

Abstract
Purpose:
PARP inhibitors (PARPi) induce synthetic lethality in homologous recombination repair (HRR)-deficient tumors and are used to treat breast, ovarian, pancreatic, and prostate cancers. Multiple PARPi resistance mechanisms exist, most resulting in restoration of HRR and protection of stalled replication forks. ATR inhibition was highlighted as a unique approach to reverse both aspects of resistance. Recently, however, a PARPi/WEE1 inhibitor (WEE1i) combination demonstrated enhanced antitumor activity associated with the induction of replication stress, suggesting another approach to tackling PARPi resistance.
Experimental Design:
We analyzed breast and ovarian patient-derived xenoimplant models resistant to PARPi to quantify WEE1i and ATR inhibitor (ATRi) responses as single agents and in combination with PARPi. Biomarker analysis was conducted at the genetic and protein level. Metabolite analysis by mass spectrometry and nucleoside rescue experiments ex vivo were also conducted in patient-derived models.
Results:
Although WEE1i response was linked to markers of replication stress, including STK11/RB1 and phospho-RPA, ATRi response associated with ATM mutation. When combined with olaparib, WEE1i could be differentiated from the ATRi/olaparib combination, providing distinct therapeutic strategies to overcome PARPi resistance by targeting the replication stress response. Mechanistically, WEE1i sensitivity was associated with shortage of the dNTP pool and a concomitant increase in replication stress.
Conclusions:
Targeting the replication stress response is a valid therapeutic option to overcome PARPi resistance including tumors without an underlying HRR deficiency. These preclinical insights are now being tested in several clinical trials where the PARPi is administered with either the WEE1i or the ATRi.
Translational Relevance.
Tumors with an underlying deficiency in DNA homologous recombination repair (HRR), such as those from germline BRCA1 or BRCA2 (gBRCA) mutation carriers, are sensitive to targeted therapies such as PARP inhibitors (PARPi). The ability to overcome PARPi resistance has become relevant for a subset of patients with breast and ovarian cancers. We argued that inhibitors of the replication stress response can overcome PARPi resistance. Using preclinical breast and ovarian cancer patient models, we identified that WEE1 inhibitor (WEE1i) response was linked to markers of replication stress and ATR inhibitor (ATRi) response was associated with ATM mutation. These preclinical insights inform several clinical trials where the PARPi is administered with either the WEE1i or the ATRi.
Introduction
Inhibitors of PARP represent the first cancer therapy to specifically target the DNA damage response (DDR; refs. 1, 2) and have the potential to be used as both monotherapy or in combination across multiple tumor types including ovarian, breast, pancreatic, and prostate cancers (3). A key tenet of DDR-based therapy is that cancers having lost one or more DDR pathway will have a greater dependency on the remaining pathways (4). The potential of PARP inhibitors (PARPi) was first highlighted in cells deficient in the tumor suppressor genes BRCA1 and BRCA2 (BRCA1/2; refs. 5, 6). PARPi sensitivity extends beyond the loss of function in BRCA1 and BRCA2 proteins to additional HRR proteins including RAD51, ATR, CHK1, PALB2, and other Fanconi anemia-associated repair factors (7–9). Clinical validation was provided using the PARPi olaparib in patients whose tumors carried BRCA1/2 mutations (10).
PARP detects and rapidly binds to DNA single-strand breaks (SSB), utilizing NAD+ to initiate repair by generating PAR chain modifications that result in chromatin remodeling and recruitment of repair factors (11). PARPis demonstrating clinical monotherapy activity prevent auto-PARylation and result in PARP trapping onto the DNA, which during replication, leads to the formation of DNA double-strand breaks (DSB; ref. 12). In the presence of functional HRR, these DNA DSBs are accurately repaired, but in cells deficient in HRR, such as those with loss-of-function BRCA1/2 mutations, the error-prone nonhomologous end joining (NHEJ) pathway is employed (13) and over multiple rounds of replication this can lead to unsupportable genomic instability and cancer cell death.
A number of different PARPi resistance mechanisms have been described in preclinical models (14–16), although many are yet to be validated in the clinic. In previous studies, we addressed which mechanisms of PARPi resistance were associated with a lack of olaparib response in a cohort of patient-derived xenograft (PDX) models from patients primarily with triple-negative breast cancer (TNBC) and enriched with BRCA1 alterations (17, 18). We observed that reactivation of HRR, determined by the presence of RAD51 foci, represents a major route to generate PARPi resistance. The aim of this study was to investigate whether additional dependencies, other than HRR, pre-exist or are generated by PARP inhibition and whether these could be targeted to reverse or overcome PARPi resistance.
One potential insight has emerged following recent publications, suggesting a link between PARP inhibition and replication stress (12, 19, 20). Replication stress can be defined as any one of a number of events that leads to the slowing or stalling of replication fork progression that, in turn, can lead to a decoupling of the replicative helicase from the polymerase and an increase in extended regions of single-stranded DNA (ssDNA). As a consequence, a replication stress response is induced (21). Trapped PARP1 on DNA, resulting from treatment with a pharmacologic inhibitor, has the potential to induce replication stress, leading to the stalling of replication forks (12). Following the binding of replication protein A (RPA) to ssDNA at stalled replication forks, a complex set of responses are initiated by the kinase Ataxia Telangiectasia and Rad3-related (ATR), and its effector Checkpoint Kinase 1 (CHK1; ref. 22). As a result, replication fork structures are stabilized, and cell-cycle progression is delayed allowing time for DNA repair, replication fork restart, and completion of DNA synthesis ahead of mitosis (23). Another important component of the RSR is the regulation of replication origin firing to prevent unscheduled DNA synthesis and rescue stalled replication forks, and ATR and CHK1 can facilitate this by negatively regulating cyclin-dependent kinase (CDK) activity through inhibition of the CDC25 family of CDK phosphatases (24). ATR can also activate an S/G2 cell-cycle checkpoint to prevent progression of cells with under-replicated DNA (23). WEE1 kinase, through phosphorylation of CDK1, is essential for regulating the G2–M checkpoint and preventing unrepaired damage being taken into mitosis, but it also plays an important role in regulating replication initiation through phosphorylation of CDK2 (25, 26). Consequently, the dependencies on ATR, CHK1, and WEE1, resulting from cancer-associated replication stress, make them potentially attractive therapeutic targets and a number are currently undergoing evaluation in the clinic (27).
It has previously been shown that PARPi-resistant BRCA1-deficient cells are increasingly dependent on ATR for survival, and that an ATR inhibitor (ATRi) can disrupt BRCA1-independent RAD51 loading to DSBs and stalled forks (28). Consequently, ATR inhibition was proposed as a unique strategy to overcome the PARPi resistance in BRCA1/2-deficient cancers. We have recently described single-agent activity of the WEE1 inhibitor (WEE1i) adavosertib (AZD1775, MK-1775) in a subpopulation of diffuse large B-cell lymphoma (DLBCL) associated with high levels of replication stress (29). In addition, the combination of adavosertib with the PARPi olaparib, enhanced antitumor activity in a patient-derived small cell lung cancer model in a manner that was associated with the induction of unsupportable levels of replication stress (30). We were therefore interested to understand whether the WEE1i in combination with olaparib could overcome PARPi resistance, and how this might compare with an ATRi /olaparib combination. Here, we have analyzed one of the largest cohorts of breast and ovarian cancer patient-derived explant models studied to date, for their WEE1i and ATRi responses as single agents, and in combination with the PARPi olaparib, the latter being of interest because this PDX cohort is enriched for tumors demonstrating both innate and acquired PARPi resistance.
Materials and Methods
Patient-derived tumor xenografts and in vivo experiments
Fresh tumor samples from patients with breast or ovarian cancer were prospectively collected for implantation into nude mice under an institutional IRB-approved protocol and the associated written informed consent. All animal procedures were approved by the Ethics Committee of Animal Research of the Vall d'Hebron Institute of Oncology and by the Catalan Government and were conformed to the principles of the WMA Declaration of Helsinki, the Department of Health and Human Services Belmont Report, and following the European Union's animal care directive (2010/63/EU). Surgical or biopsy specimens from primary tumors or metastatic lesions were immediately implanted in mice. Fragments of 30 to 60 mm3 were implanted into the mammary fat pad (surgery samples) or the lower flank (metastatic samples) of 6-week-old female athymic HsdCpb:NMRI-Foxn1nu mice (Harlan Laboratories). Animals were continuously supplemented with 1 μmol/L 17b-estradiol (Sigma-Aldrich) in drinking water. Upon growth of the engrafted tumors, the model was perpetuated by serial transplantation onto the lower flank.
