

Abstract
The coevolution of vertebrate and mammalian hosts with DNA viruses has driven the ability of host cells to distinguish viral from cellular DNA in the nucleus to induce intrinsic immune responses. Concomitant viral mechanisms have arisen to inhibit DNA sensing. At this virus-host interface, emerging evidence links cytokine responses and cellular homeostasis pathways, particularly the DNA damage response (DDR). Nuclear DNA sensors, such as the Interferon (IFN)-γ Inducible Protein 16 (IFI16), functionally intersect with the DDR regulators Ataxia Telangiectasia Mutated (ATM) and DNA-Dependent Protein Kinase (DNA-PK). Here, we discuss accumulating knowledge for the DDR-innate immunity signaling axis. Through the lens of this infection-driven signaling axis, we present host and viral molecular strategies acquired to regulate autoinflammation and antiviral responses.
Keywords: DNA Virus Infection, DNA Damage Response, Nuclear DNA Sensing, Interferon, Antiviral Response
The Emerging Interface between Intrinsic Immune Signaling and the DDR
Cytokine signaling has arisen from eons of co-evolution between vertebrates and the pathogens that infect them [1, 2]. Given that autoinflammation can damage the host, these responses are tightly regulated and are stimulated by detection of pathogen associated molecular patterns via pattern recognition receptors. Intrinsic immune signaling may also be induced downstream of self-derived, homeostatic stresses, such as DNA damage [3]. It is increasingly apparent that viral pathogens that establish life-long infections are sensed by some of the same cellular mechanisms that survey self-derived triggers of inflammation [4]. One example is when herpesviruses (see glossary) infect the nervous system. The understanding that antiviral cytokine responses are similarly elicited by normal host processes, such as DNA repair, presents a conceptual challenge for the host. How can a cell mount a cytokine response that is sufficient to suppress a pathogen or communicate cellular stress, but that is not so powerful as to inadvertently destroy host tissue? As a reciprocal challenge to pathogens, how do they dismantle cellular defenses to promote infection?
DNA damage response (DDR) pathways are increasingly linked to cytokine signaling [5, 6]. Double stranded DNA breaks (DSBs), a severe form of DNA damage, are a frequent source of cellular stress that can arise both endogenously (e.g., DNA replication) [7] and exogenously (e.g., radiation) (Fig 1). DSB repair occurs primarily via homology directed repair (HDR) and nonhomologous end joining (NHEJ), which are governed respectively by the ATM (ataxia telangiectasia mutated) and DNA-PK (DNA-dependent protein kinase) kinases (see glossary) [8]. These repair processes and kinases are appreciated already for their role in adaptive immunity for development of the T- and B- cell repertoire [9]. More recent evidence shows that DSBs can ignite cytokine responses that are directly downstream of ATM and DNA-PK. These responses seem to mirror those initiated by detection of nuclear replicating DNA viruses. Of interest is how inflammatory responses are regulated downstream of DDR kinase activation. The concept that the DDR contributes to cytokine responses is further complicated by observations that DDR activation is often necessary for DNA virus infection. Thus, several questions await explanation as we consider the interplay between the DDR and inflammatory signaling in the context of viral infection and cellular homeostasis.
Figure 1: The DNA damage response and DSB repair.
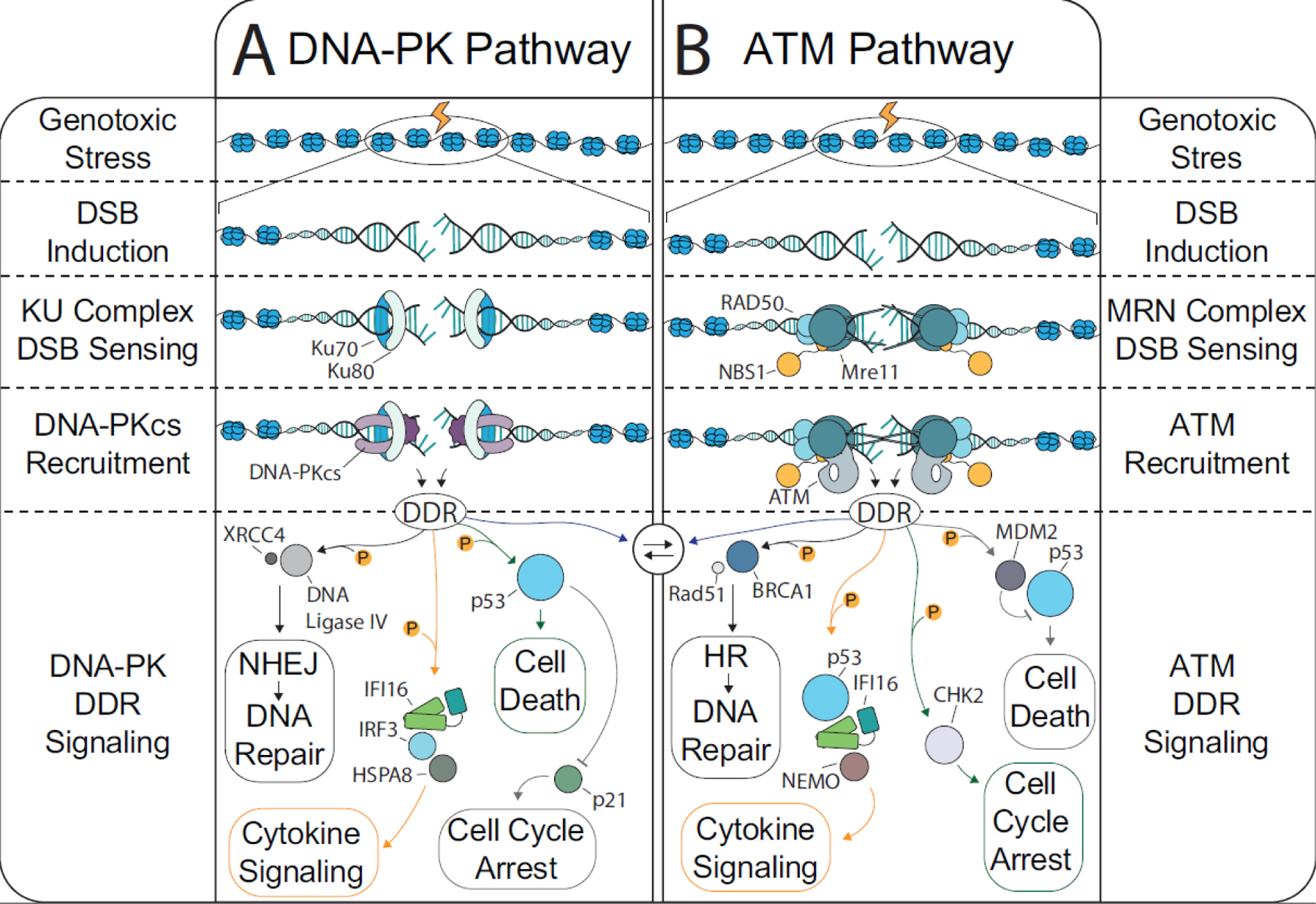
DSBs arise from endogenous (metabolism, replication fork stalling, transcription, etc.) and exogenous (irradiation, chemicals that inhibit replication proteins, drugs that directly target DNA, etc.) sources. DSBs are primarily repaired by one of four mechanisms: NHEJ, HDR, alternative- EJ, or single strand annealing. The predominant forms of DSB repair, NHEJ and HDR, are induced following a cascade of protein interactions. First, the DSB is detected by the KU (KU70 and KU80) and MRN (Mre11, Rad50, Nbs1) complexes. Afterward, the cell must decide which repair pathway to favor. A) Most of the DSB repair in the body occurs in quiescent cells where NHEJ is the preferred pathway. The KU complex recruits the catalytic subunit of the DNA-PK holoenzyme, which triggers the kinase activity of the DNA-PK complex. The DNA-PK derived DDR can trigger DNA repair, cell death, cell cycle arrest, and interferon signaling; however, the protein players involved in these processes are distinct from that involved in HDR. B) When the cell has multiple copies of DNA available, such as in S Phase, the MRN complex will trigger ATM kinase activity and induce the DDR. There are several cellular mechanisms in place to bias DNA repair either toward HDR or toward NHEJ. For example, when 53BP1 is recruited by RNF8 and RNF168 to a DSB to block end resection and thus block strand invasion, thereby promoting NHEJ. Nevertheless, there is also substantial crosstalk between ATM and DNA-PK during the DDR, particularly when end-trimming is required for NHEJ.
