

Abstract
Store operated Ca2+ entry (SOCE) is the most important Ca2+ entry pathway in non-excitable cells. However, SOCE can also play a pivotal role in excitable cells such as anterior pituitary (AP) cells. The AP gland contains five different cell types that release six major AP hormones controlling most of the entire endocrine system. AP hormone release is modulated by Ca2+ signals induced by different hypothalamic releasing hormones (HRHs) acting on specific receptors in AP cells. TRH and LHRH both induce Ca2+ release and Ca2+ entry in responsive cells while GHRH and CRH only induce Ca2+ entry. SOCE has been shown to contribute to Ca2+ responses induced by TRH and LHRH but no molecular evidence has been provided. Accordingly, we used AP cells isolated from mice devoid of Orai1 channels (noted as Orai1−/− or Orai1 KO mice) and mice lacking expression of all seven canonical TRP channels (TRPC) from TRPC1 to TRPC7 (noted as heptaTRPC KO mice) to investigate contribution of these putative channel proteins to SOCE and intracellular Ca2+ responses induced by HRHs. We found that thapsigargin-evoked SOCE is lost in AP cells from Orai1−/− mice but unaffected in cells from heptaTRPC KO mice. Conversely, while spontaneous intracellular Ca2+-oscillations related to electrical activity were not affected in the Orai1−/− mice, these responses were significantly reduced in heptaTRPC KO mice. We also found that Ca2+ entry induced by TRH and LHRH is decreased in AP cells isolated from Orai1−/−. In addition, Ca2+ responses to several HRHs, particularly TRH and GHRH, are decreased in the heptaTRPC KO mice. These results indicate that expression of Orai1, and not TRPC channel proteins, is necessary for thapsigargin-evoked SOCE and is required to support Ca2+ entry induced by TRH and LHRH in mouse AP cells. In contrast, TRPC channel proteins appear to contribute to spontaneous Ca2+-oscillations and Ca2+ responses induced by TRH and GHRH. We conclude that expression of Orai1 and TRPC channels proteins may play differential and significant roles in AP physiology and endocrine control.
Keywords: Anterior pituitary cells, Orai1, TRPC channels, Store-operated calcium entry, Hypothalamic releasing factors
1. Introduction
The anterior pituitary (AP) gland is a major endocrine modulator that regulates multiple endocrine glands and tissues by the release of different AP hormones including prolactin (PRL), growth hormone (GH), thyrotropin (TSH), adrenocorticotropin (ACTH), and the gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH). The secretory activity of the gland is under control of chemical signals derived from the hypothalamus in the form of hypothalamic-releasing hormones (HRHs) that reach the AP through the portal system vasculature. HRHs include thyrotropin-releasing hormone (TRH), growth hormone-releasing hormone (GHRH), corticotropin-releasing hormone (CRH), and gonadotropin-releasing hormone (LHRH). AP hormone secretion is limited by hypothalamic inhibitory factors such as dopamine and somatostatin as well as by negative feedback loops of peripheral hormones as well as AP hormones [1].
The effect of HRHs on AP hormone secretion is mediated by intracellular Ca2+ mobilization in target cells. In many AP cells, this Ca2+ signal is in the form of spontaneous intracellular Ca2+-oscillations that are generated by spontaneous electrical activity in the form of Ca2+ action potentials in which the depolarizing current is carried by Ca2+ instead of Na+ [2]. Inhibition of electrical activity by selected Ca2+ channel antagonists and/or physiological inhibitors like dopamine or somatostatin prevents the occurrence of Ca2+ oscillations and inhibits AP hormone secretion [2,3]. Several HRHs induce a rise in in target cells and elicit AP hormone secretion, but the mechanisms underlying these signals are not completely understood [4]. For example, TRH activates TRH receptors in thyrotropes storing TSH as well as in PRL producing cells (mammotropes), and induce a biphasic response. This biphasic response is composed of a first phase due to IP3 dependent-release of Ca2+ from intracellular stores. The second phase is complex and is associated with an increased firing of action potentials which leads to increased oscillations and enhanced entry of Ca2+ through L-type voltage-gated Ca2+ channels [3,5]. However, we reported that store-operated Ca2+ entry (SOCE) also contributed to Ca2+ entry induced by TRH [3]. At that time, the molecular players involved in SOCE were unknown and no molecular evidence for the involvement of SOCE in Ca2+ responses to TRH and other HRHs has been provided so far. Similarly, LHRH promotes gonadotropin secretion mediated by Ca2+ release in gonadotropes followed by a second phase associated to a series of intracellular oscillations. However, the occurrence and contribution of SOCE to LHRH-induced responses remain controversial. GHRH increases in somatotropes and promotes GH secretion [2,6]. Ca2+ responses elicited by GHRH are mediated by cAMP-dependent activation of protein kinase A (PKA) and a Na+ conductance that increases electrical activity of the somatotrope [7]. However, the nature of the Na+ conductance remains unknown. Finally, CRH also increases in a fraction of rat and mouse AP cells by a PKA-dependent mechanism and enhances electrical activity. In this case the mechanism is believed to be mediated by the closure of TREK-1 K+ channels involved in resting electrical activity in the corticotrope [8,9].
It is worth noting that these hormone-specific actions are not restricted/partitioned into separate, specialized cells with a single receptor type. In fact, many rat and mouse AP cells express more than one HRH receptor type [10]. These cells, termed multiresponsive cells, may contribute to AP cell plasticity and secrete AP hormones in response to several HRHs [4]. AP cells may also be polyhormonal cells storing and releasing more than one AP hormones [11]. Multiresponsive and polyhormonal AP cells are multifunctional cells present in each of the five “classic” AP cell types including mammotropes, somatotropes and gonadotropes [12] and thyrotropes [13]. Multifunctional cells are also present in human pituitary tumors [14,15] and their relative abundance is dictated by the gender and physiological status of the donor [16] as well as external influences such as stress [17]. Accordingly, signal transduction pathways are probably not partitioned into specific cell types and may contribute to hormone secretion in a complex manner, probably depending on the gender, age and physiological status.
Since TRH and LHRH induce release of Ca2+ from intracellular stores, it is plausible that SOCE contributed to evoke signals. In 1995, it was reported that SOCE contributed to entry induced by TRH in prolactin producing cells [3,18]. It was the first evidence on the occurrence and physiological role of SOCE in excitable cells. It was observed that blocking L-type, voltage-gated Ca2+ entry with dihydropyridines, PRL secretion and hormone gene expression was only partially inhibited. This data suggested the presence of another channel responsible for Ca2+ entry and hormone secretion in AP cells. In testing for the presence of SOCE in gonadotropes, the authors could find no evidence of calcium release activated calcium currents (CRAC), which underlie SOCE in non-excitable cells [19]. In addition, observations on ACTH release by rat corticotropes could find no role for SOCE [20]. As stated above, no molecular evidence for contribution of SOCE to AP cell function has been reported to date.
At the molecular level, SOCE is mediated by interactions between stromal interaction molecule 1 (STIM1) [21], a sensor of the Ca2+ concentration inside the endoplasmic reticulum (ER), with Orai1, a Ca2+ channel at the plasma membrane [22]. Related protein STIM2, as well as Orai2 and Orai3, may also contribute in some cases to SOCE but their contribution is considered minor [23]. Unfortunately, there is no information on expression of Orai and STIM proteins in AP cells.
