

Abstract
The proteasome is essential for cellular responses to various physiological stressors. However, how proteasome function impacts the stress resilience of regenerative damaged motor neurons remains unclear. Here, we develop a unique mouse model using a regulatory element of the activating transcription factor (Atf3) gene to label mitochondria in a damage‐induced manner while simultaneously genetically disrupting the proteasome. Using this model, we observed that in injury‐induced proteasome‐deficient mouse motor neurons, the increase of mitochondrial influx from soma into axons is inhibited because neurons fail to disassemble ankyrin G, an organizer of the axon initial segment (AIS), in a proteasome‐dependent manner. Further, these motor neurons exhibit amyotrophic lateral sclerosis (ALS)‐like degeneration despite having regenerative potential. Selectively vulnerable motor neurons in SOD1G93A ALS mice, which induce ATF3 in response to pathological damage, also fail to disrupt the AIS, limiting the number of axonal mitochondria at a pre‐symptomatic stage. Thus, damage‐induced proteasome‐sensitive AIS disassembly could be a critical post‐translational response for damaged motor neurons to temporarily transit to an immature state and meet energy demands for axon regeneration or preservation.
Keywords: axonal transport, nerve injury, neurodegenerative disease, organelle, stress response
Subject Categories: Molecular Biology of Disease, Neuroscience, Organelles
Proteasomes impact stress resilience by temporarily dismantling the barrier against mitochondrial axonal entry upon injury to meet energy demands for axon regeneration.
Introduction
Nerve‐injured motor neurons can regenerate through multiple stress responses, which require dynamic protein synthesis and degradation (Kiryu‐Seo & Kiyama, 2011; Patodia & Raivich, 2012). The proteasome is thought to be involved in the post‐translational stress response of damaged motor neurons; however, it remains unclear how proteasome function impacts the stress resilience of damaged motor neurons. Proteasomal dysfunction in motor neurons has been implicated in the pathology of amyotrophic lateral sclerosis (ALS) (Kabashi et al, 2012; Webster et al, 2017), a progressive neurodegenerative disease characterized by motor neuron loss, neuromuscular denervation, and axon degeneration (Taylor et al, 2016; Nijssen et al, 2017). Intriguingly, multiple stress responses observed in injured motor neurons are similarly activated in pathologically damaged motor neurons of ALS model mice (Kiryu‐Seo et al, 2005; Lobsiger et al, 2007; Sun et al, 2015; Tyzack et al, 2017; Maor‐Nof et al, 2021). We hypothesized that a proteasome‐mediated stress‐resilient mechanism exists in regenerative damaged motor neurons, which is lacking in damaged motor neurons prior to degeneration.
Alteration of axonal mitochondrial dynamics in response to damage is an early hallmark of both regeneration and degeneration (Patron & Zinsmaier, 2016; Sleigh et al, 2019). Mitochondria play pivotal roles in energy supply and metabolism that depend on cellular status (Han et al, 2020; Licht‐Mayer et al, 2020). In neurons with long axons, such as motor neurons, mitochondria must be transported from the cell body to the axon. It is evident that successful regeneration requires enhanced transport and numbers of mitochondria in injured axons (Misgeld et al, 2007; Kiryu‐Seo et al, 2010, 2016; Cartoni et al, 2016; Han et al, 2016). Increases in motile mitochondria or deletion of stationary mitochondria in injured axons can facilitate axon regeneration (Zhou et al, 2016; Han et al, 2020). Dysregulation of mitochondrial transport and quality has been observed during the very early onset of ALS degeneration (Bilsland et al, 2010; Magrane et al, 2014; Lin et al, 2017; Moller et al, 2017).
To clarify proteasome‐meditated stress‐resilient responses in damaged motor neurons, we developed a unique mouse system in which mitochondria are labeled with GFP (as a physiological indicator) and the proteasome is specifically ablated in nerve‐injured motor neurons. In addition, we established ALS mice wherein mitochondria are selectively labeled in stress‐responsive motor neurons at a pre‐symptomatic stage without injury. To ensure responsiveness to damage, we employed the activating transcription factor (Atf3) gene as a regulatory element. ATF3 is robustly induced in injured neurons to elicit transcriptional reprogramming (Nakagomi et al, 2003; Seijffers et al, 2006; Chandran et al, 2016; Palmisano et al, 2019; Lu et al, 2020; Renthal et al, 2020). Intriguingly, ATF3 is also expressed in a specific subtype of motor neurons vulnerable to degeneration in an ALS mouse model (Vlug et al, 2005; Saxena et al, 2009; Morisaki et al, 2016).
Using the newly established mouse system, we identified ankyrin G (AnkG) as a crucial target of the proteasome in damaged motor neurons. AnkG acts as an organizer of the axon initial segment (AIS) by recruiting and assembling submembrane proteins, such as voltage‐gated sodium channels and neurofascin‐186 (Huang & Rasband, 2018; Leterrier, 2018). The AIS is a transition compartment between the soma and axon that functions as a selective filter for axonal vesicles in addition to generating action potentials. Traumatic injuries dismantle the AIS, while pathogenic conditions such as tau mutation can impair AIS plasticity, leading to hyperexcitability (Schafer et al, 2009; Marin et al, 2016; Sohn et al, 2019). The cysteine protease calpain is considered to be responsible for degradation of the AIS (Schafer et al, 2009; Zhao et al, 2020), although alternative degradation systems may also contribute to this event (Hamdan et al, 2020). The physiological significance and exact mechanisms of AIS morphological changes during brain damage and disease have not been well addressed.
Here, we demonstrate a new proteasome‐mediated mechanism in vivo, in which damaged motor neurons temporarily dismantle the AIS in a proteasome‐sensitive manner. Transient disassembly of the AIS facilitates mitochondrial entry into damaged axons. This regulatory mechanism of mitochondrial logistics may be a critical post‐translational stress response that permits damaged motor neurons to produce enough energy for their regeneration or preservation of axon integrity.
Results
Mouse model of proteasome impairment specifically in nerve‐injured motor neurons
We used a hypoglossal motor nerve injury as a simple and elegant experimental model for motor neuron damage (Fig 1A). It has many experimental advantages; clear onset of damage, convenience of histological and biochemical comparison between control and injured motor neurons because hypoglossal motor neurons are densely located in bilateral neighboring hypoglossal nuclei, the regenerative ability of injured motor neurons, and numerous shared stress responses with ALS motor neurons (Nakagomi et al, 2003; Kiryu‐Seo et al, 2005, 2006; Lobsiger et al, 2007; Saxena et al, 2009; Sun et al, 2015; Maor‐Nof et al, 2021).
Figure 1. Injury‐induced Rpt3 CKO mice decrease the mitochondrial number in injured motor axons.
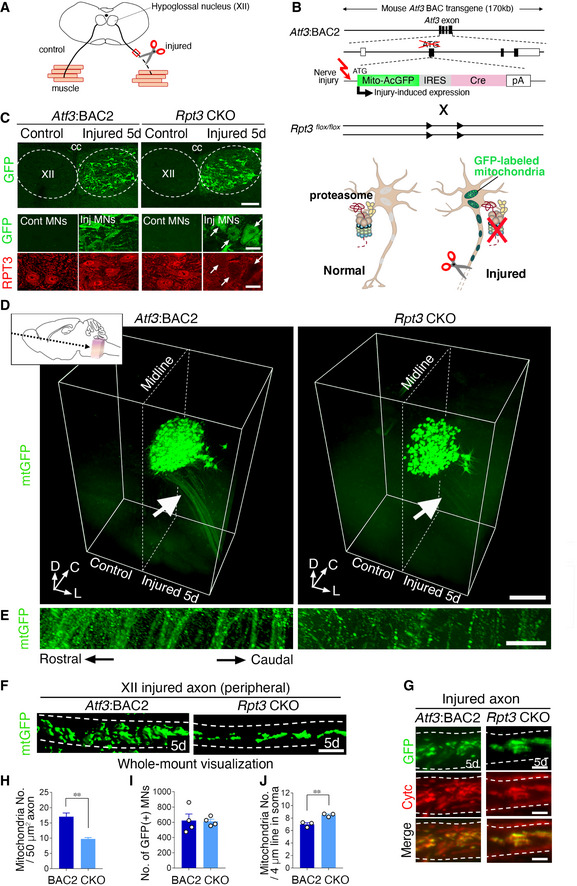
- Schematic diagram of hypoglossal nerve injury model. The axons of motor neurons in hypoglossal nucleus (XII) are projected to tongue muscle. Unilateral hypoglossal nerve is transected. Red box indicates the peripheral part of injured nerve observed in (F) and (G).
- Schematic diagram for injury‐induced deletion of proteasome subunit, Rpt3, from motor neurons. Atf3:BAC2 transgene and floxed Rpt3 gene are presented. The initiation codon of Atf3 in BAC is replaced by that of Mito‐AcGFP. Black and white boxes indicate coding and non‐coding regions of exon, respectively. Arrowheads denote loxP sites. Rpt3 deletion and mitochondrial labeling occur at the same time in injured motor neurons. See Fig EV1D–G and Materials and Methods for details.
- The immunostaining of GFP and RPT3 protein in hypoglossal nucleus (XII) of Atf3:BAC2 Tg and Rpt3 CKO mice at 5 days (5d) after axotomy. Arrows show Rpt3‐deleted injured motor neurons. cc, central canal.
- 3D images of GFP‐labeled mitochondria (mtGFP) in the transparent brainstem of Atf3:BAC2 Tg and Rpt3 CKO mice at 5 days after hypoglossal nerve injury. Inset shows the direction and portion observed by light sheet microscopy. White arrows indicate the magnified area shown in (E). (D, C, and L) indicate dorsal, caudal, and lateral directions, respectively.
- The magnified injured axons labeled by mtGFP.
- Whole‐mount visualization of mitochondria in injured hypoglossal motor axon at 5 days after injury.
- Immunostaining of injured hypoglossal nerves for GFP and cytochrome c (Cytc) at 5 days after injury.
- Number of mitochondria in injured axons of Atf3:BAC2 Tg and Rpt3 CKO mice at 5 days after injury (n = 5 mice per group).
- Total number of GFP‐positive injured motor neurons in Atf3:BAC2 Tg and Rpt3 CKO mice at 5 days after injury (n = 4 mice per group).
- Number of GFP‐positive mitochondria in soma at 5 days after injury. Data are shown as the mean ± s.e.m. (n = 3 mice). See also Fig EV2G and H.
Data information: Data are shown as the mean ± s.e.m., **P = 0.0005 in (H), no significant difference (P = 0.8915) in (I) and *P = 0.0068 in (J), determined by unpaired t‐test. Dashed lines reveal hypoglossal nucleus in (C), midline of the brain in (D) and axon in (F and G). Scale bars, 150 μm in (C, top) and 20 μm in (C, middle and bottom), 500 μm in (D), 50 μm in (E), 5 μm in (F), and 2.5 μm in (G).
Source data are available online for this figure.
To generate mouse wherein proteasome ablation occurs specifically in injured motor neurons, we used Atf3:BAC2 Tg mouse as a Cre driver mouse (Fig 1B). Atf3:BAC2 Tg mouse was designed to simultaneously express mitochondria‐targeted green fluorescent protein (GFP) and Cre recombinase in an injury‐specific manner, as driven by the Atf3 gene element within a bacterial artificial chromosome (BAC); multiple previous studies, including ours, have used ATF3 as a marker for nerve injury (Nakagomi et al, 2003; Seijffers et al, 2006; Kiryu‐Seo et al, 2008). The initiation codon of Atf3 gene in BAC was replaced by that of GFP with the mitochondrial targeting sequence, so that Atf3:BAC2 Tg mice did not express exogenous ATF3. The Atf3:BAC2 Tg mouse is a different line from the Atf3:BAC Tg mouse we previously reported (Kiryu‐Seo et al, 2016). Thus, we first examined that Atf3:BAC2 Tg mice began to express GFP in injured motor neurons at 20 h after injury and thereafter increased the number of GFP‐positive cells. In total, 60–70% of ATF3‐positive motor neurons expressed GFP following nerve injury (Fig EV1A–C).
Figure EV1. Injury‐induced Atf3:BAC2 Tg mouse and Rpt3 CKO mouse.
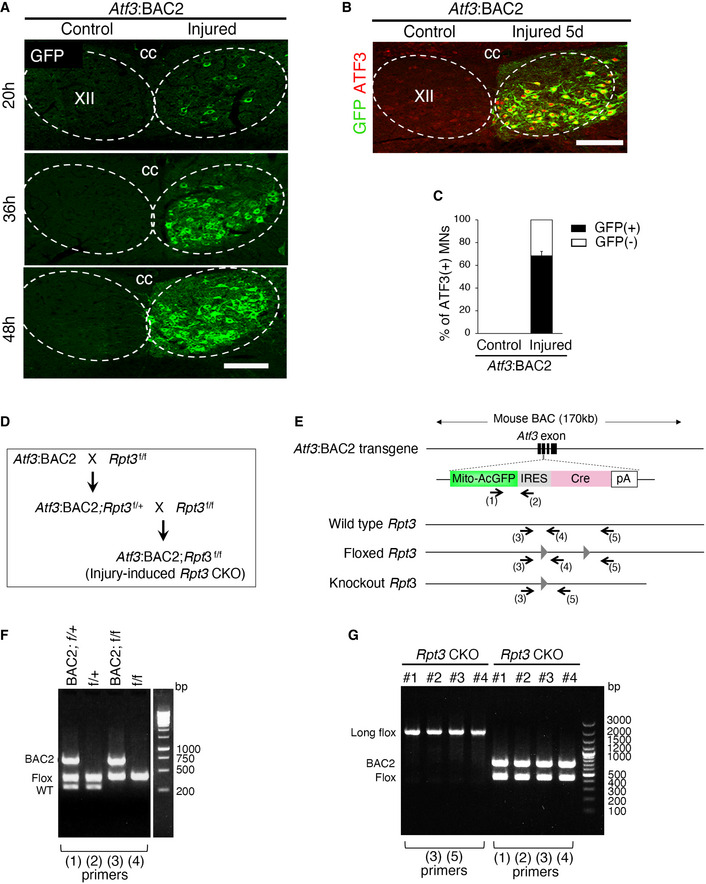
- Time‐dependent GFP expression in injured hypoglossal motor neurons of Atf3:BAC2 Tg mouse at 20, 36, and 48 h after injury.
- Representative image stained by ATF3 and GFP antibodies at 5 days following hypoglossal nerve injury of Atf3:BAC2 Tg mouse.
- The graph showing the percentage of GFP (+) or GFP (−) hypoglossal MNs in ATF3(+) MNs of control and injured sides at 5 days after injury of Atf3:BAC2 Tg mouse (n = 3 mice).
- Schematic of the breeding strategy to generate injury‐induced Rpt3 CKO mouse.
- Diagram for Atf3:BAC2 transgene and Rpt3 gene. Arrows indicate the location of the genotyping primers (1)–(5) for Atf3:BAC2 Tg and Rpt3 CKO mouse. The gray triangles show loxP sites.
