

Abstract
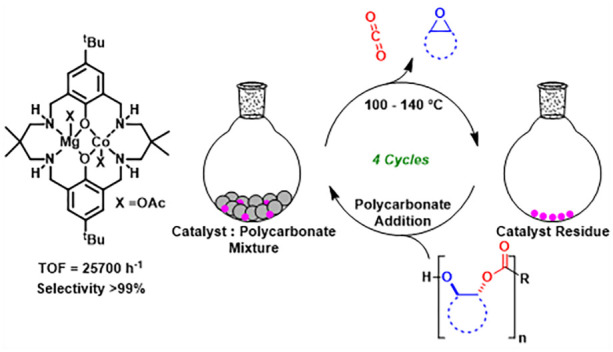
Polymer chemical recycling to monomers (CRM) could help improve polymer sustainability, but its implementation requires much better understanding of depolymerization catalysis, ensuring high rates and selectivity. Here, a heterodinuclear [Mg(II)Co(II)] catalyst is applied for CRM of aliphatic polycarbonates, including poly(cyclohexene carbonate) (PCHC), to epoxides and carbon dioxide using solid-state conditions, in contrast with many other CRM strategies that rely on high dilution. The depolymerizations are performed in the solid state giving very high activity and selectivity (PCHC, TOF = 25700 h–1, CHO selectivity >99 %, 0.02 mol %, 140 °C). Reactions may also be performed in air without impacting on the rate or selectivity of epoxide formation. The depolymerization can be performed on a 2 g scale to isolate the epoxides in up to 95 % yield with >99 % selectivity. In addition, the catalyst can be re-used four times without compromising its productivity or selectivity.
Introduction
Polymer chemical recycling to monomer (CRM) could improve sustainability by reducing wastes and limiting embedded emissions in virgin monomer production. The method forms pure monomers and allows for repolymerization to polymers that are equivalent to the original materials, thereby overcoming the deteriorating property profiles associated with other recycling technologies.1,2 All polymers have a ceiling temperature above which depolymerization to monomer is thermodynamically feasible, but there are depolymerization activation energies to overcome, and, in practice, CRM remains a major challenge. CRM catalysis needs better understanding of how to reduce depolymerization barriers, deliver high monomer selectivity, and minimize energy inputs. Depolymerizations of aliphatic polyesters/carbonates to cyclic esters/carbonates are front-runner recycling to monomer reactions.3−8 High monomer conversions and selectivity are achieved by careful manipulation of the process conditions, in particular, exploiting high dilution, and using special monomer structures, most commonly by applying low ring-strain heterocycles.9−14 The approach is to bias depolymerization equilibria: the trade-off is less favorable forward polymerization thermodynamics. Such polymers may also have low overall thermal stability, which may complicate their processing and use. An alternative strategy lies in the aliphatic polycarbonate depolymerization to epoxides and carbon dioxide, which is expected to be strongly entropically favored and driven by gaseous carbon dioxide removal.15,16 A critical factor in these depolymerizations is to limit 5-membered cyclic carbonate formation since those compounds are thermodynamically more stable than the polymers and cannot be easily repolymerized.
In 2013, Darensbourg and co-workers pioneered poly(cyclopentene carbonate) (PCPC) depolymerization to cyclopentene oxide, using a Cr(III)-salen/nBu4NN3 catalyst system, achieving 92 % selectivity.17,18 Subsequently, others reported a few more polycarbonate depolymerizations using poly(N-heterocyclic epoxide carbonates)19,20 or poly(limonene carbonate) (PLC).21,22 The CRM of poly(cyclohexene carbonate) (PCHC), the most widely studied carbon dioxide-derived polycarbonate, remained, until very recently, a challenge (Figure 1).23−25
Figure 1.

Catalysts for depolymerization of poly(cyclohexene carbonate) (PCHC) to cyclohexene oxide (CHO) and CO2.