To evaluate the sensitivity to the drugs, tumor-bearing mice were equally distributed into treatment groups with tumors ranging 100 to 300 mm3. As single agent, AZD1775 was administered orally 5 days out of 14 at 120 mg/kg in 0.5% methylcellulose. In some experiments, indicated with an *, AZD1775 was administered at 60 mg/kg 3 days out of 7. Olaparib was given at 50 mg/kg orally five times per week in 10% v/v DMSO, 10% w/v Kleptose [HP-β-CD]. AZD6738 was given at 25 mg/kg, five times per week in 10% v/v DMSO, 40% v/v PEG300. Cisplatin (6 mg/kg) was administered weekly unless RTV <0.5 or weight loss >20%. Tumor growth was measured with caliper bi-weekly from first day of treatment to end of study (typically day 21) and every 7 days in the acquired resistance setting. In all experiments, mouse weight was recorded twice weekly. The tumor volume was calculated as V = 4/3 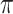 ·L·l2, “L” being the largest diameter and “l” the smallest. Mice were euthanized when tumors reached 1,500 mm3 or in case of severe weight loss, in accordance with institutional guidelines.
·L·l2, “L” being the largest diameter and “l” the smallest. Mice were euthanized when tumors reached 1,500 mm3 or in case of severe weight loss, in accordance with institutional guidelines.
The antitumor activity was determined by comparing tumor volume at 21 days to its baseline: % tumor volume change = (V21days− Vinitial)/Vinitial × 100. For sensitive PDX, the best response was defined as the minimum value of % tumor volume change sustained for at least 10 days. To classify the response of the subcutaneous implants, we modified the Response Evaluation Criteria in Solid Tumors (RECIST) criteria to be based on the % tumor volume change (31, 32): Complete Response (CR), best response <−95%; Partial Response (PR), best response <−30%; Stable Disease (SD), −30% < best response < +20%; Progressive Disease (PD), best response > +20%.
Patient-derived tumor cells and nucleoside rescue
Patient-derived tumor cells were isolated from PDXs through combination of mechanic disruption and enzymatic disaggregation following the protocol described by ref. 33. Single cells were seeded 2 × 105 cells/mL on collagen-enriched matrix Corning Matrigel growth factor reduced (GFR) basement membrane matrix (Corning) and overlaid with MEGM Mammary Epithelial Cell Growth Medium Bullet Kit (LONZA) supplemented with 2% of FBS and 10 μmol/L of ROCK inhibitor (Sigma-Aldrich). Next day, patient-derived tumor cells were treated with vehicle (DMSO), AZD1775 at 1 μmol/L plus vehicle, or AZD1775 1 μmol/L plus EmbryoMax Nucleosides 100× (Sigma-Aldrich) at 1:12.5, 1:25, or 1:50. After 72 hours, bright field pictures of ex vivo cultures were taken and cell viability was measured: Matrigel was dissolved adding PBS-EDTA 1 mmol/L and incubated at 4°C for 1 hour; spheroids were collected, rinsed once with PBS, and ATP content was measured using CellTiter-Glo (Promega) according to the manufacturer's instructions.
Metabolite analysis and tissue extraction
One hundred milligrams of tissue was homogenized and extracted twice with 1 mL ACN/MeOH/H2O 40/40/20 v/v/v using a reinforced lysis tube (CKMic50-R) on a temperature controlled Precellys 24 device following three circles of 20 seconds shake at 5,000 rpm with 30 seconds pause in between them. Clear supernatant (maximum 700 μL) was collected in a 2-mL cryovial after centrifugation (Eppendorf 5417R) at 10,600 × g for 5 minutes at 0°C. Precipitated tissue was extracted again following the same procedure described above. Both clear supernatant combined to the same cryovial and was stored at −20°C until further analysis.
Ten microliters of tissue extract was added to 90 μL HPLC grade water in a polypropylene vial and vortexed for 5 seconds to ensure homogeneity. Samples were then analyzed following U(H)PLC separation on an Ultimate RS system (Thermo Fisher Scientific) operating with Chromeleon 6.8 software. Chromatographic resolution of endogenous metabolite was obtained after a gradient solvent mixing mode of H2O spiked with 10 mmol/L tributylamine/15 mmol/L acetic acid (solvent A) and MeOH/Isopropanol 80/20 v/v (solvent B). The gradient program started at 0 to 0.5 with 100% A, 0.5 to 4 minutes from 0% to 95% A, 4 to 6 at 95% A, 6 to 6.5 changed linearly to 80% A, 6.5 to 8.5 kept at 80% A, 8.5 to 14 changed from 80% to 45% A at 14 minutes, 14 to 15 minutes steep increase to 100% B to flush the column for 2 minutes before changing to 100% A and re-equilibrate the HSS T3 2.1 mm × 100 mm 1.8 μm column for 3 minutes prior to next sample injection. Mass data were acquired on ABScienx 4000 triple quantrupole instrument (Analyst 1.6.3) following a scheduled multiple reaction monitoring experiment. Raw spectrometric data was imported to MultiQuant 2.1 software to extract peak areas for each detected metabolite. Further data normalization, scaling, univariate statistical analysis (ftest t test) and data validation was performed on Excel. For more details of the analytical platform, sample preparation and data analysis can be obtained at ref. 34.
Statistical analysis
Unless otherwise stated, statistical tests were performed with GraphPad Prism version 7.0. Paired, unpaired t test (two-tail), or two-way ANOVA with Tukey multiple comparison test were used and P values given. Error bars represent SEM of at least three biological replicates, unless otherwise stated.
Data availability
Readers can find the genomics data acquired and used in this study in Supplementary Tables S3 and S5. Further data can be provided upon request to the corresponding author. Additional methods are found as Supplementary Data.
Results
Screening of patient-derived tumor xenografts identifies a subset of tumors intrinsically sensitive to WEE1 inhibition
The WEE1i (adavosertib, AZD1775, MK-1775) was previously tested in a small number of cancer cell xenograft models using a daily dose of 120 mg/kg (35). We carried out an assessment of the WEE1i in a panel of 27 TNBC and two high-grade serous ovarian cancer (HGSOC) PDX models with detailed clinical information (Supplementary Table S1). Figure 1A shows the individual PDX models by best response in a waterfall plot and Fig. 1B indicates the mean tumor volume changes over time in a spider plot. We identified two TNBC models, PDX098 and PDX236, exhibiting a CR (Fig. 1A; Supplementary Table S2; ref. 33). Six other models showed a PR, namely the HGSOC PDX280, and five TNBCs (PDX094, PDX156, PDX060, STG139, and HBCx17). All other PDX models underwent disease stabilization (n = 6) or disease progression (n = 16). Although single-agent treatment with the WEE1i induced durable responses, tumors resumed growth upon continuous treatment in four of six models tested (Fig. 1B).
Figure 1.

Screening of patient-derived tumor xenografts identifies a subset of WEE1- inhibitor intrinsically sensitive tumors. A, Waterfall plot showing the best response to AZD1775, plotted as the percentage of tumor volume change compared with the tumor volume on day 1 after at least approximately 21 days of treatment using the 120 mg/kg schedule summarized in Supplementary Table S2 (n = 29). +20% and −30% are marked by dashed lines to indicate the range of PR, SD, and PD. Bars represent means of an average of six individual tumors and error bars represent SEM. B, Time response to AZD1775 for the PDX models shown in A. The percentage of tumor volume change during AZD1775 treatment is plotted. Shades of blue are used to label PDX models with PD, SD, PR, or CR response to the treatment.
Response biomarkers of WEE1 inhibition in TNBC
We next aimed to identify the genetic and proteomic markers associated with WEE1i sensitivity, to define candidate selection or enrichment biomarkers that could be used for patient selection. Analysis of exome sequencing indicated that STK11/LKB1 mutations, in the presence of RB1 and TP53 mutations, was the most significantly associated biomarker with response (P value = 0.004; Fig. 2A). Lack of LKB1 protein expression by IHC was also associated with WEE1i response (Supplementary Fig. S1A; Fig. 2B; P = 0.02) and this association was also significant with concomitant alterations in pRb being taken into account (P = 0.01).
Figure 2.
