

Abstract
Objective: Epilepsy is a common central nervous system disorder with pathological mechanisms including inflammation, ion channel impairment, and neurotransmitter imbalance. Despite the rapid development of current anti-epileptic drugs, epilepsy is not well controlled, so there is still a need for research on the mechanisms and new drug targets for epilepsy. CXCL14 is a member of the CXC family of chemokines, and its receptor is currently unknown. Chemokines are the third major communication mediators in the central nervous system and play a role in many diseases. Therefore, we explore the expression of CXCL14 in epilepsy and its possible mechanisms. Materials and methods: We chose the kainic acid (KA) mouse model as the epilepsy model, and studied the expression of CXCL14 in this model by western blot. Subsequently, after knocking down CXCL14, we explored the effect of CXCL14 on seizures by electrophysiology and FJB (Fluoro-Jade B) staining. Western blot and ELISA were used to explore the possible mechanism of CXCL14 affecting seizures. Results: CXCL14 expression gradually increased after a seizure until it peaked at 72 hours and then gradually decreased again. The knockdown of CXCL14 resulted in prolonged seizure latency, decreased seizure grade, and reduced degenerative necrosis of neurons in mice. Levels of GABA (γ-aminobutyric acid), GAD67 (glutamate decarboxylase 67) and GABAA receptor (γ-aminobutyric acid A receptor) were increased. Conclusion: Our results suggest that CXCL14 expression is increased after seizures and may exacerbate seizures by regulating GABA metabolism. Based on this, CXCL14 could be a new target for epilepsy treatment and antiepileptic drug development.
Keywords: CXCL14, epilepsy, chemokine, GABA, GABA metabolism, inflammation
Introduction
Epilepsy is a common, chronic central nervous system disorder that affects 70 million people worldwide [1]. It is characterized by persistent spontaneous seizures [2]. For most people with epilepsy, antiepileptic drugs are the primary form of treatment, intending to stop seizures as soon as possible without causing side effects that affect the quality of life [1]. Despite the introduction of antiepileptic drugs in recent years, a small number of patients still have difficulty controlling their seizures with drugs and eventually develop drug-refractory epilepsy [3,4]. For this reason, it is necessary to study the pathogenesis of epilepsy and discover new drug targets.
The pathologic process of seizures is associated with synaptic, receptor, and ion channel dysfunction, impaired immune system function, mitochondrial dysfunction, oxidative stress, glycogen degradation, and inflammatory responses [5]. When there is an imbalance between damaging and anti-damaging cytokines, it can lead to or further aggravate the above pathologic processes, thus exacerbating a seizure. Considerable data suggest that chemokines are the third major communication mediators in the central nervous system in addition to neurotransmitters and neuropeptides, and can act as pro-damage mediators following tissue injury and disease [6,7]. Some chemokine-chemokine receptor pairs such as CCL2-CCR2, CCL5/RANTES-CCR5 and CXCL10/IP10-CXCR3 are highly expressed in the epileptic hippocampus and are associated with exacerbation of seizures [8,9]. CXCL14, a member of the CXC chemokine family, is one of the oldest chemokines and is constitutively expressed in many regions of the brain [10], such as the cortex, basal ganglia, hippocampus, and hypothalamus [11,12]. However, CXCL14 is a chemokine with an unclear function because its receptor has not been defined [13]. Although the receptor is unclear, CXCL14 is expressed in the brain and is presumed to perform certain functions in the physiology of the brain. Several studies have confirmed that GABAergic neurons in the hippocampal dentate gyrus express CXCL14 and that synaptically released GABA (γ-aminobutyric acid) can be inhibited by CXCL14 [14,15]. Although it is well known that GABA is the main inhibitory neurotransmitter in the central nervous system and its metabolic imbalance is one of the causes of epileptic seizures [16,17], the detailed relationship between CXCL14 and epilepsy remains unclear and needs to be further explored.
Therefore, in this study, we used a kainic acid (KA)-induced mouse epilepsy model to investigate the expression of CXCL14 in epilepsy and whether it affects seizures by influencing the metabolism of GABA.
Materials and methods
Animals
To avoid the protective effect of estrogen, all experimental animals were male, weighing 20-25 g, aged 6-8 w, and were purchased from the Animal Experiment Center of North Sichuan Medical College. All animal experiments follow the guidelines of the Animal Research Committee of North Sichuan Medical College and have been approved by the North Sichuan Medical College Ethics Committee (Approval Number NSMC(A)2021(21)). During the study, experimental animals lived in a controlled environment, maintained at a constant temperature of 18-24°C with a 12-h day/night cycle and free access to water and food.
Experimental animal intervention and grouping
According to the experimental design, our animal experiments were conducted in two parts. In the first part of the experiment, we determine the expression of CXCL14 at different time points (6 h, 24 h, 72 h, 1 w, 4 w) in a mouse model of epilepsy (n = 5 per group). In the second part, we use CXCL14 adeno-associated virus (AAV) to interfere with CXCL14 in the mouse brain and next investigate whether CXCL14 affects seizures through GABA (n = 25 per group).
Acute KA-induced seizure model
The acute KA-induced seizure model is one of the classic animal epilepsy models, so we chose this model to simulate seizures. Mice were anesthetized with oxygen containing 3-4% isoflurane (RWD, Shenzhen, China), then placed on a stereotaxic apparatus (Stoelting, USA) and anesthesia was maintained intraoperatively by inhalation of oxygen containing 1-2% isoflurane through a face mask. The hair on the top of the head and the skin was cut with ophthalmic scissors, and the skull was exposed and perforated with a cranial drill (2.0 mm anterior and 1.8 mm left lateral to bregma). Then 0.3 μl KA was drawn with a 1 μl Hamilton syringe and subsequently injected at 0.06 μl/min from the previously identified bone cavity to the right hippocampus CA3 region (2.0 mm depth relative to bregma). After injection for 5 min, the needle was left for 5 min and then slowly and consistently withdrawn. Seizures occurred when mice awakened from anesthesia. Two colleagues who were unaware of the experimental subgroup rated seizure activity based on the Racine standard criteria [18].
