

Abstract
Unlike copy number variants (CNVs), inversions remain an underexplored genetic variation class. By integrating multiple genomic technologies, we discover 729 inversions in 41 human genomes. Approximately 85% of inversions <2 kbp form by twin-priming during L1 retrotransposition; 80% of the larger inversions are balanced and affect twice as many nucleotides as CNVs. Balanced inversions show an excess of common variants, and 72% are flanked by segmental duplications (SDs) or retrotransposons. Since flanking repeats promote non-allelic homologous recombination, we developed complementary approaches to identify recurrent inversion formation. We describe 40 recurrent inversions encompassing 0.6% of the genome, showing inversion rates up to 2.7×10−4 per locus per generation. Recurrent inversions exhibit a sex-chromosomal bias and co-localize with genomic disorder critical regions. We propose that inversion recurrence results in an elevated number of heterozygous carriers and structural SD diversity, which increases mutability in the population and predisposes specific haplotypes to disease-causing CNVs.
Graphical Abstract
In Brief:
Large scale analysis of haplotype-resolved inversions in human genomes unveils recurrent inversion polymorphisms and their disease relevance
Introduction
Large inversion polymorphisms play important roles in genome biology by suppressing recombination (Sturtevant, 1917) and causing disease when the events disrupt protein-coding genes or gene regulatory regions (Lakich et al., 1993; Lupiáñez et al., 2015; Puig et al., 2015). Such copy-neutral structural variants (SVs) have been challenging to discover and resolve (Abel et al., 2020; Collins et al., 2020; Ebert et al., 2021; Handsaker et al., 2015; Iafrate et al., 2004; Kidd et al., 2008; Korbel et al., 2007; Redon et al., 2006; Sebat et al., 2004; Sudmant et al., 2015) because they are often flanked by long segmental duplications (SDs) that exceed the length of sequencing reads or library inserts (Chaisson et al., 2019; Kidd et al., 2010; Sudmant et al., 2015; Vicente-Salvador et al., 2017). Investigations into select regions have shown an intimate relationship between inversions and disease-associated microdeletions and microduplications (i.e., morbid copy number variants [CNVs]) (Koolen et al., 2006; Osborne et al., 2001). Evolutionarily there is evidence that the orientation of such critical regions has changed multiple times between a direct and inverted state (Antonacci et al., 2014; Catacchio et al., 2018; Lozier et al., 2002; Maggiolini et al., 2019, 2020; Porubsky et al., 2020a; Zody et al., 2008). It is hypothesized that non-allelic homologous recombination (NAHR) between the flanking SDs increases the probability of recurrent inversions, a phenomenon we termed “inversion toggling” (Zody et al., 2008). Notably, the formation of complex SDs at the inversion flanks may make the same regions prone to recurrent morbid CNV formation as has been shown anecdotally for Williams-Beuren syndrome (WBS) (Osborne et al., 2001) and Koolen de Vries syndrome (KdVS) (Koolen et al., 2006). For other loci, the relationship has been less clear because of difficulties in inversion discovery, breakpoint definition, and haplotype ascertainment.
Here, we characterize the full spectrum of inversions ≥50 bp in size in a human diversity panel by integrating complementary genomic approaches: (1) single-cell template strand sequencing (Strand-seq) (Falconer et al., 2012); (2) haplotype-resolved de novo sequence assemblies generated from Pacific Biosciences (PacBio) high-fidelity (HiFi) and continuous long reads (Ebert et al., 2021); and (3) Bionano Genomics single-molecule optical mapping (Lam et al., 2012). We describe 40 recurrently inverted regions in humans and estimate inversion rates, ranging from 3.4×10−6 to 2.7×10−4 per locus per generation. Our analyses reveal a predominant role of SDs in inversion recurrence and provide insights into the formation of inversions associated with retrotransposition. We discover inversions mapping to the locations of well-known genomic disorders and establish a link between inversion toggling in humans and recurrent morbid CNVs.
Results
The human inversion landscape
Haplotype-resolved inversion discovery.
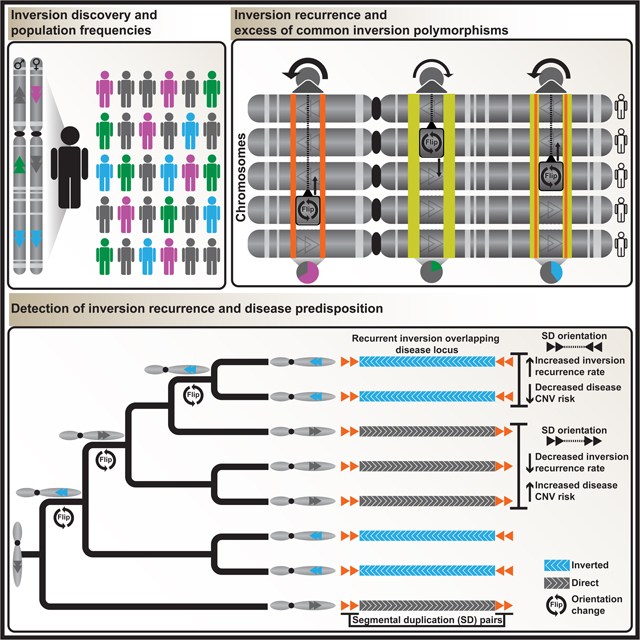
We generated our integrated callset using 41 unrelated human samples, representing 729 inversion sites after filtering (STAR Methods, Tables S1 and S2) consisting of: (i) 330 inversions internal to L1 mobile element insertion polymorphisms (discussed separately below); (ii) 292 balanced inversions; (iii) 40 inverted duplications; (iv) 29 structurally complex sites; and (v) 38 likely assembly errors in GRCh38 or rare minor alleles (Figure 1A). We devised a method for combining Strand-seq and long reads to place the inversions into full-length chromosomal haplotypes (STAR Methods) and find an average of 11.6 Mbp to be inverted, corresponding to ~0.39% (African: 0.43%, non-African: 0.34%) of a haploid genome (Figure 1B, Data S1). This is four times the number of base pairs affected by single-nucleotide polymorphisms (SNPs) (1000 Genomes Project Consortium et al., 2015) and twice the number of base pairs affected by deletion and insertion SVs seen in phased assemblies (Ebert et al., 2021). Large (>100 kbp) balanced inversions are most abundant on chromosomes 1, 2, 7, 10, 15, 16, and 17, in association with SDs (Maggiolini et al., 2020; Marques-Bonet et al., 2009; Porubsky et al., 2020a) (Figures 1C and S1AB). Strand-seq yields the largest amount of inverted base pairs, in line with its ability to discover inversions in the genome regardless of the length of the flanking repeats (Chaisson et al., 2019; Sanders et al., 2016), whereas the long-read data increases the sensitivity for events smaller than 100 kbp. Bionano technology is least sensitive but provides orthogonal support (Figure 1D, Data S1, STAR Methods).
Figure 1. Inversion discovery in a diversity panel.
A) Breakdown of inversion (Inv) classes (see Fig. 2 for L1-internal events). InvDup, inverted duplication; miso, (likely) misoriented; complex/lowconf, lower-confidence call. B) Affected bp per variant class and population. Del, deletion; Ins, insertion. C) Balanced inversion landscape (n = 292). D) Inversion discovery (n = 399 sites) by technology with affected bp (pie chart). PAV, phased assembly variant caller. E) Pericentromeric inversion on chromosome 2. Strand-seq read counts in 50 kbp bins (step size: 10 kbp) are represented as bars above (teal; Crick reads) and below (orange; Watson) the midline. SDs and morbid CNVs are annotated. Arrowhead plot reports inversions (H1, haplotype 1; H2, haplotype 2) in NA19650 and nonhuman primates. FISH probe positions shown (bottom). CEN, centromere. F) FISH confirms inversion (red) compared to control (white).
Inversion validation.
We used different methods to validate the 399 inversions outside of L1 sequences. Using three additional samples (STAR Methods), we examined events for their segregation in parent-child trios (n = 3 trios). We find Mendelian consistency for 247/260 (95.0%) inversion sites seen in the children, which increases to 99.5% (200/201) for high-confidence genotypes (genotype-likelihood ratio over reference state > 103; Table S3). We subjected 10 randomly selected, sequence-resolved balanced inversions (0.5 kbp–366 kbp) to PCR, successfully validating both breakpoints for 9/10 and one inversion breakpoint for the tenth event (Figures S2A,B). Using Oxford Nanopore Technologies (ONT) long-read data for three samples, we validated 107/202 (~53%) sites (STAR Methods). Finally, comparing to other studies (Audano et al., 2019; Chaisson et al., 2019; Giner-Delgado et al., 2019; Puig et al., 2020; Sanders et al., 2016; Sudmant et al., 2015), we find that 36.3% (145/399) of inversions have been reported previously (Figure S2C, STAR Methods). Overall, 64.7% (258/399) of these inversions, including 73.6% (215/292) of the balanced inversions, are supported by at least one orthogonal method (Figure S2D, Table S3).
Putatively novel polymorphic inversions.
Our integrated callset contains 100 previously unreported inversions based on the aforementioned reports. These inversions span ~39 Mbp, and five are >1 Mbp in size, including a ~23.2 Mbp pericentromeric inversion on chromosome 2 originating from a Mexican donor (NA19650) (Figure 1E, Table S4). We used SNP genotypes generated by PanGenie (Ebler et al., 2022) to infer the presence of this inversion in 1000 Genomes Project (1KG) samples (n = 3,202) (Byrska-Bishop et al., 2021) by looking for shared rare SNPs (Start Methods). This analysis identified the mother (NA19648) of the index donor as the only additional candidate carrier for this inversion, supporting its meiotic segregation (Data S1). We performed FISH in these two suspected inversion carriers, validating both (Figure 1F, STAR Methods). We also searched for carriers of a large (5 Mbp) 15q11-13 inversion, which revealed four likely carriers in the 1KG cohort, all of which we validated by FISH (see below). These data show that phased inversions from our callset facilitate the identification of potential carriers in whole-genome sequence (WGS) data.
Mechanisms for inversion formation
Dominant role of NAHR in balanced inversion formation.
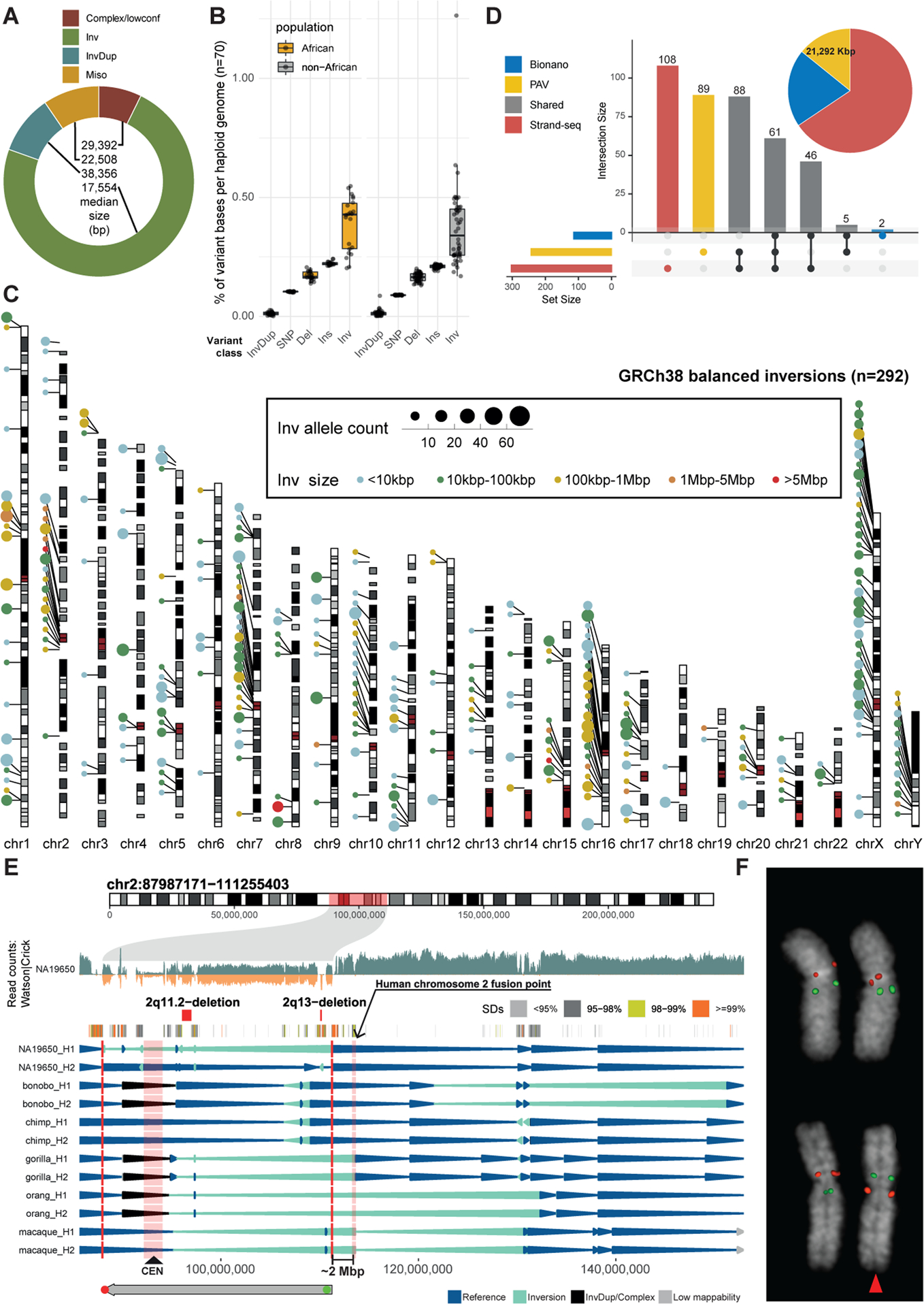
The phased assemblies fully traverse the majority of balanced inversions including their breakpoints (183/292, 63%), providing an opportunity to study mutational mechanisms. Most sequence-resolved balanced inversions (132/183; 72%) show flanking inverted repeats of at least 200 bp in length (Figures 2A and S1C), consistent with NAHR (Bailey and Eichler, 2006). This fraction is in line with prior results based on fosmid sequencing (69%) (Kidd et al., 2010) but surpasses estimates for large (≥50bp) insertions and deletions (Ebert et al., 2021) based on phased assemblies (15–25%). Out of the 132 NAHR candidate inversions, 101 (77%) showed flanking inverted SDs, whereas the remainder (23%, 31) exhibited flanking mobile element sequences (L1: n = 22, and Alu: n = 9) (Song et al., 2018). Most (21/22, 95%) inversion-flanking L1 pairs display >90% pairwise sequence identity (median: 97.2%), in sharp contrast to Alu pairs, where this is the case for only 1/9 (11%). Additionally, six out of nine Alu/Alu-flanked inversions show nearby sequence gains or losses of 35–701 bp in size (Data S1, Table S4). This suggests that Alu-flanked inversions may form through a different rearrangement process, as described for Alu-mediated deletions (Morales et al., 2015). There is a genome-wide significant correlation between the size of an inversion and the length of the flanking repeat (Pearson’s correlation: R = 0.67, p < 3×10−16) (Figures 2B and S1D). SD-mediated inversions invert more genes than other types of balanced inversions (Figure 2C) likely due to their size and the fact that mobile element insertions are biased against genes (Graham and Boissinot, 2006).
Figure 2. Inversion formation mechanisms.
A) Representation of inversions and their flanks for events <10 kbp (all sequence-resolved events are in Figure S1C). B) Size distribution for event types from (A). Unresolved: not assembled. C) Functional annotation of events. D) Depiction of twin-priming. (1) Cleavage of the first DNA strand by the L1-encoded endonuclease; (2) annealing of the L1 RNA poly(A) and initiation of reverse transcription (RT) at the free 3′OH; (3) after second strand cleavage, the derived single-stranded overhang at the 5′ TSD anneals internally to the L1 transcript, generating Junction 1 (Jct1); (4) the inverted and non-inverted cDNA products are annealed, generating Junction 2 (Jct2); both junctions are repaired by MMEJ; (5) retrotransposition finalizes with second strand synthesis and ligation. E) Size distribution for L1-associated events. IQR, interquartile range. F) Top, inversion and truncation breakpoint (BKP) density, using kernel density estimation (KDE). Bottom, likelihood of each L1 integration outcome while L1 RT progresses towards the 5′ end of L1 mRNA sequence. G) Left, fraction of full-length, 5′ deleted and inverted L1 inserts exhibiting microhomology, nucleotide insertions, and blunt joints between the 3′ end of the TSD and the 5′ end of the integrated L1. Right, size distribution (bp) for microhomologies and insertions. H) Inversion junction conformations with duplicated (Dup) and deleted (Del) pieces of L1 sequence and blunt joins.
Of the sequence-resolved balanced inversions, 28% (51/183) lack inverted repeats at their breakpoints and 23 of these are accompanied by adjacent >50 bp sized deletions or insertions, or partake in more complex SVs (Figures 2B and S1C). This complexity likely arose from a mutational process—possibly involving alternative nonhomologous end-joining, microhomology-mediated end joining (MMEJ), or microhomology-mediated break-induced replication (MMBIR) (Carvalho and Lupski, 2016; Sudmant et al., 2015)—rather than from accumulated SVs, as we do not detect corresponding intermediate events. Collectively, our data suggest NAHR as the predominant mechanism for balanced inversion formation, with a smaller fraction likely resulting from error-prone DNA repair processes.
Analysis of inversions within L1 insertions.
L1 insertions can contain inverted segments generated during retrotransposition (Ostertag and Kazazian, 2001). We therefore analyzed 93% (1,271/1,362) of the polymorphic L1 elements seen in the phased assemblies (Ebert et al., 2021), to identify and characterize compound L1 structures containing 5′ inversions (STAR Methods). These L1s are likely a result of twin-priming (Ostertag and Kazazian, 2001), an alternative mechanism for L1 integration (Figure 2D). Briefly, during twin-priming, the single-stranded 3′ end of the target site duplication (TSD) sequence (Kazazian and Moran, 1998) anneals within the L1 mRNA forming Junction 1 (denoted as Jct1, Figure 2D), priming a secondary reverse transcription reaction, which leads to the synthesis and ligation of two cDNA products in opposite orientations, generating Junction 2 (Jct2). We found that 26% (330/1,271) of the analyzed polymorphic L1s show characteristic 5′ inverted sequences, whereas the remaining are either full-length (405) or 5′ truncated (536) (Figure 2E, Table S2). The inverted segments were both 1.7 times shorter and less variable in size with respect to a random distribution of possible inversion lengths (p = 1.9×10−15; Mann-Whitney U test; Figure S3A, Data S1). The position of Jct2 is clustered towards the 3′ end of the L1, with 88% (292/330) of breakpoints occurring between base-pairs 4,000 and 6,000 (Fig. 2F). L1 5′ truncation events have a similar pattern, and there is no significant difference in the length distribution between 5′ truncated L1s and the 3′ sense orientation ends of twin-priming events (p = 0.07, Mann-Whitney U; Figure S3B–D). This suggests that the first 2 kbp are critical for successful completion of full-length L1 reverse transcription, as 73% (405/552) of L1s longer than 2 kbp are full-length.
Next, we analyzed Jct1 from 269/330 non-reference polymorphic twin-priming events, finding short insertions (n = 16; median 8 bp) and microhomologies of 1–9 bp (n = 223; median 3 bp) in 239/269 of them (Figure 2G). These data suggest that annealing precedes DNA repair of Jct1 (i.e., MMEJ) (Chandramouly et al., 2021; Kojima, 2010; Ostertag and Kazazian, 2001; Zingler et al., 2005). We also observe appreciable signatures of MMEJ at the 5′ end of truncated L1 inserts (Kojima, 2010; Zingler et al., 2005) and infrequent microhomology at this junction for full-length L1s (Figure 2G), supporting different mechanisms for integration of full-length L1s (Yamaguchi et al., 2014). We then analyzed 273/330 internal inversions of polymorphic twin-priming events and found additional sequence complexities, with frequent short deletions (61%; 166/273) and duplications (33%; 89/273) of L1 sequence at Jct2 (Figure 2H). We detect microhomology (n = 190; median 2 bp) and short insertions (n = 27; median 3 bp) at Jct 2 for 81% (217/269) of the twin-priming L1s (Figure S3E–H). Three templated insertions were adjacent to Jct2 or the TSD (Figure S3I–K), indicating that both the L1 cDNA and the flanking genomic sequence can occasionally be used as substrates for template switching during retrotransposition. Collectively, these sequence features are consistent with previous data (Kojima, 2010; Ostertag and Kazazian, 2001) and suggest a major role for MMEJ in the resolution of retrotransposition intermediates (see models in Figure 2D and Data S1), resulting in internal inversions or truncations of L1 sequences.
Inversion recurrence unveils a sex chromosomal bias in mutational toggling
Inversion discovery saturation and excess of common polymorphisms.
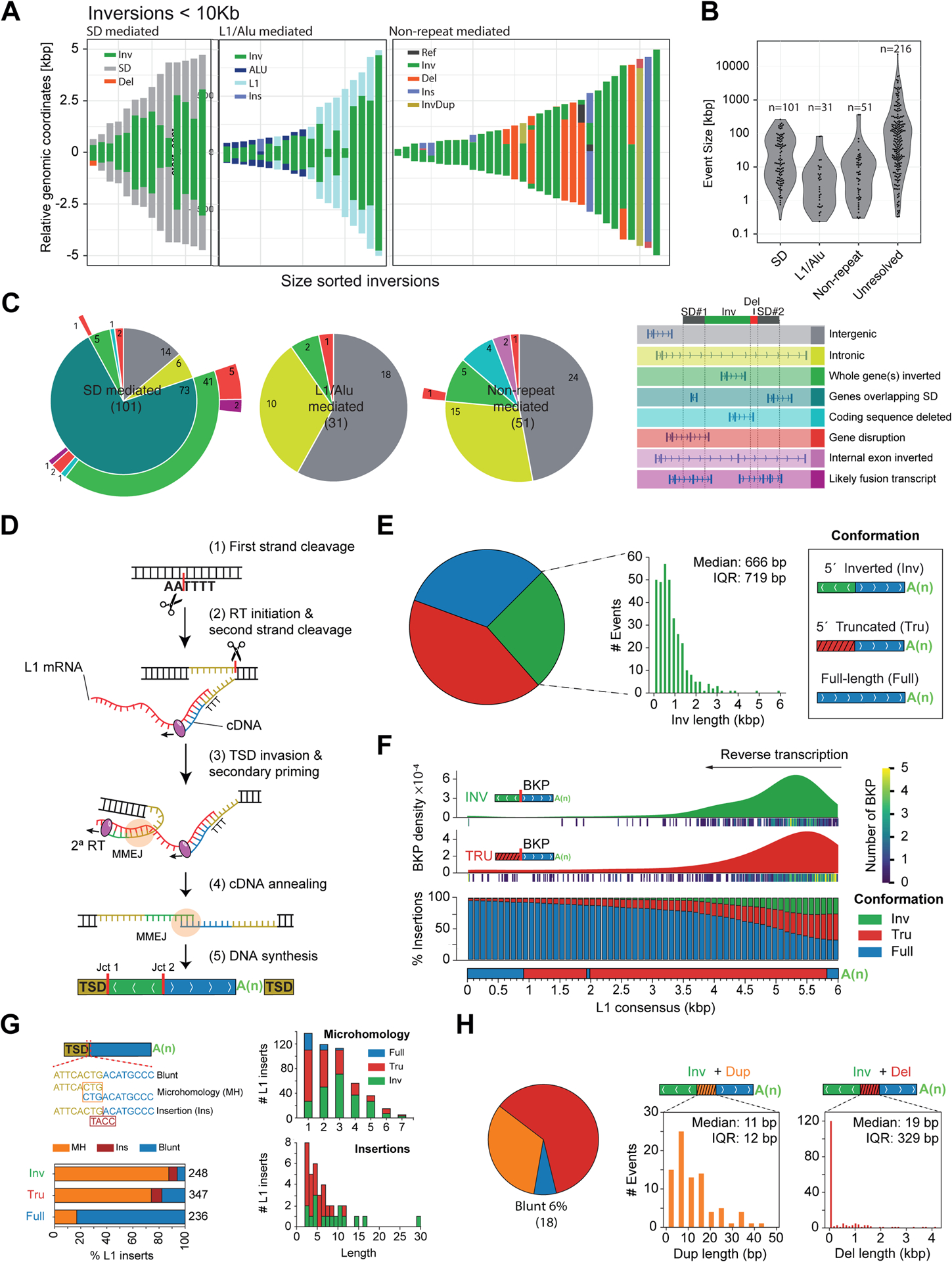
Focusing on the set of balanced inversions (n = 292), we estimated the rate of inversion discovery with each additional genome added. Remarkably, the inversion discovery rate quickly saturates with more genomes added, an effect seen in both non-African and African populations (Figure 3A), despite the fact that African populations exhibit greater genetic diversity (1000 Genomes Project Consortium et al., 2015). This represents a significant ~2.4-fold reduction in the rate of new variant discovery compared to insertion and deletion SVs (Ebert et al., 2021) (p = 1×10−24; two-sided t-test; see orange line in Figure 3A). Concomitantly, we also observe an excess of common (minor allele frequency [MAF]>5%) inversion alleles (67%) when compared to other SV classes (48%, p = 2.6×10−11, two-tailed Fisher’s exact test; STAR Methods). These observations suggest that sequencing more genomes will likely add only few more balanced polymorphic inversions without further technological advances to increase detection sensitivity in the most complex areas of the genome.
Figure 3. Recurrence of balanced inversions in the human genome.
A) Rate of balanced inversions discovered with each added genome differs from SV insertions and deletions (orange lines, right axis). Dotted lines fit logarithmic model growth. Singleton: 1 allele; polymorphic: AF < 50%; major: AF ≥ 50% (but less than 100%), putative misorient: AF = 100%. B) Inversion recurrence detection: (i) tiSNPs based, (ii) Haplotype based approach. Venn diagram depicts overlap by approach for 127 tested inversions. C–E) Evidence for single (C, 17q21) and recurrent (D, 8p23.1 [distal part chr8:8225000-8301024]; E, 11p11) loci. Left: dendrograms (centroid hierarchical clustering method) show relationships among inverted and direct-oriented haplotypes. Ancestral (blue) vs. derived (orange) SNPs, informative tiSNPs (black) and SNPs with ≥75% mappability (purple) are shown. Middle: haplotype-based principal component (PC) analysis. Right: inferred cladograms of the loci of interest. Blue dots, putative inversion events.