Some transient receptor potential (TRP) channels may contribute to SOCE as well in some cell types [24,25]. TRP channels were initially considered candidates for the channels involved in SOCE [26]. However, the controversy remains as to whether some canonical TRPs (TRPC) channels contribute to SOCE. The present view is that TRPC channels may rather modulate SOCE after their recruitment to the plasma membrane by Orai1-dependent Ca2+ influx [27]. For instance, contribution of TRPC channels to SOCE may depend on the phenotypic transformation of cells [28]. Expression of TRPCs has been assessed by qRT-PCR in rat AP cells. Results show expression of mRNA transcripts for TRPC1 ≫ TRPC6 > TRPC4 > TRPC5 > TRPC3 in rat and mouse AP cells [20,29]. In addition, in AP cells, constitutively active cation channels are stimulated further by PKA and contribute to calcium signaling indirectly by controlling the pacemaking depolarization in a sodium-dependent manner and directly by conducting calcium [29]. TRPC channel function in the different axes has not been addressed except for the case of the reproductive axis. Thus, TRPC5 has been functionally characterized in female murine gonadotropes [30]. It has been also demonstrated that TRPC5 in gonadotropes contributes to depolarization of the plasma membrane upon GnRH stimulation and increases the intracellular Ca2+ concentration via its own Ca2+ permeability and subsequent activation of voltage-gated Ca2+ channels [31]. However, conditional gene targeting experiments are required to unambiguously dissect the physiological role of TRPC channels in the different cell types of the reproductive axis in vivo.
The detection and measurement of SOCE is technically challenging in AP cells. The observed Ca2+ entry signals associated with SOCE are small in excitable cells including neurons [32] and AP cells [3] compared with the magnitude of this pathway in non-excitable cells or tumor cells [28]. In addition, Ca2+ entry signal is often contaminated with the large voltage-gated Ca2+ channels that may be active even in resting conditions when cells have not been treated with hormones. A major challenge in these studies is due to the large heterogeneity of AP cell population, containing up to five different AP cell types, which may also be further complicated by differential expression of up to seven different types of TRPC channel proteins in addition to Orai1, all of which might affect SOCE.
Here we have addressed the role of Orai1 and all seven TRPCs in SOCE and responses to all four HRHs in mouse AP cells. This was achieved by performing calcium imaging measurements on primary AP cells isolated from Orai1−/− and heptaTRPC KO mice. Results indicate that Orai1 channels, but not TRPC channels, contribute to SOCE in mouse AP cells. In addition, we show for the first time that Orai1 channels contribute to Ca2+ entry signal induced by TRH and LHRH in mouse AP cells. Conversely, TRPC channels, but not Orai1 channels, are involved in spontaneous oscillations, Finally, we also show that TRPC channels could be involved in the Ca2+ responses induced by TRH and GHRH.
2. Methods
2.1. Reagents
Hypothalamic releasing factors including thyrotropin-releasing hormone (TRH), growth-hormone releasing hormone (GHRH), gonadotropin-releasing hormone (LHRH) and corticotropin-releasing hormone (CRH) were purchased from Sigma.-Aldrich and Fura5f/AM from Setereh Biotech (now Thermo Fisher).
2.2. Animals
Mice carrying Orai1 null alleles (Orai1−/−, also noted as Orai1 KO in the text) were kindly provided by Jean-Pierre Kinet (Harvard Medical School, Boston, MA) and were generated using gene trap mutagenesis [33]. These Orai1 KO mice had a high incidence of perinatal lethality, which was improved by further outbreeding this line (C57/DBA/129 background) with Institute of Cancer Research (ICR) mice [Harlan Laboratories Inc., strain Hsd:ICR (CD-1)] and by delaying weaning in potential KO animals. Genotyping was performed on tail DNA by PCR. The following primers and reaction conditions were used to distinguish Orai1+/+ (488 bp), Orai1+/− and Orai1−/− (300 bp): 5′-TCA CGC TTG CTC TCC TCA TC-3′, 5′-TAA GGG CGA CAC GGA AAT G-3′ and 5′-AGG TTG TGG ACG TTG CTC AC-3′; cycling for 95 °C for 2 min, 35 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 45 s, followed by 72 °C for 10 min. For these studies we used five Orai1 KO mice and five WT mice (4–6 months old, all male).
Mice lacking all seven canonical TRP proteins (heptaTRPC KO mice) were originally created by Dr. Lutz Birnbaumer and maintained by the Comparative Medicine Branch at the NIEHS, Research Triangle Park, North Carolina, USA. Initially, TRPC1, TRPC3, TRPC5 and TRPC6 knockout animals (QuadKO) were generated on a mixed C57BL/6J:129SvEv background by combinatorial breeding of single knockout alleles, TRPC1, TRPC3, TRPC5, TRPC6 as reported previously [34]. Quad KO mice exhibited generally good health and TRPC3/6 double knockout mice were crossed with C57BL/6 mice to generate wildtype (WT) control animals. For these studies we used five HeptaTRPC KO mice (3 months old, 3 male and 2 female) and five WT mice (3 male and 2 female).
2.3. Primary anterior pituitary cell culture
Mouse AP cells were obtained as reported previously [11]. Mice were euthanized by cervical dislocation, and the AP glands were rapidly dissected. After isolating the neurointermediate lobe, the glands were chopped into small pieces with dissecting scissors. A cell suspension was then generated by digestion with 1 mg/ml trypsin (Sigma-Aldrich) in Hanks’ balanced salt solution (Life Technologies, Inc., Gaithersburg, MD) at 37 °C with gentle shaking for 30 min. Every 15 min, tissue pieces were triturated to release single cells by passing them repeatedly through a fire-polished siliconized Pasteur pipette. Isolated cells were then sedimented by centrifugation at 200g for 7 min, washed twice with Hanks’ balanced salt solution. This process generated about 1 × 106 cells per pituitary gland. Assessing viability by Trypan blue exclusion, dead cells were < 5%. In preparation for calcium measurements, monodispersed cells were plated onto round, 30 mm coverslips (#1.5) previously coated with 0.01 mg/ml poly-l-lysine. Plated cells were bathed in DMEM (Life Technologies, Inc.) supplemented with 10% fetal bovine serum and antibiotics, and incubated in a humidified 95% air, 5% CO2 at 37 °C and 95% CO2/O2 until used. Cells were used within 2–4 h following isolation for calcium imaging experiments.
2.4. Single cell calcium measurements
Fluorescence measurements were made in AP cells loaded with the calcium sensitive dye fura-5F (KD 400 nm) as described in detail previously [35]. Coverslips with attached cells were mounted in a Teflon chamber and incubated in DMEM with 1 μm of the acetoxymethy ester form of fura-5F (fura-5F AM, Molecular Probes, USA) for 25 min in a humidified 95% air−5% CO2 incubator at 37 °C. Before measurements were made, cells were washed 3 times and incubated for 30 min at room temperature (25 °C) in a Hepes-buffered salt solution (HBSS (mM): NaCl 120; KCl 5.4; MgCl2 0.8; Hepes 20; CaCl2 1.8 and glucose 10 mM, with pH 7.4 adjusted by NaOH). In these experiments, nominally Ca2+-free solutions were HBSS with no added CaCl2.
Fluorescence was monitored by placing the Teflon chamber with fura-5F-loaded cells onto the stage of a Nikon TS-100 inverted microscope equipped with a 20× fluor objective (0.75 NA). Fluorescence images of the cells were recorded and analyzed with a digital fluorescence imaging system (InCyt Im2, Intracellular Imaging Inc., Cincinnati, OH, USA) equipped with a light-sensitive CCD camera (Cooke PixelFly, ASI, Eugene, OR, USA). Fura-5F fluorescence was monitored by alternately exciting the dye at 340 and 380nm, and collecting the emission wavelength at 510nm. Changes in are represented as the ratio of fura-5F fluorescence due to excitation at 340nm to that due to excitation at 380nm (ratio=F340/F380). The ratio changes in fields of fura-5F-loaded cells were collected from a multiple regions of interest (ROI), with each ROI representing an individual cell. Typically, about 100 ROIs were monitored per experiment. In all cases, ratio values have been corrected for contributions by autofluorescence, which is measured after treating cells with 10 μm ionomycin and 20 mm MnCl2. Peak ratio values typically were less than 2.0 while Rmax, determined in situ, averaged 6.5, indicating that the ratio values were less than half-saturation and thus essentially proportional to Ca2+ concentration.