Virus injection
We customized CXCL14 low expression adeno-associated virus and the control virus from biotechnology companies (Obio Technology Corp, Ltd) (Shanghai), named as sh-CXCL14 and sh-NC, respectively. The sh-sequence of sh-CXCL14 is GCGCAGGGTCTACGAAGAATA, which the sh-NC is CCTAAGGTTAAGTCGCCCTCG. Both sh-CXCL14 and sh-NC express the green fluorescent protein (GFP) of adeno-associated viruses, which can be used as one of the markers of successful viral transfection. Mice were randomly distributed into the injected virus and injected virus vector groups. After the above method of anesthesia, the mouse brain was fixed on the stereotaxic apparatus (Stoelting, USA), the head hair was removed, and the bone cavity was drilled. The virus was injected into the bilateral hippocampal region (bregma as origin: posterior: -2.0 mm, medial/lateral: ±1.8 mm, and ventral: -2.0 mm) using a Hamilton Microliter Syringes at a speed of 0.03 μl/min. To prevent reflux, injections were performed slowly for 10 minutes, and upon completion the needle was left in place for 10 minutes, followed by a slow and uniform withdrawal of the needle. After the injection was completed, the mice were fed for 3 weeks until the virus transfection reached its peak for subsequent experiments.
In vivo electrophysiological recordings in the hippocampus
OmniPlex® D neural data acquisition system (Plexon, Dallas, TX, USA) was used to record the local field potential in vivo mice. We implanted microwire arrays (25 μm diameter, 4×4 channels, Yisikepu, China) for recording LFPs (local field potentials) into the right hippocampus [19]. After baseline stabilization, LFP activity was recorded continuously and digitized at a frequency of 4 kHz, followed by filtering (0.1-1,000 Hz) and pre-amplification (×1,000) for 30 min. LFPs and power spectrograms were analyzed offline by Neural Explorer® (Plexon, Dallas, TX, USA). Statistical analysis and image processing were completed by using IBM SPSS Statistics (v 25.0; IBM Corporation, USA) and GraphPad Prism (v 9.0.0; La Jolla, USA).
Western blot analysis
When the predetermined time point was reached, mice brain tissue samples were immediately obtained. Brain tissue was ground in tissue lysate, and the homogenate was centrifuged in a low temperature ultracentrifuge (12,000 rpm, 15 min, 4°C), and the supernatant was collected. The total protein concentration of each sample was measured by the BCA (Bicinchoninic acid) kit (Beyotime, China). Protein samples of the same mass (30 μg) were first electrophoresed in SDS-PAGE (sodium dodecyl sulfate-polyacrylamide; 5% spacer gel; 10% separating gel) gels and then electro-transferred onto PVDF (polyvinylidene difluoride) membranes (Millipore, USA). The membranes were sealed with 8% non-fat milk powder at room temperature for 1 hour and then incubated 16 h with a primary antibody in a 4°C refrigerator. In this experiment, we used the following primary antibodies: rabbit anti-CXCL14 polyclonal antibody (NBP1-31398, 1:1000, Novus, USA), rabbit anti-GAD67 monoclonal antibody (ab108626, 1:1000, Abcam, UK), rabbit anti-GABAAR monoclonal antibody (ab252430, 1:1000, Abcam, UK), rabbit anti-α-tubulin polyclonal antibody (1:1000, Beyotime, China), rabbit anti-GAPDH monoclonal antibody (AF0001, 1:4000, Beyotime, China). After 16 hours, the membrane was washed with TBST (Tris-buffered saline with Tween-20) (10 min, 3 times) and incubated at room temperature with anti-rabbit secondary antibody (ab97051, 1:5000, Abcam, UK) for 1 hour. Visualization and gray value analysis of protein bands were performed with chemiluminescence system (VILBER, France) and ImageJ software (US National Institute of Health), respectively.
FJB (Fluoro-Jade B) staining
Mice were anesthetized with 0.1% pentobarbital, the thorax was opened to find the heart, PBS and 4% paraformaldehyde were successively perfused and the intact brain was removed and immobilized in 4% paraformaldehyde for 24 hours. After 24 hours, the brain was sequentially dehydrated with 15% and 30% sucrose solutions in a gradient, then the brain was embedded in OCT (Sakura, Japan), frozen, and cut into coronal sections of 12 μm thickness using a frozen sectioning machine (Leica Microsystems, Germany). The slices were first dried in the oven (50°C, 30 min), immersed in a NaOH solution (0.1%) for 5 min, and then soaked in 70% alcohol for 2 min. After washing, the sections were placed in potassium permanganate solution at room temperature for 10 min, the sections were again rinsed in distilled water, FJB dye (Chemicon International, USA) was added to each section, and stained in darkness for 30 min at room temperature. The sections were washed with distilled water (1 min, 3 times) after staining, dried with a hairdryer, soaked in xylene for 5 min, and then the sections were sealed with neutral balsam. We photographed hippocampal FJB-positive cells using fluorescence microscopy. FJB-positive means that the cells have degenerated and become necrotic. The excitation wavelength is 488 nm (green) and the emitted light is detected using a wavelength of 520 nm. The number of positive cells under a 40× microscope was calculated using ImageJ software.