Cyclohexene oxide/CO2 ROCOP is the benchmark for catalyst comparisons and PCHC shows a high glass transition temperature (∼120 °C) and tensile modulus (3600 MPa).28 It has a broad processing temperature range (>100 °C), and thermal degradation begins at temperatures of >240 °C.29−31 PCHC-containing block polymers show promise as toughened plastics, elastomers, and adhesives.32 Thus, achieving its chemical recycling to monomer is important; earlier this year, we reported a Mg(II)Mg(II) catalyst, applied in para-xylene solution, which delivered very high CHO selectivity (98 %) and a turn-over-frequency (TOF) of 150 h–1 (0.33 mol % catalyst, 120 °C, [PCHC]0 = 1 M) (Figure 1).26 As this manuscript was submitted, Lu and co-workers reported PCHC pyrolysis using a Cr(III)(salen)Cl/PPNN3 catalyst system, which showed really excellent rates, albeit at high temperature (Figure 1, 0.2 mol % catalyst, TOF = 3000 h–1, 200 °C).27 A decomposed di-Zn(II) catalyst was also reported for PCHC depolymerization.33 Our focus is on improving catalytic activity, selectivity, and applicability in PCHC depolymerization. Solid-state depolymerizations were targeted since obviating organic solvents could be beneficial both for environmental impact and process scale-up. To increase rates, the principle of polymerization microscopic reversibility directs toward testing of a faster forward polymerization catalyst. In 2020, our team reported a synergic heterodinuclear Mg(II)Co(II) catalyst for CHO/CO2 ROCOP, showing significantly greater activity than the Mg(II)Mg(II) species, and thus this heterodinuclear catalyst was a candidate for investigation.34
Results and Discussion
Dihydroxy-telechelic PCHC was prepared by ROCOP of CHO and CO2 (Mn,SEC = 8000 g mol–1, ĐM = 1.07, Mn,NMR = 5400 g mol–1). Solid-state PCHC depolymerizations were achieved by mixing the polymer and catalyst with the reaction being monitored using thermogravimetric analysis (TGA). Samples were heated, under nitrogen or air flow, with either isothermal or variable temperature conditions, and PCHC mass loss was monitored over time (see SI for experimental details). First, solid samples of Mg(II)Co(II):PCHC, 1:300, were mixed using a mortar and pestle. These specimens were successfully depolymerized at 140 °C, under a nitrogen flow, resulting in an 80% PCHC mass loss in only 20 min (Table 1, entry 1). The catalyst achieved a turn-over-frequency (TOF) of 900 h–1. The mass loss data fit an exponential decay and have a pseudo first order rate coefficient for depolymerization, kobs = 7.52 h–1, indicative of a first order dependence on polymer mass (Figure 2a). Repeat experiments showed excellent reproducibility with near perfect overlay of data (Figure 2, Figure S2). Depolymerization rates were equivalently high using either nitrogen or air flow, the latter tolerance being notable as many polymerization catalysts are air-sensitive. (Figure S3). It is emphasized that these reactions are depolymerizations not merely polymer decompositions: the PCHC sample used has a significantly higher on-set for thermal decomposition (Td,5%) of 255 °C. Also, the catalyst is thermally stable, with a Td10% of 365 °C. Control samples of PCHC, without any catalyst, showed no degradation over 24 h under equivalent conditions using N2 or air (Figure 2a). To analyze the depolymerization product(s) formed under both N2 and air, a cold-trap condensed the liquid product, which by NMR spectroscopy was pure cyclohexene oxide in both cases (90% yield, Figures S4–S6). The residual mass, after depolymerization, of <3 wt % corresponds to the catalyst loading. CO2 was also detected through TGA-MS experiments, indicating that both epoxide and CO2 are extruded in the depolymerization (Figure S7).
Table 1. Data for PCHC Depolymerizations using a Mg(II)Co(II) Catalyst in the Solid State with some Solution-State Literature Benchmark Catalystsa.
| entry | catalyst | temperature (°C) | [PCHC]0:[cat]0 | time (s)b | TOF (h–1)c | mass loss rate (kg g–1 h–1)d | kobs (h–1)e |
|---|---|---|---|---|---|---|---|
| 1f | Mg(II)Co(II) | 140 | 300:1 | 1327 | 900(±50) | 0.17 (±0.09) | 7.52(±0.01) |
| 2 | Mg(II)Co(II) | 140 | 300:1 | 109 | 6000(±300) | 1.12(±0.06) | 49.9(±0.1) |
| 3 | Mg(II)Mg(II) | 140 | 300:1 | 557 | 1200(±60) | 0.22(±0.01) | 9.72(±0.01) |
| 4 | Mg(II)Co(II) | 140 | 1000:1 | 155 | 13900(±700) | 2.64(±0.1) | 38.7(±0.1) |
| 5 | Mg(II)Co(II) | 140 | 2500:1 | 241 | 22400(±1100) | 4.23(±0.2) | 22.6(±0.02) |
| 6 | Mg(II)Co(II) | 140 | 5000:1 | 420 | 25700(±1300) | 4.86(±0.2) | 13.8(±0.02) |
| 7 | Mg(II)Co(II) | 140 | 10,000:1 | 1735 | 12400(±600) | 2.35(±0.1) | 2.62(±0.02) |
| 8 | Mg(II)Co(II) | 100 | 300:1 | 2653 | 200(±10) | 0.05(±0.003) | 2.29(±0.002) |
| 9 | Mg(II)Co(II) | 110 | 300:1 | 1060 | 600(±30) | 0.11(±0.006) | 4.76(±0.001) |
| 10 | Mg(II)Co(II) | 120 | 300:1 | 370 | 1800(±90) | 0.33(±0.02) | 14.7(±0.02) |
| 11 | Mg(II)Co(II) | 130 | 300:1 | 201 | 3200(±160) | 0.61(±0.03) | 24.8(±0.1) |
| 1226 | Mg(II)Mg(II) | 120 | 300:1 | 1200 | 150 | 0.020g | 0.407 |
| 1327 | Cr(III)/PPNN3 | 200 | 1000:1 | 1200 | 3000 | 0.35h | - |
| 1427 | Cr(III)/PPNN3 | 140 | 500:1 | 36,000 | 2.5 | 0.00059h | - |
See SI for details of experimental setup, all TGA experiments run to >99% mass loss, CHO selectivity > 99%.
