

Abstract
Purpose:
To identify potential immune targets in post-neoadjuvant chemotherapy (NAC)–resistant triple-negative breast cancer (TNBC) and ER+HER2– breast cancer disease.
Experimental Design:
Following pathology review, 153 patients were identified as having residual cancer burden (RCB) II/III disease (TNBC n = 80; ER+HER2–n = 73). Baseline pre-NAC samples were available for evaluation for 32 of 80 TNBC and 36 of 73 ER+HER2– cases. Bright-field hematoxylin and eosin assessment allowed for tumor-infiltrating lymphocyte (TIL) evaluation in all cases. Multiplexed immunofluorescence was used to identify the abundance and distribution of immune cell subsets. Levels of checkpoints including PD-1/PD-L1 expression were also quantified. Findings were then validated using expression profiling of cancer and immune-related genes. Cytometry by time-of-flight characterized the dynamic changes in circulating immune cells with NAC.
Results:
RCB II/III TNBC and ER+HER2– breast cancer were immunologically “cold” at baseline and end of NAC. Although the distribution of immune cell subsets across subtypes was similar, the mRNA expression profiles were both subtype- and chemotherapy-specific. TNBC RCB II/III disease was enriched with genes related to neutrophil degranulation, and displayed strong interplay across immune and cancer pathways. We observed similarities in the dynamic changes in B-cell biology following NAC irrespective of subtype. However, NAC induced changes in the local and circulating tumor immune microenvironment (TIME) that varied by subtype and response. Specifically, in TNBC residual disease, we observed downregulation of stimulatory (CD40/OX40L) and inhibitory (PD-L1/PD-1) receptor expression and an increase in NK cell populations (especially non-cytolytic, exhausted CD56dimCD16–) within both the local TIME and peripheral white cell populations.
Conclusions:
This study identifies several potential immunologic pathways in residual disease, which may be targeted to benefit high-risk patients.
Translational Relevance.
Neoadjuvant chemotherapy (NAC) is now considered the standard of care in many early breast cancers, and obtaining a complete pathologic response has been clearly shown to improve overall survival. Conversely, cells remaining after NAC are likely to represent the cancer cell population intrinsically resistant to chemotherapy that leads to subsequent metastatic presentation. This study identifies several potential immune pathways in patients with residual disease which may be targeted. These high-risk patients might therefore benefit from immunomodulatory approaches in future clinical trials. Additionally, we show that longitudinal monitoring of tumor immune dynamics during chemotherapy is feasible and may help to predict therapeutic response early and potentially allow for tailoring of treatment prior to definitive surgery.
Introduction
Increasing evidence supports the hypothesis that breast cancer progression is influenced by the type and functionality of immune cells present within the tumor immune microenvironment (TIME). A focus on infiltrating T cells (TIL) has shown the presence of CD8+ T cells to be prognostic of ongoing remission, driven via IFNγ-dependent mechanisms (1). Similarly, CD4+ T helper-type 1 (Th1) lymphocytes activate and regulate the adaptive immune response (1, 2); and the presence of innate-like γδ T cells is associated with improved survival, likely via their capacity for tissue stress surveillance (3, 4). By contrast, FOXP3+ regulatory T lymphocytes (Treg) play key roles in inhibiting antitumor immune responses and promote disease progression (5, 6).
In highly proliferative breast tumors, e.g., triple-negative breast cancers (TNBC) and human epidermal growth factor receptor-2 (HER2)–positive breast cancers (7–11), which form the majority of reports investigating the nature of immune infiltrates and localization within the TIME, TILs are frequently observed. However, more recent studies of luminal estrogen (ER)–positive, HER2-negative breast cancers reported the presence of TILs as an adverse prognostic factor for survival, suggesting differences in the biology and prognostic significance of immunologic infiltrates across subtypes (12–14).
TNBC and HER2-positive breast cancers with high levels of TILs (>50%), often referred to as “hot”/lymphocyte-predominant breast cancers (LPBC), are more likely to be associated with increased rates of pathologic complete response (pCR) following neoadjuvant chemotherapy (NAC) and improved overall survival (OS), independent of other prognostic factors (7–9, 11, 15). The increased chemosensitivity of these breast cancers has been proposed to result from the ability of chemotherapy to support antitumor immunity by either (i) acquiring somatic mutations that produce new tumor-associated antigens recognized by TILs, (ii) modifying the TIME to enhance trafficking of effector immune cells, or (iii) expulsion of immunosuppressive cells, highlighting these breast cancer subtypes as targets for immunotherapeutic interventions (reviewed in ref. 16). In contrast, the immune cell populations and their functions within post-NAC chemotherapy-resistant disease [residual cancer burden, moderate (RCB II) or extensive (RCB III)] are underexplored. Studies in TNBC residual disease have demonstrated that an increase in TILs after NAC, compared with their pretherapeutic score, is associated with improved prognosis (17, 18), adding independent and additional prognostic information for these high-risk metastatic patients (19). In contrasting reports, an immune infiltrate characterized by high CD3+ T cells and also CD68+macrophages within post-NAC residual disease was associated with a worse disease-free survival (20). These differing observations highlight the need to better understand the immune contexture of “immune cold” tumors and how they evolve in response to chemotherapy.
We hypothesized that the prognosis of patients with residual disease whose immune infiltrate changes following NAC (low-to-high TILs) may depend on the overall net effect of pro- versus antitumor immune responses. Similarly, tumors that remain cold (low TILs) after NAC may represent tumors that have maintained/undergone immune escape, resulting in high metastatic risk disease. Thus, characterization of the TIME in these patients could identify potential immune targets that could be explored within the context of strategies for treating post-NAC chemotherapy-resistant disease.
To test this, we performed an in-depth immunopathologic characterization of chemotherapy-resistant disease (focusing primarily on high-risk RCB II and RCB III disease) following neoadjuvant sequential anthracycline and taxane (AC-T) therapy with or without platinum therapy to understand the immune drivers of TNBC and ER+HER2– residual tumors. In parallel, we performed peripheral immuno-phenotyping using cytometry by time-of-flight (CyTOF) to assess the feasibility of monitoring tumor immune dynamics longitudinally during NAC to help predict therapeutic response early, and potentially allow for tailoring of treatment for chemotherapy-resistant disease prior to definitive surgery.
Materials and Methods
Study cohorts
Retrospective data sets
A series of 298 TNBC and ER+HER2– consecutive breast cancer patient samples treated with NAC for primary breast cancer treated at the Royal Marsden Hospital, Guy's & St Thomas’ NHS trust, and The Netherlands Cancer Institute (NKI) biobank were available for evaluation. All patients were treated with sequential anthracyclines and taxane chemotherapy. For patients with TNBC, a small cohort had also received taxanes with platinum chemotherapy. Patients were identified retrospectively from prospectively maintained hospital research databases. Patients with stage IV disease were excluded, as were those without appropriate consent, or with insufficient pathologic material available for review. Following pathology review of slides and original pathology reports, 153 patients were classified as high risk with RCB II or RCB III disease (TNBC n = 80; ER+HER2–n = 73) and were included in the final data set. Additional information on the study cohort is provided in the results (Table 1).
Table 1.
Comparison of clinicopathologic characteristics of TNBC and ER+HER2– RCB II/III patients whose tumors were assessed for immune infiltrates.
| TNBC (n = 80) | ER+HER2– (n = 73) | |
|---|---|---|
| Age (median, range) | 51 (26–78) | 50 (33–75) |
| Grade (% n) | ||
| 1 | 2.5 (n = 2) | 5.5 (n = 4) |
| 2 | 25 (n = 20) | 64.5 (n = 47) |
| 3 | 72.5 (n = 58) | 30 (n = 22) |
| Pre-NAC tumor stage (% n) | ||
| 1 | 5 (n = 4) | 4.11 (n = 3) |
| 2 | 52.5 (n = 42) | 61.64 (n = 45) |
| 3 | 22.5 (n = 18) | 21.92 (n = 16) |
| 4 | 20 (n = 16) | 12.33 (n = 9) |
| Pre-NAC nodal stage (% n) | ||
| N0 | 46.25 (n = 37) | 54.79 (n = 40) |
| N1 | 45 (n = 36) | 41.1 (n = 30) |
| N2 | 2.5 (n = 2) | 1.37 (n = 1) |
| N3 | 6.25 (n = 5) | 2.74 (n = 2) |
| Pre-NAC stage (% n) | ||
| 2a | 25 (n = 20) | 45.2 (n = 33) |
| 2b | 40 (n = 32) | 26.0 (n = 19) |
| 3a | 11.2(n = 9) | 12.3 (n = 9) |
| 3b | 15 (n = 12) | 12.3 (n = 9) |
| 3c | 8.8 (n = 7) | 4.1 (n = 3) |
| Post-NAC RCB score (% n) | ||
| RCB II | 68.2 (n = 54) | 62.9 (n = 45) |
| RCB III | 31.8 (n = 25) | 37.1 (n = 27) |
Prospective data set for peripheral blood mononuclear cell (PBMC) collection
Peripheral blood samples were collected from patients with TNBC undergoing NAC treatment at Guy's and St Thomas’ Hospitals, London, after written informed consent as part of a non-interventional clinical trial (BTBC study: REC No: 13/LO/1248, IRAS ID 131133). Healthy women were recruited randomly as a control group. This study had local research ethics committee approval and was conducted adhering to the principles of the Declaration of Helsinki. Four cycles of intravenous (i.v.) epirubicin and cyclophosphamide (EC) every 2 to 3 weeks, followed by 12 weeks of i.v. paclitaxel or four cycles of i.v. docetaxel with carboplatin were administered prior to surgery. The study was initially designed to collect samples at baseline and every 3 weeks prior to a new cycle of treatment. However, as this study was carried out at the start of a global pandemic (Coronavirus disease 19, or COVID-19), research teams were unable to capture blood for each patient after every cycle. Therefore, the blood samples collected were grouped as baseline [time point 1 (TP1)], defined as blood captured just before the first EC administration, before any treatment; mid-treatment (TP2), defined as blood captured during the first four cycles of EC chemotherapy; post-NAC presurgical (TP3), defined as just after the last NAC administration; and finally post-surgical (TP4), including all samples taken after surgery, up to 8 weeks after surgery.