Immunofluorescence
As described in FJB staining, we obtained slices with a thickness of 12 μm. Sections were washed with PBS (5 min, 3 times), then infiltrated with 0.4% Triton in a 37°C incubator for 25 min, repaired with citric acid, and the sections were sealed with 10% goat serum in a 37°C incubator for 1 hour. The mixed primary antibodies were covered on the sections and incubated overnight in a 4°C refrigerator. After washing the sections three times the next day, fluorescent secondary antibodies are added and incubated for 1 hour at room temperature protected from light. After washing again three times, DAPI mounting solution is added for blocking and then photographed. Primary antibodies were as follows: rabbit anti-CXCL14 polyclonal antibody (NBP1-31398, 1:500, Novus, USA); Guinea pig monoclonal anti-MAP2 (188004, 1:200, Synaptic Systems, Germany). Secondry antibodies were as follows: Alexa Fluor 488-conjugated goat anti-rabbit IgG (A0423, 1:100, Beyotime, China); Alexa Fluor 647 Goat Anti-Guinea pig H&L (ab150187, 1:200, Abcam, UK). Images were acquired by laser confocal microscopy (Olympus, Tokyo, Japan) and confocal analysis was performed by ZEN (blue-edition) and Fv31S-SW software (Olympus, Japan). The number of co-localized cells under 40× microscopy was calculated using ImageJ software.
ELISA
The concentration of γ-aminobutyric acid (GABA) in brain tissue was measured by using a commercially available Mouse γ-Aminobuytyric (GABA) Elisa Kit (H168, Nanjing Jiancheng), and a competitive method was used. The cortical and hippocampal tissues were homogenized according to the instructions before the experiment, and the supernatant was obtained. The kit was left to equilibrate at room temperature for 30 min, and the prepared samples, standards, and biotin antigen were added sequentially according to the steps in the instruction manual, and reacted at 37°C for 30 minutes. Then we washed the plate 5 times, added HRP Conjugate Reagent, and reacted at 37°C for 30 min. We washed the plate again, repeat 5 times, added Chromogen solution A and B, and reacted at 37°C for 10 min. Finally, the stop solution was added, and the Microplate Reader was used to measure the 450 nm OD value within 10 minutes.
Statistical analysis
All data were analyzed by using IBM SPSS Statistics (v 25.0; IBM Corporation, USA). Graphs were prepared with Graphpad Prism software (v 9.0.0; La Jolla, USA). The results wre expressed as mean ± SEM (standard error of mean). Differences between the experimental and control groups were compared by using Student’s T-test (two groups) or one-way analysis of variance (ANOVA) (multiple groups). P values <0.05 were considered significant (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Results
CXCL14 expression in epileptic tissues at different time points
According to the Human Protein Altas (The Human Protein Atlas), we find that CXCL14 is widely expressed in many healthy organs and moderately expressed in our most focused area-brain, not only in mouse and pig brain, but also in human brain (Figure 1B). Neuronal cytoplasm of mouse cerebral cortex expresses CXCL14 and can co-localize with MAP-2 (Microtubule-associated protein 2), a special marker of neurons (Figure 1C). Then we first examined CXCL14 protein expression in mouse cortex and hippocampus at different time points after successful seizure modeling to assess the differences with the control group. Compared to controls, we found significantly higher protein expression of CXCL14 at 72 h (P<0.05; Figure 1E, 1F) after a seizure, while there was no difference at other time points. Therefore, we chose 72 hours as the optimal time point to study seizures in knockdown CXCL14 mice.
Figure 1.
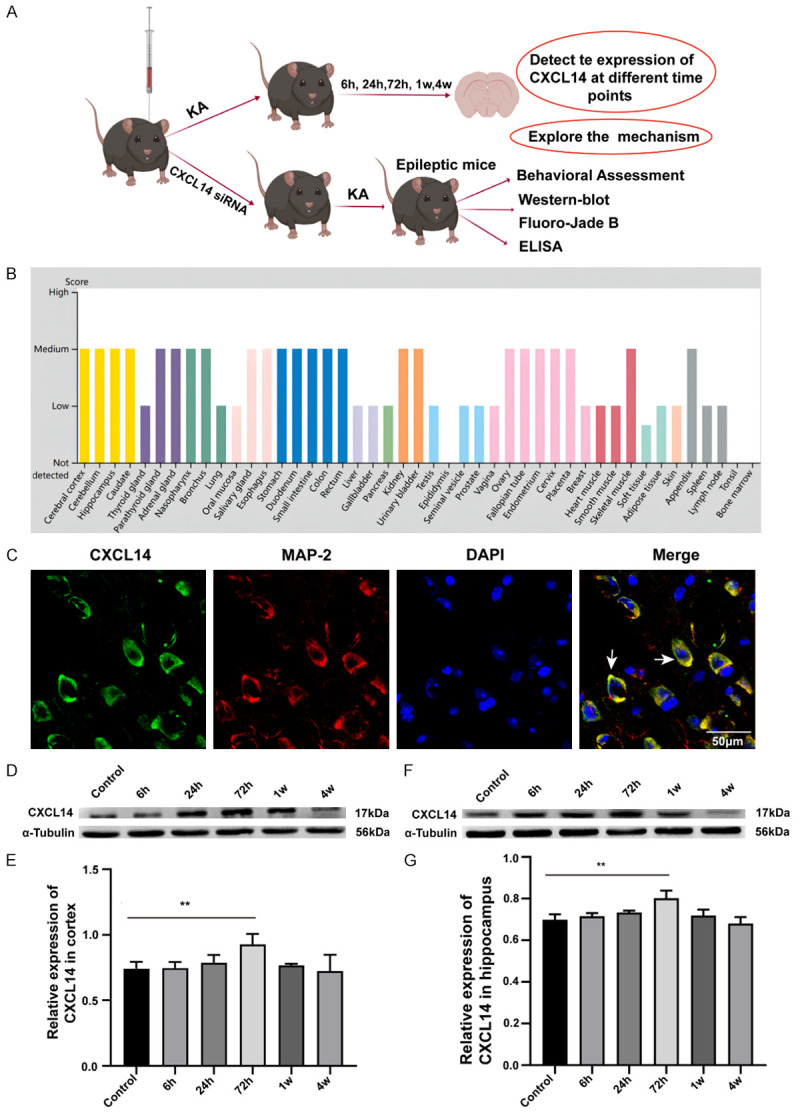
CXCL14 expression at different time points after a seizure. A. The experimental protocol diagram. B. Expression of CXCL14 in human healthy organs. CXCL14 is moderately expressed in various regions of the human brain. C. Co-localization of CXCL14 and MAP-2 in control mice cortex. Positive cells are indicated by white arrows. Scale bars: 50 µm for 400×. MAP-2, microtubule-associated protein 2. D, E. Representative western blot images of CXCL14 expression in mice cortex at different time points after seizure and quantification of intensity. F, G. Representative western blot bands of CXCL14 expression in mice hippocampus at different time points after seizure and quantification of intensity (*P<0.05, **P<0.01, means ± standard error of mean (SEM), n = 5 per group).
