent this activity upon SAR optimization is to replace the PDT nitrogen atom of the triazine core, which is reduced prior to the generation of the radical intermediates, with a carbon atom (compounds of type 24, Figure 7c). In that case the triazine ring is replaced by a pyridazine ring.
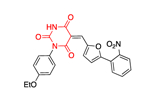
The most successful study identifying a selective PfGluPho small molecule inhibitor was that conducted by the Bode group.61 Starting with a HTS campaign that evaluated 350,000 compounds at a concentration of 20 μΜ, a promising cluster of benzothiazinone derivatives was identified, with five leading to further SAR studies. In all assays, hG6PD was used as a counter-assay to ensure specificity for PfGluPho. Essential structural features for inhibitory activity toward PfGluPho included: 1) a basic alkylamine as part of the side chain of the amide; 2) an unsubstituted nitrogen of the benzothiazinone ring; and 3) an unaltered sulfur on the benzothiazinone ring (Figure 8). A series of analogues was synthesized that included substituent variation on the styryl ring and the amido nitrogen. Further SAR on the styryl ring substituents did not lead to a dramatic shift in inhibitory potency; however, the styryl ring was a necessity for inhibitory activity. 2-Aminomethyl pyrrolidine substituents on the amido nitrogen increased the potency. Stereochemical evaluation of the 2-aminomethyl pyrrolidines showed that the (R)-enantiomer was 8-fold higher in potency than the (S)-enantiomer, delivering 25 as the most potent of all of the analogues studied (IC50: 0.9 ± 0.0 μΜ), and was specific for PfGluPhO. It exhibited mixed inhibition kinetics with NADP+ but was competitive with G6P. Given that 25 bears the Michael accepting acrylamide moiety, the authors prudently performed a GSH-based assay to measure levels of remaining compound upon exposure to GSH. The data showed that the levels of 25 remained intact upon exposure to GSH (ethacrynic acid was used as positive control). According to the protocol used, the ratio of [compound]:[GSH] was 1:5 and the final GSH concentration was 50 μΜ. The concentration ratio is a fixed ratio and does not reflect the ratio observed once the drug is internalized in the cell in all cases. The value of 50 μΜ for GSH concentration is much lower than the concentration range observed in the cytosol in cells (0.1–2 mM in most cell types, 10 mM in hepatocytes).53–54 Indeed, if 25 remains intact in the presence of a five-fold excess of GSH, the conjecture is that its levels will be not be perturbed in the presence of GSH in cellular or in in vivo studies. When bioactive compounds are Michael acceptors, a GSH-based assay is essential, as it determines their potential for further drug development, a general strategy for consideration in the field of covalent inhibitors in medicinal chemistry.62–63
Figure 8.
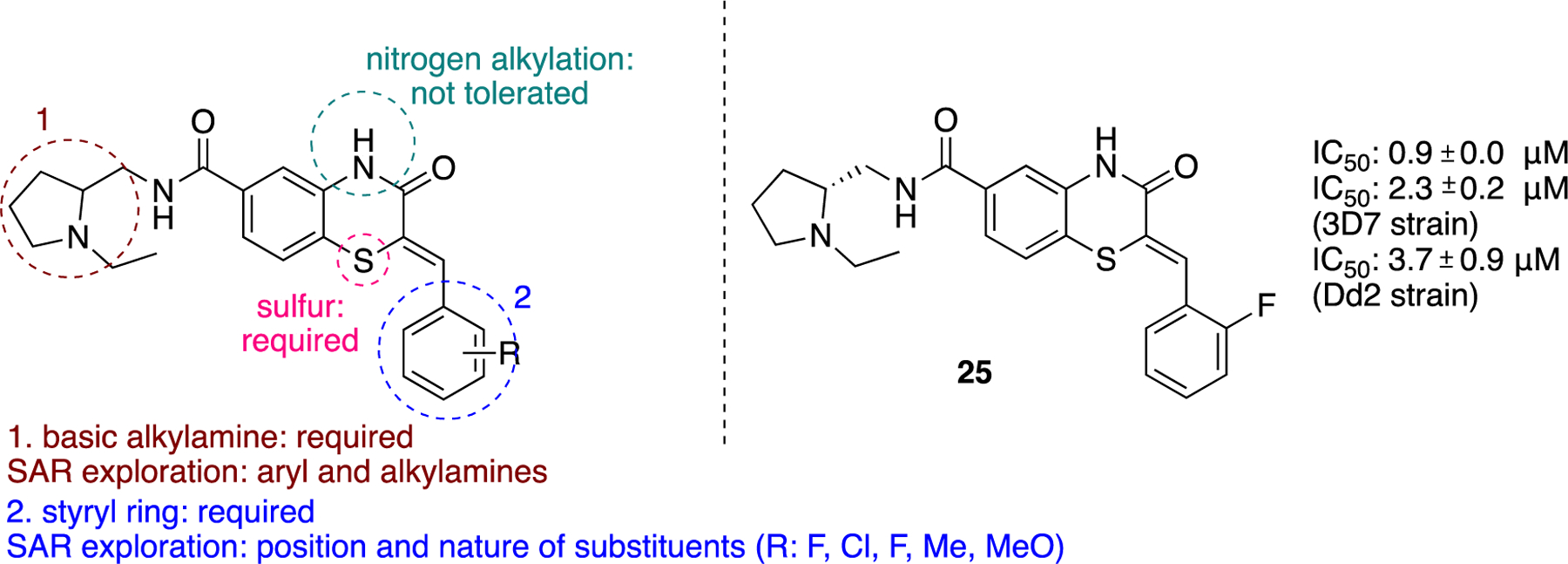
Generic benzothiazinone ring, essential features for activity, SAR exploration and specific PfGluPho compound 25.
Compound 25 was subjected to in vitro pharmacological studies to evaluate absorption, distribution, metabolism, excretion, and toxicity (ADMET). Compound 25 was highly permeable across artificial models of cell membranes. The presence of the amino groups allows for its protonation in the low pH environment of the stomach, contributing to its solubility. However, only the free base can be subsequently absorbed. A parallel artificial membrane model assay (PAMPA) was used to show its high permeability under conditions of increased pH in the donor compartment. Membrane models resembling the blood-brain barrier (BBB) were used to demonstrate high permeability. In addition, 25 was shown to be highly plasma protein bound which will influence the brain bioavailability of the compound. An assay using Fa2N-4 immortalized human hepatocytes was performed to show that 25 is not toxic under these conditions. Compound 25 was further evaluated in P. falciparum cultures in vitro using chloroquine-sensitive (3D7 strain, IC50: 2.3 ± 0.2 μΜ) and chloroquine-resistant (Dd2 strain, ΙC50: 3.7 ± 0.9 μM) strains. The same group performed studies in parasitic cultures in previous studies.51
In contrast to the Trypanosoma parasite, there are no G6PD crystal structures available for any of the Plasmodium species. However, Alencar et al., used the hG6PD structure to build a homology model for PfGluPho.64 A key difference between hG6PD and PfGluPho was the replacement of Arg365 (hG6PD) with Asp370 (PfGluPho). Given that 25 exhibits competitive inhibition for substrate G6P in biochemical assays and was selective for PfGluPho, the authors docked 25 in the G6P binding site of PfGluPho, which was superimposed to the hG6PD crystal structure (PDB ID 2BHL) with G6P bound. This led the authors to synthesize a series of G6P analogues.
Given that 25 is a competitive inhibitor of G6P and against the backdrop of the Arg365→Asp370 mutation, the authors hypothesized that the protonated amino group of 25 interacts with Asp370, an interaction that they illustrated by docking it in the G6P binding site. Building further on this hypothesis and focusing on G6P, the authors envisioned, synthesized, and evaluated a series of G6P analogues. In these G6P analogues, the phosphate group at C6 was replaced by a thioether (26) or groups acting as hydrogen bond donors or acceptors, including sulfones 27 and sulfonamides 28. In addition, a terminal amino, cyano or guanidino group was placed after the interjection of an alkyl chain. The alkyl chain filled the empty space between what would be Arg365 (hG6PD) to the existing Asp370 (PfGluPho) whereas the protonated amino or guanidino groups were projected to form ionic interactions with the carboxylate moiety of Asp370 (Figure 9).
Figure 9.
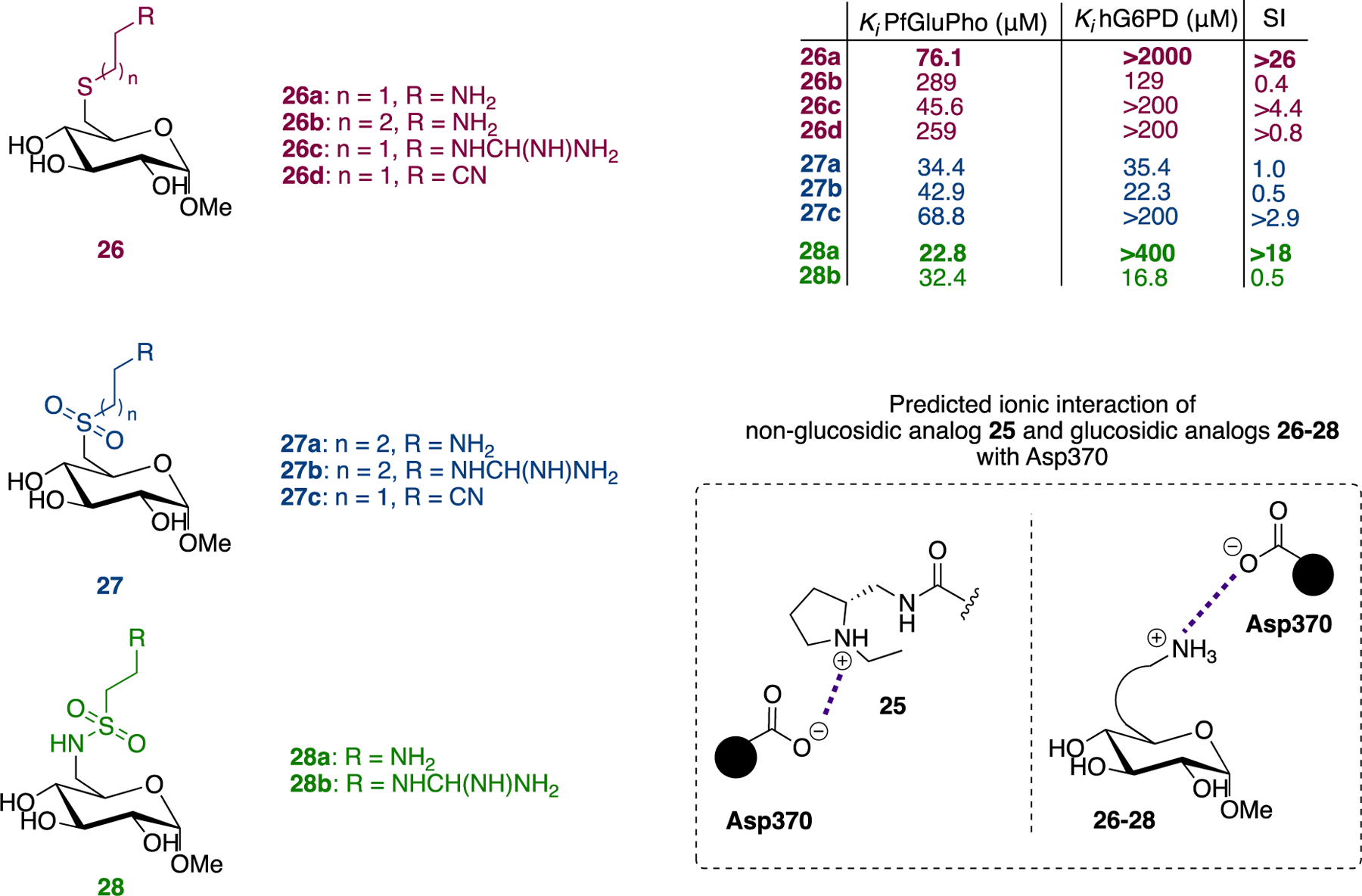
Compounds 26–28, in vitro biochemical activity Ki for G6P, and proposed interactions of non-glucosidic compound 25 and glucosidic compounds 26–28 with Asp370.