Manual stromal TIL evaluation
All assessments were conducted by experienced breast pathologists (YR and RS) and a trained pathology research scientist (PG). One of the pathologists (RS) is the founding member of a five-step standardized scoring system developed by the International Immuno-oncology Biomarker Working Group (21), along with modifications specific to the post neoadjuvant residual disease setting (22). First, we assessed RCB scores on surgical specimens of non-pCR patients using the Web-based MD Anderson RCB calculator (23). All available hematoxylin and eosin (H&E) tissue–stained sections were digitized using Hamamatsu (Nanozoomer HT) scanner, and images were used to assess the degree of lymphocytic infiltration associated with tumor stroma as per the above-mentioned internationally recognized guidelines (21) and as in previous studies of residual disease (9, 17). In brief, stromal TIL scores were defined as the percentage of the tumor–stroma area that was occupied by mononuclear inflammatory cells. TILs in tumor areas with crush artifacts and necrosis were excluded. The percentage of stromal TILs was considered a continuous parameter indicating how much of the demarcated stromal area exhibits dense mononuclear infiltrates. The mean values of the histopathologic evaluation by RS and YR were used for the analyses presented.
Multiplex IHC staining protocol
Multiplex immunofluorescent staining technology was used for simultaneous antibody‐based detection and quantification of the expression of 10 protein markers and DAPI in two staining panels. Panel 1 consisted of CD8, CD4, FOXp3, PD-1, and pCK antibodies; panel 2 consisted of CD68, CD20, CD56, PD-L1 (clone E1L3N), and pCK antibodies. Formalin-fixed, paraffin-embedded (FFPE) tumor full face and core biopsy sections were sectioned at 4 μm and subjected to the staining protocol. Details are included in Supplementary Table S1. PD-1 and PD-L1 expression was assessed (i) within invasive tumor areas and (ii) in tumor-associated stroma. It was defined as a percentage of invasive tumor/stromal areas, respectively, occupied by immunostained cells, as recommended by the International Immuno-Oncology Biomarker Working Group on Breast Cancer (TIL-WG) for quantifying tumor-infiltrating lymphocytes.
Microscopy and quantitative analysis of multiplex IHC
The H&E sections were examined by a pathologist, and regions where TILs were present (“immune hotspots”) were identified. The entire section was imaged on the Vectra system under low magnification to reveal an overarching immune context including the assessment of TIL density and distribution. Select high-powered fields (HPF) were imaged to reveal details of the immune context with resolution sufficient to describe immune subsets and precise tissue location of individual cells. Composite images were analyzed using TissueQuest to define cells as either tumor cell (CK+), T cells subsets (CD4+, CD8+, CD4+FoxP3+ (Tregs), B cell (CD20+), tumor-associated macrophages (CD68+), and natural killer (CD56+) across both TNBC and ER+HER2– breast cancers. For quantitative analysis, digital images were obtained using the Vectra 3.0 Quantitative Pathology Imaging System (PerkinElmer). HPF along the tumor–stroma interface enriched in immune cells (“hot-spots”) were chosen for analysis in each specimen. Cells expressing immune biomarkers from panels 1 and 2 were quantified using the TissueQuest cytometer, an image analysis software (TQ 4.0 TissueQuest Software, TissueGnostics). This is a multicolor tissue cytometry software permitting multicolor analysis of single cells within tissue sections similar to flow cytometry (24). The DAPI channel is the master marker used for the identification of all the immune cells. Extranuclear or nuclear masks were used as appropriate to measure staining intensity in each of the other channels. Cutoff values for all the channels were defined according to negative controls. Expressions were depicted in scattergrams of normalized gray values. The numbers of single-positive and double-positive cells were calculated as cells per mm2.
Protein digital spatial profiling (DSP) and analysis
FFPE 4-μm thick tissue sections were strategically placed onto glass slides according to the DSP protocol. The DSP workflow was carried out by NanoString Technologies as previously described (25). Briefly, FFPE tissue slides were incubated with cocktails of up to 44 unique oligonucleotide-conjugated antibodies. The compartments were identified with fluorescent imaging with antibodies targeting pan-cytokeratin (CK) to detect breast tumor compartment, CD45 for leukocyte detection, and DAPI for nuclear detection. The regions of interest (ROI) were molecularly defined using a fluorescence image of the same slide for CD45 cells. Per patient 12 ROIs were selected in TIL rich areas where possible. Areas of 600 μm in diameter were placed. Once the 12 ROIs were processed, indexing oligos were hybridized to NanoString optical barcodes for digital counting on the nCounter. Digital counts from barcodes corresponding to protein probes were then normalized to ERCC controls counts and CD45 counts. Normalized counts were log2 transformed and tested for differential protein abundance using the ROI counts for each patient separately. Differential abundance analysis was performed using the Welch t test in the R statistical environment (v3.6.1). P values from each analysis (per patient comparison between post- and pre-NAC ROIs) were combined using the Fisher method resulting in meta-P, which were subsequently adjusted for multiple comparisons using the Benjamini and Hochberg method.
Nucleic acid extraction for NanosSring nCounter profiling and analysis
Total RNA extraction was performed using an miRNeasy FFPE Kit Qiagen (cat no./ID: 217504). Procedures were performed according to the manufacturer's protocols. The quality and quantity of extracted RNA were assessed with the Bioanalyzer 6000 RNA Nano Kit (Agilent Technology; cat. #5067-1511) and purity of the sample using the NanoDrop Spectrophotometric method. The FFPE tumor tissue was sectioned at 10 μm (×3–5) and subjected to the extraction protocol. Raw NanoString data were preprocessed using R package NanoStringNorm (v1.2.1; ref. 26). Differential mRNA abundance analysis was performed using voom (TMM normalization), with R package limma (v3.40.6; ref. 27). Genes with an absolute log2 fold change of > 1 and FDR-adjusted P value of <0.1 were considered significant unless stated otherwise. Pathway scores were estimated using the singular value decompositions (SVD) on scaled data of constituent genes in each pathway. Analyses were performed in the R statistical environment (v3.6.1).
RNA-seq validation in melanoma samples
Preprocessed RNA-seq profiles of pretreatment melanoma samples (PubMed ID: 26997480) were downloaded from Gene Expression Omnibus ID: GSE78220. Differential gene abundance for selected NK cell marker genes was performed using an unpaired nonparametric Wilcox rank-sum test between the anti–PD-1 nonresponders [patients with progressive disease (PD)] and responders [patients with complete or partial response (C/PR)].
Peripheral blood sample processing
PBMCs were isolated by density centrifugation using Ficoll-Paque (Sigma-Aldrich, #17-1440-03) at 750 × g for 30 minutes at room temperature (RT). The blood mononuclear cell portion was recovered and washed three times in phosphate-buffered saline (PBS). Cells were counted and resuspended in FBS + 10% DMSO for storage at −80°C, at a density of 10 × 106 PBMCs/mL. In order to minimize run-to-run variation and to facilitate the comparison of cellular profiles over time, PBMCs were processed and cryopreserved upon collection and samples from each patient were analyzed simultaneously by CyTOF in the same run.