Extranuclear Viral DNA Sensing
DNA viruses often traffic to the nucleus to complete steps of the viral replication cycle. One advantage of nuclear viral replication is that cellular factors necessary for DNA virus gene expression, RNA metabolism, and DNA synthesis are already present within this cellular compartment. Until recently, nuclear residence of the viral genome was also viewed as an evasion strategy, as cellular mechanisms of nuclear DNA sensing were not yet known. For example, polyomavirus and papillomavirus trafficking through the secretory system bypasses cytosolic sensing of the viral DNA prior to nuclear entry (see glossary) [10]. The relatively diverse herpesvirus family has evolved a cadre of tegument associated proteins that accompany the viral capsid into the cell to immediately shut down host responses [11, 12].
From the host cell perspective, pattern recognition receptor activation is often regulated in a localization-dependent manner, thereby providing a host mechanism that allows detecting pathogenic moieties while avoiding autoinflammation. For example, approximately two decades ago it started to be understood that Toll-like receptor 9 (TLR9) detects foreign DNA in the endosome, which should be devoid of cellular DNA [13] (Fig 2). It was not until the past decade that cytosolic DNA sensors have started to be characterized, such as cyclic GMP-AMP Synthase (cGAS) [14, 15]. The cGAS-Stimulator of Interferon [IFN] Genes (STING) pathway (Fig 3A) is now well appreciated as critical for host defense response and survival following infection with nuclear-replicating DNA viruses, such as herpesviruses [16–18]. STING serves as a centralized hub to integrate the signals from multiple DNA sensors and trigger cytokine expression (Fig 2 and Fig 3). Absent in Melanoma 2 (AIM2), another cytosolic DNA sensor, is a member of the PYHIN family (see glossary) of proteins that, upon detection of DNA, triggers the formation of a caspase-1 activating inflammasome [19] (Fig 2). There are also several canonically nuclear proteins that, when localized to the cytosol, support DNA sensing upstream of STING. These proteins include DNA repair enzymes, such as DNA-PK [20], Rad50 [21], and Meiotic Recombination Protein 11 (Mre11) [22], and the RNA processing enzymes RNA Polymerase III (RNAPIII) [23] and Probable ATP-Dependent RNA Helicase 41 (DDX41) [24] (Fig 2). The capacity of these factors to contribute to cytokine induction while localized to the cytoplasm is thought to spatially separate functions to control inflammation versus other biological roles these proteins fulfill. For example, in addition to its role in sensing DSBs in the nucleus, KU70 also binds foreign DNA in the cytosol and stimulates IFN Regulatory Factor (IRF) 1 and IRF7 to produce IFN-λ via STING [25, 26]. Until recently, it was not well understood what, if any, contribution these factors have for sensing nuclear viral DNA during infection.
Figure 2: DNA sensing throughout the cell.

A) The detection of self-derived DNA and of extranuclear viral DNA can be mediated by any of several protein players: cGAS, Aim2, KU70/80, Mre11/Rad50, DNA-PK, TLR9, and others. After detecting foreign or abnormal DNA, these sensors can trigger inflammatory cytokine expression. There is mounting evidence of crosstalk between the DDR and the DNA sensors throughout the cell. Both ATM and DNA-PK can interact with IFI16 to trigger Type-I IFN responses. There is also evidence that the DDR can antagonize other nucleic acid sensors such as AIM2, cGAS, and hnRNPA2/B1 and vice-versa. B) Outside of the nucleus, the predominate triggers of DNA sensing are from endogenous sources (mitochondrial DNA or damaged nuclear DNA), bacterial DNA, DNA viruses that replicate in the cytosol (e.g., poxvirus), or from replication products encountered during the entry or egress of nuclear-replicating DNA viruses. Several cytosolic DNA sensors (cGAS, KU70/80, etc.) also have a role in nuclear DNA sensing. For simplicity, other DNA sensors that reside outside of the nucleus have not been depicted (e.g. RNAPIII, DDX41, and cytosolic IFI16). C) Several DNA sensors exist within the nucleus, being able to distinguish foreign from host DNA. The nuclear DNA sensors include IFI16, IFIX, and hnRNPA2/B1. Moreover, endogenous triggers of stress, like DNA damage, can activate ATM or DNA-PK to induce cytokine expression. There is crosstalk between the DDR and DNA sensors that can amplify or restrict innate immune signaling. For example, hnRNPA2/B1 and DNA-PK may negatively regulate each other.
Figure 3: STING- and IFI16-dependent intrinsic signaling pathways.

A) cGAS is the synthase for cyclic GMP-AMP, which serves as a messenger to activate STING (reviewed in [107]). Upon its activation, STING localizes from the cytosolic face of ER tubules to the Golgi. Thereafter, STING recruits TBK1 to phosphorylate IRF3, a transcription factor. B) Phosphorylated IRF3 dimerizes and translocates to the nucleus to trigger Type-I interferon responses via interaction with CBP/p300. C) STING activates NF-κB to further diversify the cytokine repertoire. D) Multiple routes can activate STING. Bypassing the requirement for STING translocation, recent evidence points to its activation by K63-linked ubiquitination at K150 [108]. Although not depicted, lysosomal targeting of STING, such as via interaction with Neimann-Pick type C1 (NPC1), can regulate STING responses [109]. E) IFI16 is a member of the PYHIN family of proteins that bind to DNA and then self-oligomerize via their HIN-200 and PYRIN domains, respectively. Quiescently, IFI16 resides predominantly in the nucleolus and nucleoplasm. Upon viral genome deposition, IFI16 rapidly localizes to the nuclear periphery (reviewed in [67]). IFI16 undergoes oligomeric assembly on the viral DNA. F) At the viral genome, IFI16 and DNA-PK participate in a feedback loop [5]. IFI16 recruits and/or activates DNA-PK, which in turn phosphorylates IFI16 to promote interferon expression. G) In the context of DNA damage, ATM can trigger IFI16 nuclear export with TRAF6 and p53 to induce STING ubiquitination and subsequent NF-κB activation [6].
Intranuclear Viral DNA Sensing
Several intrinsic sensors that detect foreign DNA in the nucleus have now been described—IFI16 (see glossary), IFIX, and Heterogeneous Nuclear Ribonuclear protein A2/B1 (hnRNPA2/B1) (Fig 2). By way of contrast to the cytosolic sensors discussed above, nuclear DNA sensors are localized in a compartment that is rich in a host-derived inflammatory substrate. These sensors also do not necessarily have a substrate specificity that allows them to intrinsically distinguish foreign from self-DNA. In fact, each of the described nuclear DNA sensors is capable of sequence-independent binding to viral DNA [27]. Additionally, each known sensor participates in multiple cellular pathways beyond their capacity to trigger cytokine expression. We now understand that IFI16 contributes to STING-dependent and possibly STING-independent cytokine responses following a diverse array of virus infections (Box #1) (Fig 3A). Prior to this discovery, IFI16 was found to modulate transcription and cell cycle progression via interactions with Tumor Protein 53 (p53) and Breast Cancer Type I Susceptibility Protein (BRCA1) [28, 29]. Further IFI16 protein interaction studies have uncovered extensive involvement with transcription regulation machinery, DNA repair proteins, and cell cycle modulators [30–33]. Another member of the PYHIN family, IFIX [32, 34, 35], is similarly linked to cell cycle progression [34] and colocalizes to sites of DNA damage [36]. Like IFI16, IFIX is also important for propagation of inflammatory signals after herpesvirus infection [32]. Finally, hnRNPA2/B1 was first noted for its contribution to RNA processing, and only later shown to mediate cytokine responses to nuclear viral infection [37]. Specifically, hnRNPA2/B1 binds to Herpes simplex virus 1 (HSV-1) DNA in the nucleus, then dimerizes and traffics to the cytosol to induce TANK-Binding Kinase 1 (TBK1) activation and interferon expression.
