

Abstract
Complex cellular functions within immunoinflammatory cascades are carried out by networks of interacting genes. In this study, we employed a network modeling approach to dissect and interpret global gene expression patterns in allergen-induced T-helper(Th)-cell responses which underpins human atopic disease. We demonstrate that a subnet of interconnected genes enriched for Th2 and Treg-associated signatures plus many novel genes is hard-wired into the atopic response, and is a hallmark of atopy at the systems-level. We show that activation of this subnet is stabilized via hyper-connected “hub” genes, selective disruption of which can collapse the entire network in a comprehensive fashion. Finally, we investigated gene expression in different Th-cell subsets, and show that Treg- and Th2-associated signatures partition at different stages of Th-memory cell differentiation. Moreover we demonstrate the parallel presence of a core element of the Th2-associated gene signature in bystander naive cells, which can be reproduced by recIL-4. These findings indicate that network analysis provides significant additional insight into atopic mechanisms beyond that achievable with conventional microarray analyses, predicting functional interactions between novel genes and previously recognized members of the allergic cascade. This approach provides novel opportunities for design of therapeutic strategies which target entire networks of genes rather than individual effector molecules.
Keywords: Microarray, Network Analysis, Atopic
Introduction
The general concept that complex immunological diseases such as atopy are driven via integrated pathways or molecular cascades has been accepted dogma for over two decades. The approaches to identification of relevant cascade members and in particular those which are rate limiting in the disease process driven by the cascade has not evolved greatly over this period, and generally focuses on candidate effector or regulator molecules selected stepwise on the basis of their known effector functions and their potential interactions with other perceived pathogenic factors. However, the level of clinical efficacy achieved with therapies designed to target simple linear pathways or causal chains has been disappointing (1, 2).
The development of global gene expression profiling technologies has provided opportunities for new approaches to the same problem. The conceptual leap of principal interest in this context comes from the way in which these data are analyzed, in particular the emergence of quantitative algorithms based on network theory (3). This new approach enables a systems-level characterization of the networks of interacting genes which underpin cellular function and behavior, unearthing pathways at the core of disease processes.
A fundamental organizing principle of gene networks is their scale-free topology, meaning that network connectivity is dominated by a few centralized genes designated “hubs” which are hyper-connected to a larger number of peripheral genes with few connections (3). This structural feature of gene networks coupled with additional mechanisms including feedback control, redundant wiring, and plasticity, is thought to bestow biological systems with a high degree of tolerance to perturbations such as gene-deletions (4–8). However, a scale-free organization has an inherent trade-off, because removal of hyper-connected hubs at any level of biological organization (DNA, mRNA, protein, metabolite) can result in severe or multiple phenotypes (8–14).
By inference, entire networks of immunological disease-associated genes relevant to activation of Th-memory responses may also be under the control of hubs, however this remains to be formally demonstrated, because the tools of network analysis have not yet been systematically applied broadly in the immunology field (15). In the study below, we have utilized these methodologies to re-analyze the CD4+ Th-memory responses which underpin human atopic disease, in particular responses to house dust mite (HDM) allergen which is a major trigger of atopic asthma. We focus on the early activation phase in HDM-triggered Th-memory cells isolated from blood in order to minimize distortion of the gene expression program which may result from in vitro manipulations (16). We demonstrate that gene coexpression network analysis predicts functional associations between novel and known genes and provides an additional level of insight into the operation and stabilization of the principal atopy-associated effector pathway.
Materials and Methods
Study population
The study population were healthy volunteers comprising HDM-sensitized atopics and nonatopic controls in the age range 14–45 years. Atopic status to HDM was defined by positive skin test reaction to HDM extract (wheal ≥ 3 mm) and the presence of HDM-specific serum IgE (≥ 0.7 kU/L). The study was approved by our institutional human ethics committee.
Cell preparation and culture methodologies
Peripheral blood mononuclear cells (PBMC) were thawed from cyrobanked samples and cultured in AIM-V medium (Invitrogen, Australia) in the presence or absence of 10 μg/mL HDM extract (Dermatophagoides pteronyssinus, CSL, Australia) for 24 h as detailed previously (17). At the termination of the cultures, CD8+ followed by CD4+ T-cells were isolated by positive selection employing immunomagnetic separation (Dynal Biotech, Australia). The purity of the CD4+ T-cells was not tested in every sample but was routinely 98.1 (± 0.1) %. Where specified, 1 U/ml of human recombinant IL-2 (Cetus, CA), 0.25 ng/ml of human recombinant IL-4 (ProSpec-Tany TechnoGene LTD, Israel), 5 mg/ml of anti-IL-2 (Clone MQ1–17H12; BD Pharmingen), or 5 mg/ml of anti-IL-4 (Clone MP4–25D2; BD Pharmingen) was added to the cultures. Replicate cultures containing appropriate isotype control antibodies (clones R35–95, R3–34; BD Pharmingen) were set up in parallel.
For cell sorting experiments, PBMC were labeled with anti-CD27-FITC, anti-CD45RA-PECy5, anti-CD3-APCCy7, anti-CD4-APC (BD Biosciences) and anti-CCR7-PE (R & D Systems), and sorted on a FACSAria (BD Biosciences). Postsort purities of the CD4 subpopulations (TN, TCM, TEM) ranged from 95 to 99 %, except for the TTM subset which ranged in purity from 80 to 97 % (mean 89 %).
qRT-PCR analysis
Total RNA samples were extracted employing TRIzol (Invitrogen, Australia) followed by RNeasy (QIAgen, Australia). The quality of the RNA was not tested in every sample, but the RNA integrity number was routinely 8.8 (± 0.03) as assessed on the Bioanalyzer (Agilent). Reverse transcription was performed with the Omniscript kit (QIAgen, Australia), and qRT-PCR was performed with QuantiTect SYBR Green (Qiagen, Australia) employing predeveloped assays (QIAgen, Australia) as described previously (16). Where specified, qRT-PCR data was log transformed, mean centered and scaled (see statistical analysis).
Microarray study design
A total of 90 microarrays were employed in the study to profile gene expression in paired samples of HDM-stimulated and unstimulated CD4+ T-cells from 45 subjects. The experimental design comprised two independent atopic data sets (n=15 subjects in each set) for network construction and validation, and a nonatopic data set (n=15) for statistical comparisons.
Microarray methodologies and data preprocessing
Total RNA (~100 ng) from CD4+ T-cells was labeled and hybridized to Human Gene 1.0 ST microarrays (Affymetrix, Santa Clara), employing standardized protocols and reagents from Affymetrix. The microarray data were preprocessed in Expression Consol software (Affymetrix, Santa Clara) employing the probe logarithmic intensity error algorithm (PLIER, parameters - PM-GCBG background subtraction, quantile normalization, iterPLIER summarization). The preprocessed microarray data was then imported into the R language and environment for statistical computing (http://www.r-project.org/) for further analysis. Variance stabilization was performed by adding the small constant 16 to all the data points, followed by log2 transformation. The microarray data is available in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/projects/geo/) under the accession number GSE: 14908.
Identification of the atopy transcriptome in human CD4+ Th-cell responses
The moderated t-test (18) was employed to identify genes which were significantly modulated in allergen-stimulated versus unstimulated cells from atopic (atopic transcriptome) and nonatopic (nonatopic transcriptome) subjects. The moderated t-test employs a Bayesian model to leverage information obtained from the variability across all the genes to make inferences about individual genes. To account for multiple testing, the p-values derived from the moderated t.test statistics were adjusted employing the FDR method (19). Only genes that were significantly (FDR adjusted p-value < 0.01) modulated by ≥ 1.2 fold after HDM stimulation were considered in further analyses.
Differential expression analysis of CD4+ Th-cell response patterns to allergens
To identify differences in the response profiles of atopic and nonatopic subjects, background corrected gene expression levels (i.e. level in HDM-stimulated cells relative to baseline control (HDM/ctr) on the log2 scale) for the HDM regulated genes identified above (i.e. the combined atopic and nonatopic transcriptomes) were analyzed employing the Significance Analysis of Microarrays test (SAM or S.test) (20). The S.test is a variant of the t.test which adds a small constant to the variability component of the gene-specific t.test. Differentially expressed genes are identified by comparing the S.test statistics with their null distribution, which is generated via permutation of the sample class labels. The significance level of a SAM analysis is dependent on the tuning parameter delta, and for each value of delta, the FDR is calculated as the 90th percentile of the number of false positive genes divided by the number of genes called significant. A value for delta was selected corresponding to a 90th percentile FDR < 0.01. The results of the S.test analysis were summarized on a Volcano Plot (21); a scatter plot of the S.test statistics versus the log2 fold change (atopic HDM/ctr : nonatopic HDM/ctr).
Gene coexpression network analysis
Network analysis was performed on the microarray data employing the weighted gene coexpression algorithm developed by Horvath and coworkers (10). The mathematical process involves calculating absolute Pearson correlations for all pairwise gene-gene combinations across the test samples. The correlations were transformed into connection strengths by raising them to a power β. The value for β was selected by fitting a statistical model to the data, which is based on the knowledge that the distribution of connectivity in biological networks follows a scale-free topology (10). Genes with a low connectivity to the network were removed from the analysis (bottom 20 % with low values for k.total, see below). To identify modules of interconnected genes, the topological overlap was calculated from the pairwise connection strengths and analyzed by hierarchical clustering (10, 22). The topological overlap is a measure of the degree in which each pair of genes is correlated with the same set of genes. The modules were defined from the dendrogram output of the hierarchical clustering analysis employing an automated adaptive algorithm (23).