CyTOF staining
Cells were thawed rapidly at 37°C and resuspended in RPMI. Cells were incubated with nuclease (Thermo Fisher Scientific, #88701) for 10 minutes at RT, before washing with RPMI and incubation at 37°C for 2 hours at a concentration of 2 × 106 cells/mL. PBMCs were stained with 35 metal conjugated antibodies based on the Maxpar Human Immune Monitoring Panel Kit Cell Staining (Fluidigm, #201324; Supplementary Table S2), according to the manufacturer's instructions. For viability staining, cells were washed in PBS and stained with cisplatin (Fluidigm, #201064) in serum-free RPMI at a final concentration of 5 μmol/L for 5 minutes at RT. Cells were washed in serum-containing RPMI and then Maxpar Cell Staining Buffer (Fluidigm, #201068), followed by blocking with Human TruStain FcX (FcX) block (BioLegend, #422301) in Maxpar Cell Staining Buffer at 1:11 for 10 minutes at RT. The CyTOF panel was then added as a preprepared antibody cocktail and incubated at RT for 30 minutes. Cells were washed in Maxpar Cell Staining Buffer and fixed in 1.6% PFA (Thermo Scientific, # 28906) diluted in Maxpar PBS (Fluidigm, # 201058) for 10 minutes at RT. Fixed cells were then pelleted and resuspended in 125 nmol/L Cell-ID Intercalator-Ir (Fluidigm, # 201192A), diluted in Maxpar Fix and Perm Buffer (Fluidigm, # 201067) for 1 hour at RT. Cells were then washed with Maxpar Cell Staining Buffer and resuspended in 10% DMSO/FBS and stored at –80°C.
Cells were analyzed using the Helios mass cytometer (Fluidigm). Directly before analysis, cells were washed in Maxpar Cell Acquisition Solution (CAS; Fluidigm, #201240) and then resuspended in CAS containing 0.1X EQ calibration beads (Fluidigm, #201078). Cells were diluted to a concentration of 0.5*106 cells/mL for analysis.
CyTOF data analysis
Cytobank (www.cytobank.org) was used for the initial manual gating of live, CD45+, CD66b−, single cells. High-dimensional CyTOF data analysis was performed using R version 3.6.0 (28) and library “flowCore” (package version 1.52.1; ref. 29). The pipeline implemented and used was a customized version of the workflow described in ref. 30 and included the following analytical steps.
Sample selection
Samples were assigned to four different groups defined by the treatment timescale, namely, baseline, mid-treatment, post-NAC presurgical, and post-surgery. If multiple samples of a patient were assigned to the same group, only the one taken at the latest time point was included in the analysis, to avoid sample bias. This filtering procedure removed six samples on treatment and three samples from post-surgery.
Diagnostic and cell events subsampling
Sample diagnostic analysis revealed that the range of cell events varied across samples and that three samples had less than a thousand events recorded. To maximize the statistical power and to avoid possible sample size biases, a subsampling procedure was implemented to extract 450 random cell events from each available sample. To verify the robustness of this approach, the entire pipeline analysis was run multiple times using four different seeds of the pseudorandom number generator.
In principle, this approach presented the disadvantage of being prone to low sensitivity of immune cell quantification due to the finite number of the events subsampled. However, the results obtained showed good agreement even for proportions of immune cell types making around 5% of the overall population fraction. Beyond possible biases due to differences in sample sizes, this approach had the additional advantage to allow for cell population discovery using all samples combined across treatment phases and to apply dimensionality reduction algorithms using all data available (see sections below).
Data processing
The pipeline analysis was carried out in parallel on two complementary subsets of data: the first set included 24 samples from 8 patients with TNBC for whom baseline, on-treatment, and post-surgical time points were collected; the second set included 28 samples from 14 TNBC patients at baseline and at post-surgical.
Raw CyTOF marker intensities of 26 markers were imported using the read.flowSet function and then transformed using the hyperbolic inverse sine function with cofactor 5 (31, 32) to obtain more symmetric distributions across the samples and to map them to comparable ranges of expression.
To test that marker intensities were valid features, the t-distributed stochastic neighbor embedding (t-SNE) algorithm (33) was used to project all cell events from each set of samples into two-dimensional maps and color the points by marker intensity. The distributions of expression of each marker were scaled between 0 and 1 using 1% and 99% percentiles as the boundaries and plotted for a visual diagnostic assessment.
Cell population identification
Cell population identification was conducted with a combination of hierarchical clustering and consensus clustering. Scaled intensities of markers were used to hierarchically cluster all cell events from sample subsets (14-patient analysis or 8-patient analysis) into 25 clusters using an extension of Ward's minimum variance method (34). This choice allowed us to unbiasedly obtain a level of overclustering necessary to separate rare cell types and residual debris from the main populations. The final clusters were then annotated by visual inspection of the markers’ expression distributions associated with each cluster population and assigned to one of six cell populations of interest.
Visualization of immune cell populations
All cell events from selected sample subsets (8-patient analysis or 14-patient analysis) were visualized using t-SNE (DOI: 10.5281/zenodo.5206664) via dimensionality reduction of their associated marker intensity profiles and colored according to the annotated immune cell populations. The two-dimensional maps were then stratified according to patient groups (RD and pCR) and to treatment stage (baseline and post-surgery in the 14-sample analysis; baseline, on treatment, and post-surgery in the 8-sample analysis). Because cell population identification was carried out by pooling samples together across different treatment phases, the abundance of the populations was immediately comparable between maps associated with different treatment stages and/or patient groups.
Differential abundance analysis
Differential abundance analysis of defined cell populations between patient groups (RD vs. pCR) at each time point was performed using the empirical Bayes moderated t test implemented in the package LIMMA (27) on the log-transformed cell event counts 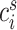 associated with cell type/family
associated with cell type/family 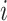 in sample
in sample 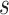 in that time point:
in that time point:
This transformation was needed to obtain Gaussian-like distributions of cell counts.
For testing the significance of DA of populations within the same group of patients between different time points (midtreatment vs. baseline and postoperation vs. baseline), a paired-samples version of the LIMMA test was used.
Data availability statement
Data are available from the authors upon reasonable request. Details of open access data are provided in DOI: 10.5281/zenodo.5206664.
Results
TNBC and ER+HER2– breast cancers with RCB II/III are immunologically “cold” at baseline and at the end of NAC
To characterize the immune microenvironment of chemotherapy-resistant breast cancers, we profiled the TIME in both posttreatment residual disease and treatment-naïve breast cancer, where available, combining bright-field H&E imaging and multiplexed IHC. In addition, we used digital spatial profiling to further characterize the expression of canonical costimulatory and inhibitory molecules on stromal TILs. Finally, we measured the expression of 1,330 cancer and immune-related genes within resected RCB II/III tumors and the baseline biopsies (Fig. 1A). A series of 298 TNBC and ER+HER2– consecutive breast cancer patient samples treated with NAC were available for evaluation. Given the primary aim of this study was to identify potential immune targets that could be further explored within the context of post-NAC chemotherapy-resistant disease, pathology review was carried out to exclude any patients who had achieved pCR/RCB I disease, known to have an excellent long-term outcome. Following the pathology review of slides and original pathology reports, 153 patients were classified as having RCB II or RCB III disease (TNBC n = 80; ER+HER2–n = 73; Fig. 1B) and were included in the final data set. Patient characteristics are summarized in Table 1. Of note, 19 cases of residual disease from patients with TNBC had also received platinum chemotherapy as part of their sequential NAC regimen. Baseline pre-NAC samples were available for evaluation for 32 of 80 TNBC and 36 of 73 and ER+HER2– cases. Consistent with previous findings (19), RCB scores were prognostic for progression-free survival (PFS) and long-term survival after NAC in both phenotypic subsets of breast cancers (Fig. 1C and D).
Figure 1.

Histopathologic evaluation of the TIME of pretreatment and residual chemotherapy-resistant samples. A, Schematic representing the four-step strategy for evaluation of immune TIME within pre- and post-NAC residual cancers. B, Flow chart of the study population selection. C, Kaplan–Meier curves for PFS in ER+HER2– and TNBC patients with RCB II and III. D, Kaplan–Meier curves for OS in ER+HER2– and TNBC patients with RCB II and III disease. E, Representative H&E images depicting increasing stromal lymphocytic infiltration within residual cancers. F, sTIL levels in pre- (baseline) and posttreatment (RCB II/III) samples within ER+HER2– and TNBC cancers. Statistical significance was calculated using the nonparametric Mann–Whitney U test. The percentage of stromal TILs is the area of stromal tissue (area occupied by mononuclear inflammatory cells over total intratumoral stromal area). G and H, Changes in stromal lymphocytic infiltration in matched pretreatment and residual disease samples. P values from two-tailed paired t tests are shown. I, Kaplan–Meier curves for (i) PFS and (ii) OS in ER+HER2– patients stratified according to sTIL infiltration (cutoff 10%). J, Kaplan–Meier curves for (i) PFS and (ii) OS in patients with TNBC stratified according to sTIL infiltration (cutoff 10%).