The efficiency of adeno-associated virus interference
3 weeks after virus injection, the expression of CXCL14 was significantly lower in the sh-CXCL14 group compared to the sh-NC group (P<0.05; Figure 2A). Meanwhile, we detected AAV-encoded self-green fluorescence in the hippocampus of mice, demonstrating successful virus transfection (Figure 2B).
Figure 2.

Expression of CXCL14 and GFP after the injection of adeno-associated virus. A, B. Western blot analysis of CXCL14 levels in the hippocampus of mice injected with adeno-associated virus (sh-CXCL14) and virus vectors (sh-NC) (**P<0.01, means ± SEM, n = 3 per group). C. Representative image shows green fluorescent protein (GFP) fluorescence in the mouse bilateral hippocampus. Scale bar = 1.6 mm.
Altered epileptic behavior and electroencephalogram in mice after CXCL14 molecular knockdown
To investigate whether CXCL14 knockout affects seizures and epileptiform discharges, behavioral tests were performed and recorded EEG at 72 hours of acute seizure onset. The results showed that compared with the sh-NC group, the seizure latency was prolonged in the sh-CXCL14 group (P<0.05; Figure 3A), and the duration of epileptic seizures above grade 3 was significantly shortened (P<0.01; Figure 3B). Seizures occurred in both the sh-NC and sh-CXCL14 groups 10 minutes after KA administration, but Racine scores were lower in the sh-CXCL14 group than in the control group, and was statistically significant in the first 40 minutes (P<0.05; Figure 3C). We recorded recurrent seizure-like events 72 hours after KA injection. As shown in Figure 3, compared to the control group, the sh-CXCL14 group exhibited lower mean energy density, which reflecting reduced energy expenditure in the brain (Figure 3D-F). In addition, the power spectral density of the sh-CXCL14 group was also lower than that of the control group (P<0.0001, Figure 3G, 3H).
Figure 3.
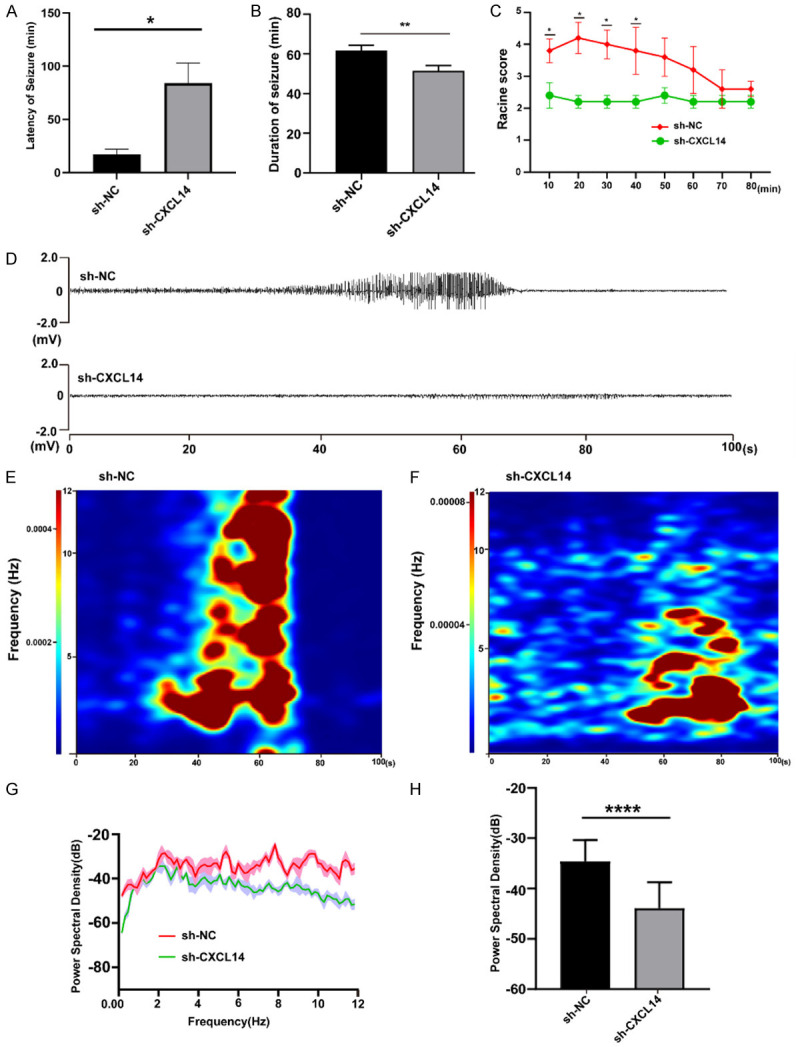
Behavioral and electrophysiological changes after CXCL14 knockdown. A. Latency period of seizures after KA injection (*P<0.05, means ± standard error of mean (SEM), n = 10 per group). B. Duration of seizure within 80 minutes (**P<0.01, means ± standard error of mean (SEM), n = 10 per group). C. Seizure rating curve within 80 minutes after seizure (*P<0.05, means ± standard error of mean (SEM), n = 10 per group). D. Traces of in vivo hippocampal electroencephalogram recordings in sh-NC and sh-CXCL14 group within 100 seconds. E, F. Heatmaps of in vivo hippocampal electroencephalogram recordings in sh-NC and sh-CXCL14 group within 100 seconds. G. Energy spectrum trend curve of the two groups. H. Statistical graph of power spectral density in sh-NC and sh-CXCL14 group (****P<0.0001, means ± standard error of mean (SEM), n = 10 per group).