![Figure 2. Response biomarkers to AZD1775. A, Summary of selected DDR genetic alterations (see Materials and Methods, for the complete gene list) identified by exome sequencing in the PDX cohort from Fig. 1A. Sensitivity (CR+PR) or resistance (SD+PD) to AZD1775 is indicated, as well as the cancer subtype. The frequency of each mutation within the PDX cohort and the P value for the association of each alteration with AZD1775 response is shown. Different colors indicate the specific type of mutation. B, Forest plot and odds ratio analysis of the response to AZD1775 according to the IHC/genetic markers LKB1, pRb/RB1 and PTEN (n = 28). Null, no expression by IHC; mut, mutant. C, Sensitivity to AZD1775 of MCF10A p53−/− Rb−/− cells upon LKB1 knockdown using two independent siRNAs separately and pooled. Bars indicate the AUC relative to the control siRNA (siCON). Error bars indicate SD of three independent experiments. P values are shown. D, Metabolite data annotation to KEGG metabolic pathways for PDX098 and PDX060 (AZD1775-sensitive), compared with PDX102 (AZD1775-resistant). Nodes represent metabolic pathways and the depicted color indicates the number of significant changes following treatment with AZD1775 for 8 or 24 hours compared with vehicle [|log2(fold change)|>0.5, P-value <0.05, QC CV < 30%]. E, REVEALER analysis for AZD1775 antitumor response in the PDX cohort. The nonlinear information coefficient (IC) and conditional information coefficient (CIC) values are provided.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9561606_4536fig2.jpg)
Response biomarkers to AZD1775. A, Summary of selected DDR genetic alterations (see Materials and Methods, for the complete gene list) identified by exome sequencing in the PDX cohort from Fig. 1A. Sensitivity (CR+PR) or resistance (SD+PD) to AZD1775 is indicated, as well as the cancer subtype. The frequency of each mutation within the PDX cohort and the P value for the association of each alteration with AZD1775 response is shown. Different colors indicate the specific type of mutation. B, Forest plot and odds ratio analysis of the response to AZD1775 according to the IHC/genetic markers LKB1, pRb/RB1 and PTEN (n = 28). mut, mutant; null, no expression by IHC. C, Sensitivity to AZD1775 of MCF10A p53−/− Rb−/− cells upon LKB1 knockdown using two independent siRNAs separately and pooled. Bars indicate the AUC relative to the control siRNA (siCON). Error bars indicate SD of three independent experiments. P values are shown. D, Metabolite data annotation to KEGG metabolic pathways for PDX098 and PDX060 (AZD1775 sensitive), compared with PDX102 (AZD1775 resistant). Nodes represent metabolic pathways and the depicted color indicates the number of significant changes following treatment with AZD1775 for 8 or 24 hours compared with vehicle [|log2(fold change)|>0.5, P value < 0.05]. E, REVEALER analysis for AZD1775 antitumor response in the PDX cohort. The nonlinear information coefficient (IC) and conditional information coefficient (CIC) values are provided.
LKB1 encodes the serine/threonine kinase, which acts as a tumor suppressor by activating the tuberous sclerosis complex protein, TSC2, and by inactivating ribosomal S6-kinase (36). In addition, LKB1-deficient cells exhibit defects in nucleotide metabolism (37, 38). A recent analysis of non–small cell lung carcinoma cell lines and a Kras/Lkb1 mutant genetically engineered mouse model, suggests that LKB1 defects might also be associated with WEE1i sensitivity, possibly via loss of an ATM/LKB1 signaling cascade required to maintain cellular fitness in response to WEE1i exposure (39). We therefore tested whether loss of LKB1 function could cause WEE1i sensitivity on the background of TP53 and RB1 defects in a breast cancer–relevant model. We found that two distinct siRNAs designed to target LKB1 caused significant WEE1i sensitivity in TP53-defective, RB1-defective MCF10A cells (Fig. 2C; Supplementary Figs. S1B and S1C). In addition, we identified a TSC2 frameshift truncation (p.915fs VAF 0.95) and RPS6KA6 amplification (6.8 copies) in PDX156 and PDX094, respectively, two WEE1i-sensitive PDXs (PR) that did not present LKB1 alterations (Fig. 2A). In total, five of six WEE1i-sensitive PDXs had activating mutations in at least one member of the LKB1/mTORC1/metabolism pathway. We also noted in our analysis of the molecular features in the PDX panel, which genetic alterations in CCNE1, encoding Cyclin E1, exhibited a trend towards being associated with WEE1i sensitivity (Fig. 2A). Cyclin E1 dysregulation has previously been linked to replication stress (40) as well as to WEE1i sensitivity (30, 41). A trend was also observed towards mutation/expression of the phosphatase-encoding gene PTEN being associated with WEE1i resistance in the PDX panel (P values = 0.09 and 0.41, respectively; Supplementary Fig. S2A). Accordingly, phosphorylation at S473 or T308 of the PTEN target AKT was statistically higher in nonresponsive PDXs than in responsive PDXs when measured by RPPA (P = 0.005 and 0.002; Supplementary Fig. S2B). No evidence of association with mTORC1/p70S6K-downstream activation markers was observed (4EBP1 pS65, S6 pS235/236, S6 pS240/244; Supplementary Fig. S2C), suggesting that LKB1/mTORC1-mediated WEE1i sensitivity is not driven via protein translation but is more likely to be linked to nucleotide metabolism.
To further establish if WEE1 inhibition resulted in intracellular nucleotide depletion in vivo, we conducted a metabolite analysis by mass spectrometry (34), in the RB1/LKB1-mutant WEE1i-sensitive model PDX098 (Fig. 2D; Supplementary Fig. S3). Treatment with WEE1i induced a profound and durable change in the purine and pyrimidine pathways in PDX098, as quantified after annotation to KEGG metabolic pathways, that resulted in perturbation of the pentose phosphate pathway. In contrast, the CCNE1-amplified PDX060 (exhibiting a PR) showed a distinct pattern of metabolic perturbation upon treatment with WEE1i, with a short-lived change in the purine and pyrimidine pathways. The WEE1i-resistant PDX102 showed minimal metabolic perturbation.
We next explored and validated a subset of hypothesis-based markers at the protein level. In addition to LKB1, we included direct targets of WEE1 (WEE1 and CDK1; Supplementary Fig. S4A), markers of early entry into S-phase (p53, p16, pRb, and pRb S807/811; Supplementary Table S3; Supplementary Fig. S4B), as well as markers of replication stress [RPA32 pS4/S8 (pRPA), c-MYC, cyclin E1, ATM, CHK1 pS345, and BRCA1 nuclear foci; Supplementary Fig. S4C; Supplementary Table S3]. Besides LKB1/pRB, only the percentage of pRPA nuclear foci–positive cells was found to be statistically higher in responders, than in nonresponders (P = 0.01; Supplementary Fig. S4C).
As well as assessing the contributions of individual biomarkers to WEE1i response in our PDX panel, we also exploited recent advances in the ability to identify coherent groups of genomic alterations and/or functional biomarkers that together were associated with WEE1i sensitivity. To do this, we used the REVEALER analysis (repeated evaluation of variables conditional entropy and redundancy; ref. 42), to identify molecular features associated with antitumor responses in PDX-bearing mice treated with AZD1175 (Supplementary Table S3). We used this approach to analyze WEE1i PDX responses both with or without the use of LKB1 expression as a “seed” feature. Without using a seed feature, we found that an elevated percentage of pRPA-positive cells provided the strongest correlation with single-agent AZD1775 sensitivity (CIC −0.51; Fig. 2E). High Cyclin E1 or high c-MYC expression were not significantly associated with WEE1i sensitivity but it improved the CIC value to −0.61 and −0.55, respectively. Using null LKB1 expression as a seed feature resulted in a less correlated biomarker solution being identified (CIC −0.49), which also included elevated pRPA.
Taken together, these analyses suggest that WEE1i sensitivity may relate to a composite of biomarkers encompassing early entry into S-phase (e.g., mutation in TP53, along with RB1 mutation or CCND1 amplification) and increased replication stress (e.g., STK11 mutation and TSC2 loss, Cyclin E1 or MYC overexpression) that result in an elevated replication stress phenotype, visualized by pRPA-positive cells (Fig. 2E).