To test individual modules for association with atopic status, background corrected gene expression levels (HDM/ctr on log2 scale) in atopics versus nonatopics were compared on a module-by-module basis employing Gene Set Analysis (24). Gene Set Analysis tests for the association of a set of genes rather than individual genes with a phenotype of interest, and employs randomization/resampling of the genes to avoid bias in the determination of the test statistic, and permutations of the samples to estimate the FDR and account for correlations between genes (24). To illustrate module enrichment, background corrected gene expression levels (HDM/ctr) in atopic versus nonatopic responses were compared employing the S.test (20), and the absolute value of the S.test statistics were plotted as box-and-whisker plots on a module-by module basis.
The interaction of each component gene to the other genes in the network is quantified by the connectivity (k). The connectivity of a gene is defined as the cumulative connectivity (i.e. sum of the pairwise connection strengths); and can be calculated with respect to the entire network (K.total) or within a specific module (K.in). The K.in values were scaled to lie between 0 and 1.
Statistical analyses
The statistical methods employed in this study were performed in the R environment including the moderated t.test (18), S.test (20), Gene Set Analysis (24), Gene coexpression network analysis (10), Wilcoxon signed-rank test, Mann Whitney U-test, paired t.test, Spearman’s rank correlation, Fisher’s Exact Test, and hierarchical clustering (10). Where specified, FDR-adjusted p-values (19) were reported to control for Type I error when performing multiple hypothesis testing. Gene Set Analysis and the S.test have built in functions based on data resampling techniques to calculate the FDR, for all other statistical methods the FDR was calculated employing the Benjamini and Hochberg method (19) in the R package multtest (available at http://www.bioconductor.org/). It is noteworthy that the Benjamini and Hochberg FDR procedure is appropriate for test statistics that are independent or positively correlated (25).
Prior to statistical analysis employing the t.test, qRT-PCR data points below the detection limit (DL) were substituted for DL/2 followed by log10 transformation to correct for data skewness and heteroscedasticity (26).
Prior to hierarchical clustering analysis, mean centering and unit variance scaling was performed on the qRT-PCR data to emphasize the more relevant variations between samples as opposed to differences in high or low abundance values (26).
Results
Identification of the atopy transcriptome in human CD4+ Th-cell responses
To characterize the patterns of gene expression in Th-cell responses to allergens, PBMC from panels of HDM-sensitized atopics (n=15) and non-sensitized nonatopic controls (n=15) were cultured in the presence or absence of HDM allergens for 24 h. At the termination of the cultures, CD4+ Th-cells were isolated by immunomagnetic separation, and gene expression was analyzed by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR). As illustrated in Fig. 1A, the atopic Th-cell response phenotype was Th2-skewed, as predicted from previous studies employing this culture system (16, 17). Treg signature genes (IL-2R, FoxP3) were also elevated in the atopic responses, however it is noteworthy that these genes are also transiently upregulated following the activation of conventional Th-cells (reviewed (27)).
Figure 1: CD4+ Th-cell response patterns to allergens in atopic and nonatopic subjects.
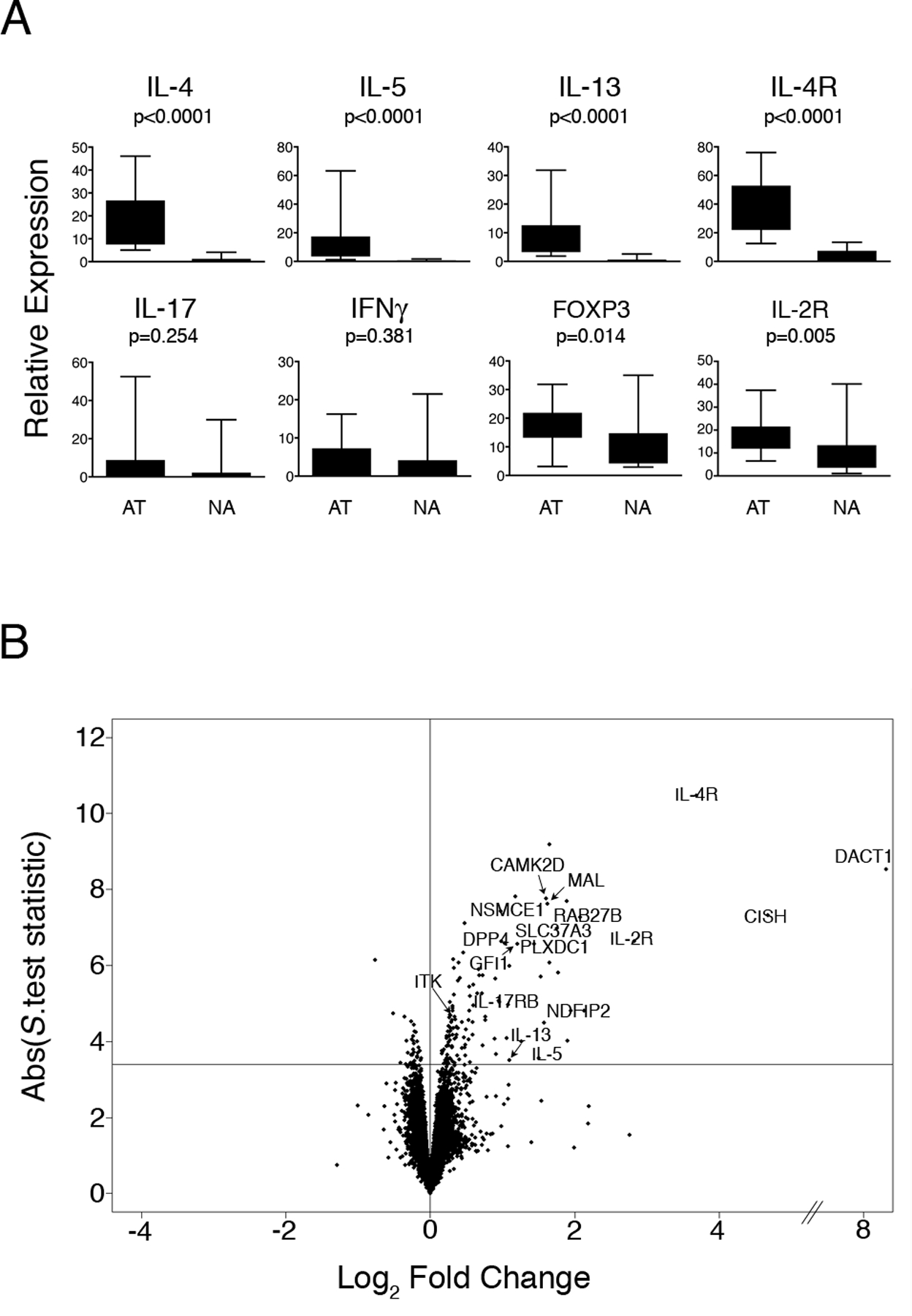
PBMC from HDM-sensitized atopics (n=15) and nonatopic controls (n=15) were cultured in the presence or absence of HDM for 24 h. At the termination of the cultures, CD4+ Th-cells were isolated and gene expression was profiled by qRT-PCR and microarray analysis.
(A) qRT-PCR analysis of selected Th-lineage signature genes demonstrates the Th2-skewed response phenotype of the atopic subjects. Data are expressed as gene expression level above baseline relative to the stably expressed gene EEF1A1 (68). Abbreviations: AT, atopic, NA, nonatopic. Statistical analysis by Mann-Whitney U.test.
(B): Microarray analysis of differential gene expression in atopic and nonatopic responses. Background corrected gene expression levels (level in HDM-stimulated cells relative to baseline control (HDM/ctr)) were compared in atopic and nonatopic responses employing the S.test (20). The data are summarized as a Volcano plot (21); which displays microarray data along axes of statistical significance (absolute S.test statistic) and differential expression magnitude (atopic HDM/ctr : nonatopic HDM/ctr on the log2 scale (Log2 Fold Change)). Genes above the horizontal line are significantly differentially expressed (FDR adjusted p-value < 0.01); positive and negative values on the horizontal axis indicate elevated expression in atopic and nonatopic responses respectively. Abbreviations: ABS, absolute.
Microarray profiling was then performed to investigate changes in global patterns of gene expression. Paired comparisons of HDM-stimulated Th-cells with unstimulated Th-cells demonstrated that a substantial gene expression program was activated in the respective responses, comprising 1442 genes in the atopic responses (i.e. the atopy transcriptome) and 1243 genes in the controls (average fold change > 1.2 and False Discovery Rate (FDR) (19) adjusted p-value < 0.01; moderated t.test (18)). Consistent with our previous findings, many of these genes were common to both groups (16), however, approximately 150 genes differed between the respective responses after accounting for multiple testing (FDR-adjusted p-value < 0.01; Significance Analysis of Microarray t.test (20); online Table S1). As illustrated in Fig. 1B, the majority of these genes were elevated in the atopic responses, including several previously recognized members of the Th2 cascade (IL-4R, IL-5, IL-13, GFI-1, ITK), as well as a cohort of novel Th2-associated genes (IL-17RB, CAMK2D, CISH, DACT1, DPP4, MAL, NDFIP2, NSMCE1, PLXDC1) reported recently by us (16) and confirmed in an independent study (28).