Side Boxes:
Box 1: IFI16 mediated nuclear sensing of viral DNA
The link between IFI16 and the antiviral response is also now clearly established; IFI16 null cell lines display decreased cytokine and interferon responses after viral infection. IFI16 responds to infections with a wide array of viral families including herpesviruses (HSV-1 [110, 111], KSHV [112], EBV [113, 114], and HCMV[74, 115]), DNA tumor viruses (polyoma-, Papilloma- and adeno- virus) [116, 117], as well as HIV-1 [118, 119]. Furthermore, IFI16 is linked to a number of autoimmune disorders, including SLE and Sjögren’s syndrome [99], highlight the need for negative regulation that prevents hyperactivation. The biochemical mechanism of IFI16 intrinsic signaling is incredibly dynamic. IFI16 is a member of the PYHIN family of proteins [19], which bind to DNA via their HIN-200 domains and then self-oligomerize via PYRIN domains [75, 120]. IFI16 oligomerization is regulated by positively charged residues within the pyrin domain [75] and underlies the capacity of IFI16 to contribute to the antiviral response. Upon binding to viral DNA, IFI16 enhances DNA-PK activation at the nuclear periphery. In turn, DNA-PK phosphorylates IFI16 at T149, a site located in a hinge region between the PYRIN and the first HIN-200 domain that contains the NLS. Phosphorylation and acetylation of residues within this region can control IFI16 subcellular localization via nuclear export. T149 phosphorylation, specifically, promotes Type-I IFN responses. As infection progresses, IFI16 oligomers change appearance, becoming filaments that accumulate in viral replication compartments [120]. It is not clear whether IFI16 must be exported from the nucleus to induce IFN-responses via STING activation or whether another mechanism regulates IFI16-dependent, but STING-independent cytokine responses. In addition to its function in intrinsic responses, IFI16 also has the capacity to suppress viral gene expression, thereby directly inhibiting viral replication [75] [120].
Box 2: Crosstalk between nuclear and cytoplasmic DNA sensing.
STING contributes to inflammation downstream of nuclear DNA sensing. For example, hnRNPA2/B1 translocates from the nucleus to activate STING during HSV-1 infection [59]. Likewise, IFI16 translocates from the nucleus to induce STING activation after DNA damage. In human keratinocytes, IFI16 is necessary for the full induction of the STING pathway via direct interaction [6]. Accordingly, STING can promote IFI16 degradation via interaction with the E3-ubiquitin ligase TRIM21, providing a mechanism to attenuate overactivation of nuclear DNA sensing [121]. IFI16 may also indirectly increase STING activation by promoting cGAS-mediated cGAMP production [122]. Conversely, it was demonstrated in fibroblasts that IFI16 and cGAS can physically interact to enhance IFI16 stability without altering cGAMP production [123]. Alternatively, IFI16 may induce the assembly of inflammasomes in the cytosol [112] as part of a STING-independent mechanism to promote expression of IL-1β and IL-18. Another PYHIN protein, AIM2, detects dsDNA in the cytosol to activate inflammasome assembly. Multiple lines of evidence suggest antagonism between AIM2 and IFI16 dependent responses. For example, an IFI16 splice variant can interact with AIM2 to block inflammasome formation [124]. AIM2 and IFI16 protein levels were negatively correlated in a fibroblast model of senescence, and DDR activation promoted IFI16 expression over AIM2 [125]. AIM2 can also suppress DNA-PK activation and DNA repair [126]. Taken together with the finding that DNA-PK can stimulate IFI16 phosphorylation and cytokine induction [5], the interplay of IFI16 and AIM2 may act to tailor the specificity, duration, and magnitude of cytokine responses.
There may be direct antagonism between cGAS and DNA repair. Depleting cGAS enhances HDR, but phosphorylation of cGAS induces its nuclear export after DNA damage [127]. These findings suggest that there is a mechanism for the DDR to exclude cGAS to promote DNA repair. A recent study also showed that DNA-PK deficiency also potentiates cGAS stimulation during viral infection and that DNA-PK directly targets cGAS for phosphorylation at T68 [128]. Mutation of cGAS T68 to a phosphomimetic glutamic acid suppressed cytokine expression after herpesvirus infection. Notably, this modification has not been observed in other cGAS AP-MS studies and therefore may be at a low level. One possibility is that, since DNA-PK also promotes cytokine responses via IFI16 modification, DNA-PK inactivates cGAS during viral infection to prevent overstimulation of innate immunity. This may be physiologically relevant given that DNA-PK is strongly stimulated during mitosis and is important for regulation of mitotic events
Several questions await explanation as we seek to understand how intranuclear intrinsic sensors are regulated to promote appropriate cytokine responses while preventing autoinflammation. Studies are also needed to understand crosstalk between nuclear DNA sensing components and with cytosolic factors (e.g., cGAS, STING, and AIM2) to fine-tune inflammatory signals (Box #2) (Fig 2). Given the above-mentioned crosstalk between IFI16, IFIX, and hnRNPA2B1 with DNA repair and cell cycle control pathways [32, 38], there is also possible functional overlap, which warrants further investigation and discussion. One possibility is that these multifunctional DNA sensors serve as regulatory hubs at the intersection of multiple previously distinct nuclear pathways (e.g., the DDR), and it is the convergence of these processes that places the DNA sensor in the correct context to induce cytokine expression.
DNA Damage Response Kinases Contribute to Cytokine Expression
DNA damage, such as following irradiation, induces inflammatory cytokine expression [39]. Moreover, chronic DNA damage is linked to autoimmune disease [40]. Yet, the connection between cytokine signaling and DNA damage has not been resolved. Does the DDR directly activate cytokine expression, or are inflammatory responses to DNA damage indirect? Two models support an indirect route of cytokine activation by the DDR. In the first, severe damage leads to cytosolic DNA called micronuclei [41]. Extranuclear DNA is sensed by either cGAS, AIM2, or another factor to activate STING or the inflammasome. Yet, DDR proteins inhibit cytosolic DNA sensor function and vice versa (Box #2) (Fig 2). The second model is that severe DNA damage leading to cell death increases extracellular DNA to activate DNA sensing in nearby cells [39]. While it is certain that both routes can stimulate inflammation, the magnitude of DNA damage required to induce these phenomena is quite severe and not representative of all contexts in which the DDR leads to cytokine expression. Rising evidence now supports that cytokine signaling can occur directly downstream of DSBs via activation of the DNA-PK and ATM kinases.
ATM responds to DSBs to promote HDR and can induce cytokine responses after many diverse types of DNA damage (Fig 1). ATM phosphorylates the NF-κB activator, NF-κB Essential Modulator (NEMO) [42, 43], after exposure to genotoxic stress such as by etoposide treatment [44] to induce its export from the nucleus. NEMO then stimulates the IκB Kinase (IKK) complex to trigger degradation of IκBα, an NF-κB inhibitor. Thereafter, NF-κB stimulates IFN-α and IFN-λ expression [42]. DNA damage also stimulates IFN-β, Interleukin 6 (IL-6) and C-C Motif Chemokine Ligand 20 (CCL20) expression in an ATM-dependent manner [6, 45]. The link between ATM and innate immunity is further reflected in antiviral responses to RNA viruses. An siRNA screen of kinases linked to the expression of responsive genes after IFN-γ treatment found that ATM is required to suppress EMCV replication [46]. ATM silencing also enhances RSV replication by reducing the expression of Type-I and -III interferons (see glossary) [47].
