

Keywords: channelopathies, developmental and epileptic encephalopathies, epileptogenesis, personalized treatment approaches, synaptopathies
Abstract
Developmental and epileptic encephalopathies (DEEs) are a heterogeneous group of disorders characterized by early-onset, often severe epileptic seizures and EEG abnormalities on a background of developmental impairment that tends to worsen as a consequence of epilepsy. DEEs may result from both nongenetic and genetic etiologies. Genetic DEEs have been associated with mutations in many genes involved in different functions including cell migration, proliferation, and organization, neuronal excitability, and synapse transmission and plasticity. Functional studies performed in different animal models and clinical trials on patients have contributed to elucidate pathophysiological mechanisms underlying many DEEs and have explored the efficacy of different treatments. Here, we provide an extensive review of the phenotypic spectrum included in the DEEs and of the genetic determinants and pathophysiological mechanisms underlying these conditions. We also provide a brief overview of the most effective treatment now available and of the emerging therapeutic approaches.
CLINICAL HIGHLIGHTS
Epilepsy is the third leading contributor to the global burden of neurological disorders and affects 65 million people worldwide. Based on clinical and EEG features, etiologies, and comorbidities, different categories of epilepsy types and syndromes are recognized. Among them, developmental and epileptic encephalopathies (DEEs) represent the most severe end of the spectrum. This review will help the readership of Physiological Reviews to increase their knowledge about the different types of DEEs, their etiologies and pathophysiological mechanisms, and available and emerging treatments.
1. INTRODUCTION TO THE CONCEPT AND PATHOPHYSIOLOGY OF DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHY
Epilepsy is the third leading contributor to the global burden of neurological disorders and affects 65 million people worldwide (1). Based on clinical and EEG features, etiologies, and comorbidities, different categories of epilepsy types and syndromes are recognized (2). Developmental encephalopathies (DEs) are a group of severe disorders with early onset of signs of developmental impairment, associated with other neurological symptoms such as autonomic dysfunction, behavioral disorders, and motor impairment (3). In DEs, developmental delay/impairment is a prominent feature, whereas the epileptic activity (seizures and EEG abnormalities) does not appear to be causally associated with developmental delay, stagnation, or regression. Epileptic encephalopathies (EEs) comprise a large, heterogeneous group of severe epilepsy syndromes characterized by several seizure types, frequent epileptiform activity on EEG, and developmental delay or regression (4). In EE, no preexisting developmental delay is observed, and the cause of delay is attributed to an interference that epilepsy has on physiological brain processes. However, when severe epilepsy has a very early onset, it is often impossible to know whether the underlying cause of the epileptic encephalopathy would not in itself have caused developmental delay even in the absence of epilepsy. For this reason, recent definitions refer to “developmental and epileptic encephalopathies” (DEEs) to designate a heterogeneous group of disorders characterized by early-onset, often severe epileptic seizures and EEG abnormalities on a background of developmental impairment that tends to worsen as a consequence of epilepsy (2). This generates a complex clinical picture in which both developmental abnormalities and severe epilepsy/EEG discharges contribute to the observed impairment, each to an extent that is hard to measure. The spectrum of DEEs by age of onset is described in TABLE 1, adopting the latest proposal for the classification of epilepsy syndromes by the International League Against Epilepsy (ILAE) Task Force on Nosology and Definitions (5–7). In certain DEEs, specific gene defects may create recognizable etiology-specific syndromes, whereas in others a variety of genetic variants may be associated with the same epilepsy syndrome. Such genetic heterogeneity is characteristic of the infantile spasms syndrome (ISS) and Lennox–Gastaut syndrome (LGS), in which genetic variants affecting distinct molecular or signaling pathways may lead to similar electroclinical syndromes (8–12). Moreover, DEEs may also result from nongenetic etiologies, including structural, toxic/metabolic, infectious, or immune, which may appear either independent of or in the setting of certain genetic etiologies (TABLE 1). Despite the advances in diagnostic tools, the underlying etiology may remain unknown in a significant portion of patients with specific syndromes. For example, in ISS, a third of the patients have unknown etiology, ∼24% have genetic or genetic-structural etiologies, and almost half have structural/metabolic or other acquired etiologies (13). Similar distributions of etiologies are also encountered in Lennox–Gastaut syndrome, although, as discussed above, increasing numbers of genetic variants and associations have emerged as use of genetic tests becomes more widespread (14).
Table 1.
Developmental and epileptic encephalopathies by age
| Age of Onset | Sex | Etiology | Electroclinical Characteristics | |
|---|---|---|---|---|
| Neonatal-infantile onset | ||||
| Early infantile DEEs (previously Ohtahara syndrome or early myoclonic epilepsy) | ≤3 mo of life | M = F | Genetic or structural/metabolic | Clinical: Abnormal neurological findings, neurodevelopmental deficits. Seizures: tonic (independent of sleep), myoclonic, epileptic spasms, sequential. |
| EEG: Burst suppression pattern; multifocal epileptiform discharges; hypsarrhythmia may appear. Seizure patterns are bilateral or focal onset, depending on seizure types. | ||||
| Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) | First year of life | M = F | Mainly genetic | Clinical: Neurodevelopmental delay, focal motor tonic or clonic seizures. |
| EEG: Migrating EEG patterns during ictal events, multifocal discharges | ||||
| Infantile spasms syndrome (ISS) | 3–12 mo (1–24 mo) | M > F | Structural/metabolic, genetic, unknown | Clinical: Epileptic spasms, other seizures may occur; neurodevelopmental disorders, intellectual disabilities. |
| EEG: Hypsarrhythmia, electrodecremental responses (ictal or interictal), multifocal epileptiform discharges. | ||||
| Dravet syndrome | 3–9 mo (1–20 mo) | M = F | Genetic | Clinical: Neurodevelopmental deficits. Prolonged hemiclonic seizure with fever in the absence of infectious/structural brain lesion; myoclonic, focal impaired awareness, atypical absences, atonic seizures, nonconvulsive status epilepticus, tonic and tonic-clonic seizures in sleep. |
| EEG: Focal or multifocal epileptiform abnormalities and seizures, photoparoxysmal responses. | ||||
| Etiology-specific syndromes: KCNQ2, CDKL5, PCDH19, SCL2A1, pyridoxine and pyridox(am)ine 5′-phosphate dependent epilepsy, glucose transporter 1 deficiency syndrome (Glu1DS), Sturge–Weber syndrome | First year of life | M = F; M < F (PCDH19, CDKL5) | Genetic | KCNQ2-DEE: Sequential or focal tonic seizures, burst suppression; autonomic symptoms, epileptic spasms. Burst suppression or multifocal EEG. |
| CDKL5-DEE: Tonic seizures, epileptic spasms. hypermotor tonic spasms; movement disorders. | ||||
| PDE, P5PDE: Intrauterine or early-life seizures; focal seizures, spasms or generalized tonic-clonic; response to pyridoxine or P5P. EEG: burst suppression or multifocal discharges. | ||||
| Glut1DS: Intellectual disability, low CSF-to-plasma glucose ratio, generalized seizures (myoclonic, myoclonic-atonic, generalized tonic-clonic, absences). EEG: 2.5–5 Hz spike wave. | ||||
| PCDH19-DEE: Intellectual disability, autism spectrum disorder; focal impaired aware to tonic or atypical absence seizures. EEG: focal onset seizures. | ||||
| Sturge–Weber: Facial port wine stain, progressive neurological course, epilepsy, hemiparesis, psychomotor delay, strokelike events, psychiatric disorders, glaucoma. Focal motor seizures, febrile seizures, infantile spasms, myoclonic-atonic, gelastic seizures. EEG: Asymmetric, focal epileptiform activities. | ||||
| Gelastic seizures with hypothalamic hamartoma | First year of life | M = F | Structural, genetic-structural | Clinical: Normal neurological exam initially, deficits appear later; precocious puberty; gelastic seizures with mirthless laughter (mandatory), gelastic and dacrystic seizures, focal impaired awareness or generalized seizures; other types of seizures may occur. |
| EEG: Focal or generalized | ||||
| Childhood onset | ||||
| Myoclonic-atonic epilepsy | 2–6 yr | M > F | Genetic | Clinical: Seizures: myoclonic-atonic (mandatory), atonic, myoclonic, absence, generalized tonic-clonic. |
| EEG: 3–6 Hz (poly)spike-slow wave discharges, generalized, activated in sleep; generalized paroxysmal fast. | ||||
| Lennox–Gastaut syndrome | 18 mo to 8 yr | M > F | Structural/metabolic, genetic | Clinical: Tonic seizures in sleep (mandatory), atypical absence, atonic, myoclonic, focal impaired awareness, generalized tonic-clonic, epileptic spasms. |
| EEG: Slow spike-wave (≤2.5 Hz), generalized; generalized paroxysmal fast; focal or multifocal slow spike-wave may be seen. | ||||
| DEE with SW activation in sleep (D/EE-SWAS) | 2–12 yr | M = F | Structural, genetic | Clinical: Neurocognitive/behavioral deficits that ameliorate/resolve with resolution of SWAS; seizure types depend on etiology: focal or focal to bilateral, typical, or atypical absences, atonic, negative myoclonus. |
| EEG: Slow background; focal or multifocal discharges; marked activation of diffuse epileptiform discharges in sleep (>50% of sleep, 1.5–2 Hz spike-wave runs). | ||||
| Febrile infection related epilepsy syndrome (FIRES) | 2–17 yr | M > F | Infectious/postinfectious | Clinical: Developmental regression, intellectual disabilities, attention or behavioral problems, motor dysfunction. Focal or multifocal seizures, superrefractory status epilepticus. |
| EEG: Slow background, multifocal epileptiform discharges; extreme delta brushes. Increasing frequency of focal onset seizures (focal >10 Hz evolving to rhythmic spike-waves). | ||||
| Hemiconvulsion-Hemiplegia-Epilepsy Syndrome (HHE) | <4 yr | M = F | Unknown, structural/metabolic, genetic | Clinical: Focal (clonic) febrile status epilepticus and persistent hemiparesis, aphasia when dominant hemisphere involved; focal or focal to bilateral motor seizures. |
| EEG: Focal or lateralized rhythmic ictal delta, focal recruiting (10 Hz) rhythms. | ||||
| Variable age onset | ||||
| Progressive myoclonus epilepsy (PME) | ||||
| Unverricht–Lundborg (EPM1) | 7–13 yr | M = F | Genetic | Clinical: progressive course; myoclonus induced by touch / photic stimulation, more pronounced upon awakening; other generalized seizures may occur. |
| EEG: Photosensitivity; generalized polyspike and wave (ictal). | ||||
| Lafora disease (EPM2) | 6–19 yr | M = F | Genetic | Clinical: Vision loss, cognitive decline, cerebellar symptoms. Myoclonic and generalized tonic-clonic seizures. |
| EEG: Photosensitivity; spike wave and polyspikes, no activation in sleep. | ||||
| Neuronal Ceroid Lipofuscinosis (NCL) | ||||
| CLN2 (late infantile) | 2–4 yr | M = F | Genetic | Clinical: Language delay, progressive course, multiple seizures febrile and afebrile, including myoclonic. |
| EEG: Photoparoxysmal response at low frequencies. | ||||
| Juvenile CLN3 | 4–10 yr | M = F | Genetic | Clinical: Visual loss progressive, macular degeneration, optic atrophy, retinitis pigmentosa. |
| EEG: Diffuse spike and wave discharges, slow background. | ||||
| Adult NCL (Kufs) | 11–50 yr | M = F | Genetic | Type A: PME with dementia and ataxia. |
| Type B: Dementia with cerebellar or extrapyramidal motor symptoms but not PME. | ||||
The list of developmental and epileptic encephalopathies (DEEs) follows the 2021 proposal of the International League Against Epilepsy (ILAE) Task Force on Nosology and Definitions for epilepsy syndromes (5–7). F, female; Glut1DS, Glucose Transporter 1 (SLC2A1) deficiency syndrome; M, male; NCL or CLN, neuronal ceroid lipofuscinosis; PDE, pyridoxine-dependent epilepsy; P5PDE, pyridox(am)ine 5′-phosphate (P5PD)-dependent epilepsy.
In DEEs, the co-occurrence of epilepsy and intellectual disability (ID) can involve at least two nonexclusive mechanisms. These mechanisms include uncontrolled frequent or prolonged seizures that can interfere with brain developing programs, resulting in inadequate construction of cortical networks and poor cognitive outcomes (15), as well as genetic mutations or adverse environmental factors that can induce both seizures and cognitive impairment. For example, genetic mutations inducing specific synaptic defects might cause seizures because of aberrant connectivity as well as intellectual disability because of altered synaptic plasticity (16). In many DEEs, epilepsy coexists with other comorbidities, both neurological and extraneurological. Neurological comorbidities differ in type and severity and span from subtle learning difficulties to psychiatric features, such as autism spectrum disorder (ASD) or depression to psychosocial concerns (2).
Despite their apparent phenotypic continuum, DEEs include a large collection of specific neurogenetic disorders. Several studies have been performed over the last two decades to identify molecular determinants and characterize pathophysiological mechanisms underlying DEEs, contributing to a greater understanding of their neurobiological and clinical aspects.
Next-generation molecular testing has boosted gene discovery for many human disorders. According to the Online Mendelian Inheritance in Man catalog (OMIM, https://www.omim.org), 172 genes have been identified as causative for epileptic encephalopathy, and among these 90 have been recognized to date as a cause of DEEs (TABLE 2 and SUPPLEMENTAL TABLE S1, available at https://doi.org/10.6084/m9.figshare.19666521.v2). However, this list may not be exhaustive, as the concept of DEEs is wide and encompasses a large number of conditions, which in up to 8% of individuals are caused by de novo copy number variants (CNVs) (17, 18).
Table 2.
Genes involved in epileptic encephalopathy and DEE pathogenesis
| Approved Symbol | Gene/Locus Name |
|---|---|
| Ion channels | |
| KCNA2 | Potassium channel, voltage-gated, Shaker-related subfamily, member 2 |
| CACNA1E | Calcium channel, voltage-dependent, alpha 1E subunit |
| KCNT2 | Potassium channel, subfamily T, member 2 |
| SCN3A | Sodium channel, voltage-gated, type III, alpha polypeptide |
| SCN2A | Sodium channel, voltage-gated, type II, alpha subunit |
| SCN1A | Sodium channel, voltage-gated, type I, alpha polypeptide |
| SCN9A | Sodium channel, voltage-gated, type IX, alpha subunit |
| CACNA2D2 | Calcium channel, voltage-dependent, alpha-2/delta subunit 2 |
| HCN1 | Hyperpolarization-activated cyclic nucleotide-gated potassium channel 1 |
| KCNQ5 | Potassium channel, voltage-gated, KQT-like subfamily, member 5 |
| KCNT1 | Potassium channel, subfamily T, member 1 |
| CACNA1B | Calcium channel, voltage-dependent, L type, alpha 1B subunit |
| SCN8A | Sodium channel, voltage gated, type VIII, alpha polypeptide |
| CACNA1G | Calcium channel, voltage-dependent, T type, alpha-1G subunit |
| CACNA1A | Calcium channel, voltage-dependent, P/Q type, alpha 1A subunit |
| SCN1B | Sodium channel, voltage-gated, type I, beta polypeptide |
| KCNB1 | Potassium voltage-gated channel, Shab-related subfamily, member 1 |
| KCNQ2 | Potassium voltage-gated channel, KQT-like subfamily, member 2 |
| CLCN4 | Chloride channel-4 |
| Receptors | |
| GABRA2 | Gamma-aminobutyric acid (GABA) A receptor, alpha-2 |
| GABRB1 | Gamma-aminobutyric acid (GABA) A receptor, beta-1 |
| GABRB2 | Gamma-aminobutyric acid (GABA) A receptor, beta-2 |
| GABRA1 | Gamma-aminobutyric acid (GABA) A receptor, alpha-1 |
| GABRG2 | Gamma-aminobutyric acid (GABA) A receptor, gamma-2 |
| NTRK2 | Neurotrophic tyrosine kinase, receptor, type 2 |
| GABBR2 | Gamma-aminobutyric acid B receptor 2 |
| GRIN1 | Glutamate receptor, ionotropic, N-methyl d-aspartate 1 |
| GABRB3 | Gamma-aminobutyric acid (GABA) A receptor, beta-3 |
| GABRA5 | Gamma-aminobutyric acid (GABA) A receptor, alpha-5 |
| GRIN2A | Glutamate receptor, ionotropic, N-methyl d-aspartate 2A |
| GRIN2D | Glutamate receptor, ionotropic, N-methyl d-aspartate 2D |
| Transporters | |
| ATP1A2 | ATPase, Na+-K+ transporting, alpha-2 polypeptide |
| SLC2A1 | Solute carrier family 2 (facilitated glucose transporter), member 1 |
| ARV1 | ARV1 homolog, fatty acid homeostasis modulator |
| SLC1A4 | Solute carrier family 1 (glutamate/neutral amino acid transporter), member 4 |
| SLC25A12 | Solute carrier family 25 (mitochondrial carrier, Aralar), member 12 |
| SLC6A1 | Solute carrier family 6 (neurotransmitter transporter, GABA), member 1 |
| SLC25A22 | Solute carrier family 25 (mitochondrial carrier, glutamate), member 22 |
| SLC1A2 | Solute carrier family 1 (glial high affinity glutamate transporter), member 2 |
| SLC13A5 | Solute carrier family 13 (sodium-dependent citrate transporter), member 5 |
| SLC25A10 | Solute carrier family 25 (mitochondrial carrier), member 10 (dicarboxylate ion carrier) |
| SLC25A42 | Solute carrier family 25, member 42 |
| ATP1A3 | ATPase, Na+-K+ transporting, alpha-3 polypeptide |
| SLC12A5 | Solute carrier family 12, (potassium-chloride transporter) member 5 |
| SLC35A2 | Solute carrier family 35 (UDP-galactose transporter), member 2 |
| SLC9A6 | Solute carrier family 9 (sodium/hydrogen exchanger), member 6 |
| Synapse related | |
| CPLX1 | Complexin 1 |
| PPP3CA | Protein phosphatase 3, catalytic subunit, alpha isoform (calcineurin A alpha) |
| SYNGAP1 | Synaptic Ras GTPase activating protein 1 |
| ADAM22 | ADAM metallopeptidase domain 22 |
| STXBP1 | Syntaxin-binding protein 1 |
| DNM1 | Dynamin-1 |
| NECAP1 | NECAP endocytosis-associated protein 1 |
| DMXL2 | DMX-like 2 |
| AP3B2 | Adaptor-related protein complex 3, beta2 subunit |
| STX1B | Syntaxin 1B |
| SYNJ1 | Synaptojanin 1 |
| NRXN1 | Neurexin 1 |
| Cell growth, division, and proliferation related | |
| MTOR | Mechanistic target of rapamycin |
| AKT3 | AKT serine/threonine kinase 3 |
| NPRL2 | NPR2-like protein, GATOR1 complex subunit |
| STAG1 | Stromal antigen 1 |
| RNF13 | RING finger protein 13 |
| PIK3CA | Phosphatidylinositol 3-kinase, catalytic, alpha |
| PPP2CA | Protein phosphatase-2 (formerly 2A), catalytic subunit, alpha isoform |
| ACTL6B | Actin-like 6B |
| RHOBTB2 | Rho-related BTB domain-containing protein 2 |
| TSC1 | Hamartin |
| AKT1 | AKT serine/threonine kinase 1 |
| NPRL3 | Nitrogen permease regulator-like 3 |
| TSC2 | Tuberin |
| PIK3R2 | Phosphatidylinositol 3-kinase, regulatory subunit 2 |
| DEPDC5 | DEP domain-containing protein 5 |
| Cell metabolism related | |
| MTHFR | Methylenetetrahydrofolate reductase |
| ST3GAL3 | ST3 beta-galactoside alpha-2,3-sialyltransferase 3 |
| PARS2 | Prolyl-tRNA synthetase 2 |
| HNRNPU | Heterogeneous nuclear ribonucleoprotein U |
| CAD | CAD trifunctional protein of pyrimidine biosynthesis |
| MDH1 | Malate dehydrogenase, soluble |
| UGP2 | Uridyl diphosphate glucose pyrophosphorylase-2 |
| BOLA3 | bolA family member 3 |
| ST3GAL5 | Sialyltransferase 9 |
| GAD1 | Glutamate decarboxylase-1, brain, 67 kDa |
| GLS | Glutaminase |
| D2HGDH | D-2-hydroxyglutarate dehydrogenase |
| UGDH | UDP-glucose dehydrogenase |
| MANBA | Mannosidase, beta A, lysosomal |
| NUS1 | NUS1 dehydrodolichyl diphosphate synthase subunit |
| MDH2 | Malate dehydrogenase, mitochondrial |
| DENND5A | DENN domain-containing protein 5A |
| NARS2 | Asparaginyl-tRNA synthetase 2 |
| ALG9 | ALG9 alpha-1,2-mannosyltransferase |
| FCSK | Fucose kinase |
| PNPO | Pyridoxamine 5′-phosphate oxidase |
| ITPA | Inosine triphosphatase-A |
| ALG13 | ALG13 UDP-N-acetylglucosaminyltransferase subunit |
| Intracellular trafficking related | |
| TRAK1 | Trafficking protein, kinesin-binding 1 |
| AP2M1 | Adaptor-related protein complex 2, mu 1 subunit |
| CAMK2G | Calcium/calmodulin-dependent protein kinase (CaM kinase) II gamma |
| TBC1D24 | TBC1 domain family, member 24 |
| NSF | N-ethylmaleimide-sensitive factor |
| CLTC | Clathrin, heavy polypeptide (Hc) |
| ARX | Aristaless-related homeobox, X-linked |
| Intracellular signaling related | |
| SZT2 | SZT2 subunit of KICSTOR complex |
| DOCK7 | Dedicator of cytokinesis 7 |
| YWHAG | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, gamma isoform |
| GNAO1 | Guanine nucleotide-binding protein (G protein), alpha-activating activity |
| PLCB1 | Phospholipase C, beta-1 |
| SIK1 | Salt-inducible kinase 1 |
| Transcription and gene expression related | |
| PUM1 | Pumilio RNA binding family member 1 |
| TSEN2 | tRNA splicing endonuclease, subunit 2 |
| PURA | Purine-rich element binding protein A |
| MEF2C | MADS box transcription enhancer factor 2, polypeptide C (myocyte enhancer factor 2C) |
| KMT2E | Lysine (K)-specific methyltransferase 2E |
| CELF2 | CUGbp- and ELAV-like family, member 2 |
| CUX2 | Cut-like homeobox 2 |
| FOXG1 | Forkhead box G1B |
| IRF2BPL | Interferon regulatory factor 2-binding protein like |
| CHD2 | Chromodomain helicase DNA binding protein-2 |
| NEUROD2 | Neurogenic differentiation 2 |
| MECP2 | Methyl-CpG-binding protein-2 |
| Protein biosynthesis/degradation related | |
| DHDDS | Dehydrodolichyl diphosphate synthase |
| ATP6V1A | ATPase, H+ transporting, V1 subunit A |
| UBA5 | Ubiquitin-like modifier activating enzyme 5 |
| GUF1 | GUF1 homolog, GTPase |
| VARS1 | Valyl-tRNA synthetase 1 |
| PLAA | Phospholipase A2-activating protein |
| CARS2 | Cysteinyl-tRNA synthetase 2 |
| AARS1 | Alanyl-tRNA synthetase 1 |
| EEF1A2 | Eukaryotic translation elongation factor-1, alpha-2 |
| PIGP | Phosphatidylinositol glycan, class P |
| PIGA | Phosphatidylinositol glycan, class A |
| Cytoskeletal proteins | |
| CYFIP2 | Cytoplasmic FMRP interacting protein 2 |
| PHACTR1 | Phosphatase and actin regulator 1 |
| SPTAN1 | Spectrin, alpha, nonerythrocytic-1 (alpha-fodrin) |
| Mitochondria proteins | |
| MFF | Mitochondrial fission factor |
| FARS2 | Phenylalanyl-tRNA synthetase 2, mitochondrial |
| RMND1 | Required for meiotic nuclear division 1 homolog |
| BRAT1 | BRCA1-associated ATM activator 1 |
| PMPCB | Peptidase, mitochondrial processing, beta |
| TWNK | Twinkle mtDNA helicase |
| DNM1L | Dynamin 1-like |
| POLG | Polymerase (DNA directed), gamma |
| GOT2 | Glutamic-oxaloacetic transaminase 2, mitochondrial |
| TIMM50 | Translocase of inner mitochondrial membrane 50 |
| Other/multiple-function proteins | |
| FBXO11 | F-box only protein 11 |
| MBD5 | Methyl-CpG-binding domain protein 5 |
| PTPN23 | Protein-tyrosine phosphatase, nonreceptor-type, 23 |
| DALRD3 | DALR anticodon-binding domain-containing protein 3 |
| SERPINI1 | Protease inhibitor 12 |
| FGF12 | Fibroblast growth factor-12 |
| CSNK2B | Casein kinase-2, beta polypeptide |
| CNPY3 | Canopy 3, zebrafish, homolog of |
| CDK19 | Cyclin-dependent kinase 19 |
| TRRAP | Transformation/transcription domain-associated protein |
| FRRS1L | Ferric-chelate reductase 1-like |
| COQ4 | Coenzyme Q4, S. cerevisiae, homolog of |
| ATN1 | Atrophin 1 |
| PACS2 | Phosphofurin acidic cluster sorting protein 2 |
| PIGB | Phosphatidylinositol glycan, class B |
| PIGQ | Phosphatidylinositol glycan anchor biosynthesis class Q protein |
| ROGDI | Rogdi atypical leucine zipper |
| WWOX | WW domain-containing oxidoreductase |
| PIGS | Phosphatidylinositol glycan, class S |
| TCF4 | Transcription factor-4 (immunoglobulin transcription factor-2) |
| PNKP | Polynucleotide kinase 3′ phosphatase |
| CDKL5 | Cyclin-dependent kinase-like 5 (serine/threonine protein kinase 9) |
| CASK | Calcium/calmodulin-dependent serine protein kinase |
| SMC1A | Segregation of mitotic chromosomes 1 (SMC1, yeast, homolog of; DXS423E; SB1.8) |
| FGF13 | Fibroblast growth factor-13 |
| ARHGEF9 | Rho guanine nucleotide exchange factor 9 |
| PCDH19 | Protocadherin 19 |
Genes gathered from OMIM (https://www.omim.org/) interrogated on October 27, 2021, with the keywords “epileptic encephalopathy” and “developmental and epileptic encephalopathy.” See SUPPLEMENTAL TABLE S1 for more details. DEE, developmental and epileptic encephalopathy.
The study of large cohorts of affected individuals with variable but related phenotypes performed with next-generation sequencing (NGS) approaches including targeted gene panels, whole exome and whole genome sequencing, has now demonstrated that 30–50% of DEEs can be attributed to de novo pathogenic variants in single genes (9, 16, 19). Low-level somatic mosaicism is observed in ∼10% of parents of probands with DEEs (20, 21), with important consequences for recurrence risk estimation. In addition to single de novo variants, DEE pathogenesis has also been associated with recessive mutations in 11–38% of patients (22, 23). Additional genetic mechanisms, which are often not detectable by standard NGS methodology, might also contribute to DEE pathophysiology, including nonexonic variants, brain mosaicism in the patient, oligogenic inheritance, and epigenetic changes.
The contribution of nonexonic variants is mainly related to “poison” exons that, when spliced into an RNA transcript, trigger nonsense-mediated decay (NMD), a surveillance system that detects and degrades RNAs harboring premature termination codons (PTCs) (FIGURE 1). An example of how such a mechanism is associated with DEEs has been described by Carvill and collaborators (24), who sequenced 11 noncoding candidate regions of the SCN1A gene in 640 individuals with unsolved DEE, selected on the basis of their evolutionary conservation and functional features. The authors identified five variants in intron 20 that promoted inclusion of a poison exon and caused a reduction of the amount of full-length SCN1A protein (24). Since transcriptome studies on purified neural progenitor cells (NPCs) have identified hundreds of differentially spliced exons (25), reduced gene expression due to inclusion of poison exons may represent an etiological mechanism of a broad range of neurological diseases, including DEEs.
FIGURE 1.
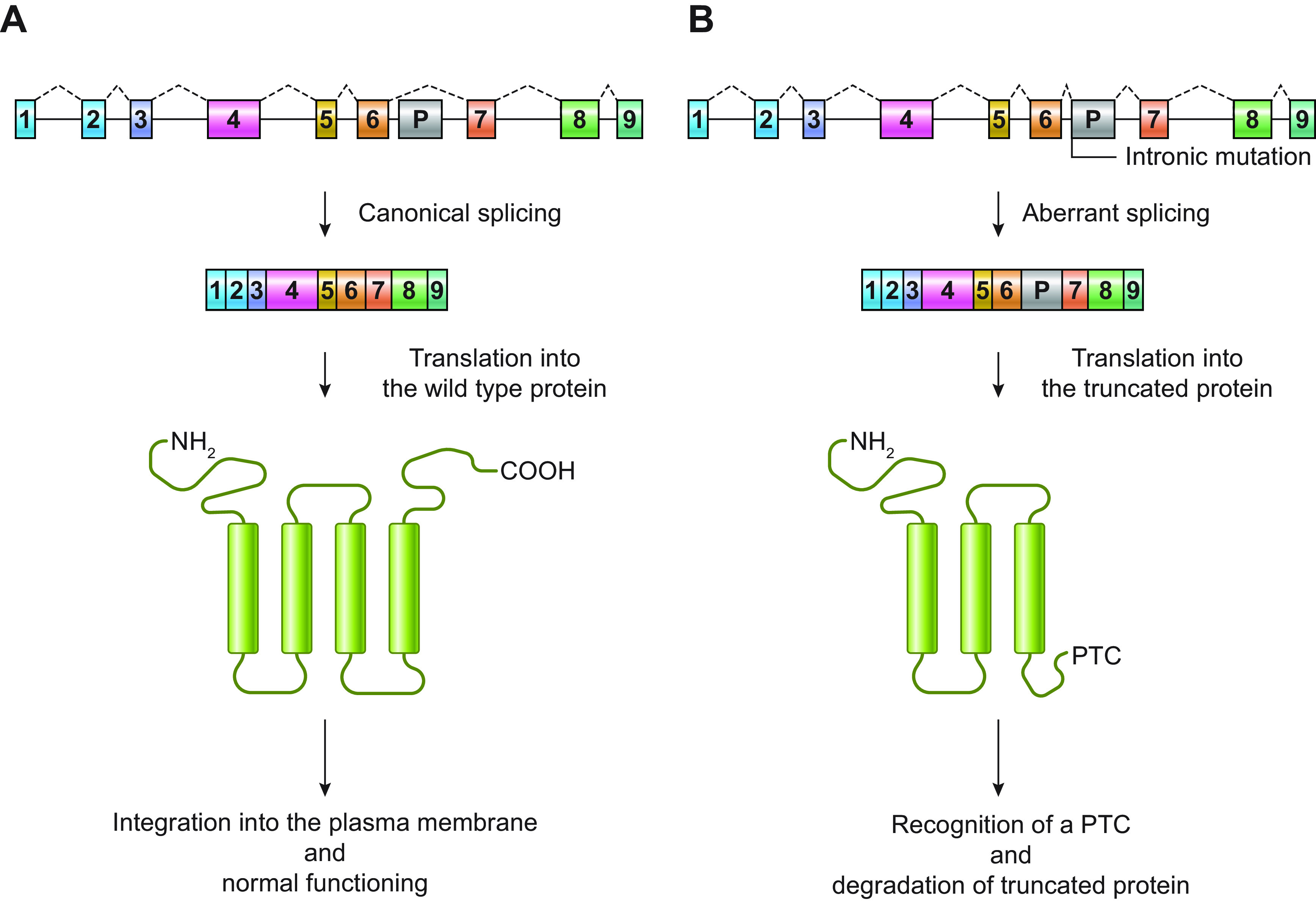
Schematic representation of an example of “poison exon”-mediated protein degradation. A shows a hypothetical gene encoding a transmembrane protein with 4 transmembrane segments. The gene comprises 9 coding exons (1–9) and a potential poison exon (P). In the canonical splicing, the poison exon is not included in the mRNA, which is translated into the wild-type protein. After translation, the protein is correctly integrated into the plasma membrane, where it exerts its normal function. In B, the presence of an intronic mutation, which can introduce a novel splicing acceptor site, activates an exonic splicing enhancer (ESE; i.e., a sequence that promotes the inclusion of an exon in an mRNA) or disrupts an exonic splicing silencer (ESS; i.e., a sequence that inhibits the inclusion of an exon in an mRNA), promoting the inclusion of the poison exon in the mRNA. The poison exon alters protein amino acid sequence and introduces a premature stop codon (PTC). The PTC is recognized by cellular surveillance systems, and the mutant protein is degraded.
Somatic mosaicism is the result of a variant arising at postzygotic level, which is inherited by daughter cells via mitotic division and results in genetically distinct subsets of cells in the same individual. Based on the timing when the variant arises, it can affect one or multiple tissues. Deep sequencing studies in dysplastic brain/blood paired samples have correlated brain-confined mutations with focal malformations of cortical development (MCDs). Mutations in the mechanistic/mammalian target of rapamycin (mTOR) pathway genes AKT1, AKT3, DEPDC5, MTOR, NPRL2, NPRL3, PIK3CA, PIK3R2, TSC1, and TSC2 can now be considered as the major known cause of focal cortical dysplasia type II (FCDII) and hemimegalencephaly (HME) (26), whereas somatic mutations in SLC35A2, encoding a UDP-galactose transporter, have been identified in a limited number of patients with FCD type I, mild malformations of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE), and nonlesional focal epilepsy (NLFE) (27–31). In addition, somatic variants, either microdeletions in SCN1A or point mutations in CDKL5, PCDH19, SCN1A, and SCN2A, have been identified in 1–3.5% of patients with DEE who were mutation negative to single gene, multigene epilepsy panel, or whole exome sequencing (WES) performed in a clinical diagnostic setting (32, 33). Despite this evidence, the impact of somatic mutations in DEEs is likely to be underestimated, as the major challenge in detecting causative somatic mutations is analyzing the right target brain tissue, which, for many DEE patients, is not easily accessible. For this reason, recent assays developed from noninvasive protocols for tumor diagnosis and progression monitoring have been attempted to identify low-level mosaicism for known or recurrent mutations associated with lesional refractory epilepsy in free DNA extracted from cerebrospinal fluid biopsies (34, 35).
Oligogenic inheritance refers to the concept that, in some cases, diseases with complex phenotypes are not inherited as simple single-gene Mendelian disorders, nor they are classic complex traits, but rather fit a model in which mutations in a small number of genes may interact genetically to manifest a phenotype (36). In oligogenic conditions, one of the genes is the major disease-causing gene, while the others act as modifiers. Some DEEs can be attributed to this type of inheritance. Indeed, various studies have demonstrated that co-occurrence of two or more mutations in distinct ion channel genes can contribute to the epileptic phenotype in both patients and animal models (37–42). Performing targeted and whole exome sequencing in a boy who presented with profound developmental delay, failure to thrive, ataxia, hypotonia, and tonic-clonic seizures that caused his death, Hasan and collaborators (40) identified a pathogenic variant in KCNJ10, encoding the inward-rectifying K+ channel Kir4.1 and a pathogenic variant in KCNT1, encoding the Na+-activated K+ channel known as Slo2.2 or SLACK. Functional studies performed in in Xenopus laevis oocytes confirmed the functional effects of the two variants. This finding revealed that, when co-occurring, pathogenic variants in Kir4.1 and SLACK result in a fatal disease (40). To gain support for the hypothesis that genetic modifiers can influence clinical presentation in patients with SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+), Hawkins and collaborators (38) combined the Scn1a-R1648H allele with either Scn2aQ54, which causes spontaneous, adult-onset partial motor seizures, or Kcnq2V182M/+, which causes increased susceptibility to induced seizures. Double-heterozygous mice exhibited early-onset, generalized tonic-clonic seizures and juvenile lethality. These results demonstrate that variants in Scn2a and Kcnq2 can dramatically worsen the phenotype of mice carrying the Scn1a-R1648H mutation (38).
A digenic inheritance based on somatic mutations in two mTOR pathway genes (MTOR and RPS6) has also been demonstrated in association with hemimegalencephaly and intractable epilepsy (43).
Aberrant chromatin states leading to altered gene expression patterns (epimutations) have been detected in several conditions. Epimutations can occur secondary to a DNA mutation in a cis- or trans-acting factor (secondary epimutations) or as “true” or primary epimutations in the absence of any DNA sequence change (44). In the brain, such alterations can impair the transfer of information that binds short-lived cellular signals to the whole neuronal activity and the global gene expression (45). Emerging findings in animal models and human brain tissue have demonstrated that DEEs could be ascribed to both classes of epimutations. A classic example of DEE associated with secondary epimutations is Rett syndrome, which can be caused by both nucleotide mutations in and duplications of MECP2, encoding a methylated DNA-binding protein (46).
Alterations in DNA methylation have not been explored much in epilepsy yet, but they represent a good example of primary epimutations related to DEEs. Indeed, in the brain this epigenetic modification regulates cell fate determination and maturation and plays a fundamental role in the induction of activity-dependent synaptic plasticity, memory, and cognition (47–49). Dynamic alterations in DNA methylation can also contribute to epileptogenesis. For example, various studies have shown that the brain-derived neurotrophic factor (BDNF), whose expression is regulated by a variety of cellular processes, including methylation of its promoter (50), results upregulated in brain areas implicated in epileptogenesis (51). In addition, in hippocampal neurons, the inhibition of DNA methyltransferases, which mediate demethylation of the BDNF promoter, results in the suppression of neuronal excitability and network activity (52).
Various nongenetic etiologies have also been associated with certain DEEs, including hypoxic-ischemic encephalopathy (HIE), cerebrovascular, infectious, or autoimmune disorders, tumors, brain trauma, and metabolic disorders (5–7, 13, 53). Although the molecular pathogenesis of these etiologies is more complex and multifactorial than genetic etiologies, they offer a setting in which questions of “common pathways or mechanisms” in disease pathogenesis, network dysfunction, and drug refractoriness can be explored to develop therapies with broader applications. For example, interneuronopathies, as well as mTOR dysregulation, have been implicated in both genetic- and nongenetic-etiology DEEs (54, 55).
Many pathophysiological mechanisms underlie epilepsy and cognitive phenotypes in DEEs, leading to either dysfunction of specific cortical networks or more generalized epileptogenic changes. These multiple and intersecting mechanisms make genotype-functional phenotype correlations difficult. Cortical and subcortical neuronal networks may interact with each other and, in turn, can cause widespread functional changes in otherwise normal cortex (56). The firing of excitatory cortical neurons is finely regulated by the interplay of sodium and potassium channel activity, which is mediated by chemical and ionic gradients across the cell membrane. If the balance between sodium and potassium gradients is perturbed (for example because of mutations in genes encoding Na+ or K+ ion channels or for components of the Na+-K+ pump) (TABLE 2), abnormal depolarization arises, which in turn causes abnormal neuronal activity and cortical excitability (57). Altered interconnections of glutamatergic neurons are another possible cause of abnormal depolarization, and mutations in various genes encoding for glutamate receptors or carriers (TABLE 2) have been associated with different DEEs (58–63). In addition to altered firing in excitatory neurons, epileptogenesis in DEEs may be related to dysfunctions in interneuron networks. An archetype of genetic lesions associated with such a mechanism is represented by SCN1A mutations, which are associated with Dravet syndrome and a broad category of other epilepsy phenotypes. A series of functional studies have highlighted how changes in membrane properties of one specific cellular population result in altered neuronal network dynamics and widespread cortical dysfunction, in turn leading to an epileptic phenotype. SCN1A mutations are mainly associated with loss of function (LoF) in a subtype of voltage-gated sodium channels, the NaV1.1 channel, mainly expressed in inhibitory neurons (64). This functional alteration would be expected to predispose to decreased neuronal activity. However, in vivo, it is actually associated with increased epileptogenicity, as it results in severely impaired sodium currents and action potential (AP) firing in γ-aminobutyric acid (GABA)ergic inhibitory neurons, without detectable effects on excitatory pyramidal neurons (64, 65). Therefore, the epileptogenic effects of SCN1A mutations are primarily mediated by an altered activity of inhibitory interneurons in the cortex rather than by abnormal firing of excitatory neurons (66). Recent single-cell RNA sequencing (RNAseq) studies performed in postmortem adult human and rodent brain tissues have confirmed that SCN1A (Scn1a) is predominantly expressed in inhibitory neurons (67). Conversely, SCN2A/3A/8A (Scn2a/3a/8a), which also encode for voltage-gated sodium channels, are preferentially expressed in excitatory neurons in multiple brain regions, suggesting that epilepsy due to mutations in these genes is mainly associated with direct alterations of the excitatory conductance (67). Expression and in vitro and in vivo electrophysiological studies are therefore crucial to establish the functional effects of channel mutations, which may differ in the same gene families or, as we shall see, even with the same gene.
Mutations in genes encoding glutamate receptors or ion channels can also cause epileptogenic structural brain abnormalities. Using the DREADD (designer receptor exclusively activated by a designer drug) approach, Hurni and collaborators (68) showed that transient embryonic activation of migrating projection neurons (PNs) induced transcriptional changes in a variety of activity-dependent receptors, including glutamatergic metabotropic, kainate, NMDA, and AMPA receptors, that were accompanied by premature branching and persistent laminar mispositioning of superficial layer PNs into deep cortical layers, without affecting expression of layer-specific molecular identity markers. These findings support the hypothesis that increased intrinsic activity during migration, a condition that can be caused also by mutations in DEE-causing genes, acts as a stop signal for migrating cortical PNs (68).
With a similar DREADD-based approach it has been demonstrated that in the developing mouse neocortex ventricular zone progenitors become more hyperpolarized as they generate successive subtypes of neurons (69). Experimental in vivo hyperpolarization shifted the transcriptional programs and division modes of these progenitors to a later developmental status, with precocious generation of intermediate progenitors and a forward shift in the laminar, molecular, morphological, and circuit features of their neuronal progeny (69). These findings indicate that, during development, altered bioelectrical processes can also affect nonexcitable cells, including neuronal progenitors.
Genetic background may also alter the genotype-phenotype associations. This is well known in humans and has also been demonstrated in mouse models from different laboratories. A striking example was presented by Glasscock and collaborators (37) whereby combination of a Kcna1 knockout and a Cacna1a missense mutation masked the absence epilepsy associated with Cacna1a and attenuated limbic seizures and death expected from the Kcna1-null mutation. The C57BL/6 background also confers a more severe phenotype in Nav1.1+/− mice with targeted deletion of the last encoding exon than the 129/SvJ background (70). In the C57BL/6 background, Nav1.1+/− mice manifest hyperthermia-induced and spontaneous seizures, cognitive and behavioral deficits, and early mortality. In contrast, in the 129/SvJ background mice have hyperthermia-induced seizures, less severe spontaneous seizures, and no cognitive deficits.
2. GENERAL CONCEPTS OF EPILEPTOGENESIS AND EPILEPSY
Epileptogenesis is the chronic process by which a brain network is functionally altered toward increased seizure susceptibility, thus having an enhanced probability of generating spontaneous and recurrent seizures. For this reason, epileptogenesis has been traditionally considered in the context of a “latent period” between the causative insult and the first clinical seizure (71). This concept, however, although applicable to several acquired conditions, especially posttraumatic, poststroke, or postinfectious epilepsy, appears to be less apt to describe what happens in the context of genetically determined DEEs, where the bases for epileptogenesis are most often imbricated with the altered dynamics of brain development and neural networking. Irrespective of its causes, however, epilepsy is defined by recurrent and unprovoked seizures and can be divided into different categories, which are defined as generalized, focal (formerly called partial), and combined generalized (57, 71) according to how the epileptogenic process is distributed in the brain. These categories are defined based on the predominant types of seizures, including generalized or focal onset (1, 2, 72).
A seizure can be conceptualized as the result of a distortion of the normal balance between excitation (E) and inhibition (I), resulting from factors that may alter brain function at many levels, from subcellular signaling cascades to widespread neuronal circuits (1). Genetic factors (i.e., mutations in specific genes) can affect brain function at any level, from ion channels to receptor function and synaptic connectivity. Acquired cerebral insults (e.g., stroke or traumatic brain injury) are mainly associated with alterations in circuit function (1). Excessive neuronal firing alone does not necessarily cause a seizure, which also requires synchronization of a network of neurons. Glutamatergic interconnections can promote such synchronization. Application of a proconvulsant agent to hippocampal slices in a feline model precipitates interictal discharges whose intracellular correlate is the paroxysmal depolarizing shift (PDS). PDS is a network-driven burst associated with a sudden depolarization of the membrane potential that lasts hundreds of milliseconds and usually triggers a series of APs on its rising phase. Since cortical pyramidal cells are richly interconnected by excitatory glutamatergic synapses, it has been proposed that the mechanism underlying PDS is a “giant” excitatory postsynaptic potential (EPSP) (73).
Gap junctions (GJs) are another possible promoter of neuron synchronization. These specialized intercellular connections allow a “low-resistance” pathway of current flow from one cell to another that, in neurons, may determine a rapid and effective synchronization. In addition, GJs located between proximal axons of principal neurons (axon-axon gap junctions) can promote epileptogenesis by providing pathways for direct spread of APs across neurons. A by-product of such spread is the ability of axonally coupled neurons to generate oscillations at very high frequencies (≥70 Hz) (74). Seizure activity, both in vivo and in vitro, begins with very high-frequency oscillations, suggesting that axon-axon GJs may play a major role in seizure initiation (74). Besides interneuronal GJs, interglial GJs can also be considered an important mechanism for seizure generation (75). Studying mice with coupling-deficient astrocytes, obtained by crossing conditional connexin-43-deficient mice (connexin-43Cx43fl/fl:hGFAP-Cre) with connexin-30−/− mice, Wallraff and collaborators (76) showed how gap junctions in the impaired astrocytes accelerated potassium clearance, limited potassium accumulation during synchronized neuronal firing, and reduced the threshold for the generation of epileptiform events. Other studies, conducted in a similar in vivo model, demonstrated that impaired glucose metabolism through astroglial networks can contribute to epileptiform activity (77).