Characterization of the coexpression network architecture of the atopy transcriptome in CD4+ Th-cell responses
The statistical analyses performed above identify a list of genes involved in Th-memory responses to allergens, but provide limited insight into how these genes interact to control the underlying T-cell activation process. To obtain more detailed information in this regard, a systems-level analysis was performed on the atopic data set, employing a weighted gene coexpression network reconstruction algorithm as detailed in Methods (10). Briefly, network analysis employs a stepwise analytical process to leverage variations in gene-by-gene correlations across the samples to describe in quantifiable terms the underlying gene networks. Pathways manifest as subnets of highly correlated genes (“modules”), and hyper-connected genes (“hubs”) are identified within pathways. The algorithm generates a branching tree-like diagram output (dendrogram), in which modules of highly correlated genes are identified as the internal branch-like structures of the dendrogram. As illustrated in Fig. 2A, network analysis resolved the atopy transcriptome into a gene coexpression network composed of a series of 7 discrete modules. Of note, the distribution of connectivity followed a scale-free topology (data not shown), as predicted from studies in other systems (3, 29). To determine if any of the modules were associated with atopy, we performed a statistical comparison of the overall expression of each module in the atopic and nonatopic responses. As illustrated in Fig. 2B, the results demonstrated that one of the modules was uniquely associated with atopic status (module-level FDR adjusted p-value < 0.001 by Gene Set Analysis (24)).
Figure 2: Modular architecture of the gene coexpression network in atopic CD4+ Th-cell responses to allergens; variations associated with allergic sensitization.
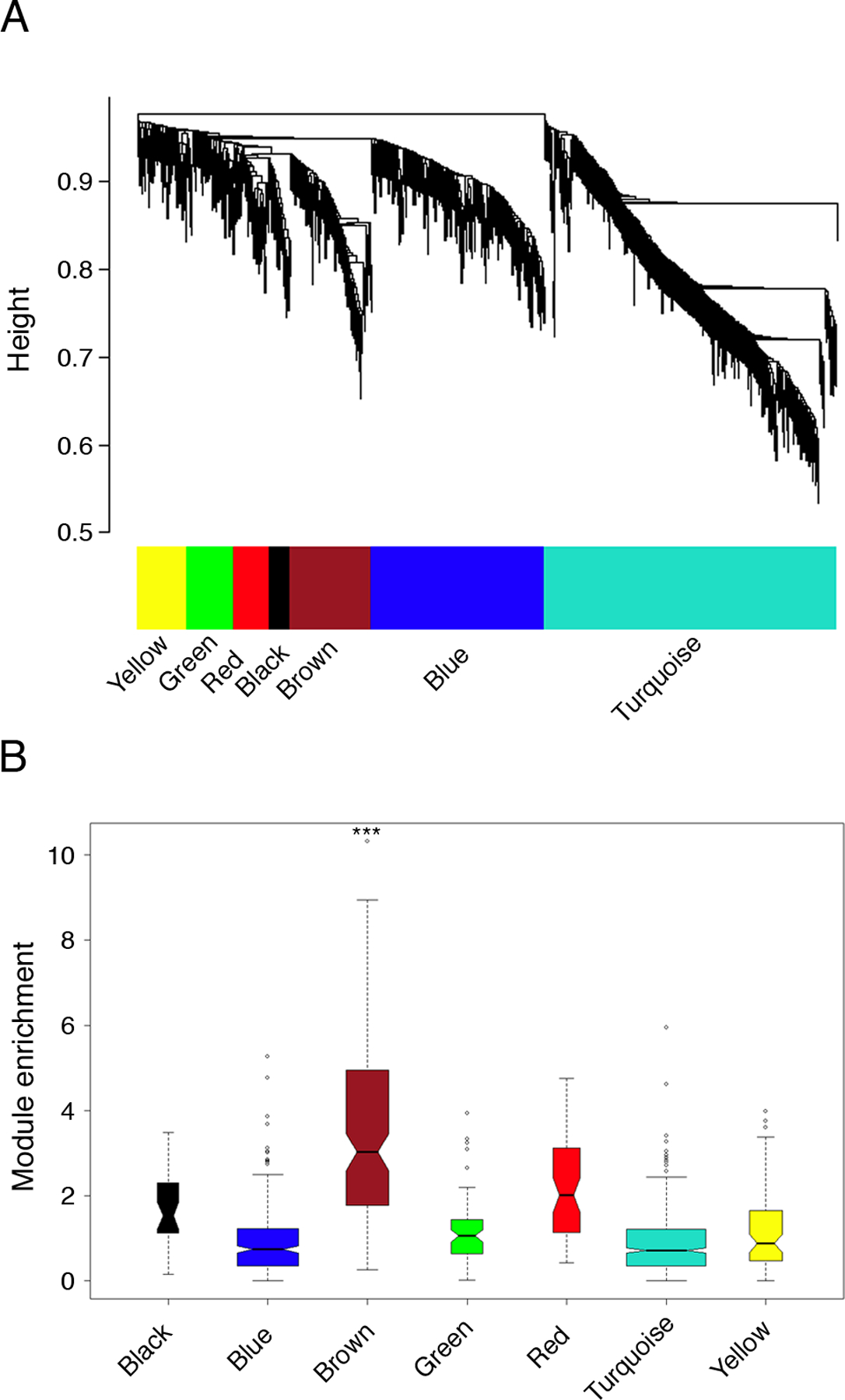
(A) Network analysis was performed on the atopic CD4+ Th-cell response microarray data set from Fig. 1B, and hierarchical clustering was employed to resolve the network into subnets (modules) of highly interconnected genes (10). The modules were defined by an automated algorithm (23), and can be visualized as the internal branch-like structures of the dendrogram output from the cluster analysis.
(B) The brown module is uniquely associated with atopic status. Background corrected gene expression levels (HDM/ctr) were compared in atopic and nonatopic responses employing the S.test (20), and the absolute value of the S.test statistics were graphed as box-and-whisker plots on a module-by module basis to visualize module enrichment. Statistical analyses were performed to compare the overall expression of each module in atopic and nonatopic responses employing Gene Set Analysis (*** FDR-adjusted p-value < 0.001) (24).
To determine if the atopy-associated module is reproducible, an additional series of microarray profiles were generated from HDM-stimulated CD4+ Th-cells from an independent panel of atopics (n=15). Network analysis of this independent data set revealed a similar coexpression network comprising discrete modules (not shown), and again a striking association was observed between a single module and atopic status (module-level FDR adjusted p-value < 0.001 by Gene Set Analysis (24)). The overlap between these two putative atopy-associated modules comprising 71 genes was highly significant (Fisher’s Exact Test p-value < 1 × 10−15), and included signatures associated with the activation of Th2 cells (IL-4R, IL-5, IL-13, ITK, GFI-1, CAMK2D, CISH, DACT1, DPP4, MAL, NDFIP2, PLXDC1, PTGER2, RAB27B) and Tregs (CISH, FoxP3, GFI-1, HIPK2, ID2, IKZF4, ITK, IL-2R, PTGER2, SOCS2, TIAM1 (30)), as well as a range of novel genes (Table I, see Table S2 online for references). This overlapping gene set was designated the “consensus atopy module”, and accordingly became the principal focus of the remainder of this study. Of note, 15 out of the 71 genes in the atopy module were not detected by the conventional statistical analyses performed above (Fig.1B), even when the FDR threshold was reduced from 0.01 to 0.05, suggesting that network analysis can unmask cryptic variations in gene expression thus revealing covert disease-associated genes (Table I).