AZD1775 treatment downregulates RRM2 and induces an S-phase DNA damage response
In addition to regulating CDK1 activity at the G2–M checkpoint, leading to early entry into mitosis, WEE1 also phosphorylates CDK2, the activity of which is critical in regulating DNA replication (25, 43). To gain further insights into the mechanism of action of AZD1775, we assessed downstream pharmacodynamic effects initially in four TNBC cell lines, MDA-MB157, BT20, MDA-MB231, and MDA-MB436 with GI50 values ranging between 268 and 406 nmol/L (Supplementary Fig. S1D) and then in a subset of 12 of our PDX tumors. Exposure to AZD1775 resulted in a marked accumulation of cells with DNA content below 4N (Supplementary Fig. S5A), consistent with observations in other breast cancer and DLBCL cell lines (29, 44). We then analyzed two of the four cell lines (MDA-MB-231 and MDA-MB-436) and demonstrated that WEE1i exposure inhibited CDK2 phosphorylation on tyrosine 15 (pY15) in a dose-dependent manner, in addition to its effect on CDK1 pY15 (Fig. 3A).
Figure 3.

WEE1 inhibition results in activation of CDK2 and subsequent DNA damage and replication stress response in breast cancer cell lines. A, Immunoblot analysis showing CDK2 and CDK1 activation 24 hours after treatment with different doses of AZD1775 in MDA-MB-231 and MDA-MB436 cells. To assess CDK2 tyrosine phosphorylation (pY15) level, total CDK2 was first immunoprecipitated (IP) and the bound fraction were eluted and analyzed. IP, immunoprecipitation; WCL, whole cell lysate. B, Immunoblot analysis of TNBC cell lines treated with DMSO (U) or AZD1775 0.5 μmol/L during different time periods. Biomarkers of target engagement, DNA damage response (DDR) and replication stress response (RSR) or mitosis were analyzed. C, Immunoblot analysis of target engagement and DDR biomarker in MDA-MB-231 cells treated with DMSO (−) or 0.5 μmol/L AZD1775 for 6 hours in the presence (+) or absence (−) of diluted EmbryoMax nucleoside solution. D, Immunoblot analysis of MDA-MB-231 cells from S-phase culture synchronized by double thymidine block treated with DMSO (−) or AZD1775 0.5 μmol/L in the presence (+) or absence (−) of RO-3306 (CDK1 inhibitor, CDK1i) or CVT-313 (CDK2 inhibitor, CDK2i). Protein samples were collected at indicated time points and the indicated biomarkers were analyzed.
An important function of CDK2 is to regulate the expression of RRM2 either directly, or through controlling the degradation of E2F1 (45, 46). Increased CDK2 activity following ATR inhibition has previously been shown to increase origin firing and downregulate RRM2 expression, events that result in nucleotide depletion, replication stress, and ultimately replication catastrophe (45). RRM2 has also been implicated as the target of a synthetic lethal interaction between the loss of histone H3 lysine 36 tri-methylation mark (H3K36me3) and WEE1 inhibition (46). We therefore examined the impact of AZD1775 exposure on RRM2 expression as a downstream marker of CDK2 activity. In MDA-MB-157, BT-20, MDA-MB-231, and MDA-MB-436 cells, we observed a marked reduction in RRM2 protein levels after WEE1i exposure (Fig. 3B). In parallel, AZD1775 exposure also resulted in the induction of DDR markers by 8 hours, specifically, phosphorylation of H2AX on S139 (γH2AX), CHK1 on S345, and RPA32 on S4/S8. In all four TNBC cell lines, we observed that AZD1775 treatment led to an increase in early mitosis, as indicated by the phospho-Histone H3 (pHH3) marker, suggesting the WEE1i was driving cells prematurely into M-phase (Fig. 3B). When cells were supplemented with exogenous nucleosides during WEE1i treatment, the activation of this DDR was partially suppressed (Fig. 3C). In addition to canonical DDR markers of replication perturbation, we observed significant phosphorylation of DNAPK on S2056 following WEE1i exposure, with delayed kinetics compared with CHK1 phosphorylation (Fig. 3B). DNAPK activation following ATR inhibition has been associated with persistent replication fork stalling, replication fork collapse, and subsequent DNA DSB formation (45). To ascertain whether the induction of the DDR in WEE1i-treated cells is a direct consequence of increased activity of CDK activity, we co-treated cells with AZD1775 and CDK inhibitors targeting either CDK2 (CVT-313) or CDK1 (RO-3306). Inhibition of CDK2 or CDK1 in MDA-MB-231 cells synchronized at the G1–S boundary resulted in restoration of RRM2 expression and the suppression of the replication stress response induced by the WEE1i (Fig. 3D). Collectively, these data demonstrate that in the TNBC cell lines tested, AZD1775 induces a replication stress response via the induction of CDK1/2 activity during S-phase.
As with the in vitro experiments, AZD1775 treatment in vivo also induced a marked accumulation of cells in early M-phase, as indicated by an increase in pHH3 S10-positive staining by immunofluorescence (Supplementary Fig. S5B). However, this increase in pHH3 upon WEE1i treatment was similar in both sensitive and resistant PDXs, suggesting that this was not the major driver of the antitumor response. We next measured levels of CDK1/2 pY15 by Western blot (WB) analysis in a representative panel of 12 PDX models (Supplementary Fig. S6A). Baseline CDK1/2 pY15 levels were higher in PDXs that exhibited tumor growth regression following treatment with WEE1i, than in PDXs exhibiting tumor growth delay or no response (P = 0.0017). Moreover, the degree of inhibition of pCDK1/2 and RRM2 downregulation achieved with WEE1i was also greater in AZD1775-responding PDXs than in resistant PDXs (Supplementary Fig. S6B). These results are consistent with those observed in cell lines and suggest that treatment with a WEE1i in sensitive PDX models could result in a reduction in dNTPs.
We conducted nucleoside rescue experiments in a 3-dimensional (3D) ex vivo setting. AZD1775 treatment resulted in significant antiproliferative activity in short-term patient-derived cultures (PDC) established from two in vivo sensitive models (PDC098 and PDC236) when compared with two resistant ones (PDC124 and PDC044; Fig. 4A). Moreover, addition of nucleosides significantly rescued the 3D ex vivo antiproliferative effect of WEE1i-sensitive models PDC098 and PDC236 by 2-fold, compared with the resistant models PDC044 and PDC124. Addition of nucleosides also attenuated the replication stress response markers in the WEE1i sensitive PDC098 and PDC236 models (Fig. 4B).
Figure 4.

Shortage of dNTP induces sensitivity to AZD1775 in PDXs. A, Bright field images and quantification of the organoid area from PDCs treated for 72 hours with DMSO or 1 μmol/L AZD1775, in the presence (+) or absence (−) of nucleosides (Embryomax, 1:12.5 dilution). B, Immunoblot analysis of DDR and RSR biomarkers in two AZD1775-sensitive models, PDC098 and PDC236, treated with 1 μmol/L AZD1775 in the presence (+) or absence (−) of nucleosides at the indicated dilution for 6 hours. C, Representative immunofluorescence images of two AZD1775-sensitive PDX models (PDX098 and PDX236) and quantification of the percentage of cells exhibiting pan-nuclear γH2AX staining in seven AZD1775-sensitive and 19 AZD1775-resistant PDXs. D, Representative immunofluorescence images of two AZD1775-sensitive PDX models (PDX098 and PDX236) and quantification of the percentage of cells in S–G2-phase (geminin-positive) with pRPA nuclear foci in seven AZD1775-sensitive and 19 AZD1775-resistant PDXs. Each dot represents the mean of at least two independent tumors per PDX model. All pictures were taken at 600× magnification. E and F, show quantifications of pan-nuclear γH2AX staining and pRPA nuclear foci in four models with acquired resistance to AZD1775.
In agreement with the observed nucleotide shortage metabolic effects, WEE1i treatment induced markers of replication stress in vivo, namely pan-nuclear phosphorylation of H2AX (γH2AX) and pRPA nuclear foci in S–G2-phase cells (geminin-positive cells; Fig. 4C and D; Supplementary Fig. S7). Consistent with increased replication stress, most of the pan-nuclear γH2AX-positive cells were also positive for pRPA or pDNAPK S2056. Moreover, these markers were reduced upon tumor regrowth under treatment, that is, with acquired resistance, further arguing that the induction of replication stress drives the antitumor response (Fig. 4E and F). A higher percentage of double-positive cells for pan-nuclear γH2AX and pHH3 S10, indicative of increased DNA damage associated with chromatin condensation, were also found in WEE1-treated cells, albeit both in AZD1775-sensitive and -resistant models, like the pHH3 S10 staining (Supplementary Figs. S7A and S7B). Together, these data suggest that WEE1i-sensitive tumors exhibit features of high baseline replication stress that is further enhanced following WEE1i treatment.