Interval of time from 20 to 80% mass loss of the polymer.
TOF = moles of PCHC consumed (20–80% conversion)/moles of catalyst/time (see SI). Average error taken from repeat runs (<5%).
Mass loss rate = mass PCHC consumed (20–80% conversion)/catalyst mass/time.
kobs = gradient of linear fitting of the logarithm of %polymer mass vs time (Figures S8–18).
Polymer:catalyst mixed by pestle and mortar.
Values calculated from ref (26).
Values calculated from ref (27).
Figure 2.

(a) Solid-state PCHC depolymerizations using Mg(II)Co(II) (1:300), at 140 °C, showing mass loss over time. Black = PCHC, red = Mg(II)Co(II):PCHC, 1:300, mixed with a mortar and pestle; blue = Mg(II)Co(II):PCHC, 1:300, film (solvent cast), CHO selectivity > 99%. Depolymerization rate coefficients, kobs, are obtained by exponential fits to the data. (b) Plot showing depolymerization rate coefficients, kobs, vs nitrogen flow rate (mL min–1) at 140 °C for polymer films (i.e., Mg(II)Co(II):PCHC, 1:300, film).
Sample mixing was investigated with catalyst/polymer mixtures dissolved in minimum solvent (THF or toluene) and the solvent removed in-vacuo to form a polymer film (see SI). These samples, which have the same catalyst loading (Mg(II)Co(II):PCHC, 1:300), showed much faster rates, achieving 80% PCHC depolymerization in just 3.5 mins corresponding to a TOF of 6000 h–1. The data also fit exponential depolymerization decays, and the rate coefficient, kobs = 49.9 h–1, is ∼7 times greater than samples mixed with a mortar and pestle (Table 1, Entry 2, Figure 2). Furthermore, the film depolymerizations occurred with improved initiation times as compared with both the blended solid-state and solution-phase reactions (<1 min vs >10 mins).26 It is proposed that the faster rates arise from better catalyst dispersion in the polymer and the high film surface area. The nitrogen flow rate also influenced the catalysis with the best rates arising from moderate flows (Figure 2b and Table S2). At nitrogen flows from 25 to 100 mL min–1, the activity was consistently high and data showed exponential decays, with rate constants (kobs) of ∼50 ± 1 h–1. At higher flow rates, 100–500 mL min–1, rates slowed perhaps due to CO2 and/or CHO release inhibition. This is attributed to the high flow rates causing overpressure of the reaction furnace and preventing release of the co-monomers from the TGA crucible.
The Mg(II)Co(II) shows 5× greater activity than the Mg(II)Mg(II) catalyst, when tested under the solid-state conditions. The Mg(II)Mg(II) catalyst achieved a TOF = 1200 h–1, while the Mg(II)Co(II) catalyst had a TOF of 6000 h–1 (Table 1, entry 3). This finding suggests that faster polymerization catalysts are faster depolymerization catalysts. It also indicates that depolymerization catalysis can be accelerated by exploiting heterodinuclear synergy.34,35 To understand the limits of catalyst tolerance, the loading of Mg(II)Co(II) was decreased from 1:300 to 1:10000. At a 1:5000 catalyst:PCHC loading, complete depolymerization was achieved corresponding to a TOF of 25700 h–1 (kobs = 13.8 h–1, Figure S20). Such an activity is remarkable since it would correspond to a mass loss rate of 5 kg PCHC per gram catalyst per hour. At the lowest loadings, depolymerization still occurred although with slightly lower activity, (1:10000, TOF = 12400 h–1), which is attributed to some catalyst decomposition. Compared with the next best other depolymerization catalyst (Cr(salen)/PPNCl) under as close to equivalent conditions as possible,27 the Mg(II)Co(II) catalyst shows >5000× greater activity at one tenth the catalyst loading (Table 1, entry 14).