CXCL14 knockdown attenuates hippocampal neuronal degenerative necrosis
We assessed the effect of CXCL14 on hippocampal neuronal necrosis after seizures by FJB staining. As shown in the figure, epilepsy causes severe neuronal necrosis in the hippocampal region. FJB-positive cells in the hippocampal region of the sh-CXCL14 group were significantly lower than those in the sh-NC group (P<0.0001; Figure 4A, 4B). Thus, the knockdown of CXCL14 attenuates neuronal degenerative necrosis caused by acute seizures. CXCL14 also reduced acute seizure-induced microglial activation in the cortex (P<0.05; Figure S1A) and hippocampus (P<0.01; Figure S1A).
Figure 4.

FJB staining of 72 hours after seizure. A. FJB-positive cells observed by fluorescence microscopy 72 hours after seizure (green; AlexaFluor-488 staining). FJB positive neurons are indicated by white arrows. Scale bars: 200 µm for 100×, 100 µm for 200×, 50 µm for 400×. B. Statistical graph of FJB positive cells in sh-NC and sh-CXCL14 groups (****P<0.0001, means ± standard error of mean (SEM), n = 3 per group).
Knockdown of CXCL14 increases GAD67, GABA, and GABAA receptor expression and attenuates seizures
Neuronal excitation and inhibition may be regulated by many different neurotransmitters and GABA is the main inhibitory neurotransmitter in the cerebral cortex [20]. GABAergic inhibition is thought to be the main brake preventing the generation and propagation of paroxysmal activity in neuronal networks. GABA production rate-limiting enzyme GAD67 (glutamate decarboxylase 67) dominates the synthesis of GABA and plays an important role after neuronal injury. Therefore, ELISA and weestern blots were used to detect the expression of GABA and GAD67, and GABAA receptors in the brain, respectively. Prior to viral intervention of CXCL14 expression, we found significantly lower levels of GABA (P<0.001, Figure 5F) at 72 hours of seizure compared to controls, as well as lower levels of GAD67 (P<0.05, Figure 5B; the image can be seen in Figure 5A left panel) and GABAA receptor (P<0.01, Figure 5D; the image can be seen in Figure 5A left panel). However, after the CXCL14 intervention, we can see that the contents of GABA (P<0.01, Figure 5G), GAD67 (P<0.05, Figure 5C, the image can be seen in Figure 5A right panel), and GABAA receptor (P<0.01, Figure 5E, the image can be seen in Figure 5A right panel) in the sh-CXCL14 group were significantly higher than those of the sh-NC group. Our results suggest that the metabolism of GABA may be related to the expression of CXCL14.
Figure 5.
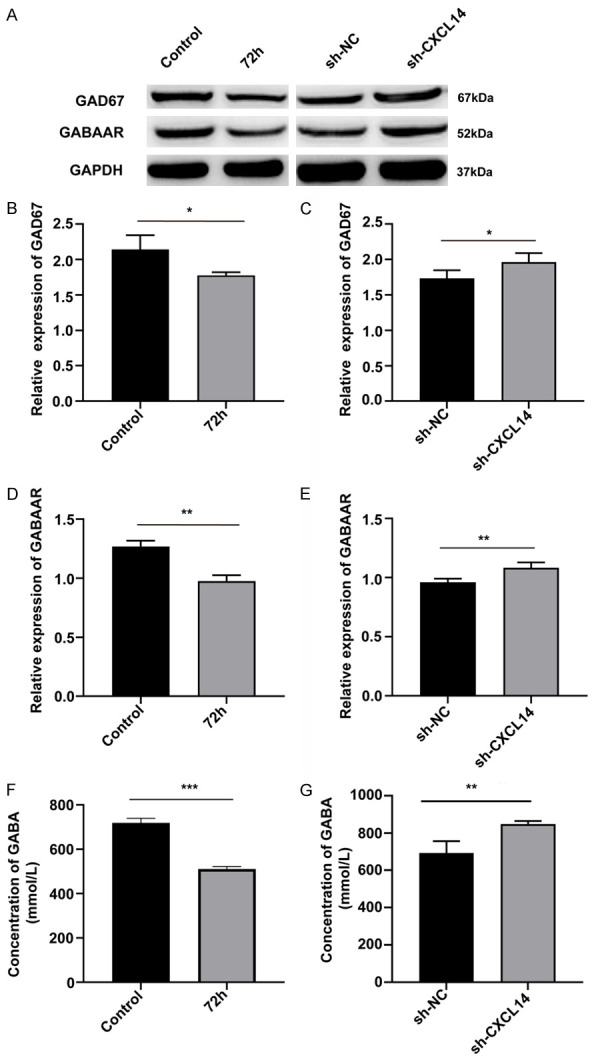
The knockdown of CXCL14 can increase the content of GAD67, GABAAR and GABA. To explore the effect of CXCL14 on GABA metabolism after seizures, we examined the expression of GAD67, GABAAR and GABA before and after knockdown of CXCL14. A. Left panel: Representative western blot images of GAD67, GABAAR before CXCL14 knockdown. Right panel: Representative western blot images of GAD67, GABAAR after CXCL14 knockdown. B, C. GAD67 expression quantification of intensity before/after CXCL14 knockdown (*P<0.05, means ± standard error of mean (SEM), n = 3 per group). D, E. GABAAR expression quantification of intensity before/after CXCL14 knockdown (**P<0.01, means ± standard error of mean (SEM), n = 3 per group). F, G. GABA levels in the brain before/after CXCL14 knockdown (***P<0.001, **P<0.01 means ± standard error of mean (SEM), n = 6 per group).