Like ATM, DNA-PK also stimulates innate immune responses in addition to coordinating DNA repair (Fig 1). In response to CpG-DNA, DNA-PK and TLR9 promote IFN-α, IFN-β, and IL-6 expression [20, 48]. DNA-PKcs −/− mice are deficient in IL-10 production [20]. The DNA-PK complex itself can sense poxvirus DNA in the cytosol and induce IFN-β responses in a STING-dependent manner [20]. Moreover, IRF3 is a substrate of direct DNA-PK kinase activity [49]. Finally, DNA-PK kinase activity stimulates IFN-β and granulocyte-macrophage colony stimulating factor (GM-CSF) secretion after sensing DSBs in human fibroblast [5]. Taken together, DDR kinase activation is clearly connected to cytokine expression. It is not yet understood how or why DDR activation triggers inflammation. Moreover, in many cases DNA viruses implement mechanisms that use members of the DDR to enhance viral replication; thus, the roles of DDR kinases in cytokine signaling following recognition of infections with DNA viruses remains to be fully understood.
Nuclear DNA Sensors and the DDR Coordinate Nuclear Intrinsic Responses
Several lines of evidence now link ATM and DNA-PK to IFI16-dependent cytokine responses. The Unterholzner group showed that IFI16 and p53 interact with and activate STING following etoposide treatment [6] (Fig 3G). ATM activation promotes IFI16 and p53 translocation from the nucleus to bind STING at the ER membrane (Fig 3G). Thereafter, TRAF6, a polyubiquitin ligase, catalyzes K63-linked ubiquitin chains on STING to trigger cytokine expression. The enzyme poly(ADP-ribose) polymerase 1 (PARP-1) was shown to be involved in promoting the IFI16-STING interaction and the resulting signaling. These findings add to the accumulating knowledge of the roles of the PARP family of proteins at the intersection between the DDR and innate immune response (reviewed in [50]). The observation that IFI16 is downstream of ATM activation is consistent with an established role for IFI16 in cell cycle regulation via interaction with p53 and BRCA1, a DNA repair protein that is important for homology directed repair [28, 51]. As will be discussed below, ATM is proviral. Therefore, it is not yet clear how or if ATM contributes to cytokine stimulation after DNA virus infection. IFI16 also interacts with DNA-PK following HSV-1 infection of primary fibroblasts [5] (Fig 3F). This interaction occurs at early time points of viral infection, at sites of viral genome deposition at the nuclear periphery. DNA-PK inhibition decreases IFN-β secretion following both viral infection and DNA damage. Moreover, DNA-PK directly phosphorylates IFI16 at T149, a residue within the nuclear localization signal region of IFI16, where posttranslational modifications are already known to regulate IFI16-dependent antiviral responses [52]. IFI16 T149 mutagenesis decreases IFN-β expression following HSV-1 infection. Thus, IFI16 and DNA-PK work together to coordinate intrinsic responses to HSV-1 infection. Taken together with the ATM-IFI16 axis and the observation that both ATM and DNA-PK share extensive substrate overlap, it is possible that IFI16 serves as a key intermediate linking the DDR master kinases to cytokine expression. However, many questions await explanation before the interplay of these responses can be reconciled. For example, IFI16 knockout does not affect yH2AX activation (a signal of DDR stimulation) [6], yet IFI16 is required for DNA-PK activation at HSV-1 genomes as measured by DNA-PK autophosphorylation [5]. More specifically, is IFI16 upstream or downstream of DNA recognition by ATM and/or DNA-PK?
IFIX is another PYHIN protein that is linked to cell cycle arrest and functionally similar to IFI16 given that it resides in the nucleus, contributes to antiviral innate immunity [32, 35], promotes p53 stability [53], and acts as a tumor suppressor [54]. Affinity purification (AP)-mass spectrometry (MS) characterization of IFIX interacting partners revealed significant associations with DNA repair proteins [32]. When compared within the same study, the IFIX interactions with DDR proteins were more pronounced than for IFI16. Conversely, the IFI16 interactome revealed more extensive connections with RNA processing, splicing, and RNA transport machinery than IFIX, which is consistent with functions of IFI16 in depressing viral transcription [32]. Interestingly, IFIX undergoes dynamic relocalization from the nucleolus to sites of DNA damage following exposure to ionizing radiation (IR) [36]. Given the extensive and ubiquitous interactions that IFI16 and IFIX share with members of the DNA damage surveillance complexes, such as the BRCA1 Associated Genome Surveillance complex (MSH2, MSH6, MLH1, ATM, BLEM, RAD50, BRCA1, etc.) [55] and the Ku complex (see glossary) it is possible that these proteins link DSB surveillance to innate immunity [5, 32] (Fig 2).
hnRNPA2/B1 appears to negatively regulate the DDR. The best-described function of hnRNPA2/B1 is its association with nascent pre-mRNAs to control hnRNP particle assembly. These particles are important for mRNA maturation and transport to the cytosol for translation [56, 57]. Like both IFI16 and IFIX, hnRNPA2/B1 interacts directly with all members of the DNA-PK heterocomplex, but the prevailing notion is that hnRNPA2/B1 directly inhibits the NHEJ pathway [38, 58]. Although it is not clear how or whether hnRNPA2/B1 impacts cytokine signaling following DNA damage, there is a clear role for hnRNPA2/B1-driven cytokine induction after viral infection [59]. One possibility is that there is mutual antagonism between the DNA-PK and hnRNPA2/B1 responses to regulate the balance of DNA repair versus transcription, which may help control the timing of these events (Fig 2). Although it is not clear how hnRNPA2/B1 may interact with the DDR to promote cytokine expression after DNA damage, a handoff between these two responses could help sustain inflammation during viral infection, since DNA-PK is broadly inhibited by viral strategies (Fig 4).
Figure 4: Viral strategies to subvert the DDR and nuclear DNA sensors.

Nuclear replicating DNA viruses have acquired many mechanisms to overcome the link between DDR and intrinsic immunity. A) Papillomaviruses replication requires ATM pathway components as well as the KU-complex. The papillomavirus oncogenes, E6 and E7, inhibit DNA-PK and IFI16. B) Polyomaviruses replication also utilizes ATM pathway components to amplify the viral DNA. Simultaneously, ATM activation inhibits DNA-PK localization to the viral DNA and the agnoprotein induces KU-complex nuclear export. It is not known if polyomaviruses usurp IFI16. C) Adenoviruses broadly inhibit the DDR via expression of the E1A, E4 and E1B proteins. Adenoviruses also suppress IFI16 expression through an unknown mechanism. Interestingly, the KU-complex, but not DNA-PK, localizes to the Adenoviruses replication complex. D). The α-herpesvirus, HSV-1, extensively reorganizes the DDR. ATM and the MRN complex support HSV-1 replication, but the DNA-PK pathway components are suppressed. The viral E3-ubiquitin ligase, ICP0, promotes both IFI16 and DNA-PK proteolytic degradation. E) Relatively less is understood about how β-herpesviruses, such as HCMV, interact with the DDR. DNA-PK kinase activity is suppressed during HCMV infection. There is conflicting evidence regarding the role and status of ATM during infection. It is possible that the DDR is relevant during HCMV infection given the potent and immediate inhibition of IFI16 by the HCMV tegument protein, pUL83. E) γ-herpesviruses, like EBV and KSHV, suppress DNA-PK kinase activity, while ATM increases viral replication. Interestingly, DNA-PK can inhibit ATM activation during KSHV infection. KSHV lytic infection suppresses IFI16 via an unknown mechanism.
Virus inhibition of DDR-dependent intrinsic responses
DNA damage response proteins are frequently modulated during infections with DNA viruses (Fig 4). As we now understand that DDR activation directly leads to the expression of inflammatory cytokines, it is relevant to consider how DNA viruses disrupt this response. Nuclear-replicating DNA viruses are primarily divided into two families: circular (e.g., polyomavirus and papillomavirus) and linear (e.g., adenovirus and herpesvirus) (see glossary). ATM is noted to drive viral amplification by both classes of DNA viruses, including polyoma-, papilloma-, and herpesviruses [60–63]. Given that ATM activation is frequently proviral, the leading hypothesis is that the kinase aids in repair events needed for viral DNA replication [64]. For example, during polyomavirus infection, ATM prevents rolling circle DNA replication and promotes viral DNA amplification [41, 64]. ATM is also linked to recombination-dependent herpesvirus genome replication [65]. The HSV-1 UL12 exonuclease directly interacts with the MRN complex (see glossary) [66], which is upstream of ATM activation. Given that these DNA viruses employ ATM in a proviral capacity, there are likely viral strategies to functionally separate the ability of ATM to promote cytokines from its role in DNA repair. For example, ATM promotes IFI16 translocation to STING following DNA damage [6]. Accordingly, IFI16 is targeted for proteolytic degradation during HSV-1 infection by ICP0, a viral immediate early gene [67]. Many DNA viruses also employ NF-κB, a downstream component of the ATM signaling pathway [47], to promote viral gene expression [68], which may mislocalize this host transcription factor from its targets on the host genome.