Successful depolymerization catalysis was achieved at temperatures above 100 °C, yielding CHO as the sole product in all cases (Figure 3a). As expected, the catalytic activity increased with temperature from TOF = 200 h–1 (kobs 0.05 h–1) at 100 °C to TOF = 6000 h–1 (kobs 49.9 h–1) at 140 °C. Depolymerizations were also feasible at higher temperatures but could not be monitored as the kinetics were too fast. The depolymerization activation barrier, attributed to the catalyst-alkoxide backbiting reaction to form cyclohexene oxide, was determined using an Arrhenius analysis. Plots of the logarithm of the observed rate constant (kobs) against inverse temperature (1/T) are linear (Figure 3b). The PCHC depolymerization barrier is 100.2 ± 5.8 kJ mol–1, which is higher than the PCHC polymerization activation energy (80.7 kJ mol–1) using the same Mg(II)Co(II) catalyst. It is expected that forward polymerization, i.e., CHO + CO2, would have a high enthalpic driving force due to release of an epoxide ring strain. It is remarkable that the depolymerization to epoxide has a sufficiently low barrier to be achievable, and the reaction can be favored by removal of the gaseous carbon dioxide and epoxide products.
Figure 3.

(a) Solid state PCHC depolymerizations using Mg(II)Co(II) catalyst (1:300) at temperatures from 100 to 140 °C. (b) Arrhenius plot for PCHC depolymerizations (ln(kobs) vs 1/T) using data collected from 100 to 140 °C.
Other CO2-derived polycarbonates were also successfully chemically recycled to epoxides using the Mg(II)Co(II) catalyst. These included poly(cyclopentene carbonate) (PCPC), poly(4-vinylcylcohexene carbonate) (PVCHC), and poly(limonene carbonate) (PLC) (Table 2, entries 1–4, Figure S21). All samples were subjected to the standard depolymerization conditions (catalyst:polycarbonate, 1:300, 140 °C), and in most cases, >80% mass loss occurred in ∼4 min (kobs = 49.9–66.6 h–1). PLC took significantly longer to depolymerize, with an 80 % mass loss requiring 30 min (kobs = 6.9 h–1), likely due to the higher boiling point of limonene oxide. All epoxides were isolated after the depolymerization and purity confirmed using 1H and 13C NMR spectroscopy (Figures S25–S30).
Table 2. Depolymerization Data for PCHC, PVCHC (poly(vinyl cyclohexene carbonate)), PCPC (poly(cyclopentene carbonate)), and PLC (poly(limonene carbonate)) using Mg(II)Co(II)a.

| entry | polymer | time (s)b | TOF (h–1)c | mass loss rate(kg g–1 h–1)d | kobs (h–1)e | selectivity(%)f |
|---|---|---|---|---|---|---|
| 1 | PCHC | 109 | 6000 (±300) | 1.12(±0.06) | 49.9(±0.1) | >99 |
| 2 | PVCHC | 88 | 7300 (±70) | 1.63(±0.01) | 57.4(±0.05) | >99 |
| 3 | PCPC | 68 | 9500 (±800) | 1.61(±0.1) | 66.6(±0.2) | >99 |
| 4 | PLC | 635 | 1000 (±40) | 0.27(±0.01) | 6.86(±0.01) | >99 |
Polymer films ([Mg(II)Co(II)]:[polycarbonate]00 = 1:300) were depolymerized at 140 °C using a N2 flow of 25 mL min–1 (see SI for further details).
Interval of time from 20 to 80% mass loss of the polymer.
TOF = PCHC conversion (20–80%)/moles of catalyst/time (see SI). Error taken from repeat reactions.
Mass loss rate = mass polycarbonate consumed (20–80% conversion)/catalyst mass/time.
kobs = gradient of linear fits to plots of ln(polycarbonate conversion) vs time (Figures S6 and S22–S24)
Determined by 1H NMR spectroscopy.