Discussion
We demonstrated that CXCL14 expression is upregulated in epilepsy and may influence seizures by regulating GABA metabolism. CXCL14 was first identified from mammary gland and kidney tissues, is highly conserved in vertebrates, and is widely expressed in normal tissues, as well as in some pathologic tissues [21]. The current study suggested that CXCL14 has the ability to promote the renewal of Langerhans cells in the skin, inhibit angiogenesis, and promote or inhibit tumor growth [22,23]. We found in epileptic tissues at different time points (6 h, 24 h, 72 h, 1 w, 4 w) that CXCL14 levels in brain tissue gradually increased after seizures, peaked at 72 hours, and then gradually decreased during the chronic phase (Figure 1). This may suggest that CXCL14, like some other chemokines such as CXCL2 and CXCL13, may play a role in seizures [24-26]. Therefore, we inhibited CXCL14 expression in the mouse brain with adeno-associated virus and found that the seizure latency was prolonged and the duration of seizures was reduced in the sh-CXCL14 group of mice, as well as the seizure class. The field potential results indicated that the sh-CXCL14 group seemed to have fewer abnormal seizure waves and less energy expenditure in the brain (Figure 3). The FJB experiment also confirmed that the sh-CXCL14 group had fewer necrotic neurons in the CA3 region of the hippocampus (Figure 4). In addition, CXCL14 is expressed in the cell membrane of neurons (Figure 1). Based on this, it can be concluded that the increased expression of CXCL14 in epileptic tissues aggravates seizures and the degenerative necrosis of neurons after seizures.
Present studies suggested that the receptor for CXCL14 is obscure, but it was previously reported that CXCL14 is thought to have high affinity for binding to G protein-coupled receptors such as CXCR4 and CXCR7 [27,28]. Hippocampal GABAergic and inhibitory neurons both can express CXCL14 [11]. Interestingly, possibly because of its high affinity for CXCR4, CXCL14 has a completely opposite effect to CXCL12, which amplifies the effects of GABA released in GABAergic neurons, stimulates the release of glutamate inastrocytes, and affects synaptic activity inneurons, in addition to reducing calcium oscillations in hippocampal neurons [14,29]. In contrast, in GABAergic neurons, CXCL14 suppressed the tonic and phasic effects of synaptically released GABA [11]. It has been shown that CXCL14 induces intracellular signal transduction and increases neuronal excitability through G protein-coupled cell surface receptors [30].
Gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the cerebral cortex, discovered in 1950, and plays a major inhibitory role in 20% to 44% of cortical neurons [31]. Within the epileptic brain, GABAergic neurons in the cortex are reduced, and GABA concentrations in the hippocampus and cerebrospinal fluid are decreased [32]. Our study confirmed this, with significantly lower levels of GABA 72 hours after seizure compared to mice without seizures. In addition to playing a major inhibitory role in epilepsy, GABA also affects the maturation of neural stem cells during nervous system development, as well as the maturation of the subventricular zone (SVZ) and subgranular zone (SGZ) of the adult hippocampus [33,34]. GABA metabolism begins with α-ketoglutarate, an intermediate product of the tricarboxylic acid cycle, which produces L-glutamate through glutamate dehydrogenase or transaminase, then L-glutamate converted to GABA by the rate-limiting enzyme glutamic acid decarboxylase (GAD67 and GAD65) and released into the synaptic gap to act on pre- or postsynaptic GABAA receptors and GABAB receptors (γ-aminobutyric acid B receptor) [35]. GABA content is influenced by a variety of factors in the metabolic process, and when abnormalities in GABA synthesis, release, uptake and GABA receptors occur, the balance of neuronal electrical activity is altered, leading to the development of epilepsy [36]. In vitro experiments have proven that activation of the CXCR4 signaling pathway by CXCL12 induces GAD67 expression in embryonic hippocampal neurons [37]. Since CXCL14 has completely opposite effects to CXCL12, it is speculated that CXCL14 may inhibit the expression of GAD67 in neurons. Besides, the vast majority of neurons expressing CXCL14 in the hippocampal DG region also express GAD [11]. In our study, before CXCL14 intervention, the levels of GABA, GABAA receptor, and GAD67 were all decreased compared to the mice without seizures. Then, we further verified that there was increased expression of GAD67 in the cerebral cortex and hippocampus of mice in the sh-CXCL14 group compared to the sh-NC group. Along the metabolic process of GABA, we found that the content of GABA and GABAA receptor was also increased. GABA performs most of its functions through GABAA receptors, and the α1 subunit we examined is the most abundant subunit, expressed in almost all parts of the brain [38]. Therefore, these results demonstrate that CXCL14 may play a role in the metabolism of GABA. As we know that seizures can be suppressed by increasing GABA levels in the brain, and also by suppressing GABA levels and thus promoting seizures [39-41]. Thus, CXCL14 may be a new target for epilepsy treatment by regulating GABA.
Limitations
This study has some limitations that may need to be addressed by further research. Our findings suggest that CXCL14 may exacerbate seizures by inhibiting the production pathway of GABA, but we did not study glutamate aminotransferase, succinic semialdehyde dehydrogenase, and the final metabolite succinic semialdehyde, which are required for the GABA uptake pathway [42]. The effect of CXCL14 on the GABA uptake pathway still deserves further investigation. Secondly, we only explored the effect of CXCL14 on the acute phase of epilepsy, not on the chronic phase, and our experimental results suggest that CXCL14 may be reduced in expression during the chronic phase of epilepsy, but the exact role remains uncertain. Thirdly, we explored only the effect of low expression of CXCL14 on seizures in this article, but to elaborate on the role of CXCL14 in epilepsy, we should explore the effect of overexpression of CXCL14 on seizures.