Table I:
Functional annotation of the consensus atopy module
| Function/Pathway | Gene symbol | Chromosome | Gene name |
|---|---|---|---|
| Apoptosis | BCL2 | 18q21.33 | B-cell CLL/lymphoma 2 |
| BTG1a | 12q22 | B-cell translocation gene 1 | |
| CASP3 | 4q34 | Caspase 3 | |
| STK17B | 2q32.3 | Serine/threonine kinase 17b; DRAK2 | |
|
| |||
| Inflammation and | CD200R1 | 3q13.2 | CD200 receptor 1 |
| immunoregulation | CISH | 3p21.3 | Cytokine inducible SH2-containing protein |
| CSF1 | 1p21-p13 | Colony stimulating factor 1 | |
| IL-1F6a, b | 2q12-q14.1 | Interleukin 1 family, member 6 (epsilon) | |
| LIF | 22q12.2 | Leukemia inhibitory factor | |
| SOCS2 | 12q | Suppressor of cytokine signaling 2 | |
|
| |||
| Leukotriene and | ALOX5 | 10q11.2 | Arachidonate 5-lipoxygenase |
| Prostaglandin signaling | HPGD | 4q34-q35 | Hydroxyprostaglandin dehydrogenase |
| PTGER2 | 14q22 | Prostaglandin E receptor 2 (EP2 receptor) | |
|
| |||
| Protease activity and | PITRM1a | 10p15.2 | Pitrilysin metallopeptidase 1 |
| regulation | SPINT2 | 19q13.1 | Serine peptidase inhibitor, Kunitz type, 2 |
| TPP2a, b | 13q32-q33 | Tripeptidyl peptidase II | |
|
| |||
| Protein trafficking | RAB19Ba | 7q34 | GTP-binding protein RAB19B (RAB19) |
| RAB27B | 18q21.2 | RAB27B, member RAS oncogene family | |
| RAB30 | 11q12-q14 | RAB30, member RAS oncogene family | |
|
| |||
| Signal transduction | ARRDC2a, b | 19p13.11 | Arrestin domain containing 2 |
| CNKSR2a | Xp22.12 | Connector enhancer of kinase suppressor of Ras 2 | |
| KPNA6a | 1p35.1-p34.3 | Karyopherin alpha 6 (importin alpha 7) | |
| NCOA3 | 20q12 | Nuclear receptor coactivator 3 | |
| NDFIP2 | 13q31.1 | Nedd4 family interacting protein 2 | |
| NSMCE1 | 16p12.1 | Non-SMC element 1 homolog | |
| PGM1a | 1p31 | Phosphoglucomutase 1 | |
| RAP2Bb | 3q25.2 | RAP2B, member of RAS oncogene family | |
| RASL11Aa | 13q12.2 | RAS-like, family 11, member A | |
| SOCS2 | 12q | Suppressor of cytokine signaling 2 | |
| TBC1D1a | 4p14 | TBC1 domain family, member 1 | |
|
| |||
| Solute carrier activity | SLC26A11a | 17q25.3 | Solute carrier family 26, member 11 |
| SLC37A3 | 7q34 | Solute carrier family 37, member 3 | |
| SLC39A8 | 4q22-q24 | Solute carrier family 39, member 8 | |
|
| |||
| TcR signaling | APBB1IPa, b | 19p13.11 | Arrestin domain containing 2 |
| CEACAM1 | 19q13.2 | Carcinoembryonic antigen-related cell adhesion 1 | |
| CISH | 3p21.3 | Cytokine inducible SH2-containing protein | |
| DPP4 | 2q24.3 | Dipeptidyl-peptidase 4 (CD26) | |
| IL-2R | 10p15-p14 | Interleukin 2 receptor, alpha | |
| ITK | 5q31-q32 | IL2-inducible T-cell kinase | |
| LCP2b | 5q33.1-qter | Lymphocyte cytosolic protein 2 (SLP-76) | |
| MAL | 2cen-q13 | Mal, T-cell differentiation protein | |
| RAP1A | 1p13.3 | RAP1A, member of RAS oncogene family | |
|
| |||
| Th2 regulation and | CCL1 | 17q12 | C-C chemokine ligand 1, I-309 |
| function | GFI1 | 1p22 | Growth factor independent 1 |
| ID2 | 2p25 | Inhibitor of DNA binding 2 | |
| IL-13 | 5q31 | Interleukin 13 | |
| IL-17RB | 3p21.1 | Interleukin 17 receptor B (IL-25 receptor) | |
| IL-4R | 16p11.2–12.1 | Interleukin 4 receptor | |
| IL-5 | 5q31.1 | Interleukin 5 | |
| ITK | 5q31-q32 | IL2-inducible T-cell kinase | |
| RRS1b | 8q13.1 | RRS1 ribosome biogenesis regulator homolog | |
|
| |||
| Treg expression and | CISH | 3p21.3 | Cytokine inducible SH2-containing protein |
| function | FOXP3 | Xp11.23 | Forkhead box P3 |
| GFI1 | 1p22 | Growth factor independent 1 | |
| HIPK2 | 7q32-q34 | Homeodomain interacting protein kinase 2 | |
| IKZF4a, b | 12q13 | IKAROS family zinc finger 4 (Eos) | |
| IL-2R | 10p15-p14 | Interleukin 2 receptor, alpha | |
| ITK | 5q31-q32 | IL2-inducible T-cell kinase | |
| PTGER2 | 14q22 | Prostaglandin E receptor 2 (EP2 receptor) | |
| SOCS2 | 12q | Suppressor of cytokine signaling 2 | |
| TIAM1a | 21q22.1 | T-cell lymphoma invasion and metastasis 1 | |
|
| |||
| Transcriptional regulation | BATFa, b | 14q24.3 | Basic leucine zipper transcription factor, ATF-like |
| ENO1a, b | 1p36.3-p36.2 | Enolase 1, (alpha) | |
| HIPK1a | 1p13.2 | Homeodomain interacting protein kinase 1 | |
| ID2 | 2p25 | Inhibitor of DNA binding 2 | |
| IKZF1b | 7p13-p11.1 | IKAROS family zinc finger 1 (Ikaros) | |
| IKZF4a, b | 12q13 | IKAROS family zinc finger 4 (Eos) | |
| KLF3b | 4p14 | Kruppel-like factor 3 (basic) | |
| LASS6a | 2q24.3 | LAG1 homolog, ceramide synthase 6 | |
| PCGF5b | 10q23.32 | Polycomb group ring finger 5 | |
| TSC22D3b | Xq22.3 | TSC22 domain family, member 3; GILZ | |
|
| |||
| Wnt/β-catenin signaling | BCL9La, b | 11q23.3 | B-cell CLL/lymphoma 9-like |
| CAMK2D | 4q26 | Calcium-dependent protein kinase II delta | |
| DACT1 | 14q23.1 | Dapper homolog 1 | |
| HIPK2 | 7q32-q34 | Homeodomain interacting protein kinase 2 | |
| ID2 | 2p25 | Inhibitor of DNA binding 2 | |
| MEOX1a | 17q21 | Mesenchyme homeobox 1 | |
| TIAM1a | 21q22.1 | T-cell lymphoma invasion and metastasis 1 | |
|
| |||
| Unclassified or unknown | ETNK1a | 12p12.1 | Ethanolamine kinase 1 |
| function | FAM102Ba | 1p13.3 | Family with sequence similarity 102, member B |
| GNPDA1a | 5q21 | Glucosamine-6-phosphate deaminase 1 | |
| IMMT | 2p11.2 | Inner membrane protein, mitochondrial (mitofilin) | |
| OBFC2Aa | 2q32.3 | Oligonucleotide-binding fold containing 2A | |
| PLXDC1 | 17q21.1 | Plexin domain containing 1 | |
| SNTB1 | 8q23-q24 | Syntrophin, beta 1 | |
Genes were assigned to functional classes (NB: some genes in more than one class) based on classifications from the Gene Ontology consortium (http://www.geneontology.org/), as well as manual curation based on information from the literature and other sources. See Table S2 for additional information and references.
Novel genes which have not been previously reported in the context of atopy or Th2 regulation.
Genes which were specifically detected as atopy-associated based on their patterns of interconnectivity with other members of the atopy module, but were not detected as atopy-associated by differential expression analyses in Fig.1B.
The consensus atopy-associated module is enriched with functionally coherent genes
Modules execute high level biological functions by mobilizing sets of coexpressed genes which function in the same pathway (29, 31). As detailed in Table I, the atopy-associated module was enriched for functionally coherent genes involved in TcR and Wnt/β-catenin signaling, signal transduction, transcriptional regulation, Th2 regulation and function, inflammation, and Treg function. Although a generic function has been assigned to most of the genes in the module by the gene ontology consortium, for the vast majority of genes there is no information available about their regulation and/or function specifically in T-cells. Moreover, the module contained 25 genes which have not previously been reported in the context of atopy (Table 1).
Validation of constituent genes in the consensus atopy-associated module
Microarray detection of thousands of molecules in a single assay can result in false positive signals due to non-specific background and cross-hybridization (32). Therefore we sought confirmation of expression of the module genes in atopic Th-cell responses to allergens via more quantitative qRT-PCR methodology, employing the RNA samples from the independent groups of study subjects used for the microarray experiments. At least 90 % of the genes tested validated in these analyses (online Table S3), which incorporated FDR controls for multiple hypothesis testing (19).
Additional validation across microarray platforms was provided via meta-analysis of in house data sets on mixed CD4+ and CD8+ atopic T-cell responses to HDM which were generated as reported earlier (16) on the previous series Affymetrix microarrays. These analyses identified a module which significantly overlapped with the consensus atopy module (Fisher’s Exact Test p-value = 5.5 × 10−7). In addition to the core Th2 cluster on chromosome 5q31.1, these analyses also identified a broad range of the novel genes illustrated in Table 1 including BATF, CAMK2D, CEACAM1, DACT1, NDFIP2 and RAB27B (data not shown).
Variations in connectivity within the atopy-associated module reveals hyper-connected “hubs” which are essential for overall module functionality
Systematic studies in other systems have reported that the connectivity of individual genes within biological networks is correlated with essential functions (8–13). Thus by inference, hyper-connected hubs within an individual module may be indispensable to regulation and function of the module as a whole, and we wished to test this concept in relation to the atopy-associated module. To identify putative atopy-associated hyper-connected hubs, we first calculated the cumulative connectivity of each gene within the atopy-associated module (i.e. intramodular connectivity (k.in), see Methods). Genes which are highly correlated with many genes in the module will have large values for k.in, and vice versa. The statistical reliability of the network measure k.in is unknown in the current experimental setting, hence we calculated the k.in separately for both independent atopic data sets. As illustrated in Fig. 3A, the analyses demonstrated that variations in connectivity within the atopic module are highly reproducible (Spearman rho = 0.84, p-value < 1 × 10−15). Moreover, several hyper-connected hubs were identified; and these appear in the upper right region of Fig. 3A. To illustrate the wiring diagram of the atopy module, the pairwise interaction data (i.e. pairwise correlation data) were submitted to a bioinformatics tool for network visualization (33). As shown in Fig. 3B, the hubs manifest as the large nodes at the centre of the network depiction which are hyper-connected to peripheral nodes of low connectivity.
Figure 3: Visualization of the atopy-associated module and identification of putative hubs.