Overcoming PDX PARPi resistance by targeting WEE1
We were interested in exploring whether WEE1 inhibition could overcome PARPi resistance in our cohort of PDX models, and more specifically in tumors harboring BRCA1-alterations that had restored HRR. Our PDX model cohort included 17 models harboring BRCA1/2 alterations of which 11 exhibited PARPi resistance (refs. 17 and 18; Supplementary Table S1). The PDXs’ response showed a high concordance with the patients’ response to a platinum/PARPi-based therapy, namely in 16 of 17 models (94%; Supplementary Table S1). Olaparib in combination with AZD1775 increased the response rate (% of CR+PR) up to 59% (17 of 29) compared with single agent AZD1775 (28%; 8 of 29) or olaparib (17%; 5 of 29; Figs. 1B and 5A). Interestingly, sensitivity to olaparib (CR+PR) was not overlapping with sensitivity to AZD1775, demonstrating that WEE1i sensitivity does not rely on the tumor's deficiency in HRR (Fig. 5B). Moreover, combination responses resulting in CR/PR were observed in those PDXs, where treatment with single-agent AZD1775 or olaparib resulted in tumor growth delay but not regression (exemplified by Fig. 5C; Supplementary Fig. S8A). In the context of two BRCA1-altered models, PDX124 and STG201, combination of AZD1775 with olaparib delayed the acquisition of PARPi resistance (Supplementary Fig. S8B). In a laboratory model of acquired resistance to olaparib PDX230OR2, the combination with AZD1775 induced tumor regression (Supplementary Fig. S8C). Compared with a platinum agent (a relevant standard of care chemotherapy), olaparib plus AZD1775 demonstrated similar levels of antitumor activity in 16 models (50%), greater activity in 9 models (28%), and a worse response in only 2 models (6%) out of 32 analyzed (Fig. 5D).
Figure 5.

WEE1 inhibition sensitizes PARPi-resistant tumors. A, Waterfall plots showing the best response to the indicated drugs as percentage of tumor volume change after at least 21 days of treatment, compared with the tumor volumes on day 1 (n = 29). +20% and −30% are marked by dashed lines to indicate the range of PR, SD, and PD. Bars represent means and error bars SEM. B, Radar plot comparatively showing the percentage of tumor volume change upon treatment with AZD1775, olaparib, and the combination as in Figs. 1A and 5A. C, Relative tumor volume during treatment with vehicle, AZD1775, olaparib, or combination in PDX252. D, Comparative analysis of the antitumor activity of AZD1775 plus olaparib versus cisplatin (n = 32). AZD1775 was administered at 120 mg/kg (5 days on / 9 days off) or at 60 mg/kg (5 days on / 2 days off, **). When both doses were tested in the same model, we observed that: *, the model exhibited a reduction of antitumor response when treated with the low dose of AZD1775, compared with the high dose, or #, the model exhibited the same antitumor response upon treatment with the low dose of AZD1775, compared with the high dose. E, REVEALER analysis for AZD1775 plus olaparib antitumor response in the PDX cohort. The nonlinear information coefficient (IC) and conditional information coefficient (CIC) values are provided. F, Quantification of cells with pan-nuclear γH2AX staining (top) and cells in S–G2-phase of the cell cycle (geminin positive) with pRPA nuclear foci (bottom) following treatment with vehicle, olaparib, AZD1775, or the combination (Olap+1775) in PDXs showing (n = 5) or not (n = 10) antitumor response upon combination (Combo) treatment. Each dot represents the mean of at least two independent tumors per PDX model. P values, Tukey multiple comparison test.
No single-biomarker correlated with response to the olaparib combination with AZD1775. However, REVEALER identified the features that associated with sensitivity to the combination of olaparib and AZD1775 that differed to those for single-agent WEE1i sensitivity. Specifically, in the absence of a seed feature, we found that the combination of high c-MYC expression, high Cyclin E1, and low nuclear p16 correlated with sensitivity to the AZD1775/olaparib combination together with LKB1 mutation for AZD1775, or lack of RAD51 nuclear foci for olaparib response (Supplementary Fig. S8D). Seeding on Cyclin E1 and p16 provided the best correlation with AZD1775/olaparib sensitivity together with c-MYC (CIC −0.68; Fig. 5E). In agreement with the observed WEE1i single-agent phenotypic and response biomarkers, we observed that combination-sensitive tumors also exhibited higher induction of replication stress than combination-resistant ones, in terms of pan-nuclear γH2AX and pRPA in geminin-positive cells (Fig. 5F; P < 0.0001).
Comparison of ATR with WEE1 inhibition highlights differences in biomarker association
Given the role of ATR in the replication stress response, as well as the dependency of PARPi-resistant BRCA1-deficient cell lines on ATR (28), we envisaged that targeting ATR in combination with olaparib might also overcome PARPi resistance in our PDX panel. Initial investigation into the single-agent activity of the ATRi AZD6738 demonstrated tumor regression/stabilization in three models (PDX094, PDX098, and PDX196; Fig. 6A; Supplementary S8E; Supplementary Table S4). Interestingly, ATM missense mutations (identified by exome sequencing) associated with AZD6738-induced stabilization or regression (P = 0.0046; Fig. 6B). A synthetic lethal interaction between ATM and ATRi treatment in cell line models of various cancer types has been described (47–49). Here, we provide further validation for ATRi sensitivity in ATM-deficient backgrounds by testing AZD6738 in A549 and FaDu cells, where ATM has been deleted using CRISPR/Cas9 knockout (Supplementary Fig. S8F). When AZD6738 was combined with olaparib (Fig. 6A; Supplementary S8G), we also observed a more select number of responsive models than the olaparib combination with AZD1775. Moreover, with the exception of PDX127, all AZD6738/olaparib-sensitive models were also sensitive to the AZD1775 combination with olaparib (Fig. 6C). Consistent with the single-agent biomarker observation, three out of five PDXs that showed tumor regression/stabilization to the AZD6738/olaparib combination exhibited ATM alterations, of which two were predicted to be deleterious by in silico analysis (P = 0.17). FaDu ATM knockout cells also showed response to olaparib plus AZD6738 in vivo (Supplementary Fig. S8G). REVEALER further identified ATRi biomarkers of single-agent response (ATM mutations or lack of RAD51 foci) to be enriched in tumors responding to the ATRi/olaparib combination (Fig. 6D; Supplementary Table S5). Mechanistically, the combination of PARP plus ATR inhibition resulted in significant RRM2 downmodulation and induction of the replication stress markers pRPA and γH2AX, similar to WEE1i monotherapy (Fig. 6E; Supplementary S8I). In summary, the antitumor activity of the ATRi/olaparib combination appears more selective than that of the WEE1i, and is primarily associated with ATM alterations.
Figure 6.

ATR inhibition sensitizes PARPi-resistant tumors. A, Waterfall plots showing the best response to the indicated drugs as percentage of tumor volume change after at least 21 days of treatment, compared with the tumor volumes on day 1 (n = 31). +20% and −30% are marked by dashed lines to indicate the range of PR, SD, and PD. Bars represent means and error bars SEM. B, Summary of selected DDR genetic alterations identified by exome sequencing in the PDX cohort from A. Sensitivity (tumor regression) or resistance (tumor progression) to AZD6738 is indicated, as well as the cancer subtype. The frequency of each mutation within the PDX cohort and the P value for the association of each alteration with AZD6738 response is shown. *, To perform this statistical analysis and given the low number of CR/PR AZD6738 responders, the two models that exhibited SD with AZD6738 were also considered responders. Different colors indicate the specific type of mutation. C, Comparative analysis of the antitumor activity of olaparib plus AZD1775 versus olaparib plus AZD6738 (n = 31). AZD1775 was administered at 120 mg/kg (5 days on / 9 days off) or at 60 mg/kg (5 days on / 2 days off, **). When both doses were tested in the same model, we observed that: *, the model exhibited a reduction of antitumor response when treated with the low dose of AZD1775, compared with the high dose, or #, the model exhibited the same antitumor response upon treatment with the low dose of AZD1775, compared with the high dose. D, REVEALER analysis for AZD6738 plus olaparib antitumor response in the PDX cohort. The nonlinear information coefficient (IC) and conditional information coefficient (CIC) values are provided. E, Expression levels of the indicated proteins by immunoblot. Each dot represents individual tumors. Bars represent means and error bars SEM. P values, Tukey multiple comparison test.