To probe the solid-state depolymerization mechanism, samples of cyclic carbonate (trans-CHC) were subjected to similar conditions. Thus, films comprising Mg(II)Co(II) and trans-CHC (1:300) were analyzed using TGA (Figure S31). Under these reaction conditions, trans-CHC also degrades to form CHO and CO2, but its rate is an order of magnitude slower than for PCHC (TOF = 800 h–1). In the absence of Mg(II)Co(II), a mass loss of trans-CHC is also observed, although at a slower rate than when a catalyst is present. Furthermore, in depolymerizations conducted with Mg(II)Co(II) and PCHC (1:300), trans-CHC was not detected by 1H NMR spectroscopy (Figure S32) or MS (Figure S7) in the crucible or in the evolved gases throughout the reaction. These observations, coupled with the significant rate differences between degradation of PCHC and trans-CHC, suggest that trans-CHC is not an intermediate in PCHC depolymerization. To understand whether polyethers are relevant intermediates, poly(cyclohexene oxide) (PCHO) was subjected to the same depolymerization conditions (Figure S33). There was no mass loss over 60 min at 140 °C and no depolymerization. This result indicates that the depolymerization mechanism requires a good leaving group in the ring-closing to epoxide step. The poor leaving ability of an alkoxide in comparison to a carbonate may rationalize the failure to depolymerize PCHO. Next, to probe whether depolymerization occurred via a chain-scission or chain-end backbiting mechanism, the depolymerization of an acetyl-end capped PCHC (PCHC-OAc, 1:300, solvent cast) was attempted. At 140 °C, no mass loss was observed using PCHC-OAc for up to 2 h (Figure S34). This contrasts with the hydroxyl end-capped PCHC, which under analogous conditions degrades completely in <5 min. Lastly, in a depolymerization conducted at 140 °C with Mg(II)Co(II):PCHC loadings of 1:30, acetic acid was detected in the condensate by 1H NMR spectroscopy (Figure S35). The depolymerization mechanism is thus proposed to occur through a PCHC chain backbiting reaction. Accordingly, the PCHC hydroxyl end-groups react with the catalyst to form an alkoxide intermediate, thereby liberating an equivalent of acetic acid. The alkoxide intermediate may then back-bite upon its own chain. This reaction forms an equivalent of epoxide (CHO) and a catalyst-carbonate intermediate. The catalyst-carbonate intermediate rapidly decarboxylates, with carbon dioxide extrusion favored by the moderate gas flow, to (re)form a chain-shortened catalyst-alkoxide intermediate (Figure 4). A related mechanism was proposed for the Mg(II)Mg(II) catalyst.26
Figure 4.
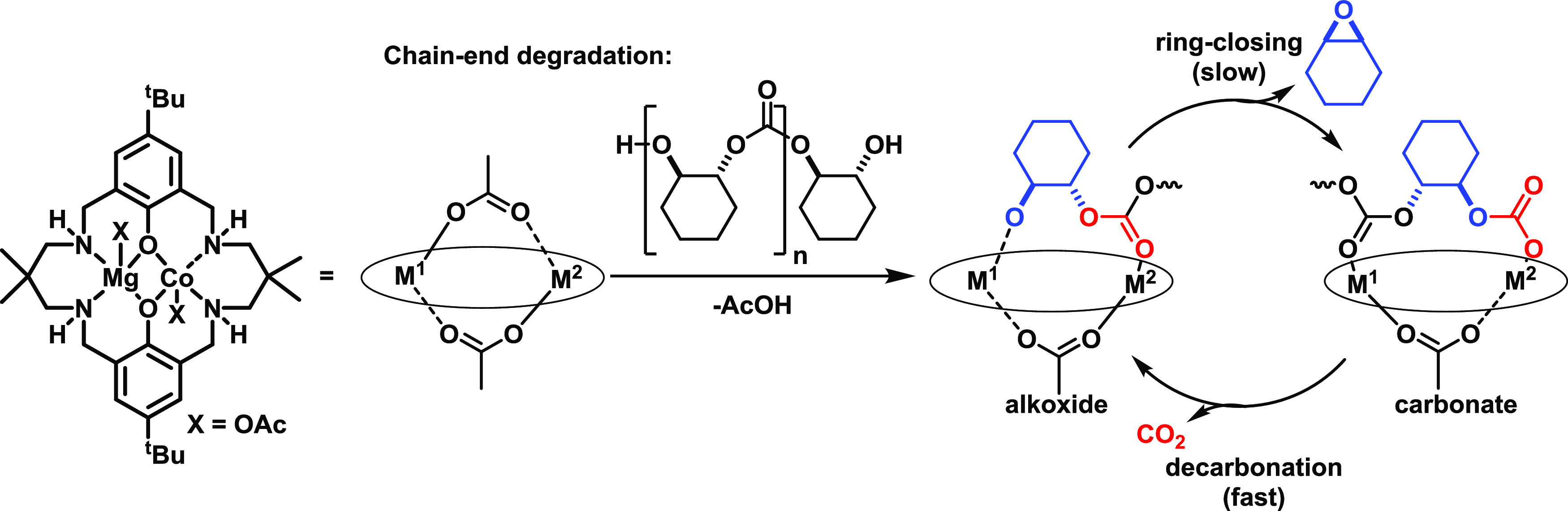
Proposed mechanism for the depolymerization of PCHC catalyzed by Mg(II)Co(II).