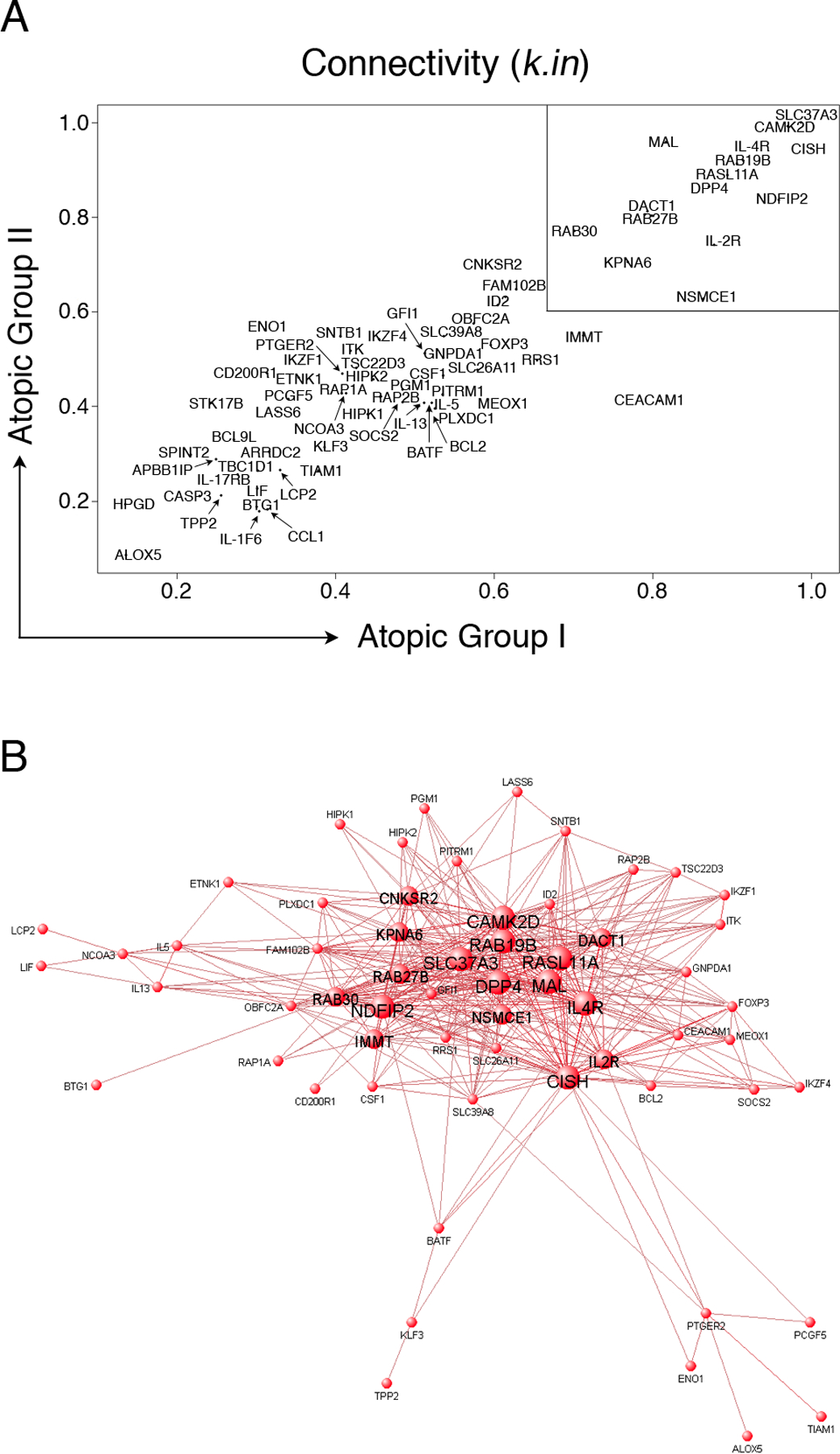
(A): Network analysis was performed separately on the two independent atopic CD4+ Th-cell response microarray data sets and variations in the patterns of connectivity within the atopy module were investigated by calculating the k.in (see methods). Hyper-connected hub genes have a k.in approaching 1.0, and are therefore located in the upper right region of the scatter diagram (box). The k.in values were highly correlated across the two independent data sets (Spearman rho = 0.84, p-value < 1 × 10−15).
(B): A graphical representation of the atopy-associated module. The microarray data from the two independent atopic data sets was pooled and network analysis was performed. The top gene-gene interaction data (i.e. all pairwise connection strengths > 0.25; corresponding to the top ~ 500 interactions) within the module were submitted to VisANT software (http://visant.bu.edu/) for network visualization. To illustrate the hubs, progressively larger font and node sizes were selected based on the connectivity data, which was partitioned into four categorical bins of > 31 links, 21 – 30 links, 11 – 20 links, and < 10 links. The hubs appear as the large central nodes in the network diagram.
As detailed above the initial identification of these putative atopy-associated hub genes was based solely on the strength of underlying statistical associations, and additional levels of (more direct) proof are required to determine the level of biological plausibility inherent in the information obtained via this approach. To explore this question we focused on two hub genes (IL-2R, IL-4R) in pathways whose functionality in relation to the Th2 cascade is well established (34, 35), and for which blocking reagents are readily accessible. The novelty of the new information on the allergen-triggered Th2 cascade provided via this approach (Fig 3B) concerns the additional range of genes putatively “networked” with IL-2R and IL-4R in this module. In particular, if the linkages identified exist in vivo and are biologically meaningful, then blocking these hubs alone or in combination may affect the overall structural integrity of the module, including the expression of these previously unrecognized members of the cascade. To test this proposition, positive and negative perturbation experiments were performed by supplementing the PBMC cultures with exogenous recombinant IL-2 or IL-4, or relevant neutralizing antibodies. At the termination of the cultures, CD4+ Th-cells were isolated and expression of the module was profiled by qRT-PCR. As illustrated in Fig. 4, hierarchical clustering analysis of these data segregated the genes into two distinct parallel pathways/clusters. The top cluster contained IL-4R, and genes within this cluster were highly induced by treatment with rIL-4, and strongly abrogated by anti-IL-4. In contrast, the bottom cluster contained IL-2R, and genes within this cluster were highly induced by treatment with rIL-2 and strongly abrogated by anti-IL-2. Strikingly, the combination of anti-IL-2 plus anti-IL-4 silenced expression of almost every gene in the module that was investigated (see online Table S4 for detailed statistical analyses). It should be emphasized that many genes within the module are totally novel (Table I), thus their regulation by IL-2 and IL-4 signaling was predicted solely on the basis of their interconnectivity with IL-2R/IL-4R.
Figure 4: IL-2R and IL-4R are principal hubs driving the expression of the atopy-associated module.
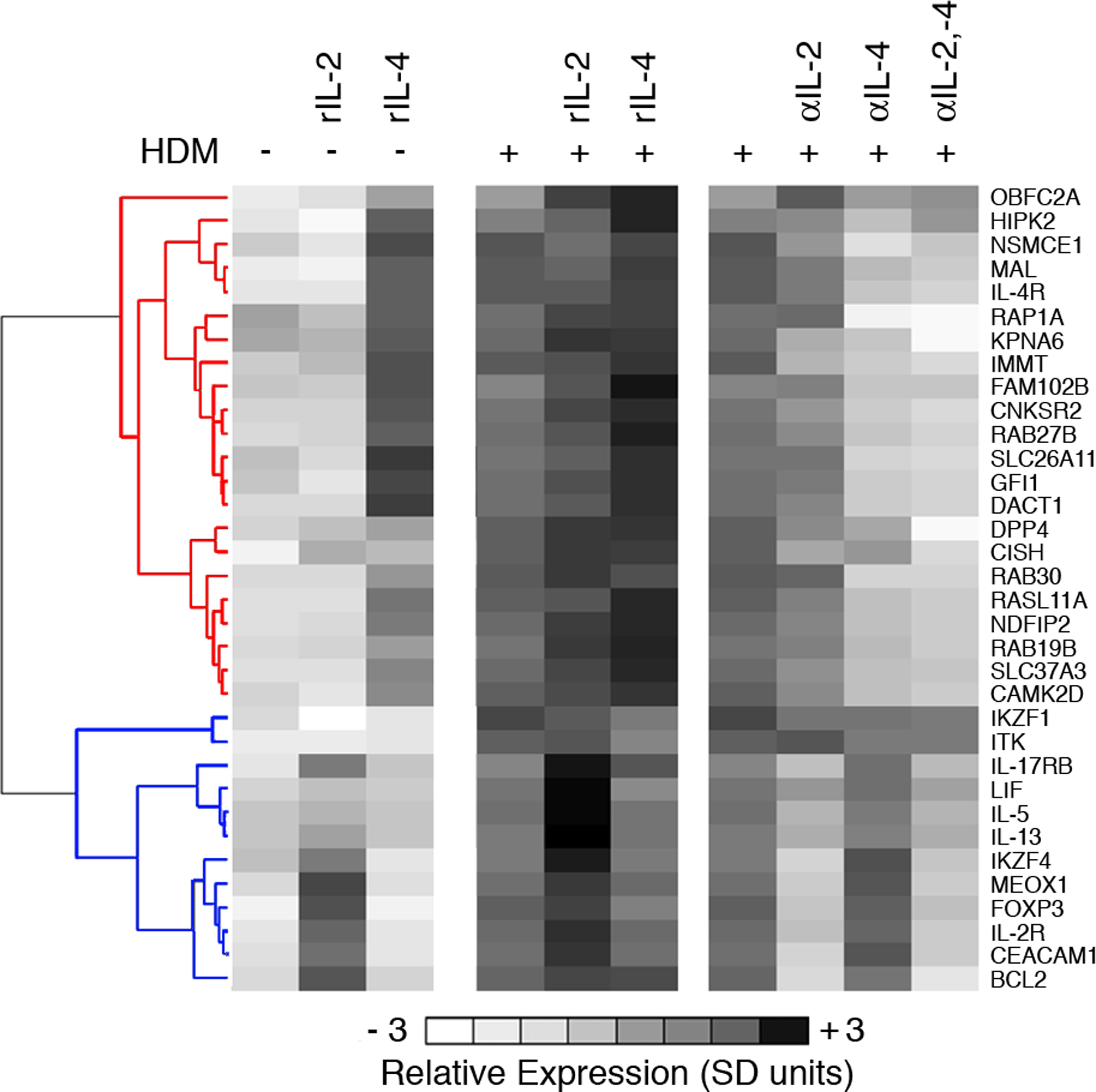
PBMC from HDM-sensitized atopics (n=8) were cultured in the presence (+) or absence (–) of HDM, recombinant IL-2 (rIL-2), recombinant IL-4 (rIL-4), or neutralizing antibodies against IL-2 (αIL-2), or IL-4 (αIL-4), or both (αIL-2, −4). At the termination of the 24 h cultures, CD4+ Th-cells were purified and expression of a subset of genes from the atopy-associated module was profiled by qRT-PCR. The qRT-PCR data were normalized to the stably expressed gene EEF1A1 (68), averaged across the subjects, mean centered (26), and scaled for unit variance (26). Hierarchical clustering (10) was performed on the genes to partition them into clusters of coexpressed genes. Two major expression patterns were identified; corresponding to genes that were regulated by IL-4 (red cluster dendrogram) or IL-2 (blue cluster dendrogram) genes. Detailed statistical analyses were also performed and are presented online in Table S4. Additional cultures were set up with appropriate isotype control antibodies and these did not substantively affect expression profiles (not shown). Abbreviations; SD, standard deviations.
