

Abstract
Motivation
Machine learning models for predicting cell-type-specific transcription factor (TF) binding sites have become increasingly more accurate thanks to the increased availability of next-generation sequencing data and more standardized model evaluation criteria. However, knowledge transfer from data-rich to data-limited TFs and cell types remains crucial for improving TF binding prediction models because available binding labels are highly skewed towards a small collection of TFs and cell types. Transfer prediction of TF binding sites can potentially benefit from a multitask learning approach; however, existing methods typically use shallow single-task models to generate low-resolution predictions. Here, we propose NetTIME, a multitask learning framework for predicting cell-type-specific TF binding sites with base-pair resolution.
Results
We show that the multitask learning strategy for TF binding prediction is more efficient than the single-task approach due to the increased data availability. NetTIME trains high-dimensional embedding vectors to distinguish TF and cell-type identities. We show that this approach is critical for the success of the multitask learning strategy and allows our model to make accurate transfer predictions within and beyond the training panels of TFs and cell types. We additionally train a linear-chain conditional random field (CRF) to classify binding predictions and show that this CRF eliminates the need for setting a probability threshold and reduces classification noise. We compare our method’s predictive performance with two state-of-the-art methods, Catchitt and Leopard, and show that our method outperforms previous methods under both supervised and transfer learning settings.
Availability and implementation
NetTIME is freely available at https://github.com/ryi06/NetTIME and the code is also archived at https://doi.org/10.5281/zenodo.6994897.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide modeling of non-coding DNA sequence function is among the most fundamental and yet challenging tasks in biology. Transcriptional regulation is orchestrated by transcription factors (TFs), whose binding to DNA initiates a series of signaling cascades that ultimately determine both the rate of transcription of their target genes and the underlying DNA functions. Both the cell-type-specific chromatin state and the DNA sequence affect the interactions between TFs and DNA in vivo (Ching et al., 2018). Experimentally determining cell-type-specific TF binding sites is made possible through high-throughput techniques such as chromatin immunoprecipitation followed by sequencing (ChIP-seq) (Johnson et al., 2007). Due to experimental limitations, however, it is infeasible to perform ChIP-seq (or related single-TF-focused experiments) on all TFs across all cell types and organisms (Ching et al., 2018). Therefore, computational methods for accurately predicting in vivo TF binding sites are essential for studying TF functions, especially for less well-known TFs and cell types.
Multiple community crowdsourcing challenges have been organized by the DREAM Consortium (http://dreamchallenges.org/about-dream/) to find the best computational methods for predicting TF binding sites in both in vitro and in vivo settings (Kundaje et al., 2017; Weirauch et al., 2013). These challenges also set the community standard for both processing data and evaluating methods. However, top-performing methods from these challenges have revealed key limitations in the current TF binding prediction community. Generalizing predictions beyond the training panels of cell types and TFs can potentially benefit from multitask learning and increased prediction resolution. However, many existing methods still use shallow single-task models. Predictions generated from these methods typically have low resolution, and they cannot achieve competitive performance for the prediction of binding regions shorter than 50 base pairs (bp), although the actual TF binding sites are considerably shorter (Stewart et al., 2012).
1.1 Related work
Early TF binding prediction methods such as MEME (Bailey and Elkan, 1994; Bailey et al., 2006) focused on deriving interpretable TF motif position weight matrices that characterize TF sequence specificity. Amid rapid advancement in machine learning, however, growing evidence has suggested that sequence specificity can be more accurately captured through more abstract feature extraction techniques. For example, a method called DeepBind (Alipanahi et al., 2015) used a convolutional neural network to extract TF binding patterns from DNA sequences. Several modifications to DeepBind subsequently improved model architecture (Hassanzadeh and Wang, 2016) as well as prediction resolution (Salekin et al., 2018). Yuan et al. (2019) developed BindSpace, which embeds TF-bound sequences into a common high-dimensional space. Embedding methods like BindSpace belong to a class of representation learning techniques commonly used in natural language processing (Mikolov et al., 2013) and genomics (Asgari and Mofrad, 2015; Yi et al., 2019) for mapping entities to vectors of real numbers. New methods also explicitly model protein binding sites with multiple binding mode predictors (Gfeller et al., 2011), and the effect of sequence variants on non-coding DNA functions at scale (Kelley et al., 2018; Zhou et al., 2018; Zhou and Troyanskaya, 2015).
In general, the DNA sequence at a potential TF binding site is just the beginning of the full DNA-function picture, and the state of the surrounding chromosome, the TF and TF-cofactor expression and other contextual factors play an equally large role. This multitude of factors changes substantially from cell type to cell type. In vivo TF binding site prediction, therefore, requires cell-type-specific data such as chromatin accessibility and histone modifications. Convolutional neural networks as well as TF- and cell-type-specific embedding vectors have both been used to learn cell-type-specific TF binding profiles from DNA sequences and DNase-seq data (Qin and Feng, 2017). The DREAM Consortium also initiated the ENCODE-DREAM challenge to systematically evaluate methods for predicting in vivo TF binding sites (Kundaje et al., 2017). Apart from carefully designed model architectures, top-ranking methods in this challenge also rely on extensively curated feature sets. One such method, called Catchitt (Keilwagen et al., 2019), achieves top performance by leveraging a wide range of features including DNA sequences, genome annotations, TF motifs, DNase-seq, and RNA-seq.
1.2 Current limitations
Compendium databases such as ENCODE (Moore et al., 2020) and Remap (Chèneby et al., 2020) have collected ChIP-seq data for a large collection of TFs in a handful of well-studied cell types and organisms (Ching et al., 2018). Within a single organism, however, the ENCODE TF ChIP-seq collection is highly skewed towards only a few TFs in a small collection of well-characterized cell lines and primary cell types (Supplementary Fig. S1). Transfer learning from well-known cell types and TFs is crucial for understanding less-studied cell types and TFs. One way to achieve transfer learning is by reusing information from a previously learned task to improve the learning efficiency of a related task (Torrey and Shavlik, 2010). For example, pretraining machine learning models with data from multiple TFs allows the models to learn common binding characteristics among TFs and thus, improves fine-tuning performance on a single TF of interest (Novakovsky et al., 2021; Zheng et al., 2021). Existing methods that adopt the above transfer learning approach (Novakovsky et al., 2021; Zheng et al., 2021) do not yet include model components that account for the TF and cell-type identities in an integrated fashion, which makes the fine-tuning step necessary for predicting binding preferences for a particular TF of interest. In contrast, multitask learning models that can account for TF and cell-type identities eliminate the necessity of fine-tuning when learning to predict binding preferences for new TFs in new cell types (this in turn enables a more meaningful integration of much larger training sets). Moreover, as different TFs have different binding mechanisms under various cellular conditions (Smith and Matthews, 2016), models that can account for TF and cell-type identities are potentially more effective at transfer learning compared to models, such as Novakovsky et al. (2021) and Zheng et al. (2021), which do not have a proper strategy for recognizing binding data of different TF and cell-type origins.