A third mechanism of enhanced synchronization is based on impaired inhibition. Individual GABAergic interneurons can effectively phase spontaneous firing in hippocampal pyramidal cells due to the interaction between GABAA receptor-mediated hyperpolarizing synaptic events and intrinsic oscillatory mechanisms in the pyramidal cells. Since interneurons make numerous connections to pyramidal cells in local areas, a single discharging interneuron can synchronously hyperpolarize a large number of pyramidal cells. Once GABAergic inhibition ceases, there is activation of voltage-dependent currents in pyramidal cells, resulting in a synchronous depolarization of a number of cells that might be high enough to trigger seizures (78).
Certain signature seizures and EEG patterns in DEEs also highlight the importance of the corticothalamic network [spike-wave discharge (SWD)/atypical absences, generalized seizures] or brain stem structures (tonic seizures, spasms) in the generation or development of generalized seizure activity.
The thalamus serves as a gate in trafficking sensory information to and from the cerebral cortex, and its activity is controlled by the basal ganglia (79). The sensory relay thalamic (TC) neurons form reciprocal glutamatergic connections with the cortex but also project to the GABAergic neurons of the nucleus thalami reticularis (nRT). The nRT neurons receive excitatory input from both the cortex and TC neurons and send inhibitory projections to TC neurons (FIGURE 2A). Additional inhibitory input to the TC relay neurons also stems from the local GABAergic interneurons (FIGURE 2A). The SWDs generated after a burst of excitatory postsynaptic potentials (EPSPs) from the TC neurons excites the nRT interneurons, which in turn send bursts of inhibitory input to the TC neurons causing pronounced inhibitory postsynaptic potentials (IPSPs) (GABAAR and GABABR IPSPs; FIGURE 2A) leading to activation of hyperpolarization-activated cyclic nucleotide-gated cation (HCN) channels and low-threshold calcium (T) channels, which cause a calcium spike that triggers a burst of APs. The reexcitation of TC neurons gives rise to a new cycle by eventually also engaging cortical neurons. The typical SWD in humans is ∼3 Hz, whereas in models of absence and in many control rodents, typical SWDs are 7–8 Hz (79).
FIGURE 2.

Schematic representation of the cortical and subcortical zones and mechanisms involved in epileptic seizure generation and spreading. A: schematic model of networks involved in spike and wave generation in generalized epilepsies. Thalamic relay neurons in the thalamocortical circuit can activate cortical pyramidal neurons and vice versa. Thalamus-mediated cortical activation is largely controlled by thalamic reticular neurons. They hyperpolarize thalamic relay neurons through γ-aminobutyric acid type B (GABAB)-mediated signals and are themselves inhibited by neighboring reticular neurons through GABA type A (GABAA)-mediated signals. Cortical pyramidal neurons can, in turn, activate thalamic reticular neurons in a glutamate-mediated feedforward loop. The neuronal basis of the EEG spike and wave in this reverberating loop derives from an alternance of the summated outside-negative excitatory membrane events (each spike) and the summated outside-positive inhibitory membrane events (each slow wave). Spike and waves appear as negative (upward going) events because of a dipole effect as the soma and apical dendrites maintain opposite polarity. B: in the epileptic brain, focal epileptic seizures are generated in the epileptogenic zone (EZ), whereas clinical seizures are generated in the seizure onset zone (SOZ). If the EZ is larger than the SOZ, as in the case depicted, its complete removal is required to guarantee seizures disappearing, as multiple SOZs with different thresholds may coexist in the same EZ. Complete EZ disconnection or removal is also required to ensure that seizures do not spread to other areas connected to it via cortico-cortical and subcortical (i.e. thalamocortical) connections (purple arrows), which can cause secondary generalization. Additional specific cortical areas that can be identified in the epileptic brain are the epileptogenic lesion (EL), which may correspond to either a macroscopic epileptogenic lesion (e.g. focal cortical dysplasia, as shown) or hyperexcitable adjacent cortex, the irritative zone (IZ), representing the area of the normal cortex generating interictal spikes, and the functional deficit zone (FDZ), representing the area of the cortex that does not function normally in the interictal period.
Atypical absence seizures (AASs) are slower (<2.5 Hz in humans, usually 3–6 Hz in rodents), may have atypical morphology, and may not necessarily impair awareness. AASs also utilize the corticothalamic network, although there may be more inputs from the limbic structures that result in augmented cortical excitation. Certain forms of atypical SWD may also be generated from isolated cortex or thalamus (80, 81). The utilization of existing circuitry and networks for the generation of these seizures allows for transitional states, during which SWDs may arise from physiological rhythms such as sleep spindles, when cortical neurons become hyperresponsive to the thalamocortical excitatory input (82).
Although our knowledge about the functional bases of generalized epileptogenesis and seizures is largely based on experimental models, studies on focal epileptogenesis have taken advantage of intracranial recordings in the setting of neurosurgery for epilepsy patients. These studies have contributed to shaping the now widely accepted concept that clinical seizures in focal epilepsies originate in the “seizure-onset zone” (SOZ), whereas epileptic seizure activity is generated in the epileptogenic zone (EZ), i.e., the cortical area that is indispensable for seizure generation independent of their clinical manifestation (83). Additional specific cortical areas that can be identified in the epileptic brain are the irritative zone (IZ), representing the area of the cortex generating interictal spikes, the epileptogenic lesion (EL), which may correspond to either a macroscopic epileptogenic lesion (e.g., focal cortical dysplasia) or the hyperexcitable adjacent cortex, and the functional deficit zone (FDZ), representing the area of cortex that does not function normally in the interictal period (84) (FIGURE 2B). The EZ may be either more or less extensive than the SOZ. If the EZ is smaller than the SOZ, even its partial resection or disconnection may result in seizure disappearance, as the remaining SOZ would not be sufficient to generate further epileptic seizures. Conversely, if the EZ is larger than the SOZ, total removal of the SOZ cannot ensure seizure disappearance because multiple SOZs with different thresholds may coexist in the same EZ. Indeed, in this case, after the resection of the first SOZ, another SOZ with higher threshold may become clinically evident (83). However, this picture is simplistic. Although seizures tend to have preferential spreading patterns, cortical connections spread in all directions, from any given cortical point, through cortico-cortical and subcortical connections (FIGURE 2). Consequently, disconnection of specific networks does not guarantee that seizure activity does not progress via alternative pathways, resulting in a modified clinical semiology but not in their disappearance (84). Only the complete disconnection or removal of all the potential SOZs can guarantee that seizures disappear.
Some of the seizure types observed in epileptic encephalopathies are almost exclusively seen within this category of severe epilepsies and do not fall within the categorization of generalized or focal epileptogenesis. Epileptic spasms and tonic seizures, which are some of the signature seizures in ISS and LGS, are considered to be bilateral seizures. The possible involvement of the brain stem in generating these seizures was demonstrated in 1958 by Kreindler and collaborators (85), who reported bilateral tonic convulsions in cats and rats when stimulating the reticular substance and periaqueductal substance. These certainly may explain reports of epileptic spasms in infants with hydranencephaly (86), although they do not certainly exclude the contribution of other higher structures that could activate a broader network generating these tonic seizures. Animal models of ISS, for example, have provided evidence that cortical or cortico-hippocampal lesions may suffice to trigger spasms (87, 88). Seizures and epileptic activity may also be multifocal, as in epilepsy of infancy with migrating focal seizures (EIMFS), as an expression of a widespread, genetically determined epileptogenesis in immature brains with incomplete myelination and poorly functioning connections (89).
3. MAIN (KNOWN) DETERMINANTS OF NORMAL AND ABNORMAL BRAIN DEVELOPMENT THAT INFLUENCE EPILEPTOGENESIS
Normal brain development is a dynamic process that proceeds asynchronously and at different tempos and trajectories across brain regions, cell types, and sexes and is further modified by biological or environmental factors (FIGURE 3). This asynchronous and timed maturation also extends to key developmental processes that potentially control the susceptibility to seizures and epileptogenesis: migration and differentiation of progenitors of excitatory and inhibitory neurons, neurogenesis and synaptogenesis of excitatory and inhibitory synapses, morphological changes of various brain regions (FIGURE 3A), signaling modes of molecular or electrophysiological signaling cascades that control neuronal excitability, differentiation, function, or communication, and survival (FIGURE 3B) (92).
FIGURE 3.
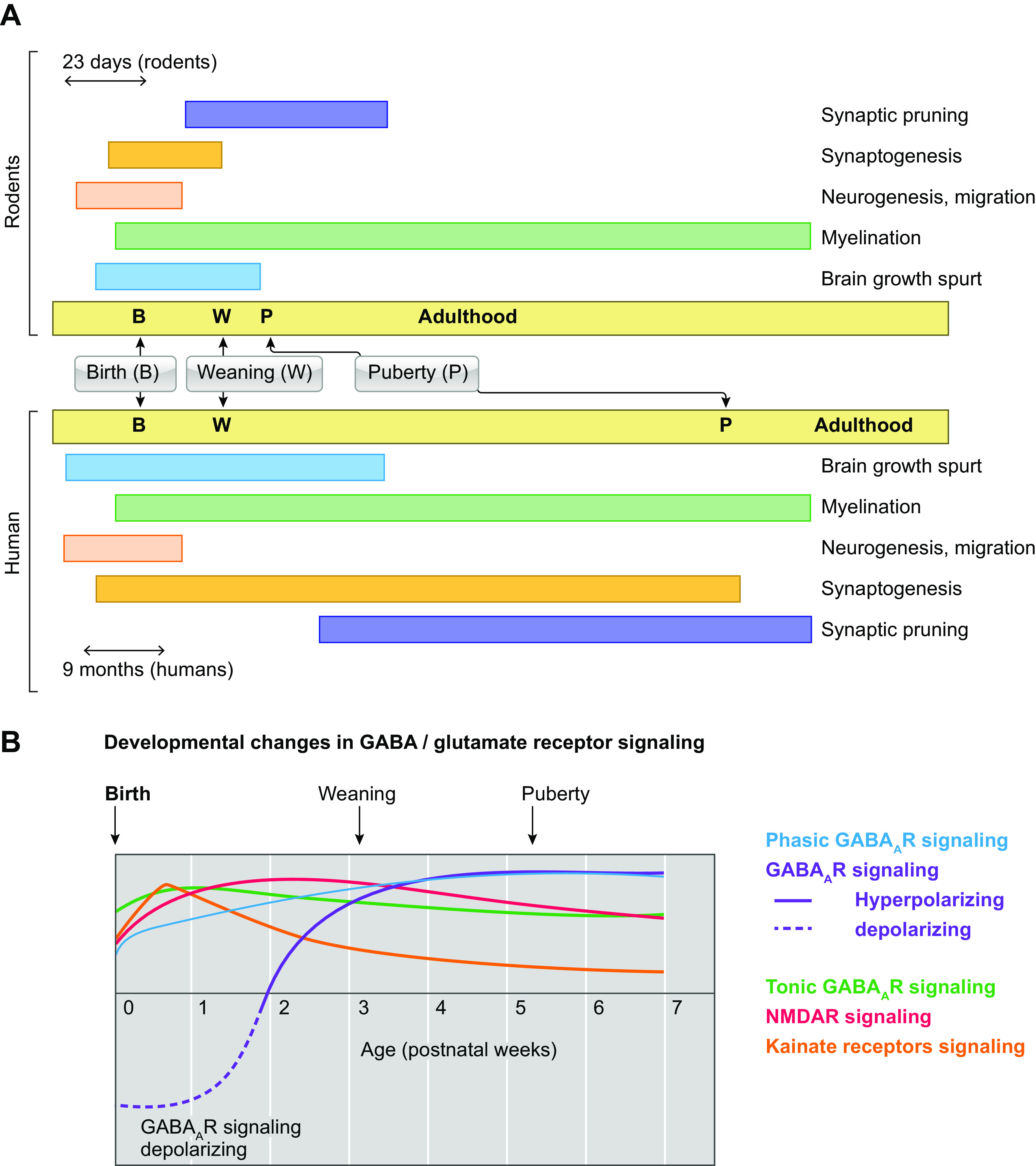
Trajectories of developmental processes in normal brain development. A: the temporal trajectories of selected developmental processes that are important for normal brain development in rodents (top) and humans (bottom) are shown. Birth (B), weaning (W), puberty (P), and adulthood are indicated separately in each panel for rodent or human development. The different timescales used across species (23 days in rodents, 9 mo in human) highlight the significant differences in the speed of maturation across species. The time of brain growth spurt in humans (full-term birth) and rodents [around postnatal day (PN)10] has been used to indicate the ages across species that correspond to a full-term newborn human baby (90). Brain growth spurt in these studies included gross brain growth, DNA, cholesterol, and water content. Puberty onset occurs around PN32–36 in female rats and PN35–45 in male rats, whereas in humans it starts around 10–11 yr in girls and 11–12 yr in boys (91). Distinct processes, such as neurogenesis and migration, synaptogenesis, and synaptic pruning, myelination follow different time courses (92–100). However, the staging of the equivalence of developmental stages across species is only approximate, and each developmental process needs to be considered individually. B: significant changes occur during development in the expression or function or various signaling processes. A schematic depiction of the age-related changes in GABA type A receptor (GABAAR) and glutamatergic signaling in rats is presented here; however cell type-, region-, and sex-specific differences also exist (92, 101,102). Early in development, there is less effective GABAAR-mediated inhibition, because of the presence of depolarizing GABAAR signaling (see also FIGURE 4), more tonic and less phasic GABAAR inhibition. In contrast, glutamatergic receptors, such as NMDA receptor (NMDAR) or kainate receptors, also show age-related expression patterns.
It is generally considered that around postnatal day (PN)10, a rodent is equivalent to a full-term human neonate, an assumption based on studies of brain growth spurt (90). Brain growth spurt in these studies included gross brain growth, DNA, cholesterol, and water content. Puberty onset occurs around PN32–36 in female rats and PN35–45 in male rats, whereas in humans it starts around 10–11 yr in girls and 11–12 yr in boys (91). Distinct processes, such as neurogenesis and migration, synaptogenesis and synaptic pruning, and myelination follow different time courses (FIGURE 3B) (92–100). For studies that specifically target or relate to specific developmental processes it is therefore important to consider the maturational trajectories of the specific developmental processes of interest across species, when these are known.
There are extensive and continuous molecular, morphological, or functional changes through development that significantly diversify the effects of epileptogenic processes in the brain, thus rendering the immature brain more amenable to developing the intense and sometimes multifocal epileptic activity associated with DEEs (92). Ontogenetic studies with kindling, a method of repetitive electrical stimulations of limbic structures that results in a progressively more severe seizure phenotype, demonstrated that postictal refractoriness to manifesting another seizure is shorter in developing animals, which is likely a factor that predisposes them to develop clusters of seizures (103). In addition, kindling antagonism, whereby kindling stimulation at one limbic structure inhibits the kindling development in another, is not operative in immature pups (104). Although the immature brain is more prone to seizures and seizure clusters than that of adults, it is also more resilient, demonstrating no or less severe injury after prolonged seizures (105, 106).
The trajectories of maturation of these developmental processes often follow age-, sex-, and region-specific patterns. These have been extensively studied for GABAA receptors (GABAARs) and include changes in receptor composition and kinetics of depolarizing/hyperpolarizing postsynaptic GABAAR responses or network effects (101, 107–112) (FIGURE 4). GABAAR signaling is usually depolarizing in immature neurons with relatively high intracellular Cl− content and elicits hyperpolarizing responses in mature neurons that have low intracellular Cl− (121–126). The polarity of GABAAR responses depends upon the relative abundance of cation-chloride cotransporters and channels that import [e.g., NKCC1, abundant early on, decreases later in life (127–129)] or export [e.g., KCC2, expression increases postnatally (117, 130)] Cl−, in a process that requires energy generated by Na+-K+-ATPase (FIGURE 4). Depolarizing GABA can have neurotrophic effects in immature neurons and is a normal physiological phenomenon needed for normal development (113, 118, 131). The developmental shift of GABAAR signaling from depolarizing in immature neurons to hyperpolarizing in mature neurons has been proposed to follow a rostro-caudal gradient, with earlier maturation in the most caudal regions. In reality, there are significant cell type-, region-, and sex-specific factors that create a more complex temporo-spatial pattern of maturation not only of GABAARs but also of other neurotransmitter and signaling pathways. For example, hyperpolarizing GABAAR responses may occur earlier in rat CA1 pyramidal neurons than in substantia nigra pars reticulata (SNR) GABAergic neurons (112, 119); GABAAR-mediated hyperpolarizing responses are already seen in neonatal thalamic neurons, in contrast to cortical neurons, which mature later on (132). The maturation to hyperpolarizing GABAAR signaling may occur earlier in females than in males in certain brain regions (hippocampus, substantia nigra) (109, 112, 118, 119, 133), whereas it may emerge later in females in other regions (cerebellar Purkinje cells) (134). Premature cessation of depolarizing GABAAR signaling may disrupt the excitatory synapse formation and dendritic arborization of cortical neurons, leading to neurodevelopmental deficits (114, 115, 135). Disruptions of the GABAAR-sensitive patterns of communication across cells or brain regions may also occur with seizures or epileptogenesis, stressors, genetic variants, drugs, or metabolic disorders, altering threshold for ictogenesis or epileptogenesis and predisposing to behavioral or cognitive impairments (110, 118–120). Depolarizing GABA can also be seen under pathological conditions, e.g., after axonal injury, hypoxia/hypoglycemia, during prolonged seizures, or in epileptic tissue, and its significance there can be dual: partly protective to promote neuronal healing but also potentially epileptogenic, as it may promote neuronal excitability and epileptogenesis (120). Furthermore, KCC2 effects are not strictly under Cl− regulation; the COOH terminus of KCC2 may promote synapse formation (136) and the NH2 terminus can affect neuroprotection (137, 138). Loss of function mutations in KCC2 have been found in epilepsy syndromes, including epilepsy of infancy with migrating focal seizures (EIMFS) (139–141).
FIGURE 4.
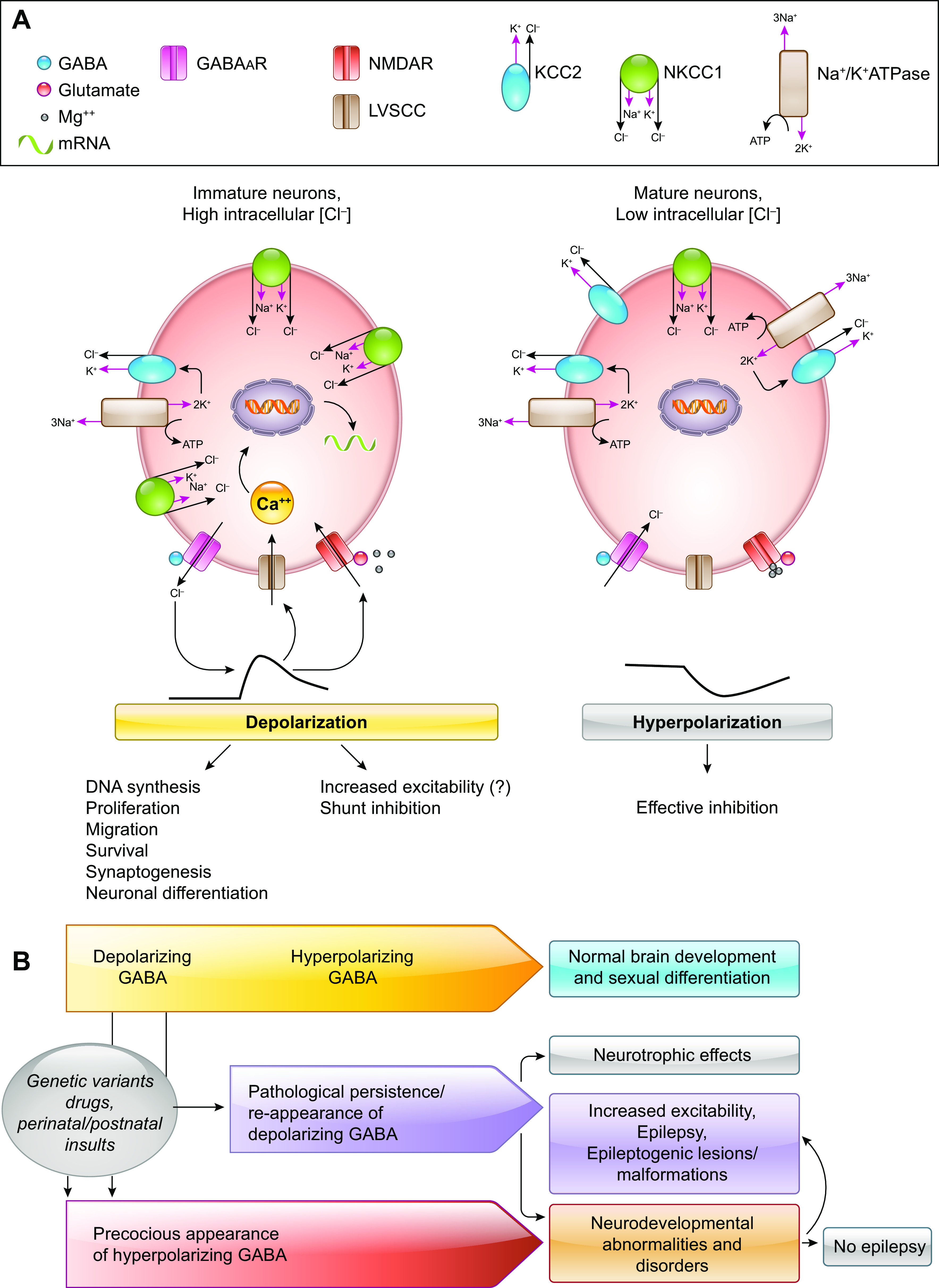
Depolarizing and hyperpolarizing GABA type A receptor (GABAAR) signaling in normal development and disease. A: GABAAR signaling is depolarizing early in life because of the higher intracellular Cl− concentrations ([Cl−]i) that force Cl− efflux upon GABAAR activation. Although the GABAAR depolarizations render GABA inhibition less efficient, as it relies upon shunt inhibition, they are critical for normal brain development. GABAAR depolarizations may activate L-type voltage-sensitive calcium (LVSCC) channels and may release the Mg2+ block of NMDA receptors (NMDARs), triggering intracellular Ca2+ rises that are important for neuronal survival, migration, differentiation and integration (113–116). The [Cl−]i in immature neurons is a result of increased expression and/or activity of Cl− importers, like NKCC1 (a Na+-K+-Cl− cotransporter) over Cl− exporters, like KCC2 (a K+-Cl− cotransporter). The Na+-K+-ATPase provides the energy to maintain the cation-chloride cotransporter function. During development, there is a gradual switch in the relative dominance of these cation-chloride cotransporters at specific time points that follows cell type-, region-, and sex-specific patterns (109, 117–119). As a result, mature neurons demonstrate hyperpolarizing GABAAR responses that allow for effective inhibition to occur. B: normal brain development depends upon the age-, cell type-, region-, and sex-appropriate presence of depolarizing and hyperpolarizing GABAAR signaling. Genetic variants, drugs, and perinatal or postnatal insults that trigger precocious presence of hyperpolarizing GABAAR signaling may result in neurodevelopmental deficits or abnormalities that could increase the risk for epilepsy (114–116). Conversely, pathological persistence or reappearance of depolarizing GABA has been described in epileptogenic pathologies and may predispose to increased excitability (120).
GABA signaling is also an important regulator of cortical-subcortical networks that control fundamental physiological functions, such as learning and memory, but also seizures (111, 142–144). The modus operandi of these networks undergoes significant age- and sex-dependent changes through development. The substantia nigra pars reticulata (SNR), largely composed of GABAergic interneurons, acts as a gate through which the cortico-striatal input may activate or inhibit the activity of thalamo-cortical neurons. In adults, activation of GABAARs in the SNR exerts anticonvulsant effects in a variety of seizure models (145–147). However, in developing rodents, the outcome of GABAAR-sensitive SNR-mediated seizure control (anticonvulsant vs. proconvulsant vs. no effect) varies by region, age, and sex (111, 143, 144). All this knowledge remains too fragmentary, but it represents a promising field to fully understand the human pathology.
Excitatory neurotransmitters also undergo significant developmental changes in expression and subunit composition (FIGURE 3), which modify their biological effects in regard to both seizure susceptibility and control (111, 148) and neuronal survival (149).
An important concept in developmental epileptogenesis is the existence of critical or sensitive periods for the development of specific traits. The effects of biological factors or exogenous insults can be time-locked to specific developmental periods that render the brain sensitive, as shown for the hormonal regulation of the differentiation of seizure-controlling subcortical networks (150). The biological roles of channels may also be age dependent, as demonstrated, for example, for M channels (151). M-channel activity is essential for the normal morphological development of hippocampus but only during the first postnatal weeks of murine development. Loss of M-channel activity at later periods does not have overt morphological sequalae in the hippocampus, although cognitive deficits as well as increased neuronal excitability can still be observed. The therapeutic effects of a treatment can be specific for certain developmental periods when it can modify its desired targets, as shown for neonatal estradiol given to prevent interneuronopathy in an Arx knockin mouse model of epileptic spasms (152). The effects of genetic mutations may differ depending upon the developmental period when these are expressed, as shown for the GABAARγ2(R43Q) epilepsy mutation (153).
Beyond gene effects, critical developmental periods are also important in determining the severity of dysfunction conferred by the abnormal excitability. Using bicuculline and penicillin application in the visual cortex of rabbits to induce sporadic epileptiform discharges, Chow and colleagues (154) showed that the ensuing epileptic activity disrupted the appearance of complex and oriented-directional type cells at the lateral geniculate nucleus in developing, but not adult, rabbits. More recent studies also demonstrated that the induction of epileptiform discharges by electrical stimulation of the hippocampus in developing animals disrupts place cell formation (155, 156).
4. NONGENETIC DETERMINANTS OF THE EPILEPTOGENIC RISK
Inflammation and cytotoxic injury may trigger a chronic and evolving ISS phenotype in the multiple-hit model, suggesting that the cortical-subcortical network disruption due to the structural lesion, in tandem with neuroinflammation, may trigger spasms and epileptogenesis with neurodevelopmental deficits (88, 157–163). In the same model, dysregulation of the mTOR (mechanistic target of rapamycin) pathway was a critical pathogenic feature, and restoration of its activity with rapamycin resulted in partial improvement of the cognitive deficits and reduced epilepsy development, lending further support for the central role of mTOR in epileptogenesis (157, 163). This model also provides evidence for sleep-epilepsy interplay, with most adult motor seizures emerging from sleep, as well as evolution to a slow spike-wave EEG, reminiscent of Lennox–Gastaut syndrome (LGS) in adulthood (157).
The deficit in parvalbumin (PV)-positive interneurons from the contralateral cortex (161) is reminiscent of the interneuronopathy seen in ARX-related ISS (152, 164, 165), although the quality of the interneuronopathy under these conditions is different. Mechanistically, these also seem different, since the neonatal estradiol treatment that corrected the ARX-related interneuronopathy and epilepsy (152, 165) did not improve the phenotype of the multiple-hit rats (159).
Stress has been long advocated as a key pathogenic mechanism of ISS (166, 167). Administration of corticotropin-releasing hormone (CRH) in the cortex or hippocampus of pups induced seizures but not spasms (168). Exposure to conditions that mimic aspects of early-life stressors or stress response, such as prenatal betamethasone or stress (169–171), or that disrupt adrenal function aggravated NMDA-induced spasms (172), although some of these conditions (such as prenatal betamethasone) enhanced responsiveness of NMDA-induced spasms to ACTH (170). A more recent model of chronic early-life stress due to fragmented nurturing behaviors was reported to manifest a mild spasm phenotype (173).
5. GENETIC DETERMINANTS OF DEE: EXPERIMENTAL MODELS
DEE-causing genes can be grouped into broad functional categories involved in different cellular processes including ion/transmitter/small molecule transport, regulation of synaptic function, cell growth, division, and proliferation, cell metabolism, intracellular trafficking and signaling, gene transcription, and protein biosynthesis/degradation. (TABLE 2 and SUPPLEMENTAL TABLE S1).
We focus here on gene products that have been extensively studied for their effects on neuronal excitability and epileptogenesis in in vitro and in vivo studies, using relevant natural or genetically engineered models. They offer clearer targets to design rational therapies to restore normal function in dysfunctional networks that can be easily assessed with physiological tests. We also discuss more succinctly other genes that emerged as genes of interest for certain DEEs, particularly those associated with migration disorders; however, their mechanisms of action are more complex, involving multiple cellular processes that need to be disentangled before designing safe and effective therapies.
Neuronal circuits are formed by principal glutamatergic excitatory neurons and inhibitory GABAergic neurons (FIGURE 5). It is thought that in cortical circuits glutamatergic neurons perform computational tasks, whereas GABAergic neurons control and organize the activity of the network and are important for the generation of rhythms of activities, which are the substrate of brain rhythms. Glial cells not only are important for neuron homeostasis and protection but are implicated in synaptic functions. In neurons, the somato-dendritic compartment receives most of the synaptic inputs that are computed in the dendritic tree and integrated in the axon initial segment, where APs are generated. Forward-propagating APs reach presynaptic terminals, where they evoke neurotransmitter release. Back-propagating APs in the somato-dendritic compartment are implicated in dendritic computation and modulation of synaptic inputs. Ion channels are essential in all neuronal subcompartments and for neuronal signaling. Thus, it is not surprising that channelopathies are implicated in numerous DEEs (174) (FIGURE 5).
FIGURE 5.
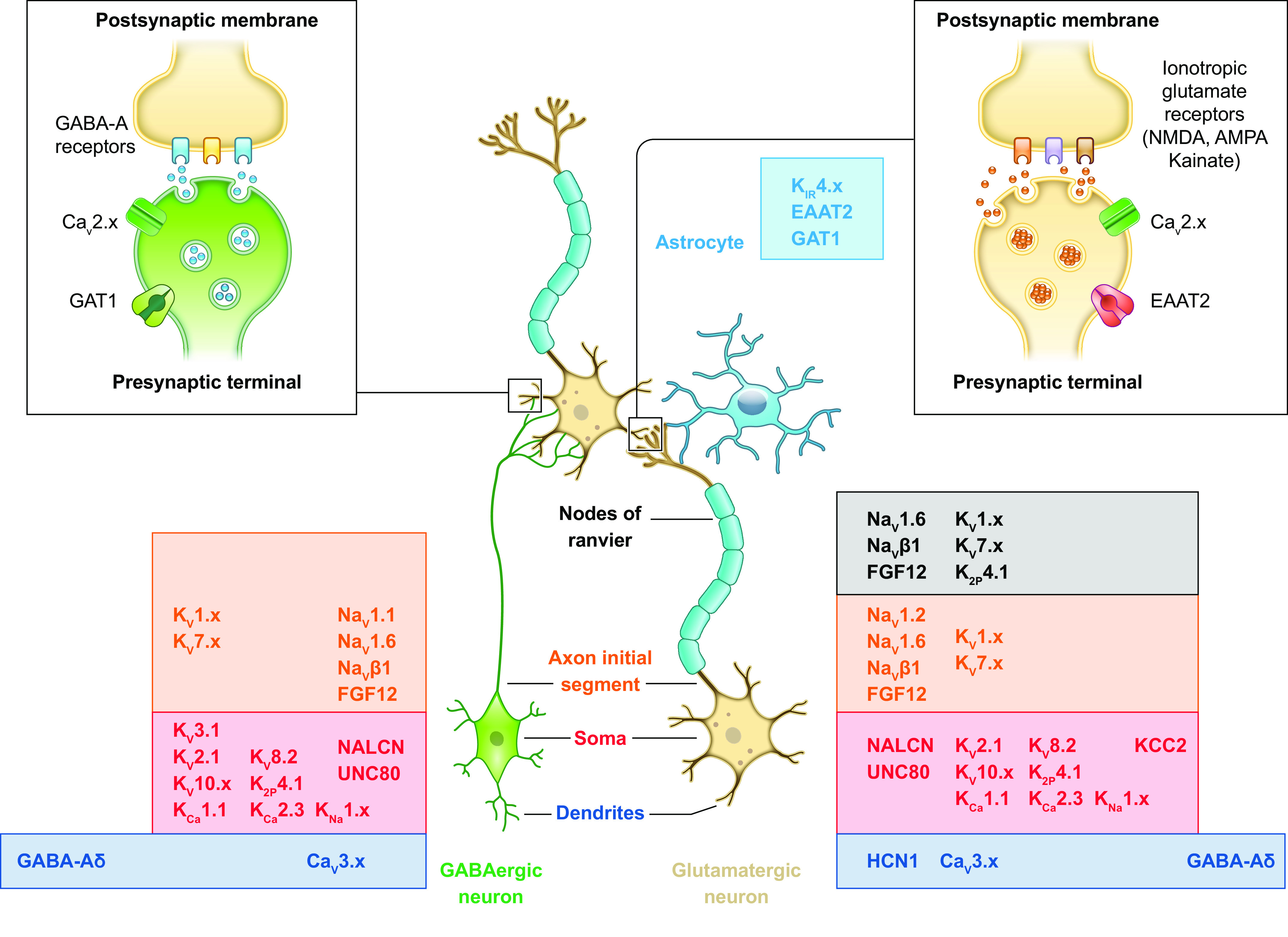
Simplified diagram of a cortical microcircuit with interconnected glutamatergic and GABAergic neurons, an astrocyte, and cellular/subcellular distribution of ion channels and transporters. A cortical neuronal microcircuit is illustrated as a presynaptic GABAergic neuron (green) and a presynaptic myelinated glutamatergic neuron (ocher) that form synaptic connections on the dendrites of a myelinated glutamatergic neuron (ocher). Glial cells are displayed as an astrocyte (light blue) in proximity of the glutamatergic synapses and as the myelin sheets around the axons of the glutamatergic neurons formed by oligodendrocytes (violet; the soma is not displayed), allowing saltatory conduction at the nodes of Ranvier. The insets at top show in more detail a GABAergic (left) and a glutamatergic (right) synapse. The ion channels and transporters targeted by DEE mutations are indicated with their protein name (see text for details) and their known cellular/subcellular distribution, according to the neuronal subcompartments (dendrites, soma, axon initial segment, nodes of Ranvier of the myelinated axon, presynaptic terminal, and postsynaptic membrane).
Functional analysis of mutations using a variety of experimental systems is essential for shedding light on the detailed pathomechanisms and clarifying genotype-phenotype correlations, which can in turn facilitate diagnosis, genetic counseling, management, and development of therapeutic approaches. Functional analysis using electrophysiological techniques identifies even subtle modifications in the properties of ion channels. Experimental methods include both in vitro and in vivo systems (175, 176) (FIGURE 6).
FIGURE 6.
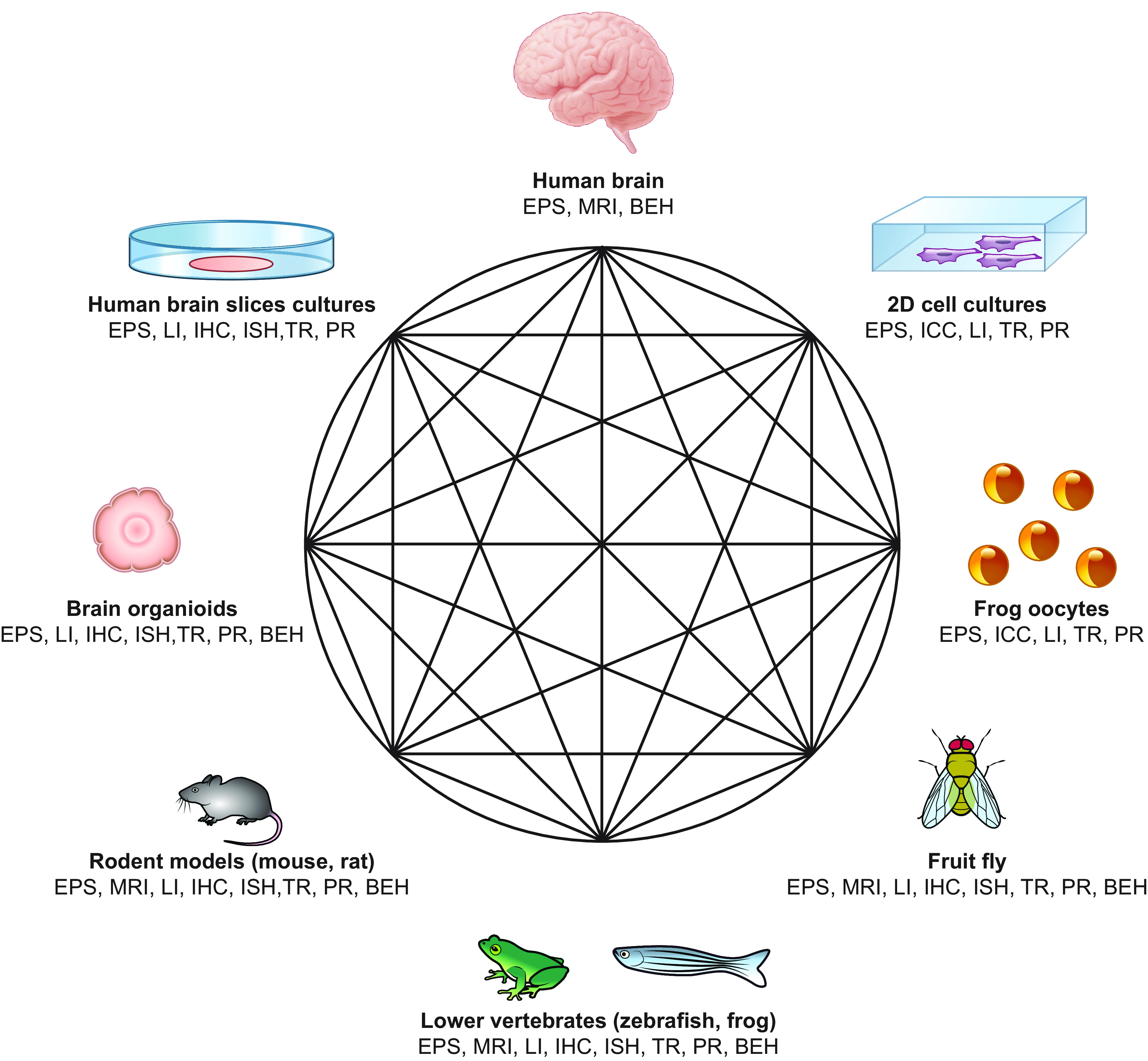
Schematic representation of the different in vitro and in vivo models that can be used to study functional effects of mutations affecting developmental and epileptic encephalopathies (DEEs) causative genes. Regardless of the starting point, researchers can move from one model to another based on the type of functional assay they want to apply and the physiological process they want to study. BEH, behavioral studies; EPS, electrophysiological studies; ICC, immunocytochemistry; IHC, immunohistochemistry; ISH, in situ hybridization; LI, live imaging; MRI, magnetic resonance imaging; PR, proteomics; TR, transcriptomics; 2D, 2-dimensional.
In vitro experimental systems often use cells that do not endogenously express the protein of interest, thus simplifying the functional analysis of its properties. They are in general human cell lines [e.g., transfected human embryonic kidney (HEK) cells] or, less frequently, oocytes of the clawed frog Xenopus laevis injected with the cRNA of interest that allow massive expression, although a human cellular background is generally preferable. Transfected/transduced neurons in primary cultures are a further in vitro system that provides a true neuronal cellular background to evaluate effects on neuronal and network properties. In vivo/ex vivo systems are organisms or preparations obtained from them (e.g., brain slices), which should better model the complexity of brain circuits and the actual pathophysiological conditions as well as provide information of in vivo phenotypes. The animals more frequently used for generating in vivo models of genetic variants are mice and rats (TABLE 3), because early site-directed mutagenesis techniques exploiting homologous recombination in embryonic stem cells allows easy genetic manipulation of these mammals. The mouse is still the organism of choice for generating animal models of genetic variants, although more recent methods of genome editing [e.g., clustered regularly interspaced palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9)] can be used to generate mutant models with other mammals. It is not possible to perform high-throughput studies with mammalian models, either for studying functional effects of variants or for drug screens. Simpler animal models make it possible to perform relatively large screens, in particular the zebrafish, which has vertebrate features (227). However, findings obtained with these simple systems need to be validated in mammalian models. Neurons differentiated from induced pluripotent stem cells (iPSCs) generated from patient biopsies are increasingly used to study mutations in human neurons as they bear the patient’s genetic background. They can be used for investigations of neuronal properties at the single-cell level or can be induced to generate in vitro miniature organs resembling the brain (brain organoids) that represent an excellent integrated experimental system to study brain development (228). However, large variability in the properties of these neurons makes studies difficult, and reproducibility is still an issue, as observed in studies of SCN1A mutations (229–236).
Table 3.