In vitro evaluation of the compounds showed that indeed compounds bearing either the amino or guanidino group (26a, 26c, 28a, n = 1, Figure 9) had higher affinity for PfGluPho compared to the compounds with the cyano group. In addition, based on the SI measured for the Ki for G6P [SI: ratio Ki hG6PD/Ki PfGluPho], the same compounds were more selective for PfGluPho. Notably, 26a exhibited 26-fold selectivity for PfGluPho compared to hG6PD. The internal substitution at C6 also affects affinity, with the compounds bearing sulfonamides showing the highest affinity and thioethers the lowest (28a>27a>26a, 28b>27b>26c).
Despite the broad therapeutic index for a few of these compounds, the IC50 values obtained from treatment of parasitic cultures were in the range of sub- to low millimolar, presumably owing to the high polarity of the compounds as sugar analogues. The compounds were also tested in the human hepatoma HepG2 cell line at a concentration ranging from 0.2 to 2 mM. Even at the highest concentration of 2 mM, none of the compounds impacted cell viability by more than 30%.
Another avenue of research would be to develop small molecules which are not analogues of G6P but are based on the projected pharmacophore between 25 and the PfGluPho; non-glucoside molecules may have an enhanced profile in the phenotypic assays due to their greater lipophilic character. Nonetheless, the authors projected a potential pharmacophoric interaction of 25 with the target and a plausible rationale for its selectivity toward PfGluPhO compared to hG6PD. Overall, the data for the non-glucosidic 25 and glucosidic analogues 26–28 provide an anchor for their optimization or even for de novo design of other analogues.
GSSG, the oxidized form of GSH, inhibits PfGluPho.57 Both the G6PD and 6PGLase domains of PhGluPho were inhibited as a function of S-glutathionylation of cysteine residues present in both domains. Except for Cys144, all of other cysteines reside in the G6PD domain of PfGluPho. In contrast, GSH (the reduced form of GSSG) and DTT increases the activity of the G6PD enzyme, but not the 6PGLase. DTT reverses the inhibition caused by S-glutathionylation of PhGluPho, indicating that the inhibition is reversible. Regarding the respective enzymes in humans (hG6PD and h6PGLase), it was shown that only h6PGLase was inhibited by GSSG. A plausible explanation for this phenomenon is that the PfGluPho has a region which is very similar to the GSH-binding site in GSH transferase.47 This discrepancy in effect of GSSG suggests that it is a selective inhibitor of the G6PD domain of PhGluPho and hG6PD, and it could be considered as a potential therapeutic. However, as a hexapeptide, it is susceptible to proteolytic degradation when delivered into the blood. Notably, the monomeric GSH is not susceptible to peptidases/proteases, compared to other peptides, because the peptide bond between the glutamate and the amino group of cysteine is formed at the γ-COOH instead of α-COOH. However, this bond can only be hydrolyzed by γ-glutamyltranspeptidase.65 This resistance may prove beneficial for GSSG delivery but may need additional optimization. As well as being a peptide, GSSG is a disulfide and, thus, it can participate in thiol exchange reactions with off-target free cysteines.66 This poses a second limitation for its consideration as a therapeutic agent. The sequence difference identified recently between the human (Arg) and the parasite (Glu) has allowed for the design of scaffolds, using molecular modeling methods, which selectively inhibit PhGluPho.67 The authors reported that they are currently examining how to capitalize on this information.
In addition to S-glutathionylation, S-nitrosation mediated by nitrosated GSH is another modification that can inhibit the activity of the enzyme. In contrast to what was shown in the previous work by Jortzik et al., Haeussler et al. showed that the G6PD part of PfGluPho and PvGluPho (Pv: Plasmodium vivax, P. vivax) are hardly affected by these post-translational modifications, with S-nitrosation leading to only minor changes in enzyme activity.68
As well as examining the effect of these post-translational modifications on enzymatic activity, the authors determined the effect of amino acid substitution of the PfGluPho sequence on activity. Three of these mutations are naturally occurring within the PfGluPho gene (S315Y, L395F, F507L) whereas two of them included serine substitution at positions 899 and 900 to examine effect of phosphorylation, another post-translational modification, on enzymatic activity. Interestingly, these substitutions have minimal effects on enzymatic activity and substrate affinities compared to wild-type PfGluPho. Notably the double serine sequence at positions of 899–900 is specific and highly conserved to Plasmodium species. Taking advantage of the differences in amino acid residues across different species (parasitic vs. human) through protein engineering, may be a strategy for developing selective chemical probes for a given species, as was the case for 25. This strategy is known as site-directed drug discovery.69
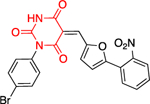
In addition to 25, which selectively inhibits PfGluPho over hG6PD, 29 had the same selectivity profile for PfGluPho, was even more potent (IC50: ~190 nM)70 and was also potent against PvG6PD (IC50: 15.3 ± 0.9 μM) (Figure 10).68 This study highlights the importance of achieving two aims at once: the discovery of a compound selective for the G6PD belonging to different genera (Plasmodium vs. human) and the possibility of one compound to be efficacious across the species of the same genus (P. falciparum and P. vivax). Superimposition of 25 and 29 validates the stringent requirement for the (R)-stereochemistry on the side chain of amido nitrogen (Figure 10). The benzothiazinone ring has been expanded by one carbon to benzothiazepinone in 29 and fused with the aromatic ring instead of being connected with it via an alkylidene bridge as in 25. This alters the relative orientation of the aromatic ring resulting in a more planar structure for 25 and a folded arrangement between thiazepinone and the aromatic ring for 29. This second chemical probe validates the SAR and opens the road for further modifications in the benzothiazinone or benzothiazepinone ring. Since both compounds are competitive inhibitors for G6P, this can be guided by docking both at the substrate binding site and thoroughly examining the pharmacophore with G6PD.
Figure 10.
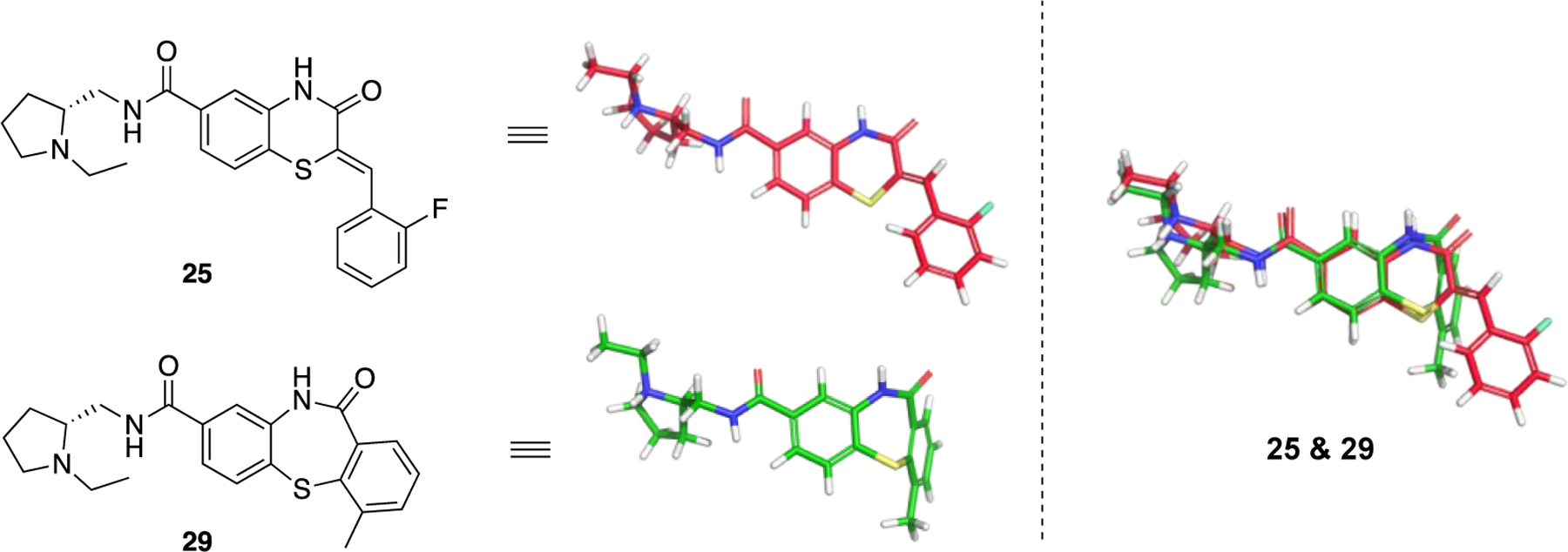
Compounds 25 (ML276) and compound 29 (ML304). 3D alignment performed with Schrödinger 2021–3, Maestro, using largest Bemis-Murcko scaffold. ML276 and ML304 as taken from the original paper.
Cancer
Except for 25 and 29, which showed promising selectivity, the lack of selectivity observed for the rest of the compounds between the parasitic and hG6PD has inspired research groups to shift their focus to hG6PD inhibitors which target cancer and inflammation. Bode and co-workers screened 50,000 compounds at a concentration of 5 μg/mL and identified 107 that inhibit hG6PD by 50%.71 The five hG6PD inhibitors 12a-b, 14a, 30, 31 (Table 2) demonstrated IC50 values of < 4 μΜ and were selected for follow up studies. Among these five compounds, four of them (12a-b, 14a, 31) were previously reported to inhibit PfGLuPho. In Table 2, we present the IC50 values for PfGluPho and hG6PD for comparison.
Table 2.
Comparison of IC50 values for PfGluPho and hG6PD; shift of focus from antimalarial to anticancer agents by the Bode group; Numbers in parenthesis as taken from the original paper. For 14a 1: (w/ pre-incubation and post-dilution), 2: (only pre-incubation).
| Compound | Structure | PfGluPho IC50 (μΜ) | IC50 Comparison | hG6PD IC50 (μΜ) |
|---|---|---|---|---|
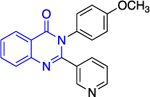
| 12a (CB104) |
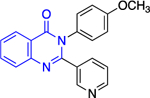
|
7.6 ± 4.0 | > | 3.0 ± 1.2 |
| 12b (CB70) |
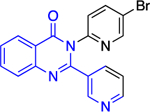
|
4.5 ± 1.6 | > | 2.6 ± 0.1 |
| 14a (CB83) |
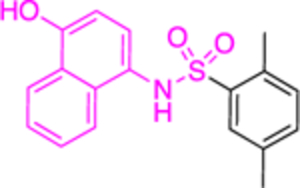
|
22.0 ± 0.8 | > | 0.2 ± 0.01 or 0.4 ± 0.02 |
| 30 (CB72) |
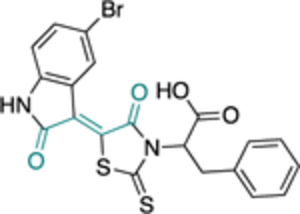
|
- | 3.1 ± 0.8 | |
| 31 (CB63) |
|
1.7 ± 0.2 | > | 0.6 ± 0.0 |
Compounds 12a and 12b incorporate the common quinazolinone nucleus highlighted in blue. In a prior study by Bode, 12b was tested in both biochemical and parasitic assays but was found to be a potent inhibitor of PfGluPho only in the biochemical assay. The other three compounds 14a, 30, 31 are structural alerts, either because they may engage in redox cycling activity (14a) or act as a Michael acceptor (30 and 31 where the 1,4-conjugated moiety is shown in teal). Compound 14a can generate the phenoxy radical which can react with superoxide ion (as in Figure 7b) and be oxidized to a benzoquinone imine of type 16 which is a Michael acceptor (Figure 6).
The forms of inhibition by these five compounds were then determined. Initial dose-response experiments to calculate IC50 values, in which pre-incubation was used without post-dilution (to examine reversibility), placed 14a as the most potent inhibitor with an IC50 of 0.4 ± 0.0 μM and 30 as the least potent inhibitor with an IC50 of 3.1 ± 0.8 μM (Table 2). Inhibition of hG6PD by sulfonamide 14a was irreversible; its effect was not diminished following post-dilution.51 Furthermore, the IC50 was 100-fold lower after pre-incubation relative to no pre-incubation conditions. Compounds 12a, 12b and 31 are competitive inhibitors for G6P and showed a mixed type of inhibition for NADP+, with 31 showing both mixed type and non-competitive inhibition for NADP+. Compound 30 is also an inhibitor that is competitive for both G6P and NADP+. Notably, 12a and 12b inhibited hG6PD with a different kinetic signature compared to PfGluPho; for PfGluPho these inhibitors exhibited a mixed-type of inhibition with respect to G6P and were non-competitive with NADP+ (see under non-steroidal inhibitors, infectious diseases). This difference in inhibitory signature can serve as anchor to develop selective competitive inhibitors based on 12a and 12b for G6P for hG6PD.
