

Abstract
SARS-CoV-2 is the causative agent behind the COVID-19 pandemic. The main protease (Mpro, 3CLpro) of SARS-CoV-2 is a key enzyme that processes polyproteins translated from the viral RNA. Mpro is therefore an attractive target for the design of inhibitors that block viral replication. We report the diastereomeric resolution of the previously designed SARS-CoV-2 Mpro α-ketoamide inhibitor 13b. The pure (S,S,S)-diastereomer, 13b-K, displays an IC50 of 120 nM against the Mpro and EC50 values of 0.8–3.4 μM for antiviral activity in different cell types. Crystal structures have been elucidated for the Mpro complexes with each of the major diastereomers, the active (S,S,S)-13b (13b-K), and the nearly inactive (R,S,S)-13b (13b-H); results for the latter reveal a novel binding mode. Pharmacokinetic studies show good levels of 13b-K after inhalative as well as after peroral administration. The active inhibitor (13b-K) is a promising candidate for further development as an antiviral treatment for COVID-19.
Introduction
An outbreak of a new coronavirus, then called 2019-nCoV, was detected in December 2019 in Wuhan, China.1−3 The virus was later renamed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and as of May 2022 has caused >520 million cumulative cases registered by the World Health Organization and >6 million deaths worldwide.4 Symptoms displayed by individuals infected with SARS-CoV-2 include fever, dry cough, breathing difficulties (dyspnoea), and loss of taste or smell. Severe cases can result in pneumonia, acute respiratory distress syndrome, and death.2,5 In addition, up to 84% of severe COVID-19 cases show neurological symptoms such as confusion, loss of consciousness, or epileptic seizures.6 SARS-CoV-2 was genetically identified as a betacoronavirus with 79.6% sequence identity shared with the causative agent of the 2003 SARS epidemic, SARS-CoV.2 Intensive research efforts have led to the present availability of a number of vaccines.7 However, a substantial number of mutations have emerged and continue to emerge within the SARS-CoV-2 spike protein, including escape mutations against which some of the vaccines offer less protection. This development, together with the sub-100% efficacy of available vaccines and the challenge of global vaccine administration, makes the development of effective antivirals an important precondition for the effective control and treatment of SARS-CoV-2 and related viral infections.
The coronavirus main protease (Mpro, also known as 3C-like protease, 3CLpro) is a cysteine protease responsible (together with the papain-like protease) for processing the two polyproteins (pp1a and pp1ab) that are translated from the viral RNA. Cleavage of the viral polyproteins by Mpro generates 12 nonstructural proteins (Nsp5–Nsp16) including proteins essential for viral replication, such as the RNA-dependent RNA polymerase (RdRp) Nsp12 with its associated processivity factors Nsp7 and Nsp8, the helicase Nsp13, and the two methyltransferases Nsp14 and Nsp16.8 The SARS-CoV-2 Mpro also cleaves important signaling proteins of the host cell such as NEMO, the essential modulator of nuclear factor-κB, thereby causing some of the neurologic symptoms of COVID-199 and blocking type-I interferon production.10 Because of its important role in viral polyprotein and host protein processing, inhibition of Mpro has proven to be a viable strategy for the development of a coronavirus therapeutic.11
There is a body of literature disclosing the development of peptidomimetic and small-molecule covalent and non-covalent inhibitors of the main protease of coronaviruses.12−19 The crystal structure of the SARS-CoV-2 main protease was reported in 2020, revealing structural features that have helped guide the design of potent SARS-CoV-2 peptidomimetic inhibitors.20 One of these inhibitors, 13b, displays potent inhibition of SARS-CoV-2 Mpro and possesses favorable pharmacokinetic properties, providing a potential route toward an antiviral treatment for COVID-19.20
Herein, we build on the discovery of 13b by reporting a highly potent α-ketoamide inhibitor, 13b-K, through diastereomeric resolution of the previously disclosed compound. We describe the crystal structures of (S,S,S)-13b (13b-K), and its near-inactive diastereomer (R,S,S)-13b (13b-H), in complex with the SARS-CoV-2 Mpro. Furthermore, we assess the antiviral activity of 13b-K in various SARS-CoV-2-infected cell types. Moreover, we report the detailed pharmacokinetic characterization of 13b-K and demonstrate peroral bioavailability.
Results
Synthesis of 13b was conducted according to a previously described route,20 summarized in Scheme 1. The final compound 11, obtained following column chromatography, had a 1H NMR spectrum matching the published spectrum for 13b(20) (Figure S1). There were two sets of resonances, most clearly evident at 5.6–5.7 ppm (corresponding to the methine proton at the P2 position) and 8.9–9.0 ppm (corresponding to the amide proton adjacent to the P2 position), that subsequently condensed into a single set of peaks at 5.6 ppm (t, 1H) and 8.9 ppm (d, 1H) upon crystallization of 11. The product from the mother liquor was also isolated and purified. This purification sequence afforded two separate compounds 12 and 13, exhibiting 1H NMR spectra that were broadly consistent with the structure of the parent compound but most clearly distinct from each other at the regions highlighted in Figure S1.
Scheme 1. Synthesis and structures of 11–13.

Reagents and conditions: (a) H2SO4, NaNO2, H2O, 0–5 °C; (b) SOCl2, MeOH, 0 °C; (c) Tf2O, 2,6-lutidine, DCM, 0 °C; (d) tert-butyl (2-oxo-1,2-dihydropyridin-3-yl)carbamate, NaH, THF, 0 °C; (e) LiOH.H2O, MeOH, H2O, RT; (f) methyl (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoate hydrochloride, EDC·HCl, HOBt, TEA, DCM, 0 °C; (g) NaBH4, MeOH, RT; (h) DMP, NaHCO3, DCM, RT; (i) benzyl isocyanide, AcOH, DCM, RT; (j) LiOH.H2O, MeOH, H2O, RT; (k) DMP, NaHCO3, DCM, RT.
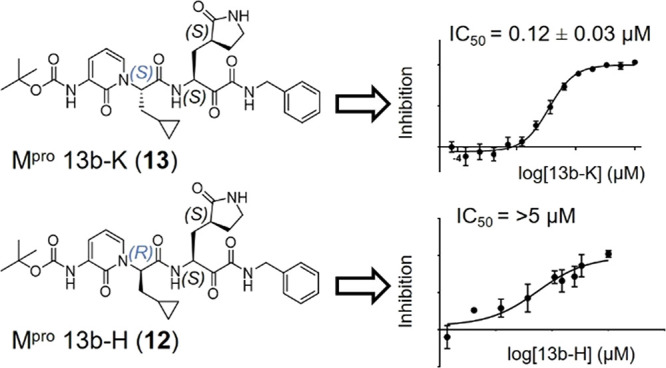
The complexity in the published 13b1H NMR spectrum (and matched by 11 synthesized here) had previously been ascribed to the existence of rotamers in the solution. However, assignment of the 1H NMR spectra for the two compounds 12 and 13 that were isolated following crystallization of 11 suggested that they were diastereomers differing at a single chiral center (Figure 1a). To confirm this hypothesis, we solved the structure of 12 by X-ray crystallography, establishing the absolute stereochemical configuration as (R) at the P2 center (Figure 1b and Figure S2).
Figure 1.

Stereochemical configuration of 13b diastereomers. (A) 1H NMR spectra of 13b-H (12) (upper) and 13b-K (13) (lower). Peaks that differ most clearly by chemical shift between the two compounds are highlighted and correspond to the highlighted protons on the chemical structures (B) for each diastereomer. (B) Structures of 13b-H (12) and 13b-K (13) with absolute stereochemistry indicated.
Initially, the loss of optical integrity at this center was rationalized to have happened in the reaction between triflate 3 and the pyridone during the construction of 4 (Scheme 1) as chiral HPLC analysis has unequivocally revealed 5 to in fact have been a 1:1 mixture of enantiomers. The displacement reaction itself would be expected to be stereospecific, with complete inversion of the stereocenter being invoked in an SN2 reaction mechanism. Nevertheless, racemization could still be induced at this stage by the presence of an excess of the strong base sodium hydride with an extended reaction time. During subsequent investigations employing sub-stoichiometric amounts of base, although we observed a varyingly reduced degree of racemization, complete stereochemical control could still not readily be obtained. We therefore developed an efficient alternative route to 4 by implementing a Mitsunobu reaction under neutral conditions. That notwithstanding, racemization was still not avoided as isolation of 6 after saponification and amide coupling was found to be a mixture of diastereomers (Scheme S1). This at least implies that racemization must also occur during the saponification reaction, a conclusion supported by the finding in our parallel synthetic work without the pyridone functionality that coupling of hydroxy acid 14 with the same γ-lactam afforded 15 as the expected single diastereomer (Scheme S2), which could then be cleanly deprotected to give analogue 16 ready for further elaboration. Thus, we hypothesize that the pyridone moiety facilitates racemization under basic conditions through stabilization of the enolate via an aza-ylide resonance, as depicted in Figure 2. In summary, the studies infer that defined stereoisomers like 12 or 13 either need to be prepared by a base-free stereospecific route or by a separation approach like the one described above.
Figure 2.

Stabilization of enolate via aza-ylide resonance.
The biochemical activity of both diastereomers, as well as a 1:1 mixture of the two, was assessed against purified recombinant SARS CoV-2 Mpro using a Förster resonance energy transfer (FRET)-based cleavage assay. Compounds 12 and 13 inhibited SARS-CoV-2 Mpro with IC50 values of >5 μM and 0.12 ± 0.03 μM, respectively, while the 1:1 mixture of the two had an IC50 of 0.30 ± 0.09 μM (Figure S3). Compound 13 ((S,S,S)-13b, hereinafter 13b-K) was therefore significantly more potent than the mixture of diastereomers previously annotated as 13b (IC50 = 0.67 ± 0.18 μM20 and IC50 = 0.38 ± 0.04 μM after improving the standard conditions for the assay; see the Experimental Section). Compound 12 ((R,S,S)-13b, hereinafter 13b-H) is at least ∼50-fold less active than 13b-K in this assay.
The series of α-ketoamides from which compound 13b is derived display a reversible covalent mechanism of action. Nucleophilic attack onto the inhibitor α-ketoamide group by Mpro Cys145 results in formation of a thiohemiketal (with the (S) configuration at the α-carbon) at the protease’s catalytic center.20 The P2-cyclopropylmethyl group of 13b occupies the S2 pocket of the protease in the (S) configuration.20,21 In order to rationalize the differential activity of 13b-K and 13b-H, we obtained co-crystal structures of each in complex with SARS-CoV-2 Mpro by X-ray crystallography. The structures reveal that 13b-K binds to the Mpro in the canonical way, with the cyclopropylmethyl group occupying the S2 pocket, the glutamine-derived γ-lactam inserted into the S1 site, and the P1′ benzyl group being partly in the S1′ site but actually extending beyond this very small pocket of the enzyme (Figure 3a; see the Supporting Information for structural details). 13b-H, in contrast, displays a very different and highly unusual binding mode (Figure 3b). Being in the (R)-configuration, its P2-cyclopropylmethyl moiety is unable to bind to the S2 pocket and instead is oriented toward the periphery of the complex. The P1-γ-lactam (with S,S-stereochemistry at the Cα atom and in the lactam ring) binds to the S1 pocket as expected, but at the site of covalent interaction with Cys145 of the Mpro, the compound reverts its orientation so that the P1′-benzyl group is inserted into the hydrophobic S2 pocket of the enzyme. Furthermore, after formation of the covalent bond between the nucleophilic thiol (Cys145) and the Cα of the 13b-H α-ketoamide warhead, the latter is in the (R)-configuration, whereas this atom is in the (S)-configuration in the complex between 13b-K and the main protease.