Selected animal models of DEEs
| Models | Species | Induction Method | Spasms, Age of Onset | Subsequent Epilepsy | Behavioral/Neurodevelopmental Deficits | Response to ACTH/Vigabatrin | Model of: | References |
|---|---|---|---|---|---|---|---|---|
| Models of ES | ||||||||
| Acute | ||||||||
| NMDA | Rat, mouse C57 | NMDA ip, PN7–18 | PN7–18 | NR | Rat: Learning and coordination deficits | High-dose ACTH1-39; vigabatrin | Spasms | (169, 172, 177–181) |
| 3–7 days after NMDA (mouse): Increased anxiety, impaired motor coordination and poor memory retention | ||||||||
| NMDA variants | ||||||||
| Prenatal betamethasone/postnatal NMDA | Rat | Betamethasone ip G15; NMDA ip, PN12–15 | PN12-15 | NR | NR after spasm induction | Low-dose ACTH1-39; Vigabatrin | Spasms, ACTH sensitivity | (170, 182–184) |
| Prenatal stress/postnatal NMDA | Rat | Forced restraint (FR) (G15) or forced swim test (FST) (G1–parturition); NMDA ip, PN15 | PN15 | NR | NR after spasm induction | FST/NMDA: Responds to ACTH1-39 | Spasms, stress | (169, 171) |
| FR/NMDA: Responds to repeated low dose ACTH1-39 | ||||||||
| Prenatal MAM/postnatal NMDA | Rat | MAM (2 doses, G15); NMDA ip, PN12–15 (1 or 3 doses) | PN12–15 | NR | NR after spasm induction | No effect of low-dose ACTH | Spasms, dysplasias | (185) |
| Responds to vigabatrin | ||||||||
| Adrenalectomy/postnatal NMDA | Rat | Adrenalectomy (PN10); NMDA ip (PN11) | PN11 | NR | NR after spasm induction | High-dose ACTH1-39 | Spasms | (172) |
| Tsc1gfap −/+ mouse, postnatal NMDA | Mouse, Tsc1flox/flox-GFAP-Cre knockout C57BL/6 and SV129 | As in Tsc1gfap−/+ | NR after spasm induction | NR | Spasms, induced on a genetic background | (186) | ||
| Down syndrome/GBL | Mouse (Ts65Dn), C57BL/6JEiXC3H/HesnJ | γ-Butyrolactone (GBL) ip | 1 wk to 2 mo | NR | NR after spasm induction | Responds to ACTH1-24 but not to ACTH1-39 | Spasms, induced on a genetic background | (187–189) |
| Responds to vigabatrin | ||||||||
| Chronic | ||||||||
| Tetrodotoxin (TTX) | Rat | Intrahippocampal or intracortical TTX, unilateral (PN10–38) | ≥PN21 | Yes | NR | Sensitive to ACTH, vigabatrin | ISS structural, hypsarrhythmia; drug-sensitive | (87, 190–193) |
| Multiple hit | Rat | PN3: Right intracortical LPS, right intraventricular doxorubicin | PN4–13 | Yes | Sociability deficits, learning/memory deficits, stereotypies | Resistant to ACTH; partial/transient response to vigabatrin | ISS structural, drug resistant | (88, 157–163) |
| PN5: PCPA ip | ||||||||
| Arx cKO | Mouse, CD1 and C57BL/6 | Arx deletion from ganglionic eminence neuronal progenitors | Adulthood | Yes | NR | NR | ISS, genetic | (164) |
| Arx KI [Arx (GCG)10+7] | Mouse, 75% C57BL/6; 25% 129S5/SvEvBrd | Expansion of 1st polyalalanine tract repeat (PA1) of Arx | PN7–11 | Yes | Low anxiety, impaired learning and sociability | NR | ISS, genetic | (152, 165, 194) |
| Arx with PA1 or PA2 expansion | Mouse, C57BL/6N-Hsd | PA1 or PA2 expansion of Arx | ≥PN10 to adulthood (myoclonic seizures) | NR | 1–2 mo: Sociability, neuromuscular strength deficits, anxiety and fear | NR | ISS, genetic | (195) |
| Adenomatous polyposis (Apc) | Mouse | Apc gene knockout from excitatory CamKII neurons | Peak at PN9 | Yes | Reduced social interest, increased repetitive behaviors | NR | ISS, genetic | (196) |
| Tsc1+/− | Mouse, C57BL/6 | Heterozygous Tsc1−/+ | PN12–16; observed for 1 day/pup | NR | NR | NR | ISS, genetic | (197) |
| Aged CDKL5, heterozygous females | Mouse, C57BL/6J | Cdkl5R59X/+ or Cdkl5KO/+ | >PN300 female | No (only spasms seen) | NR in females | NR | ISS, genetic | (198) |
| Sociability deficits in males | ||||||||
| Chronic early-life stress | Rat | Unpredictable and fragmented nurturing behaviors in dams (PN2–9 period) | PN17–35; last for 1 or several days | NR | NR | NR | ISS, unknown | (173) |
| Models of Lennox–Gastaut syndrome, atypical absence seizures | ||||||||
| AY9944 | Rat | AY9944 7.5 mg/kg sc (PN2, 8, 14, PN20) | NR | Slow SWD | Cognitive deficits, hyperactivity, anxiety, spatial learning, olfactory recognition deficits | Responsive to DZP, ETH, CGP35348; Worse with CZP, baclofen, γ-OH-butyrate | Chronic atypical absence seizures | (199–205) |
| MAM-AY9944 | Rat | Prenatal MAM/postnatal AY9944 | NR | Slow SWD | NR | Refractory to ETH, VPA, CGP35348, CBZ | Refractory atypical absence seizures | (206) |
| PValb-Dnm1Ftfl/flox | Mouse, C57BL/6J | Dnm1Ftfl/flox in PV cells | NR | PN19–50: SWD, lethal seizures | Tremor | NR | LGS | (207) |
| GABABR1a | Mouse, C57BL/6 | GABABR1a overexpression in forebrain | NR | Slow SWD | Impairment in learning, spatial memory | Responsive to ETH, CGP35348 | Chronic atypical absence seizures | (208) |
| GABABR1b | Mouse | GABABR1b overexpression in forebrain | NR | Slow SWD | Mild impairment in learning, spatial memory | Responsive to ETH, CGP35348 | Chronic atypical absence seizures | (209) |
| NHE1 | Mouse, SJL/J and C57BL/6J | Na+/H+ exchanger null | NR | Slow SWD (3 Hz) (at 4–5 wk); lethal tonic or tonic-clonic seizures | Ataxia;early mortality | NR | Chronic atypical absence seizures | (210) |
| Multiple hit | Rat | PN3: Right intracortical LPS, right intraventricular doxorubicin PN5: PCPA ip | PN4–13 | Adulthood: slow SWD (5–6 Hz), motor seizures in sleep | Sociability deficits, learning/memory deficits, stereotypies | NR | ISS with LGS features | (157) |
| Models of Dravet syndrome | ||||||||
| Scn1a KO | Mouse | Exon 26 deletion, global constitutive | Convulsive seizures, hyperthermia seizures, mortality | Hyperactivity, stereotypies, sociability, and spatial memory deficits | Tested | Dravet | (64) | |
| Scn1a CKO | Mouse, C57BL/6 and 129Sv | Exon 25 deletion, forebrain GABAergic interneurons | Motor seizures, hyperthermia seizures | NR | Dravet | (211) | ||
| Scn1a CKO | Mouse, B6.SJL-Tg(ACTFLPe)9205Dym/J and C57BL/6 | Conditional deletion of exon 7 | Inhibitory neurons | ≥PN16 seizures, occasional death | Hypoactive, jerks, death | Dravet | (212) | |
| Forebrain excitatory neurons | No | NR | (212) | |||||
| Forebrain excitatory neurons and haploinsufficiency in inhibitory neurons | Improved lethality from seizures | NR | Dravet | (212) | ||||
| PV interneurons | ≥PN14 seizures, death | Ataxia (PN10) | Dravet | (212) | ||||
| Scn1a KI, R1407X | Mouse, 129/SvJ and C57BL/6J | Human R1407X nonsense mutation | ≥1 mo: Seizures | Hyperactivity, stereotypies, sociability and spatial memory deficits | Dravet | (65, 213, 214) | ||
| Scn1a KI, S1231R | Drosophila | S1231R mutation, loss of function | Seizures | NR | Dravet | (215) | ||
| Scn1Lab (didys552) | Zebrafish | Scn1Lab mutation (low expression) | Increased locomotor activity, epileptiform activity, seizures | Impaired exploration, decreased mobility | Tested | Dravet | (216) | |
| Scn1Lab−/− | Zebrafish | Scn1Lab null | Increased locomotor activity and epileptiform activity | NR | Tested | Dravet | (217) | |
| Scn1a-A1783V/WT KI | Mouse, C57BL/6J | Scn1a-A1783V/WT KI | Hyperthermia seizures | NR | Tested | Dravet | (218) | |
| Scn1a R1648H KI (after induction of short seizures) | Mouse | Knock-in R1658H missense mutation, global constitutive | Convulsive seizures, hyperthermia seizures, mortality | Hyperactivity, stereotypies, sociability, and spatial memory deficits | Dravet and GEFS+ | (219) | ||
| Other etiology-specific models of DEE | ||||||||
| Kcnq2 KI | Mouse, C57BL/6J | Kcnq2-Y284C/+, Kcnq2-A306T/+ | NR | NR | Retigabine reduces KA-seizures | KCNQ2 DEE | (220) | |
| Kcnq2-Thr274Met/+ | Mouse, 129Sv, C57BL/6N | Kcnq2-Thr274Met/+ | Yes (>PN20) | Spatial learning and memory deficits | NR | KCNQ2 DEE | (221) | |
| Death by 3rd mo (25%) | ||||||||
| Kcna1−/− | Mouse | Kcna1−/− | Yes | NR | Retigabine reduces spontaneous seizures | KCNA1 epilepsy | (222) | |
| Kcnq1-A340E/A340E | Mouse | Kcnq1-A340E/A340E | Rare spontaneous seizures | NR | Retigabine: adverse cardiac effects | KCNQ1 epilepsy | (222) | |
| Pcdh19 KO and heterozygous females | Mouse, 129S5.C57BL/6 | Pcdh19 KO and heterozygous females | NR; increased susceptibility to 6 Hz and flurothyl seizures | NR | PCDH19 DEE | (223) | ||
| Pcdh19-HET | Mouse, C57BL/6N | Pcdh19-HET | Mossy fiber deficits | NR | Pattern completion and separation deficits | NR | PCDH19 DEE | (224) |
| Pcdh19 KO | Mouse, C57BL/6N | Pcdh19 KO | NR | Increased exploratory behavior, reduced anxiety | NR | PCDH19 DEE | (223) | |
| Cdkl5 CKO | Mouse, CD1 | Cdkl5 CKO in glutamatergic or GABAergic neurons | Defective dendritic arborization and spine maturation | Yes when deleted in glutamatergic neurons. | Autistic symptomatology, motor coordination, memory and breathing abnormalities | Epigallatocathechin-3-gallate (EGCG) corrects synaptic deficits | CDKL5 DEE | (225, 226) |
ACTH, adrenocorticotropic hormone; apc, adenomatous polyposis colon; arx, aristaless X-linked homeobox protein; AY9944, trans-1,4-bis-cyclohexane dihydrochloride, cholesterol biosynthesis inhibitor; CDKL5, cyclin-dependent kinase-like 5; CGP35348, GABAB receptor inhibitor; DEE, developmental and epileptic encephalopathy; ES, epileptic spasms; CKO, conditional knockout; CZP, clonazepam; Dnm1, dynamin 1; DZP, diazepam; EGCG, epigallatocathechin-3-gallate; ETH, ethosuximide; GBL, gamma butyrolactone; FR, forced restraint; FST, forced swim test; G, gestational day; GABABR: GABAB receptor; gfap, glial acidic fibrillary protein; HET, heterozygous; ISS, infantile spasms syndrome; Kcna1, potassium voltage-gated channel subfamily a member 1; KCNQ, potassium voltage-gated channel subfamily Q; KI, knockin; KO, knockout; LGS, Lennox–Gastaut syndrome; LPS, lipopolysaccharide; MAM, methyl-azoxy-methanol acetate; NHE1, Na+/H+ exchanger; NMDA, N-methyl-d-aspartate; NR, not reported; PA, polyalanine; PCPA, p-chlorophenylalanine (inhibits serotonin synthesis); PN, postnatal; PV or Pvalb, parvalbumin; Scn1a, sodium channel 1alpha; SWD, spike and slow-wave discharge; tsc, tuberous sclerosis complex; TTX, tetrodotoxin; VPA, valproic acid.
6. DYSFUNCTIONS IN NEURAL CELL MIGRATION, PROLIFERATION, AND SYNAPTOGENESIS
Mutations in distinct DEE genes can trigger different molecular and biochemical alterations that, depending on the developmental stage involved and the type of alteration, may result in a brain with a grossly abnormal morphology or structurally normal but functionally abnormal (FIGURE 7). Disruption of any of the overlapping steps that contribute to the development of the human cerebral cortex that can be recognized as malformations in studies of brain imaging is designated as “malformation of cortical development” (MCD). MCDs can be broadly classified into three major groups that recapitulate the main developmental steps, i.e., malformations of cell proliferation, neuronal migration, or postmigrational cortical organization (237).
FIGURE 7.

Brain MRI of patients with different malformations of cortical development. A: T1-weighted (T1W) coronal section. Lissencephaly in a boy with ARX mutation. The ventricles are severely dilated, the corpus callosum is absent, and the basal ganglia are severely hypoplastic. B and C: coronal T1W and axial T2-weighted (T2W) sections of a brain with posterior > anterior pachygyria and increased cortical thickness. Boy with LIS1 mutation. White asterisk in B indicates the point of more severe cortical thickening. White arrows in C point to areas of more severely smooth and thick cortex. D: T1W coronal section. Diffuse subcortical band heterotopia in a girl with DCX mutation. White circle surrounds the subcortical laminar heterotopia, which forms an almost continuous band beneath the cortex, separated from it by white matter. E: axial T2W section. Right occipital cortical dysplasia (surrounded by a white circle) in a girl with a very low-level mosaic mutation in AKT3 (0.67% in brain, not detectable in blood). F and G: axial fluid-attenuated inversion recovery (FLAIR) and coronal T1W sections in 2 patients carrying mosaic mutations in the MTOR gene with different % of mosaicism (F: p.Thr1977Ile, 20% of mosaicism in blood; G: p.Ser2215Phe, 5.5% of mosaicism in the surgically removed dysplastic brain tissue). In F, the patient has megalencephaly with large ventricles and multiple areas of abnormal cortex alternating infoldings with smooth surface. This pattern is suggestive of polymicrogyria (white arrows). In G, white circle highlights an area of cortical dysplasia with increased volume of the brain parenchyma, blurring of the gray-white matter junction, and irregular cortical folding. H: T2W coronal section. Left parieto-temporal focal cortical dysplasia in a girl with NPRL2 mutation. Circle surrounds the parietal portion of the cortical abnormality. I and J: T1W axial sections in 2 patients carrying the p.Gly373Arg PIK3R2 gene mutation with different % of mosaicism (I: 13% of mosaicism in blood, 43% in saliva; J: 10% of mosaicism in blood, 29% in saliva). Both patients have bilateral perisylvian polymicrogyria (white circles). K and L: axial FLAIR and coronal T1W sections showing right posterior quadrantic dysplasia (white circle) in a boy with a constitutional PTEN mutation. M and N: T2W coronal and sagittal sections in 2 patients with constitutional TSC2 mutations (M: p.Thr1623Ile; N: p.Pro1202His) showing right posterior quadrantic dysplasia caused by a large cortical tuber (M, white circle) and an extensive dysplastic area involving most of the right frontal lobe (N, white arrowheads). O and P: T2W axial and T1W sagittal sections. Lissencephaly with normally thick cortex and cerebellar hypoplasia (P, asterisk) in a girl with RELN mutation. White circle surrounds a hypoplastic brain stem. Q and R: axial and sagittal T1W sections. Thickened cortex with simplified gyral pattern and cerebellar hypoplasia in a boy with TUBA1A mutation. Circles surround the hypoplastic cerebellum and brain stem. Asterisk indicates the area below a hypoplastic cerebellar vermis, and black arrow points to a hypoplastic corpus callosum lacking its most posterior part. S: T1W axial section. Diffusely simplified gyral pattern with prominent thickening and infolding of the sylvian fissures in a boy with TUBB2B mutation. Arrows point to an area of smooth cortex. T: T2W axial section. Severe dysgyria with simplified gyral pattern in a girl with SCN3A mutation. U: T1W axial section. Classical bilateral periventricular nodular heterotopia in a girl with FLNA mutation. Bilateral nodules of subependymal heterotopia (white arrowheads) are contiguous, extensively lining the ventricular walls. V: T1W axial section. Diffuse polymicrogyria, more prominent posteriorly (white arrows) in a boy with ATP1A2 mutation. W: T2W coronal section. Polymicrogyria with abnormal cortical infoldings and packed microgyria (black arrows), combined with abnormal sulcation in a boy with ATP1A3 mutation. X: T2W axial section. Bilateral frontoparietal cortical thickening and diffusely abnormal cortical pattern in a boy with biallelic GPR56 mutations. Y and Z: T1W axial and sagittal sections. Pachygyria and perisylvian polymicrogyria in a girl with DYNC1H1 mutation. Asterisks in Y are located where there is maximum cortical thickening, in the posterior cortex. Asterisk in Z is located beneath a hypoplastic cerebellar vermis. AA: T2W axial section. Diffuse polymicrogyria in a boy with a GRIN2B mutation. BB: T1W axial section. Diffuse abnormality of the cortical pattern with smooth cortex and areas of abnormal infolding, suggestive of polymicrogyria in a boy with biallelic WDR62 mutations.
Among brain morphological abnormalities, MCDs are those most frequently associated with recurrent seizures. In MCDs, altered processes of development involve cells that, under normal circumstances, would participate in the formation of the normal cerebral cortex, and seizures can arise because of neuronal malpositioning, abnormal proliferation or differentiation, or abnormal cortical organization (238).
Neuronal malpositioning is the result of altered neuronal migration during brain development. A classic example of DEE associated with neuronal migration deficit is caused by mutations in the aristaless-related homeobox (ARX) gene, located on chromosome Xp21, which can result in a phenotypic spectrum comprising a nearly continuous series of neurodevelopmental disorders including lissencephaly with ambiguous genitalia (XLAG) (FIGURE 7A), Proud syndrome, Partington syndrome, infantile spasms without brain malformations, and syndromic and nonsyndromic intellectual disability with epilepsy.
The ARX protein belongs to the Aristaless-related subset of the paired (Prd) class of homeodomain proteins. Homeodomain transcription factors play crucial roles in cerebral development and patterning (239). In particular, ARX is involved in the normal tangential migration of GABAergic neurons, and the occurrence of seizures in most patients carrying mutations in this gene can be ascribed to mislocalization or malfunction of this class of neurons and loss of inhibitory neurotransmission (240). For this reason, ARX-related DEE is considered a “developmental interneuronopathy,” a term coined to differentiate developmental brain disorders caused by impaired development, migration, or function of interneurons from functional deficits or secondary loss of interneurons and from the more common channelopathies (241). Developmental interneuronopathies may also include Dravet syndrome, as discussed above, and classical lissencephaly due to mutations in PAFAH1B1, also known as LIS1 (FIGURE 7, B and C), or DCX (FIGURE 7D). The PAFAH1B1 gene encodes for the regulatory β-subunit of the cytosolic type I platelet-activating factor (PAF) acetylhydrolase, which is involved in interneuronal migration and survival, whereas DCX encodes for a microtubule-associated protein essential for both radial and nonradial interneuronal migration into the cerebral cortex (55). Interneuronopathies have also been described in models of acquired DEEs, such as the multiple-hit model of ISS (161), although the underlying interneuronal deficits and mechanisms are likely distinct, requiring different targeted therapeutic approaches (159).
The phenotypic continuum of MCDs, which includes focal cortical dysplasia type II (FCDII), hemimegalencephaly (HME), megalencephaly (MEG), and dysplastic megalencephaly (DMEG), is mainly caused by constitutional and somatic mutations in mTOR pathway genes, i.e., AKT1 and AKT3 (FIGURE 7E), DEPDC5 and MTOR (FIGURE 7, F and G), NPRL2 (FIGURE 7H), NPRL3, PIK3CA, and PIK3R2 (FIGURE 7, I and J), PTEN (FIGURE 7, K and L), and TSC1 and TSC2 (FIGURE 7, M and N), and represents the paradigm of DEEs caused by abnormal neuronal proliferation or differentiation in addition to abnormal neuronal migration. FCD is the cause of seizures in almost half of children referred for surgical treatment of medically refractory epilepsy, roughly estimated as near 400,000 people in the United States (Wolters Kluwer UpToDate website, https://www.uptodate.com/contents/evaluation-and-management-of-drug-resistant-epilepsy). In addition to abnormal cortical lamination, FCDII also features large dysmorphic neurons without (type IIa) or with (type IIb) balloon cells (242). HME, a condition in which one hemisphere is abnormally larger than the contralateral, and DMEG, a condition in which cortical dysplasia is associated with segmental brain overgrowth, exhibit histopathological features similar to FCDII (237). Tuberous sclerosis complex (TSC), a congenital syndrome characterized by the development of benign tumors (hamartomas) in multiple organs including the brain and a neurological phenotype consisting mainly of early-onset seizures, intellectual disability, and at times autism, is also caused by mTOR pathway dysregulation due to mutations in the tumor suppressor genes TSC1 or TSC2 (243).
Balloon cells in FCDIIb and TSC exhibit features of both neurons and glia. On histopathological analysis, dysmorphic or cytomegalic neurons are often associated with increased proliferation of normal-appearing glia or abnormal reactive astrocytes, which overexpress specific intermediate filaments and other immature molecular markers. Taken together, these findings strongly implicate primary defects in cellular proliferation and differentiation in the pathogenesis of mTORopathies (244).
Electrophysiological recordings in neocortical samples surgically removed from patients with FCD have demonstrated that, under reduced K+ conductance, dysmorphic cytomegalic neurons, but not balloon cells, generate large Ca2+ currents when stimulated, indicating that this aberrant cell type plays an important role in epileptogenesis (245). Both the morphology and size of dysmorphic cytomegalic neurons have been implicated with the origin of aberrant electrical discharges. By using electrophysiology, calcium imaging, morphological analyses, and modeling studies, Williams and collaborators (246) demonstrated that Pten (an mTOR pathway inhibitor)-knockout neurons exhibit rapid-onset hypertrophy and higher density of synapses proximal to the soma and that these phenotypic abnormalities promote firing at more hyperpolarized membrane potentials, with greater peak spike rates, and higher sensitivity to depolarizing synaptic input.
Using mouse models with cell type-specific mTOR pathway activation obtained by electroporating the Pik3ca p.H1047R mutation, which is associated with both FCD and HME, D’Gama and collaborators (247) demonstrated that mTOR pathway activation in excitatory neurons and glia, but not in interneurons, is sufficient to cause abnormal cortical lamination and overgrowth. In FCD, epileptic seizures appear to be triggered by a peculiar mechanism that we detail below. By treating in vitro-maintained organotypic slice cultures of resected FCD lesions with 4-aminopyridine, a potent convulsant agent acting as potassium channel blocker, Avoli and collaborators (248) demonstrated intrinsically generated ictallike epileptiform events. These epileptiform events were triggered by an amino acid receptor-mediated mechanism that was glutamatergic independent and mainly attributable to a GABAA receptor-mediated conductance (248). These findings are in line with the observation that, in some cases, dysplastic tissue of FCDIIb and cortical tubers retain features of immature tissue (249, 250).
7. DYSFUNCTIONS IN INTRINSIC EXCITABILITY
7.1. Voltage-Gated Na+ Channels
Na+ channels are clustered at high density at the axonal initial segment (AIS), which is, for this reason, the primary site for generation of APs in neurons (FIGURE 5). In myelinated axons, Na+ channels are clustered at high density also at the nodes of Ranvier to allow saltatory axonal conduction. Voltage-gated Na+ channels are essential for the generation of neuronal excitability because they generate the depolarizing Na+ currents that initiate and propagate APs. These Na+ currents have fast activation and inactivate within a few milliseconds after opening, although a small fraction of slowly inactivating (persistent) current remains for longer periods during depolarizations (251, 252). NaVs are composed by a principal pore-forming α-subunit (9 isoforms: NaV1.1–NaV1.9 for the proteins, SCN1A–SCN11A for the genes) and auxiliary β-subunits (4 isoforms: β1–β4 for the proteins, SCN1B–SCN4B for the genes) (251, 253). The primary sequence of the α-subunits contains four homologous domains (DI–DIV), each comprising six predicted transmembrane segments (S1–S6) that form voltage-sensing modules (S1–S4; S4 is the voltage sensor) and pore modules (S5 and S6 and their connecting extracellular loop) in each domain. The β-subunits contain a single transmembrane segment. SCNs/NaVs are targets of clinically used sodium channel blockers, among them several antiseizure medications (ASMs) (254). Mutations in SCN1A/NaV1.1, SCN2A/NaV1.1, SCN3A/NaV1.1, and SCN8A/NaV1.6 as well as of SCN1B/β1, which are expressed in the central nervous system (CNS), are important causes of DEEs (251, 255–257). Patients carrying variants in voltage-gated Na+ channels also exhibit an increased risk of sudden unexpected death in epilepsy (SUDEP) (258). Mutations implicated in DEEs have been found also in genes encoding proteins that can modulate NaV properties, like FGF12/FHF1 (259, 260).
7.1.1. NaV1.1 channels (SCN1A).
SCN1A, encoding the NaV1.1 α-subunit, is one of the most clinically relevant epilepsy genes, with hundreds of mutations reported thus far, whose associated phenotypes range from Dravet syndrome, a severe form of DEE, to milder GEFS+ and other diseases such as hemiplegic migraine (4, 261, 262).
Dravet syndrome is caused by often de novo heterozygous SCN1A mutations (263, 264), of which approximately half are missense and half are predicted to give rise to a truncated nonfunctional protein. The clinical spectrum of Dravet syndrome does not have firmly established boundaries, but the core phenotype is characterized by intractable seizures precipitated by increased body temperature with onset between 6 mo and 1 yr of age and subsequent appearance of multiple hyperthermia-induced and hyperthermia-independent seizures. Development is normal in the first year of life but plateaus rapidly, with most patients showing cognitive impairment. In patients with Dravet syndrome, SCN1A mutations can also be inherited from a parent with less severe clinical manifestations, at times carrying somatic mosaicism (265). It has been proposed that mutations in other genes can cause Dravet syndrome-like phenotypes (including SCN1B, HCN1, KCN2A, GABRA1, GABRG2, and STXBP1), but with specific clinical pictures (see below). Thus, the association between SCN1A mutations and Dravet syndrome is highly specific. The familial epilepsy GEFS+ syndrome can be also caused by heterozygous missense SCN1A mutations and is characterized by febrile seizures plus (FS+: febrile/hyperthermic seizures that extend beyond 6 yr of age) and afebrile generalized tonic-clonic seizures (GTCS), at times including absence, myoclonic, atonic, or focal seizures, and Dravet syndrome. Missense SCN1A mutations can also cause sporadic/familial hemiplegic migraine type 3 (S/FHM3), a rare form of migraine with aura and onset in adolescence, characterized by hemiparesis as part of the aura phase. Moreover, two missense mutations with gain of function (GoF) have been associated with an extremely severe early infantile DEE with earlier seizure onset, profound cognitive and motor impairments, and a hyperkinetic movement disorder (266–268).
The functional effect of truncating mutations causing Dravet syndrome has been thought since their identification to be haploinsufficiency: 50% reduction of functional NaV1.1 protein in heterozygotes, with complete loss of function (LoF) (263), which has been verified in functional studies (269). The effect of missense mutations studied in transfected cell lines has been more controversial, but most of the results point to a LoF, whose severity tends to correlate with that of the phenotype (251, 270). Thus, the severity spectrum of SCN1A-related epilepsies could be a continuum and depend on the amount of LoF of the mutant. Some SCN1A missense mutations cause LoF because of folding/trafficking defects that lead to channel degradation (271) that can at least partially be rescued by interacting proteins stabilizing the correct folding conformation (272–275).
NaV1.1 is the predominant Na+ channel of GABAergic interneurons whose decreased excitability induced by SCN1A epileptogenic mutations reduces GABAergic inhibition and causes network hyperexcitability (64). A study with a knockin model expressing a truncating nonsense DS mutation reported a similar phenotype, showing that Nav1.1 localizes to the axon initial segment of GABAergic interneurons, in particular fast-spiking parvalbumin (PV)-positive ones (65). Subsequent studies have shown that these mice also display comorbidities including cognitive and behavioral deficits, ataxia, SUDEP, dysregulated circadian rhythms, and sleep dysfunctions (213, 214, 251, 270). Several other studies, including those performed with conditional mouse models that express the mutations in specific neuronal subtypes (251, 270) and with a knockin model of a GEFS+ mutation (Scn1aR1648H/+) (276, 277), have confirmed that hypoexcitability of GABAergic neurons is the initial pathological mechanism in Dravet syndrome models and that the amount of LoF can determine the severity of the phenotype. Notably, Scn1a mouse models have also allowed disclosure of genetic modifiers and cellular remodeling induced by the initial effect of the Scn1a mutation, which can modulate the phenotype, implementing homeostatic or propathological modifications, including homeostatic upregulation of Na+ channels in GABAergic neurons and seizure-induced hyperexcitability of glutamatergic neurons (251, 270). For instance, the interaction between seizures and the genetic mutation can induce remodeling in the Scn1aR1648H/+ mouse model, transforming a basically asymptomatic phenotype into a Dravet syndrome-like one, because of increased excitability of excitatory glutamatergic neurons (219). Overall, these results show that the initial pathological mechanism in mouse models of human epileptogenic SCN1A mutations is LoF and hypoexcitability of at least some subtypes of GABAergic neurons. However, this initial dysfunction triggers both homeostatic and pathological remodeling, depending on the neuronal types, age, genetic background, and interactions between SCN1A mutations and experienced seizures.
Although some drugs like stiripentol (278), fenfluramine (279) or cannabidiol (280) have been shown to be partially effective in some patients, Dravet syndrome still remains drug resistant for most patients. Scn1a gene-targeted mice have been used for testing therapeutic approaches, both classical pharmacological treatments and novel methods, including gene therapy, in some cases obtaining significant amelioration of the phenotype (251, 281). Some novel approaches have shown particularly effective results and have led to clinical trials. For example, an antisense oligonucleotide-based targeted augmentation of nuclear gene output (TANGO) approach has been used to increase the expression of functional Nav1.1 channels in Dravet syndrome mice, observing a substantial decrease in spontaneous seizures and SUDEP when specific antisense oligonucleotides were injected in newborn mice (282).
Mutations causing sporadic/familial hemiplegic migraine (S/FHM) cause NaV1.1 gain of function (GoF) by inducing gating modifications and increasing the slowly inactivating “persistent” Na+ current (251, 270). Gene-targeted mouse models of S/FHM mutations have been generated, and they show facilitated/spontaneous generation of cortical spreading depression (CSD), a proposed pathological mechanism of migraine with aura but no seizures (283, 284). Genetic and acute models have shown that these mutations induce hyperexcitability of GABAergic neurons, leading to increase of extracellular K+ and clamped depolarizing block of neuronal activity (285). A very mild GoF has also been observed for the p.T226M mutation, causing an extremely severe early infantile SCN1A DEE (268), but it is not clear yet how such a mild change could induce the severe phenotype.
7.1.2. NaV1.2 channels (SCN2A).
SCN2A encodes the NaV1.2 sodium channel, which is widely expressed in the central nervous system, particularly in cortical and hippocampal glutamatergic neurons (251). These are the main Na+ channels of the axonal initial segment (AIS) and the nodes of Ranvier in the first 10 days of postnatal life in rodents; then they are partially replaced by SCN8A/NAV1.6. It is thought that NaV1.2 channels in the proximal AIS promote backpropagation to the soma. In the adult brain, NaV1.2 is also present in thin processes, presumably distal unmyelinated portions of preterminal axons.
The first epilepsy syndrome clearly associated with SCN2A mutations was benign familial neonatal/infantile seizures (BFNIS) (286), characterized by a mild phenotype. Subsequently, it has been shown that mutations in SCN2A cause a wide range of neurodevelopmental disorders, including DEE of varying severity. DEE mutations generally arise de novo, and ∼80% are missense. About 60% of SCN2A DEE have onset in the first 3 mo of life (287, 288). All neonatal-early infantile DEE patients display intellectual disability. About 50% of them have variable seizure types, whereas the others have Ohtahara syndrome (neonatal-onset spasms or tonic seizures and EEG with burst suppression pattern), sometimes evolving into West syndrome, Lennox–Gastaut phenotypes, or epilepsy with early infantile migrating focal seizures (EIMFS). DEEs with infantile and childhood onset are ∼40% of the total (287, 288). The phenotype can be correlated to the age of onset. Patients with onset between 3 mo and 1 yr of age in general have West syndrome, with possible evolution into a Lennox–Gastaut phenotype. Patients with onset after 1 yr of age often show variable seizure phenotypes with developmental/cognitive delay and autistic traits that can appear before seizure onset.
Functional analysis in transfected cell lines has shown some genotype-phenotype correlations (251, 289). GoF mutations are related to mild benign neonatal/infantile epilepsy and to neonatal/early infantile DEE, which can be responsive to treatment with Na+ channel blockers (288), whereas LoF mutations are linked to infantile/childhood epileptic encephalopathy or neurodevelopmental disorders (autism and intellectual disability) without seizures. However, identifying genotype-phenotype correlations within these GoF and LoF mutants is difficult because of partially overlapping phenotypes.
Recordings in brain slices of heterozygous knockout Scn2a (Scn2a+/−) mice have shown that Nav1.2, besides its established axonal role, has important dendritic functions in pyramidal neurons of the prefrontal cortex, where its haploinsufficiency impairs synaptic plasticity and synaptic strength, even when Scn2a expression is reduced in single neurons late in postnatal development (290). However, these studies have shown that Scn2a+/− mice, differently from Scn1a models, have a relatively mild phenotype, including short absence-like seizures, spatial memory deficits but enhanced fear memory, plus autistic traits (251, 291). A Scn2a mouse model carrying the severe hypomorphic mutation Δ1898 shows a more severe phenotype, with robust autistic-like features (292). Two additional studies have shown that either complete deletion (293) or reduced expression (293, 294) of Scn2a can paradoxically induce hyperexcitability of glutamatergic neurons. Overall, these findings suggest that reduction of SCN2A to more than haploinsufficiency could be needed to induce severe phenotypes. A knockin mouse model of the recurrent GOF mutation R1882Q has been generated (295), showing that heterozygous mice display hyperexcitability of cortical pyramidal neurons and develop spontaneous seizures at PN1 and premature death between PN13 and PN30. Interestingly, reduction of NaV1.2 expression by specific antisense oligonucleotides reduced seizures and extended life span.
7.1.3. NaV1.3 channels (SCN3A).
SCN3A encodes the NaV1.3 Na+ channel, whose functions have not been completely determined yet. SCN3A is widely expressed in the brain at high levels during embryonic development and is downregulated afterward (251).
SCN3A-related clinical phenotypes comprise a wide spectrum, including mild epilepsy with intellectual dysfunction, early infantile DEE often associated with polymicrogyria, as well as speech and oral motor dysfunctions associated with polymicrogyria without epilepsy. Relatively mild neonatal-childhood onset focal epilepsy (296) would be the mildest phenotype in the spectrum (297, 298). Disrupted cerebral cortical folding and neuronal migration were recapitulated in ferrets expressing the mutant SCN3A (298). Functional studies in transfected human cell lines have shown that most of the SCN3A mutations associated with severe human phenotypes exhibit prominent GoF, inducing, in particular, large increases of persistent Na+ current (297–299). LoF caused by reduced current density has been reported for some SCN3A variants. Heterozygous adult Scn3a+/− knockout mice were investigated as a model of SCN3A LoF mutations, but they do not show features of DEE (300).
7.1.4. NaV1.6 channels (SCN8A).
SCN8A encodes NaV1.6, the main Na+ channel in the axon initial segment and nodes of Ranvier of myelinated axons in mature excitatory neurons (251).
De novo heterozygous mutations in SCN8A have been identified in a range of phenotypes. Most patients show a DEE with multiple seizure types and early onset, severe intellectual disability, and movement disorders and have an increased risk of SUDEP (301). Other patients exhibit milder phenotypes, including benign familial infantile seizures (BFIS) and epilepsies with intermediate phenotypes, or generalized epilepsy with absence seizures (302, 303). Some patients show intellectual disability, autism, or movement disorders without epilepsy (304, 305). About 20% of patients have recurrent mutations.
Mutations that cause DEE or milder epilepsy induce GoF with negative shifts of voltage dependence of activation, positive shifts of voltage dependence of inactivation, slowed channel inactivation, or increased persistent or resurgent current. These functional changes are all consistent with neuronal hyperexcitability, which has been confirmed in transfected cultured neurons (304, 306). Other mutations causing intellectual disability, autism, or movement disorders without epilepsy induce LoF, which can be complete (304, 305). Massive GoF can induce reduced generation of APs, mimicking LoF mutations (304). LoF mutations have been identified in patients with absence seizures (303), a phenotype reproduced by heterozygous LoF mouse models.
A standard global knockin (307) and a conditional floxed knockin (308) mouse model of SCN8A DEE mutations are available. Overall, knockin mice indicate that SCN8A GoF mutations are sufficient to induce hyperexcitability of some subtypes of excitatory neurons, generating severe seizures and a lethal phenotype. In contrast, spontaneous mouse models carrying LoF SCN8A mutations (309) show a phenotype similar to that of patients with intellectual disability and/or movement disorders without epilepsy.
Some patients with SCN8A GoF mutations respond to high doses of sodium channel blockers (303, 310), but available blockers are not isoform specific and chronic therapy at high doses can induce adverse effects. A recent study has used antisense oligonucleotides that reduce Scn8a expression by 25–50%, showing a delay of seizure onset and lethality in a knockin Scn8a mouse model (311). These genetic approaches are highly specific for SCN8A, but problems of oligonucleotide delivery and half-life need to be solved before a translational use.
7.1.5. Auxiliary subunits of Na+ channels.
SCN1B encodes the “auxiliary” β1 Na+ channel subunit that is widely expressed in different organs, including the central nervous system. β-Subunits were originally called auxiliary because they do not directly form the channel but modulate the properties and the membrane delivery of α-subunits. We now know that they are multifunctional molecules implicated, besides direct modulation of α-subunits, in diverse and essential roles in multiple tissues, including cell adhesion and migration, neuronal pathfinding, fasciculation, and neurite outgrowth (256). Mutations in β-subunits can alter numerous functions, including modulations of all the α-subunits, and thereby they are involved in epilepsy, neurodegenerative disorders, neuropathic pain, cardiac arrhythmias, and cancer (256). Mutations in SCN1B have been identified in a range of epilepsy phenotypes. Homozygous SCN1B mutations have been identified in DEE patients that were initially included in the Dravet syndrome spectrum (312). However, it is now clear that their clinical features are distinct from Dravet syndrome, showing earlier-onset seizures and more severe neurodevelopmental phenotype, including psychomotor, stagnation or regression, and microcephaly (313, 314).
Functional analysis of SCN1B DEE mutations in transfected cell lines have identified either LoF modulation of coexpressed α-subunits or induction of complex gating modifications in different coexpressed α-subunits (312–314). Homozygous knockout null Scn1b−/− mice show spontaneous seizures and a high mortality rate (315). Cortical neurons in brain slices obtained from these mice have shown dysfunctions in the excitability of both pyramidal excitatory neurons and GABAergic fast-spiking interneurons. GABAergic interneurons were hypoexcitable, whereas dysfunctions of pyramidal neurons were more complex, with subsets of them exhibiting hyperexcitability at low current injections as well as hypoexcitability at high stimulation intensities.
7.2. Voltage-Gated K+ Channels
K+ channels are the most diverse group of ion channels and can be classified into different families depending on the number of transmembrane domains in each subunit and the gating mechanisms. Voltage-gated K+ (KV) channels are composed of four subunits, each with six transmembrane segments, a voltage sensor module formed by segments S1 to S4, and the pore region formed by S5, S6, and their connecting loop, as in NaV channels. They generate repolarizing currents that oppose the action of depolarizing currents and are involved in different epileptic encephalopathies.
7.2.1. M-current K+ channels (KV7, KCNQ).
Five KCNQ genes have been identified that encode KV7 K+ channels, which can form both homo- and heterotetramers (316, 317). Of the four channels expressed in the nervous system (KCNQ2–5), three (particularly KCNQ2 but also KCNQ3 and KCNQ5) host DEE mutations. KCNQ2 and KCNQ3 are expressed in many brain areas, whereas expression of KCNQ5 is more limited (318). They generate the M (muscarinic receptor inhibited) current, a slow noninactivating K+ current that activates at subthreshold membrane potentials (319). KCNQ2-linked DEE has its onset in early infancy and features motor impairment and variable intellectual disability (320). Often, the EEG initially shows a burst suppression pattern that may later evolve to multifocal epileptiform activity. KCNQ3 and KCNQ5 DEEs display a variable degree of severity, and some patients show intellectual disability apparently without epilepsy (9, 316). Mutations in the three KCNQ genes can also cause “benign familial neonatal seizures” (BFNS), which is mild and self-limited (317).
According to their subcellular localization, KV channels can regulate specific features of neuronal excitability. Somato-dendritic channels are strongly activated by back-propagating APs, attenuate repetitive firing, and contribute to the medium and slow components of the afterhyperpolarization (AHP), determining the refractory period and regulating spiking frequency. In the perisomatic region, slow activation of the M current decreases neuronal firing in response to sustained stimuli, inducing spike-frequency adaptation (318, 321, 322). Axonal KV channels mostly function by stabilizing the resting membrane potential, leading to increased activation of axonal NaV channels (323–325) and of presynaptic Ca2+ channels, modulating synaptic release (326, 327). Both KCNQ2 and KNCQ3 are also expressed in GABAergic interneurons (328).
Numerous in vitro studies have investigated functional effects of KCNQ DEE mutations in cell lines or Xenopus oocytes (316). Most studies agree that LoF, often leading to haploinsufficiency, is the pathophysiological mechanism of mild and self-limited BFNS (316, 317). DEE mutations can induce more severe LoF consistent with a dominant-negative effect. A single report showed that the recurrent p.A294V KV7.2 pore mutation does not modify the properties of the current but induces LoF altering the subcellular localization of KV channels (329). DEE can also be caused by GoF, in which case disease severity correlates with that of functional alteration (316). The first transgenic mouse model expressing a KCNQ2 dominant-negative mutant (151) showed spontaneous focal and generalized tonic-clonic seizures, impaired hippocampus-related memory, and pronounced hyperactivity, but also cell loss in the hippocampus, which is not observed in patients. The first knockin model of KCNQ2-DEE (221) carrying the p.T274M mutation displayed an overall 70–80% reduction of the M current (330). KCNQ2T274M/+ mice show spontaneous generalized seizures and impairment of spatial learning and memory and do not show major structural brain defects or neuronal mortality, which is consistent with the clinical features observed in patients. The selective deletion of KCNQ2 and KCNQ3 in parvalbumin-positive interneurons increased their firing, leading to homeostatic potentiation of excitatory synaptic activity. Consistently, this model shows reduced latency for picrotoxin-induced seizures (331).
7.2.2. Delayed-rectifier K+ channels (KV1.1/KCNA1; KV1.2/KCNA2; KV2.1/KCNB1; KV8.2/KCNV2).
7.2.2.1. KV1.1 (KCNA1) AND KV1.2 (KCNA2).
KCNA2 encodes the KV1.2 shaker-type voltage-gated K+ channel subunit, which is highly expressed in both excitatory and inhibitory neurons, particularly in the axon (332–334). The KV1.2 channel generates the delayed-rectifier K+ current, an important component of the repolarizing currents during APs. KV1.2 channels have high homology to KV1.1 channels but require stronger depolarization to activate. KV1.1 and KV1.2 subunits can produce heteromeric potassium channels with intermediate properties between the respective homomers (335). KV1.1 and KV1.2 subunits are associated with cell adhesion molecules (CAMs), including LGI1, which contributes to their subcellular localization (336). KCNA2-DEE clinical features have been correlated with effects of mutations studied in vitro and can result in strong GoF, mixed GoF and LoF, and LoF with negative dominance (337, 338). Patients with strong GoF variants have onset between 5 and 15 mo of age with intellectual disability, ataxia, and cerebellar atrophy, often with generalized seizures. Patients with variants showing mixed GoF and LoF effects have onset from the neonatal period to 6 mo of age with refractory generalized and focal seizures and developmental delay. Patients with dominant-negative LoF variants can have less severe features. The LoF mechanism is consistent with the classical role of K+ channels and the phenotype of KV1.2-mutant mice. Homozygous knockout mice show severe seizures and early mortality, and heterozygous knockout mice are more sensitive to convulsants, although they do not show spontaneous seizures (339). Both heterozygous and homozygous “Pingu” mice, which carry a chemically induced LoF missense mutation, show motor abnormalities (340). Since KV1.2 channels are expressed in both excitatory and inhibitory neurons, the cellular mechanism of KCNA2 mutations is not completely clear. Their repolarizing role can help in both sustaining high firing rates and setting the resting membrane potential. Recordings from LoF mouse models have shown contrasting modifications of neuronal firing, with hypoexcitability of glycinergic neurons in Kcna2 knockout (339) and hyperexcitability of cerebellar basket cells in Pingu mice (340). Although the detailed pathological mechanism is still elusive, the identification of GoF or LoF effects can provide important information for orienting the therapy in a precision medicine approach. In fact, it has been recently shown that the K+ channel blocker 4-aminopyridine antagonized GoF defects caused by variants in KCNA2 in vitro and was effective in reducing symptoms in patients carrying GoF KCNA2 mutations (341). KCNA1 LoF mutations have also been identified in some DEE patients (342, 343), extending the pathological spectrum of this gene to severe epilepsy.
7.2.2.2. KV2.1 (KCNB1) AND KV8.2 (KCNV2).
KCNB1 encodes the KV2.1 pore-forming and voltage-sensing α-subunit, which contributes to generate the delayed-rectifier K+ current (317). It is expressed in both excitatory and inhibitory neurons of the mammalian brain and is localized to the soma, proximal dendrites, and axon initial segments (344). KV2.1 is important for sensing and homeostatically regulating excitability, because its activity can be inhibited by phosphorylation; increased neuronal activity induces dephosphorylation, which results in increased delayed-rectifier current leading to reduced neuronal excitability (345, 346). Mutations in KCNB1 have been recently reported in patients with early-onset DEE (347–349). KCNB1-related DEEs encompass a wide spectrum of neurodevelopmental disorders with different types of epileptic seizures, predominant language difficulties, and behavioral impairment. Most variants occur de novo and mainly consist of missense variants located on the voltage sensor and the pore domain. Truncating variants in the COOH-terminal domain are associated with a less severe epileptic phenotype, although cognitive/behavioral impairment is still severe. Functional studies in transfected cells have shown a variety of defects, including loss of ion selectivity, reduced conductance, and dominant-negative effects, as well as milder effects on gating properties (347, 350, 351), which induce reduced function and increased neuronal excitability. Kcnb1−/− mice show hyperexcitable hippocampal circuits but do not show spontaneous seizures, although they exhibit increased seizure susceptibility (346). The voltage-gated K+ channel subunit KV8.2, encoded by the KCNV2 gene, is a silent subunit when expressed as a homotetramer, but it increases the KV2 current when coassembled as a heterotetramer with other KV2 channels, for example in hippocampal pyramidal neurons, in which it colocalizes with Kv2.1 and contributes to the generation of the delayed-rectifier K+ current (352, 353). De novo LoF variants in the KCNV2 gene, encoding the voltage-gated K+ channel subunit KV8.2, have been identified in a few DEE patients and contribute to epilepsy susceptibility in mice (353).
7.2.3. Fast K+ channels (KV3.1/KCNC1 and KV4.1–4.3/KCND).
KCNC1 encodes the KV3.1 channel, which is preferentially expressed in neurons that generate high-frequency firing, including parvalbumin-containing GABAergic interneurons in the cerebral cortex, hippocampus and amygdala, auditory brain stem neurons, cerebellar granule cells, and neurons of the reticular nucleus of the thalamus (354). In fast spiking GABAergic neurons, KV3.1 channels are localized to the proximal dendrites, somata, axon hillock, and synaptic terminals but are not found in distal dendrites (354, 355). Specific functional properties that distinguish KV3.1 channels from other KV channels are the very fast kinetics of activation and deactivation and the voltage dependence of activation shifted toward depolarized potentials (354). These properties are optimized for promoting the generation of high firing rates, up to hundreds of hertz (356). Mutations in KCNC1 have been identified in early-onset DEE and in patients with intellectual disability without seizures (357, 358). Mutants have been functionally characterized in Xenopus laevis oocytes. All characterized mutations resulted in partial or complete LoF, with some of them inducing negative dominance leading to >50% reduction of current in heterozygosis (357–359). These results are consistent with hypoexcitability of fast-spiking neurons as the main pathogenic mechanism of KCNC1 DEE. Knockout Kcnc1−/− mice have a subtle phenotype (360), and there are no mouse models engineered to carry human KCNC1 mutations.
KCND1–3 encode the KV4.1–3 α-subunits of the Shal family of the A-type voltage-gated K+ channels, which generate a rapidly inactivating outward K+ current and are important in membrane repolarization in excitable cells (361). The de novo heterozygous missense variant p.V404M of KCND2 has been associated with DEE with onset at 2 mo (362). Functional analysis in X. laevis oocytes showed modified current kinetics and reduced inactivation, consistent with GoF, but effects on neuronal excitability were not investigated (362).