- Representative genotyping results using primers (1)–(4) to obtain Rpt3 CKO mouse. PCR products on the gel are amplified using indicated primer pairs.
- Representative genotyping using tail samples of adult Rpt3 CKO mice from #1 to #4 shows that the floxed Rpt3 alleles are not deleted.
Data information: Dashed lines outline hypoglossal nucleus in (A and B). XII, hypoglossal nucleus; cc, central canal. Scale bars, 150 μm in (A and B).
Source data are available online for this figure.
By crossing Atf3:BAC2 Tg mouse with mouse floxed for Rpt3, an essential subunit of 26S proteasome for the protein degradation, we generated injury‐induced proteasome‐deficient mouse (Rpt3 CKO) (Figs 1B and EV1D–G). Injury‐induced Rpt3‐deficient mouse is a uniquely advantageous experimental tool to examine proteasome‐mediated early stress responses in damaged motor neurons. Rpt3 CKO mice reached adulthood, whereas the previously reported Rpt3 knockout mouse died during early gestation (Sakao et al, 2000). We used Atf3:BAC2 Tg mice instead of Atf3:BAC Tg mice in this study, because the progeny of Rpt3 CKO mice crossed with Atf3:BAC Tg mice had a shorter life span. Atf3:BAC2 Tg mice and Rpt3 CKO mice exhibited identical numbers of choline acetyltransferase (ChAT)‐positive hypoglossal motor neurons under the normal condition (Appendix Fig S1A and B). The expression of endogenous RPT3 protein was specifically abolished in GFP‐positive injured motor neurons of Rpt3 CKO mice by 5 days after injury, whereas RPT3 protein expression was maintained in control motor neurons of Rpt3 CKO mice and both control and injured motor neurons of Atf3:BAC2 Tg mice (Fig 1C).
Injury‐induced Rpt3‐ablated motor neurons decrease numbers of mitochondria in axons
To clarify the cellular responses affected by proteasome deficiency, we acquired three‐dimensional (3D) images of the transparent brains of Atf3:BAC2 Tg and Rpt3 CKO mice at 5 days after hypoglossal nerve injury, because these mice enabled us to observe GFP‐labeled mitochondria in nerve‐injured motor neurons, which are a good indicator of the cellular status (Fig 1D). Day 3–7 is the time point when dramatic change in protein expression occurs after injury. Three‐dimensional images acquired by lightsheet microscopy clearly demonstrated global views of injured hypoglossal motor neuron morphologies in mice (Fig 1D; Movies EV1 and EV2). We found that the intensity of GFP signal was reduced throughout injured motor axons of Rpt3 CKO mice (Fig 1D and E). 3D images provided us the useful information, which ordinary histological sections missed. We further observed whole‐mount peripheral injured axons of both Atf3:BAC2 Tg and Rpt3 CKO mice using a confocal microscopy with higher‐resolution, to count numbers of mitochondria in injured axons more precisely (Figs 1F and EV2). The number of mitochondria in injured axons of Rpt3 CKO mice was significantly reduced compared with Atf3:BAC2 Tg mice (Fig 1H). Notably, this result was not due to unlabeled mitochondria in Rpt3 CKO mice because the results shown in Fig 1G confirmed that staining of cytochrome c (a mitochondrial marker) in injured axons of both mice was completely consistent with GFP‐labeled mitochondria. When we examined lysosomes as one of the other cargos in injured axons, the distribution was not affected by Rpt3 deletion at least 5 days after injury (Fig EV2B–D), suggesting that mitochondria and lysosomes move separately by different mechanisms in response to damage. Thereafter, lysosomes in Rpt3‐deficient injured axons were significantly increased at 10 days after injury, probably reflecting the impairment of axonal transport or endolysosomal‐autophagic systems in severely degenerating axons. The total number of GFP‐positive injured motor neurons was similar in Atf3:BAC2 Tg and Rpt3 CKO mice (Figs 1I and EV2E and F). Super‐resolution microscopy further showed that mitochondrial density within soma of injured motor neurons seemed to be higher in Rpt3 CKO mice (Figs 1J and EV2G and H). Thus, our findings indicate the possibility that proteasome deficiency affects mitochondrial dynamics in injured axons at early time points after injury.
Figure EV2. GFP‐labeled mitochondria in injured hypoglossal motor neurons of Atf3:BAC2 Tg and Rpt3 CKO mice.
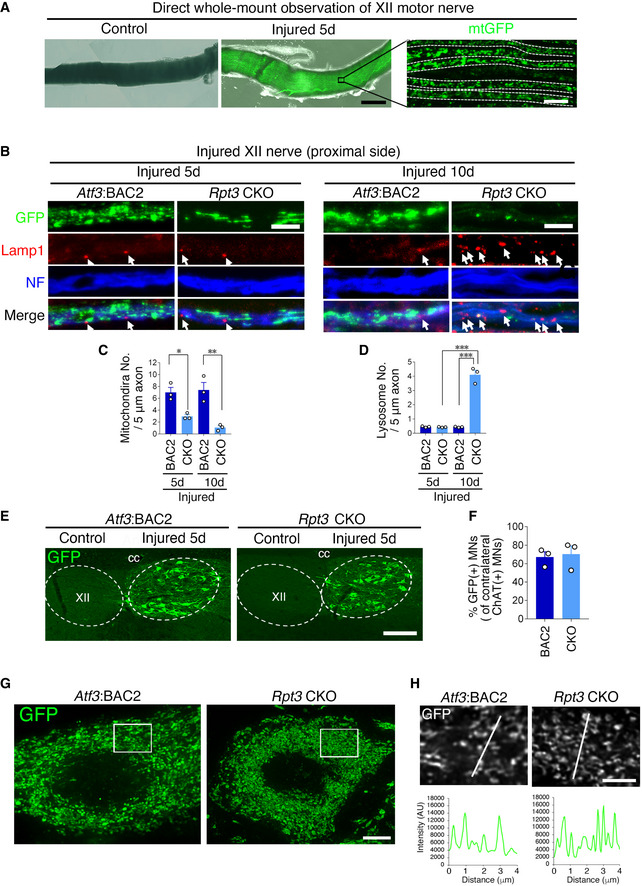
- Whole‐mount observation of hypoglossal nerve of Atf3:BAC2 Tg mouse without staining.
- Immunostaining of hypoglossal nerve at 5 and 10 days after injury, using anti‐GFP, Lamp1, and neurofilament (NF) antibodies. Arrows show Lamp1 positive signals in injured nerve.
- Graph shows mitochondria number in hypoglossal (XII) axon at 5 days and 10 days after injury (n = 4 mice).
- Graph shows lysosome number in hypoglossal (XII) axon at 5 days and 10 days after injury (n = 4 mice).
- The GFP expression of injured hypoglossal motor neurons at 5 days following injury. XII, hypoglossal nucleus; cc, central canal.
- The percentage of GFP‐positive injured motor neurons compared with ChAT‐positive uninjured motor neurons (n = 3 mice per group).
- Super‐resolution images of GFP‐stained mitochondria in injured hypoglossal motor neuron. Stacked image of 2 μm thickness.
- Single layer image corresponding to boxed area in (G) and plot profile of the fluorescence intensity corresponding to the line in the image.
Data information: Dashed lines outline axon in (A) and hypoglossal nucleus in (E). Data are shown as the mean ± s.e.m. *P = 0.0247, **P = 0.0011 in (C), ***P < 0.0001 in (D), P = 0.7854 in (F), determined by one‐way ANOVA followed by Tukey post hoc test. Scale bars, 300 μm in (A; left two panels) and 10 μm in (A; right panel), 5 μm in (B), 150 μm in (E), 4 μm in (G), and 2 μm in (H).
Source data are available online for this figure.
Injury‐induced Rpt3‐ablated motor neurons exhibit ALS‐relevant degeneration, despite their regenerative potential
To further examine the cellular condition at 5 days after axotomy, we performed quantitative reverse‐transcription PCR (qRT–PCR) for the expression of regeneration‐associated genes (RAGs) using control and injured hypoglossal nuclei. Unexpectedly, the alteration of mRNA expression of RAGs after injury did not show any significant difference between Atf3:BAC2 Tg and Rpt3 CKO mice (Fig 2A). In addition, the expression patterns of representative regeneration‐associated proteins, which were up‐ and downregulated, were similar in injured motor neurons of Atf3:BAC2 Tg mice and Rpt3 CKO mice at 5 days after injury (Fig 2B). Downregulation of ChAT and NeuN proteins in Rpt3‐deficient injured motor neurons is probably due to compensatory autophagy system. We next examined the fate of motor neurons after hypoglossal nerve injury. Thionine staining showed that motor neurons of Rpt3 CKO mice survived at 7 days after injury, similar to those of Atf3:BAC2 Tg mice; thereafter, those in Rpt3 CKO mice began to degenerate and died completely by 17 days after injury, while around 85% of injured motor neurons of Atf3:BAC2 Tg survived (Fig 2C and D). Microglial hyper‐activation but not astrocyte was also observed at the proximity of the Rpt3‐deleted injured motor neurons at 7 days after injury (Appendix Fig S1C and D). These results suggest that proteasome‐mediated stress pathways are located at a critical point for further regeneration to proceed after regenerative programming. Rpt3‐deleted injured motor neurons accumulated TDP‐43 and p62 around 7 days after injury (Fig 2E). The accumulation of TDP‐43 was shown in nucleus in our study, although it was generally exported and accumulated in the cytoplasm in ALS motor neurons. Its accumulation in nucleus may be a stress response rather than pathology, as previously described (Udan‐Johns et al, 2014). Aggregation of p62, an autophagic marker, could indicate the compensation for proteasome impairment in injured motor neurons of Rpt3 CKO mice, although the ubiquitin‐proteasome system (UPS) is a dominant protein degradation system in motor neurons (Tashiro et al, 2012). The injured axons of Atf3:BAC2 Tg mice, in which GFP‐labeled mitochondria were transported from soma upon injury, reinnervated the muscle in tongue and initiated to reform neuromuscular junctions (NMJs) at 28 days after injury, while those of Rpt3 CKO mice did not (Fig 2F). Injured motor neurons in Atf3:BAC2 Tg mice also survived at 28 days after injury (Fig 2G). Collectively, Rpt3‐deleted motor neurons have competence to regenerate during the initial stage of nerve injury response, but likely degenerate. Dysfunction of proteasome‐mediated response during the initial stage may be involved.
Figure 2. Injury‐induced Rpt3‐ablated motor neurons show regenerative responses but degenerate.
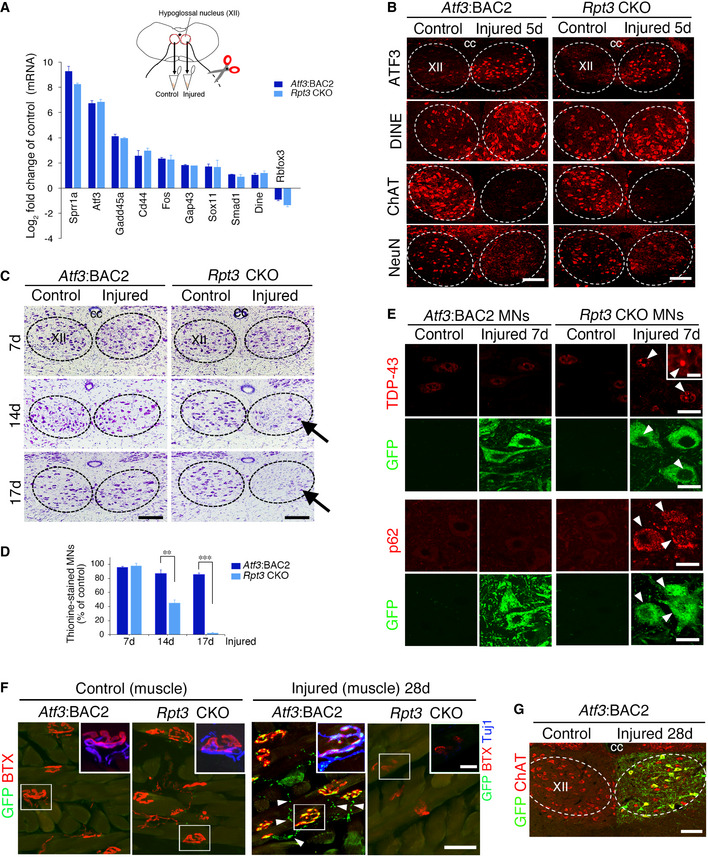
- The fold change in the mRNA expression of regeneration‐associated genes (RAGs) in injured hypoglossal nuclei compared with control hypoglossal nuclei of Atf3:BAC2 Tg and Rpt3 CKO mice at 5 days following nerve injury. Data are shown as the mean ± s.e.m. Fold changes in RAGs before and after injury have no significance between Atf3:BAC2 Tg and Rpt3 CKO mice, determined by unpaired t‐test (3 independent experiments).
- The expression of representative regeneration‐associated proteins is properly up‐ or downregulated in injured motor neurons of Rpt3 CKO mice at 5 days following hypoglossal nerve injury.
- Thionine‐stained motor neurons of Atf3:BAC2 Tg and Rpt3 CKO mice at 7, 14, and 17 days after hypoglossal nerve axotomy. Arrows indicate the loss of motor neurons.
- The graph showing the percentage of surviving motor neurons on the injured side compared with that on the control side. Data are shown as the mean ± s.e.m. (**P = 0.0002, ***P < 0.0001, determined by unpaired t‐test, n = 5 mice per group at each time point).
- The P62‐ and TDP‐43 positive immunostaining in control and injured motor neurons (MNs) of Atf3:BAC2 Tg and Rpt3 CKO mice at 7 days after hypoglossal nerve axotomy. Arrowheads show injured MNs with the accumulated proteins.
- Regenerating axons and neuromuscular junction (NMJ) in tongue of Atf3:BAC2 Tg and Rpt3 CKO mice. Arrowheads show GFP‐labeled mitochondria in regenerating motor axons to reform NMJ. The boxed region is magnified in inset, showing GFP‐labeled mitochondria, axon (Tuj1) and α‐Bungarotoxin (BTX).
- Immunostaining of ChAT and GFP‐positive motor neurons of Atf3:BAC2 Tg mouse at 28 days after hypoglossal nerve injury.
Data information: Dashed lines outline hypoglossal nucleus in (B, C, and G). XII, hypoglossal nucleus; cc, central canal. Scale bars, 150 μm in (B and C), 100 μm in (G), 20 μm in (E and F), 10 μm in (F inset), and 5 μm in (E inset).
Source data are available online for this figure.