Figure 3.

X-ray co-crystal structures of (A) 13b-K (PDB entry: 8A4T) and (B) 13b-H (PDB entry: 8A4Q) in the substrate-binding cleft of the Mpro. The orientation of the view was rotated ∼45° in the case of (B) for clarity (see Figure S4 for the alternate orientation). The carbon atoms of the compounds are depicted in yellow and those of the protein in cyan, with the exception of the Ser1 of the other monomer (green). Blue symbols (S1 and S2) indicate the canonical binding pockets for moieties P1–P4 (red symbols) of the inhibitors. The 2Fo–Fc map carved around the inhibitor is depicted as a blue mesh (contoured at 1.0). Polar contacts are represented as red dashed lines.
The antiviral activity of 13b-K against SARS-CoV-2 infection was investigated in different cellular infection models. Treatment with 13b-K of human bronchial epithelial cells (Calu 3 cells) infected with SARS-CoV-2 revealed a dose-dependent antiviral effect on the release of infectious viral particles, with an EC50 of 2.4 ± 0.7 μM (see Figure 4a for details). The reported EC50 for the diastereomeric mixture 13b in Calu 3 cells infected with SARS-CoV-2 is 4–5 μM,20 which is consistent with our findings that only one of the diastereomers is active. 13b-K was also tested for its antiviral activity using a fully replication-competent SARS-CoV-2 strain in A549-ACE2-TMPRSS2, Huh7, and VeroE6 cell cultures. Since VeroE6 cells show a high efflux of some chemotypes, this latter antiviral assay was performed in the presence of the MDR1-inhibitor CP-100356 (0.5 μM).22 The dose–response curves in these model systems are shown in Figure 4b, and nonlinear fitting results in EC50 values (95% CI) of 1.3 μM (1.16–1.55), 3.4 μM (1.58–5.03), and 0.84 μM (0.64–1.08) in the VeroE6 + CP/SARS2, Huh7/SARS2, and A549-ACE2-TMPRSS2 systems, respectively, and CC50 values >100 μM.
Figure 4.

Antiviral activity of 13b-K in human bronchial epithelial cells and other cell lines. (A) Calu3 cells were infected with SARS-CoV-2 using an MOI of 0.1, treated with different concentrations of 13b-K (0, 0.1, 2.5, 5, 10, and 20 μM), and analyzed at 24 hpi. The amount of infectious viral particles released was determined by the TCID50 method from a cell-free culture supernatant. Three biological experiments, each with double replicates, were performed. The amount of infectious viral particles was calculated by means of the TCID method on Vero E6 cells. (B) Similarly, the anti-SARS-CoV-2 activity was determined on different cell lines. Cells were treated with different concentrations of 13b-K and infected with SARS-CoV-2 using an MOI of 0.001 (VeroE6), 0.003 (Huh7), or 0.005 (A549) TCID50/cell. Several days after infection, the cell viability was determined. High cell viability indicates inhibition from the virus-induced cytopathogenic effect and thus antiviral activity. In the experiment with VeroE6, we included the MDR1-inhibitor CP-100356 to block compound efflux. (C) In a parallel experiment, the toxicity of different concentrations of 13b-K was determined under the same conditions but without adding the virus. At least two independent dose–response curves were investigated in each model system, and the average and standard deviations are shown.
The pharmacokinetic (PK) properties of 13b-K were tested using both inhalative and peroral routes. After inhalative administration of 25 mg/kg 13b-K using an Aeroneb nebulizer device (KentScientific), concentrations in the broncheoalveolar lavage (BALF), plasma, urine, and lung tissue were determined at different time points (Figure 5 and Figure S5). BALF levels were above the IC50 for at least 8 h (Figure S5). Furthermore, determination of the concentrations of 13b-K in different compartments after inhalative administration over 24 h suggests that the compound is distributed from the epithelial lining fluid (ELF) to the lung and then toward plasma and, finally, excreted via urine as it appears already 15 min post administration in plasma as well as in urine. 13b-K was found in plasma as well as lung tissue up to 24 h after a single inhalative dose administration. ELF concentrations were determined from BALF samples using the urea method.23 Interestingly, ELF concentrations were more than 100-fold above the cellular EC50 of 13b-K for more than 8 h. Finally, the BALF/plasma exposure ratio suggested a favorable lung and, especially, BALF/ELF bioavailability that renders pulmonary administration of 13b-K a viable alternative to local administration in SARS-CoV-2 infection models (Table 1).
Figure 5.

Levels of 13b-K in ELF, plasma, and lung tissue after 25 mg/kg inhalative and 100 mg/kg peroral administration.
Table 1. PK Parameters after 25 mg/kg Inhalative Administration and 100 mg/kg Peroral Administrationa.
| 25 mg/kg inhalative | 100 mg/kg peroral | |
|---|---|---|
| plasma Tmax [h] | 0.33 ± 0.1 | 2.5 ± 0.7 |
| plasma Cmax [ng/ml] | 236.47 ± 29.8 | 357.00 ± 171.8 |
| plasma AUC 0-t [ng/ml·h] | 2896.63 ± 269.6 | 1061.49 ± 290.1 |
| plasma Vz/F_obs [l/kg] | 141.07 ± 48.1 | 131.34 |
| plasma Cl/F_obs [ml/min/kg] | 83.92 ± 52.1 | 1162.74 |
| plasma t1/2 [h] | 1.3 | |
| plasma MRT [h] | 2.57 | |
| BALF Cmax [ng/ml] | 2330.33 ± 1034.4 | |
| BALF AUC 0-t [ng/ml·h] | 5162.20 ± 1163.6 | |
| ratio BALF AUC 0-t/Plasma AUC 0-t | 1.7 | |
| ELF Cmax [μg/ml] | 433.52 ± 222.6 | |
| ELF AUC 0-t [μg/ml·h] | 1042.34 ± 586.9 | |
| ratio ELF AUC 0-t/plasma AUC 0-t | 359 | |
| lung tissue Cmax [ng/g] | 261,748 ± 1294.4 |
t1/2: half-life, Tmax: time point at maximal concentration, Cmax: maximal concentration, AUC 0-t: area under the curve/exposure from time 0-t, MRT: mean residence time, Vz/F_obs: fractionated observed volume of distribution, and Cl/F_obs: fractionated observed clearance.
The peroral bioavailability of 13b-K was also evaluated. Inspired by the fact that liposomes have already been used in the context of novel antivirals,24 we used a 1,2,-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS)-based large unilamellar vesicle (LUV) formulation to dissolve 13b-K at higher concentrations. Moreover, LUV facilitate absorption as they fuse more easily with membranes and, thereby, enhance peroral bioavailability, especially in the case of peptidomimetic molecules like 13b-K, as the gastrointestinal epithelium frequently represents not only a physical but also a biochemical barrier for proteins and peptides.25−27 We administered the 13b-K LUV formulation at 100 mg/kg perorally and determined plasma and urine concentrations at designated time points. Moreover, we collected terminal lung tissue and BALF and calculated ELF concentrations. Liposomal formulation of 13b-K resulted in a peroral bioavailability of around 17%. The Tmax was reached after 2.5 h, and plasma concentrations were above the IC50 for around 3 h (Table 1 and Figure 5). Surprisingly, BALF concentrations at 5 h post administration were in the same range as after inhalative administration of 25 mg/kg. This is also applicable to the lung tissue as well as ELF concentrations. Thus, the pronounced lung tropism of 13b-K is also detected after systemic, peroral administration. Nevertheless, PK data illustrate that multiple dosing is necessary for 13b-K to reach sufficient levels in the lung as well as plasma for more than 24 h to exert an effect in efficacy models.
Discussion
The COVID-19 pandemic is a global health emergency with far-reaching social and economic consequences. In addition to remdesivir, a phosphoramidate prodrug of the RdRp inhibitor GS-441524 that received FDA approval for the treatment of adult and pediatric (aged >12 years) SARS-CoV-2 patients (the clinical efficacy of which is a matter of debate28), the nucleoside prodrug molnupiravir29 and the Mpro inhibitor PF-07321332 (nirmatrelvir)17 have shown to be clinically effective specific antiviral drugs. Experience from human immunodeficiency virus therapy30 though suggests that these antiviral drugs may lead to the emergence of drug-resistant mutations, ultimately requiring the application of cocktails of several antiviral drugs. Additional antiviral treatments for SARS-CoV-2 are therefore desirable. The main protease of SARS-CoV-2 is an attractive drug target since it has an essential role in the processing of viral polyproteins and host–cell signaling proteins and it has no human homolog. Previous works have demonstrated the feasibility of this approach.17−21,31
In this work, we report a highly potent α-ketoamide inhibitor of the SARS-CoV-2 Mpro, 13b-K, obtained through diastereomeric resolution of the previously disclosed inhibitor 13b. In early 2020, Zhang et al. published the Mpro inhibitor 13b and demonstrated that this compound has potent activity in vitro and in cellulo with favorable pharmacokinetic properties.20 We discovered that the compound previously reported as 13b is in fact a mixture of two diastereomers, only one of which has substantial inhibitory activity against Mpro. This is a significant finding since the active diastereomer, 13b-K, is substantially more potent against SARS-CoV-2 Mpro than the mixture. This has important implications for researchers looking to reproduce the results published by Zhang et al. and furthers the progress toward a clinical candidate compound based on this series.
Compound 13b-K shows modest antiviral activity in Huh7 cells infected with SARS-CoV-2 and better activity in VeroE6 cells (in the presence of the MDR1 inhibitor CP-100356) and in Calu3 cells. The best antiviral activity, however, was observed in A549 cells engineered to express the ACE2 receptor and the TMPRSS2 protease that process the spike protein of the virus.32 It has been proposed by Steuten et al.32 that viral protease inhibitors showing good antiviral activity in Vero E6 cells but poor activity in cells expressing TMPRSS2 actually target cathepsin L (rather than the coronaviral main protease). Cathepsin L takes over the task of spike protein processing in cells lacking TMPRSS2. We note that this hypothesis does not apply to our inhibitor 13b-K since its antiviral activity is the highest in A549 cells expressing TMPRSS2.
To further characterize the active diastereomer 13b-K, we performed additional pharmacokinetic studies (beyond those that were carried out with the mixture of diastereomers, 13b(20)) with two different routes of administration. We demonstrate that local administration via nebulization and systemic peroral administration result in good compound levels in the lung tissue as well as in BALF and ELF. The half-life in plasma after systemic administration remains similar compared to 13b, the mixture of diastereomers.20 The liposomal formulation of 13b-K now opens avenues to administer the compound via the peroral route as well as the pulmonary route. Further optimization of the liposomal formulation will most likely enhance bioavailability, which is already close to that of the clinically approved hepatitis C drug boceprevir.33 In addition, pulmonary liposomal delivery has already been proven to be efficacious in the context of antibacterial therapy.34 Liposomes are biocompatible and can enhance the therapeutic index as well as the retention at the desired target, especially in the lung.34 Consequently, pulmonary liposomal delivery of 13b-K represents a viable way to reach high concentrations to target SARS-CoV-2 lung infections.
