

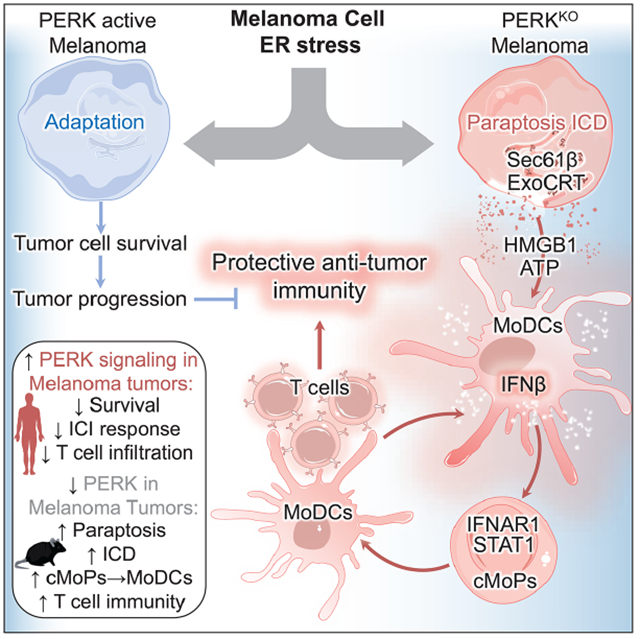
Summary
Activation of unfolded protein responses (UPR) in cancer cells undergoing endoplasmic reticulum (ER) stress promotes survival. However, how UPR in tumor cells impacts anti-tumor immune responses remains poorly described. Here, we investigate the role of the UPR mediator pancreatic ER kinase (PKR)-like ER kinase (PERK) in cancer cells in the modulation of anti-tumor immunity. Deletion of PERK in cancer cells or pharmacological inhibition of PERK in melanoma-bearing mice incites robust activation of anti-tumor T cell immunity and attenuates tumor growth. PERK elimination in ER stressed-malignant cells triggers SEC61β-induced paraptosis, thereby promoting immunogenic cell death (ICD) and systemic anti-tumor responses. ICD induction in PERK-ablated tumors stimulates type I interferon production in dendritic cells (DCs), which primes CCR2-dependent tumor trafficking of common-monocytic precursors and their intra-tumor commitment into monocytic-lineage inflammatory Ly6C+CD103+ DCs. These findings identify how tumor cell-derived PERK promotes immune evasion and highlight the potential of PERK-targeting therapies in cancer immunotherapy.
Keywords: Tumor immunity, PERK, Unfolded protein responses, Type I IFN, Immunogenic cell death
Graphical Abstract
eTOC/“In Brief” Paragraph
How adaptation to endoplasmic reticulum (ER) stress in cancer cells modulates anti-tumor immunity remains elusive. Mandula et al. demonstrate that elimination of the ER stress-related kinase, PERK, in melanoma cells activates protective T cell responses through paraptosis-mediated immunogenic cell death that primes expansion of monocytic-lineage inflammatory DCs via type-I IFN-STAT1.
Introduction
The altered immune function occurring in most individuals with tumors has emerged as a primary driver of malignant cell progression and metastasis, as well as a major limitation to promising anticancer therapies (Binnewies et al., 2018). Exposure of immune cells to reactive compounds and metabolic conditions present in the tumor microenvironment (TME) primes distinct stress-induced signaling pathways that can intrinsically restrict their anti-tumor potential (Cubillos-Ruiz et al., 2017). Cancer cells are also continually subjected to similar stresses and must likewise compensate. The interplay between stress-induced signaling in cancer cells and the regulation of protective anti-tumor immunity remains poorly understood.
Endoplasmic reticulum (ER) stress is induced in cells as a consequence of multiple stressors, including the accumulation of misfolded proteins in the ER lumen, nutrient deprivation, hypoxia, therapeutic agents, and inflammatory factors (Chen and Cubillos-Ruiz, 2021). To cope with ER stress, cancer cells activate an integrated signaling network known as the unfolded protein response (UPR), which is characterized by the activation of the pancreatic ER kinase (PKR)-like ER kinase (PERK), the inositol-requiring enzyme-1α (IRE1α), and the activating transcription factor 6 (ATF6) (Urra et al., 2016). During ER stress, auto-phosphorylated PERK dissociates from its negative regulator and phosphorylates several targets, including the translation initiation factor 2α (eIF2α) (Pytel et al., 2016), which triggers the expression of the activating transcription factor 4 (ATF4) that transiently contributes to cancer cell survival (Chevet et al., 2015). Our previous reports have elucidated the immunoinhibitory role of the intrinsic activation of PERK in tumor-associated T cells and myeloid-derived suppressor cells (MDSCs) (Cao et al., 2019; Mohamed et al., 2020). However, the endogenous contribution of PERK in cancer cells in controlling immune responses in tumor-bearing hosts remains unknown.
The processes whereby tumors undergo cell death have emerged as key regulators of anti-tumor immunity (Kroemer et al., 2022). Cancer cell death processes that unleash anti-tumor immune responses both locally and remotely are grouped under the term immunogenic cell death (ICD), which is characterized by the release of intracellular ATP and high mobility group box protein 1 (HMGB1), externalization of calreticulin to the cell membrane (ExoCRT), and production of type I interferons (IFN) (Galluzzi et al., 2020). Although ICD remains an attractive strategy for overriding tumor-linked immune evasion (Kepp et al., 2014), strategies for reliably inducing ICD and the mechanisms of how ICD enables protective anti-tumor immunity remain unclear.
Here, we aim to elucidate the mechanistic crosstalk between PERK activation in cancer cells and immune evasion in tumor-bearing hosts. Deletion or inhibition of PERK in melanoma cells limits their capacity to survive after ER stress and results in paraptosis-mediated ICD that stimulates the production of type I IFN in intra-tumor dendritic cells (DCs), which thereby provokes tumor trafficking and differentiation of common monocyte precursors (cMoPs) into Ly6C+CD103+ Monocytic-lineage inflammatory DCs (MoDCs) and T cell immunity. These results support the immunotherapeutic potential of targeting PERK in melanoma and elucidate mechanisms whereby ICD reprograms myelopoiesis and promotes protective anti-tumor T cell immunity.
Results
PERK in cancer cells restricts protective anti-tumor T cell immunity
Whether PERK signaling in melanoma tumors impacts clinical outcome and anti-tumor immunity remains unknown. To evaluate the relationship between PERK activity and clinical outcome in the absence of PERK phosphorylation information, we developed a PERK activity mRNA signature based on the top-250 transcripts reduced after EIF2AK3 (PERK encoding gene) silencing in the A375 melanoma cell line from the Connectivity Map platform (Subramanian et al., 2017) (Figure S1A). Analyses in different melanoma datasets, including The Cancer Genome Atlas (TCGA, 457 patients), Moffitt Cancer Center (117 patients), and a previously reported cohort of patients treated with checkpoint immunotherapy (ICI) (69 patients) (Gide et al., 2019; Hugo et al., 2016; Riaz et al., 2017), showed that patients with tumors having high PERK signaling mRNA signature had reduced overall survival compared to patients with low tumor PERK signaling (Figure 1A). Also, we tested whether PERK signaling in melanoma tumors impacts the efficacy of ICI. Retrospective analyses in a cohort of 68 melanoma patients who received anti-PD-1 and/or anti-CTLA-4 showed an unfavorable ICI response in patients with tumors exhibiting high PERK signaling mRNA score compared to counterparts with low PERK signaling (Figure 1B). Thus, PERK signaling in human melanoma tumors limits clinical outcome and responses to ICI. Next, we assessed whether PERK activation in human melanoma cells affects the expansion of intra-tumor T cells. Expression of phospho-PERK in melanoma cells (SOX10+) was stratified in 133 human metastatic melanoma tumors and results correlated with intra-tumor T cell frequency (Figure 1C, Figure S1B). Higher expression of phospho-PERK in SOX10+ melanoma cells correlated with a reduced proportion of CD4+ and CD8+ T cells in tumor beds (Figure 1D), suggesting that PERK phosphorylation in the melanoma cell compartment constrains T cell expansion in human tumors.
Figure 1: PERK in melanoma cells limits anti-tumor T cell immunity.
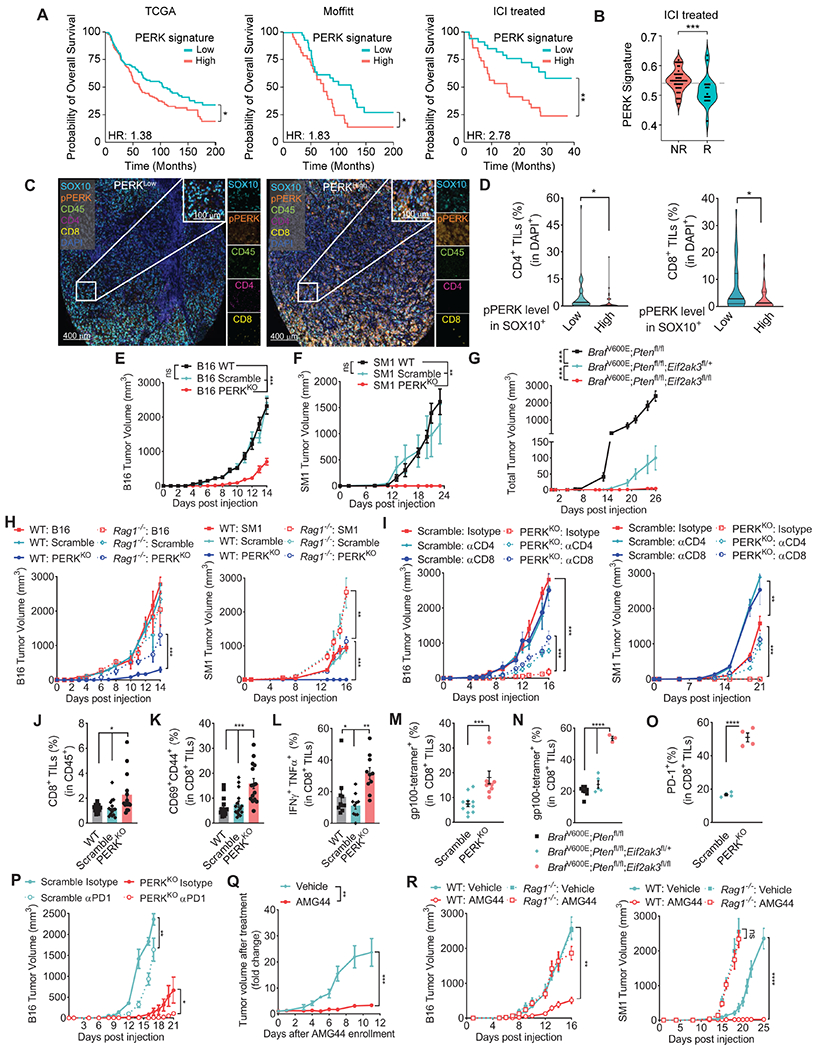
(A) Overall survival in melanoma patients from Skin Cutaneous Melanoma datasets (TCGA, Moffitt, and ICI treated) stratified by PERK mRNA signature scores calculated by GSVA. Samples were categorized based on a median split of the signature score: (TCGA: n= 228 above and n= 229 below; Moffitt n=59 above and n= 58 below; ICI treated n=35 above and n=34 below).
(B) PERK signaling score was applied into ICI treated melanoma patients and categorized into Responders (R, n=19) or Non-Responders (NR, n=49) as annotated by the CRI iAtlas portal. Data are the median (gray dashed line) from the min to max and width is the population frequency at that interval.
(C) Illustrative images by Automated Multispectral Imaging (400μm and 100μm) of 133 metastatic melanoma tumors showing SOX10 (Cyan), phospho-PERK (Orange), CD45 (Green), CD4 (Magenta), CD8 (Yellow) and DAPI (Blue). Samples were ranked as PERKhigh or PERKlow as in the Methods.
(D) Proportion of intra-tumor CD4+ (left) and CD8+ (right) T cells in DAPI+ cells in the stratified groups of phospho-PERK in SOX10+ cells from (C). Data are the median (bold horizontal line) ± quartiles (light horizontal line) with n= 33 (Low) and 36 (High).
(E-F) Tumor volume ± SEM in C57BL/6 mice bearing wildtype (WT), Scramble or PERKKO B16 (E, n=29/group), or SM1 (F, n=9/group) tumors.
(G) Total tumor volume ± SEM in tamoxifen-treated Eif2ak3fl/+ or Eif2ak3fl/fl BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice vs. BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice (n=12/group).
(H) Tumor volume ± SEM of B16, B16 Scramble, and B16 PERKKO cells (left), as well as SM1 counterparts (right) injected into C57BL/6 (Wild Type, WT) or Rag1−/− mice. n=8/group.
(I) Tumor growth ± SEM in mice bearing B16 Scramble or B16 PERKKO tumors (left), or SM1 counterparts (right), treated with isotype, anti-CD4, or anti-CD8. n=5/group.
(J-M) Percentage ± SEM of intra-tumor CD8+ T cells in CD45+ cells (J), and CD69+CD44+ (K), IFNγ+TNFα+ (L), and gp 100-reactive EGSRNQDWL-H-2Db-tetramer+ cells in intra-tumor CD8+ T cells (M) from WT, Scramble and PERKKO B16 tumors at day 17 post-injection. n=10-15/group.
(N) Proportion (mean ± SEM) of EGSRNQDWL-H-2Db-tetramer+ intra-tumor CD8+ T cells from tamoxifen-exposed Eif2ak3fl/+ or Eif2ak3fl/fl BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice vs. BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice. n=3-8/group.
(O) Proportion ± SEM of CD8+PD-1+ in CD45+ cells from WT, Scramble or PERKKO B16 tumors.
(P) Tumor volume ± SEM in mice bearing Scramble or PERKKO B16 tumors and treated with isotype or anti-PD-1. n=4/group.
(Q) Tamoxifen-exposed BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice carrying ~150 mm3 tumors were treated with vehicle or AMG44. Tumor volume normalized to initial treatment volume. n=9/group.
(R) Tumor volume ± SEM in WT and Rag1−/− mice bearing B16 (left) or SM1 (right) tumors and treated after day 6 post-tumor injection with AMG44 (12.5 mg/kg). n=10/group.
Statistics applied using one-way ANOVA (E – L, N, P – R), Student’s t-test (D, M, O), Mann-Whitney t-test (B), or log-rank test (A). *, p<0.05; **, p<0.01; ***, p < 0.001; ****, p < 0.0001. Please also see Figure S1.