We first assessed H&E slides of resected post-NAC ER+HER2– and TNBC residual disease and matched pre-NAC core biopsies via bright-field microscopy for stromal lymphocytic infiltration (Fig. 1E). TILs were scored in 153 cases of residual disease where tissue was available. We found that most chemotherapy-resistant ER+HER2– and TNBC breast cancers were immune “cold” at baseline with comparably low mean stromal TIL (sTIL) scores of 4.2% (SD 6.27) and 6.9% (SD 7.98), respectively (Fig. 1F). By contrast, post-NAC sTILs in TNBC RCB II/III disease were significantly higher than those in ER+HER2– RCB II/III disease [median 14% (SD 16.9) vs. 5.1% (SD 6.86), respectively]. In the 36 of 73 ER+/HER2− tumors and 32 of 80 TNBC tumors where we had paired pre-NAC biopsies, there was no significant change in the sTIL counts following NAC (Fig. 1G and H).
Next, we assessed the prognostic influence of the level of immune infiltrates in the residual disease. Kaplan–Meier analysis was performed using the median value for RD sTILs in ER+HER2– (3%) and TNBC (8.8%) residual disease to dichotomize groups, and there was no significant difference in either PFS or OS between patients based on median RD sTILs in either breast cancer subtypes (Supplementary Fig. S1A and S1B). A TIL cutoff of 10% is considered to be clinically relevant and recently proposed to define “immune-enriched” breast cancers (35). We, therefore, hypothesized there might be a smaller subpopulation of patients with high TILs scores that have a differential outcome. To test this, we performed Kaplan–Meier analyses by dichotomizing TILs scores at the value of 10% in the ER+HER2– patient population for both PFS (Fig. 1Ii) and OS (Fig. 1Iii). Tumors with stromal TILS >10% had a significantly poorer PFS (log-rank P = 0.0013; median 4.183 vs. undefined years; Fig. 1Ii) and OS (log-rank P = 0.0042; median 10.13 vs. 5.811years; Fig. 1Iii) compared with tumors with stromal TILs <10%. This did not hold true for TNBC residual cancers at cutoffs of 10% (Fig. 1Ji and ii).
Chemotherapy-resistant residual breast cancers are characterized by low B-cell infiltrates and downregulation of immune-checkpoint receptor expression
Given the different prognostic implications of sTILs between breast cancer subtypes with residual cancers, we hypothesized that the immune cell compositions of TNBC and ER+HER2– RCB II/III disease are also likely to be qualitatively different. We used H&E and multiplexed immunofluorescence panels to further characterize immune TIME of RCB II and RCB III residual disease and its matched baseline pre-NAC cores (Fig. 2A). Cells were classified using canonical surface markers as either tumor cells (CK+), T-cell subsets [CD4+, CD8+, CD4+FoxP3+ (Tregs)], B cells (CD20+), tumor-associated macrophages (CD68+), or natural killer cells (CD56+). Although no significant difference in the distribution of these immune cell subsets between TNBC and ER+HER2– residual disease (Fig. 2B) was observed, a significant decrease in B cells was detected in ER+HER2– residual disease compared with pre-NAC biopsies (Fig. 2Ci). This was further supported by a significant decrease in total immune cell infiltrate seen in ER+/HER2− residual disease (Supplementary Fig. S2Ai). Likewise, a significant decrease in B cells was apparent in TNBC residual disease compared with pre-NAC biopsies (Fig. 2Cii). This was also accompanied by increased NK cell infiltration in post-NAC samples (Fig. 2Cii); however, there was no overall change in total immune cell infiltrates after NAC in TNBCs (Supplementary Fig. S2Aii). It is also noteworthy that in TNBC RCB II/III disease, a trend for decrease in immunosuppressive Tregs but an increase in the CD68+ tumor-associated macrophages was observed.
Figure 2.

Immune context of immunologically cold chemotherapy-resistant ER+HER2– and TNBC breast cancers. A, Representative multiplexed IHC images showing staining of key lineage markers and immune-checkpoint expression in tissue sections. B, Bar charts showing immune composition of ER+HER2– (n = 16, blue) and TNBC (n = 15, red) residual disease samples. Statistical analysis to compare immune composition between subtypes was performed using the two-stage Mann–Whitney test. C, Log2 FC in immune cell types in residual disease samples compared with pretreatment samples in (i) ER+HER2– disease (n = 10 matched-paired samples) and (ii) TNBC (n = 10 matched paired samples; Wilcoxon matched-pairs signed rank test; *, P ≤ 0.05; **, P ≤ 0.01). D (i–ii), Percentage of tumoral and stromal PD-L1 expression within the TIME of TNBC RCB II/III disease. D (iii–iv), Percentage of PD-1 expression on immune cells found within the invasive tumor (iii) and tumor stroma (iv) in TNBC RCB II/III disease (Mann–Whitney test; *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001). E, Representative image of a selected ROI for digital spatial profiling based on CD45 staining. Per patient 12 ROIs were selected in TIL-rich areas where possible. Areas of 600 μm in diameter were placed. F and G, Quantification of protein expression of immune cell lineage markers and immune checkpoints in selected ROIs from ER+HER2– and TNBC residual disease samples. H, Heatmap showing differential protein expression of significantly upregulated or downregulated immune-checkpoint markers within TNBC RCB II/III tumors compared with pre-NAC baseline samples. Only those immune-checkpoint receptors where they were significant in at least 2 of 3 patients with FC in the same direction are indicated (P < 0.05; see also Supplementary Table S3).
The recent FDA approval of pembrolizumab (anti–PD-L1) immunotherapy in combination with chemotherapy for high-risk early TNBC highlights the need to understand the role of chemotherapy in modulating the PD-1/PD-L1 axis (36), especially in chemotherapy-refractory disease where immunotherapies may play a vital role. We therefore also evaluated the expression of PD-L1 (cutoff for positivity ≥1%) on both tumor (tumoral PD-L1) and immune cells (stromal PD-L1) in pre-NAC biopsies (baseline) and post-NAC RCB II/III disease. The majority of the TNBC and ER+HER2– residual tumors were PD-L1-negative (Supplementary Fig. S2B). For TNBC, this represented a significant decrease in the tumoral and stromal PD-L1 expression following NAC in RCB II/III disease when compared with baseline samples (Fig. 2Di and ii). This was specific to TNBC, as no change was detected in ER+HER2– RCB II/III disease from baseline (Supplementary Fig. S2Ci and ii). We also evaluated the expression of PD-1 on immune infiltrates within the invasive tumor and those within the tumor stroma (Fig. 2Diii and iv; Supplementary Fig. S2Ciii and iv). As with PD-L1, a significant decrease in the PD-1 expression in TNBC residual disease was measured, whereas no significant change was observed in ER+HER2– RCB II/III disease (Fig. 2Diii–iv; Supplementary Fig. S2Cii and iv).
To validate these findings and further characterize the expression of costimulatory and inhibitory immune checkpoints within chemotherapy-resistant disease, we used the NanoString DSP technology (Fig. 2E) on a subset of samples. In keeping with immunofluorescence findings (Fig. 2B), DSP confirmed a similar distribution of canonical immune cell markers across both TNBC and ER+HER2– RCB II/III disease, with markers of CD4 and CD8 T cells and monocyte/macrophages (CD14/CD68/CD163) being most abundant and those of Tregs (CD25/FOXP3) and NK cells (CD56) being the least abundant (Fig. 2F). Immune-checkpoint receptor expression was also mirrored across TNBC and ER+HER2– RCB II/III disease (Fig. 2G). In TNBC cancers, stimulatory and inhibitory immune-checkpoint receptors including CD86 and OX40L were significantly downregulated in post-NAC residual disease compared with pre-NAC core biopsies (FDR-adjusted P < 0.05; Fig. 2H; Supplementary Table S3 and DOI: 10.5281/zenodo.5206664). These data highlight the complexity of immune regulatory networks, and careful dissection of the stimulatory and inhibitory pathways activated following NAC will be required for the design of strategies to overcome resistance, relapses, and to improve response rates.
Circulating immune cell phenotype dynamics within patients with TNBC are comparable to the post-NAC RCB II/III TIME
Peripheral blood immune parameters such as circulating T-cell repertoire, proliferation, expansion, and immune-checkpoint expression are well-known correlates of chemotherapy and immunotherapy response (37–40). We, therefore, asked whether NAC-induced changes in the TIME of TNBC residual disease could be detected in the peripheral blood. We prospectively recruited 14 patients with TNBC and profiled their PBMCs before, during, and after NAC treatment using CyTOF (Fig. 3A). Of these, 7 patients achieved a pCR or minimal disease burden after NAC (RCB I), whereas 7 had poor response with RCB II/III disease (Table 2). Samples were collected from all 14 patients at baseline and post-surgery and, of these, 8 patients (4 with residual disease and 4 with pCR) had blood captured during NAC treatment. Across both outcome groups, patients had tumors of comparable size and stage before starting NAC and were treated with a combination of anthracycline–cyclophosphamide and taxane-based chemotherapy, although a higher proportion of patients who achieved pCR were treated with additional platinum-based therapy (Table 2).