The corresponding matched-pair diastereomeric compound 13b-H is at least ∼50-fold less active than 13b-K in the Mpro inhibition assay and as such could provide the research community with a control compound to strengthen their experimental design.35 Co-crystallization of both 13b-K and 13b-H with the target enzyme revealed useful information on the respective binding modes. 13b-K binds largely in the canonical way previously described for the structure obtained with the diastereomeric mixture 13b.(20) This supports the assumption that the enzyme selected the highly active (S,S,S)-diastereomer from the mixture during the crystallization process in the original experiment. The much-less active diastereomer, 13b-H, binds to the Mpro in a highly unusual, semi-circular conformation, which allows some important conclusions for the further optimization of this compound series. Because its P2-Cα atom is in the (R)-configuration, the cyclopropyl methyl moiety is unable to enter the S2 pocket of the protease. Instead, it is oriented away from the substrate-binding site (Figure 3b). In contrast, the P1 moiety, the Cα carbon of which has the correct configuration, (S), in 13b-H, binds to the S1 pocket in the canonical way. As in the active diastereomer, it forms two hydrogen bonds with the main-chain oxygen of Phe140 and the Nε2 of His163. The P1′ benzyl group of 13b-H does not bind to the S1′ site of the enzyme but instead folds back and penetrates the S2 pocket, which is left empty because the P2-cyclopropyl methyl is prevented from entering it due to the incorrect stereochemistry at the P2 Cα. This can be taken as an indication that filling the hydrophobic S2 pocket with a hydrophobic ligand is of prime importance in contrast to having a hydrophobic moiety at the S1′ site. The P1’ benzyl group is too large for the latter site. Hence, we are currently preparing derivatives of 13b-K that carry a small group such as methyl or even no substituent at P1′ to further explore this finding.
Conclusions
In summary, we report a highly potent inhibitor of the SARS-CoV-2 Mpro, 13b-K, that was revealed through diastereomeric resolution of a previously reported inhibitor. 13b-K demonstrates peroral as well as inhalative bioavailability and represents a promising candidate for further development as an antiviral treatment for COVID-19.
Experimental Section
Chemistry General Methods
All reagents and solvents were purchased from commercial sources and used without further purification. Nuclear magnetic resonance spectra were recorded on a Bruker Avance III HD spectrometer operating at 400 MHz for 1H NMR, 100 MHz for 13C NMR, and 376 MHz for 19F, unless otherwise stated. 1H NMR and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) and are referenced to residual protium in the solvent and to the carbon resonances of the residual solvent peak, respectively. DEPT and correlation spectra were run in conjunction to aid assignment. 19F NMR chemical shifts are reported in ppm and are uncorrected. Coupling constants (J) are quoted in Hertz (Hz), and the following abbreviations were used to report multiplicity: s = singlet, d = doublet, dd = doublet of doublets, ddd = double doublet of doublets, t = triplet, q = quartet, m = multiplet, and br s = broad singlet. Purification by flash column chromatography was carried out using Fisher Scientific silica gel 60 Å (35–70 μm) or Supelco silica gel (technical grade, pore size: 60 Å, 40–63 μm, 230–400 mesh). Analytical thin-layer chromatography was performed on glass plates precoated with silica gel (Analtech, UNIPLATE 250 μm/UV254 or Merck TLC silica gel 60 F254), with visualization being achieved using UV light (254 nm) and/or by staining with alkaline potassium permanganate dip. Reaction monitoring LC–MS analyses were conducted using either Agilent InfinityLab LC/MSD systems or Agilent 1260 HPLC System with a DAD detector and an Agilent 6130 quadrupole mass detector with electro spray ionization (ESI) (MeCN/H2O + 0.1% formic acid). Chiral HPLC analysis was conducted using an Agilent 1100 series LC equipped with a DAD (G1315B), column oven (G1316A), autosampler (G1313A) with a 100 μL loop, quaternary pump (G1311A), and an Agilent 1200 series degasser (G1322A). Data collection and processing were conducted using an Agilent Technologies Chemstation B.04.03. SFC purity analyses were conducted using a Waters ACQUITY UPC2 system with a binary solvent manager (K17C2B854M), sample manager (L17C2S782M), convergence manager (K17C2M838M), column manager (E18AZ3268M), and PDA detector (M17C2P349A). Data were collected and processed using Empower 3 Build 3471 software. Optical rotations were recorded on a Bellingham & Stanley ADP450 polarimeter. High-resolution mass spectral (HRMS) data were collected in the laboratories of the University of Bath Chemistry Department using an Agilent 6545 LC/Q-TOF system. Single-crystal X-ray measurements were recorded in the laboratories of Newcastle University Chemistry Department. Purity is greater than 95% for all compounds tested biologically.
Synthesis of (S,S,S)-MPro 13b-K (13) and Its Diastereomer (R,S,S)-MPro 13b-H (12)
(R)-3-Cyclopropyl-2-hydroxypropanoic Acid (1)
To a stirred solution of d-cyclopropylalanine (45.00 g, 0.35 mol) in 2 M H2SO4 (675 mL) at 0 °C was added a solution of sodium nitrite (120.00 g, 1.71 mol) in water (270 mL) in a dropwise fashion over 50 min, maintaining the temperature below 5 °C over the course of the addition. The resulting reaction mixture was stirred for 3 h at 0–5 °C, and then allowed to warm to ambient temperature, and stirred for 16 h. The reaction mixture was extracted with TBME (600 mL), and the aqueous phase was further extracted with TBME (5 × 200 mL). The combined organic extracts were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to afford the title compound as pale-yellow oil (21.20 g, 47%). 1H NMR (CDCl3) δ: 4.36 (dd, J1 = 7.0 Hz, J2 = 4.8 Hz, 1H), 1.76–1.66 (m, 2H), 0.95–0.82 (m, 1H), 0.60–0.44 (m, 2H), 0.20–0.06 (m, 2H). 13C NMR (CDCl3) δ: 179.90, 70.86, 39.08, 6.70, 4.56, 3.94.
Methyl (R)-3-Cyclopropyl-2-hydroxypropanoate (2)
To a stirred solution of (R)-3-cyclopropyl-2-hydroxypropanoic acid (1) (21.10 g, 162.13 mmol) in methanol at 0 °C was added thionyl chloride (23.65 mL, 324.27 mol) in a dropwise fashion over 20 min. The resulting solution was allowed to warm to ambient temperature over 2 h and was subsequently concentrated under reduced pressure. Purification by flash column chromatography, eluting with 25% EtOAc/petroleum ether (40:60), afforded the title compound as colorless oil (12.54 g, 54%). 1H NMR (CDCl3) δ: 4.28 (dd, J1 = 6.4 Hz, J2 = 4.6 Hz, 1H), 3.79 (s, 3H), 1.74–1.58 (m, 3H), 0.92–0.80 (m, 1H), 0.53–0.43 (m, 2H), 0.15–0.04 (m, 2H). 13C NMR (CDCl3) δ: 175.69, 70.93, 52.40, 39.39, 6.62, 4.43, 3.88.
Methyl (R)-3-Cyclopropyl-2-(((trifluoromethyl)sulfonyl)oxy)propanoate (3)
To a stirred solution of methyl (R)-3-cyclopropyl-2-hydroxypropanoate (2) (12.50 g, 86.70 mmol) in DCM (163 mL) at 0 °C was added 2,6-lutidine (25.10 mL, 215.89 mmol) followed by triflic anhydride (32.50 mL, 193.35 mmol) in a dropwise fashion, maintaining the temperature at 0 °C over the course of the addition and thereafter for a further 40 min. The reaction mixture was treated with 1 M HCl (aq) (200 mL) and brine (600 mL), and the organic phase was separated. The aqueous component was extracted with DCM (2 × 100 mL), and the combined organic extracts were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give brown oil. The oil was taken up in diethyl ether (300 mL) and further washed with 1 M HCl (aq) and brine before being dried over anhydrous magnesium sulfate and concentrated under reduced pressure. This afforded the title compound as pale brown oil (21.74 g, 91%), which was used directly in the next step with no further purification. 1H NMR (CDCl3) δ: 5.20 (dd, J1 = 7.8 Hz, J2 = 4.5 Hz, 1H), 3.85 (s, 3H), 1.99–1.85 (m, 2H), 0.92–0.81 (m, 1H), 0.65–0.51 (m, 2H), 0.25–0.11 (m, 2H). 13C NMR (CDCl3) δ: 167.73, 118.63 (d, J = 320 Hz), 83.97, 53.29, 37.09, 6.67, 5.08, 3.90. 19F NMR (CDCl3) δ: −74.95.
tert-Butyl (2-Oxo-1,2-dihydropyridin-3-yl)carbamate
To a stirred solution of 3-amino-2-hydroxypyridine (25.00 g, 227.04 mmol) in THF (530 mL) at ambient temperature was portion-wise added Boc anhydride (49.55 g, 227.04 mmol). The reaction mixture was heated to reflux, and after 4 h, a further portion of Boc anhydride (39.60 g, 181.63 mmol) was added, with reflux being continued for a further 18 h. The reaction mixture was cooled to ambient temperature and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 2% MeOH/DCM, afforded the title compound as an off-white solid (25.65 g, 54%). 1H NMR (CDCl3) δ: 12.75 (br s, 1H), 8.14 (d, J = 6.7 Hz, 1H), 7.57 (s, 1H), 7.03 (dd, J1 = 6.6 Hz, J2 = 1.7 Hz, 1H), 6.36 (t, J = 7.0 Hz, 1H), 1.54 (s, 9H). 13C NMR (CDCl3) δ: 158.96, 152.84, 129.84, 125.49, 122.08, 108.00, 80.96, 28.39.
Methyl 2-(3-((tert-Butoxycarbonyl)amino)-2-oxopyridin-1(2H)-Yl)-3-cyclopropylpropanoate (4)
To a stirred solution of tert-butyl (2-oxo-1,2-dihydropyridin-3-yl)carbamate (15.90 g, 75.63 mmol) in THF (475 mL) at 0 °C was portion-wise added sodium hydride (4.69 g, 117.23 mmol, 60% dispersion in mineral oil), and after completion of the addition, the reaction mixture was stirred at 0 °C for a further 30 min. A solution of methyl (R)-3-cyclopropyl-2-(((trifluoromethyl)sulfonyl)oxy)propanoate (3) (21.58 g, 78.13 mmol) in THF (225 mL) was then added dropwise at such a rate as to maintain the temperature at 0 °C, with further stirring thereafter being maintained at this temperature for a total of 43 h. The reaction mixture was concentrated under reduced pressure. Purification by flash column chromatography, eluting with 20% EtOAc/petroleum ether (40:60), afforded the title compound as thick yellow oil (12.31 g, 48%). 1H NMR (DMSO-d6) δ: 7.85 (dd, J1 = 7.5 Hz, J2 = 1.1 Hz, 1H), 7.80 (s, 1H), 7.40 (dd, J1 = 7.0 Hz, J2 = 1.7 Hz, 1H), 6.34 (t, J = 7.2 Hz, 1H), 5.23 (dd, J1 = 9.8 Hz, J2 = 5.2 Hz, 1H), 3.64 (s, 3H), 2.08–1.92 (m, 2H), 1.47 (s, 9H), 0.57–0.43 (m, 1H), 0.39–0.23 (m, 2H), 0.11–0.04 (m, 1H), −0.08 to −1.15 (m, 1H). 13C NMR (DMSO-d6) δ: 169.54, 156.37, 152.14, 130.44, 128.48, 120.30, 105.48, 80.08, 61.80, 52.21, 33.07, 27.85, 7.36, 4.30, 3.35.