Methods for characterizing mutational toggling of inversions.
We hypothesized that the excess of common balanced inversions may be due to recurrent mutations in humans (Aguado et al., 2014; Zody et al., 2008), mediated through NAHR between inverted repeats. To test this hypothesis, we devised two complementary methods to infer inversion toggling (Figure 3B, STAR Methods). We first developed a toggling-indicating SNP (tiSNP) based approach to identify SNPs discrepant with a single inversion origin, based on haplotype-resolved Strand-seq reads. Signals of tiSNPs were aggregated for each inversion to find support for inversion toggling. In addition, we developed a haplotype-based approach to infer toggling based on the fully integrated set of phased genetic variants (generated by integrating Strand-seq and PacBio data). We apply coalescent-based methods to the phased SNPs to find evidence in support of inversion recurrence. The two approaches are complementary as the first evaluates each SNP independently, being largely unaffected by recombination, while the second leverages linkage and variation patterns to provide estimates on the number of recurrent inversion events as well as inversion rates per generation.
Inferred inversion recurrence and inversion rates.
We tested and applied both methods on two previously studied inversions as controls. As a negative control, we tested the well-known 706 kbp 17q21.31 inversion (allele frequency [AF] = 11%; Figure 3C) that was hypothesized to have formed once in the last 2.3 million years (Koolen et al., 2006; Stefansson et al., 2005; Zody et al., 2008). As a positive control, we compared the results to the 5.3 Mbp 8p23.1 inversion (AF = 50%; Figure 3D) thought to be subject to limited recurrence (Mohajeri et al., 2016; Salm et al., 2012). Using the first method we find 0% (0/3,834) and 9.2% (1,366/14,801) tiSNPs for the 17q21.31 and 8p23.1 inversion polymorphisms, respectively (Table 1). The tiSNPs are seen across the whole length of the 8p23.1 inversion (Figure 3D, Figure S4A). In agreement with these findings, the haplotype-based approach demonstrates clear evidence for multiple recurrences of the 8p23.1 inversion at several levels, in stark contrast to the single origin of the 17q21.31 inversion. Our haplotype-based principal component and hierarchical clustering-based tree analyses show that while all inverted haplotypes at 17q21.31 form a cluster distinct from the directly oriented haplotypes, the 8p23.1 locus exhibits inverted and directly oriented haplotypes in the same clusters (Figure 3C–D, Figure S4A–B). In addition, we observe a wide distribution of identity by state among haplotypes at 8p23.1, in contrast to the distinct identity by state clusters seen for 17q21.31 haplotypes (Figure S4C–D). Together, these analyses confirm that the 8p23.1 inversion arose independently on different genetic backgrounds, in contrast to a single origin of 17q2.31 inverted haplotypes.
Table 1.
Recurrent inversions in the human genome.
| Locus | Position | Size (kbp) | AF | FIR size (kbp) | FIR identity | Morbid CNVs | tiSNPs | Recurrent events [.95 CI] | Inversion rate (x10−4) [.95 CI] | Evidence for recurrence |
|---|---|---|---|---|---|---|---|---|---|---|
| 1p36.21 | chr1:13104252-13122521 | 18.30 | 0.69 | 60.00 | 95% | - | 4 (2.47%) | 13 [7.00, 13.75] | 1.02 [0.272, 1.21] | tiSNPs & Hb |
| 10q11.22 | chr10:46983451-47468232 | 484.80 | 0.09 | 0.42 | 61% | - | 41 (4.64%) | 7 [5.00, 7.00] | 0.59 [0.326, 0.799] | tiSNPs & Hb |
| 11p11.12 | chr11:50154999-50324102 | 169.10 | 0.40 | 41.72 | 95% | - | 54 (13.88%) | 8 [6.15, 9.00] | 0.4 [0.328, 0.571] | tiSNPs & Hb |
| 15q13.2-15q13.3 | chr15:30618103-32153204 | 1,535.10 | 0.11 | 0.34 | 74% | 15q11.2, 15q13.3, 15q26 | 6 (0.17%) | 4 [2.00, 7.00] | 0.278 [0.0895, 0.6] | tiSNPs & Hb |
| 15q25.2 | chr15:84373375-84416696 | 43.30 | 0.56 | 34.22 | 99% | 15q26 | 5 (3.45%) | 9 [5.30, 10.00] | 0.529 [0.301, 0.693] | tiSNPs & Hb |
| 16p12.3 | chr16:16721273-18073542 | 1,352.30 | 0.08 | 0.37 | 66% | ATR-16 | 5 (0.13%) | 4 [3.00, 5.00] | 0.287 [0.15, 0.484] | tiSNPs & Hb |
| 16p12.1-16p11.2 | chr16:28471892-28637651 | 165.80 | 0.36 | 23.53 | 98% | ATR-16, 16p11.2-p12.2 | 4 (1.19%) | 6 [3.27, 6.00] | 0.484 [0.264, 0.661] | tiSNPs & Hb |
| 2p11.1 | chr2:91832040-92012663 | 180.60 | 0.41 | 48.31 | 99% | - | 10 (6.8%) | 19 [10.62, 19.38] | 1.41 [0.931, 1.85] | tiSNPs & Hb |
| 2q11.1-2q11.2 | chr2:95800191-96024403 | 224.20 | 0.08 | 49.02 | 96% | 2q11.2-deletion | 3 (1.59%) | 4 [2.38, 5.00] | 0.408 [0.234, 0.681] | tiSNPs & Hb |
| 3q29 | chr3:195749463-195980207 | 230.70 | 0.26 | 0.36 | 73% | 3p25.3, 3q29 | 34 (4.22%) | 5 [3.00, 9.00] | 0.422 [0.229, 0.837] | tiSNPs & Hb |
| 7p22.1 | chr7:5989046-6735643 | 746.60 | 0.10 | 60.04 | 98% | - | 33 (1.75%) | 7 [6.00, 8.00] | 0.506 [0.314, 0.815] | tiSNPs & Hb |
| 7q11.1 | chr7:60911891-61578023 | 666.10 | 0.52 | 33.66 | 99% | - | 100 (13.77%) | 16 [14.10, 20.00] | 0.654 [0.49, 0.869] | tiSNPs & Hb |
| 7q11.21 | chr7:65219157-65531823 | 312.70 | 0.33 | 15.02 | 97% | - | 1 (0.13%) | 5 [3.00, 8.00] | 0.318 [0.167, 0.663] | tiSNPs & Hb |
| 7q11.23 | chr7:73113989-74799029 | 1,685.00 | 0.05 | 0.75 | 80% | WBS | 19 (0.93%) | 3 [2.00, 4.00] | 0.262 [0.136, 0.433] | tiSNPs & Hb |
| 7q11.23 | chr7:74869950-75058098 | 188.10 | 0.10 | 43.32 | 95% | - | 1 (0.53%) | 6 [1.90, 6.00] | 0.57 [0.126, 0.779] | tiSNPs & Hb |
| 8p23.2 | chr8:2343351-2378385 | 35.00 | 0.51 | 55.88 | 99% | 8p23.1 | 32 (12.36%) | 17 [3.40, 17.00] | 1.13 [0.33, 1.53] | tiSNPs & Hb |
| 8p23.1 | chr8:7301024-12598379 | 5,297.40 | 0.50 | 1.04 | 86% | 8p23.1 | 1366 (9.23%) | 15 [4.75, 17.00] | 1.11 [0.228, 1.6] | tiSNPs & Hb |
| 1p13.3 | chr1:108310642-108383736 | 73.10 | 0.57 | 60.01 | 99% | 1p36 | 3 (1.44%) | 5 [5.02, 5.97] | 0.184 [0.184, 0.194] | tiSNPs & Hb |
| 11q14.3 | chr11:89920623-89923848 | 3.20 | 0.53 | 48.70 | 99% | - | 3 (25%) | 5 [5.05, 6.95] | 0.336 [0.338, 0.411] | tiSNPs & Hb |
| 16p13.11 | chr16:14954790-15100859 | 146.10 | 0.77 | 33.43 | 79% | ATR-16, 16p13.11 | 5 (2.23%) | 3 [3.00, 8.00] | 0.264 [0.191, 0.832] | tiSNPs & Hb |
| 7q11.21 | chr7:62290674-62363143 | 72.50 | 0.42 | 19.58 | 96% | - | 12 (5.08%) | 10 [5.50, 10.90] | 0.892 [0.598, 0.896] | tiSNPs & Hb |
| 7q11.21 | chr7:62408486-62456444 | 48.00 | 0.57 | 2.90 | 71% | - | 12 (5.91%) | 18 [9.12, 19.00] | 0.942 [0.458, 1.24] | tiSNPs & Hb |
| Xq22.2 | chrX:103989434-104049428 | 60.00 | 0.63 | 49.52 | 94% | - | 2 (2.67%) | 5 [2.22, 5.00] | 0.58 [0.308, 0.651] | tiSNPs & Hb |
| Xq28 | chrX:149599490-149655967 | 56.50 | 0.08 | 0.12 | 62% | - | 3 (5.17%) | 3 [2.00, 3.00] | 0.351 [0.234, 0.47] | tiSNPs & Hb |
| Xq28 | chrX:149681035-149722249 | 41.20 | 0.61 | 28.37 | 98% | - | 7 (15.56%) | 9 [7.25, 9.88] | 0.85 [0.78, 1.21] | tiSNPs & Hb |
| Xq28 | chrX:153149748-153250226 | 100.50 | 0.60 | 42.88 | 99% | - | 46 (20%) | 6 [6.00, 6.00] | 0.573 [0.401, 0.624] | tiSNPs & Hb |
| Xq28 | chrX:154347246-154384867 | 37.60 | 0.44 | 11.39 | 98% | Xq28 | 1 (2.13%) | 4 [4.00, 4.92] | 0.613 [0.542, 0.936] | tiSNPs & Hb |
| Xq28 | chrX:154591327-154613096 | 21.80 | 0.43 | 35.74 | 99% | Xq28 | 1 (5.56%) | 3 [3.00, 5.85] | 0.495 [0.475, 0.785] | tiSNPs & Hb |
| Xq28 | chrX:155386727-155453982 | 67.30 | 0.15 | 50.58 | 98% | - | 1 (1.25%) | 5 [5.00, 5.00] | 0.577 [0.447, 0.659] | tiSNPs & Hb |
| Xp11.22 | chrX:52077120-52176974 | 99.90 | 0.36 | 36.41 | 99% | SHOX, Xp11.22-p11.23 | 14 (4.71%) | 6 [5.40, 11.00] | 0.542 [0.233, 1.03] | tiSNPs & Hb |
| Xq13.1-Xq13.2 | chrX:72997772-73077479 | 79.70 | 0.20 | 9.60 | 98% | SHOX, STS | 16 (13.11%) | 6 [2.60, 6.00] | 0.548 [0.288, 0.598] | tiSNPs & Hb |
| Xq28 | chrX:152729753-152738707 | 9.00 | 0.40 | 51.16 | 98% | - | 5 (12.5%) | 5 [5.05, 6.95] | 0.559 [0.352, 0.553] | tiSNPs & Hb |
| Yp11.2 | chrY:6452942-9763793 | 3,310.90 | 0.10 | 6.81 | 66% | - | NA | 2 | 1.07 [0.95, 1.22] | Y phylogeny |
| Yp11.2 | chrY:9797298-9817138 | 19.80 | 0.43 | 1.90 | 71% | - | NA | 2 | 1.07 [0.95, 1.22] | Y phylogeny |
| Yq11.221 | chrY:14019657-14023071 | 3.40 | 0.67 | 45.63 | 94% | - | NA | 4 | 2.15 [1.89, 2.43] | Y phylogeny |
| Yq11.221 | chrY:16269646-16315779 | 46.10 | 0.37 | 59.92 | 99% | - | NA | 5 | 2.68 [2.37, 3.04] | Y phylogeny |
| Yq11.222 | chrY:17949447-17956300 | 6.90 | 0.70 | 69.77 | 96% | AZFb+AZFc | NA | 5 | 2.68 [2.37, 3.04] | Y phylogeny |
| Yq11.222 | chrY:18640355-18667145 | 26.80 | 0.13 | 46.67 | 98% | AZFb+AZFc | NA | 2 | 1.07 [0.95, 1.22] | Y phylogeny |
| Yq11.223 | chrY:21021692-21063744 | 42.10 | 0.60 | 1.09 | 63% | AZFb+AZFc | NA | 4 | 2.15 [1.89, 2.43] | Y phylogeny |
| Yq11.223 | chrY:22204071-22384088 | 180.00 | 0.17 | 64.67 | 99% | AZFb+AZFc | NA | 5 | 2.68 [2.37, 3.04] | Y phylogeny |
FIR, flanking inverted repeat, Hb, haplotype-based approach, CI, central interval (confidence intervals are given for rate estimates on the Y chromosome).
To reconstruct the evolutionary history of these balanced inversions, we inferred the underlying genealogical relationship among haplotypes (STAR Methods). Because of the massive size of the 8p23.1 inversion, we focused our analysis on a 100 kbp region located at the distal portion of this locus (Figure 3D). Our analysis shows bootstrap support for both global and marginal trees (Figure S4E), suggesting that the underlying genealogical relationship among haplotypes is well recapitulated. Given the inferred tree, our method parsimoniously infers 15 independent inversions occurring at the 8p23.1 locus in humans (95% central interval: 4.75–17; STAR Methods) and estimates 1.11×10−4 inversions per generation (95% central interval: 2.28×10−5 – 1.60×10−4). This is in contrast to the 17q21.31 inversion where our method predicts a single event (Figure 3C), with an inversion rate of 3.47×10−6 (95% central interval: 2.71×10−6–1.03×10−5). Thus, inversion rates can vary by as much as two orders of magnitude.
Rates and genetic architecture of inversion toggling on autosomes and X chromosome.
To understand the extent of inversion toggling in humans, we applied both approaches to a subset of 127 balanced inversion sites across the autosomes and chromosome X that passed a series of QC filters (STAR Methods). We find that 52% (66/127) of inversions show evidence for inversion recurrence by at least one of the two approaches (Figure 3B, Table S5), suggesting extensive inversion toggling in humans. Among a “consensus” set of 93 inversions where both approaches agree, we find 32 consensus recurrent (34% [32/93] toggling inversions, Table 1) and 61 consensus single-event inversions. Among the consensus inversions, we estimate inversion rates ranging between 3.4×10−6 and 1.4×10−4 (median: 1.2×10−5). Notably, analysis of the chromosomal origin of these inversions shows a significant excess of recurrent inversions on the X chromosome compared to the autosomes (odds ratio: 27.2, 95% C.I.: [2.55, 142.4]; p = 1.2×10−4, chi-squared test), suggesting X-biased recurrence of inversions.
Among the 32 consensus recurrent inversions, we find that six overlap a set of 23 inversions previously suggested to be recurrent in the great apes (Porubsky et al., 2020a). Additionally, we detect a 38 kbp recurrent inversion on the X chromosome (AF = 44%; Table 1, Fig. S4F–G) encompassing the genes FLNA and EMD (Small et al., 1997), which was previously demonstrated as recurrently inverted over the evolution of eutherian mammals (Cáceres et al., 2007). We predict four independent inversion events (95% central interval: 4.0 − 4.92) in the past 200,000 years of human evolution, with an inversion rate of 6.13×10−5 (95% central interval: 5.42×10−5 – 9.36×10−5). We additionally analyzed a recurrent inversion at chromosome 11p11 in more detail. We identify 54 tiSNPs (14% of 389 detected SNPs contained in the inversion), which are distributed across the inverted region (Figure 3E). Our haplotype-based approach shows that eight independent inversion events occurred at 11p11 (95% central interval: 6.15 − 9), with an estimated inversion rate of 4.0×10−5 (95% central interval: 3.28×10−5 – 5.71×10−5).
From a mechanistic perspective, we find that both the length of the flanking inverted repeat (Pearson’s correlation: 0.51; p = 1.7×10−7) and its sequence identity (Pearson’s correlation: 0.39; p = 1.3×10−4) positively correlate with inversion recurrence (Figure S4H). A multivariate logistic regression analysis shows that the major driver for the inversion status is flanking inverted repeat length (p = 7.2×10−3). Furthermore, the majority (72%, 23/32) of recurrent inversions on the autosomes and X chromosome exhibited ≥10 kbp long flanking inverted SDs with high (≥79%) sequence identity (Table 1). Combined, these analyses strongly implicate NAHR as the primary driver for inversion recurrence, helping to explain the intimate association of high MAF, recurrent inversions, and flanking SDs.
Inversion toggling affects 6% of Y chromosome.
The lack of meiotic recombination outside of pseudoautosomal regions of the Y chromosome has the benefit of unambiguous phylogeny, which facilitates recurrence analyses (STAR Methods). The 16 male samples in our study carry 15 inversions on the Y chromosome (sizes: 3.4 kbp–3.3 Mbp; median: 26.7 kbp), 8 of which were previously reported (Hallast et al., 2013; Lange et al., 2009; Repping et al., 2002, 2006; Shi et al., 2019a, 2019b). The majority (13/15; 87%) are flanked by SDs and invert 10 protein-coding genes and 14 transcribed pseudogenes (Figure 4A, Table S3). Out of 11 balanced inversions passing genotype quality filters, we classified 8 as recurrent, displaying two up to five occurrences in the Y phylogenetic tree (Table 1, STAR Methods). These recurrent inversions span ~3.6 Mbp, which corresponds to ~6% of the Y chromosomal sequence, and we estimate inversion rates ranging from 1.07 × 10−4 (95% C.I.: 0.95 × 10−4 to 1.22 × 10−4) to 2.68 × 10-4 (95% C.I.: 2.37 × 10−4 to 3.04 × 10−4) per father-to-son Y transmission (Table S3). These rates correspond to one recurrent inversion per 642 (95% C.I.: 567 – 728) father-to-son Y transmissions. The relative proportion of toggling inversions compared to single-event inversions on the Y chromosome is ~7-fold higher when compared to the autosomes (p = 0.0066, chi-squared test; Figure 4B), consistent with a sex chromosomal bias for inversion recurrence.
Figure 4. Recurrence on chromosome Y.
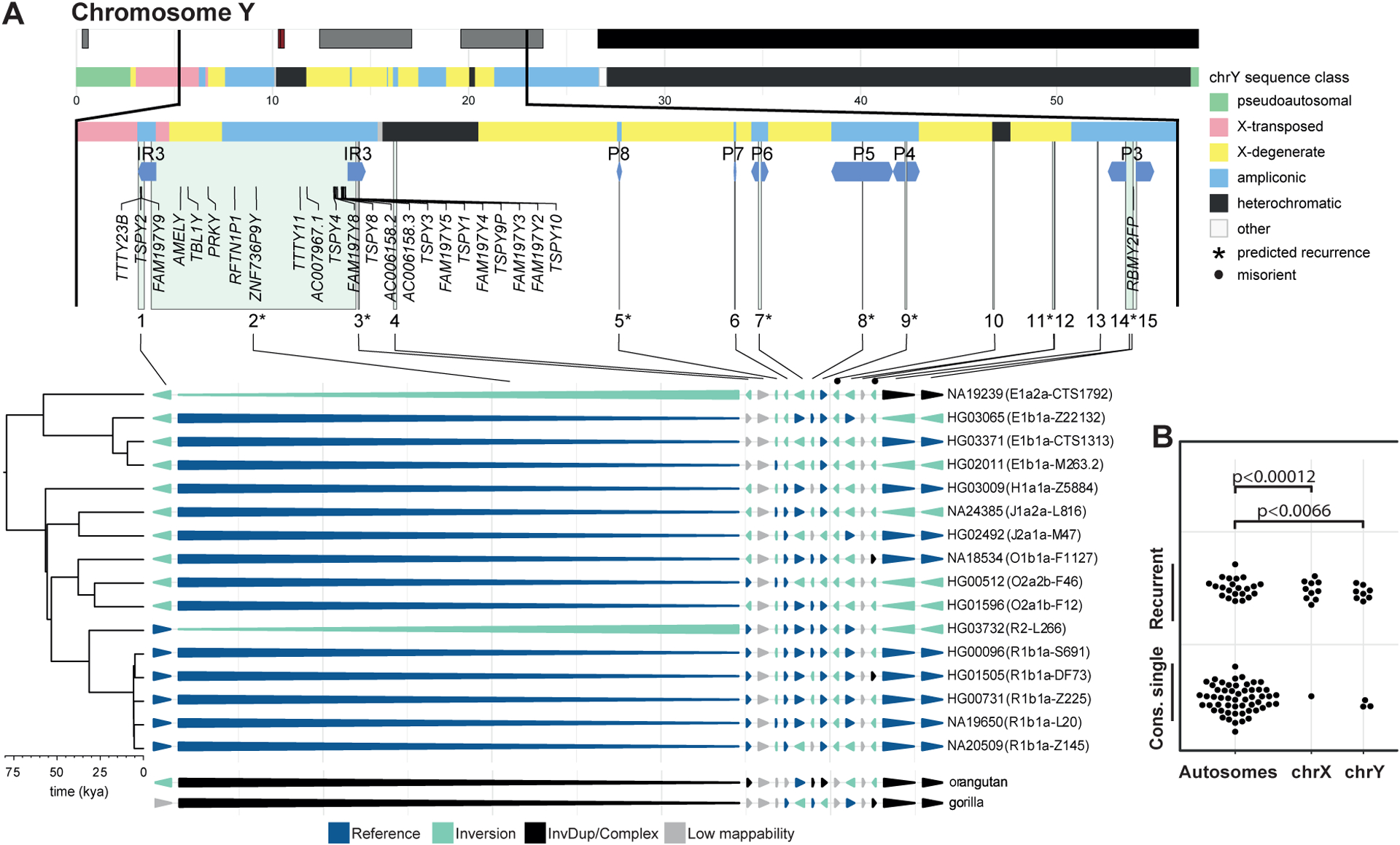
A) Annotated chromosome Y (top) and sites of inversion (enumerated 1–15) projected onto haplotypes. Phylogeny (left) with estimated divergence times (kya, 1000 years ago). B) Sex chromosome enrichment of recurrent inversions (cons. single, consensus single-event).
Relationship of polymorphic inversions with morbid CNV regions
Recurrent inversions are hotspots for morbid CNV formation.
More than 30 genomic regions have been identified where recurrent microdeletions and microduplications have been associated with pediatric developmental delay and neuropsychiatric disorders (Bragin et al., 2014; Coe et al., 2014; Cooper et al., 2011; Lupski, 1998). We tested whether inversion polymorphisms are associated with such known morbid CNVs (Antonacci et al., 2014; Koolen et al., 2006; Osborne et al., 2001), using genome-wide permutation analysis (STAR Methods). We find a significant co-localization between morbid CNVs and balanced inversions in our callset (14%, 40/292, p = 0.0029, twofold excess; Figure 5A). In addition to WBS and KdVS, this includes several well-known genomic disorders, such as Prader-Willi/Angelman syndrome (PWAS), Smith-Magenis/Potocki-Lupski syndrome (SMPLS), as well as the 15q13 and 16p11.2 regions associated with autism. Remarkably, most of the association is driven by recurrent inversions, for which the enrichment is fivefold (31%, 10/32, p = 0.0001, Figure 5A). This suggests a relationship between the mutational toggling of inversions in humans and recurrent CNVs associated with disease.
Figure 5. Association of toggling inversions with morbid CNVs.
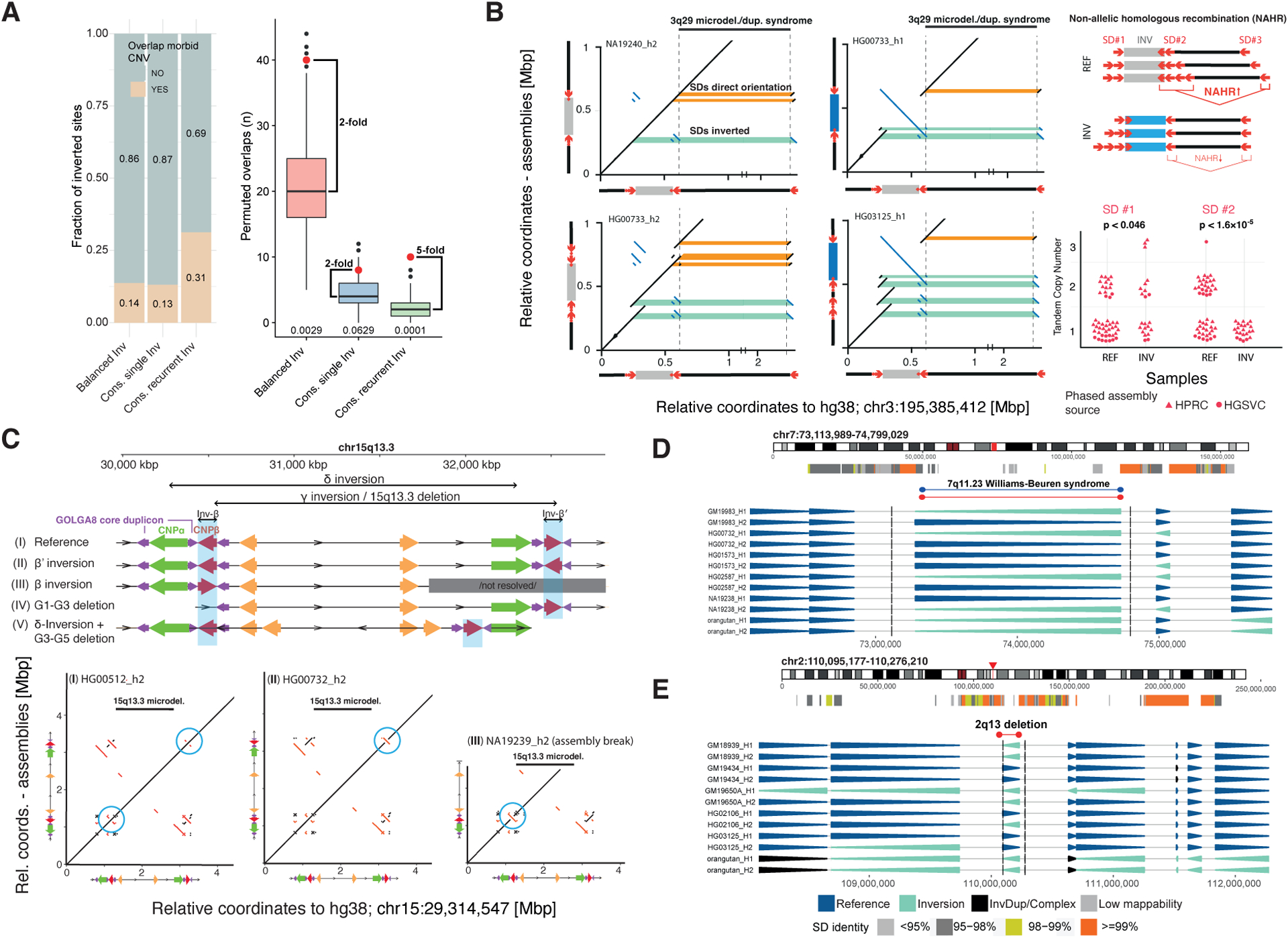
A) Left: Overlap of balanced inversions with a redundant list (n = 155) of morbid CNVs. Cons., consensus. Right: permuted overlaps, p-values (bottom). B) Left: Dot plots of representative assembled haplotypes at 3q29. SD pairs are highlighted in orange (direct) and green (inverse). Tandem duplications of at least one inversion-mediating SD (2nd row) are observed in 43/68 (63%) haplotypes. Right: Direct duplications (SD #2), increasing risk of morbid CNV formation, are common in direct and absent in inverted haplotypes (p-values, Fisher’s exact test). C) Structural haplotypes at 15q13.3, where INV-β and INV-β′ configurations potentially promote recurrent inversions or morbid CNVs. Additional haplotypes (IV, V) containing deletions putatively protect against inversions and morbid CNVs (see also Data S2). D, E) Inversions at 7q11.23 and 2q13.