Except for 12b, the remaining 4 compounds were tested in cell-based assays. Two cell lines were used: MCF10-A, used as a control, and the tumorigenic lMCF10-AT1 cell line. Compounds 12a, 30 and 31 had no effect on cell growth, with this being attributed to low membrane permeability or adhesion to other biomolecules. Sulfonamide 14a was the sole compound to demonstrate a measurable IC50 of 25 μΜ for the tumorigenic cell line compared to the >50 μΜ observed for the control cell line. This is another example where the GSH-based assay should have been performed,61 to validate that the inhibitory effect of 14a is not due to direct reaction between 16 with thiolates from free cysteines via a Michael addition, as shown in Figure 6.
Up to this point, we have discussed the selectivity of the identified inhibitors for G6PD from different species. In the context of cancer, another selectivity issue to consider is the selectivity towards the malignant cells relative to healthy cells. For these inhibitors to be further developed as anticancer agents, they must selectively act on malignant cells otherwise there is a risk of triggering hemolytic anemia to subjects. There are several means to improve such selectivity, including nano-formulation-based delivery using either passive targeting, which relies on the enhanced retention and permeability of cancerous tissues, or active targeting.72 Active targeting employs the functionalization of the nanoparticles surface for targeted delivery to the desired tissue.
In addition to targeting G6PD in humans for the treatment of cancer, another enzyme which is distinct from the cytosolic G6PD is hexose-6-phosphate dehydrogenase (H6PD). H6PD resides in the endoplasmic reticulum and catalyzes both the first and second step observed in the PPP; that is the conversion of G6P to 6-phosophogluconate (6PG) (Figure 1).73 Like PfGluPho, H6PD contains a fusion protein of G6PD and 6PGLase. It is found predominantly in the liver and other tissues, but it is absent in red blood cells. It shares homology with hG6PD, is dimeric, and uses a different pool of NADP+ as a cofactor compared to the cytosolic variant.74–76 It was recently shown that H6PD plays an important role in breast and lung cancer by genetic inhibition77 and, thereby, it can be considered for modulation using small molecules. Given that H6PD is absent in red blood cells, selective small molecule inhibitors for H6PD over hG6PD are compelling in that they will not cause hemolytic crises. However, the need for selective targeting of cancerous over healthy cells in all other tissues remains.
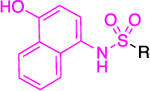
Inflammation
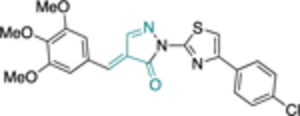
The work of Cordeiro44 that led to the identification of TrG6PD inhibitors served as an inspiration for the discovery of a hG6PD inhibitor by the Rabinowitz group.78 This hG6PD inhibitor, G6PDi (32) (Figure 11), reduced NADPH levels in a variety of immune cell types, including T-cells, macrophages, and neutrophils. This corresponded to a decrease in cytokine production and oxidative bursts, in T-lymphocytes and neutrophils, respectively, revealing a role for G6PD in immune cells. G6PD is critical for normal immune cell function.79 G6PD and the PPP have been associated with neuroinflammation,80 and adipose tissue inflammation in obesity,81 suggesting G6PD is a viable target for pathological inflammation.
Figure 11.
Generic structures of non-steroidal G6PD inhibitors used in infectious diseases, inflammation, and cancer. Main core observed in all cases: quinazolinone. Ligand alignment performed with Schrödinger 2021–3, Maestro, using largest Bemis-Murcko scaffold. M: mixed-type, NC: non-competitive, C: competitive, MOI: modality of inhibition. Biorender.com was used in part for Figure 11.
In the context of drug discovery, 32 is an analogue of the quinazolinone 10c (Figure 11), which was previously identified as a trypansomal G6PD inhibitor,44 and extensive chemistry optimization effort by Rabinowitz et al. resulted in the G6PD inhibitor 32 (Figure 11). The 3-methylaniline in 10c was replaced by a 2-cyano-4-aminothiophene, while the cyclohexanone moiety has been replaced by cycloheptanone. Notably, the replacement of 2-cyano-4-aminothiophene by 3-aminobenzonitrile led to 33, which had no effect on immune cells. Thus, 33 was used as a negative control in the studies. Compounds 32 and 33 differ in their 3D shape as shown by the structural overlay depicted in Figure 11. In addition, the substitution of H with a CH3 on the nitrogen atom between the heterocycle and phenyl rings diminishes the hydrogen bond donating features in 33. Therefore, even minimal structural changes can lead to dramatic activity shifts. In addition to the shift observed for species selectivity between 10c and 32, there was a shift in MOI. Although both agents exhibited reversible type of inhibition in the dilution assays, 10c was shown to be uncompetitive for TrG6PD whereas 32 non-competitive for hG6PD. On the contrary, in the case of 12a and 12b, replacement of the methoxy group by a bromine and the benzene by a pyridine ring did not alter the selectivity for hG6PD over parasitic G6PD but 12b was found to be more potent than 12a for each species (Figure 11 & Table 2). In addition, the MOI was different for each species, as already addressed in this perspective. Overall, the preference for hG6PD informs SAR for the “hit to lead” phase to improve both preference and pharmacological effect.
The non-androgenic quinazolinones are a frequently encountered scaffold; they are found in inhibitors of inflammasomes associated with diabetes, cardiovascular and neurodegenerative diseases.82–83 The quinazolinones also have anticancer agents, due to their inhibitory effects on inflammasomes formation82–83 and tubulin polymerization.84–85 Recently quinazolinones were found to be inhibitors of epoxide hydrolase, an enzyme targeted for blood pressure reduction, insulin sensitivity improvement and inflammation decrease.86 Such a broad scope of applications suggests lower selectivity toward G6PD.
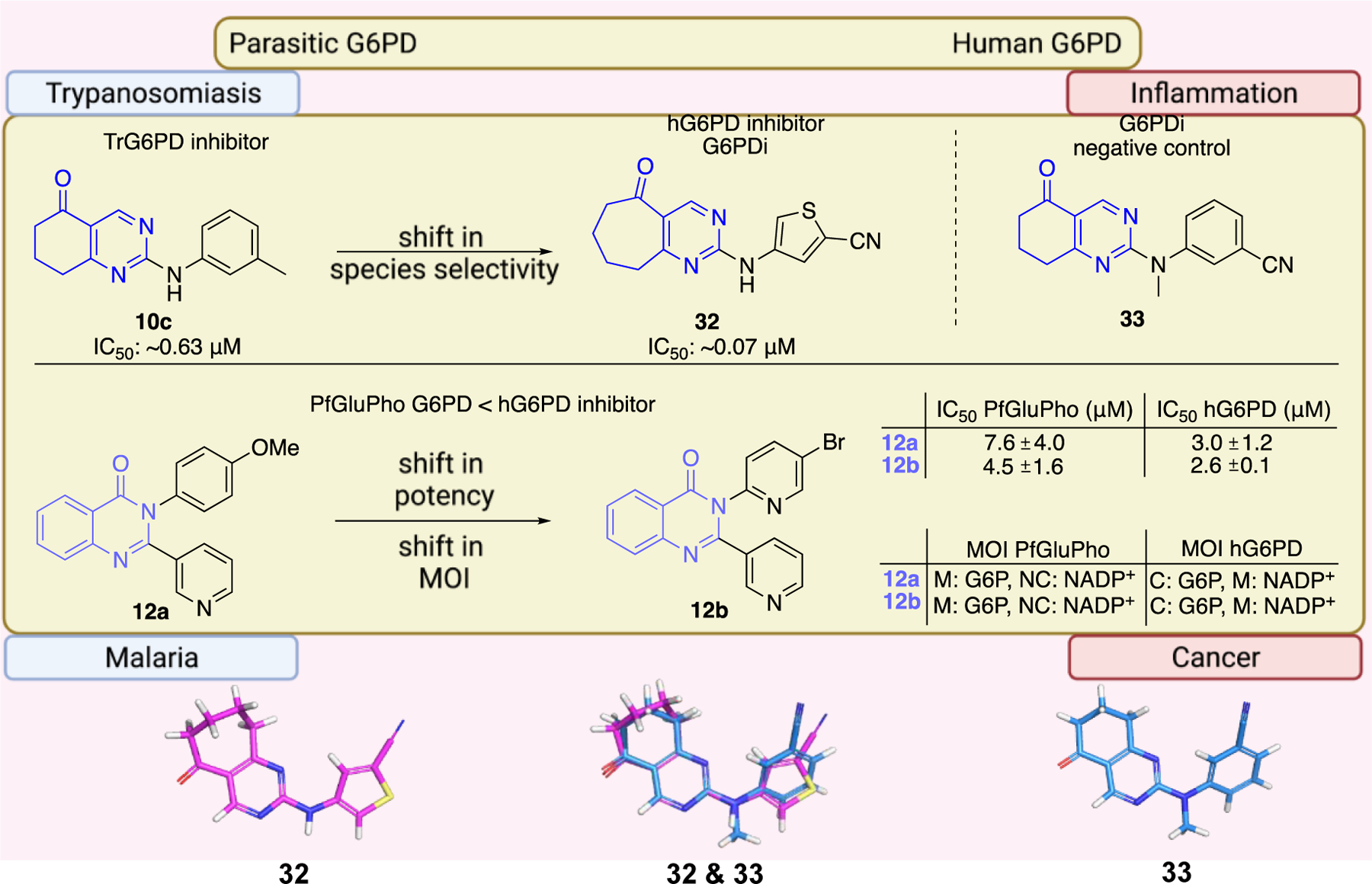
The work of Ramírez-Nava et al. identified four non-quinazolinone compounds with moderate inhibitory activity toward hG6PD.87 Initial HTS led to 55 candidate compounds, four of which inhibited hG6PD activity by 40% at a concentration of 400 μΜ (Figure 12a). Two of the four compounds, 34a and 34b, are homologues that contain a biphenyl ring and a phenylalkanoic group. The other two compounds, 35a and 35b, differ in the substitution of the aromatic ring (chloro for 35a vs butoxy ring for 35b). Although they share the 2-nitrothiazole ring with anti-protozoal nitazoxanide 35, nitazoxanide 35 does not act through G6PD inhibition. In addition, the bridge between the headgroups in compounds 35a and 35b is a urea whereas in nitazoxanide the linker is an amide bond, a difference which impacts the 3D shape between them (Figure 12a). For all three compounds 35, 35a, and 35b, the preferred conformation would be anticipated to involve the carbonyl oxygen from the amide or urea being oriented toward the sulfur of the nitrothiazole. As such, the non-bonding lone pair of the oxygen is able to engage with the σ* antibonding orbital of sulfur, an interaction which confers stability to the system (Figure 12b).88 This non-covalent interaction between sulfur and oxygen can be considered when further designing and optimizing drugs carrying this motif. Dose-response experiments showed that 35a had the greatest inhibitory activity among the four compounds toward the recombinant hG6PD (IC50 = 121 μΜ). Despite the structural similarities for the compound pairs of type 34 and 35, the type of inhibition varied. Compounds 34a, 34b, and 35b are non-competitive or uncompetitive inhibitors with respect to both G6P and NADP+, whereas the most potent compound, 35a, is a non-competitive inhibitor with respect to G6P and an uncompetitive inhibitor with respect to NADP+. These differences in mechanism of inhibition suggest differences in the amino acids with which each of these compounds interact.
Figure 12.
a. Compounds 34a, 34, 35a, 35b showed inhibitory activity against hG6PD and used for blind molecular docking; Codes in parenthesis as taken from the original paper. Ligand alignment performed with Schrödinger 2021–3, Maestro, using largest Bemis-Murcko scaffold for 34a & 34b and sample reference 35a for 35a, 35b & 35; b. Preferred conformation of the nitro compounds 35, 35a & 35b: overlap of the non-bonding lone pair of oxygen with the σ* orbital of sulfur.