2-(3-((tert-Butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanoic Acid (5)
To a stirred solution of methyl 2-(3-((tert-butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanoate (4) (12.30 g, 36.57 mmol) in methanol (346 mL) at ambient temperature was added water (69.2 mL) followed by lithium hydroxide monohydrate (3.07 g, 73.13 mmol), and the resulting reaction mixture was stirred for an hour. The reaction mixture was acidified to pH 6 with 1 M HCl (aq) and then partially concentrated under reduced pressure to remove the bulk of the methanol. The resulting aqueous solution was initially extracted with diethyl ether (5 × 200 mL), which, after drying and concentration of the extracts, afforded a small initial crop of product. The aqueous phase was subsequently extracted with DCM and further with 10% MeOH/DCM; these extracts were also being dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give further crops. All crops of the product were combined and purified by flash column chromatography, eluting with 5% MeOH/DCM increased to 10% MeOH/DCM. This afforded the title compound as a pale yellow solid (8.80 g, 73%). 1H NMR (DMSO-d6) δ: 13.08 (br s, 1H), 7.82 (dd, J1 = 7.4 Hz, J2 = 1.4 Hz, 1H), 7.77 (s, 1H), 7.37 (dd, J1 = 7.0 Hz, J2 = 1.7 Hz, 1H), 6.31 (t, J = 7.2 Hz, 1H), 5.24 (dd, J1 = 10.6 Hz, J2 = 4.9 Hz, 1H), 2.08–1.89 (m, 2H), 1.47 (s, 9H), 0.55–0.43 (m, 1H), 0.37–0.24 (m, 2H), 0.10–0.03 (m, 1H), −0.05 to −0.13 (m, 1H). 13C NMR (DMSO-d6) δ: 171.34, 156.80, 152.13, 129.78, 127.93, 119.36, 104.50, 79.95, 60.77, 35.56, 27.86, 7.97, 4.27, 3.56. m/z (ES+): 343.3 [M + Na+]+. [α]D24 = −0.6° (c = 1, MeOH); 1:1 mixture of enantiomers determined by chiral HPLC (Chiralpak AD-H, 250 × 4.6 mm, 5 μm, 90% hexane + 0.1% TFA isocratic).
Dimethyl (2S,4R)-2-((tert-butoxycarbonyl)amino)-4-(cyanomethyl)pentanedioate
To a stirred solution of N-Boc l-glutamic acid dimethyl ester (40.00 g, 145.30 mmol) in THF (440 mL) at −70 °C was dropwise added LHMDS (313.84 mL, 313.84 mmol, 1 M solution in THF) for over 15 min, maintaining the temperature below −63 °C during the course of the addition. After stirring the resulting reaction mixture at −70 °C for 1 h, bromoacetonitrile (10.83 mL, 155.47 mmol) was added dropwise via a syringe pump over 1 h, maintaining the temperate at or below −70 °C during the course of the addition. After stirring at −70 °C for a further 3.75 h, cold methanol (29 mL) was added in one portion. After 30 min of further stirring, a cold mixture of acetic acid (29 mL) in THF (175 mL) was added in one portion, and the reaction mixture was allowed to warm to ambient temperature with stirring overnight. The reaction mixture was poured into brine (720 mL), and the organic phase was separated. The aqueous component was extracted with ethyl acetate (2 × 500 mL), and the combined organic extracts were dried over anhydrous magnesium sulfate and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 25% EtOAc/petroleum ether (40:60), afforded the title compound as pale yellow oil (39.36 g, 86%). 1H NMR (CDCl3) δ: 5.13 (d, J = 7.9 Hz, 1H), 4.44–4.33 (m, 1H), 3.76 (s, 3H), 3.75 (s, 3H), 2.91–2.81 (m, 1H), 2.78 (m, 2H), 2.23–2.10 (m, 2H), 1.44 (s, 9H). 13C NMR (CDCl3) δ: 172.44, 172.18, 155.72, 117.27, 80.74, 52.90, 52.83, 51.17, 38.36, 34.07, 28.37, 19.15.
Methyl (S)-2-((tert-Butoxycarbonyl)amino)-3-((S)-2-oxopyrrolidin-3-yl)propanoate
A stirred solution of dimethyl (2S,4R)-2-((tert-butoxycarbonyl)amino)-4-(cyanomethyl)pentanedioate (39.00 g, 124.07 mmol) in methanol (730 mL) was treated with cobalt chloride hexahydrate (14.76 g, 62.04 mmol) and then cooled to 0 °C. Sodium borohydride (18.78 g, 496.29 mmol) was added portion-wise over 1 h, maintaining the temperature below +2 °C during the course of the addition. The reaction mixture was allowed to slowly warm to ambient temperature and was stirred for 22 h before being concentrated under reduced pressure. The resulting brown oil was partitioned between ethyl acetate (1500 mL), brine (720 mL), and 2 M HCl (aq) (360 mL). The organic phase was separated, and the aqueous component was extracted with ethyl acetate (2 × 400 mL). The combined organic extracts were washed with brine (200 mL), dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 4% MeOH/DCM, and subsequent trituration with petroleum ether (40:60) afforded the title compound as an off-white solid (14.98 g, 52.32 mmol, 42%). 1H NMR (CDCl3) δ: 5.76 (br s, 1H), 5.48 (d, J = 7.9 Hz, 1H), 4.38–4.28 (m, 1H), 3.75 (s, 3H), 3.41–3.31 (m, 2H), 3.54–3.42 (m, 2H), 2.20–2.09 (m, 1H), 1.94–1.80 (m, 2H), 1.45 (s, 9H). 13C NMR (CDCl3) δ: 179.76, 173.02, 155.88, 80.05, 52.55, 52.38, 40.51, 38.23, 34.29, 28.42, 28.25. m/z (ES+): 309.2 [M + Na+]+.
Methyl (S)-2-Amino-3-((S)-2-oxopyrrolidin-3-yl)propanoate Hydrochloride
Methyl (S)-2-((tert-butoxycarbonyl)amino)-3-((S)-2-oxopyrrolidin-3-yl)propanoate (12.96 g, 45.26 mmol) was treated with hydrogen chloride (226 mL, 905.27 mmol, 4 M solution in 1,4-dioxane), and the resulting mixture was stirred at ambient temperature for 2 h before being concentrated under reduced pressure. The residue was triturated with diethyl ether to afford the title compound as a colorless solid (10.08 g, quant.). 1H NMR (DMSO-d6) δ: 8.81 (br s, 3H), 7.96 (s, 1H), 4.17 (m, 1H), 3.76 (s, 3H), 3.26–3.14 (m, 2H), 2.66–2.56 (m, 1H), 2.35–2.25 (m, 1H), 2.14–2.04 (m, 1H), 1.92–1.83 (m, 1H), 1.73–1.61 (m, 1H). 13C NMR (DMSO-d6) δ: 178.03, 169.76, 66.37, 52.83, 50.99, 38.10, 31.77, 27.68. [α]D24 = + 45.6° (c = 1.0, MeOH).
Methyl (S)-2-(2-(3-((tert-Butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanamido)-3-((S)-2-oxopyrrolidin-3-yl)propanoate (6)
To a stirred solution of 2-(3-((tert-butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanoic acid (5) (8.55 g, 26.52 mmol) in DCM (200 mL) at 0 °C was added HOBt (4.93 g, 29.18 mmol) and EDC·HCl (5.59 g, 29.18 mmol). After stirring at 0 °C for 1 h, a solution containing a mixture of methyl (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoate hydrochloride (6.19 g, 27.85 mmol) and triethylamine (2.95 g, 29.18 mmol) in dichloromethane (120 mL) was added dropwise. Furthermore, triethylamine was added dropwise to adjust the reaction mixture to pH 9, after which point stirring was maintained at 0 °C overnight. After 18 h, the reaction mixture was warmed to ambient temperature over 6 h. TLC indicated that the reaction mixture contained a mixture of the acid starting material and the product. Additional portions of EDC·HCl (1.53 g, 7.98 mmol) and HOBt (1.34 g, 7.98 mmol) were added followed by (S)-2-amino-3-((S)-2-oxopyrrolidin-3-yl)propanoate hydrochloride (2.43 g, 7.98 mmol) and triethylamine (0.85 g, 8.40 mmol). After 42 h, the reaction mixture was treated with water (200 mL) and the organic phase was separated. The aqueous component was extracted with DCM (100 mL), and the combined organic extracts were washed with saturated sodium bicarbonate solution (2 × 200 mL), dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 3% MeOH/DCM, and subsequent trituration with petroleum ether (40:60) afforded the title compound as a white solid (8.87 g, 68%). 1H NMR (DMSO-d6) δ: 8.88 (dd, J1 = 19.2 Hz, J2 = 7.3 Hz, 1H), 7.79 (d, J = 7.4 Hz, 1H), 6.29 (t, J = 7.2 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H), 7.66 (d, J = 10.1 Hz, 1H), 6.29 (t, J = 7.1 Hz, 1H), 5.72–5.56 (m, 1H), 4.34–4.24 (m, 1H), 3.61 (d, J = 10.0 Hz, 3H), 3.19–3.03 (m, 2H), 2.30–2.17 (m, 1H), 2.16–1.87 (m, 3H), 1.85–1.71 (m, 1H), 1.67–1.53 (m, 2H), 1.46 (s, 9H), 0.57–0.41 (m, 1H), 0.36–0.27 (m, 2H), 0.17–0.09 (m, 1H), 0.03 to −0.05 (m, 1H). 13C NMR (DMSO-d6) δ: 177.85, 177.84, 172.08, 172.06, 169.39, 169.21, 156.83, 156.78, 152.18, 128.84, 128.80, 127.98, 127.97, 119.94, 119.87, 80.09, 58.06, 57.57, 51.98, 51.97, 50.58, 50.53, 37.64, 37.58, 35.66, 35.19, 32.37, 32.11, 27.88, 27.19, 27.10, 22.48, 19.87, 19.27, 18.64, 14.16, 11.24, 7.52, 7.49, 4.58, 4.43, 3.58, 3.51. m/z (ES+): 513.3 [M + Na+]+.
tert-Butyl (1-(-3-Cyclopropyl-1-(((S)-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (7)
To a stirred solution of methyl (S)-2-(2-(3-((tert-butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanamido)-3-((S)-2-oxopyrrolidin-3-yl)propanoate (6) (8.70 g, 17.73 mmol) in methanol (180 mL) at ambient temperature was portion-wise added sodium borohydride (3.35 g, 88.68 mmol) for over 20 min at such a rate so as to maintain the temperature around 20 °C. The reaction mixture was stirred for 1.5 h and then treated first slowly with water (20 mL) and then adjusted to pH 9 by dropwise addition of 1 M hydrochloric acid. The reaction mixture was partially concentrated to a volume of about 50 mL and then extracted with dichloromethane (3 ×x 30 mL). The combined organic extracts were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give the title compound as a white solid (8.13 g, 99%). 1H NMR (DMSO-d6) δ: 8.10 (dd, J1 = 20.8 Hz, J2 = 8.6 Hz, 1H), 7.78 (d, J = 6.8 Hz, 1H), 7.73 (s, 1H), 7.53 (d, J = 15.7 Hz, 1H), 7.44–7.37 (m, 1H), 6.28 (td, J1 = 7.2 Hz, J2 = 2.2 Hz, 1H), 5.62–5.49 (m, 1H), 4.67 (m, 1H), 3.76 (m, 1H), 3.39–3.20 (m, 2H), 3.19–1.98 (m, 2H), 2.21–1.86 (m, 3H), 1.85–1.69 (m, 2H), 1.64–1.28 (m, 2H), 1.46 (s, 9H), 0.55–0.40 (m, 1H), 0.37–0.24 (m, 2H), 0.19–0.07 (m, 1H), 0.03 to −0.05 (m, 1H). 13C NMR (DMSO-d6) δ: 178.73, 178.69, 168,87, 168.77, 156.79, 152.18, 129.02, 128.71, 127.96, 127.93, 119.86, 104.81, 104.74, 80.09, 63.67, 63.62, 58.33, 58.10, 49.17, 37.72, 37.51, 35.78, 35.48, 32.34, 32.23, 27.90, 27.62, 27.54, 7.57, 4.51, 4.46, 3.67, 3.53. m/z (ES+): 485.3 [M + Na+]+.
tert-Butyl (1-(3-Cyclopropyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)propan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (8)
To a stirred solution of tert-butyl (1-(3-cyclopropyl-1-(((S)-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (7) (8.00 g, 17.30 mmol) in DCM (800 mL) at ambient temperature were added NaHCO3 (0.509 g, 6.05 mmol) and Dess–Martin periodinane (9.17 g, 216.20 mmol), and the solution was stirred for 1 h. The reaction mixture was treated with a saturated aqueous solution of NaHCO3 (200 mL), and the reaction mixture was filtered through Celite. The organic phase was separated, and the aqueous component was extracted with DCM (2 × 50 mL). The combined organic extracts were dried over anhydrous magnesium sulfate and concentrated under reduced pressure to a yellow solid. Purification by flash column chromatography, eluting with 4% MeOH/DCM, afforded the title compound as a white solid (7.17 g, 90%). 1H NMR (DMSO-d6) δ: 9.40 (d, J = 10.1 Hz, 1H), 8.81 (dd, J1 = 12.9 Hz, J2 = 7.3 Hz, 1H), 7.86–7.67 (m, 2H), 7.67–7.50 (m, 1H), 7.43–7.37 (m, 1H), 6.33–6.25 (m, 1H), 5.68–5.54 (m, 1H), 4.37–4.13 (m, 1H), 3.24–2.96 (m, 3H), 2.31–1.28 (m, 6H), 1.46 (s, 9H), 0.57–0.39 (m, 1H), 0.37–0.23 (m, 2H), 0.20–0.07 (m, 1H), 0.04 to −0.08 (m, 1H). m/z (ES−): 459.3 [M-H+]−.