Top-performing methods from the ENCODE-DREAM Challenge typically adopt the single-task learning approach. For example, Catchitt (Keilwagen et al., 2019) trains one model per TF and cell type. Cross cell-type transfer predictions are achieved by providing a trained model with input features from a new cell type. This approach can be highly unreliable as the chromatin landscapes between the trained and predicted cell types can be drastically different (Calderon et al., 2019) and these differences can be functionally important (Sijacic et al., 2018). Alternatively, each model can be trained on one TF using cell-type-specific data across multiple cell types of interest (Quang and Xie, 2019). Without proper mechanisms to distinguish cell types, however, such models tend to assign high-binding probabilities to common binding sites among training cell types. A few methods have adopted the multitask learning approach in which data from multiple cell types and TFs are trained jointly in order to improve the overall model performance (Avsec et al., 2021a,b; Kelley et al., 2018; Quang and Xie, 2016; Schreiber et al., 2020; Zhou et al., 2018; Zhou and Troyanskaya, 2015). The multitask solution adopted by DeepSea (Zhou and Troyanskaya, 2015) and several other methods (Avsec et al., 2021b; Kelley et al., 2018; Quang and Xie, 2016; Schreiber et al., 2020; Zhou et al., 2018) involves training a multiclass classifier on DNA sequences, where each class represents the occurrence of binding sites for one TF in one cell type. This solution is suboptimal as it cannot generalize predictions beyond the training TF and cell-type pairs.
Sequence context affects TF binding affinity (Siggers and Gordân, 2014), and increasing context size can improve TF binding site prediction (Zhou and Troyanskaya, 2015). TF binding sites are typically only 4–20 bp long (Stewart et al., 2012); TF binding models that can achieve base-pair prediction resolution are therefore beneficial for experimental validation as well as de novo motif discovery. However, instead of identifying precise TF binding locations, existing methods mainly focus on determining the presence of binding sites. Predictions from these models suffer from either low resolution or low context size, depending on the input sequence length. Leopard (Li and Guan, 2021) and BPNet (Avsec et al., 2021a) are two recently proposed base-pair resolution binding prediction methods for predicting cell-type-specific TF binding sites. Leopard uses both DNA sequences and DNase-seq chromatin accessibility data as input, whereas BPNet predicts binding sites solely from DNA sequences. However, Leopard is a single-task learning model that requires training one model per TF and per cell type. Although BPNet uses multitask learning, the model does not include any task-specific components for distinguishing different TF and cell-type identities, and it’s performance when training on more than four conditions [described in Avsec et al. (2021a)] has not been evaluated.
In this work, we address the above challenges by introducing NetTIME (Network for TF binding Inference with Multitask-based condition Embeddings), a multitask learning framework for base-pair resolution prediction of cell-type-specific TF binding sites. NetTIME jointly trains multiple cell types and TFs, and effectively distinguishes different conditions using cell-type-specific and TF-specific embedding vectors. It achieves base-pair resolution and accepts input sequences up to 1 kb.
2 Approach
2.1 Feature and label generation
The ENCODE Consortium has published a large collection of TF ChIP-seq data, all of which are generated and processed using the same standardized pipelines (Moore et al., 2020). We therefore collect our TF binding target labels from ENCODE to minimize data heterogeneity. Each replicated ENCODE ChIP-seq experiment has two biological replicates, from which two sets of peaks—conserved and relaxed—are derived; peaks in both sets are highly reproducible between replicates (https://www.encodeproject.org/about/experiment-guidelines). Compared to the relaxed peak set, the conserved peak set is generated with a more stringent threshold and is generally used to provide target labels. However, the conserved peak set usually contains too few peaks to train the model efficiently. Therefore, we use both conserved and relaxed peak sets to provide target labels for training, and the conserved peak set alone for evaluating model performance.
To collect target labels for a representative set of conditions that cover a wide range of cellular conditions and binding patterns, we first select 7 cell types and 22 TFs for which ENCODE has available binding data. The seven cell types include three cancer cell types, three normal cell types and one stem cell type. The 22 TFs include 17 TFs from 7 TF protein families as well as 5 functionally related TFs. Conserved and relaxed peak sets are collected from 71 ENCODE replicated ChIP-seq experiments conducted on our cell types and TFs of interest. Each of these TF ChIP-seq experiment is henceforth referred to as a condition. All peaks from these conditions form a set of information-rich regions where at least one TF of interest is bound. We generate samples by selecting non-overlapping L-bp genomic windows from this information-rich set, where L is the context size. We set the context size L = 1000 as it was previously shown to improve TF binding prediction performance (Zhou and Troyanskaya, 2015).
In vivo TF binding sites are affected by DNA sequences and the cell-type-specific chromatin landscapes. In addition to using DNase-seq, which maps chromatin accessibility, we collect ChIP-seq data for 3 types of histone modifications to form our cell-type-specific feature set. The histone modifications we include are H3K4me1, H3K4me3 and H3K27ac, which are often associated with enhancers (Rada-Iglesias, 2018), promoters (Benayoun et al., 2014) and active enhancers (Creyghton et al., 2010), respectively.
2.2 Methods
NetTIME performs TF binding predictions in three steps: (i) generating the feature vector given a TF label p, a cell type label q and a sample DNA sequence where each , (ii) training a neural network to predict base-pair resolution binding probabilities and (iii) converting binding probabilities to binary binding decisions of p in q by either setting a probability threshold or additionally training a conditional random field (CRF) classifier (Fig. 1).