The study of Ramírez-Nava et al. also included molecular docking experiments. Although the projected inhibitors are weak, this is the first example of an in silico study in the history of non-steroidal G6PD inhibitors. For steroidal inhibitors, in silico studies of 6 and its analogues provided support for the selective effect of these compounds on the trypanosomal over hG6PD,89–90 a trend observed by the group of Cordeiro in biochemical and cell-based assays. Using a blind docking protocol, Ramírez-Nava et al. the ΔG free energies are consistent with the biochemical activities and suggested that the docking site for the compounds was either close to the catalytic or structural NADP+, or near the binding of G6P. Compound 35a, which was the most potent in the biochemical assays, had a ΔG of −6.91 kcal/mol at the structural NADP+ site, which was slightly higher when compared to the site near the G6P-binding site (-7.51 kcal/mol). However, it was considered more likely for inhibitor 35a to enter the structural NADP+ region as it interacts with five out of the twelve amino acid residues involved in the binding of the structural NADP+, despite the higher ΔG observed for this binding. Such compounds that interact with the structural NADP+ are important for selective inhibitory activity on hG6PD, as the structural NADP+ site is absent in other dehydrogenases or parasitic G6PDs. Such in silico docking information by Ramírez-Nava et al. now provides potential grid boxes for future computational studies, as opposed to blind docking where the whole target represents the grid box.
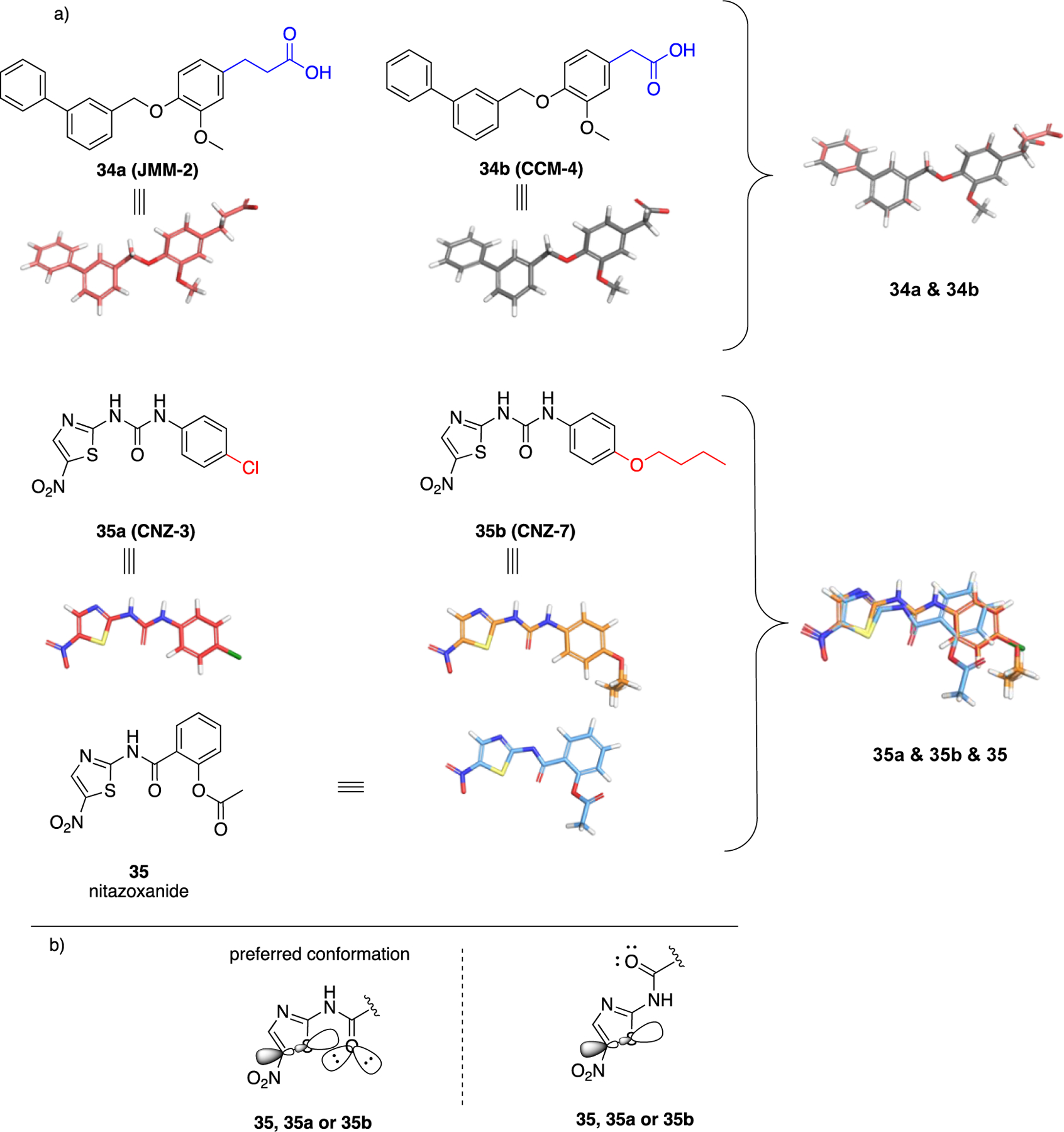
A remarkable transition from steroidal to non-steroidal small molecules was the discovery of tamoxifen. The synthetic form of estradiol (36) is diethylstilbestrol (DES 37), a derivative of trans-stilbene (Figure 13). The two phenolic oxygens in 37 are disposed at the same distance that phenolic oxygen at C3 and the hydroxy group at C17 exist in 36 and, like the prototype, 37 is an agonist.91 This SAR subtended a rational approach for transitioning from the steroidal 36 to non-steroidal 37 which, at that time, was conducted in the absence of co-crystal structures. Tamoxifen 38 is a selective estrogen receptor modulator that acts as an antagonist in breast cancer cells but acts as an agonist in the uterus.92–93 Both 37 and 38 possess the trans-stilbene core and the original synthetic agonist served as a scaffold to envision antagonists. Agonists such as 37, stabilize helix 12 in the ligand binding domain and are fully engulfed in it. On the other hand, antagonists such as 38 that bear an additional bulky side chain cave into helices 3 and 11. This interjection prevents helix 12 in the receptor from adopting the agonist conformation and blocks recruitment of the coactivator to the ligand-binding site.94–95 Similarly, it is expected upon unveiling the pharmacophoric interactions between G6PD and 1 or 2 that more knowledge will be available to refine the structure of small molecules obtained from HTS. The fact that similar structures arose from different screening strategies and cause similar biological activities adds to the confidence for the observed SAR to date.
Figure 13.
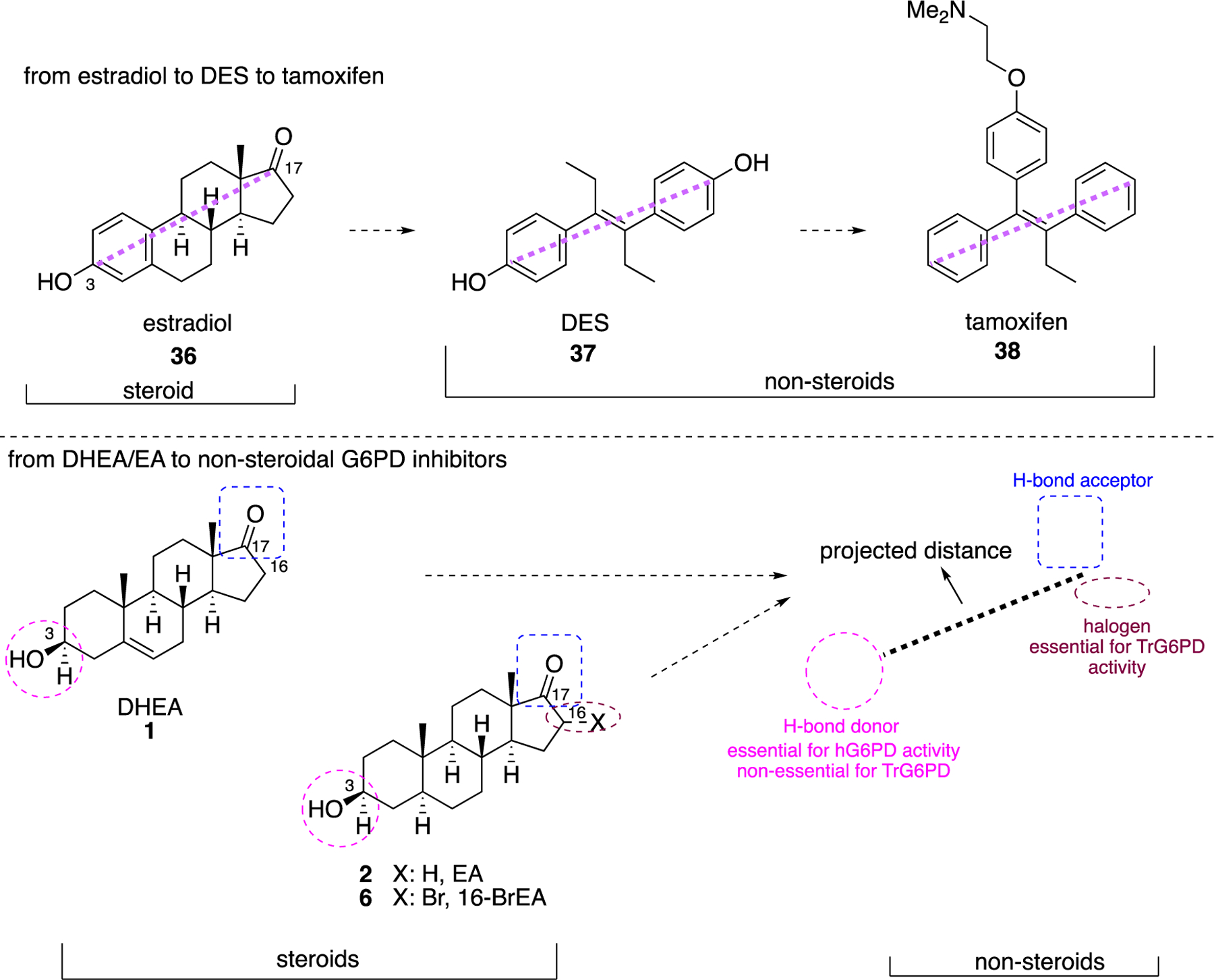
Top: from estradiol (36) to DES (37) to tamoxifen (38); bottom; perspective for transition of steroidal 1, 2, 6 to non-steroidal analogues.
The groups of Hamilton32 and Cordeiro41 have shown that hydrogen bond donors at C3 of the steroids are essential, and studies by Hamilton support the contention that hydrogen bond acceptors are necessary at C17. These data bring the discovery of non-steroidal G6PD molecules in the same perspective as for the identification of synthetic, non-steroidal estradiol agonists and antagonists, where groups at C3 and C17 were retained as part of the pharmacophore in the non-steroidal analogues (Figure 13).
We discussed earlier the interactions of hG6PD with co-factors and substrates, based on existing co-crystal structures that were used, guide structure-based drug design. In addition, we have highlighted the existing small molecule inhibitors that incorporate structural alerts which render them potentially unattractive candidates for lead optimization. These structural alerts include, but are not limited to, Michael acceptors and redox-cycling compounds. For Michael acceptors, promiscuity would be of concern but there are many Michael acceptors that are quite selective for their targets. In structure-based drug design, such moieties can be exploited under the premise that molecular recognition has been achieved between the target and the ligand structure. Molecular recognition, a concept and principle encountered primarily in supramolecular chemistry,96 takes place via non-covalent interactions; these interactions are secondary interactions and assist with the localization of the drug in the target. Next, a Michael acceptor can be incorporated as a functional group for primary interaction to advance activity. Indeed, covalent drugs have gained ground in the recent years,97–99 using this dual strategy of molecular recognition and incorporation of a highly reactive, yet discriminating, moiety. A notable successful example based on this strategy includes the allosteric KRAS inhibitor, AMG510 39, which has an acrylamide moiety acting as Michael acceptor, yet it reacts specifically with Cys12 of the KRAS binding pocket (Figure 14).100–101 This cysteine appears solely in mutated KRAS proteins present in cancerous cells and not in wild type KRAS (in wild type KRAS protein, position 12 is occupied by a glycine). Not only is pocket specificity achieved, but also selectivity between mutant and wild type isoforms, offering selectivity between cancerous and healthy cells, respectively.
Figure 14.