To assess SOCE, AP cells were initially bathed in HBSS lacking extracellular calcium and containing 200 μM of the calcium chelator BAPTA and 5 μM of nisoldipine to prevent Ca2+ entry and activation of L-type voltage-gated Ca2+ channels. The cells were then treated with the sarcoplasmic and endoplasmic reticulum Ca2+ ATPase (SERCA) pump blocker thapsigargin (TG, 1 μM). This treatment results in a transient rise in intracellular and the complete depletion the ER calcium stores. After 15 min, the external HBSS bathing cells exchanged to include 5 mM Ca2+ in the presence of nisoldipine. This condition will permit the observation of any rise in intracellular due to TG-induced, store-operated calcium entry (SOCE). The magnitude of the TG-induced calcium release and calcium entry (SOCE) was expressed as a ΔRatio, and represents the peak Ratio with the baseline Ratio subtracted.
To estimate the size of the releasable Ca2+ store, we monitored the magnitude of the signal evoked by the Ca2+ ionophore ionomycin (1 μM), which was added to AP cells in the absence of extracellular Ca2+ [3]. A ΔRatio for the resulting transient response was calculated as described above. Observations of spontaneous oscillations were performed over a period of at least 10 min. Cells displaying significant changes in during the recording period were pooled as a fraction of oscillating cells. When measuring the responses of AP cells to HRHs, a protocol was followed whereby cells were stimulated sequentially with mouse TRH, GHRH, LHRH and CRH. All HRHs were tested at concentrations of 10 nM or 1 μM. For analysis of Ca2+ responses in WT and Orai1−/− mice we performed 57 independent experiments and analyzed about 5.000 individual cells. For analysis of Ca2+ responses in WT and heptaTRPC KO mice we performed 45 independent experiments and analyzed more than 4.000 individual cells.
2.5. Analysis of intracellular calcium signals
An R package developed by Elena Hernando-Pérez and Enrique Pérez-Riesgo in our lab and named CimageD4 v1.10.1 is available at https://github.com/emodoro/CimageD4. This software permits the analysis of many parameters elicited from calcium imaging experiments. These include measuring the peak and areas of responses within each individual AP cell in a particular experiment, and the identification and removal of ‘outliers’ within each experimental under a multivariate criterion. The software package summarizes the peak and areas values for each experiment. As spontaneous oscillations in AP cells are quite variable, it is not simple to measure amplitude and/or frequency of oscillations. We have developed previously a parameter termed oscillations index [3,36]. This parameter is computed as the average of all changes in (in absolute values) during a period of time. OI, therefore, is a parameter influenced by both the amplitude and the frequency of oscillations. Cells showing no oscillations had OI values lower than 0.05.
2.6. Statistics
Data are presented as mean ± SEM. When only two means were compared, Student’s t-test was used. For more than two groups, statistical significance of the data was assessed by one-way ANOVA. Differences were considered significant at p < 0.05. To compare the effects of channel removal on Ca2+ responses to HRHs, a Completely Randomized Design (CRD) has been set up. Regarding the assignment of treatments to experimental units, in order to control the experimental error as much as possible, all experiments were carried out on a pairwise basis, such that each stimulus is applied to both WT and KO samples at the same time, and experimental day is used as blocking factor (Complete Block Designs). Error rates were controlled using the Bonferroni method which controls the Family-Wise Error Rate, or the Benjamini-Hochberg method which controls the False Discovery Rate. The signification level from which we reject the null hypothesis is 0.05. Outlier detection was carried out by Multivariate Criteria employing Mahalanobis distance which takes into account the correlation structure between different variables, which are the height and areas of all different response variables measured from each experiment (hypothalamic factors, or calcium stimulus), in addition to basal fluorescence. After outlier elimination, the median response of all measurement units within the experimental unit is employed as summary response for each experimental unit.
3. Results
3.1. Contribution of Orai1 to store-operated Ca2+ entry and spontaneous Ca2+ oscillations in mouse anterior pituitary cells
The presence of SOCE was tested in AP cells isolated from WT and Orai1 KO mice by use of the SERCA blocker thapsigargin (TG). As described in the Methods section, AP cells isolated from WT mice were treated with TG in the absence of extracellular Ca2+, resulting in a transient rise in due to passive leak of Ca2+ from the ER (Fig. 1A). After 15 min, extracellular Ca2+ (5 mM) was restored to the extracellular medium and resulted in a sustained increase in , which was inhibited by 2 μM Gd3+ (Fig. 1A). The sustained signal induced by TG, and blocked by Gd3+, is indicative of SOCE. We repeated the same experiment in AP cells isolated from Orai1KO mice. In these experiments, while TG induced a similar (Fig. 1A & B), transient rise in the absence of extracellular calcium, the rise in observed in the presence of extracellular Ca2+ was lost (Fig. 1A & C). Indeed, the residual entry signal observed was similar to that observed in control experiments with Orai1 KO cells not treated with TG.
Fig. 1.

Contribution of Orai1 channels to store-operated Ca2+ entry and Ca2+ store content in mouse AP cells.
Isolated AP cells from male WT and Orai1−/− mice were subjected to fluorescence imaging for monitoring SOCE and calcium store content. A. Cells were treated with thapsigargin 1 μM (TG) in Ca2+ free medium containing 1 μM nisoldipine. Then, medium containing 5 mM Ca2+ (Ca5) was added to estimate SOCE. Finally, medium containing 2 μM Gd3+ was added to prevent SOCE. Shown are average (mean ± SEM) recordings of (Ratio F340/F380) (black trace, 100 cells wt; gray trace 95 cells Orai1−/−) from two representative experiments. B. Bars show average (mean ± SEM) values of ΔRatio increases induced by addition of thapsigargin in Ca2+ free medium. C. Bars show average (mean ± SEM) values of ΔRatio increases induced by addition of extracellular Ca2+ to cells treated with thapsigargin (TG) or untreated cells (−TG). Data in B and C are from n = 5 experiments in WT mice (237 cells) and n = 6 experiments in KO mice (332 cells). D. Cells were treated with 1 μM ionomycin (TG) in Ca2+ free medium to assess Ca2+ store content. Average recordings (mean ± SEM) of (Ratio F340/F380) of AP cells from WT (black trace, 90 cells) and Orai1−/− (gray trace, 81 cells) mice in two representative experiments. E. Average (mean ± SEM) values of ΔRatio increases induced by ionomycin in Ca2+ free medium in AP cells from wt and Orai1−/− (KO) mice. n = 6 experiments (wt, 331 cells) and n = 6 experiments (Orai1−/−, 313 cells). *p < 0.05.
The transient response induced by TG was similar in AP cells from WT and Orai1KO mice (Fig. 1C) suggesting that Ca2+ store content is similar in AP cells derived from these mice. To confirm this data, we measured the effects of the Ca2+ ionophore ionomycin in Ca2+ free medium, the transient nature of which is indicative of Ca2+ store (Fig. 1D). Consistently, we found that release of Ca2+ from intracellular stores evoked by ionomycin was similar in AP cells from WT and Orai1KO cells (Fig. 1E). These results indicate that while the expression of Orai1 is required for SOCE in mouse AP cells, the lack of Orai1 does not reduce Ca2+ store content.
Mouse AP cells, just like rat and human AP cells, display spontaneous Ca2+-oscillations that are caused by spontaneous firing of Ca2+ action potentials and opening of voltage-gated Ca2+ channels (VOCCs), particularly L-type VOCCs. We tested whether the expression of Orai1 channels could be involved in the firing of spontaneous Ca2+ oscillations in mouse AP cells. Fig. 2A shows representative recordings of spontaneous Ca2+ oscillations in single AP cells from WT and Orai1KO mice, respectively. We found that a fraction of mouse AP cells display no spontaneous oscillations in WT and Orai1−/− animals. The remaining cells displayed different types of oscillations with variable frequency and amplitude (Fig. 2A). Both oscillating and non-oscillating cells are observed in the same cell cultures. The fraction of cells showing spontaneous oscillations was similar in cell cultures from WT and Orai1KO animals (Fig. 2B). To quantify oscillations, we calculated the oscillations index (OI) for oscillating cells as reported previously [3,36]. This parameter reflects the amplitude and/or frequency of oscillations. We found that OI was similar for oscillating AP cells from WT and Orai1−/− mice suggesting that lack of Orai1 channels does not contribute to spontaneous oscillations in mouse AP cells.