Conclusion
Our results confirm that CXCL14 may exacerbate seizures by inhibiting the GABA production pathway and, to our knowledge, this is the first article to investigate the role of CXCL14 in epilepsy. Our results further confirm the role of chemokines in epilepsy and their possible role as one of the molecules that cause seizures. In addition, CXCL14 may serve as a new target for the development of new epilepsy drugs, providing a fundamental theoretical basis for the development of new drugs and the search for new biomarkers.
Acknowledgements
This work was supported by funding from Nanchong City School Science and Technology Strategic Cooperation Special (No. 20SXQT0259 and No. 19SXHZ0110).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Thijs RD, Surges R, O’Brien TJ, Sander JW. Epilepsy in adults. Lancet. 2019;393:689–701. doi: 10.1016/S0140-6736(18)32596-0. [DOI] [PubMed] [Google Scholar]
- 2.Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA, Glynn M, Hesdorffer DC, Lee BI, Mathern GW, Moshé SL, Perucca E, Scheffer IE, Tomson T, Watanabe M, Wiebe S. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–482. doi: 10.1111/epi.12550. [DOI] [PubMed] [Google Scholar]
- 3.Billakota S, Devinsky O, Kim KW. Why we urgently need improved epilepsy therapies for adult patients. Neuropharmacology. 2020;170:107855. doi: 10.1016/j.neuropharm.2019.107855. [DOI] [PubMed] [Google Scholar]
- 4.Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657–678. doi: 10.1111/j.1528-1167.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 5.He LY, Hu MB, Li RL, Zhao R, Fan LH, He L, Lu F, Ye X, Huang YL, Wu CJ. Natural medicines for the treatment of epilepsy: bioactive components, pharmacology and mechanism. Front Pharmacol. 2021;12:604040. doi: 10.3389/fphar.2021.604040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler MW, Geller EB, Chen X, Rogers TJ. Viewing chemokines as a third major system of communication in the brain. AAPS J. 2006;7:E865–870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerri C, Caleo M, Bozzi Y. Chemokines as new inflammatory players in the pathogenesis of epilepsy. Epilepsy Res. 2017;136:77–83. doi: 10.1016/j.eplepsyres.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 8.van Gassen KL, Netzeband JG, de Graan PN, Gruol DL. The chemokine CCL2 modulates Ca2+ dynamics and electrophysiological properties of cultured cerebellar Purkinje neurons. Eur J Neurosci. 2005;21:2949–2957. doi: 10.1111/j.1460-9568.2005.04113.x. [DOI] [PubMed] [Google Scholar]
- 9.Sanz P, Garcia-Gimeno MA. Reactive Glia inflammatory signaling pathways and epilepsy. Int J Mol Sci. 2020;21:4096. doi: 10.3390/ijms21114096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz RD. The GABAA receptor-gated ion channel: biochemical and pharmacological studies of structure and function. Biochem Pharmacol. 1988;37:3369–3375. doi: 10.1016/0006-2952(88)90684-3. [DOI] [PubMed] [Google Scholar]
- 11.Banisadr G, Bhattacharyya BJ, Belmadani A, Izen SC, Ren D, Tran PB, Miller RJ. The chemokine BRAK/CXCL14 regulates synaptic transmission in the adult mouse dentate gyrus stem cell niche. J Neurochem. 2011;119:1173–1182. doi: 10.1111/j.1471-4159.2011.07509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto T, Yamashita A, Yamada K, Hata R. Immunohistochemical localization of chemokine CXCL14 in rat hypothalamic neurons. Neurosci Lett. 2011;487:335–340. doi: 10.1016/j.neulet.2010.10.051. [DOI] [PubMed] [Google Scholar]
- 13.Frederick MJ, Henderson Y, Xu X, Deavers MT, Sahin AA, Wu H, Lewis DE, El-Naggar AK, Clayman GL. In vivo expression of the novel CXC chemokine BRAK in normal and cancerous human tissue. Am J Pathol. 2000;156:1937–1950. doi: 10.1016/S0002-9440(10)65067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya BJ, Banisadr G, Jung H, Ren D, Cronshaw DG, Zou Y, Miller RJ. The chemokine stromal cell-derived factor-1 regulates GABAergic inputs to neural progenitors in the postnatal dentate gyrus. J Neurosci. 2008;28:6720–6730. doi: 10.1523/JNEUROSCI.1677-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge S, Pradhan DA, Ming GL, Song H. GABA sets the tempo for activity-dependent adult neurogenesis. Trends Neurosci. 2007;30:1–8. doi: 10.1016/j.tins.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Trevelyan AJ, Schevon CA. How inhibition influences seizure propagation. Neuropharmacology. 2013;69:45–54. doi: 10.1016/j.neuropharm.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Abdullahi AT, Adamu LH. Neuronal network models of epileptogenesis. Neurosciences (Riyadh) 2017;22:85–93. doi: 10.17712/nsj.2017.2.20160455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 19.Kase D, Inoue T, Imoto K. Roles of the subthalamic nucleus and subthalamic HCN channels in absence seizures. J Neurophysiol. 2012;107:393–406. doi: 10.1152/jn.00937.2010. [DOI] [PubMed] [Google Scholar]
- 20.Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42(Suppl 3):8–12. doi: 10.1046/j.1528-1157.2001.042suppl.3008.x. [DOI] [PubMed] [Google Scholar]