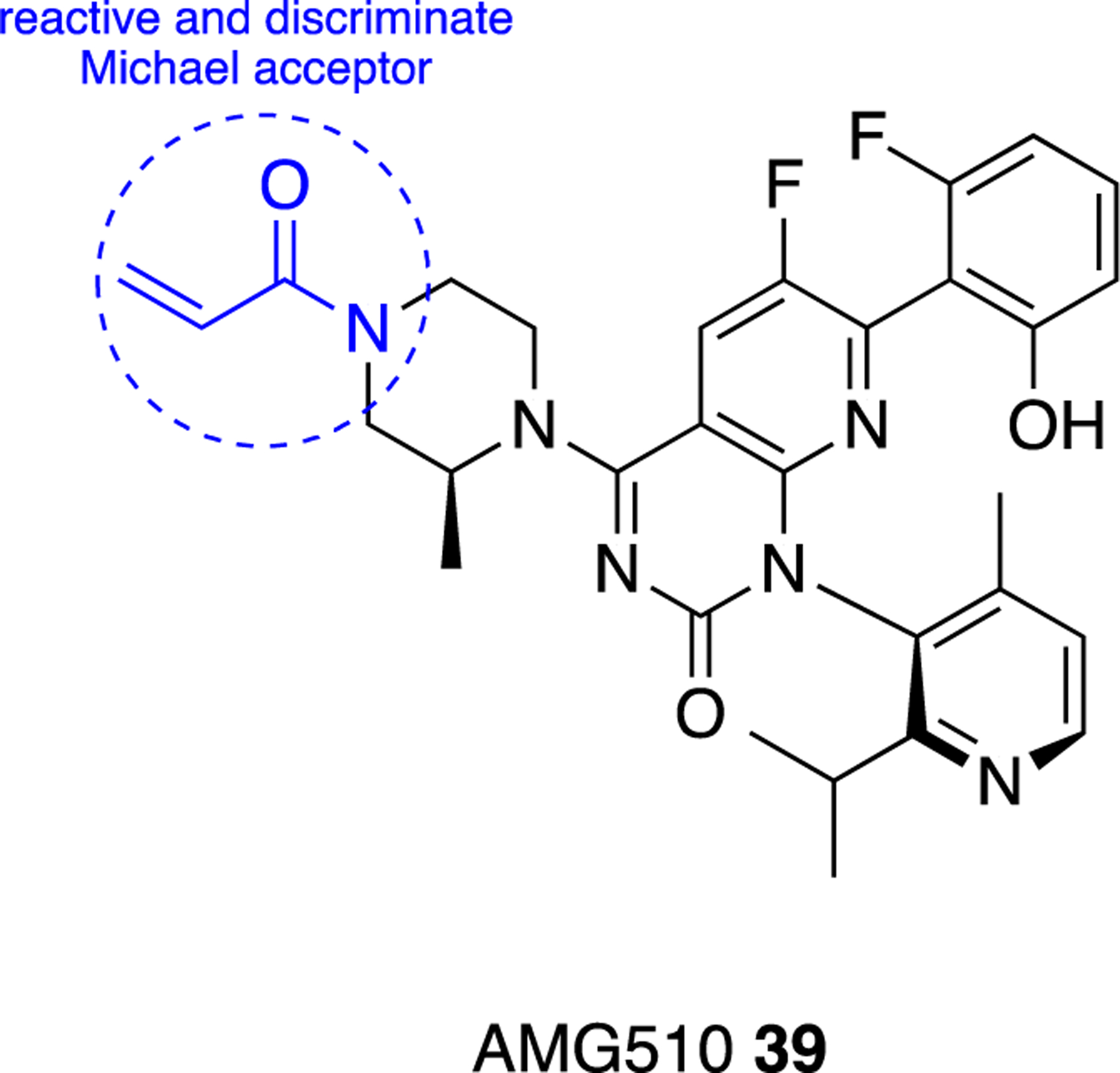
AMG510 39, selective KRAS inhibitor for cancerous cells; designed for molecular recognition and for specific reaction with Cys12 in mutated malignant cells.
Finally, although the crystal structure of the bacterial L. mesenteroides G6PD has been determined,30 no agents or small molecules targeting the bacterial G6PD have been developed. This approach could open the road for new anti-bacterial agents and replace the current antibiotics, whose major liability is the resistance development. Suffice is to mention that selectivity for the bacterial G6PD over hG6PD will be essential for such inhibitors to be considered as antibiotics.
Competitive G6PD Inhibitors for FBDD: Advantages and Disadvantages
In FBDD, the number of compounds which may be used for screening is in the range of a few thousands in contrast to HTS, where the number may reach many millions. Molecular weights of the compounds in FBDD are 150–200 Da and even millimolar affinities can prove useful in early stage of such a screen. Positive fragments need to then be grown (up to 500 Da) to optimize the compound and obtain nanomolar affinity. The translation of the fragment hit with weak affinities into the final drug lead can rely on in silico information (when crystal structure is available), but can achieved also in its absence.102 Initial fragments can also be grown through tethering of the fragments to the protein-binding site. This method introduces a disulfide bond between the fragment and a cysteine in a protein to stabilize the interactions, with the cysteine introduced in the target protein near the presumed binding site via mutagenesis.69, 103
The above methodologies can be applied, for example, on 6-aminonicotinamide (6AN, 40), which inhibits G6PD competitively and its molecular weight is 137.14 (Figure 15).104 However, 40 is not specific for G6PD since it competes for the binding of NAD+ or NADP+ of other dehydrogenases. Both a highly specific screen and computational docking of this fragment can identify a specific and high affinity lead.
Figure 15.
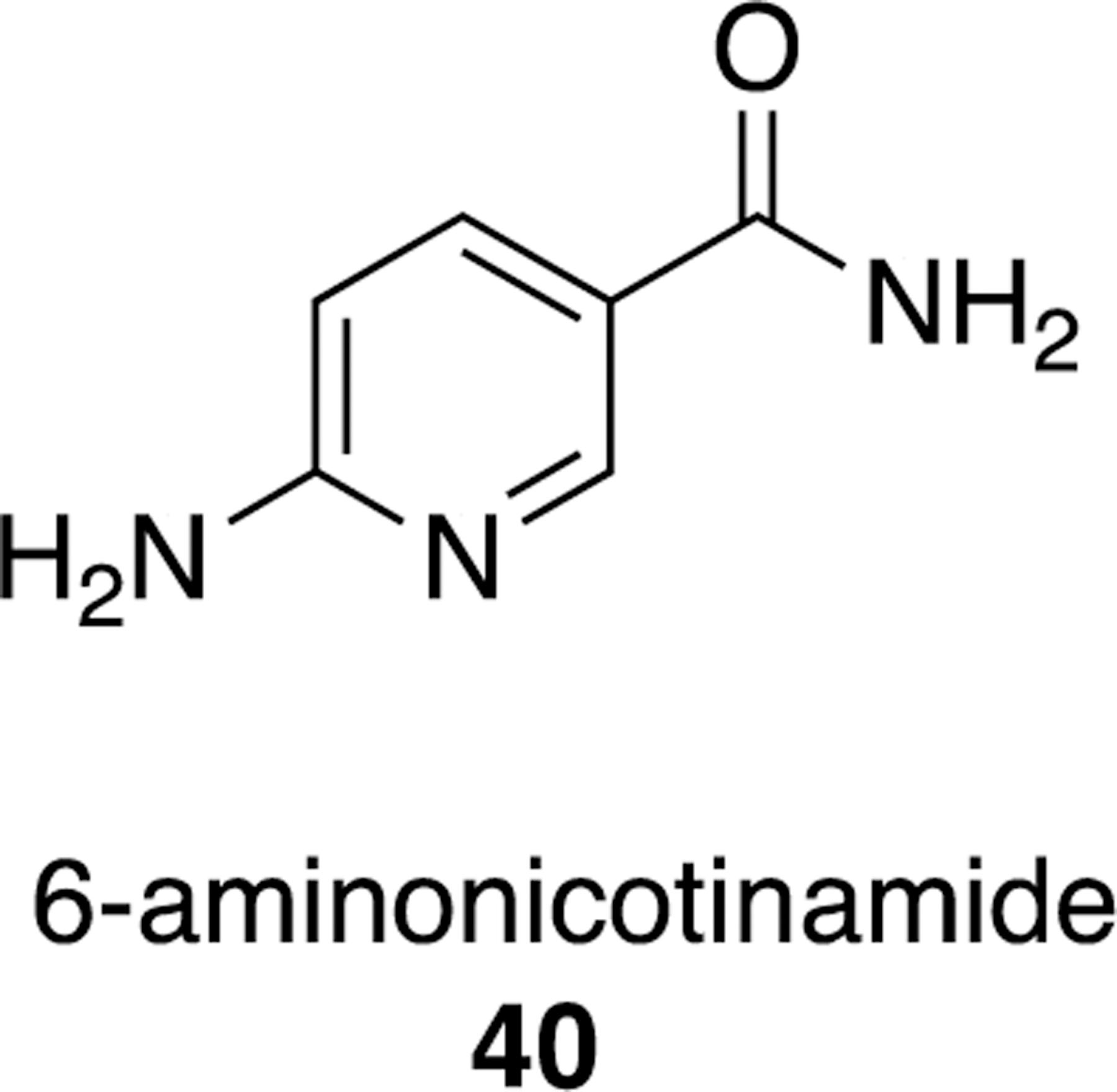
6-Aminonicotinamide 40, a potential fragment for FBDD of competitive G6PD inhibitors.
Advantages of FBDD include the use of a smaller library and therefore the ability to use a more complicated assay, which might not be suitable for HTS. Following fragment screen, bioisosteric replacement leads to molecules with the improved and desired properties that are to be characterized and confirmed by orthogonal assays. A second advantage to begin with FBDD is that as the medicinal chemistry campaign begins with a fragment, there is a better chance of generating a small enough molecule with the desired pharmacological features. A disadvantage for FBDD is the reduced chemical space that is searched. However, the number of possible compounds that can constructed from the original fragment is exponential, creating sufficient exploration of diverse chemical space. Another disadvantage is the very weak interaction of the fragment with the target (Kd up to 5000 μM) requiring assays that are much more sensitive than those use in HTS. Although tethering, through disulfide bind to the target overcomes this limitation, it requires that all compounds in the fragment library to have a -SH group. It also requires quite extensive mutagenesis campaign of the target to identify the optimal cysteine mutant for the screen. Nevertheless, the lack of optimal and selective inhibitors for G6PD argues that more efforts in the field, including the use of FBDD should be employed.
Summary and Outlook
In this perspective, we discussed small molecules which have been discovered and optimized for G6PD inhibition in the context of cancer, infectious diseases, and inflammation. Candidate G6PD inhibitors can be classified into two major structural groups: steroidal and non-steroidal. Hamilton et al. and Cordeiro et al. presented their work on steroids with potential anticancer and trypanocidal activity, respectively. Their data suggest that hydrogen bond donors and acceptors at C3 and C17 of the steroids govern the SAR for potency and species selectivity. Despite the encouraging results regarding potency and species selectivity, the off-target androgenic effects pose a limitation for the development of these molecules as G6PD-targeting therapeutics.
In the case of non-steroidal small molecule G6PD inhibitors, the lack of species selectivity between the microorganism and hG6PD has served as an impetus for hit modification as an approach to the identification of species-selective G6PD antagonists. Three compounds stand out from the drug discovery efforts conducted to date and all are derived from HTS campaigns: the antimalarial compounds 25 and 29 and the anti-inflammatory agent 32. Notably, inhibitor 32 evolved from optimization of trypanocidal agents that were introduced by the Cordeiro group. Although all three of these compounds have reached a degree of translatability in cellular and in vivo studies, their use in the clinic has yet to occur.
Non-steroidal G6PD inhibitors have been studied in the context of cancer using both biochemical and cellular assays; however, the lead inhibitors are structural alerts for either being Michael acceptors or for their redox cycling activity. p-Aminophenols of type 14 and PDT 19 are such examples. In addition, false positive hits can also stem from the conditions employed in biochemical assays; in the presence of super stoichiometric amounts of reductants and oxidants in buffer solutions, functional groups can be reduced or oxidized, respectively. Under physiological conditions these modifications would not occur. In cellular assays, enzyme metabolism and off-target cellular effects may account for false positive results. On the other hand, the polarity of the compounds and their consequent poor permeability creates a barrier for translation in cells or in in vivo studies and, thus, false negative results are obtained for in cellulo activity.
Regarding the structural alerts, there are in vitro assays that can evaluate the promiscuity of these ligands as Michael acceptors or redox cycling molecules; these assays measure the ability of the ligands to remain intact in the presence of free thiols, deplete GSH levels or produce H2O2, respectively. There are two solutions to address this substrate promiscuity during SAR optimization. The first one involves the structural modification in the part of the molecule responsible for reactivity (for example removal of the 1,4-conjugated system in Michael acceptors or replacement of the triazine ring with diazine in flavone derivatives). Contrastingly, in the second strategy a highly reactive site is intentionally embedded in the molecule; however, promiscuity will need to be attenuated by judicious decoration of a molecule such that it is recognized by a specific pocket in the target.