Fig. 2.

Contribution of Orai1 channels to spontaneous oscillations in mouse AP cells.
Isolated AP cells from wt and Orai1−/− mice were subjected to fluorescence imaging for monitoring spontaneous oscillations. A. Representative recordings of resting Ratio F340/F380 in 6 individual AP cells from WT (left) and 6 individual AP cells from Orai1−/− (right) mice. Paired recordings are representative examples of individual cells present in the same microscopic field showing no oscillations (flat recordings) and different oscillations. B. Bars show average (mean ± SEM) fraction of AP cells (%) displaying spontaneous oscillations. C. Bars show average values (mean ± SEM) of oscillations index (average changes in in absolute values during the recording period) in the fraction of oscillating AP cells from wt and KO mice. Data in B and C are from n = 9 experiments in WT mice (711 cells) and n = 10 experiments in Orai1−/− mice (932 cells).
3.2. Contribution of TRPC channels to store-operated Ca2+ entry and spontaneous Ca2+ oscillations in mouse anterior pituitary cells
TRPC channels have been suggested to be involved in SOCE in different cell types. Most data have been derived from studies in which a single TRPC channel is silenced or knocked-down. However, since multiple TRPC channels are often expressed in the same cell, interpretation of such results is complex. In this study, we have utilized the heptaTRPC KO mice in which all seven TRPC channels (from TRPC1 to TRPC7) were knocked-down. This would allow us to unambiguously investigate the contribution of TRPC channels to SOCE in mouse AP cells.
As described for the experiments in Fig. 1, we used fura-5F loaded AP cells isolated from WT and heptaTRPC KO mice to observe the effects of TG in the absence and presence of extracellular Ca2+ (Fig. 3). As can be seen in Fig. 3A, the pattern of responses induced by TG-treatment was indistinguishable between AP cells isolated from WT or heptaTRPC KO mice.
Fig. 3.

Contribution of TRPC channels to store-operated Ca2+ entry and Ca2+ store content in mouse AP cells.
Isolated AP cells from male WT and heptaTRPC KO mice were subjected to fluorescence imaging for monitoring SOCE and calcium store content. A. Cells were treated with thapsigargin 1 μM (TG) in Ca2+ free medium containing 1 μM nisoldipine. Then, medium containing 5 mM Ca2+ (Ca5) was added to estimate SOCE. Finally, medium containing 2 μM Gd3+ was added to prevent SOCE. Shown are average (mean ± SEM) recordings of (Ratio F340/F380) (black trace, 90 cells wt; gray trace 89 cells heptaTRPC KO) from two representative experiments. B. Bars show average (mean ± SEM) values of ΔRatio increases induced by addition of thapsigargin in Ca2+ free medium. C. Bars show average (mean ± SEM) values of ΔRatio increases induced by addition of extracellular Ca2+ to cells treated with thapsigargin (TG) or untreated cells (−TG). Data in B and C are from n = 6 experiments in WT mice (343 cells) and n = 6 experiments in KO mice (501 cells). D. Cells were treated with 1 μM ionomycin (TG) in Ca2+ free medium to assess Ca2+ store content. Average recordings (mean ± SEM) of (Ratio F340/F380) of AP cells from WT (black trace, 62 cells) and heptaTRPC KO (gray trace, 80 cells) mice in two representative experiments. E. Average (mean ± SEM) values of ΔRatio increases induced by ionomycin in Ca2+ free medium in AP cells from wt and heptaTRPC KO mice. n = 4 experiments (wt, 147 cells) and n = 4 experiments (heptaTRPC KO, 262 cells). *p < 0.05.
Analysis of the transient TG-induced response in the absence of extracellular Ca2+ is summarized in Fig. 3B, and the TG-induced Ca2+ entry response is summarized in Fig. 3C. The Ca2+ entry signal observed in the absence of TG was also similar in both AP cell populations (Fig. 3C). Moreover, Ca2+ store content assessed by ionomycin-induced transients (Fig. 3D) was also similar in AP cells from WT and heptaTRPC KO mice (summarized in Fig. 3E). These results suggest that expression of TRPC proteins do not contribute neither to SOCE nor the Ca2+ store content in mouse AP cells.
We tested next whether the presence or absence of TRPC channel proteins could be involved in the firing of spontaneous oscillations in mouse AP cells. As in the case of Orai1−/− mice, a fraction of WT and heptaTRPC KO cells display no spontaneous oscillations while the remaining cells displayed different types of oscillations with variable frequency and amplitude (Fig. 4A). However, the fraction of AP cells showing no spontaneous oscillations was significantly lower in the heptaTRPC KO mice than in the WT mice (Fig. 4B). In addition, average OI values for oscillating cells are significantly lower in AP cells from the heptaTRPC KO mice than in AP cells from WT animals. These data suggest that, in contrast to Orai1 channels, TRPC channels could contribute to spontaneous oscillations in mouse AP cells.
Fig. 4.

Contribution of TRPC channels to spontaneous oscillations in mouse AP cells.
Isolated AP cells from wt and heptaTRPC KO mice were subjected to fluorescence imaging for monitoring spontaneous oscillations. A. Representative recordings of resting Ratio F340/F380 in 6 individual AP cells from WT mice (left panels) and 6 AP cells from KO mice (right panels). Paired recordings are representative examples of individual cells present in the same microscopic field showing no oscillations (flat recordings) and different oscillations. B. Bars show average (mean ± SEM) fraction of AP cells (%) displaying spontaneous oscillations. C. Bars show average values (mean ± SEM) of oscillations index (average changes in in absolute values during the recording period) in the fraction of oscillating AP cells from wt and KO mice. Data in B and C are from n = 23 experiments in WT mice (2275 cells) and n = 22 experiments in KO mice (1338 cells). *p < 0.05.
3.3. Effect of Orai1 expression on responses induced by hypothalamic releasing hormones in mouse anterior pituitary cells
We assessed the effects of hypothalamic releasing hormones (HRHs) on signals in AP cells isolated from WT and Orai1−/− mice. Fura-5f-loaded AP cells were stimulated sequentially with the four HRHs in standard medium devoid of dihydropyridines. Each HRH increases in a fraction of responsive cells that may depend on age, sex and physiological status of the AP donor as reported previously [11,17]. Fig. 5A and B show representative single-cell recordings of responses to individual HRHs in mouse AP cells from WT and Orai1−/− mice, respectively. The fraction of cells responsive to each HRH is shown in Fig. 5C. The data indicate that the fraction of AP cells responsive to TRH, CRH and LHRH was similar in AP cells from WT and Orai1−/− mice. However, the percent of mouse AP cells responsive to GHRH was slightly but significantly lower in the Orai1−/− mice compared to WT mice.
Fig. 5.