To define whether tumor-cell intrinsic PERK affected anti-tumor immune actions, we developed two PERK-null (PERKKO) melanoma cell lines, SM1 (BrafV600E;Cdkn2a−/−) and B16 tumors, via CRISPR/Cas9-based ablation of the Eif2ak3 gene (Figure S1C). This approach did not alter cellular proliferation under regular culture conditions, but forestalled induction of PERK signaling targets phospho-eIF2α or CHOP without affecting IRE1α signaling after treatment with the ER stressor Thapsigargin (Thaps) (Figure S1D–E). In vivo, we found a marked delay in tumor growth and complete tumor rejection in C57BL/6 mice implanted with PERKKO B16 or SM1 tumors, respectively, compared to Scramble or wildtype controls (Figure 1E–F). Moreover, we introduced Eif2ak3 gene deletion in a model of autochthonous melanoma induced after exposure to topical tamoxifen in BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice. Tumor cell-selective loss of one or two alleles of Eif2ak3 delayed tumor growth after exposure of BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice to tamoxifen (Figure 1G). Also, the in vivo anti-tumor actions induced by PERK ablation in melanoma cells translated in an extended mice survival (Figure S1F). To test whether lymphocyte activity was required for the delayed progression of PERKKO tumors, we implanted wildtype, Scramble or PERKKO B16 or SM1 cells into immunodeficient Rag1−/− mice; or depleted CD4+ or CD8+ T cells using antibody-based approaches. The anti-tumor effect triggered by PERK deletion in B16 or SM1 cells in control mice was partially prevented in Rag1−/− mice, or in wildtype mice treated with anti-CD4 or anti-CD8 antibodies (Figure 1H–I), suggesting the anti-tumor role of T cells in PERKKO tumors. Consistently, tumors from mice bearing PERKKO B16 cells compared to control tumors, showed higher proportion of CD8+ and CD4+ T cells, primed CD69+CD44+CD8+ T cells, polyfunctional IFNγ+TNFα+ CD8+ T cells, and melanoma antigen gp100-specific EGSRNQDWL-H-2Db-tetramer+ CD8+ T cells, but not macrophages, B cells or NK cells (Figure 1J–M; Figure S1G–H). A similar elevation of spontaneously expanded gp100-reactive CD8+ T cells was found in lesions from tamoxifen-treated BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice lacking one or two alleles of Eif2ak3, compared to PERK-competent controls (Figure 1N). Moreover, in agreement with the development of effector T cell immunity, higher PD-1 levels were detected in intra-tumor CD8+ T cells from PERKKO tumors compared to controls (Figure 1O). Also, treatment of PERKKO B16-bearing mice with anti-PD-1 resulted in augmented anti-tumor effects compared to isotype-treated PERKKO tumors or anti-PD1-treated controls (Figure 1P). Thus, PERK deletion in melanoma tumors promotes protective T cell immunity and expression of PD-1 in T cells, which can be therapeutically addressed. Furthermore, we noticed reduced tumor blood vessel density, as indicated by a lower frequency of CD31+ vasculature structures, in B16 and BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 tumors lacking PERK compared to controls (Figure S1I–J). Also, reduced levels of the vascularization driver VEGF were detected in PERKKO tumor suspensions compared to Scramble counterparts (Figure S1K), indicating that in addition to inciting protective T cell immunity, deletion of PERK in melanoma cells promoted tumor vascularization processes.
To recapitulate the deletion of PERK in therapeutic models, we next tested the anti-tumor effects of AMG44, a potent and selective PERK inhibitor (Smith et al., 2015). Inhibition of PERK impacted the growth of established autochthonous melanomas developed after topical tamoxifen exposure of BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice (Figure 1Q). AMG44 treatment also delayed B16 tumor progression and induced rejection of SM1 tumors, while identically treated Rag1−/− mice and vehicle-treated counterparts had progressive tumor growth (Figure 1R). The lack of tumor tissue in SM1-bearing mice treated with AMG44 limited the access to this compartment. However, spleens from SM1-bearing mice and tumors from B16-carrying mice treated with AMG44 showed higher frequency of gp100-specific EGSRNQDWL-H-2Db-tetramer+ CD8+ T cells (Figure S1L–M). Thus, PERK inhibition blunted tumor growth and induced expansion of tumor-specific T cells.
PERK ablation in ER-stressed tumor cells provokes paraptosis cell death
To understand the immune-stimulatory effects induced by the elimination of PERK in cancer cells, we interrogated the role of PERK in tumor cell survival after ER stress. PERKKO tumors exhibited heightened susceptibility to ER stress-induced cell death compared to controls, as indicated by the higher binding of Annexin V and the depolarization of mitochondrial membrane potential detected via DiOC2(3) after treatment with Thaps (Figure 2A, Figure S2A). Elevated Annexin V was also observed in B16 and SM1 cells pre-treated with AMG44 prior to Thaps exposure (Figure S2B). Moreover, increased mitochondrial membrane depolarization was noted in PERKKO B16 tumors after exposure to physiological stress conditions such as serum or glucose starvation (Figure S2C). Next, we sought to identify the programs driving cell death in PERKKO tumors undergoing ER stress. Pharmacological inhibition of apoptosis, necroptosis, ferroptosis, pyroptosis, autophagy, or PARPoptosis failed to prevent Annexin V elevation in Thaps-treated PERKKO B16 cells (Figure S2D–I), suggesting that these cell death programs did not play an obligatory role in ER stressed PERKKO tumor cell death.
Figure 2: PERK ablation sensitizes melanoma cells to ER stress-induced paraptosis.
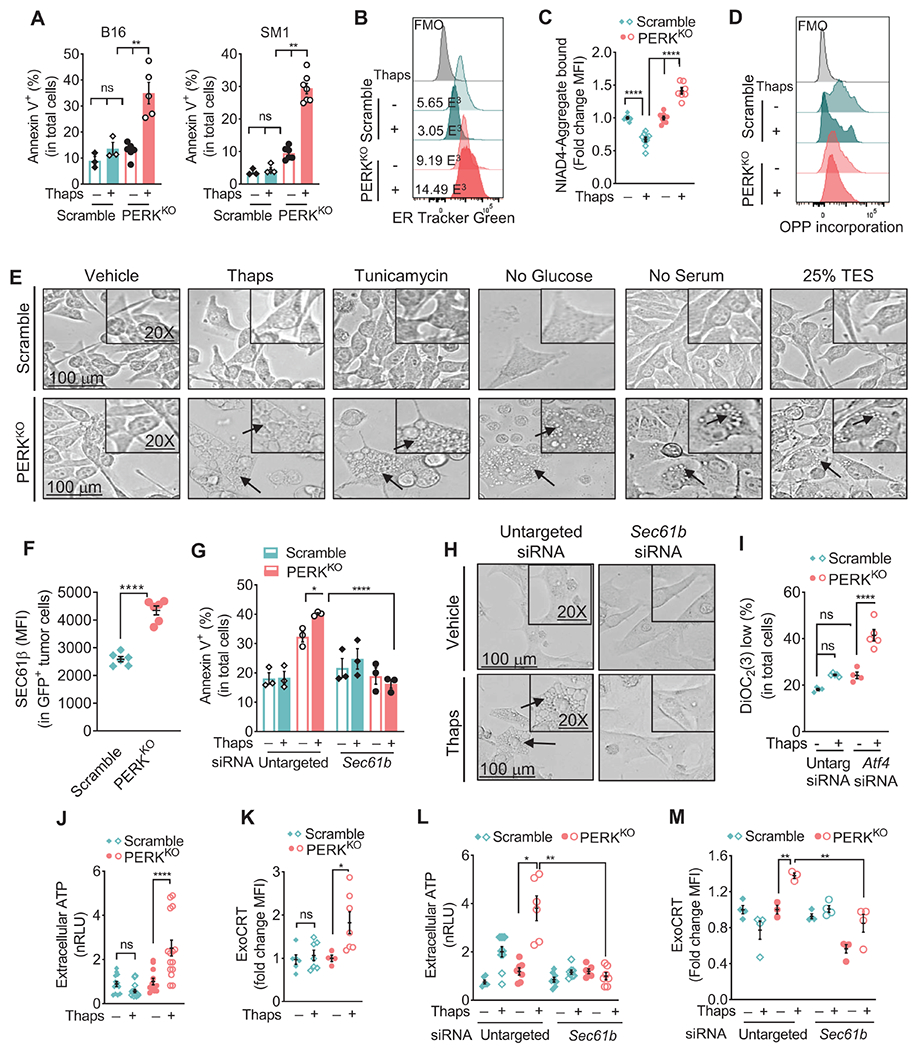
(A) Annexin V+ mean percentage ± SEM in Scramble and PERKKO B16 (left) or SM1 (right) tumors cultured for 24 hours with or without Thaps. n=3-7 of 3 independent repeats.
(B) Representative histograms from 3 distinct repeats showing ER Tracker Green in Scramble and PERKKO B16 cells treated as in (A).
(C) Mean fluorescence intensity (MFI) via flow cytometry ± SEM of NIAD4-bound protein aggregates in Scramble and PERKKO B16 cells treated as in (A). n=4-7/group of 3 independent repeats.
(D) Representative histograms from 3 repeats for fluorescent click chemistry-based detection of O-propargyl-puromycin (OPP) incorporation in Scramble and PERKKO B16 cells treated as in (A).
(E) Light microscopy morphology (100μm and 20X digital magnification) of Scramble and PERKKO B16 cells cultured in the presence of vehicle, Thaps, or Tunicamycin; in the absence of Glucose or Serum; or with 25% TES.
(F) SEC61β (MFI ± SEM) in eGFP+ Scramble and PERKKO B16 tumors from mice. n=6/group.
(G) Annexin V+ percentage ± SEM in Scramble and PERKKO B16 cells transfected with untargeted control or Sec61b siRNA and treated as in (A). n=3/group.
(H) Light microscopy morphology in PERKKO B16 cells transfected with mock or Sec61b siRNA prior to Thaps exposure.
(I) Percentage ± SEM of DiOC2 low cells in B16 cells transfected with untargeted or Atf4 siRNA and treated as in (A). n=4-5/group.
(J) ATP in supernatants from Scramble or PERKKO B16 cells incubated or not with Thaps. Data are the normalized relative light units (nRLU) ± SEM of n=12-15/group.
(K) ExoCRT fold change (MFI ± SEM) in Scramble or PERKKO B16 cells cultured as in (A). n=4-7/group.
(L-M) Extracellular ATP (L) and ExoCRT (M) ± SEM in Scramble or PERKKO B16 cells carrying mock or Sec61b siRNA and cultured with or without Thaps. n=6-10/group (L); n=3-4/group (M).
Statistics applied using one-way ANOVA (A, C, F, G, I – M) or Student’s t-test (F), *, p<0.05; **, p<0.01; ****, p < 0.0001. Please also see Figure S2.
Notably, PERKKO melanoma cells underwent ER mass enlargement, expanded misfolded protein accumulation, maintained active translation, and had augmented reactive oxygen species (ROS) levels after treatment with Thaps, compared to identically treated Scramble controls (Figure 2B–D; Figure S2J). A similar increase in ER tracker and ROS was noted in PERKKO B16 cells after culture in 25% tumor explant supernatants (TES) or under serum or glucose starvation (Figure S2K–L). Moreover, in response to ER stressors Thaps or Tunicamycin, starvation of glucose or serum, or TES, PERKKO B16 cells showed a distinct cellular morphology characterized by an extensive cytoplasmic vacuolization, intact nuclei, and retention of cellular adhesion (Figure 2E). A similar morphology was noted in Thaps-treated SM1 cells (Figure S2M). These alterations are consistent with the induction of paraptosis, a de novo translation-dependent form of cell death characterized by extensive cytoplasmic vacuolation and ER swelling in response to disrupted proteostasis (Sperandio et al., 2000). Treatment with de novo translation inhibitor cycloheximide prevented the Annexin V increase and paraptosis morphology in Thaps-treated PERKKO B16 cells, without impacting controls (Figure S2N–O). Although the signals regulating paraptosis remain unclear, impaired peptide transport through the ER via SEC61 translocon, a complex regulated by member SEC61β, has emerged as a key contributor (Lang et al., 2017). Higher levels of SEC61β were found in PERKKO B16 tumors from mice, or after treatment in vitro with different stressors, including Thaps, TES, or serum starvation (Figure 2F, Figure S2P). Moreover, Sec61b silencing prevented the Annexin V increase and paraptosis-related morphology in PERKKO tumors treated with Thaps compared to identically treated mock controls (Figure 2G–H; Figure S2Q), suggesting a role of SEC61β in PERKKO tumor cell death after ER stress. Next, we studied the role of ATF4 in the paraptosis occurring in PERKKO tumors. Lower ATF4 levels were noted in PERKKO tumors undergoing ER stress or collected from mice, compared to Scramble controls (Figure S2R–S). Also, similar to Thaps-exposed PERKKO tumors, Atf4 silencing in Scramble B16 cells treated with Thaps resulted in augmented mitochondrial membrane depolarization, paraptosis morphology, and elevated levels of SEC61β compared to untargeted siRNA treated cells (Figure 2I; S2T–V), indicating a potential role of ATF4 in the SEC61β-mediated cell death induced by unresolved ER stress in PERKKO tumors.
Because the immunostimulatory nature of the tumor cell death induced by PERK deletion, we next tested the role of SEC61β in the expression of ICD drivers. After Thaps treatment, PERKKO B16 cells showed higher production of ICD hallmarks, including the release of ATP and HMGB1, and ExoCRT induction, compared to controls (Figure 2J–K, Figure S2W). Furthermore, Sec61b siRNA abrogated the ATP release and ExoCRT induction in Thaps-treated PERKKO B16 cells (Figure 2L–M), indicating an upstream effect of SEC61β-driven paraptosis in the release of ICD mediators in ER stressed PERKKO tumors.
PERK ablation in melanoma drives ICD abscopal anti-tumor immunity
The induction of ICD mediators in cultured PERKKO cells motivated us to determine if PERKKO tumors underwent ICD in vivo. Analysis of PERKKO B16 cells from mice showed increased ER enlargement and protein aggregation, as well as higher production of extracellular ATP, HMGB1, and ExoCRT compared to controls (Figure 3A–E). In addition, elevation of type I IFN-linked transcripts, Ifnb1, Isg15, and Ifit3, but not Ifna1, Ifna7, Ifna13, Ifna14, and Ifna16, were detected in bulk PERKKO tumors compared to controls (Figure 3F, Figure S3A–B). Consistently, PERKKO tumor suspensions from mice exhibited higher levels of IFNβ1 protein, but not IFNα1, compared to Scramble counterparts (Figure S3C). Next, we tested the clinical impact of ICD in melanoma tumors from TCGA dataset by profiling an ICD metagene signature (Garg et al., 2016). Extended overall, progression free, and disease specific survival was found in patients with tumors having elevated ICD mRNA signature compared to those with reduced ICD score (Figure 3G, Figure S3D–E), suggesting an impact of ICD in human melanoma. To confirm the activation of ICD in PERKKO tumor-bearing mice, we tested the development of systemic anti-tumor effects at distant sites. Briefly, mice implanted with wildtype or PERKKO SM1 tumors for 10 days, were injected with a second wildtype or PERKKO SM1 tumor on the opposing flank (Figure 3H). Injection with control SM1 tumors did not confer growth delay to a challenge with wildtype SM1 tumors (Group 1) nor affected the rejection of PERKKO SM1 tumors (Group 2). In addition, initial implantation with PERKKO SM1 tumors promoted the elimination of contralaterally injected wildtype SM1 tumors (Group 3) and did not alter the spontaneous rejection of SM1 PERKKO tumors (group 4), indicating that PERK deletion in tumors triggered anti-tumor abscopal effects (Figure 3H). Next, we evaluated the development of memory responses against PERKKO tumors. C57BL/6 mice that had previously rejected PERKKO SM1 tumors were fully resistant to wildtype SM1 cells, as compared to tumor-naïve mice (Figure 3I). However, this protective effect did not occur against unrelated Lewis lung carcinoma (LLC) tumors (Figure 3J), indicating the induction of protective tumor-specific immune memory.
Figure 3: PERK deletion in cancer cells provokes ICD-driven anti-tumor immunity.