Non-selective competitive inhibitors that inhibit several NADP+-dependent dehydrogenases, such as 40, are also useful leads as they can provide a basis for in silico design, FBDD and medicinal chemistry efforts to increase their specificity for G6PD. The bar for selectivity is high in this case since there is already selectivity for a whole family of enzymes depending on the specific co-factor 6AN competes with (NADP+ or NAD+). For that, in the case of FBDD, highly sensitive assays are required as opposed to HTS drug discovery, where specificity is the major factor governing subsequent SAR optimization.
A general challenge in the reports on the efforts to generate G6PD inhibitors is that IC50 and not Ki values are provided. IC50 values are useful for biological evaluation; however, the values depend on the concentration of the enzyme used in the assay and, therefore, does not allow a comparison across other studies. In contrast, Ki is an intrinsic parameter of each inhibitor; it depends on the equilibrium between the bound and unbound inhibitor, and, unlike IC50, it is independent of enzyme concentration. The shift from providing IC50 to Ki values is currently a practice but must become a mandate.
As the critical role of G6PD in a variety of human diseases becomes apparent, the field is now poised to address challenges and fill in gaps for the discovery of small molecule G6PD inhibitors. We anticipate this perspective will provide a platform to boost the discovery of target-and species-selective G6PD inhibitors for cancer, infectious diseases, inflammation, and other diseases in which G6PD plays a crucial role which is currently known or remains to be identified.
ACKNOWLEDGMENT
A.K and D.M-R are supported by NIH R01 HD08442 awarded to D.M-R. A.A.G thanks the National Science Foundation for a Graduate Research Fellowship (NSF-GRFP) (Grant DGE 1656518) and National Institutes of Health (NIH) for a training grant (Grant 5T32GM113854).
Funding Sources
The work was supported by R01 HD08442 to D.M-R.. NSF-GRFP; A.A.G was supported in part by DGE 1656518 and 5T32GM113854.
ABBREVIATIONS
- 6PG
6-phosophogluconate
- 6PGL
6-phsopsoglucono-δ-lactone
- 6PGLase
6-phsopsoglucono-δ-lactonase
- 16-BrEA
16-bromoepiandrosterone
- 17BHDSD5
17-β-hydroxysteroid dehydrogenase 5
- BBB
blood-brain barrier
- C
competitive
- Caco-2
colorectal adenocarcinoma cell line 2
- DES
diethylstilbestrol
- DHEA
dehydroepiandrosterone
- DMF
dimethylfumarate
- DTT
dithiothreitol
- EA
epiandrosterone
- Fa2N-4
immortalized human hepatocytes line
- FBDD
fragment-based drug discovery
- GABAA
gamma-amino-butyric acid-A
- G6PD
glucose-6-phosphate dehydrogenase
- G6PDi
glucose-6-phosphate dehydrogenase inhibitor
- GSH
glutathione
- GSSG
glutathione disulfide
- h
hour
- H6PD
hexose-6-phosphate dehydrogenase
- H9C2
rat heart myocytes
- HepG2
human hepatoma cells
- HEK293T
human embryonic kidney cell line expressing antigen T
- hG6PD
human glucose-6-phosphate dehydrogenase
- h6PGLase
human 6-phsopsoglucono-δ-lactonase
- HSD3B2
3-β-hydroxysteroid dehydrogenase 2
- hTERT
human telomerase reverse transcriptase
- HTS
high-throughput screening
- IC50
half maximal inhibitory concentration
- IDH
isocitrate dehydrogenase
- Inc.
incorporated
- LD50
median lethal dose
- M
mixed-type
- MCF10-A
non-malignant breast epithelial cell line
- MCF10-AT1
premalignant breast epithelial cell line
- MOI
modality of inhibition
- NC
non-competitive
- NADP+
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- PAMPA
parallel artificial membrane permeability assay
- PDT
pyrimidinetriazinedione
- PfGluPho
Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase
- P.falciparum
Plasmodium falciparum
- P.vivax
Plasmodium vivax
- PPP
pentose phosphate pathway
- SAR
structure-activity relationship
- SI
selectivity index
- R-5P
ribose-5-phosphate
- Ru-5-P
ribulose-5-phosphate
- ROS
reactive oxygen species
- T. cruzi
Trypanosoma cruzi
- TrG6PD
trypanosomal glucose-6-phosphate dehydrogenase
- vs.
versus
BIOGRAPHICAL SKETCHES
Ana Koperniku obtained her BSc in Pharmacy in 2011 and MSc in Pharmaceutical Chemistry in 2013 from National and Kapodistrian University of Athens. In 2013 she started at the University of British Columbia (UBC) as a PhD student and her doctoral thesis focused on the chemistry of N-silylated amines as precursors to diheteroarylamides and as substrates in transition metal catalyzed reactions to generate novel alkyl substituted amines in the groups of Profs. David Grierson and Laurel Schafer, respectively. She was an NSERC CREATE SusSyn and a Four-Year Doctoral Fellow at UBC. She is currently a PDF in the Mochly-Rosen laboratory at Stanford University, where she is working on the identification of small molecule activators to correct enzymopathies.
Adriana A. Garcia received a B.S. in biochemistry from San Francisco State University. She is currently a graduate student in the department of Chemical and Systems Biology at Stanford University School of Medicine, in the lab of Daria Mochly-Rosen. She is a National Science Foundation – Graduate Research Fellowship Program (NSF-GRFP) fellow and her research uses biochemical and biophysical methods to study the structure-function relationship of clinically relevant mutations of a particular human enzymopathy. The goal of her studies is to identify therapeutic strategies that may correct protein structure and reduced enzymatic function caused by these mutations.
Daria Mochly-Rosen is a protein chemist in the Chemical and Systems Biology department at Stanford and leads a multi-disciplinary research lab focusing on the molecular mechanisms of diseases. She has published >300 articles and dozens of patents, was the Chair of her department, and the Senior Associate Dean for Research at Stanford University, School of Medicine. She is an expert in ‘translational research’ and through SPARK, a program she founded at Stanford in 2006, she helps shepherd scores of academic discoveries to licensing and clinical trials; >50% of the ~150 projects were licensed and/or entered clinical studies. Her lab’s research is focused on identifying peptide regulators of pathological protein-protein interactions and small molecules activators that correct common human enzymopathies; all as leads for drug discoveries.
Footnotes
We declare no competing interests.
REFERENCES
- 1.Stanton RC, Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB life 2012, 64 (5), 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian W-N; Braunstein LD; Pang J; Stuhlmeier KM; Xi Q-C; Tian X; Stanton RC, Importance of Glucose-6-phosphate Dehydrogenase Activity for Cell Growth. J. Biol. Chem 1998, 273 (17), 10609–10617. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal PK; Canvin DT, The pentose phosphate pathway in relation to fat synthesis in the developing castor oil seed. Plant Physiol 1971, 47 (5), 672–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutchings D; Rawsthorne S; Emes MJ, Fatty Acid Synthesis and the Oxidative Pentose Phosphate Pathway in Developing Embryos of Oilseed Rape (Brassica napus L.). J. Exp. Bot 2004, 56 (412), 577–585. [DOI] [PubMed] [Google Scholar]
- 5.Kornberg A; Horecker BL; Horecker BL; Smyrniotis PZ, [42] Glucose-6-phosphate dehydrogenase 6-Phosphogluconic Dehydrogenase. In Methods in Enzymology, Academic Press: 1955; Vol. 1, pp 323–327. [Google Scholar]
- 6.Gao J; Aksoy BA; Dogrusoz U; Dresdner G; Gross B; Sumer SO; Sun Y; Jacobsen A; Sinha R; Larsson E; Cerami E; Sander C; Schultz N, Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2013, 6 (269), pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerami E; Gao J; Dogrusoz U; Gross BE; Sumer SO; Aksoy BA; Jacobsen A; Byrne CJ; Heuer ML; Larsson E; Antipin Y; Reva B; Goldberg AP; Sander C; Schultz N, The cBio Cancer Genomics Portal: an Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov 2012, 2 (5), 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li R; Wang W; Yang Y; Gu C, Exploring the Role of Glucose‑6‑phosphate Dehydrogenase in Cancer. Oncol. Rep 2020, 44 (6), 2325–2336. [DOI] [PubMed] [Google Scholar]
- 9.Khan A; Siddiqui S; Husain SA; Mazurek S; Iqbal MA, Phytocompounds Targeting Metabolic Reprogramming in Cancer: An Assessment of Role, Mechanisms, Pathways, and Therapeutic Relevance. J. Agric. Food Chem 2021, 69 (25), 6897–6928. [DOI] [PubMed] [Google Scholar]
- 10.Cho ES; Cha YH; Kim HS; Kim NH; Yook JI, The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther 2018, 26 (1), 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beutler E, G6PD Deficiency. Blood 1994, 84 (11), 3613–36. [PubMed] [Google Scholar]
- 12.Gaskin RS; Estwick D; Peddi R, G6PD Deficiency: its Role in the High Prevalence of Hypertension and Diabetes Mellitus. Ethn. Dis 2001, 11 (4), 749–754. [PubMed] [Google Scholar]
- 13.Heymann AD; Cohen Y; Chodick G, Glucose-6-Phosphate Dehydrogenase Deficiency and Type 2 Diabetes. Diabetes Care 2012, 35 (8), e58–e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vimercati C; Qanud K; Mitacchione G; Sosnowska D; Ungvari Z; Sarnari R; Mania D; Patel N; Hintze TH; Gupte SA; Stanley WC; Recchia FA, Beneficial Effects of Acute Inhibition of the Oxidative Pentose Phosphate Pathway in the Failing Heart. Am. J. Physiol. Heart Circ. Physiol 2014, 306 (5), H709–H717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meloni L; Manca MR; Loddo I; Cioglia G; Cocco P; Schwartz A; Muntoni S; Muntoni S, Glucose-6-phosphate Dehydrogenase Deficiency Protects Against Coronary Heart Disease. J. Inherit. Metab. Dis 2008, 31 (3), 412–417. [DOI] [PubMed] [Google Scholar]
- 16.Hecker PA; Lionetti V; Ribeiro RF Jr.; Rastogi S; Brown BH; O'Connell KA; Cox JW; Shekar KC; Gamble DM; Sabbah HN; Leopold JA; Gupte SA; Recchia FA; Stanley WC, Glucose 6-phosphate Dehydrogenase Deficiency Increases Redox Stress and Moderately Accelerates the Development of Heart Failure. Circ. Heart Fail 2013, 6 (1), 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chhabra A; Mishra S; Kumar G; Gupta A; Keshri GK; Bharti B; Meena RN; Prabhakar AK; Singh DK; Bhargava K; Sharma M, Glucose-6-phosphate Dehydrogenase is Critical for Suppression of Cardiac Hypertrophy by H2S. Cell Death Discov 2018, 4 (1), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang BL, Neuroprotection by Glucose-6-phosphate Dehydrogenase and the Pentose Phosphate Pathway. J. Cell. Biochem 2019, 120 (9), 14285–14295. [DOI] [PubMed] [Google Scholar]