Axon injury‐induced disappearance of the AIS is suppressed in Rpt3 CKO mice
Based on 3D imaging results shown in Fig 1, we considered that proteasome dysfunction might affect the entry of mitochondria from the soma to injured axons at early time points after injury, a time when motor neuron death was not apparent (Fig 2). A transition compartment between the soma and axon is known as the AIS. Previous studies have reported the AIS changes its morphology in response to neuronal activity and neuronal insults (Yamada & Kuba, 2016; Huang & Rasband, 2018). We examined morphological changes in the AIS before and after hypoglossal nerve injury using higher‐resolution cellular and subcellular 2D confocal images (Fig 3A and B). Immunohistochemistry for AnkG, an organizer of the AIS, demonstrated that the AIS disappeared in injured motor neurons of Atf3:BAC2 Tg mice at 5 days after injury, while the AIS remained in those of Rpt3 CKO mice (Fig 3C). As shown in Fig 3D and E, the difference of AIS number and length in injured motor neurons between Atf3:BAC2 Tg mice and Rpt3 CKO mice at day 5 was statistically significant. The number and length of the AIS were then recovered in injured motor neurons of Atf3:BAC2 Tg mice at 28 days after nerve injury, suggesting the reassembly of the AIS structure. AIS and GFP‐positive injured motor neurons were not detected in Rpt3 CKO mice at 28 days after injury, because the injured motor neurons degenerated as shown in Fig 2C and D and F. The number of AIS in Atf3:BAC2 Tg mice was not completely recovered to the control level at 28 days after injury (Fig 3D), because all of the injured motor neurons in Atf3:BAC2 Tg mice did not regenerate in this model. These findings suggest that temporal disassembly of the AIS in injured motor neurons is a positive stress response mediated by the proteasome.
Figure 3. Injury‐induced ablation of Rpt3 affects the disassembly of the AIS in injured motor neuron.
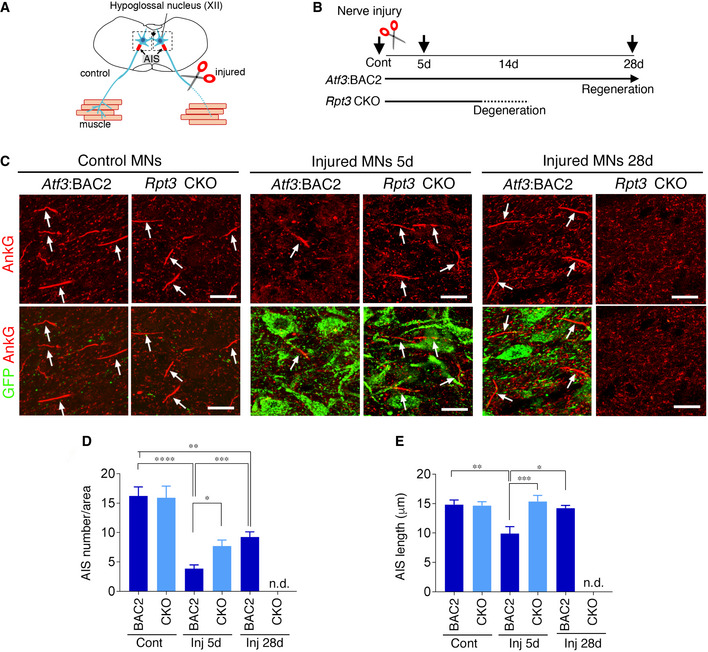
- Schematic diagram of the AIS in hypoglossal nerve injury model.
- Schematic showing the time course of motor neurons after nerve injury. Arrows indicate the time point at which the experiments were performed.
- Hypoglossal motor neurons of Atf3:BAC2 Tg and Rpt3 CKO mice before and at 5 days (5d) and 28 days (28d) after injury, immunostained by AnkG and GFP. Arrows indicate the AIS stained by AnkG. Scale bars, 20 μm.
- Quantification of the number of the AIS in control (Cont) and injured (Inj) hypoglossal motor neurons of Atf3:BAC2 and Rpt3 CKO mice. Data are expressed as mean ± s.e.m. *P = 0.043, **P = 0.0162, ***P = 0.008, ****P < 0.0001, determined by one‐way ANOVA followed by Tukey post hoc analysis, n.d., not detected, n = 5 mice for each group.
- Quantification of the AIS length in control (Cont) and injured (Inj) hypoglossal motor neurons of Atf3:BAC2 and Rpt3 CKO mice. Data are expressed as mean ± s.e.m. *P = 0.0087, **P = 0.0024, ***P = 0.0007, determined by one‐way ANOVA followed by Tukey post hoc analysis, n.d., not detected, n = 5 mice for each group.
Source data are available online for this figure.
AnkG is degraded by the ubiquitin‐proteasome system
We next attempted to clarify whether AnkG can be degraded by the UPS upon injury. We performed qRT–PCR using control and injured hypoglossal nuclei of wild‐type mice collected at 5 days after axotomy, to examine transcriptional regulation of AnkG before and after injury (Fig 4A). The results showed that mRNA expression of AnkG was unaltered before and after injury, while that of Atf3 and Chat as positive and negative RAGs was dramatically up‐ and downregulated, respectively. The longer AnkG splicing variant called giant AnkG, which is considered to be responsible for AIS formation, was also unchanged before and after injury. Thus, the regulation of AnkG at protein level is critical after nerve injury.
Figure 4. Proteasome is responsible for the AIS‐specific degradation of AnkG in injured motor neurons.
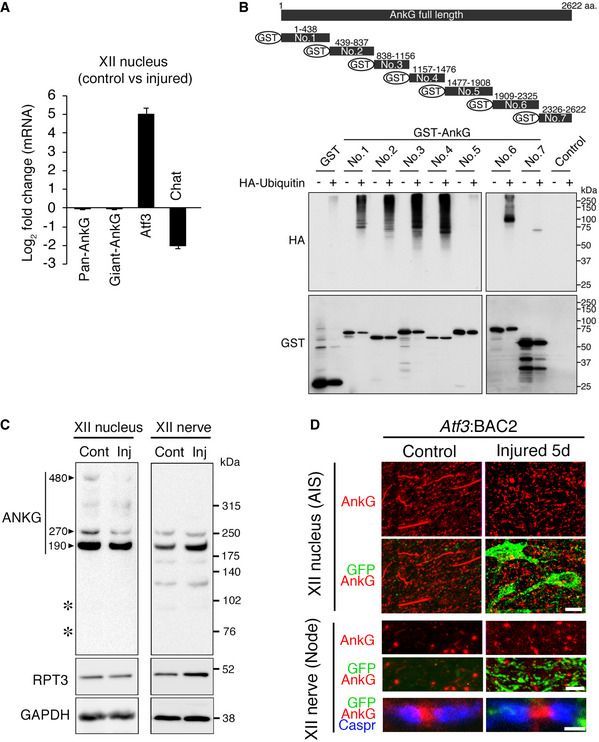
- Graph represents fold change in mRNAs in injured hypoglossal (XII) nuclei compared with control hypoglossal nuclei of wild‐type mice at 5 days after injury. Data are shown as the mean ± s.e.m. (n = 3 independent experiments).
- Ubiquitination assay of AnkG protein. The lysates of COS‐7 cells expressing the indicated truncated AnkG tagged with GST and HA‐Ubiquitin were pulled down with glutathione Sepharose beads, followed by western blotting with anti‐HA and anti‐GST antibodies.
- Western blot of hypoglossal nuclei (left) and nerves (right) at 5 days after injury with anti‐AnkG, RPT3 and GAPDH antibodies. AnkG products of 190/270/480 kDa are indicated by arrowheads. The positions of the degraded products by calpain are shown by asterisks at ~ 95 kDa and 72 kDa.
- The expression of AnkG (red) and GFP (green) in the AIS of hypoglossal (XII) motor neurons and that of AnkG, GFP, and Caspr (blue) in nodes of hypoglossal motor nerves at 5 days after injury. Scale bars, 15, 5, and 1.5 μm from the top.
Source data are available online for this figure.
A computer search such as UbPred predicted that AnkG protein has multiple ubiquitination sites. To examine ubiquitination of AnkG, we performed a ubiquitination assay using cell lysates in which sequentially fragmented AnkG constructs were transfected into COS‐7 cells (Fig 4B). A fragmented construct of AnkG was used for the ubiquitination assay because full‐length AnkG was too long for this experiment. The results showed that most AnkG fragments were experimentally poly‐ubiquitinated, suggesting that AnkG protein could be degraded by the UPS. Immunoblots of control and injured hypoglossal nuclei showed that both 480 and 270‐kDa forms of AnkG were downregulated after injury, while injured nerves exhibited no signals for 480‐kDa of AnkG and no obvious changes of 270‐kDa and major 190‐kDa AnkG between control and injured nerves (Fig 4C). A previous study reported that AnkG was degraded by the calcium‐dependent protease calpain into ~ 95 and 72‐kDa forms (Schafer et al, 2009). However, injured hypoglossal nuclei did not exhibit breakdown products, suggesting the possibility that proteasome is dominantly involved in this injury model. Immunohistochemical staining revealed that the broadly scattered dot‐like signals of AnkG were still present in the injured side of the hypoglossal nucleus, while AnkG‐positive AIS was lost in injured motor neurons of Atf3:BAC2 Tg mice (Fig 4D). These dot‐like signals were assumed to be nodes of Ranvier (node), which exhibit similar protein components to the AIS. When we examined the nodes of hypoglossal motor nerves, both control and injured motor axons similarly exhibited signals for AnkG and Caspr, indicators of nodes and paranodes, respectively. Collectively, these results indicate that proteasome‐mediated AnkG degradation occurred specifically in the AIS, but not in nodes of injured motor neurons.
Nerve injury‐induced disassembly of AIS enhances mitochondrial influx into axons
We next evaluated the physiological significance of proteasome‐dependent disassembly of the AIS in injured motor neurons. We hypothesized that the absence of the AIS was beneficial for mitochondrial entry into injured axons. To evaluate this hypothesis, we focused on mitochondrial localization in the AIS region before and after hypoglossal nerve injury (Fig 5A). Atf3:BAC2 Tg mice exhibited mutually exclusive distribution of AnkG‐positive AIS and mitochondria (Fig 5B). The AIS of Atf3:BAC2 Tg mice was gradually disassembled in response to injury and subsequently reassembled at 28 days after injury along with regeneration. In contrast, numerous GFP‐labeled mitochondria appeared in the AnkG‐disappeared region of the AIS after injury, while a limited number of mitochondria remained in the recovered AIS region at 28 days after injury. This was not an artificial effect of Atf3:BAC2 Tg mice because the widely described Thy1‐Mito mouse, in which mitochondria are labeled by CFP in neurons under control of the Thy1 promoter, showed a similar relationship between the AIS and mitochondria (Fig EV3A–E). Unlike injured motor neurons in Atf3:BAC2 Tg mice, Rpt3‐deficient injured motor neurons preserved the AIS at 5 days after injury and exhibited few GFP‐labeled mitochondria in it (Fig 5C–E). Correlatively, the number of mitochondria in axotomized motor nerves of Atf3:BAC2 Tg mice was significantly increased compared with normal nerves of Thy1‐Mito mice and subsequently returned to a control level along with the AIS recovery (Fig 5F and G). However, numbers of mitochondria in Rpt3‐deficient nerves did not show any injury‐induced changes, suggesting that the presence or absence of the AIS is involved in regulating mitochondrial influx into injured axons.
Figure 5. Absence of the AIS facilitates mitochondrial influx to injured axons.
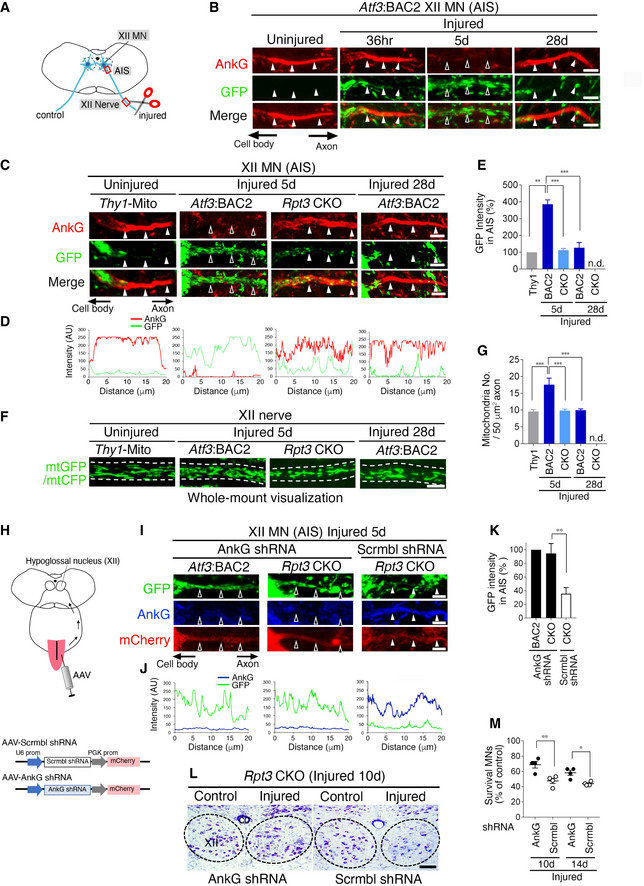
- Schematic diagram of hypoglossal nerve injury model. The areas shown by the red boxes (the AIS and hypoglossal (XII) nerve) are observed in this fig.
- Time‐dependent localization of AnkG and GFP‐labeled mitochondria in motor neuron (MN) of Atf3:BAC2 Tg mouse before and after hypoglossal nerve injury.
- The localization of AnkG and GFP‐labeled mitochondria in the AIS of uninjured MN of Thy1‐Mito Tg mice and injured motor neurons of Atf3:BAC2 Tg and Rpt3 CKO mice.
- Graphs show the GFP and AnkG fluorescent intensity scans corresponding to (C) over a 20 μm line running from the soma to the axon.
- Graph represents the percentage (%) of GFP intensity in the AIS, compared with that in the AIS of Thy1‐Mito mouse (n = 5 mice per group).
- Whole‐mount observation of GFP‐ or CFP‐labeled mitochondria (mtGFP or mtCFP) in the proximal region of peripheral hypoglossal (XII) nerve as shown in (A). Dashed lines outline the axon.
- Graph shows mitochondrial number in hypoglossal (XII) axon (n = 5 mice).
- Schematic diagram demonstrating the AAV infection to tongue and the AAV constructs.
- Altered localization of the GFP‐labeled mitochondria in the AIS of injured motor neurons of AnkG shRNA AAV‐infected Rpt3 CKO mice at 5 days after injury.
- Fluorescent intensity scans of GFP and AnkG corresponding to (I).
- Graph represents the percentage (%) of GFP intensity in the AIS, compared with that in the AIS of Atf3:BAC2 Tg mouse treated with AnkG shRNA (n = 5 mice per group).
- Thionine‐stained hypoglossal motor neurons of Rpt3 CKO mice at 10 days after injury. Rpt3 CKO mice are infected by AnkG shRNA or scramble shRNA AAVs. Dashed lines outline hypoglossal nucleus. XII, hypoglossal nucleus; cc, central canal.
- The percentage of surviving motor neurons on the injured side compared with that on the control side in Rpt3 CKO mice after infection of AnkG shRNA or scramble shRNA AAVs (n = 4 mice per group).
Data information: For (E, G, K, and M), data are shown as the mean ± s.e.m., **P = 0.0003, ***P < 0.0001 in (E and G), **P = 0.0064 in (K), *P = 0.0405, **P = 0.003 in (M), determined by one‐way ANOVA followed by Tukey post hoc test, n.d., not detected. In (B, C, and I), closed arrowheads denote the presence of the AIS, while open arrowheads show disappeared AIS. Scale bars, 5 μm in (B, C, F, and I) and 100 μm in (L).