7.2.4. Noninactivating K+ channels.
The ether-a-go-go (EAG) K+ channel family is formed by KCNH1 and KCNH5, which encode the KV10.1 (EAG1) and KV10.2 (EAG2) channels, respectively. They generate noninactivating voltage-dependent K+ currents and in expression systems can form heteromeric channel complexes, in which the slow activation of KV10.1 is dominant (363). KCNH1 heterozygous missense mutations have been identified in patients with Zimmermann–Laband and Temple–Baraitser syndromes as well as in patients with unclassified syndromes and a broad phenotypic spectrum with intellectual disability and epilepsy (364–368). Epilepsy is a key phenotypic feature in most patients with KCNH1-related syndromes, who show both generalized and focal tonic-clonic seizures (367, 368). Functional studies of KCNH1 mutants in cell lines have shown left-shifted voltage dependence of activation and slower deactivation kinetics, consistent with GoF (364, 365), although it is not clear how this effect could cause the observed phenotypes. De novo heterozygous missense variants of KCNH5 have been identified in few DEE patients (369, 370). Functional analysis in transfected cell lines has shown a strong hyperpolarizing shift of voltage dependence of activation and an acceleration of activation, consistent with GoF.
7.2.5. Ca2+-activated K+ channels.
KCNMA1 encodes the pore-forming α-subunit of the large-conductance KCa1.1 Ca2+ activated K+ channel (“Big K+,” BK), which is activated by depolarizations and intracellular Ca2+ (371). KCa1.1 is widely distributed in excitable and nonexcitable cells. Expression levels are highest in brain and muscle, where BK channels are critical regulators of neuronal excitability and muscle contractility. In both excitatory and inhibitory neurons, BK channels are implicated in AP repolarization and afterhyperpolarization, influencing the shape, frequency, and propagation of APs (371). Heterozygous de novo missense mutations in KCNMA1 are associated with a wide phenotypic spectrum primarily defined by brain and muscle dysfunction (372–374). KCNMA1-linked DEE is characterized by a heterogeneous combination of epilepsy, dyskinesia, and intellectual disability.
Functional analysis in cell lines transfected with KCNMA1 mutations has shown both LoF and GoF effects that correlate with phenotypic features (373). Although seizures do not show differential distribution between patients carrying GoF and LoF variants, neurodevelopmental and structural brain abnormalities are prevalent in patients with LoF mutations. There are no animal models carrying mutations identified in patients. Kcnma1−/− mice show ataxia, tremor, and impaired coordination and spatial learning (375, 376). Pharmacological inhibition of KCa1.1 channels induces tremor and ataxia in animals (377) but might be used for treatment of GoF mutations.
7.2.6. Na+-activated K+ channels.
KCNT1 encodes the KNa1.1 subunit, which has a classical six-transmembrane segment structure and forms a tetrameric Na+-activated K+ channel (also called Slack, KCa4.1, or Slo2.2). KNa1.1 is activated by both intracellular Na+ and voltage and is expressed in different organs, including the nervous system, heart, and kidney. In the central nervous system, it has a distinct expression pattern with respect to KNa1.2 (encoded by the gene KCNT2), but the two subunits can colocalize and form heteromeric channels (378, 379). KNa channels can modulate intrinsic excitability and firing properties of different types of neurons. In particular, they contribute to generate the slow afterhyperpolarization that follows AP discharges, which induce Na+ influx through NaV channels (380).
Mutations in KCNT1 cause different phenotypes, including DEEs. De novo heterozygous mutations were first identified in epilepsy of infancy with migrating focal seizures (EIMFS), also called malignant migrating partial seizures of infancy (MMPSI) (381). EIMFS is characterized by refractory focal seizures arising and status epilepticus with onset in the first 6 mo of life. The neurological status progressively deteriorates, with progressive hypotonia and severe development arrest. De novo heterozygous mutations have been also identified in severe autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), sleep-related hypermotor epilepsy (SHE) (382), and other less common DEE phenotypes (383).
The identified mutations induce large GoF, increasing the KNa1.1 current amplitude (381, 384), whereas one heterozygous LoF variant (p.F932I) from a patient with severe generalized seizures and delayed myelination causes impaired KNa1.1 trafficking (385). Knockin mice of the p.Y796H GoF KCNT1 mutation show early-onset seizures (386). Although KNa1.1 increases KNa currents in both excitatory and inhibitory cortical neurons in primary cultures, an increase of the current in the subthreshold voltages is only found in inhibitory neurons, resulting in inhibitory neuron-specific impairments in excitability and AP generation. GoF KCNT2 mutations also cause DEE (387). Blocking KCNT GoF mutants with quinidine, a class I antiarrhythmic drug, has shown variable success in patients because of dose-limiting off-target effects, poor blood-brain barrier (BBB) penetration, and low potency (383).
7.3. Ca2+ Channels
Voltage-gated Ca2+ channels are important for numerous physiological functions; in neurons, they contribute to neuronal excitability and, at the synaptic level, mediate synchronous transmitter release. The voltage-dependent Ca2+ currents are generated by monomeric α1-subunits that have the canonical four domains of six transmembrane segments (388, 389). The 10 cloned α1-subunits can be grouped into three families according to the membrane potential range at which the channel is activated and the sensitivity to drugs: the high-voltage-activated dihydropyridine-sensitive channels (L type; CACNA1S/CaV1.1, CACNA1C/CaV1.2, CACNA1/CaV1.3, and CACNA1F/CaV1.4), the high/moderate-voltage-activated dihydropyridine-insensitive channels (P/Q, N, and R types; CACNA1A/CaV2.1, CACNA1B/CaV2.2, and CACNA1E/CaV2.3), and the low-voltage-activated channels (T type; CACNA1G/CaV3.1, CACNA1H/CaV3.2, and CACNA1I/CaV3.3). It has been proposed that L-type channels may be implicated in the generation of epileptiform paroxysmal depolarization shifts (390) and that they may be therapeutic targets for epilepsy (391). However, besides Timothy syndrome, a severe multiorgan disorder caused by CACNA1A/CaV2.1 GoF mutations (in which surviving patients can develop epilepsy and autism) (392), there are no reports of their direct involvement in DEE. At least for high-voltage-activated channels, functional properties and intracellular trafficking of α1-subunits can be modulated by accessory subunits, including the β-, α2δ-, and γ-subunits (393).
7.3.1. T-type channels.
T-type channels (CACNA1G/H/I, CaV3.1–3) are widely expressed throughout the nervous system (394). Voltage-dependent opening of T-type channels occurs at comparatively negative membrane potentials and, depolarizing the cell membrane, facilitates the generation of APs. This function is especially relevant in several central neurons in which T-type channels are particularly abundant in dendrites, where they enhance subthreshold postsynaptic potentials and facilitate the propagation of the electrotonic signal to the cell body (395). They are also involved in the firing of rebound bursts of APs that support various forms of neuronal pacemaking, particularly in the thalamocortical network, whose dysfunction is implicated in absence seizures (396). Polymorphisms in CACNA1G/CaV3.1 and CACNA1H/CaV3.2 causing in general mild GoF have been identified as risk factors for idiopathic generalized epilepsy (397). De novo heterozygous missense variants in CACNA1G/CaV3.1 and CACNA1I/CaV3.1 have been recently identified in patients with infantile-onset DEE (398, 399) and variable neurodevelopmental phenotypes including DEE with cognitive impairment, hypotonia, and epilepsy (400). Functional effects are consistent with GoF (including persistent Ca2+ current at resting membrane potential for CACNA1I/CaV3.1 variants) or mixed GoF and LoF. Evidence for the involvement of CACNA1H/CaV3.2 in DEE is more limited. Gene-targeted animal models of T-type channel DEE mutations are not available yet.
7.3.2. P/Q-type Ca2+ channels.
CACNA1A encodes the Cav2.1 Ca2+ channel, which generates the high-voltage-activated P/Q-type Ca2+ current with moderate voltage-dependent inactivation. CACNA1A is widely expressed in the central nervous system and is essential for fast and synchronous neurotransmitter release in numerous neuronal subtypes (401). CACNA1A mutations have been associated with episodic ataxia type 2 (EA2), spinocerebellar ataxia type 6 (SCA6), and familial hemiplegic migraine type 1 (FHM1) (402–404) and, more recently, with severe intellectual disability, motor impairment, and episodic ataxia (61, 405). Functional characterization of CACNA1A DEE mutations performed in transfected cell lines has generated controversial results because both LoF (decreased channel targeting at the cell membrane) and GoF (hyperpolarized shift of the activation curve) effects were reported (406). A mouse model of CACNA1A DEE has not been generated yet.
7.3.3. N-type Ca2+ channels.
CACNA1B encodes the Cav2.2 N-type Ca2+ channel, which generates the high-voltage-activated Ca2+ current with moderate voltage-dependent inactivation. CACNA1B is expressed throughout the central nervous system and acts synergistically or complementarily with CACNA1A for generating presynaptic Ca2+ fluxes that mediate neurotransmitter release (401). Expression of CACNA1B is thought to be crucial for neurotransmission in the early postnatal period, as Cav2.2 channels are replaced by Cav2.1 channels in most mature synapses (401). In particular, Cav2.2 is implicated in asynchronous synaptic release, which occurs up to tens of seconds after the AP (401). Cav2.2 may also have a role in synaptic plasticity, synaptogenesis, gene transcription, neuronal survival, and migration of immature neurons (388). CACNA1B variants have been identified in a severe form of autosomal recessive DEE featuring developmental delay, microcephaly, inability to walk or speak, early-onset refractory seizures, myoclonus/dyskinesia, frequent feeding difficulties, and high risk of premature demise (407). The identified variants are predicted to truncate the Cav2.2 protein, leading to LoF and haploinsufficiency, although the genotype-phenotype relationships are not clear. There are no gene-targeted mouse models of the identified human variants, but Cacna1b−/− mouse models show overt neurodevelopmental abnormalities, including abnormal locomotor activity and memory impairment (408).
7.3.4. R-type Ca2+ channels.
CACNA1E encodes the moderate-voltage-activated CaV2.3 Ca2+ channel, which generates the R-type Ca2+ current with fast voltage-dependent inactivation. It is highly expressed in the central nervous system and involved in generating the Ca2+ influx in synaptic terminals that initiates rapid release of neurotransmitters. Mutations in CACNA1E cause severe autosomal-dominant DEE with macrocephaly, hypotonia, early-onset refractory seizures, profoundly impaired neurodevelopment, and hyperkinesia (409). Functional analysis of CACNA1E variants in transfected cell lines mostly showed GoF effects with facilitated activation and slowed inactivation (409), consistent with increased neurotransmitter release and network hyperexcitability. Other variants were predicted to generate truncated nonfunctional proteins and haploinsufficiency. Cacna1e−/− null knockout mice do not show overt neurological phenotypes (410).
7.4. Other Cation Channels
7.4.1. Voltage-independent Na+ channels.
The sodium leak NALCN channel (NaVI2.1) is predominantly expressed in neurons, where it is important for setting resting potential and controlling neuronal excitability. NALCN encodes for a voltage-independent, noninactivating cation channel permeable to Na+, K+, and Ca2+ that generates the background TTX-resistant Na+ leak current (411, 412). Autosomal-recessive missense and nonsense NALCN mutations have been identified in a DEE with onset at birth or in early infancy, characterized by variable degrees of hypotonia, speech impairment, and intellectual disability (infantile hypotonia with psychomotor retardation and characteristic facies, IHPRF) (413). A subsequent study found heterozygous de novo NALCN missense variants to cause congenital contractures of the limbs and face, hypotonia, and developmental delay (CLIFAHDD) (414, 415). Functional studies in transfected cell lines have generated controversial results, showing GoF, LoF, or dominant-negative effects for both syndromes (415, 416). Nalcn–/– knockout mice show severely disrupted respiratory rhythm and die within 24 h of birth.
7.4.2. Hyperpolarization-activated cyclic nucleotide-gated channels.
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are Na+/K+-permeable channels that are activated by hyperpolarization at voltages more negative than −50 mV (417). cAMP and cGMP directly bind to the intracellular cyclic nucleotide-binding domain and shift the activation curve of HCN channels to more positive voltages, increasing channel activity. The four known HCN isoforms (HCN1–4) have secondary structure that resembles that of K+ channels. HCN1 and HCN2 are the main isoforms expressed in the brain, with a predominant somato-dendritic expression. The hyperpolarization-activated currents generated by HCN channels in neurons, named Ih or Iq, are depolarizing and contribute to set the resting membrane potential, shape synaptic inputs, and generate rhythmic and synchronized neuronal activity implicated in pacemaking and somato-dendritic oscillations (418).
De novo dominant missense HCN1 variants were initially identified in DEE patients with a Dravet-like phenotype (419) but have subsequently been related to a wider phenotypic spectrum ranging from mild generalized epilepsy or GEFS+ to severe early-onset DEE (420). Functional studies in cell lines identified GoF as the major pathophysiological mechanism and a LoF with dominant-negative effect for a limited number of variants (419). Knockout HCN−/− mice show deficits in motor learning and modifications of spatial memory but not a DEE phenotype (421), whereas the knockin mouse for the p.M305L mutation shows DEE (422). Functional analysis in CHO cells and pyramidal neurons demonstrated that the p.M305L mutation causes a constitutive activation of the channel (420).
7.5. The Na+-K+-ATPase Pump
The Na+-K+-ATPase (NKA) ion pump is a ubiquitous transmembrane enzyme responsible for active exchange of Na+ and K+ ions across the plasma membranes in higher eukaryotic cells and is composed of a large catalytic α-subunit and smaller β- and γ-subunits that modulate the membrane exposure and activity of α-subunits (423, 424). In neural tissue, NKA generates an outward current that contributes to the resting membrane potential and powers secondary active transports, including Na+/H+ and Na+/Ca2+ exchanges, K+-Cl− cotransport, and Na+-dependent neurotransmitter uptake (423).
The four known human isoforms of the α-subunit (α1–4), encoded by four paralogous genes (ATP1A1-4), have developmental and tissue expression specificity. The α2- and α3-isoforms, encoded by ATP1A2 and ATP1A3, are predominantly expressed in the central nervous system. Constitutional heterozygous mutations in ATP1A2/A3 have been associated with several autosomal dominant neurological disorders including familial hemiplegic migraine (FHM), rapid-onset dystonia-parkinsonism (RDP), alternating hemiplegia of childhood (AHC), cerebellar ataxia-areflexia-progressive optic atrophy (CAPOS), and relapsing encephalopathy with cerebellar ataxia (RECA) (425–427). Epilepsy and intellectual disability may co-occur with FHM and AHC, and severe epilepsies have been described in rare patients with ATP1A2 or ATP1A3 mutations (428, 429). Early lethal hydrops fetalis, intrauterine growth restriction, arthrogryposis, microcephaly, polymicrogyria, and lack of respiratory drive have been associated with homozygous truncating mutations in ATP1A2 (430, 431). Investigating the genetic causes of DEEs variably associated with malformations of cortical development in a large cohort of patients, Vetro and collaborators (432) identified 22 patients carrying de novo or inherited heterozygous ATP1A2/A3 mutations. Most patients manifested early, often neonatal, onset seizures with a multifocal or migrating pattern. A distinctive, “profound” phenotype, featuring polymicrogyria or progressive brain atrophy and epilepsy, resulted in early lethality. Neuropathological analysis of the whole brain in two individuals with polymicrogyria revealed a mainly neural pathogenesis, compounded by vascular and leptomeningeal abnormalities (432). Functional studies performed in COS-1 cells transfected with different mutations demonstrated a LoF mechanism, with a wide continuum of severity distributed across mutations that variably impaired NKA pump activity. Mutations associated with most severe phenotypes cause lack of COS-1 cell-survival (432). Interestingly, PRRT2, a gene whose deficiency is associated with paroxysmal disorders including epilepsy (see sect. 8), is coexpressed with and is a physiological activator of α3-NKA in several brain areas (433).
8. DYSFUNCTIONS IN SYNAPTIC TRANSMISSION AND PLASTICITY
Although the most straightforward causes of hyperexcitability have been historically and widely associated with mutations in ion channel genes, dysfunctions of synaptic transmission are emerging as primary causes of epilepsy and DEE. That is why the term “synaptopathies” was coined, inherently meaning that anomalies of information transfer at the synapse can profoundly affect network excitability, excitatory/inhibitory (E/I) balance, and nervous system development, triggering epileptogenesis.
In the last 30 years, the machinery of synaptic transmission has been largely clarified in the most subtle molecular details. Several physiologically important molecular actors orchestrate neurotransmitter (NT) release at the presynaptic side and decode its message into a biological response at the postsynaptic side. Biology teaches that the more important a process is, the higher the complexity involved in its control. Thus, there is a relatively large probability that gene mutations hit some of these numerous actors.
Neurotransmitter release evoked by APs reaching nerve terminals is based on the exocytotic fusion of small organelles, synaptic vesicles (SVs), real nanomachines that sense Ca2+ entry through presynaptic voltage-dependent channels and trigger exocytosis by activating a SNARE-mediated fusion machine that incorporates the SV in the presynaptic membrane and releases its content into the synaptic cleft. The SV-competent membrane patch is eventually retrieved by distinct endocytotic mechanisms that take place at distinct activity patterns. Endocytosis regenerates new SVs that are loaded by NT by an active proton gradient generated by the SV-associated vacuolar H+-adenosine triphosphatase (vATPase) (434, 435). Synaptic vesicles are organized into distinct functional pools, including 1) the readily releasable pool (RRP), comprising SVs immediately recruited for exocytosis, 2) a recycling pool (RP) made by SVs that can be rapidly recruited to replenish the RRP depleted upon activity, and 3) a resting pool (ResP) representing a reserve of SVs that are not immediately releasable upon activity (436, 437).
When referring to alterations in synaptic transmission and plasticity, some issues should be considered. Except for postsynaptic receptors and scaffolding proteins, the presynaptic components of excitatory and inhibitory synapses share most of the actuators of NT release and, in principle, should be equally affected by mutation-induced protein dysfunction, without net changes in the excitatory/inhibitory balance. However, differentially expressed proteins or the different impact of protein dysfunction in excitatory and inhibitory neurons very often implies an overt phenotype affecting network excitability and/or the excitatory/inhibitory balance. For example, a protein involved in SV recycling and refilling of RRP will be more essential for the proper functionality of high-frequency firing neurons (e.g., the PV-positive interneurons) than for excitatory neurons.
Another important issue is the respective weight of basal synaptic strength and short-term plasticity mechanisms. Short-term plasticity is a fundamental determinant of network activity and excitability and thereby plays a central role in epileptogenesis. As neurons do not fire single action potentials (APs) but rather trains of APs, short-term plasticity phenomena, such as depression or facilitation, have a strong impact on the network computational activities, including frequency band filtering of synaptic inputs or pattern detection activities (438, 439).
Synaptic transmission is a major determinant of excitability at the neuronal network level. Intrinsic excitability at the single-neuron level is mostly affected by the size of the neuron and its input resistance and by the level of expression and functionality of voltage-dependent ion channels, particularly at the axon initial segment, the AP initiation spot. However, at the level of a population of synaptically connected neurons, intrinsic excitability is not the only factor defining the firing and bursting behavior of the network as well as its synchrony. Whereas inhibitory synapses act to confine the excitation waves temporally and spatially, excitatory synaptic connections play a prominent role in establishing the activity of the network, owing to their short-term plasticity properties (440).
Mutations in numerous genes affect synaptic functions, and the number of so-called “synaptopathies” is continuously rising. Strong evidence, accumulated in the last two decades, has shown that mutations in fundamental actors of the complex process of NT release can result in neurodevelopmental disorders with epilepsy (FIGURE 8). Here we only consider mutations that affect purely synaptic genes, leaving mutations in the genes of presynaptic and postsynaptic ion channels to sects. 8.1 and 8.2, respectively. Among genes encoding for bona fide synaptic proteins, a distinction should be made between expression/function alterations affecting fundamental proteins for the synaptic transmission machinery and genes that play a modulatory action on the process by impacting more on synaptic plasticity than on the essential synaptic machinery. In addition, the targets of mutations can participate in distinct processes contributing to synaptic transmission. According to the view of the tetrapartite synapse, four distinct entities are involved in the physiological regulation of synaptic transmission: the presynaptic neuron, the target postsynaptic neuron, the extracellular matrix, and the perisynaptic astrocyte. Although the impact of mutations in astrocyte-specific genes is still poorly understood, we can consider three main classes of synaptopathies, namely 1) presynaptic synaptopathies, including defects of the postdocking SV priming/fusion processes, as well as defects in the processes regulating SV trafficking or NT synthesis and loading into SVs; 2) postsynaptic synaptopathies, including defects in postsynaptic receptors and their scaffold/transduction systems; and finally 3) extracellular synaptopathies, including defects in transsynaptic and extracellular matrix (ECM) proteins at the synaptic cleft and secreted synaptic proteins (FIGURE 8). For all these classes of synaptic pathologies, the identification of gene mutations in patients has catalyzed extensive research activity that often contributed to clarifying the pathomechanism of the disease and elucidating the physiological role of the synapse-specific gene products. A substantial contribution to physiopathology of synapse-based DEEs came from the exploitation of murine knockout or knockin experimental models, recapitulating at least some traits of the clinical phenotypes, as well as from mutation studies in transfected cell lines and primary neurons, up to the studies performed in iPSC-derived neurons either containing the patient’s mutation in the patient’s own genetic background or in which the mutation under study has been reproduced by CRISPR-Cas9 gene editing in wild-type cells. There is a wide spectrum of phenotypic DEE features stemming from alterations of synaptic function, although genotype-phenotype correlations are not always straightforward (441). We follow the aforementioned classification to list the main DEE-related genes.
FIGURE 8.
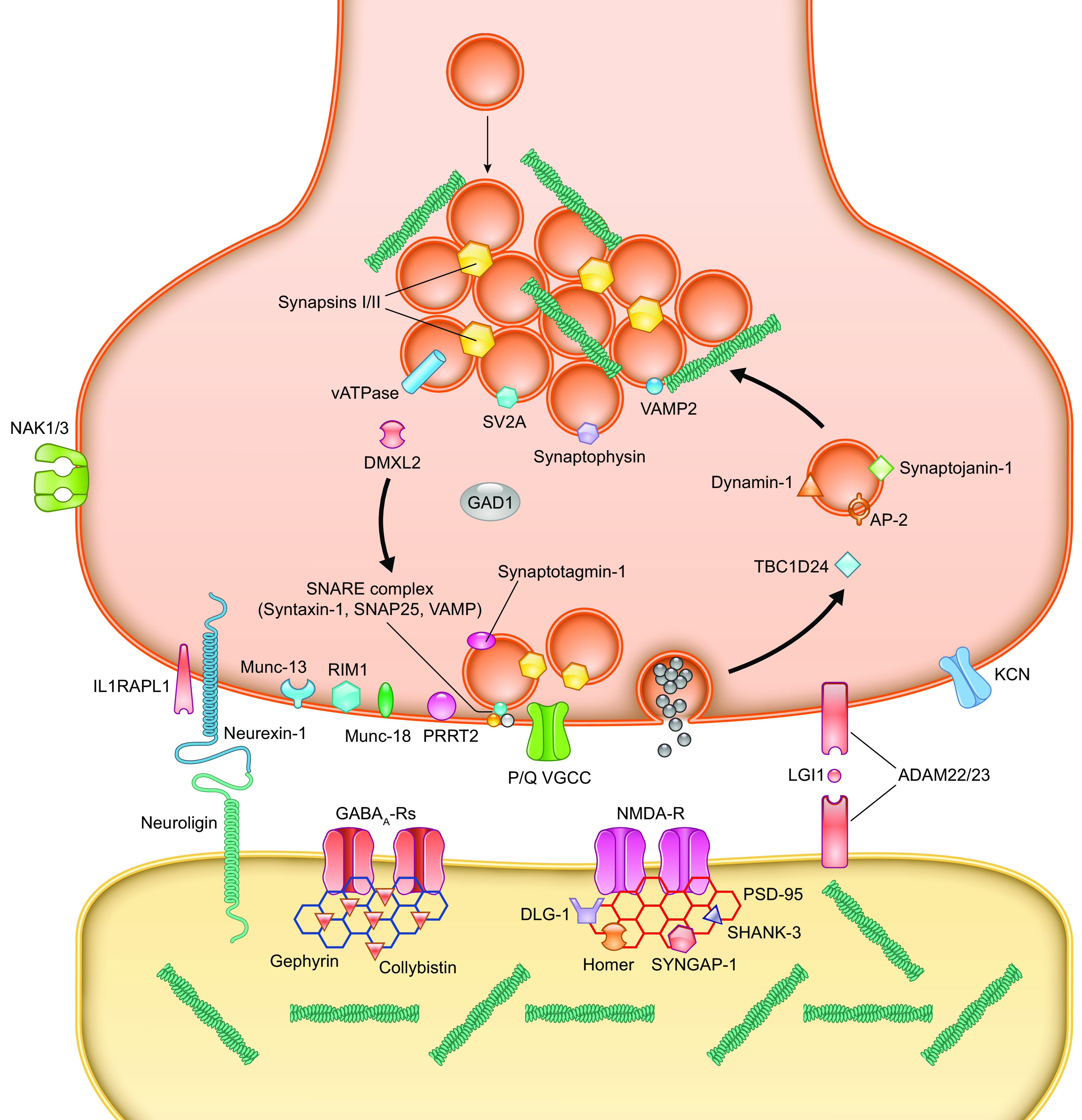
Main actors of synaptic transmission and mapping of the synaptic gene products causing synaptic encephalopathies with epilepsy. Schematic representation of a symbolic synapse containing excitatory and inhibitory synaptic components. The main targets of synaptopathies are as follows: 1) At the presynaptic level, gene products involved in the postdocking synaptic vesicle (SV) priming/fusion processes (SNAREs and SNARE-associated proteins: Munc-13, IM1, Munc-18, PRRT2, SNARE proteins, synaptotagmin-1/2, voltage-gated Ca2+ channels), SV trafficking (trafficking proteins: Synapsins I/II, vATPase, synaptophysin, SV2A, VAMP2, synaptojanin-1, AP-2, dynamin-1, TBC1D24), and neurotransmitter (NT) synthesis and loading into SVs (transport proteins: GAD1, vATPase). 2) At the postsynaptic level, postsynaptic receptors and their scaffold/transduction systems (GABAA and NMDA receptors, gephyrin, collybistin, PSD-95, Homer, Shank-3, SynGAP-1, DLG-1). 3) At the synaptic cleft level, transsynaptic and extracellular matrix proteins and their receptors (neurexin-1, neuroligin, IL1RAPL1, ADAM 22/23, LGI1), as well as secreted proteins (SRPX2, reelin). Other presynaptic voltage-gated channels that affect the dynamics of nerve terminal activation and NT release are also shown. Green: the actin-based cytoskeleton that regulates trafficking and maintenance of SV pool in the nerve terminal and concentrates postsynaptic receptors on the postsynaptic side.
8.1. Presynaptic Synaptopathies
Mutations in many genes encoding proteins involved in the multistep process of NT release cause epilepsy. Starting from the machinery essential for regulated exocytosis, mutations in genes encoding t-SNARE proteins Syntaxin 1B, SNAP25b, and VAMP/synaptobrevin, the SNARE-associated proteins STXBP1 (Munc18-1) and PRRT2, the a-subunit of the P/Q type voltage-dependent Ca2+ channel, and the critical SV Ca2+ sensor synaptotagmin have been identified and associated with epilepsy and DEEs (9, 406, 407, 442–448). In mice, constitutive knockout of these genes greatly hinders AP-driven NT release, with a compensatory increase of spontaneous release and synaptic facilitation (449–455). However, it remains unclear how an impairment of ubiquitous players of evoked NT release can trigger epileptogenesis, presumably by their different impact on distinct neuronal populations, resulting in E/I imbalance and circuit instability. Similar mechanisms are likely to take place in the case of mutations in genes encoding proteins not directly actuating NT release but supporting trafficking, maintenance, and integrity of the SV pools. Loss of function in these proteins, which include the SV proteins synapsins and SV2 and the proteins actuating the endocytotic retrieval of SVs after each round of exocytosis, such as dynamin or amphiphysin, may induce network hyperexcitability by affecting, to a larger extent, neurons undergoing high-frequency activity, such as parvalbumin (PV)-positive inhibitory neurons.
8.1.1. Perturbation of priming/fusion event and of Ca2+ sensitivity.
8.1.1.1. SNARE PROTEINS.
Mutations in the large family of genes encoding membrane proteins mediating vesicle fusion, the so-called SNARE proteins SNAP-25, Syntaxin-1, and VAMP2, constituting the fusion machine actuating exocytosis have been reported. Mutations in the two SNARE motifs of SNAP-25, particularly in its splicing isoform SNAP-25b, cause a combination of epilepsy and cognitive deficits that are reproduced by the selective knockout (KO) of SNAP-25b in mice (443, 456–458). The second presynaptic SNARE protein, syntaxin-1, has been less implicated in epilepsy and DEE. However, some mutations associated with febrile epilepsy have been mapped to the Habc region of the Syntaxin-1b isoform (STX1B) that controls its open/closed conformation permitting the assembly of the SNARE complex. In mice, the a and b isoforms of Syntaxin-1 are redundant, so that the single-KO mice are viable. However, the genetic “freezing” of the b isoform in the open conformation in the syntaxin-1a KO generates severe seizure activity and premature death (450, 459). Mutations in the SNARE motif of VAMP2 were also identified in patients with intellectual disability, epilepsy, and hyperkinetic movement disorder (444). Although it is conceivable that inactivation of SNARE proteins, profoundly altering neuronal signaling at the synapse, can cause neurodevelopmental deficits, the mechanism of the frequently associated hyperexcitability and epilepsy is more elusive. Notably, the three SNARE proteins are the specific targets of tetanus and botulinum neurotoxins (460), and either toxin-mediated or genetic inactivation of either SNARE protein irreversibly blocks evoked neurotransmitter release. The different sensitivities of excitatory and inhibitory neurons to SNARE inactivation is believed to generate an E/I imbalance resulting in the developmental deficits.
8.1.1.2. CALCIUM SENSING/TRIGGERING MACHINERY.
Synaptotagmin1/2 is the fundamental Ca2+-binding sensor for evoked fast and synchronous NT release. Synaptotagmin binds to SNARE proteins together with complexins limiting spontaneous release and, bearing two C2 domains, acts as a low-affinity Ca2+ sensor that is exposed to high concentrations of Ca2+ entering through activated voltage-gated Ca2+ channels (VGCCs) and upon Ca2+ binding triggers membrane deformation and fusion (461, 462). Five heterozygous mutations were reported in Synaptotagmin-1 located at the Ca2+ and phospholipid binding motifs of the second C2 domain (C2B) in patients presenting with various degrees of developmental delay and EEG abnormalities but no epilepsy (463). Synaptic transmission was impaired in neurons expressing the synaptotagmin mutants, with graded dominant-negative effects that could be rescued by K+ channel antagonists (448). Presynaptic VGCCs Cav2.1 and Cav2.2, corresponding to the P/Q- and N-type VGCCs, are the essential electrochemical transducers concentrated in the nano- and microdomains of the active zones and converting APs into synchronous exocytosis of SVs.
PRRT2 is a neuron-specific, type 2 membrane protein with a COOH-terminal anchor that concentrates in synaptic and axonal domains, where it interacts with key components of the fusion/Ca2+ sensing machinery (synaptotagmin 1/2, SNAP-25, and VAMP2), boosting the Ca2+ sensitivity of NT release and suggesting a function in the Ca2+-dependent transition from SV priming to fusion (446). As a consequence, the probability of release at excitatory synapses is dramatically decreased, with a parallel marked increased synaptic facilitation that results in an E/I imbalance and lack of network stability (454, 464, 465). This role of PRRT2 has recently been supported by the discovery of a direct interaction of PRRT2 and P/Q VGCCs that contributes to concentrate them at the nanodomain where the machinery for synchronous release is assembled. In the absence of PRRT2, membrane targeting and concentration of P/Q channels at active zones is impaired, decreasing Ca2+ influx in response to APs (466). PRRT2 has been identified as the causative gene for several paroxysmal neurological disorders including epilepsy, paroxysmal kinesigenic dyskinesia, but also migraine and ataxia. These disturbances have been associated with a severe encephalopathic phenotype with intellectual disability in the few patients with homozygous or compound heterozygous mutations (446).
Another SV protein playing an important role in regulating the initial release probability of SVs and neural network synchronous activity is Synaptic Vesicle glycoprotein-2 (SV2). SV2 is encoded by three paralog genes (SV2A, SV2B, SV2C) with distinct patterns of expression in neuronal populations. SV2 plays multiple roles as a catalyzer of evoked NT release: it accelerates SV priming, increases the size of the RRP, and regulates the stability and trafficking of synaptotagmin, boosting the Ca2+ sensitivity of release. Interestingly, SV2A is essentially expressed in inhibitory neurons, and its loss of function impairs synaptic inhibition. Accordingly, individuals bearing point mutations in the SV2A gene experience epilepsy and cognitive deficits (467, 468).
8.1.1.3. SV PRIMING MACHINERY.
The preparation of SVs for fusion after docking is an essential mechanism for allowing fast and synchronous release. Two proteins are essential for this progression, Munc-18 (STXBP1) and Munc-13 (UNC13A), whereas other nerve terminal proteins such as RIM1, SV2, and synapsin I have a modulatory role. The Munc-18 and Munc-13 tandem is needed for a correct assembly of the SNARE complex, whereby Munc-18 regulates the participation of syntaxin-1 to the complex and Munc-13 activates syntaxin-1, stabilizes the active zone-SNARE-SV assembly and protects the SNARE complex from disassembly by the ATPase N-ethylmaleimide-sensitive factor (NSF) (469, 470). Whereas a nonsense mutation in UNC13A was identified in a single patient with microcephaly and interictal multifocal epileptiform EEG activity (471), mutations in STXBP1 were repeatedly identified in a series of epileptic encephalopathies including Ohtahara, West, and Rett syndromes. Over 85 distinct STXBP1 mutations are known, and de novo STXBP1 mutations are among the most frequent causes of DEEs of synaptic origin with severe intellectual disability (472).
8.1.1.4. ORGANELLE ACIDIFICATION AND NT LOADING.
As mentioned above, vATPase plays a fundamental role in the active loading of SVs with the surprisingly reproducible amount of NT that constitutes the “quantum.” Quantal reproducibility relies on the fact that the amount of NT is an important determinant of the probability of release, so that only fully filled SVs are likely to be released (473, 474). This mechanism is based on the dissociation of the V1 catalytic cytosolic domain from the V0 transmembrane proton transfer domain upon complete buildup of the proton gradient and corresponding NT loading. The synaptic protein encoded by the DMXL2 gene, rabconnectin-3a, is strictly involved in the assembly and incorporation of vATPase into SVs (475, 476). The role of vATPase in pH homeostasis and intracellular signaling pathways is ubiquitous, with a prominent role in the nervous system. In humans, mutations in 16 of the 22 genes encoding for vATPase subunits are associated with a variety of congenital disorders with neurological impairment. Recently, four different de novo missense mutations in ATPV1A have been associated with a clinical spectrum of DEE ranging from rapidly progressive early encephalopathies to mild intellectual disability with epilepsy (477). Unexpectedly, rather than NT loading, the identified pathomechanism involves dysfunctions of lysosomal homeostasis impacting on neuronal connectivity. Missense pathogenic variants of ATPV0A1, which participates in the transmembrane proton-pumping machinery of vATPase, are also associated with DEE (478). Heterozygous copy number variations and loss-of-function biallelic mutations in DMLX2, regulating the trafficking and activity of v-ATPase, are associated with a severe DEE (Ohtahara syndrome) with a superimposable pathomechanism consisting of defective endolysosomal homeostasis and autophagy resulting in synaptic loss (479).
8.1.2. Perturbation of synaptic vesicle trafficking within nerve terminals.
Synaptic vesicles undergo a restless cycle within nerve terminals aimed at preserving their availability in a sufficient amount in the RRP and maintaining synaptic transmission also during high-frequency activity. Thus, loss of function of any of the vast cohort of nerve terminal proteins operating SV endocytosis and maintaining the recycling pool of SVs is predicted to impact on SV availability and impair NT release in the tonic high-frequency discharging neurons (435). Since neurons with these characteristics are often inhibitory interneurons (such as the PV-positive neurons) a diffuse impairment in SV endocytosis/recycling often results in an E/I imbalance.
A large array of gene products mediating various steps of the endocytosis process has been associated with epilepsy and cognitive impairment. These include the membrane adaptor AP-2, synaptophysin (SYP), dynamin-1 (DNM1), the coat protein clathrin (CLTC), as well as proteins involved in activity-dependent bulk endocytosis such as Rab11, TBS1D24, and AP-1. In the case of AP-2, four patients bearing a point mutation in the µ2-subunit of the protein presented with epilepsy and developmental delay (480). Synaptophysin is an integral SV protein interacting with VAMP2 and regulating its availability and retrieval during endocytosis (481). Nonsense and missense mutations in the X-linked SYP gene have been associated with epilepsy and cognitive defects (482, 483). De novo mutations in the DNM1 gene result in DEEs including West and Lennox–Gastaut syndromes (484). Dynamin-1 is the essential GTPase that mediates SV fission by assembling in a collar around the neck of the budding vesicle and inducing constriction thanks to the mechanical force produced by GTP hydrolysis (485). Most of the DNM1 mutations identified thus far cluster within the GTPase domain (484). A series of missense and nonsense mutations in the CLTC gene encoding for clathrin, the building block of the endocytic coat that allows increase of the curvature of the vesicle bud to form the endocytic vesicle, have been identified in patients with epilepsy and intellectual disability (486, 487). Impairment in bulk endocytosis also occurs with mutations in AP-1, Rab11, and TBC1D24. This gene, whose mutants are associated with epilepsy and DOORS (deafness, onychodystrophy, osteodystrophy, mental retardation and seizures) syndrome, affects several processes linked to neural development and synaptic transmission, such as endocytosis, endosome/lysosome flux, and neurite growth (488–492). Synapsins (Syns) constitute a family of three synaptic genes (SYN1, SYN2, and SYN3) that encode for SV-associated proteins regulating SV formation, SV pool maintenance, and SV trafficking between the functional SV pools. Whereas Syn3 is a developmental isoform and is downregulated after birth, Syn1 and Syn2 represent the adult isoforms (493–496). Knockout mice for Syn1 (homozygous females or hemizygous males), Syn2, Syn1/Syn2, and Syn1/Syn2/Syn3 all exhibit an epileptic and autistic-like phenotype with impairment of social interactions and cognitive functions (497, 498). As observed in other mouse models (e.g., Grin1 and Grin2b), heterozygous mice do not show an overt phenotype. An array of nonsense and missense mutations have been identified in patients with epilepsy and/or ASD. Most nonsense mutations in the X-linked gene SYN1 have been identified in epilepsy patients, whose seizures were typically triggered by contact with water regardless of the water temperature. Developmental delay of a variable degree was frequently associated (499–504). Owing to their role in regulating network activity and stability, presynaptic proteins can also represent a target for antiepileptic drugs, such as levetiracetam that binds to SV2 on SVs (505).
8.2. Postsynaptic and Extracellular Synaptopathies
Neurotransmitters released by the presynaptic terminals diffuse in the extracellular space and transfer information downstream by interacting with specific postsynaptic receptors. Mutations have been identified in genes encoding receptors for the main excitatory (glutamate) and inhibitory (GABA) neurotransmitters, as well as in transporters involved in ionic homeostasis and neurotransmitter reuptake, receptor-associated scaffolding proteins, and extracellular transsynaptic proteins.
8.2.1. Ionotropic neurotransmitter receptors.
8.2.1.1. NMDA GLUTAMATE RECEPTORS.
NMDA glutamate receptor subunits are encoded by the GRIN gene family, formed by seven genes: GRIN1 (encoding the GluN1 subunit), GRIN2A–D (encoding the GluN2A to D subunits), and GRIN3A–B (encoding the GluN3A and B subunits). Receptor activation requires binding of both glutamate and glycine, which are often considered coagonists. Functional NMDA receptors are heterotetramers composed of two GRIN1 subunits binding glycine and two GRIN2 or GRIN3 subunits binding glutamate. They are essential for numerous physiological functions, including neuronal migration, synaptic connectivity, neuronal pruning and survival, and synaptic plasticity. They are Ca2+ permeable and generate a slow voltage-dependent synaptic current.
Among NMDA receptor subunits, GRIN1, GRIN2A, and GRIN2B are the target of most variants identified in patients with neurodevelopmental disorders. Epilepsy in patients carrying GRIN mutations has onset from birth to few years of early childhood. Mutations can be either inherited or de novo and generate a spectrum of phenotypes ranging from mild intellectual disability to severe DEE (506). The GRIN1 spectrum includes LoF of varying severity with a dominant-negative effect and is characterized by severe intellectual disability with absent speech, seizures (in ∼65% of patients), hypotonia, dyskinesia, cortical blindness, and generalized cerebral atrophy (63). GRIN2A mutations have either LoF or GoF effects and are associated with epilepsy and intellectual disability with normal brain imaging (58, 507, 508). Seizures often originate in the temporo-rolandic regions, and EEGs often show centrotemporal spike-wave discharges, which may be continuous during slow-wave sleep (CSWS or CSWSS). Affected individuals exhibit a range of language/speech problems and, at times, complete aphasia (506). The GRIN2B spectrum, characterized by various GoF and LoF mechanisms, is similar to the GRIN1 spectrum and includes developmental delay, hypotonia, epilepsy (in ∼50% of patients), movement disorders and, at times, cortical malformations (509). The phenotypic range of GRIN2D missense GoF variants includes developmental delay with failure to thrive, intellectual disability, hypotonia, and hyperreflexia (59).
Heterozygous and homozygous knockout (null) mouse models have been generated for each of the seven GRIN genes (506). Mice carrying homozygous null mutations for Grin1 and Grin2b are postnatal lethal, whereas heterozygous mice survive normally, but they have not been characterized in detail. Also, knockin mice carrying mutations identified in patients have been generated (510–512). Grin2a+S/644G and Grin2a+/N615K mice show perinatal lethality in homozygosis, increased seizure propensity, and behavioral and cognitive deficits in heterozygosis. Grin2b+/C456Y mice show anxiety-like behavior with strongly reduced Grin2b levels and NMDA currents.
8.2.1.2. AMPA GLUTAMATE RECEPTORS.
AMPA glutamate receptors are composed by four types of subunits: GluA1–GluA4 subunits (also named GluR1–4) encoded by the GRIA1–GRIA4 genes. Most AMPA receptors (AMPARs) are heterotetrameric, consisting of a symmetric “dimer of dimers” of GRIA2 and either GRIA1, GRIA3, or GRIA4 (513, 514). AMPA receptors interact with multiple accessory proteins (e.g., TARP and cornichon). Glutamate released from the presynaptic terminal triggers the rapid opening of AMPA receptor channels that generate an inward cation current. The GRIA2 mRNA is often edited at the p.Q607 residue that confers Ca2+ impermeability to mature receptors containing the edited GluA2 subunit. The AMPA current underlies most of the excitatory synaptic signaling in the central nervous system. It is typically brief (on the order of a few milliseconds) because glutamate rapidly unbinds and is removed from the synaptic cleft and leads to a brief depolarization of the postsynaptic neuron. De novo heterozygous GRIA2 mutations have been found in patients with different associations of seizures, speech impairment, intellectual disability, and autistic features (515) with both GoF and LoF mechanisms. The most severe phenotypes were associated with the p.A639S mutation, which caused DEE with death in infancy. There are no mouse models available for human mutations. Gria2−/− mice exhibit increased mortality, impaired motor coordination, and behavioral abnormalities, whereas heterozygous mice do not show an overt phenotype (516). De novo heterozygous missense mutations in GRIA3, as well as genomic deletions, have been initially identified in a form of X-linked intellectual disability with dysmorphic features, a relatively mild phenotype due to a LoF mechanism (517). However, GRIA3 mutations have been associated with a larger phenotypic spectrum that also includes severe early-onset DEE (518). There are no animal models that carry human mutations. Finally, de novo heterozygous variants in GRIA4 have been identified in a range of phenotypes characterized by variable developmental delay, ranging from mild to severe, with absent speech, epilepsy, gait abnormalities, and behavioral problems (519).
8.2.1.3. GABA RECEPTORS.
Ionotropic GABA (GABAA) receptors are pentameric assemblies of up to 3 of 19 subunits encoded by distinct GABR genes (GABRA1–6, GABRB1–3, GABRG1–3, GABRR1–3, GABRD, GABRE, GABRP, and GABRQ). The combination of two α-subunits (GABRA), two β-subunits (GABRB), and one γ-subunit (GABRG) is the most common functional receptor in the brain. GABAA receptors are cys-loop ligand-gated chloride/anion channels that, at low intracellular chloride, implement an outward inhibitory current generating hyperpolarization and decrease in membrane impedance by a shunting effect. The GABAergic synaptic action is essential for reducing excitability in neuronal networks and generating rhythms of activity. The synaptic GABAA receptors provide brief but strong phasic inhibition, whereas those extrasynaptic receptors can induce long-lasting tonic effects in response to ambient GABA. GABRA1, GABRA2, GABRA3, GABRA5, GABRB1, GABRB2, GABRB3, and GABRG2 genes have been identified as targets of de novo heterozygous DEE mutations (520). Mutations in GABRA2, GABRA3, GABRA5, and GABRB1 have been mostly associated with severe phenotypes, whereas variants in other GABAA genes include, in addition to DEE, phenotypes associating moderate/mild intellectual disability with epilepsy and familial epilepsy without intellectual disability (16, 521–523). The severe phenotypes of GABRA1 DEE mutations share some of the clinical features of Dravet syndrome (524). Most GABAA mutations induce LoF (with dominant-negative effects), by decreasing membrane targeting or modifying gating properties or GABA sensitivity (522, 523, 525–527). Overall, LoF of GABAA receptors reduces the inhibitory tone in neuronal networks and thus generates hyperexcitability; the effect has some similarity to that of SCN1A mutations that reduce the intrinsic excitability of GABAergic neurons. A different functional effect has recently been proposed for mutations in GABRD, which encodes the GABAA δ-subunit found in extrasynaptic receptors generating tonic GABAA currents (528). DEE patients carrying mutations in this gene have generalized epilepsy, intellectual disability, and behavioral problems.