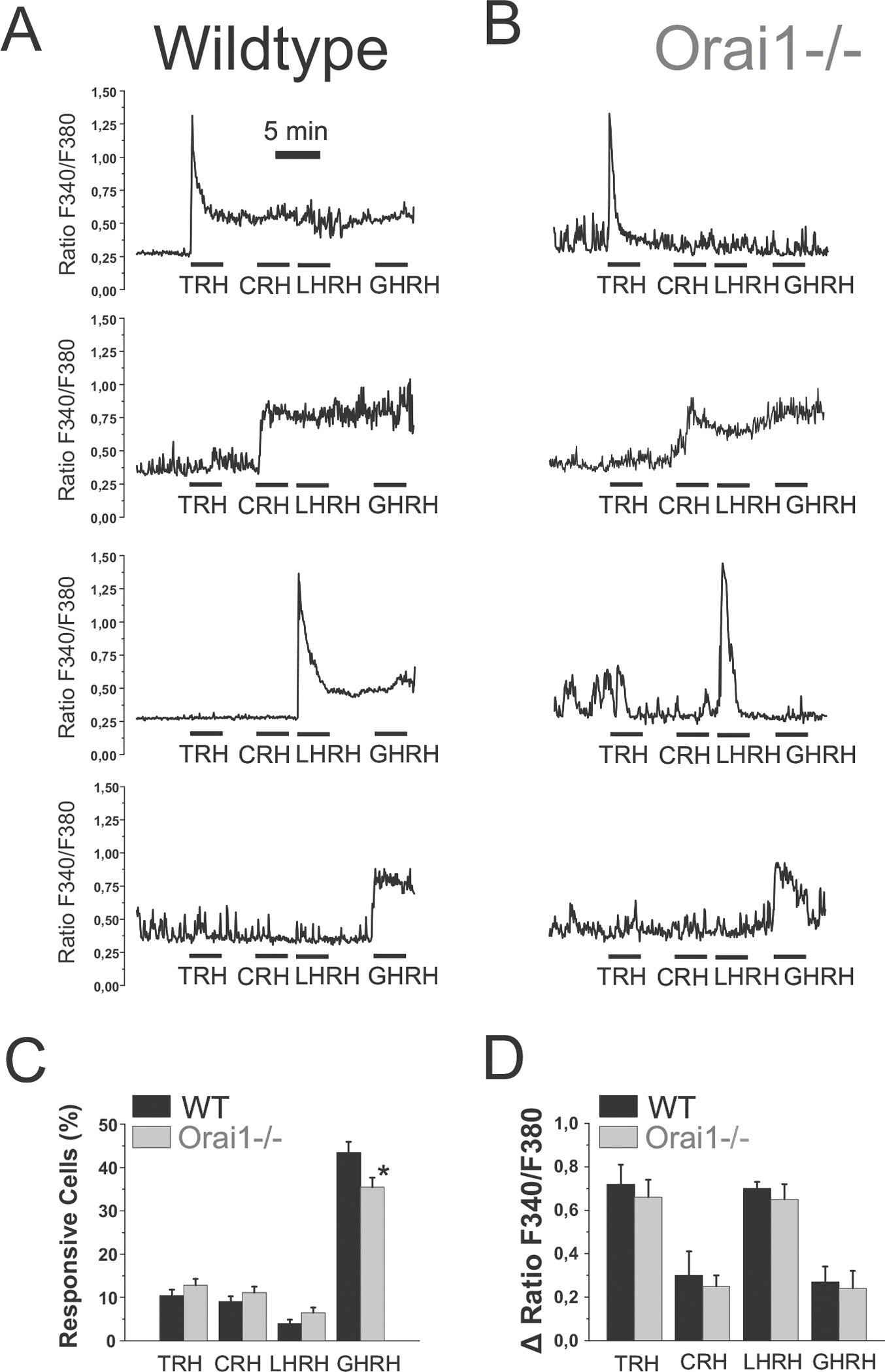
Contribution of Orai1 channels to Ca2+ responses induced by hypothalamic releasing hormones in mouse AP cells.
Isolated AP cells from wt and Orai1−/− mice were subjected to fluorescence imaging for monitoring Ca2+ responses to HRHs. Shown are representative recordings of individual AP cells from WT (A) and Orai1−/− (B) mice responsive to TRH, CRH, LHRH and GHRH. C. Average (mean ± SEM) fraction of AP cells (%) showing clear cut responses to each HRH for WT and Orai1−/− mice. Data are from n = 6 experiments (WT, 564 cells) and n = 6 experiments (KO, 562 cells). D. Average (mean ± SEM) values of the increases in (ΔRatio) induced by each HRH in responsive cells from wt and Orai1−/− mice. Data are from n = 6 experiments (wt) and n = 6 experiments (Orai1−/−). *p < 0.05.
To compare the magnitude of responses to the four HRHs in AP cells from WT and Orai1KO, we calculated the peak response induced by each HRH as a ΔRatio (Fig. 5D). Indeed, the response for all HRHs was similar for WT and Orai1KO cells.
We have previously reported that responses to TRH and LHRH are biphasic corresponding to Ca2+ release (first phase) followed by enhanced Ca2+ entry (second phase) [3,11]. We tested and compared the biphasic effects of TRH and LHRH in AP cells isolated from WT and Orai1−/− mice (Fig. 6). The peak response of each agonist primarily reflects intracellular Ca2+ release, and was similar in WT and Orai1KO cells as shown above. However, responses after 1 to 2 min of stimulation depend on Ca2+ entry, and differences could be observed between WT and Orai1KO AP cells. In summary, for both TRH and LHRH, the number of AP cells exhibiting a sustained Ca2+ entry signal was reduced in Orai1KO cells (Fig. 6C), and the magnitude of the observed Ca2+ entry signal was significantly reduced in Orai1KO cells compared to WT cells (Fig. 6D). These data indicate that expression of Orai1 channels contributes to sustained Ca2+ entry induced by TRH and LHRH in mouse AP cells. In addition, it is known that second phase in response to TRH and LHRH are also associated to increased oscillations. This effect is best appreciated at low agonist concentrations. Consistently, we found that, at low concentrations, TRH induced or increased oscillations and this effect is similar in AP cells from wt and Orai−/− mice (data not shown).
Fig. 6.

Contribution of Orai1 channels to Ca2+ entry induced by TRH and LHRH in mouse AP cells.
Isolated AP cells from WT and Orai1−/− mice were subjected to fluorescence imaging for monitoring Ca2+ entry induced by TRH and LHRH. Shown are average (mean ± SEM) recordings of ΔRatio in AP cells responsive to TRH (A, 15 cells WT, 12 cells Orai1−/−) and LHRH (B, 7 cells WT and 10 cells Orai1−/−), respectively, from two representative experiments. C. Average (mean ± SEM) fraction of HRH responsive AP cells (%) showing second phase in response to TRH and LHRH. Data are from n = 6 experiments in WT mice (55 cells TRH; 19 cells LHRH) and n = 6 experiments in Orai1−/− mice (68 cells TRH; 31 cells LHRH). D. Average (mean ± SEM) values of the increases in (ΔRatio) induced by TRH and LHRH during the second phase of Ca2+ entry in responsive cells from WT and Orai1−/− mice. Data are from the same experiments as in C. *p < 0.05.
3.4. Effect of TRPC expression on responses induced by hypothalamic releasing hormones in mouse anterior pituitary cells
We also assessed the contribution of TRPC channels to responses induced by HRHs in mouse AP cells. Using the protocols described for Fig. 6, we assessed the effects of HRHs in AP cells isolated from WT and heptaTRPC KO mice. As before, AP cells were stimulated sequentially with the four HRHs and representative recordings are shown in Fig. 7A and B. The fraction of AP cells responsive to CRH and LHRH was similar in WT and heptaTRPC KO mice. However, the percent of cells responsive to TRH and GHRH were reduced significantly in heptaTRPC KO mice (Fig. 7C). The magnitude (ΔRatio) of agonist-induced responses was also assessed (Fig. 7D). The responses induced by TRH and GHRH were similar for WT and TRPC KO cells. The increase induced by LHRH was slightly, but significantly lower in heptaTRPC KO cells. However, surprisingly, the response induced by CRH was significantly larger in heptaTRPC KO cells compared to WT.
Fig. 7.

Contribution of TRPC channels to Ca2+ responses induced by hypothalamic releasing hormones in mouse AP cells.