Source data are available online for this figure.
Figure EV3. Expression of CFP‐labeled mitochondria in Thy1‐mito‐CFP Tg (Thy1‐Mito) mouse before and after motor nerve injury.
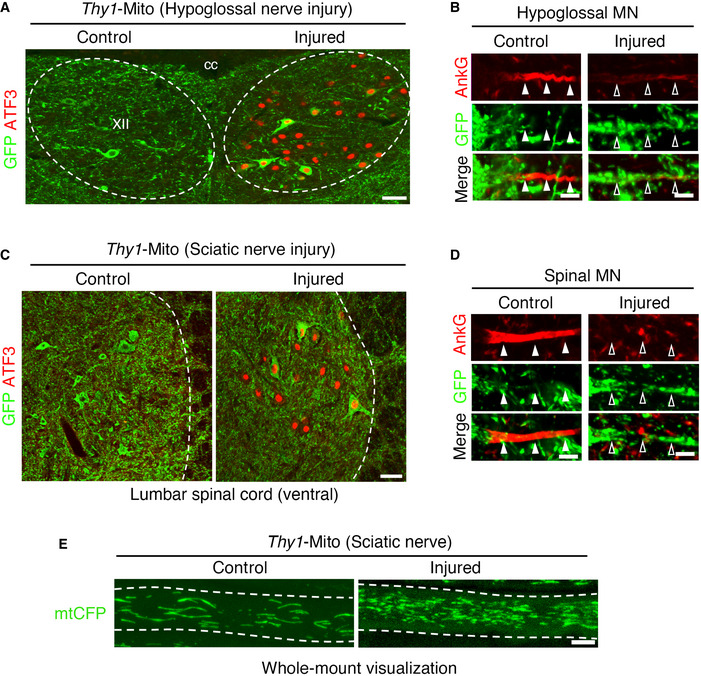
- Immunostaining of hypoglossal nucleus (XII) of Thy1‐Mito mouse using ATF3 (red) and GFP (green) antibodies at 5 days following hypoglossal nerve injury. XII, hypoglossal nucleus; cc, central canal.
- The localization of AnkG and CFP‐labeled mitochondria in the AIS of uninjured and injured hypoglossal motor neurons of Thy1‐Mito mouse.
- Immunostaining of the lumbar spinal motor neurons of Thy1‐Mito mouse using ATF3 (red) and GFP (green) antibodies at 5 days following sciatic nerve injury.
- The localization of AnkG and CFP‐labeled mitochondria in the AIS of uninjured and injured spinal motor neurons of Thy1‐Mito mouse.
- Whole‐mount observation of CFP‐labeled mitochondria (mtCFP) in sciatic nerve.
Data information: Dashed lines outline hypoglossal nucleus (A), spinal cord (C) and axon (E). Closed arrowheads indicate the AIS, while open arrowheads indicate the region where the AIS is disrupted in (B and D). Scale bars, 50 μm (A and C) and 5 μm (B, D and E).
To further address whether the loss of the AIS is directly involved in the increased number of mitochondria observed in injured axons or whether the two events are independent, we forced disruption of the preserved AIS structures in injured motor neurons of Rpt3 CKO mice. An AnkG shRNA was designed to suppress the larger splicing variant of AnkG. After injecting an AAV carrying AnkG shRNA into the tongue, it was retrogradely transported into motor neurons, whereby it suppressed AnkG expression (Figs 5H and EV4A and B). Our results showed that infection with AAV‐AnkG shRNA disrupted the AIS, and numbers of GFP‐labeled mitochondria in Rpt3‐ablated injured motor neurons were increased (Fig 5I–K). Infection with AAV‐scramble shRNA affected neither the appearance of the AIS nor the localization of GFP‐labeled mitochondria in Rpt3‐deficient injured motor neurons. Infection with AAV‐AnkG shRNA in uninjured motor neurons of Thy1‐Mito mouse did not increase the number of GFP‐labeled mitochondria in axons, suggesting that injury‐induced driving forces are necessary to increase mitochondrial density in axons (Fig EV4C and D). Furthermore, we examined whether the knockdown of AnkG influences the survival of Rpt3‐deficient injured motor neurons or not. The infection of AAV‐AnkG shRNA delayed the death of Rpt3‐deficient injured motor neurons, compared with that of AAV‐scramble shRNA (Fig 5L and M). These results suggest that the AIS disassembly is necessary for injury‐induced increase in mitochondrial influx into the axons to promote regeneration.
Figure EV4. Suppression of AnkG in uninjured hypoglossal motor neurons.
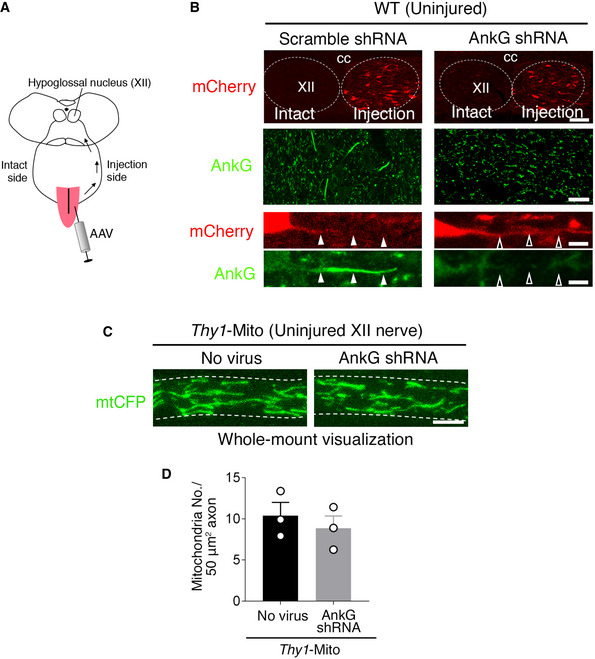
- Schematic diagram for AAV injection to tongue.
- Hypoglossal motor neurons labeled by mCherry (red) in injection side of AAVs. The decrease of AnkG (green) immunoreactivity is observed after the infection of AAV‐AnkG shRNA but not AAV‐scramble shRNA. Dashed lines show hypoglossal nucleus (XII). cc, central canal. Closed arrowheads indicate the AIS, while open arrowheads indicate the region where the AIS is disrupted.
- CFP‐labeled mitochondria in uninjured hypoglossal nerve of Thy1‐Mito mouse after the infection of AAV‐AnkG shRNA to tongue. Dashed lines show axon.
- Graph shows the number of CFP‐labeled mitochondria in uninjured hypoglossal (XII) axon of Thy1‐Mito mouse before and after AAV infection. Data are shown as the mean ± s.e.m. (no significant difference (P = 0.519), determined by two‐tailed Student's t‐test, n = 3 mice).
Data information: Scale bars = 100 μm (top), 20 μm (middle), and 5 μm (bottom) in (B) and 5 μm in (C).
Source data are available online for this figure.
Mitochondrial motility in injured axons is reduced in Rpt3 CKO mice
Because two types of mitochondria, motile and stationary, are present in axons, we examined which type was responsible for the observed change in mitochondrial numbers in injured axons. For live imaging of mitochondrial flow, we used another nerve injury model: sciatic nerve injury (Fig 6A). Injured motor neurons after sciatic nerve injury showed similar responses of the AIS and mitochondria to those of hypoglossal nerve injury (Fig EV3C–E; Appendix Fig S2A and B). We performed live imaging of CFP‐ or GFP‐labeled mitochondria in uninjured sciatic nerves of Thy1‐Mito mice and injured sciatic nerves of both Atf3:BAC2 Tg mice and Rpt3 CKO mice (Fig 6B and C). Atf3:BAC2 Tg mice exhibited increased numbers of motile mitochondria in injured nerves compared with Thy1‐Mito mice and Rpt3 CKO mice (Fig 6D and E). Increased numbers of motile mitochondria in injured sciatic nerves of Atf3:BAC2 Tg mice returned to control levels along with regeneration. Notably, stationary mitochondria were not significantly altered in any of these mice (Fig 6F). Nerve injury induces proteasome‐dependent AIS degradation in motor neurons, which could be coupled with the increase in mitochondrial transport into axons under the presence of driving forces.
Figure 6. Mitochondrial motility in injured axons is up‐regulated in Atf3:BAC2 Tg mice but not in Rpt3 CKO mice.
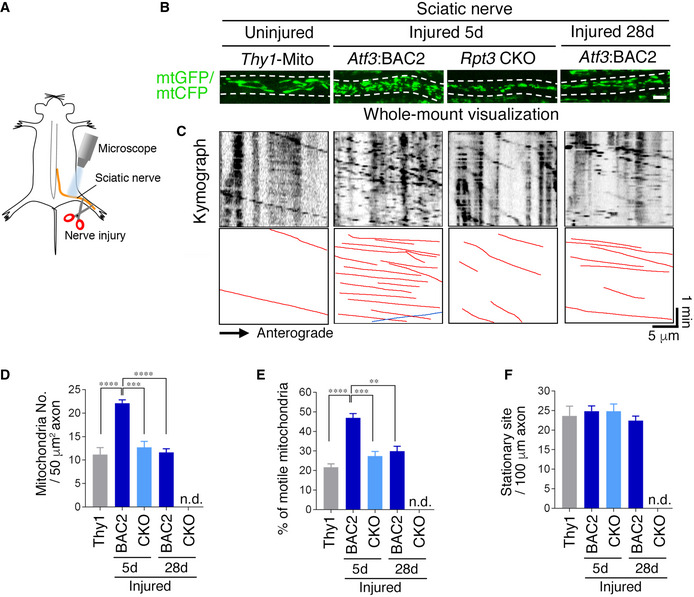
-
ASchematic diagram demonstrating live imaging of sciatic nerve.
-
BWhole‐mount visualization of GFP‐ or CFP‐labeled mitochondria (mtGFP or mtCFP) in sciatic nerve. Dashed lines indicate the boundary of axon. Scale bar, 5 μm.
-
CRepresentative kymographs showing the behavior of mtCFP in uninjured sciatic nerves of Thy1‐Mito Tg and mtGFP in injured sciatic nerves of Atf3;BAC2 and Rpt3 CKO mice.
-
D–FThe number of mitochondria (D), percentage of motile mitochondria (E), and stationary mitochondrial site (F) in axons.
Data information: Data are shown as the mean ± s.e.m. ***P = 0.0001, ****P < 0.0001 in (D), **P = 0.0006, ***P = 0.0001, ****P < 0.0001 in (E), determined by one‐way ANOVA followed by Tukey post hoc test, n.d., not detected, n = 5 mice for each group.
Source data are available online for this figure.
Failure of AIS disassembly in ATF3‐expressing ALS motor neurons at a pre‐symptomatic stage
ATF3‐expressing spinal motor neurons exist in ALS mice without injury (Vlug et al, 2005; Saxena et al, 2009; Morisaki et al, 2016). We crossed Atf3:BAC Tg mice with SOD1G93A (SOD1) ALS mice and generated Atf3:BAC;SOD1G93A (Atf3;SOD1) mice to selectively label ATF3‐expressing motor neurons (Figs 7A and EV5A). We used Atf3:BAC Tg mice here, because Atf3:BAC Tg mice mimic the expression pattern of endogenous ATF3 more effectively than Atf3:BAC2 Tg mice. Atf3;SOD1 mice showed similar life span and age‐dependent motor neuron death with SOD1 mice, suggesting that the Atf3:BAC transgene does not affect the disease phenotype of SOD1 mice (Fig EV5B–D). 3D image of transparent spinal cord in Atf3;SOD1 mouse clearly showed GFP‐labeled motor neurons, while that of Atf3:BAC Tg mouse (littermate control) did not (Fig 7B; Movie EV3). The expression of GFP was co‐localized well with that of ATF3 in motor neurons of Atf3;SOD1 mouse (Fig 7C). Of ChAT‐positive spinal motor neurons, GFP‐expressing motor neurons were < 20% at P70 (pre‐symptomatic stage; Fig 7D). These GFP‐positive motor neurons appeared around P60, increased in number, and then degenerated along with age in Atf3;SOD1 mouse (Fig EV5E and F). Among the heterogenous spinal motor neuron pool, most of GFP‐positive motor neurons in Atf3;SOD1 mice expressed ATF3 (Fig 7E). They also expressed matrix metalloproteainase‐9 (MMP‐9) or osteopontin (OPN), which are reportedly localized in vulnerable motor neurons (Figs 7E and EV5G) (Kaplan et al, 2014; Morisaki et al, 2016), suggesting that stress‐responsive vulnerable motor neurons are selectively labeled by GFP in Atf3;SOD1 mice.
Figure 7. Failure of the AIS disassembly and mitochondrial influx in stress‐responsive GFP motor neurons of Atf3;SOD1 mouse.
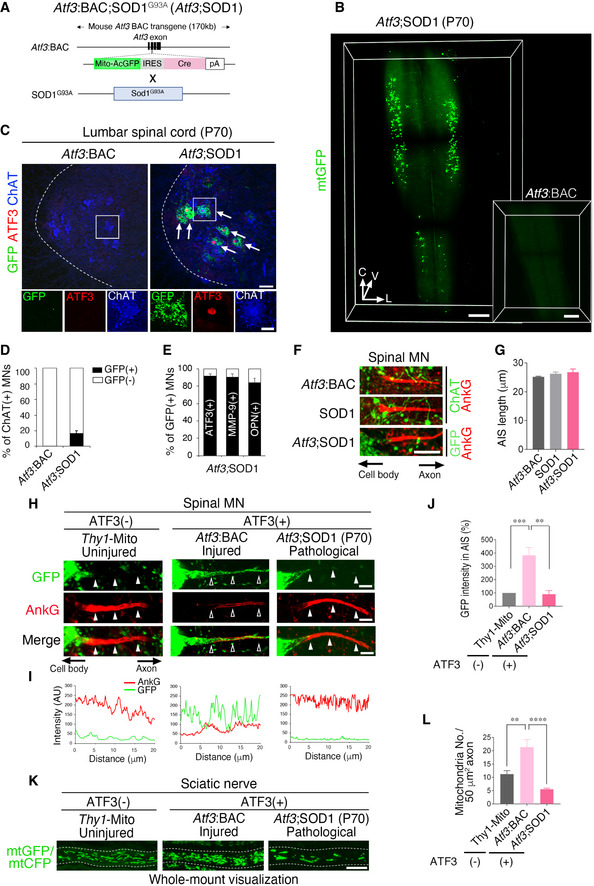
- Breeding schemes to obtain Atf3:BAC; SOD1G93A (Atf3;SOD1) mouse.
- 3D images of GFP‐labeled mitochondria (mtGFP) in the transparent spinal cord without injury of Atf3;SOD1 mouse at pre‐symptomatic stage (P70, 70‐day‐old) and Atf3:BAC Tg mouse (inset). V, C, and L indicate ventral, caudal, and lateral directions, respectively.