Recurrent inversions affect the SD architecture at the 3q29 and 15q13.3 critical regions.
We investigated the architecture of inverted haplotypes in more detail, in order to identify genomic features that may predispose to de novo CNVs. We searched for pairs of homologous SDs on the same haplotype that change their relative orientation through inversion, such that the inversion or reference (direct) orientation of a segment might represent a pre-mutational state (Zody et al., 2008) for morbid CNVs (STAR Methods; Data S2). We find 79 balanced inversions affecting the relative orientation of altogether 1,094 SD pairs (Table S6), with 86% (68/79) of inversions changing the relative orientation of several (up to 112) SD pairs at once. We focused on those SD pairs affected by a single inversion site and considered only those sites where more than 90% of SD pairs (weighted by length) were flipped into a direct or inverse orientation, respectively—thus avoiding more complex SD regions. Using this approach, we isolate 20 ‘potential CNV pre-mutational state inducing’ and 9 ‘potentially CNV protective’ inversions (Table S6).
For example, we characterized a recurrent inversion flanking the 3q29 microdeletion syndrome (Ballif et al., 2008; Willatt et al., 2005), which reorients a 21 kbp SD at one critical region flank (Figure 5B). On the inverted haplotypes, this SD is in inverted orientation relative to the corresponding homologous SD at the distal end of the critical region, whereas non-inverted haplotypes exhibit this SD pair in a direct orientation. We further find directly oriented duplications of an SD homologous with the distal breakpoint region, which are common in directly oriented haplotypes (>50% of cases), but entirely absent in inverted haplotypes (Figure 5B). These data suggest that a recurrent inversion flanking the 3q29 microdeletion critical region may be protective with respect to morbid CNV formation.
We also analyzed the architecture of a 1.5 Mbp recurrent inversion overlapping the 15q13.3 microdeletion region (Antonacci et al., 2014). We find two independent inversions ~210 kbp in size (denoted INV-β and β′), which encompass either copy of the CNPβ repeat previously implicated (Antonacci et al., 2014) in the formation of the 15q13.3 microdeletion as well as the 1.5 Mbp inversion (INV-γ) (Figure 5C and Data S2). We hypothesize that either of the β and β′ inversions, when occurring in isolation, create a pre-mutational state for morbid CNV formation. Other configurations of β and β′, by comparison, may instead mediate INV-γ recurrence. We also find deletions involving the CNPα and β duplicons in two haplotype structures, which potentially protect both against morbid CNVs and recurrent inversions (Figure 5C).
Inversions at the WBS and juvenile nephronophthisis critical regions.
Encouraged by these findings, we performed more in-depth analysis of inversions intersecting sites of genomic disorders. For example, we find that the 7q11-23 inversion (Figure 5D, Table 1, Data S1), associated with WBS, has undergone toggling with three recurrent inversion events (central interval: 2, 4) across the critical region (chr7:73,113,989-74,799,029), translating to a rate of 2.62 × 10−5 [central interval: 1.36 × 10−5, 4.33 × 10−5] inversion events per generation. It was previously proposed that an inversion spanning this region predisposes to morbid CNV formation (Osborne et al., 2001); future studies in patient cohorts may address whether a subset of the 7q11-23 inversion haplotypes act as a pre-mutational state for WBS. Interestingly, we also find two nested polymorphic inversions at the boundary of this critical region, which might exhibit protective or pre-mutational properties with respect to WBS (Data S2).
Furthermore, we observe a putatively recurrent inversion at 2q13, overlapping morbid CNVs implicated in juvenile nephronophthisis and autism (Figure 5E, Data S1) (Chen et al., 2017; Parisi et al., 2004; Yasuda et al., 2014; Yuan et al., 2015), although recurrence of this region was inferred by the haplotype-based coalescent approach only (Table S5). Two SD pairs are predicted to change their relative orientation as a result of the inversion (Table S6). One of the inverted haplotypes harbors a deletion spanning this SD pair, which may confer a protective role with respect to de novo morbid CNVs (Data S2).
Manual investigation of HiFi-based assemblies from 28 haplotypes reveals further examples of complexity and diversity of SDs flanking common inversion, with the majority of such polymorphisms (11/15, 73%) appearing adjacent to recurrent inversions (Table S4). To illustrate this breakpoint complexity, we focused on chromosome 1p36.13, which is associated with both interstitial and terminal deletions and for which we find inversion polymorphisms of the flanking SDs (Aagaard Nolting et al., 2020; Shapira et al., 1997). Manual analysis of optical maps of this region reveal an extraordinary level of structural complexity, with 46 distinct haplotype structures 723 kbp–1.2 Mbp in size, which arose through inverted duplication, balanced inversion, and CNV events (Figure 6A,B, Data S1, STAR Methods). These haplotypes contain the NBPF1 core duplicon (Jiang et al., 2007). The NBPF1 gene encodes tandemly repeated Olduvai domains, which have expanded during primate and especially human evolution potentially in association with brain size (O’Bleness et al., 2012; Popesco, 2006; Sikela and van Roy, 2017; Uddin et al., 2011; Zimmer and Montgomery, 2015). Notably, a flanking SD thought to mediate morbid CNV formation (Aagaard Nolting et al., 2020) (yellow arrow in Figure 6A,B) exists in different copy number states and orientations, leaving the possibility that some structural haplotypes may predispose to differential susceptibility to 1p36.13 rearrangements.
Figure 6. Complex inverted haplotypes and inversions at sites of morbid CNVs.
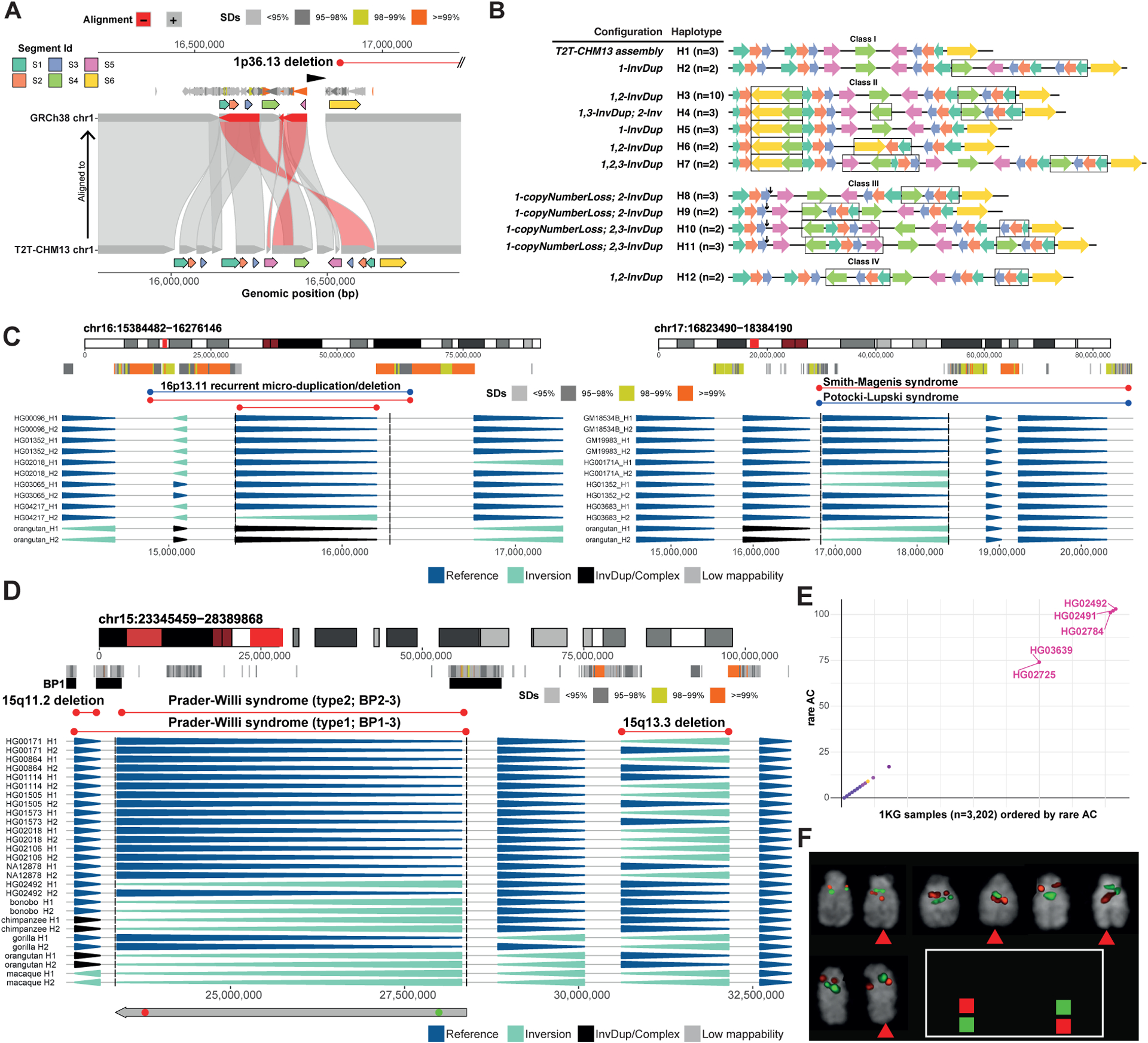
A) The 1p36.13 region differs between the T2T-CHM13 and GRCh38 references. B) Optical mapping reveals four haplotype classes (I-IV), with 12 (H1-H12) seen at least twice at 1p36.13. Colored arrows represent genomic segments, and black arrows deletions. Black rectangle outlines variants relative to T2T-CHM13. C) Inversions at 16p13.11 and 17p11.2. D) An inversion overlapping the PWAS type II region (recurrent CNV breakpoints denoted as BP1, 2 and 3). FISH probe positions shown (bottom). E) Scatterplot depicting shared rare SNPs within the 1KG data for the locus in (D). AC, allele count. F) FISH validation of the locus in panel D. CEN, centromere.
Our analysis also discovered inversions overlapping well-known morbid CNV regions not previously known to be polymorphically inverted (Figure 6C, Data S1). This includes an inversion corresponding to the 16p13-11 microduplication and microdeletion syndrome critical region, which we detected in a single individual of Telugu ancestry. We identify one inversion proximal to the critical region that results in the reorientation of an SD pair showing >90% reciprocal overlap with the respective morbid CNV, potentially conferring a protective effect (Table S6). We also identify an inversion at 17p11-2 partially overlapping the well-known SMPLS region, seen in two unrelated carriers (Figure 6C). This inversion is predicted to lead to a reorientation of mostly directly oriented SD pairs and, as such, could potentially have protective effects with respect to 17p11-2 CNV formation (Table S6).
Lastly, we highlight a 5 Mbp 15q11.2-13.1 inversion, identified from a single sample of Punjabi ancestry. The inversion overlaps the well-known PWAS type II critical region (Coe et al., 2014) and has been postulated to predispose to disease (Gimelli et al., 2003). This critical region shows a complex SD architecture at the flanks and underwent evolutionary inversion toggling (Maggiolini et al., 2020; Porubsky et al., 2020a) (Figure 6D). We set out to predict other samples in the 1KG panel carrying this inversion by analyzing rare SNP alleles after genotyping using PanGenie. We detected four additional carriers (including the mother of the index sample), all of which are of Punjabi ancestry, suggesting a potential founder inversion event (Figure 6E). FISH experiments verified all (5/5; 100%) predicted carriers (Figure 6F, Data S1). Since the inversion is thought to be enriched in parents of Angelman syndrome patients (Gimelli et al, 2003), this technique could be used to identify families at-risk.
Discussion
Extensive inversion recurrence in the genome.
Our analysis suggests that inversion toggling is one of the most common mutational processes. We estimate recurrence rates of 3.4×10−6–2.7×10−4 per site per generation and identify 40 regions of inversion toggling corresponding to ~0.6% of the human genome. The toggled segments are gene-rich, and often hundreds of kilobase pairs in length. Interestingly, 6/40 regions have also toggled between human and nonhuman ape species (Porubsky et al., 2020a). This suggests that toggling has been a long-standing and persistent property over the last 15 million years. The propensity for certain regions to toggle may in fact be even more ancient as originally reported for the emerin-filamin (EMD-FLN) inversion, which toggled at least 10 times during eutherian mammal evolution (Cáceres et al., 2007).
From a genetic perspective, toggling inversions are more likely to complicate, or be missed by, standard eQTL mapping (STAR Methods, Data S1) and genome-wide association studies because they arise independently on diverse haplotype backgrounds. While rare SNPs can point towards potential carriers (Figure 6E), the inversion status cannot be directly determined using short reads alone. Going forward, large-scale screens, such as using pooled Strand-seq in 1KG cell lines, could provide a cost-effective means of identifying and genotyping inversions (STAR Methods, Figure S5).
A sex chromosome bias in mutational toggling.
We observe an enrichment for recurrent inversions on the sex chromosomes, with 45% of toggling inversions residing on the X or Y (Table 1). It has been hypothesized that X chromosome hemizygosity and limited homologous recombination may promote intrachromosomal NAHR within the unpaired portion sex chromosomes (Cáceres et al., 2007). While most autosomal double-strand breaks (Lange et al., 2016) are repaired early during meiotic prophase, double-strand breaks persist on the unpaired X chromosome where they remain associated with recombinases Rad51 and Dmc1 (Enguita-Marruedo et al., 2019; Moens et al., 1997). At later stages of meiosis, factors for NHEJ co-localize to the XY bivalent (Goedecke et al., 1999). Furthermore, research in yeast has demonstrated the involvement of inverted repeat sequences in generating hairpin structures that can lead to DNA double-strand breaks or ectopic recombination between regions within the hairpin (Mizuno et al., 2013; Nag and Kurst, 1997; Nasar et al., 2000); these mechanisms may also be relevant to SDs flanking inversions in an inverted orientation. One possibility then is that breaks arising in nonhomologous XY regions are repaired by inter-sister recombination or NHEJ after crossovers have been formed, facilitating recurrent inversion formation. In this model, the male germline would be a key driver for inversion recurrence.
Association of recurrent inversions and hotspots of disease-causing rearrangements.
While an intimate relationship between inversions and some recurrent morbid CNVs has been known for decades (Antonacci et al., 2009, 2014; Koolen et al., 2006; Maggiolini et al., 2019; Osborne et al., 2001), our study establishes genome-wide association of disease-causing CNVs with hotspots of recurrent inversion. There are at least two possible explanations. First, inversion recurrence increases the MAF (theoretically converging to 0.5 if no other evolutionary forces counteract) leading to more heterozygous inversion carriers in the population, which promotes genomic instability and disease-causing CNVs: In a heterozygous inverted state, homologous recombination is suppressed (Sturtevant, 1917). In the case of meiotic double-strand break initiation, resection is likely to occur at the site of DNA breaks when homologous recombination is suppressed, and error-prone non-homologous repair may subsequently result in increased deletion and ectopic recombination events. If homologous recombination does occur between homologs within a heterozygous inverted segment, the resulting dicentric and acentric chromosomal segments may be recovered leading to subsequent rearrangements (Hermetz et al., 2014), including inverted duplications and terminal deletions as has been proposed for the chromosome 8p23.1 locus (Ciccone et al., 2006; Giglio et al., 2001; Giorda et al., 2007). Interestingly, we find that 8p23.1 is among the most recurrent inversions, estimated to have arisen at least 15 times in human history. It is also among the most common inversions with an allele frequency of 0.5 and, therefore, predicted to be heterozygous in 50% of all meioses.
An alternative, but not mutually exclusive, scenario may be that recurrent inversions lead to SD architectural diversity at their boundaries, creating pre-mutational states for de novo CNVs. We observed, for example, changes in copy number and orientation of SDs especially among structural haplotypes associated with inversion recurrence. Certain structural haplotypes, thus, may “switch” from protective (i.e., predisposing to recurrent inversion) to at-risk (i.e., predisposing to recurrent morbid CNVs). This is the case for the chromosome 17q2.31 inversion associated with KdVS, where the more complex, directly oriented SDs associated with the European H2 haplotype predispose to microdeletion making the syndrome largely European-specific (Steinberg et al., 2012). Our sequence-level analysis supports considerable structural diversity for chromosome 7q11.23 (WBS), 15q13, and 2q13, as well as the 3q29 microdeletion/duplication region, creating potential pre-mutation or protective haplotypes for CNV formation. Notably, in the case of the 3q29 inversion, our prediction that the inverted state may confer a protective role is consistent with the observation, reported in a current preprint, of 3q29 microdeletions forming on patient haplotypes in direct, rather than the inverted, genomic orientation (Yilmaz et al., 2021). At the genome-wide level, we observe reorganization of >1,000 SD pairs mediated by a total of 79 polymorphic inversions. These observations suggest a vast potential of inversions to prime or protect against morbid CNV formation, thereby shaping the human landscape of repeat-mediated mutation that is yet to be fully explored.
Insights into L1-internal inversions.
Amongst smaller inverted sequences (<2 kbp), we report frequent inversions of L1-internal sequences. Sequence analysis of 5′ junctions suggests MMEJ is involved in both 5′ truncation and twin-priming, and contrasts with full-length L1 insertions. This supports the annealing of microhomologous sequences leading to premature truncation and internal inversion of L1 sequences (Yamaguchi et al., 2014; Zingler et al., 2005) and is consistent with the involvement of DNA repair in the truncation of L1s and the frequency of retrotransposition (Coufal et al., 2011; Suzuki et al., 2009). Sporadic templated nucleotide insertions at Jct1–2 for twin-priming events and at the 5′ end of truncated L1s suggest the participation of polymerase θ mediated end joining, although further research is needed to elucidate the role of alternative polymerases in internal priming and reverse transcription (Chandramouly et al., 2021).
Limitations of the study.
With the exception of the Y chromosome, we inferred inversion toggling by requiring confirmation of two independent approaches and as a result may actually underestimate inversion recurrence, especially if selective forces are operating (Steinberg et al., 2012). The study is limited to 82 unrelated human haplotypes and more genomes will be required to distinguish single-origin inversions from recurrent events. Many SD regions flanking the inversions are not yet fully sequence resolved (Table 1), and this is a critical next step to understand the mechanisms responsible for inversion formation (Antonacci et al., 2014) (Figure S2E–G, STAR Methods). Resolving the flanking sequences fully will reveal whether there are particular genomic signatures or “scars” at their breakpoints (Figure S2H) associated with recurrent and nonrecurrent events, and whether at certain loci inversion toggling results from partially overlapping inversions with distinct breakpoints (STAR Methods, Data S1). In our study, Strand-seq was critical for the discovery and genotyping of most large (>100 kbp) inversions, especially those flanked by SDs (Data S1). Orthogonal optical mapping data in concert with the long-read and Strand-seq data helped, for example, to validate more complex haplotype structures (e.g., at 1p36.13). Characterization of the complete spectrum of SVs at the sequence level remains an important goal that is likely to be unattainable outside of a multi-platform approach.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact Jan O. Korbel (jan.korbel@embl.org).
Materials Availability
This study did not generate any new unique reagents or materials to report. All cell lines used are commercially available.
Data and Code Availability
The full inversion callset is available in Table S2. Raw genomic datasets are available through the International Nucleotide Sequence Database Collaboration (INSDC) including Illumina WGS, RNA-seq, Bionano Genomics, PacBio, and Strand-seq data, and are publicly available as of the date of publication Accession numbers are listed in the key resources table. The URL is listed in the key resources table. Phased assemblies of the 82 haplotypes were obtained from Ebert et al. 2021 (PGAS v12 assemblies) and Ebler et al. 2022 (PGAS v13 hifiasm assemblies) and are listed in the key resource table. Select loci were also examined from Human Pangenome Reference Consortium (HPRC); https://github.com/human-pangenomics/HPP_Year1_Assemblies. Dot plot visualizations of several recurrent inversion loci can we obtained as described in the key resource table. Other publicly available data (PacBio and ONT) used in this study are reported in Table S1. VCF files with integratively phased single-nucleotide variants (SNVs) and inversion genotypes have been deposited at IGSR FTP. The URL is listed in the key resources table. Data previously generated as part of Ebert et al. 2021 study are publicly available from the IGSR (www.internationalgenome.org/data-portal/data-collection/hgsvc2).
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| Deposited data | ||
| Strand-seq: NA19036 | This paper | NCBI: PRJEB39750 |
| Strand-seq: NA19434 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG00268 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG01352 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG01573 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG02018 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG02059 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG02106 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG04217 | This paper | NCBI: PRJEB39750 |
| Strand-seq: LCL pools | This paper | NCBI: PRJEB39750 |
| Strand-seq: trio samples (n=9) | (Chaisson et al., 2019) | NCBI: PRJEB12849 |
| Strand-seq: other samples (n=34) | (Ebert et al., 2021) | NCBI: PRJEB39750 |
| Strand-seq: HG002/NA24385 | Public HPRC data (https://github.com/human-pangenomics/HG002_Data_Freeze_v1.0) | https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=NHGRI_UCSC_panel/HG002/hpp_HG002_NA24385_son_v1/Strand_seq/ |
| Strand-seq: NA12878 | (Porubský et al., 2016) | NCBI: PRJEB14185. |
| WGS Illumina data (1KG panel) | (Byrska-Bishop et al.) | NCBI: PRJEB37677 |
| RNA-seq data | (Ebert et al., 2021) | NCBI SRA: ERP123231 |
| PacBio data 1 | (Ebert et al., 2021) | NCBI: PRJEB36100 |
| PacBio data 2 | (Ebert et al., 2021) | EBI/ENA: ERP125611 |
| PacBio data 3 | (Ebert et al., 2021) | NCBI: PRJNA698480 |
| PacBio data: HG00268 | N/A | NCBI: PRJNA558774 |
| PacBio data: HG01352 | N/A | NCBI: PRJNA339719 |
| PacBio data: HG02059 | N/A | NCBI: PRJNA339726 |
| PacBio data: HG02106 | N/A | NCBI: PRJNA480858 |
| PacBio data: HG04217 | N/A | NCBI: PRJNA481794 |
| PacBio data: NA19434 | N/A | NCBI: PRJNA385272 |
| Oxford Nanopore: HG00733 ultra-log | (Logsdon et al., 2021) | NCBI: PRJNA686388 |
| Oxford Nanopore: HG00733 | (Shafin et al., 2020) | NCBI: PRJEB37264 |
| Oxford Nanopore: NA19240 | N/A | NCBI: PRJEB26791 |
| Oxford Nanopore: HG002/NA24385 | (Shafin et al., 2020) | https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/UCSC_Ultralong_OxfordNanopore_Promethion/ |
| BioNano data | (Ebert et al., 2021) | EBI/ENA: ERP124807 |
| 1KG phased genotypes for 3,202 samples | (Byrska-Bishop et al.) | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_phased/ |
| Phased SNVs (VCFs) and inversion genotype tables | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/working/20210917_SSEQplusWHintegrativePhasing_inversionCallset/ |
| HGSVC phased assemblies (PGAS v12) | (Ebert et al., 2021) | https://www.internationalgenome.org/data-portal/data-collection/hgsvc2 |
| HGSVC phased assemblies (PGAS v13) | (Ebler et al. 2022) | DOI:10.5281/zenodo.5607680 |
| Dot plot visualizations of several recurrent inversion loci | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/working/20220209_recurrent_inversions_resolved/ |
| Software and algorithms | ||
| primatR | (Porubsky et al., 2020) | https://github.com/daewoooo/primatR |
| breakpointR | (Porubsky et al., 2019) | https://github.com/daewoooo/breakpointR |
| StrandPhaseR | (Porubsky et al.,2017), New functionalities added in this paper | https://github.com/daewoooo/StrandPhaseR, branch=devel |
| ArbiGent | This paper | DOI:10.5281/zenodo.6405196 |
| PAV | (Ebert et al., 2021) | https://github.com/EichlerLab/pav |
| MEIGA-PAV | (Ebert et al., 2021), New functionalities added in this paper | DOI:10.5281/zenodo.6077336 |
| ti-SNPs detection | This paper | DOI:10.5281/zenodo.6405152 |
| Detection of altered SD organization | This paper | DOI:10.5281/zenodo.6411308 |
| Mendelian consistency analysis | This paper | DOI:10.5281/zenodo.6411714 |
| BWA aligner (v0.7.15–0.7.17) | (Li and Durbin, 2010) | http://bio-bwa.sourceforge.net/ |
| SAMtools (v1.3.1–1.10) | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| sambamba (v1.0) | (Tarasov et al., 2015) | https://lomereiter.github.io/sambamba/ |
| RTG tool (v3.11) | Copyright (c) 2018 Real Time Genomics Ltd | https://www.realtimegenomics.com/products/rtg-tools |
| Relate (v1.1.7) | (Speidel et al., 2019) | https://myersgroup.github.io/relate/ |
| IQ-TREE (v2.1.3) | (Minh et al., 2020) | http://www.iqtree.org/ |
| SV-Pop | (Audano et al., 2019; Ebert et al., 2021) | https://github.com/EichlerLab/svpop |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Detailed descriptions of all cell lines used in this study can be obtained from Table S1.
METHOD DETAILS
Human diversity panel.