Fig. 1.

Schematic method overview. (a) Constructing feature vector w from input sequence x, TF label p and cell type label q. w consists of the sequence one-hot encoding, and a set of cell-type-specific features—DNase-seq signals, and H3K4me1, H3K4me3 and H3K4ac histone ChIP-seq signals—in cell type q. (b) Feature vector w, TF label p and cell type label q are provided to the NetTIME neural network to predict base-pair resolution binding probability z. An additional CRF classifier is trained to predict binary binding event y from z
2.2.1 Feature representation
We construct the feature vector from , where K represents the number of features. Different types of features are independently stacked along the first dimension. For each element in w, wl is the concatenation of the one-hot encoding of the DNA sequence , and the cell-type-specific feature (Fig. 1a).
| (1) |
High-dimensional embedding vectors can be trained to distinguish different conditions as well as implicitly learning condition-specific features and are therefore preferred by many machine learning models over one-dimensional condition labels (Qin and Feng, 2017; Yi et al., 2019; Yuan et al., 2019). Given TF label p and cell-type label q, NetTIME learns the TF- and cell-type-specific embeddings and , where .
2.2.2 Binding probability prediction
NetTIME adopts an encoder–decoder structure similar to that of neural machine translation models (Cho et al., 2014b; Sutskever et al., 2014; Vaswani et al., 2017) (Fig. 1b, Supplementary Fig. S2):
Encoder: the model encoder maps the input feature w to a hidden vector . The main structure of the encoder, called the Basic Block, consists of a convolutional neural network (CNN) followed by a recurrent neural network (RNN). CNN uses multiple short convolution kernels to extract local binding motifs, whereas bi-directional RNN is effective at capturing long-range TF-DNA interactions (Cho et al., 2014a; Hochreiter and Schmidhuber, 1997). We choose the ResBlock structure introduced by ResNet (He et al., 2016) as our CNN, as it has become a standard approach for training deep neural networks (Huang et al., 2017; Vaswani et al., 2017). Traditional RNNs are challenging to train due to the vanishing gradient problem (Hochreiter and Schmidhuber, 1997). We therefore use the bi-directional gated recurrent unit (bi-GRU) (Cho et al., 2014a), a variant of RNN proposed to address the above challenge. The hidden state of bi-GRU is initialized by concatenating the embedding vectors and .
Decoder: the model decoder converts the hidden vector h to binding probabilities z. The conversion is achieved through a fully connected feed-forward network, as the relationship between h and z may not be trivial. A softmax function subsequently transforms the decoder output to the binding probabilities.
2.2.3 Training
We train the model by minimizing the negative conditional log-likelihood of z:
| (2) |
where N denotes the number of training samples. The loss function is optimized by the Adam optimizer (Kingma and Ba, 2015) (also see Supplementary Section S1.2).
2.2.4 Binding event classification
Binary binding events y can be directly derived from z by setting a probability threshold such that
| (3) |
This approach has been used by many existing TF binding predictions models to admit exact inference (Li et al., 2019; Li and Guan, 2021). Alternatively, a linear-chain CRF classifier can be trained to achieve the same goal. It computes the conditional probability of y given z, defined as the following:
| (4) |
where
is a normalization factor,
is a transition matrix, where p denotes the number of classes of the classification problem and each represents the transition probability from class label i to j,
calculates the likelihood of yl given zl, and
measures the likelihood of given yl.
In CRF, the class label at position l affects the classification at position l + 1 (Sutton and McCallum, 2012). This is potentially beneficial for TF binding site classification as positions adjacent to a putative binding site are also highly likely to be occupied by TFs. We train the CRF by minimizing over all training samples. The Adam optimizer (Kingma and Ba, 2015) is used to update the parameter V.
2.3 Model selection
We follow the guideline provided by the ENCODE-DREAM Challenge (Kundaje et al., 2017) to perform data split as well as model selection whenever possible. Training, validation and test data are split according to chromosomes (Supplementary Table S1). We use the area under the Precision-Recall Curve (auPRC) score to select the best neural network model checkpoint.
To access how well our model predictions recover the positive binding sites in the truth target labels, we evaluate classifiers’ performance according to Intersection Over Union (IOU) score. Suppose P and T are sets of predicted and target binding sites, respectively. Then
| (5) |
We test 300 random probability thresholds and select the best threshold, i.e. the threshold that achieves the highest IOU score in the validation set. We also train a CRF using predictions generated from the best neural network checkpoint. The best CRF checkpoint is selected according to the average loss on the validation set. Model performance reported here is evaluated using the test set.
3 Results
3.1 Multitask learning improves performance by increasing data availability
NetTIME can be trained using data from a single condition (single-task learning) or multiple conditions (multitask learning). Jointly training multiple conditions allows the model to use data more efficiently and improves model generalization (Caruana, 1997). Multitask learning is particularly suitable for learning cell-type-specific TF binding preferences because a TF has common binding sites across different cell types, and functionally related TFs share similar binding sites (Spitz and Furlong, 2012). We therefore evaluate the effectiveness of multitask learning when jointly training multiple related conditions. For this analysis, we choose three TFs from the JUN family that exhibit overlapping functions: JUN, JUNB and JUND (Mechta-Grigoriou et al., 2001). Combining multiple cell types of JUND allows the multitask learning model to significantly outperform the single-task learning models, each of which is trained with one JUND condition (Fig. 2a). Jointly training multiple JUN family TFs further improves performance compared to training each JUN family TF separately (Fig. 2b). However, we observe decreased performance when subsampling the multitask models’ training data to match the number of samples in the corresponding single-task models (Fig. 2).
Fig. 2.