- Immunostaining of spinal cord in Atf3:BAC Tg and Atf3;SOD1 mice at P70. The boxed regions are magnified in lower panels. Arrows indicate GFP‐ and ATF3‐positive motor neurons (MNs). Dashed lines outline the edge of the gray matter in spinal cord.
- The percentage (%) of GFP‐positive MNs (black bar) in ChAT‐positive MNs of Atf3:BAC and Atf3;SOD1 mice at P70 (n = 5 mice per each group).
- The percentage (%) of ATF3, MMP‐9 or osteopontin (OPN)‐positive MNs (black bar) in GFP‐positive MNs of Atf3;SOD1 mice at P65‐70 (n = 5–7 mice per group).
- Immunostaining of AnkG in ChAT‐positive spinal MN of Atf3:BAC and SOD1 and in GFP‐positive spinal MN of Atf3;SOD1 mouse at P70.
- Quantification of the AIS length (n = 5 mice per group).
- The localization of AnkG and GFP‐labeled mitochondria in uninjured spinal MN of Thy1‐Mito (without ATF3 induction), traumatically sciatic nerve‐injured MN of Atf3:BAC (with ATF3 induction) and pathologically damaged MN of Atf3;SOD1 mice (with ATF3 induction). Closed arrowheads denote the presence of the AIS, while open arrowheads show disappeared AIS.
- Fluorescent intensity scan of GFP and AnkG in corresponding to (H).
- The percentage (%) of GFP intensity in the AIS, compared with that in the AIS of uninjured MNs in Thy1‐Mito mice (n = 5 mice per group).
- Whole‐mount observation of mtGFP or mtCFP in sciatic nerve. Dashed lines outline the axon.
- The number of mtGFP or mtCFP in sciatic nerve (n = 5 mice).
Data information: For (D, E, G, J, and L), data are shown as the mean ± s.e.m., no significant difference in (G), **P = 0.0008, ***P = 0.0007 in (J), **P = 0.0036, ****P < 0.0001 in (L), determined by one‐way ANOVA followed by Tukey post hoc test, n.d., not detected. Scale bars, 700 μm in (B), 1,000 μm in (B; inset), 50 μm in (C; upper panel), 20 μm in (C; lower), 10 μm in (F and K) and 5 μm in (H). See also Fig EV5.
Source data are available online for this figure.
Figure EV5. Characterization of Atf3:BAC;SOD1G93A (Atf3;SOD1) mouse.
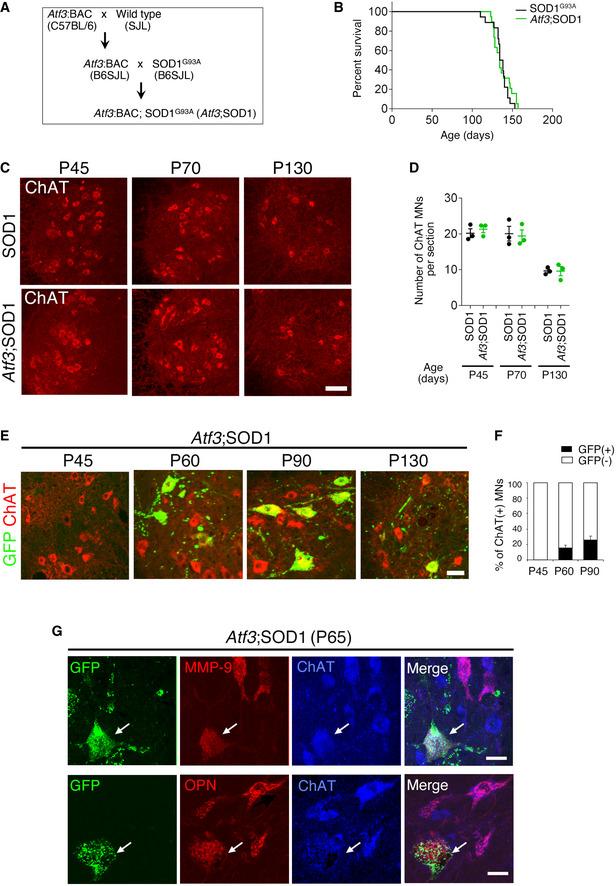
- Schematic of the breeding strategy to generate Atf3;SOD1 mouse.
- Kaplan–Meier survival analysis (long‐rank test, P = 0.4674). The Atf3:BAC transgene has no significant effect on life span of SOD1G93A.
- Representative image of spinal motor neurons at 45, 70, and 130 days, immunostained with ChAT.
- Quantification of age‐dependent ChAT‐positive MN in the ventral horn of lumbar spinal cord. Data are shown as the mean ± s.e.m., no significant differences at same age, determined by unpaired t‐test, n = 3 mice per group.
- The expression of GFP and ChAT in spinal motor neurons of Atf3;SOD1 at 45, 60, 90 and 130 days.
- The percentage (%) of GFP‐positive MNs (black bar) of ChAT‐positive MNs in Atf3; SOD1 mice. Data are shown as the mean ± s.e.m. n = 5 mice at each age.
- Immunostaining of spinal motor neurons in Atf3;SOD1 at P65 using GFP, MMP‐9, OPN, and ChAT antibodies. Arrows denote GFP‐positive motor neurons co‐localized with MMP‐9 or OPN.
Data information: Scale bar, 100 μm in (C), 50 μm in (E), and 20 μm in (G).
Source data are available online for this figure.
Given that ALS motor neurons reduce proteasome activity (Jenkins et al, 2020) and initiate to induce ATF3 at pre‐symptomatic stage, we wondered whether the failure of AIS disassembly in injured motor neurons of Rpt3 CKO mice may occur in GFP‐positive ALS motor neurons. We found that GFP‐positive spinal motor neurons in Atf3;SOD1 mice retained the AIS and that the length of the AIS did not show any significant difference with that in the ChAT‐positive spinal motor neurons in Atf3:BAC and SOD1 mice (Fig 7F and G). We next evaluated mitochondrial localization in the AIS region. Both traumatically damaged GFP‐positive motor neurons in Atf3:BAC and pathologically damaged GFP‐positive motor neurons in Atf3;SOD1 induce ATF3. However, pathologically damaged motor neurons failed to dismantle the AIS and exhibited few GFP‐labeled mitochondria in it (Fig 7H–J). Concomitantly, GFP‐positive motor neurons of Atf3;SOD1 mice did not increase the number of axonal mitochondria despite inducing ATF3. (Fig 7K and L).
We further confirmed that ATF3‐inducing ALS motor neurons failed to disassemble the AIS using hypoglossal nerve injury model (Appendix Fig S3). The number of GFP‐positive motor neurons of Atf3;SOD1 mice was originally much lower in the hypoglossal nucleus than in the spinal cord, probably because cranial motor neurons retained higher proteasomal activity than spinal motor neurons (An et al, 2019; Appendix Fig S3A–C). Almost all hypoglossal motor neurons in Atf3;SOD1 mice were labeled by GFP following nerve injury because of ATF3 induction. However, they did not disassemble the AIS nor increase mitochondria (Appendix Fig S3D and E). Collectively, the ATF3‐positive ALS motor neurons lack or reduce proteasome‐mediated stress responses, resembling injury‐induced Rpt3‐deficient motor neurons.
Discussion
In this study, the combination of motor nerve injury model with unique genetically engineered mice has clarified a new proteasome‐mediated stress‐resilient mechanism that is lacking in damaged motor neurons prior to degeneration. Injury‐induced Rpt3‐deficient motor neurons fail to activate proteasome‐dependent AIS disassembly and exhibit ALS‐relevant degeneration, despite remaining regenerative capacity. The functional consequence of the transient AIS disassembly after neuron damage has become clear in this study. Upon damage, proteasome‐mediated disruption of the AIS facilitates mitochondrial entry into axons, perhaps to compensate for the increased energy expenditure associated with axon regeneration. Intriguingly, the failure of this proteasome‐mediated mechanism against pathological damage might be associated with the vulnerability of ALS motor neurons (Fig 8).
Figure 8. Schematic diagram of the proteasome‐sensitive AIS disassembly and mitochondrial supply upon damage.
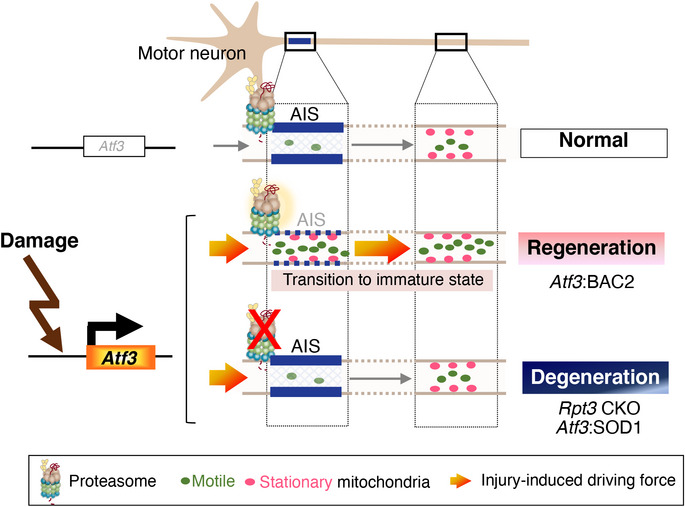
Damaged motor neurons induce stress‐sensitive transcription factor, ATF3, to initiate regeneration program. ATF3‐positive injured motor neurons temporarily dismantle the AIS in a proteasome‐dependent manner and increase mitochondrial entry into injured axons under the injury‐induced driving force to meet the energy demand for axon regeneration. However, proteasome‐deficient injured motor neurons and stress‐responsive vulnerable ALS motor neurons with proteasome dysfunction cannot activate this mechanism despite inducing ATF3.
Transient AIS disassembly is critical for injured motor neurons to accelerate mitochondrial influx into axons
Here, we defined that the presence or absence of the AIS alters mitochondrial density in injured axons. It has been established that mitochondrial supply is critical for injured axons and growth cones to acquire energy for reconstruction (Chamberlain & Sheng, 2019). However, the mechanism underlying enhanced mitochondrial transport has remained elusive. Furthermore, the physiological significance of AIS dismantling in injury and disease has been unclear, although AIS plasticity has been implicated in changes in neuronal excitability. This study is the first to examine the association between damage‐induced disruption of the AIS and mitochondrial entry in vivo by taking advantage of a unique mouse model. Under normal conditions, the AIS acts as a selective barrier in which nuclear distribution element‐like 1 (NDEL1) excludes non‐axonal cargo (Farias et al, 2015; Jenkins et al, 2015; Kuijpers et al, 2016; Gumy et al, 2017). Strikingly, suppression of AnkG does not always influence axonal transport under normal conditions, partly because the proximal axonal region contributes to selective cargo transport in addition to the AIS (Farias et al, 2015; Jenkins et al, 2015). They are consistent with our result that the suppression of AnkG did not show any obvious change in mitochondrial number in axon under normal condition. However, the situation seems to be different upon injury, whereby the activation of mitochondrial transport‐associated molecules such as dual‐leucine zipper kinase 1 (Dlk1), armadillo repeat‐containing X‐linked 1 (Armcx1), mitochondrial Rho GTPases (Miro), and dynamin‐related protein 1 (Drp1) and the modification of microtubules enhance mitochondrial localization in injured axons (Hellal et al, 2011; Benned‐Jensen et al, 2016; Cartoni et al, 2016; Kiryu‐Seo et al, 2016; Kalinski et al, 2019; Han et al, 2020). Under the presence of these powerful driving forces upon injury, the breakdown of the selective filter is efficient for enhanced mitochondrial entry into injured axons.
The absence of the AIS is usually observed in immature neurons. The AIS dismantling in response to injury may contribute to the temporal transition of injured motor neurons to an immature state at post‐translational level, in order to acquire regenerative potential. Because immature neurons require higher mitochondrial motility in axon during development and progressively reduce it along with development (Lewis et al, 2016). Recent progresses have clarified that the transcriptional and epigenetic reprogramming makes injured neurons revert to immature state for regeneration (Chandran et al, 2016; Palmisano et al, 2019; Bradke et al, 2020; Lu et al, 2020; Renthal et al, 2020). In line with this, the proteasome‐mediated disassembly of the AIS would be a post‐translational mechanism after the transcriptional and epigenetic reprogramming. Injured motor neurons likely use this mechanism to meet urgent energy demand for regeneration. As we show in this study, the AIS is reassembled at appropriate timing along with regeneration. The regulation mechanism of the AIS reassembly would also contribute to complete regeneration. Conclusively, the AIS disassembly and reassembly could be one of the key mitochondrial logistic mechanisms under emergency situations.
Proteasomal degradation is the major mechanism for AIS disassembly in damaged motor neurons
Using injury‐induced Rpt3 CKO mice, we found that AnkG, the organizing component of the AIS, could be a proteasomal target upon injury. We showed that AnkG protein was indeed poly‐ubiquitinated. The data are also supported by previous study showing that 26S proteasome dysfunction in neurons caused global accumulation of ubiquitinated proteins including AnkG (Ugun‐Klusek et al, 2017). We do not rule out the possibility that other protein degradation systems, such as calpain and autophagy, participate in AIS loss in injured motor neurons or play a compensatory role in Rpt3‐deficient injured motor neurons. Most notably, calpain is considered to be responsible for degradation of the AIS after brain damage (Schafer et al, 2009; Benned‐Jensen et al, 2016; Zhao et al, 2020). One possible explanation is the different dominancy of protein degradation systems among neurons, because motor neurons are susceptible to proteasomal degradation rather than autophagy (Tashiro et al, 2012; Rudnick et al, 2017). Alternatively, differences in regenerative capacity between the central and peripheral nervous systems may reflect differences in degradation systems for the AIS. In central nervous system traumatic injury models, such as optic nerve injury and ischemia, both the AIS and nodes, which are composed of identical membranous proteins, seem to be disrupted (Marin et al, 2016); however, injured motor neurons in our injury model did not exhibit deteriorated nodes, perhaps because apparent demyelination did not occur. Taken together with these differences, it could be concluded that the proteasome is responsible for degradation of the AIS in damaged motor neurons. To regulate proteasome activity in an AIS‐specific manner in damaged motor neurons, intrinsic or extrinsic local signaling such as signaling kinases and glial contact could be activated following injury (Yoshimura & Rasband, 2014; Baalman et al, 2015; Tamada et al, 2021). Otherwise, AIS‐residing ubiquitin ligases, such as TRIM46, may participate in AIS‐specific breakdown; although, thus far, TRIM46 reportedly has microtubule crosslinking activity, but not E3 ligase activity (van Beuningen et al, 2015).
Apart from disruption of the AIS, there likely exist further proteasome‐mediated stress responses in injured motor neurons. Our previous study demonstrated that mitochondrial fission is a temporally adaptive response for nerve regeneration (Kiryu‐Seo et al, 2016). Because mitochondrial proteins responsible for fusion/fission are regulated by degradation, the proteasome may be also involved in processes in injured motor neurons. Recently, Kong et al (2020) identified an interaction between AMPK and the proteasome in axoplasm to promote degradation and drive regeneration (Kong et al, 2020). Considering that uninjured motor neurons maintain the AIS in the presence of the proteasome, Rpt3‐deficient uninjured motor neurons which were previously reported would degenerate in different mechanisms from the failure of the AIS disassembly (Tashiro et al, 2012). Therefore, other multiple proteasome‐dependent stress responses or injury‐independent proteasome functions would also participate in resilience of damaged motor neurons.