Finally, the potential to scale-up the solid-state depolymerization was investigated by mixing the catalyst and polymer and heating them under static vacuum with product condensation (Figure S36). The reagents were mixed, using a pestle and mortar and Mg(II)Co(II):PCHC 1:300, heated to 140 °C under a static vacuum (∼10–2 mbar) with a collection flask. Using this process, 2 g of PCHC was depolymerized and 1.27 g of CHO was isolated, corresponding to a 92 % isolated yield with the epoxide purity being determined by NMR spectroscopy and boiling point determination (Table S3, entry 1). After the reaction, a fresh quantity of PCHC was added to the residual catalyst and the “second batch” was also successfully depolymerized to CHO, with equivalent selectivity. Further catalyst recycles, up to four in total, were achieved, and in all cases, the catalyst productivity and selectivity for CHO were maintained (Figure S37). MS (MALDI-TOF) and IR spectroscopic analysis of the catalyst residue showed that the complex remained intact post depolymerization (Figures S38 and S39, respectively). These results are encouraging since they demonstrate the feasibility both to scale the reaction and to reuse the catalyst, without requiring any intermediary isolation/purification steps. The larger-scale solid-state depolymerizations were conducted using 2 g of each of the polycarbonates, i.e., PVCHC, PCPC, and PLC, affording the pure epoxides as colorless liquids in up to 95 % isolated yield (Table S3). All these recycled epoxides were used in ROCOP reactions with carbon dioxide to re-form polycarbonates of equivalent molar mass to equivalent samples made using “virgin” epoxide, demonstrating the chemical recycling to monomer concept (Table S4 and Figures S40–43).
Conclusions
In conclusion, a highly active and selective catalyst, applied in solid-state depolymerizations, for polycarbonate chemical recycling to epoxide and carbon dioxide was reported. The catalytic activity is 26000 h–1 or 5 kg polycarbonate/g catalyst/h, and it is tolerant to low loadings, operating efficiently at 0.01 mol %. The depolymerization process was monitored both in situ at a small scale, using TGA, and at a larger scale using 2 g polymer batches. The catalyst can be “recycled” up to four times without compromise to its activity or selectivity. The catalyst and solid state depolymerization methods presented here are expected to be generally useful for other chemical recycling to monomer processes. They open the door to applying CO2-derived polymers in sectors where waste polymer recycling would be especially beneficial such as packaging, consumer goods, and automotive sectors, to name a few.
Acknowledgments
The EPSRC ((EP/S018603/1, EP/R027129/1, EP/V003321/1), Oxford Martin School (Future of Plastics), The Royal Society (UF/160021, Fellowship to A.B.), and the UK Catalysis Hub (EP/R027129/1) are acknowledged for research funding. We thank the Material and Chemical Characterization Facility (MC2) at the University of Bath for assistance with TGA-MS experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c06937.
Experimental methods, characterization, and polymer/monomer purity evaluation data (PDF)
The authors declare the following competing financial interest(s): CKW is a director of Econic Technologies.
Supplementary Material
References
- Coates G. W.; Getzler Y. D. Y. L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. 10.1038/s41578-020-0190-4. [DOI] [Google Scholar]
- Hong M.; Chen E. Y. X. Chemically recyclable polymers: a circular economy approach to sustainability. Green Chem. 2017, 19, 3692–3706. 10.1039/C7GC01496A. [DOI] [Google Scholar]
- Zhang W.; Dai J.; Wu Y.-C.; Chen J.-X.; Shan S.-Y.; Cai Z.; Zhu J.-B. Highly Reactive Cyclic Carbonates with a Fused Ring toward Functionalizable and Recyclable Polycarbonates. ACS Macro Lett. 2022, 11, 173–178. 10.1021/acsmacrolett.1c00653. [DOI] [PubMed] [Google Scholar]