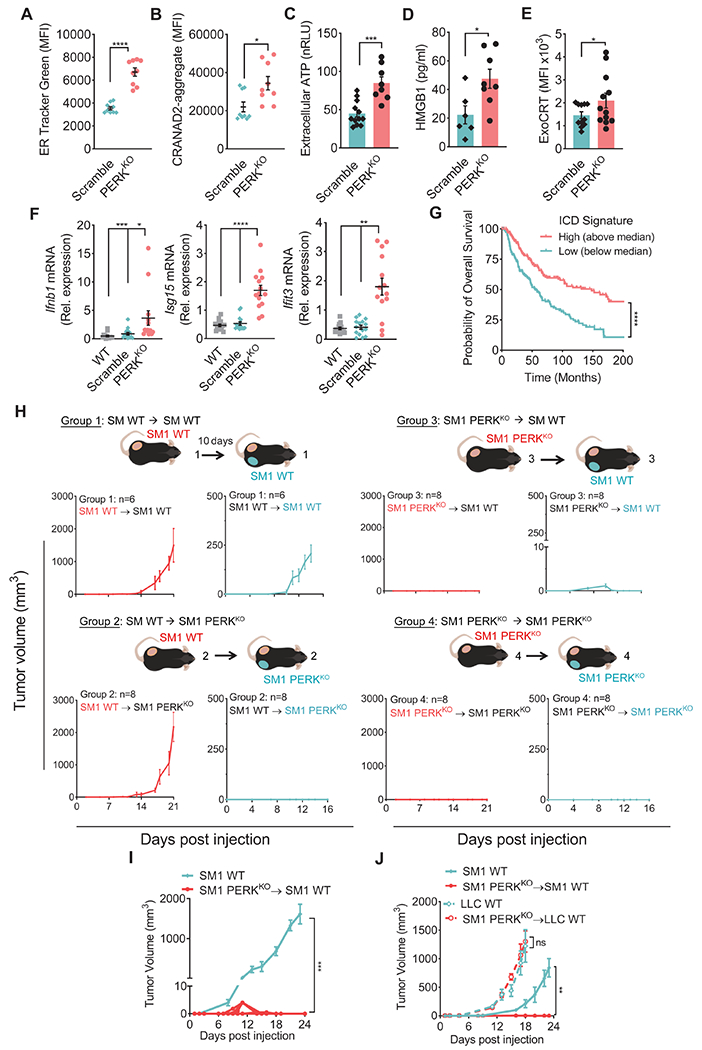
(A-B) ER Tracker Green (A) and CRANAD-labeled protein aggregates (B) by flow cytometry (MFI ± SEM) in CD45− cells from Scramble and PERKKO B16 tumors from mice. n=9/group.
(C-D) ATP (C) and HMGB1 (D) (mean ± SEM) in supernatants from resected Scramble or PERKKO B16 tumors cultured for 24 hours. n=8-12/group (C); n=6-8/group (D).
(E) ExoCRT (MFI ± SEM) in CD45− cells from freshly isolated Scramble or PERKKO B16 tumors. n=12/group.
(F) Relative expression of Ifnb1, Isg15 and Ifit3 mRNA ± SEM in bulk wildtype, Scramble or PERKKO B16 tumors from mice. n=13-15/group.
(G) Overall survival for Skin Cutaneous Melanoma patients (TCGA, PanCancer Atlas) stratified by the median ICD metagene signature score. Kaplan-Meier curves of overall survival for ICD high (above the median; n = 228) and low (below the median; n = 229). Gene signature calculation by GSVA.
(H) Mice injected with WT SM1 tumors on the left flank for 10 days later, were implanted with WT (Group 1) or PERKKO (Group 2) SM1 cells on the right flank. Mice were injected with SM1 PERKKO cells on the left flank, followed by injection 10 days later with WT (Group 3) or PERKKO (Group 4) SM1 tumors on the right flank. Tumor growth ± SEM. n=10/group.
(I) WT SM1 tumor growth ± SEM in naïve mice or mice that had rejected PERKKO SM1 tumors. n=10/group.
(J) Volume ± SEM for SM1 (left flank; SM1 WT) and LLC (right flank; LLC WT) tumors in naïve mice and mice that previously rejected PERKKO SM1 tumors. n=5/group.
Statistics applied by one-way ANOVA (F, I, J), Student’s t-test (A – E), or log-rank test (G). *, p<0.05; **, p<0.01; ***, p < 0.001; ****, p < 0.0001. Please also see Figure S3.
PERK ablation in tumors incites the expansion of MoDCs
Aberrant myelopoiesis in tumor-bearing hosts promotes expansion of MDSCs and restricts the maturation and function of immunostimulatory DCs (Ugel et al., 2021; Veglia et al., 2021). Reduced proportion and T cell-suppressive activity of MDSCs concomitant with higher DC frequency were found in B16 PERKKO tumors, compared to Scramble and wildtype (WT) controls (Figure 4A–B, Figure S4A). Next, we investigated the role of CD11c+ DCs in directing the anti-tumor responses in PERKKO tumors. CD11c+-DCs deletion in ItgaxDTR/EGFP mice upon treatment with diphtheria toxin (DT) restored B16 PERKKO tumor growth, while it delayed growth in Scramble controls (Figure 4C; Figure S4B). Next, we tested the potential of DCs from PERKKO B16 tumors to spontaneously activate gp100-specific Pmel T cells. In agreement with the ability of DCs from PERK-null B16 tumors to present engulfed tumor antigens to T cells, DCs from PERKKO tumors induced higher expression of activation markers CD44 and CD69 in co-cultured naïve Pmel CD8+ T cells, compared to DCs from Scramble controls (Figure 4D). These results suggest the key role of DCs in promoting anti-tumor responses against PERKKO tumors.
Figure 4: PERK in cancer cells regulates accumulation of MoDCs.
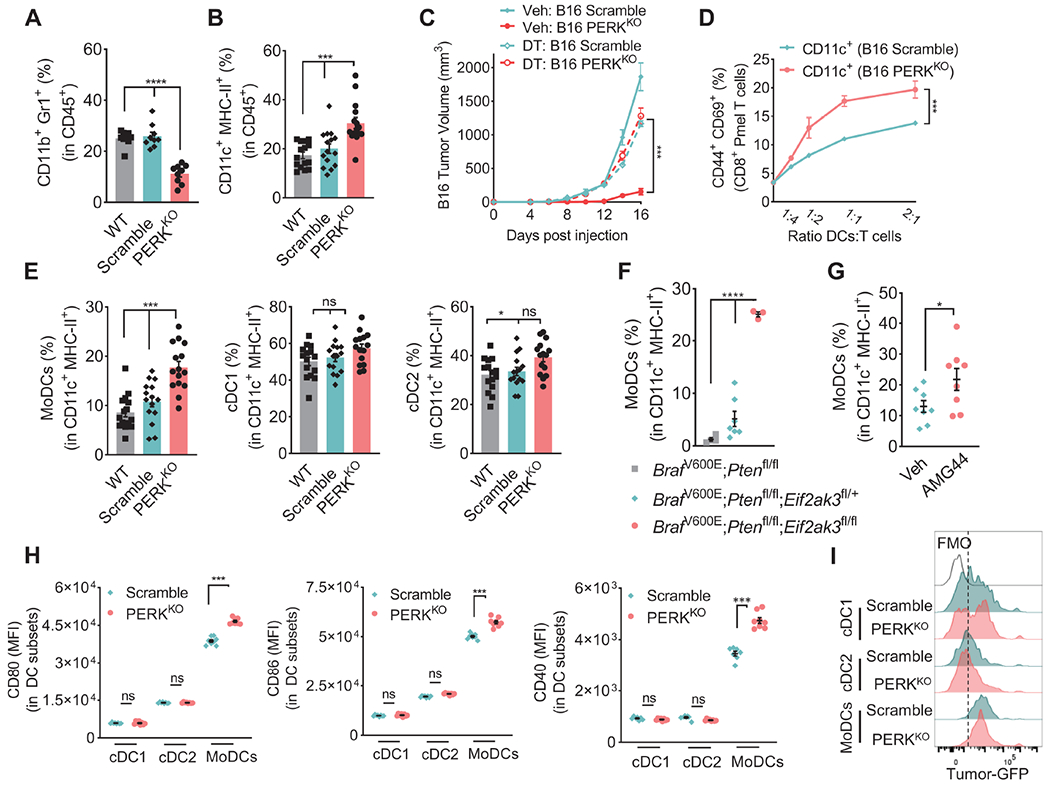
(A-B) Percentage of CD11b+Gr1+ (A) and CD11c+MHC-II+ (B) cells ± SEM in CD45+ cells in tumors from mice bearing wildtype (WT), Scramble or PERKKO B16 tumors for 17 days. n=9/group (A); n=15/group (B).
(C) Volume ± SEM of Scramble and PERKKO B16 tumors implanted into ItgaxDTR/EGFP mice treated with vehicle or diphtheria toxin. n=8/group.
(D) Percentage of CD44+CD69+ ± SEM in CD8+ Pmel T cells co-cultured for 48 hours with sorted CD11c+ cells from Scramble or PERKKO B16 tumors (ratio 1:1/4). n=3/group.
(E) Proportion ± SEM of MoDCs, cDC1, and cDC2 in CD11c+MHC-II+ cells from WT, Scramble, or PERKKO B16 tumors. n=15/group.
(F-G) MoDCs percentage ± SEM within CD11c+MHC-II+ cells in tumors from tamoxifen-treated Eif2ak3fl/+ or Eif2ak3fl/fl BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice vs. BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice (F) or from tamoxifen-exposed BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice treated with vehicle (Veh) or AMG44 (G). n=3-7/group (F); n=8/group (G).
(H) Levels of CD80, CD86, or CD40 (MFI ± SEM) in cDC1, cDC2, and MoDCs from Scramble and PERKKO B16 tumors. n=8/group.
(I) Representative histograms of eGFP in intra-tumor-MoDCs, cDC1 or cDC2 from B16-eGFP+ Scramble and B16-eGFP PERKKO-bearing mice. Representative of n=5/group.
Statistics applied using one-way ANOVA (A – C, E, F) or Student’s t-test (D, G, H), *, p<0.05; ***, p < 0.001; ****, p < 0.0001. Please also see Figure S4.
Next, we compared the expansion of DC subsets in PERKKO and Scramble tumors (Figure S4C). Augmented proportion of MoDCs (Coillard and Segura, 2021; Sharma et al., 2018), but not conventional DCs (cDC1 and cDC2) were detected in PERKKO B16 tumors compared to Scramble or WT controls (Figure 4E). Increased expansion of MoDCs was also found in melanoma tumors from tamoxifen-exposed BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice lacking one or two alleles of Eif2ak3 or treated with AMG44, compared to PERK-intact counterparts (Figure 4F–G). Moreover, PERK deletion in B16 tumor cells increased the expression of co-stimulatory molecules CD80, CD86, and CD40 in intra-tumor MoDCs, without altering their expression on cDC1 and cDC2 subsets (Figure 4H). Next, we tested the capacity of DC subsets to engulf tumor products after implantation with eGFP-expressing Scramble or PERKKO B16 tumors. Compared to cDC1 or cDC2, MoDCs showed higher eGFP expression within the TME, which was similar in Scramble and PERKKO tumors (Figure 4I). These results show the expansion of MoDCs in PERKKO tumors, which carry higher capacity to engulf tumor products and immunostimulatory potential.
PERK in tumor cells controls differentiation of myeloid precursors into MoDCs
MoDCs have been reported to arise from cMoPs expanding in bone marrow, spleen, and tumors (Cortez-Retamozo et al., 2012; Sharma et al., 2021; Sharma et al., 2018; Swirski et al., 2009). Although no changes were found in bone marrow, we detected a higher frequency of cMoPs in spleens and tumors of mice carrying PERKKO B16 tumors compared to Scramble controls (Figure 5A; Figure S5A). A similar elevation in tumor-cMoPs was found in tamoxifen-treated BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 tumor-bearing mice lacking Eif2ak3 or receiving AMG44, compared to PERK-active controls (Figure 5B). Also, increased proportions of cMoPs expressing MoDC markers were detected in tumors and spleens of mice carrying PERKKO B16 tumors compared to controls (Figure 5C), as well as in tumors from BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 tumor-bearing mice treated with AMG44 (Figure S5B). To determine whether PERK deletion in tumor cells primes differentiation of cMoPs into MoDCs, fluorescently-labeled splenic cKit+ precursors from tumor-free mice were transferred into Scramble or PERKKO tumors established in mice. Elevated frequency of transferred cKit+ cells acquiring MoDCs markers was detected in PERKKO tumors compared to the same precursors delivered into Scramble controls (Figure 5D). Moreover, higher development of MoDCs was noticed in GM-CSF-treated splenic cKit+ cells from PERKKO-bearing mice compared to counterparts from controls (Figure 5E), suggesting the commitment of cMoPs into MoDCs in PERKKO tumor-bearing mice. Next, we assessed the contribution of spleen-originated cMoPs in the anti-tumor effects induced by PERK deletion. Splenectomy of mice prior to tumor injection overcame the anti-tumor effects and blunted the expansion of tumor-related cMoPs and MoDCs induced by PERK deletion in B16 tumors (Figure 5F–H). Also, transfer of splenic cKit+ precursors into splenectomized mice bearing PERKKO tumors restored the anti-tumor phenotype, while it did not impact tumor progression in Sham surgery control mice bearing Scramble or PERKKO tumors (Figure 5F). Transfer of splenic cKit+ cells additionally increased the expansion of intra-tumor cMoPs and MoDCs in splenectomized mice bearing PERKKO tumors (Figure 5G–H). Thus, results suggest the role of spleen-originated cMoPs in the expansion of tumor-MoDCs and in the anti-tumor effects observed in PERKKO tumors.
Figure 5: PERK in tumor cells controls cMoP to MoDC ontogeny.
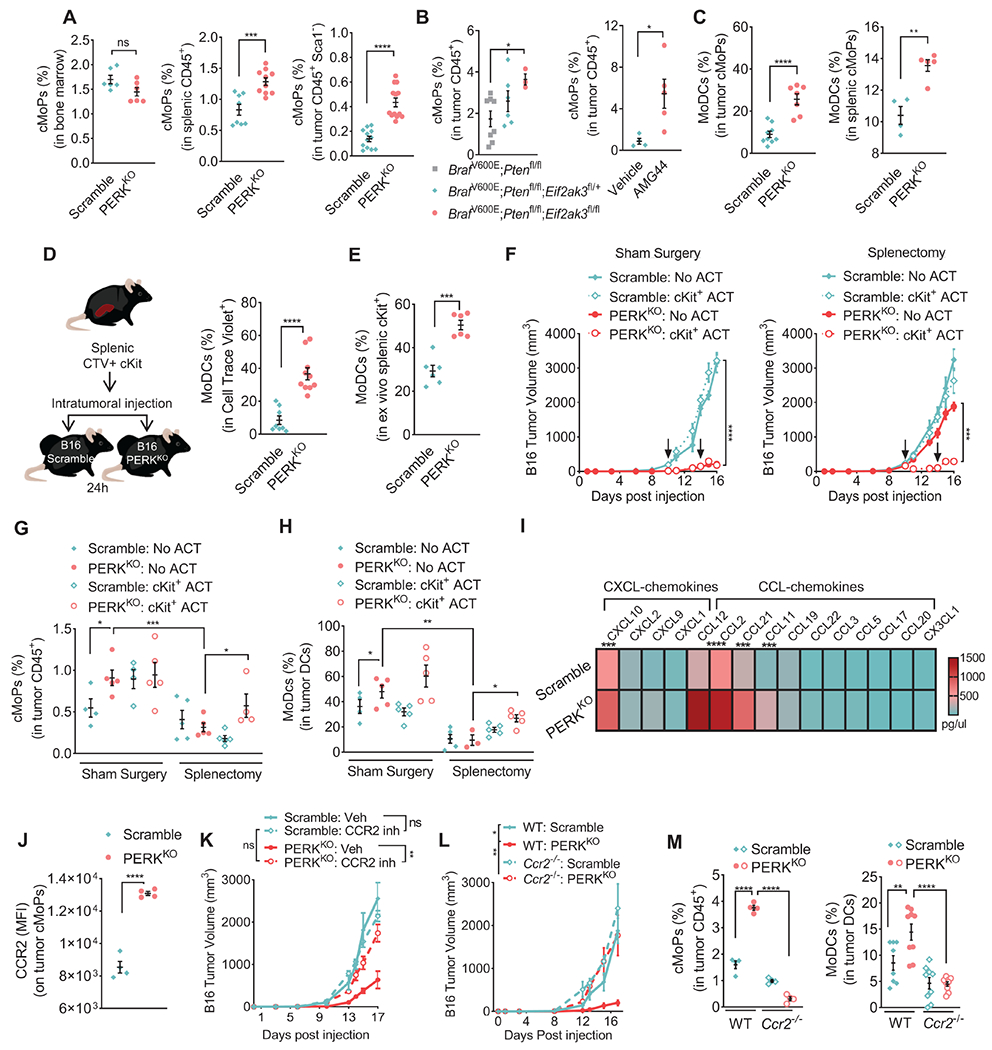
(A) Percentage ± SEM of cMoPs (cKit/CD117+CSFR1/CD115LowCD135−Ly6C+) in bone marrows (left), spleens (middle), and tumors (right) from mice implanted for 17 days with Scramble or PERKKO B16 tumors. n=7/group (left), n=8-10/group (middle), n=12-14 (right).