Failure of AIS disassembly and subsequent mitochondrial insufficiency in vulnerable type of ALS motor neuron
Given that injury‐induced Rpt3‐ablated motor neurons exhibit ALS‐like degeneration and that ALS motor neurons reduce the proteasome activity (Jenkins et al, 2020), we assumed that ALS motor neurons lack proteasome‐mediated stress responses at pre‐symptomatic stage, similar to injury‐induced Rpt3‐deficient motor neurons. Previous reports demonstrated that pathological ALS motor neurons show similar multiple stress responses to injured motor neurons, including ATF3 induction (Lobsiger et al, 2007). Notably, ATF3‐positive motor neurons increase in number along with disease progression, or under the manipulation of disease‐associated genes such as TANK‐binding kinase 1 (TBK1), although it is unclear whether ATF3 drives survival or cell death mechanisms in pathological damaged motor neurons (Vlug et al, 2005; Seijffers et al, 2014; Morisaki et al, 2016; Gerbino et al, 2020). In this study, Atf3;SOD1 mice permitted selective visualization of ATF3‐positive motor neurons by GFP at the pre‐symptomatic stage. ATF3 is required for axotomy‐induced reprogramming of injured neurons (Chandran et al, 2016; Renthal et al, 2020). Therefore, GFP‐inducing SOD1 motor neurons might attempt to initiate regeneration programs but failed it. One of the reasons of the failure would be the lack of proteasome‐mediated stress responses, which was demonstrated in this study.
We describe here that the proteasome‐mediated disassemble of the AIS could be a stress response, which is somehow missing in GFP‐positive SOD1 motor neurons. We did not find a significant change in AIS lengths in SOD1 motor neurons, although a previous study showed slightly shorter AIS structures in ALS motor neurons (Bonnevie et al, 2020). Instead, we found that GFP‐positive SOD1 motor neurons had fewer mitochondria in the AIS and a concomitantly reduced number of axonal mitochondria, similar to injury‐induced Rpt3‐ablated motor neurons. Previous study using electron microscopy suggested the possibility that mitochondrial transport was impaired at the proximal axon in SOD1G93A mice and ALS patients (Sasaki & Iwata, 1996; Sasaki et al, 2005). Defective axonal transport of mitochondria is the earliest phenomena observed in ALS models and has been proposed to be causative in this disease (Magrane et al, 2014; Moller et al, 2017). Atf3;SOD1 mice and injury‐induced Rpt3 CKO mice showed the possibility that the failure of proteasome‐mediated AIS disruption would cause a defective supply of mitochondria from the cell body to the axon, at least in ATF3‐positive motor neurons. Lin et al (2017) have reported that mitochondria‐anchoring protein syntaphilin is involved in the removal of damaged mitochondria from axon at early pre‐symptomatic stage (Lin et al, 2017). Therefore, ALS motor neurons might require the replenishment of healthy mitochondria to axons via AIS disassembly, in cooperation with the removal of damaged mitochondria.
Overall, our mouse model system using unique character of Atf3 gene is a powerful tool for expanding our understanding of the molecular basis of damaged neurons prior to neurodegeneration. Using the system, we uncover the fundamental mechanism that the proteasome‐dependent temporal breakdown of the AIS would be beneficial for traumatically or pathologically damaged motor neurons to supply mitochondrial energy to damaged axon through the transition to an immature state. This transient proteasome‐mediated AIS disassembly could be a new machinery in the mitochondrial logistics system to keep the integrity of damaged motor neurons, and this system could be a new therapeutic target in ALS.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| C57BL/6NCrl (Mus Musculus) | SLC | N/A |
| SJL/J (Mus Musculus) | Charles River | N/A |
| Atf3:BAC Tg (Mus Musculus) | Kiryu‐Seo et al (2016) | N/A |
| Atf3:BAC2 Tg (Mus Musculus) | This paper | N/A |
| Rpt3‐flox (Mus Musculus) | Tashiro et al (2012) | N/A |
| B6SJL‐Tg(SOD1*G93A)1Gur/J (Mus Musculus) | The Jackson Laboratory | JAX stock # 002726; RRID: IMSR_JAX:002726 |
| B6.Cg‐Tg(Thy1‐CFP/COX8A)S2Lich/J (Mus Musculus) | The Jackson Laboratory | IMSR Cat# JAX:007967, RRID:IMSR_JAX:007967 |
| COS‐7 cell | ATCC | Cat# CRL‐1651 |
| Recombinant DNA | ||
| Unmodified BAC clone | The BACPAC Resources Center at the Children's Hospital Oakland Research Institute | RP24‐318C6 |
| Rat AnkG (1‐438, 439‐837, 838‐1156, 1157‐1476, 1477‐1908, 1909‐2325, 2326‐2622 aa.) in pEF‐BOS‐GST | This paper | N/A |
| HA‐Ubiquitin in pcDNA3 | Addgene | Addgene plasmid 18712 |
| MitoGFP‐ires‐Cre in pL451 | Kiryu‐Seo et al (2016) | N/A |
| Antibodies | ||
| Rabbit anti‐GFP polyclonal (1:1,000) | MBL | Cat# 598, RRID:AB_591819 |
| Rat anti‐GFP monoclonal (1:1,000) | Nacalai Tesque | Cat# 04404‐84, RRID:AB_10013361 |
| Rabbit anti‐Ank‐G (Ankirin‐G) polyclonal (1:500) | Frontier Institute | Cat# AnkG‐Rb, RRID:AB_2571661 |
| Mouse anti‐Ankyrin G Monoclonal (4G3F8) (1:300) | Thermo Fisher Scientific | Cat# 33‐8800, RRID:AB_2533145 |
| Rabbit anti‐RPT3 polyclonal (1:500) | Enzo Life Sciences | Cat# BML‐PW8175, RRID:AB_10541231 |
| Rabbit anti‐TARDBP polyclonal (1:500) | Proteintech | Cat# 10782‐2‐AP, RRID:AB_615042 |
| Mouse anti‐TARDBP monoclonal (1:500) | Novus | Cat# NBP1‐92695, RRID:AB_11005586 |
| Guinea pig anti‐p62 polyclonal (1:500) | Progen | Cat# GP62‐C, RRID:AB_2687531 |
| Goat anti‐ECEL1 (N‐20) polyclonal (1:1,000) | Santa Cruz Biotechnology | Cat# sc‐11338, RRID:AB_2097871 |
| Mouse anti‐NeuN monoclonal (1:1,000) | Millipore | Cat# MAB377, RRID:AB_2298772 |
| Rabbit anti‐ATF‐3 (C‐19) polyclonal (1:1,000) | Santa Cruz Biotechnology | Cat# sc‐188, RRID:AB_2258513 |
| Rabbit anti‐ATF3 monoclonal (1:1,000) | Abcam | Abcam Cat# ab207434, RRID:AB_2734728 |
| Goat anti‐ChAT polyclonal (1:1,000) | Millipore | Cat# AB144P, RRID:AB_2079751 |
| Rabbit anti‐ChAT polyclonal (1:5,000) | Ichikawa et al (1997) | |
| Rabbit anti‐Iba1 polyclonal (1:1,000) | Wako | Cat# 019‐19741, RRID:AB_839504 |
| Goat anti‐GFAP polyclonal (1:1,000) | Abcam | Cat# ab53554, RRID:AB_880202 |
| Rabbit anti‐Caspr polyclonal (1:1,000) | Abcam | Cat# ab34151, RRID:AB_869934 |
| Mouse anti‐Neurofilament H monocolonal (1:1,000) | Covance | Cat# SMI‐32P‐100, RRID:AB_10719742 |
| Mouse anti‐cytochrome c monoclonal (1:500) | Thermo Fisher Scientific | Cat# 45‐6100, RRID:AB_2533821 |
| Goat anti‐MMP‐9 polyclonal (1:1,000) | Sigma‐Aldrich | Cat# M9570, RRID:AB_1079397 |
| Goat anti‐Osteopontin polyclonal (1:1,000) | R and D Systems | Cat# AF808, RRID:AB_2194992 |
| Rat ant‐Lamp‐1 monoclonal (1:1,000) | Santa Cruz Biotechnology | Cat# sc‐19992, RRID:AB_2134495 |
| Mouse anti‐GAPDH Monoclonal (6C5) (1;5,000) | Thermo Fisher Scientific | Cat# AM4300, RRID:AB_2536381 |
| α‐Bungarotoxin, Alexa Fluor™ 594 conjugate (1:300) | Thermofisher | B13423 |
| GST‐Tag Polyclonal (1:1,000) | MBL | Cat# PM013, RRID:AB_591789 |
| Mouse Anti‐HA.11 Monoclonal (1:1,000) | Covance | Cat# MMS‐101P‐1000, RRID:AB_291259 |
| Alexa Donkey anti‐rat 488 (1:500) | Thermo Fisher Scientific | Cat# A‐21208, RRID:AB_2535794 |
| Alexa Donkey anti‐rabbit 488 (1:500) | Thermo Fisher Scientific | Cat# A‐21206, RRID:AB_2535792 |
| Alexa Donkey anti‐rabbit 594 (1:500) | Thermo Fisher Scientific | Cat# A‐21207, RRID:AB_14163 |
| Alexa Donkey anti‐mouse 594 (1:500) | Thermo Fisher Scientific | Cat# A‐21203, RRID:AB_141633 |
| Alexa Donkey anti‐goat 647 (1:500) | Thermo Fisher Scientific | Cat# A‐21447, RRID:AB_2535864 |
| Alexa Goat anti‐rat 488 (1:500) | Thermo Fisher Scientific | Cat# A‐11006, RRID:AB_2534074 |
| Alexa Goat anti‐rabbit 594 (1:500) | Thermo Fisher Scientific | Cat# A‐11012, RRID:AB_2534079 |
| Alexa goat anti‐mouse 594 (1:500) | Thermo Fisher Scientific | A‐11005, RRID:AB_2534073 |
| Alexa goat anti‐guina pig 594 (1:500) | Thermo Fisher Scientific | Cat# A‐11076, RRID:AB_2534120 |
| Oligonucleotides and sequence‐based reagents | ||
| See Appendix Table S1 | ||
| Chemicals, enzymes and other reagents | ||
| ECL™ Prime Western Blotting System | Amersham | Cat# RPN2232 |
| SuperSepTMAce | WAKO | Cat# 197‐15011 |
| RNeasy lipid tissue midi kit | QIAGEN | Cat# 74804 |
| SuperScript® III RNase H‐Reverse Transcriptase | Invitrogen | Cat# 18080‐085 |
| CUBIC‐L | Tokyo Chemical Industry Co., Ltd. | Cat# T3740 |
| CUIBC‐R | Tokyo Chemical Industry Co., Ltd. | Cat# T3741 |
| Mounting solution | Tokyo Chemical Industry Co., Ltd. | Cat# M3294 |
| Fast SYBRTM Green Master Mix | Thermo Fisher Scientific | Cat# 4385616 |
| Software | ||
| Fiji | Image J | Fiji NIH http://fiji.sc/ |
| GraphPad Prism version7.0 | Graphpad Software | GraphPad Prism version 7.0 GraphPad Software, Inc https://www.graphpad.com/scientificsoftware/ |
| Imaris | Bitplane | Bitplane |
| Zen black and blue | Carl Zeiss | Zeiss |
| Other | ||
| AAV9‐AnkG shRNA‐mCherry ‐ shRNA sequence 5'‐GCGTCTCCTATTAGATCTTTC‐3′ | Vector builder | N/A |
| AAV9‐scramble shRNA | Vector builder | N/A |
| Fv10i | Olympus | |
| Lightsheet 7 | Carl Zeiss | |
| SpinSR | Olympus | |
Methods and Protocols
Animals
All animal protocols were carried out in accordance with the Nagoya university animal committee guidelines for the care and use of laboratory animals and were approved by the Nagoya university institutional animal care and use committee. All possible efforts were made to minimize suffering. Mice were housed in standard cages with ad libitum access to food and water under a 12‐h light/dark cycle.
Atf3:BAC2 Tg mice were generated according to general bacterial artificial chromosome (BAC) modification protocols as previously described (Kiryu‐Seo et al, 2016). The BAC clone RP24‐318C6, containing the Atf3 locus, was purchased from the BACPAC Resources Center at the Children's Hospital Oakland Research Institute (CHORI) (BPRC, California, USA). Briefly, a gene fragment of GFP from Aequorea coerulescens (AcGFP) attached to the mitochondrial targeting sequence of cytochrome c oxidase subunit VIII, followed by an IRES, cre recombinase and SV40‐polyA, was inserted into PL451 in front of the FRT‐Neo‐FRT cassette. The initiation codon of Atf3 gene in BAC was replaced by that of GFP with the mitochondrial targeting sequence. BAC DNA was purified and injected into pronuclei from C57BL/6 oocytes (Institute of Immunology Co., Ltd., Tochigi, Japan) to generate Tg mice. The Atf3:BAC2 Tg mice were crossed with C57BL/6Ncr mice for at least seven generations. The hemizygous mice were fertile and viable without neurological abnormalities.
To generate injury‐induced Rpt3 CKO mice (Atf3:BAC2;Rpt3 flox/flox ), we carefully optimized cell‐type‐specific recombination (Luo et al, 2020). We crossed Atf3:BAC2 Tg mice with Rpt3 flox/flox mice and maintained the colony as heterozygous Atf3:BAC2;Rpt3 flox/+ mice. Rpt3 flox/flox mice were provided by Drs. Tashiro and Takahashi (Kyoto University). Heterozygous Atf3:BAC2;Rpt3 flox/+ mice were bred with homozygous Rpt3 flox/flox mice to generate injury‐induced Rpt3 CKO mice (Atf3:BAC2;Rpt3 flox/flox ; Fig EV1E–H). Homozygous Rpt3 flox/flox mice were always maintained by crossing with Rpt3 flox/flox , which had not crossed with cre driver mice to avoid unexpected germline recombination.
Thy1‐Mito mice were purchased from Jackson Laboratory (Bar Harbor, ME; B6.Cg‐Tg (Thy1‐CFP/COX8A) S2Lich/J Stock No: 007967) and were maintained as a hemizygous colony.
To generate Atf3:BAC; SOD1G93A (Atf3;SOD1) mice, we initially crossed male SJL/J mice with female Atf3:BAC Tg mice (C57BL/6 background) which we reported previously (Kiryu‐Seo et al, 2016) and obtained Atf3:BAC Tg mice with B6SJL background. The SOD1G93A mice (B6SJL‐Tg(SOD1*G93A)1Gur/J, stock number 002726, B6SJL background, Jackson Laboratory, Bar Harbor, ME) were crossed with Atf3:BAC Tg mice with B6SJL background to generate Atf3;SOD1 mice (Fig EV5A).