Functional analysis of the identified mutations showed GoF of GABAA receptors containing the δ-subunit with increased GABAA current. Thus, increased tonic GABAA-evoked current may be a novel pathological mechanism in DEE and neurodevelopmental diseases. Electroclinical findings in these patients resembled those reported in patients carrying LoF mutations in the GABA uptake transporter SLC6A1/GAT1 (529), consistent with a similar pathogenic mechanism (see below). Heterozygous Gabra1 knockout mice show spontaneous electrographic spike-wave discharges with behavioral absencelike seizures and develop myoclonic seizures later in life, consistent with a relatively mild idiopathic generalized epilepsy (IGE) phenotype (530). Gabrb2 knockout mice do not show spontaneous epilepsy but are more susceptible to seizures and exhibit behavioral disturbances (531). Heterozygous Gabrb3 knockout mice show epileptic seizures, EEG abnormalities, and a range of behavioral deficits (532, 533). Homozygous Gabra3 knockout mice do not show seizures (534). The mouse line carrying the human GABRG2 p.Q390X DEE mutation, which has a dominant-negative effect, shows a more severe phenotype compared with Gabrg2−/− mice (535). Mutations in GABBR2 that induce LoF with reduced slow GABAergic inhibition (536) have been identified in DEE (11, 16).
8.2.2. Synaptic transporters.
DEE variants have also been identified in genes that although not directly implicated in synaptic transmission are involved in synaptic functions.
8.2.2.1. SLC12A5 AND CLCN4.
Cl− is essential for numerous physiological functions, including GABAergic inhibition. It is actively transported and its concentration tightly regulated in neurons and virtually all cell types. Cation-chloride cotransporters are postsynaptic plasma membrane proteins that determine the intracellular Cl− homeostasis and are thereby directly implicated in GABAA current generation (537). The gene family includes the Na+-Cl− cotransporter (NCC), the Na+-K+-2Cl− cotransporters (NKCCs), and the K+-Cl− cotransporters (KCCs). The SLC12A5 gene, exclusively expressed in the central nervous system, encodes the neuronal KCC2 K+-Cl− cotransporter, which is the major extruder of intracellular chloride in mature neurons. Low KCC2 activity can lead to increased intracellular Cl− and to depolarizing GABAergic transmission (538).
SLC12A5 recessive de novo mutations have been identified in a spectrum of epileptic disorders (539, 540). The most severe phenotype is a DEE with features of epilepsy of infancy with migrating focal seizures (EIMFS) caused by LoF mutations that decrease KCC2 surface expression and reduce protein glycosylation (141).
The CLCN gene family contains nine members in mammals, four of which encode plasma membrane chloride channels (CLCN1, CLCN2, CLCNKA, CLCNKB) and five intracellular 2Cl−/H+ exchangers (CLCN3–7) (541). Their function is not completely clear, but CLCN Cl− channels are involved in the regulation of excitability by controlling extra- and intracellular ion homeostasis. Dysfunction of some CLCN genes leads to severe neurological disorders; in particular, LoF mutations in CLCN4 cause a spectrum of phenotypes including severe DEE with drug-resistant seizures and cognitive and behavioral disorders (542, 543).
8.2.2.2. SLC1A2 AND SLC6A1.
SLC1A are plasma membrane glutamate transporters expressed by glial cells and/or glutamatergic presynaptic terminals. They are essential for the removal and termination of action of glutamate released from the synapses. Mutations in SLC1A2, encoding the astrocytic EAAT2 glutamate transporter selectively expressed in astrocytes, have been identified in DEE patients (544). Functional studies in transfected cell lines showed LoF and negative dominance (544), consistent with impaired clearance of extracellular glutamate. Slc1a2 knockout mice show a severe epilepsy phenotype only in homozygosity (545).
The SLC6A1 gene, encoding the GAT1 GABA transporter, is responsible for the reuptake of GABA into presynaptic terminals and astrocytes, emerging as a common cause of DEE (546, 547). Functional studies have shown that SLC6A1 DEE mutations induce LoF of GAT1 possibly associated with negative dominance and reduced GABA reuptake (529). Slc6a1−/− mice recapitulate some features of the human phenotypes, including motor and cognitive impairment, whereas heterozygous Slc6a1+/− mice do not show an overt phenotype (546). Overall, the mechanism may be similar to that of GABRD mutations, which increase the extrasynaptic GABAA current (528).
8.2.3. Other postsynaptic proteins.
Other mutations in genes encoding for receptor-associated proteins, such as the GTPase SynGAP1 (548) and Stargazin (549), are also associated with dysfunctions in circuit excitability and epileptic encephalopathies. Mutations in transsynaptic adhesion protein genes, including neurexin-1, ILRAPL1, and Caspr2, were also reported to cause epilepsy in a limited number of cases (550–552).
8.2.4. Extracellular synaptic proteins.
An interesting group of epilepsy-related genes encodes for proteins that are secreted at the synaptic cleft and contribute to transsynaptic communication and synapse maintenance. Loss-of-function mutations in LGI1 gene (or LGI1-neutralizing autoantibodies) are associated with epilepsy with cognitive impairment (553). LGI1 is a protein secreted by both presynaptic and postsynaptic neurons that is part of a transsynaptic structural/functional bridge linking the integral proteins ADAM22 and ADAM23, exposed on the opposite sides of the synaptic cleft, that regulates the assembly and organization of AMPA glutamate receptors by the scaffold protein PSD95 (FIGURE 8) (142, 554). Other genes encoding for the synaptically secreted proteins SRPX2 and Reelin can cause epilepsy if mutated. These proteins play a role in synaptogenesis and in the neuronal development of the cerebral cortex by acting as extracellular synaptic organizers (555–558). In case of Reelin, the mutation impairs reelin secretion, and the reelin variant is retained and eventually degraded through the autophagic way.
9. DYSFUNCTIONS IN NEURONAL HOUSEKEEPING: mTOR AND AUTOPHAGY
Mature neurons are postmitotic cells that do not replicate and have a very high workload throughout life. The housekeeping of their structure/function is therefore of paramount importance. Autophagy, a highly conserved structural turnover process that directs dysfunctional macromolecules and organelles to lysosomal degradation, is an indispensable means for neurons to maintain their integrity and functionality over time (559–561). Besides neuronal survival, autophagy plays a key role in neural development and in the formation and maintenance of synaptic connectivity. It is therefore conceivable that dysfunctional autophagy, due to mutations in genes controlling its multistep processes, can cause severe neurodevelopmental disorders including DEEs.
One of the main pathways controlling autophagy and cell homeostasis is the mTOR (mammalian/mechanistic target of rapamycin) pathway. MTOR is an atypical serine/threonine protein kinase that is activated by a very complex pathway integrating intra- and extracellular signals and strictly controlling matter and energy balance within the cell. The most important mTOR effectors are the p70 ribosomal S6 protein kinase-1 (p70S6K1) and the eukaryotic initiation factor 4E-binding proteins (4E-BPs) that transduce extracellular signals, such as neurotransmitters, growth factors, and hormones, into an activation of translation. In neurons, these targets implicate mTOR in cell growth, neurite outgrowth, and synaptic formation, all fundamental activities for neuronal functions and plasticity. The final synergistic effectors mTOR cascade are two mTOR complexes, mTORC1, mainly implicated in cell growth and proliferation, and mTORC2, mainly regulating cytoskeleton and dendrite growth (562, 563). Here we focus on mTORC1, since virtually all pathogenic mutations identified thus far affect the mTORC1 cascade, and the implication of mTORC2 in DEEs is still limited. The activation of mTORC1 is tightly controlled by two inhibitory switches, the tuberous sclerosis complex (TSC) and the GATOR1 complex (FIGURE 9). Both complexes are under the inhibitory control of two upstream complexes, GATOR2 that inhibits GATOR1 and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway that inhibits TSC. The canonical mTOR pathway starts with the activation of the PI3K/Akt pathway in response to extracellular signals (which is subjected to a further inhibitory control by PTEN) that relieves the mTOR complex from the tonic TSC inhibition. To be active, mTOR needs to attach to intracellular organelles, which requires the G proteins Rheb and Rag (part of the Ragulator complex) to be in an active, GTP-bound state. The Ragulator complex, present on the membrane through an interaction with the proton pump vATPase, is a guanine nucleotide exchange factor (GEF) that activates Rag and allows mTOR membrane binding and activation. On the other hand, TSC and GATOR1 inhibitory complexes act as GTPase-activating proteins (GAPs) to deactivate Rheb and Rag, respectively, and thereby block mTOR activation. In addition to the stimulation of protein synthesis, activated mTOR inhibits autophagy by inhibiting the ULK1 complex that, through the sequential activation of the Beclin complex and the WD-repeat phosphoinositide-interacting protein 4 (WIPI4), activates the maturation and fusion of autophagosomes with acidic lysosomes to form autolysosomes (560). A central role in the regulation of mTOR activation, as well as in the autophagy progression, is played by the proton pump vATPase and its ancillary proteins, which allows docking of Ragulator to the organelle membrane, which in turn recruits and activates the mTORC1 complex (FIGURE 9).
FIGURE 9.
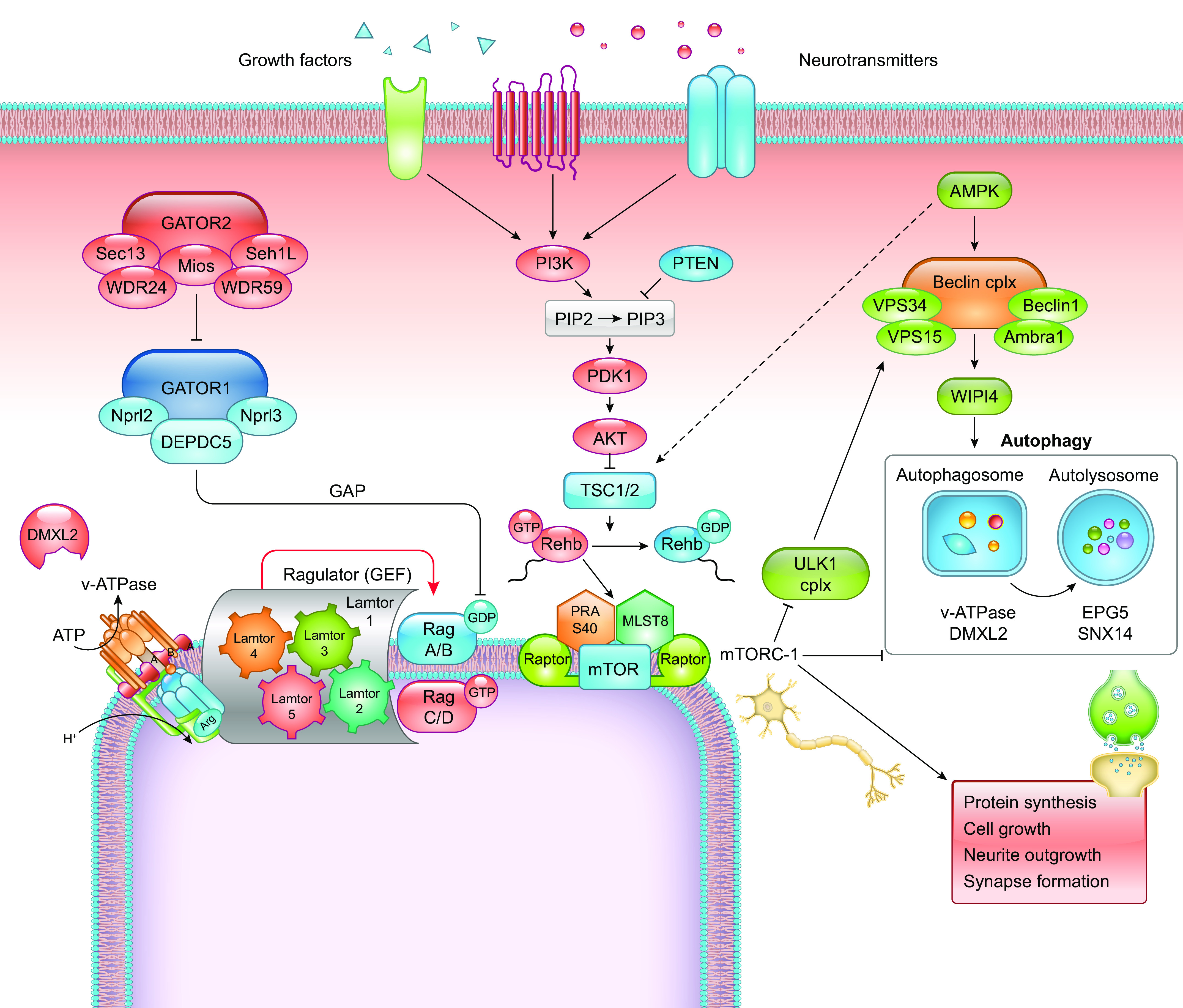
Schematic representation of the mechanistic/mammalian target of rapamycin (mTOR) and autophagy intracellular cascades and their interrelationships. The complex regulatory cascade triggering the activation of the mTORC1 complex is initiated by extracellular signals (growth factors, neurotransmitter, hormones). To be activated, mTORC1 needs to bind to the organelle membrane, a process that depends on the active form of the small G protein Rheb and by the presence of a docking complex on the membrane formed by the guanine nucleotide exchange factor (GEF) Ragulator and an appropriate combination of GTP- and GDP-bound Rag G proteins. The membrane location of the latter complex depends on the presence of the vacuolar H+-adenosine triphosphatase (vATPase) on the membrane, which is favored by DMXL2. Since the small G proteins are the molecular switches for mTOR activation, they are also the targets of the 2 main upstream inhibitory complexes that act as GTPase activating proteins (GAPs), namely the TSC and Gator1 complexes that inactivate Rheb and Rags, respectively. These inhibitory TSC and Gator1 complexes are in turn subjected to inhibition by the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, activated by extracellular signals and Gator 2, respectively, that therefore catalyze release of mTORC1 from inhibition. Activation of mTORC1 favors anabolism, protein synthesis, cell growth, and, in neurons, outgrowth of neuronal processes and formation of synaptic connections. On the other hand, activation of mTORC1 silences the autophagy chain by inhibiting the ULK1 complex, which is required to recruit the Beclin complex to the phagophore for its activation by AMPK. This results in the following steps of autophagy flux, including LC3 conversion and binding, formation of the autophagosome and subsequent fusion with lysosomes to form autolysosomes. In these processes, the proton gradient established by vATPase is essential, as well as the activity of accessory proteins such as DMXL2, EPG5, and SNX14.
Not surprisingly, dysregulation, particularly hyperactivation, of the mTOR pathway caused by mutations in the various components of the PI3K-mTOR pathway and regulatory cascades results in DEEs, the so-called mTORopathies, that are often associated with malformations of cortical development. mTORopathies include a spectrum of drug-resistant epilepsy syndromes ranging from apparently nonlesional focal epilepsy to tuberous sclerosis complex (TSC), FCDII, and HME that are associated with brain malformations (563, 564). The complexity of the architecture of the mTOR pathway, while it guarantees a full control over key processes for cell survival and adaptation, also offers several genes whose mutations impact on critical neuronal processes and are causative of DEEs (565). The first identified, prototypical mTORopathy is the TSC linked to germline/somatic mutations in TSC1 or TSC2 genes that remove the inhibitory brake of TSC on the downstream mTOR complex 1 (mTORC1) and determine mTOR hyperactivity in several tissues including the brain. Subsequently, high-throughput sequencing has discovered an array of germline and de novo somatic mutations causing mTOR hyperactivity that involved either loss of function of inhibitory complexes (PI3K, PTEN, AKT3, TSC1, TSC2, RHEB, DEPDC5, NPRL2, and NPRL3) or gain-of-function hyperactive variants of MTOR. Animal models of mTORopathies are constantly characterized by an epileptic phenotype. Pten, Tsc1, Tsc2, and Depdc5 knockout mice, as well as Mtor knockin mice, recapitulate the human phenotype of mTORopathies by displaying dysplastic cortical areas with enlarged cortex, ectopic and hypertrophic neurons, and epilepsy. Constitutive and generalized mTOR hyperactivity is embryonic lethal, so that conditional models have been used to effectively reproduce human pathology. In addition to the dysplastic phenotype, mTOR hyperactivity causes hyperexcitability (563, 566, 567). This is not only attributable to the intrinsic instability of dysplastic circuits with dendritic and somatic hypertrophy and formation of reverberant connections but also depends on an augmented intrinsic excitability of principal cortical neurons and potentiation of excitatory synaptic transmission at both pre- and postsynaptic levels promoting E/I imbalance. Another interesting feature of mTORopathies regards the relative frequency of somatic mutations that are likely to occur predominantly during brain development and that establish a condition of mosaicism in the neuronal populations of the cerebral cortex. The intersection between inherited germline mutations and acquired somatic ones (“second hit”) causes loss of heterozygosity or mosaic compound heterozygosity that can convert a latent deficit into an overt phenotype in restricted brain areas. This phenomenon was clearly demonstrated for DEPDC5 mutations both in humans and in experimental models in which the “somatic” knockdown of Depdc5 was reproduced by in utero electroporation and/or RNA interference, generating focal cortical dysplasia (29, 568, 569). One of the mechanisms by which mTOR hyperactivity causes epileptic encephalopathies is through a dysregulation of autophagy, a process strongly implicated in neuronal survival and plasticity (559, 560, 568). As mentioned above, neuronal autophagy is a highly regulated process in which mTOR inhibition releases the ULK1 activator, which, together with AMPK, activates the autophagy initiator Beclin complex (composed of VPS34, VPS15, Ambra1, and Beclin1), which is recruited to the phagophore, stimulates the production of phosphatidylinositol 3-phosphate (PI3P) and the binding of WIPI proteins. This allows the conversion of LC3-I into phosphatidylethanolamine-bound LC3-II that is the fundamental signal activating autophagy (FIGURE 9). mTOR and AMPK represent a yin-yang mechanism controlling autophagy: not only do the two pathways exert opposite controls on the underlying autophagic process, but AMPK also stimulates the TSC complex, keeping mTOR inactive during autophagy activation (570). The final common pathway of autophagy is the fusion of mature autophagosomes with lysosomes to form the autolysosomes, where the vesicular content is finally degraded and released into the cytoplasm. This event is regulated by the autophagic proteins EPG5 and SNX14 and requires internal acidification of the organelle to allow for proteolysis. Biallelic mutations of EPG5 and SNX14 cause respectively the Vici syndrome, a rare and severe congenital multisystem disorder characterized by failure of the corpus callosum, cataracts, oculocutaneous hypopigmentation, cardiomyopathy, and combined immunodeficiency (571), and a form of cerebellar ataxia and intellectual disability (572). Acidification is provided by vATPase, a multifunctional proton pump that regulates multiple cellular processes including membrane trafficking, receptor-mediated endocytosis, SV cycling, and NT loading. Acidification is necessary for autophagy progression and drugs or vATPase variants impairing the buildup of the proton gradient block autophagy (573). vATPase is composed of a V1 cytosolic domain that hydrolyzes ATP and a V0 transmembrane domain that transfers protons to the organelle’s interior (574). Although the proton-pumping activity of vATPase occurs in all tissues, the brain is particularly vulnerable to vATPase defects for its multiple functions in exocytosis (474, 575) and autophagy. In humans, as many as 22 genes encode for the multiple and redundant subunits of the V1 and V2 complexes, allowing the composition of different vATPase complexes with specific properties and tissue expression.
Several, heterozygous or biallelic mutations in ATP6V1A coding for the V1 subunit A have been recently described in patients with DEEs of variable severity, ranging from moderate intellectual disability with seizures to an early-onset DEE with premature lethality (477, 576). ATP6V1A variants affect lysosomal homeostasis and autophagy and, when expressed in postsynaptic neurons, impair neurite development and the formation/maintenance of excitatory synaptic connections (477). ATP6V1A deficits have also been recently associated with neuronal impairment and neurodegeneration. ATP6V1A-silenced neurons show reduced network activity and alteration of synaptic proteins consistent with a key role of ATP6V1A in neuronal maturation and activity (577).
In addition, mutations in ancillary proteins of the vATPase complex, known to regulate its function and trafficking to the organelles, also cause severe neurodevelopmental disorders with epilepsy and impair brain development when modeled in mice (478, 578). A de novo variant of ATP6AP2, a vATPase accessory protein, was identified in a patient with neurodevelopmental disorder and fulminant degeneration. In murine models and patient neurons, loss of function of ATP6AP2 results in lysosomal and autophagic defects with impaired neuronal survival, revealing a key role of this vATPase modulator in brain function (578). Homozygous recessive and compound heterozygous mutations in the DMXL2 gene were recently found to cause a severe and rapidly progressing DEE associated with Ohtahara syndrome and premature death (479). The gene encodes for the vesicular protein DMXL2 (also known as rabconnectin-3a), a member of the WD40 repeat (WDR) protein family that is highly expressed in brain tissue, regulates the trafficking and activity of vATPase, and interacts with the SV-associated G-protein Rab3A (579, 580). Altered lysosomal homeostasis and defective autophagy were recapitulated in Dmxl2-silenced mouse hippocampal neurons that exhibit impaired neurite elongation and synaptic loss (479). Dmxl2−/− mice are embryonic lethal (581), whereas heterozygous Dmxl2 mice show brain malformations, uncovering a penetrant role of Dmxl2 in brain development (582).
These data confirm a primary role of autophagy dysregulation in alterations of cortical development and epileptogenesis and suggest that the severity of the clinical phenotypes and the extent of neurodegeneration depend on the stage of neuronal development and on the specific consequences of the impairment of synaptic transmission and neuronal survival due to the stressful condition of impaired autophagy (583).
10. THE SEARCH FOR PERSONALIZED TREATMENT APPROACHES
Currently, most therapies for DEEs target individual symptoms such as seizures, and not the underlying disease mechanisms. For many individuals with DEEs, seizure control is not achieved, even when antiseizure medications (ASMs) are optimized for the underlying etiology, whereas in patients in whom seizure control is achieved, developmental impairments and other comorbidities often remain severe (4, 445). In addition, since patients with the same type of clinical seizures may respond differently to ASMs, the pathophysiological events that underlie epileptic seizures apparently not only differ between unique seizure syndromes and specific etiologies but may also be multifactorial for the same types (584).The remarkable growth of animal models of DEEs has enabled preclinical studies that tested several experimental drugs have been tested in different animal models of DEEs (TABLE 4). Both acute and chronic models of epileptic spasms have been used to study effects on spasm prevention with protocols that administer drugs before the induction (acute models and certain chronic models) (177, 182–184, 187, 585–587), spasm control/cessation (chronic models: multiple hit, TTX) (152, 157–160, 162, 163, 195), hypsarrhythmia (TTX model) (190), and disease modification or antiepileptogenesis (chronic models: multiple hit, ARX KI) (152, 157). Among the existing models, resistance to existing therapies for ISS (ACTH, vigabatrin) is shown in the multiple-hit rat model of ISS due to structural lesions (88). The only model that has been used to study effects on hypsarrhythmia is the TTX model, which allows testing of effects on the EEG with multiple electrodes in postpubertal animals. This is technically challenging in rodent models that manifest spasms only during restricted early developmental periods, when the skull is small and fragile. The prenatal betamethasone/postnatal NMDA model suggested that prenatal betamethasone increases the sensitivity to ACTH, even though it increases the severity of NMDA-induced spasms and has been used to explore pharmacologically therapeutic effects of pathways thought to mediate the ACTH effects, i.e., mineralocorticoid receptor signaling (183). Among the treatments tested in these preclinical trials, two drugs eventually acquired orphan status for ISS (carisbamate, CPP-115). CPP-115 has also been tested in a case report on an infant with drug-resistant ISS with significant improvement in spasms and seizure control compared with vigabatrin (603). In support of the promising effects of the mTOR inhibitor rapamycin on spasms in the multiple-hit model, recent clinical case reports suggested a possible benefit in infants with tuberous sclerosis-related ISS treated with the mTOR inhibitor everolimus (604).
Table 4.
Experimental drugs tested in models of DEE
| Drug | Mechanism | Model/Species | Treatment Protocol | Effect | Reference |
|---|---|---|---|---|---|
| ES models | |||||
| CGP35348 | GABABR antagonism | Mouse, Down/GBL | Pretreatment | Shortens EDRs | (183) |
| Baclofen | GABABR agonist | Mouse, Down/GBL | Pretreatment | Prolongs EDRs | (183) |
| 5-OH-tryptophan | Serotonin increase | Mouse, Down/GBL | Pre-treatment | Prolongs EDRs | (183) |
| GIRK2 knockout | Deletion of GABABR associated inward-rectifying potassium channel | GIRK2 KO mouse, Down/GBL | GIRK2 knockout mice | GIRK2 knockout confers resistance to GBL-induced spasms | (187) |
| Rapamycin | mTOR inhibitor | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment | No effect, no evidence of target relevance | (182) |
| Ganaxolone | Allosteric activator of GABAAR, synaptic and extrasynaptic | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment | Reduces number, delays onset of NMDA spasms | (585) |
| Acton prolongatum® | Synthetic ACTH based on porcine ACTH | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment | Reduces number, delays onset of NMDA spasms after 2 doses but not after single dose | (586) |
| AQB-565 | ACTH1–24 linked to melanocyte stimulating hormone, acts on MC3, MC4 melanocortin receptors | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment | Reduces number of NMDA spasms after 8 doses | (183) |
| Estradiol, diethylstilbestrol | Gonadal hormone, estrogen analogue | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment (PN3–10) | No effect on spasms; increased GAD67 cells in neocortex | (184) |
| β-OH-butyrate | Ketoacid | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment | Reduces spasms and delays latency to NMDA spasms after repeat but not single dose administration | (177) |
| β-OH-butyrate | Ketoacid | Rat, Prenatal betamethasone / postnatal NMDA | Pretreatment (200 mg/kg ip) | No effect | (587) |
| 2-Deoxyglucose | Metabolic inhibition of glycolysis | Rat, prenatal betamethasone/postnatal NMDA | Pretreatment | No effect | (587) |
| ISS models | |||||
| Rapamycin, pulse | mTOR inhibitor | Rat, multiple hit | Treatment after spasm onset, 3 days | Decreases spasms within 2 h; stops spasms with repeat dosing; improves spatial learning; prevents adult epilepsy; reverses mTOR dysregulation | (157, 163) |
| CPP-115 | High-affinity vigabatrin analog | Rat, multiple hit | Treatment after spasm onset, repeated (PN4–12) | Reduces spasms from the first hour and for up to 3 days; better efficacy and tolerability than vigabatrin | (158) |
| Carisbamate | Unknown; effect on spasms not due to sodium channel blockade | Rat, multiple hit | Treatment after spasms onset, single dose (PN4 or PN6–7) | Reduces spasms within the first hour | (162) |
| NAX-5055 | Galanin receptor 1 (GalR1) agonist | Rat, multiple hit | Treatment after spasms onset, single dose (PN4 or PN6–7) | No effect; low expression of GalR1 in pups | (160) |
| VX-765 | Caspase 1 inhibitor | Rat, multiple hit | Treatment after spasms onset, single dose (PN4) | No effect | (159) |
| CGP 35348 | GABABR antagonism | Rat, multiple hit | Treatment after spasms onset, single dose (PN4) | No effect | (159) |
| 17β-Estradiol | Gonadal hormone | Rat, multiple hit | Treatment started after induction (PN3–10) | No effect | (159) |
| 17β-Estradiol | Gonadal hormone | Mouse, Arx KI [Arx (GCG10+7)] | Pretreatment (PN3–10) | Prevents spasms and other seizures, restores interneuronopathy | (152) |
| 17β-estradiol | Gonadal hormone | Mouse, Arx with PA1 or PA2 expansion | Pre-treatment (PN3-10) | Reduces seizures but not the abnormal behavior | (195) |
| Dravet syndrome models | |||||
| Soticlestat | Cholesterol 24-hydroxylase inhibitor | Mouse, Scn1atm1Kea with exon 1 deletion | Treatment after hyperthermia priming | Reduced seizures, protected against hyperthermia seizures, prevented SUDEP | (588) |
| Medium chain triglyceride diet (decanoic C10, octanoic acid C8 mix) | Ketogenic diet metabolite | Mouse, Scn1a KI, R1407X | 4-wk treatment prior to hyperthermia | C10/C8 (80:20) reduce both seizures and mortality; C10 reduces mortality | (589) |
| Gabra2 repair | GABRA2 expression restoration (increase) | Mouse, Scn1a+/− | Genetic repair of Gabra2 | Rescues epilepsy phenotype | (590) |
| Trpv1 receptor deletion | Trpv1 receptor deletion | Mouse, Scn1a+/− | Trpv1 receptor deletion | No effects on hyperthermia seizures, frequency of spontaneous seizures or survival | (591) |
| SB-705498 | Trpv1-selective antagonist | Mouse, Scn1a+/- | No effect on seizures or survival | (591) | |
| Cannabigerolic acid | Phytocannabinoid | Mouse, Scn1a+/- | Pretreatment | Potentiated clobazam effects on hyperthermia-induced and spontaneous seizures, anticonvulsant in MES, proconvulsant in 6-Hz test. | (592) |
| Cannabichromene, 5-fluoro- cannabichromene | Phytocannabinoid | Mouse, Scn1a+/− | Pretreatment | Anticonvulsant | (593) |
| Ketogenic diet | Ketogenic diet | Mouse, Scn1a KI, R1407X | Ketogenic diet, 14 days | Decreases SUDEP, protects against seizure-induced respiratory arrest | (594) |
| SCN1A transfer in the brain (adenoviral) | SCN1A expression in the brain | Mouse, SCN1A-A1783V KI | Adenovirus expressing SCN1A, intracerebral injection | Protected from death, attenuation of epilepsy; hyperactivity persisted; cognitive effects variable | (595) |
| Naltrexone | Opioid antagonist | Zebrafish, scn1Lab | Pretreatment, 30 min before PTZ | Anticonvulsant effects | (596) |
| Fenfluramine, Norflenfluramine enantiomers | Reuptake inhibitor of 5-OH-tryptamine | Zebrafish, scn1Lab-/- | Treatment exposure | Anticonvulsant effects | (217) |
| Fenfluramine | Reuptake inhibitor of 5-OH-tryptamine | Zebrafish, scn1Lab | Treatment exposure | Anticonvulsant effects | (597) |
| Dimethadione | Calcium channel blocker | Zebrafish, scn1Lab | Treatment exposure | Anticonvulsant effects | (597) |
| Clemizole | Serotoninergic | Zebrafish, scn1Lab | Treatment exposure | Anticonvulsant effects | (598) |
| PK11195 | Pck1 activator and translocator protein ligand | Zebrafish, scn1Lab | Treatment exposure | Anticonvulsant effects | (599) |
| GR-46611 | 5HT1D receptor agonist | Mouse, 129S-Scn1Atm1Kea/Mmjax | Pretreatment | Protects from hyperthermia seizures, decreases seizure severity, improves survival | (600) |
| ASO, increases Scn1a | ASO, increases Scn1a (TANGO method) | Mouse, F1:129S-Scn1a+/− Å∼ C57BL/6J | PN2 or 14, intracerebroventricular injection | Reduces seizures and SUDEP | (282) |
| CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons | CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons | Mouse, Scn1aRx/+, floxed dCas9-VPRVPR/+, Vgat-CreCre/+, | Intravenous AAV injection, 4 wk | Improved behavioral deficits, ameliorated febrile seizures | (601) |
| MV1369, MV1312 | NaV1.6 inhibitor | Zebrafish, Scn1Lab KO | Treatment | Reduced spontaneous burst movements and seizures | (602) |
| AA43279 | NaV1.1 activator | Zebrafish, Scn1Lab KO | Treatment | Reduced spontaneous burst movements and seizures | (602) |
AQB-565, ACTH1–24 linked to melanocyte stimulating hormone; ASO, antisense oligonucleotides; CPP-115, high-affinity GABA aminotransferase inhibitor; CRISPR, clustered regularly interspaced short palindromic repeats; EDRs, electrodecremental responses; GABRA2, GABAAR A2 subunit; CGP35348, GABAB receptor (GABABR) inhibitor; DEE, developmental and epileptic encephalopathy; ES, epileptic spasms; GalR, galanin receptor; GBL, gamma butyrolactone; GIRK, inward-rectifying potassium channel; 5-HT, serotonin; KI, knockin; MC, melanocortin receptor; mTOR, mechanistic target of rapamycin; Nav, sodium channel; NMDA, N-methyl-d-aspartate; PCK1, phosphoenolpyruvate carboxykinase 1; PN, postnatal; Scn1a, sodium channel 1; SUDEP, sudden death in epilepsy; Trpv1, transient receptor potential cation channel subfamily V member 1; vgat, vesicular GABA transporter.
There has also been remarkable progress in screening for new treatments for Dravet syndrome, using both rodents (282, 432, 588–595, 600, 601) as well as high-throughput studies in zebrafish (217, 439, 596–599, 602). A noteworthy development has been the recent approval by the Food and Drug Administration of fenfluramine, a reuptake inhibitor of 5-OH-tryptamine, for the treatment of Dravet syndrome, based on evidence provided by both preclinical (217, 597) and clinical (279, 605–609) trials.
A challenge in these preclinical drug trials has been the comparison of findings across models, given the different species (rats vs. other animal models), different induction protocols, developmental periods of exposure to the drugs, and treatment protocols (pre- vs. posttreatments) (152, 157, 159, 163, 182, 184, 195). Hopefully, identification of biomarkers guiding treatment implementation would help de-risk the process of selecting promising candidates for transitioning in clinical trials, as well as optimizing treatment protocols and designs.
The choice of ASM for DEE in clinical practice is mainly influenced by cumulative experience in open-label studies and only a limited number of drug trials in etiologically homogeneous conditions (610). For most DEEs, treatment choices rely on the hypothesized drug action on clinical and EEG phenomenology and remain confined to symptom relief and general principles of epilepsy management. Commercially available ASMs are typically grouped by their principal mode of action (drugs that affect voltage-dependent sodium channels, calcium currents, GABA activity, the glutamate receptors, and drugs with other mechanisms of action; TABLE 5), although for many drugs the precise mechanism of action remains unknown or multiple actions are hypothesized. As for other conditions, treatments for DEEs may include conventional drugs or repurposed therapies (i.e., with specific actions that may have been used in entirely unrelated conditions).
Table 5.
Main drugs used to treat DEEs and target pathways/molecules
| Excitatory Transmission | Na+ Channels | Ca2+ Channels | K+ Channels | Glutamate Receptors | 5-HT Receptors | Sigma-1 Receptors | Inhibitory Transmission | GABA-A Receptors | GABA Turnover | GABA-B Receptors | SV2A Binding | Carbonic Anhydrase | Clinical Efficacy | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Barbiturates | YES | YES | BS | |||||||||||
| Benzodiazepines | YES | YES | BS | |||||||||||
| Brivaracetam | YES | FS | ||||||||||||
| Cannabidiol | YES | LGS, DS | ||||||||||||
| Carbamazepine | YES | YES | YES | FS, GTCS | ||||||||||
| Cenobamate | YES | YES | YES | FS (adult) | ||||||||||
| Clobazam | YES | YES | YES | LGS, FS | ||||||||||
| Eslicarbazepine | YES | YES | FS | |||||||||||
| Ethosuximide | YES | YES | ABS | |||||||||||
| Felbamate | YES | YES | YES | YES | YES | YES | BS | |||||||
| Fenfluramine | YES | YES | YES | DS | ||||||||||
| Gabapentin | YES | YES | YES | YES | YES | FS, GTCS | ||||||||
| Lacosamide | YES | YES | YES | FS, SGS | ||||||||||
| Lamotrigine | YES | YES | YES | FS, GCTs, ABS | ||||||||||
| Levetiracetam | YES | YES | YES | YES | YES | YES | BS | |||||||
| Oxcarbazepine | YES | YES | YES | YES | FS, GTCS | |||||||||
| Perampanel | YES | YES | FS, GTCS | |||||||||||
| Phenobarbital | YES | YES | YES | YES | YES | FS | ||||||||
| Phenytoin | YES | YES | YES | FS, GTCS | ||||||||||
| Pregabalin | YES | YES | YES | FS | ||||||||||
| Primidone | YES | YES | YES | YES | FS, GTCS | |||||||||
| Retigabine | YES | YES | FS | |||||||||||
| Rufinamide | YES | YES | LGS | |||||||||||
| Stiripentol | YES | YES | DS | |||||||||||
| Tiagabine | YES | YES | FS | |||||||||||
| Topiramate | YES | YES | YES | YES | YES | YES | YES | YES | BS | |||||
| Valproate | YES | YES | YES | YES | YES | YES | BS | |||||||
| Vigabatrin | YES | YES | FS, ES | |||||||||||
| Zonisamide | YES | YES | YES | YES | FS, GTCs, MYO |
ABS, absence; BS, broad spectrum; DEE, developmental and epileptic encephalopathy; DS, Dravet syndrome; ES: epileptic spasms; FS, focal seizures; GABA, γ-aminobutyric acid; GTCS, generalized tonic-clonic seizures; 5-HT, 5-hydroxytryptamine; LGS, Lennox–Gastaut syndrome; MYO: myoclonic, SGS: secondary generalized seizures; SV2A, synaptic vesicle protein 2. Adapted from https://www.uptodate.com/contents/antiseizure-drugs-mechanism-of-action-pharmacology-and-adverse-effects.
Only recently have the enormous amount of knowledge generated by molecular genetic findings in the DEE and increased knowledge of the underlying disease mechanisms allowed designing etiology-specific trials for genetic DEEs (https://clinicaltrials.gov/ct2/home) (see TABLE 6 and SUPPLEMENTAL TABLE S2, available at https://doi.org/10.6084/m9.figshare.17694728.v2, for examples).
Table 6.
Clinical trials on DEEs
| 1 | Study Title: Study to Evaluate NBI-921352 as Adjunctive Therapy in Subjects With SCN8A Developmental and Epileptic Encephalopathy Syndrome (SCN8A-DEE) |
| Recruiting: Not yet | |
| Has results: No | |
| Conditions: SCN8A DEE syndrome | |
| Interventions: Drug: NBI-921352, placebo | |
| Locations: Neurocrine Clinical Site, Washington, DC | |
| 2 | Study Title: XEN496 (Ezogabine) in Children With KCNQ2 Developmental and Epileptic Encephalopathy |
| Recruiting: Yes | |
| Has results: No | |
| Conditions: Epilepsy, epilepsy in children, epilepsy; seizure disease, brain diseases, central nervous system diseases, nervous system diseases. epileptic syndromes | |
| Interventions: Drug: XEN496, placebo | |
| Locations: Children’s Hospital of Colorado, Aurora, CO; Northwest Florida Clinical Research Group, Gulf Breeze, FL; Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL; Columbia University Irving Medical Center, New York, NY; The Cleveland Clinic Foundation, Cleveland, OH; Oregon Health and Science University, Portland, OR; MultiCare Health System-Mary Bridge Pediatrics-Tacoma, Tacoma, WA; Sydney Children’s Hospital, Sydney, NSW, Australia; Children’s Health Queensland Hospital and Health Service, South Brisbane, QLD, Australia; Austin Health, Heidelberg, VIC, Australia | |
| 3 | Study Title: An Open-Label Extension of the Study XEN496 (Ezogabine) in Children With KCNQ2-DEE |
| Recruiting: Yes | |
| Has results: No | |
| Conditions: Epilepsy, epilepsy in children, seizure disease, brain diseases, central nervous system diseases, nervous system diseases, epileptic syndromes | |
| Interventions: Drug: XEN496, placebo | |
| Locations: MultiCare Health System-Mary Bridge Pediatrics-Tacoma, Tacoma, WA | |
| 4 | Study Title: Study of TAK-935 as an Adjunctive Therapy in Participants With Developmental and/or Epileptic Encephalopathies |
| Recruiting: No | |
| Has results: Yes | |
| Conditions: Developmental and/or epileptic encephalopathies | |
| Interventions: Drug: TAK-935, placebo | |
| Locations: Xenoscience, Phoenix, AZ; Medsol Clinical Research Center, Port Charlotte, FL; University of South Florida, Tampa, FL; Center for Integrative Rare Disease Research, Atlanta, GA; Bluegrass Epilepsy Research, Lexington, KY; Mid-Atlantic Epilepsy and Sleep Center, Bethesda, MD; The Comprehensive Epilepsy Care Center for Children and Adults, St. Louis, MO; Northeast Regional Epilepsy Group, Hackensack, NJ; Thomas Jefferson University, Philadelphia, PA; Medical University of South Carolina, Charleston, SC; University of Virginia Health Sciences Center, Charlottesville, VA | |
| 5 | Study Title: A Phase 2, Multicenter, Randomized, Double-blind, Placebo-controlled Study to Evaluate the Efficacy, Safety, and Tolerability of TAK-935 (OV935) as an Adjunctive Therapy In Pediatric Participants With Developmental and/or Epileptic Encephalopathies |
| Recruiting: No | |
| Has results: Yes | |
| Conditions: Epilepsy, Dravet syndrome, Lennox–Gastaut syndrome | |
| Interventions: Drug: TAK-935, placebo | |
| Locations: Phoenix Children's Hospital, Phoenix, AZ; Children’s Hospital Los Angeles, Los Angeles, CA: Colorado Children’s Hospital, Aurora, CO. Nicklaus Children’s Hospital, Miami, FL; Pediatric Neurology PA, Orlando, FL; Rare Disease Research, LLC, Atlanta, GA; Center for Rare Neurological Diseases, Norcross, GA; Ann and Robert H Lurie Childrens Hospital of Chicago, Chicago, IL; Mayo Clinic-PPDS, Rochester, MN; Northeast Regional Epilepsy Group, Hackensack, NJ; Children’s Hospital at Saint Peter’s University Hospital, New Brunswick, NJ; Columbia University Medical Center, New York, NY; Wake Forest Baptist Medical Center, Winston-Salem, NC; Medical University of South Carolina, Charleston, SC; Cook Children’s Medical Center, Fort Worth, TX; Monash Children’s Hospital, Clayton, VIC, Australia; Austin Hospital, Heidelberg West, VIC, Australia; Hospital For Sick Children, Toronto, ON, Canada; Peking University First Hospital, Beijing, China; Capital Medical University (CMU)-Beijing Children's Hospital, Beijing, China; Beijing Children’s Hospital, Capital Medical University, Beijing, China. Xiangya Hospital Central South University, Changsha, China; Children’s Hospital of Fudan University, Shanghai, China; Shenzhen Children’s Hospital, Shenzhen, China; Sheba Medical Center-PPDS, Tel Hashomer, Ramat Gan, Israel; Soroka University Medical Centre, Bear Sheva, Israel; Bnai Zion Medical Center, Haifa, Israel; Edith Wolfson Medical Center, Holon, Israel; Hadassah Medical Center, Jerusalem, Israel; Schneider Childrens Medical Center of Israel, Petach Tikva, Israel; Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; Uniwersyteckie Centrum Kliniczne PPDS, Gdansk, Pomorskie, Poland; NZOZ Centrum Neurologii Dzieciecej i Leczenia Padaczki, Kielce, Swietokrzyskie, Poland; Szpital Kliniczny im. H. Swiecickiego Uniwersytetu Medycznego im. Karola Marcinkowskiego w Poznaniu, Poznan, Wielkopolskie, Poland; Centrum Medyczne Plejady, Krakow, Poland; Samodzielny Publiczny Dzieciecy Szpital Kliniczny w Warszawie, Warsaw, Poland; Instytut Pomnik Centrum Zdrowia Dziecka, Warsaw, Poland; Centro Hospitalar Lisboa Central-Hospital Dona Estefania, Lisboa, Portugal; Centro Hospitalar Lisboa Norte, E.P.E. Hospital de Santa Maria, Lisboa, Portugal; Largo da Maternidade de Julio Dinis Centro Materno Infantil do Norte, Porto, Portugal; Clinica Universidad Navarra, Pamplona, Navarra, Spain; Hospital Vithas La Salud, Granada, Spain. Hospital Ruber Internacional, Madrid, Spain; Hospital Universitari i Politecnic La Fe de Valencia, Valencia, Spain |
Clinical trials gathered from ClinicalTrials.gov (https://clinicaltrials.gov/ct2/home) interrogated on October 27, 2021 with the keyword “developmental and epileptic encephalopathy.”
When clinical, EEG, and imaging findings suggest focal localization of the epileptogenic zone, and if any resulting neurological deficit is not more severe than epilepsy itself, surgical treatment of epilepsy remains one of the best options. For example, for patients with TSC epilepsy surgery is considered after failure of two ASMs even when multiple cortical lesions are present, and TSC operated patients, including those with infantile spasm, have a 50–60% chance of long-term seizure freedom after surgery for epilepsy (610).
A promising alternative approach to treat DEEs is represented by gene therapies. The growth in gene therapies in medicine has partly been realized through the development of safe and effective means of gene delivery using viral vectors. These vectors are engineered to avoid their replication in human cells and to deliver the wild-type form of the gene mutated in a given patient into the right cell target. The expression of the gene product delivered by viral vectors in the cells where the genetic defect needs to be corrected is ensured with a specific promoter. For DEEs, this treatment approach has some drawbacks, as most viral vectors can only carry a limited amount of DNA (and thus cannot be used to replace big genes such as SCN1A). In addition, to bypass the blood-brain barrier (BBB), they need to be injected in the brain through intraventricular injections. Gene therapy based on vectors derived from adeno-associated virus (AAV) has the potential to overcome the latter limitation, since it has been demonstrated that different AAV serotypes are able to cross the BBB and thus can be used to implement gene delivery strategies based on them to treat CNS diseases (611). Gene therapy, including AAV for DEE, carries several major technical limitations (612). Delivery would need to target the whole brain, as most DEEs are related to widespread brain dysfunction. Diffusely and irreversibly altering the genetic makeup of neurons may be a worrisome perspective, since some of the genetic defects causing DEE result from both loss- and gain-of-function effects and overdosing can cause its own pathology. Dosing and distribution are further complicated by the X-linked or somatic mosaic genetic alteration that underlies the disorder, with only half or small percentages of cells needing the supplemental transgene delivery. Additional limitations are related to the fact that delivery should be realized in a critical period, before mature networks are established.