(B) Percentage ± SEM of cMoPs within CD45+ cells in tumors from tamoxifen-exposed Eif2ak3fl/+ or Eif2ak3fl/fl BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice vs. BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 controls (left) or in tumors from tamoxifen-exposed BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice treated with vehicle or AMG44 (right). n=3-8/group (left), n=4-5/group (right).
(C) Percentage ± SEM of MoDCs (CD11b+CD11c+MHC-II+Ly6C+CD103+) within cMoPs from tumors (left) and spleens (right) of mice bearing Scramble and PERKKO B16 tumors for 17 days. n=7-9/group (left), n=4-5/group (right).
(D) Cell trace violet (CTV)-labeled splenic cKit+ cells were transferred into Scramble or PERKKO B16 tumors (left), and percentage of MoDCs ± SEM in CTV+ cells tested in tumors after 24 hours by flow cytometry (right). n=8-10/group (left), n=6/group (right).
(E) Splenic cKit+ cells from Scramble or PERKKO B16-bearing mice were cultured for 48 hours with mGM-CSF and tested for MoDCs differentiation. Data show percentage of MoDCs ± SEM in cKit+ cells.
(F-H) Mice received Sham Surgery or splenectomy and 14 days later, Scramble or PERKKO B16 tumors were injected on the opposite flank. A group of mice additionally received adoptive transfer (ACT) of splenic cKit+ cells on days 9 and 14 post-tumor injection (arrows). Tumor volume ± SEM (F), and percentage ± SEM of intra-tumor cMoPs (G) or MoDCs (H) were then tested. n=5/group.
(I) Chemokine multiplex heatmap from Scramble or PERKKO B16 tumor homogenates from 5 mice/group.
(J) CCR2 MFI ± SEM in intra-tumor cMoPs from Scramble and PERKKO B16 tumors. n=4/group.
(K) Tumor growth ± SEM in mice bearing Scramble or PERKKO B16 tumors and treated with vehicle or BMS-CCR2-22 (0.5 mg/kg) daily starting since tumor inoculation. n=5/group.
(L) Tumor volume ± SEM in Scramble or PERKKO B16 tumors injected into wildtype or Ccr2−/− mice. n=5/group.
(M) Percentage ± SEM of intra-tumor cMoPs (in CD45+, left) and MoDCs (in CD11c+MHC-II+, right) in wildtype and Ccr2−/− mice bearing Scramble or PERKKO B16 tumors. n=4/group (left), n=8-10/group (right).
Statistics were applied using one-way ANOVA (B left, F – H, K – M) or Student’s t-test (A, B right, C, D, E, I, J) *, p<0.05; **, p<0.01; ***, p < 0.001; ****, p < 0.0001. Please also see Figure S5.
We also sought to identify the mediators regulating the migration of cMoPs into PERKKO tumors. Increased CCL2, CCL12, CXCL10, CCL21, and CCL11 levels were noted in PERKKO tumor homogenates compared to controls (Figure 5I; Figure S5C). Because CCL2 and CCL12 bind to CCR2 and direct chemotaxis of cells into tumors (Flores-Toro et al., 2020), we next studied the role of CCR2 in the trafficking of cMoPs into tumors. cMoPs from PERKKO tumors exhibited higher CCR2 expression compared to those from controls (Figure 5J). Also, treatment of PERKKO B16-bearing mice with CCR2 antagonist, BMS-CCR2-22 (Cherney et al., 2008), restored tumor growth and impaired intra-tumor expansion of cMoPs and MoDCs (Figure 5K, Figure S5D). Similarly, Ccr2 deletion in mice rescued PERKKO tumor growth and blunted the accumulation of cMoPs and MoDCs in PERKKO tumors (Figure 5L–M), without altering splenic cMoPs expansion (Figure S5E), suggesting the role of CCR2 in the homing of cMoPs into PERKKO tumors.
Type I IFN signaling drives anti-tumor immunity to PERKKO tumors
To study differences between myeloid precursors in Scramble and PERKKO tumors, intra-tumoral cKit+ cells were compared via RNA-seq (Figure S6A). Differential gene expression and gene set enrichment analysis (GSEA) found distinctly induced transcripts related to MoDC commitment and type I IFN signaling in cKit+ precursors from PERKKO B16 bearing mice compared to controls (Figure 6A–B, Figure S6B). To substantiate the role of type I IFN signaling in the actions induced by PERK deletion in tumors, we next tested the effects of an anti-type I IFN receptor 1 (IFNAR1) blocking antibody. Treatment with anti-IFNAR1 restored tumor growth and fully prevented the expansion of cMoPs and MoDCs in PERKKO tumors compared to isotype-treated counterparts (Figure 6C–D), suggesting the major role of IFNAR1 in PERKKO anti-tumor effects.
Figure 6: Host-derived Type I IFN drives anti-tumor immune responses in PERKKO tumors.
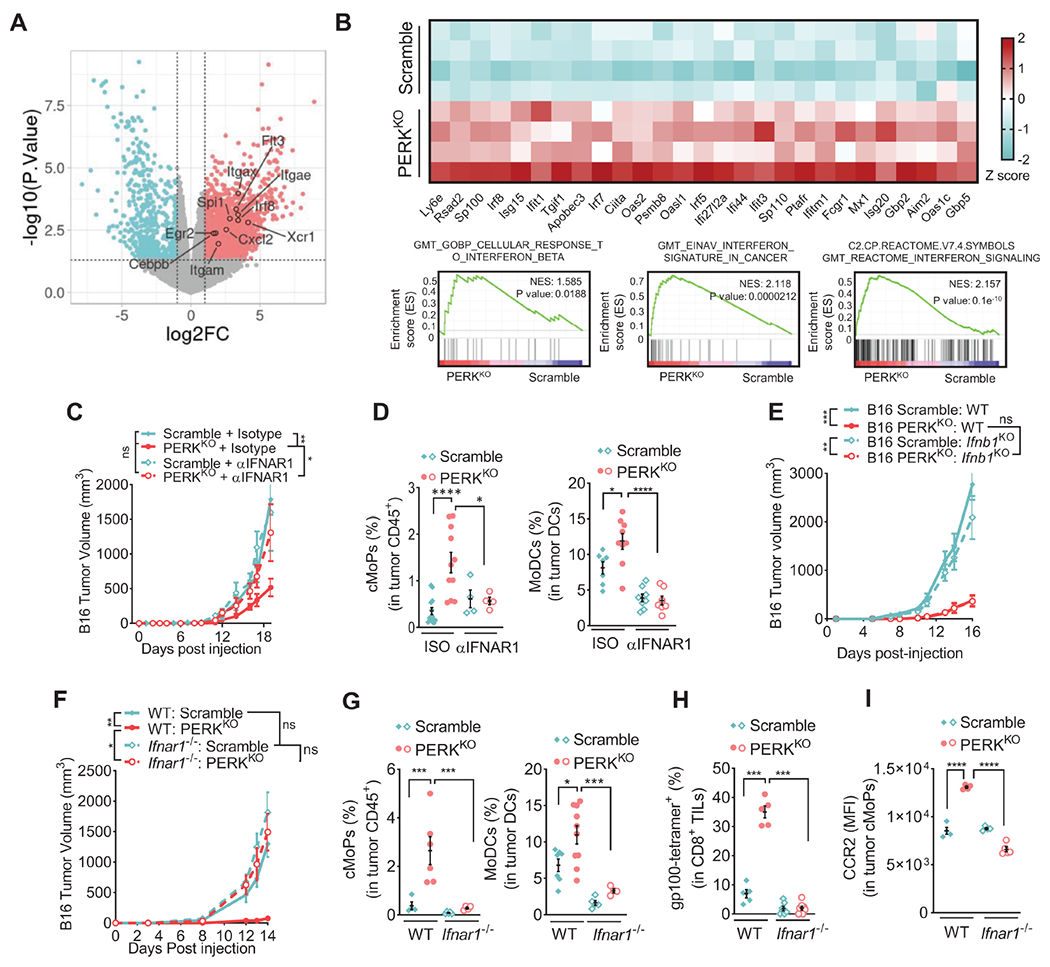
(A) Volcano plot indicating MoDC-linked transcripts from RNA-seq on cKit+ cells from control or PERKKO B16 tumors. Transcripts differentially (p < 0.05) elevated after PERK deletion are shown in red (log2 fold change > 1), and downregulated transcripts are in teal (log2 fold change < −1). n=4/group.
(B) Selected RNA-seq transcripts of cKit+ cells from Scramble and PERKKO B16 tumors. Heatmap (top) showing differentially expressed type I IFNs-regulated transcripts (log2 transformed then z-score normalized). GSEA of IFN pathways from MSigDB (bottom).
(C) Tumor volume ± SEM in mice bearing Scramble or PERKKO B16 tumors and treated with αIFNAR1 antibody or isotype (ISO). n=10/group.
(D) Percentage ± SEM of cMoPs in tumor-CD45+ (left) and MoDCs in tumor-CD11c+MHC-II+ cells (right) from (C). n=4-13/group (left), n=7-9/group (right).
(E) Tumor growth ± SEM in Scramble, PERKKO, Scramble Ifnb1KO and PERKKO;Ifnb1KO B16 tumors implanted into mice. n=5/group.
(F) Volume ± SEM of Scramble or PERKKO B16 tumors injected into wildtype (WT) or Ifnar1−/− mice. n=7-10/group.
(G) Percentage ± SEM of tumor-linked cMoPs (left) and MoDCs (right) in Scramble or PERKKO B16 tumors implanted into WT or Ifnar1−/− mice. n=4-7/group.
(H) Proportion ± SEM of EGSRNQDWL-H-2Db-tetramer+ CD8+ T cells in tumors from WT or Ifnar1−/− mice injected with Scramble or PERKKO B16 tumors. n=4-7/group.
(I) CCR2 (MFI ± SEM) in tumor-cMoPs from Scramble or PERKKO B16 tumors implanted into WT or Ifnar1−/− mice. n=4-5/group.
Statistics applied using one-way ANOVA (C – I), *, p<0.05; **, p<0.01; ***, p<0.001; ****, p < 0.0001. Please also see Figure S6.
Considering that dying tumor cells could represent the source of type I IFNs, we next tested the production and signaling of type I IFN in PERKKO tumor cells. Comparable IFNAR1 and IFNβ1 protein levels were found in PERKKO B16 tumor cells from mice or treated with Thaps, compared to controls (Figure S6C–D). Also, blockade of IFNAR1 did not alter the further elevation in Annexin V noticed in Thaps-treated PERKKO B16 cells (Figure S6E). Moreover, CRISPR/Cas9-based deletion of Ifnb1 (IFNβ1KO) failed to restore growth of PERKKO B16 tumors in mice (Figure 6E, Figure S6F), indicating that intrinsic IFNβ1 production did not play an obligatory role in the delayed growth of PERKKO tumors. To alternatively study the contribution of host-derived type I IFN signaling, we implanted Scramble and PERKKO tumors into Ifnar1-null mice. Restored tumor growth, as well as abrogated expansion of cMoPs, MoDCs, and gp100-reactive CD8+ T cells were found in PERKKO tumors injected into Ifnar1-deficient mice compared to counterparts injected into wildtype mice (Figure 6F–H, Figure S6G). Also, lower expression of cMoP-migration driver CCR2 and reduced Ccl2 mRNA levels were detected in PERKKO tumors injected into Ifnar1-null mice compared to controls (Figure 6I, Figure S6H). Thus, our results demonstrate the role of host-derived IFNAR1 signaling in the anti-tumor immune responses raised in PERKKO tumors.
Type I IFN in DCs primes commitment of cMoPs into MoDCs via STAT1
Given that multiple stromal subsets could secrete type I IFN, we tested as a proxy of type I IFN production, the cellular source of IFNβ1 in PERKKO tumors using IFNβ1-EYFP reporter mice. While macrophages, MDSCs, and cMoPs had similar IFNβ1-EYFP expression within Scramble and PERKKO tumors, we found higher IFNβ1-EYFP in DCs from PERKKO tumors compared to controls (Figure 7A). Within DCs, MoDCs from PERKKO B16 tumors exhibited higher IFNβ1-EYFP expression compared to cDC1 or cDC2 (Figure 7B, Figure S7A). Also, splenic MoDCs from mice bearing Scramble and PERKKO tumors did not express substantial IFNβ1 levels (Figure 7C, Figure S7B), indicating that IFNβ1 induction occurs preferentially in intra-tumor MoDCs. Next, we determined whether exposure to ICD mediators released from PERKKO tumors provoked the production of IFNβ1 in DCs. Higher IFNβ1-EYFP expression was found in DCs from spleen, or generated bone marrow, after exposure to supernatants from PERKKO B16 cells pre-treated with Thaps, compared to those cultured with explants from Thaps or vehicle treated Scramble controls (Figure 7D; Figure S7C). To evaluate the specific role of ICD drivers in the induction of IFNβ1, we neutralized signaling related with DNA, HMGB1 or ATP. Exposure of IFNβ1-EYFP DCs to media pre-treated with DNAse failed to prevent induction of IFNβ1-EYFP (Figure S7D), ruling out an effect of DNA-linked signals. In agreement, conditional deletion of the DNA sensor STING in myeloid cells (Tmem173fl/flLyz2-cre mice) failed to restore growth of PERKKO tumors (Figure S7E). Notably, treatment with antagonists of the ATP responsive P2X purinoreceptors (P2XRs), Evan’s Blue (EvB) or pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS), as well as anti-HMGB1 impaired IFNB1-EYFP induction in DCs exposed to supernatants from ER-stressed PERKKO tumors (Figure 7E; Figure S7F). Thus, ER stressed PERKKO tumors release ICD-related ATP and HMGB1, which trigger IFNβ1 expression in MoDCs.
Figure 7: Type I IFN from DCs promotes migration and commitment of cMoPs into MoDCs in PERK-null tumors via STAT1.