Unlike for many other DNA viruses, ATM activation is deleterious to adenovirus amplification due to antiviral DNA repair [69]. The adenoviruses E4 proteins inhibit ATM pathway activation by degrading the upstream sensing complex, MRN [60] (Fig 4C). Mutant adenoviruses that cannot degrade the MRN complex have high ATM activation and experience end-joining of the linear viral genome that impedes proper viral replication. This stands in contrast to herpesviruses that also have a linear DNA genome, but instead utilize the ATM and MRN pathways for viral replication [6, 65]. One possibility is that adenoviruses did not acquire mechanisms to repurpose ATM and, therefore, instead inhibit its activation to avoid cytokine induction.
As mentioned above, ATM can drive cytokine expression following DNA damage. However, ATM-dependent cytokine signaling may be highly context dependent or may be modulated by viral infection. Orthogonal evidence suggests that ATM may suppress Type-I IFN responses in certain contexts. For example, ATM mutations in humans cause ataxia telangiectasia, which is a cancer-prone neurodegenerative disease with autoimmune and inflammatory symptoms [70]. Cells from ATM-deficient mice that are infected with the murine γ-herpesvirus 68 (MHV68) produce higher levels of the IFN-stimulated genes (ISGs), ubiquitin specific peptidase 18 (USP18) and melanoma differentiation-associated protein 5 (MDA5) [71]. A complicated facet to the relationship between DNA viruses and ATM occurs in the case of infection by human cytomegalovirus (HCMV), a β-herpesvirus. As with other herpesviruses, ATM contributes to HCMV replication [72]. During HCMV infection, the expression of pUL76 stimulates ATM to drive NF-κB activation and IL-8 expression [73]. In this context, ATM-linked cytokine expression may recruit myeloid lineage cells to the site of primary HCMV infection to promote viral latency. As such, HCMV may be the exception that proves the rule: the virus retains the ATM-cytokine axis as a feature of infection. HCMV also encodes a tegument protein, pUL83, that blocks IFI16 oligomerization [74]—a key step for IFI16-dependent intrinsic immune responses [75]. IFI16 is a downstream component of the ATM pathway that is required for IFN-β expression after DNA damage [6]. Taking these observations together, it is tempting to speculate that HCMV has acquired the capacity to fine-tune the cytokine repertoire to promote chemoattractant (IL-8) expression while inhibiting antiviral cytokine responses (IFN-β).
In contrast to ATM, DNA-PK is unambiguously linked to cytokine responses following DNA virus infection. Burleigh et al. found that DNA-PK initiates IFN-β expression in a STING-independent manner [76]. This DNA-PK-dependent IFN expression is suppressed by the adenovirus protein E1A [76], possibly through direct interaction with the KU-complex [77]. DNA-PK also primes STING to respond to γ-herpesvirus infection (Kaposi’s sarcoma herpesvirus [KSHV]) by forming a cGAS stimulating ribonucleoprotein complex [78] and promoting IFN-α and IFN-β expression. Finally, DNA-PK drives IFN-β, GM-CSF, and C-X-C motif chemokine ligand 10 (CXCL10) expression during α-herpesvirus infection (HSV-1) by phosphorylating IFI16 [5, 20] (Fig 4). Taken together, it is not surprising that DNA-PK inactivation is a conserved milestone during infection with many DNA virus families.
The circular DNA virus families have multiple strategies to inhibit DNA-PK responses (Fig 4B). SV40, the model polyomavirus, excludes DNA-PK from viral replication compartments in an ATM-dependent manner [79] downstream of T Antigen expression. The JC polyomavirus agnoprotein mislocalizes Ku70, an upstream component of DNA-PK activation, to the cytosol [80]. The papillomavirus E6 and E7 proteins directly interact with DNA-PK and NHEJ cofactors to block their activity (Fig 4A) [81–83]. The prevailing hypothesis is that circular DNA viruses suppress DNA-PK to block the concatenation of non-unit length genomes by NHEJ [64]. Although these viruses have circular DNA, and thus no DNA ends to ligate, programmed DSBs are induced by topoisomerase to separate the circular genomes after replication (Cairns intermediates). Yet, this form of programmed DNA damage is not typically susceptible to end-joining. The Fanning group proposed that collapsed viral replication forks are the substrate for concatenation, but DNA-PK inhibition did not increase viral replication in ATM deficient cells [64].
Nuclear-replicating linear DNA virus families also have multiple mechanisms in place to block DNA-PK activation and subsequent interferon signaling (Fig 4C). The adenovirus E4 protein binds to DNA-PK to block NHEJ [84, 85] and the E1A protein impairs DNA-PK driven cytokine signaling as discussed above (Fig 4C). The KSHV PF-8 protein binds to the KU-complex members and blocks DSB repair by NHEJ [86] (Fig 4F). The Epstein-Barr virus (EBV)(γ-herpesvirus) LMP1 protein inhibits DNA-PK kinase activity [87] (Fig 4F). The HSV-1 E3-ubiquitin ligase, ICP0, induces proteolytic degradation of DNA-PKcs [88, 89] (Fig 4D). HSV-1 also targets key component of the DDR machinery to fine-tune the DDR proteome at viral DNA [90]. Specifically, HSV-1 promotes MDC1 and γH2AX accumulation at the viral genome while suppressing the recruitment of core DNA repair factors such as p53 binding protein (53BP1) and BRCA1. Other studies have found that HSV-1 can also degrade KU complex components that are necessary for DNA-PK activation [91].
In the context of linear DNA viruses, such as adenoviruses, the prevailing hypothesis is that DNA-PK mediates an antiviral modality of DNA repair that inhibits proper amplification of the viral genome [85, 92]. It should be noted that a prevailing model of herpesvirus replication is that, soon after nuclear entry, the virus circularizes to amplify the viral DNA via rolling circle replication. A contrasting model of herpesvirus replication suggests that the circularization of the viral DNA is antiviral. Instead, the virus is shown to undergo a recombination-driven replication of the linear DNA genome [92]. Accordingly, the mechanism underlying the antiviral function of DNA-PK during herpesvirus replication is contested. As discussed above, the DDR kinases are increasingly linked to intrinsic immune responses via interaction with IFI16. IFI16 is targeted for proteolytic degradation early during HSV-1 infection by ICP0 [30, 93]. Given that IFI16 and DNA-PK interact to coordinate cytokine responses to HSV-1, decreasing IFI16 protein levels may be yet another viral strategy to uncouple the DNA-PK response from innate immune signaling (Fig 4). This is supported by the observation that IFI16 is also targeted during other viral infections. For example, IFI16 protein levels are decreased during KSHV infection [94], the papillomavirus E7 protein employs a cellular E3-ubiquitin ligase (Tripartite Motif Containing 21 [TRIM21]) to degrade IFI16 [95], and adenovirus infection suppresses IFI16 gene expression [96]. Taken together, it is likely that viruses have acquired mechanisms to target the DDR during infection for multiple reasons: first, to fine-tune viral DNA repair events, and second, to bypass the DDR-Cytokine Signaling axis by destroying key signaling components that drive this response.