Expression of the atopy-associated module varies across the CD4+ Th-memory compartment
The network analyses presented above demonstrate that IL-2R and IL-4R are hyper-connected within the atopy-associated module, and that dual inhibition of these parallel pathways destabilizes gene expression programs in the overall Th-memory response in a comprehensive fashion. However, it is not clear why the atopy module contains gene expression signatures associated with the early differentiation of Th2 cells (28), Th2 memory responses (16), and Treg function (30). One possibility is that because the Th-memory compartment is phenotypically and functionally heterogeneous (36), the above analyses on total CD4 T-cells are likely to have provided an oversimplified picture. A series of experimental observations support a relationship between the phenotype, function, and stage of CD4+ Th-cell memory differentiation (reviewed (37)). One simple model to account for the origin of these diverse subsets proposes that there is a stepwise differentiation of naive Th-cells (TN) -> central memory (TCM) -> transitory memory (TTM) -> effector memory (TEM) (36, 38). To investigate expression of the module across the differentiation pathway during a Th-memory response, PBMC from six HDM-allergic subjects were stimulated with HDM for 20 h. At the termination of the cultures, CD4+ Th-cell subsets were isolated by multiparametric cell sorting employing the markers CD4, CD45RA, CCR7, and CD27 (38), and gene expression was profiled by qRT-PCR.
As illustrated in Fig. 5C, analysis of the qRT-PCR data by hierarchical clustering demonstrated that expression of the module partitioned unevenly across the differentiation pathway (see online Table S5 for statistical analyses). For instance, IL-4R-associated genes (eg. DACT1, GFI1, MAL, RAB30, RASL11A, SLC26A11) were highly expressed in naive and to a lesser extent in central memory, whereas Treg-associated genes (IL-2R, FoxP3) were highly expressed in central memory relative to naive, but peaked together with several other genes (CEACAM1, CISH, DPP4, IKZF4) within transitory memory (Fig. 5C/D). The effector memory compartment was characterized by high level expression of genes associated with effector functions (IL-2, IL-4, IL-5, IL-13, IL-17RB, LIF), together with the NFκB regulator NDFIP2 and the inhibitory receptor CD200R1 (Fig. 5C/D). These data demonstrate that naive Th-cells participate in the Th-memory response, and further show that the Treg and Th2-associated signatures segregate in the TTM and TEM compartments respectively.
Figure 5: Expression of the atopy-associated module varies across naive and memory CD4+ Th-cell subsets.
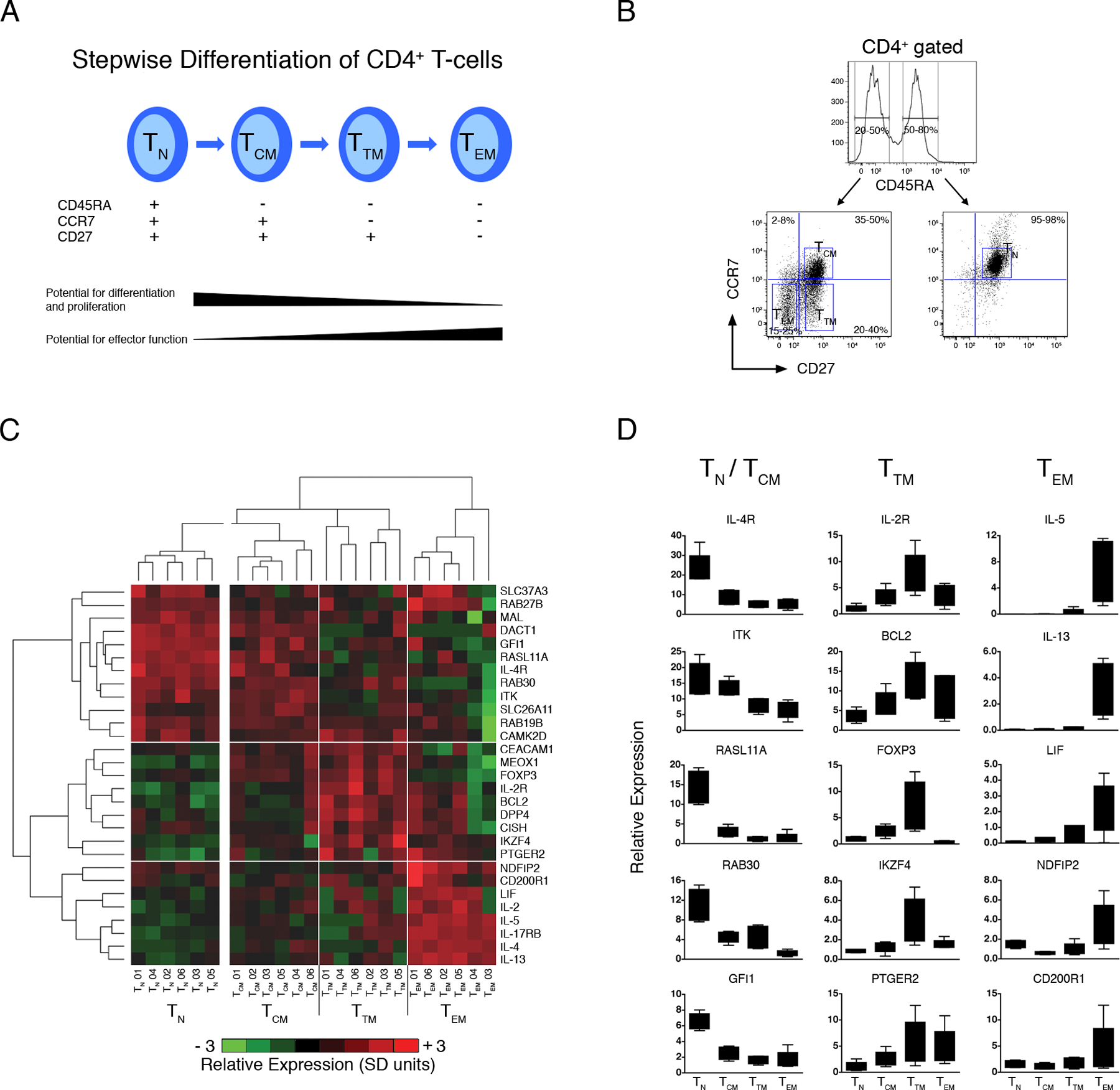
(A) Stepwise model of CD4+ Th-cell memory differentiation (38).
(B) Multiparametric cell sorting strategy employed to isolate CD4+ Th-cell subsets.
(C) PBMC from HDM-sensitized atopics (n=6) were stimulated with HDM for 20 h. At the termination of the cultures, multiparametric cell sorting was employed to isolate naive (TN), central memory (TCM), transitory memory (TTM) and effector memory (TEM) subpopulations. Expression of a subset of genes from the atopy-associated module was profiled by qRT-PCR. The qRT-PCR data was normalized to the stably expressed gene EEF1A1 (68), log2 transformed, mean centered (26), and scaled for unit variance (26). Mean centering and unit variance was performed separately for the naive and memory compartments to emphasize variations across the latter compartment. Hierarchical clustering (10) was employed to identify clusters of coexpressed genes, and clusters of samples with similar expression profiles. Four major sample clusters and three major gene clusters were identified, as shown by the respective vertical and horizontal white lines. Detailed statistical analyses were performed and are presented online in Table S5. Abbreviations; SD, standard deviations.
(D) Box-and-whisker plots of the qRT-PCR data (centering, scaling, and log transformation was not performed) for selected genes from (C). See Table S6 for complete data set.
Discussion
Our findings demonstrate that the hallmark of the atopic phenotype at the systems-level is the activation of an interconnected module enriched for Th2 and Treg-associated expression signatures, networked together with an additional series of 25 novel genes not previously associated with atopy. Of note, application of network analysis techniques to the microarray data not only confirmed the importance of the bulk of Th2-associated genes identified by more conventional analyses in previous studies (16, 28), but also added a second tier of previously unrecognized Th2-associated genes identified purely on the basis of their patterns of interconnectivity with other members of the atopy module (Table I). These novel genes encode proteins mainly involved in generic functions such as transcriptional regulation and signal transduction, however their precise function in allergen-driven Th-cell responses remains to be investigated. In the parlance of this emerging field, genes in the network displaying highest overall connectivity strengths are designated as “hubs” which are ascribed key roles in overall network functionality (8–13). Confirmation that the gene interconnectivity measures employed to reconstruct this atopy-associated module have a biological as opposed to purely statistical basis was provided via blocking experiments targeting the hubs IL-2R and IL-4R. Notably, disruption of IL-2 and IL-4 signaling during stimulation of Th-memory cells with specific allergen collapsed the overall atopy module including expression of these novel genes. This finding justifies further pursuit of this overall approach, in particular studies related to the network regulatory functions of other “novel” hub genes. The most direct approach would be to employ siRNA-mediated knockdown of hub(s) as has been performed in Th-cell lines (28), however effective methodology is not available to achieve this in primary human Th-memory responses.