Discussion
Three WEE1 inhibitors have recently entered into clinical development: Debio 0123, ZN-c3, and adavosertib (AZD1775) (NCT03968653, NCT04158336; ref. 27). Prior to this study, only a small number of trials had investigated the monotherapy opportunity that adavosertib might provide (refs. 50, 51; NCT01748825, NCT00648648, NCT02482311) and these were phase I trials. The three main goals of the study presented here were to identify biomarkers of sensitivity to single-agent WEE1i, understand if a WEE1i in combination with a PARPi could overcome resistance to PARP inhibition and if so, to compare PARPi/WEE1i with the PARPi/ATRi combination.
Cell line–based studies have previously identified alterations in either p53 (52), PKMYT1 (35), SETD2, KDM4A (46), LKB1 (39), MYC, or CDKN2A deletion, that is, deletion of INK4a and ARF (29), as all being associated with WEE1i sensitivity. In addition, in vitro RNA interference screens suggested defects in HRR or Fanconi anemia-associated genes could also induce WEE1i sensitivity (53), but in vivo, no single biomarker alone adequately explains drug response. More recently, a study of four patient-derived in vivo explant models of small cell lung cancer, identified a sensitive patient-derived explant model (CDX3) with multiple cell cycle and DDR alterations (Cyclin E1, pRb, CDK6, and PALB2), which also demonstrated high basal levels of replication stress (indicated by high levels of pRPA; ref. 30). One of the challenges in determining the main biological drivers of WEE1 dependency using panels of in vitro cancer cell lines is the relatively small dynamic range of response to AZD1775. The largest study to date assessed over 500 cell lines from 16 different tumor types, where the mean single-agent EC50 range was only 4-fold (0.28–1.16 μmol/L; ref. 35). In contrast, in vivo studies have demonstrated a full spectrum of response.
The only single-gene alteration in this study showing a statistically relevant correlation with WEE1i treatment was STK11/LKB1 and in all cases, the STK11 mutation was found in tumors with TP53 and RB1 mutations, with or without BRCA1 mutation. STK11/LKB1 is a tumor suppressor gene and the third most common alteration found in lung adenocarcinomas, where it is most frequently co-mutated along with KRAS (54). A previous study linking LKB1 to WEE1i sensitivity in non–small cell lung cancer (NSCLC), was on a background of KRAS mutation (39). In our study, the breast and ovarian responsive tumors did not have accompanying KRAS mutations (these being relatively rare in these cancer types), but instead were associated with an accompanying RB1 mutation, suggesting the link between WEE1i sensitivity and LKB1 deficiency might be broader than KRAS-mutant NSCLC alone. Although LKB1 deficiency is linked to pleotropic biological effects, we provide evidence suggesting that WEE1i sensitivity is associated with the reduction of the dNTP pool and a concomitant increase replication stress. Previous studies have linked WEE1 inhibition with the downregulation of RRM2, a key enzyme in dNTP synthesis (45, 46). Although we observed this effect in both WEE1i-responsive and -nonresponsive TNBC in vitro models, RRM2 reduction was exclusive to WEE1i in vivo PDX responses. Moreover, metabolomic analysis showed that a WEE1i-responsive model with LKB1 alteration exhibited decreased purine and pyrimidine levels upon treatment and furthermore, WEE1i sensitivity could be abrogated in PDCs treated with nucleosides.
The majority of PDX models in this cohort exhibiting tumor regression could be linked to STK11/LKB1 mutations or additional members of the LKB1/mTORC1/metabolism pathway. To understand drivers of response across all sensitive models, a bioinformatic analysis comparing 27 different biomarkers (both genomic and protein-based) across the PDX cohort revealed the top basal biomarker predicting WEE1i response was pRPA, an observation consistent with elevated replication stress.
Bringing together the findings from this study, along with our previous studies (29, 30) and those of others (35, 39, 41, 52), a pattern emerges for WEE1i single-agent sensitivity being driven by a number of different alterations, each of which is associated with replication stress or the replication stress response (Supplementary Fig. S10A). The first group contains alterations abrogating the G1–S cell-cycle checkpoint and driving cells into and through S-phase with a suboptimal dNTP pool. In the majority of WEE1i-sensitive models, there were usually two or more “hits” in this category. The second group consists of alterations that increase replication stress, either through replication fork stalling as a consequence of oncogenic drivers such as mutant KRAS or MYC overexpression, and/or replication fork stalling resulting from an imbalance in the ratio of available dNTP production to active replication origins. The third category contains DDR proteins associated with HRR, but in the context of WEE1i sensitivity, it is their roles in replication fork stabilization and restart that are most likely to be relevant. Tumor indications with an enrichment of alterations captured by these three groups represent an opportunity for testing adavosertib monotherapy in the clinic. In this respect, the recent clinical study to assess adavosertib monotherapy in uterine serous cancer has provided very promising results in a tumor indication with multiple biomarkers being present in >50% of the tumors treated, as highlighted in Supplementary Fig. S10A (55).
Inducers of replication stress and the DDR proteins involved in the replication stress response are shown in Supplementary Fig. S10B, which highlights the two main mechanisms behind replication fork stalling, namely the shortage of dNTPs and the physical blocking of replication fork progression. The latter can occur as a consequence of transcription–replication collision, due to oncogenic drivers such as mutant KRAS and MYC overexpression. However, other physical blocks to replication fork progression can be generated by proteins bound to DNA. As an example, topoisomerase I inhibitors covalently cross-link TOP1 to the DNA and trapped PARP has the potential to induce replication fork stalling and a replication stress response. In this sense, the combination of the PARPi olaparib with the WEE1i more than doubled the response rate in our PDX cohort. The combination effect did not appear to be driven by an underlying HRR deficiency, because there was no overlap of olaparib and AZD1775 monotherapy activity and the combination was effective in a substantial proportion of olaparib-resistant, RAD51 foci–positive tumors. Rather, the driver of combination response correlated with biomarkers of replication stress, both genetic (MYC, Cyclin E1) as well as phenotypic (pRPA and pan-γH2AX). These results were also consistent with high basal replication stress influencing the therapeutic index of WEE1i plus olaparib in vitro (56). We believe these observations therefore support the notion that PARPi resistance can be overcome by targeting the replication stress response roles of WEE1.
We have also conducted a direct comparison of a PARPi combination with WEE1i versus ATRi. Either WEE1i or ATRi combinations with the PARPi olaparib were able to induce tumor regression in >50% of the gBRCA PARPi-resistant PDXs (8 of 15). Importantly, combination activity was also observed in PDXs without a gBRCA mutation. These preclinical insights are now being tested clinically in various arms of a number of trials where the PARPi olaparib is being given concurrently with either an ATRi (AZD6738) including OLAPCO (NCT02576444) and CAPRI (NCT03462342; refs. 57–59), or with a WEE1i (AZD1775; ref. 60). The RP2D and schedules for the two olaparib combinations indicate an exposure equal to or greater than achievable in preclinical models for the ATRi in combination with olaparib (61). Emerging results from the EFFORT trial (NCT03579316) have demonstrated encouraging results for both the monotherapy adavosertib arm as well as the olaparib combination arm in the post-PARPi ovarian cancer setting (60). Recently published preclinical data have also demonstrated that alternating sequencing of the WEE1i combination with olaparib maintains efficacy in tumor cells while decreasing the impact on normal cells (56) and this new scheduling approach is now being tested in the clinic in the STAR trial (NCT04197713).
Given the increasing importance of PARPi in the treatment of multiple tumor types, and their use in earlier lines of therapy, PARPi resistance will become relevant to a new patient population. The ability to address PARPi resistance through combinations of WEE1i and ATRi that target the replication stress response, will therefore also become increasingly important for patients.