Isolated AP cells from WT and heptaTRPC KO mice were subjected to fluorescence imaging for monitoring Ca2+ responses to HRHs. Shown are representative recordings of individual AP cells from WT (A) and heptaTRPC KO (B) mice responsive to GHRH, TRH, LHRH and CRH. C. Average (mean ± SEM) fraction of AP cells (%) showing clear cut responses to each HRH. Data are from n = 11 experiments (WT, 949 cells) and n = 11 experiments (KO, 914 cells). D. Average (mean ± SEM) values of the increases in (ΔRatio) induced by each HRH in responsive cells from WT and heptaTRPC KO mice. Data are from n = 11 experiments (WT) and n = 11 experiments (heptaTRPC KO). *p < 0.05.
Finally, we also compared the effects of TRH and LHRH on responses in AP cells from WT and heptaTRPC KO mice (Fig. 8A and B, respectively). As can be seen in representative experiments, the peak response to each agonist was similar in AP cells from WT and heptaTRPC KO cells. However, the second phase response to each agonist was differentially affected by lack of TRPC channels. Specifically, Ca2+ entry induced by TRH was reduced in the heptaTRPC KO cells while the entry of Ca2+ induced by LHRH was unaffected (Fig. 8A and B). Nevertheless, as illustrated in Fig. 8C, the fraction of cells showing a sustained Ca2+ entry in response to LHRH was slightly but significantly reduced in AP cells isolated from heptaTRPC KO cells compared to WT. In contrast, the fraction of cells showing a sustained Ca2+ entry in response to TRH was similar in WT and heptaTRPC KO cells.
Fig. 8.

Contribution of TRPC channels to Ca2+ entry induced by TRH and LHRH in mouse AP cells.
Isolated AP cells from wildtype (WT) and heptaTRPC KO mice were subjected to fluorescence imaging for monitoring of Ca2+ entry induced by TRH and LHRH. Shown are average (mean ± SEM) recordings of ΔRatio increases in AP cells responsive to TRH (A, 10 cells WT, 6 cells heptaTRPC KO) and LHRH (B, 6 cells WT and 5 cells heptaTRPC KO), respectively, from two representative experiments. C. Average (mean ± SEM) fraction of HRH responsive AP cells (%) showing second phase in response to TRH and LHRH. Data in WT mice are from n = 11 experiments for TRH (150 cells) and n = 10 experiments for LHRH (45 cells). Data in heptaTRPC KO mice are from n = 11 experiments for TRH (63 cells) and n = 8 experiments of LHRH (55 cells). D. Average (mean ± SEM) values of the increases in (ΔRatio) induced by TRH and LHRH during the second phase of Ca2+ entry in responsive cells from WT and Orai1−/− mice. Data are from the same experiments as in C. *p < 0.05.
To ascertain contribution of TRPC channels to Ca2+ entry induced by each agonist, we have computed the magnitude of the Ca2+ entry response induced by TRH and LHRH in all cells responsive to each agonist (Fig. 8D). The data illustrate that TRH-induced Ca2+-entry is significantly smaller in TRPC KO mice compared to WT. In contrast, no difference is observed for LHRH-induced Ca2+ entry between WT and heptaTRPC KO cells. These results suggest that expression of TRPC channels may play a role in supporting sustained Ca2+ entry induced by TRH. Consistently, oscillatory activity induced by low concentrations of TRH is decreased in mouse AP cells from TRPCKO animals relative to wt mice (data not shown). In contrast, contribution of TRPC channels to Ca2+ entry induced by LHRH is minor or negligible.
4. Discussion
The AP gland plays a pivotal in control of the entire endocrine system by the release of important AP hormones, a process primarily dictated by hypothalamic factors, most particularly HRHs that reach the AP via the hypophyseal portal system [1]. These peptides activate specific receptors in target cells leading to Ca2+ responses and release of specific AP hormones [3,5–8]. It has been previously suggested that store-operated channels (SOCs) and non-selective channels may contribute to Ca2+ responses to HRHs but the molecular evidence has remained elusive. Here we show that Orai1 channels, but not TRPC channels, are essential for thapsigargin-evoked SOCE in mouse AP cells. This does not mean necessarily that TRPC channels play no role in SOCE in AP cells. In fact, TRPC channels are believed to modulate SOCE in some cell types [27]. In addition, we do not know whether removal of TRPC channels might influence expression of other ion channels and regulatory proteins involved in SOCE. Further research is required to address whether this is also the case in AP cells. However, our results clearly indicate that Orai1 channels are mandatory for SOCE in mouse AP cells while TRPC channels are not.
One of the physiological roles of SOCE is to provide a Ca2+ influx pathway for store refilling after Ca2+ release. However, Ca2+ store content is similar in AP cells from Orai1−/− and WT mice suggesting that other Ca2+ entry pathways contribute to store refilling in AP cells. We have no information on whether other Orai channels (Orai2, Orai3) are expressed differentially in mouse AP cells from Orai1 −/− mice. However, the single removal of Orai1 is enough to abolish thapsigargin-evoked SOCE without affecting Ca2+ store content. Nevertheless, removal of Orai1 could influence the kinetics of Ca2+ refilling after stimulation.
About 50 to 60% of all mouse AP cells display spontaneous oscillations that are related to electric activity and VOCCs, thus providing alternative means for store refilling. However, our results show that inhibition of VOCCs with nisoldipine does not affect Ca2+ store content, thus suggesting that VOCCs are also dispensable for Ca2+ store refilling.
Our results indicate a role for TRPC channels, but not Orai1 channels, in spontaneous oscillations in AP cells. Thus, loss of Orai1 channels has no influence on spontaneous oscillations in mouse AP cells or on Ca2+ oscillations induced by TRH. Consistently, we have reported previously that thapsigargin-evoked SOCE does not increase oscillations in GH3 pituitary cells [3]. In contrast, lack of TRPC channels decreases both the percent of AP cells showing spontaneous oscillations and also the frequency and/or amplitude of oscillations without decreasing Ca2+ store content. These data suggest that, in contrast to Orai1 channels, TRPC channels may contribute to AP cell excitability. Whether lack of TRPC channels promotes overexpression of VOCCs or other ion channels involved in AP cell excitability remains to be established. In summary, our results indicate that Orai1 channels are critical for SOCE but do not contribute to spontaneous oscillations in mouse AP cells. Conversely, TRPC channels are dispensable for SOCE but may contribute to spontaneous Ca2+ oscillations in the same cells.
We have investigated also whether Orai1 and TRPC channels contribute to Ca2+ responses elicited by HRHs in mouse AP cells. There are four main HRHs including TRH, LHRH, GHRH and CRH. It is well known that TRH and LHRH evoked a biphasic Ca2+ response. The first phase corresponds to agonist-evoked synthesis of IP3 and the ensuing release of intracellular Ca2+ stores. The second phase corresponds to enhanced Ca2+ entry associated to enhanced electric activity. As TRH and LHRH induce Ca2+ release, it has been proposed that SOCE may contribute as well to the second phase of Ca2+ entry in response to these stimuli. In contrast, GHRH and CRH do not induce Ca2+ release and SOCE should not play a role in Ca2+ responses to these agonists.
TRH is involved in control of TSH and PRL secretion from thyrotropes and mammotropes, respectively. As stated above, TRH induces Ca2+ release followed by enhanced electric activity and Ca2+ entry (second phase) in mamotropes [5]. We proposed long ago that TRH activated not only VOCCs but also SOCE in mammotropes [3]. Here, we provide molecular evidence that both Orai1 and TRPC channels contribute also to Ca2+ entry (second phase) induced by TRH in mouse AP cells. Specifically, removal of Orai1 channels prevents partially Ca2+ entry induced by TRH without influencing the fraction of AP cells responsive to TRH or the release of Ca2+ induced by TRH (first phase). In turn, lack of TRPC channels also decreases Ca2+ entry induced by TRH without influencing Ca2+ release. Therefore, both Orai1 channels and TRPC channels are involved in Ca2+ entry and Ca2+ responses to TRH, thus contributing together with VOCCs to Ca2+ entry induced by TRH. Since Orai1 channels are activated by store-depletion, and TRPC channels also contribute to TRH-induced Ca2+ entry, it is possible that TRPC channels will modulate SOCE evoked by TRH-induced Ca2+ release in mouse AP cells.