Figure 3.
![Figure 3. Longitudinal CyTOF profiling of patient PBMCs reveals NAC-induced enrichment of NK cell populations associated with clinical response. A, Schematic of CyTOF study. B(i), t-SNE analysis of matched PBMC samples from patients with RD (n = 4) and pCR (n = 4) at baseline, on-treatment, and post-surgery showing the identification of major immune cell families. B(ii and iii), Log2 FC in the abundance of each cell population on treatment (n = 8) and post-surgery (n = 14), compared with baseline, among patients with B(ii) pCR and B(iii) RCB II/III disease; LIMMA test. P values represent those which showed statistical significance in at least 3 of the 4 seeds tested, with a |log2 fold change (FC)| > 1.5. B(iv) and B(v), Log2FC in proportions of major immune cell families on-treatment [B(iv)) (n = 8)] and post-surgery [(B(v)) (n = 14)], normalized to baseline proportions (LIMMA). C, Proportion of CD16+ and CD16- NK cell clusters at baseline among patients with RD and pCR (n = 14). LIMMA test. D, Proportion of CD16+ and CD16– NK cell clusters on-treatment among patients with RD and pCR (n = 14). LIMMA test. E, FACS gating strategy of manually gated CD3– CD19–CD123–CD56+ NK cells showing identification of 4 NK cell subsets based on CD56 and CD16 expression. F, Baseline peripheral NK cell subsets among patients who went on to achieve pCR or RD after NAC and healthy donors (Mann–Whitney U test; ***, P ≤ 0.0001). G, Peripheral NK cell subsets at on-treatment time point from patients who went on to achieve pCR or RD after NAC (Mann–Whitney U test; *, P ≤ 0.05). H, Peripheral NK cell subsets at the post-surgical timepoint among patients who achieved pCR or RD after NAC (Mann–Whitney U test; ns P > 0.05).](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9561554_4494fig3.jpg)
Longitudinal CyTOF profiling of patient PBMCs reveals NAC-induced enrichment of NK cell populations associated with clinical response. A, Schematic of CyTOF study. B (i), t-SNE analysis of matched PBMC samples from patients with RD (n = 4) and pCR (n = 4) at baseline, on-treatment, and post-surgery showing the identification of major immune cell families. B (ii and iii), Log2 FC in the abundance of each cell population on treatment (n = 8) and post-surgery (n = 14), compared with baseline, among patients with B (ii) pCR and B (iii) RCB II/III disease; LIMMA test. P values represent those which showed statistical significance in at least 3 of the 4 seeds tested, with a |log2 fold change (FC)| > 1.5. B (iv) and B (v), Log2FC in proportions of major immune cell families on-treatment [B (iv); n = 8] and post-surgery [(B (v); n = 14], normalized to baseline proportions (LIMMA). C, Proportion of CD16+ and CD16- NK cell clusters at baseline among patients with RD and pCR (n = 14). LIMMA test. D, Proportion of CD16+ and CD16– NK cell clusters on-treatment among patients with RD and pCR (n = 14). LIMMA test. E, FACS gating strategy of manually gated CD3– CD19–CD123–CD56+ NK cells showing identification of 4 NK cell subsets based on CD56 and CD16 expression. F, Baseline peripheral NK cell subsets among patients who went on to achieve pCR or RD after NAC and healthy donors (Mann–Whitney U test; ***, P ≤ 0.0001). G, Peripheral NK cell subsets at “on-treatment” time point from patients who went on to achieve pCR or RD after NAC (Mann–Whitney U test; *, P ≤ 0.05). H, Peripheral NK cell subsets at the “post-surgical” timepoint among patients who achieved pCR or RD after NAC (Mann–Whitney U test; ns P > 0.05).
Table 2.
Comparison of clinicopathologic characteristics of patients with TNBC achieving pCR/RCB I or RCD II/III after NAC whose peripheral blood was analyzed by liquid CyTOF.
| pCR/RCB I (n = 7) | RCB II/III (n = 7) | |
|---|---|---|
| Age (median, range) | 46 (37–66) | 50 (32–57) |
| Grade at consent (n) | ||
| 2 | 1 | 1 |
| 3 | 5 | 6 |
| Unknown | 1 | 0 |
| Stage at consent (n) | ||
| IA | 2 | 1 |
| IIA | 4 | 3 |
| IIB | 1 | 1 |
| IIIA | 0 | 1 |
| IIIB | 0 | 1 |
| Nodes (n) | ||
| Positive | 2 | 2 |
| Negative | 5 | 5 |
| Pre-NAC tumor size (mm) | ||
| Median | 38 | 25 |
| Range | 21–64 | 8–50 |
| Chemotherapy (n) | ||
| Anthracycline–ccyclophosphamide | 6 | 7 |
| Platinum | 5 | 1 |
| Taxane | 5 | 7 |
We carried out high-dimensional analysis to first identify relevant lineage markers (CD3, CD4, CD8, CD56, CD19, and CD11c; Fig. 3Bi; Supplementary Tables S4 and S5; DOI: 10.5281/zenodo.5206664). Mirroring the depletion of B cells observed at the site of residual disease (Fig. 2C), a significant reduction in the proportion of circulating B cells on treatment could be detected in both outcome groups (Fig. 3Bii and iii; on-treatment green bars). Utilizing an unsupervised clustering approach (t-SNE), 16 distinct immune cell clusters were identified (Supplementary Fig. S3A; DOI: 10.5281/zenodo.5206664). A deeper analysis of the B-cell clusters confirmed a decrease in memory B cells following NAC across all patient groups (Supplementary Fig. S3B). These findings are consistent with other recent studies (41) reporting on the selective influence of chemotherapy on the B-cell subsets.
Specific to patients with TNBC with RCB II/III disease, and not observed in patients with TNBC who achieved pCR to NAC, we observed an increase in the proportion of peripheral NK cells following treatment (Fig. 3Biii). This was comparable with the observation in TNBC post-NAC RCB II/III TIME in situ (Fig. 2C). In fact, NK cells represented the only population with significant temporal changes on chemotherapy or following resection between the two outcome groups (pCR/RCB I vs. RCB II/III disease; Fig. 3Biv and v).
CD16+ NK cells possess potent cytolytic capacity, a function often associated with effective tumor rejection (42). Analysis of the NK cell cluster revealed that patients who achieved a complete response/minimal residual disease with NAC had significantly more circulating CD16+ NK cells at baseline compared with those patients who had RCB II/III (Fig. 3C). Moreover, analysis of the CD16+ and CD16– NK cells in the 8 patients with available on-treatment samples revealed higher proportions of cytotoxic CD16+ NK cells in patients on track to achieve a pCR (Fig. 3D). This was in stark contrast to patients on RCB II/III disease trajectory at surgery, who instead exhibited higher proportions of CD16– NK cells pre- and- on-treatment (Fig. 3D). Thus, a lack of cytotoxic NK cells prior to NAC may contribute to chemotherapy resistance.
Since NK cells were enriched in both the TIME and periphery of patients with high-risk resistant disease, we further investigated the NK cell phenotypes, based on the expression of CD16 and CD56 (Fig. 3E; refs. 43, 44). CD56brightCD16−cells exhibit superior cytokine production, whereas CD56dimCD16+ cells primarily demonstrate enhanced cytotoxicity. It has been demonstrated that upon activation, CD56dimCD16+ NK cells lose the expression of CD16 through metalloprotease-mediated shedding and become CD56dimCD16− (referred to as activation-induced shedding; ref. 45). Here, patients destined to achieve a complete response had baseline profiles of NK cell subsets comparable with healthy controls (Fig. 3F). In contrast, patients with residual tumor at surgery had significantly lower proportions of cytotoxic peripheral NK cells (CD56dimCD16+) at baseline (Fig. 3F). We observed a further decrease in this subset among the residual disease patient group during NAC (Fig. 3G). This was accompanied by an enrichment of CD56dimCD16– NK cells, which became the dominant subtype in high-risk residual disease patients. Following removal of the residual disease, circulating NK phenotypes were comparable across both patient groups (Fig. 3H), supporting the notion that tumor cells directly alter the NK cell phenotype as opposed to the other way around.