Another possible way to correct the effects of a mutation at intracellular level with genetic material is the use of antisense oligonucleotides (ASOs), single-stranded deoxyribonucleotides that are complementary to a specific target mRNA and that upon binding it alter its splicing, impede its translation, or promote its degradation. ASOs are usually administered intrathecally. Studies in a conditional mouse model with Cre-dependent expression of the pathogenic patient SCN8A mutation p.Arg1872Trp (R1872W) have recently demonstrated that administration of an ASO directed against the Scn8a mutant transcript delayed seizure onset and increased survival, suggesting that reduction of SCN8A transcript is a promising approach to treatment of intractable childhood epilepsies (311). The effectiveness of ASOs has been confirmed in an additional study carried out by Li and collaborators (295). These authors demonstrated that targeted reduction of Scn2a mRNA expression by central administration of gapmer ASOs in Scn2a Q/+ mice reduced spontaneous seizures and significantly extended life span in treated animals. These results suggest that human SCN2A gapmer ASOs could likewise impact the lives of patients with SCN2A gain-of-function DEE (295). Because ASOs can regulate splicing, it is possible to use them to increase the production of translated mRNA. The TANGO (targeted augmentation of nuclear gene output) approach, which modulates naturally occurring, nonproductive splicing events to increase target gene and protein expression, has recently been used in animal models of DS to increase Scn1a transcript and Nav1.1 protein expression. In these models, a single intracerebroventricular dose of a lead ASO at postnatal day 2 or 14 reduced the incidence of electrographic seizures and sudden unexpected death in epilepsy (SUDEP), suggesting that TANGO may provide a unique, gene-specific approach for the treatment of DS (282).
ASOs can also be used to regulate the activity of miRNAs, which are “multipathway” regulatory molecules. Mature miRNAs are generated via a multistep process. They are initially transcribed as relatively large (even more than 1 kb) hairpin structures known as pri-miRNA. This undergoes cleavage in the nucleus by the enzyme Drosha to produce a 60- to 70-nucleotide (nt) stem loop pre-miRNA, which is subsequently transported from the nucleus to the cytoplasm. The enzyme Dicer recognizes pre-miRNA and cleaves the stem loop, leaving an imperfect ∼21- to 23-nt miRNA duplex with an ∼2-nt 3′ overhang at each end. The less thermodynamically stable end of the pre-miRNA duplex is then uploaded to a binding pocket within an Argonaute (Ago) protein to form the RNA-induced silencing complex (RISC). The miRNA-loaded RISC then traffics along mRNAs searching for complementary binding sites and, upon finding mRNA targets containing an ∼7- to 8-nt seed match [typically within the 3′ untranslated region (UTR)], triggers either target degradation or translational repression (613). Since there have been >300 studies on miRNA and epilepsy, and >100 different miRNAs have found to be altered in experimental models and human samples [EpimiRBase (614)], miRNA could represent a novel class of molecules to be targeted using ASOs to treat epilepsy in DEEs.
Gene therapy can be also pursued by correcting a specific genetic defect in patients’ cells through endogenous gene editing. This approach is not based on expressing the wild-type form of a given gene in cells that do not express it properly but on the substitution of a specific portion of the endogenous mutated gene (e.g., that flanking a mutation) with the wild-type sequence. Clustered regularly interspaced palindromic repeats (CRISPR)-Cas9 is a gene-editing technology that makes it possible to correct errors in the genome and turn on or off genes in cells and organisms quickly, cheaply, and with relative ease. This gene-editing technology has a number of laboratory applications including rapid generation of cellular and animal models, functional genomic screens, live imaging of the cellular genome, and gene therapy (615, 616). CRISPR-Cas9 involves two essential components: a guide RNA to match a desired target gene, and Cas9 (CRISPR-associated protein 9), an endonuclease that causes a double-stranded DNA break, allowing modifications of the genome. One of the most exciting applications of CRISPR-Cas9 is its potential use to treat genetic disorders caused by single-gene mutations. Examples of such diseases include cystic fibrosis (CF), Duchenne muscular dystrophy (DMD) and hemoglobinopathies (616). A modified version of the CRISPR-Cas9 system, called dead Cas9 (dCas9), can be tailored to obtain a robust and highly specific activation of the Scn1a gene both in cultured neurons and in the brain tissue of a DS mouse model, suggesting that dCas9 may be an effective and targeted approach to DS and other disorders resulting from altered gene dosage (281). A number of challenges remain before the potential of CRISPR-Cas9 can be translated to effective treatments at the bedside. Indeed, its clinical translation has been hampered by varying efficiency, off-target effects, and, on occasion, insufficient vector size for the necessary genetic material (610). In addition, a suitable vector is needed to safely deliver Cas9-nuclease-encoding genes and guide RNAs in vivo without any associated toxicity (616). To overcome this problem, the use of AAV vectors has been proposed. However, this delivery system may be too small to allow efficient transduction of the Cas9 gene. A smaller Cas9 gene could be used, but this has additional implications for efficacy (615).
11. CONCLUSIONS
Multiple genetically determined or, at times, acquired etiologies may severely alter the balance between excitatory and inhibitory neuronal activity and result in widespread epileptogenesis in the developing brain. If the causative defect imposes serious consequences in physiological brain function, the superimposed epilepsy will add a clinical burden to the already compromised neurodevelopmental processes. If the causative defect only mildly impacts physiological brain development and function, any superimposed severe epileptogenic process will cause considerable neurodevelopmental deterioration (617). Development of higher cortical functions is the most complex and vulnerable process and will be the most severely affected. Impairment may be relatively selective, in the context of DEE in which, for example, language, memory, attention or executive functions are impaired to a different extent. Generalized severe cognitive impairment or autistic features may be the result of a widespread epileptogenic process. Although DEEs have a multitude of causes and variable clinical patterns, with many genes involved whose altered expression may affect different aspects of neural cell functioning, mechanisms whereby epileptic activity may interfere with brain function and produce such patterns tend to be relatively limited. Moderate levels of hyperexcitability can disrupt cortical processing, with temporal and anatomic specificity (618). Disruption is time-locked with the EEG event accompanying hyperexcitability and specific to the modality represented in the anatomic area involved (619–621). Redundant and frequent EEG abnormalities as observed in DEE may cause widespread cortical dysfunction that can be manifested with signs of early cognitive regression even when seizure activity is not yet overly manifested (622, 623). The timing of onset, network distribution, and duration of the epileptogenic process influence how the DEE will be manifested, at times in close relationship with its specific etiology, at times irrespective from it. Similar anatomoclinical backgrounds may be accompanied by very different forms of DEE in different individuals. In this perspective, although efforts toward development of precision treatment approaches in DEE will ideally target the causative mechanisms, traditional approaches with antiseizure medications addressed at mitigating the consequences of redundant epileptic activity on physiological brain function still have a relevant role.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.19666521.v2.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.17694728.v2.
GRANTS
R. Guerrini acknowledges grant support by the Tuscany Region Call for Health 2018 (Project DECODE-EE) and by Fondazione Cassa di Risparmio di Firenze (Project BRAIN). A. S. Galanopoulou acknowledges grant support by NIH U54 NS100064, R01 NS091170, American Epilepsy Society seed grant, the Heffer Family and the Segal Family Foundations, and the Abbe Goldstein/Joshua Lurie and Laurie Marsh/Dan Levitz families. F. Benfenati acknowledges grant support by Era-Net Neuron 2017 Snaropathies, IRCCS Ospedale Policlinico San Martino (Ricerca Corrente and “5x1000”), the EU Joint Programme on Neurodegenerative Disease Research 2020 Project “Neurophage,” and the Italian Ministry of University and Research (PRIN 2015-H4K2CR and 2017-A9MK4R). The support of Telethon-Italy (Grant GGP19120 to F. Benfenati) is also acknowledged. M. Mantegazza acknowledges grant support by the Laboratory of Excellence “Ion Channel Science and Therapeutics” (LabEx ICST, ANR-11-LABX-0015-01, France) and the IDEX UCA-Jedi (University Côte d’Azur ANR-15-IDEX-01, France).
DISCLOSURES
A.S. Galanopoulou is the Editor-in-Chief of Epilepsia Open and receives royalties from Elsevier, Medlink, and Morgan and Claypool for publications. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
R.G., V.C., M.M., A.S.G., and F.B. prepared figures; R.G., V.C., M.M., A.S.G., and F.B. drafted manuscript; R.G., V.C., S.B., A.S.G., and F.B. edited and revised manuscript; R.G., V.C., M.M., S.B., A.S.G., and F.B. approved final version of manuscript.
ACKNOWLEDGMENTS
Graphical abstract was created with BioRender.com.
REFERENCES
- 1. Stafstrom CE, Carmant L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med 5: a022426, 2015. doi: 10.1101/cshperspect.a022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58: 512–521, 2017. doi: 10.1111/epi.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cutri-French C, Armstrong D, Saby J, Gorman C, Lane J, Fu C, Peters SU, Percy A, Neul JL, Marsh ED. Comparison of core features in four developmental encephalopathies in the Rett Natural History Study. Ann Neurol 88: 396–406, 2020. doi: 10.1002/ana.25797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 15: 304–316, 2016. doi: 10.1016/S1474-4422(15)00250-1. [DOI] [PubMed] [Google Scholar]
- 5. Riney K, Bogacz A, Somerville E, Hirsch E, Nabbout R, Scheffer IE, et al. ILAE Classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions (Online). 2021. http://files/1501/Riney_VariableAges_7Apr21.pdf. [DOI] [PubMed]
- 6. Specchio N, Wirrel EC, Scheffer IE, Nabbout R, Riney K, Samia P, et al. ILAE Classification and definition of epilepsy syndromes with onset in childhood: position paper by the ILAE Task Force on Nosology and Definitions (Online). 2021. http://files/1503/Specchio. [DOI] [PubMed]
- 7. Zuberi SM, Wirrel EC, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE Classification & definition of epilepsy syndromes in the neonate and infant: position statement by the ILAE Task Force on Nosology and Definitions (Online). 2021. http://files/1505/Zuberi. [DOI] [PubMed]
- 8. Bayat A, Bayat M, Rubboli G, Møller RS. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes 12: 1051, 2021. doi: 10.3390/genes12071051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature 501: 217–221, 2013. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epilepsy Phenome/Genome Project Epi4K Consortium. Copy number variant analysis from exome data in 349 patients with epileptic encephalopathy. Ann Neurol 78: 323–328, 2015. doi: 10.1002/ana.24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 95: 360–370, 2014. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol 45: 355–367, 2011. doi: 10.1016/j.pediatrneurol.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wirrell EC, Shellhaas RA, Joshi C, Keator C, Kumar S, Mitchell WG; Pediatric Epilepsy Research Consortium. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Epilepsia 56: 617–625, 2015. doi: 10.1111/epi.12951. [DOI] [PubMed] [Google Scholar]
- 14. Asadi-Pooya AA. Lennox-Gastaut syndrome: a comprehensive review. Neurol Sci 39: 403–414, 2018. doi: 10.1007/s10072-017-3188-y. [DOI] [PubMed] [Google Scholar]
- 15. Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol 5: 1055–1063, 2006. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- 16. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet 101: 664–685, 2017. doi: 10.1016/j.ajhg.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mefford HC, Yendle SC, Hsu C, Cook J, Geraghty E, McMahon JM, Eeg-Olofsson O, Sadleir LG, Gill D, Ben-Zeev B, Lerman-Sagie T, Mackay M, Freeman JL, Andermann E, Pelakanos JT, Andrews I, Wallace G, Eichler EE, Berkovic SF, Scheffer IE. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol 70: 974–985, 2011. doi: 10.1002/ana.22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma Y, Chen C, Wang Y, Wu L, He F, Chen C, Zhang C, Deng X, Yang L, Chen Y, Wu L, Yin F, Peng J. Analysis copy number variation of Chinese children in early-onset epileptic encephalopathies with unknown cause: CNVs analysis in EOEEs. Clin Genet 90: 428–436, 2016. doi: 10.1111/cge.12768. [DOI] [PubMed] [Google Scholar]
- 19. Happ HC, Carvill GL. A 2020 view on the genetics of developmental and epileptic encephalopathies. Epilepsy Curr 20: 90–96, 2020. doi: 10.1177/1535759720906118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Myers CT, Hollingsworth G, Muir AM, Schneider AL, Thuesmunn Z, Knupp A, King C, Lacroix A, Mehaffey MG, Berkovic SF, Carvill GL, Sadleir LG, Scheffer IE, Mefford HC. Parental mosaicism in “de novo” epileptic encephalopathies. N Engl J Med 378: 1646–1648, 2018. doi: 10.1056/NEJMc1714579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Lange IM, Koudijs MJ, van ’t Slot R, Sonsma AC, Mulder F, Carbo EC, van Kempen MJ, Nijman IJ, Ernst RF, Savelberg SM, Knoers NV, Brilstra EH, Koeleman BP. Assessment of parental mosaicism in SCN1A-related epilepsy by single-molecule molecular inversion probes and next-generation sequencing. J Med Genet 56: 75–80, 2019. doi: 10.1136/jmedgenet-2018-105672. [DOI] [PubMed] [Google Scholar]
- 22. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, Tang S, Helbig I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med 18: 898–905, 2016. doi: 10.1038/gim.2015.186. [DOI] [PubMed] [Google Scholar]
- 23. Papuc SM, Abela L, Steindl K, Begemann A, Simmons TL, Schmitt B, Zweier M, Oneda B, Socher E, Crowther LM, Wohlrab G, Gogoll L, Poms M, Seiler M, Papik M, Baldinger R, Baumer A, Asadollahi R, Kroell-Seger J, Schmid R, Iff T, Schmitt-Mechelke T, Otten K, Hackenberg A, Addor MC, Klein A, Azzarello-Burri S, Sticht H, Joset P, Plecko B, Rauch A. The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur J Hum Genet 27: 408–421, 2019. doi: 10.1038/s41431-018-0299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carvill GL, Engel KL, Ramamurthy A, Cochran JN, Roovers J, Stamberger H, Lim N, Schneider AL, Hollingsworth G, Holder DH, Regan BM, Lawlor J, Lagae L, Ceulemans B, Bebin EM, Nguyen J, Barsh GS, Weckhuysen S, Meisler M, Berkovic SF, De Jonghe P, Scheffer IE, Myers RM, Cooper GM, Mefford HC; EuroEPINOMICS Rare Epilepsy Syndrome, Myoclonic-Astatic Epilepsy, and Dravet Working Group. Aberrant inclusion of a poison exon causes Dravet syndrome and related SCN1A-associated genetic epilepsies. Am J Hum Genet 103: 1022–1029, 2018. doi: 10.1016/j.ajhg.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, Black DL, Kharchenko PV, Sharp PA, Walsh CA. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166: 1147–1162.e15, 2016. doi: 10.1016/j.cell.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. D’Gama AM, Walsh CA. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci 21: 1504–1514, 2018. doi: 10.1038/s41593-018-0257-3. [DOI] [PubMed] [Google Scholar]
- 27. Winawer MR, Griffin NG, Samanamud J, Baugh EH, Rathakrishnan D, Ramalingam S, et al. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol 83: 1133–1146, 2018. doi: 10.1002/ana.25243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sim NS, Seo Y, Lim JS, Kim WK, Son H, Kim HD, Kim S, An HJ, Kang HC, Kim SH, Kim DS, Lee JH. Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurol Genet 4: e294, 2018. doi: 10.1212/NXG.0000000000000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baldassari S, Ribierre T, Marsan E, Adle-Biassette H, Ferrand-Sorbets S, Bulteau C, Dorison N, Fohlen M, Polivka M, Weckhuysen S, Dorfmüller G, Chipaux M, Baulac S. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol 138: 885–900, 2019. doi: 10.1007/s00401-019-02061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller KE, Koboldt DC, Schieffer KM, Bedrosian TA, Crist E, Sheline A, Leraas K, Magrini V, Zhong H, Brennan P, Bush J, Fitch J, Bir N, Miller AR, Cottrell CE, Leonard J, Pindrik JA, Rusin JA, Shah SH, White P, Wilson RK, Mardis ER, Pierson CR, Ostendorf AP. Somatic SLC35A2 mosaicism correlates with clinical findings in epilepsy brain tissue. Neurol Genet 6: e460, 2020. doi: 10.1212/NXG.0000000000000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bonduelle T, Hartlieb T, Baldassari S, Sim NS, Kim SH, Kang HC, Kobow K, Coras R, Chipaux M, Dorfmüller G, Adle-Biassette H, Aronica E, Lee JH, Blumcke I, Baulac S. Frequent SLC35A2 brain mosaicism in mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE). Acta Neuropathol Commun 9: 3, 2021. doi: 10.1186/s40478-020-01085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakayama T, Ishii A, Yoshida T, Nasu H, Shimojima K, Yamamoto T, Kure S, Hirose S. Somatic mosaic deletions involving SCN1A cause Dravet syndrome. Am J Med Genet A 176: 657–662, 2018. doi: 10.1002/ajmg.a.38596. [DOI] [PubMed] [Google Scholar]
- 33. Stosser MB, Lindy AS, Butler E, Retterer K, Piccirillo-Stosser CM, Richard G, McKnight DA. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med 20: 403–410, 2018. doi: 10.1038/gim.2017.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ye Z, Chatterton Z, Pflueger J, Damiano JA, McQuillan L, Harvey AS, Malone S, Do H, Maixner W, Schneider A, Nolan B, Wood M, Lee WS, Gillies G, Pope K, Wilson M, Lockhart PJ, Dobrovic A, Scheffer IE, Bahlo M, Leventer RJ, Lister R, Berkovic SF, Hildebrand MS. Cerebrospinal fluid liquid biopsy for detecting somatic mosaicism in brain. Brain Commun 3: fcaa235, 2021. doi: 10.1093/braincomms/fcaa235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim S, Baldassari S, Sim NS, Chipaux M, Dorfmüller G, Kim DS, Chang WS, Taly V, Lee JH, Baulac S. Detection of brain somatic mutations in cerebrospinal fluid from refractory epilepsy patients. Ann Neurol 89: 1248–1252, 2021. doi: 10.1002/ana.26080. [DOI] [PubMed] [Google Scholar]
- 36. Eichers ER, Lewis RA, Katsanis N, Lupski JR. Triallelic inheritance: a bridge between Mendelian and multifactorial traits. Ann Med 36: 262–272, 2004. doi: 10.1080/07853890410026214. [DOI] [PubMed] [Google Scholar]
- 37. Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci 10: 1554–1558, 2007. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 38. Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis 41: 655–660, 2011. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, Morgan M, Muzny D, Gibbs R, Noebels J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145: 1036–1048, 2011. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hasan S, Balobaid A, Grottesi A, Dabbagh O, Cenciarini M, Rawashdeh R, Al-Sagheir A, Bove C, Macchioni L, Pessia M, Al-Owain M, D’Adamo MC. Lethal digenic mutations in the K+ channels Kir4.1 (KCNJ10) and SLACK (KCNT1) associated with severe-disabling seizures and neurodevelopmental delay. J Neurophysiol 118: 2402–2411, 2017. doi: 10.1152/jn.00284.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calhoun JD, Hawkins NA, Zachwieja NJ, Kearney JA. Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia 58: e111–e115, 2017. doi: 10.1111/epi.13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fernández-Marmiesse A, Roca I, Díaz-Flores F, Cantarín V, Pérez-Poyato MS, Fontalba A, Laranjeira F, Quintans S, Moldovan O, Felgueroso B, Rodríguez-Pedreira M, Simón R, Camacho A, Quijada P, Ibanez-Mico S, Domingno MR, Benito C, Calvo R, Pérez-Cejas A, Carrasco ML, Ramos F, Couce ML, Ruiz-Falcó ML, Gutierrez-Solana L, Martínez-Atienza M. Rare variants in 48 genes account for 42% of cases of epilepsy with or without neurodevelopmental delay in 246 pediatric patients. Front Neurosci 13: 1135, 2019. doi: 10.3389/fnins.2019.01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pelorosso C, Watrin F, Conti V, Buhler E, Gelot A, Yang X, Mei D, McEvoy-Venneri J, Manent JB, Cetica V, Ball LL, Buccoliero AM, Vinck A, Barba C, Gleeson JG, Guerrini R, Represa A. Somatic double-hit in MTOR and RPS6 in hemimegalencephaly with intractable epilepsy. Hum Mol Genet 28: 3755–3765, 2019. doi: 10.1093/hmg/ddz194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horsthemke B. Epimutations in human disease. Curr Top Microbiol Immunol 310: 45–59, 2006. doi: 10.1007/3-540-31181-5_4. [DOI] [PubMed] [Google Scholar]
- 45. Henshall DC, Kobow K. Epigenetics and epilepsy. Cold Spring Harb Perspect Med 5: a022731, 2015. doi: 10.1101/cshperspect.a022731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 8: 1056–1072, 2009. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 47. Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature 447: 178–182, 2007. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 48. Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci 6: 108–118, 2005. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 49. Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281: 15763–15773, 2006. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 50. Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and ratBDNF gene structure and expression revisited. J Neurosci Res 85: 525–535, 2007. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Binder DK, The role of BDNF in epilepsy and other diseases of the mature nervous system. In: Recent Advances in Epilepsy Research, edited by Binder DK, Scharfman HE. Boston, MA: Springer US, 2004, p. 34–56. [DOI] [PubMed] [Google Scholar]
- 52. Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci 28: 395–406, 2008. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Salar S, Moshé SL, Galanopoulou AS. Metabolic etiologies in West syndrome. Epilepsia Open 3: 134–166, 2018. doi: 10.1002/epi4.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Galanopoulou AS, Gorter JA, Cepeda C. Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia 53: 1119–1130, 2012. doi: 10.1111/j.1528-1167.2012.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Katsarou AM, Moshé SL, Galanopoulou AS. Interneuronopathies and their role in early life epilepsies and neurodevelopmental disorders. Epilepsia Open 2: 284–306, 2017. doi: 10.1002/epi4.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lado FA, Rubboli G, Capovilla G, Capovilla P, Avanzini G, Moshé SL. Pathophysiology of epileptic encephalopathies. Epilepsia 54: 6–13, 2013. doi: 10.1111/epi.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scharfman HE. The neurobiology of epilepsy. Curr Neurol Neurosci Rep 7: 348–354, 2007. doi: 10.1007/s11910-007-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carvill GL, Regan BM, Yendle SC, O’Roak BJ, Lozovaya N, Bruneau N, Burnashev N, Khan A, Cook J, Geraghty E, Sadleir LG, Turner SJ, Tsai MH, Webster R, Ouvrier R, Damiano JA, Berkovic SF, Shendure J, Hildebrand MS, Szepetowski P, Scheffer IE, Mefford HC. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet 45: 1073–1076, 2013. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li D, Yuan H, Ortiz-Gonzalez XR, Marsh ED, Tian L, McCormick EM, Kosobucki GJ, Chen W, Schulien AJ, Chiavacci R, Tankovic A, Naase C, Brueckner F, von Stülpnagel-Steinbeis C, Hu C, Kusumoto H, Hedrich UB, Elsen G, Hörtnagel K, Aizenman E, Lemke JR, Hakonarson H, Traynelis SF, Falk MJ. GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am J Hum Genet 99: 802–816, 2016. doi: 10.1016/j.ajhg.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Molinari F, Raas-Rothschild A, Rio M, Fiermonte G, Encha-Razavi F, Palmieri L, Palmieri F, Ben-Neriah Z, Kadhom N, Vekemans M, Attie-Bitach T, Munnich A, Rustin P, Colleaux L. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am J Hum Genet 76: 334–339, 2005. doi: 10.1086/427564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Myers CT, McMahon JM, Schneider AL, Petrovski S, Allen AS, Carvill GL, Zemel M, Saykally JE, LaCroix AJ, Heinzen EL, Hollingsworth G, Nikanorova M, Corbett M, Gecz J, Coman D, Freeman J, Calvert S, Gill D, Carney P, Lerman-Sagie T, Sampaio H, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Johnson MR, Kuzniecky R, Marson AG. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet 99: 287–298, 2016. doi: 10.1016/j.ajhg.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, James VM, Pepler A, Steiner I, Hörtnagel K, Neidhardt J, Ruf S, Wolff M, Bartholdi D, Caraballo R, Platzer K, Suls A, De Jonghe P, Biskup S, Weckhuysen S. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol 75: 147–154, 2014. doi: 10.1002/ana.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lemke JR, Geider K, Helbig KL, Heyne HO, Schütz H, Hentschel J, et al. Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86: 2171–2178, 2016. doi: 10.1212/WNL.0000000000002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9: 1142–1149, 2006. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 65. Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27: 5903–5914, 2007. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy: NaV1.1 channels and epilepsy. J Physiol 588: 1849–1859, 2010. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Du J, Simmons S, Brunklaus A, Adiconis X, Hession CC, Fu Z, Li Y, Shema R, Møller RS, Barak B, Feng G, Meisler M, Sanders S, Lerche H, Campbell AJ, McCarroll S, Levin JZ, Lal D. Differential excitatory vs inhibitory SCN expression at single cell level regulates brain sodium channel function in neurodevelopmental disorders. Eur J Paediatr Neurol 24: 129–133, 2020. doi: 10.1016/j.ejpn.2019.12.019. [DOI] [PubMed] [Google Scholar]
- 68. Hurni N, Kolodziejczak M, Tomasello U, Badia J, Jacobshagen M, Prados J, Dayer A. Transient cell-intrinsic activity regulates the migration and laminar positioning of cortical projection neurons. Cereb Cortex 27: 3052–3063, 2017. doi: 10.1093/cercor/bhx059. [DOI] [PubMed] [Google Scholar]
- 69. Vitali I, Fièvre S, Telley L, Oberst P, Bariselli S, Frangeul L, Baumann N, McMahon JJ, Klingler E, Bocchi R, Kiss JZ, Bellone C, Silver DL, Jabaudon D. Progenitor hyperpolarization regulates the sequential generation of neuronal subtypes in the developing neocortex. Cell 174: 1264–1276.e15, 2018. doi: 10.1016/j.cell.2018.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rubinstein M, Westenbroek RE, Yu FH, Jones CJ, Scheuer T, Catterall WA. Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol Dis 73: 106–117, 2015. doi: 10.1016/j.nbd.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pitkänen A, Lukasiuk K, Dudek FE, Staley KJ. Epileptogenesis. Cold Spring Harb Perspect Med 5: a022822, 2015. doi: 10.1101/cshperspect.a022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, Lagae L, Moshé SL, Peltola J, Roulet Perez E, Scheffer IE, Zuberi SM. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58: 522–530, 2017. doi: 10.1111/epi.13670. [DOI] [PubMed] [Google Scholar]
- 73. Johnston D, Brown TH. The synaptic nature of the paroxysmal depolarizing shift in hippocampal neurons. Ann Neurol 16: S65–S71, 1984. doi: 10.1002/ana.410160711. [DOI] [PubMed] [Google Scholar]
- 74. Traub RD, Michelson-Law H, Bibbig AE, Buhl EH, Whittington MA. Gap junctions, fast oscillations and the initiation of seizures. In: Recent Advances in Epilepsy Research, edited by Binder DK, Scharfman HE.. Boston, MA: Springer US, 2004, p. 110–122. [DOI] [PubMed] [Google Scholar]
- 75. Mylvaganam S, Ramani M, Krawczyk M, Carlen PL. Roles of gap junctions, connexins, and pannexins in epilepsy. Front Physiol 5: 172, 2014. doi: 10.3389/fphys.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26: 5438–5447, 2006. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322: 1551–1555, 2008. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- 78. Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378: 75–78, 1995. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- 79. Pinault D, O’Brien TJ. Cellular and network mechanisms of genetically-determined absence seizures. Thalamus Relat Syst 3: 181–203, 2005. doi: 10.1017/S1472928807000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Blumenfeld H. Cellular and network mechanisms of spike-wave seizures. Epilepsia 46, Suppl 9: 21–33, 2005. doi: 10.1111/j.1528-1167.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- 81. van Luijtelaar G, Onat FY, Gallagher MJ. Animal models of absence epilepsies: what do they model and do sex and sex hormones matter? Neurobiol Dis 72: 167–179, 2014. doi: 10.1016/j.nbd.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kostopoulos GK. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clin Neurophysiol 111: S27–S38, 2000. doi: 10.1016/S1388-2457(00)00399-0. [DOI] [PubMed] [Google Scholar]
- 83. Rosenow F, Lüders H. Presurgical evaluation of epilepsy. Brain 124: 1683–1700, 2001. doi: 10.1093/brain/124.9.1683. [DOI] [PubMed] [Google Scholar]
- 84. Lüders HO, Najm I, Nair D, Widdess-Walsh P, Bingman W. The epileptogenic zone: general principles. Epileptic Disord 8: S1–S9, 2006. [PubMed] [Google Scholar]
- 85. Kreindler A, Zuckermann E, Steriade M, Chimion D. Electro-clinical features of convulsions induced by stimulation of brain stem. J Neurophysiol 21: 430–436, 1958. doi: 10.1152/jn.1958.21.5.430. [DOI] [PubMed] [Google Scholar]
- 86. Neville BG. The origin of infantile spasms: evidence from a case of hydranencephaly. Dev Med Child Neurol 14: 644–647, 1972. doi: 10.1111/j.1469-8749.1972.tb02647.x. [DOI] [PubMed] [Google Scholar]
- 87. Lee CL, Frost JD Jr, Swann JW, Hrachovy RA. A new animal model of infantile spasms with unprovoked persistent seizures. Epilepsia 49: 298–307, 2008. doi: 10.1111/j.1528-1167.2007.01377.x. [DOI] [PubMed] [Google Scholar]
- 88. Scantlebury MH, Galanopoulou AS, Chudomelova L, Raffo E, Betancourth D, Moshé SL. A model of symptomatic infantile spasms syndrome. Neurobiol Dis 37: 604–612, 2010. doi: 10.1016/j.nbd.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Burgess R, Wang S, McTague A, Boysen KE, Yang X, Zeng Q, Myers KA, Rochtus A, Trivisano M, Gill D, Sadleir LG, Specchio N, Guerrini R, Marini C, Zhang YH, Mefford HC, Kurian MA, Poduri AH, Scheffer IE; EIMFS Consortium. The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann Neurol 86: 821–831, 2019. doi: 10.1002/ana.25619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev 3: 79–83, 1979. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 91. Ojeda SR, Advis JP, Andrews WW. Neuroendocrine control of the onset of puberty in the rat. Fed Proc 39: 2365–2371, 1980. [PubMed] [Google Scholar]
- 92. Galanopoulou AS, Moshé SL. In search of epilepsy biomarkers in the immature brain: goals, challenges and strategies. Biomark Med 5: 615–628, 2011. doi: 10.2217/bmm.11.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Buyanova IS, Arsalidou M. Cerebral white matter myelination and relations to age, gender, and cognition: a selective review. Front Hum Neurosci 15: 662031, 2021. doi: 10.3389/fnhum.2021.662031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience 105: 7–17, 2001. doi: 10.1016/S0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- 95. Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus 16: 1032–1060, 2006. doi: 10.1002/hipo.20225. [DOI] [PubMed] [Google Scholar]
- 96. Lenroot RK, Giedd JN. Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci Biobehav Rev 30: 718–729, 2006. doi: 10.1016/j.neubiorev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 97. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106-107: 1–16, 2013. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron 89: 248–268, 2016. doi: 10.1016/j.neuron.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Steiner P. Brain fuel utilization in the developing brain. Ann Nutr Metab 75, Suppl 1: 8–18, 2019. doi: 10.1159/000508054. [DOI] [PubMed] [Google Scholar]
- 100. Zeiss CJ. Comparative milestones in rodent and human postnatal central nervous system development. Toxicol Pathol 49: 1368–1373, 2021. doi: 10.1177/01926233211046933. [DOI] [PubMed] [Google Scholar]
- 101. Akman O, Moshé SL, Galanopoulou AS. Sex-specific consequences of early life seizures. Neurobiol Dis 72: 153–166, 2014. doi: 10.1016/j.nbd.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Insel TR, Miller LP, Gelhard RE. The ontogeny of excitatory amino acid receptors in rat forebrain—I. N-methyl-D-aspartate and quisqualate receptors. Neuroscience 35: 31–43, 1990. doi: 10.1016/0306-4522(90)90117-M. [DOI] [PubMed] [Google Scholar]
- 103. Moshé SL, Albala BJ. Maturational changes in postictal refractoriness and seizure susceptibility in developing rats. Ann Neurol 13: 552–557, 1983. doi: 10.1002/ana.410130514. [DOI] [PubMed] [Google Scholar]
- 104. Haas KZ, Sperber EF, Moshé SL. Kindling in developing animals: interactions between ipsilateral foci. Brain Res Dev Brain Res 68: 140–143, 1992. doi: 10.1016/0165-3806(92)90257-W. [DOI] [PubMed] [Google Scholar]
- 105. Haas KZ, Sperber EF, Opanashuk LA, Stanton PK, Moshé SL. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus 11: 615–625, 2001. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- 106. Sperber EF, Stanton PK, Haas K, Ackermann RF, Moshé SL. Developmental differences in the neurobiology of epileptic brain damage. Epilepsy Res Suppl, 9: 67–81, 1992. [PubMed] [Google Scholar]
- 107. Chudomel O, Hasson H, Bojar M, Moshé SL, Galanopoulou AS. Age- and sex-related characteristics of tonic GABA currents in the rat substantia nigra pars reticulata. Neurochem Res 40: 747–757, 2015. doi: 10.1007/s11064-015-1523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chudomel O, Herman H, Nair K, Moshé SL, Galanopoulou AS. Age- and gender-related differences in GABAA receptor-mediated postsynaptic currents in GABAergic neurons of the substantia nigra reticulata in the rat. Neuroscience 163: 155–167, 2009. doi: 10.1016/j.neuroscience.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Galanopoulou AS. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res 80: 99–113, 2008. doi: 10.1016/j.eplepsyres.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Galanopoulou AS, Moshé SL. The epileptic hypothesis: developmentally related arguments based on animal models. Epilepsia 50, Suppl 7: 37–42, 2009. doi: 10.1111/j.1528-1167.2009.02217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Giorgi FS, Galanopoulou AS, Moshé SL. Sex dimorphism in seizure-controlling networks. Neurobiol Dis 72: 144–152, 2014. doi: 10.1016/j.nbd.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kyrozis A, Chudomel O, Moshé SL, Galanopoulou AS. Sex-dependent maturation of GABAA receptor-mediated synaptic events in rat substantia nigra reticulata. Neurosci Lett 398: 1–5, 2006. doi: 10.1016/j.neulet.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 113. Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 3: 728–739, 2002. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 114. Cancedda L, Fiumelli H, Chen K, Poo M. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci 27: 5224–5235, 2007. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang DD, Kriegstein AR. Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex 21: 574–587, 2011. doi: 10.1093/cercor/bhq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang DD, Kriegstein AR. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci 28: 5547–5558, 2008. doi: 10.1523/JNEUROSCI.5599-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem 271: 16245–16252, 1996. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 118. Galanopoulou AS. GABAA receptors in normal development and seizures: friends or foes? Curr Neuropharmacol 6: 1–20, 2008. doi: 10.2174/157015908783769653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Galanopoulou AS. Dissociated gender-specific effects of recurrent seizures on GABA signaling in CA1 pyramidal neurons: role of GABA(A) receptors. J Neurosci 28: 1557–1567, 2008. doi: 10.1523/JNEUROSCI.5180-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Galanopoulou AS, Moshé SL. Does epilepsy cause a reversion to immature function? Adv Exp Med Biol 813: 195–209, 2014. doi: 10.1007/978-94-017-8914-1_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416: 303–325, 1989. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Brown DA, Scholfield CN. Depolarization of neurones in the isolated olfactory cortex of the guinea-pig by gamma-aminobutyric acid. Br J Pharmacol 65: 339–345, 1979. doi: 10.1111/j.1476-5381.1979.tb07835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Galvan M, Scholfield CN, Brown DA. Depolarizing actions of gamma-aminobutyric acid (GABA) on mammalian brain slices. Br J Pharmacol 62: 410P, 1978. [PMC free article] [PubMed] [Google Scholar]
- 124. LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15: 1287–1298, 1995. doi: 10.1016/0896-6273(95)90008-X. [DOI] [PubMed] [Google Scholar]
- 125. Luhmann HJ, Prince DA. Postnatal maturation of the GABAergic system in rat neocortex. J Neurophysiol 65: 247–263, 1991. doi: 10.1152/jn.1991.65.2.247. [DOI] [PubMed] [Google Scholar]
- 126. Mueller AL, Chesnut RM, Schwartzkroin PA. Actions of GABA in developing rabbit hippocampus: an in vitro study. Neurosci Lett 39: 193–198, 1983. doi: 10.1016/0304-3940(83)90076-9. [DOI] [PubMed] [Google Scholar]
- 127. Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem 269: 25677–25683, 1994. doi: 10.1016/S0021-9258(18)47302-4. [DOI] [PubMed] [Google Scholar]
- 128. Payne JA, Forbush B 3rd.. Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc Natl Acad Sci USA 91: 4544–4548, 1994. doi: 10.1073/pnas.91.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Xu JC, Lytle C, Zhu TT, Payne JA, Benz E Jr, Forbush B 3rd.. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci USA 91: 2201–2205, 1994. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gillen CM, Brill S, Payne JA, Forbush B 3rd.. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem 271: 16237–16244, 1996. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- 131. Galanopoulou AS. GABA receptors as broadcasters of sexually differentiating signals in the brain. Epilepsia 46, Suppl 5: 107–112, 2005. doi: 10.1111/j.1528-1167.2005.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron 63: 657–672, 2009. doi: 10.1016/j.neuron.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Galanopoulou AS, Kyrozis A, Claudio OI, Stanton PK, Moshé SL. Sex-specific KCC2 expression and GABAA receptor function in rat substantia nigra. Exp Neurol 183: 628–637, 2003. doi: 10.1016/S0014-4886(03)00213-9. [DOI] [PubMed] [Google Scholar]
- 134. Roux S, Lohof A, Ben-Ari Y, Poulain B, Bossu JL. Maturation of GABAergic transmission in cerebellar Purkinje cells is sex dependent and altered in the valproate model of autism. Front Cell Neurosci 12: 232, 2018. doi: 10.3389/fncel.2018.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Horn Z, Ringstedt T, Blaesse P, Kaila K, Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci 31: 2142–2155, 2010. doi: 10.1111/j.1460-9568.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- 136. Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56: 1019–1033, 2007. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 137. Mavrovic M, Uvarov P, Delpire E, Vutskits L, Kaila K, Puskarjov M. Loss of non-canonical KCC2 functions promotes developmental apoptosis of cortical projection neurons. EMBO Rep 21: e48880, 2020. doi: 10.15252/embr.201948880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Winkelmann A, Semtner M, Meier JC. Chloride transporter KCC2-dependent neuroprotection depends on the N-terminal protein domain. Cell Death Dis 6: e1776, 2015. doi: 10.1038/cddis.2015.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Saito T, Ishii A, Sugai K, Sasaki M, Hirose S. A de novo missense mutation in SLC12A5 found in a compound heterozygote patient with epilepsy of infancy with migrating focal seizures. Clin Genet 92: 654–658, 2017. doi: 10.1111/cge.13049. [DOI] [PubMed] [Google Scholar]
- 140. Saitsu H, Watanabe M, Akita T, Ohba C, Sugai K, Ong WP, Shiraishi H, Yuasa S, Matsumoto H, Beng KT, Saitoh S, Miyatake S, Nakashima M, Miyake N, Kato M, Fukuda A, Matsumoto N. Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci Rep 6: 30072, 2016. doi: 10.1038/srep30072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Stödberg T, McTague A, Ruiz AJ, Hirata H, Zhen J, Long P, Farabella I, Meyer E, Kawahara A, Vassallo G, Stivaros SM, Bjursell MK, Stranneheim H, Tigerschiöld S, Persson B, Bangash I, Das K, Hughes D, Lesko N, Lundeberg J, Scott RC, Poduri A, Scheffer IE, Smith H, Gissen P, Schorge S, Reith ME, Topf M, Kullmann DM, Harvey RJ, Wedell A, Kurian MA. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat Commun 6: 8038, 2015. doi: 10.1038/ncomms9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 313: 1792–1795, 2006. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- 143. Velísková J, Moshé SL. Sexual dimorphism and developmental regulation of substantia nigra function. Ann Neurol 50: 596–601, 2001. doi: 10.1002/ana.1248. [DOI] [PubMed] [Google Scholar]
- 144. Velísková J, Moshé SL. Update on the role of substantia nigra pars reticulata in the regulation of seizures. Epilepsy Curr 6: 83–87, 2006. doi: 10.1111/j.1535-7511.2006.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Garant DS, Gale K. Substantia nigra-mediated anticonvulsant actions: role of nigral output pathways. Exp Neurol 97: 143–159, 1987. doi: 10.1016/0014-4886(87)90289-5. [DOI] [PubMed] [Google Scholar]
- 146. Iadarola MJ, Gale K. Substantia nigra: site of anticonvulsant activity mediated by gamma-aminobutyric acid. Science 218: 1237–1240, 1982. doi: 10.1126/science.7146907. [DOI] [PubMed] [Google Scholar]
- 147. Sperber EF, Wurpel JN, Zhao DY, Moshé SL. Evidence for the involvement of nigral GABAA receptors in seizures of adult rats. Brain Res 480: 378–382, 1989. doi: 10.1016/0006-8993(89)90211-4. [DOI] [PubMed] [Google Scholar]
- 148. Wurpel JN, Sperber EF, Moshé SL. Age-dependent differences in the anticonvulsant effects of 2-amino-7-phosphono-heptanoic acid or ketamine infusions into the substantia nigra of rats. Epilepsia 33: 439–443, 1992. doi: 10.1111/j.1528-1157.1992.tb01688.x. [DOI] [PubMed] [Google Scholar]
- 149. Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, Tenkova T, Dikranian K, Olney JW. Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol 62: 401–405, 2001. doi: 10.1016/S0006-2952(01)00696-7. [DOI] [PubMed] [Google Scholar]
- 150. Giorgi FS, Velísková J, Chudomel O, Kyrozis A, Moshé SL. The role of substantia nigra pars reticulata in modulating clonic seizures is determined by testosterone levels during the immediate postnatal period. Neurobiol Dis 25: 73–79, 2007. doi: 10.1016/j.nbd.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 8: 51–60, 2005. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- 152. Olivetti PR, Maheshwari A, Noebels JL. Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Sci Transl Med 6: 220ra12, 2014. doi: 10.1126/scitranslmed.3007231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Chiu C, Reid CA, Tan HO, Davies PJ, Single FN, Koukoulas I, Berkovic SF, Tan SS, Sprengel R, Jones MV, Petrou S. Developmental impact of a familial GABAA receptor epilepsy mutation. Ann Neurol 64: 284–293, 2008. doi: 10.1002/ana.21440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Baumbach HD, Chow KL. Visuocortical epileptiform discharges in rabbits: differential effects on neuronal development in the lateral geniculate nucleus and superior colliculus. Brain Res 209: 61–76, 1981. doi: 10.1016/0006-8993(81)91172-0. [DOI] [PubMed] [Google Scholar]
- 155. Shatskikh TN, Raghavendra M, Zhao Q, Cui Z, Holmes GL. Electrical induction of spikes in the hippocampus impairs recognition capacity and spatial memory in rats. Epilepsy Behav 9: 549–556, 2006. doi: 10.1016/j.yebeh.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 156. Zhou JL, Lenck-Santini PP, Zhao Q, Holmes GL. Effect of interictal spikes on single-cell firing patterns in the hippocampus. Epilepsia 48: 720–731, 2007. doi: 10.1111/j.1528-1167.2006.00972.x. [DOI] [PubMed] [Google Scholar]