Our results invite speculation on the role of Orai1 and TRPC channels in TSH and PRL secretion evoked by TRH. It has been previously reported that non-selective blockers of SOCs inhibit resting electric activity of AP cells and PRL release [37]. In addition, Orai1 channel has been previously involved in lactation and breast cancer [38,39]. During lactation, milk is ejected by way of pulsatile contractions of alveolar units. Interestingly, in mammary glands of Orai1−/− mice, these contractions are infrequent and poorly coordinated and this effect has been attributed to defects of SOCE in the mammary epithelial cells [40]. Whether defective TRH signaling in Orai1KO mice may influence TSH and PRL secretion as well deserves further consideration.
Hypothalamic LHRH activates LHRH receptors in a fraction of AP cells (gonadotropes) promoting gonadotropin release for control of the reproductive axis. LHRH induces Ca2+ release (first phase) followed by oscillations associated to entry of Ca2+ (second phase) [41]. Ca2+ entry during LHRH-induced oscillations involves several ion channels. However, whether Orai1 and TRPC channels contribute to this entry remains unknown. We show here that lack of Orai1 channels decreases partially Ca2+ entry induced by LHRH, but not Ca2+ release or the fraction of AP cells responsive to LHRH. In striking contrast, removal of TRPC channels has no effect on Ca2+ entry induced by LHRH (second phase) but it reduces slightly the fraction of AP cells responsive to LHRH and the fraction of cells showing LHRH-induced second phase. These results indicate that Orai1 channels are involved in Ca2+ entry induced by LHRH in mouse gonadotropes. However, the role of TRPC channels in Ca2+ entry induced by LHRH is minor if any.
There is presently no information on whether Orai1 is involved in gonadotropin release. However, it has been also recently reported that male (but not female) mice lacking Orai1 channels show defective fertility [40]. Specifically, Orai1−/− male mice are sterile and have severe defects in spermatogenesis, with prominent deficiencies in mid- to late-stage elongating spermatid development. These data suggest an essential in vivo role for Orai1 channels in male reproductive function and identify Orai1 channels as potential non-steroidal regulators of male fertility [40]. The suggested mechanism includes SOCE downregulation in mouse testis cells and lack of proper sperm development or spermatogenesis. Whether defective Ca2+ signaling to LHRH in mouse gonadotropes contributes to male fertility defects warrants also further research.
Hypothalamic GHRH is involved in GH secretion by somatotropes. In contrast to TRH and LHRH, activation of GHRH receptors does not induce release of Ca2+ from intracellular stores. Instead, GHRH enhances electrical activity and Ca2+ entry by a not fully understood mechanism [2,7]. Here we show that lack of Orai1 channels does not influence Ca2+ entry induced by GHRH in mouse AP cells. However, it decreased slightly, but significantly, the fraction of AP cells responsive to GHRH. It has been previously reported that Orai1−/− mice show short stature that has been attributed to dysfunction of skeletal muscle development [42]. Whether decreased responsiveness to GHRH contributes to growth deficits in the Orai1−/− mice remains to be established. More importantly, lack of TRPC channels decreased largely the fraction of AP cells responsive to GHRH without changing the rise in induced by this agonist in the responsive population. These data suggest an important role of TRPC channels in control of GHRH receptor expression and/or signaling in mouse somatotropes.
The secretion of ACTH from corticotropes is a key component in the endocrine response to stress. The resting potential of corticotropes is set by the basal activities of TWIK-related K+ (TREK)-1 channels. CRH, the major ACTH secretagogue, closes background TREK-1 channels via the cAMP-dependent pathway, resulting in depolarization and a sustained rise in [9]. We show here that removal of Orai1 channels does not influence either the fraction of CRH responsive cells or the rise in induced by CRH, thus excluding any role for Orai1 channels in ACTH secretion induced by CRH. Consistently, blockers of SOCs do not affect ACTH release induced by CRH [20]. However, these results do not exclude that Orai1 channels may contribute to ACTH secretion induced by Ca2+-releasing agonists like arginine vasopressin [20]. Nevertheless, in contrast to previous reports [20], our results indicate that SOCE in corticotropes involves Orai1 channels but not TRPC channels. Surprisingly, we found that lack of TRPC channels increased slightly but significantly the rise in induced by CRH without affecting the fraction of cells responsive to this agonist. Further research is required to address this intriguing point.
In summary, these results indicate the critical role of Orai1 channels in SOCE and Ca2+ entry induced by TRH and LHRH in mouse AP cells. In addition, our results also suggest a role for TRPC channels in spontaneous oscillations in mouse AP cells and Ca2+ responses to several HRHs, particularly TRH and GHRH. The present study focuses solely on AP cells, and therefore we cannot extrapolate it to the more general question of the relative importance of TRPC and Orai channels in Ca2+ entry mechanisms elsewhere. Additional work will be necessary to establish the roles of these two channel types on store-operated and receptor-operated channels in other tissues. Hopefully, the availability of these and other mouse models will facilitate the important identification of channels underlying Ca2+ signaling in a variety of important physiological systems.
Transparency document
The Transparency document associated with this article can be found, in online version.
Supplementary Material
Acknowledgements
This work was supported by grants BFU2012–37146 and BFU2015–70131R from MINECO, (Spain), the Salvador de Madariaga visiting fellowship research program (Spain) and the Intramural Research Program of the National Institutes of Health (NIH) (USA).
We are very grateful to Dr. Lutz Birnbaumer (Institute for Biomedical Research (BIOMED), School of Medical sciences, Catholic University of Argentina, Buenos Aires, Argentina) for providing the HeptaKO TRPC mice, which were produced in his laboratory at the National Institute of Environmental Health Sciences in Research Triangle Park, NC USA.
Footnotes
This article is part of a Special Issue entitled: ECS Meeting edited by Claus Heizmann, Joachim Krebs and Jacques Haiech.