Subtype- and chemotherapy-specific immune profiles exist in post-NAC chemotherapy-resistant ER+HER2– and TNBCs
To further characterize immune and cancer pathways active in TNBC and ER+HER2– RCB II/III disease following NAC, we quantified bulk mRNA expression using the NanoString nCounter immunology and pancancer panels. Ninety-five immune genes were significantly differentially expressed [DE; FDR-adjusted P < 0.1 and |log2 fold change (FC)| of >1] across TNBC and ER+HER2– residual disease (41 upregulated and 54 downregulated in TNBC disease; Fig. 4A; Supplementary Tables S6 and S7). Additionally, from 730 genes in the PanCancer Pathway set, 132 genes were found to be significantly DE (FDR-adjusted P < 0.1 and |log2FC| of >1) across TNBC and ER+HER2– residual disease samples (Fig. 4B; Supplementary Tables S8 and S9). Interestingly, in line with previous reports (46, 47), the most significantly DE immune genes in TNBC residual disease (defined as having a log2FC >2 with an FDR-adjusted P < 0.1) were enriched for pathways related to neutrophil degranulation and IL17 signaling (LCN2, CCL23, SI00A8, S100A7, and S100B; Fig. 4A; Supplementary Table S6). By contrast, the enrichment of genes involved in extracellular matrix organization (CTSG, TPSAB1, CXCL14, and CCL14) was seen in ER+HER2– disease (Fig. 4A; Supplementary Table S7).
Figure 4.

Subtype- and chemotherapy-specific immune profiles observed in post-NAC chemotherapy-resistant breast cancers. A, Heatmap showing expression of immune profiling gene set in ER+HER2– and TNBC residual disease samples. DE genes with FDR-adjusted P < 0.1 and a |log2FC| of >2 are highlighted in the heatmap. Red represents genes that are highly enriched in TNBC residual disease, and blue represents genes highly enriched in ER+HER2– disease. B, Heatmap showing expression of the pan-cancer profiling gene set in ER+HER2– and TNBC residual disease samples. C, GZMM, GZMA, and KLRC2 represent downregulated genes in carboplatin-treated residual disease. D, Heatmap showing Spearman rank correlation coefficient between pan-cancer and immune-related genes in ER+HER2– residual disease samples. E, Heatmap showing Spearman rank correlation coefficient between pan-cancer and immune-related genes in TNBC residual disease samples from patients who received AC-T only (i) or with (ii) additional platinum-based chemotherapy.
In addition to being subtype specific, the post-NAC profiles of RCB disease were also chemotherapy specific (Fig. 4C; Supplementary Fig. S4A; Supplementary Table S10). TNBC samples treated with the addition of carboplatin exhibited 41 DE genes [18 immune (Fig. 4C) and 23 pan-cancer (Supplementary Fig. S4A)]. Specifically, we observed that in carboplatin-treated residual disease, the GZMM, GZMA, and KLRC2 genes, all related to innate effector cells such as NK−, CD8, or γδ T cells, were downregulated compared with residual disease treated with sequential anthracyclines and taxanes alone (Fig. 4C).
Next, we evaluated if specific changes in the immune pathways were correlated with changes in pan-cancer pathways. We observed a weak to modest correlation between immune and cancer pathways in ER+HER2– RCB II/III disease, except macrophage functions (Fig. 4D). By contrast, a strong correlation between immune and cancer pathways existed in TNBC residual disease for both AC-T–treated and AC-T– and carboplatin-treated cancers (Fig. 4Ei and ii), consistent with a greater degree of tumor–immune cross-talk within TNBCs.
Immune gene-expression changes after chemotherapy in ER+HER2– and TNBC RCB II/III disease
Next, we evaluated the changes in the gene-expression profiles (cancer and immune) of ER+HER2– and TNBC RCB II/III disease compared with their matched pre-NAC baseline tumor tissue. It is well recognized that immunosuppressive cells such as Tregs, M2 macrophages, and myeloid-derived suppressor cells can support protumoral immunity within the TIME. We therefore specifically looked at how genes known to be markers of suppressor cells changed with NAC in chemotherapy-refractory disease (Supplementary Table S11). We did not identify any significant hits; however, consistent with previous reports (48), we noted upregulation in several genes associated with the MAPK pathway in both ER+HER2– and TNBC residual cancers (Supplementary Fig. S4B and S4C). Specifically, immune profiling revealed significant changes in 30 and 41 immune genes in ER+HER2– and TNBC RCB II/II disease, respectively (FDR-adjusted P < 0.1 and |log2FC| > 1; Fig. 5A and B). The majority of genes were upregulated following NAC in both subtypes; however, minimal overlapping DE genes were observed between ER+HER2– and TNBC, indicating subtype-specific responses (Fig. 5C). Consistent with earlier findings (Figs. 2C and 3B), the “top” downregulated genes in residual tumors irrespective of the subtype were related to B-cell biology (CD19, CD79B, TNF, MS4A1, PAX5, and CD22; Fig. 5D and E). Note that in TNBC RCB II/III disease, downregulation of CD209, a marker of dendritic cells, was one of the “top” hits, potentially providing another mechanism through which resistant tumors promote a tolerogenic TIME phenotype. Additionally, the increased NK cell count seen in TNBC RCB II/III disease (Figs. 2C and 3B) was further validated; many of the upregulated genes were associated with NK cell biology (KLRC1, KLRC2, KIR3DL3, KLRD1, and EOMES; Fig. 5F) and were particularly associated with NK cell inhibition or exhaustion (49).
Figure 5.

Changes in immune gene expression after chemotherapy in ER+HER2– and TNBC RCB II/III disease. A, Heatmap showing expression of immune profiling gene set in matched pretreatment and residual disease ER+HER2– samples. B, Heatmap showing expression of the immune profiling gene set in matched pretreatment and residual disease TNBC samples. C, Venn diagram showing upregulated or downregulated genes after chemotherapy treatment in ER+HER2– and TNBC samples. D, STRING analysis of known interactions between hit genes downregulated in ER+HER2– residual disease samples, compared with pretreatment. E, STRING analysis of known interactions between hit genes downregulated in TNBC residual disease samples, compared with pretreatment. F, STRING analysis of known interactions between hit genes upregulated in TNBC residual disease samples, compared with pretreatment. G, Expression of NK-related marker genes in PD-L1/PD-1–low melanoma tumors stratified by response to anti–PD-1 therapy.
To bring all our data together, we summarized that the post-NAC chemotherapy-refractory TNBC tumors are PD-1/PD-L1 low (Fig. 2D and H), but also show enrichment for an exhausted NK cell phenotype both locally and peripherally (Figs. 2C, 3D, 3E, and 5F). To investigate if this post-NAC chemotherapy-refractory disease could benefit from an immunomodulatory approach, we interrogated a publicly available data set of PD-1/PD-L1–low melanoma tumors with pretreatment biopsies identifying factors that may influence innate sensitivity or resistance to anti–PD-1 therapy (50). We first identified all marker genes that are expressed stably and specifically in NK cells within the immune profiling panel, as per the method described by Danaher and colleagues (51). These act as reference genes specific to individual cell types, as they are expressed only in their nominal cell type. Within the melanoma cohort, we identified 13 patients (8 nonresponders and 5 responders) with extremely low expression of PD-1/PD-L1 tumors (defined as having log2 mRNA abundance of ≤1). Within these 13 patients, we examined the expression of the above-mentioned NK-related genes (RRAD, PLA2G6, ZNF205, BCL2, NCR1, MPPED1, GZMB, FOXJ1, GTF3C1, IL21R, and FUT5) between the nonresponders (NR) and those who exhibited a complete or partial response (R) to anti–PD-1 therapy (Fig. 5G). Although we observed no statistically significant differences in the expression of these genes across Rs or NRs in PD-1/PD-L1–low tumors, several markers (PLA2G6, ZNF205, BCL2, GZMB, and GTF3C1) are moderately expressed in these tumors. It is plausible that checkpoint inhibition with antagonistic antibodies (e.g., anti-KIR mAbs targeting KIR2DL3) could potentially rescue NK cell exhaustion (as observed in Fig. 4) and thereby generate an antitumor capacity.
Discussion
Immune cells constitute an important component of the tumor microenvironment, and a strong association between lymphocytic infiltrate and chemotherapy response in early breast cancers has been described (23). Here, we present multidimensional immunologic characterization of chemotherapy-resistant (defined as RCB II and RCB III) disease following neoadjuvant sequential anthracycline and taxane therapy with or without platinum therapy and begin to understand some of the immune drivers of subtype-specific differences observed in TNBC and ER+HER2– residual tumors. We were also able to identify associations between circulating immune cells, particularly NK populations, and response to NAC in patients with early triple-negative disease.
Consistent with the correlation between high immune infiltrates and pCR, we found that irrespective of breast cancer subtype, RCB II and III tumors were predominantly immunologically cold (non-LPBCs) at baseline and end of NAC. This suggested a poor immune cell engagement that was not enhanced by chemotherapy via mechanisms such as chemotherapy-induced immunogenic death response. Previous studies have described heterogeneous post-NAC immune responses in residual cancer tissue, with one study reporting that 48% of patients showed an increase in the level of TILs in residual disease and 47% experienced a decrease (52). We focused specifically on the high metastatic risk RCB II and III disease, where no significant increase in TILs scores in matched pre- and post-NAC cases in ER+ HER2– and TNBC disease was observed. TILs are increasingly being recognized as a continuous variable, obviating the need to determine binary cutoffs (21). We observed that even within defined immunologic cold tumors, TNBC RCB II/III disease was associated with higher levels of immune infiltration compared with ER+HER2– residual disease. However, the immunophenotyping of TIME of these “cold” TNBC and ER+HER2– RCB II and III disease revealed no significant differences in the distribution of the immune infiltrates, or the immune-checkpoint receptor expression across the differing subtypes. Importantly, residual disease across subtypes was associated with high proportions of CD68+ macrophages and CD4+ and CD8+ T cells, and an improved understanding of their mechanistic role in tumoricidal or protumoral functions within high metastatic risk cancers will be crucial for therapeutic manipulation of the TIME.