All mice were matched for age and sex. Experiments were performed on both sexes, with the exception of experiments involving Atf3;SOD1 mice. Male Atf3;SOD1 mice and the male littermates carrying only Atf3:BAC transgene were used for experiment. Primer sequences for genotyping of Atf3:BAC Tg, Atf3:BAC2 Tg and Rpt3 flox/flox mice are described in Appendix Table S1. Primer sequences and protocol for genotyping of Thy1‐Mito and SOD1G93A mice are available through the Jackson Laboratory.
Surgical procedures
Animals of either sex at 8–10 weeks old were anesthetized under isoflurane. For hypoglossal nerve injury, the right hypoglossal nerve was carefully exposed under the digastric muscle and transected with a pair of scissors. The skin was closed with sutures. For sciatic nerve injury, the right sciatic nerve was exposed in the mid‐thigh level. The nerve was carefully freed from surrounding connective tissue and then cut with a pair of scissors. The skin was closed with sutures.
Immunohistochemistry
Mice were perfused transcardially with 1% paraformaldehyde (PFA) in PB containing 0.2% picric acid. Brains, spinal cords, nerves, and tongues were removed, post‐fixed, and then transferred to 30% sucrose solution. For immunohistochemistry using DINE antibody, mice were decapitated and the brains were freshly frozen in powdered dry ice. Both perfused and fresh frozen samples were embedded in OCT compound and cut into serial 16‐μm‐thick sections on a cryostat. Fresh frozen sections were fixed in 4% PFA for 10 min before immunohistochemical procedures. Both sections were washed in 0.01 M PBS, blocked in 0.01 M PBS containing 1% BSA and 0.3% Triton, and incubated with following primary antibodies; anti‐ATF3 (Santa Cruz, 1:1,000, Cat# sc‐188, RRID:AB_2258513), anti‐RPT3 (Enzo Life Sciences, 1:500, Cat# BML‐PW8175, RID:AB_10541231), anti‐GFP (Nacalai, 1:1,000, Cat# 04404‐84, RRID:AB_10013361 or MBL,1:1,000, Cat# 598, RRID:AB_591819), anti‐Iba1 (WAKO, 1:1,000, Cat# 019‐19741, RRID:AB_839504), anti‐p62 (Progen, 1:500, Cat# GP62‐C, RRID:AB_2687531), anti‐TDP‐43 (Proteintech, 1:500, Cat# 10782‐2‐AP, RRID:AB_615042 or Novus, 1:500, Cat# NBP1‐92695, RRID:AB_11005586), anti‐AnkG (Frontier Institute, 1:500, Cat# AnkG‐Rb, RRID:AB_2571661 or Thermo Fisher Scientific, 1:300, Cat# 33‐8800, RRID:AB_2533145), anti‐Caspr (Abcam, 1:1,000, Cat# ab34151, RRID:AB_869934), anti‐NeuN (Millipore, 1:1,000, Cat# MAB377, RRID:AB_2298772), anti‐DINE/ ECEL1 (1:1,000, Cat# sc‐11,338, RRID:AB_2097871), anti‐cytochrome c (Thermo Fisher Scientific, 1:500, Cat# 45‐6100, RRID:AB_2533821), anti‐GFAP (Abcam, 1:1,000, Cat# ab53554, RRID:AB_880202), anti‐MMP‐9 (Sigma‐Aldrich, 1:500, Cat# M9570, RRID:AB_1079397), anti‐Osteopontin (R and D systems, 1:1,000, Cat# AF808, RRID:AB_2194992), anti‐Neurofilament (Covance, 1:1,000, Cat# SMI‐32P‐100, RRID:AB_10719742), anti‐Lamp1 (Santa Cruz Biotechnology, 1:1,000, Cat# sc‐19992, RRID:AB_2134495), anti‐ChAT antibody (Millipore, 1:1,000, Cat# AB144P, RRID:AB_2079751) and Rabbit anti‐ChAT antibody provided by Dr. Misawa (Keio University) (Ichikawa et al, 1997). After washing with PBS, sections were incubated with secondary antibodies: AlexaFluor 488, 594, or 647 (Life Technologies, 1:500) or AlexaFluor 594‐conjugated α‐Bungarotoxin (Thermo Fisher, 1:300, cat# B13423). Images were acquired on a confocal laser scanning microscope (Olympus FV10i, Tokyo, Japan) using x10 objective or ×60 water‐immersion objective (NA1.2).
Tissue clearing
After mice were perfused transcardially with 4% PFA‐PBS, brain and spinal cord were dissected, washed with PBS, and processed according to CUBIC protocol (Tainaka et al, 2018). The samples were immersed in 1:1 water‐diluted CUBIC‐L (Tokyo Chemical Industry, M0374) for 1 day and then incubated with CUBIC‐L with gentle shaking at 37°C for 4–5 days. After washing with PBS, the samples were immersed in 1:1 water‐diluted CUBIC‐R for 1 day and then in CIBUC‐R with gentle shaking at room temperature for 1–2 days.
Lightsheet microscopy
The images are acquired with lightsheet 7 microscope (Zeiss, Germany). For CUBIC treated brain and spinal cord, images were captured using a 5 × objective lens with digital zoom from 0.8 × to 2.5 × with digital zoom equipped with laser emitting 488 nm. The RI‐matched sample was immersed in an oil mixture (RI = 1.525). Raw data were handled using a ZEISS Zen blue and black. 3D images were created in Imaris (Bitplane, Oxford Instruments, Belfast, UK).
Counting of motor neurons
For quantification of cell survival after axotomy, sections were stained with thionine. Thionine‐stained hypoglossal motor neurons in injured and control sides were counted separately and at the identical level between animals as previously described (Kiryu‐Seo et al, 2005). Data were presented as the percentage of surviving neurons on the injured side, compared with neurons on control side. For cell survival of GFP‐positive injured motor neurons, the number of GFP‐positive injured hypoglossal motor neurons at 5 days after injury was counted while that of ChAT‐positive uninjured hypoglossal motor neurons was counted at the identical level between animals. Data were presented as the percentage of GFP‐positive injured motor neurons compared with ChAT‐positive uninjured motor neurons. For the cell number of uninjured hypoglossal motor neurons, ChAT‐positive motor neurons in hypoglossal nucleus were counted at the identical level between animals. Data were shown as the cell number of unilateral hypoglossal nucleus per section. For counting GFP‐positive hypoglossal motor neurons in transparent 3D images, serial 2D slices were used and motor neurons were carefully identified. Data were shown as the total number of GFP‐positive motor neurons in hypoglossal nucleus.
RNA isolation and quantitative reverse‐transcription PCR
Contralateral and ipsilateral hypoglossal nuclei at 5 days after injury were dissected from 7–10 mice. Total RNA was extracted using an RNeasy Lipid Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. cDNA was synthesized using SuperscriptIII (Thermo Fisher Scientific, Massachusetts, USA) following the standard protocols. Quantitative PCR was performed on the StepOnePlus™ Real‐Time PCR System using primer pairs and SYBR Green PCR Master Mix (Thermo Fisher Scientific). Data were normalized to the GAPDH levels and calculated using the comparative threshold cycle method. Primer sequences are described in Appendix Table S1.
Immunoblotting
Contralateral and ipsilateral hypoglossal nuclei and nerves from 5–7 mice were dissected and homogenized in lysis buffer A containing 150 mM NaCl, 20 mM Tris–HCl (pH 7.5), 10 mM EDTA, 1% NP‐40, 0.5% deoxycholate, 0.1% SDS, 5 μg/ml aprotinin, 1 mM PMSF, and 1 μg/ml leupeptin and centrifuged to collect the supernatant. Proteins were resolved by SDS–PAGE using 4–12% Bis‐Tris gels (FUJIFILM Wako pure chemical corporation, Japan) and were transferred to PVDF membranes. Anti‐AnkG (Frontier Institute, Cat# AnkG‐Rb, RRID:AB_2571661), anti‐RPT3 (Enzo Life Sciences, Cat# BML‐PW8175, RRID:AB_10541231) and anti‐GAPDH (Thermo Fisher Scientific, Cat# AM4300, RRID:AB_2536381) primary antibodies and were diluted 1:1,000–1:5,000. Signals were detected using ECL (GE Healthcare, Chicago, USA).
Ubiquitination assay
The cDNA fragments encoding truncated forms of rat AnkG were subcloned into pEF‐BOS vector. Each GST‐tagged AnkG plasmid and HA‐tagged ubiquitin in pcDNA3 (addgene18712) were cotransfected into COS‐7 cells. At 18 h after transfection, the cells were treated with 25 μM ALLN (Calbiochem, Merck, Darmstadt, Germany) for 6 h. The cells were lysed in lysis buffer B (20 mM Tris–HCl, pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.5% NP‐40 substitute, 1 mM DTT, 25 μM ALLN, 0.5 mM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml antipain, and 10 μg/ml benzamidine). The cell lysates were incubated with Glutathione Sepharose 4B beads (GE Healthcare, Chicago, USA) for 2 h at 4°C, and the beads were subsequently washed three times with lysis buffer B. After washing, the beads were resuspended with SDS–PAGE sample buffer, and the samples were subjected to immunoblot analysis using anti‐HA (Covance, Cat# MMS‐101P‐1000, RRID:AB_291259) and anti‐GST (MBL, Cat# PM013, RRID:AB_591789) antibodies.
Image analysis of the fluorescently labeled AIS and mitochondria
All image analysis studies including length and number of the AIS, intensity ratio of GFP in the AIS, and fluorescent line intensity plots were performed by using Fiji software (NIH, Bethesda, MD, USA). Images were acquired on a confocal laser scanning microscope (Olympus FV10i, Tokyo, Japan) using a ×60 water‐immersion objective (NA1.2). The length and number of AIS stained by AnkG was measured using z‐stacked images. The number of the AIS per area with 1,024 × 1,024 pixel was counted. 20–30 neurons for AIS length and 3–6 areas for AIS number were measured in each mouse. For fluorescent intensity of GFP in the AIS region, we traced the line along the AIS region and subtracted the random fluorescence intensity in these images, to control for background signals. The mean intensities of GFP in the AIS were averaged and presented as the percentage of the intensity from Thy1‐Mito mouse. 10–20 neurons from each mouse were measured. For fluorescent line intensity, the fluorescent intensity of GFP and AnkG in the AIS was plotted from the cell body to the axon along 1‐μm‐wide lines. Representative images and their corresponding plots are shown in figures. The disappeared AIS region after injury was identified when injured motor neurons retained very weak or fragmented signal of AnkG or when injured motor neuron extended the neurite, which exited hypoglossal nucleus and formed hypoglossal nerve bundle.
Super‐resolution images of mitochondria in injured motor neurons were acquired by SpinSR10 (Olympus, Tokyo, Japan) using a x100 oil‐immersion objective and deconvoluted. A single layer with highest resolution in the images was selected, and the fluorescent intensity of GFP in cell body was plotted along the line. Each peak represents the existence of mitochondria on a line within soma. The number of peaks on a line was counted as mitochondrial number from 5–7 neurons from each mouse.
In vivo imaging
Mice of either sex at 8–10 weeks old were anesthetized by i.p. injection of pentobarbital (45 mg/kg). To image axonal mitochondria in the sciatic nerve, we surgically exposed the uninjured and injured sciatic nerve without causing damage and positioned the mouse on an inverted laser scanning confocal microscope (Olympus FV10i) equipped with a temperature‐controlled chamber at 37°C. Time‐lapse movies were acquired on a confocal microscope equipped with a temperature‐controlled stage at 37°C and with a ×60 water‐immersion objective (NA1.2) (Olympus FV10i). Images were captured every 5 s and 100 frames were acquired in total for each recording, with laser power set to < 30% to minimize damage. Time‐lapse images of mitochondrial dynamics were collected in a single focal plane at 1,024 × 1,024 pixel.
Quantification of axonal mitochondria
For whole‐mount observation of mitochondria in nerves, hypoglossal and sciatic nerves were removed from mice, which were perfused transcardially with 4% paraformaldehyde (PFA) in PB. Subsequently, images of whole‐mount axon were acquired on a confocal laser scanning microscope using ×60 water‐immersion objective (Olympus FV10i). Digital Z stack images were obtained with sequential acquisition setting. The number of mitochondria in a single axon was counted using Fiji software. Data were presented as mitochondrial number in 50 μm2 of axon. A 5–8 axons from each mouse were used for counting. To quantify mobile and stationary mitochondria, kymographs were generated using the Fiji software (NIH, Bethesda, MD, USA) as described previously (Kiryu‐Seo et al, 2010). Motile mitochondria appeared as diagonal lines. Each mobile mitochondrion confirmed to be moving for six or more consecutive frames within the area was measured. Stationary mitochondrial sites were identified as vertical lines on the kymographs. The percentage of motile mitochondria of total mitochondria is defined in a given distance for 180 s per axons. Stationary mitochondria were counted per given distance of axon.
Adeno‐associated viruses (AAVs)
The shRNA of AnkG was cloned into pAAV vector with the sequence, GCGTCTCCTATTAGATCTTTC, targeting a serine‐rich region shared by both the 270‐kDa and 480‐kDa isoforms of mouse AnkG (Ye et al, 2020), followed by PGK promoter and mCherry (VectorBuilder, Chicago, USA). The scrambled shRNA followed by PGK promoter and mCherry was purchased from VectorBuilder. The plasmids were transfected into 293 cells to produce AAVs carrying serotype 9. The titers of all viral preparations were at least 1.0 × 1013 GC/ml. The AAVs were injected into the tongue of 6–8‐week‐old mice using Hamilton syringe at 3–4 weeks before hypoglossal nerve injury.
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed using Prism7 (GraphPad software). Unpaired t‐test was used to compare two groups. One‐way ANOVA was used for multiple comparisons, followed by Tukey post hoc analysis. Survival data were analyzed by long‐rank test. P‐values < 0.05 were considered significant.
Author contributions
Sumiko Kiryu‐Seo: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; writing – original draft; writing – review and editing. Reika Matsushita: Data curation; formal analysis; investigation. Yoshitaka Tashiro: Resources; writing – review and editing. Takeshi Yoshimura: Data curation; formal analysis; investigation; writing – review and editing. Yohei Iguchi: Resources; writing – review and editing. Masahisa Katsuno: Resources; writing – review and editing. Ryosuke Takahashi: Resources; writing – review and editing. Hiroshi Kiyama: Conceptualization; supervision; funding acquisition; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Source Data for Expanded View and Appendix
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgments
We appreciate Ayako Asano and Yoshiko Itai for managing the project. We thank Naomi Tawarayama and Kyoko Muraki for excellent technical assistance. We thank Chenmin Li and Hiromi Tamada for sharing data. We thank Hideo Misawa (Keio University) for providing rabbit ChAT antibody. We acknowledge staffs of Division of Experimental Animals, Nagoya University Graduate School of Medicine and staffs of the Nagoya University Equipment Sharing system for their technical support. We are also grateful to Hiroshi Kuba and Rei Yamada (Nagoya University, Japan) for their useful comments, and Matthew N. Rasband (Baylor College of Medicine, USA) for critical reading of our manuscript. We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript. This work was supported by JSPS KAKENHI (25430036, 18H02524 and 21K19310) to S. K.‐S., TOYOAKI SCHOLARSHIP FOUNDATION to S. K.‐S. and JSPS KAKENHI (16H05117 and 22H03437) to H.K.