- 157. Akman O, Briggs SW, Mowrey WB, Moshé SL, Galanopoulou AS. Antiepileptogenic effects of rapamycin in a model of infantile spasms due to structural lesions. Epilepsia 62: 1985–1999, 2021. doi: 10.1111/epi.16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Briggs SW, Mowrey W, Hall CB, Galanopoulou AS. CPP-115, a vigabatrin analogue, decreases spasms in the multiple-hit rat model of infantile spasms. Epilepsia 55: 94–102, 2014. doi: 10.1111/epi.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Galanopoulou AS, Mowrey WB, Liu W, Li Q, Shandra O, Moshé SL. Preclinical screening for treatments for infantile spasms in the multiple hit rat model of infantile spasms: an update. Neurochem Res 42: 1949–1961, 2017. doi: 10.1007/s11064-017-2282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Jequier Gygax M, Klein BD, White HS, Kim M, Galanopoulou AS. Efficacy and tolerability of the galanin analog NAX 5055 in the multiple-hit rat model of symptomatic infantile spasms. Epilepsy Res 108: 98–108, 2014. doi: 10.1016/j.eplepsyres.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Katsarou AM, Li Q, Liu W, Moshé SL, Galanopoulou AS. Acquired parvalbumin-selective interneuronopathy in the multiple-hit model of infantile spasms: a putative basis for the partial responsiveness to vigabatrin analogs? Epilepsia Open 3: 155–164, 2018. doi: 10.1002/epi4.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ono T, Moshé SL, Galanopoulou AS. Carisbamate acutely suppresses spasms in a rat model of symptomatic infantile spasms. Epilepsia 52: 1678–1684, 2011. doi: 10.1111/j.1528-1167.2011.03173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Raffo E, Coppola A, Ono T, Briggs SW, Galanopoulou AS. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis 43: 322–329, 2011. doi: 10.1016/j.nbd.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, Christian SL, Mancini G, Labosky P, Dobyns W, Brooks-Kayal A, Golden JA. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 132: 1563–1576, 2009. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Siehr MS, Massey CA, Noebels JL. Arx expansion mutation perturbs cortical development by augmenting apoptosis without activating innate immunity in a mouse model of X-linked infantile spasms syndrome. Dis Model Mech 13: dmm042515, 2020. doi: 10.1242/dmm.042515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Baram TZ. Pathophysiology of massive infantile spasms: perspective on the putative role of the brain adrenal axis. Ann Neurol 33: 231–236, 1993. doi: 10.1002/ana.410330302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Brunson KL, Eghbal-Ahmadi M, Baram TZ. How do the many etiologies of West syndrome lead to excitability and seizures? The corticotropin releasing hormone excess hypothesis. Brain Dev 23: 533–538, 2001. doi: 10.1016/S0387-7604(01)00312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Baram TZ, Schultz L. Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Brain Res Dev Brain Res 61: 97–101, 1991. doi: 10.1016/0165-3806(91)90118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Shi XY, Yang XF, Tomonoh Y, Hu LY, Ju J, Hirose S, Zou LP. Development of a mouse model of infantile spasms induced by N-methyl-D-aspartate. Epilepsy Res 118: 29–33, 2015. doi: 10.1016/j.eplepsyres.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 170. Velísek L, Chachua T, Yum MS, Poon KL, Velísková J. Model of cryptogenic infantile spasms after prenatal corticosteroid priming. Epilepsia 51, Suppl 3: 145–149, 2010. doi: 10.1111/j.1528-1167.2010.02630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Yum MS, Chachua T, Velíšková J, Velíšek L. Prenatal stress promotes development of spasms in infant rats. Epilepsia 53: e46–e49, 2012. doi: 10.1111/j.1528-1167.2011.03357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wang YJ, Zhang Y, Liang XH, Yang G, Zou LP. Effects of adrenal dysfunction and high-dose adrenocorticotropic hormone on NMDA-induced spasm seizures in young Wistar rats. Epilepsy Res 100: 125–131, 2012. doi: 10.1016/j.eplepsyres.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 173. Dubé CM, Molet J, Singh-Taylor A, Ivy A, Maras PM, Baram TZ. Hyper-excitability and epilepsy generated by chronic early-life stress. Neurobiol Stress 2: 10–19, 2015. doi: 10.1016/j.ynstr.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol Rev 70: 142–173, 2018. doi: 10.1124/pr.117.014456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mantegazza M, Rusconi R, Scalmani P, Avanzini G, Franceschetti S. Epileptogenic ion channel mutations: from bedside to bench and, hopefully, back again. Epilepsy Res 92: 1–29, 2010. doi: 10.1016/j.eplepsyres.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 176. Guerrini R, Marini C, Mantegazza M. Genetic epilepsy syndromes without structural brain abnormalities: clinical features and experimental models. Neurotherapeutics 11: 269–285, 2014. doi: 10.1007/s13311-014-0267-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Yum MS, Lee M, Woo DC, Kim DW, Ko TS, Velíšek L. β-Hydroxybutyrate attenuates NMDA-induced spasms in rats with evidence of neuronal stabilization on MR spectroscopy. Epilepsy Res 117: 125–132, 2015. doi: 10.1016/j.eplepsyres.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 178. Kábová R, Liptáková S, Slamberová R, Pometlová M, Velísek L. Age-specific N-methyl-D-aspartate-induced seizures: perspectives for the West syndrome model. Epilepsia 40: 1357–1369, 1999. doi: 10.1111/j.1528-1157.1999.tb02006.x. [DOI] [PubMed] [Google Scholar]
- 179. Mares P, Velísek L. N-methyl-D-aspartate (NMDA)-induced seizures in developing rats. Brain Res Dev Brain Res 65: 185–189, 1992. doi: 10.1016/0165-3806(92)90178-Y. [DOI] [PubMed] [Google Scholar]
- 180. Stafstrom CE, Sasaki-Adams DM. NMDA-induced seizures in developing rats cause long-term learning impairment and increased seizure susceptibility. Epilepsy Res 53: 129–137, 2003. doi: 10.1016/S0920-1211(02)00258-9. [DOI] [PubMed] [Google Scholar]
- 181. Velísek L, Jehle K, Asche S, Velísková J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol 61: 109–119, 2007. doi: 10.1002/ana.21082. [DOI] [PubMed] [Google Scholar]
- 182. Chachua T, Yum MS, Velíšková J, Velíšek L. Validation of the rat model of cryptogenic infantile spasms. Epilepsia 52: 1666–1677, 2011. doi: 10.1111/j.1528-1167.2011.03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chern CR, Chern CJ, Velíšková J, Velíšek L. AQB-565 shows promise in preclinical testing in the model of epileptic spasms during infancy: head-to-head comparison with ACTH. Epilepsy Res 152: 31–34, 2019. doi: 10.1016/j.eplepsyres.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Chachua T, Di Grazia P, Chern CR, Johnkutty M, Hellman B, Lau HA, Shakil F, Daniel M, Goletiani C, Velíšková J, Velíšek L. Estradiol does not affect spasms in the betamethasone-NMDA rat model of infantile spasms. Epilepsia 57: 1326–1336, 2016. doi: 10.1111/epi.13434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kim EH, Yum MS, Lee M, Kim EJ, Shim WH, Ko TS. A new rat model of epileptic spasms based on methylazoxymethanol-induced malformations of cortical development. Front Neurol 8: 271, 2017. doi: 10.3389/fneur.2017.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Rensing N, Johnson KJ, Foutz TJ, Friedman JL, Galindo R, Wong M. Early developmental electroencephalography abnormalities, neonatal seizures, and induced spasms in a mouse model of tuberous sclerosis complex. Epilepsia 61: 879–891, 2020. doi: 10.1111/epi.16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Blichowski M, Shephard A, Armstrong J, Shen L, Cortez MA, Eubanks JH, Snead OC 3rd.. The GIRK2 subunit is involved in IS-like seizures induced by GABA(B) receptor agonists. Epilepsia 56: 1081–1087, 2015. doi: 10.1111/epi.13034. [DOI] [PubMed] [Google Scholar]
- 188. Cortez MA, Shen L, Wu Y, Aleem IS, Trepanier CH, Sadeghnia HR, Ashraf A, Kanawaty A, Liu CC, Stewart L, Snead OC 3rd.. Infantile spasms and Down syndrome: a new animal model. Pediatr Res 65: 499–503, 2009. doi: 10.1203/PDR.0b013e31819d9076. [DOI] [PubMed] [Google Scholar]
- 189. Joshi K, Shen L, Michaeli A, Salter M, Thibault-Messier G, Hashmi S, Eubanks JH, Cortez MA, Snead OC. Infantile spasms in Down syndrome: rescue by knockdown of the GIRK2 channel. Ann Neurol 80: 511–521, 2016. doi: 10.1002/ana.24749. [DOI] [PubMed] [Google Scholar]
- 190. Le JT, Frost JD Jr, Swann JW. Acthar®Gel (repository corticotropin injection) dose-response relationships in an animal model of epileptic spasms. Epilepsy Behav 116: 107786, 2021. doi: 10.1016/j.yebeh.2021.107786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Frost JD Jr, Le JT, Lee CL, Ballester-Rosado C, Hrachovy RA, Swann JW. Vigabatrin therapy implicates neocortical high frequency oscillations in an animal model of infantile spasms. Neurobiol Dis 82: 1–11, 2015. doi: 10.1016/j.nbd.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Frost JD Jr, Lee CL, Hrachovy RA, Swann JW. High frequency EEG activity associated with ictal events in an animal model of infantile spasms. Epilepsia 52: 53–62, 2011. doi: 10.1111/j.1528-1167.2010.02887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Frost JD Jr, Lee CL, Le JT, Hrachovy RA, Swann JW. Interictal high frequency oscillations in an animal model of infantile spasms. Neurobiol Dis 46: 377–388, 2012. doi: 10.1016/j.nbd.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Price MG, Yoo JW, Burgess DL, Deng F, Hrachovy RA, Frost JD Jr, Noebels JL. A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10+7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci 29: 8752–8763, 2009. doi: 10.1523/JNEUROSCI.0915-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Jackson MR, Lee K, Mattiske T, Jaehne EJ, Ozturk E, Baune BT, O’Brien TJ, Jones N, Shoubridge C. Extensive phenotyping of two ARX polyalanine expansion mutation mouse models that span clinical spectrum of intellectual disability and epilepsy. Neurobiol Dis 105: 245–256, 2017. doi: 10.1016/j.nbd.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 196. Pirone A, Alexander J, Lau LA, Hampton D, Zayachkivsky A, Yee A, Yee A, Jacob MH, Dulla CG. APC conditional knock-out mouse is a model of infantile spasms with elevated neuronal β-catenin levels, neonatal spasms, and chronic seizures. Neurobiol Dis 98: 149–157, 2017. doi: 10.1016/j.nbd.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Gataullina S, Lemaire E, Wendling F, Kaminska A, Watrin F, Riquet A, Ville D, Moutard ML, de Saint Martin A, Napuri S, Pedespan JM, Eisermann M, Bahi-Buisson N, Nabbout R, Chiron C, Dulac O, Huberfeld G. Epilepsy in young Tsc1+/- mice exhibits age-dependent expression that mimics that of human tuberous sclerosis complex. Epilepsia 57: 648–659, 2016. doi: 10.1111/epi.13325. [DOI] [PubMed] [Google Scholar]
- 198. Mulcahey PJ, Tang S, Takano H, White A, Davila Portillo DR, Kane OM, Marsh ED, Zhou Z, Coulter DA. Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp Neurol 332: 113388, 2020. doi: 10.1016/j.expneurol.2020.113388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Chan KF, Burnham WM, Jia Z, Cortez MA, Snead OC 3rd.. GABAB receptor antagonism abolishes the learning impairments in rats with chronic atypical absence seizures. Eur J Pharmacol 541: 64–72, 2006. doi: 10.1016/j.ejphar.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 200. Chan KF, Jia Z, Murphy PA, Burnham WM, Cortez MA, Snead OC 3rd.. Learning and memory impairment in rats with chronic atypical absence seizures. Exp Neurol 190: 328–336, 2004. doi: 10.1016/j.expneurol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 201. Han HA, Cortez MA, Snead OC. GABAB receptor and absence epilepsy (Online). doi: 10.1093/med/9780199746545.003.0019. [DOI] [PubMed]
- 202. Li H, Kraus A, Wu J, Huguenard JR, Fisher RS. Selective changes in thalamic and cortical GABAA receptor subunits in a model of acquired absence epilepsy in the rat. Neuropharmacology 51: 121–128, 2006. doi: 10.1016/j.neuropharm.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 203. Smith KA, Bierkamper GG. Paradoxical role of GABA in a chronic model of petit mal (absence)-like epilepsy in the rat. Eur J Pharmacol 176: 45–55, 1990. doi: 10.1016/0014-2999(90)90130-X. [DOI] [PubMed] [Google Scholar]
- 204. Smith KA, Fisher RS. The selective GABAB antagonist CGP-35348 blocks spike-wave bursts in the cholesterol synthesis rat absence epilepsy model. Brain Res 729: 147–150, 1996. doi: 10.1016/S0006-8993(96)00174-6. [DOI] [PubMed] [Google Scholar]
- 205. Stewart LS, Cortez MA, Snead OC 3rd.. Environmental enrichment improves behavioral outcome in the AY-9944 model of childhood atypical absence epilepsy. Int J Neurosci 122: 449–457, 2012. doi: 10.3109/00207454.2012.677881. [DOI] [PubMed] [Google Scholar]
- 206. Serbanescu I, Cortez MA, McKerlie C, Snead OC 3rd.. Refractory atypical absence seizures in rat: a two hit model. Epilepsy Res 62: 53–63, 2004. doi: 10.1016/j.eplepsyres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 207. Asinof S, Mahaffey C, Beyer B, Frankel WN, Boumil R. Dynamin 1 isoform roles in a mouse model of severe childhood epileptic encephalopathy. Neurobiol Dis 95: 1–11, 2016. doi: 10.1016/j.nbd.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wu Y, Chan KF, Eubanks JH, Wong CG, Cortez MA, Shen L, Liu CC, Velazquez JP, Wang YT, Jia Z, Snead OC 3rd.. Transgenic mice over-expressing GABA(B)R1a receptors acquire an atypical absence epilepsy-like phenotype. Neurobiol Dis 26: 439–451, 2007. doi: 10.1016/j.nbd.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 209. Stewart LS, Wu Y, Eubanks JH, Han H, Leschenko Y, Perez Velazquez JL, Cortez MA, Snead OC 3rd.. Severity of atypical absence phenotype in GABAB transgenic mice is subunit specific. Epilepsy Behav 14: 577–581, 2009. doi: 10.1016/j.yebeh.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 210. Cox GA, Lutz CM, Yang CL, Biemesderfer D, Bronson RT, Fu A, Aronson PS, Noebels JL, Frankel WN. Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 91: 139–148, 1997. doi: 10.1016/S0092-8674(01)80016-7. [DOI] [PubMed] [Google Scholar]
- 211. Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51: 1650–1658, 2010. doi: 10.1111/j.1528-1167.2010.02640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA 109: 14646–14651, 2012. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489: 385–390, 2012. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ito S, Ogiwara I, Yamada K, Miyamoto H, Hensch TK, Osawa M, Yamakawa K. Mouse with Nav1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis 49: 29–40, 2013. doi: 10.1016/j.nbd.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 215. Schutte RJ, Schutte SS, Algara J, Barragan EV, Gilligan J, Staber C, Savva YA, Smith MA, Reenan R, O’Dowd DK. Knock-in model of Dravet syndrome reveals a constitutive and conditional reduction in sodium current. J Neurophysiol 112: 903–912, 2014. doi: 10.1152/jn.00135.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun 4: 2410, 2013. doi: 10.1038/ncomms3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Li J, Nelis M, Sourbron J, Copmans D, Lagae L, Cabooter D, de Witte PA. Efficacy of fenfluramine and norfenfluramine enantiomers and various antiepileptic drugs in a zebrafish model of Dravet syndrome. Neurochem Res 46: 2249–2261, 2021. doi: 10.1007/s11064-021-03358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Pernici CD, Mensah JA, Dahle EJ, Johnson KJ, Handy L, Buxton L, Smith MD, West PJ, Metcalf CS, Wilcox KS. Development of an antiseizure drug screening platform for Dravet syndrome at the NINDS contract site for the Epilepsy Therapy Screening Program. Epilepsia 62: 1665–1676, 2021. doi: 10.1111/epi.16925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Salgueiro-Pereira AR, Duprat F, Pousinha PA, Loucif A, Douchamps V, Regondi C, Ayrault M, Eugie M, Stunault MI, Escayg A, Goutagny R, Gnatkovsky V, Frassoni C, Marie H, Bethus I, Mantegazza M. A two-hit story: seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol Dis 125: 31–44, 2019. doi: 10.1016/j.nbd.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 220. Ihara Y, Tomonoh Y, Deshimaru M, Zhang B, Uchida T, Ishii A, Hirose S. Retigabine, a Kv7.2/Kv7.3-channel opener, attenuates drug-induced seizures in knock-in mice harboring Kcnq2 mutations. PLoS One 11: e0150095, 2016. doi: 10.1371/journal.pone.0150095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Milh M, Roubertoux P, Biba N, Chavany J, Spiga Ghata A, Fulachier C, Collins SC, Wagner C, Roux JC, Yalcin B, Félix MS, Molinari F, Lenck-Santini PP, Villard L. A knock‐in mouse model for KCNQ2‐related epileptic encephalopathy displays spontaneous generalized seizures and cognitive impairment. Epilepsia 61: 868–878, 2020. doi: 10.1111/epi.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Vanhoof-Villalba SL, Gautier NM, Mishra V, Glasscock E. Pharmacogenetics of KCNQ channel activation in 2 potassium channelopathy mouse models of epilepsy. Epilepsia 59: 358–368, 2018. doi: 10.1111/epi.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Rakotomamonjy J, Sabetfakhri NP, McDermott SL, Guemez-Gamboa A. Characterization of seizure susceptibility in Pcdh19 mice. Epilepsia 61: 2313–2320, 2020. doi: 10.1111/epi.16675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Hoshina N, Johnson-Venkatesh EM, Hoshina M, Umemori H. Female-specific synaptic dysfunction and cognitive impairment in a mouse model of PCDH19 disorder. Science 372: eaaz3893, 2021. doi: 10.1126/science.aaz3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Trovò L, Fuchs C, De Rosa R, Barbiero I, Tramarin M, Ciani E, Rusconi L, Kilstrup-Nielsen C. The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo. Neurobiol Dis 138: 104791, 2020. doi: 10.1016/j.nbd.2020.104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang HT, Zhu ZA, Li YY, Lou SS, Yang G, Feng X, Xu W, Huang ZL, Cheng X, Xiong ZQ. CDKL5 deficiency in forebrain glutamatergic neurons results in recurrent spontaneous seizures. Epilepsia 62: 517–528, 2021. doi: 10.1111/epi.16805. [DOI] [PubMed] [Google Scholar]
- 227. Gawel K, Langlois M, Martins T, van der Ent W, Tiraboschi E, Jacmin M, Crawford AD, Esguerra CV. Seizing the moment: zebrafish epilepsy models. Neurosci Biobehav Rev 116: 1–20, 2020. doi: 10.1016/j.neubiorev.2020.06.010. [DOI] [PubMed] [Google Scholar]
- 228. Shcheglovitov A, Peterson RT. Screening platforms for genetic epilepsies—zebrafish, iPSC-derived neurons, and organoids. Neurotherapeutics 18: 1478–1489, 2021. doi: 10.1007/s13311-021-01115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Xie Y, Ng NN, Safrina OS, Ramos CM, Ess KC, Schwartz PH, Smith MA, O’Dowd DK. Comparisons of dual isogenic human iPSC pairs identify functional alterations directly caused by an epilepsy associated SCN1A mutation. Neurobiol Dis 134: 104627, 2020. doi: 10.1016/j.nbd.2019.104627. [DOI] [PubMed] [Google Scholar]
- 230. Kearney J. The more, the better: modeling Dravet syndrome with induced pluripotent stem cell-derived neurons. Epilepsy Curr 14: 33–34, 2014. doi: 10.5698/1535-7597-14.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O’Malley HA, Patino GA, O’Brien JE, Rusconi R, Gupta A, Thompson RC, Natowicz MR, Meisler MH, Isom LL, Parent JM. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol 74: 128–139, 2013. doi: 10.1002/ana.23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. McTague A, Rossignoli G, Ferrini A, Barral S, Kurian MA. Genome editing in iPSC-based neural systems: from disease models to future therapeutic strategies. Front Genome Ed 3: 630600, 2021. doi: 10.3389/fgeed.2021.630600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Kimura Y, Tanaka Y, Shirasu N, Yasunaga S, Higurashi N, Hirose S. Establishment of human induced pluripotent stem cells derived from skin cells of a patient with Dravet syndrome. Stem Cell Res 47: 101857, 2020. doi: 10.1016/j.scr.2020.101857. [DOI] [PubMed] [Google Scholar]
- 234. Tanaka Y, Higurashi N, Shirasu N, Yasunaga S, Moreira KM, Okano H, Hirose S. Establishment of a human induced stem cell line (FUi002-A) from Dravet syndrome patient carrying heterozygous R1525X mutation in SCN1A gene. Stem Cell Res 31: 11–15, 2018. doi: 10.1016/j.scr.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 235. Sun Y, Paşca SP, Portmann T, Goold C, Worringer KA, Guan W, Chan KC, Gai H, Vogt D, Chen YJ, Mao R, Chan K, Rubenstein JL, Madison DV, Hallmayer J, Froehlich-Santino WM, Bernstein JA, Dolmetsch RE. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet syndrome patients. Elife 5: e13073, 2016. doi: 10.7554/eLife.13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Freel BA, Sheets JN, Francis KR. iPSC modeling of rare pediatric disorders. J Neurosci Methods 332: 108533, 2020. doi: 10.1016/j.jneumeth.2019.108533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol 13: 710–726, 2014. doi: 10.1016/S1474-4422(14)70040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135: 1348–1369, 2012. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, Ben Jeema L, Zemni R, Vinet MC, Francis F, Couvert P, Gomot M, Moraine C, van Bokhoven H, Kalscheuer V, Frints S, Gecz J, Ohzaki K, Chaabouni H, Fryns JP, Desportes V, Beldjord C, Chelly J. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 11: 981–991, 2002. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- 240. Poirier K, Van Esch H, Friocourt G, Saillour Y, Bahi N, Backer S, Souil E, Castelnau-Ptakhine L, Beldjord C, Francis F, Bienvenu T, Chelly J. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Brain Res Mol Brain Res 122: 35–46, 2004. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 241. Kato M, Dobyns WB. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol 20: 392–397, 2005. doi: 10.1177/08830738050200042001. [DOI] [PubMed] [Google Scholar]
- 242. Mirzaa GM, Campbell CD, Solovieff N, Goold C, Jansen LA, Menon S, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 73: 836–845, 2016. doi: 10.1001/jamaneurol.2016.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Holmes GL, Stafstrom CE; Tuberous Sclerosis Study Group. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia 48: 617–630, 2007. doi: 10.1111/j.1528-1167.2007.01035.x. [DOI] [PubMed] [Google Scholar]
- 244. Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia 49: 8–21, 2008. doi: 10.1111/j.1528-1167.2007.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Cepeda C, Hurst RS, Flores-Hernández J, Hernández-Echeagaray E, Klapstein GJ, Boylan MK, Calvert CR, Jocoy EL, Nguyen OK, André VM, Vinters HV, Ariano MA, Levine MS, Mathern GW. Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia: abnormal cells in human cortical dysplasia. J Neurosci Res 72: 472–486, 2003. doi: 10.1002/jnr.10604. [DOI] [PubMed] [Google Scholar]
- 246. Williams MR, DeSpenza T Jr, Li M, Gulledge AT, Luikart BW. Hyperactivity of newborn pten knock-out neurons results from increased excitatory synaptic drive. J Neurosci 35: 943–959, 2015. doi: 10.1523/JNEUROSCI.3144-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. D’Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, Najm I, Ying Z, Yang E, Barkovich AJ, Kwiatkowski DJ, Vinters HV, Madsen JR, Mathern GW, Blümcke I, Poduri A, Walsh CA. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep 21: 3754–3766, 2017. doi: 10.1016/j.celrep.2017.11.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Avoli M, Bernasconi A, Mattia D, Olivier A, Hwa GG. Epileptiform discharges in the human dysplastic neocortex: in vitro physiology and pharmacology. Ann Neurol 46: 816–826, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 249. Cepeda C, André VM, Levine MS, Salamon N, Miyata H, Vinters HV, Mathern GW. Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav 9: 219–235, 2006. doi: 10.1016/j.yebeh.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 250. Talos DM, Kwiatkowski DJ, Cordero K, Black PM, Jensen FE. Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol 63: 454–465, 2008. doi: 10.1002/ana.21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Mantegazza M, Cestèle S, Catterall WA. Sodium channelopathies of skeletal muscle and brain. Physiol Rev 101: 1633–1689, 2021. doi: 10.1152/physrev.00025.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr 7: 15–22, 2007. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004: re15, 2004. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- 254. Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol 9: 413–424, 2010. doi: 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- 255. Meisler MH, Hill SF, Yu W. Sodium channelopathies in neurodevelopmental disorders. Nat Rev Neurosci 22: 152–166, 2021. doi: 10.1038/s41583-020-00418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Bouza AA, Isom LL. Voltage-gated sodium channel β subunits and their related diseases. In: Voltage-Gated Sodium Channels: Structure, Function and Channelopathies, edited by Chahine M. Cham, Switzerland: Springer International Publishing, 2018, p. 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Brunklaus A, Lal D. Sodium channel epilepsies and neurodevelopmental disorders: from disease mechanisms to clinical application. Dev Med Child Neurol 62: 784–792, 2020. doi: 10.1111/dmcn.14519. [DOI] [PubMed] [Google Scholar]
- 258. Rochtus AM, Goldstein RD, Holm IA, Brownstein CA, Pérez‐Palma E, Haynes R, Lal D, Poduri AH. The role of sodium channels in sudden unexpected death in pediatrics. Mol Genet Genomic Med 8: e1309, 2020. doi: 10.1002/mgg3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Siekierska A, Isrie M, Liu Y, Scheldeman C, Vanthillo N, Lagae L, de Witte PA, Van Esch H, Goldfarb M, Buyse GM. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology 86: 2162–2170, 2016. doi: 10.1212/WNL.0000000000002752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Oda Y, Uchiyama Y, Motomura A, Fujita A, Azuma Y, Harita Y, Mizuguchi T, Yanagi K, Ogata H, Hata K, Kaname T, Matsubara Y, Wakui K, Matsumoto N. Entire FGF12 duplication by complex chromosomal rearrangements associated with West syndrome. J Hum Genet 64: 1005–1014, 2019. doi: 10.1038/s10038-019-0641-1. [DOI] [PubMed] [Google Scholar]
- 261. Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia 60: S2–S7, 2019. doi: 10.1111/epi.16054. [DOI] [PubMed] [Google Scholar]
- 262. Scheffer IE, Nabbout R. SCN1A‐related phenotypes: epilepsy and beyond. Epilepsia 60: S17–S24, 2019. doi: 10.1111/epi.16386. [DOI] [PubMed] [Google Scholar]
- 263. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68: 1327–1332, 2001. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Dravet C. Dravet syndrome history. Dev Med Child Neurol 53: 1–6, 2011. doi: 10.1111/j.1469-8749.2011.03964.x. [DOI] [PubMed] [Google Scholar]
- 265. Depienne C, Arzimanoglou A, Trouillard O, Fedirko E, Baulac S, Saint-Martin C, Ruberg M, Dravet C, Nabbout R, Baulac M, Gourfinkel-An I, LeGuern E. Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy. Hum Mutat 27: 389, 2006. doi: 10.1002/humu.9419. [DOI] [PubMed] [Google Scholar]
- 266. Sadleir LG, Mountier EI, Gill D, Davis S, Joshi C, DeVile C, Kurian MA, Mandelstam S, Wirrell E, Nickels KC, Murali HR, Carvill G, Myers CT, Mefford HC, Scheffer IE; DDD Study. Not all SCN1A epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology 89: 1035–1042, 2017. doi: 10.1212/WNL.0000000000004331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Gorman KM, Peters CH, Lynch B, Jones L, Bassett DS, King MD, Ruben PC, Rosch RE. Persistent sodium currents in SCN1A developmental and degenerative epileptic dyskinetic encephalopathy. Brain Commun 3: fcab235, 2021. doi: 10.1093/braincomms/fcab235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, Petrou S. SCN1A gain of function in early infantile encephalopathy. Ann Neurol 85: 514–525, 2019. doi: 10.1002/ana.25438. [DOI] [PubMed] [Google Scholar]
- 269. Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome NaV1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 53: 87–100, 2012. doi: 10.1111/j.1528-1167.2011.03346.x. [DOI] [PubMed] [Google Scholar]
- 270. Mantegazza M, Broccoli V. SCN1A/NaV1.1 channelopathies: mechanisms in expression systems, animal models, and human ipsc models. Epilepsia 60: S25–S38, 2019. doi: 10.1111/epi.14700. [DOI] [PubMed] [Google Scholar]
- 271. Terragni B, Scalmani P, Franceschetti S, Cestèle S, Mantegazza M. Post-translational dysfunctions in channelopathies of the nervous system. Neuropharmacology 132: 31–42, 2018. doi: 10.1016/j.neuropharm.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 272. Bechi G, Rusconi R, Cestèle S, Striano P, Franceschetti S, Mantegazza M. Rescuable folding defective NaV1.1 (SCN1A) mutants in epilepsy: properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol Dis 75: 100–114, 2015. doi: 10.1016/j.nbd.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 273. Rusconi R, Combi R, Cestèle S, Grioni D, Franceschetti S, Dalprà L, Mantegazza M. A rescuable folding defective Nav1.1 ( SCN1A ) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum Mutat 30: E747–E760, 2009. doi: 10.1002/humu.21041. [DOI] [PubMed] [Google Scholar]
- 274. Rusconi R, Scalmani P, Cassulini RR, Giunti G, Gambardella A, Franceschetti S, Annesi G, Wanke E, Mantegazza M. Modulatory Proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J Neurosci 27: 11037–11046, 2007. doi: 10.1523/JNEUROSCI.3515-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Thompson CH, Porter JC, Kahlig KM, Daniels MA, George AL. Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J Biol Chem 287: 42001–42008, 2012. doi: 10.1074/jbc.M112.421883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Martin MS, Dutt K, Papale LA, Dubé CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, Goldin AL, Escayg A. Altered function of the SCN1A voltage-gated sodium channel leads to γ-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem 285: 9823–9834, 2010. doi: 10.1074/jbc.M109.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Hedrich UB, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihné M, Beck H, Mantegazza M, Lerche H. Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human NaV1.1 mutation. J Neurosci 34: 14874–14889, 2014. doi: 10.1523/JNEUROSCI.0721-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Chiron C, Marchand MC, Tran A, Rey E, d’Athis P, Vincent J, Dulac O, Pons G. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. Lancet 356: 1638–1642, 2000. doi: 10.1016/S0140-6736(00)03157-3. [DOI] [PubMed] [Google Scholar]
- 279. Ceulemans B, Schoonjans AS, Marchau F, Paelinck BP, Lagae L. Five‐year extended follow‐up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia 57: e129–e134, 2016. doi: 10.1111/epi.13407. [DOI] [PubMed] [Google Scholar]
- 280. Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, Scheffer IE, Thiele EA, Wright S; Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med 376: 2011–2020, 2017. doi: 10.1056/NEJMoa1611618. [DOI] [PubMed] [Google Scholar]
- 281. Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, Valassina N, Bido S, Ricci R, Castoldi V, Marenna S, Church T, Massimino L, Morabito G, Benfenati F, Schorge S, Leocani L, Kullmann DM, Broccoli V. dCas9-based Scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol Ther 28: 235–253, 2020. doi: 10.1016/j.ymthe.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, Liu C, Leiser SC, Meena, Aznarez I, Liau G, Isom LL. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med 12: eaaz6100, 2020. doi: 10.1126/scitranslmed.aaz6100. [DOI] [PubMed] [Google Scholar]
- 283. Jansen NA, Dehghani A, Linssen MML, Breukel C, Tolner EA, Maagdenberg AM. First FHM3 mouse model shows spontaneous cortical spreading depolarizations. Ann Clin Transl Neurol 7: 132–138, 2020. doi: 10.1002/acn3.50971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Auffenberg E, Hedrich UB, Barbieri R, Miely D, Groschup B, Wuttke TV, Vogel N, Lührs P, Zanardi I, Bertelli S, Spielmann N, Gailus-Durner V, Fuchs H, Hrabě de Angelis M, Pusch M, Dichgans M, Lerche H, Gavazzo P, Plesnila N, Freilinger T. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J Clin Invest 131: e142202, 2021. doi: 10.1172/JCI142202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Chever O, Zerimech S, Scalmani P, Lemaire L, Pizzamiglio L, Loucif A, Ayrault M, Krupa M, Desroches M, Duprat F, Léna I, Cestèle S, Mantegazza M. Initiation of migraine-related cortical spreading depolarization by hyperactivity of GABAergic neurons and NaV1.1 channels. J Clin Invest 131: e142203, 2021. doi: 10.1172/JCI142203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, Shevell M, Mercho S, Seni MH, Guiot MC, Mulley JC, Berkovic SF, Scheffer IE. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 360: 851–852, 2002. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- 287. Wolff M, Brunklaus A, Zuberi SM. Phenotypic spectrum and genetics of SCN2A‐related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 60: S59–S67, 2019. doi: 10.1111/epi.14935. [DOI] [PubMed] [Google Scholar]
- 288. Wolff M, Johannesen KM, Hedrich UB, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140: 1316–1336, 2017. doi: 10.1093/brain/awx054. [DOI] [PubMed] [Google Scholar]
- 289. Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, Bernier RA, Catterall WA, Chung WK, Empfield JR, George AL Jr, Hipp JF, Khwaja O, Kiskinis E, Lal D, Malhotra D, Millichap JJ, Otis TS, Petrou S, Pitt G, Schust LF, Taylor CM, Tjernagel J, Spiro JE, Bender KJ. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci 41: 442–456, 2018. doi: 10.1016/j.tins.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Spratt PW, Ben-Shalom R, Keeshen CM, Burke KJ Jr, Clarkson RL, Sanders SJ, Bender KJ. The autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron 103: 673–685.e5, 2019. doi: 10.1016/j.neuron.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, Atapour N, Miura E, Mazaki E, Ernst SJ, Cao D, Ohtani H, Itohara S, Yanagawa Y, Montal M, Yuzaki M, Inoue Y, Hensch TK, Noebels JL, Yamakawa K. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol 1: 96, 2018. doi: 10.1038/s42003-018-0099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Wang HG, Bavley CC, Li A, Jones RM, Hackett JE, Bayleyen Y, Lee FS, Rajadhyaksha AM, Pitt GS. Scn2a severe hypomorphic mutation decreases excitatory synaptic input and causes autism-associated behaviors. JCI Insight 6: e150698, 2021. doi: 10.1172/jci.insight.150698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Spratt PW, Alexander RP, Ben-Shalom R, Sahagun A, Kyoung H, Keeshen CM, Sanders SJ, Bender KJ. Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep 36: 109483, 2021. doi: 10.1016/j.celrep.2021.109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Zhang J, Chen X, Eaton M, Wu J, Ma Z, Lai S, Park A, Ahmad TS, Que Z, Lee JH, Xiao T, Li Y, Wang Y, Olivero-Acosta MI, Schaber JA, Jayant K, Yuan C, Huang Z, Lanman NA, Skarnes WC, Yang Y. Severe deficiency of the voltage-gated sodium channel NaV1.2 elevates neuronal excitability in adult mice. Cell Rep 36: 109495, 2021. doi: 10.1016/j.celrep.2021.109495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Li M, Jancovski N, Jafar-Nejad P, Burbano LE, Rollo B, Richards K, Drew L, Sedo A, Heighway J, Pachernegg S, Soriano A, Jia L, Blackburn T, Roberts B, Nemiroff A, Dalby K, Maljevic S, Reid CA, Rigo F, Petrou S. Antisense oligonucleotide therapy reduces seizures and extends life span in an SCN2A gain-of-function epilepsy model. J Clin Invest 131: e152079, 2021. doi: 10.1172/JCI152079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Vanoye CG, Gurnett CA, Holland KD, George AL Jr, Kearney JA. Novel SCN3A variants associated with focal epilepsy in children. Neurobiol Dis 62: 313–322, 2014. doi: 10.1016/j.nbd.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Zaman T, Helbig I, Božović IB, DeBrosse SD, Bergqvist AC, Wallis K, Medne L, Maver A, Peterlin B, Helbig KL, Zhang X, Goldberg EM. Mutations in SCN3A cause early infantile epileptic encephalopathy. Ann Neurol 83: 703–717, 2018. doi: 10.1002/ana.25188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, Hill RS, Shin T, Chen AY, Doan RN, Anttonen AK, Ignatius J, Medne L, Bönnemann CG, Hecht JL, Salonen O, Barkovich AJ, Poduri A, Wilke M, de Wit MC, Mancini GM, Sztriha L, Im K, Amrom D, Andermann E, Paetau R, Lehesjoki AE, Walsh CA, Lehtinen MK. Sodium Channel SCN3A (NaV1.3) Regulation of human cerebral cortical folding and oral motor development. Neuron 99: 905–913.e7, 2018. doi: 10.1016/j.neuron.2018.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Zaman T, Helbig KL, Clatot J, Thompson CH, Kang SK, Stouffs K, et al. SCN3A‐related neurodevelopmental disorder: a spectrum of epilepsy and brain malformation. Ann Neurol 88: 348–362, 2020. doi: 10.1002/ana.25809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Lamar T, Vanoye CG, Calhoun J, Wong JC, Dutton SB, Jorge BS, Velinov M, Escayg A, Kearney JA. SCN3A deficiency associated with increased seizure susceptibility. Neurobiol Dis 102: 38–48, 2017. doi: 10.1016/j.nbd.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Gardella E, Marini C, Trivisano M, Fitzgerald MP, Alber M, Howell KB, Darra F, Siliquini S, Bölsterli BK, Masnada S, Pichiecchio A, Johannesen KM, Jepsen B, Fontana E, Anibaldi G, Russo S, Cogliati F, Montomoli M, Specchio N, Rubboli G, Veggiotti P, Beniczky S, Wolff M, Helbig I, Vigevano F, Scheffer IE, Guerrini R, Møller RS. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 91: e1112–e1124, 2018. doi: 10.1212/WNL.0000000000006199. [DOI] [PubMed] [Google Scholar]
- 302. Johannesen KM, Gardella E, Encinas AC, Lehesjoki AE, Linnankivi T, Petersen MB, et al. The spectrum of intermediate SCN 8A ‐related epilepsy. Epilepsia 60: 830–844, 2019.doi: 10.1111/epi.14705. [DOI] [PubMed] [Google Scholar]
- 303. Johannesen KM, Liu Y, Koko M, Gjerulfsen CE, Sonnenberg L, Schubert J, et al. Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain 145: 2991–3009, 2022. doi: 10.1093/brain/awab321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Liu Y, Schubert J, Sonnenberg L, Helbig KL, Hoei-Hansen CE, Koko M, Rannap M, Lauxmann S, Huq M, Schneider MC, Johannesen KM, Kurlemann G, Gardella E, Becker F, Weber YG, Benda J, Møller RS, Lerche H. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 142: 376–390, 2019. doi: 10.1093/brain/awy326. [DOI] [PubMed] [Google Scholar]
- 305. Wagnon JL, Barker BS, Ottolini M, Park Y, Volkheimer A, Valdez P, Swinkels ME, Patel MK, Meisler MH. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet 3: e170, 2017. doi: 10.1212/NXG.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 90: 502–510, 2012. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 24: 506–515, 2015. doi: 10.1093/hmg/ddu470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Bunton-Stasyshyn RK, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, Bhatia K, Jones JM, Maniaci MR, Parent JM, Goodkin HP, Patel MK, Meisler MH. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142: 362–375, 2019. doi: 10.1093/brain/awy324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Meisler MH, Plummer NW, Burgess DL, Buchner DA, Sprunger LK. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica 122: 37–45, 2004. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- 310. Boerma RS, Braun KP, van den Broek MP, van de Broek MP, van Berkestijn FM, Swinkels ME, Hagebeuk EO, Lindhout D, van Kempen M, Boon M, Nicolai J, de Kovel CG, Brilstra EH, Koeleman BP. Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 13: 192–197, 2016. doi: 10.1007/s13311-015-0372-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Lenk GM, Jafar-Nejad P, Hill SF, Huffman LD, Smolen CE, Wagnon JL, Petit H, Yu W, Ziobro J, Bhatia K, Parent J, Giger RJ, Rigo F, Meisler MH. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann Neurol 87: 339–346, 2020. doi: 10.1002/ana.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O’Malley HA, Gray CB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29: 10764–10778, 2009. doi: 10.1523/JNEUROSCI.2475-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Aeby A, Sculier C, Bouza AA, Askar B, Lederer D, Schoonjans AS, Vander Ghinst M, Ceulemans B, Offord J, Lopez-Santiago LF, Isom LL. SCN1B‐linked early infantile developmental and epileptic encephalopathy. Ann Clin Transl Neurol 6: 2354–2367, 2019. doi: 10.1002/acn3.50921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Scala M, Efthymiou S, Sultan T, De Waele J, Panciroli M, Salpietro V, Maroofian R, Striano P, Van Petegem F, Houlden H, Bosmans F. Homozygous SCN1B variants causing early infantile epileptic encephalopathy 52 affect voltage‐gated sodium channel function. Epilepsia 62: e82–e87, 2021. doi: 10.1111/epi.16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Hull JM, O’Malley HA, Chen C, Yuan Y, Denomme N, Bouza AA, Anumonwo C, Lopez-Santiago LF, Isom LL. Excitatory and inhibitory neuron defects in a mouse model of Scn1b‐linked EIEE52. Ann Clin Transl Neurol 7: 2137–2149, 2020. doi: 10.1002/acn3.51205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Nappi P, Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Taglialatela M. Epileptic channelopathies caused by neuronal Kv7 (KCNQ) channel dysfunction. Pflugers Arch 472: 881–898, 2020. doi: 10.1007/s00424-020-02404-2. [DOI] [PubMed] [Google Scholar]
- 317. Maljevic S, Lerche H. Potassium channel genes and benign familial neonatal epilepsy. Prog Brain Res 213: 17–53, 2014. doi: 10.1016/B978-0-444-63326-2.00002-8. [DOI] [PubMed] [Google Scholar]
- 318. Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci USA 107: 10232–10237, 2010. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6: 850–862, 2005. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- 320. Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LR, Deprez L, Smets K, Hristova D, Yordanova I, Jordanova A, Ceulemans B, Jansen A, Hasaerts D, Roelens F, Lagae L, Yendle S, Stanley T, Heron SE, Mulley JC, Berkovic SF, Scheffer IE, de Jonghe P. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 71: 15–25, 2012. doi: 10.1002/ana.22644. [DOI] [PubMed] [Google Scholar]
- 321. Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells: M- and h-channel functions in hippocampal pyramidal cells. J Physiol 566: 689–715, 2005. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci USA 105: 19974–19979, 2008. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Battefeld A, Tran BT, Gavrilis J, Cooper EC, Kole MH. Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J Neurosci 34: 3719–3732, 2014. doi: 10.1523/JNEUROSCI.4206-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci 26: 2599–2613, 2006. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci 24: 1236–1244, 2004.doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Martire M, Castaldo P, D’Amico M, Preziosi P, Annunziato L, Taglialatela M. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci 24: 592–597, 2004. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Peretz A, Sheinin A, Yue C, Degani-Katzav N, Gibor G, Nachman R, Gopin A, Tam E, Shabat D, Yaari Y, Attali B. Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol 97: 283–295, 2007. doi: 10.1152/jn.00634.2006. [DOI] [PubMed] [Google Scholar]
- 328. Lawrence JJ, Saraga F, Churchill JF, Statland JM, Travis KE, Skinner FK, McBain CJ. Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. J Neurosci 26: 12325–12338, 2006. doi: 10.1523/JNEUROSCI.3521-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Abidi A, Devaux JJ, Molinari F, Alcaraz G, Michon FX, Sutera-Sardo J, Becq H, Lacoste C, Altuzarra C, Afenjar A, Mignot C, Doummar D, Isidor B, Guyen SN, Colin E, De La Vaissière S, Haye D, Trauffler A, Badens C, Prieur F, Lesca G, Villard L, Milh M, Aniksztejn L. A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol Dis 80: 80–92, 2015. doi: 10.1016/j.nbd.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 330. Orhan G, Bock M, Schepers D, Ilina EI, Reichel SN, Löffler H, Jezutkovic N, Weckhuysen S, Mandelstam S, Suls A, Danker T, Guenther E, Scheffer IE, De Jonghe P, Lerche H, Maljevic S. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy: KCNQ2 defects in EE. Ann Neurol 75: 382–394, 2014.doi: 10.1002/ana.24080. [DOI] [PubMed] [Google Scholar]
- 331. Soh H, Park S, Ryan K, Springer K, Maheshwari A, Tzingounis AV. Deletion of KCNQ2/3 potassium channels from PV+ interneurons leads to homeostatic potentiation of excitatory transmission. Elife 7: e38617, 2018. doi: 10.7554/eLife.38617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci 14: 4588–4599, 1994. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Monaghan MM, Trimmer JS, Rhodes KJ. Experimental localization of Kv1 family voltage-gated K+ channel α and β subunits in rat hippocampal formation. J Neurosci 21: 5973–5983, 2001. doi: 10.1523/JNEUROSCI.21-16-05973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci 28: 14329–14340, 2008. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. D’Adamo MC, Imbrici P, Sponcichetti F, Pessia M. Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K+ channel function. FASEB J 13: 1335–1345, 1999. doi: 10.1096/fasebj.13.11.1335. [DOI] [PubMed] [Google Scholar]