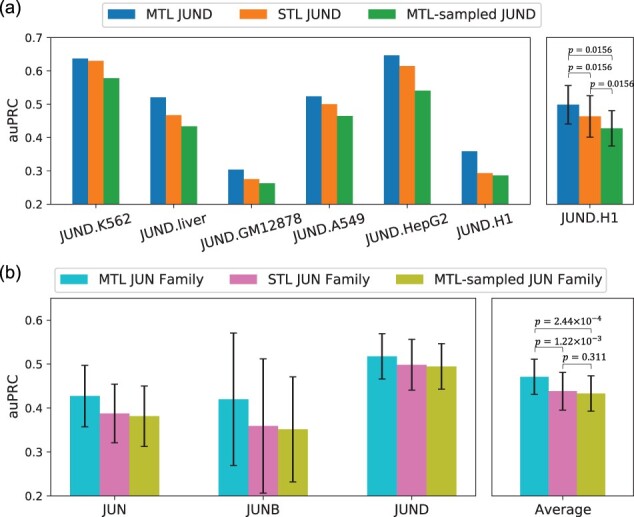
Performance comparison between multitask learning and single-task learning approaches using JUN family TFs. Models are trained with datasets from (a) JUND across multiple cell types, and (b) multiple TFs in the JUN family across multiple cell types. MTL, multitask learning; MTL-sampled, multitask learning training data that has been subsampled to match the number of samples in the corresponding single-task models; STL, single-task learning. The right panels in (a) and (b) are the averaged auPRC of the models shown in the corresponding left panels. Error bars represent standard error of the mean across all training conditions. P-values are calculated using the Wilcoxon signed-rank test using auPRC scores across all conditions
This indicates that the multitask learning strategy is more efficient due to the increased data available to the multitask models rather than to the increased data diversity. Similar results are also observed when the same analysis is performed on three unrelated TFs (Supplementary Fig. S3).
3.2 Supervised predictions made by NetTIME outperforms existing baseline methods
Our complete feature set includes DNA sequence, and cell-type-specific features including DNase-seq and three types of histone ChIP-seq. In practice, however, data for these features are not always available for the conditions of interest. Additionally, TF motif enrichment has often been used by existing methods to provide TF binding sequence specificity information (Keilwagen et al., 2019; Quang and Xie, 2019). We therefore evaluate the quality of our model predictions when we vary the types of input features available during training.
We first train separate models after removing cell-type-specific features using training data from all conditions mentioned in Section 2.1. Model prediction accuracy is evaluated in the supervised fashion using the test data from the same set of conditions. The addition of cell-type-specific features significantly improves NetTIME performance. However, adding TF motif enrichment features (Supplementary Section S1.1), either in addition to DNA sequence features or in addition to both sequence and cell-type features, does not introduce significant performance improvement (Fig. 3a). Despite exhibiting high sequence specificity in vitro, TF binding sites in vivo correlate poorly with TF motif enrichment (Chen et al., 2017). Motif qualities in TF motif databases vary significantly depending on the available binding data and motif search algorithms. Nevertheless, TF motifs have been the gold standard for TF binding site analyses due to their interpretability and scale. However, TF motif enrichment features are likely redundant when our model can effectively capture TF binding sequence specificity, though it’s possible our protocol for generating TF motif enrichment features is suboptimal.
Fig. 3.

NetTIME significantly improves predictive performance against state-of-the-art baseline methods. (a) Comparing NetTIME performance under different input feature settings. Seq, DNA sequence feature; CT, cell-type-specific features including DNase-seq, and H3K4me1, H3K4me3 and H3K27ac histone ChIP-seq data; TF, TF motif enrichment feature. (b) Comparing NetTIME performance (b) against Catchitt and Leopard under 200-bp and 1000-bp resolutions, and (c) against Leopard under 1-bp resolution. DNase, cell-type-specific DNase-seq feature. (d) Comparing NetTIME Seq + DNase model performance against Catchitt and Leopard under 200-bp resolution using the ENCODE-DREAM Challenge data
We further compare NetTIME predictive performance with that of Catchitt (Keilwagen et al., 2019) and Leopard (Li and Guan, 2021). As Catchitt and Leopard use only DNase-seq data as their cell-type-specific input feature, we train a separate NetTIME model using DNA sequences and DNase-seq data as input. Additionally, because Catchitt is evaluated under the 200-bp resolution for the ENCODE-DREAM Challenge, we reduce the NetTIME and Leopard prediction resolution by taking the maximum prediction probability across the center 200-bp regions for all the example sequences in our test set. Performance of these three methods is further compared under 1000-bp resolution to evaluate per-sample prediction accuracy. Prediction auPRC scores consistently increase for all three methods as we decrease the prediction resolution from 200 to 1000 bp. Nevertheless, NetTIME outperforms both baseline methods under both prediction resolutions (Fig. 3b). Furthermore, NetTIME significantly outperforms Leopard when predictions are evaluated on the per-base-pair level (Fig. 3c). Although the TF motifs are not used by Leopard as a type of input feature, Leopard derives target binding labels by subsetting TF ChIP-seq peaks with regions that show TF motif enrichment (Li and Guan, 2021). This data generation procedure potentially introduces unwanted biases and contributes to the reduced performance when the model is evaluated on the complete set of TF ChIP-seq peaks.
Both Catchitt and Leopard can only be trained using examples derived genome-wide. To ensure a fair comparison, we train additional Seq + DNase NetTIME models using DNase seq data and ChIP-seq labels provided by the ENCODE-DREAM Challenge. All three methods are benchmarked against the 13 test conditions in the ENCODE-DREAM Challenge, and their model performance is evaluated at 200-bp resolution using examples generated by sliding a 200-bp window across all test chromosomes with a 50-bp overlap between adjacent examples. Predictions at 200-bp resolution from NetTIME and Leopard are generated by taking the maximum probability across each 200-bp region from the 1-bp resolution predictions generated by these two methods. NetTIME improves the mean prediction auPRC score by 11.8% and 6.3% over Catchitt and Leopard, respectively (Fig. 3d).
3.3 TF- and cell-type-specific embeddings are crucial for an effective multitask learning strategy
Here, we evaluate the relative contributions of different model components to our predictive accuracy. We use the TF and cell-type embedding vectors to learn condition-specific features and biases, and a combination of CNNs and RNNs to learn the non-condition-specific TF-DNA interaction patterns. TF and cell-type embedding vectors can be replaced with random vectors at prediction time and at training time to evaluate the contribution of each component individually.
To evaluate the model’s sensitivity to different TF and cell-type labels, TF and cell-type embedding vectors are replaced with random vectors at prediction time (Fig. 4). When NetTIME is trained with both TF and cell-type embeddings, the model learns to use both pieces of condition-specific information in order to make accurate predictions. As a result, substituting both types of embeddings with random vectors reduces our model performance by 69.1% on average. Replacing either TF or cell-type embeddings with random vectors also drastically reduce auPRC scores. This indicates that cell-type-specific chromatin landscape, in addition to TF identity, is important for defining in vivo TF binding sites, which explains the redundancy of TF motif features and the lack of correlation between TF ChIP-seq signals and TF motif enrichment mentioned in Section 3.2 and Chen et al. (2017).