We selected 44 samples from the 1KG (1000 Genomes Project Consortium et al., 2015) for inversion discovery. These included individuals with ancestry from Africa (n = 13), America (n = 8), East Asia (n = 9), Europe (n = 8), and South Asia (n = 6). We performed Strand-seq in nine samples and combined these data with previously generated data from three orthogonal platforms (Strand-seq, long-read assemblies, and Bionano; Table S1) available for 35 samples (Chaisson et al., 2019; Ebert et al., 2021). Excluding three related family members (children in family trios), inversions were discovered in 41 unrelated individuals (82 haplotypes). The three parent-child trios were subsequently used to test Mendelian segregation of inversions in families.
Strand-seq data generation and data processing.
Strand-seq data were generated as follows. EBV-transformed lymphoblastoid cell lines from the 1KG (Coriell Institute; Table S1) were cultured in BrdU (100 uM final concentration; Sigma, B5002) for 18 or 24 hours, and single isolated nuclei (0.1% NP-40 lysis buffer (Sanders et al., 2017)) were sorted into 96-well plates using the BD FACSMelody cell sorter. In each sorted plate, 94 single cells plus one 100-cell positive control and one 0-cell negative control were deposited. Strand-specific single-cell DNA sequencing libraries were generated using the previously described Strand-seq protocol (Falconer et al., 2012; Sanders et al., 2017) and automated on the Beckman Coulter Biomek FX P liquid handling robotic system (Sanders et al., 2020). Following 15 rounds of PCR amplification, 288 individually barcoded libraries (amounting to three 96-well plates) were pooled for sequencing on the Illumina NextSeq5000 platform (MID-mode, 75 bp paired-end protocol). The demultiplexed FASTQ files were aligned to the GRCh38 reference assembly (GCA_000001405.15) using BWA aligner (version 0.7.15–0.7.17) for standard library selection. Aligned reads were sorted by genomic position using SAMtools (version 1.10) and duplicate reads were marked using sambamba (version 1.0). Low-quality libraries were excluded from future analyses if they showed low read counts (<50 reads per Mbp), uneven coverage, or an excess of ‘background reads’ (reads mapped in opposing orientation for chromosomes expected to inherit only Crick or Watson strands) yielding noisy single-cell data, as previously described (Sanders et al., 2017). Aligned BAM files were used for inversion discovery as described below. On average, there are 68 (median: 57) single-cell Strand-seq libraries per sample (n = 44, including newly and previously published Strand-seq data included in the study), with an average 964,021 (median: 820,090) BWA-aligned reads (mapq ≥10) per cell.
Strand-seq-based inversion discovery.
To detect inversions using Strand-seq data, directional composite files were generated for each sample as previously described (Chaisson et al., 2019; Sanders et al., 2016). For this we used the breakpointR function ‘synchronizeReadDir’ (Porubsky et al., 2020b), which locates Watson-Watson (WW) and Crick-Crick (CC) regions in each chromosome and for each cell before building these into the sample-specific composite files. Composite file generation is designed to work well with inversions up to ~4 Mbp. We detected two large-scale inversions (chr2:87987171-111255403 in NA19650; chr15:23345459-28389868 in HG02492) that were not represented in our composite file but were visible in individual Strand-seq libraries. While these inversions are very rare, we set to recover such events by locally correcting our composite files. We used the primatR function ‘synchronizeReadDirRegion’ to create a regional composite file around the inverted region. Next, we replaced reads in the original composite file with the reads from the regional composite file. This allowed us to correctly genotype and visualize these inverted regions.
Segmental changes in composite file orientation, suggestive of an inverted allele, were identified using breakpointR (Porubsky et al., 2020b). To detect both larger and smaller strand-state changes, we used breakpointR in two settings—applying either a window size length of 5 kbp or 20 reads per bin. In both cases, we scaled an initial bin size by multiples of 2, 3, 4, 5, 10 and 20. This resulted in a redundant dataset with putative inversions detected per sample (mean and median: 144 per sample).
To construct a nonredundant set of Strand-seq inversions, we merged and filtered all detected strand-state changes (putative inversions) in multiple stages as follows: We started out by cropping out inversion flanks that overlap with highly identical SDs (≥98% identity) or gaps defined in GRCh38. Second, we iteratively merged inversion ranges with ≥50% reciprocal overlap until no more ranges could be merged. Such collapsed ranges were then subjected to a re-genotyping step using the ‘genotypeRegions’ function of the primatR package (Porubsky et al., 2020a). Each region in each sample was assigned a genotype: ‘HET’ - approximately equal mixture of plus and minus reads, ‘HOM’ - majority of minus reads, ‘REF’ - majority of plus (reference) reads and ‘lowReads’ - less than 20 reads in a region. Ranges that genotype only as a reference (‘REF’) orientation or have less than 20 (‘lowReads’) reads across all samples were filtered out. Next, we collapsed ranges that share the same genotype across all samples and are embedded with respect to one another. Lastly, for regions that were genotyped only as a ‘HET’ or ‘lowReads’ across all samples, we retained only unique (nonoverlapping) genomic ranges. The same procedure was repeated for window sizes defined by the reads per bin (20 reads per bin as mentioned above), which allowed adding smaller inversions, missed by the larger bin size, to the final Strand-seq inversion callset. Finally, we merged the nonredundant set of inversions created by both window sizes (5 kbp and 20 reads per bin) into a final nonredundant set (n = 341) created by the automated procedure described above (Data S1).
We then manually curated the resultant Strand-seq calls to increase the overall accuracy of the callset, as was done in previous studies (Chaisson et al., 2019; Porubsky et al., 2020a). We projected sample-specific composite files onto the UCSC Genome Browser in order to evaluate the mapping of Strand-seq reads inside complex regions of the genome. This procedure led to the addition of 26 inversions and divided 31 regions into more than one inverted event with respect to the automated nonredundant callset (n = 341). This resulted in the final manually curated nonredundant Strand-seq based inversion discovery callset (n = 419). Among those 419, 39 inversions were marked as false positives mostly caused by a single sample (NA19239) likely due to the extent of background (i.e., strand-unspecified) reads in sample-specific composite files. The manually curated Strand-seq inversion calls were subsequently expanded into the redundant callset (n = 6,642) and used in the inversion merging process along with assembly- and Bionano-specific inversion callsets (as described below) (Data S1).
Inversion discovery using the Phased Assembly Variant Caller (PAV).
Haplotype-phased assemblies (Data and Code Availability) were used to generate a long-read-based inversion callset, using the PAV (Ebert et al., 2021) tool, and these assemblies were further utilized to perform sequence-level characterization of inverted sequences. PAV was run on 32/44 samples (64/88 haplotypes) with available phased assemblies. Briefly, PAV aligns each assembled haplotype (2 per sample) with minimap2 (Li, 2018) and finds evidence of inversions by analyzing fragmented alignments and aberrant SV patterns created when alignments traverse through an inversion breakpoint. As part of our previous work, variants in assembly collapses were identified using SDA (Vollger et al., 2018) and removed.
Long-read assembly-based discovery of L1-internal inversions mediated by twin-priming.
Non-reference L1 insertion calls previously generated by the HGSVC (Ebert et al., 2021) were subjected to a refined version of the MEIGA-PAV annotation pipeline in order to identify and characterize twin-priming events. First, in order to have all L1 inserts in forward orientation, the reverse complement sequence for every L1 insertion occurring in the minus strand was obtained. Then, poly (A) tails were detected and trimmed for every insert, requiring poly (A) monomers to be at least 10 bp in size, have a minimum purity of 80%, and be located at a maximum distance of 30 bp relative to the insert end. The resulting trimmed inserts were aligned using BWA-MEM 0.7.17-r1188 into a consensus L1 sequence derived from the 632 FL-L1 insertions included in the HGSVC callset. In order to maximize sensitivity for particularly short L1 events a minimum seed length (-k) of 8 bp and a minimum score (-T) of 0 were used. Alignment hits over the L1 consensus were chained based on complementarity in order to identify the minimum set of nonoverlapping alignments that span the maximum percentage of the inserted sequence. A second targeted alignment with BWA-MEM is applied to insert ends that failed to align in the initial alignment round. Based on the alignment chains, L1s are classified as full-length (single hit spanning >99% of the consensus L1), 5′ truncated (single hit spanning ≤99% of the consensus L1), and 5′ inverted (two hits with the first in reverse while the second in forward orientation). Then, the inversion junction conformation for every twin-priming event is determined based on the alignment position over the consensus for the inverted and non-inverted L1 pieces. Blunt joints are characterized by perfect complementary alignments, while overlapping and discontinuous alignments define duplications and deletions at joints, respectively.
Reference L1s were processed similarly as non-reference with two additional preprocessing steps prior to annotation with MEIGA-PAV. RepeatMasker annotations for the GRCh38 genome build were downloaded from the UCSC Table Browser (Karolchik et al., 2004). We noticed that the existence of 5′ inversions frequently led to fragmented annotations, whereby the inverted and non-inverted sequences are erroneously annotated as independent L1s in the reference genome. To correct for this, we merged pairs of L1 annotations adjacent to each other, if they were in opposite orientation and complemented one another at the sequence level. Then, reference L1s were intersected with deletion calls previously generated by the HGSVC (Ebert et al., 2021) in order to select polymorphic L1s that were deleted in at least one of the 64 haplotypes. After doing the reverse complement for insertions in the minus strand and trimming poly (A) tails, these L1 elements were further analyzed using MEIGA-PAV.
We successfully inferred the configuration for 93% (1,271/1,362) of L1 polymorphisms, finding that 26% (330/1,271) of them show characteristic 5′ inverted sequences, whereas the remaining are either full-length (405) or 5′ truncated (536). For 7% of L1 polymorphisms (n = 91) there was uncertainty regarding the insertion configuration, and these elements were therefore not analyzed.
Simulations and evaluation of L1 annotation pipeline.
We generated a simulated dataset, including 9,000 synthetic L1 inserts (Data S1), evenly distributed among the three possible insertion configurations: full-length (FL), 5′ deleted, or inverted L1. All inserts derived from the same consensus L1 sequence were used as reference for L1 annotation. While the complete consensus was included for FL-L1, random breakpoint positions were sampled for the generation of truncated and inverted events. A single breakpoint located between 10 and 6,013 positions was sampled for 5′ deletions, ensuring a minimum deletion and insertion size of 10 bp. To simulate 5′ inversions, the inversion junction structure was randomly selected among three possible configurations: blunt, deletion, and duplication. Duplication and deletion sizes at the junctions were sampled between 1 and a maximum length of 100 bp and 500 bp, respectively. Similarly, the inversion size was determined based on a random distribution using a minimum inversion length of 10 bp. Then, a random 3′ breakpoint position compatible with the inversion length and junction structure was sampled and the position of the 5′ breakpoint was determined relative to the 3′ breakpoint while taking into account insertion features. Microhomologies and nucleotide insertions at the junction between the TSD and the 5′ end of full-length, truncated, and inverted L1s were characterized based on a search of complementary DNA sequences between the 3′ end of the TSD and the L1 sequence adjacent to the insertion breakpoint.
In order to account for potential sequencing errors, a single mismatch was allowed at the microhomology patches. The same approach was applied for the characterization of the junction between the inverted and non-inverted L1 fragments for twin-priming events. Templated insertions were detected based on manual inspection and alignment of the inserted sequences to the genomic sequences within 50 bp of the integration breakpoint or to the L1 consensus sequence. Insertions shorter than 8 bp were excluded from this analysis because they could not be reliably aligned (typically not mapping or producing multiple possible alignments). Based on these analyses we determined that 7% (5/71) of the detected insertions seen in association with twin-priming events are templated.
The 9,000 simulated L1 inserts were annotated using MEIGA-PAV and annotations were systematically evaluated using the simulated insertion features as a reference. The predicted insertion configurations were highly consistent with expectations (Data S1), with only 18 misannotated insertions, which correspond to insertions with short 5′ deletions misclassified as full-length. Junction conformations were also accurately ascertained (Data S1), with 98% (1,016/1,034) duplications, 97% (974/1,008) deletions, and 91% (870/958) blunt joints being concordant. Predicted lengths for inversions, duplications, and deletions at inversion joints were strongly correlated with the expected sizes (Data S1). Finally, 75% (4,483/5,940) of all inversion breakpoints were accurately detected, with inaccurate breakpoints having a median deviation of 1 bp (max = 22 bp) (Data S1).
Bionano Genomics–based inversion discovery.
We analyzed Bionano Genomics Optical Mapping data by using Saphyr 2nd generation instruments (Part # 60325) and Instrument Control Software (ICS) version 4.9.19316.1. De novo assemblies of each sample were obtained using the Bionano Solve v3.5 De Novo Assembly pipeline with haplotype-aware arguments (optArguments_haplotype_DLE1_saphyr_human_downSampleLongestMole.xml) as described previously (Ebert et al., 2021). Using the Overlap-Layout-Consensus paradigm, pairwise comparisons of DNA molecules at least 250 kbp in length, contributing to a coverage of 250X, were generated to create a layout overlap graph and produce initial consensus genome maps. By realigning molecules to the genome maps (alignment confidence cutoff of Bionano p-value < 1×10−12) (Anantharaman et al., 2004) and by using only the best matching molecules, a refinement step was applied to label positions on the genome maps and to remove chimeric joins. Next, during an extension step, molecules were aligned to genome maps (Bionano p-value < 1×10−12), and the maps were extended based on the molecules aligning past the map ends. Overlapping genome maps were then merged (Bionano p-value < 1×10−16). These extension and merge steps were repeated five times before a final refinement was applied to “finish” all genome maps. To identify all alleles, clusters of molecules that were aligned to genome maps with unaligned ends >30 kbp in the extension step were re-assembled to identify potential alternate alleles. To identify alternate alleles with smaller size differences from the assembled allele, clusters of molecules that aligned to genome maps with internal alignment gaps of size <50 kbp were identified, in which case, the genome maps were converted into two haplotype maps. Inversions were identified using the Bionano Solve v3.5 De Novo Assembly pipeline, in which the final genome maps were aligned (Bionano p-value < 1×10−12) to GRCh38. Manual curation of inversions was performed using Bionano Access (v1.5.2). Optical maps of samples were visually evaluated for inversions not automatically detected by the pipeline. Molecule support for each inversion was evaluated by using the molecule data of each contig containing the inversion. Inversions without molecule support (molecules that span either the entire inversion or anchored to unique labels in proximal and distal inversion breakpoints) were excluded. In addition, inversions identified in centromeric regions and in tandem repeat regions without distinct labeling pattern (direct and inverted configurations of a region of interest show the same labeling pattern) were excluded.
Inversion merging into a provisional integrated callset.
To create a final nonredundant inversion callset outside of L1 insertions, we merged inversion calls based on different technologies using SV-Pop (Audano et al., 2019; Ebert et al., 2021). Merging of overlapping inversion calls was done in the following priority order: phased assembly-based calls (based on PAV), Strand-seq, and Bionano manual callsets. This means that the PAV range is considered first in case two or more inversion calls overlap. In addition, to prevent removing manually curated Strand-seq calls from more complex regions, such as centromeres, we switched off any filtering (applied in (Ebert et al., 2021)) during the merging step. This merging procedure resulted in a provisional merged inversion callset with 613 genomic regions.
However, in this procedure, a small number of inversion calls made manually using Strand-seq in complex regions of the genome may have been lost, because a PAV inversion call based on an incomplete assembly takes precedence, and thus may lead to a loss of a valid Strand-seq-based call. We thus recovered Strand-seq manual calls with less than 50% reciprocal overlap with the merged callset. By doing so, we ended up adding two simple inversions (chr6-26738711-INV-24388; chr10-79542902-INV-674513) and three inverted duplications (chr16-55798460-invDup-32830; chr17-19240629-invDup-2318213; chrX-141585258-invDup-102910) to the final provisional merged callset (n = 618) (Data S1). We subsequently continued with the re-genotyping of all regions using ArbiGent as described below. Note that for any PAV call, we genotyped inner breakpoints reported by PAV whenever inner breakpoints were completely embedded within outer breakpoints.
Inversion genotyping and phasing with ArbiGent.
We devised a Strand-seq-based inversion genotyping method, termed ArbiGent, which we employed for three purposes: 1) to unify inversion calls across samples, 2) to verify inversion calls made with other platforms, and 3) to integrate information about inversion loci across samples and accordingly improve the individual callsets. ArbiGent determines inversion genotype likelihoods for genomic loci containing at least 500 bp of sequence uniquely mappable with 75 bp paired-end reads (Data S1), using strand-specific reads as an input. ArbiGent utilizes an adapted statistical framework previously used for subclonal SV calling in cancer (Sanders et al., 2020). We extended this framework to allow estimating SV genotype likelihoods for DNA segments of choice using Strand-seq data. Based on a Bayesian probability framework that models strand- and haplotype-specific read counts using negative Binomial distributions (Sanders et al., 2020), ArbiGent computes inversion genotype likelihoods for inversions and copy number changes. SV genotype likelihoods derived from individual cells from the same sample are concatenated by summing up log likelihoods across cells, to result in a combined genotype likelihood estimate per sample and genomic locus of interest. We consider genotype calls made by ArbiGent as ‘high confidence’ if they display a likelihood ratio over a reference state of >103. Integrating genotype labels across samples, ArbiGent additionally assigns labels at the locus level, including ‘potentially FalsePositive (FP)’ if the locus is never seen inverted in any sample, or ‘AlwaysComplex’ if the locus was not seen in a non-complex (or reference) state in any sample. In GRCh38, 489 out of 615 (79.5%) inversions in the initial discovery set were assigned a ‘passing’ label (‘Pass’,’InvDup’,’NoReads’,’LowConf’,’Misorient’), with the ratio increasing for inversion loci called by at least two independent techniques (75 out of 77, 97.4%) (Figure S1E,F).
ArbiGent supports haplotype phasing of events based on the StrandPhaseR method, which infers phase information from Strand-seq reads (Porubský et al., 2016; Porubsky et al., 2017). We additionally synchronized the haplotype assignments per chromosome (H1 vs. H2) post-hoc to the long-read-based phased genome assemblies (in the subset of samples [35/44] where such assemblies were available). This is done by comparing heterozygous SNP sites phased by StrandPhaseR (Porubsky et al., 2017) to those called by PAV (in phased assemblies) and adjusting the ArbiGent phase accordingly on chromosomes where the PAV and Strand-seq SNPs are phased orthogonally. This procedure was applied to all inversions on 796/805 chromosomes (35 independent samples × 23 chromosome sets), while the remaining nine displayed potential errors in phased assemblies in which case ArbiGent calls were reported using their original phase (Data S1). Using this procedure, we genotyped all 618 inversions, of which 615 were used for subsequent analysis (three inversions from unassigned fragments of the GRCh38 reference were dropped: chr14_GL000225v1_random-107057-INV-5515, chr17_GL000205v2_random-160765-INV-1685, and chrUn_KI270743v1-150894-INV-11414; Data S1).
Inversion filtering and generation of the final integrated inversion callset.
We applied a number of filters to remove low-quality inversion calls and generate the final integrated callset for inversions outside of L1 sequences. First, we removed calls that were genotyped by ArbiGent as ‘Alwayscomplex’, ‘Alwayscomplex-InvDup’ or ‘FP’ (false positive). Second, we removed any remaining inversions unique to the Strand-seq callset that were flagged as either ‘FP’ (false positive) or SD (‘segmental duplication’) during initial inversion discovery. Third, we removed any unique automated Bionano call (reported by automated Bionano procedure) with less than 90% reciprocal overlap with the manually curated Bionano inversion callset. Lastly, we dropped inversion calls with 90% or more reciprocal overlap with another inversion call within the callset, which brought the number of inversions to 418 (Data S1). We marked putative reference assembly misorientations (misorients) as regions defined as ‘miso’ by ArbiGent or marked as putative misorient during the manual curation of Strand-seq callset (all carriers show homozygous inversions). Any call that showed at least one clear heterozygous genotype was not marked as a putative misorient.
To finalize the callset we manually evaluated dot plot alignments for all 418 inversions, using the phased assemblies (STAR Methods). We identified contigs spanning both breakpoints for 183/418 (44%), predominated by smaller inversions below 200 kbp (Figure S1C). We used the alignments to verify inversion status and to annotate repeats at the breakpoints as well as other SVs at the flanks, such as insertions, deletions, and duplications. We used a 50 bp lower cutoff in reporting homology and SVs. Finally, we intersected all homologous repeats with mobile elements present in RepeatMasker v4.1.2 (Tarailo-Graovac and Chen, 2009). Using this approach, we identified inversions (n = 31) likely driven by mobile elements (both inversely oriented homologous repeats displayed >80% reciprocal overlap with an individual mobile element of the same class). Using the phased assemblies, we then adjusted inversion breakpoint positions. After breakpoint adjustment, we re-genotyped all inverted regions once more using ArbiGent. We then manually checked inversions for potentially redundant calls, which were removed from the callset. This led to the removal of six redundant calls (variant IDs: chr6-167197698-INV-159879, chr8-2235761-INV-247546, chrX-52472287-INV-68115, chrX-155384040-INV-73061, chr10-46986413-INV-193930, chr10-79526936-INV-290309). We also removed calls flagged as false positives during the manual dot plot evaluation and with no support by Strand-seq data (n = 13), resulting in the final callset of 399 inversions. Finally, to avoid reporting low-confidence genotypes for small inversions discovered only by PAV, we replaced the ArbiGent genotypes with PAV genotypes for these small events if the ArbiGent genotype was labeled as “low confidence”.
We categorized 399 inversions that occurred outside of L1 insertions into 292 balanced inversions (‘Inv’) - at least one sample shows a confident balanced inversion of genomic sequence; 40 inverted duplications (‘InvDup’) - marked based on ArbiGent prediction; 29 structurally complex sites (‘Complex/lowconf’) - inverted segment contains CNVs or estimated inversion genotype likelihoods (Table S2, Figure S1G) are of low confidence (threshold for confident genotypes: likelihood ratio over reference state > 103) based on ArbiGent genotypes followed by manual curation; and 38 likely assembly errors in GRCh38 or rare minor alleles (‘Miso’) - where all human haplotypes are inverted with respect to the reference. We emphasize that such extensive manual curation of our callset is crucial to deliver accurate and confident inversion callset because many inversions lie within the most complex repeat-rich regions of the human genome.
Inversion refinement with dot plots and global genome alignments.
We manually reviewed all loci from our initial GRCh38 inversion callset using continuous long-read– and circular consensus sequence–based haplotype-resolved genomes previously reported in (Ebert et al., 2021) and (Chaisson et al., 2019) for 35 samples overlapping with our sample set. To this end, we first extracted 10 kbp long sequence ‘anchors’ from 200 kbp upstream of each 5′ inversion breakpoint in the GRCh38 reference and used Minimap2 (Li, 2018) to identify the corresponding region in each assembled haplotype. By expanding downstream from this ‘anchor’ region, we then proceeded to extract the full sequence of each inversion locus, including the surrounding regions in each assembled haplotype. Minimap2 was then used a second time to create pairwise sequence alignments between these extracted regions and their reference counterpart in GRCh38. Alignments were finally visualized as dot plots using a modified version of dotPlotly (Poorten, 2017). Manual curation of inversion breakpoints and associated SDs, insertions, deletions, and other rearrangements was then performed using breakpoints identified in these alignments, with dot plots used for visual guidance. Using this approach, we curated 183/419 (44%) inversions, primarily smaller events below 200 kbp in size (Figure S1C, Data S1). We note that the majority of larger inversions were not accessible to this approach, consistent with breaks in the haplotype assemblies caused by long SDs.
Phasing and correction of chromosome-length inversion haplotypes.
Chromosome-length haplotypes fully containing inversions are an important prerequisite for an accurate prediction of inversion toggling in humans. While Strand-seq is by design well suited to detect inversions and can be used for long-range (chromosome-length) haplotyping, the haplotypes constructed by StrandPhaseR (Porubský et al., 2016; Porubsky et al., 2017) over the inverted region are not in the correct phase with the rest of the chromosome. This is because inversions change the directionality status (plus or minus) of Strand-seq reads with respect to the surrounding regions. The scope of the problem differs for homozygous and heterozygous inversions. Homozygous inversions appear as a complete switch of haplotypes in comparison to the haplotypes from uninverted regions. This is caused by a switch in directionality of Crick (plus) and Watson (minus) reads with respect to the uninverted regions. Such a switch in haplotypes inside a homozygous inversion can be corrected by ‘flipping’ such haplotypes. In contrast, heterozygous inversions are more difficult to correct as only one strand is inverted, and thus, either the Watson or Crick strand changes its directionality over the inverted region (Data S1). This creates a mixing of alleles in heterozygous state so the heterozygous inversions need to be phased de novo using Strand-seq cells that inherited either only Watson or Crick strands from each parent for a given chromosome. In such cells, heterozygous inversions appear as an equal mixture of Crick and Watson reads as only one strand (one haplotype) is inverted. Such cells are informative and can be used for unambiguous phasing for a given inverted region, since the Crick and Watson reads are coming from different parental homologs. Inverted haplotypes were assigned to a respective parental homolog based on the phasing of reads from Strand-seq cells that inherited either Watson or Crick strands from each parent. We implemented the functionalities for correcting the phase of inverted sequenced in the R package StrandPhaseR (v0.99), in a function called ‘correctInvertedRegionPhasing’. We supplied this function with the sample-specific inverted regions reported in this study, in order to correct each of them using the given function parameters (recall.phased = TRUE, het.genotype = ‘lenient’, pairedEndReads = TRUE, min.mapq = 10, background = 0.1, lookup.bp = 1000000, bsGenome = BSgenome.Hsapiens.UCSC.hg38, assume.biallelic = TRUE).
After inversion phase correction, chromosome-length haplotypes based on Strand-seq data were used to guide phasing of the long-read assemblies, by executing a previously described integrative phasing framework (Porubsky et al., 2017). Integrative phasing was completed using WhatsHap (version 0.18) and subsequently linkage disequilibrium was calculated using PLINK (version 1.9), with a window size of 200 kbp. For integrative phasing, we used a defined set of variant positions (available at http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_phased/). Finally, we re-genotyped the sample VCFs based on the long-read BAM files.
Mendelian consistency analysis.