Concluding Remarks
Origins of the DDR-Cytokine Signaling Axis
As discussed above, it is not clear what selective pressure encouraged integration of the DDR with cytokine signaling. It is possible that the mechanisms acquired by viruses to exploit the DDR drove this feature. This possibility agrees with the observation that the DDR imposes a barrier for a wide variety of DNA viruses via inflammation. Alternatively, given the relationship between DDR and DNA replication, DNA viruses may have evolved a capacity to repurpose the DDR as a mechanism to block a preexisting link between the DDR and innate immunity. There are several potential mechanisms that may support a connection between the DDR and cytokine expression outside of the context of viral infection. For example, warning nearby cells of a severe genotoxic threat may slow down the cell cycle progression in neighboring cells, limiting the extent of tissue destruction caused by programmed cell death. In the case of cytokine production after less pervasive DNA damage, such as that caused by a single DSB, autocrine signaling to reinforce a cell cycle delay of the afflicted cell may maximize the window for DNA repair. Whatever its origin, it is now clear that DNA viruses must overcome the link between the DDR and interferon expression to promote viral spread. Understanding the interplay between ATM and DNA-PK during DNA virus infections may help to uncover strategies for therapeutic interventions.
Cellular Regulation of the DDR-Cytokine Signaling Axis
Integration of foreign nucleic acid sensing in the nucleus with intrinsic immune sensing of non-pathogenic stimuli represents a unique regulatory challenge to the cell and organism. Given the substantial crosstalk between nuclear pathways that converge upon cytokine signaling, perhaps it is at the interface of these pathways that intrinsic signals are given the correct context to lead to inflammation. For example, perhaps it is the context of DDR activation concomitant with IFI16 or IFIX expression that controls when the DDR can trigger cytokine expression. This hypothesis is supported by the observations that IFI16 is dynamically regulated during both embryogenesis and in the germinal center during B-cell maturation [97, 98], which are both circumstances when the cell undergoes programmed DNA breaks with repair mediated by ATM and DNA-PK. Thus, programmed regulation of IFI16 may be scheduled to “turn off” a capacity of DNA repair to transmit cytokine signals. Then, like a toggle, IFI16 can be restored in differentiated cells in which any DNA damage would be unplanned and thus would necessitate the transmission of warning signals. This notion is further supported by the observation that many viral infections inhibit IFI16 by either proteolytic degradation or sequestration. Evidence is also mounting that posttranslational modification of nuclear DNA sensors is central to their activation. It is in this manner that a cell may “choose” when, for example, DNA damage is inflammatory or not.
The DDR-Cytokine Signaling Axis in Autoimmunity and Cancer
Exploring the interplay of DNA sensors, the DDR, and innate immunity may yield insight into autoinflammatory diseases linked to these factors. For example, there is substantial evidence that links IFI16 to the etiology of systemic lupus erythematosus (SLE) [99]. Aberrant DNA repair is also linked to the progression of SLE, specifically through defective base excision and DSB repair. Several relevant DDR proteins are upregulated in the SLE disease state including DNA-PKcs and BRCA1 [100, 101]. Thus, an interplay between IFI16 and the DDR in the context of SLE should be explored. Specifically, does hyperactivation of ATM or DNA-PK leads to phosphorylation and stimulation of IFI16? If so, there are numerous high-quality DDR kinase inhibitors that could alleviate this interferonopathy.
In the context of cancer, IFI16 is often considered a tumor suppressor given its contribution to cell cycle arrest and apoptosis via interactions with p53 [28, 102–104]. Recent findings show that IFI16 expression is elevated following treatment with chemotherapeutic agents that cause DNA damage, and that IFI16 can inhibit DNA repair by suppressing ATM activation [105]. Thus, IFI16 may play a nuanced role in the development and progression of cancer by linking cytokine responses to mutagenesis. It should be noted that an elevated level of DNA repair is necessary to accommodate the high levels of replication stress present in cancer cells [106]. As such, targeting DDR kinases (e.g., ATM, DNA-PK) is a contemporary strategy to enhance radiosensitivity. As we now understand that DDR activation can suppress cytokine responses from cGAS and AIM2 (Box #2), it is tempting to speculate that the negative regulation of IFI16 may allow cancer cells to maintain high DDR activation without inducing cytokine expression. However, it is not yet known whether cancer cells acquire a capacity to modulate of IFI16 levels or whether this is a by-product of normal cellular homeostasis. Specifically, is IFI16 dynamically regulated throughout the cell cycle in normal cells to account for periods of anticipated replication stress? As is often the case, understanding the mechanisms that viruses have acquired to avoid these nucleus-associated intrinsic sensors and responses will be a key step to explore this avenue of cell biology and human health.
Outstanding Questions.
How do DNA viruses functionally disassemble the DNA sensing network that connects the DDR to intrinsic and innate immunity? Are these mechanisms recapitulated during normal development and cellular homeostasis to fine-tune the inflammatory capacity of the DDR?
What specific cellular factors control the distinct involvement of the DDR in cell cycle arrest, DNA repair, or cytokine expression?
Does the DDR-DNA sensing circuit contribute to the pathology of interferonopathies such as those experienced following chronic infection with viruses or persistent DNA damage? Can the DDR or DNA sensors be pharmacologically targeted to alleviate these disease symptoms?
Can the inflammatory capacity of the DDR be reestablished or amplified during viral infection and in cancer cells? Alternatively, given that the DDR can suppress cytosolic DNA sensors and is chronically activated during infection with most DNA viruses, can DDR pathways be targeted to stimulate intrinsic immune signaling?
Highlights.
Upon viral infection, DNA damage response kinases modulate Type-I Interferon expression.
DDR-dependent cytokine expression is linked to DNA sensors, like IFI16, that can distinguish viral DNA from cellular DNA in the nucleus.
Insights into the connection between the DDR and nuclear DNA sensors have unraveled viral mechanisms to evade intrinsic immune defenses and inhibit cytokine secretion.
The DDR-DNA sensing interface provides a platform to understand how cells fine-tune cytokine expression downstream of programmed DDR activation.
DDR activation can negatively regulate cytosolic intrinsic immune responses, while positively regulating nuclear DNA sensing, highlighting cellular strategies to suppress viral infection while maintaining homeostasis.
Acknowledgments
Funding was provided by the NIH grant NIGMS R01-GM114141, the Stand Up To Cancer Convergence 3.1416, and the Princeton Catalysis Initiative. We thank Peter Metzger for the critical reading of the manuscript.
Glossary
- Adenovirus
A family of viruses with linear DNA genomes of intermediate size. The model virus for this family is Ad5. Adenoviruses cause respiratory illness and other common-cold like diseases. Type-I and Type-III IFN responses to adenovirus infection are important to prevent disseminated infection.
- ATM
Ataxia Telangiectasia Mutated. A DNA damage response serine/threonine kinase of the Phosphatidylinositol 3-kinase-related kinases family. It is activated by the MRN complex.
- DNA-PK
A heterocomplex composed of three subunits. KU70 and KU80 form the KU complex which senses DNA breaks. The DNA-PKcs catalytic subunit is a serine/threonine kinase of the Phosphatidylinositol 3-kinase-related kinases family.
- Herpesvirus
A diverse family of linear dsDNA viruses with large genomes. This family has several members that are categorized by the cell type in which they establish latency. The α-herpesviruses subfamily latently infects sensory neurons and incudes HSV-1 and varicella-zoster virus. The β-herpesvirus subfamily latently infects leukocytes and contains HCMV and MHV68. The γ-herpesvirus family latently infects B- and T- cells and includes KSHV and EBV.
- IFI16
Interferon (IFN)-γ Inducible Protein 16. This is a member of the PY-HIN family of proteins that regulates DNA sensing in the nucleus. IFI16 is capable of inducing Type-I interferons through STING. In certain cell types, IFI16 can also induce inflammasome formation.
- KU Complex
Composed of KU70 and KU80 the proteins encoded by the XRCC6 and XRCC5 genes, respectively. This complex detects DNA breaks and recruits DNA-PKcs.
- MRN Complex
composed of Rad50, Nbs1, and Mre11. Senses DNA breaks and activates ATM.
- PYHIN proteins
a family of proteins that contain a pyrin domain, which mediates oligomerization and at least one HIN200 domain, which can bind DNA in a sequence independent manner. Members include IFI16, IFIX, and Aim2.
- Papillomavirus
a family of small circular DNA viruses. These viruses frequently infect the skin and can induce cellular proliferation through expression of their oncogenes. Failure of the immune system to control this family can result in tumor growth.