Although we acknowledge that the immunophenotyping of the TIME lacked comparative samples from patients achieving pCR/RCB I or all four IHC breast cancer subtypes, we had intentionally designed the study to understand the immune drivers of TNBC RCB II/III disease representing the worst prognostic subgroup following NAC. ER+HER2– residual cancers represent the other extreme of the prognostic spectrum. Longitudinal studies with on-treatment biopsies to better understand dynamic changes in the TIME over the course of NAC are currently planned.
In this study, deeper immunophenotyping through gene-expression profiling and assessment of changes in the immune profiles within matched cases revealed subtype-specific profiles of TNBC and ER+HER2– residual disease. These are in line with other studies (53). Broadly speaking, we found that residual disease after NAC in TNBC demonstrated a strong interplay between immune and cancer pathways as compared with ER+HER2– residual cancers. We speculate that cancer cell–specific mechanisms primarily drive progression in ER+HER2– disease as opposed to TNBC, where the interplay between cancer cells and the immune infiltrate is active and can influence long-term outcomes. These data support the notion that immune modulation is likely to have only minimal efficacy in ER+HER2– chemotherapy-resistant disease, and these strategies may be more meaningful in TNBC RCB II/III disease.
More specifically, our analyses identified several findings that require further investigation in the post-NAC residual disease setting. First, we observed downregulation of several stimulatory and inhibitory immune-checkpoint receptor expression (including PD-L1 and PD-1) in post-NAC TNBC RCB II/III disease, highlighting the complexity of therapeutic interventional with immunotherapies in the post-NAC adjuvant setting. Second, we observed an enrichment of neutrophil-attracting chemokines in post-NAC residual disease in TNBC. Tumor-infiltrating neutrophils are known to possess both tumor-promoting and antitumor immune functions (N1 vs. N2; ref. 54). Moreover, CD62Ldim neutrophils have been implicated in the development of lung metastasis, specifically within TNBC via release of tumor-derived HMGB1 (55). Given that HMGB1 is also released in response to chemotherapy treatment (56), further exploration of this pathway and neutrophil recruitment in chemotherapy-resistant TNBC is warranted. Third, we observed an increase in NK cell populations in patients with post-NAC residual disease in TNBC within both the local tumor microenvironment and peripheral white cell populations. Breast cancers have been shown to evade NK cell–mediated antitumor activity by decreasing the NK cell cytotoxic potential (57). NK cells present in the breast TIME are predominantly nonfunctional and immature, with enrichment of the CD56dimCD16– and CD56brightCD16– subsets (44, 57), and, consistent with this, we observed CD56dimCD16– as the dominant subtype in circulation during NAC in high-risk residual disease patients. Interestingly, following removal of the residual disease, the NK phenotypes were once again comparable across those patients with pCR and residual disease, supporting the notion that tumor cells directly alter the NK cell phenotype in situ. The increase in the NK cell population in the TIME of TNBC-resistant cancers may represent expansion/infiltration of these nonfunctional cells, and follow-up studies on the functional characterization of the NK cell population within TNBC residual tumors are indicated.
Finally, assessment of the peripheral immune response has the potential to provide valuable longitudinal information in a minimally invasive manner in clinical practice. Other recent work has identified cytotoxic gene signatures detectable in peripheral blood that are associated with residual disease after NAC (37). We show here that chemotherapy-induced changes in the tumor microenvironment of TNBC residual disease, namely the depletion of B cells and enrichment of NK cells, are also reflected in the peripheral blood. Our work has further identified features of peripheral immunity at baseline that are related to clinical outcomes after NAC treatment. Importantly, at baseline, patients with TNBC destined for a poor response to chemotherapy (RCB II/III at surgery) were found to have lower levels of the cytotoxic CD16+ NK cell population, which continued to remain depleted during chemotherapy. Given the specific effects of NAC on NK cell phenotype, our observations support further investigations into the potential for effects of an additive NK cell–dependent immunotherapy for patients with chemotherapy-resistant TNBCs evidenced by the presence of RCB II/III residual disease.
Taken together, we present a detailed assessment of chemotherapy-resistant, immunologically cold TNBC and ER+HER2– cancers identifying a number of potential immunologic targets that require further exploration within this context, as these high-risk patients might benefit from immunomodulatory approaches that could be tested in future clinical trials. Additionally, monitoring of tumor immune dynamics longitudinally during chemotherapy is feasible and mirrors events within the tumor, and therefore may help to predict therapeutic response early, and potentially allow for tailoring of treatment prior to definitive surgery.
Supplementary Material
Acknowledgments
This research was supported by grants from Cancer Research UK to S. Irshad (C56773/A24869) and program grants from Breast Cancer Now to A. Tutt at King's College London and to the Breast Cancer Now Toby Robins Research Center at the Institute of Cancer Research, London. The work was also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London and/or the NIHR Clinical Research Facility. We thank members of the GSTT trial team who contributed to patient recruitment for the BTBC study at Guy's Hospital and clinical colleagues at GSTT for assisting with patient identification and sample collection. We thank the patients and the families of those who contributed to this research. We thank the team members at the BRC Flow Cytometry Core based at Guy's and St Thomas’ NHS Foundation Trust and King's College London for their support with the CyTOF panel design and data acquisition, with specific thanks to Dr. R. Ellis, R. Wester, C. Bishop, and Dr. K. Todd. We thank members of the Kordasti lab who provided training for team members, specifically Dr. J. Timms and P. Nocerino. We thank members of the biobank team, based at Guys Cancer Centre, for sample processing and management, specifically N. Woodman and J. Owen.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
R. Buus reports personal fees from NanoString Technologies outside the submitted work. T. Alaguthurai reports grants from CRUK during the conduct of the study. R. Graham reports grants from CRUK during the conduct of the study. J. Wesseling reports research funding for Cancer Grand Challenge from Cancer Research UK and KWF Dutch Cancer Society, focused on Ductal Carcinoma In Situ. M. Cheang reports personal fees from Advisory, grants from Eli Lilly, and grants from AstraZeneca outside the submitted work; in addition, M. Cheang has a patent for PAM50/Bioclassifier with royalties paid from Veracyte. R. Salgado reports other support from BMS, Exact Sciences, and Merck outside the submitted work. M. Dowsett reports personal fees from AstraZeneca, Lilly, Roche, Radius, H3 Biomedicine, and G1 outside the submitted work. A.N. Tutt reports personal fees from Pfizer, Vertex, MD Anderson, Merck KGAA, Artios, Prime Oncology, Medscape Education, EMPartners, Inbiomotion, Gilead, GBCC, Cancer Panel, Research to practice, SABCS, and Penn Medicine, other support from CRUK, AZ Symposium at ESMO, GE Healthcare, VJ Oncology, Gilead, AstraZeneca, and Innovation in Breast Cancer Symposium, and AACR, personal fees and other support from AstraZeneca, grants and other support from AstraZeneca, and grants from Myriad genetic, MERCK KGAA, Medivation, CRUK, and Breast Cancer Now outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
P. Gazinska: Formal analysis, investigation, methodology. C. Milton: Formal analysis, investigation, methodology, writing–review and editing. J. Iacovacci: Data curation, formal analysis, investigation, methodology, writing–review and editing. J. Ward: Investigation, methodology, writing–review and editing. R. Buus: Investigation, methodology, writing–review and editing. T. Alaguthurai: Investigation, methodology, writing–review and editing. R. Graham: Investigation, methodology, writing–review and editing. A. Akarca: Investigation, methodology, writing–review and editing. E. Lips: Resources, writing–review and editing. K. Naidoo: Investigation, methodology, writing–review and editing. J. Wesseling: Resources, supervision, methodology, writing–review and editing. T. Marafioti: Resources, supervision, methodology, writing–review and editing. M. Cheang: Resources, methodology, writing–review and editing. C. Gillett: Resources, writing–review and editing. Y. Wu: Investigation, writing–review and editing. A. Khan: Supervision, investigation, methodology, writing–review and editing. A. Melcher: Supervision, methodology, writing–review and editing. R. Salgado: Conceptualization, supervision, investigation, methodology, writing–review and editing. M. Dowsett: Conceptualization, resources, supervision, writing–review and editing. A. Tutt: Conceptualization, resources, supervision, investigation, methodology, writing–review and editing. I. Roxanis: Conceptualization, formal analysis, supervision, investigation, methodology, writing–review and editing. S. Haider: Conceptualization, data curation, formal analysis, supervision, investigation, visualization, writing–review and editing. S. Irshad: Conceptualization, supervision, funding acquisition, investigation, visualization, methodology, writing–original draft, project administration.