We tested the inversion genotypes reported for events outside of L1-internal sequences for Mendelian consistency using previously generated parent-child trio-based Strand-seq data (Chaisson et al., 2019). Cases were flagged as ‘complex’ if at least one member of a trio had evidence for a non-balanced event, such as deletions and inverted duplications: 260/399 inversions showed exclusively simple inversion genotypes in all trios, out of which 95% (247/260) were Mendelian consistent (99.5% (200/201) when considering confident inversion genotypes only (genotype-likelihood ratio over reference state > 103). Sites showing modest SV diversity beyond balanced inversions, i.e., those labeled ‘complex’ in at least one but not all of the trios, passed Mendelian consistency in 53/56 (95%) of cases (100% (37/37) of high-confidence genotypes). The remaining inversions (n = 83), marked ‘complex’ in all three trios, were not tested for Mendelian consistency. See Table S3 for a tabular view of the results.
PCR validation.
As a further line of validation for the callset, we subjected 10 randomly selected breakpoint-resolved inversions (length: 0.8–366 kbp) outside of L1-internal sequences to site and genotype validation via PCR. Primers were designed using a computational SV validation primer design pipeline, as previously described (Sudmant et al., 2015), which is in turn based on the Primer3 method (Koressaar and Remm, 2007) and available at https://github.com/zichner/primerDesign. Briefly, the pipeline uses an iterative approach to extract two uniquely mapping, inversely oriented primers pairs per inversion, with each pair mapping to opposite sides of one inversion breakpoint. Utilizing sequences generated with primerDesign, combinations of primer pairs were then systematically tested for PCR amplification in supposed inversion carriers and non-carriers to validate genotypes. We designed four primers per inversion: primer 1 (P1) and primer 3 (P3) are designed flanking the leftmost breakpoint of the inversion site and would amplify a region spanning the breakpoint in a reference (direct) orientation. Primer 2 (P2) and primer 4 (P4) are analogously designed flanking the rightmost breakpoint and would amplify a region spanning the breakpoint in a reference orientation. In an inverted locus, P2 and P3 would switch orientation and the combinations P1+P3 or P2+P4 would no longer yield a PCR product, as both primers would be in the same orientation. In an inverted orientation, P1+P2 and P3+P4 combinations would be productive and yield an amplification around the breakpoint. PCR primers were obtained from Sigma-Aldrich at 100 μM concentration in H2O. Genomic DNA was either ordered from Coriell or extracted from cultured cell lines with Qiagen’s QIAamp DNA Blood Mini Kit and set to 10 ng/μl concentration. PCR was done with ThermoFisher’s Phusion Human Specimen Kit using the 20 μl reaction volume consisting of 10 μl of 2x reaction buffer, 0.4 μl Phusion polymerase, 7.6 μl of H2O, 1 μl of 10 μM primers (final concentration 1 μM), and 1 μl of 10 ng/μl genomic DNA. Cycling conditions were: 98C 5min, 34x cycles of 98C 1sec, 63C 5sec, 72C 2min, 72C 5min, 4C hold. Amplified fragments were visualized in 2% agarose gels containing SybrSafe (ThermoFisher).
Inversion site verification using ONT reads.
We used publicly available ONT reads from three samples—HG002, HG00733 and NA19240 (Data and Code Availability)—in order to verify inverted regions in our integrated inversion callset outside of L1 insertion sequences (n = 399). First, we mapped ONT reads onto the reference genome (GRCh38) with minimap2 (Li, 2016) (version 2.20-r1061) using the following parameters: --secondary = no -z 400,0 -r 100,1k. All alignments were reported in the PAF format. We processed each alignment that was no further than 1 kbp from the predicted inversion breakpoints. For further analysis we kept only those ONT reads that showed a split alignment with at least one plus and one minus oriented alignment. Next, we calculated the fraction of bases contributed by inverted and direct alignments inside and outside of the inversion range. We considered an inversion to be supported by ONT reads if there were at least three split-read mappings. We also required that the fraction of inverted base pairs inside an inverted region is at least 0.5 higher than the fraction of inverted base pairs outside of the inverted region (Data S1). The ONT data validated inversions at 107 (~53%) sites, with a bias for orthogonally supporting small inversions consistent with the reduced accessibility of larger inversions to long reads (Figure 1D).
Intersection of our inversion discovery set with prior inversion studies.
To seek additional support for our inversion calls outside of L1-internal sequences, we compiled inversion callsets from six recent studies (Audano et al., 2019; Chaisson et al., 2019; Giner-Delgado et al., 2019; Puig et al., 2020; Sanders et al., 2016; Sudmant et al., 2015). Inversions by Giner-Delgado et al. and Puig et al. were merged into a single dataset (denoted ‘Caceres’ based on the shared last author). We transferred inversion coordinates from Giner-Delgado et al. from hg18 to hg38 using the UCSC Genome Browser hosted liftOver tool, requiring a minimum ratio of bases that must remap of 0.5. At the merging step we removed two redundant inversions (HsInv0241 and HsInv0389) into a final set of 63 inversions. Next, we preprocessed the inversion callset by Sanders et al. by lifting hg19 coordinates to hg38 using the same procedure as described above. In this study inversions are divided into three groups: polymorphic inversions, male invertome, and female invertome. We successfully managed to lift inversions for the majority of inversions except for two polymorphic events – one male, and one female inversion. Two additional inversions were removed as their coordinates were lifted to alternative contigs. We merged all remaining inversions from Sanders et al. into a single callset (n = 251). Lastly, we preprocessed the inversion callset by Sudmant et al., which contains 272 sites with support by another orthogonal technology (short reads). All coordinates were lifted from hg19 to hg38 as described above. We then compared our inversion callset (n = 399; for inversions outside of L1-internal sequence) separately to each study by reporting for each inversion site the site from the published callset with the highest reciprocal overlap using the primatR (Porubsky et al., 2020a) function ‘getReciprocalOverlaps’. We assumed an inversion site is orthogonally supported if it shares 50% reciprocal overlap with an inversion site in a prior study that was reported using an orthogonal genomic technology. We assumed that an inversion site is novel if it has no or less than 10% reciprocal overlap with a reported inversion site from prior studies (Audano et al., 2019; Chaisson et al., 2019; Giner-Delgado et al., 2019; Puig et al., 2020; Sanders et al., 2016; Sudmant et al., 2015).
Identifying potential inversion carriers using PanGenie.
In order to find other potential carriers of the pericentromeric inversion on chromosome 2 detected in NA19650 and the 5 Mbp inversion on chromosome 15 detected in HG02492, we considered SNP alleles present in the inversion haplotypes of these two samples as follows. We used the HGSVC freeze 4 SNP genotypes (Ebert et al., 2021) available for all 3,202 1KG samples (Byrska-Bishop et al., 2021) (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/release/v2.0/PanGenie_results/) to determine which of these SNPs are rare (allele frequency ≤0.01 across unrelated samples). In HG02492 we identified 103, and for NA19650 we detected 333 such rare SNPs within the respective inverted segments. We then counted these rare alleles in the genotypes of all 3,202 samples. Those samples that share a high number of rare SNPs with the respective inversion haplotype were considered potential carriers of the inversion. For the inversion on chromosome 15, we identified four samples sharing a high number of rare SNP alleles with HG002492: HG002491 (102/103 alleles in common; 99%; mother of HG02492), HG02784 (101/103; 98%), HG02725 (74/103; 72%), and HG03639 (74/103; 72%). For the pericentromeric inversion on chromosome 2, sample NA19648 (mother of NA19650) shared 330/333 (99%) rare alleles with the respective haplotype-resolved segment in NA19650.
FISH validation.
Metaphases were obtained from eight human lymphoblast cell lines (NA19648, NA19650A, HG03639, HG02784, HG02725, HG02491, HG02492 and NA12878 as control). Two-color FISH experiments were performed using human fosmid (n = 2) or BAC (n = 2) clones directly labeled by nick-translation with Cy3-dUTP (PerkinElmer) and fluorescein-dUTP (Enzo) as previously described (Lichter et al., 1990), with minor modifications. Briefly, 300 ng of labeled probe was used for the FISH experiments; hybridization was performed at 37°C in 2xSSC, 50% (v/v) formamide, 10% (w/v) dextran sulfate, and 3 mg sonicated salmon sperm DNA in a volume of 10 mL. Post-hybridization washing was at 60°C in 0.1xSSC (three times, high stringency). Metaphases were simultaneously DAPI stained. Digital images were obtained using a Leica DMRXA2 epifluorescence microscope equipped with a cooled CCD camera (Princeton Instruments). DAPI, Cy3, Cy5, and fluorescein fluorescence signals, detected with specific filters, were recorded separately as grayscale images. Pseudocoloring and merging of images were performed using Adobe Photoshop software. Since the tested inversions were >2 Mbp, two-color FISH on metaphase chromosomes was performed using two probes within the inverted region and the centromere as anchor. In Figure 1F and Data S1, probe ABC8-2121940H19 (red) maps at chr2:88223569-88269173, and probe WI2-1849B17 (green) maps at chr2:110712025-110745244. Probes ABC8-41788900G7 (red) and RP11-640H21 (green) mapping at chr15:23751929-23796236 and chr15:27894428-28091240, respectively, were used for the experiments in Figure 6F and Data S1.
Identification of inversions flanked by repeats and mobile elements.
Inverted repeats flanking inversions were extracted from the dot plot alignments, with a lenient 50bp cutoff to define breakpoint homology. Inversions were further considered as mobile element insertion (MEI)-flanked if >90% of each flanking repeat sequence was overlapped by exactly one mobile element annotated in RepeatMasker 4.1.2 (Tarailo-Graovac and Chen, 2009). In 39% of MEI-flanked inversions (12/31), the balanced inversion breakpoints map to full-length mobile elements (6/22 (27%) for L1 pairs, and 6/9 (67%) for Alu pairs on both flanks), whereas for the remainder (19/31) at least one inversion breakpoint maps to a truncated element. Assembled sequences of Alu/Alu flanked inversions (n = 9) were further aligned to the hg38 reference genome, revealing nearby sequence gains or losses in 6/9 cases (35–701 bp in size). We find that L1-flanked inversions are, on average, 8.1-fold larger (median: 5.6 kbp) than Alu-flanked inversions (685 bp; p = 0.038, two-sided t-test). In contrast, the fully assembled SD-mediated inversions (n = 101) are larger than either class of mobile element mediated event (median: 9.0 kbp).
Focussing on 28 HiFi-based haplotype assemblies, we manually investigated all identified SD-associated inversion loci for signs of additional complexity and diversity near the inversion flanks. Such additional complexities include polymorphic insertions or deletions, tandem duplications of SDs or unique sequences or combinations of those. The majority of such polymorphisms (11/15, 73%, Fisher’s test for enrichment n.s.) appear adjacent to recurrent inversions (Table S4).
Excess in common inversion polymorphisms in the genome.
We compared the growth rate of our balanced inversion callset (n=292 events) to insertion and deletion SVs. Since we were evaluating inversions excluding putative misorients (inversions found in all haplotypes), we also excluded variants found in all haplotypes from the SV and SNP callsets to avoid biasing allele frequency and growth rate comparisons. Singletons are defined as variant calls with an allele count of 1. Statistics were computed per haplotype (two haplotypes per sample). Callset growth rate was computed for each haplotype by taking the number of singletons and dividing by the callset size less the number of singletons, which is the proportion of growth if the whole callset was constructed and that haplotype added. This was computed for each haplotype, and the mean growth rate was reported. The supporting p-value was computed over the growth rate for all samples, computed for each variant type, and testing two variant types with a two-tailed Student’s t-test assuming independence. Currently, we estimate an increase of only 0.2% for each new human haplotype added for balanced inversions. By comparison, we find a 0.48% callset growth for SV insertions and deletions, which does not differ significantly from the SNP growth rate (p = 0.45, t-test). Although African genomes exhibit the greatest diversity (1000 Genomes Project Consortium et al., 2015), we do not observe a noticeable increase in the inversion discovery rate for the 13 African samples, which indicates that adding new African genomes would not yield significant numbers of new inversions. Note that for visualization purposes, putative misorients (n = 38) were included in Figure 3A, however, these misorients are not considered in the analysis described above.
We noticed an excess in common (MAF>5%) inversion alleles (67%) compared to other SV classes (48%) and SNPs (47%), which likely explains the callset growth rate. A test of significance of allele frequency cutoffs was conducted by splitting the callset by allele frequency (<0.05 and ≥0.05) and comparing the counts with a two-tailed Fisher’s exact test. For this test, we corrected the SV insertion and deletion (SV INS/DEL) callset variant sizes from Ebert et al. to eliminate differing distributions from smaller SVs, and we eliminated all shared SV INS/DEL variants to maintain compatibility with the inversion callset. Before correction, 52% of the SV INS/DEL calls (n: inversion (INV) = 292, INS/DEL = 105,913) had an allele frequency ≥5% with a p-value of 7.85×10−8 (SV INV vs SV INS/DEL split at 5% allele frequency, two-tailed Fisher’s exact test). When SV INS/DEL variants were restricted to events 300 bp or greater (to better match the callsets by size) as the minimum inversion size (n: INV = 292, INS/DEL = 38,734), we observed 48% with allele frequency ≥5% (p = 2.63×10−11), which becomes 29% (p = 1.83×10−8) for SV INS/DEL ≥10 kbp (n: INV = 203, INS/DEL = 906). This strongly supports the observation that uncommon inversion alleles (<5% AF) are significantly depleted when compared to SV insertions and deletions of similar size and that the results remain significant whether or not they are corrected for similar SV size.
Toggling indicating SNP-based analysis of inversion recurrence.
We developed a statistical tiSNP-based approach that detects, using the haplotype-resolved Strand-seq data, evidence of inversion recurrence by individually considering the occurrence of biallelic SNPs within an inverted locus (Figure 3B). The decision of whether a SNP suggests inversion recurrence is made based on how often each of its alleles occur in an inverted/non-inverted haplotype across all samples. On the basis of aggregated evidence across all SNPs, the inversion is then termed as ‘recurrent’ or ‘single-event’. We based this analysis on the set of 279 balanced inversions from the autosomes and chromosome X.
The analysis steps are as follows:
For each biallelic SNP within an inversion, the number of Strand-seq reads in Watson (W) and Crick (C) orientation were recorded. The reads were further filtered for quality by removing secondary alignments, duplicates, and reads with mapping quality lesser than ‘10’. The read counts were maintained individually for each single cell per sample. For this analysis, we only consider biallelic SNPs with allele frequency ≥5% because with rarer SNPs the method does not have sufficient power to detect evidence of recurrence. This SNP filtering led to the removal of 27 inversions, leaving us with 252 that could be further tested. Using the background/normal cell state, these strand notations were translated to ‘forward/non-inverted’ and ‘reverse/inverted’ notation. For example, if the background cell state is ‘CC’, all the Watson reads mapping to the SNP would be termed as ‘inverted’ while all the Crick reads would be termed as ‘non-inverted’. For ease of comparison to the normal cell state, only ‘WW’ and ‘CC’ cells from each sample were considered for further analysis.
At this point, we had a record for each ‘within inversion’ SNP per single cell, indicating how often we observed each SNP allele in the ‘inverted’ and ‘non-inverted’ state. These occurrence counts were then aggregated first across all single cells and then across all samples, resulting in a table that stored for each SNP the occurrence of each possible SNP-inversion haplotype configuration.
The next step was to identify tiSNPs. Theoretically, observing each SNP allele in both ‘inverted’ and ‘non-inverted’ haplotype at least once indicates that the inversion recurred, but to account for the ‘background’ reads in the wrong orientation (~5% in Strand-seq data), a SNP was termed as a ‘toggling-indicating SNP’ or tiSNP, only if each of the four possible SNP-inversion configurations was seen at least thrice, i.e., each of its alleles had at least ‘3’ ‘inverted’ and ‘non-inverted’ reads mapped to them. For a quantitative assessment, for each inversion, a record of the fraction of tiSNPs compared to the total number of considered SNPs was maintained.
For 49/252 inversions, we observed at least one tiSNP. To make sure our approach is detecting true evidence of recurrence, the analysis was applied to a control set consisting of random, non-inverted regions of the human genome with the same size distribution as the inversion callset. Only 0.02% of the randomized intervals showed evidence of recurrence with the fraction of tiSNPs being extremely low (≤0.002). Using this approach, there appears to be an extremely low possibility of seeing a recurrence signal randomly, which supports the claim that it enables inferring ‘true’ signals of inversion recurrence. For visualization purposes (see Figures 3C–E) we distinguish ancestral and derived SNP alleles determined using a chimpanzee genome (PanTro6).
Quality control of recurrent inversions detected by the tiSNP-based method.
In order to provide further evidence for the reliability of the tiSNP method, we aimed to confirm that the detected inversions are consistent with mechanistic models of mutational recurrence. Mechanistically, inversion recurrence is likely to be mediated by homologous inverted sequences flanking both inversion breakpoints (i.e., NAHR) and we hence focused our attention on the flanking sequences of each genomic locus in question. In this scenario, we assumed that the longer the flanking inverted repeat, the higher the chance that an inversion is found to be recurrent. To test whether our analysis is consistent with this model, inversions labeled as ‘recurrent’ were compared to the ones showing no tiSNPs, in terms of length of the longest flanking inverted repeat sequence. Only the repeats with one end lying within 20 kbp (−10 kbp to +10 kbp around each annotated breakpoint) and extending up to 70 kbp flanking region were considered. The inversions showing evidence of recurrence turned out to be clearly enriched for longer flanking inverted repeats with fraction of tiSNPs increasing with increasing length, while the ones where we did not observe any recurrence signal showed enrichment for shorter flanking inverted repeats (Data S1).
Another quality check was to make sure that the analysis is not driven by inversion length, because a recurrence signal (i.e., tiSNPs) is more likely to be observed in longer inversions with more SNPs as compared to small inversions with fewer SNPs. Additionally, the length of flanking inverted repeats is directly associated with inversion length (Fig. S1D). Therefore, to confirm that the observed relationship between length of flanking inverted repeat and the fraction of tiSNPs does not involve inversion length as a confounder, a multiple linear regression model was used. The model expressed the fraction of tiSNPs as a function of inversion length, length of the longest flanking inverted repeat, and MAF (we considered MAF, since low MAF is likely to decrease the statistical power of detecting recurrence). The regression results clearly suggested that the recurrence signal detected by the analysis is primarily influenced by the length of flanking inverted repeats and MAF (p= 7.5×10−4 and p= 2.65×10−6, respectively), with inversion length having no significant influence (p= 0.835). The results of the tiSNP-based method are therefore consistent with the model that NAHR drives the recurrence of inversions at loci comprising long inverted SDs at their flanks.
Haplotype-based coalescent approach for detecting inversion recurrence.
As a second approach, we developed a haplotype-based coalescent approach that considers four lines of empirical evidence for inversion recurrence, using phylogenetics and population genetics methods: 1) haplotype-based principal component analysis (PCA), 2) haplotype identity by state, 3) window-based phylogenetic tree reconstruction, followed by bootstrap analysis, and 4) reconstruction of ancestral recombination graphs using Relate (Speidel et al., 2019). Input VCFs were created using the procedure described in Methods section ‘Phasing and correction of chromosome-length inversion haplotypes.’ Individuals who we failed to unambiguously assign an inverted haplotype were removed from any analysis. In addition, as a QC filter we excluded SNVs mapping to SDs (sequence identity >98%) and heterochromatic satellites including centromeres and telomeres. To ensure the quality of our analysis, we focused on 127 inversions from chromosome X and the autosomes showing sufficient unique sequence and at least 10 SNVs within an inversion to construct haplotypes. We describe each method separately and provide a detailed description of the procedures as follows:
PCA: We performed a haplotype-based PCA following the description of Browning et al. (Browning et al., 2016). Briefly, each haplotype base was encoded numerically, where 0 and 1 (and 2 if needed) represent ancestral (using the chimpanzee assembly as the outgroup (Kronenberg et al., 2018)) and derived alleles, respectively. We used the R package irlba (v2.3.3) to calculate and visualize principal components and keep track of direct and inverted alleles.
Haplotype identity by state: Identity by state is defined as the proportion of matches between a certain pair of haplotypes. Pairwise differences between haplotypes were visualized and grouped based on inverted versus direct orientations.
Ancestral recombination graph reconstruction: We used the software Relate (Speidel et al., 2019) to reconstruct the local genealogy of an inversion locus. The method infers underlying coalescent events consistent with the observed data under the infinite-sites model, taking into account recombination, and thus, recapitulates the multi-locus genealogy of the genomic region. We used a mutation rate of 1.25 × 10−8 per base per generation and set the haploid effective population sizes as 10,000 (Gutenkunst et al., 2009) and 6,900 (Veeramah et al., 2014) for autosomal and X chromosome loci, respectively. We projected the inversion genotype onto the resulting trees and inferred the number of inversion events using Fitch’s algorithm for homoplasy (Fitch, 1971). The rate of inversion at each inversion locus is defined as the estimated number of inversion events divided by the total tree length in generations. Note that for a given tree, a monophyletic inversion group indicates a single origin of these inverted haplotypes, while a polyphyletic group of inverted haplotypes suggests that these inverted haplotypes are derived from more than one common ancestor and, thus, recurrent. To evaluate the consistency of topology across inferred trees, we used generalized Robinson-Foulds distances implemented in the R package TreeDist (v2.1.1).
Phylogenetic tree reconstruction: For phylogenetic tree reconstruction, we applied the maximum likelihood-based software IQ-TREE (v2.1.3) using the following options: ‘-keep-ident -redo -bb 1000 -m MFPMERGE --date date.file --date-options “-u 0” --clock-sd 0.4 --date-tip 0 ‘. We assumed six million years of divergence between human and chimpanzee (specified in the date.file). Firstly, for each inversion locus, we inferred phylogenetic trees for the entire locus and for 100 block-bootstraps and estimated the number of inversion events for individual trees to assess confidence. Secondly, we sliced the region into 2,000 and 20,000 bp windows for loci smaller and greater than 20,000 bp, respectively, and inferred a local phylogenetic tree for each window. We again used the Robinson-Foulds distances to evaluate the consistency of tree topology among inferred trees across the locus and computed the number of independent inversion events for each local tree. For each tree, the number of events was divided by the total tree length to obtain inversion rates.
Measuring uncertainty: Finally, for each inversion under consideration, we computed three intervals to measure the uncertainty of the inferred inversion rates using 1) a 95% central interval, computed based on the 2.5 and 97.5 percentiles of the estimates from all marginal trees inferred by Relate at a locus; 2) a similar 95% central interval computed from the local trees built using the window-based phylogenetic tree reconstruction; and 3) a 95% confidence interval constructed using the 100 block-bootstrap trees for the entire inversion locus. We determined that an inversion is recurrent if all three intervals indicate at least two independent origins of the inversion. Unless mentioned otherwise, for each inversion tested, we reported the 95% central interval computed based on the inference of Relate throughout the paper as this interval tends to be wider and therefore more conservative.
Analysis of the genetic architecture of recurrent inversion loci.
Out of the subset of 127 balanced inversion sites mapping to autosomes and the X chromosome that passed QC filters, there were 93 inversions where both approaches for identifying inversion toggling agreed. Of those, 32 were labeled as recurrent and 61 as consensus single-event inversions. The remaining 34 out of 127 inversions were only called as recurrent by one of the two approaches, likely due to differences in sensitivity and specificity in detecting recurrence between the two approaches (Figure 3B). Among the recurrent sites, we observed a significant excess of toggling inversions on the X chromosome compared to the autosomes (odds ratio: 27.2, 95% C.I.: [2.55, 142.4]; p = 1.2×10−4, chi-squared test; Figure 4B), suggesting X-biased recurrence of inversions; however, among the consensus set of 32 recurrent inversions, we detected no significant difference in inversion rates between the X chromosome and the autosomes (p = 0.43; Mann-Whitney U test).
Next, we used the consensus set of recurrent inversions to revisit the relationship of the recurrence status to the architecture of flanking sequences (see also Quality control of recurrent inversions detected by the tiSNP-based method): 70% (23/32) of recurrent inversions exhibit ≥10 kbps of flanking inverted repeat sequences with high (≥79%) sequence identity (Table 1). Flanking inverted repeat length and repeat sequence identity are themselves strongly correlated (Pearson’s correlation: 0.63, p = 1×10−11). However, a multivariate logistic regression analysis performed using the full consensus set (93 inversions, 32 recurrent and 61 single events) confirms that the major driver for inversion recurrence status is flanking inverted repeat length (p = 0.0072), while neither repeat sequence identity nor inversion length have any significant influence (p = 0.31 and p = 0.86, respectively).
Furthermore, inversion recurrence within human population history may either (i) affect the same segment along the same lineage multiple times leading to a segment toggling back and forth in orientation (‘serial’ toggling), or (ii) affect the same segment in different parts of the tree – whereby all detected events invert segments into the same (non-reference) orientation, which may imply irreversibility (non-serial toggling). Based on our analysis of the trees inferred through our study, we find that 23 out of the 32 recurrent inversions we describe on the autosomes and chromosome X show evidence for serial toggling, while the remaining 9 loci exclusively harbor non-serial toggling events (Table S5). As an example, the recurrent chromosome 8p23 and 11p11 inversions, highlighted in the main text (Figure 3D–E), show evidence for serial toggling events in part of the trees (Data S1). By comparison, inversions at chromosomes 2q11 and 7p22 represent examples for non-serial toggling events (Data S1). However, we caution that our sampling of diversity is still incomplete; as we begin to sample more haplotypes, it is likely that more inversions will show evidence of serial toggling.
Chromosome Y inversion genotyping.
ArbiGent, at present, is not well tailored to genotype haploid chromosomes such as chromosome Y in males. Because of this caveat we decided to re-genotype reported chromosome Y inversions (n = 15) using Strand-seq data only based on the binomial distribution of Crick (plus) and Watson (minus) reads. For this purpose we used the R function ‘genotypeRegions’ implemented in R package primatR (Porubsky et al., 2020a). We required a minimum of five Strand-seq reads (min.reads = 5), in order to report a genotype and allowed for 10% of background reads (alpha = 0.1). Genotypes for chromosome Y inversions are reported in a supplementary table (Table S3).
Construction and dating of Y phylogeny and Y inversion rate estimation.
We called the genotypes of 17 samples (16 males included in the current study plus NA19384 used to root the Y phylogenetic tree) jointly from the 1KG high-coverage WGS data (n = 3,202 samples) using the ~10.3 Mbp of chromosome Y sequence previously defined as accessible to short-read sequencing (Poznik et al., 2013). BCFtools (v1.9) was used with minimum base quality and mapping quality 20, defining ploidy as 1, followed by filtering out SNVs within 5 bp of an indel call (SnpGap) and removal of indels. Additionally, we filtered for a minimum read depth of 3. If multiple alleles were supported by reads, then the fraction of reads supporting the called allele should be ≥0.85; otherwise, the genotype was converted to missing data. Sites with ≥6% of missing calls across samples were removed using VCFtools (v0.1.16). After filtering, a total of 10,407,641 sites remained, including 5,494 variant sites.