References
- [1].Perez-Castro C, Renner U, Haedo MR, Stalla GK, Arzt E, Cellular and molecular specificity of pituitary gland physiology, Physiol. Rev. 92 (2012) 1–38. [DOI] [PubMed] [Google Scholar]
- [2].Thorner MO, Holl RW, Leong DA, The somatotrope: an endocrine cell with functional calcium transients, J. Exp. Biol. 139 (1988) 169–179. [DOI] [PubMed] [Google Scholar]
- [3].Villalobos C, García-Sancho J, Capacitative Ca2+ entry contributes to the calcium influx induced by thyrotropin-releasing hormone (TRH) in GH3 pituitary cells, Pflugers Arch. 430 (1995) 923–935. [DOI] [PubMed] [Google Scholar]
- [4].Villalobos C, Núñez L, Frawley LS, García-Sancho J, Sánchez A, Multi-responsiveness of single anterior pituitary cells to hypothalamic releasing hormones: a cellular basis for paradoxical secretion, Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 14132–14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barros F, Villalobos C, García-Sancho J, del Camino D, de la Peña P, The role of the inwardly rectifying K+ current in resting potential and thyrotropin-releasing-hormone-induced changes in cell excitability of GH3 rat anterior pituitary cells, Pflugers Arch. 426 (1994) 221–230. [DOI] [PubMed] [Google Scholar]
- [6].Rawlings SR, Hoyland J, Mason WT, Calcium homeostasis in bovine somatotrophs: calcium oscillations and calcium regulation by growth hormone-releasing hormone and somatostatin, Cell Calcium 12 (1991) 403–414. [DOI] [PubMed] [Google Scholar]
- [7].Naumov AP, Herrington J, Hille B, Actions of growth-hormone-releasing hormone on rat pituitary cells: intracellular calcium and ionic currents, Pflugers Arch. 427 (1994) 414–421. [DOI] [PubMed] [Google Scholar]
- [8].Zemkova H, Tomić M, Kucka M, Aguilera G, Stojilkovic SS, Spontaneous and CRH-induced excitability and calcium signaling in mice corticotrophs involves sodium, calcium, and cation-conducting channels, Endocrinology 157 (2016) 1576–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tse AK, Tse Lee FW, Ca2+ signaling and exocytosis in pituitary corticotropes, Cell Calcium 51 (2012) 253–259. [DOI] [PubMed] [Google Scholar]
- [10].Villalobos C, García-Sancho Núñez J, Functional glutamate receptors in a subpopulation of anterior pituitary cells, FASEB J. 10 (1996) 654–660. [DOI] [PubMed] [Google Scholar]
- [11].Núñez L, Villalobos C, Senovilla L, García-Sancho J, Multifunctional cells of mouse anterior pituitary reveal a striking sexual dimorphism, J. Physiol. Lond. 549 (2003) 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Villalobos C, Núñez L, García-Sancho J, Phenotypic characterization of multifunctional somatotropes, mammotropes and gonadotropes of the mouse anterior pituitary, Pflugers Arch. 449 (2004) 257–264. [DOI] [PubMed] [Google Scholar]
- [13].Villalobos C, Núñez L, García-Sancho J, Anterior pituitary thyrotropes are multifunctional cells, Am. J. Phys. 287 (2004) E1166–E1170. [DOI] [PubMed] [Google Scholar]
- [14].Senovilla L, Núñez L, de Campos JM, de Luis DA, Romero E, Sánchez A, García-Sancho J, Villalobos C, Multifunctional cells in human pituitary adenomas: implications for paradoxical secretion and tumorigenesis, J. Clin. Endocrinol. Metab. 89 (2004) 4545–4552. [DOI] [PubMed] [Google Scholar]
- [15].Senovilla L, Núñez L, de Campos JM, de Luis DA, Romero E, García-Sancho J, Villalobos C, Single-cell phenotypic characterization of human pituitary GHomas and non-functioning adenomas based on hormone content and calcium responses to hypothalamic releasing hormones, Front. Oncol. 5 (2015) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Senovilla L, García-Sancho J, Villalobos C, Changes in expression of hypothalamic releasing hormone receptors in individual rat anterior pituitary cells during maturation, puberty and senescence, Endocrinology 146 (2005) 4627–4634. [DOI] [PubMed] [Google Scholar]
- [17].Senovilla L, Núñez L, Villalobos C, García-Sancho J, Rapid changes in anterior pituitary cell phenotypes in male and female mice after acute cold stress, Endocrinology 149 (2008) 2159–2167. [DOI] [PubMed] [Google Scholar]
- [18].Carew MA, Mason WT, Control of Ca2+ entry into rat lactotrophs by thyrotrophin-releasing hormone, J. Physiol. Lond. 486 (1995) 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li YX, Stojilković SS, Keizer J, Rinzel J, Sensing and refilling calcium stores in an excitable cell, Biophys. J. 72 (1997) 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yamashita M, Oki Y, Iino K, Hayashi C, Yogo K, Matsushita F, Sasaki S, Nakamura H, The role of store-operated Ca2+ channels in adrenocorticotropin release by rat pituitary cells, Regul. Pept. 156 (2009) 57–64. [DOI] [PubMed] [Google Scholar]
- [21].Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., T. Meyer, STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx, Curr. Biol. 15 (2005) 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A, A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature 441 (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [23].Hoth M, Niemeyer BA, The neglected CRAC proteins: Orai2, Orai3, and STIM2, Curr. Top. Membr. 71 (2013) 237–271. [DOI] [PubMed] [Google Scholar]
- [24].Hardie RC, Minke B, Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: implications for phosphoinositide-mediated Ca2+ mobilization, Trends Neurosci. 16 (1993) 371–376. [DOI] [PubMed] [Google Scholar]
- [25].Liao Y, Abramowitz J, Birnbaumer L, The TRPC family of TRP channels: roles inferred (mostly) from knockout mice and relationship to ORAI proteins, Handb. Exp. Pharmacol. 223 (2014) 1055–1075. [DOI] [PubMed] [Google Scholar]
- [26].Parekh AB, Putney JW Jr., Store-operated calcium channels, Physiol. Rev. 85 (2005) (2005) 757–810. [DOI] [PubMed] [Google Scholar]
- [27].Ong HL, de Souza LB, Ambudkar IS, Role of TRPC channels in store-operated calcium entry, Adv. Exp. Med. Biol. 898 (2016) 87–109. [DOI] [PubMed] [Google Scholar]
- [28].Sobradillo D, Hernández-Morales M, Ubierna D, Moyer MP, Núñez L, Villalobos C, A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells, J. Biol. Chem. 289 (2014) 28765–28782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tomić M, Kucka M, Kretschmannova K, Li S, Nesterova M, Stratakis CA, Stojilkovic SS, Role of nonselective cation channels in spontaneous and protein kinase A-stimulated calcium signaling in pituitary cells, Am. J. Physiol. Endocrinol. Metab 301 (2011) E370–E379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Beck V, Götz S, Qiao P, Weissgerber V, Flockerzi M, Boehm Freichel U, Functional characterization of transient receptor potential (TRP) channel C5 in female murine gonadotropes, Endocrinology 158 (2017) 887–902. [DOI] [PubMed] [Google Scholar]
- [31].Götz V, Qiao S, Beck A, Boehm U, Transient receptor potential (TRP) channel function in the reproductive axis, Cell Calcium 67 (2017) 138–147. [DOI] [PubMed] [Google Scholar]
- [32].Calvo-Rodríguez M, García-Durillo M, Villalobos C, Núñez L, In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry in rat hippocampal neurons, Biochim. Biophys. Acta, Mol. Cell Res 1863 (2016) 2637–2649. [DOI] [PubMed] [Google Scholar]
- [33].Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP, Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels, Nat. Immunol. 9 (2008) 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sexton JE, Desmonds T, Quick K, Taylor R, Abramowitz J, Forge A, Kros CJ, Birnbaumer L, Wood JN, The contribution of TRPC1, TRPC3, TRPC5 and TRPC6 to touch and hearing, Neurosci. Lett. 610 (2016) 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bird GS, Putney JW Jr., Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells, J. Physiol. Lond. 562 (2005) 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Núñez L, Sánchez A, Fonteriz RI, García-Sancho J, Mechanisms for synchronous calcium oscillations in cultured rat cerebellar neurons, Eur. J. Neurosci. 8 (1996) 192–201. [DOI] [PubMed] [Google Scholar]
- [37].Kučka M, Kretschmannová K, Stojilkovic SS, Zemková H, Tomić M, Dependence of spontaneous electrical activity and basal prolactin release on nonselective cation channels in pituitary lactotrophs, Physiol. Res 61 (2012) 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, Monteith GR, ORAI1-mediated calcium influx in lactation and in breast cancer, Mol. Cancer Ther. 10 (2011) 448–460. [DOI] [PubMed] [Google Scholar]
- [39].Davis FM, Janoshazi A, Janardhan KS, Steinckwich N, D’Agostin DM, Petranka JG, Desai PN, Roberts-Thomson SJ, Bird GS, Tucker DK, Fenton SE, Feske S, Monteith GR, Putney JW Jr., Essential role of Orai1 store-operated calcium channels in lactation, Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 5827–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Davis FM, Goulding EH, D’Agostin DM, Janardhan KS, Cummings CA, Bird GS, Eddy EM, Putney JW Jr., Male infertility in mice lacking the store-operated Ca2+ channel Orai1, Cell Calcium 59 (2016) 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stojilkovic SS, Bjelobaba I, Zemkova H, Ion channels of pituitary gonadotrophs and their roles in signaling and secretion, Front. Endocrinol. 8 (2017) 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lyfenko AD, Dirksen RT, Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1, J. Physiol. Lond. 586 (2008) 4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.