(3S)-1-(Benzylamino)-3-(2-(3-((tert-butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanamido)-1-oxo-4-((S)-2-oxopyrrolidin-3-yl)butan-2-yl Acetate (9)
To a stirred solution of tert-butyl (1-(3-cyclopropyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)propan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (8) (7.10 g, 15.42 mmol) and acetic acid (1.85 g, 30.83 mmol) in DCM (710 mL) at ambient temperature was added benzyl isocyanide (1.81 g, 15.42 mmol), and the reaction mixture was stirred for 22 h before being concentrated under reduced pressure. Purification by flash column chromatography, eluting with 3% MeOH/DCM, afforded the title compound as an off-white solid (8.37 g, 85%). 1H NMR (DMSO-d6) δ: 8.68–8.23 (m, 2H), 7.81 (m, 1H), 7.73 (d, J = 17.6 Hz, 1H), 7.60 (m, 1H), 7.47–7.16 (m, 6H), 6.35–6.26 (m, 1H), 5.65–5.52 (m, 1H), 5.16–4.85 (m, 1H), 4.35–4.14 (m, 3H), 3.16–2.98 (m, 2H), 2.22–1.58 (m, 8H), 1.47 (s, 9H), 1.57–1.02 (m, 2H), 0.55–0.42 (m, 1H), 0.37–0.27 (m, 2H), 0.19–0.09 (m, 1H), 0.05 to −0.07 (m, 1H). m/z (ES+): 660.4 [M + Na+]+.
tert-Butyl (1-(1-(((2S)-4-(benzylamino)-3-hydroxy-4-oxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (10)
A stirred solution of (3S)-1-(benzylamino)-3-(2-(3-((tert-butoxycarbonyl)amino)-2-oxopyridin-1(2H)-yl)-3-cyclopropylpropanamido)-1-oxo-4-((S)-2-oxopyrrolidin-3-yl)butan-2-yl acetate (9) (28.25 g, 44.30 mmol) in methanol (2000 mL) and water (400 mL) was treated with lithium hydroxide monohydrate (3.72 g, 88.60 mmol), and the resulting solution was stirred at ambient temperature for 1.5 h. The reaction mixture was acidified to pH 6–7 with 1 M hydrochloric acid and then partially concentrated under reduced pressure to remove the bulk of the methanol. The resulting solution was extracted with DCM (3 × 500 mL). The combined organic extracts were washed with brine (250 mL), dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to afford the title compound (a mixture of diastereomers) as an off-white foam (24.25 g, 92%). 1H NMR (DMSO-d6) δ: 8.40–7.70 (m, 4H), 7.58–7.15 (m, 7H), 6.31–6.26 (m, 1H), 5.86–5.78 (m, 1H), 5.75 (s, 1H), 5.70–5.54 (m, 1H), 4.32–3.88 (m, 4H), 3.19–2.96 (m, 2H), 2.26–0.91 (m, 6H), 1.45 (s, 9H), 0.52–0.36 (m, 1H), 0.34–0.23 (m, 2H), 0.18–0.09 (m, 1H), 0.01 to −0.11 (m, 1H). m/z (ES−): 594.3 [M-H+]−.
tert-Butyl (1-(-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (11)
To a stirred solution of tert-butyl (1-(1-(((2S)-4-(benzylamino)-3-hydroxy-4-oxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (10) (3.60 g, 6.04 mmol) in DCM (436 mL) was added sodium hydrogen carbonate (0.22 g, 2.60 mmol) and Dess–Martin periodinane (3.33 g, 7.86 mmol), and the resulting mixture was stirred at ambient temperature for 1 h. Following the addition of saturated aqueous NaHCO3 solution (150 mL), the organic phase was separated, and the aqueous component was extracted with DCM (2 × 150 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. Purification by flash column chromatography, eluting with 2% MeOH/DCM, afforded the title compound as an off-white solid (3.59 g, quant.).
tert-Butyl (1-((R)-1-(((S)-4-(Benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate, (R,S,S)-Mpro 13b-H (12)
Recrystallization from acetonitrile afforded the title compound as an off-white solid. 1H NMR (DMSO-d6) δ: 9.22 (t, 1H), 9.03 (d, J = 6.6 Hz, 1H), 7.78 (d, J = 7.2 Hz, 1H), 7.72 (d, J = 18.8 Hz, 2H), 7.35–7.19 (m, 6H), 6.27 (t, J = 7.0 Hz, 1H), 5.74–5.66 (m, 1H), 5.03–4.93 (m, 1H), 4.36–4.22 (m, 2H), 3.22–3.08 (m, 1H), 2.41–2.29 (m,1H), 2.23–2.12 (m, 1H), 1.98–1.84 (m, 2H), 1.81–1.57 (m, 3H), 1.46 (s, 9H), 0.52–0.41 (m, 1H), 0.34–0.26 (m, 2H), 0.16–0.07 (m, 1H), 0.01 to −0.06 (m, 1H). 13C NMR (DMSO-d6) δ: 196.06, 177.94, 169.23, 160.74, 156.76, 152.17, 138.47, 128.76, 128.26, 127.94, 127.32, 126.92, 119.86, 104.80, 80.10, 57.34, 52.62, 42.05, 37.81, 35.66, 31.14, 27.91, 27.17, 7.51, 4.40, 3.52. HRMS (ES−) calculated for [C31H39N5O7-H+]− found: 592.2771. [α]D24 = +86.2° (c = 0.5, MeOH); >98% purity determined by SFC (Trefoil AMY1 (150 × 3.0 nm, 2.5 μm), IPA:CO2 (34.5% IPA isocratic)).
tert-Butyl (1-((S)-1-(((S)-4-(Benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate, (S,S,S)-Mpro 13b-K (13)
Concentration of the filtrate and purification by preparative HPLC afforded the title compound as a pale pink solid. 1H NMR (DMSO-d6) (120 °C) δ: 8.63 (m, 1H), 8.51 (d, J = 6.6 Hz, 1H), 7.77 (dd, J1 = 7.7 Hz, J2 = 1.7 Hz, 1H), 7.57 (s, 1H), 7.34–7.21 (m, 6H), 7.16 (s, 1H), 6.25 (t, J = 7.3 Hz, 1H), 5.60 (dd, J1 = 8.9 Hz, J2 = 6.0 Hz, 1H), 5.05–4.99 (m, 1H), 4.36 (d, J = 6.6 Hz, 1H), 3.20–3.09 (m, 2H), 2.36–2.26 (m, 1H), 2.21–2.12 (m, 1H), 2.06–1.92 (m, 2H), 1.86–1.63 (m, 3H), 1.50 (s, 9H), 0.66–0.55 (m, 1H), 0.41–0.33 (m, 1H), 0.15–0.09 (m, 1H), 0.04 to −0.02 (m 1H). 13C NMR (DMSO-d6) δ: 196.20, 177.88, 169.40, 160.73, 156.82, 152.17, 138.46, 128.75, 128.28, 128.08, 128.97, 127.40, 127.30, 126.93, 119.93, 104.85, 80.08, 57.79, 52.74, 42.02, 37.78, 35.42, 30.91, 27.89, 27.09, 7.48, 4.61, 3.55. HRMS (ES−) calculated for [C31H39N5O7-H+]− found: 592.2771. [α]D26 = −73.1° (c = 0.5, MeOH); >97% purity determined by SFC (Trefoil AMY1 (150 × 3.0 nm, 2.5 μm), IPA:CO2 (20–60% IPA isocratic)).
Single-Crystal X-ray Diffraction Analysis of Compound 13b-H
Crystals of 12 (compound 13b-H) were grown from hexafluoro-2-isopropanol in PDMSO via the encapsulated nanodroplet crystallization method,36 allowing the absolute stereochemistry to be determined by single-crystal X-ray diffraction analysis. A small colorless single crystal with a needle morphology was mounted onto a Bruker D8 Venture diffractometer equipped with a Photon 2 detector and a copper X-radiation microfocus source. Data were collected at a temperature maintained at 150 K in an open flow of nitrogen gas provided by an Oxford Cryosystems cryostream. All data were processed using the APEX 3 software suite; structure solution and refinement were completed using the OLEX2 interface to the SHELX suite of programs, namely, SHELXT and SHELXL.
Crystal Structure of 13b-H
The compound crystallizes in the orthorhombic space group P212121, with one complete molecule in the asymmetric unit. The structure provides clear validation for the proposed molecular entity, with confirmation of the stereochemical centers (Flack: −0.06). Absolute stereochemistry was determined to be (S) for the lactam carbon (C10), (S) for the P1 α-carbon (C12), and (R) for the P2 α-carbon (C17). The principal intermolecular interactions in the structure provide a classic N–H...O hydrogen bonding network, with these interactions predominantly oriented along the crystallographic a axis. Some disorder is observable in the tert-butyl groups due to a low-energy barrier to rotation.
Recombinant Protein Production
The protocol for recombinant production of SARS-CoV-2 Mpro was described previously.20 In brief, the target gene of Mpro was cloned into the PGEX-6p-1 vector. Nsp4-Nsp5 and PreScission cleavage sites were designed at the N- and C-termini, respectively, to generate the authentic target protein. The gene of the target protein was expressed in the BL21-Gold (DE3) (Novagen) strain of Escherichia coli. Purification of the Mpro was performed by using HisTrap FF (GE Healthcare) and ion-exchange (Q FF, GE Healthcare) columns. In the last step, the Mpro at high purity was subjected to a buffer exchange (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.5) for further experiments.