References
- 1. Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 2005;174:2591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 2005;201:1061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vdelta1(+) gammadelta T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med 2019;11:eaax9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Craven KE, Gokmen-Polar Y, Badve SS. CIBERSORT analysis of TCGA and METABRIC identifies subgroups with better outcomes in triple-negative breast cancer. Sci Rep 2021;11:4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565–70. [DOI] [PubMed] [Google Scholar]
- 6. Ghebeh H, Barhoush E, Tulbah A, Elkum N, Al-Tweigeri T, Dermime S. FOXP3+ Tregs and B7-H1+/PD-1+ T lymphocytes co-infiltrate the tumor tissues of high-risk breast cancer patients: Implication for immunotherapy. BMC Cancer 2008;8:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol 2014;32:2959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010;28:105–13. [DOI] [PubMed] [Google Scholar]
- 9. Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol 2013;31:860–7. [DOI] [PubMed] [Google Scholar]
- 10. Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest 2013;123:2873–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, et al. Tumor-infiltrating lymphocytes are prognostic in triple-negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol 2014;25:1544–50. [DOI] [PubMed] [Google Scholar]
- 12. Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 2018;19:40–50. [DOI] [PubMed] [Google Scholar]
- 13. Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc cooperates with ras by programming inflammation and immune suppression. Cell 2017;171:1301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quigley D, Silwal-Pandit L, Dannenfelser R, Langerod A, Vollan HK, Vaske C, et al. Lymphocyte invasion in IC10/basal-like breast tumors is associated with wild-type TP53. Mol Cancer Res 2015;13:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heppner B Ingold, Untch M, Denkert C, Pfitzner BM, Lederer B, Schmitt W, Eidtmann H, et al. Tumor-infiltrating lymphocytes: a predictive and prognostic biomarker in neoadjuvant-treated HER2-positive breast cancer. Clin Cancer Res 2016;22:5747–54. [DOI] [PubMed] [Google Scholar]
- 16. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol 2008;8:59–73. [DOI] [PubMed] [Google Scholar]
- 17. Dieci MV, Criscitiello C, Goubar A, Viale G, Conte P, Guarneri V, et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: a retrospective multicenter study. Ann Oncol 2015;26:1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dieci MV, Criscitiello C, Goubar A, Viale G, Conte P, Guarneri V, et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: a retrospective multicenter study. Ann Oncol 2014;25:611–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luen SJ, Salgado R, Dieci MV, Vingiani A, Curigliano G, Gould RE, et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple-negative breast cancer patients after neoadjuvant chemotherapy. Ann Oncol 2019;30:236–42. [DOI] [PubMed] [Google Scholar]
- 20. Garcia-Martinez E, Gil GL, Benito AC, Gonzalez-Billalabeitia E, Conesa MA, Garcia Garcia T, et al. Tumor-infiltrating immune cell profiles and their change after neoadjuvant chemotherapy predict response and prognosis of breast cancer. Breast Cancer Res 2014;16:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an international TILs working group 2014. Ann Oncol 2015;26:259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dieci MV, Radosevic-Robin N, Fineberg S, van den Eynden G, Ternes N, Penault-Llorca F, et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: a report of the international immuno-oncology biomarker working group on breast cancer. Semin Cancer Biol 2018;52(Pt 2):16–25. [DOI] [PubMed] [Google Scholar]
- 23. Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol 2007;25:4414–22. [DOI] [PubMed] [Google Scholar]
- 24. Ecker RC, Steiner GE. Microscopy-based multicolor tissue cytometry at the single-cell level. Cytometry A 2004;59:182–90. [DOI] [PubMed] [Google Scholar]
- 25. Toki MI, Merritt CR, Wong PF, Smithy JW, Kluger HM, Syrigos KN, et al. High-plex predictive marker discovery for melanoma immunotherapy-treated patients using digital spatial profiling. Clin Cancer Res 2019;25:5503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Waggott D, Chu K, Yin S, Wouters BG, Liu FF, Boutros PC. NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics 2012;28:1546–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. R: A language and environment for statistical computing. https://www.R-project.org/.
- 29. Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, et al. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinf 2009;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nowicka M, Krieg C, Crowell HL, Weber LM, Hartmann FJ, Guglietta S, et al. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Res 2017;6:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011;332:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruggner RV, Bodenmiller B, Dill DL, Tibshirani RJ, Nolan GP. Automated identification of stratifying signatures in cellular subpopulations. Proc Natl Acad Sci U S A 2014;111:E2770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 34. Szekely GJ, Rizzo ML. Hierarchical clustering via joint between-within distances: extending Ward's minimum variance method. J Classif 2005;22:151–83. [Google Scholar]
- 35. Loi S, Michiels S, Adams S, Loibl S, Budczies J, Denkert C, et al. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: clinical utility in an era of checkpoint inhibition. Ann Oncol 2021;32:1236–44. [DOI] [PubMed] [Google Scholar]
- 36. Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med 2020;382:810–21. [DOI] [PubMed] [Google Scholar]
- 37. Axelrod ML, Nixon MJ, Gonzalez-Ericsson PI, Bergman RE, Pilkinton MA, McDonnell WJ, et al. Changes in peripheral and local tumor immunity after neoadjuvant chemotherapy reshape clinical outcomes in patients with breast cancer. Clin Cancer Res 2020;26:5668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valpione S, Galvani E, Tweedy J, Mundra PA, Banyard A, Middlehurst P, et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer 2020;1:210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nabet BY, Esfahani MS, Moding EJ, Hamilton EG, Chabon JJ, Rizvi H, et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell 2020;183:363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020;579:274–8. [DOI] [PubMed] [Google Scholar]
- 41. Waidhauser J, Schuh A, Trepel M, Schmalter AK, Rank A. Chemotherapy markedly reduces B cells but not T cells and NK cells in patients with cancer. Cancer Immunol Immunother 2020;69:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Desbois M, Rusakiewicz S, Locher C, Zitvogel L, Chaput N. Natural killer cells in non-hematopoietic malignancies. Front Immunol 2012;3:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mamessier E, Pradel LC, Thibult ML, Drevet C, Zouine A, Jacquemier J, et al. Peripheral blood NK cells from breast cancer patients are tumor-induced composite subsets. J Immunol 2013;190:2424–36. [DOI] [PubMed] [Google Scholar]
- 45. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013;121:3599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee HJ, Lee JJ, Song IH, Park IA, Kang J, Yu JH, et al. Prognostic and predictive value of NanoString-based immune-related gene signatures in a neoadjuvant setting of triple-negative breast cancer: relationship to tumor-infiltrating lymphocytes. Breast Cancer Res Treat 2015;151:619–27. [DOI] [PubMed] [Google Scholar]
- 47. Geng SK, Fu SM, Ma SH, Fu YP, Zhang HW. Tumor-infiltrating neutrophil might play a major role in predicting the clinical outcome of breast cancer patients treated with neoadjuvant chemotherapy. BMC Cancer 2021;21:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov 2014;4:232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol 2019;16:430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2017;168:542. [DOI] [PubMed] [Google Scholar]
- 51. Danaher P, Warren S, Dennis L, D'Amico L, White A, Disis ML, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Criscitiello C, Bayar MA, Curigliano G, Symmans FW, Desmedt C, Bonnefoi H, et al. A gene signature to predict high tumor-infiltrating lymphocytes after neoadjuvant chemotherapy and outcome in patients with triple-negative breast cancer. Ann Oncol 2018;29:162–9. [DOI] [PubMed] [Google Scholar]
- 53. O'Meara T, Marczyk M, Qing T, Yaghoobi V, Blenman K, Cole K, et al. Immunological differences between immune-rich estrogen receptor-positive and immune-rich triple-negative breast cancers. JCO Precis Oncol 2020;4:PO.19.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009;16:183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Z, Yang C, Li L, Jin X, Zhang Z, Zheng H, et al. Tumor-derived HMGB1 induces CD62L(dim) neutrophil polarization and promotes lung metastasis in triple-negative breast cancer. Oncogenesis 2020;9:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guerriero JL, Ditsworth D, Catanzaro JM, Sabino G, Furie MB, Kew RR, et al. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol 2011;186:3517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mamessier E, Sylvain A, Bertucci F, Castellano R, Finetti P, Houvenaeghel G, et al. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res 2011;71:6621–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the authors upon reasonable request. Details of open access data are provided in DOI: 10.5281/zenodo.5206664.