- Polyomavirus
a family of small circular DNA viruses. Consist of the prototype virus, SV40, and several human polyomaviruses, including JC polyomavirus, BK polyomavirus, and Merkel Cell Polyomavirus. Failure of the immune system to control this family of viruses can cause cancer, kidney nephropathy, and neurological disease.
- Type-I Interferons
consist of several IFN-α subtypes, IFN-β, IFN-ε, IFN-κ, and IFN-ω. Secreted interferons are sensed by Interferon- α/ β receptor (IFNAR) to activate STAT (signal transducer and activator of transcription) signaling that leads the production of interferon stimulated genes (ISGs). ISGs can mediate antiviral effects, such as shutting down translation or priming the expression of DNA sensors like IFI16.
- Type-II Interferon
Consists of IFN-γ and is sensed by the IFN-γ receptors, IFNGR1 and IFNGR2.
- Type-III Interferons
Consist of four IFN-λ subtypes. Contributes to epithelial innate responses following recognition by the IFN-λ receptor, IFNL1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Works Cited:
- 1.Majzoub K, et al. (2019) The Innate Antiviral Response in Animals: An Evolutionary Perspective from Flagellates to Humans. Viruses 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallucci S and Maffei ME (2017) DNA Sensing across the Tree of Life. Trends Immunol 38, 719–732 [DOI] [PubMed] [Google Scholar]
- 3.Härtlova A, et al. (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42, 332–343 [DOI] [PubMed] [Google Scholar]
- 4.Reinert LS, et al. (2016) Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun 7, 13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Justice JL, et al. (2021) Systematic profiling of protein complex dynamics reveals DNA-PK phosphorylation of IFI16 en route to herpesvirus immunity. Sci Adv 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunphy G, et al. (2018) Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol Cell 71, 745–760 e745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tubbs A and Nussenzweig A (2017) Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giglia-Mari G, et al. (2011) DNA damage response. Cold Spring Harb Perspect Biol 3, a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jolly CJ, et al. (2008) Fixing DNA breaks during class switch recombination. J Exp Med 205, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uhlorn BL, et al. (2020) Vesicular trafficking permits evasion of cGAS/STING surveillance during initial human papillomavirus infection. PLoS Pathog 16, e1009028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu YZ, et al. (2017) Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe 21, 231–243 [DOI] [PubMed] [Google Scholar]
- 12.Wu JJ, et al. (2015) Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 18, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22, 240–273, Table of Contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao TS and Fitzgerald KA (2013) The cGAS-STING pathway for DNA sensing. Mol Cell 51, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L, et al. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinert LS, et al. (2021) Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mørk N, et al. (2015) Mutations in the TLR3 signaling pathway and beyond in adult patients with herpes simplex encephalitis. Genes Immun 16, 552–566 [DOI] [PubMed] [Google Scholar]
- 18.Li XD, et al. (2013) Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connolly DJ and Bowie AG (2014) The emerging role of human PYHIN proteins in innate immunity: implications for health and disease. Biochem Pharmacol 92, 405–414 [DOI] [PubMed] [Google Scholar]
- 20.Ferguson BJ, et al. (2012) DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roth S, et al. (2014) Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat Immunol 15, 538–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo T, et al. (2013) DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A 110, 2969–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiu YH, et al. (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, et al. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, et al. (2011) Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol 186, 4541–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sui H, et al. (2017) STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA. Sci Signal 10 [DOI] [PubMed] [Google Scholar]
- 27.Dempsey A and Bowie AG (2015) Innate immune recognition of DNA: A recent history. Virology 479–480, 146–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aglipay JA, et al. (2003) A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene 22, 8931–8938 [DOI] [PubMed] [Google Scholar]
- 29.Ouchi M and Ouchi T (2008) Role of IFI16 in DNA damage and checkpoint. Front Biosci 13, 236–239 [DOI] [PubMed] [Google Scholar]
- 30.Diner BA, et al. (2015) Interactions of the antiviral factor IFI16 mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol Cell Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diner BA, et al. (2016) Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. MBio 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diner BA, et al. (2015) The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol Syst Biol 11, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy A, et al. (2019) IFI16, a nuclear innate immune DNA sensor, mediates epigenetic silencing of herpesvirus genomes by its association with H3K9 methyltransferases SUV39H1 and GLP. Elife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding Y, et al. (2004) Antitumor activity of IFIX, a novel interferon-inducible HIN-200 gene, in breast cancer. Oncogene 23, 4556–4566 [DOI] [PubMed] [Google Scholar]
- 35.Crow MS and Cristea IM (2017) Human Antiviral Protein IFIX Suppresses Viral Gene Expression during Herpes Simplex Virus 1 (HSV-1) Infection and Is Counteracted by Virus-induced Proteasomal Degradation. Mol Cell Proteomics 16, S200–S214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howard TR, et al. (2021) The DNA Sensor IFIX Drives Proteome Alterations To Mobilize Nuclear and Cytoplasmic Antiviral Responses, with Its Acetylation Acting as a Localization Toggle. mSystems 6, e0039721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, et al. (2019) Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 365 [DOI] [PubMed] [Google Scholar]
- 38.Iwanaga K, et al. (2005) Heterogeneous nuclear ribonucleoprotein B1 protein impairs DNA repair mediated through the inhibition of DNA-dependent protein kinase activity. Biochem Biophys Res Commun 333, 888–895 [DOI] [PubMed] [Google Scholar]
- 39.Nastasi C, et al. (2020) DNA Damage Response and Immune Defense. Int J Mol Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Souliotis VL, et al. (2019) DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. Int J Mol Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Justice JL, et al. (2019) BK Polyomavirus Activates the DNA Damage Response To Prolong S Phase. J Virol 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu ZH, et al. (2006) Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 311, 1141–1146 [DOI] [PubMed] [Google Scholar]
- 43.Wu ZH, et al. (2010) ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell 40, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyamoto S (2011) Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res 21, 116–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodier F, et al. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11, 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watling D, et al. (2008) Multiple kinases in the interferon-gamma response. Proc Natl Acad Sci U S A 105, 6051–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang L, et al. (2015) Ataxia telangiectasia mutated kinase mediates NF-κB serine 276 phosphorylation and interferon expression via the IRF7-RIG-I amplification loop in paramyxovirus infection. J Virol 89, 2628–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma C, et al. (2015) Involvement of DNA-PKcs in the type I IFN response to CpG-ODNs in conventional dendritic cells in TLR9-dependent or -independent manners. PLoS One 10, e0121371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karpova AY, et al. (2002) Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc Natl Acad Sci U S A 99, 2818–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu H, et al. (2021) The Critical Role of PARPs in Regulating Innate Immune Responses. Front Immunol 12, 712556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dutta D, et al. (2015) BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-β Responses. PLoS Pathog 11, e1005030. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Li T, et al. (2012) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109, 10558–10563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding Y, et al. (2006) Interferon-inducible protein IFIXalpha1 functions as a negative regulator of HDM2. Mol Cell Biol 26, 1979–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang S, et al. (2021) Interferon-inducible protein, IFIX, has tumor-suppressive effects in oral squamous cell carcinoma. Sci Rep 11, 19593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, et al. (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 14, 927–939 [PMC free article] [PubMed] [Google Scholar]
- 56.Haley B, et al. (2009) Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int J Radiat Biol 85, 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He Y and Smith R (2009) Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci 66, 1239–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han J, et al. (2008) [Effects of hnRNP B1 on DNA-PK activity, cell cycle and apoptosis in human lung adenocarcinoma cell line A549]. Sichuan Da Xue Xue Bao Yi Xue Ban 39, 815–818 [PubMed] [Google Scholar]