Inhibition Assay against Recombinant SARS-CoV-2 Mpro
IC50 values for inhibition of the recombinant SARS-CoV-2 main protease were determined by an FRET assay using the peptide Dabcyl-KTSAVLQ↓SGFRKM-E(Edans)-NH2 (Biosyntan) as the substrate (↓ indicates the cleavage site). The assay has been previously described,20 but we optimized both enzyme and substrate concentrations, as well as the buffer composition, to achieve higher sensitivity. Mpro (50 nM) was dissolved in buffer containing 20 mM HEPES, 120 mM NaCl, 0.4 mM EDTA, 20% glycerol, pH 7.0. Then, 4 mM DTT was added just prior to the measurements. The concentration of compounds 13b-K and 13b-H (in DMSO) was varied between 0 and 100 μM. After incubation of the enzyme and compound for 10 min at 37 °C, the reaction was initiated by adding the FRET substrate at a final concentration of 10 μM to each well (final volume: 100 μL/well). A Tecan Spark fluorescence plate reader was used, with an excitation wavelength of 360 nm and an emission wavelength of 460 nm. GraphPad Prism 9.2.0 software (GraphPad) was used for the calculation of the IC50 values. Measurements of inhibitory activities of the compounds were performed in triplicate and are presented as the mean ± SD.
Crystallization of Peptidomimetic Ligands in Complex with SARS-CoV-2 Mpro
The purified Mpro was concentrated to 13 mg/mL and mixed with 13b-H or 13b-K at a molar ratio of 1:5. The mixture was incubated in the cold room overnight. The next day, centrifugation (10,000g) was applied to remove precipitate from the Mpro–compound mixture. The supernatant was subjected to a basic co-crystallization screen with commercially available kits: PACT premier HT-96 (Molecular Dimensions) and Morpheus (Molecular Dimensions) for 13b-H and PEG Rx 1 & 2 and PACT premier HT-96 for 13b-K. The vapor diffusion sitting-drop method was employed at 20 °C with 0.15 μL of the protein–compound mixture and 0.15 μL of mother liquor in the drop, equilibrating against 40 μL of the reservoir. Crystals were observed in several different conditions for the two compounds in the presence of Mpro. Crystals were fished from different drops with the corresponding cryoprotectant. Liquid nitrogen was employed for flash-cooling of the fished crystals prior to diffraction data collection.
Diffraction Data Collection, Phase Determination, Model Building, and Refinement
A diffraction dataset for the structure of the Mpro in complex with 13b-H was collected from a crystal grown under the condition of PACT premier HT-96 number F7 (0.2 M sodium acetate trihydrate, 0.1 M Bis-Tris propane, pH 6.5, 20% w/v PEG 3350), with a cryoprotectant consisting of mother liquor plus 25% glycerol and 5 mM 13b-H. Data for the structure of the Mpro in complex with 13b-K was collected from a crystal fished from a drop prepared with the PEG Rx 2 kit (Hampton Research), condition number 21 (20% v/v 2-Propanol, 0.1 M MES monohydrate, pH 6.0, 20% PEG monomethyl ether 2000). Crystals were cryoprotected with mother liquor plus 15% glycerol and 2 mM 13b-K. All diffraction data sets were collected at the PETRA III P11 beamline of DESY, Hamburg, using a Pilatus 6M detector, at 100 K and a wavelength of 1.0332 Å.
Diffraction data sets were processed by using the programs XDSapp37 and Pointless38−40 and scaled by Scala.38,40 The molecular replacement method was used for determination of the structures, employing the Molrep program of the CCP4i suite,40,41 and the structure of the SARS-CoV-2 Mpro free enzyme (6Y2E20) as a search model. Geometric restraints for 13b-K and 13b-H were prepared by using the JLigand program,40,42 and the compounds were built into Fo–Fc difference density using the Coot software.43 The Refmac5 program40,44 was used for the refinement of the structures.
Detailed Description of the Crystal Structures
The Mpro complex with 13b-K yielded crystals of space group H3, with two complex units (A and B) per asymmetric unit (Table S1). The crystal structure was determined at 2.4 A resolution. While the electron density is well defined for molecule A, it is somewhat poor for protomer B. Because the pH of crystallization was 6.0 instead of 8.5 as for the crystals obtained from the mixture of 13b diastereomers, there is a deviation from the structure derived from the latter.20 Residue Glu166 appears to be protonated on its OE1 atom in molecule A of the Mpro dimer (but not in B), preventing it from forming the salt bridge with the N-terminal residue of molecule B (Ser1B) that is usually observed.20 Instead, it can only accept hydrogen bonds from the NH3+ and the side-chain OH of Ser1B, as well as from His172, whereas its (protonated) OE1 is devoid of any H-bonding interaction. As a consequence, the side chain of Glu166A has a somewhat different orientation from Glu166B. The conformation found in molecule A is actually very similar to that observed in a neutron structure of the free SARS-CoV-2 Mpro, crystals for which were grown at pH 6.6. Hydrogen has not been attached to Glu166 OE1 in the model for the neutron structure, but an Fo–Fc difference map suggests that there may be a partially occupied proton.
SARS-CoV-2 Antiviral Assay in Calu3 Cells
Infection studies with Calu3 cells were performed at the BSL4 laboratory of the Institute of Virology, Marburg, Germany. The human bronchial epithelial cell line Calu3 (HTB55; ATCC) was cultured in DMEM-F12 (Gibco cat. no. 31331-028) supplemented with 10% v/v heat-inactivated FBS, 1% penicillin–streptomycin (10,000 U mL–1) (Gibco cat. no. 15140122), and l-glutamine (200 mM) (100×, Gibco cat. no. 25030-024) in a humidified 5% CO2 incubator at 37 °C. Calu3 cells were seeded in 12-well plates (Greiner Bio-One, Cellstar 12-well culture plate cat. no. 665180) at a density of 1,000,000 cells per well in a culture medium. Twenty-four hours later, cells were infected with SARS-CoV-2 (BavPat1/2020 isolate, European Virus Archive Global #026 V-03883) using an MOI of 0.1 for 1 h in DMEM-F12 without FCS supplemented with penicillin–streptomycin and l-glutamine. Then, the infection medium was removed and culture media with different concentrations of 13b-K (0, 0.1, 2.5, 5, 10, and 20 μM) were added. The infected and treated cells were incubated (37 °C, 5% CO2) overnight, and 24 h post infection (pi) supernatants were collected. Viral releases into supernatants of infected cells were determined using the TCID50 method on VeroE6 cells as described previously.45 Briefly, VeroE6 cells (Vero C1008, ATCC, cat no. CRL-1586, RRID: CVCL_0574) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco cat. no. 21969-035) supplemented with 10% FBS, 1% glutamine, and 1% penicillin/streptomycin and seeded on 96-well plates. Serial dilutions of the infectious supernatants of treated and infected Calu3 cells were performed on sub-confluent VeroE6 cells for calculation of TCID50/mL.
SARS-CoV-2 Antiviral Assay in VeroE6 Cells
The SARS-CoV-2 antiviral assay was derived from the previously established SARS-CoV assay.46 In this assay, fluorescence of VeroE6-eGFP cells (provided by Dr. M. van Loock, J&JPRD; Beerse, Belgium) declines after infection with SARS-CoV-2 due to the cytopathogenic effect of the virus. In the presence of an antiviral compound, the cytopathogenicity is inhibited and the fluorescent signal is rescued. VeroE6-EGFP cells are maintained in DMEM (Gibco cat no 41965-039) supplemented with 10% v/v heat-inactivated fetal bovine serum (FBS) and 500 μg/mL G418 (Gibco cat no 10131-027). On day 1, compound 13b-K was serially diluted in 100 μL of assay medium (DMEM, supplemented with 2% v/v FCS) in 96-well black view plates (Greiner Bio-One, Vilvoorde). In the next step, 50 μL of VeroE6-eGFP cells (25,000 cells/well) was added together with the MDR1-inhibitor CP-100356 (final concentration: 0.5 μM). The plates were incubated (37 °C, 5% CO2 and 95% relative humidity) overnight. On day 0, 50 μL of SARS-CoV-2-GHB-03021/2020 was added at 20 TCID50/well and the plates were stored in a humidified incubator at 37 °C and 5% CO2. At 4 days pi, the wells were examined for eGFP expression using an argon laser-scanning microscope with excitation at 488 nm and emission at 510 nm, and the fluorescence images of the wells were converted into signal values. The results were expressed as EC50 values defined as the concentration of the compound achieving 50% rescue from the virus-reduced eGFP signals as compared to the untreated virus-infected control cells. Toxicity of compounds in the absence of virus was evaluated in a standard MTS assay as described previously.47
SARS-CoV-2 Antiviral Assay in Huh7 Cells
The human hepatoma cell line Huh7 (kindly provided by R. Bartenschlager, University of Heidelberg, Germany) was maintained in DMEM (Gibco cat no. 41965-039) supplemented with 10% v/v heat-inactivated FBS, 2% HEPES 1 M (Gibco cat no. 15630106), 5 mL of a solution of non-essential amino acids (NEAA Gibco cat no. 11140050), and 1% penicillin–streptomycin 10,000 U mL–1 (Gibco cat no. 15140148) in a humidified 5% CO2 incubator at 37 °C. The assay medium, used for producing virus stocks and antiviral testing, was prepared similarly but with 4% FBS. To quantify antiviral activity on Huh7 cells, we selected a SARS-CoV-2 virus strain that produces sufficient cytopathogenic effect (CPE) on this cell line. We started from passage 6 of the SARS-CoV-2 strain BetaCoV/Belgium/GHB-03021/2020 (EPI ISL 407976, 3 February 2020) that has been described previously48 and passaged this three additional times on Huh7 cells while selecting those cultures that showed the most CPE. This resulted in a virus stock (passage 9) that confers a full CPE on Huh7 (5.6 × 104 median tissue culture infectious dose (TCID50) per ml) as well as on VeroE6 cells (1.8 × 107 TCID50 per ml). The genotype of this virus stock shows four nucleotide changes as compared with the mother virus stock (P6), and these are currently being analyzed. None of the nucleotide changes occur in the part of the genome that encodes the main protease, validating this virus stock for testing protease inhibitors.
For antiviral testing, Huh7 cells were seeded in 96-well plates (Corning CellBIND 96-well Microplate cat no. 3300) at a density of 6000 cells per well in an assay medium. After overnight growth, cells were treated with the indicated compound concentrations and infected with a multiplicity of infection of 0.005 TCID50 per cell of the P9 virus (final volume of 200 μL per well in assay medium). On day 4 post infection, differences in cell viability caused by the virus-induced CPE or by compound-specific side effects were analyzed using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) as described previously.47 Cytotoxic effects caused by compound treatment alone were monitored in parallel plates containing mock-infected cells.
SARS-CoV-2 Antiviral Assay in A549-ACE2-TMPRSS2 Cells
A SARS-CoV-2 isolate alpha B.1.1.7 (derived from hCoV-19/Belgium/rega-12211513/2020; EPI_ISL_791333, 2020-12-21) was cultured in-house from nasopharyngeal swabs taken from a traveler returning to Belgium in December 2020. Virus stocks were first grown on VeroE6 and then on Calu-3 cells, and the tissue culture infectious doses (TCID50) were defined by end-point titration.49 A549-ACE2-TMPRSS2 cells (A549-Dual-hACE2-TMPRSS2) were obtained from InvivoGen (Toulouse, France). Cells were propagated in a growth medium prepared by supplementing DMEM (Gibco cat no 41965-039) with 10% v/v heat-inactivated FCS and 10 μg/mL blasticidin (InvivoGen ant-bl-05), 100 μg/mL hygromycin (InvivoGen ant-hg-1), 0.5 μg/mL puromycin (InvivoGen ant-pr-1), and 100 μg/mL zeocin (InvivoGen ant-zn-05). For antiviral testing, cells were seeded in an assay medium (DMEM supplemented with 2% v/v FCS) at a density of 15,000 cells/well in 96 well plates. The next day, the compound, serially diluted in an assay medium, was added and cells were infected with the SARS-CoV-2 B.1.1.7 isolate at a MOI of approximately 0.003 TCID50/cell in a final volume of 200 μL. On day 4 pi, differences in cell viability caused by virus-induced CPE or by compound-specific side effects were analyzed using MTS as described previously.47 Cytotoxic effects caused by compound treatment alone were monitored in parallel plates containing mock-infected cells.