Fig. 4.

Properties of trained embedding vectors. Evaluating the contribution of condition-specific network components (a) at prediction time and (b) at training time by replacing trained TF and cell-type embeddings with random vectors. CT + TF embeddings, trained TF and cell-type embeddings; TF embeddings only, trained TF embeddings and random cell-type vectors; CT embeddings only, trained cell-type embeddings and random TF vectors; No trained embeddings, random TF and cell-type vectors. (c) t-SNE visualization of the TF embedding vectors. Orange circles indicate related TFs that are in close proximity in t-SNE projection space: solid circles illustrate TFs from the same protein family, and dashed circles illustrate TFs having similar functions (A color version of this figure appears in the online version of this article.)
We additionally swap either or both types of embedding vectors during training to evaluate the contribution of the non-condition-specific network component. Replacing both types of embedding vectors during training results in a 26.2% drop in the mean auPRC score across all training conditions (Fig. 4b). However, the significant performance decrease is mainly due to the removal of TF embeddings—separately removing TF embeddings and cell-type embeddings result in a 25.7% and a 0.5% drop in the mean auPRC, respectively. Under the current model input feature setting, TF identity can only be learned through the TF embedding vectors. In contrast, cell-type-specific chromatin landscape can be learn from the cell-type-specific input features in addition to cell-type embeddings. In the presence of cell-type-specific input features, cell-type embeddings are used by the model to capture residual cell-type-specific information, and therefore only introduce marginal performance improvement (Fig. 4 and Supplementary Fig. S4).
Visualizing the trained TF embedding vectors in two dimensions using t-distributed stochastic neighbor embedding (t-SNE, Van der Maaten and Hinton, 2008) reveals that a subset of embedding vectors also reflects the TF functional similarities. Some TFs that are in close proximity in t-SNE space are from the same TF families, including FOXA1 and FOXA2, HNF4A and HNF4G, ATF2, ATF3 and ATF7, and JUN, JUNB, and JUND (Fig. 4c, solid circles). Functionally related TFs such as IRF3 and STAT3 (Mogensen, 2019) are also adjacent to each other in t-SNE space (Fig. 4c, dashed circle). However, these TF embedding vectors are explicitly trained to learn the biases introduced by TF labels. Available data for TFs of the same protein family are not necessarily from the same set of cell-types. As a result, not all functionally related TFs are close in the t-SNE space, such as IRF (IRF3, IRF4 and IRF5) family proteins and TFs associated with c-Myc proteins (MAX and MAZ).
3.4 TF and cell-type embeddings allow more reliable transfer predictions
Transfer learning allows models to make cross-TF and cross-cell-type predictions beyond training conditions. Existing single-task learners such as Catchitt achieve transfer learning by providing input features from a new cell type to a model trained on a different cell type. If multiple trained cell types are available for the same TF, the final cross-cell-type predictions are generated by averaging predictions from all trained cell types (Fig. 5a, Average Train). A different transfer learning strategy proposed by AgentBind (Zheng et al., 2021) involves pretraining a multi-TF model that does not distinguish different TF identities before fine-tuning the model on a single TF of interest (No Embedding Transfer, Fig. 5a). The former strategy cannot take advantage of the additional information introduced by other functionally related TFs, whereas the latter does not distinguish different TF identities in the multitask pretraining step. Since TF binding prediction can benefit from the multitask learning paradigm (Section 3.1), and a multitask learning model performance is highly influenced by the TF identity (Section 3.3), we hypothesize that NetTIME’s transfer learning strategy (Fig. 5a, Embedding Transfer) is superior for cross-TF and cross-cell-type binding prediction.
Fig. 5.

Comparison of different transfer learning strategies. (a) Detailed overview of the training scheme and prediction generation procedure using three transfer learning strategies implemented by NetTIME, Catchitt and AgentBind. and denote the particular TF (t or p) and cell type (c or q) combinations. S refers to the set of all conditions in our training dataset. T is the set of 10 leave-out conditions. Cp is the set of cell types that satisfy . is the input data from . is the per-base-pair binding probability predictions for . F and M can be any machine learning models. To avoid performance differences introduced by the model architecture, we use NetTIME model with TF and cell-type embeddings (F) and with no embedding (M). (b) Transfer predictions using different transfer learning strategies are generated for 10 leave-out TF and cell-type combinations within the training panels of TFs and cell types
To evaluate the prediction quality of these three approaches, we pretrain a NetTIME model by leaving out 10 conditions for transfer learning. Transfer learning predictions are generally less accurate compared to supervised predictions (Supervised). For each transfer condition , we use the pretrained model to directly derive predictions for each transfer learning strategy (Fig. 5a). However, transfer predictions generated by Embedding Transfer still significantly outperform those of the Average Train and the No Embedding Transfer (Fig. 5b). Transfer predictions derived from NetTIME also achieve considerably higher accuracy compared to those from Catchitt and Leopard (Supplementary Fig. S5a and c). We additionally investigate whether different transfer learning strategies can benefit from fine-tuning by fine tuning all models using all conditions from TF p excluding . This fine-tuning step additionally improves performance for Embedding and No Embedding Transfer approaches, whereas Average Train performance after fine-tuning remains low compared to two other approaches (Supplementary Fig. S5b and d).
Using trained TF and cell-type embeddings additionally allows models to perform binding predictions beyond the training panels of TFs and cell types. We therefore test our model’s robustness when making predictions on unknown conditions using four conditions from four new TFs in three new cell types. Starting from a NetTIME model pretrained on all original training conditions (Section 2.1), we fine-tune the pretrained model for each transfer condition separately by collecting available ENCODE datasets from all conditions from TF and all conditions in cell type excluding . Transfer predictions generated from models trained with TF and cell-type embeddings (Trained Embedding Transfer) significantly outperform those from models trained with no embeddings (No Embedding Transfer) that cannot distinguish different TF and cell-type identities (Fig. 6a). TF binding motifs derived from predicted binding sites also show a strong resemblance to those derived from conserved ChIP-seq peaks (Fig. 6b).