- 59.Zhang X, et al. (2019) hnRNPA2B1: a nuclear DNA sensor in antiviral immunity. Cell Res 29, 879–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pancholi NJ, et al. (2017) Take your PIKK: tumour viruses and DNA damage response pathways. Philos Trans R Soc Lond B Biol Sci 372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarakanova VL, et al. (2007) Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anacker DC and Moody CA (2017) Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res 231, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiaofei E and Kowalik TF (2014) The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 6, 2155–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sowd GA, et al. (2013) ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog 9, e1003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alekseev O, et al. (2014) Inhibition of ataxia telangiectasia mutated (ATM) kinase suppresses herpes simplex virus type 1 (HSV-1) keratitis. Invest Ophthalmol Vis Sci 55, 706–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balasubramanian N, et al. (2010) Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J Virol 84, 12504–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Howard TR and Cristea IM (2020) Interrogating Host Antiviral Environments Driven by Nuclear DNA Sensing: A Multiomic Perspective. Biomolecules 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Santoro MG, et al. (2003) NF-kappaB and virus infection: who controls whom. EMBO J 22, 2552–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shah GA and O’Shea CC (2015) Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell 162, 987–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song X, et al. (2019) Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J Neurosci 39, 6378–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Darrah EJ, et al. (2017) ATM supports gammaherpesvirus replication by attenuating type I interferon pathway. Virology 510, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.E X, et al. (2011) An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog 7, e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Costa H, et al. (2013) Human cytomegalovirus gene UL76 induces IL-8 expression through activation of the DNA damage response. PLoS Pathog 9, e1003609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li T, et al. (2013) Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lum KK, et al. (2019) Charge-Mediated Pyrin Oligomerization Nucleates Antiviral IFI16 Sensing of Herpesvirus DNA. mBio 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burleigh K, et al. (2020) Human DNA-PK activates a STING-independent DNA sensing pathway. Sci Immunol 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frost JR, et al. (2017) The interaction of adenovirus E1A with the mammalian protein Ku70/XRCC6. Virology 500, 11–21 [DOI] [PubMed] [Google Scholar]
- 78.Morchikh M, et al. (2017) HEXIM1 and NEAT1 Long Non-coding RNA Form a Multi-subunit Complex that Regulates DNA-Mediated Innate Immune Response. Mol Cell 67, 387–399.e385 [DOI] [PubMed] [Google Scholar]
- 79.Sowd GA, et al. (2014) SV40 utilizes ATM kinase activity to prevent non-homologous end joining of broken viral DNA replication products. PLoS Pathog 10, e1004536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Darbinyan A, et al. (2004) Role of JC virus agnoprotein in DNA repair. J Virol 78, 8593–8600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hu C, et al. (2020) Beta Human Papillomavirus 8E6 Attenuates Non-Homologous End Joining by Hindering DNA-PKcs Activity. Cancers (Basel) 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leeman JE, et al. (2019) Human papillomavirus 16 promotes microhomology-mediated end-joining. Proc Natl Acad Sci U S A 116, 21573–21579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sitz J, et al. (2019) Human papillomavirus E7 oncoprotein targets RNF168 to hijack the host DNA damage response. Proc Natl Acad Sci U S A 116, 19552–19562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen C and Bridge E (2022) DNA-PK phosphorylation at Ser2056 during adenovirus E4 mutant infection is promoted by viral DNA replication and independent of the MRN complex. Virology 565, 82–95 [DOI] [PubMed] [Google Scholar]
- 85.Boyer J, et al. (1999) Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263, 307–312 [DOI] [PubMed] [Google Scholar]
- 86.Xiao Y, et al. (2013) Lytic infection of Kaposi’s sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J Gen Virol 94, 1870–1875 [DOI] [PubMed] [Google Scholar]
- 87.Lu J, et al. (2016) EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett 380, 191–200 [DOI] [PubMed] [Google Scholar]
- 88.Parkinson J, et al. (1999) Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73, 650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lees-Miller SP, et al. (1996) Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol 70, 7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lilley CE, et al. (2011) The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog 7, e1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Chiara G, et al. (2016) Herpes Simplex Virus-Type1 (HSV-1) Impairs DNA Repair in Cortical Neurons. Front Aging Neurosci 8, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith S and Weller SK (2015) HSV-I and the cellular DNA damage response. Future Virol 10, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Orzalli MH, et al. (2016) Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J Virol 90, 8351–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roy A, et al. (2016) Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J Virol 90, 8822–8841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song Y, et al. (2020) HPV E7 inhibits cell pyroptosis by promoting TRIM21-mediated degradation and ubiquitination of the IFI16 inflammasome. Int J Biol Sci 16, 2924–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao H, et al. (2017) Posttranscriptional Regulation in Adenovirus Infected Cells. J Proteome Res 16, 872–888 [DOI] [PubMed] [Google Scholar]
- 97.Meistermann D, et al. (2021) Integrated pseudotime analysis of human pre-implantation embryo single-cell transcriptomes reveals the dynamics of lineage specification. Cell Stem Cell 28, 1625–1640.e1626 [DOI] [PubMed] [Google Scholar]
- 98.Piccaluga PP, et al. (2015) IFI16 Expression Is Related to Selected Transcription Factors during B-Cell Differentiation. J Immunol Res 2015, 747645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mondini M, et al. (2007) Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann N Y Acad Sci 1110, 47–56 [DOI] [PubMed] [Google Scholar]
- 100.Meas R, et al. (2017) DNA repair and systemic lupus erythematosus. DNA Repair (Amst) 56, 174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Souliotis VL, et al. (2016) Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus. Arthritis Res Ther 18, 182. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102.Song LL, et al. (2008) Expression of an IFN-inducible cellular senescence gene, IFI16, is up-regulated by p53. Mol Cancer Res 6, 1732–1741 [DOI] [PubMed] [Google Scholar]
- 103.Kim EJ, et al. (2005) IFI16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/JAK/STAT pathway in medullary thyroid carcinoma cells. J Biol Chem 280, 4913–4920 [DOI] [PubMed] [Google Scholar]
- 104.Choubey D, et al. (2008) Interferon-inducible IFI16 protein in human cancers and autoimmune diseases. Front Biosci 13, 598–608 [DOI] [PubMed] [Google Scholar]
- 105.Ka NL, et al. (2021) IFI16 inhibits DNA repair that potentiates type-I interferon-induced antitumor effects in triple negative breast cancer. Cell Rep 37, 110138. [DOI] [PubMed] [Google Scholar]
- 106.Yang H, et al. (2020) Beyond DNA Repair: DNA-PKcs in Tumor Metastasis, Metabolism and Immunity. Cancers (Basel) 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Motwani M, et al. (2019) DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20, 657–674 [DOI] [PubMed] [Google Scholar]
- 108.Li Q, et al. (2018) TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discov 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chu TT, et al. (2021) Tonic prime-boost of STING signalling mediates Niemann-Pick disease type C. Nature 596, 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Unterholzner L, et al. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Orzalli MH, et al. (2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109, E3008–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kerur N, et al. (2011) IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ansari MA, et al. (2013) Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol 87, 8606–8623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pisano G, et al. (2017) Interferon-γ-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency. Virol J 14, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gariano GR, et al. (2012) The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 8, e1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lo Cigno I, et al. (2015) The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters. J Virol 89, 7506–7520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Krump NA, et al. (2021) Merkel Cell Polyomavirus Infection Induces an Antiviral Innate Immune Response in Human Dermal Fibroblasts. J Virol 95, e0221120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Monroe KM, et al. (2014) IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jakobsen MR, et al. (2013) IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A 110, E4571–4580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Merkl PE and Knipe DM (2019) Role for a Filamentous Nuclear Assembly of IFI16, DNA, and Host Factors in Restriction of Herpesviral Infection. MBio 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li D, et al. (2019) STING-Mediated IFI16 Degradation Negatively Controls Type I Interferon Production. Cell Rep 29, 1249–1260.e1244 [DOI] [PubMed] [Google Scholar]
- 122.Almine JF, et al. (2017) IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8, 14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Orzalli MH, et al. (2015) cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112, E1773–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang PH, et al. (2018) Inhibition of AIM2 inflammasome activation by a novel transcript isoform of IFI16. EMBO Rep 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Duan X, et al. (2011) Differential roles for the interferon-inducible IFI16 and AIM2 innate immune sensors for cytosolic DNA in cellular senescence of human fibroblasts. Mol Cancer Res 9, 589–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wilson JE, et al. (2015) Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med 21, 906–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu H, et al. (2018) Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 [DOI] [PubMed] [Google Scholar]
- 128.Sun X, et al. (2020) DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat Commun 11, 6182. [DOI] [PMC free article] [PubMed] [Google Scholar]