All virus-related work with Vero E6, Huh7, and A549-ACE2-TMPRSS2 cells was conducted in the high-containment BSL3+ facilities of the KU Leuven Rega Institute (3CAPS) under licenses AMV 30112018 SBB 21920180892 and AMV 23102017 SBB 2192017 0589 according to institutional guidelines.
Pharmacokinetics
For pharmacokinetic experiments, outbred male CD-1 mice (Charles River, Germany), 4 weeks old, were used. The animal studies were conducted in accordance with the recommendations of the European Community (Directive 86/609/EEC, 24 November 1986). All animal procedures were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science and the European Health Law of the Federation of Laboratory Animal Science Associations. Animals were excluded from further analysis if sacrifice was necessary according to the human endpoints established by the ethical board. All experiments were approved by the ethical board of the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany. 13b-K was dissolved in a liposomal formulation containing a final concentration of 1 mM DOPS (1,2-dioleoyl-sn-glycero-3-phospho-l-serine sodium salt, Avanti Polar Lipids). Liposomes were prepared as described previously.5013b-K was administered at 25 mg/kg using an Aeroneb Nebulizer (KentScientific). At the time points 0.25, 0.5, 1, 2, 4, 8, and 24 h post administration, mice were euthanized to collect blood from the heart as well as to perform broncheoalveolar lavage (BALF) and to remove lungs aseptically. Furthermore, 13b-K was administered at 100 mg/kg peroral using intragastric gavage. At the time points 0.5, 1, and 3 post administration, up to 25 μL of blood were collected from the lateral tail vein. At 5 h post administration, mice were euthanized to collect blood from the heart as well as to perform broncheoalveolar lavage (BALF) and to remove lungs aseptically. Whole blood was collected into Eppendorf tubes coated with 0.5 M EDTA and immediately spun down at 15870g for 10 min at 4 °C. Then, plasma was transferred into a new Eppendorf tube and lungs were homogenized using a Polytron tissue homogenizer. BALF, lung, and plasma samples were stored at −80 °C until analysis. PK samples from the lung, BALF, and plasma were prepared as described previously.20 Furthermore, epithelial lining fluid (ELF) concentrations were calculated using the urea method as described previously.51 PK parameters and the AUC of BALF samples were determined using a non-compartmental analysis with PKSolver.52
Acknowledgments
G.P.M. and H.J.M. are grateful to Dr. Alex Moloney for review of the manuscript, Dr. Michael J Hall (Indicatrix Crystallography Ltd., UK) for small-molecule crystallization work, and Dr. Michael R Probert (Newcastle University, UK) for X-ray analysis of 12. R.H. is grateful to Dr. Haifa El Kilani for discussion. K.R. thanks Andrea Ahlers, Kimberley Vivien Sander, and Janine Schreiber for excellent technical assistance. R.H. and K.R. acknowledge funding from the German Center for Infection Research (DZIF; ALPHA-Keto, grant number TTU01.930). This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under grant no. 10100362 (the SCORE project). R.H. is also supported by the Government of Schleswig-Holstein through its Structure and Excellence Fund as well as by a close partnership between the Possehl Foundation (Lübeck) and the University of Lübeck. K.R. receives support from the German Center for Infection Research (DZIF, TTU 09.719) for operating the DZIF Pharmacokinetics/Pharmacodynamics Unit.
Glossary
ABBREVIATIONS
- Boc2O
Boc anhydride, di-tert-butyl dicarbonate
- DEAD
diethyl azodicarboxylate
- DIPEA
diisopropylethylamine
- DMP
Dess-Martin Periodinane
- EDC·HCl
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- FRET
Förster resonance energy transfer
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HOBt
1-hydroxybenzotriazole hydrate
- TEA
triethylamine
- TBME
tert-butyl methyl ether
- Tf2O
triflic anhydride, trifluoromethanesulfonic anhydride
- TIPSCl
triisopropylsilyl chloride
- TMSCl
trimethylsilyl chloride
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01131.
Molecular strings for all compounds, with associated biochemical and biological data (CSV)
Associated synthetic procedures, 1H NMR spectrum of compound 11 (13b), single-crystal X-ray diffraction data for 12 (13b-H), dose–response curves from the FRET-based assay, X-ray co-crystal structures of 13b-K and 13b-H in the substrate-binding cleft of the Mpro, levels of 13b-K in urine and BALF, alternative synthetic sequence toward 6, a stereo-controlled approach to analogue 16, SFC purity trace for 12 (13b-H), SFC purity trace for 13 (13b-K), diffraction data, and model refinement statistics (PDF)
Accession Codes
Crystallographic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) with accession codes 8A4Q (Mpro co-crystalized with 13b-H) and 8A4T (Mpro co-crystalized with 13b-K). CCDC 2113703 contains the supplementary crystallographic data for compound 12.
Author Contributions
◆ M.S.C. and L.Z. are co-first authors, having contributed equally. Conceptualization was done by M.S.C, G.P.M., H.J.M., and R.H. The investigation was contributed to by all authors. The manuscript was written by H.J.M., G.P.M., R.H., M.B., L.Z., L.S., S.B., and D.J. Funding acquisition was done by R.H., S.B., J.N., and K.R.
The authors declare the following competing financial interest(s): The University of Lubeck has filed a patent application covering compound 13b as well as related compounds, with LZ, Daizong Lin, and RH as inventors. Tocris Bioscience (part of Bio-Techne) have made (S, S, S)-Mpro 13b-K and (R, S, S)-Mpro 13b-H commercially available to researchers.
Notes
Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; Yuan M.-L.; Zhang Y.-L.; Dai F.-H.; Liu Y.; Wang Q.-M.; Zheng J.-J.; Xu L.; Holmes E. C.; Zhang Y.-Z. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.; Yang X.-L.; Wang X.-G.; Hu B.; Zhang L.; Zhang W.; Si H.-R.; Zhu Y.; Li B.; Huang C.-L.; Chen H.-D.; Chen J.; Luo Y.; Guo H.; Jiang R.-D.; Liu M.-Q.; Chen Y.; Shen X.-R.; Wang X.; Zheng X.-S.; Zhao K.; Chen Q.-J.; Deng F.; Liu L.-L.; Yan B.; Zhan F.-X.; Wang Y.-Y.; Xiao G.-F.; Shi Z.-L. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N.; Zhang D.; Wang W.; Li X.; Yang B.; Song J.; Zhao X.; Huang B.; Shi W.; Lu R.; Niu P.; Zhan F.; Ma X.; Wang D.; Xu W.; Wu G.; Gao G. F.; Tan W. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Coronavirus Disease (COVID-19) Dashboard (Available at Https://Covid19.Who.Int).
- Huang C.; Wang Y.; Li X.; Ren L.; Zhao J.; Hu Y.; Zhang L.; Fan G.; Xu J.; Gu X.; Cheng Z.; Yu T.; Xia J.; Wei Y.; Wu W.; Xie X.; Yin W.; Li H.; Liu M.; Xiao Y.; Gao H.; Guo L.; Xie J.; Wang G.; Jiang R.; Gao Z.; Jin Q.; Wang J.; Cao B. Clinical Features of Patients eInfected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms J.; Kremer S.; Merdji H.; Clere-Jehl R.; Schenck M.; Kummerlen C.; Collange O.; Boulay C.; Fafi-Kremer S.; Ohana M.; Anheim M.; Meziani F. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. 10.1056/NEJMc2008597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; Shao W.; Chen X.; Zhang B.; Wang G.; Zhang W. Real-World Effectiveness of COVID-19 Vaccines: A Literature Review and Meta-Analysis. Int. J. Infect. Dis. 2022, 114, 252–260. 10.1016/j.ijid.2021.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- V’kovski P.; Kratzel A.; Steiner S.; Stalder H.; Thiel V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J.; Lampe J.; Müller-Fielitz H.; Schuster R.; Zille M.; Müller K.; Krohn M.; Körbelin J.; Zhang L.; Özorhan Ü.; Neve V.; Wagner J. U. G.; Bojkova D.; Shumliakivska M.; Jiang Y.; Fähnrich A.; Ott F.; Sencio V.; Robil C.; Pfefferle S.; Sauve F.; Coêlho C. F. F.; Franz J.; Spiecker F.; Lembrich B.; Binder S.; Feller N.; König P.; Busch H.; Collin L.; Villaseñor R.; Jöhren O.; Altmeppen H. C.; Pasparakis M.; Dimmeler S.; Cinatl J.; Püschel K.; Zelic M.; Ofengeim D.; Stadelmann C.; Trottein F.; Nogueiras R.; Hilgenfeld R.; Glatzel M.; Prevot V.; Schwaninger M. The SARS-CoV-2 Main Protease M(pro) Causes Microvascular Brain Pathology by Cleaving NEMO in Brain Endothelial Cells. Nat. Neurosci. 2021, 24, 1522–1533. 10.1038/s41593-021-00926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeldt C. J.; Cerikan B.; Cortese M.; Frankish J.; Lee J.-Y.; Plociennikowska A.; Heigwer F.; Prasad V.; Joecks S.; Burkart S. S.; Zander D. Y.; Subramanian B.; Gimi R.; Padmanabhan S.; Iyer R.; Gendarme M.; El Debs B.; Halama N.; Merle U.; Boutros M.; Binder M.; Bartenschlager R. SARS-CoV-2 Infection Induces a pro-Inflammatory Cytokine Response through CGAS-STING and NF-ΚB. Commun. Biol. 2022, 5, 45. 10.1038/s42003-021-02983-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford B. The Path to Paxlovid. ACS Cent. Sci. 2022, 8, 405–407. 10.1021/acscentsci.2c00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Yang J. A Review of the Latest Research on Mpro Targeting SARS-COV Inhibitors. RSC Med. Chem. 2021, 12, 1026–1036. 10.1039/D1MD00066G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Liang C.; Xin L.; Ren X.; Tian L.; Ju X.; Li H.; Wang Y.; Zhao Q.; Liu H.; Cao W.; Xie X.; Zhang D.; Wang Y.; Jian Y. The Development of Coronavirus 3C-Like Protease (3CLpro) Inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711 10.1016/j.ejmech.2020.112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.-H.; Spasov K. A.; Reilly R. A.; Hollander K.; Stone E. A.; Ippolito J. A.; Liosi M.-E.; Deshmukh M. G.; Tirado-Rives J.; Zhang S.; Liang Z.; Miller S. J.; Isaacs F.; Lindenbach B. D.; Anderson K. S.; Jorgensen W. L. Optimization of Triarylpyridinone Inhibitors of the Main Protease of SARS-CoV-2 to Low-Nanomolar Antiviral Potency. ACS Med. Chem. Lett. 2021, 12, 1325–1332. 10.1021/acsmedchemlett.1c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. H.; Goins C. M.; Arya T.; Shin W.-J.; Maw J.; Hooper A.; Sonawane D. P.; Porter M. R.; Bannister B. E.; Crouch R. D.; Lindsey A. A.; Lakatos G.; Martinez S. R.; Alvarado J.; Akers W. S.; Wang N. S.; Jung J. U.; Macdonald J. D.; Stauffer S. R. Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). J. Med. Chem. 2022, 65, 2880–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W.; Groborz K.; Zhang L.; Sun X.; Zmudzinski M.; Pawlik B.; Wang X.; Jochmans D.; Neyts J.; Młynarski W.; Hilgenfeld R.; Drag M. SARS-CoV-2 M(pro) Inhibitors and Activity-Based Probes for Patient-Sample Imaging. Nat. Chem. Biol. 2021, 17, 222–228. 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An Oral SARS-CoV-2 M(pro) Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Qiao J.; Li Y.-S.; Zeng R.; Liu F.-L.; Luo R.-H.; Huang C.; Wang Y.-F.; Zhang J.; Quan B.; Shen C.; Mao X.; Liu X.; Sun W.; Yang W.; Ni X.; Wang K.; Xu L.; Duan Z.-L.; Zou Q.-C.; Zhang H.-L.; Qu W.; Long Y.-H.-P.; Li M.-H.; Yang R.-C.; Liu X.; You J.; Zhou Y.; Yao R.; Li W.-P.; Liu J.-M.; Chen P.; Liu Y.; Lin G.-F.; Yang X.; Zou J.; Li L.; Hu Y.; Lu G.-W.; Li W.-M.; Wei Y.-Q.; Zheng Y.-T.; Lei J.; Yang S. SARS-CoV-2 M(pro) Inhibitors with Antiviral Activity in a Transgenic Mouse Model. Science 2021, 371, 1374–1378. 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; Zhang B.; Jiang X.-M.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Peng J.; Liu F.; Li C.; Li Y.; Bai F.; Wang H.; Cheng X.; Cen X.; Hu S.; Yang X.; Wang J.; Liu X.; Xiao G.; Jiang H.; Rao Z.; Zhang L.-K.; Xu Y.; Yang H.; Liu H. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved a-Ketoamide Inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Kusov Y.; Nian Y.; Ma Q.; Wang J.; von Brunn A.; Leyssen P.; Lanko K.; Neyts J.; de Wilde A.; Snijder E. J.; Liu H.; Hilgenfeld R. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J.; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiem S.; Schentag J. J. Interpretation of Antibiotic Concentration Ratios Measured in Epithelial Lining Fluid. Antimicrob. Agents Chemother. 2008, 52, 24–36. 10.1128/AAC.00133-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A.; Akhtar M. F.; Mushtaq M. F.; Saleem M.; Muhammad S. T.; Akhtar B.; Sharif A.; Peerzada S. Current Trends in the Treatment of Hepatitis C: Interventions to Avoid Adverse Effects and Increase Effectiveness of Anti-HCV Drugs. EXCLI J. 2016, 15, 578–588. 10.17179/excli2016-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daeihamed M.; Dadashzadeh S.; Haeri A.; Akhlaghi M. F. Potential of Liposomes for Enhancement of Oral Drug Absorption. Curr. Drug Delivery 2016, 13, 289–303. 10.2174/1567201813666160115125756. [DOI] [PubMed] [Google Scholar]
- Martins S.; Sarmento B.; Ferreira D. C.; Souto E. B. Lipid-Based Colloidal Carriers for Peptide and Protein Delivery-Liposomes versus Lipid Nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [PMC free article] [PubMed] [Google Scholar]
- Renukuntla J.; Vadlapudi A. D.; Patel A.; Boddu S. H. S.; Mitra A. K. Approaches for Enhancing Oral Bioavailability of Peptides and Proteins. Int. J. Pharm. 2013, 447, 75–93. 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; Lopez de Castilla D.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.-D.; Ruiz-Palacios G. M.; Benfield T.; Fätkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C.; Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W. A. 2nd; Eron J. J. J.; Holman W.; Cohen M. S.; Fang L.; Szewczyk L. J.; Sheahan T. P.; Baric R.; Mollan K. R.; Wolfe C. R.; Duke E. R.; Azizad M. M.; Borroto-Esoda K.; Wohl D. A.; Coombs R. W.; James Loftis A.; Alabanza P.; Lipansky F.; Painter W. P. A Phase 2a Clinical Trial of Molnupiravir in Patients with COVID-19 Shows Accelerated SARS-CoV-2 RNA Clearance and Elimination of Infectious Virus. Sci. Transl. Med. 2022, 14, eabl7430 10.1126/scitranslmed.abl7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer S. M.; Squires K. E.; Hughes M. D.; Grimes J. M.; Demeter L. M.; Currier J. S.; Eron J. J. J.; Feinberg J. E.; Balfour H. H. J.; Deyton L. R.; Chodakewitz J. A.; Fischl M. A.; Phair J. P.; Pedneault L.; Nguyen B. Y.; Cook J. C. A Controlled Trial of Two Nucleoside Analogues plus Indinavir in Persons with Human Immunodeficiency Virus Infection and CD4 Cell Counts of 200 per Cubic Millimeter or Less. AIDS Clinical Trials Group 320 Study Team. N. Engl. J. Med. 1997, 337, 725–733. 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- Yang H.; Xie W.; Xue X.; Yang K.; Ma J.; Liang W.; Zhao Q.; Zhou Z.; Pei D.; Ziebuhr J.; Hilgenfeld R.; Yuen K. Y.; Wong L.; Gao G.; Chen S.; Chen Z.; Ma D.; Bartlam M.; Rao Z. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuten K.; Kim H.; Widen J. C.; Babin B. M.; Onguka O.; Lovell S.; Bolgi O.; Cerikan B.; Neufeldt C. J.; Cortese M.; Muir R. K.; Bennett J. M.; Geiss-Friedlander R.; Peters C.; Bartenschlager R.; Bogyo M. Challenges for Targeting SARS-CoV-2 Proteases as a Therapeutic Strategy for COVID-19. ACS Infect. Dis. 2021, 7, 1457–1468. 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote B. S.; Spooner L. M.; Belliveau P. P. Boceprevir: A Protease Inhibitor for the Treatment of Chronic Hepatitis C. Ann. Pharmacother. 2011, 45, 1085–1093. 10.1345/aph.1P744. [DOI] [PubMed] [Google Scholar]
- Bassetti M.; Vena A.; Russo A.; Peghin M. Inhaled Liposomal Antimicrobial Delivery in Lung Infections. Drugs 2020, 80, 1309–1318. 10.1007/s40265-020-01359-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P.; Collins I. Probing the Probes: Fitness Factors For Small Molecule Tools. Chem. Biol. 2010, 17, 561–577. 10.1016/j.chembiol.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler A. R.; Ragbirsingh R.; McMonagle C. J.; Waddell P. G.; Heaps S. E.; Steed J. W.; Thaw P.; Hall M. J.; Probert M. R. Encapsulated Nanodroplet Crystallization of Organic-Soluble Small Molecules. Chem 2020, 6, 1755–1765. 10.1016/j.chempr.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug M.; Weiss M. S.; Heinemann U.; Mueller U. XDSAPP: A Graphical User Interface for the Convenient Processing of Diffraction Data Using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. 10.1107/S0021889812011715. [DOI] [Google Scholar]
- Evans P. Scaling and Assessment of Data Quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Evans P. R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular Replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Lebedev A. A.; Young P.; Isupov M. N.; Moroz O. V.; Vagin A. A.; Murshudov G. N. JLigand: A Graphical Tool for the CCP4 Template-Restraint Library. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 431–440. 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halwe S.; Kupke A.; Vanshylla K.; Liberta F.; Gruell H.; Zehner M.; Rohde C.; Krähling V.; Gellhorn Serra M.; Kreer C.; Klüver M.; Sauerhering L.; Schmidt J.; Cai Z.; Han F.; Young D.; Yang G.; Widera M.; Koch M.; Werner A.; Kämper L.; Becker N.; Marlow M. S.; Eickmann M.; Ciesek S.; Schiele F.; Klein F.; Becker S. Intranasal Administration of a Monoclonal Neutralizing Antibody Protects Mice against SARS-CoV-2 Infection. Viruses 2021, 13, 1498. 10.3390/v13081498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens T.; Van den Eynde C.; Van Acker K.; Nijs E.; Dams G.; Bettens E.; Ohagen A.; Pauwels R.; Hertogs K. Development of a Homogeneous Screening Assay for Automated Detection of Antiviral Agents Active against Severe Acute Respiratory Syndrome-Associated Coronavirus. J. Virol. Methods 2005, 129, 56–63. 10.1016/j.jviromet.2005.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochmans D.; Leyssen P.; Neyts J. A Novel Method for High-Throughput Screening to Quantify Antiviral Activity against Viruses That Induce Limited CPE. J. Virol. Methods 2012, 183, 176–179. 10.1016/j.jviromet.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudewijns R.; Thibaut H. J.; Kaptein S. J. F.; Li R.; Vergote V.; Seldeslachts L.; Van Weyenbergh J.; De Keyzer C.; Bervoets L.; Sharma S.; Liesenborghs L.; Ma J.; Jansen S.; Van Looveren D.; Vercruysse T.; Wang X.; Jochmans D.; Martens E.; Roose K.; De Vlieger D.; Schepens B.; Van Buyten T.; Jacobs S.; Liu Y.; Martí-Carreras J.; Vanmechelen B.; Wawina-Bokalanga T.; Delang L.; Rocha-Pereira J.; Coelmont L.; Chiu W.; Leyssen P.; Heylen E.; Schols D.; Wang L.; Close L.; Matthijnssens J.; Van Ranst M.; Compernolle V.; Schramm G.; Van Laere K.; Saelens X.; Callewaert N.; Opdenakker G.; Maes P.; Weynand B.; Cawthorne C.; Vande Velde G.; Wang Z.; Neyts J.; Dallmeier K. STAT2 Signaling Restricts Viral Dissemination but Drives Severe Pneumonia in SARS-CoV-2 Infected Hamsters. Nat. Commun. 2020, 11, 5838. 10.1038/s41467-020-19684-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnabi R.; Boudewijns R.; Foo C. S.; Seldeslachts L.; Sanchez-Felipe L.; Zhang X.; Delang L.; Maes P.; Kaptein S. J. F.; Weynand B.; Vande Velde G.; Neyts J.; Dallmeier K. Comparing Infectivity and Virulence of Emerging SARS-CoV-2 Variants in Syrian Hamsters. EBioMedicine 2021, 68, 103403 10.1016/j.ebiom.2021.103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blockus S.; Sake S. M.; Wetzke M.; Grethe C.; Graalmann T.; Pils M.; Goffic R. L.; Galloux M.; Prochnow H.; Rox K.; Hüttel S.; Rupcic Z.; Wiegmann B.; Dijkman R.; Rameix-Welti M.-A.; Eléouët J.-F.; Duprex W. P.; Thiel V.; Hansen G.; Brönstrup M.; Haid S.; Pietschmann T. Labyrinthopeptins as Virolytic Inhibitors of Respiratory Syncytial Virus Cell Entry. Antiviral Res. 2020, 177, 104774 10.1016/j.antiviral.2020.104774. [DOI] [PubMed] [Google Scholar]
- Schütz C.; Ho D.-K.; Hamed M. M.; Abdelsamie A. S.; Röhrig T.; Herr C.; Kany A. M.; Rox K.; Schmelz S.; Siebenbürger L.; Wirth M.; Börger C.; Yahiaoui S.; Bals R.; Scrima A.; Blankenfeldt W.; Horstmann J. C.; Christmann R.; Murgia X.; Koch M.; Berwanger A.; Loretz B.; Hirsch A. K. H.; Hartmann R. W.; Lehr C.-M.; Empting M. A New PqsR Inverse Agonist Potentiates Tobramycin Efficacy to Eradicate Pseudomonas Aeruginosa Biofilms. Adv. Sci. 2021, 8, 2004369. 10.1002/advs.202004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Huo M.; Zhou J.; Xie S. PKSolver: An Add-in Program for Pharmacokinetic and Pharmacodynamic Data Analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.