Supplementary Material
Acknowledgments
We would like to thank Caroline Hirst, James Yates, Jennifer Moss, Chris Vellano, Cristina Bernadó, Martine Roudier, and Caroline Hirst for their technical support; Maurizio Scaltriti (MSKCC), Dejan Juric (MGH), Massimo Broggini, and Elisa Caiola (Mario Negri IRCCS) for providing study materials; and Emma Dean and Susan Galbraith for feedback on the manuscript. This work was supported by the Spanish Instituto de Salud Carlos III (ISCIII), an initiative of the Spanish Ministry of Economy and Innovation partially supported by European Regional Development FEDER Funds (FIS PI17/01080 to V. Serra, PI12/02606 to J. Balmaña); European Research Area-NET, Transcan-2 (AC15/00063), Asociación Española contra el Cáncer (AECC; LABAE16020PORTT), the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR; 2017 SGR 540), La Marató TV3 (654/C/2019), and ERAPERMED2019–215 to V. Serra. We also acknowledge the GHD-Pink program, the FERO Foundation, and the Orozco Family for supporting this study (to V. Serra). V. Serra was supported by the Miguel Servet Program (ISCIII; CPII19/00033); M. Castroviejo-Bermejo and C. Cruz (AIOC15152806CRUZ) by AECC; A. Herencia-Ropero by Generalitat de Catalunya-PERIS (SLT017/20/000081); M. Palafox by Juan de la Cierva (FJCI-2015–25412); A. Lau by AECC and Generalitat de Catalunya-PERIS (INVES20095LLOP, SLT002/16/00477); A. Gris-Oliver by FI-AGAUR (2015 FI_B 01075). This work was supported by Breast Cancer Research Foundation (BCRF-19–08), Instituto de Salud Carlos III Project Reference number AC15/00062, and the EC under the framework of the ERA-NET TRANSCAN-2 initiative co-financed by FEDER, Instituto de Salud Carlos III (CB16/12/00449 and PI19/01181), and Asociación Española Contra el Cáncer (to J. Arribas). The xenograft program in the Caldas laboratory was supported by Cancer Research UK and also received funding from an EU H2020 Network of Excellence (EuroCAN). The RPPA facility is funded by NCI #CA16672.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
V. Serra reports grants from Spanish Instituto de Salud Carlos III (ISCIII), European Research Area-NET, Transcan-2, Asociación Española contra el Cáncer (AECC), Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR), La Marató TV3, and FERO Foundation, as well as grants and personal fees from AstraZeneca during the conduct of the study. V. Serra also reports grants from Tesaro, Genentech, and Novartis, as well as personal fees from AbbVie outside the submitted work; in addition, V. Serra has a patent for PCT/EP2018/086759 pending. V. Serra also acknowledges the GHD-Pink program and the Orozco Family for supporting this study. A.T. Wang reports other support from AstraZeneca during the conduct of the study, and is an employee/shareholder of AstraZeneca. M. Castroviejo-Bermejo reports a patent for PCT/EP2018/086759 pending. U.M. Polanska reports ownership of AstraZeneca stock. G.N. Jones reports personal fees and other support from AstraZeneca during the conduct of the study. A. Llop-Guevara reports grants from Asociación Española Contra el Cáncer (AECC) and AGAUR-PERIS during the conduct of the study; in addition, A. Llop-Guevara has a patent for PCT/EP2018/086759 pending. K.C. Bulusu reports employment with AstraZeneca and ownership of AstraZeneca stock. J. Nikkilä reports personal fees from AstraZeneca during the conduct of the study, as well as personal fees from AstraZeneca outside the submitted work. A. Hughes reports ownership of AstraZeneca stock. A. Gris-Oliver reports grants from Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) during the conduct of the study. J. Arribas reports grants from Breast Cancer Research Foundation, Instituto de Salud Carlos III, ERA-NET TRANSCAN-2, Asociación Española Contra el Cáncer, Roche, Synthon, and Molecular Partners, as well as grants and personal fees from Menarini outside the submitted work; in addition, J. Arribas has a patent for EP22382294.1 issued, a patent for EP20382457.8 licensed to Mnemo Therapeutics, and a patent for EP2360187B1 licensed to Innovent. J. Cortés reports personal fees from Roche, Celgene, Cellestia, AstraZeneca, Seattle Genetics, Daiichi Sankyo, Erytech, Athenex, Polyphor, Lilly, Merck Sharp & Dohme, GSK, Leuko, Bioasis, Clovis Oncology, Boehringer Ingelheim, Ellipses, Hibercell, BioInvent, Gemoab, Gilead, Menarini, Zymeworks, Reveal Genomics, Roche, Novartis, Eisai, Pfizer, Daiichi, Sankyo, AstraZeneca, and Gilead, as well as grants from Roche, Ariad Pharmaceuticals, AstraZeneca, Baxalta GMBH/Servier Affaires, Bayer Healthcare, Eisai, F. Hoffman-La Roche, Guardant Health, Merck Sharp & Dohme, Pfizer, Piqur Therapeutics, Puma C, and Queen Mary University of London outside the submitted work; in addition, J. Cortés has a patent for Pharmaceutical combinations of a Pi3k inhibitor and a microtubule destabilizing agent 2014/199294 A issued and a patent for Her2 as a predictor of response to dual HER2 blockade in the absence of cytotoxic therapy 2019/0338368 A1 licensed. C. Saura reports personal fees and other support from AstraZeneca, AX'Consulting, Byondis B.V., Daiichi Sankyo, Eisai, Exact Sciences, Exeter Pharma, F. Hoffmann - La Roche Ltd., MediTech, Merck Sharp & Dohme, Novartis, Pfizer, Philips, Pierre Fabre, PintPharma, Puma, Roche Farma, SeaGen, Zymeworks, and Gilead outside the submitted work. A. Lau reports other support from AstraZeneca during the conduct of the study, as well as other support from AstraZeneca outside the submitted work; in addition, A. Lau reports employment with AstraZeneca and ownership of AstraZeneca stock. S. Critchlow reports employment with AstraZeneca and ownership of AstraZeneca stock. B. Dougherty reports personal fees, nonfinancial support, and other support from AstraZeneca during the conduct of the study. C. Caldas reports grants from Cancer Research UK, EU H2020 Network of Excellence, and AstraZeneca during the conduct of the study, and is a member of AstraZeneca's iMED External Science Panel and Illumina's Scientific Advisory Board; in addition, C. Caldas is a recipient of research grants (administered by the University of Cambridge) from Genentech, Roche, AstraZeneca, Servier, and Varsity Therapeutics. G.B. Mills reports grants, personal fees, nonfinancial support, and other support from Amphista, AstraZeneca, Chrysallis Biotechnology, GSK, ImmunoMET, Ionis, Lilly, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda, Turbine, Zentalis Pharmaceuticals, Catena, HRD assay to Myriad Genetics DSP to Nanostring, Adelson Medical Research Foundation, Breast Cancer Research Foundation, Komen Research Foundation, Ovarian Cancer Research Foundation, Prospect Creek Foundation, Nanostring Center of Excellence, Ionis (provision of tool compounds), and Genentech during the conduct of the study. J.C. Barrett reports other support from AstraZeneca during the conduct of the study. J.V. Forment reports other support from AstraZeneca during the conduct of the study, as well as other support from AstraZeneca outside the submitted work. C.J. Lord reports personal fees from Syncona, Sun Pharma, Gerson Lehrman Group, Merck KGaA, Vertex, AstraZeneca, Tango Therapeutics, Third Rock, Ono Pharma, Artios, Abingworth, Tesselate, Dark Blue, and Neophore, as well as grants from AstraZeneca, Merck KGaA, and Artios outside the submitted work; in addition, C.J. Lord reports ownership of Tango, Ovibio, Enedra Tx., Hysplex, and Tesselate stock. C.J. Lord also has patents describing the use of DNA repair inhibitors and stands to gain from their development and use as part of the ICR “Rewards to Inventors” scheme. C. Cruz reports grants from Asociación Española Contra el Cáncer (AECC) during the conduct of the study; in addition, C. Cruz has a patent for WO2019122411A1 issued. J. Balmaña reports grants from Instituto de Salud Carlos III and AstraZeneca during the conduct of the study, as well as personal fees from AstraZeneca and Pfizer outside the submitted work; in addition, J. Balmaña has a patent for PCT/EP2018/086759 issued. M.J. O'Connor reports employment with AstraZeneca and ownership of AstraZeneca stock. No disclosures were reported by the other authors.