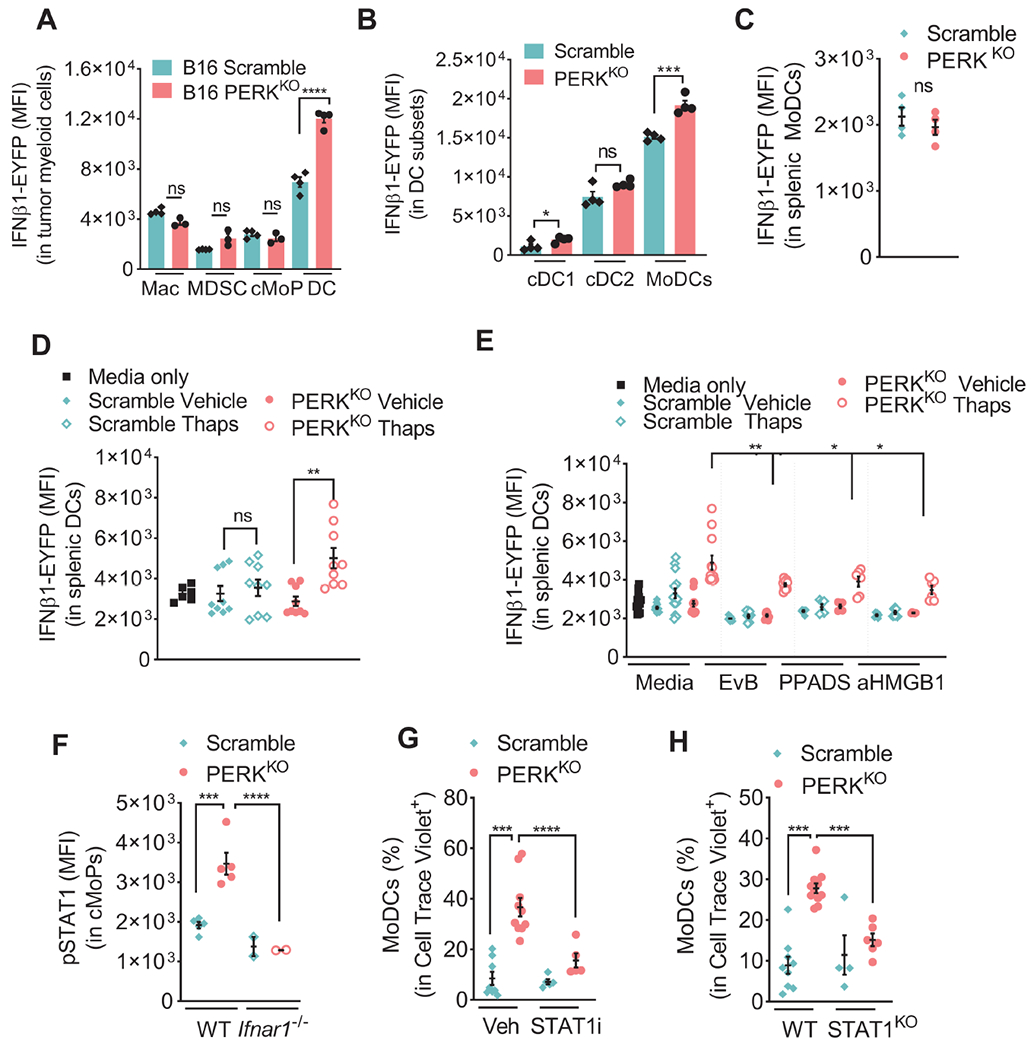
(A-B) IFNβ1-EYFP (MFI ± SEM) in Macrophages, MDSCs, cMoPs, and DCs (A), and cDC1, cDC2, or MoDCs (B) from Scramble or PERKKO B16 tumors from mice. n=3-4/group.
(C) IFNβ1-EYFP (MFI ± SEM) in splenic MoDCs from Scramble or PERKKO B16 tumor-bearing mice. n=4/group.
(D) IFNβ1-EYFP in splenic CD11c+ cells (MFI ± SEM) cultured with supernatants from Scramble or PERKKO B16 cells previously treated or not with Thaps, extensively washed, and cultured for 18 hours in regular media. n=6-9/group.
(E) IFNβ1-EYFP MFI ± SEM in splenic CD11c+ cells exposed as in (D) and pretreated with EvB, PPADS or anti-HMGB1. n=6/group.
(F) pSTAT1 (MFI ± SEM) in intra-tumor cMoPs from wildtype (WT) or Ifnar1−/− mice bearing Scramble or PERKKO B16 tumors. n=2-5/group.
(G-H) Cell trace violet (CTV)-labeled splenic cKit+ cells from tumor-free mice were treated with vehicle or fludarabine (STAT1i, 100 μM) (G), or collected from WT or STAT1-null mice (H) and transferred into Scramble or PERKKO B16 tumors. Percentage of MoDCs in CTV+ cells tested in tumors 24 hours later. n=5-10/group (G); n=4-11/group (H).
Statistics were applied using one-way ANOVA (E – H) or a Student’s t-test (A – D), *, p<0.05; **, p<0.01; ***, p < 0.001; ****, p < 0.0001. Please also see Figure S7.
To further test the postulate that differentiation of cMoPs into MoDCs is regulated by type I IFN-linked signals in PERKKO tumors, we focused on STAT1, which is primed by IFNAR1 signaling and impacts DC ontogeny (Gautier et al., 2005). Heightened phospho-STAT1 levels were found in cMoPs from PERKKO B16 tumors compared to those from Scramble controls, which also depended on IFNAR1 expression (Figure 7F). Moreover, GSEA of cKit+ cells from PERKKO B16 tumors identified higher mRNA expression of genes regulated by STAT1 compared to Scramble controls (Figure S7G). To test if active STAT1 promoted cMoPs to MoDCs commitment, we treated fluorescently labeled cKit+ precursors with STAT1 inhibitor fludarabine, followed by their transfer into Scramble or PERKKO B16 tumors. Similarly, we transferred fluorescently labeled cKit+ precursors from STAT1KO mice and evaluated their differentiation into MoDCs in tumors. STAT1 inhibition or deletion impaired the differentiation of the transferred precursors into MoDCs in PERKKO tumors (Figure 7G–H). These results substantiate the role of IFNAR1 signaling-linked STAT1 in the commitment of cMoPs into MoDCs in PERKKO tumors.
Discussion
Here, we elucidated the mechanistic role of PERK in malignant cells in the evasion of anti-tumor immunity. PERK protects ER-stressed cancer cells from paraptosis-induced ICD, thereby limiting type I IFN-driven commitment of cMoPs into MoDCs and subsequent protective T cell responses. Therefore, PERK inhibition could represent a promising therapeutic strategy to restore protective immunity in tumors and to augment the effects of cancer immunotherapy.
Activation of PERK signaling transiently promotes survival in ER stressed cancer cells through the induction of cytoprotective autophagy (Hart et al., 2012; Ma et al., 2014). Additional reports highlighted a role of PERK in tumor cell dormancy, malignant cell proliferation, and tumor vascularization processes (Bobrovnikova-Marjon et al., 2010; Ranganathan et al., 2008; Wang et al., 2012). Less known is the processes whereby PERK ablation promotes tumor cell death. We report that PERK deletion primes paraptosis-associated ICD in stressed melanoma cells. Paraptosis is triggered by proteostasis alterations, a process heavily dependent on SEC61 translocon complex (Elia et al., 2019; Monel et al., 2017). Dysregulation of SEC61 complex by regulatory subunit SEC61β drives paraptosis by altering the trafficking of proteins through the ER (Lang et al., 2017). Our data show that PERK and ATF4 serve as upstream negative regulators of SEC61β-mediated paraptosis. However, the mechanistic insights of the PERK, ATF4, and SEC61β crosstalk in the regulation of anti-tumor immunity remain to be elucidated.
Transmissible ER stress from cancer cells into myeloid cells has been reported to predominantly impair anti-tumor immune responses (Mahadevan et al., 2011). Here, we show that PERK ablation in cancer cells undergoing unresolved ER stress provoked transmissible activation of protective immunity through the release of ICD drivers, production of type I IFN by DCs, CCR2-dependent intra-tumor migration and STAT1-driven differentiation of cMoPs into MoDCs, and activation of anti-tumor T cells. Complementarily, our previous reports demonstrated that intrinsic elimination of PERK in tumor-associated MDSCs reprogrammed them into cells that prime T cell responses (Mohamed et al., 2020), while PERK deletion in T cells additionally increased their anti-tumor potential (Cao et al., 2019; Hurst et al., 2019). Also, PERK signaling in splenic myeloid precursors regulates their differentiation into immunosuppressive subsets (Liu et al., 2022). Thus, inhibition of PERK in tumor beds is likely to: 1) drive direct anti-tumor effects (Atkins et al., 2013; Bi et al., 2005); 2) reprogram MDSCs into immune stimulatory cells (Mohamed et al., 2020); 3) boost cytotoxic activity in intra-tumor T cells (Cao et al., 2019); and 4) restrict tumor vascularization.
Activation of ICD in cancer cells undergoing unresolved stress has been identified to depend on eIF2α phosphorylation (Galluzzi et al., 2020; Kepp et al., 2015), which can be triggered by PERK. Instead, we observed that release of HMGB1 and ATP from dying ER stressed PERK-null tumor cells correlated with lower phospho-eIF2α and activated the production of type I IFN by DCs. Thus, while the release of ICD drivers under specific cell death conditions could depend on phospho-eIF2α, the induction of protective immunity in PERKKO tumors undergoing ICD occurs through a complex system that integrates stromal populations, including cMoPs, MoDCs, and T cells. Consistently, phospho-eIF2α independent induction of ICD has been reported in multiple contexts, including anthracycline treatment in colon cancer (Obeid et al., 2007), oncolytic peptide-induced ICD in sarcoma (Pasquereau-Kotula et al., 2018), and oxaliplatin or mitoxantrone directed ICD (Bezu et al., 2018). Elucidation of the immune effects induced by ICD mediated through phospho-eIF2α dependent and independent mechanisms could enable the discovery of impactful strategies for improving immunotherapy in cancer patients.
Collectively, our results demonstrate the key role of PERK signaling in tumor cells in the evasion of protective immunity. Although these findings predominantly pertain to melanoma, other tumor types which exhibit UPR activation may possess a similar susceptibility to PERK-targeted therapy. Also, interventions able to intercept adaptation to stress may sensitize cancer cells to intrinsic and extrinsic stressors, including those triggered by radiation therapy and chemotherapy.
STAR METHODS text
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paulo C. Rodriguez, PhD (Paulo.Rodriguez@Moffitt.org).
Materials Availability
The authors declare that all the results supporting the findings of this study are available within the paper and its Supplemental Figures.
Data and Code Availability
RNA-seq from cMoPs are publicly available at the GEO repository with accession GSE206783. Datasets analyzed were TCGA SKCM, an internal Moffitt melanoma cohort (doi: 10.1158/1055-9965.EPI-20-0307), and multiple datasets from ICI treated patients: PRJEB23709, GSE78220, GSE91061.
For survival analysis, we used the coding tool DRPPM-PATH-SURVEIOR: https://github.com/shawlab-moffitt/DRPPM-PATH-SURVEIOR.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Experiments using mice were developed through an approved Institutional Animal Care and Use Committee (IACUC) protocol (IS00008833) and an active Institutional Biosafety Committee (IBC) study (#PROT02020-043), both reviewed by the Integrity and Compliance board at the University of South Florida and Moffitt Cancer Center. Thus, presented work has complied with all the relevant ethical regulations for animal testing and research. Wildtype C57BL/6J mice (6 to 8 weeks) were from Envigo (Huntingdon, UK). Rag1−/− mice (NOD.129S7 (B6)-Rag1tm1Mom/J), Ccr2−/− mice (B6.129S4-CCR2tm1lfc/J), Stat1−/− mice (B6.129S(Cg)-Stat1tm1Dlv/J), Ifnar1−/− mice (B6(Cg)-Ifnar1tm1.2Ees/J), Ifnar1fl/fl mice (B6(Cg)-Ifnar1tm1.1Ees/J), Lyz2-cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J), ItgaxDTR/EGFP mice (B6.FVB-1700016L21RikTg (Itgax-DTR/EGFP)57Lan/J), Tmem173fl/fl mice (B6;SJL-STING1tm1.1Camb/J), EYFP-Ifnb reporter mice (B6.129-Ifnb1tm1Lky/J), Eif2ak3fl/fl mice (Eif2ak3tm1.2Drc/J), and BrafV600E;Ptenfl/fl tamoxifen driven Tyrosinase-Cre mice (B6.Cg-Tg(Tyr-Cre/ERT2)13Bos Braftm1Mmcm;Ptentm1Hwu/BosJ) mice were from the Jackson Laboratories (Bar Harbor, ME). Eif2ak3fl/+;BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice were created after breeding Eif2ak3fl/+ mice with BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice. Tmem173fl/fl;Lyz2-cre mice were developed after breeding Tmem173fl/fl and Lyz2-cre mice. Mice of both sexes were randomly assigned to experimental groups and maintained under specific pathogen-free conditions prior and during to use between 6-10 weeks of age.
Human Materials
All human materials and studies were covered through approval by the Institutional Review Board (IRB) exempt protocol #19223, previous reviewed by the Regulatory Affairs Committee Board at Moffitt cancer Center. Two tissue microarrays (TMA) containing a total of 133 metastatic melanoma tumor cores were used. Tissue Microarray Melanoma 3 (MCC-50368, n=96) was obtained from Moffitt Cancer Center Biorepository and Tissue Microarray Melanoma SKU: 69572925-2925 was from TriStar Technology Group (n=37). All de-identified patients signed approved consent forms. TMAs were composed of 4 μm section of primary and metastatic melanoma patient samples on positively charged slides. Survival analyses were accomplished in different melanoma datasets, including TCGA (n=457 patients), Moffitt Cancer Center (118, but 1 patient without survival information; n= 117), and a cohort of patients receiving immunotherapy (n=75 patients, but 6 patients without survival information, n= 69; and 7 patients without annotated information of ICI response, n=68) (Gide et al., 2019; Hugo et al., 2016; Riaz et al., 2017).
Cell Lines
B16-F10 (#CRL-6475), Lewis lung carcinoma (LLC; #CRL-1642), and SM1 (Koya et al., 2012) provided by Dr. R. Koya (University of Chicago) were cultured in RPMI-1640 supplemented with 2 mM L-glutamine, 25 mM HEPES, 100 U/ml Penicillin/Streptomycin, 5 μM β-mercaptoethanol and 10% heat-inactivated Fetal bovine serum (FBS), and maintained at 37°C in a humidified incubator with 5% CO2. Cell lines were routinely screened and validated to be mycoplasma-free using the Universal Mycoplasma Detection Kit (#30-1012K, ATCC) and maintained in culture for fewer than 10 passages. For genetic editing of Eif2ak3 and Ifnb1, B16 and SM1 cells were transduced with lentivirus containing three targeting guide RNAs, spCas9, and a puromycin resistance cassette. After puromycin selection, singe cell clones were generated and screened for efficient deletion of target genes via western blot and qRT-PCR. To control for effects of transduction, we generated scramble cell lines transduced with lentivirus containing non-targeted gRNA. To generate eGFP expressing cells, Scramble or PERKKO cell lines were transduced with pre-established eGFP vectors. For siRNA mediated silencing of Sec61b, B16 cells were transfected with 100 nM Sec61b siRNA (Eupheria Biotech; Cat# EMU162401-20UG) or untargeted control (ThemoFisher; Cat# AM4620), 72 hours prior to assay. Microscopy images were obtained in an Advance Microscopy Group Evos fluorescent digital inverted microscope.