- Saxon D. J.; Gormong E. A.; Shah V. M.; Reineke T. M. Rapid Synthesis of Chemically Recyclable Polycarbonates from Renewable Feedstocks. ACS Macro Lett. 2021, 10, 98–103. 10.1021/acsmacrolett.0c00747. [DOI] [PubMed] [Google Scholar]
- Olsén P.; Odelius K.; Albertsson A.-C. Ring-Closing Depolymerization: A Powerful Tool for Synthesizing the Allyloxy-Functionalized Six-Membered Aliphatic Carbonate Monomer 2-Allyloxymethyl-2-ethyltrimethylene Carbonate. Macromolecules 2014, 47, 6189–6195. 10.1021/ma5012304. [DOI] [Google Scholar]
- Olsén P.; Undin J.; Odelius K.; Keul H.; Albertsson A.-C. Switching from Controlled Ring-Opening Polymerization (cROP) to Controlled Ring-Closing Depolymerization (cRCDP) by Adjusting the Reaction Parameters That Determine the Ceiling Temperature. Biomacromolecules 2016, 17, 3995–4002. 10.1021/acs.biomac.6b01375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiderman D. K.; Vanderlaan M. E.; Mannion A. M.; Panthani T. R.; Batiste D. C.; Wang J. Z.; Bates F. S.; Macosko C. W.; Hillmyer M. A. Chemically Recyclable Biobased Polyurethanes. ACS Macro Lett. 2016, 5, 515–518. 10.1021/acsmacrolett.6b00193. [DOI] [PubMed] [Google Scholar]
- Li C.; Wang L.; Yan Q.; Liu F.; Shen Y.; Li Z. Rapid and Controlled Polymerization of Bio-sourced δ-Caprolactone toward Fully Recyclable Polyesters and Thermoplastic Elastomers. Angew. Chem., Int. Ed. 2022, 61, e202201407 10.1002/anie.202201407. [DOI] [PubMed] [Google Scholar]
- Zhu J. B.; Watson E. M.; Tang J.; Chen E. Y.-X. A synthetic polymer system with repeatable chemical recyclability. Science 2018, 360, 398–403. 10.1126/science.aar5498. [DOI] [PubMed] [Google Scholar]
- Hong M.; Chen E. Y. X. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of γ-butyrolactone. Nat. Chem. 2016, 8, 42–49. 10.1038/nchem.2391. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wu J.; Hu X.; Zhu N.; Guo K. Advances, Challenges, and Opportunities of Poly(γ-butyrolactone)-Based Recyclable Polymers. ACS Macro Lett. 2021, 10, 284–296. 10.1021/acsmacrolett.0c00813. [DOI] [PubMed] [Google Scholar]
- Tang X.; Hong M.; Falivene L.; Caporaso L.; Cavallo L.; Chen E. Y. X. The Quest for Converting Biorenewable Bifunctional α-Methylene-γ-butyrolactone into Degradable and Recyclable Polyester: Controlling Vinyl-Addition/Ring-Opening/Cross-Linking Pathways. J. Am. Chem. Soc. 2016, 138, 14326–14337. 10.1021/jacs.6b07974. [DOI] [PubMed] [Google Scholar]
- Zhu J.-B.; Chen E. Y.-X. Living Coordination Polymerization of a Six-Five Bicyclic Lactone to Produce Completely Recyclable Polyester. Angew. Chem., Int. Ed. 2018, 57, 12558–12562. 10.1002/anie.201808003. [DOI] [PubMed] [Google Scholar]
- Cederholm L.; Wohlert J.; Olsén P.; Hakkarainen M.; Odelius K. “Like Recycles Like:” Selective Ring-Closing Depolymerization of Poly(L-Lactic Acid) to L-Lactide. Angew. Chem., Int. Ed. 2022, 61, e202204531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darensbourg D. J. Comments on the depolymerization of polycarbonates derived from epoxides and carbon dioxide: A mini review. Polym. Degrad. Stab. 2018, 149, 45–51. 10.1016/j.polymdegradstab.2018.01.019. [DOI] [Google Scholar]
- Xu G.; Wang Q. Chemically recyclable polymer materials: polymerization and depolymerization cycles. Green Chem. 2022, 24, 2321–2346. 10.1039/D1GC03901F. [DOI] [Google Scholar]
- Darensbourg D. J.; Wei S.-H.; Yeung A. D.; Ellis W. C. An Efficient Method of Depolymerization of Poly(cyclopentene carbonate) to Its Comonomers: Cyclopentene Oxide and Carbon Dioxide. Macromolecules 2013, 46, 5850–5855. 10.1021/ma401286x. [DOI] [Google Scholar]
- Darensbourg D. J.; Yeung A. D.; Wei S.-H. Base initiated depolymerization of polycarbonates to epoxide and carbon dioxide co-monomers: a computational study. Green Chem. 2013, 15, 1578–1583. 10.1039/c3gc40475g. [DOI] [Google Scholar]
- Liu Y.; Zhou H.; Guo J.-Z.; Ren W.-M.; Lu X.-B. Completely Recyclable Monomers and Polycarbonate: Approach to Sustainable Polymers. Angew. Chem., Int. Ed. 2017, 56, 4862–4866. 10.1002/anie.201701438. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Fang L.-M.; Liu Y.; Lu X.-B. Chemical Synthesis of CO2-Based Polymers with Enhanced Thermal Stability and Unexpected Recyclability from Biosourced Monomers. ACS Catal. 2021, 11, 8349–8357. 10.1021/acscatal.1c01376. [DOI] [Google Scholar]