Authors' Contributions
V. Serra: Conceptualization, resources, formal analysis, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing. A.T. Wang: Formal analysis, methodology, writing–review and editing. M. Castroviejo-Bermejo: Formal analysis, methodology. U.M. Polanska: Formal analysis, methodology, writing–review and editing. M. Palafox: Formal analysis, methodology. A. Herencia-Ropero: Formal analysis, writing–review and editing. G.N. Jones: Formal analysis, methodology. Z. Lai: Formal analysis, methodology. J. Armenia: Formal analysis, methodology. F. Michopoulos: Formal analysis, methodology. A. Llop-Guevara: Formal analysis, supervision, writing–review and editing. R. Brough: Formal analysis, methodology. A. Gulati: Formal analysis, methodology. S.J. Pettitt: Supervision, writing–review and editing. K.C. Bulusu: Formal analysis. J. Nikkilä: Methodology. Z. Wilson: Methodology. A. Hughes: Formal analysis. P.W.G. Wijnhoven: Methodology. A. Ahmed: Methodology. A. Bruna: Methodology. A. Gris-Oliver: Methodology. M. Guzman: Methodology. O. Rodríguez: Methodology. J. Grueso: Formal analysis. J. Arribas: Resources. J. Cortés: Resources. C. Saura: Resources. A. Lau: Resources, supervision, writing–review and editing. S. Critchlow: Resources, supervision, writing–review and editing. B. Dougherty: Resources. C. Caldas: Resources, writing–review and editing. G.B. Mills: Resources, writing–review and editing. J.C. Barrett: Resources. J.V. Forment: Supervision. E. Cadogan: Supervision. C.J. Lord: Resources, supervision, writing–review and editing. C. Cruz: Supervision. J. Balmaña: Conceptualization, resources, supervision, funding acquisition, project administration, writing–review and editing. M.J. O'Connor: Conceptualization, resources, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci 2014;105:370–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell 2015;60:547–60. [DOI] [PubMed] [Google Scholar]
- 3. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 2019;16:81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009;461:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farmer H, Mccabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21. [DOI] [PubMed] [Google Scholar]
- 6. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7. [DOI] [PubMed] [Google Scholar]
- 7. Mccabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006;66:8109–15. [DOI] [PubMed] [Google Scholar]
- 8. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer 2016;16:110–20. [DOI] [PubMed] [Google Scholar]
- 9. Niraj J, Färkkilä A, D'andrea AD. The Fanconi anemia pathway in cancer. Annu Rev Cancer Biol 2019;3:457–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361:123–34. [DOI] [PubMed] [Google Scholar]
- 11. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012;13:411–24. [DOI] [PubMed] [Google Scholar]
- 12. Pommier Y, O'Connor MJ, De Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps17. [DOI] [PubMed] [Google Scholar]
- 13. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A 2011;108:3406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. D'Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 2018;71:172–6. [DOI] [PubMed] [Google Scholar]
- 15. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov 2015;5:1137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med 2013;19:1381–8. [DOI] [PubMed] [Google Scholar]
- 17. Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol 2018;29:1203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutiérrez-Enríquez S, Ducy M, Ibrahim YH, et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med 2018;10:e9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colicchia V, Petroni M, Guarguaglini G, Sardina F, Sahún-Roncero M, Carbonari M, et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017;36:4682–91. [DOI] [PubMed] [Google Scholar]
- 20. Parsels LA, Karnak D, Parsels JD, Zhang Q, Vélez-Padilla J, Reichert ZR, et al. PARP1 trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res 2018;16:222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014;16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 2017;18:622–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018;361:806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res 2012;40:477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parker L, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992;257:1955–7. [DOI] [PubMed] [Google Scholar]
- 26. Beck H, Nähse-Kumpf V, Larsen MSY, O'hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 2012;32:4226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forment JV, O'Connor MJ. Targeting the replication stress response in cancer. Pharmacol Ther 2018;188:155–67. [DOI] [PubMed] [Google Scholar]
- 28. Yazinski SA, Comaills V, Buisson R, Genois M-M, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 2017;31:318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Young LA, O'connor LO, De Renty C, Veldman-Jones MH, Dorval T, Wilson Z, et al. Differential activity of ATR and WEE1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res 2019;79:3762–75. [DOI] [PubMed] [Google Scholar]
- 30. Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW, et al. The combination of the PARP inhibitor olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res 2018;24:5153–64. [DOI] [PubMed] [Google Scholar]
- 31. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318–25. [DOI] [PubMed] [Google Scholar]
- 32. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 2000;92:205–16. [DOI] [PubMed] [Google Scholar]
- 33. Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 2016;167:260–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Michopoulos F, Whalley N, Theodoridis G, Wilson ID, Dunkley TPJ, Critchlow SE. Targeted profiling of polar intracellular metabolites using ion-pair-high performance liquid chromatography and -ultra high performance liquid chromatography coupled to tandem mass spectrometry: applications to serum, urine and tissue extracts. J Chromatogr A 2014;1349:60–8. [DOI] [PubMed] [Google Scholar]
- 35. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther 2013;12:1442–52. [DOI] [PubMed] [Google Scholar]
- 36. Corradetti MN, Inoki K, Bardeesy N, Depinho RA, Guan K-L. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 2004;18:1533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 2010;468:659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y, Marks K, Cowley GS, Carretero J, Liu Q, Nieland TJF, et al. Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov 2013;3:870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Richer AL, Cala JM, O'brien K, Carson VM, Inge LJ, Whitsett TG. WEE1 kinase inhibitor AZD1775 has preclinical efficacy in LKB1-deficient non-small cell lung cancer. Cancer Res 2017;77:4663–72. [DOI] [PubMed] [Google Scholar]
- 40. Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol 2015;10:425–48. [DOI] [PubMed] [Google Scholar]
- 41. Chen X, Low K-H, Alexander A, Jiang Y, Karakas C, Hess KR, et al. Cyclin E overexpression sensitizes triple-negative breast cancer to wee1 kinase inhibition. Clin Cancer Res 2018;24:6594–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim JW, Botvinnik OB, Abudayyeh O, Birger C, Rosenbluh J, Shrestha Y, et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat Biotechnol 2016;34:539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hughes BT, Sidorova J, Swanger J, Monnat RJ, Clurman BE. Essential role for Cdk2 inhibitory phosphorylation during replication stress revealed by a human Cdk2 knockin mutation. Proc Natl Acad Sci U S A 2013;110:8954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012;2:524–39. [DOI] [PubMed] [Google Scholar]
- 45. Buisson R, Boisvert JL, Benes CH, Zou L. Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol Cell 2015;59:1011–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, et al. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell 2015;28:557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reaper PM, Griffiths MR, Long JM, Charrier J-D, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011;7:428–30. [DOI] [PubMed] [Google Scholar]
- 48. Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun 2016;7:13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Min A, Im S-A, Jang H, Kim S, Lee M, Kim DK, et al. AZD6738, a novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol Cancer Ther 2017;16:566–77. [DOI] [PubMed] [Google Scholar]
- 50. Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 2015;33:3409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leijen S, van Geel RMJM, Pavlick AC, Tibes R, Rosen L, Razak ARA, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol 2016;34:4371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 2009;8:2992–3000. [DOI] [PubMed] [Google Scholar]
- 53. Aarts M, Bajrami I, Herrera-Abreu MT, Elliott R, Brough R, Ashworth A, et al. Functional genetic screen identifies increased sensitivity to WEE1 inhibition in cells with defects in fanconi anemia and HR pathways. Mol Cancer Ther 2015;14:865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu JF, Xiong N, Campos SM, Wright AA, Krasner C, Schumer S, et al. Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. J Clin Oncol 2021;39:1531–9. [DOI] [PubMed] [Google Scholar]
- 56. Fang Y, Mcgrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell 2019;35:851–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eder JP, Sohal D, Mahdi H, Do K, Keedy V, Hafez N, et al. Olaparib and the ATR inhibitor AZD6738 in relapsed, refractory cancer patients with homologous recombination (HR) repair mutations – OLAPCO. Mol Cancer Ther 2019;18:12s (Suppl. abstr. A080). [Google Scholar]
- 58. Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 2020;11:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shah PD, Zarrin H, Wethington S, Latif N, Martin L, Rodriguez D, et al. Abstract A72: combination ATR and PARP inhibitor (CAPRI) for recurrent, platinum-resistant ovarian cancer. Clin Cancer Res 2020;26:13s (Suppl. abstr. A72). [Google Scholar]
- 60. Westin SN, Coleman RL, Fellman BM, Yuan Y, Sood AK, Soliman PT, et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: a randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J Clin Oncol 2021;39:5505. [Google Scholar]
- 61. Berges A, Cheung SYA, Pierce AJ, Dean E, Felicetti B, Standifer N, et al. PK-biomarker-safety modelling aids choice of recommended phase II dose and schedule for AZD6738 (ATR inhibitor). Cancer Res 2018;78:13s (abstr. CT118). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Readers can find the genomics data acquired and used in this study in Supplementary Tables S3 and S5. Further data can be provided upon request to the corresponding author. Additional methods are found as Supplementary Data.