An alternative approach to probe further into the function of the gene network which we are following involves more precise cellular localization of relevant expression signals during the Th-memory response. Recent literature indicates that reactivation of Th-memory cells sets in train a stepwise differentiation process giving rise to functionally distinct memory subpopulations (36–38), and the expression signals we are detecting is likely to represent a summation of the activity of these subsets. We hypothesized that resolving the allergen-activated CD4+ Th-cell population into discrete memory subsets prior to expression analysis may provide some clarity to this complex picture. This approach has resulted in a series of novel observations, and in particular has facilitated partitioning of key elements of the overall atopy module expression signal to different stages of the Th-memory response. Our findings demonstrated that IL-4 associated genes were predominantly expressed at early stages of differentiation (naïve Th-cells, TCM), whereas IL-2 regulated genes were expressed at early (TCM) and later stages (TTM, TEM). Moreover, we found that Treg and Th2 effector signatures partitioned into the TTM and TEM compartments respectively. Whilst we did not perform functional assays to determine if the TTM (CD27+) subset had suppressive activity, CD27 has been previously reported to discriminate regulatory from effector T-cells (39). Moreover, we detected a bona fide Treg signature in the TTM compartment, which includes genes such as IKZF4 (Eos) that are highly specific for Treg (30).
The nature of the relationship between Treg and conventional T-cells is a contentious issue. Several laboratories have demonstrated that expression of FoxP3 is upregulated as a normal consequence of human CD4 T-cell activation (40–42), and it has been suggested that regulatory activity may be a reversible peripheral state of differentiation (reviewed (27)). However, the interpretation of these data is confounded by the knowledge that FoxP3 expression does not necessarily confer suppressive activity (41–43). Moreover, concerns have been raised about the specificity of FoxP3 staining in activated T-cells (41, 44). One possible interpretation of our data is that the TCM compartment gives rise to both TEM and Treg, either via parallel or sequential pathways which are programmed by IL-2. Indeed, an IL-2-dependent mechanism drives the development of antigen-specific effector and regulatory T-cells from the same naïve precursors in animal models of autoimmune disease (45), and we observed that both Th2 effector and Treg-associated signatures are dependent on IL-2 (Fig. 4). Data from human studies suggest that Treg may be derived from the allergen-driven expansion of memory T-cells (46, 47). In human T-cells, IL-2 driven FoxP3 expression does not bestow de novo immunoregulatory properties until at least four days of stimulation (42). This delayed acquisition of regulatory function provides a plausible mechanism to generate adaptive Treg that can switch off acute inflammation thus preventing chronic disease (45, 48, 49).
The synchronous activation of the Treg and Th2 machinery during recall responses to allergens may explain the inherent plasticity of the CD4 compartment, which has recently been documented at the epigenetic level (50). Studies in animal models have shown that in some circumstances Treg can be converted into Th2 effectors, via internal regulation of their gene expression program (43, 51). In this context it is also noteworthy that PTGER2 was highly expressed in the TTM and TEM compartments (Table S6), because signaling via this pathway can convert conventional T-cells into Treg via induction of FoxP3 (52, 53). Further studies are warranted to investigate this possibility in allergy.
The presence of a parallel IL-4 dependant activation signal in the naïve Th-cell compartment was striking. Although it has been reported that the role of IL-4 is redundant in some (54) but not all models (55) of Th2 differentiation in vivo, IL-4 is essential for the collateral priming of naïve Th-cells to other bystander allergens (56). Bystander sensitization is a recognized feature of persistent allergic disease in humans (reviewed (49)), and it is also noteworthy that successful allergen-specific immunotherapy against single allergens prevents sensitization to new allergens (reviewed (57)).
The identification of IL-2R as a T-cell activation-associated, atopy-associated hub is intriguing. Systematic studies of disease-gene relationships across multiple disorders have suggested that common pathways may underlie susceptibility to many diseases (58), especially inflammatory diseases (59, 60). IL-2R may represent an archetypal example of a hub which is relevant to multiple inflammatory diseases, because disruption of IL-2 signaling in mice leads to autoimmunity (61). Moreover, genetic variants in IL-2R differentially confer risk to type I diabetes and multiple sclerosis in humans (62). The relevance of this pathway to the pathogenesis of human asthma was recently demonstrated in a clinical trial, where blocking IL-2R improved lung function and asthma control in patients with moderate to severe disease (63). Further studies are now warranted to determine if genetic variation in the IL-2R locus is important in disease risk and severity.
This study has significant limitations which require addressing in follow-up investigations. In particular, for logistical reasons we focused on a single time-point post Th-cell activation, however biological systems are best modeled in dynamic terms (64). Moreover, we have shown in previous kinetic studies that the 24 h time point favors identification of Th2 activation/differentiation-associated genes in contrast to late effector genes such as IFN-γ (16) and TNF-α which also contribute to asthma pathogenesis (17). This approach also cannot reproduce microenvironmental conditions at the asthma lesional site where T-cell triggering is controlled by highly differentiated mucosal dendritic cells (65). Additionally, as noted in Results, the focus on IL-2R and IL-4R genes was dictated by the availability of relevant inhibitors, and more systematic study of the role of other potential hubs in the Th2 network will require utilization of a wider range of blocking reagents. Such functional studies are important because unlike the situation in protein interaction networks, which are based on detection of protein-protein interactions in functional assays, coexpression networks represent more general relationships between genes which are simply based on statistical correlations and not functional data. Thus the biological significance of the other putative hubs is unknown in the absence of functional data. Notwithstanding these limitations, this study has provided novel insight into the operation of the Th2 cascade in human atopics, and in doing so illustrates some important principles that are readily applicable to research into a broad range of other immunoinflammatory diseases. Notably, our findings demonstrate how inferences derived from network analysis can be used to identify novel disease-associated genes within causal pathways and place them within a testable functional context. This approach thus has the potential to accelerate drug discovery programs aimed at destabilization of inflammatory gene networks rather than inhibiting individual effector molecules. The latter approach is currently the dominant paradigm in pharmacology and has shown only limited success in the treatment of allergic diseases (66, 67). Finally, network analysis may also increase capacity to identify subtle gene/gene regulatory interactions that escape detection via conventional microarray methodology, and thus may also enhance the precision of basic mechanistic studies.
Supplementary Material
Acknowledgements
The authors thank Matt Wikstrom, Debbie Strickland, Jenny Thomas, Rob Shackleton, and Barb Holt for help and advice regarding cell sorting and handling techniques.
This work was supported by the National Health & Medical Research Council of Australia and via NIH grant #1R21AI78511–1.