- Li C.; Sablong R. J.; van Benthem R. A. T. M.; Koning C. E. Unique Base-Initiated Depolymerization of Limonene-Derived Polycarbonates. ACS Macro Lett. 2017, 6, 684–688. 10.1021/acsmacrolett.7b00310. [DOI] [PubMed] [Google Scholar]
- Carrodeguas L. P.; Chen T. T. D.; Gregory G. L.; Sulley G. S.; Williams C. K. High elasticity, chemically recyclable, thermoplastics from bio-based monomers: carbon dioxide, limonene oxide and ε-decalactone. Green Chem. 2020, 22, 8298–8307. 10.1039/D0GC02295K. [DOI] [Google Scholar]
- Childers M. I.; Longo J. M.; Van Zee N. J.; LaPointe A. M.; Coates G. W. Stereoselective Epoxide Polymerization and Copolymerization. Chem. Rev. 2014, 114, 8129–8152. 10.1021/cr400725x. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Darensbourg D. J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. 10.1016/j.ccr.2018.06.004. [DOI] [Google Scholar]
- Scharfenberg M.; Hilf J.; Frey H. Functional Polycarbonates from Carbon Dioxide and Tailored Epoxide Monomers: Degradable Materials and Their Application Potential. Adv. Funct. Mater. 2018, 28, 1704302. 10.1002/adfm.201704302. [DOI] [Google Scholar]
- Singer F. N.; Deacy A. C.; McGuire T. M.; Williams C. K.; Buchard A. Chemical Recycling of Poly(Cyclohexene Carbonate) Using a Di-Mg(II) Catalyst. Angew. Chem., Int. Ed. 2022, 61, e2022017 10.1002/anie.202201785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Gao B.; Liu Y.; Lu X.-B. Efficient and Selective Chemical Recycling of CO2-based Alicyclic Polycarbonates via Catalytic Pyrolysis. Angew. Chem., Int. Ed. 2022, 61, e202204492. [DOI] [PubMed] [Google Scholar]
- Koning C.; Wildeson J.; Parton R.; Plum B.; Steeman P.; Darensbourg D. J. Synthesis and physical characterization of poly(cyclohexane carbonate), synthesized from CO2 and cyclohexene oxide. Polymer 2001, 42, 3995–4004. 10.1016/S0032-3861(00)00709-6. [DOI] [Google Scholar]
- Spyridakou M.; Gardiner C.; Papamokos G.; Frey H.; Floudas G. Dynamics of Poly(cyclohexene carbonate) as a Function of Molar Mass. ACS Appl. Polym. Mater. 2022, 4, 3833–3843. 10.1021/acsapm.2c00299. [DOI] [Google Scholar]
- Paul S.; Zhu Y.; Romain C.; Brooks R.; Saini P. K.; Williams C. K. Ring-opening copolymerization (ROCOP): synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459–6479. 10.1039/C4CC10113H. [DOI] [PubMed] [Google Scholar]
- Guerin W.; Diallo A. K.; Kirilov E.; Helou M.; Slawinski M.; Brusson J.-M.; Carpentier J.-F.; Guillaume S. M. Enantiopure Isotactic PCHC Synthesized by Ring-Opening Polymerization of Cyclohexene Carbonate. Macromolecules 2014, 47, 4230–4235. 10.1021/ma5009397. [DOI] [Google Scholar]
- Sulley G. S.; Gregory G. L.; Chen T. T. D.; Peńa Carrodeguas L.; Trott G.; Santmarti A.; Lee K.-Y.; Terrill N. J.; Williams C. K. Switchable Catalysis Improves the Properties of CO2-Derived Polymers: Poly(cyclohexene carbonate-b-ε-decalactone-b-cyclohexene carbonate) Adhesives, Elastomers, and Toughened Plastics. J. Am. Chem. Soc. 2020, 142, 4367–4378. 10.1021/jacs.9b13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X.; Cui F.; He J.-H.; Ren W.-M.; Lu X.-B.; Zhang Y.-T. Sustainable Approach for Synthesis and Completely Recycle of Cyclic CO2-based Polycarbonates. Chem. Sci. 2022, 13, 6283–6290. 10.1039/D2SC01387H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacy A. C.; Kilpatrick A. F. R.; Regoutz A.; Williams C. K. Understanding metal synergy in heterodinuclear catalysts for the copolymerization of CO2 and epoxides. Nat. Chem. 2020, 12, 372–380. 10.1038/s41557-020-0450-3. [DOI] [PubMed] [Google Scholar]
- Reis N. V.; Deacy A. C.; Rosetto G.; Durr C. B.; Williams C. K. Heterodinuclear Mg(II)M(II) (M=Cr, Mn, Fe, Co, Ni, Cu and Zn) Complexes for the Ring Opening Copolymerization of Carbon Dioxide/Epoxide and Anhydride/Epoxide. Chem. – Eur. J. 2022, 28, e202104198 10.1002/chem.202104198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.