METHOD DETAILS
In vivo Tumor models
Mice were subcutaneously injected (s.c.) with 150,000 B16 or SM1 cells/mouse to generate flank localized tumors. For models with tumor rechallenge, mice demonstrating full regression (no regrowth after 4 weeks) of SM1 PERKKO tumors were reinjected with SM1 wildtype on one flank and LLC on the opposing flank. For abscopal models, mice were injected with wildtype or PERKKO SM1 tumors on one flank, following by a second spatially separate injection of SM1 wildtype or PERKKO on the opposing flank ten days later. For the topical tamoxifen inducible BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 melanoma model, bare skin was topically treated with 2.5 μl 4-hydroxytamoxifen (1.9 mg/ml) for three consecutive days. Tumors were measured with digital calipers and tumor volumes calculated with the formula: [((smallest tumor diameter)2 x (largest tumor diameter) x 0.5)]. Tumor-bearing mice were humanely terminated when tumors reached 2000 mm3. B16 bearing-mice treated with PERK inhibitor received i.p. injections with AMG44 (12.5 mg/kg) every other day starting at day 6 post-tumor injection. For BrafV600E/+;Ptenfl/fl;Tyrosinase-CreERT2 mice treated with AMG44, treatment was initiated when tumors reached on average 150mm2. B16-bearing mice receiving CCR2 antagonist treatment received daily injections with BMS-CCR2-22 (0.5 mg/kg, Tocris) starting the day of B16 inoculation. For CD11c+ cells depletion, ItgaxDTR/EGFP mice received i.p. DT injections (100 ng/mouse, Sigma; D0564) on 3 days prior to tumor inoculation. Subsequent DT i.p. injections were delivered every other day at 50 ng/mouse to maintain CD11c+ DC depletion. Depletion efficiency was confirmed via detection of CD11c+ DTR-eGFP+ cells by flow cytometry. For neutralization of IFNAR1, tumor-bearing mice were injected i.p. with 1 mg/mouse anti-IFNAR1 (clone MAR1-5A3, BioXcell) at day 0 followed by every 3rd day treatments until experimental endpoint. For mice treated with T cell depletion antibodies, mice were injected i.p. every third day with anti-CD4, anti-CD8 or isotype antibody (400 μg/dose) starting on day 0 of tumor injection. Splenectomy or sham surgery procedures were performed two weeks prior to tumor initiation. In mice that received sham surgery and splenectomy, tumors were inoculated on the opposite side of the surgical incision. For mice receiving adoptive transfer of cKit+ precursors, isolation of cKit+ (CD117+) cells was achieved through positive enrichment (Miltenyi Biotec) and MojoSort Magnets (Biolegend). Enrichment for cKit+ cells was confirmed via flow cytometry. Precursors were injected intra-tumor and administered at 5X105/mouse on days 10 and 14 post tumor injection. For survival analysis, mice which achieved tumor volumes of 3000 mm3 were considered at endpoint.
Tumor digestions
Resected and minced tumors were digested with DNase I and Liberase (Roche USA) prior to lysis of red blood cells with ammonium-chloride-potassium buffer. Samples were strained with 100 μM sterile mesh filters and utilized in single cell suspension.
Immunoblot and ELISA
Protein isolates were quantitated with the Pierce BCA protein assay kit (ThermoFisher) and equal amounts of total and phospho-lysates electrophoresed in 8 or 10% Tris-Glycine gels (Novex-Invitrogen), transferred to PVDF membranes with an iBlot Gel Transfer Device (ThermoFisher) and blotted with indicated primary and secondary antibodies detailed in Key Resources Table. Imagining of membrane-bound immune complexes was performed with a ChemiDoc Imaging System (BioRad, #17001401). Detection of IFNB1 and IFNA1 was performed via murine IFNβ1 ELISA (R&D Systems; Cat# DY8234-05) and IFNα1 ELISA (Abcam; Cat# ab252352) according to the manufacturer’s protocol. HMGB1 detection in tumors suspensions was assessed using an HMGB1 detection kit (Novus; Cat#NBP2-62767) using the manufacturer’s recommendations.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD117 (c-kit) BV421 | Biolegend | Cat# 105827; RRID: AB_10898120 |
| Anti-mouse CD117 (c-kit) Pe/Dazzle 594 | Biolegend | Cat #105834; RRID: AB_ 2564055 |
| Anti-mouse CD115 (CSF-1R) PE | Biolegend | Cat# 135505; RRID: AB_1937254 |
| Anti-mouse IFNAR-1 APC | Biolegend | Cat# 127313; RRID: AB_2122746 |
| Anti-mouse Ly-6A/E (Sca-1) BV785 | Biolegend | Cat# 108139; RRID: AB_2565957 |
| Anti-mouse CD45 AF700 | Biolegend | Cat# 103128; RRID: AB_493715 |
| Anti-mouse CD45 FITC | Invitrogen | Cat# 11-0451-85; RRID: AB_465050 |
| Anti-mouse CD45 BV421 | Biolegend | Cat# 103133; RRID: AB_10899570 |
| Anti-mouse CD45 BV785 | Biolegend | Cat# 103149; RRID: AB_2564590 |
| Anti-mouse CD45 APC | Biolegend | Cat# 103112; RRID: AB_312977 |
| Anti-mouse I-A/I-E (MHCII) BV421 | Biolegend | Cat# 107632; RRID: AB_26508 |
| Anti-mouse I-A/I-E (MHCII) FITC | Invitrogen | Cat# 11-5321-82; RRID: AB_465232 |
| Anti-mouse MHC Class II (I-A/I-E) PE | Tonbo | Cat# 50-5321-U100; RRID: AB_2621796 |
| Anti-mouse H-2kb PE | BD Biosciences | Cat# 553570; RRID: AB_394928 |
| Anti-mouse H-2Db (KH95) FITC | BD Biosciences | Cat# 553573; RRID: AB_394931 |
| Anti-mouse CD80 PE | eBioscience | Cat# 12-0801-82; RRID: AB_465752 |
| Anti-mouse CD40 PE | BD Biosciences | Cat# 553791; RRID: AB_395055 |
| Anti-mouse CD86 PE | eBiosceince | Cat# 12-0862-82; RRID: AB_465768 |
| Anti-mouse CD86 APC | Biolegend | Cat# 105011; AB_493343 |
| Anti-mouse Ly-6C BV785 | Biolegend | Cat# 12804; RRID: AB_2565852 |
| Anti-mouse Ly-6C PE | BD Biosciences | Cat# 560592; RRID: AB_1727556 |
| Anti-mouse Ly-6C APC | BD Biosciences | Cat# 560595; RRID: AB_1727554 |
| Anti-mouse Ly-6C AF700 | Biolegend | Cat# 128023; RRID: AB_10640119 |
| Anti-mouse Ly-6C FITC | BD Biosciences | Cat# 553104; RRID: AB_394628 |
| Anti-mouse Ly-6G PE | BD Biosciences | Cat# 551461; RRID: AB_394208 |
| Anti-mouse Ly-6G Pe/Dazzle 594 | Biolegend | Cat# 127648; RRID: AB_2566319 |
| Anti-mouse Ly-6G FITC | BD Biosciences | Cat# 551460; RRID: AB_394207 |
| Anti-mouse Ly-6G AF700 | Biolegend | Cat# 127622; RRID: AB_10643269 |
| Anti-mouse Ly-6G APC | Tonbo | Cat# 20-1276-U100; RRID: AB_2621589 |
| Anti-mouse Ly-6G/Ly-6C (Gr-1) FITC | Biolegend | Cat# 108406; RRID: AB_313371 |
| Anti-mouse Ly-6G/Ly-6C (Gr-1) PE | BD Biosciences | Cat# 553128; RRRID: AB_394644 |
| Anti-mouse Ly-6G/Ly-6C (Gr-1) PE/Dazzle 594 | Biolegend | Cat# 108452; RRID: AB_2564249 |
| Anti-mouse Ly-6G/Ly-6C (Gr-1) APC | Biolegend | Cat# 108412; RRID: AB_313377 |
| Anti-mouse CD11c VioletFluor 450 | Tonbo | Cat# 75-0114-U025; RRID: AB_2621937 |
| Anti-mouse CD11c BV421 | Biolegend | Cat# 117329; RRID: AB_10897814 |
| Anti-mouse CD11c APC | Tonbo | Cat# 20-0114-U100; RRID: AB_2621557 |
| Anti-mouse CD11c PE | eBiosciences | Cat# 12-0114-82; RRID: AB_465552 |
| Anti-mouse CD11c BV785 | Biolegend | Cat# 117336; RRID: AB_2565268 |
| Anti-mouse CD11b PE/Dazzle 594 | Biolegend | Cat# 101256; RRID: AB_2563648 |
| Anti-mouse CD11b PE | BD Biosciences | Cat# 553311; RRID: AB_ 394775 |
| Anti-mouse CD11b BV421 | Biolegend | Cat# 101251; RRID: AB_2562904 |
| Anti-mouse CD11b APC | Tonbo | Cat# 20-0112-U100; RRID: AB_2621556 |
| Anti-mouse CD11b FITC | eBiosciences | Cat# 11-0112-85; RRID: AB_464936 |
| Anti-mouse CD11c biotin | Tonbo | Cat# 30-0114-U100 |
| Anti-mouse CD103 BV421 | Biolegend | Cat# 121421; RRID: AB_10900074 |
| Anti-mouse CD103 APC | Biolegend | Cat# 121414; RRID: AB_1227502 |
| Anti-mouse CCR2 PE | Biolegend | Cat# 150609; RRID: AB_2616981 |
| Anti-CD4 FITC | Tonbo | Cat# 35-0042-U500; RRID: AB_2621666 |
| Anti-CD4 APC | Tonbo | Cat# 20-0042-U100; RRID: AB_2621544 |
| Anti-CD4 AF700 | Biolegend | Cat# 100430; RRID: AB_493699 |
| Anti-CD4 PE | BD Biosciences | Cat# 553652; RRID: AB_394972 |
| Anti-CD8a FITC | BD Biosciences | Cat# 553031; RRID: AB_394569 |
| Anti-CD8a BV785 | Biolegend | Cat# 100749; RRID: AB_11218801 |
| Anti-CD8a PE | BD Biosciences | Cat# 553033; RRID: AB_394571 |
| Anti-CD8a BV421 | Biolegend | Cat# 100753; RRID: AB_2562558 |
| Anti-CD8a APC | Biolegend | Cat# 100712; RRID: AB_312751 |
| Anti-CD44 Pe/dazzle 594 | Biolegend | Cat# 103055; RRID: AB_2564043 |
| Anti-CD44 BV785 | Biolegend | Cat# 103041; RRID: AB_11218802 |
| Anti-CD44 FITC | Biolegend | Cat# 103022; RRID: AB_493685 |
| Anti-CD44 APC | Invitrogen | Cat# 17-0441-82; RRID: AB_469390 |
| Anti-CD69 PE | BD Biosciences | Cat# 553237; RRID: AB_394726 |
| Anti-CD69 PE/dazzle 594 | Biolegend | Cat# 104536; RRID: AB_2565583 |
| Anti-CD274 APC | Biolegend | Cat# 124311; RRID: AB_10612935 |
| Anti-CD274 BV421 | Biolegend | Cat# 124315; RRID: AB_10897097 |
| Anti-TNFα APC | Biolegend | Cat# 506307; RRID: AB_315428 |
| Anti-TNFα PE | Biolegend | Cat# 506306; RRID: AB_315427 |
| Anti-IFNγ APC | Biolegend | Cat# 505810; RRID: AB_315404 |
| Anti-IFNγ PE | BD Biosciences | Cat# 554412; RRID: AB_395376 |
| Anti-IFNγ BV421 | Biolegend | Cat# 505829; RRID: AB_10897937 |
| Anti-Phospho STAT1 (Ser727), Recombinant rabbitt monoclonal APC | Invitrogen | Cat# MA5-28056; RRID: AB_2745055 |
| Anti-Mouse Calreticulin (1G6A7) APC | Novus | Cat# NBP1-47518APC; RRID: AB_10010469 |
| Purified rat anti-mouse CD16/32 | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-mouse Ki67 APC | Biolegend | Cat# 652406; RRID: AB_2561930 |
| Anti-mouse Ki67 PE | Biolegend | Cat# 652403; RRID: AB_2561524 |
| Anti-SEC61b APC | LSBio | Cat# LS-C268919 |
| Anti-p84 | Abcam | Cat #ab487; RRID: AB_304696 |
| Anti-PERK (C33E10) Rb mAb | Cell Signaling | Cat# 3192; RRID: AB_2095847 |
| Anti-Phospho-PERK (Thr980) Rb mAb | Cell Signaling | Cat# 3179; RRID: AB_2095853 |
| Anti-IRE1α (14C10) Eb mAb | Cell Signaling | Cat# 3294; RRID: AB_823545 |
| Anti-Phospho-IRE1α (S274) Rb polyAb | Abcam | Cat# 48187; RRID: AB_873899 |
| Anti-Chop Rb polyAb | SantaCruz | Cat# Sc-793; RRID: AB_631364 |
| Anti-Caspase-3 (8G10) Rabbit mAb | Cell Signaling | Cat# 9665; RRID: AB_2069872 |
| Anti-eIf2α antibody Rabbit polyclonal | Cell Signaling | Cat# 9722; RRID: AB_2230924 |
| Anti-Phospho-eIf2α Rabbit | Cell Signaling | Cat# 9721; RRID: AB_330951 |
| Anti-XBP-1s (E9V3E) | Cell Signaling | Cat# 40435; RRID: AB_2891025 |
| Anti-BiP (C50B12) | Cell Signaling | Cat# 3177; RRID: AB_2119845 |
| Anti-B-actin mAb | Sigma | Cat# A2228-100ul; RRID: AB_476697 |
| Anti-Vinculin mAb | Sigma | Cat# V9131-2ML; RRID: AB_477629 |
| Anti-ATF4 | Cell signaling | Cat# 11815; RRID: AB_2616025 |
| Anti-CD31 | Abcam | Cat# ab28364; RRID: AB_726362 |
| InVivoMAb Anti-mouse CD8a | BioXCell | Cat# BE0004-1; RRID: AB_1107671 |
| InVivoMAb Anti-mouse CD4 | BioXCell | Cat# BE0003-1; RRID: AB_1107636 |
| InVivoMAb Anti-mouse IFNAR-1 | BioXCell | Cat# BE0241; RRID: AB_2689923 |
| InVivoMAb polyclonal rat IgG | BioXCell | Cat# BE0094; RRID: AB_1107795 |
| InVivoPlus Anti-mouse PD-1 (CD279) | BioXCell | Cat# BP0146; RRID: AB_10949053 |
| InVivoMAb mouse IgG1 isotype control | BioXCell | Cat# BE0083; RRID: AB_1107784 |
| InVivoMAb polyclonal human IgG | BioXCell | Cat# BE0092; RRID: AB_1107779 |
| Ultra-LEAF Purified Anti-HMGB1 blocking antibody | Biolegend | Cat# 651413; RRID: AB_2728487 |
| IFNAR1 blocking antibody: MAR1-5A3 Mouse IgG1κ | BioXCell | Cat# B10241 |
| Mouse IgG1 isotype control | BioXCell | Cat# BE0083 |
| Bacterial and virus strains | ||
| Ifnb1 sgRNA CRISPR/spCas9 All-in-One Lentivector set (3 gRNA) (Mouse) | ABMGOOD | Cat# 242661140595 |
| CRISPR/spCas9 Scrambled sgRNA All-in-One Lentivector | ABMGOOD | Cat# K010 |
| Eif2ak3 sgRNA CRISPR/spCas9 All-in-One Lentivector set (3 gRNA) (Mouse) | ABMGOOD | Cat# 190761140895 |
| Biological samples | ||