The EMBO Journal (2022) 41: e110486.
Contributor Information
Sumiko Kiryu‐Seo, Email: skiryu@med.nagoya-u.ac.jp.
Hiroshi Kiyama, Email: kiyama@med.nagoya-u.ac.jp.
Data availability
This study includes no data deposited in external repositories.
References
- An D, Fujiki R, Iannitelli DE, Smerdon JW, Maity S, Rose MF, Gelber A, Wanaselja EK, Yagudayeva I, Lee JY et al (2019) Stem cell‐derived cranial and spinal motor neurons reveal proteostatic differences between ALS resistant and sensitive motor neurons. Elife 8: e44423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baalman K, Marin MA, Ho TS, Godoy M, Cherian L, Robertson C, Rasband MN (2015) Axon initial segment‐associated microglia. J Neurosci 35: 2283–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benned‐Jensen T, Christensen RK, Denti F, Perrier JF, Rasmussen HB, Olesen SP (2016) Live imaging of Kv7.2/7.3 cell surface dynamics at the axon initial segment: high steady‐state stability and calpain‐dependent excitotoxic downregulation revealed. J Neurosci 36: 2261–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beuningen SFB, Will L, Harterink M, Chazeau A, van Battum EY, Frias CP, Franker MAM, Katrukha EA, Stucchi R, Vocking K et al (2015) TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88: 1208–1226 [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G (2010) Deficits in axonal transport precede ALS symptoms in vivo . Proc Natl Acad Sci U S A 107: 20523–20528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnevie VS, Dimintiyanova KP, Hedegaard A, Lehnhoff J, Grondahl L, Moldovan M, Meehan CF (2020) Shorter axon initial segments do not cause repetitive firing impairments in the adult presymptomatic G127X SOD‐1 amyotrophic lateral sclerosis mouse. Sci Rep 10: 1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke F, Di Giovanni S, Fawcett J (2020) Neuronal maturation: challenges and opportunities in a nascent field. Trends Neurosci 43: 360–362 [DOI] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, He Z (2016) The mammalian‐specific protein Armcx1 regulates mitochondrial transport during axon regeneration. Neuron 92: 1294–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain KA, Sheng ZH (2019) Mechanisms for the maintenance and regulation of axonal energy supply. J Neurosci Res 97: 897–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L et al (2016) A systems‐level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89: 956–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Guardia CM, Britt DJ, Guo X, Bonifacino JS (2015) Sorting of dendritic and axonal vesicles at the pre‐axonal exclusion zone. Cell Rep 13: 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbino V, Kaunga E, Ye J, Canzio D, O'Keeffe S, Rudnick ND, Guarnieri P, Lutz CM, Maniatis T (2020) The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 106: 789–805.e785 [DOI] [PubMed] [Google Scholar]
- Gumy LF, Katrukha EA, Grigoriev I, Jaarsma D, Kapitein LC, Akhmanova A, Hoogenraad CC (2017) MAP2 defines a pre‐axonal filtering zone to regulate KIF1‐ versus KIF5‐dependent cargo transport in sensory neurons. Neuron 94: 347–362.e7 [DOI] [PubMed] [Google Scholar]
- Hamdan H, Lim BC, Torii T, Joshi A, Konning M, Smith C, Palmer DJ, Ng P, Leterrier C, Oses‐Prieto JA et al (2020) Mapping axon initial segment structure and function by multiplexed proximity biotinylation. Nat Commun 11: 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M (2016) Mitochondria localize to injured axons to support regeneration. Neuron 92: 1308–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Xie Y, Ordaz JD, Huh AJ, Huang N, Wu W, Liu N, Chamberlain KA, Sheng ZH, Xu XM (2020) Restoring cellular energetics promotes axonal regeneration and functional recovery after spinal cord injury. Cell Metab 31: 623–641.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J et al (2011) Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331: 928–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Rasband MN (2018) Axon initial segments: structure, function, and disease. Ann N Y Acad Sci 1420: 46–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa T, Ajiki K, Matsuura J, Misawa H (1997) Localization of two cholinergic markers, choline acetyltransferase and vesicular acetylcholine transporter in the central nervous system of the rat: In situ hybridization histochemistry and immunohistochemistry. J Chem Neuroanat 13: 23–39 [DOI] [PubMed] [Google Scholar]
- Jenkins PM, Kim N, Jones SL, Tseng WC, Svitkina TM, Yin HH, Bennett V (2015) Giant ankyrin‐G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci U S A 112: 957–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins EC, Shah N, Gomez M, Casalena G, Zhao D, Kenny TC, Guariglia SR, Manfredi G, Germain D (2020) Proteasome mapping reveals sexual dimorphism in tissue‐specific sensitivity to protein aggregations. EMBO Rep 21: e48978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Agar JN, Strong MJ, Durham HD (2012) Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13: 367–371 [DOI] [PubMed] [Google Scholar]
- Kalinski AL, Kar AN, Craver J, Tosolini AP, Sleigh JN, Lee SJ, Hawthorne A, Brito‐Vargas P, Miller‐Randolph S, Passino R et al (2019) Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol 218: 1871–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE (2014) Neuronal matrix metalloproteinase‐9 is a determinant of selective neurodegeneration. Neuron 81: 333–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu‐Seo S, Kiyama H (2011) The nuclear events guiding successful nerve regeneration. Front Mol Neurosci 4: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu‐Seo S, Hirayama T, Kato R, Kiyama H (2005) Noxa is a critical mediator of p53‐dependent motor neuron death after nerve injury in adult mouse. J Neurosci 25: 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu‐Seo S, Gamo K, Tachibana T, Tanaka K, Kiyama H (2006) Unique anti‐apoptotic activity of EAAC1 in injured motor neurons. EMBO J 25: 3411–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu‐Seo S, Kato R, Ogawa T, Nakagomi S, Nagata K, Kiyama H (2008) Neuronal injury‐inducible gene is synergistically regulated by ATF3, c‐Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J Biol Chem 283: 6988–6996 [DOI] [PubMed] [Google Scholar]
- Kiryu‐Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD (2010) Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci 30: 6658–6666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu‐Seo S, Tamada H, Kato Y, Yasuda K, Ishihara N, Nomura M, Mihara K, Kiyama H (2016) Mitochondrial fission is an acute and adaptive response in injured motor neurons. Sci Rep 6: 28331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong G, Zhou L, Serger E, Palmisano I, De Virgiliis F, Hutson TH, McLachlan E, Freiwald A, La Montanara P, Shkura K et al (2020) AMPK controls the axonal regenerative ability of dorsal root ganglia sensory neurons after spinal cord injury. Nat Metab 2: 918–933 [DOI] [PubMed] [Google Scholar]
- Kuijpers M, van de Willige D, Freal A, Chazeau A, Franker MA, Hofenk J, Rodrigues RJ, Kapitein LC, Akhmanova A, Jaarsma D et al (2016) Dynein regulator NDEL1 controls polarized cargo transport at the axon initial segment. Neuron 89: 461–471 [DOI] [PubMed] [Google Scholar]
- Leterrier C (2018) The axon initial segment: An updated viewpoint. J Neurosci 38: 2135–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr, Turi GF, Kwon SK, Losonczy A, Polleux F (2016) Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo . Curr Biol 26: 2602–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht‐Mayer S, Campbell GR, Canizares M, Mehta AR, Gane AB, McGill K, Ghosh A, Fullerton A, Menezes N, Dean J et al (2020) Enhanced axonal response of mitochondria to demyelination offers neuroprotection: Implications for multiple sclerosis. Acta Neuropathol 140: 143–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, Sheng ZH (2017) Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94: 595–610.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger CS, Boillee S, Cleveland DW (2007) Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci U S A 104: 7319–7326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Brommer B, Tian X, Krishnan A, Meer M, Wang C, Vera DL, Zeng Q, Yu D, Bonkowski MS et al (2020) Reprogramming to recover youthful epigenetic information and restore vision. Nature 588: 124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Ambrozkiewicz MC, Benseler F, Chen C, Dumontier E, Falkner S, Furlanis E, Gomez AM, Hoshina N, Huang WH et al (2020) Optimizing nervous system‐specific gene targeting with Cre driver lines: Prevalence of germline recombination and influencing factors. Neuron 106: 37–65.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Cortez C, Gan WB, Manfredi G (2014) Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 23: 1413–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor‐Nof M, Shipony Z, Lopez‐Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, Blum JA, Castruita PA, Linares GR, Ruan K et al (2021) p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 184: 689–708.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin MA, de Lima S, Gilbert HY, Giger RJ, Benowitz L, Rasband MN (2016) Reassembly of excitable domains after CNS axon regeneration. J Neurosci 36: 9148–9160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW (2007) Imaging axonal transport of mitochondria in vivo . Nat Methods 4: 559–561 [DOI] [PubMed] [Google Scholar]
- Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ (2017) Amyotrophic lateral sclerosis‐associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet 26: 4668–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki Y, Niikura M, Watanabe M, Onishi K, Tanabe S, Moriwaki Y, Okuda T, Ohara S, Murayama S, Takao M et al (2016) Selective expression of Osteopontin in ALS‐resistant motor neurons is a critical determinant of late phase neurodegeneration mediated by matrix Metalloproteinase‐9. Sci Rep 6: 27354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi S, Suzuki Y, Namikawa K, Kiryu‐Seo S, Kiyama H (2003) Expression of the activating transcription factor 3 prevents c‐Jun N‐terminal kinase‐induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J Neurosci 23: 5187–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijssen J, Comley LH, Hedlund E (2017) Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol 133: 863–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmisano I, Danzi MC, Hutson TH, Zhou L, McLachlan E, Serger E, Shkura K, Srivastava PK, Hervera A, Neill NO et al (2019) Epigenomic signatures underpin the axonal regenerative ability of dorsal root ganglia sensory neurons. Nat Neurosci 22: 1913–1924 [DOI] [PubMed] [Google Scholar]
- Patodia S, Raivich G (2012) Role of transcription factors in peripheral nerve regeneration. Front Mol Neurosci 5: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron LA, Zinsmaier KE (2016) Mitochondria on the road to power axonal regeneration. Neuron 92: 1152–1154 [DOI] [PubMed] [Google Scholar]
- Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, Geschwind DH, Woolf CJ (2020) Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron 108: 128–144.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM, Maniatis T (2017) Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc Natl Acad Sci U S A 114: E8294–E8303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakao Y, Kawai T, Takeuchi O, Copeland NG, Gilbert DJ, Jenkins NA, Takeda K, Akira S (2000) Mouse proteasomal ATPases Psmc3 and Psmc4: Genomic organization and gene targeting. Genomics 67: 1–7 [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M (1996) Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology 47: 535–540 [DOI] [PubMed] [Google Scholar]
- Sasaki S, Warita H, Abe K, Iwata M (2005) Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol 110: 48–56 [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P (2009) A role for motoneuron subtype‐selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12: 627–636 [DOI] [PubMed] [Google Scholar]
- Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN (2009) Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci 29: 13242–13254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Allchorne AJ, Woolf CJ (2006) The transcription factor ATF‐3 promotes neurite outgrowth. Mol Cell Neurosci 32: 143–154 [DOI] [PubMed] [Google Scholar]
- Seijffers R, Zhang J, Matthews JC, Chen A, Tamrazian E, Babaniyi O, Selig M, Hynynen M, Woolf CJ, Brown RH Jr (2014) ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc Natl Acad Sci U S A 111: 1622–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Rossor AM, Fellows AD, Tosolini AP, Schiavo G (2019) Axonal transport and neurological disease. Nat Rev Neurol 15: 691–703 [DOI] [PubMed] [Google Scholar]
- Sohn PD, Huang CT, Yan R, Fan L, Tracy TE, Camargo CM, Montgomery KM, Arhar T, Mok SA, Freilich R et al (2019) Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104: 458–470.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis‐Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S et al (2015) Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1‐mediated ALS. Proc Natl Acad Sci U S A 112: E6993–E7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tainaka K, Murakami TC, Susaki EA, Shimizu C, Saito R, Takahashi K, Hayashi‐Takagi A, Sekiya H, Arima Y, Nojima S et al (2018) Chemical landscape for tissue clearing based on hydrophilic reagents. Cell Rep 24: 2196–2210.e9 [DOI] [PubMed] [Google Scholar]
- Tamada H, Kiryu‐Seo S, Sawada S, Kiyama H (2021) Axonal injury alters the extracellular glial environment of the axon initial segment and allows substantial mitochondrial influx into axon initial segment. J Comp Neurol 529: 3621–3632 [DOI] [PubMed] [Google Scholar]
- Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H et al (2012) Motor neuron‐specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem 287: 42984–42994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539: 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzack GE, Hall CE, Sibley CR, Cymes T, Forostyak S, Carlino G, Meyer IF, Schiavo G, Zhang SC, Gibbons GM et al (2017) A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat Commun 8: 1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan‐Johns M, Bengoechea R, Bell S, Shao J, Diamond MI, True HL, Weihl CC, Baloh RH (2014) Prion‐like nuclear aggregation of TDP‐43 during heat shock is regulated by HSP40/70 chaperones. Hum Mol Genet 23: 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugun‐Klusek A, Tatham MH, Elkharaz J, Constantin‐Teodosiu D, Lawler K, Mohamed H, Paine SM, Anderson G, John Mayer R, Lowe J et al (2017) Continued 26S proteasome dysfunction in mouse brain cortical neurons impairs autophagy and the Keap1‐Nrf2 oxidative defence pathway. Cell Death Dis 8: e2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlug AS, Teuling E, Haasdijk ED, French P, Hoogenraad CC, Jaarsma D (2005) ATF3 expression precedes death of spinal motoneurons in amyotrophic lateral sclerosis‐SOD1 transgenic mice and correlates with c‐Jun phosphorylation, CHOP expression, somato‐dendritic ubiquitination and Golgi fragmentation. Eur J Neurosci 22: 1881–1894 [DOI] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Shaw PJ, De Vos KJ (2017) Protein homeostasis in amyotrophic lateral sclerosis: Therapeutic opportunities? Front Mol Neurosci 10: 123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada R, Kuba H (2016) Structural and functional plasticity at the axon initial segment. Front Cell Neurosci 10: 250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Li J, Ye F, Zhang Y, Zhang M, Wang C (2020) Mechanistic insights into the interactions of dynein regulator Ndel1 with neuronal ankyrins and implications in polarity maintenance. Proc Natl Acad Sci U S A 117: 1207–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Rasband MN (2014) Axon initial segments: diverse and dynamic neuronal compartments. Curr Opin Neurobiol 27: 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Chen X, Li J, Tian C, Chen J, Xiao C, Zhong G, He S (2020) Calcineurin signaling mediates disruption of the axon initial segment cytoskeleton after injury. iScience 23: 100880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH (2016) Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol 214: 103–119 [DOI] [PMC free article] [PubMed] [Google Scholar]