- 19.Jeng W; Loniewska MM; Wells PG, Brain Glucose-6-phosphate Dehydrogenase Protects against Endogenous Oxidative DNA Damage and Neurodegeneration in Aged Mice. ACS Chem. Neurosci 2013, 4 (7), 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Webb SJ; Geoghegan TE; Prough RA; Michael Miller KK, The Biological Actions of Dehydroepiandrosterone Involves Multiple Receptors. Drug. Metab. Rev 2006, 38 (1–2), 89–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turcu A; Smith JM; Auchus R; Rainey WE, Adrenal Androgens and Androgen Precursors-definition, Synthesis, Regulation and Physiologic Actions. Compr. Physiol 2014, 4 (4), 1369–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marks PA; Banks J, Inhibition of Mammalian Glucose-6-Phosphate Dehydrogenase by Steroids. Proc. Natl. Acad. Sci 1960, 46 (4), 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon G; Mackow MC; Levy HR, On the Mechanism of Interaction of Steroids with Human Glucose-6-phosphate Dehydrogenase. Arch. Biochem. Biophys 1995, 318 (1), 25–29. [DOI] [PubMed] [Google Scholar]
- 24.Beutler E; Kuhl W, Characteristics and Significance of the Reverse Glucose-6-phosphate Dehydrogenase Reaction. J. Lab. Clin. Med 1986, 107 (6), 502–507. [PubMed] [Google Scholar]
- 25.Bautista JM; Mason PJ; Luzzatto L, Purification and Properties of Human Glucose-6-phosphate Dehydrogenase Made in E. coli. Biochim. Biophys. Acta 1992, 1119 (1), 74–80. [DOI] [PubMed] [Google Scholar]
- 26.Cornish-Bowden A, Why is Uncompetitive Inhibition so Rare? FEBS Lett 1986, 203 (1), 3–6. [DOI] [PubMed] [Google Scholar]
- 27.Au SWN; Gover S; Lam VMS; Adams MJ, Human Glucose-6-phosphate Dehydrogenase: The Crystal Structure Reveals a Structural NADP+ Molecule and Provides Insights into Enzyme Deficiency. Structure 2000, 8 (3), 293–303. [DOI] [PubMed] [Google Scholar]
- 28.Kotaka M; Gover S; Vandeputte-Rutten L; Au SW; Lam VM; Adams MJ, Structural Studies of Glucose-6-phosphate and NADP+ Binding to Human Glucose-6-phosphate Dehydrogenase. Acta Crystallogr. D 2005, 61 (5), 495–504. [DOI] [PubMed] [Google Scholar]
- 29.Mercaldi GF; Dawson A; Hunter WN; Cordeiro AT, The Structure of a Trypanosoma Cruzi Glucose-6-phosphate Dehydrogenase Reveals Differences from the Mammalian Enzyme. FEBS Lett 2016, 590 (16), 2776–2786. [DOI] [PubMed] [Google Scholar]
- 30.Rowland P; Basak AK; Gover S; Levy HR; Adams MJ, The Three-dimensional Structure of Glucose 6–phosphate Dehydrogenase from Leuconostoc mesenteroides Refined at 2.0 Å Resolution. Structure 1994, 2 (11), 1073–1087. [DOI] [PubMed] [Google Scholar]
- 31.Cosgrove MS; Loh SN; Ha J-H; Levy HR, The Catalytic Mechanism of Glucose 6-Phosphate Dehydrogenases: Assignment and 1H NMR Spectroscopy pH Titration of the Catalytic Histidine Residue in the 109 kDa Leuconostoc mesenteroides Enzyme. Biochemistry 2002, 41 (22), 6939–6945. [DOI] [PubMed] [Google Scholar]
- 32.Hamilton NM; Dawson M; Fairweather EE; Hamilton NS; Hitchin JR; James DI; Jones SD; Jordan AM; Lyons AJ; Small HF; Thomson GJ; Waddell ID; Ogilvie DJ, Novel Steroid Inhibitors of Glucose 6-Phosphate Dehydrogenase. J. Med. Chem 2012, 55 (9), 4431–4445. [DOI] [PubMed] [Google Scholar]
- 33.Freilich D; Ferris S; Wallace M; Leach L; Kallen A; Frincke J; Ahlem C; Hacker M; Nelson D; Hebert J, 16α-Bromoepiandrosterone, a Dehydroepiandrosterone (DHEA) Analogue, Inhibits Plasmodium falciparum and Plasmodium berghei Growth. Am. J. Trop. Med 2000, 63 (5), 280–283. [PubMed] [Google Scholar]
- 34.Rasmussen KR; Arrowood MJ; Healey MC, Effectiveness of Dehydroepiandrosterone in Reduction of Cryptosporidial Activity in Immunosuppressed Rats. Antimicrob. Agents Chemother 1992, 36 (1), 220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morales-Montor J; Baig S; Mitchell R; Deway K; Hallal-Calleros C; Damian RT, Immunoendocrine Interactions During Chronic Cysticercosis Determine Male Mouse Feminization: Role of IL-6. J. Immunol 2001, 167 (8), 4527–4533. [DOI] [PubMed] [Google Scholar]
- 36.dos Santos CD; Toldo MPA; Júnior J. C. d. P., Trypanosoma cruzi: The Effects of Dehydroepiandrosterone (DHEA) Treatment during Experimental Infection. Acta Trop 2005, 95 (2), 109–115. [DOI] [PubMed] [Google Scholar]
- 37.Vargas-Villavicencio JA; Larralde C; Morales-Montor J, Treatment with Dehydroepiandrosterone in vivo and in vitro Inhibits Reproduction, Growth and Viability of Taenia crassiceps metacestodes. Int. J. Parasitol 2008, 38 (7), 775–781. [DOI] [PubMed] [Google Scholar]
- 38.Carrero JC; Cervantes C; Moreno-Mendoza N; Saavedra E; Morales-Montor J; Laclette JP, Dehydroepiandrosterone Decreases while Cortisol Increases in vitro Growth and Viability of Entamoeba histolytica. Microb. Infect 2006, 8 (2), 323–331. [DOI] [PubMed] [Google Scholar]
- 39.Cordeiro AT; Thiemann OH; Michels PAM, Inhibition of Trypanosoma brucei Glucose-6-phosphate Dehydrogenase by Human Steroids and their Effects on the Viability of Cultured Parasites. Bioorg. Med. Chem 2009, 17 (6), 2483–2489. [DOI] [PubMed] [Google Scholar]
- 40.Cordeiro AT; Thiemann OH, 16-Bromoepiandrosterone, an Activator of the Mammalian Immune System, Inhibits Glucose 6-phosphate Dehydrogenase from Trypanosoma Cruzi and Is Toxic to These Parasites Grown in Culture. Bioorg. Med. Chem 2010, 18 (13), 4762–4768. [DOI] [PubMed] [Google Scholar]
- 41.Fredo Naciuk F; do Nascimento Faria J; Gonçalves Eufrásio A; Torres Cordeiro A; Bruder M, Development of Selective Steroid Inhibitors for the Glucose-6-phosphate Dehydrogenase from Trypanosoma cruzi. ACS Med. Chem. Lett 2020, 11 (6), 1250–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eberling P; Koivisto VA, Physiological Importance of Dehydroepiandrosterone. Lancet 1994, 343 (8911), 1479–1481. [DOI] [PubMed] [Google Scholar]
- 43.O’Shaughnessy PJ; Antignac JP; Le Bizec B; Morvan M-L; Svechnikov K; Söder O; Savchuk I; Monteiro A; Soffientini U; Johnston ZC; Bellingham M; Hough D; Walker N; Filis P; Fowler PA, Alternative (backdoor) Androgen Production and Masculinization in the Human Fetus. PLoS Biol 2019, 17 (2), e3000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mercaldi GF; Ranzani AT; Cordeiro AT, Discovery of New Uncompetitive Inhibitors of Glucose-6-Phosphate Dehydrogenase. J. Biomol. Screen 2014, 19 (10), 1362–1371. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Irujo JJ; Villahermosa ML; Mercapide J; Cabodevilla JF; Santiago E, Analysis of the Combined Effect of Two Linear Inhibitors on a Single Enzyme. Biochem. J 1998, 329 (3), 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Theorell H; Yonetani T, Studies on Liver Alcohol Dehydrogenase Complexes: IV. Spectrophotometric Observations on the Enzyme Complexes. Arch. Biochem. Biophys 1964, 106, 252–258. [DOI] [PubMed] [Google Scholar]
- 47.O'Brien E; Kurdi-Haidar B; Wanachiwanawin W; Carvajal J-L; Vulliamy TJ; Cappadoro M; Mason PJ; Luzzatto L, Cloning of the Glucose 6-phosphate Dehydrogenase Gene from Plasmodium falciparum. Mol. Biochem. Parasitol 1994, 64 (2), 313–326. [DOI] [PubMed] [Google Scholar]
- 48.Clarke JL; Scopes DA; Sodeinde O; Mason PJ, Glucose-6-phosphate Dehydrogenase-6-phosphogluconolactonase. A Novel Bifunctional Enzyme in Malaria Parasites. Eur. J. Biochem 2001, 268 (7), 2013–2019. [DOI] [PubMed] [Google Scholar]
- 49.Scopes DA; Bautista JM; Vulliamy TJ; Mason PJ, Plasmodium falciparum Glucose-6-phosphate Dehydrogenase (G6PD) — the N-terminal Portion is Homologous to a Predicted Protein Encoded near to G6PD in Haemophilus influenzae. Mol. Microbiol 1997, 23 (4), 847–848. [DOI] [PubMed] [Google Scholar]
- 50.Clarke JL; Sodeinde O; Mason PJ, A Unique Insertion in Plasmodium berghei Glucose-6-phosphate Dehydrogenase-6-phosphogluconolactonase: Evolutionary and Functional Studies. Mol. Biochem. Parasitol 2003, 127 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- 51.Preuss J; Hedrick M; Sergienko E; Pinkerton A; Mangravita-Novo A; Smith L; Marx C; Fischer E; Jortzik E; Rahlfs S; Becker K; Bode L, High-Throughput Screening for Small-Molecule Inhibitors of Plasmodium falciparum Glucose-6-Phosphate Dehydrogenase 6-Phosphogluconolactonase. J. Biomol. Screen 2012, 17 (6), 738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shafiq N; Arshad U; Zarren G; Parveen S; Javed I; Ashraf A, A Comprehensive Review: Bio-Potential of Barbituric Acid and its Analogues. Curr. Org. Chem 2020, 24 (2), 129–161. [Google Scholar]
- 53.Meister A, Glutathione Metabolism and its Selective Modification. J. Biol. Chem 1988, 263 (33), 17205–17208. [PubMed] [Google Scholar]
- 54.Forman HJ; Zhang H; Rinna A, Glutathione: Overview of its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med 2009, 30 (1–2), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lauterburg BH; Corcoran GB; Mitchell JR, Mechanism of Action of N-acetylcysteine in the Protection against the Hepatotoxicity of Acetaminophen in Rats in Vivo. J. Clin. Invest 1983, 71 (4), 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guiguemde WA; Shelat AA; Bouck D; Duffy S; Crowther GJ; Davis PH; Smithson DC; Connelly M; Clark J; Zhu F; Jiménez-Díaz MB; Martinez MS; Wilson EB; Tripathi AK; Gut J; Sharlow ER; Bathurst I; El Mazouni F; Fowble JW; Forquer I; McGinley PL; Castro S; Angulo-Barturen I; Ferrer S; Rosenthal PJ; Derisi JL; Sullivan DJ; Lazo JS; Roos DS; Riscoe MK; Phillips MA; Rathod PK; Van Voorhis WC; Avery VM; Guy RK, Chemical Genetics of Plasmodium falciparum. Nature 2010, 465 (7296), 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jortzik E; Mailu Boniface M.; Preuss J; Fischer M; Bode L; Rahlfs S; Becker K, Glucose-6-phosphate Dehydrogenase–6-phosphogluconolactonase: A Unique Bifunctional Enzyme from Plasmodium falciparum. Biochem. J 2011, 436 (3), 641–650. [DOI] [PubMed] [Google Scholar]
- 58.Rana P; Naven R; Narayanan A; Will Y; Jones LH, Chemical Motifs that Redox Cycle and their Associated Toxicity. MedChemComm 2013, 4 (8), 1175–1180. [Google Scholar]
- 59.Johnston PA, Redox Cycling Compounds Generate H2O2 in HTS buffers Containing Strong Reducing Reagents--Real Hits or Promiscuous Artifacts? Curr. Opin. Chem. Biol 2011, 15 (1), 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lor LA; Schneck J; McNulty DE; Diaz E; Brandt M; Thrall SH; Schwartz B, A Simple Assay for Detection of Small-molecule Redox Activity. J Biomol. Screen 2007, 12 (6), 881–890. [DOI] [PubMed] [Google Scholar]