| TMA slides | Moffitt Cancer Center, Biomax, TriStar Technology | Tissue Core Moffitt Cancer Center. Also, commercial vendors Biomax and TriStar Technology |
| Chemicals, peptides, and recombinant proteins | ||
| Mouse GM-CSF | Gemini | Cat# 300-308P |
| Mouse G-CSF | Gemini | Cat# 300-207P |
| APC Annexin V | BD Pharmingen | Cat# 550475; RRID: AB_2868885 |
| T-select MHC Class I mouse gp100 tetramer: KVPRNQDWL-PE | Medical & Biological Laboratories | Cat# TS-M505-1 |
| AMG-PERK-44 | Tocris | Cat# 5517 |
| PERK Inhibitor I, GSK2606414 | Sigma Aldrich | Cat# 516535-5MG |
| Thapsigargin | Sigma Aldrich | Cat# T9033-5MG |
| BMS-CCR2-22; CCR2 inhibitor | Tocris | Cat# 3129 |
| Z-Vad(OH)-FMK; Pan-Caspase inhibitor | Cayman Chemicals | Cat# 14467 |
| Ferrostatin-1; Ferroptosis inhibitor | Cayman Chemicals | Cat# 17729 |
| Necrostatin-1; Necroptosis inhibitor | Cayman Chemicals | Cat# 11658 |
| Dapansutrile; NLRP3/Pyroptosis inhibitor | Cayman Chemicals | Cat# 24671 |
| MKC-3946; IRE1a inhibitor | Cayman Chemicals | Cat# 19152 |
| Hydroxychloroquine (sulfate) | Cayman Chemicals | Cat# 17911 |
| PPADS tetrasodiun salt | Tocris | Cat# 0625 |
| Evans blue tetrasodium salt | Tocris | Cat# 0845 |
| Cycloheximide | Cayman Chemicals | Cat# 14126 |
| Fludarabine phosphate | Cayman Chemicals | Cat# 14251 |
| DNase I | Roche | Cat# 10104159001 |
| Liberase | Roche | Cat# 05401127001 |
| Ionomycin | Sigma Aldrich | Cat# I3909-1ML |
| Golgi stop | BD Biosciences | Cat# 554724; RRID: AB_2869012 |
| Perm/Wash buffer | BD Bioscience | Cat# 554723; RRID: AB_2869011 |
| Cytofix/Cytoperm | Fisher Scientific | Cat# BDB554714 |
| ACK RBC lysis buffer | Gibco | Cat# A10492-01 |
| H2DCFDA (H2-DCCF, DCF) | Invitrogen | Cat# D399 |
| 5(6)-CFDA,SE; CFSE | Invitrogen | Cat# V12883A |
| Propidium Iodide solution | BD Bioscience | Cat# 556463; RRID: AB_2869075 |
| Zombie NIR Fixable Viability Kit | Invitrogen | Cat# 423105 |
| ER-Tracker Green (BODIPY FL Glibenclamide) | Cell Signaling | Cat# 8787S |
| NIAD4 | Cayman Chemicals | Cat# 18520 |
| CRANAD2 | Cayman Chemicals | Cat# 19814 |
| Pierce Concentrator, PES, 10K MWCO 0.5ml | Thermo Scientific | Cat# 88513 |
| Mouse Cytokine Array / Chemokine Array 44-Plex (MD44) | Eve Technologies | Cat# MD44 |
| DNase I recombinant, RNAse-free | Roche | Cat# 04716728001 |
| iTaq universal SYBR green super mix | Bio Rad | Cat# 1725121 |
| Verso cDNA synthesis Kit | ThermoFisher | Cat# AB-1453/B |
| Ovalbumin peptide (OVA257-264) | Anaspec Peptide | Cat# 60193 |
| Critical commercial assays | ||
| MitoProbe DiOC2(3) Assay | Molecular Probes | Cat# M34150 |
| CellTrace Violet cell proliferation kit | ThermoFisher | Cat# C34571 |
| Anti-CD117 Microbeads, mouse | Miltenyi Biotec | Cat# 130-091-224 |
| Mojosort Streptavidin Nanobeads | Biolegend | Cat# 76447 |
| Mouse HMGB1/HMG-1 ELISA Kit (colorimetric) | Novus | Cat# NBP2-62767 |
| Mouse IFNα ELISA | Abcam | Cat# ab252352 |
| Mouse IFNβ DuoSet ELISA | R&D Systems | Cat# DY8234-05 |
| ADP/ATP Ratio assay kit | Sigma | Cat# MAK135 |
| Deposited data | ||
| RNAseq from cMoPs | GEO accession #GSE206783 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE206783 |
GSE206783 |
| TCGA SKCM | https://www.cancer.gov/tcga | |
| Moffitt melanoma cohort | Doi: 10.1158/1055-9965.EPI-20-0307 | |
| ICI treated Melanoma cohorts | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJEB23709, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE78220, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE91061 | PRJEB23709, GSE78220, GSE91061 |
| Experimental models: Cell lines | ||
| B16-F10 | ATCC | CRL-6475; RRID: CVCL_0159 |
| SM1 | NA | NA |
| Lewis lung carcinoma (LLC) | ATCC | #CRL-1642; RRID: CVCL_4358 |
| Experimental models: Organisms/strains | ||
| C57BL/6 Mice | Wildtype | Envigo |
| NOD.129S7 (B6)-Rag1 tm1Mom/J | RAG1 | Jackson Laboratory |
| B6.129P2-Lyz2 tm1(Cre)Ifo/J | Lyz2-Cre | Jackson Laboratory |
| B6(Cg)-Sting1tm1.2Camb/J | STING flox/flox | Jackson Laboratory |
| B6(Cg)-Ifnar1 tm1.1Ees/J | IFNAR1 flox/flox | Jackson Laboratory |
| Eif2ak3tm1.2Drc/J | PERK flox/flox | Jackson Laboratory |
| B6(Cg)-Ifnar1tm1.2Ees/J | Total body IFNAR1 knockout | Jackson Laboratory |
| B6.129S4-CCR2 tm1lfc/J | Total body CCR2 knockout | Jackson Laboratory |
| B6.129S(Cg)-Stat1tm1Dlv/J | Total body STAT1 knockout | Jackson Laboratory |
| B6.FVB-1700016L21RikTg(Itgax-DTR/EGFP)57Lan/J | Diphtheria toxin receptor-CD11c GFP (DTR-CD11c-GFP) | Jackson Laboratory |
| B6.Cg-Gt(ROSA)26Sor tm1.4(CAG-TdTomato)Hze/J | TdTomato | Jackson Laboratory |
| B6.129-Ifnb1 tm1Lky/J | Type I interferon reporter mouse; EYFP-IFNB1 | Jackson Laboratory |
| B6.Cg-Tg(Tyr-Cre/ERT2)13Bos Braf tm1Mmcm Pten tm1Hwu/BosJ | Inducible melanoma, ERT-Tyrosinase-Cre BrafV600E Ptenfl/fl (TBP) | Jackson Laboratory |
| Oligonucleotides and Primers | ||
| Sec61b siRNA | Sigma | EMU162401 |
| Atf4 siRNA | Sigma | EMU079511 |
| Mock siRNA | Sigma | SIC001-10NMOL |
| Ccl2 Forward | IDT | CAGGTCCCTGTCATGCTTCT |
| Ccl2 Reverse | IDT | GTGGGGCGTTAACTGCATCT |
| Ccl12 Forward | IDT | ACACTGGTTCCTGACTCCTCT |
| Ccl12 Reverse | IDT | ACCTGAGGACTGATGGTGGT |
| Ifnb1 Mm_Ifnb1_1_SG | Qiagen | Cat# QT00249662 |
| Isg15 Forward | IDT | CTAGAGCTAGAGCCTGCAG |
| Isg15 Reverse | IDT | AGTTAGTCACGGACACCAG |
| Ifit3 Forward | IDT | TTCCCAGCAGCACAGAAAC |
| Ifit3 Reverse | IDT | AAATTCCAGGTCAAATGGCA |
| Cxcl10 Forward | IDT | CCAAGTGCTGCCGTCATTTTC |
| Cxcl10 Reverse | IDT | GGCTCGCAGGGATGATTTCAA |
| Actin-b | Qiagen | Cat# QT00095242 |
| Ifna1 Forward | IDT | GGATGTGACCTTCCTCAGACTC |
| Ifna1 Reverse | IDT | ACCTTCTCCTGCGGGAATCCAA |
| Ifna14 Forward | IDT | AGCAGGTAGAGATACAGGCACC |
| Ifna14 Reverse | IDT | TTTCAGGCTGGTCAGCAACTT |
| Ifna13 Forward | IDT | GGCTCAAGCCATCCCTTTTG |
| Ifna13 Reverse | IDT | TGAAACATGTAGGCAGGTTGATTG |
| Ifna7 Forward | IDT | GTCCTGGTGGTGTTGAGCTA |
| Ifna7 Reverse | IDT | TGCAGAACACAGAGGGCTTG |
| Ifna16 Forward | IDT | AGGATGTGACCTGCCTCAGACT |
| Ifna16 Reverse | IDT | AGGGTATCCACCTTCTCCTGGG |
| Atf4 primers | Qiagen | PPM04670E-200 |
| Sec61b Forward SET1 | IDT | GATTCCCCAGGGCTCAAGT |
| Sec61b Reverse SET1 | IDT | AGCCCAATCTATGATCGCGT |
| Sec61b Forward SET2 | IDT | CCCAGTGCTGGTGATGAGTC |
| Sec61b Reverse SET2 | IDT | GCCCAATCTATGATCGCGTG |
| Software and algorithms | ||
| Prism 7 | GraphPad | |
| FlowJo | FlowJo | |
| Wave | Agilent Technologies | |
| Aperio Image scope | Leica Biosystems | |
| Zen 2.3 (blue edition) | Carl Zeiss AG | |
| Definiens Tissue Studio software v4.7 | Definiens AG | |
| Halo | Indica labs | |
| Other | ||
Protein Multiplex
Measurement of cytokines and chemokines in tumor suspension protein extracts were monitored using the Mouse Cytokine/Chemokine 44-Plex Discovery Assay® Array assay (Cat #MD44, Eve-Technologies). Values were normalized based on protein concentration. Tumor homogenates were prepared from equal weights of resected tumor tissue subjected to three rounds of 60 seconds homogenization with 1 mm glad beads (Sigma #1002619844) in Benchmark Bead Blaster homogenizer (3000 RPM) in homogenate buffer. Homogenate buffer was composed of 20 mM Tris, 150 mM NaCl, 1% NP40 and 100U/ml Leupeptin (Roche #91058027), aprotinin (Roche #10236614001), trypsin/chymotrypsin inhibitor (Sigma #TP777-50MG), phosphatase inhibitor cocktail 2 (Sigma #P0044-1ML) and phosphatase inhibitor cocktail 3 (Sigma #P576-1ML).
Quantitative RT-PCR
Total RNA was isolated from bulk tumor or isolated cell samples via TRIzol (Life Technologies). Reverse transcription was performed with the Verso cDNA synthesis kit (Thermo Scientific). Quantitative PCR reactions were done with Bio-Rad SYBR green master mix and performed on an Applied Biosystems Thermocycler (7900 HT) using primers detailed in Key Resources Table.
DC development and culture of cKit+ precursor
Bone marrow-derived DCs (BM-DC) from EYFP-IFNβ1 mice were developed from bone marrow cells treated with mGM-CSF (20 ng/ml) for 7 days. Splenic DC counterparts were obtained after CD11c+ selection with MojoSort (Biolegend) followed by cultured in the presence of mGM-CSF for 48 hours. BM-DCs and splenic DCs were exposed for 24 hours to supernatants from Scramble or PERKKO B16 and SM1 cells previously treated with Thaps (1μM) for 4 hours, washed with fresh media, and cultured for additional 18 hours in regular media. Then, EYFP-IFNβ1 analyzed via flow cytometry. Isolation of cKit+ (CD117+) cells was achieved through positive enrichment (Miltenyi Biotec; Cat# 130-091-224) and MojoSort Magnets (Biolegend). Enrichment for cKit+ cells was confirmed via flow cytometry. cKit+ precursors labeled with CellTrace Violet (CTV; ThermoFisher; Cat# C34571) and intratumorally transferred into Scramble or PERKKO B16 tumor-bearing mice. In some studies, cKit+ precursors were treated with STAT1 inhibitor fludarabine (100uM; Cayman; Cat# 14251) followed by injection into tumors. A day later, tumors were harvested and phenotypes of CTV+ cells analyzed by flow cytometry.
Ex vivo Antigen presentation
DCs were enriched from tumors of mice bearing Scramble or PERKKO B16 tumors via CD11c+ enrichment (Tonbo; Cat#30-0114-U100). CD11c+ cells were co-cultured at varying concentrations with CD8+ pmel T cells labeled with 5 μM cell trace violet (Thermo Fisher; Cat #C34571). CD8+ T cells were enriched using mouse T cell negative selection kits (MagniSort, Invitrogen) from the spleen and lymph nodes of pmel mice. Purity ranged between 95% and 99% as tested by flow cytometry. CD44 and CD69 levels were tested in CD8+ T cells 48 hours later by flow cytometry.
Flow cytometry phenotyping and sorting
Conjugated antibodies and probes used for flow cytometry are listed in the Key Resources Table. For surface staining, cells were labelled with appropriate antibodies in the presence of Fc blocker. For intracellular staining, surface-labeled cells were fixed with Cytofix/Cytoperm™ Solution (BD Biosciences), washed in Perm/Wash™ IX solution, and labelled with intracellular antibodies. Cells were then washed in Perm/Wash™ IX and PBS. For intra-cellular detection of IFNγ and TNFα in tumor-infiltrating T cells, tumors were treated with phorbol myristate acetate (PMA, 750 ng/mL, Sigma Aldrich) and ionomycin (50 μg/mL, Sigma-Aldrich) for 5 hours in the presence of Golgi stop 0.8 μl/ml, BD Biosciences). Next, cells underwent extracellular staining followed by fixation and permeabilization with Cytofix/Cytoperm Solution. For intracellular detection of phospho-STAT1, cells underwent surface staining followed by fixation and permeabilization with Cytofix/Cytoperm Solution (BD Biosciences) followed by washing with 1x Perm/Wash and re-permeabilization with 90% ice cold methanol. For ER-tracker staining, cells were probed with 100 nM of ER tracker green (Invitrogen) in Hank’s Buffered Saline Solution (HBSS) at 37°C for 30 min and then stained for surface markers. For detection of cell death after exposure to Thaps, tumor cells were labeled with the Annexin-V-APC Apoptosis detection kit (BD Pharmingen) according to manufacturer’s details. ROS were detected by DCFDA (10 μM). For determination of translation rate, OPP click chemistry labeling (Click Chemistry Tools; Cat#1391) was performed according to manufacturer’s protocol. Fluorescent labeling of protein aggregates was monitored with probes NIAD4 (Cayman; Cat#8520) and CRANAD2 (Cayman; Cat#19814) at 400 nM in PBS and 10 μM in HBSS, respectively, for 30 min at 37°C, followed by staining with extracellular antibodies. Data acquisition was performed in a CytoFLEX II (Beckman Coulter), and analysis performed with FlowJo version 11 software. Flow cytometric cell sorting was performed on a FACSAria-II SORP (BD) under sterile conditions.