- 21.Liu M, Zhang SB, Luo YX, Yang YL, Zhang XZ, Li B, Meng Y, Chen YJ, Guo RX, Xiong YC, Xin WJ, Li D. NFATc2-dependent epigenetic upregulation of CXCL14 is involved in the development of neuropathic pain induced by paclitaxel. J Neuroinflammation. 2020;17:310. doi: 10.1186/s12974-020-01992-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaerli P, Willimann K, Ebert LM, Walz A, Moser B. Cutaneous CXCL14 targets blood precursors to epidermal niches for Langerhans cell differentiation. Immunity. 2005;23:331–342. doi: 10.1016/j.immuni.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Westrich JA, Vermeer DW, Colbert PL, Spanos WC, Pyeon D. The multifarious roles of the chemokine CXCL14 in cancer progression and immune responses. Mol Carcinog. 2020;59:794–806. doi: 10.1002/mc.23188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu H, Zhu T, Gong L, Zhao Y, Shao Y, Li S, Sun Z, Ling Y, Tao Y, Ying Y, Lan C, Xie Y, Jiang P. Transient receptor potential melastatin 2 contributes to neuroinflammation and negatively regulates cognitive outcomes in a pilocarpine-induced mouse model of epilepsy. Int Immunopharmacol. 2020;87:106824. doi: 10.1016/j.intimp.2020.106824. [DOI] [PubMed] [Google Scholar]
- 25.Kim JE, Ryu HJ, Yeo SI, Kang TC. P2X7 receptor regulates leukocyte infiltrations in rat frontoparietal cortex following status epilepticus. J Neuroinflammation. 2010;7:65. doi: 10.1186/1742-2094-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, Liu X, Xiong H, Wang W, Liu Y, Yin L, Tu C, Wang H, Xiang X, Xu J, Duan B, Tao A, Zhao Z, Mei Z. CXCL13/CXCR5 signaling contributes to diabetes-induced tactile allodynia via activating pERK, pSTAT3, pAKT pathways and pro-inflammatory cytokines production in the spinal cord of male mice. Brain Behav Immun. 2019;80:711–724. doi: 10.1016/j.bbi.2019.05.020. [DOI] [PubMed] [Google Scholar]
- 27.Lu J, Chatterjee M, Schmid H, Beck S, Gawaz M. CXCL14 as an emerging immune and inflammatory modulator. J Inflamm (Lond) 2016;13:1. doi: 10.1186/s12950-015-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sand LG, Scotlandi K, Berghuis D, Snaar-Jagalska BE, Picci P, Schmidt T, Szuhai K, Hogendoorn PC. CXCL14, CXCR7 expression and CXCR4 splice variant ratio associate with survival and metastases in Ewing sarcoma patients. Eur J Cancer. 2015;51:2624–2633. doi: 10.1016/j.ejca.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 29.Ohshima Y, Kubo T, Koyama R, Ueno M, Nakagawa M, Yamashita T. Regulation of axonal elongation and pathfinding from the entorhinal cortex to the dentate gyrus in the hippocampus by the chemokine stromal cell-derived factor 1 alpha. J Neurosci. 2008;28:8344–8353. doi: 10.1523/JNEUROSCI.1670-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hydén H, Rapallino MV, Cupello A. Unraveling of important neurobiological mechanisms by the use of pure, fully differentiated neurons obtained from adult animals. Prog Neurobiol. 2000;60:471–499. doi: 10.1016/s0301-0082(99)00035-0. [DOI] [PubMed] [Google Scholar]
- 32.Petroff OA. GABA and glutamate in the human brain. Neuroscientist. 2002;8:562–573. doi: 10.1177/1073858402238515. [DOI] [PubMed] [Google Scholar]
- 33.Ben-Ari Y, Holmes GL. The multiple facets of gamma-aminobutyric acid dysfunction in epilepsy. Curr Opin Neurol. 2005;18:141–145. doi: 10.1097/01.wco.0000162855.75391.6a. [DOI] [PubMed] [Google Scholar]
- 34.Bordey A. Enigmatic GABAergic networks in adult neurogenic zones. Brain Res Rev. 2007;53:124–134. doi: 10.1016/j.brainresrev.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Kim HY, Suh PG, Kim JI. The role of phospholipase C in GABAergic inhibition and its relevance to epilepsy. Int J Mol Sci. 2021;22:3149. doi: 10.3390/ijms22063149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akyuz E, Polat AK, Eroglu E, Kullu I, Angelopoulou E, Paudel YN. Revisiting the role of neurotransmitters in epilepsy: an updated review. Life Sci. 2021;265:118826. doi: 10.1016/j.lfs.2020.118826. [DOI] [PubMed] [Google Scholar]
- 37.Luo Y, Lathia J, Mughal M, Mattson MP. SDF1alpha/CXCR4 signaling, via ERKs and the transcription factor Egr1, induces expression of a 67-kDa form of glutamic acid decarboxylase in embryonic hippocampal neurons. J Biol Chem. 2008;283:24789–24800. doi: 10.1074/jbc.M800649200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jembrek MJ, Vlainic J. GABA receptors: pharmacological potential and pitfalls. Curr Pharm Des. 2015;21:4943–4959. doi: 10.2174/1381612821666150914121624. [DOI] [PubMed] [Google Scholar]
- 39.Curtis DR, Felix D, McLellan H. GABA and hippocampal inhibition. Br J Pharmacol. 1970;40:881–883. doi: 10.1111/j.1476-5381.1970.tb10663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- 41.Khazipov R. GABAergic synchronization in epilepsy. Cold Spring Harb Perspect Med. 2016;6:a022764. doi: 10.1101/cshperspect.a022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarasa SB, Mahendran R, Muthusamy G, Thankappan B, Selta DRF, Angayarkanni J. A brief review on the non-protein amino acid, Gamma-amino Butyric Acid (GABA): its production and role in microbes. Curr Microbiol. 2020;77:534–544. doi: 10.1007/s00284-019-01839-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.