The Y haplogroups of each sample were predicted from the all-site vcf file with yHaplo software (https://github.com/23andMe/yhaplo) using a version where the Y marker coordinates in the relevant input files had been replaced to correspond to the GRCh38 assembly (Bergström et al., 2020). The identified terminal marker SNV for each sample was used to update the haplogroup name to correspond to the International Society of Genetic Genealogy nomenclature (ISOGG, https://isogg.org, v15.73) (Table S3). We used the coalescence-based method implemented in BEAST (v1.10.4 (Drummond and Rambaut, 2007) to estimate the ages of internal nodes in the Y phylogeny. A starting maximum likelihood phylogenetic tree for BEAST was constructed with RAxML (v8.2.10 (Stamatakis, 2014)) with the GTRGAMMA substitution model using all sites. Markov chain Monte Carlo samples were based on 100 million iterations, logging every 1000 iterations. The first 10% of iterations were discarded as burn-in. A constant-sized coalescent tree prior, the HKY substitution model, accounting for site heterogeneity (gamma) and a strict clock with a substitution rate of 0.76 × 10−9 (95% confidence interval: 0.67 × 10−9 – 0.86 × 10−9) single-nucleotide mutations per bp per year was used (Fu et al., 2014). A prior with a normal distribution based on the 95% confidence interval of the substitution rate was applied. A summary tree was produced using TreeAnnotator (v1.10.4) and visualized using the FigTree software.
In order to estimate the inversion rate, we counted the minimum number of inversion events that would explain the observed genotype patterns in the Y phylogeny. A total of 4,419 SNVs called in the set of 16 analyzed males and Y chromosomal substitution rate from above was used. A total of 126.4 years per SNV mutation was then calculated (0.76 × 10−9 × 10,407,641 bp)−1, which was converted into generations assuming a 30-year generation time (Fenner, 2005). Each SNV thus corresponds to 4.21 generations, translating into a total branch length of 18,623 generations for the 16 samples. For a single inversion event in the phylogeny this yields a rate of 5.37 × 10−5 (95% CI: 4.73 × 10−5 to 6.08 × 10−5) mutations per father-to-son Y transmission. The confidence interval of the inversion rate was obtained using the confidence interval of the SNV rate. For the inversion recurrence analysis we focused on a subset of 11 balanced inversions—excluding two inversions seen on all 16 Y chromosomes, which represent minor alleles in GRCh38 or misorientations, and excluding two events with low genotype quality exhibiting too few mapped reads (Table S5).
In support of our measurements, we estimated a genotype concordance of 100% for four chromosome Y inversions identified and genotyped by both Strand-seq and Bionano (Table S3). For example, the ~3.3 Mbp IR3/IR3 inversion was previously reported to toggle at least 12 times in recent human history, with an estimated rate of ≥2.3 × 10−4 per father-to-son Y transmission (Repping et al., 2006). We identified this inversion in an African (NA19239) and a Southeast Asian (HG03732) individual carrying Y lineages E1a2a1a1a-CTS1792 and R2-L266, respectively, closely related to those previously reported to be inverted (Figure 4A, Data S1). Our estimated inversion rate based on two events across 16 male samples is 1.07 × 10−4 (95% C.I.: 0.95 × 10−4–1.22 × 10−4) per generation, which is close to the published estimate. Our analyses show particularly extensive inversion toggling among large inverted SDs with >99.9% sequence identity (also referred to as Y palindromes in the literature), elements previously thought to be prone to inversion formation (Lange et al., 2009; Repping et al., 2002). The extent and rates of inversion recurrence within these structures were previously incompletely understood. We find inversions for six of the eight Y palindromes, out of which five show mutational toggling, with two (P4; ~190 kbp long SDs) up to five (P3, P5 and P6 – 110 kbp to 495 kbp long SDs) identified inversion recurrences (Figure 4A). Previously, a per-generation rate of 1.36–1.72 × 10−5 was estimated for P6, but due to the technical limitations, only inversions with breakpoints in the outer ~16% of palindrome arms could be identified and therefore this rate has been considered an underestimate (Hallast et al., 2013). An inversion of palindrome P3, containing some of the copy number variable RBMY1 genes, was previously reported as a single inversion event based on fiber FISH experiments in a panel of 14 male samples (Shi et al., 2019a). In contrast, we identify a ~180 kbp long recurrent inversion at P3 (~284 kbp long flanking arms) with five separate inversion events. We estimate an inversion rate at P3 of 2.68 × 10−4 (95% C.I.: 2.37 × 10−4 to 3.04 × 10−4) per father-to-son Y transmission. We further observe an inverted duplication in an African male (NA19239), affecting a ~118 kbp segment overlapping with this region (Data S1), consistent with extensive structural variability of this genomic region (Shi et al., 2019a).
Detection of nested inversions and events with imprecise breakpoint reuse.
We also identified imprecise breakpoint reuse in further support of inversion recurrence. SDs that drive NAHR occur in large blocks with multiple substrates for unequal crossover (Antonacci et al., 2014), which facilitates recurrent inversion formation with disparate breakpoints, leading to a shift in inversion coordinates on different haplotypes; such loci, therefore, are not necessarily classified as recurrent by our tiSNP- and haplotype-based coalescent approaches. We first determined inversions that are completely embedded within another inverted range in our callset (n = 33). Additionally, we compared genotypes of all possible pairs of inversions and kept those that consistently share the same genotype across all samples (n = 17). We compiled these two sets of inversions into a nonredundant candidate list of inversions with potentially shifted breakpoints (n = 19). We removed any sites that involve putative misorients. We manually inspected binned read counts of Strand-seq data over each candidate region and selected three of the most confident regions where an inversion breakpoint shift is plausible. These three regions (2q21, 12q24 and 16p12) show distinct, albeit largely overlapping, inversions (Data S1), suggesting that a given region was subject to recurrent change in orientation. All three regions are flanked by large SDs supporting that the underlying SD architecture was disparately used to create distinct inversions with shifted breakpoints (Data S1).
We additionally identify two nearby inversions on the X chromosome with highly correlated genotypes, with an additional inversion residing in between (Data S1). Manual inspection revealed that these events comprise a small (41 kbp) inversion fully nested within a larger (165 kbp) inversion (Data S1). The nested segment was identified as toggling using our tiSNP- and haplotype-based approaches (Table 1) and, additionally, was subject to inversion during primate genome evolution (Data S1). These data suggest that inversion recurrence is occasionally associated with disparate, partially overlapping DNA rearrangements in regions of high SD density.
Finally, we clustered the balanced inversion breakpoints by genomic location and, doing so, identified 30 inversion hotspots (Table S2), six of which map adjacently to centromeric satellite regions (<1 Mbp). Chromosomes 1, 2, 7, 10, 16, 20, X and Y appear particularly enriched, showing 10 or more inversion breakpoints in a single hotspot (Figure S1B), consistent with extensive clustering of inversions at genomic sites rich in SDs.
Inversions affecting the orientation of SD pairs.
We devised a computational approach that systematically scans all identified inversions for their potential to change the relative orientation of pairs of SDs in the genome, by inverting one SD out of a pair. Starting from an annotated set of 69,906 annotated SDs of >90% sequence identity obtained through the UCSC Table Browser (Karolchik et al., 2004), we selected SDs longer than 10 kbp and with homologous partners on the same chromosome (Mefford and Eichler, 2009), resulting in 7,672 SDs, representing 3,795 one-to-one pairs. We then identified SD pairs in which one but not both partners are embedded within the same inversion, yielding 2,265 SDs (1,094 SD pairs). Filtering on the level of inversions next, we identified 79 inversions flipping the orientation of at least one SD pair, with a median of eight pairs reoriented. Out of these, we identified 29 inversions that predominantly (>90% of flipped SD pairs weighted by length) affect SD pairs in direct or in inverse orientation. We classify these inversions as ‘potentially protective’ (n = 9) and ‘potential pre-mutational state’ (n = 20), respectively. Morbid CNVs from the decipher database (Bragin et al., 2014) were additionally intersected with this set of 29 inversions, identifying 6 inversions overlapping with 8 distinct morbid CNVs (Table S6).
eQTL analysis.
Utilizing deep transcriptomic data available for 33/44 of the samples (Data and Code Availability), the set of 399 inversions outside of L1-internal sequences was tested for association with expression in nearby genes together with a set of 41,833 deletions, 66,825 insertions, and 16.4 million SNPs as previously reported in Ebert et al. (2021). Before RNA-seq read mapping, adapters and low-quality reads and bases were removed using Trim Galore (Andrews et al., 2015). The remaining reads were mapped to GRCh38 using STAR aligner in 2-pass mode (Dobin et al., 2013). Leveraging sample-specific SNP calls based on GRCh38 as reported previously in Ebert et al. (2021), alignment to this genome reference was performed with WASP filtering (van de Geijn et al., 2015) to mitigate allelic mapping bias. To prepare the data for haplotype-unaware eQTL analysis, reads were quantified with respect to GENCODE v35 genome annotation (Frankish et al., 2019) using featureCounts (Liao et al., 2014). Read counts were normalized using the weighted trimmed mean of M-values (TMM) normalization implemented in edgeR (Robinson et al., 2010) and transformed to the transcripts-per-million (TPM) metric.
We next identified expressed genes (TPM > 0.5 in at least 5 samples) located in a window of 2 Mbp centered on each inversion breakpoint. This resulted in 4,469 genes to be tested for association with all variants overlapping a 2 Mbp window centered around the gene. All variants with MAF ≥1% were considered, resulting in a total of 13.4 million gene-variant pairs to test. eQTL tests were performed using a pipeline based on nonlinear mixed models, implemented in LIMIX (Lippert et al., 2014). PLINK (v.1.90) was used to estimate genetic ‘kinship’-matrices between samples based on SNP and indel variants, which served as a basis for population principal components used as latent factors in the model. As additional cofactors, we used the principal components of the gene expression matrix to account for remaining systematic expression biases. We found n = 4 covariates for bias correction (Expression-PC1, Population-PC1&2, Sex) to maximize the number of discovered eQTLs, with larger numbers of covariates showing signs of overfitting due to the relatively low sample size. The results of the eQTL mappings were initially corrected for multiple testing both on the level of tested variants per gene and the level of number of genes tested, as described in Ebert et al. (2021). Variant-level correction was performed via genotype permutations as implemented in LIMIX, while the number of genes was corrected for using a Storey Q-value–based procedure. Using this rigorous global correction, we find 166 globally significant eQTLs (1 INV, 1 DEL, 164 SNVs). In parallel to this all-variant approach, we also analyze inversion eQTLs separately, allowing us to reduce the number of tests to correct for by a factor of ~6,600 and recover significant inversion eQTLs that would otherwise be overshadowed by the large number of variants tested. We used Benjamini-Hochberg correction on the level of inversion-gene tests, yielding 11 globally significant (FDR < 0.2) inversion eQTLs (11/2,007 tested sites), compared to 0.09% (56/59,464) of deletion-gene pairs and 0.04% (33/85,122) of insertion-gene pairs (using SV INS/DEL calls as previously reported in (Ebert et al., 2021). Lead eQTLs in all cases were determined by comparing raw p-values of all variants of a given gene.
Analysis of the inversion eQTLs showed that six inversion eQTLs are associated with the common 17q21.31 inversion, previously reported as an inversion eQTL using targeted genotyping (Giner-Delgado et al., 2019), and this includes two eQTLs where the inversion is the lead variant (MAPK8IP1P2, AC126544.2) surpassing other genetic variants in significance. We further find significant associations of inversions with the expression of ATP13A2, OR4C6, MAGEH1, and RP11-460N20.4.2 (Data S1), again consistent with the aforementioned study. The sample set per population used in this study is relatively small for eQTL mapping yet shows the possibility of associating balanced inversion polymorphisms with gene expression phenotypes to study their functional consequences.
Enrichment analysis of inversions intersecting morbid CNV regions.
We tested whether various subclasses of inversions outside of L1-internal sequences (n = 399), namely balanced inversions (n = 292), consensus single-inversion events (n = 61), and consensus recurrent inversion events (n = 32) are enriched in the vicinity of known pathogenic CNVs (redundant set, n = 155) (Bragin et al., 2014; Coe et al., 2014; Cooper et al., 2011). We used the R package regioneR (Gel et al., 2016) with its function ‘permTEST’ to perform permutation testing (n = 10,000 permutations). At each permutation, we randomized the position of each inversion in every tested subgroup using regioneR’s function ‘circularRandomizeRegions’. This way the relative distance of each inversion is kept, as inversion occurrence on each chromosome is not completely random and highly depends on the underlying SD architecture. At each permutation, we counted the number of inversions overlapping with the redundant set of morbid CNVs, allowing for a 50 kbp gap between each set of coordinates (to account for the fact that recurrent inversion as well as sites of recurrent microdeletions and microduplications are often flanked by an extensive, hard-to-penetrate SD architecture).
Bionano Genomics analysis of 1p36 complex region.
We analyzed the 1p36.13 region by visual inspection of labeling patterns from optical maps. Segment copies were additionally analyzed in the phased assemblies using BLASTN (version 2.9.0+). Bionano Genomics optical maps were manually evaluated to determine the haplotype in each sample. Single molecules were evaluated using Bionano Access (v1.5.2) to determine whether molecules containing SVs (inversions and CNVs) (n≥3) were anchored to proximal and distal unique regions. Samples with PGAS (v13) hifiasm-phased assemblies (Cheng et al., 2021; Ebler et al., 2022) (n = 11) were used as orthogonal support to confirm the haplotypes identified by optical mapping data (see ‘Concordance between Bionano optical maps and phased assemblies in 1p36.13’). To highlight the complexity of the 1p36.13 region, we also visualized differences between the GRCh38 and T2T-CHM13 reference genomes (Nurk et al., 2021) at this locus in a Miropeats-style plot (Figure 6A), using minimap2 alignments.
Concordance between Bionano optical maps and phased assemblies in 1p36.13.
Bionano Genomics optical maps of 35 samples were manually evaluated to determine the haplotype in each sample (n = 35). Contigs aligning to 1p36.13 (16.5–17 Mbp GRCh38, 16–17 Mbp T2T-CHM13) of each sample were used to characterize the structure of the 1p36.13 region. The orientation and copy number of each 1p36.13 segment, which were represented as colored arrows, were determined by using the structure in the T2T-CHM13 genome assembly as a reference. Next, single-molecule data of each contig were evaluated using Bionano Access (v1.5.2). Molecules with a confidence score of less than 30 were not included in the manual evaluation. Molecules were visually evaluated using Bionano Access v1.5.2 to check if the structure of the 1p36.13 region observed on the contig was the same as the haplotype observed in the single molecules. Molecules were also evaluated to check if they were anchored to the proximal or distal unique region of 1p36.13. Haplotypes were considered as true positives if at least three molecules fit the criteria described above. If the molecules were inconclusive to identify the haplotype of the 1p36.13 region, local molecules aligning to the region of interest were extracted, and then local de novo assembly was performed with pipelineCL.py, which is part of the scripts package provided by Bionano Solve v3.5.1.
python2.7 Solve3.5.1_01142020/Pipeline/1.0/pipelineCL.py -T 64 -U -j 64 -jp 64 -N 6 -f 0.25 -i 5 -w -c 1 \ -y \
-b ${bionano_bnx} \
-l ${output_dir} \
-t Solve3.5.1_01142020/RefAligner/1.0/ \
-a Solve3.5.1_01142020/RefAligner/1.0/optArguments_haplotype_DLE1_saphyr_human.xml \
-r ${reference_genome}
We assessed the concordance between the Bionano-guided manual annotation of copy number polymorphic segments and phased assemblies of genomic sequences in 11 samples (22 haplotypes) (HG00512, HG00513, HG00514, HG00731, HG00732, HG00733, NA19238, NA19239, NA19240, NA12878, HG002) for the 1p36.13 locus relative to the T2T-CHM13 v1.1 reference genome (with the reasoning that the GRCh38 1p36.13 structural haplotype could not be found amongst the samples in our diversity panel). Starting from the annotation of copy number polymorphic segments (N = 432) for 11 samples created on the basis of Bionano optical maps, we computed a concordance estimate between this annotation and the corresponding set of phased assemblies in a similar way as described previously (Ebert et al., 2021). For each phased assembly, we cut out those parts of the contig sequences that were identified as a putative segment in the Bionano optical maps. Each segment was then aligned to the T2T-CHM13 1p36.13 locus with minimap2 v2.22 (preset “map-hifi”) and scored as follows: an alignment between assembly segment and reference in the annotated orientation and matching in segment color was counted as fully concordant; alignments matching only in color were counted as “orientation errors” (41.4% of segments) but otherwise concordant; in the case of a split alignment of the segment (9.3% of segments), the alignment with the lowest sequence divergence as reported by minimap2 was scored as described above. This strategy led to a concordance estimate of 89.6% (48.1% fully concordant segments) between phased assemblies and the 1p36.13 haplotypes identified in the Bionano optical maps.
Hardy-Weinberg equilibrium test and multi-allelic sites.
With the inclusion of complex genotypes, several inversion sites in our inversion callset were predicted to be ‘multi-allelic’ – that is, they appeared to involve more than two different allelic conformations across samples, for example, an inverted duplication in one sample and balanced inversion in another. Before testing the genotypes for Hardy-Weinberg equilibrium, the multi-allelic sites were converted into ‘pseudo bi-allelic’ ones. To achieve this, firstly, for each variant, a list of unique alleles and their occurrence count across all samples was generated. Next, all the sample genotypes were transformed by encoding the major allele (the one with highest allele frequency) as ‘1’ and others as ‘0’. The heterozygosity for each variant site was thus determined in terms of the major allele, i.e., all genotypes carrying the major allele and exactly one (any) other allele were termed as heterozygous (0/1). VCFtools (--hardy) was then used to perform Hardy-Weinberg equilibrium test on all inversion sites where no sample genotypes were missing. Furthermore, for multiple testing correction of the resulting p-value for each inversion, Benjamini-Hochberg correction was applied. For the GRCh38 callset (Data S1), 60/399 inversions belonging to sex chromosomes and 64/339 inversions belonging to autosomal chromosomes had at least one missing sample genotype. Therefore, 275/399 inversions were tested for Hardy-Weinberg equilibrium, 224 (81.45%) of which passed (Table S3).
Genotyping Strand-seq libraries from cell pools.
To provide additional support for our inversion callset we designed a pooled Strand-seq experiment, where we analyzed 1KG samples pooled together by mixing cell lines. We pooled together multiple different 1KG lymphoblastoid cell line samples (n = 40 per pool, for a total of n = 120 samples from three pools) of diverse population origin, followed by subjecting these pooled samples to Strand-seq (STAR Methods, Figure S5A). We initiated data processing by alignment of single cell fastq files to the reference genome (GRCh38) to generate cell-specific BAM files as described above. In order to detect SNVs, we first merge BAM files in each pool using the SAMtools (version 1.3.1) function ‘merge’. Such merged BAM files are then processed by the RTG tool (version 3.11) to detect SNV positions using following parameters: snp --min-mapq 10 --min-base-quality 10 --snps-only --no-calibration --machine-errors illumina --max-coverage 30. Next, in each single cell we define haplotype-informative regions (so called Watson-Crick regions) as those where variants from each parental homologue can be defined (Porubský et al., 2016). Such regions can be extracted using the breakpointR function ‘exportRegions’ with the following parameters: collapseInversions = TRUE, collapseRegionSize = 5000000, minRegionSize = 5000000, state = ‘wc’. These regions then serve as an input for StrandPhaseR function ‘genotypeStrandScells’ along with VCF produced by the RTG tool and a set of phased SNVs from 1KG samples. We downloaded a set of phased SNVs for all 1KG samples from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_phased/. The last required input is the path to the folder where aligned BAM files for each pool are stored. We used the StrandPhaseR function ‘genotypeStrandScells’ with the following parameters: min.snv.cov = 2, max.snv.cov = 30, max.snv.per.chr = 10000, blacklist = <Genomic Ranges object containing coordinates of segmental duplication in GRCh38>. In this function we loaded a set of variable positions detected in each pool using the RTG tool and filtered them based on minimum and maximum allowed SNV coverage. In order to speed up the analysis, we allow a maximum of 10000 SNVs per chromosome. We used these SNV positions to extract maternally and paternally inherited alleles in previously defined Watson-Crick regions in each pool. These alleles are extracted from aligned reads stored in cell-specific BAM files. Next, we compared the agreement of alleles in each single-cell Strand-seq library against phased alleles in the 1KG panel per chromosome. Then for each single cell we calculated the fraction of alleles that agree with maternal and paternal haplotype against all 1KG samples. On this basis, for each single cell, we defined the original sample as the 1KG sample with the highest proportion of alleles matching the alleles defined in the given single cell library (Figure S5B–F).
Subsequently, to infer inversion genotypes, we used our Bayesian probability framework, ArbiGent (STAR Methods). Prior to genotyping in the additional samples, we performed a benchmarking experiment to determine ArbiGent’s performance given a small number of single cells per sample. We randomly downsampled single cells (to 1, 2, 3, and 12 single cells) belonging to sample ‘HG00733’ from our diversity panel. For this experiment, we focused on 249 autosomal inversions predicted to be ‘simple’ (balanced) in sample ‘HG00733’ from our callset. For large inversions (containing >50 kbp of uniquely mappable inverted sequence), we observed genotypes from downsampled single cells to be highly consistent with those detected in high-coverage Strand-seq libraries (Figure S5G). Encouraged by this observation, we performed ArbiGent genotyping in all 1KG samples included in the pools. We obtained additional support for 74 inversions containing >50 kbp of uniquely mappable inverted sequence from our callset. Estimated inversion allele frequencies based on the pools matched well with those from the original diversity panel (Figure 5H). The full set of inversion genotypes generated in the pooled samples is available in Table S7.
Inversion breakpoint analysis in high-identity flanking SDs.
We selected two inversions on chromosome 2 to test inversion breakpoint detection within highly identical flanking SDs. One example represents an inversion (ID: chr2-95496991-INV-82806) flanked by short and simple SDs (~82 kbp). The other example is represented by an inversion (ID: chr2-110095179-INV-181032) flanked by large and complex SDs (~315 kbp). For each inverted region we selected one direct and one inverted haplotype using open access phased assemblies from the Human Pangenome Reference Consortium (HPRC) (Data and Software Availability). We defined flanking SDs in each selected region based as highly identical self-alignments. Next, we extracted the sequence for proximal and distal SDs separately for the direct and the inverted haplotype (Figure S2E). In order to be able to create a multiple sequence alignment (MSA), we created a reverse complement of distal SDs both for the direct and the inverted haplotype. MSAs were created using the R package DECIPHER (version 2.22.0). In each MSA, we selected positions where at least two SDs share the same paralogous sequence variant (PSV). We labeled each PSV according to whether it corresponded to the proximal or distal SD. Lastly, we manually defined the change points between PSVs specific for proximal and distal SDs (Figure S2F,G).
Generation of short-read based copy number profiles.
Copy number estimates around the inverted region on chromosome 2 (2q13) and chromosome 16 (16p13-11). Reads were extracted from alignment files generated by the NYGC as part of the 1KG high-coverage sequencing effort (Byrska-Bishop et al., 2021), parsed into 36 bp segments, and mapped with mrsFAST (Hach et al., 2010) allowing an edit distance of 2 to a hardmasked GRCh38 reference genome. Read-depth-based copy number estimates were generated using the FastCN (Pendleton et al., 2018) software package, which uses known copy number stable regions to correct for Illumina sequencing GC bias and convert read depth to diploid copy number over 1,000 base-pair windows.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis related to inversions within L1 insertions.
Mann-Whitney U test was performed to compare the length of inversions within L1 insertions to a background distribution derived from the random sampling of inversion breakpoint positions from a consensus L1 sequence. The same test was applied to evaluate the level of significance in the differences between the length distribution for 5′ truncated L1s and the 3′ sense orientation ends of twin-priming events. Further details of these analyses are provided in STAR Methods (Simulations and evaluation of L1 annotation pipeline).
Analysis of inversion flanked by repeats and mobile elements.
We used a two-sided t-test to determine if there exists a significant difference between the size of L1-flanked and Alu-flanked inversions. Fisher’s exact test was used to test for enrichment of complexities including polymorphic insertions or deletions, tandem duplications of SDs or unique sequences or combinations of those adjacent to recurrent inversions. Further details of these analyses are provided in STAR Methods (Identification of inversions flanked by repeats and mobile elements).
Analysis of excess in common inversion polymorphisms.
We used a two-tailed Student’s t-test assuming independence to compare growth rate between different variant types. Moreover, a test of significance of allele frequency cutoffs was conducted by splitting the callset by allele frequency (<0.05 and ≥0.05) and comparing the counts with a two-tailed Fisher’s exact test. Further details of these analyses are provided in STAR Methods (Excess in common inversion polymorphisms in the genome) and Results section (Inversion discovery saturation and excess of common polymorphisms).
Analysis of influence of flanking inverted repeat length on inversion recurrence status.
A multiple linear regression model expressing the fraction of tiSNPs as a function of inversion length, length of the longest flanking inverted repeat and MAF was used to confirm that the fraction of tiSNPs is being influenced by length of flanking inverted repeat instead of the inversion length. We additionally used Pearson’s correlation to confirm the positive correlation between flanking inverted repeat length and repeat sequence identity and a multivariate logistic regression analysis to confirm that the major driver for inversion recurrence status is flanking inverted repeat length while neither repeat sequence identity nor inversion length have any significant influence. Further details of this analysis are provided in STAR Methods (Quality control of recurrent inversions detected by the tiSNP-based method; Analysis of the genetic architecture of recurrent inversion loci).
Analysis involved in haplotype-based approach for detecting inversion recurrence.
Haplotype-based PCA following the description of Browning et al. (Browning et al., 2016) was performed using the R package irlba (v2.3.3) to calculate and visualize principal components and keep track of direct and inverted alleles. For phylogenetic tree reconstruction, we applied the maximum likelihood-based software IQ-TREE (v2.1.3). Further details of these analyses are provided in STAR Methods (Haplotype-based coalescent approach for detecting inversion recurrence).
Analysis comparing toggling inversions on sex chromosomes and autosomes.