Detection of immunogenic cell death markers
Extracellular ATP was measured in media of cell lines treated with ER stressors or in media of resected tumors cultured ex vivo for 24 hours via the ADP/ATP ratio assay kit (Millipore Sigma). ExoCRT was measured via flow cytometry in Thaps-treated cell lines or bulk tumor samples via surface labeling with fluorophore conjugated anti-calreticulin antibody (Novus Bio). Extracellular release of HMGB1 was detected in tumor samples with a mouse specific HMGB1 ELISA (Novus). Also, HMGB1 release was detected in media of cell lines exposed to Thaps via immunoblot of size fractionated supernatants (10kDa; Pierce Concentrator PERK 10K MWCO, Thermo).
Multispectral imagining
Formalin fixed paraffin embedded melanoma TMA sections were stained using an automated OPAL-IHC system (PerkinElmer) in a BOND RX (Leica Biosystems). Briefly, slides were treated with the PerkinElmer blocking buffer for 10 min and incubated with the specific primary antibodies, followed by OPAL-HRP polymer and one OPAL fluorophore. Individual antibody complexes were stripped after each round of detection and DAPI applied as the last staining. Auto-fluorescence slides (negative control) included primary and secondary antibodies, omitting the OPAL fluorophores. Slides were imaged with a Vectra Automated Quantitative Pathology Imaging System. Multi-layer TIFF images were exported from InForm (PerkinElmer) into HALO (Indica Labs) for quantitative image analysis. Each fluorophore was assigned to a dye color and positivity thresholds determined visually per marker based on nuclear or cytoplasmic staining patterns, and by intensity thresholds normalized for exposure (counts/2bit depth x exposure time x gain x binning area). Cell segmentation results from each core were analyzed using FCS Express 6 Image Cytometry (De Novo software). After excluding normal skin control samples and samples with poor SOX10 staining or incomplete sample cores, samples were stratified by the percentage of SOX10 positive cells exhibiting expression of pPERK with “High pPERK” (n=5) defined as samples with values greater than two standard deviations above the mean, “Intermediate pPERK” (n=26) defined as samples with values above the mean but less than two standard deviations above the mean, and “Low pPERK” (n = 37) defined as samples with values below the mean.
RNA sequencing
RNA sequencing was performed on mRNA isolated via Qiagen RNeasy micro kit (#74004) with inline DNase treatment (Qiagen #1023460) from cKit+ cells isolated from Scramble and PERKKO B16 tumors via magnetic bead enrichment and completed by the Molecular Genomics core at Moffitt Cancer Center. Extracted RNA was quantitated with the Qubit Fluorometer (ThermoFisher Scientific, Waltham, MA) and screened for quality on the Agilent TapeStation 4200 (Agilent Technologies, Santa Clara, CA). The samples were then processed for RNA-sequencing using the NuGEN Universal RNA-Seq with NuQuant, Mouse AnyDeplete kit (Tecan Genomics, Redwood City, CA). Briefly, 50 ng of RNA was used to generate cDNA and a strand-specific library following the TruSeq Stranded Total RNA protocol. Quality control steps were performed, including TapeStation size assessment and quantification using the Kapa Library Quantification Kit (Roche, Wilmington, MA). The final libraries were normalized, denatured, and sequenced on the Illumina NextSeq 2000 sequencer with the P3-200 cycle reagent kit to generate at least 50M million 105-base read pairs per sample (Illumina, Inc., San Diego, CA). Read adapters were detected using BBMerge (v37.88) (Bushnell et al., 2017) and subsequently removed with cutadapt (v1.8.1). Processed raw reads were then aligned to mouse MM10 using STAR (v2.5.3a) (Dobin et al., 2013). Gene expression was evaluated as read count at gene level with HTSeq (v0.6.1) (Anders et al., 2015) and Gencode gene model vM21. Gene express data were then normalized to counts per million (CPM) and differential expression between experimental groups were evaluated using LIMMA (Ritchie et al., 2015) with Pval < 0.05 and FDR < 10% as cutoffs. Unsupervised heierarchical clustering based on the top 100 variable genes (MAD ranked) was performed using the DRPPM Expression Analysis ShinyApp (https://github.com/shawlab-moffitt/DRPPM-ExprAnalysisShinyApp). Gene set enrichment analyses were conducted using GSEA v4.1.0 with MSigDB v7.4. Murine gene symbols were converted to human gene symbols using the Mouse_Gene_Symbol_Remapping_Human_Orthologs_MSIGBv7.4.chip. The GSEA parameters used were “-metric signal2noise -set_min 4 -permute gene_set”. Significant gene sets were defined as an FDR < 10%. RNA-seq data is in the process to be deposited into GEO.
Gene Activity Estimation and Survival Analysis
We downloaded the TCGA-SKCM expression data and clinical outcome from cBioportal (Cerami et al., 2012). Expression and clinical outcome data of patients treated with Immune Checkpoint Blockade were downloaded from the CRI iAtlas Portal (Eddy et al., 2020). A separate cohort from the Moffitt and Bristol-Myers Squibb clinical trial (George et al., 2016) was curated and derived from the Moffitt Cancer Analytics Platform, with its expression and clinical data serving as validation. The immunogenic cell death signature was derived from (Garg et al., 2016). A PERK-driven gene signature was derived from the Connectivity Map platform (Subramanian et al., 2017) based on transcripts reduced after PERK knockdown in the A375 melanoma cell line. The top-250 transcripts reduced after PERK knockdown were ranked based on the Z-score after restricting candidates with p-value = 0.0 and self-correlation > 0.7. Gene signature activities were profiled in the melanoma expression data by single-sample GSEA as implemented in the gene set variation analysis package (GSVA) (Hanzelmann et al., 2013). The influence of each gene signature was examined to see if the overall survival of the two-group formed by a median-split of the gene signature score were statistically significantly different. The two-sided log-rank test was used to calculate P values. Hazard ratios were derived from the Cox proportional hazards model.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism 7 (San Diego, CA). For comparison of two groups in datasets with normally distributed with equal standard deviations, group means were compared by two-tailed unpaired Student’s t test; for sample with significantly different standard deviations, unpaired T tests with Welch’s correction were performed. For data sets with multiple groups and equal standard deviations, One-Way ANOVA with Tukey’s multiple comparisons test was performed; for samples with unequal standard deviations, One-Way ANOVA with Dunnett’s multiple comparisons test was performed. P values of < 0.05 were considered statistically significant. Specific statistical test results are indicated in each figure with *, p < 0.05; **, p < 0.01 ***, p < 0.001.
Supplementary Material
Highlights.
Kinase PERK in Melanoma cells restricts protective tumor-specific T cell immunity.
PERK targeting drives immunogenic melanoma cell death via SEC61β-linked paraptosis.
PERK-null tumors promote the expansion of immune competent Monocyte-derived DCs.
Stroma-originated type I IFN reprograms myelopoiesis in PERK-null tumors via STAT1.
Acknowledgments
Authors thank the Flow Cytometry; Biostatistics and Bioinformatics; and Advanced Analytical and Digital Pathology Cores from the Moffitt Cancer Center, which are partially funded by P30-CA076292.
Financial support:
This study was supported in part by the National Institutes of Health (NIH) grants: R01-CA184185, R01-CA233512; R01-CA262121; P01-CA250984 Project #4; and P30-CA076292; and Florida Department of Health grant #20B04 to PCR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of conflict of interest
The authors declare no competing interests.
References:
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B , Goetz A, Gaul N, et al. (2013). Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002. [DOI] [PubMed] [Google Scholar]
- Bezu L, Sauvat A, Humeau J, Gomes-da-Silva LC, Iribarren K, Forveille S, Garcia P, Zhao L, Liu P, Zitvogel L, et al. (2018). eIF2alpha phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ 25, 1375–1393. 10.1038/s41418-017-0044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. (2005). ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 24, 3470–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D , and Diehl JA (2010). PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29, 3881–3895. 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B, Rood J, and Singer E (2017). BBMerge - Accurate paired shotgun read merging via overlap. PLoS One 12, e0185056. 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Trillo-Tinoco J, Sierra RA, Anadon C, Dai W, Mohamed E, Cen L, Costich TL, Magliocco A, Marchion D, et al. (2019). ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat Commun 10, 1280. 10.1038/s41467-019-09263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Cubillos-Ruiz JR (2021). Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer 21, 71–88. 10.1038/s41568-020-00312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney RJ, Mo R, Meyer DT, Nelson DJ, Lo YC, Yang G, Scherle PA, Mandlekar S, Wasserman ZR, Jezak H, et al. (2008). Discovery of disubstituted cyclohexanes as a new class of CC chemokine receptor 2 antagonists. J Med Chem 51, 721–724. 10.1021/jm701488f. [DOI] [PubMed] [Google Scholar]
- Chevet E, Hetz C, and Samali A (2015). Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov 5, 586–597. 10.1158/2159-8290.CD-14-1490. [DOI] [PubMed] [Google Scholar]
- Coillard A, and Segura E (2021). Antigen presentation by mouse monocyte-derived cells: Re-evaluating the concept of monocyte-derived dendritic cells. Mol Immunol 135, 165–169. 10.1016/j.molimm.2021.04.012. [DOI] [PubMed] [Google Scholar]
- Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, Ryan RJ, Iwamoto Y, Marinelli B, Gorbatov R, et al. (2012). Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A 109, 2491–2496. 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz JR, Mohamed E, and Rodriguez PC (2017). Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J Immunother Cancer 5, 5. 10.1186/s40425-016-0203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy JA, Thorsson V, Lamb AE, Gibbs DL, Heimann C, Yu JX, Chung V, Chae Y, Dang K, Vincent BG, et al. (2020). CRI iAtlas: an interactive portal for immuno-oncology research. F1000Res 9, 1028. 10.12688/f1000research.25141.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia F, Yadhanapudi L, Tretter T, and Romisch K (2019). The N-terminus of Sec61p plays key roles in ER protein import and ERAD. PLoS One 14, e0215950. 10.1371/journal.pone.0215950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, Singh R, Schall TJ, Datta M, Jain RK, et al. (2020). CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U S A 117, 1129–1138. 10.1073/pnas.1910856117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, Chan TA, Coukos G, Demaria S, Deutsch E, et al. (2020). Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer 8. 10.1136/jitc-2019-000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, De Ruysscher D, and Agostinis P (2016). Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: A large-scale meta-analysis. Oncoimmunology 5, e1069938. 10.1080/2162402X.2015.1069938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, Trinchieri G, Caux C, and Garrone P (2005). A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med 201, 1435–1446. 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Motzer RJ, Hammers HJ, Redman BG, Kuzel TM, Tykodi SS, Plimack ER, Jiang J, Waxman IM, and Rini BI (2016). Safety and Efficacy of Nivolumab in Patients With Metastatic Renal Cell Carcinoma Treated Beyond Progression: A Subgroup Analysis of a Randomized Clinical Trial. JAMA Oncol 2, 1179–1186. 10.1001/jamaoncol.2016.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, Lim SY, Velickovic R, Wongchenko M, et al. (2019). Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238–255 e236. 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al. (2012). ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 122, 4621–4634. 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, and Thaxton JE (2019). Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8(+) T Cells. Cancer Immunol Res 7, 476–486. 10.1158/2326-6066.CIR-18-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buqué A, Huang X, Zhou H, Senovilla L, Kroemer G, and Galluzzi L (2015). eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol 33, 86–92. 10.1016/j.semcancer.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. (2014). Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3, e955691. 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, and Ribas A (2012). BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res 72, 3928–3937. 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galassi C, Zitvogel L, and Galluzzi L (2022). Immunogenic cell stress and death. Nat Immunol 23, 487–500. 10.1038/s41590-022-01132-2. [DOI] [PubMed] [Google Scholar]
- Lang S, Pfeffer S, Lee PH, Cavalie A, Helms V, Forster F, and Zimmermann R (2017). An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front Physiol 8, 887. 10.3389/fphys.2017.00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wu C, Luo S, Hua Q, Chen HT, Weng Y, Xu J, Lin H, Wang L, Li J, et al. (2022). PERK reprograms hematopoietic progenitor cells to direct tumor-promoting myelopoiesis in the spleen. J Exp Med 219. 10.1084/jem.20211498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. (2014). Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 124, 1406–1417. 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, and Zanetti M (2011). Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc.Natl.Acad.Sci.U.S.A 108, 6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, de Mingo Pulido A, Mandula J, Zhang S, Thevenot P, et al. (2020). The Unfolded Protein Response Mediator PERK Governs Myeloid Cell-Driven Immunosuppression in Tumors through Inhibition of STING Signaling. Immunity 52, 668–682 e667. 10.1016/j.immuni.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monel B, Compton AA, Bruel T, Amraoui S, Burlaud-Gaillard J, Roy N, Guivel-Benhassine F, Porrot F, Genin P, Meertens L, et al. (2017). Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. EMBO J 36, 1653–1668. 10.15252/embj.201695597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau-Kotula E, Habault J, Kroemer G, and Poyet JL (2018). The anticancer peptide RT53 induces immunogenic cell death. PLoS One 13, e0201220. 10.1371/journal.pone.0201220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pytel D, Majsterek I, and Diehl JA (2016). Tumor progression and the different faces of the PERK kinase. Oncogene 35, 1207–1215. 10.1038/onc.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan AC, Ojha S, Kourtidis A, Conklin DS, and Aguirre-Ghiso JA (2008). Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res 68, 3260–3268. 10.1158/0008-5472.CAN-07-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e916. 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Pacholczyk R, Shi H, Berrong ZJ, Zakharia Y, Greco A, Chang CS, Eathiraj S, Kennedy E, Cash T, et al. (2021). Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity 54, 2354–2371 e2358. 10.1016/j.immuni.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, Shimoda M, Pacholczyk R, Shi H, Lee EJ, et al. (2018). Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c(+)CD103(+) Monocytic Antigen-Presenting Cells in Tumors. Immunity 48, 91–106 e106. 10.1016/j.immuni.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Andrews KL, Beckmann H, Bellon SF, Beltran PJ, Booker S, Chen H, Chung YA, D’Angelo ND, Dao J, et al. (2015). Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J Med Chem 58, 1426–1441. 10.1021/jm5017494. [DOI] [PubMed] [Google Scholar]
- Sperandio S, de Belle I, and Bredesen DE (2000). An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A 97, 14376–14381. 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417. 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. (2009). Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616. 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S, Cane S, De Sanctis F, and Bronte V (2021). Monocytes in the Tumor Microenvironment. Annu Rev Pathol 16, 93–122. 10.1146/annurev-pathmechdis-012418-013058. [DOI] [PubMed] [Google Scholar]
- Urra H, Dufey E, Avril T, Chevet E, and Hetz C (2016). Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2, 252–262. 10.1016/j.trecan.2016.03.007. [DOI] [PubMed] [Google Scholar]
- Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 21, 485–498. 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, and Polverini PJ (2012). The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res 72, 5396–5406. 10.1158/0008-5472.CAN-12-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq from cMoPs are publicly available at the GEO repository with accession GSE206783. Datasets analyzed were TCGA SKCM, an internal Moffitt melanoma cohort (doi: 10.1158/1055-9965.EPI-20-0307), and multiple datasets from ICI treated patients: PRJEB23709, GSE78220, GSE91061.
For survival analysis, we used the coding tool DRPPM-PATH-SURVEIOR: https://github.com/shawlab-moffitt/DRPPM-PATH-SURVEIOR.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.