- 61.Preuss J; Maloney P; Peddibhotla S; Hedrick MP; Hershberger P; Gosalia P; Milewski M; Li YL; Sugarman E; Hood B; Suyama E; Nguyen K; Vasile S; Sergienko E; Mangravita-Novo A; Vicchiarelli M; McAnally D; Smith LH; Roth GP; Diwan J; Chung TDY; Jortzik E; Rahlfs S; Becker K; Pinkerton AB; Bode L, Discovery of a Plasmodium falciparum Glucose-6-phosphate Dehydrogenase 6-phosphogluconolactonase Inhibitor (R,Z)-N-((1-Ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide (ML276) That Reduces Parasite Growth in Vitro. J. Med. Chem 2012, 55 (16), 7262–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaminskyy D; Kryshchyshyn A; Lesyk R, Recent Developments with Rhodanine as a Scaffold for Drug Discovery. Expert Opin. Drug. Discov 2017, 12 (12), 1233–1252. [DOI] [PubMed] [Google Scholar]
- 63.Sutanto F; Konstantinidou M; Dömling A, Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem 2020, 11 (8), 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alencar N; Sola I; Linares M; Juárez-Jiménez J; Pont C; Viayna A; Vílchez D; Sampedro C; Abad P; Pérez-Benavente S; Lameira J; Bautista JM; Muñoz-Torrero D; Luque FJ, First Homology Model of Plasmodium falciparum Glucose-6-phosphate dehydrogenase: Discovery of Selective Substrate Analog-based Inhibitors as Novel Antimalarial Agents. Eur. J. Med. Chem 2018, 146, 108–122. [DOI] [PubMed] [Google Scholar]
- 65.Lu SC, Glutathione Synthesis. Biochim. Biophys. Acta Gen. Subj 2013, 1830 (5), 3143–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alkarakooly Z; Kilaparty S; Al-Anbaky Q; Khan M; Ali N, Dichloroacetic Acid (DCA)-Induced Cytotoxicity in Human Breast Cancer Cells Accompanies Changes in Mitochondrial Membrane Permeability and Production of Reactive Oxygen Species. J. Canc. Ther 2014, 5, 1234–1248. [Google Scholar]
- 67.Viayna A; Vílchez D; Vázquez J; Pérez-Benavente S; Bautista JM; Luque FJ, Searching for Selective Scaffolds against Plasmodium falciparum Glucose-6-Phosphate Dehydrogenase 6-Phosphogluconolactonase. Proc 2019, 22 (1), 28. [Google Scholar]
- 68.Haeussler K; Berneburg I; Jortzik E; Hahn J; Rahbari M; Schulz N; Preuss J; Zapol'skii VA; Bode L; Pinkerton AB; Kaufmann DE; Rahlfs S; Becker K, Glucose 6-phosphate Dehydrogenase 6-Phosphogluconolactonase: Characterization of the Plasmodium vivax Enzyme and Inhibitor Studies. Malar. J 2019, 18 (1), 22–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erlanson DA; Braisted AC; Raphael DR; Randal M; Stroud RM; Gordon EM; Wells JA, Site-Directed Ligand Discovery. Proc. Natl. Acad. Sci 2000, 97 (17), 9367–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maloney P; Hedrick M; Peddibhotla S; Hershberger P; Milewski M; Gosalia P; Li L; Preuss J; Sugarman E; Hood B; Suyama E; Nguyen K; Vasile S; Sergienko E; Salanawil S; Stonich D; Su Y; Dahl R; Mangravita-Novo A; Vicchiarelli M; McAnally D; Smith LH; Roth G; Diwan J; Chung TDY; Pinkerton AB; Bode L; Becker K, A 2nd Selective Inhibitor of Plasmodium falciparum Glucose-6-Phosphate Dehydrogenase (PfG6PDH) - Probe 2. In Probe Reports from the NIH Molecular Libraries Program, National Center for Biotechnology Information (US): Bethesda (MD), 2010. [PubMed] [Google Scholar]
- 71.Preuss J; Richardson AD; Pinkerton A; Hedrick M; Sergienko E; Rahlfs S; Becker K; Bode L, Identification and Characterization of Novel Human Glucose-6-Phosphate Dehydrogenase Inhibitors. J. Biomol. Screen 2013, 18 (3), 286–297. [DOI] [PubMed] [Google Scholar]
- 72.Torchilin VP, Passive and Active Drug Targeting: Drug Delivery to Tumors as an Example. Handb. Exp. Pharmacol 2010, (197), 3–53. [DOI] [PubMed] [Google Scholar]
- 73.Senesi S; Csala M; Marcolongo P; Fulceri R; Mandl J; Banhegyi G; Benedetti A, Hexose-6-phosphate Dehydrogenase in the Endoplasmic Reticulum. Biol. Chem 2010, 391 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- 74.Hewitt KN; Walker EA; Stewart PM, Minireview: Hexose-6-phosphate dehydrogenase and Redox control of 11{beta}-Hydroxysteroid Dehydrogenase Type 1 Activity. Endocrinology 2005, 146 (6), 2539–43. [DOI] [PubMed] [Google Scholar]
- 75.Mason PJ; Stevens D; Diez A; Knight SW; Scopes DA; Vulliamy TJ, Human Hexose-6-phosphate Dehydrogenase (Glucose 1-Dehydrogenase) Encoded at 1p36: Coding Sequence and Expression. Blood Cells Mol. Dis 1999, 25 (1), 30–7. [DOI] [PubMed] [Google Scholar]
- 76.Beutler E; Morrison M, Localization and Characteristics of Hexose 6-Phosphate Dehydrogenase (Glucose Dehydrogenase). J. Biol. Chem 1967, 242 (22), 5289–93. [PubMed] [Google Scholar]
- 77.Cossu V; Bonanomi M; Bauckneht M; Ravera S; Righi N; Miceli A; Morbelli S; Orengo AM; Piccioli P; Bruno S; Gaglio D; Sambuceti G; Marini C, Two High-rate Pentose-phosphate Pathways in Cancer Cells. Sci. Rep 2020, 10 (1), 22111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghergurovich JM; García-Cañaveras JC; Wang J; Schmidt E; Zhang Z; TeSlaa T; Patel H; Chen L; Britt EC; Piqueras-Nebot M; Gomez-Cabrera MC; Lahoz A; Fan J; Beier UH; Kim H; Rabinowitz JD, A Small Molecule G6PD Inhibitor Reveals Immune Dependence on Pentose Phosphate Pathway. Nat. Chem. Biol 2020, 16, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zamani S; Hoseini AZ; Namin AM, Glucose-6-phosphate Dehydrogenase (G6PD) Activity Can Modulate Macrophage Response to Leishmania Major Infection. Int. Immunopharmacol 2019, 69, 178–183. [DOI] [PubMed] [Google Scholar]
- 80.Tu D; Gao Y; Yang R; Guan T; Hong JS; Gao HM, The Pentose Phosphate Pathway Regulates Chronic Neuroinflammation and Dopaminergic Neurodegeneration. J. Neuroinflammation 2019, 16 (1), 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park YJ; Choe SS; Sohn JH; Kim JB, The Role of Glucose-6-phosphate Dehydrogenase in Adipose Tissue Inflammation in Obesity. Adipocyte 2017, 6 (2), 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abdullaha M; Mohammed S; Ali M; Kumar A; Vishwakarma RA; Bharate SB, Discovery of Quinazolin-4(3H)-ones as NLRP3 Inflammasome Inhibitors: Computational Design, Metal-free Synthesis, and in vitro Biological Evaluation. J. Org. Chem 2019, 84 (9), 5129–5140. [DOI] [PubMed] [Google Scholar]
- 83.Zahid A; Li B; Kombe AJK; Jin T; Tao J, Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol 2019, 10, 2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hour M-J; Huang L-J; Kuo S-C; Xia Y; Bastow K; Nakanishi Y; Hamel E; Lee K-H, 6-Alkylamino- and 2,3-Dihydro-3‘-methoxy-2-phenyl-4-quinazolinones and Related Compounds: Their Synthesis, Cytotoxicity, and Inhibition of Tubulin Polymerization. J. Med. Chem 2000, 43 (23), 4479–4487. [DOI] [PubMed] [Google Scholar]
- 85.Ceramella J; Caruso A; Occhiuzzi MA; Iacopetta D; Barbarossa A; Rizzuti B; Dallemagne P; Rault S; El-Kashef H; Saturnino C; Grande F; Sinicropi MS, Benzothienoquinazolinones as New Multi-target Scaffolds: Dual Inhibition of Human Topoisomerase I and Tubulin Polymerization. Eur. J.Med. Chem 2019, 181, 111583. [DOI] [PubMed] [Google Scholar]
- 86.Hejazi L; Rezaee E; Tabatabai SA, Quinazoline-4(3H)-one Derivatives as Novel and Potent Inhibitors of Soluble Epoxide Hydrolase: Design, Synthesis and Biological Evaluation. Bioorg. Chem 2020, 99, 103736. [DOI] [PubMed] [Google Scholar]
- 87.Ramírez-Nava EJ; Hernández-Ochoa B; Navarrete-Vázquez G; Arreguín-Espinosa R; Ortega-Cuellar D; González-Valdez A; Martínez-Rosas V; Morales-Luna L; Martínez-Miranda J; Sierra-Palacios E; Rocha-Ramírez LM; De Franceschi L; Marcial-Quino J; Gómez-Manzo S, Novel Inhibitors of Human Glucose-6-phosphate Dehydrogenase (HsG6PD) Affect the Activity and Stability of the Protein. Biochim. Biophys. Acta Gen. Subj 2021, 1865 (3), 129828. [DOI] [PubMed] [Google Scholar]
- 88.Beno BR; Yeung K-S; Bartberger MD; Pennington LD; Meanwell NA, A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem 2015, 58 (11), 4383–4438. [DOI] [PubMed] [Google Scholar]
- 89.Ortiz C; Moraca F; Medeiros A; Botta M; Hamilton N; Comini MA, Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules 2016, 21 (3), 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ortíz C; Moraca F; Laverriere M; Jordan A; Hamilton N; Comini MA, Glucose 6-Phosphate Dehydrogenase from Trypanosomes: Selectivity for Steroids and Chemical Validation in Bloodstream Trypanosoma brucei. Molecules 2021, 26 (2), 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dodds EC; Goldberg L; Lawson W; Robinson R, OEstrogenic Activity of Certain Synthetic Compounds. Nature 1938, 141 (3562), 247–248. [Google Scholar]
- 92.Jordan VC; Morrow M, Tamoxifen, Raloxifene, and the Prevention of Breast Cancer. Endocr. Rev 1999, 20 (3), 253–278. [DOI] [PubMed] [Google Scholar]
- 93.Bentrem DJ; Craig Jordan V, Tamoxifen, Raloxifene and the Prevention of Breast Cancer. Minerva Endocrinol 2002, 27 (2), 127–139. [PubMed] [Google Scholar]
- 94.Nettles KW; Bruning JB; Gil G; O'Neill EE; Nowak J; Guo Y; Kim Y; DeSombre ER; Dilis R; Hanson RN; Joachimiak A; Greene GL, Structural plasticity in the Oestrogen Receptor Ligand-binding Domain. EMBO Rep 2007, 8 (6), 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shiau AK; Barstad D; Loria PM; Cheng L; Kushner PJ; Agard DA; Greene GL, The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95 (7), 927–937. [DOI] [PubMed] [Google Scholar]
- 96.Sikder A; Chakraborty S; Rajdev P; Dey P; Ghosh S, Molecular Recognition Driven Bioinspired Directional Supramolecular Assembly of Amphiphilic (Macro)molecules and Proteins. Acc.Chem. Res 2021, 54 (11), 2670–2682. [DOI] [PubMed] [Google Scholar]
- 97.Singh J; Petter RC; Baillie TA; Whitty A, The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov 2011, 10 (4), 307–317. [DOI] [PubMed] [Google Scholar]
- 98.Bauer RA, Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies. Drug Discov. Today 2015, 20 (9), 1061–1073. [DOI] [PubMed] [Google Scholar]
- 99.Bian Y; Jun JJ; Cuyler J; Xie X-Q, Covalent Allosteric Modulation: An Emerging Strategy for GPCRs Drug Discovery. Eur. J. Med. Chem 2020, 206, 112690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM, K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503 (7477), 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ostrem JML; Shokat KM, Direct Small-molecule Inhibitors of KRAS: From Structural Insights to Mechanism-based Design. Nat. Rev. Drug Discov 2016, 15 (11), 771–785. [DOI] [PubMed] [Google Scholar]
- 102.Erlanson DA; Davis B; Jahnke W, Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol 2019, 26 1, 9–15. [DOI] [PubMed] [Google Scholar]
- 103.Erlanson DA; Wells JA; Braisted AC, Tethering: Fragment-based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct 2004, 33, 199–223. [DOI] [PubMed] [Google Scholar]
- 104.Köhler E; Barrach H; Neubert D, Inhibition of NADP+ Dependent Oxidoreductases by the 6-Aminonicotinamide Analogue of NADP+. FEBS Lett 1970, 6 (3), 225–228. [DOI] [PubMed] [Google Scholar]