Fig. 6.
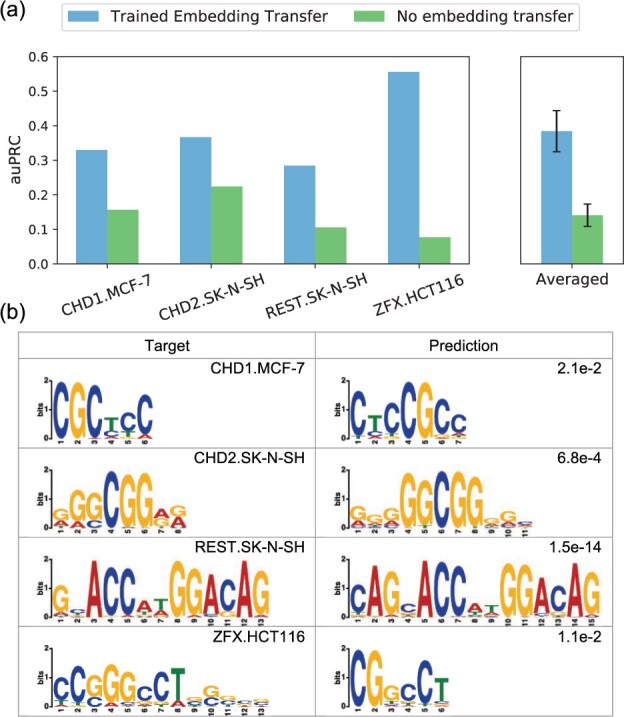
Transfer predictions to new TF and cell-type combinations beyond the training panels of TFs and cell types. (a) Transfer predictions using models trained with either TF and cell-type embedding vectors (Trained Embedding Transfer) or no trained embeddings (No embedding transfer). (b) Comparison of predicted TF binding motifs to those derived from target ChIP-seq conserved peaks. Predicted motifs are derived from Trained Embedding Transfer predictions. De novo motif discovery is conducted using STREME (Bailey, 2020) software. Motif similarity P-values shown in the top right corner of the Prediction column are derived by comparing predicted and target motifs using TOMTOM (Gupta et al., 2007)
3.5 A CRF classifier post-processing step effectively reduces prediction noise
Summarizing the binding strength, or probability, along the chromosome at each discrete binding site is an important step for several downstream tasks ranging from visualization to validation. Deriving binary binding decision from binding probabilities are typically done by finding a probability threshold that achieves the best prediction accuracy (Li et al., 2019; Li and Guan, 2021; Yuan et al., 2019). We test this baseline approach by evaluating the model’s predictive performance at 300 randomly selected probability thresholds. We find that at threshold 0.1302, the model achieves the highest IOU score of 36% (Fig. 7a).
Fig. 7.
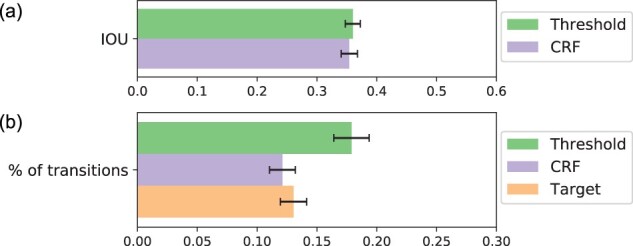
Binary classification performance using the probability threshold and CRF. Performance evaluated by (a) the mean IOU score and (b) the percentage of class label transitions per sequence (bottom), both calculated over all training conditions
We alternatively train a CRF classifier, as a manually selected probability threshold is poorly generalizable to unknown datasets. These two approaches achieve comparable predictive performance as evaluated by IOU scores (Fig. 7a). However, prediction noises manifested as high probability spikes are likely to be classified as bound using the probability threshold approach. To evaluate the effectiveness of reducing prediction noises using the probability threshold and the CRF approaches, we calculate the percentage of class label transitions per sequence within the target labels and within each of the predicted labels generated by these two approaches. The transition percentage using CRF is comparable to that of the true target labels and is also significantly lower than the percentage obtained using the probability threshold approach (Fig. 7b). This indicates that CRF is more effective at reducing prediction noise, and therefore CRF predictions exhibit a higher degree of resemblance to target labels.
4 Conclusions
In this work, we address several challenges facing existing methods for TF binding site predictions by introducing a multitask learning framework, called NetTIME, which learns base-pair resolution TF binding sites using embeddings. We show that our multitask learning approach improves prediction accuracy by increasing the data available to the model. Both the condition-specific and non-condition-specific components in our multitask framework are important for making accurate condition-specific binding predictions. The use of TF and cell-type embedding vectors additionally allows us to make accurate transfer learning predictions within and beyond the training panels of TFs and cell types. Our method also significantly outperforms previous methods under both supervised and transfer learning settings, including Catchitt and Leopard.
Although DNA sequencing currently can achieve base-pair resolution, the resolution of ChIP-seq data is still limited by the size of DNA fragments obtained through random clipping. A considerable fraction of the fragments are therefore false positives, whereas many transient and low-affinity binding sites are missed (Park, 2009). Additionally, ChIP-seq requires suitable antibodies for proteins of interest, which can be difficult to obtain for rare cell types and TFs. Alternative assays have been proposed to improve data resolution (He et al., 2015; Rhee and Pugh, 2011; Rossi et al., 2018) as well as to eliminate the requirement for antibodies (Southall et al., 2013; van Steensel and Henikoff, 2000). However, datasets generated from these techniques are rare or missing in data consortiums such as ENCODE (Moore et al., 2020) and ReMap (Chèneby et al., 2020). NetTIME can potentially provide base-pair resolution solutions to more complex DNA sequence problems as labels generated from these alternative assays become more widely available in the future.