References
- 1.Schadt EE, Sachs A, and Friend S. 2005. Embracing complexity, inching closer to reality. Sci STKE 2005:pe40. [DOI] [PubMed] [Google Scholar]
- 2.Van Regenmortel MH 2004. Reductionism and complexity in molecular biology. Scientists now have the tools to unravel biological and overcome the limitations of reductionism. EMBO Rep 5:1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barabasi AL, and Oltvai ZN. 2004. Network biology: understanding the cell’s functional organization. Nat Rev Genet 5:101–113. [DOI] [PubMed] [Google Scholar]
- 4.Kitano H 2004. Biological robustness. Nat Rev Genet 5:826–837. [DOI] [PubMed] [Google Scholar]
- 5.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, and Johnston M. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. [DOI] [PubMed] [Google Scholar]
- 6.Bergman A, and Siegal ML. 2003. Evolutionary capacitance as a general feature of complex gene networks. Nature 424:549–552. [DOI] [PubMed] [Google Scholar]
- 7.Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, and Gerstein M. 2004. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431:308–312. [DOI] [PubMed] [Google Scholar]
- 8.Albert R, Jeong H, and Barabasi AL. 2000. Error and attack tolerance of complex networks. Nature 406:378–382. [DOI] [PubMed] [Google Scholar]
- 9.Davierwala AP, Haynes J, Li Z, Brost RL, Robinson MD, Yu L, Mnaimneh S, Ding H, Zhu H, Chen Y, Cheng X, Brown GW, Boone C, Andrews BJ, and Hughes TR. 2005. The synthetic genetic interaction spectrum of essential genes. Nat Genet 37:1147–1152. [DOI] [PubMed] [Google Scholar]
- 10.Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, and Nelson SF. 2006. Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics 7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeong H, Mason SP, Barabasi AL, and Oltvai ZN. 2001. Lethality and centrality in protein networks. Nature 411:41–42. [DOI] [PubMed] [Google Scholar]
- 12.Lee I, Lehner B, Crombie C, Wong W, Fraser AG, and Marcotte EM. 2008. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet 40:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong H, Tombor B, Albert R, Oltvai ZN, and Barabasi AL. 2000. The large-scale organization of metabolic networks. Nature 407:651–654. [DOI] [PubMed] [Google Scholar]
- 14.Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, and Vidal M. 2008. High-quality binary protein interaction map of the yeast interactome network. Science 322:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benoist C, Germain RN, and Mathis D. 2006. A plaidoyer for ‘systems immunology’. Immunol Rev 210:229–234. [DOI] [PubMed] [Google Scholar]
- 16.Bosco A, McKenna KL, Devitt CJ, Firth MJ, Sly PD, and Holt PG. 2006. Identification of novel Th2-associated genes in T memory responses to allergens. J Immunol 176:4766–4777. [DOI] [PubMed] [Google Scholar]
- 17.Heaton T, Rowe J, Turner S, Aalberse RC, de Klerk N, Suriyaarachchi D, Serralha M, Holt BJ, Hollams E, Yerkovich S, Holt K, Sly PD, Goldblatt J, Le Souef P, and Holt PG. 2005. An immunoepidemiological approach to asthma: identification of in-vitro T-cell response patterns associated with different wheezing phenotypes in children. Lancet 365:142–149. [DOI] [PubMed] [Google Scholar]
- 18.Smyth GK 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology 3:Article 3. [DOI] [PubMed] [Google Scholar]
- 19.Benjamini Y, and Hochberg Y. 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc Ser B 57:289–300. [Google Scholar]
- 20.Tusher VG, Tibshirani R, and Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui X, and Churchill GA. 2003. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol 4:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, and Barabasi AL. 2002. Hierarchical organization of modularity in metabolic networks. Science 297:1551–1555. [DOI] [PubMed] [Google Scholar]
- 23.Langfelder P, Zhang B, and Horvath S. 2008. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut library for R. Bioinformatics 24:719–720. [DOI] [PubMed] [Google Scholar]
- 24.Efron B, and Tibshirani R. 2007. On testing the significance of sets of genes Annals of Applied Statistics 1:107–109. [Google Scholar]
- 25.Benjamini Y, and Yekutieli D. 2001. The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188. [Google Scholar]
- 26.van den Berg RA, Hoefsloot HC, Westerhuis JA, Smilde AK, and van der Werf MJ. 2006. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 7:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pillai V, and Karandikar NJ. 2007. Human regulatory T cells: a unique, stable thymic subset or a reversible peripheral state of differentiation? Immunol Lett 114:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund RJ, Loytomaki M, Naumanen T, Dixon C, Chen Z, Ahlfors H, Tuomela S, Tahvanainen J, Scheinin J, Henttinen T, Rasool O, and Lahesmaa R. 2007. Genome-wide identification of novel genes involved in early Th1 and Th2 cell differentiation. J Immunol 178:3648–3660. [DOI] [PubMed] [Google Scholar]
- 29.Schadt EE, and Lum PY. 2006. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res 47:2601–2613. [DOI] [PubMed] [Google Scholar]
- 30.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, and Benoist C. 2007. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27:786–800. [DOI] [PubMed] [Google Scholar]
- 31.Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, Stichweh D, Blankenship D, Li L, Munagala I, Bennett L, Allantaz F, Mejias A, Ardura M, Kaizer E, Monnet L, Allman W, Randall H, Johnson D, Lanier A, Punaro M, Wittkowski KM, White P, Fay J, Klintmalm G, Ramilo O, Palucka AK, Banchereau J, and Pascual V. 2008. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 29:150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chuaqui RF, Bonner RF, Best CJ, Gillespie JW, Flaig MJ, Hewitt SM, Phillips JL, Krizman DB, Tangrea MA, Ahram M, Linehan WM, Knezevic V, and Emmert-Buck MR. 2002. Post-analysis follow-up and validation of microarray experiments. Nat Genet 32 Suppl:509–514. [DOI] [PubMed] [Google Scholar]
- 33.Hu Z, Mellor J, Wu J, Yamada T, Holloway D, and Delisi C. 2005. VisANT: data-integrating visual framework for biological networks and modules. Nucleic Acids Res 33:W352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, and Paul WE. 1990. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Exp Med 172:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swain SL, Weinberg AD, English M, and Huston G. 1990. IL-4 directs the development of Th2-like helper effectors. J Immunol 145:3796–3806. [PubMed] [Google Scholar]
- 36.Sallusto F, Lenig D, Forster R, Lipp M, and Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712. [DOI] [PubMed] [Google Scholar]
- 37.Harari A, Dutoit V, Cellerai C, Bart PA, Du Pasquier RA, and Pantaleo G. 2006. Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol Rev 211:236–254. [DOI] [PubMed] [Google Scholar]
- 38.Fritsch RD, Shen X, Sims GP, Hathcock KS, Hodes RJ, and Lipsky PE. 2005. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol 175:6489–6497. [DOI] [PubMed] [Google Scholar]
- 39.Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, and Sallusto F. 2005. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med 201:1793–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, and Ziegler SF. 2003. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest 112:1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran DQ, Ramsey H, and Shevach EM. 2007. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood 110:2983–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng Y, Manzotti CN, Burke F, Dussably L, Qureshi O, Walker LS, and Sansom DM. 2008. Acquisition of suppressive function by activated human CD4+ CD25- T cells is associated with the expression of CTLA-4 not FoxP3. J Immunol 181:1683–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wan YY, and Flavell RA. 2007. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445:766–770. [DOI] [PubMed] [Google Scholar]
- 44.Pillai V, and Karandikar NJ. 2008. Attack on the clones? Human FOXP3 detection by PCH101, 236A/E7, 206D, and 259D reveals 259D as the outlier with lower sensitivity. Blood 111:463–464; author reply 464–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knoechel B, Lohr J, Kahn E, Bluestone JA, and Abbas AK. 2005. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med 202:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vukmanovic-Stejic M, Zhang Y, Cook JE, Fletcher JM, McQuaid A, Masters JE, Rustin MH, Taams LS, Beverley PC, Macallan DC, and Akbar AN. 2006. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest 116:2423–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vukmanovic-Stejic M, Agius E, Booth N, Dunne PJ, Lacy KE, Reed JR, Sobande TO, Kissane S, Salmon M, Rustin MH, and Akbar AN. 2008. The kinetics of CD4Foxp3 T cell accumulation during a human cutaneous antigen-specific memory response in vivo. J Clin Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strickland DH, Stumbles PA, Zosky GR, Subrata LS, Thomas JA, Turner DJ, Sly PD, and Holt PG. 2006. Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. J Exp Med 203:2649–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, and Lafaille JJ. 2008. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29:114–126. [DOI] [PubMed] [Google Scholar]
- 50.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O’Shea JJ, and Zhao K. 2009. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joetham A, Matsubara S, Okamoto M, Takeda K, Miyahara N, Dakhama A, and Gelfand EW. 2008. Plasticity of regulatory T cells: subversion of suppressive function and conversion to enhancement of lung allergic responses. J Immunol 180:7117–7124. [DOI] [PubMed] [Google Scholar]
- 52.Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc’h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, and Dubinett SM. 2005. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol 175:1483–1490. [DOI] [PubMed] [Google Scholar]
- 53.Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, and Dubinett SM. 2005. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res 65:5211–5220. [DOI] [PubMed] [Google Scholar]
- 54.van Panhuys N, Tang SC, Prout M, Camberis M, Scarlett D, Roberts J, Hu-Li J, Paul WE, and Le Gros G. 2008. In vivo studies fail to reveal a role for IL-4 or STAT6 signaling in Th2 lymphocyte differentiation. Proc Natl Acad Sci U S A 105:12423–12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sokol CL, Barton GM, Farr AG, and Medzhitov R. 2008. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol 9:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eisenbarth SC, Zhadkevich A, Ranney P, Herrick CA, and Bottomly K. 2004. IL-4-dependent Th2 collateral priming to inhaled antigens independent of Toll-like receptor 4 and myeloid differentiation factor 88. J Immunol 172:4527–4534. [DOI] [PubMed] [Google Scholar]
- 57.Holgate ST, and Polosa R. 2008. Treatment strategies for allergy and asthma. Nat Rev Immunol 8:218–230. [DOI] [PubMed] [Google Scholar]
- 58.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, and Barabasi AL. 2007. The human disease network. Proc Natl Acad Sci U S A 104:8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, and Trent JM. 1998. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci U S A 95:9979–9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamada R, and Ymamoto K. 2005. Recent findings on genes associated with inflammatory disease. Mutat Res 573:136–151. [DOI] [PubMed] [Google Scholar]
- 61.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, and Alt FW. 1995. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3:521–530. [DOI] [PubMed] [Google Scholar]
- 62.Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, Clark PM, Healy B, Walker N, Aubin C, Oksenberg JR, Hauser SL, Compston A, Sawcer S, De Jager PL, Wicker LS, Todd JA, and Hafler DA. 2009. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet 5:e1000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Busse WW, Israel E, Nelson HS, Baker JW, Charous BL, Young DY, Vexler V, and Shames RS. 2008. Daclizumab improves asthma control in patients with moderate to severe persistent asthma: a randomized, controlled trial. Am J Respir Crit Care Med 178:1002–1008. [DOI] [PubMed] [Google Scholar]
- 64.Kitano H 2002. Systems biology: a brief overview. Science 295:1662–1664. [DOI] [PubMed] [Google Scholar]
- 65.Holt PG, Strickland DH, Wikstrom ME, and Jahnsen FL. 2008. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol 8:142–152. [DOI] [PubMed] [Google Scholar]
- 66.Hopkins AL 2008. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4:682–690. [DOI] [PubMed] [Google Scholar]
- 67.Kay A 2006. The role of T lymphocytes in asthma. Chem Immunol Allergy 91:59–75. [DOI] [PubMed] [Google Scholar]
- 68.Hamalainen HK, Tubman JC, Vikman S, Kyrola T, Ylikoski E, Warrington JA, and Lahesmaa R. 2001. Identification and validation of endogenous reference genes for expression profiling of T helper cell differentiation by quantitative real-time RT-PCR. Anal Biochem 299:63–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.