We performed a chi-squared test to determine significant enrichment of toggling inversions on the X chromosome compared to the autosomes. Additionally, we used Mann-Whitney U test to determine the significance of difference in inversion rates between the X chromosome and the autosomes. We used a chi-squared test also to determine the difference between the relative proportion of toggling inversions compared to single-event inversions on the Y chromosome compared to autosomes. Further details of these analyses are provided in STAR Methods (Analysis of the genetic architecture of recurrent inversion loci) and Results section (Rates and genetic architecture of inversion toggling on autosomes and X chromosome).
Tests used in eQTL analysis.
To test association of inversions with expression in nearby genes a pipeline based on nonlinear mixed models, implemented in LIMIX (Lippert et al., 2014) was used. PLINK (v.1.90) was used to estimate genetic ‘kinship’-matrices between samples based on SNP and indel variants, which served as a basis for population principal components used as latent factors in the model. We used Benjamini-Hochberg correction on the level of inversion-gene tests, to determine globally significant (FDR < 0.2) inversion eQTLs. Further details of these analyses are provided in STAR Methods (eQTL analysis).
Analysis of inversions intersecting morbid CNV regions.
In order to find enrichment for different subclasses of inversions in the vicinity of known pathogenic CNVs, we performed permutation testing using the R package regioneR (Gel et al., 2016) with its function ‘permTEST’, followed by randomizing the position of each inversion in every tested subgroup using regioneR’s function ‘circularRandomizeRegions’. Further details of these analyses are provided in STAR Methods (Enrichment analysis of inversions intersecting morbid CNV regions).
Hardy-Weinberg equilibrium test.
We used the ‘hardy’ function available in VCFtools to test our inversion genotypes for Hardy-Weinberg equilibrium. This function assesses sites for Hardy-Weinberg Equilibrium using an exact test. For multiple testing correction of the resulting p-value for each inversion, Benjamini-Hochberg correction was applied. Further details of these analyses are provided in STAR Methods (Hardy-Weinberg equilibrium test and multi-allelic sites).
Supplementary Material
Data S1. Supporting data related to main figures, STAR Methods, and discussion points.
Figure S1. Inversion genotyping and callset overview. A) The percentage of balanced inversions ≥100 kbp (top) out of the total inversion count (n = 292), and distributions of inversion sizes (bottom) are shown for each chromosome. Red lines depict median. Chromosomes with the highest proportion of large (≥100 kbp) balanced inversions are highlighted by red bars. B) A genome-wide distribution of inversion breakpoint hotspots (colored rectangles) based on all 292 balanced inversions. BP, breakpoints per hotspot. The bottom of each chromosomal bar depicts positions of SDs in orange rectangles. C) Structural conformations for balanced inversions and their flanks. Inversions are grouped by likely formation mechanism (left-to-right: SD-mediated, mobile element-mediated, non-repeat-mediated) and vertically by total event length (top-to-bottom: <40 kbp, ≥40 kbp in size). Inv. Transloc; inverted translocation; D) Flanking inverted repeat length correlates with event size. E) ArbiGent inversion classification stratified by technology used for discovery. Loci were rejected from the final inversion callset if they showed evidence for absence (‘FP’, false positive) or were classified as ‘Complex’ in all samples, indicative of a low quality call. Inversion loci were retained if they were classified as either ‘Passing’, ‘InvDup’, ‘Misorient’, ‘LowConf’, or ‘noReads’. Non-concordance with Mendelian inheritance was noted (“Mendelfail”), but was not used as a criterion for exclusion. F) ArbiGent classifications stratified by event length and discovery technique. The distributions mirror expected technological biases, such as an enrichment of ‘FP’ calls in long PAV-based and short Bionano-based calls. G) ArbiGent genotype confidence on the set of 399 inversions outside of L1-internal sequence, stratified by length. Genotypes were considered “high confidence” if genotype-likelihood ratios over reference state were above 103 in >90% of samples. Median inversion widths for confidence subgroups are indicated by vertical lines (orange: low-confidence, blue: high-confidence, black: all inversions).
Figure S2. Inversion validation and breakpoint refinement. A) Schematic view of PCR primer positions and orientation in reference and inverted alleles. B) PCR products obtained from one randomly selected sequence-resolved balanced inversion. Molecular weight markers are indicated with an orange asterisk. Further PCR results are in Data S1. C) Bar plot representing the fraction of events from our callset (n = 399) that meet the required reciprocal overlaps (‘seen’, green) and those that do not (‘unique’ - brown). D) Fraction of inversion sites (out of n=399) with orthogonal support (i.e., orthogonal technology from this study, orthogonal technology from a prior study, long ONT reads, or Hardy-Weinberg equilibrium). E) A depiction of the expected conformation of SDs flanking a direct and inverted haplotype. When an inversion forms, we expect repositioning of paralogous sequence variants (PSVs) specific either to proximal or distal SDs such that the resulting proximal and distal SDs represent hybrids of both. In order to detect such PSVs, we generated a multiple sequence alignment (MSA) using proximal and distal SDs from both direct and inverted haplotypes. F) An example of an inversion flanked by relatively short and simple SDs (~82 kbp). We compared flanking SDs from a direct haplotype assembled as part of the CHM13-T2T assembly. As depicted in (A), PSVs specific to the proximal (SD1, bright green) and distal (SD2, dark yellow) SD are coloured separately. Gaps in the MSA are white and alleles not present in the proximal and distal SDs are shown in black and red, respectively. Vertical dashed lines depict detected change points, with numbers showing the change point position with respect to the SD. We predict that the inversion breakpoints lie between the 21,668–67,040 bp coordinates of the interval shown. G) An inversion flanked by long SDs (~315 kbp). Different from (G), here, we are unable to pinpoint a range corresponding to the likely inversion breakpoints. Phased sample-specific sequence data underlying panels (G) and (H) are from the HPRC. H) Dotplots for a chromosome 16 recurrent inversion locus shown for four samples. A subset of haplotypes display a ~11 kbp deletion (red circle) in one of the inversion-flanking SDs. The presence and absence of this deletion on haplotypes displaying inverted and directly oriented segments is consistent with the recurrent nature of the inversion. Haplotype assignments depicted (h1/h2) to visualize these assemblies are from hifiasm algorithm (Cheng et al. 2021), and these were not synchronized with our haplotype assignments.
Figure S3. Inversion internal to L1 sequences due to twin-priming. A–C) Size distribution for 5′ inversions likely mediated by twin-priming (A), non-inverted (B), and non-truncated (C) 3′ ends. Observed (OBS) versus expected (EXP), based on a random model (STAR Methods), with p-values based on a two-tailed Mann–Whitney U-test. Line, median; box, 25th to 75th percentile (interquartile range, IQR); whiskers, data within 1.5 times the IQR. D) Comparison of the observed size distributions in A–C. E) Schematic for blunt, microhomology and insertions at the inversion junctions. F) Proportion of inversion junctions being blunt, subjected to microhomology (MH) or insertions (INS). G) Length distribution for microhomologies. H) Length distribution for insertions. I–K) Templated insertion events (pink) identified at breakpoint junction 1–2 for twin-priming events and the junction between the target site duplication (TSD, gold) and the 5’ end of truncated L1s. L1 sequence coloured in blue with inverted sequence in green. For each sequence-resolved insertion a model (not scaled) of the insertion conformation is provided above the sequence.
Figure S4. Inversion recurrence analysis. A) Hierarchical clustering-based phylogenetic trees constructed independently for the first, second, and last third of the 8p23.1 genomic locus. Inverted and non-inverted haplotypes clustered together across the whole inversion, with tiSNPs distributed across the whole locus. B) Representation as in (A) for the 17q21.31 locus. C) Distinct distributions of pairwise identity by state among haplotypes at the 8p23.1 inversion locus. Identity by state is defined as the percentage of identity between a pair of haplotypes with the same orientation (inverted/inverted or direct/direct orientations) or in the opposite direction (inverted/direct orientations). D) Representation as in (C) for the 17q21.31 locus. E) Ancestry tree reconstruction analysis of the 100 kbp region (chr8:8201024-8301024) located at the distal end of the 8p23.1 locus using population genetics and phylogenetics-based approaches. This region shows little evidence for recombination. Ancestral recombination graph-based tree reconstruction using Relate (v1.1.7); 36 marginal genealogical trees (blue) inferred by Relate were overlaid on the plot (top-left). Triangles denote inverted orientation, while colors denote population origin. A heatmap of tree topological similarity among the 36 marginal trees is in the bottom-left panel. Each square on the heatmap is a comparison in topology between two marginal trees, while colors indicate the similarity between the trees measured using the Robinson-Foulds distance. Squares are listed in distal to proximal order for the 8p23.1 locus. The warmer the color is, the more similar tree topologies are. Top-right panel shows phylogenetic tree reconstruction of 8p23.1 locus using 100 random bootstraps and IQ-TREE (v2.1.3). Heatmap of tree topological similarity among the 100 random bootstrap trees inferred by IQ-TREE is shown in the bottom-right panel. F) Inferred cladogram of the Xp28 locus based on maximal likelihood software (IQ-Tree (v2.1.3)). G) Hierarchical clustering of haplotypes (rows) at Xp28 locus. Columns represent SNVs (polarized using chimpanzee (panTro6); STAR Methods). Of note, this inversion is not in Hardy-Weinberg equilibrium (Fisher’s exact test p = 5×10−3) and shows a significantly higher than expected allele frequency in African samples (AF = 87.5%; global AF = 42%; p ≤ 1×10−5; 100,000 permutations). H) Relationship between inversion status classified as ‘recurrent’ or ‘single-event’ and flanking inverted repeat length and sequence identity, for the consensus set of 93 balanced inversion loci. Blue lines and gray areas are regression lines and their 95% confidence areas, respectively (p-value based on Pearson’s correlation).
Figure S5. Pooled Strand-Seq experiment. A) Inversion genotyping by pooled Strand-seq (schematic). B) Number of samples per continental population in each cell line pool (Pools A, B and C). C) Numbers of good-quality Strand-seq (SSEQ) libraries per pool. D) Population of origin of Strand-seq libraries, inferred by SNP genotyping (STAR Methods). E) Recovery fraction relative to the original number of pooled samples (n = 40) represented in each pool. F) Numbers of high-quality libraries assigned to unique 1KG samples by SNP genotyping. G) Randomized cell sampling using the HG00733 sample indicates that accurate inversion genotypes can be obtained from single cells. Confusion matrices with rows representing ground truth genotypes and columns representing predicted genotypes (using ArbiGent) obtained from randomly subsampled Strand-seq (single cell) libraries (in sets of N = 1, 2, 3, and 12 cells, respectively). Ground truth genotypes are based on our inversion resource. We assessed all inversions with >50 kbp of mappable sequence (showing >75% mappability). G) A scatter plot comparing inversion AFs from our resource (based on n = 44 samples) compared to AF estimates generated by genotyping the same inversion sites in the sequenced cell pools (y-axis, based on n = 60 non-overlapping 1KG samples). Recurrent inversions are depicted in red and single-event inversions in green. Depicted 95% confidence intervals are the ±2 standard errors (SE), computed using AF p and total number of alleles observed n, We removed a predicted misoriented region (misorient; chr20-28556078-INV-648848) from the analysis, which is embedded in a repetitive region with multiple gaps in GRCh38.
Table S1. Details relevant to data sets and cell lines used in this study, related to Figure 1 and STAR Methods.
Table S2. Inversion callsets along with relevant details and inversion hotspots for GRCh38, related to Figure 1.
Table S3. Inversion callset validations, reciprocal overlap with published data and details specifically relevant to chromosome Y inversions, related to STAR Methods.
Table S4. Inversion flank complexity and likely novel inversions, related to results and STAR Methods.
Table S5. Recurrence analysis results for all balanced inversions, related to Figure 3 and STAR Methods.
Table S6. Inversions flipping SD pairs overlapping with morbid CNVs. related to results and STAR Methods.
Table S7. ArbiGent inversion genotypes based on Strand-seq pooled samples, related to STAR Methods.
Multi-platform characterization of inversion polymorphisms from 82 human haplotypes
Mechanisms of inversion formation implicate segmental duplications and retrotransposons
Excess of common balanced inversions reveals hotspots of inversion recurrence
Recurrent inversions associate with de novo disease-causing CNVs
Acknowledgements
The authors thank Simone Köhler for comments on the manuscript, Tonia Brown for editing, Marc Jan Bonder for advice on eQTL mapping, the EMBL Genomics Core Facility, IT Services and Data Science Centre for technical assistance, and the Centre for Information and Media Technology at Heinrich Heine University Düsseldorf for providing computational infrastructure and support. Funding was provided by: National Institutes of Health (NIH) grants U24HG007497 (to C.L., E.E.E., J.O.K., T.M.), U01HG010973 (to T.M., E.E.E., and J.O.K.), and R01HG002385 and R01HG010169 (to E.E.E.); the German Federal Ministry for Research and Education (BMBF 031L0184 to J.O.K. and T.M.); the German Research Foundation (DFG 391137747 to T.M.), the German Human Genome-Phenome Archive (DFG (NFDI 1/1) to J.O.K.); and the European Research Council (ERC Consolidator grant 773026 to J.O.K.). The EMBL (J.O.K, P.Hasenfeld, E.B.) and the EMBL International PhD Programme (W.H.) provided support. Computational work was supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A532B, 031A533A, 031A533B, 031A534A, 031A535A, 031A537A, 031A537B, 031A537C, 031A537D, 031A538A). E.E.E. is an investigator of the Howard Hughes Medical Institute. B.R.M. is supported by a Bridging Excellence Fellowship provided by the Life Science Alliance. P.Hsieh is supported by the NIH Pathway to Independence Award (NHGRI, K99HG011041). C.R.B. and P.A.A. receive support from NIH NIGMS R35GM133600 and NCI P30CA034196. F.Y., P.Hallast. and Q.Z. are supported by NIH U24HG007497. WGS data for the full set of 1KG samples (n = 3,202) were generated at the New York Genome Center with funds provided by NHGRI Grants 3UM1HG008901-03S1 and 3UM1HG008901-04S2. We further acknowledge the National Human Genome Research Institute (NHGRI) for funding the following grants in support of creating the human pangenome reference: 1U41HG010972, 1U01HG010971, 1U01HG010961, 1U01HG010973, 1U01HG010963, and the Human Pangenome Reference Consortium (HPRC, https://humanpangenome.org). We thank the many people who were generous with contributing their samples to the 1KG project, which formed the basis of this study.
Inclusion and diversity
We worked to ensure diversity in experimental samples through the selection of the cell lines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. C.L. is a scientific advisory board (SAB) member of Nabsys, Inc. The following authors have previously disclosed a patent application (no. EP19169090) relevant to Strand-seq: A.D.S., J.O.K., T.M., and D.P.; the other authors declare no competing interests.
Supplementary Figures Description
Figure S1. Inversion genotyping and callset summary, related to STAR methods and Figure 1
Figure S2. Inversion validation and breakpoint refinement, related to Figure 1
Figure S3. Inversions internal to L1 sequences due to twin-priming, related to Figure 2
Figure S4. Inversion recurrence analysis, related to Figure 3
Figure S5. Pooled Strand-seq experiment, related to STAR Methods
References
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aagaard Nolting L, Brasch-Andersen C, Cox H, Kanani F, Parker M, Fry AE, Loddo S, Novelli A, Dentici ML, Joss S, et al. (2020). A new 1p36.13-1p36.12 microdeletion syndrome characterized by learning disability, behavioral abnormalities, and ptosis. Clin. Genet 97, 927–932. [DOI] [PubMed] [Google Scholar]
- Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, Layer RM, Neale BM, Salerno WJ, Reeves C, et al. (2020). Mapping and characterization of structural variation in 17,795 human genomes. Nature 583, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado C, Gayà-Vidal M, Villatoro S, Oliva M, Izquierdo D, Giner-Delgado C, Montalvo V, García-González J, Martínez-Fundichely A, Capilla L, et al. (2014). Validation and genotyping of multiple human polymorphic inversions mediated by inverted repeats reveals a high degree of recurrence. PLoS Genet. 10, e1004208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman TS, Mysore V, and Mishra B (2004). Fast and cheap genome wide haplotype construction via optical mapping. Biocomputing 2005. [DOI] [PubMed] [Google Scholar]
- Andrews S, Krueger F, Segonds-Pichon A, Biggins L, Virk B, Dalle-Pezze P, Wingett S, Saadeh H, and Ahlfors H (2015). Trim Galore. Trim Galore. [Google Scholar]
- Antonacci F, Kidd JM, Marques-Bonet T, Ventura M, Siswara P, Jiang Z, and Eichler EE (2009). Characterization of six human disease-associated inversion polymorphisms. Hum. Mol. Genet 18, 2555–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, Miroballo M, Graves TA, Vives L, Malig M, et al. (2014). Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat. Genet 46, 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. (2019). Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, and Eichler EE (2006). Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet 7, 552–564. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, Madan-Khetarpal S, Schmidt KR, Tervo R, Escobar LF, Friedrich CA, et al. (2008). Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol. Cytogenet 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström A, McCarthy SA, Hui R, Almarri MA, Ayub Q, Danecek P, Chen Y, Felkel S, Hallast P, Kamm J, et al. (2020). Insights into human genetic variation and population history from 929 diverse genomes. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin E, Chatzimichali EA, Wright CF, Hurles ME, Firth HV, Bevan AP, and Swaminathan GJ (2014). DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 42, D993–D1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Grinde K, Plantinga A, Gogarten SM, Stilp AM, Kaplan RC, Avilés-Santa ML, Browning BL, and Laurie CC (2016). Local Ancestry Inference in a Large US-Based Hispanic/Latino Study: Hispanic Community Health Study/Study of Latinos (HCHS/SOL). G3 6, 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. (2021). High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. [DOI] [PMC free article] [PubMed]
- Cáceres M, National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program, Sullivan RT, and Thomas JW (2007). A recurrent inversion on the eutherian X chromosome. Proc. Natl. Acad. Sci. U. S. A 104, 18571–18576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, and Lupski JR (2016). Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet 17, 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catacchio CR, Maggiolini FAM, D’Addabbo P, Bitonto M, Capozzi O, Lepore Signorile M, Miroballo M, Archidiacono N, Eichler EE, Ventura M, et al. (2018). Inversion variants in human and primate genomes. Genome Res. 28, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. (2019). Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, Zhao J, McDevitt S, Rusanov T, Hoang T, Borisonnik N, Treddinick T, Lopezcolorado FW, Kent T, Siddique LA, et al. (2021). Polθ reverse transcribes RNA and promotes RNA-templated DNA repair. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-P, Lin S-P, Lee C-L, Chern S-R, Wu P-S, Chen Y-N, Chen S-W, and Wang W (2017). Recurrent 2q13 microduplication encompassing MALL, NPHP1, RGPD6, and BUB1 associated with autism spectrum disorder, intellectual disability, and liver disorder. Taiwan. J. Obstet. Gynecol 56, 98–101. [DOI] [PubMed] [Google Scholar]
- Cheng H, Concepcion GT, Feng X, Zhang H, and Li H (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccone R, Mattina T, Giorda R, Bonaglia MC, Rocchi M, Pramparo T, and Zuffardi O (2006). Inversion polymorphisms and non-contiguous terminal deletions: the cause and the (unpredicted) effect of our genome architecture. J. Med. Genet 43, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LELM, et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet 46, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. (2020). A structural variation reference for medical and population genetics. Nature 581, 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. (2011). A copy number variation morbidity map of developmental delay. Nat. Genet 43, 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, and Boeke JD (1998). Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry 37, 18081–18093. [DOI] [PubMed] [Google Scholar]
- Cost GJ, Feng Q, Jacquier A, and Boeke JD (2002). Human L1 element target-primed reverse transcription in vitro. EMBO J. 21, 5899–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Marchetto MCN, Muotri AR, Mu Y, Carson CT, Macia A, Moran JV, and Gage FH (2011). Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. U. S. A 108, 20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, and Rambaut A (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol 7, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, Sulovari A, Ebler J, Zhou W, Serra Mari R, et al. (2021). Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372(6537), eabf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebler J, Ebert P, Clarke WE, Rausch T, Audano PA, Houwaart T, Mao Y, Korbel JO, Eichler EE, Zody MC, et al. (2022). Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat Genet in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enguita-Marruedo A, Martín-Ruiz M, García E, Gil-Fernández A, Parra MT, Viera A, Rufas JS, and Page J (2019). Transition from a meiotic to a somatic-like DNA damage response during the pachytene stage in mouse meiosis. PLoS Genet. 15, e1007439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer E, Hills M, Naumann U, Poon SSS, Chavez EA, Sanders AD, Zhao Y, Hirst M, and Lansdorp PM (2012). DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 9, 1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner JN (2005). Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. American Journal of Physical Anthropology 128, 415–423. [DOI] [PubMed] [Google Scholar]
- Fitch WM (1971). Toward Defining the Course of Evolution: Minimum Change for a Specific Tree Topology. Syst. Biol 20, 406–416. [Google Scholar]
- Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, Johnson PLF, Aximu-Petri A, Prüfer K, de Filippo C, et al. (2014). Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B, McVicker G, Gilad Y, and Pritchard JK (2015). WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gel B, Díez-Villanueva A, Serra E, Buschbeck M, Peinado MA, and Malinverni R (2016). regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, et al. (2001). Olfactory Receptor–Gene Clusters, Genomic-Inversion Polymorphisms, and Common Chromosome Rearrangements. Am. J. Hum. Genet 68, 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, et al. (2003). Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum. Mol. Genet 12, 849–858. [DOI] [PubMed] [Google Scholar]
- Giner-Delgado C, Villatoro S, Lerga-Jaso J, Gayà-Vidal M, Oliva M, Castellano D, Pantano L, Bitarello BD, Izquierdo D, Noguera I, et al. (2019). Evolutionary and functional impact of common polymorphic inversions in the human genome. Nat. Commun 10, 4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorda R, Ciccone R, Gimelli G, Pramparo T, Beri S, Bonaglia MC, Giglio S, Genuardi M, Argente J, Rocchi M, et al. (2007). Two classes of low-copy repeats comediate a new recurrent rearrangement consisting of duplication at 8p23.1 and triplication at 8p23.2. Hum. Mutat 28, 459–468. [DOI] [PubMed] [Google Scholar]
- Goedecke W, Eijpe M, Offenberg HH, van Aalderen M, and Heyting C (1999). Mre11 and Ku70 interact in somatic cells, but are differentially expressed in early meiosis. Nat. Genet 23, 194–198. [DOI] [PubMed] [Google Scholar]
- Graham T, and Boissinot S (2006). The genomic distribution of L1 elements: the role of insertion bias and natural selection. J. Biomed. Biotechnol 2006, 75327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, and Bustamante CD (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5, e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hach F, Hormozdiari F, Alkan C, Hormozdiari F, Birol I, Eichler EE, and Sahinalp SC (2010). mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods 7, 576–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallast P, Balaresque P, Bowden GR, Ballereau S, and Jobling MA (2013). Recombination dynamics of a human Y-chromosomal palindrome: rapid GC-biased gene conversion, multi-kilobase conversion tracts, and rare inversions. PLoS Genet. 9, e1003666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, and McCarroll SA (2015). Large multiallelic copy number variations in humans. Nat. Genet 47, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermetz KE, Newman S, Conneely KN, Martin CL, Ballif BC, Shaffer LG, Cody JD, and Rudd MK (2014). Large inverted duplications in the human genome form via a fold-back mechanism. PLoS Genet. 10, e1004139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, and Lee C (2004). Detection of large-scale variation in the human genome. Nat. Genet 36, 949–951. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, and Eichler EE (2007). Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat. Genet 39, 1361–1368. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, and Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH Jr, and Moran JV (1998). The impact of L1 retrotransposons on the human genome. Nat. Genet 19, 19–24. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. (2008). Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, and Eichler EE (2010). A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima KK (2010). Different integration site structures between L1 protein-mediated retrotransposition in cis and retrotransposition in trans. Mob. DNA 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Vissers LELM, Pfundt R, de Leeuw N, Knight SJL, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, et al. (2006). A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet 38, 999–1001. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. (2007). Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koressaar T, and Remm M (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291. [DOI] [PubMed] [Google Scholar]
- Kronenberg ZN, Fiddes IT, Gordon D, Murali S, Cantsilieris S, Meyerson OS, Underwood JG, Nelson BJ, Chaisson MJP, Dougherty ML, et al. (2018). High-resolution comparative analysis of great ape genomes. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakich D, Kazazian HH Jr, Antonarakis SE, and Gitschier J (1993). Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat. Genet 5, 236–241. [DOI] [PubMed] [Google Scholar]
- Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. (2012). Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol 30, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Skaletsky H, van Daalen SKM, Embry SL, Korver CM, Brown LG, Oates RD, Silber S, Repping S, and Page DC (2009). Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell 138, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Yamada S, Tischfield SE, Pan J, Kim S, Zhu X, Socci ND, Jasin M, and Keeney S (2016). The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 167, 695–708.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lichter P, Tang CJ, Call K, Hermanson G, Evans GA, Housman D, and Ward DC (1990). High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 247, 64–69. [DOI] [PubMed] [Google Scholar]
- Lippert C, Casale FP, Rakitsch B, and Stegle O (2014). LIMIX: genetic analysis of multiple traits. [DOI] [PubMed]
- Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC, Rhie A, et al. (2021). The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozier JN, Dutra A, Pak E, Zhou N, Zheng Z, Nichols TC, Bellinger DA, Read M, and Morgan RA (2002). The Chapel Hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc. Natl. Acad. Sci. U. S. A 99, 12991–12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 161, 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, and Eickbush TH (1993). Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 72, 595–605. [DOI] [PubMed] [Google Scholar]
- Lupski JR (1998). Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 14, 417–422. [DOI] [PubMed] [Google Scholar]