ATAC-seq [Assay for Transposase-Accessible Chromatin using sequencing (Buenrostro et al., 2013)] has overtaken DNase-seq as the preferred assay to profile chromatin accessibility, as it requires fewer steps and input materials. However, these two techniques each offer unique insights into the cell-type-specific chromatin states (Calviello et al., 2019), and it is therefore potentially beneficial to incorporate both data types for TF binding predictions. In fact, extensive feature engineering has been the focus of many recent in vivo TF binding prediction methods (Chen et al., 2017; Keilwagen et al., 2019; Quang and Xie, 2019). It is also important to note that, without strategies for handling missing features, increasing feature requirements significantly restricts models’ scope of application (Supplementary Fig. S1). A comprehensive evaluation of data imputation methods (Amodio et al., 2019; Howie et al., 2009; Troyanskaya et al., 2001; Van Dijk et al., 2018) can be difficult due to the lack of knowledge of the true underlying data distribution. We plan to extend our model’s ability to learn from a more diverse set of features and investigate more efficient ways to handle missing data. We also plan to explore other neural network architectures to improve model performance while reducing the model’s feature requirement.
NetTIME is extensible and can be adapted to improve solutions to other biology problems, such as transcriptional regulatory network (TRN) inference. TRN inference identifies genome-wide functional regulations of gene expressions by TFs. TFs control the expression patterns of target genes by first binding to regions containing promoters, distal enhancers and/or other regulatory elements. However, functional interactions between TFs and target genes are further complicated by TF concentrations and co-occurrence of other TFs. A series of methods have been proposed for inferring TRNs from gene expression data and prior knowledge of the network structure (Greenfield et al., 2013; Irrthum et al., 2010; Yuan and Bar-Joseph, 2019). Prior knowledge can be obtained by identifying open chromatin regions close to gene bodies that are also enriched with TF motifs (Miraldi et al., 2019). However, this method is problematic for identifying TF functional regulations towards distal enhancers and binding sites without motif enrichment. In vivo predictions of TF binding profiles, however, can serve as a more flexible approach to generating prior network structure as it bypasses the aforementioned unnecessary constraints. In future work, we hope to adapt the NetTIME framework to explore more efficient approaches for generating prior knowledge for more biophysically motivated TRN inference.
Supplementary Material
Acknowledgements
We thank members of the Bonneau and Cho labs for providing helpful suggestions on the project and on the manuscript. We thank the NYU High Performance Computing team and the Flatiron Institute Scientific Computing team for their excellent high performance computing support.
Funding
R.B. and R.Y. thank the following sources for research support: National Science Foundation (NSF) [IOS-1546218], National Institutes of Health [R35GM122515, R01HD096770 and R01NS116350], New York University and Simons Foundation. K.C. is partly supported by Samsung Advanced Institute of Technology (Next Generation Deep Learning: from pattern recognition to AI) and Samsung Research (Improving Deep Learning using Latent Structure). K.C. also thanks Naver, eBay, NVIDIA and NSF Award 1922658 for support.
Conflict of Interest: none declared.
Contributor Information
Ren Yi, Department of Computer Science, New York University, New York, NY 10011, USA.
Kyunghyun Cho, Department of Computer Science, New York University, New York, NY 10011, USA; Center for Data Science, New York University, New York, NY 10011, USA; Prescient Design, a Genentech accelerator, New York, NY 10010, USA.
Richard Bonneau, Department of Computer Science, New York University, New York, NY 10011, USA; Center for Data Science, New York University, New York, NY 10011, USA; Prescient Design, a Genentech accelerator, New York, NY 10010, USA; Department of Biology, New York University, New York, NY 10003, USA.
Data Availability
The data used in this manuscript are downloaded from the ENCODE project website: https://www.encodeproject.org/. The assession numbers are provided in Supplementary Data 1-4. The ENCODE-DREAM Challenge data are downloaded from the Challenge website (Kundaje et al., 2017).
References
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. [DOI] [PubMed] [Google Scholar]
- Amodio M. et al. (2019) Exploring single-cell data with deep multitasking neural networks. Nat. Methods, 16, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari E., Mofrad M.R. (2015) Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One, 10, e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsec Ž. et al. (2021a) Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet., 53, 354–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsec Ž. et al. (2021b) Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods, 18, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L. and Elkan,C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In: Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, USA. AAAI Press, pp. 28–36. [PubMed]
- Bailey,T.L. (2021) STREME: accurate and versatile sequence motif discovery. Bioinformatics, 37, 2834–2840. 10.1093/bioinformatics/btab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L. et al. (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res., 34, W369–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun B.A. et al. (2014) H3k4me3 breadth is linked to cell identity and transcriptional consistency. Cell, 158, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D. et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon D. et al. (2019) Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet., 51, 1494–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calviello A.K. et al. (2019) Reproducible inference of transcription factor footprints in ATAC-seq and DNase-seq datasets using protocol-specific bias modeling. Genome Biol., 20, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana R. (1997) Multitask learning. Mach. Learn., 28, 41–75. [Google Scholar]
- Chen X. et al. (2017) Mocap: large-scale inference of transcription factor binding sites from chromatin accessibility. Nucleic Acids Res., 45, 4315–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chèneby J. et al. (2020) Remap 2020: a database of regulatory regions from an integrative analysis of human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res., 48, D180–D188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching T. et al. (2018) Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface, 15, 20170387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K. et al. (2014a) Learning phrase representations using RNN encoder-decoder for statistical machine translation. In: Proceedings of the 2014 Conferenceon Empirical Methods in Natural Language Processing (EMNLP). Association for Computational Linguistics, Doha, Qatar, pp. 1724–1734.
- Cho K. et al. (2014b) On the properties of neural machine translation: encoder-decoder approaches. In: Proceedings of SSST-8, Eighth Workshop on Syntax, Semantics and Structurein Statistical Translation. Association for Computational Linguistics, Doha, Qatar, pp. 103–111.
- Creyghton M.P. et al. (2010) Histone h3k27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA, 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje,A. et al. (2017) ENCODE-DREAM in vivo Transcription Factor Binding Site Prediction Challenge. Synapse. https://www.synapse.org/#!Synapse:syn6131484/wiki/402026 (24 November 2020, date last accessed).
- Gfeller D. et al. (2011) The multiple-specificity landscape of modular peptide recognition domains. Mol. Syst. Biol., 7, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield A. et al. (2013) Robust data-driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics, 29, 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. et al. (2007) Quantifying similarity between motifs. Genome Biol., 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanzadeh H.R., Wang M.D. (2016) Deeperbind: enhancing prediction of sequence specificities of DNA binding proteins. In: 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) Shenzhen, China. IEEE, pp. 178–183. [DOI] [PMC free article] [PubMed]
- He K. et al. (2016) Deep residual learning for image recognition. In: Proceedings of the IEEE conference on computer vision and pattern recognition, Las Vegas, NV, USA, pp. 770–778.
- He Q. et al. (2015) Chip-nexus enables improved detection of in vivo transcription factor binding footprints. Nat. Biotechnol., 33, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter S., Schmidhuber J. (1997) Long short-term memory. Neural Comput., 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- Howie B.N. et al. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G. et al. (2017) Densely connected convolutional networks. In: Proceedings of the IEEE conference on computer vision and pattern recognition, Honolulu, HI, USA, pp. 4700–4708.
- Irrthum A. et al. (2010) Inferring regulatory networks from expression data using tree-based methods. PLoS One, 5, e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.S. et al. (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science, 316, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Keilwagen J. et al. (2019) Accurate prediction of cell type-specific transcription factor binding. Genome Biol., 20, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D.R. et al. (2018) Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res., 28, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Ba J. (2015) Adam: a method for stochastic optimization. In: 3rd International Conference on Learning Representations, ICLR 2015, San Diego, CA, USA, May 7-9, 2015, Conference Track Proceedings.
- Li H., Guan Y. (2021) Fast decoding cell type-specific transcription factor binding landscape at single-nucleotide resolution. Genome Res., 31, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2019) Anchor: trans-cell type prediction of transcription factor binding sites. Genome Res., 29, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechta-Grigoriou F. et al. (2001) The mammalian Jun proteins: redundancy and specificity. Oncogene, 20, 2378–2389. [DOI] [PubMed] [Google Scholar]
- Mikolov T. et al. (2013) Efficient estimation of word representations in vector space. In: 1st International Conference on Learning Representations, ICLR 2013, Scottsdale,Arizona, USA, May 2-4, 2013, Workshop Track Proceedings.
- Miraldi E.R. et al. (2019) Leveraging chromatin accessibility for transcriptional regulatory network inference in t helper 17 cells. Genome Res., 29, 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen T.H. (2019) IRF and STAT transcription factors-from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front. Immunol., 9, 3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J.E. et al. ; ENCODE Project Consortium. (2020) Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature, 583, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovsky G. et al. (2021) Biologically relevant transfer learning improves transcription factor binding prediction. Genome Biol., 22, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P.J. (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet., 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q., Feng J. (2017) Imputation for transcription factor binding predictions based on deep learning. PLoS Comput. Biol., 13, e1005403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D., Xie X. (2016) DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res., 44, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D., Xie X. (2019) FactorNet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods, 166, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A. (2018) Is H3K4me1 at enhancers correlative or causative? Nat. Genet., 50, 4–5. [DOI] [PubMed] [Google Scholar]
- Rhee H.S., Pugh B.F. (2011) Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell, 147, 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M.J. et al. (2018) Simplified chip-exo assays. Nat. Commun., 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salekin S. et al. (2018) Base-pair resolution detection of transcription factor binding site by deep deconvolutional network. Bioinformatics, 34, 3446–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber J. et al. (2020) Avocado: a multi-scale deep tensor factorization method learns a latent representation of the human epigenome. Genome Biol., 21, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggers T., Gordân R. (2014) Protein-DNA binding: complexities and multi-protein codes. Nucleic Acids Res., 42, 2099–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijacic P. et al. (2018) Changes in chromatin accessibility between Arabidopsis stem cells and mesophyll cells illuminate cell type-specific transcription factor networks. Plant J., 94, 215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N.C., Matthews J.M. (2016) Mechanisms of DNA-binding specificity and functional gene regulation by transcription factors. Curr. Opin. Struct. Biol., 38, 68–74. [DOI] [PubMed] [Google Scholar]
- Southall T.D. et al. (2013) Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev. Cell., 26, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F., Furlong E.E. (2012) Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet., 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Stewart A.J. et al. (2012) Why transcription factor binding sites are ten nucleotides long. Genetics, 192, 973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutskever I. et al. (2014) Sequence to sequence learning with neural networks. In: Advances in neural information processing systems, Montréal, QC, Canada, pp. 3104–3112.
- Sutton,C and McCallum,A. (2012) An introduction to conditional random fields. FNT. in Machine Learning, 4, 267–373. 10.1561/2200000013. [DOI] [Google Scholar]
- , Torrey L., Shavlik J. (2010) Transfer learning. In: Handbook of Research on Machine Learning Applications and Trends: Algorithms, Methods, and Techniques. IGI global, pp. 242–264. [Google Scholar]
- Troyanskaya O. et al. (2001) Missing value estimation methods for DNA microarrays. Bioinformatics, 17, 520–525. [DOI] [PubMed] [Google Scholar]
- Van der Maaten L., Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Van Dijk D. et al. (2018) Recovering gene interactions from single-cell data using data diffusion. Cell, 174, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B., Henikoff S. (2000) Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat. Biotechnol., 18, 424–428. [DOI] [PubMed] [Google Scholar]
- Vaswani A. et al. (2017) Attention is all you need. In: Advances in Neural Information Processing Systems, Long Beach, CA, USA, pp. 5998–6008.
- Weirauch M.T. et al. ; DREAM5 Consortium. (2013) Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol., 31, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R. et al. (2019) Learning from data-rich problems: a case study on genetic variant calling. arXiv preprint. 10.48550/arXiv.1911.05151. [DOI]
- Yuan H. et al. (2019) Bindspace decodes transcription factor binding signals by large-scale sequence embedding. Nat. Methods, 16, 858–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Bar-Joseph Z. (2019) Deep learning for inferring gene relationships from single-cell expression data. Proc. Natl. Acad. Sci. USA, 116, 27151–27158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng A. et al. (2021) Deep neural networks identify sequence context features predictive of transcription factor binding. Nat. Mach. Intell., 3, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Troyanskaya O.G. (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods, 12, 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. et al. (2018) Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet., 50, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this manuscript are downloaded from the ENCODE project website: https://www.encodeproject.org/. The assession numbers are provided in Supplementary Data 1-4. The ENCODE-DREAM Challenge data are downloaded from the Challenge website (Kundaje et al., 2017).