- Maggiolini FAM, Cantsilieris S, D’Addabbo P, Manganelli M, Coe BP, Dumont BL, Sanders AD, Pang AWC, Vollger MR, Palumbo O, et al. (2019). Genomic inversions and GOLGA core duplicons underlie disease instability at the 15q25 locus. PLoS Genet. 15, e1008075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiolini FAM, Sanders AD, Shew CJ, Sulovari A, Mao Y, Puig M, Catacchio CR, Dellino M, Palmisano D, Mercuri L, et al. (2020). Single-cell strand sequencing of a macaque genome reveals multiple nested inversions and breakpoint reuse during primate evolution. Genome Res. 30, 1680–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Kidd JM, Ventura M, Graves TA, Cheng Z, Hillier LW, Jiang Z, Baker C, Malfavon-Borja R, Fulton LA, et al. (2009). A burst of segmental duplications in the genome of the African great ape ancestor. Nature 457, 877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, and Eichler EE (2009). Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev 19, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, and Lanfear R (2020). IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol 37, 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K. ’ichi, Miyabe I, Schalbetter SA, Carr AM, and Murray JM (2013). Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 493, 246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens PB, Chen DJ, Shen Z, Kolas N, Tarsounas M, Heng HH, and Spyropoulos B (1997). Rad51 immunocytology in rat and mouse spermatocytes and oocytes. Chromosoma 106, 207–215. [DOI] [PubMed] [Google Scholar]
- Mohajeri K, Cantsilieris S, Huddleston J, Nelson BJ, Coe BP, Campbell CD, Baker C, Harshman L, Munson KM, Kronenberg ZN, et al. (2016). Interchromosomal core duplicons drive both evolutionary instability and disease susceptibility of the Chromosome 8p23.1 region. Genome Res. 26, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monot C, Kuciak M, Viollet S, Mir AA, Gabus C, Darlix J-L, and Cristofari G (2013). The specificity and flexibility of l1 reverse transcription priming at imperfect T-tracts. PLoS Genet. 9, e1003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales ME, White TB, Streva VA, DeFreece CB, Hedges DJ, and Deininger PL (2015). The contribution of alu elements to mutagenic DNA double-strand break repair. PLoS Genet. 11, e1005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag DK, and Kurst A (1997). A 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics 146, 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasar F, Jankowski C, and Nag DK (2000). Long palindromic sequences induce double-strand breaks during meiosis in yeast. Mol. Cell. Biol 20, 3449–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. (2021). The complete sequence of a human genome. [DOI] [PMC free article] [PubMed]
- O’Bleness MS, Michael Dickens C, Dumas LJ, Kehrer-Sawatzki H, Wyckoff GJ, and Sikela JM (2012). Evolutionary History and Genome Organization of DUF1220 Protein Domains. G3 Genes|Genomes|Genetics 2, 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, et al. (2001). A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat. Genet 29, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, and Kazazian HH Jr (2001). Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 11, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DWW, McDonald R, Eddy A, Chance PF, and Glass IA (2004). The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet 75, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton AL, Shen F, Taravella AM, Emery S, Veeramah KR, Boyko AR, and Kidd JM (2018). Comparison of village dog and wolf genomes highlights the role of the neural crest in dog domestication. BMC Biol. 16, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorten T (2017). dotPlotly (Github)
- Popesco MC (2006). Human Lineage-Specific Amplification, Selection, and Neuronal Expression of DUF1220 Domains. Science 313, 1304–1307. [DOI] [PubMed] [Google Scholar]
- Porubský D, Sanders AD, van Wietmarschen N, Falconer E, Hills M, Spierings DCJ, Bevova MR, Guryev V, and Lansdorp PM (2016). Direct chromosome-length haplotyping by single-cell sequencing. Genome Res. 26, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Garg S, Sanders AD, Korbel JO, Guryev V, Lansdorp PM, and Marschall T (2017). Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun 8, 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Sanders AD, Höps W, Hsieh P, Sulovari A, Li R, Mercuri L, Sorensen M, Murali SC, Gordon D, et al. (2020a). Recurrent inversion toggling and great ape genome evolution. Nat. Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porubsky D, Sanders AD, Taudt A, Colomé-Tatché M, Lansdorp PM, and Guryev V (2020b). breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics 36, 1260–1261. [DOI] [PubMed] [Google Scholar]
- Poznik GD, Henn BM, Yee M-C, Sliwerska E, Euskirchen GM, Lin AA, Snyder M, Quintana-Murci L, Kidd JM, Underhill PA, et al. (2013). Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 341, 562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, Casillas S, Villatoro S, and Cáceres M (2015). Human inversions and their functional consequences. Brief. Funct. Genomics 14, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, Lerga-Jaso J, Giner-Delgado C, Pacheco S, Izquierdo D, Delprat A, Gayà-Vidal M, Regan JF, Karlin-Neumann G, and Cáceres M (2020). Determining the impact of uncharacterized inversions in the human genome by droplet digital PCR. Genome Res. 30, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. (2006). Global variation in copy number in the human genome. Nature 444, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repping S, Skaletsky H, Lange J, Silber S, Van Der Veen F, Oates RD, Page DC, and Rozen S (2002). Recombination between palindromes P5 and P1 on the human Y chromosome causes massive deletions and spermatogenic failure. Am. J. Hum. Genet 71, 906–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repping S, van Daalen SKM, Brown LG, Korver CM, Lange J, Marszalek JD, Pyntikova T, van der Veen F, Skaletsky H, Page DC, et al. (2006). High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat. Genet 38, 463–467. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salm MPA, Horswell SD, Hutchison CE, Speedy HE, Yang X, Liang L, Schadt EE, Cookson WO, Wierzbicki AS, Naoumova RP, et al. (2012). The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 22, 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AD, Hills M, Porubský D, Guryev V, Falconer E, and Lansdorp PM (2016). Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res. 26, 1575–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AD, Falconer E, Hills M, Spierings DCJ, and Lansdorp PM (2017). Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc 12, 1151–1176. [DOI] [PubMed] [Google Scholar]
- Sanders AD, Meiers S, Ghareghani M, Porubsky D, Jeong H, van Vliet MACC, Rausch T, Richter-Pechańska P, Kunz JB, Jenni S, et al. (2020). Single-cell analysis of structural variations and complex rearrangements with tri-channel processing. Nat. Biotechnol 38, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Månér S, Massa H, Walker M, Chi M, et al. (2004). Large-scale copy number polymorphism in the human genome. Science 305, 525–528. [DOI] [PubMed] [Google Scholar]
- Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, Armstrong J, Tigyi K, Maurer N, Koren S, et al. (2020). Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol 38, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SK, McCaskill C, Northrup H, Spikes AS, Elder FF, Sutton VR, Korenberg JR, Greenberg F, and Shaffer LG (1997). Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am. J. Hum. Genet 61, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Louzada S, Grigorova M, Massaia A, Arciero E, Kibena L, Ge XJ, Chen Y, Ayub Q, Poolamets O, et al. (2019a). Evolutionary and functional analysis of RBMY1 gene copy number variation on the human Y chromosome. Hum. Mol. Genet 28, 2785–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Massaia A, Louzada S, Handsaker J, Chow W, McCarthy S, Collins J, Hallast P, Howe K, Church DM, et al. (2019b). Birth, expansion, and death of VCY-containing palindromes on the human Y chromosome. Genome Biol. 20, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikela JM, and van Roy F (2017). Changing the name of the NBPF/DUF1220 domain to the Olduvai domain. F1000Res. 6, 2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small K, Iber J, and Warren ST (1997). Emerin deletion reveals a common X-chromosome inversion mediated by inverted repeats. Nat. Genet 16, 96–99. [DOI] [PubMed] [Google Scholar]
- Song X, Beck CR, Du R, Campbell IM, Coban-Akdemir Z, Gu S, Breman AM, Stankiewicz P, Ira G, Shaw CA, et al. (2018). Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 28, 1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel L, Forest M, Shi S, and Myers SR (2019). A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet 51, 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, et al. (2005). A common inversion under selection in Europeans. Nat. Genet 37, 129–137. [DOI] [PubMed] [Google Scholar]
- Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M, et al. (2012). Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat. Genet 44, 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant AH (1917). Genetic Factors Affecting the Strength of Linkage in Drosophila. Proc. Natl. Acad. Sci. U. S. A 3, 555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH-Y, et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Yamaguchi K, Kajikawa M, Ichiyanagi K, Adachi N, Koyama H, Takeda S, and Okada N (2009). Genetic evidence that the non-homologous end-joining repair pathway is involved in LINE retrotransposition. PLoS Genet. 5, e1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, and Chen N (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4, Unit 4.10. [DOI] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M, Sturge M, Peddle L, O’Rielly DD, and Rahman P (2011). Genome-wide signatures of “rearrangement hotspots” within segmental duplications in humans. PLoS One 6, e28853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, Gutenkunst RN, Woerner AE, Watkins JC, and Hammer MF (2014). Evidence for increased levels of positive and negative selection on the X chromosome versus autosomes in humans. Mol. Biol. Evol 31, 2267–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Salvador D, Puig M, Gayà-Vidal M, Pacheco S, Giner-Delgado C, Noguera I, Izquierdo D, Martínez-Fundichely A, Ruiz-Herrera A, Estivill X, et al. (2017). Detailed analysis of inversions predicted between two human genomes: errors, real polymorphisms, and their origin and population distribution. Hum. Mol. Genet 26, 567–581. [DOI] [PubMed] [Google Scholar]
- Vollger MR, Dishuck PC, Sorensen M, Welch AE, Dang V, Dougherty ML, Graves-Lindsay TA, Wilson RK, Chaisson MJP, and Eichler EE (2018). Long-read sequence and assembly of segmental duplications. Nat. Methods in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, FitzPatrick DR, Maher E, Martin H, Parnau J, et al. (2005). 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am. J. Hum. Genet 77, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Kajikawa M, and Okada N (2014). Integrated mechanism for the generation of the 5’ junctions of LINE inserts. Nucleic Acids Res. 42, 13269–13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Hashimoto R, Fukai R, Okamoto N, Hiraki Y, Yamamori H, Fujimoto M, Ohi K, Taniike M, Mohri I, et al. (2014). Duplication of the NPHP1 gene in patients with autism spectrum disorder and normal intellectual ability: a case series. Ann. Gen. Psychiatry 13, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz F, Gurusamy U, Mosley TJ, Mostovoy Y, Shaikh TH, Zwick ME, Kwok P-Y, Lee C, and Mulle JG (2021). Multi-modal investigation of the schizophrenia-associated 3q29 genomic interval reveals global genetic diversity with unique haplotypes and segments that increase the risk for non-allelic homologous recombination. medRxiv 10.1101/2021.11.10.21266197. [DOI] [Google Scholar]
- Yuan B, Liu P, Gupta A, Beck CR, Tejomurtula A, Campbell IM, Gambin T, Simmons AD, Withers MA, Harris RA, et al. (2015). Comparative Genomic Analyses of the Human NPHP1 Locus Reveal Complex Genomic Architecture and Its Regional Evolution in Primates. PLoS Genet. 11, e1005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer F, and Montgomery SH (2015). Phylogenetic Analysis Supports a Link between DUF1220 Domain Number and Primate Brain Expansion. Genome Biology and Evolution 7, 2083–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingler N, Willhoeft U, Brose H-P, Schoder V, Jahns T, Hanschmann K-MO, Morrish TA, Löwer J, and Schumann GG (2005). Analysis of 5’ junctions of human LINE-1 and Alu retrotransposons suggests an alternative model for 5′-end attachment requiring microhomology-mediated end-joining. Genome Res. 15, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung H-C, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, et al. (2008). Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet 40, 1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting data related to main figures, STAR Methods, and discussion points.
Figure S1. Inversion genotyping and callset overview. A) The percentage of balanced inversions ≥100 kbp (top) out of the total inversion count (n = 292), and distributions of inversion sizes (bottom) are shown for each chromosome. Red lines depict median. Chromosomes with the highest proportion of large (≥100 kbp) balanced inversions are highlighted by red bars. B) A genome-wide distribution of inversion breakpoint hotspots (colored rectangles) based on all 292 balanced inversions. BP, breakpoints per hotspot. The bottom of each chromosomal bar depicts positions of SDs in orange rectangles. C) Structural conformations for balanced inversions and their flanks. Inversions are grouped by likely formation mechanism (left-to-right: SD-mediated, mobile element-mediated, non-repeat-mediated) and vertically by total event length (top-to-bottom: <40 kbp, ≥40 kbp in size). Inv. Transloc; inverted translocation; D) Flanking inverted repeat length correlates with event size. E) ArbiGent inversion classification stratified by technology used for discovery. Loci were rejected from the final inversion callset if they showed evidence for absence (‘FP’, false positive) or were classified as ‘Complex’ in all samples, indicative of a low quality call. Inversion loci were retained if they were classified as either ‘Passing’, ‘InvDup’, ‘Misorient’, ‘LowConf’, or ‘noReads’. Non-concordance with Mendelian inheritance was noted (“Mendelfail”), but was not used as a criterion for exclusion. F) ArbiGent classifications stratified by event length and discovery technique. The distributions mirror expected technological biases, such as an enrichment of ‘FP’ calls in long PAV-based and short Bionano-based calls. G) ArbiGent genotype confidence on the set of 399 inversions outside of L1-internal sequence, stratified by length. Genotypes were considered “high confidence” if genotype-likelihood ratios over reference state were above 103 in >90% of samples. Median inversion widths for confidence subgroups are indicated by vertical lines (orange: low-confidence, blue: high-confidence, black: all inversions).
Figure S2. Inversion validation and breakpoint refinement. A) Schematic view of PCR primer positions and orientation in reference and inverted alleles. B) PCR products obtained from one randomly selected sequence-resolved balanced inversion. Molecular weight markers are indicated with an orange asterisk. Further PCR results are in Data S1. C) Bar plot representing the fraction of events from our callset (n = 399) that meet the required reciprocal overlaps (‘seen’, green) and those that do not (‘unique’ - brown). D) Fraction of inversion sites (out of n=399) with orthogonal support (i.e., orthogonal technology from this study, orthogonal technology from a prior study, long ONT reads, or Hardy-Weinberg equilibrium). E) A depiction of the expected conformation of SDs flanking a direct and inverted haplotype. When an inversion forms, we expect repositioning of paralogous sequence variants (PSVs) specific either to proximal or distal SDs such that the resulting proximal and distal SDs represent hybrids of both. In order to detect such PSVs, we generated a multiple sequence alignment (MSA) using proximal and distal SDs from both direct and inverted haplotypes. F) An example of an inversion flanked by relatively short and simple SDs (~82 kbp). We compared flanking SDs from a direct haplotype assembled as part of the CHM13-T2T assembly. As depicted in (A), PSVs specific to the proximal (SD1, bright green) and distal (SD2, dark yellow) SD are coloured separately. Gaps in the MSA are white and alleles not present in the proximal and distal SDs are shown in black and red, respectively. Vertical dashed lines depict detected change points, with numbers showing the change point position with respect to the SD. We predict that the inversion breakpoints lie between the 21,668–67,040 bp coordinates of the interval shown. G) An inversion flanked by long SDs (~315 kbp). Different from (G), here, we are unable to pinpoint a range corresponding to the likely inversion breakpoints. Phased sample-specific sequence data underlying panels (G) and (H) are from the HPRC. H) Dotplots for a chromosome 16 recurrent inversion locus shown for four samples. A subset of haplotypes display a ~11 kbp deletion (red circle) in one of the inversion-flanking SDs. The presence and absence of this deletion on haplotypes displaying inverted and directly oriented segments is consistent with the recurrent nature of the inversion. Haplotype assignments depicted (h1/h2) to visualize these assemblies are from hifiasm algorithm (Cheng et al. 2021), and these were not synchronized with our haplotype assignments.
Figure S3. Inversion internal to L1 sequences due to twin-priming. A–C) Size distribution for 5′ inversions likely mediated by twin-priming (A), non-inverted (B), and non-truncated (C) 3′ ends. Observed (OBS) versus expected (EXP), based on a random model (STAR Methods), with p-values based on a two-tailed Mann–Whitney U-test. Line, median; box, 25th to 75th percentile (interquartile range, IQR); whiskers, data within 1.5 times the IQR. D) Comparison of the observed size distributions in A–C. E) Schematic for blunt, microhomology and insertions at the inversion junctions. F) Proportion of inversion junctions being blunt, subjected to microhomology (MH) or insertions (INS). G) Length distribution for microhomologies. H) Length distribution for insertions. I–K) Templated insertion events (pink) identified at breakpoint junction 1–2 for twin-priming events and the junction between the target site duplication (TSD, gold) and the 5’ end of truncated L1s. L1 sequence coloured in blue with inverted sequence in green. For each sequence-resolved insertion a model (not scaled) of the insertion conformation is provided above the sequence.
Figure S4. Inversion recurrence analysis. A) Hierarchical clustering-based phylogenetic trees constructed independently for the first, second, and last third of the 8p23.1 genomic locus. Inverted and non-inverted haplotypes clustered together across the whole inversion, with tiSNPs distributed across the whole locus. B) Representation as in (A) for the 17q21.31 locus. C) Distinct distributions of pairwise identity by state among haplotypes at the 8p23.1 inversion locus. Identity by state is defined as the percentage of identity between a pair of haplotypes with the same orientation (inverted/inverted or direct/direct orientations) or in the opposite direction (inverted/direct orientations). D) Representation as in (C) for the 17q21.31 locus. E) Ancestry tree reconstruction analysis of the 100 kbp region (chr8:8201024-8301024) located at the distal end of the 8p23.1 locus using population genetics and phylogenetics-based approaches. This region shows little evidence for recombination. Ancestral recombination graph-based tree reconstruction using Relate (v1.1.7); 36 marginal genealogical trees (blue) inferred by Relate were overlaid on the plot (top-left). Triangles denote inverted orientation, while colors denote population origin. A heatmap of tree topological similarity among the 36 marginal trees is in the bottom-left panel. Each square on the heatmap is a comparison in topology between two marginal trees, while colors indicate the similarity between the trees measured using the Robinson-Foulds distance. Squares are listed in distal to proximal order for the 8p23.1 locus. The warmer the color is, the more similar tree topologies are. Top-right panel shows phylogenetic tree reconstruction of 8p23.1 locus using 100 random bootstraps and IQ-TREE (v2.1.3). Heatmap of tree topological similarity among the 100 random bootstrap trees inferred by IQ-TREE is shown in the bottom-right panel. F) Inferred cladogram of the Xp28 locus based on maximal likelihood software (IQ-Tree (v2.1.3)). G) Hierarchical clustering of haplotypes (rows) at Xp28 locus. Columns represent SNVs (polarized using chimpanzee (panTro6); STAR Methods). Of note, this inversion is not in Hardy-Weinberg equilibrium (Fisher’s exact test p = 5×10−3) and shows a significantly higher than expected allele frequency in African samples (AF = 87.5%; global AF = 42%; p ≤ 1×10−5; 100,000 permutations). H) Relationship between inversion status classified as ‘recurrent’ or ‘single-event’ and flanking inverted repeat length and sequence identity, for the consensus set of 93 balanced inversion loci. Blue lines and gray areas are regression lines and their 95% confidence areas, respectively (p-value based on Pearson’s correlation).
Figure S5. Pooled Strand-Seq experiment. A) Inversion genotyping by pooled Strand-seq (schematic). B) Number of samples per continental population in each cell line pool (Pools A, B and C). C) Numbers of good-quality Strand-seq (SSEQ) libraries per pool. D) Population of origin of Strand-seq libraries, inferred by SNP genotyping (STAR Methods). E) Recovery fraction relative to the original number of pooled samples (n = 40) represented in each pool. F) Numbers of high-quality libraries assigned to unique 1KG samples by SNP genotyping. G) Randomized cell sampling using the HG00733 sample indicates that accurate inversion genotypes can be obtained from single cells. Confusion matrices with rows representing ground truth genotypes and columns representing predicted genotypes (using ArbiGent) obtained from randomly subsampled Strand-seq (single cell) libraries (in sets of N = 1, 2, 3, and 12 cells, respectively). Ground truth genotypes are based on our inversion resource. We assessed all inversions with >50 kbp of mappable sequence (showing >75% mappability). G) A scatter plot comparing inversion AFs from our resource (based on n = 44 samples) compared to AF estimates generated by genotyping the same inversion sites in the sequenced cell pools (y-axis, based on n = 60 non-overlapping 1KG samples). Recurrent inversions are depicted in red and single-event inversions in green. Depicted 95% confidence intervals are the ±2 standard errors (SE), computed using AF p and total number of alleles observed n, We removed a predicted misoriented region (misorient; chr20-28556078-INV-648848) from the analysis, which is embedded in a repetitive region with multiple gaps in GRCh38.
Table S1. Details relevant to data sets and cell lines used in this study, related to Figure 1 and STAR Methods.
Table S2. Inversion callsets along with relevant details and inversion hotspots for GRCh38, related to Figure 1.
Table S3. Inversion callset validations, reciprocal overlap with published data and details specifically relevant to chromosome Y inversions, related to STAR Methods.
Table S4. Inversion flank complexity and likely novel inversions, related to results and STAR Methods.
Table S5. Recurrence analysis results for all balanced inversions, related to Figure 3 and STAR Methods.
Table S6. Inversions flipping SD pairs overlapping with morbid CNVs. related to results and STAR Methods.
Table S7. ArbiGent inversion genotypes based on Strand-seq pooled samples, related to STAR Methods.
Data Availability Statement
The full inversion callset is available in Table S2. Raw genomic datasets are available through the International Nucleotide Sequence Database Collaboration (INSDC) including Illumina WGS, RNA-seq, Bionano Genomics, PacBio, and Strand-seq data, and are publicly available as of the date of publication Accession numbers are listed in the key resources table. The URL is listed in the key resources table. Phased assemblies of the 82 haplotypes were obtained from Ebert et al. 2021 (PGAS v12 assemblies) and Ebler et al. 2022 (PGAS v13 hifiasm assemblies) and are listed in the key resource table. Select loci were also examined from Human Pangenome Reference Consortium (HPRC); https://github.com/human-pangenomics/HPP_Year1_Assemblies. Dot plot visualizations of several recurrent inversion loci can we obtained as described in the key resource table. Other publicly available data (PacBio and ONT) used in this study are reported in Table S1. VCF files with integratively phased single-nucleotide variants (SNVs) and inversion genotypes have been deposited at IGSR FTP. The URL is listed in the key resources table. Data previously generated as part of Ebert et al. 2021 study are publicly available from the IGSR (www.internationalgenome.org/data-portal/data-collection/hgsvc2).
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| Deposited data | ||
| Strand-seq: NA19036 | This paper | NCBI: PRJEB39750 |
| Strand-seq: NA19434 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG00268 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG01352 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG01573 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG02018 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG02059 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG02106 | This paper | NCBI: PRJEB39750 |
| Strand-seq: HG04217 | This paper | NCBI: PRJEB39750 |
| Strand-seq: LCL pools | This paper | NCBI: PRJEB39750 |
| Strand-seq: trio samples (n=9) | (Chaisson et al., 2019) | NCBI: PRJEB12849 |
| Strand-seq: other samples (n=34) | (Ebert et al., 2021) | NCBI: PRJEB39750 |
| Strand-seq: HG002/NA24385 | Public HPRC data (https://github.com/human-pangenomics/HG002_Data_Freeze_v1.0) | https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=NHGRI_UCSC_panel/HG002/hpp_HG002_NA24385_son_v1/Strand_seq/ |
| Strand-seq: NA12878 | (Porubský et al., 2016) | NCBI: PRJEB14185. |
| WGS Illumina data (1KG panel) | (Byrska-Bishop et al.) | NCBI: PRJEB37677 |
| RNA-seq data | (Ebert et al., 2021) | NCBI SRA: ERP123231 |
| PacBio data 1 | (Ebert et al., 2021) | NCBI: PRJEB36100 |
| PacBio data 2 | (Ebert et al., 2021) | EBI/ENA: ERP125611 |
| PacBio data 3 | (Ebert et al., 2021) | NCBI: PRJNA698480 |
| PacBio data: HG00268 | N/A | NCBI: PRJNA558774 |
| PacBio data: HG01352 | N/A | NCBI: PRJNA339719 |
| PacBio data: HG02059 | N/A | NCBI: PRJNA339726 |
| PacBio data: HG02106 | N/A | NCBI: PRJNA480858 |
| PacBio data: HG04217 | N/A | NCBI: PRJNA481794 |
| PacBio data: NA19434 | N/A | NCBI: PRJNA385272 |
| Oxford Nanopore: HG00733 ultra-log | (Logsdon et al., 2021) | NCBI: PRJNA686388 |
| Oxford Nanopore: HG00733 | (Shafin et al., 2020) | NCBI: PRJEB37264 |
| Oxford Nanopore: NA19240 | N/A | NCBI: PRJEB26791 |
| Oxford Nanopore: HG002/NA24385 | (Shafin et al., 2020) | https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/UCSC_Ultralong_OxfordNanopore_Promethion/ |
| BioNano data | (Ebert et al., 2021) | EBI/ENA: ERP124807 |
| 1KG phased genotypes for 3,202 samples | (Byrska-Bishop et al.) | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_phased/ |
| Phased SNVs (VCFs) and inversion genotype tables | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/working/20210917_SSEQplusWHintegrativePhasing_inversionCallset/ |
| HGSVC phased assemblies (PGAS v12) | (Ebert et al., 2021) | https://www.internationalgenome.org/data-portal/data-collection/hgsvc2 |
| HGSVC phased assemblies (PGAS v13) | (Ebler et al. 2022) | DOI:10.5281/zenodo.5607680 |
| Dot plot visualizations of several recurrent inversion loci | This paper | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/HGSVC2/working/20220209_recurrent_inversions_resolved/ |
| Software and algorithms | ||
| primatR | (Porubsky et al., 2020) | https://github.com/daewoooo/primatR |
| breakpointR | (Porubsky et al., 2019) | https://github.com/daewoooo/breakpointR |
| StrandPhaseR | (Porubsky et al.,2017), New functionalities added in this paper | https://github.com/daewoooo/StrandPhaseR, branch=devel |
| ArbiGent | This paper | DOI:10.5281/zenodo.6405196 |
| PAV | (Ebert et al., 2021) | https://github.com/EichlerLab/pav |
| MEIGA-PAV | (Ebert et al., 2021), New functionalities added in this paper | DOI:10.5281/zenodo.6077336 |
| ti-SNPs detection | This paper | DOI:10.5281/zenodo.6405152 |
| Detection of altered SD organization | This paper | DOI:10.5281/zenodo.6411308 |
| Mendelian consistency analysis | This paper | DOI:10.5281/zenodo.6411714 |
| BWA aligner (v0.7.15–0.7.17) | (Li and Durbin, 2010) | http://bio-bwa.sourceforge.net/ |
| SAMtools (v1.3.1–1.10) | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| sambamba (v1.0) | (Tarasov et al., 2015) | https://lomereiter.github.io/sambamba/ |
| RTG tool (v3.11) | Copyright (c) 2018 Real Time Genomics Ltd | https://www.realtimegenomics.com/products/rtg-tools |
| Relate (v1.1.7) | (Speidel et al., 2019) | https://myersgroup.github.io/relate/ |
| IQ-TREE (v2.1.3) | (Minh et al., 2020) | http://www.iqtree.org/ |
| SV-Pop | (Audano et al., 2019; Ebert et al., 2021) | https://github.com/EichlerLab/svpop |