- 336. Hivert B, Marien L, Agbam KN, Faivre-Sarrailh C. ADAM22 and ADAM23 modulate the targeting of the Kv1 channel-associated protein LGI1 to the axon initial segment. J Cell Sci 132: jcs219774, 2019. doi: 10.1242/jcs.219774. [DOI] [PubMed] [Google Scholar]
- 337. Pena SD, Coimbra RL. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: proposal for a new channelopathy: ataxia and myoclonic epilepsy due to a heterozygous new mutation. Clin Genet 87: e1–e3, 2015. doi: 10.1111/cge.12542. [DOI] [PubMed] [Google Scholar]
- 338. Masnada S, Hedrich UB, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype–phenotype associations of KCNA2-related encephalopathies. Brain 140: 2337–2354, 2017. doi: 10.1093/brain/awx184. [DOI] [PubMed] [Google Scholar]
- 339. Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, Robbins CA, McKee-Johnson J, Chiu SY, Messing A, Tempel BL. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J Neurophysiol 98: 1501–1525, 2007. doi: 10.1152/jn.00640.2006. [DOI] [PubMed] [Google Scholar]
- 340. Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem 285: 32160–32173, 2010. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Hedrich UB, Lauxmann S, Wolff M, Synofzik M, Bast T, Binelli A, Serratosa JM, Martínez-Ulloa P, Allen NM, King MD, Gorman KM, Zeev BB, Tzadok M, Wong-Kisiel L, Marjanovic D, Rubboli G, Sisodiya SM, Lutz F, Ashraf HP, Torge K, Yan P, Bosselmann C, Schwarz N, Fudali M, Lerche H. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci Transl Med 13: eaaz4957, 2021. doi: 10.1126/scitranslmed.aaz4957. [DOI] [PubMed] [Google Scholar]
- 342. Rogers A, Golumbek P, Cellini E, Doccini V, Guerrini R, Wallgren-Pettersson C, Thuresson AC, Gurnett CA. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am J Med Genet A 176: 1748–1752, 2018.doi: 10.1002/ajmg.a.38840. [DOI] [PubMed] [Google Scholar]
- 343. Verdura E, Fons C, Schlüter A, Ruiz M, Fourcade S, Casasnovas C, Castellano A, Pujol A. Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J Med Genet 57: 132–137, 2020. doi: 10.1136/jmedgenet-2019-106373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Trimmer JS. Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 85: 238–256, 2015. doi: 10.1016/j.neuron.2014.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Mohapatra DP, Park KS, Trimmer JS. Dynamic regulation of the voltage-gated Kv2.1 potassium channel by multisite phosphorylation. Biochem Soc Trans 35: 1064–1068, 2007. doi: 10.1042/BST0351064. [DOI] [PubMed] [Google Scholar]
- 346. Speca DJ, Ogata G, Mandikian D, Bishop HI, Wiler SW, Eum K, Wenzel HJ, Doisy ET, Matt L, Campi KL, Golub MS, Nerbonne JM, Hell JW, Trainor BC, Sack JT, Schwartzkroin PA, Trimmer JS. Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability: Kv2.1 deletion and hyperexcitability. Genes Brain Behav 13: 394–408, 2014. doi: 10.1111/gbb.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Torkamani A, Bersell K, Jorge BS, Bjork RL Jr, Friedman JR, Bloss CS, Cohen J, Gupta S, Naidu S, Vanoye CG, George AL Jr, Kearney JA. De novo KCNB1 mutations in epileptic encephalopathy. Ann Neurol 76: 529–540, 2014. doi: 10.1002/ana.24263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Marini C, Romoli M, Parrini E, Costa C, Mei D, Mari F, Parmeggiani L, Procopio E, Metitieri T, Cellini E, Virdò S, De Vita D, Gentile M, Prontera P, Calabresi P, Guerrini R. Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurol Genet 3: e206, 2017. doi: 10.1212/NXG.0000000000000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Bar C, Barcia G, Jennesson M, Le Guyader G, Schneider A, Mignot C, et al. Expanding the genetic and phenotypic relevance of KCNB1 variants in developmental and epileptic encephalopathies: 27 new patients and overview of the literature. Hum Mutat 41: 69–80, 202 0. doi: 10.1002/humu.23915. [DOI] [PubMed] [Google Scholar]
- 350. Thiffault I, Speca DJ, Austin DC, Cobb MM, Eum KS, Safina NP, Grote L, Farrow EG, Miller N, Soden S, Kingsmore SF, Trimmer JS, Saunders CJ, Sack JT. A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J Gen Physiol 146: 399–410, 2015. doi: 10.1085/jgp.201511444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Calhoun JD, Vanoye CG, Kok F, George AL Jr, Kearney JA. Characterization of a KCNB1 variant associated with autism, intellectual disability, and epilepsy. Neurol Genet 3: e198, 2017. doi: 10.1212/NXG.0000000000000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Czirják G, Tóth ZE, Enyedi P. Characterization of the heteromeric potassium channel formed by Kv2.1 and the retinal subunit Kv8.2 in Xenopus oocytes. J Neurophysiol 98: 1213–1222, 2007. doi: 10.1152/jn.00493.2007. [DOI] [PubMed] [Google Scholar]
- 353. Jorge BS, Campbell CM, Miller AR, Rutter ED, Gurnett CA, Vanoye CG, George AL Jr, Kearney JA. Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility. Proc Natl Acad Sci USA 108: 5443–5448, 2011. doi: 10.1073/pnas.1017539108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Kaczmarek LK, Zhang Y. Kv3 channels: enablers of rapid firing, neurotransmitter release, and neuronal endurance. Physiol Rev 97: 1431–1468, 2017. doi: 10.1152/physrev.00002.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Xu M, Cao R, Xiao R, Zhu MX, Gu C. The axon dendrite targeting of Kv3 (Shaw) channels is determined by a targeting motif that associates with the T1 domain and ankyrin G. J Neurosci 27: 14158–14170, 2007. doi: 10.1523/JNEUROSCI.3675-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci 24: 517–526, 2001. doi: 10.1016/S0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- 357. Cameron JM, Maljevic S, Nair U, Aung YH, Cogné B, Bézieau S, Blair E, Isidor B, Zweier C, Reis A, Koenig MK, Maarup T, Sarco D, Afenjar A, Huq AH, Kukolich M, Billette de Villemeur T, Nava C, Héron B, Petrou S, Berkovic SF. Encephalopathies with KCNC1 variants: genotype‐phenotype‐functional correlations. Ann Clin Transl Neurol 6: 1263–1272, 2019.doi: 10.1002/acn3.50822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Park J, Koko M, Hedrich UB, Hermann A, Cremer K, Haberlandt E, Grimmel M, Alhaddad B, Beck-Woedl S, Harrer M, Karall D, Kingelhoefer L, Tzschach A, Matthies LC, Strom TM, Ringelstein EB, Sturm M, Engels H, Wolff M, Lerche H, Haack TB. KCNC1 ‐related disorders: new de novo variants expand the phenotypic spectrum. Ann Clin Transl Neurol 6: 1319–1326, 2019. doi: 10.1002/acn3.50799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Muona M, Berkovic SF, Dibbens LM, Oliver KL, Maljevic S, Bayly MA, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 47: 39–46, 2015. doi: 10.1038/ng.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Espinosa F, McMahon A, Chan E, Wang S, Ho CS, Heintz N, Joho RH. Alcohol hypersensitivity, increased locomotion, and spontaneous myoclonus in mice lacking the potassium channels Kv3.1 and Kv3.3. J Neurosci 21: 6657–6665, 2001. doi: 10.1523/JNEUROSCI.21-17-06657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Isbrandt D, Leicher T, Waldschütz R, Zhu X, Luhmann U, Michel U, Sauter K, Pongs O. Gene structures and expression profiles of three human KCND (Kv4) potassium channels mediating A-type currents ITO and ISA. Genomics 64: 144–154, 2000. doi: 10.1006/geno.2000.6117. [DOI] [PubMed] [Google Scholar]
- 362. Lee H, Lin MC, Kornblum HI, Papazian DM, Nelson SF. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 23: 3481–3489, 2014. doi: 10.1093/hmg/ddu056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Schönherr R, Gessner G, Löber K, Heinemann SH. Functional distinction of human EAG1 and EAG2 potassium channels. FEBS Lett 514: 204–208, 2002. doi: 10.1016/S0014-5793(02)02365-7. [DOI] [PubMed] [Google Scholar]
- 364. Kortüm F, Caputo V, Bauer CK, Stella L, Ciolfi A, Alawi M, Bocchinfuso G, Flex E, Paolacci S, Dentici ML, Grammatico P, Korenke GC, Leuzzi V, Mowat D, Nair LD, Nguyen TT, Thierry P, White SM, Dallapiccola B, Pizzuti A, Campeau PM, Tartaglia M, Kutsche K. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet 47: 661–667, 2015. doi: 10.1038/ng.3282. [DOI] [PubMed] [Google Scholar]
- 365. Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, Ru K, Baillie GJ, Alanay Y, Jacquinet A, Debray FG, Verloes A, Shen J, Yesil G, Guler S, Yuksel A, Cleary JG, Grimmond SM, McGaughran J, King GF, Gabbett MT, Taft RJ. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet 47: 73–77, 2015. doi: 10.1038/ng.3153. [DOI] [PubMed] [Google Scholar]
- 366. Bramswig NC, Ockeloen CW, Czeschik JC, van Essen AJ, Pfundt R, Smeitink J, Poll-The BT, Engels H, Strom TM, Wieczorek D, Kleefstra T, Lüdecke HJ. ‘Splitting versus lumping’: Temple–Baraitser and Zimmermann–Laband syndromes. Hum Genet 134: 1089–1097, 2015. doi: 10.1007/s00439-015-1590-1. [DOI] [PubMed] [Google Scholar]
- 367. Mastrangelo M, Scheffer IE, Bramswig NC, Nair LD, Myers CT, Dentici ML, Korenke GC, Schoch K, Campeau PM, White SM, Shashi V, Kansagra S, Van Essen AJ, Leuzzi V. Epilepsy in KCNH1-related syndromes. Epileptic Disord 18: 123–136, 2016. doi: 10.1684/epd.2016.0830. [DOI] [PubMed] [Google Scholar]
- 368. von Wrede R, Jeub M, Ariöz I, Elger CE, von Voss H, Klein HG, Becker AJ, Schoch S, Surges R, Kunz WS. Novel KCNH1 mutations associated with epilepsy: broadening the phenotypic spectrum of KCNH1-associated diseases. Genes 12: 132, 2021. doi: 10.3390/genes12020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, Barth-Maron A, Greenberg ME, Stuhlmann T, Weinert S, Jentsch TJ, Pazzi M, Restifo LL, Talwar D, Erickson RP, Hammer MF. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 54: 1270–1281, 2013. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Yang Y, Vasylyev DV, Dib-Hajj F, Veeramah KR, Hammer MF, Dib-Hajj SD, Waxman SG. Multistate structural modeling and voltage-clamp analysis of epilepsy/autism mutation Kv10.2-R327H demonstrate the role of this residue in stabilizing the channel closed state. J Neurosci 33: 16586–16593, 2013. doi: 10.1523/JNEUROSCI.2307-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Gonzalez-Perez V, Lingle CJ. Regulation of BK channels by beta and gamma subunits. Annu Rev Physiol 81: 113–137, 2019. doi: 10.1146/annurev-physiol-022516-034038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Zhang ZB, Tian MQ, Gao K, Jiang YW, Wu Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov Disord 30: 1290–1292, 2015. doi: 10.1002/mds.26216. [DOI] [PubMed] [Google Scholar]
- 373. Miller JP, Moldenhauer HJ, Keros S, Meredith AL. An emerging spectrum of variants and clinical features in KCNMA1-linked channelopathy. Channels (Austin) 15: 447–464, 2021. doi: 10.1080/19336950.2021.1938852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Liang L, Li X, Moutton S, Schrier Vergano SA, Cogné B, Saint-Martin A, Hurst AC, Hu Y, Bodamer O, Thevenon J, Hung CY, Isidor B, Gerard B, Rega A, Nambot S, Lehalle D, Duffourd Y, Thauvin-Robinet C, Faivre L, Bézieau S, Dure LS, Helbling DC, Bick D, Xu C, Chen Q, Mancini GMS, Vitobello A, Wang QK. De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet 28: 2937–2951, 2019. doi: 10.1093/hmg/ddz117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem 279: 36746–36752, 2004. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 376. Typlt M, Mirkowski M, Azzopardi E, Ruettiger L, Ruth P, Schmid S. Mice with deficient BK channel function show impaired prepulse inhibition and spatial learning, but normal working and spatial reference memory. PLoS One 8: e81270, 2013. doi: 10.1371/journal.pone.0081270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Imlach WL, Finch SC, Dunlop J, Meredith AL, Aldrich RW, Dalziel JE. The molecular mechanism of “ryegrass staggers,” a neurological disorder of K+ channels. J Pharmacol Exp Ther 327: 657–664, 2008. doi: 10.1124/jpet.108.143933. [DOI] [PubMed] [Google Scholar]
- 378. Rizzi S, Knaus HG, Schwarzer C. Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J Comp Neurol 524: 2093–2116, 2016. doi: 10.1002/cne.23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Mao X, Bruneau N, Gao Q, Becq H, Jia Z, Xi H, Shu L, Wang H, Szepetowski P, Aniksztejn L. The epilepsy of infancy with migrating focal seizures: identification of de novo mutations of the KCNT2 gene that exert inhibitory effects on the corresponding heteromeric KNa1.1/KNa1.2 potassium channel. Front Cell Neurosci 14: 1, 2020. doi: 10.3389/fncel.2020.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Franceschetti S, Lavazza T, Curia G, Aracri P, Panzica F, Sancini G, Avanzini G, Magistretti J. Na+-activated K+. J Neurophysiol 89: 2101–2111, 2003. doi: 10.1152/jn.00695.2002. [DOI] [PubMed] [Google Scholar]
- 381. Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 44: 1255–1259, 2012. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, Scheffer IE, Dibbens LM. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 44: 1188–1190, 2012. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- 383. Cole BA, Clapcote SJ, Muench SP, Lippiat JD. Targeting KNa1.1 channels in KCNT1-associated epilepsy. Trends Pharmacol Sci 42: 700–713, 2021. doi: 10.1016/j.tips.2021.05.003. [DOI] [PubMed] [Google Scholar]
- 384. Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, Kaczmarek LK. Human Slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep 9: 1661–1672, 2014. doi: 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Evely KM, Pryce KD, Bhattacharjee A. The Phe932Ile mutation in KCNT1 channels associated with severe epilepsy, delayed myelination and leukoencephalopathy produces a loss-of-function channel phenotype. Neuroscience 351: 65–70, 2017. doi: 10.1016/j.neuroscience.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Shore AN, Colombo S, Tobin WF, Petri S, Cullen ER, Dominguez S, Bostick CD, Beaumont MA, Williams D, Khodagholy D, Yang M, Lutz CM, Peng Y, Gelinas JN, Goldstein DB, Boland MJ, Frankel WN, Weston MC. Reduced GABAergic neuron excitability, altered synaptic connectivity, and seizures in a KCNT1 gain-of-function mouse model of childhood epilepsy. Cell Rep 33: 108303, 2020. doi: 10.1016/j.celrep.2020.108303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Ambrosino P, Soldovieri MV, Bast T, Turnpenny PD, Uhrig S, Biskup S, Döcker M, Fleck T, Mosca I, Manocchio L, Iraci N, Taglialatela M, Lemke JR. De novo gain‐of‐function variants in KCNT2 as a novel cause of developmental and epileptic encephalopathy. Ann Neurol 83: 1198–1204, 2018. doi: 10.1002/ana.25248. [DOI] [PubMed] [Google Scholar]
- 388. Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82: 24–45, 2014. doi: 10.1016/j.neuron.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 389. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. Voltage-gated calcium channels in GtoPdb v.2021.2. IUPHAR/BPS Guide to Pharmacol CITE 2021, 2021. doi: 10.2218/gtopdb/F80/2021.2. [DOI]
- 390. Laryushkin DP, Maiorov SA, Zinchenko VP, Gaidin SG, Kosenkov AM. Role of L-type voltage-gated calcium channels in epileptiform activity of neurons. Int J Mol Sci 22: 10342, 2021. doi: 10.3390/ijms221910342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Striessnig J, Ortner NJ, Pinggera A. Pharmacology of L-type calcium channels: novel drugs for old targets? Curr Mol Pharmacol 8: 110–122, 2015. doi: 10.2174/1874467208666150507105845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119: 19–31, 2004. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 393. Campiglio M, Flucher BE. The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J Cell Physiol 230: 2019–2031, 2015. doi: 10.1002/jcp.24998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83: 117–161, 2003. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 395. Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci 30: 15419–15429, 2010. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape H. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci 15: 3110–3117, 1995. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Lory P, Nicole S, Monteil A. Neuronal Cav3 channelopathies: recent progress and perspectives. Pflugers Arch 472: 831–844, 2020. doi: 10.1007/s00424-020-02429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Kunii M, Doi H, Hashiguchi S, Matsuishi T, Sakai Y, Iai M, Okubo M, Nakamura H, Takahashi K, Katsumoto A, Tada M, Takeuchi H, Ishikawa T, Miyake N, Saitsu H, Matsumoto N, Tanaka F. De novo CACNA1G variants in developmental delay and early-onset epileptic encephalopathies. J Neurol Sci 416: 117047, 2020. doi: 10.1016/j.jns.2020.117047. [DOI] [PubMed] [Google Scholar]
- 399. Berecki G, Helbig KL, Ware TL, Grinton B, Skraban CM, Marsh ED, Berkovic SF, Petrou S. Novel missense CACNA1G mutations associated with infantile-onset developmental and epileptic encephalopathy. Int J Mol Sci 21: 6333, 2020. doi: 10.3390/ijms21176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. El Ghaleb Y, Schneeberger PE, Fernández-Quintero ML, Geisler SM, Pelizzari S, Polstra AM, van Hagen JM, Denecke J, Campiglio M, Liedl KR, Stevens CA, Person RE, Rentas S, Marsh ED, Conlin LK, Tuluc P, Kutsche K, Flucher BE. CACNA1I gain-of-function mutations differentially affect channel gating and cause neurodevelopmental disorders. Brain 144: 2092–2106, 2021. doi: 10.1093/brain/awab101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Dolphin AC, Lee A. Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat Rev Neurosci 21: 213–229, 2020. doi: 10.1038/s41583-020-0278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87: 543–552, 1996. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 403. Catterall WA, Lenaeus MJ, Gamal El-Din TM. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu Rev Pharmacol Toxicol 60: 133–154, 202 0. doi: 10.1146/annurev-pharmtox-010818-021757. [DOI] [PubMed] [Google Scholar]
- 404. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat Genet 15: 62–69, 1997. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 405. Indelicato E, Boesch S. From genotype to phenotype: expanding the clinical spectrum of CACNA1A variants in the era of next generation sequencing. Front Neurol 12: 639994, 2021. doi: 10.3389/fneur.2021.639994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Jiang X, Raju PK, D’Avanzo N, Lachance M, Pepin J, Dubeau F, Mitchell WG, Bello-Espinosa LE, Pierson TM, Minassian BA, Lacaille JC, Rossignol E. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 60: 1881–1894, 2019. doi: 10.1111/epi.16316. [DOI] [PubMed] [Google Scholar]
- 407. Gorman KM, Meyer E, Grozeva D, Spinelli E, McTague A, Sanchis-Juan A, et al. Bi-allelic loss-of-function CACNA1B mutations in progressive epilepsy-dyskinesia. Am J Hum Genet 104: 948–956, 2019. doi: 10.1016/j.ajhg.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Nakagawasai O, Onogi H, Mitazaki S, Sato A, Watanabe K, Saito H, Murai S, Nakaya K, Murakami M, Takahashi E, Tan-No K, Tadano T. Behavioral and neurochemical characterization of mice deficient in the N-type Ca2+ channel α1B subunit. Behav Brain Res 208: 224–230, 2010. doi: 10.1016/j.bbr.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 409. Helbig KL, Lauerer RJ, Bahr JC, Souza IA, Myers CT, Uysal B, et al. De novo pathogenic variants in CACNA1E cause developmental and epileptic encephalopathy with contractures, macrocephaly, and dyskinesias. Am J Hum Genet 103: 666–678, 2018. doi: 10.1016/j.ajhg.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Saegusa H, Kurihara T, Zong S, Minowa O, Kazuno A, Han W, Matsuda Y, Yamanaka H, Osanai M, Noda T, Tanabe T. Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proc Natl Acad Sci USA 97: 6132–6137, 2000. doi: 10.1073/pnas.100124197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 129: 371–383, 2007. doi: 10.1016/j.cell.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 412. Cochet-Bissuel M, Lory P, Monteil A. The sodium leak channel, NALCN, in health and disease. Front Cell Neurosci 8: 132, 2014. doi: 10.3389/fncel.2014.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Al-Sayed MD, Al-Zaidan H, Albakheet A, Hakami H, Kenana R, Al-Yafee Y, Al-Dosary M, Qari A, Al-Sheddi T, Al-Muheiza M, Al-Qubbaj W, Lakmache Y, Al-Hindi H, Ghaziuddin M, Colak D, Kaya N. Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am J Hum Genet 93: 721–726, 2013. doi: 10.1016/j.ajhg.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Bramswig NC, Bertoli-Avella AM, Albrecht B, Al Aqeel AI, Alhashem A, Al-Sannaa N, et al. Genetic variants in components of the NALCN–UNC80–UNC79 ion channel complex cause a broad clinical phenotype (NALCN channelopathies). Hum Genet 137: 753–768, 2018. doi: 10.1007/s00439-018-1929-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Chong JX, McMillin MJ, Shively KM, Beck AE, Marvin CT, Armenteros JR, et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet 96: 462–473, 2015. doi: 10.1016/j.ajhg.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Bouasse M, Impheng H, Servant Z, Lory P, Monteil A. Functional expression of CLIFAHDD and IHPRF pathogenic variants of the NALCN channel in neuronal cells reveals both gain- and loss-of-function properties. Sci Rep 9: 11791, 2019. doi: 10.1038/s41598-019-48071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Rivolta I, Binda A, Masi A, DiFrancesco JC. Cardiac and neuronal HCN channelopathies. Pflugers Arch 472: 931–951, 2020. doi: 10.1007/s00424-020-02384-3. [DOI] [PubMed] [Google Scholar]
- 418. Combe CL, Gasparini S. Ih from synapses to networks: HCN channel functions and modulation in neurons. Prog Biophys Mol Biol 166: 119–132, 2021. doi: 10.1016/j.pbiomolbio.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Nava C, Dalle C, Rastetter A, Striano P, de Kovel CG, Nabbout R, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet 46: 640–645, 2014. doi: 10.1038/ng.2952. [DOI] [PubMed] [Google Scholar]
- 420. Marini C, Porro A, Rastetter A, Dalle C, Rivolta I, Bauer D, et al. HCN1 mutation spectrum: from neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain 141: 3160–3178, 2018. doi: 10.1093/brain/awy263. [DOI] [PubMed] [Google Scholar]
- 421. Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115: 551–564, 2003. doi: 10.1016/S0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- 422. Bleakley LE, McKenzie CE, Soh MS, Forster IC, Pinares-Garcia P, Sedo A, Kathirvel A, Churilov L, Jancovski N, Maljevic S, Berkovic SF, Scheffer IE, Petrou S, Santoro B, Reid CA. Cation leak underlies neuronal excitability in an HCN1 developmental and epileptic encephalopathy. Brain 144: 2060–2073, 2021. doi: 10.1093/brain/awab145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Friedrich T, Tavraz NN, Junghans C. ATP1A2 mutations in migraine: seeing through the facets of an ion pump onto the neurobiology of disease. Front Physiol 7: 239, 2016. doi: 10.3389/fphys.2016.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem 71: 511–535, 2002. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 425. Bassi MT, Bresolin N, Tonelli A, Nazos K, Crippa F, Baschirotto C, Zucca C, Bersano A, Dolcetta D, Boneschi FM, Barone V, Casari G. A novel mutation in the ATP1A2 gene causes alternating hemiplegia of childhood. J Med Genet 41: 621–628, 2004. doi: 10.1136/jmg.2003.017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Swoboda KJ, Kanavakis E, Xaidara A, Johnson JE, Leppert MF, Schlesinger-Massart MB, Ptacek LJ, Silver K, Youroukos S. Alternating hemiplegia of childhood or familial hemiplegic migraine?: a novelATP1A2 mutation. Ann Neurol 55: 884–887, 2004. doi: 10.1002/ana.20134. [DOI] [PubMed] [Google Scholar]
- 427. Dard R, Mignot C, Durr A, Lesca G, Sanlaville D, Roze E, Mochel F. Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Dev Med Child Neurol 57: 1183–1186, 2015. doi: 10.1111/dmcn.12927. [DOI] [PubMed] [Google Scholar]
- 428. Paciorkowski AR, McDaniel SS, Jansen LA, Tully H, Tuttle E, Ghoneim DH, Tupal S, Gunter SA, Vasta V, Zhang Q, Tran T, Liu YB, Ozelius LJ, Brashear A, Sweadner KJ, Dobyns WB, Hahn S. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 56: 422–430, 2015. doi: 10.1111/epi.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Liu J, Tong L, Song S, Niu Y, Li J, Wu X, Zhang J, Zai CC, Luo F, Wu J, Li H, Wong AH, Sun R, Liu F, Li B. Novel and de novo mutations in pediatric refractory epilepsy. Mol Brain 11: 48, 2018. doi: 10.1186/s13041-018-0392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Chatron N, Cabet S, Alix E, Buenerd A, Cox P, Guibaud L, Labalme A, Marks P, Osio D, Putoux A, Sanlaville D, Lesca G, Vasiljevic A. A novel lethal recognizable polymicrogyric syndrome caused by ATP1A2 homozygous truncating variants. Brain 142: 3367–3374, 2019. doi: 10.1093/brain/awz272. [DOI] [PubMed] [Google Scholar]
- 431. Monteiro FP, Curry CJ, Hevner R, Elliott S, Fisher JH, Turocy J, Dobyns WB, Costa LA, Freitas E, Kitajima JP, Kok F. Biallelic loss of function variants in ATP1A2 cause hydrops fetalis, microcephaly, arthrogryposis and extensive cortical malformations. Eur J Med Genet 63: 103624, 2020. doi: 10.1016/j.ejmg.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 432. Vetro A, Nielsen HN, Holm R, Hevner RF, Parrini E, Powis Z, Møller RS, Bellan C, Simonati A, Lesca G, Helbig KL, Palmer EE, Mei D, Ballardini E, Van Haeringen A, Syrbe S, Leuzzi V, Cioni G, Curry CJ, Costain G, Santucci M, Chong K, Mancini GM, Clayton-Smith J, Bigoni S, Scheffer IE, Dobyns WB, Vilsen B, Guerrini R; ATP1A2/A3-collaborators. ATP1A2- and ATP1A3- associated early profound epileptic encephalopathy and polymicrogyria. Brain 144: 1435–1450, 2021. doi: 10.1093/brain/awab052. [DOI] [PubMed] [Google Scholar]
- 433. Sterlini B, Romei A, Parodi C, Aprile D, Oneto M, Aperia A, Valente P, Valtorta F, Fassio A, Baldelli P, Benfenati F, Corradi A. An interaction between PRRT2 and Na+/K+ ATPase contributes to the control of neuronal excitability. Cell Death Dis 12: 292, 2021. doi: 10.1038/s41419-021-03569-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Chanaday NL, Cousin MA, Milosevic I, Watanabe S, Morgan JR. The synaptic vesicle cycle revisited: new insights into the modes and mechanisms. J Neurosci 39: 8209–8216, 2019. doi: 10.1523/JNEUROSCI.1158-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Fassio A, Fadda M, Benfenati F. Molecular machines determining the fate of endocytosed synaptic vesicles in nerve terminals. Front Synaptic Neurosci 8: 10, 2016. doi: 10.3389/fnsyn.2016.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Alabi AA, Tsien RW. Synaptic vesicle pools and dynamics. Cold Spring Harb Perspect Biol 4: a013680, 2012. doi: 10.1101/cshperspect.a013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Rizzoli SO. Synaptic vesicle recycling: steps and principles. EMBO J 33: 788–822, 2014. doi: 10.1002/embj.201386357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405, 2002. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 439. Abbott LF, Regehr WG. Synaptic computation. Nature 431: 796–803, 2004. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- 440. Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci 14: 337–349, 2013. doi: 10.1038/nrn3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Fukata Y, Fukata M. Epilepsy and synaptic proteins. Curr Opin Neurobiol 45: 1–8, 2017. doi: 10.1016/j.conb.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 442. Wolking S, May P, Mei D, Møller RS, Balestrini S, Helbig KL, et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology 92: e1238–e1249, 2019. doi: 10.1212/WNL.0000000000007089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Fukuda H, Imagawa E, Hamanaka K, Fujita A, Mitsuhashi S, Miyatake S, Mizuguchi T, Takata A, Miyake N, Kramer U, Matsumoto N, Fattal-Valevski A. A novel missense SNAP25b mutation in two affected siblings from an Israeli family showing seizures and cerebellar ataxia. J Hum Genet 63: 673–676, 2018. doi: 10.1038/s10038-018-0421-3. [DOI] [PubMed] [Google Scholar]
- 444. Salpietro V, Malintan NT, Llano-Rivas I, Spaeth CG, Efthymiou S, Striano P, et al. Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. Am J Hum Genet 104: 721–730, 2019. doi: 10.1016/j.ajhg.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 encephalopathy: Aa neurodevelopmental disorder including epilepsy. Neurology 86: 954–962, 2016. doi: 10.1212/WNL.0000000000002457. [DOI] [PubMed] [Google Scholar]
- 446. Valtorta F, Benfenati F, Zara F, Meldolesi J. PRRT2: from paroxysmal disorders to regulation of synaptic function. Trends Neurosci 39: 668–679, 2016. doi: 10.1016/j.tins.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 447. Baker K, Gordon SL, Melland H, Bumbak F, Scott DJ, Jiang TJ, Owen D, Turner BJ, Boyd SG, Rossi M, Al-Raqad M, Elpeleg O, Peck D, Mancini GM, Wilke M, Zollino M, Marangi G, Weigand H, Borggraefe I, Haack T, Stark Z, Sadedin S, Tan TY, Jiang Y, Gibbs RA, Ellingwood S, Amaral M, Kelley W, Kurian MA, Cousin MA; Broad Center for Mendelian Genomics. SYT1-associated neurodevelopmental disorder: a case series. Brain 141: 2576–2591, 2018. doi: 10.1093/brain/awy209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Bradberry MM, Courtney NA, Dominguez MJ, Lofquist SM, Knox AT, Sutton RB, Chapman ER. Molecular basis for synaptotagmin-1-associated neurodevelopmental disorder. Neuron 107: 52–64.e7, 2020. doi: 10.1016/j.neuron.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19–26, 2002. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- 450. Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, Verhage M, Rosenmund C, Südhof TC. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 321: 1507–1510, 2008. doi: 10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294: 1117–1122, 2001. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- 452. Kovacevic J, Maroteaux G, Schut D, Loos M, Dubey M, Pitsch J, Remmelink E, Koopmans B, Crowley J, Cornelisse LN, Sullivan PF, Schoch S, Toonen RF, Stiedl O, Verhage M. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain 141: 1350–1374, 2018. doi: 10.1093/brain/awy046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Michetti C, Castroflorio E, Marchionni I, Forte N, Sterlini B, Binda F, Fruscione F, Baldelli P, Valtorta F, Zara F, Corradi A, Benfenati F. The PRRT2 knockout mouse recapitulates the neurological diseases associated with PRRT2 mutations. Neurobiol Dis 99: 66–83, 2017. doi: 10.1016/j.nbd.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Valente P, Romei A, Fadda M, Sterlini B, Lonardoni D, Forte N, Fruscione F, Castroflorio E, Michetti C, Giansante G, Valtorta F, Tsai JW, Zara F, Nieus T, Corradi A, Fassio A, Baldelli P, Benfenati F. Constitutive inactivation of the PRRT2 gene alters short-term synaptic plasticity and promotes network hyperexcitability in hippocampal neurons. Cereb Cortex 29: 2010–2033, 2019. doi: 10.1093/cercor/bhy079. [DOI] [PubMed] [Google Scholar]
- 455. Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79: 717–727, 1994. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 456. Rohena L, Neidich J, Truitt Cho M, Gonzalez KD, Tang S, Devinsky O, Chung WK. Mutation in SNAP25 as a novel genetic cause of epilepsy and intellectual disability. Rare Dis 1: e26314, 2013. doi: 10.4161/rdis.26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 83: 2247–2255, 2014. doi: 10.1212/WNL.0000000000001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Johansson JU, Ericsson J, Janson J, Beraki S, Stanić D, Mandic SA, Wikström MA, Hökfelt T, Ogren SO, Rozell B, Berggren PO, Bark C. An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS Genet 4: e1000278, 2008. doi: 10.1371/journal.pgen.1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 46: 1327–1332, 2014. doi: 10.1038/ng.3130. [DOI] [PubMed] [Google Scholar]
- 460. Megighian A, Pirazzini M, Fabris F, Rossetto O, Montecucco C. Tetanus and tetanus neurotoxin: from peripheral uptake to central nervous tissue targets. J Neurochem 158: 1244–1253, 2021. doi: 10.1111/jnc.15330. [DOI] [PubMed] [Google Scholar]
- 461. Rizo J. Mechanism of neurotransmitter release coming into focus. Protein Sci 27: 1364–1391, 2018. doi: 10.1002/pro.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Brunger AT, Choi UB, Lai Y, Leitz J, White KI, Zhou Q. The pre-synaptic fusion machinery. Curr Opin Struct Biol 54: 179–188, 2019. doi: 10.1016/j.sbi.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Baker K, Gordon SL, Grozeva D, van Kogelenberg M, Roberts NY, Pike M, Blair E, Hurles ME, Chong WK, Baldeweg T, Kurian MA, Boyd SG, Cousin MA, Raymond FL. Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. J Clin Invest 125: 1670–1678, 2015. doi: 10.1172/JCI79765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Valente P, Castroflorio E, Rossi P, Fadda M, Sterlini B, Cervigni RI, Prestigio C, Giovedì S, Onofri F, Mura E, Guarnieri FC, Marte A, Orlando M, Zara F, Fassio A, Valtorta F, Baldelli P, Corradi A, Benfenati F. PRRT2 is a key component of the Ca2+-dependent neurotransmitter release machinery. Cell Rep 15: 117–131, 2016. doi: 10.1016/j.celrep.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Fruscione F, Valente P, Sterlini B, Romei A, Baldassari S, Fadda M, Prestigio C, Giansante G, Sartorelli J, Rossi P, Rubio A, Gambardella A, Nieus T, Broccoli V, Fassio A, Baldelli P, Corradi A, Zara F, Benfenati F. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain 141: 1000–1016, 2018. doi: 10.1093/brain/awy051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Ferrante D, Sterlini B, Prestigio C, Marte A, Corradi A, Onofri F, Tortarolo G, Vicidomini G, Petretto A, Muià J, Thalhammer A, Valente P, Cingolani LA, Benfenati F, Baldelli P. PRRT2 modulates presynaptic Ca2+ influx by interacting with P/Q-type channels. Cell Rep 35: 109248, 2021. doi: 10.1016/j.celrep.2021.109248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Ciruelas K, Marcotulli D, Bajjalieh SM. Synaptic vesicle protein 2: a multi-faceted regulator of secretion. Semin Cell Dev Biol 95: 130–141, 2019. doi: 10.1016/j.semcdb.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Harper CB, Small C, Davenport EC, Low DW, Smillie KJ, Martínez-Mármol R, Meunier FA, Cousin MA. An epilepsy-associated SV2A mutation disrupts synaptotagmin-1 expression and activity-dependent trafficking. J Neurosci 40: 4586–4595, 2020. doi: 10.1523/JNEUROSCI.0210-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Rizo J, Südhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—guilty as charged? Annu Rev Cell Dev Biol 28: 279–308, 2012. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- 470. Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N, Rhee J, Brunger AT. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 95: 591–607.e10, 2017. doi: 10.1016/j.neuron.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Engel AG, Selcen D, Shen XM, Milone M, Harper CM. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol Genet 2: e105–e106, 2016. doi: 10.1212/NXG.0000000000000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Lanoue V, Chai YJ, Brouillet JZ, Weckhuysen S, Palmer EE, Collins BM, Meunier FA. STXBP1 encephalopathy: connecting neurodevelopmental disorders with α-synucleinopathies. Neurology 93: 114–123, 2019. doi: 10.1212/WNL.0000000000007786. [DOI] [PubMed] [Google Scholar]
- 473. Rost BR, Schneider F, Grauel MK, Wozny C, Bentz C, Blessing A, Rosenmund T, Jentsch TJ, Schmitz D, Hegemann P, Rosenmund C. Optogenetic acidification of synaptic vesicles and lysosomes. Nat Neurosci 18: 1845–1852, 2015. doi: 10.1038/nn.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Bodzęta A, Kahms M, Klingauf J. The presynaptic v-ATPase reversibly disassembles and thereby modulates exocytosis but is not part of the fusion machinery. Cell Rep 20: 1348–1359, 2017. doi: 10.1016/j.celrep.2017.07.040. [DOI] [PubMed] [Google Scholar]
- 475. Gowrisankaran S, Milosevic I. Regulation of synaptic vesicle acidification at the neuronal synapse. IUBMB Life 72: 568–576, 2020. doi: 10.1002/iub.2235. [DOI] [PubMed] [Google Scholar]
- 476. Jaskolka MC, Winkley SR, Kane PM. RAVE and rabconnectin-3 complexes as signal dependent regulators of organelle acidification. Front Cell Dev Biol 9: 698190, 2021. doi: 10.3389/fcell.2021.698190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Fassio A, Esposito A, Kato M, Saitsu H, Mei D, Marini C, Conti V, Nakashima M, Okamoto N, Olmez Turker A, Albuz B, Semerci Gündüz CN, Yanagihara K, Belmonte E, Maragliano L, Ramsey K, Balak C, Siniard A, Narayanan V, Ohba C, Shiina M, Ogata K, Matsumoto N, Benfenati F, Guerrini R; C4RCD Research Group. De novo mutations of the ATP6V1A gene cause developmental encephalopathy with epilepsy. Brain 141: 1703–1718, 2018. doi: 10.1093/brain/awy092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Aoto K, Kato M, Akita T, Nakashima M, Mutoh H, Akasaka N, Tohyama J, Nomura Y, Hoshino K, Ago Y, Tanaka R, Epstein O, Ben-Haim R, Heyman E, Miyazaki T, Belal H, Takabayashi S, Ohba C, Takata A, Mizuguchi T, Miyatake S, Miyake N, Fukuda A, Matsumoto N, Saitsu H. ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice. Nat Commun 12: 2107, 2021. doi: 10.1038/s41467-021-22389-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Esposito A, Falace A, Wagner M, Gal M, Mei D, Conti V, Pisano T, Aprile D, Cerullo MS, De Fusco A, Giovedì S, Seibt A, Magen D, Polster T, Eran A, Stenton SL, Fiorillo C, Ravid S, Mayatepek E, Hafner H, Wortmann S, Levanon EY, Marini C, Mandel H, Benfenati F, Distelmaier F, Fassio A, Guerrini R. Biallelic DMXL2 mutations impair autophagy and cause Ohtahara syndrome with progressive course. Brain 142: 3876–3891, 2019. doi: 10.1093/brain/awz326. [DOI] [PubMed] [Google Scholar]
- 480. Helbig I, Lopez-Hernandez T, Shor O, Galer P, Ganesan S, Pendziwiat M, Rademacher A, Ellis CA, Hümpfer N, Schwarz N, Seiffert S, Peeden J, Shen J, Štěrbová K, Hammer TB, Møller RS, Shinde DN, Tang S, Smith L, Poduri A, Krause R, Benninger F, Helbig KL, Haucke V, Weber YG; GRIN Consortium. A recurrent missense variant in AP2M1 impairs clathrin-mediated endocytosis and causes developmental and epileptic encephalopathy. Am J Hum Genet 104: 1060–1072, 2019. doi: 10.1016/j.ajhg.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481. Gordon SL, Leube RE, Cousin MA. Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J Neurosci 31: 14032–14036, 2011. doi: 10.1523/JNEUROSCI.3162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Gordon SL, Cousin MA. X-linked intellectual disability-associated mutations in synaptophysin disrupt synaptobrevin II retrieval. J Neurosci 33: 13695–13700, 2013. doi: 10.1523/JNEUROSCI.0636-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483. Harper CB, Mancini GM, van Slegtenhorst M, Cousin MA. Altered synaptobrevin-II trafficking in neurons expressing a synaptophysin mutation associated with a severe neurodevelopmental disorder. Neurobiol Dis 108: 298–306, 2017. doi: 10.1016/j.nbd.2017.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. von Spiczak S, Helbig KL, Shinde DN, Huether R, Pendziwiat M, Lourenço C, Nunes ME, Sarco DP, Kaplan RA, Dlugos DJ, Kirsch H, Slavotinek A, Cilio MR, Cervenka MC, Cohen JS, McClellan R, Fatemi A, Yuen A, Sagawa Y, Littlejohn R, McLean SD, Hernandez-Hernandez L, Maher B, Møller RS, Palmer E, Lawson JA, Campbell CA, Joshi CN, Kolbe DL, Hollingsworth G; EuroEPINOMICS-RES NLES Working Group. DNM1 encephalopathy: a new disease of vesicle fission. Neurology 89: 385–394, 2017. doi: 10.1212/WNL.0000000000004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485. Morlot S, Roux A. Mechanics of dynamin-mediated membrane fission. Annu Rev Biophys 42: 629–649, 2013. doi: 10.1146/annurev-biophys-050511-102247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Robinson MS. Forty years of clathrin-coated vesicles. Traffic 16: 1210–1238, 2015. doi: 10.1111/tra.12335. [DOI] [PubMed] [Google Scholar]
- 487. DeMari J, Mroske C, Tang S, Nimeh J, Miller R, Lebel RR. CLTC as a clinically novel gene associated with multiple malformations and developmental delay. Am J Med Genet A 170A: 958–966, 2016. doi: 10.1002/ajmg.a.37506. [DOI] [PubMed] [Google Scholar]
- 488. Balestrini S, Milh M, Castiglioni C, Lüthy K, Finelli MJ, Verstreken P, et al. TBC1D24 genotype–phenotype correlation. Neurology 87: 77–85, 2016. doi: 10.1212/WNL.0000000000002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Falace A, Filipello F, La Padula V, Vanni N, Madia F, De Pietri Tonelli D, de Falco FA, Striano P, Dagna Bricarelli F, Minetti C, Benfenati F, Fassio A, Zara F. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am J Hum Genet 87: 365–370, 2010. doi: 10.1016/j.ajhg.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490. Falace A, Buhler E, Fadda M, Watrin F, Lippiello P, Pallesi-Pocachard E, Baldelli P, Benfenati F, Zara F, Represa A, Fassio A, Cardoso C. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc Natl Acad Sci USA 111: 2337–2342, 2014. doi: 10.1073/pnas.1316294111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Finelli MJ, Aprile D, Castroflorio E, Jeans A, Moschetta M, Chessum L, Degiacomi MT, Grasegger J, Lupien-Meilleur A, Bassett A, Rossignol E, Campeau PM, Bowl MR, Benfenati F, Fassio A, Oliver PL. The epilepsy-associated protein TBC1D24 is required for normal development, survival and vesicle trafficking in mammalian neurons. Hum Mol Genet 28: 584–597, 2019. doi: 10.1093/hmg/ddy370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Aprile D, Fruscione F, Baldassari S, Fadda M, Ferrante D, Falace A, Buhler E, Sartorelli J, Represa A, Baldelli P, Benfenati F, Zara F, Fassio A. TBC1D24 regulates axonal outgrowth and membrane trafficking at the growth cone in rodent and human neurons. Cell Death Differ 26: 2464–2478, 2019. doi: 10.1038/s41418-019-0313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Fornasiero EF, Bonanomi D, Benfenati F, Valtorta F. The role of synapsins in neuronal development. Cell Mol life Sci C 67: 1383–1396, 2010. doi: 10.1007/s00018-009-0227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91: 313–348, 2010. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 495. Fassio A, Raimondi A, Lignani G, Benfenati F, Baldelli P. Synapsins: from synapse to network hyperexcitability and epilepsy. Semin Cell Dev Biol 22: 408–415, 2011. doi: 10.1016/j.semcdb.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 496. Longhena F, Faustini G, Brembati V, Pizzi M, Benfenati F, Bellucci A. An updated reappraisal of synapsins: structure, function and role in neurological and psychiatric disorders. Neurosci Biobehav Rev 130: 33–60, 2021. doi: 10.1016/j.neubiorev.2021.08.011. [DOI] [PubMed] [Google Scholar]
- 497. Greco B, Managò F, Tucci V, Kao HT, Valtorta F, Benfenati F. Autism-related behavioral abnormalities in synapsin knockout mice. Behav Brain Res 251: 65–74, 2013. doi: 10.1016/j.bbr.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Michetti C, Caruso A, Pagani M, Sabbioni M, Medrihan L, David G, Galbusera A, Morini M, Gozzi A, Benfenati F, Scattoni ML. The knockout of synapsin II in mice impairs social behavior and functional connectivity generating an ASD-like phenotype. Cereb Cortex 27: 5014–5023, 2017. doi: 10.1093/cercor/bhx207. [DOI] [PubMed] [Google Scholar]
- 499. Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, Pozzi D, Messa M, Defranchi E, Fadda M, Corradi A, Baldelli P, Lapointe L, St-Onge J, Meloche C, Mottron L, Valtorta F, Khoa Nguyen D, Rouleau GA, Benfenati F, Cossette P. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet 20: 2297–2307, 2011. doi: 10.1093/hmg/ddr122. [DOI] [PubMed] [Google Scholar]
- 500. Giannandrea M, Guarnieri FC, Gehring NH, Monzani E, Benfenati F, Kulozik AE, Valtorta F. Nonsense-mediated mRNA decay and loss-of-function of the protein underlie the X-linked epilepsy associated with the W356× mutation in synapsin I. PLoS One 8: e67724, 2013. doi: 10.1371/journal.pone.0067724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Corradi A, Fadda M, Piton A, Patry L, Marte A, Rossi P, Cadieux-Dion M, Gauthier J, Lapointe L, Mottron L, Valtorta F, Rouleau GA, Fassio A, Benfenati F, Cossette P. SYN2 is an autism predisposing gene: loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum Mol Genet 23: 90–103, 2014. doi: 10.1093/hmg/ddt401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Nguyen DK, Rouleau I, Sénéchal G, Ansaldo AI, Gravel M, Benfenati F, Cossette P. X-linked focal epilepsy with reflex bathing seizures: characterization of a distinct epileptic syndrome. Epilepsia 56: 1098–1108, 2015. doi: 10.1111/epi.13042. [DOI] [PubMed] [Google Scholar]
- 503. Guarnieri FC, Pozzi D, Raimondi A, Fesce R, Valente MM, Delvecchio VS, Van Esch H, Matteoli M, Benfenati F, D’Adamo P, Valtorta F. A novel SYN1 missense mutation in non-syndromic X-linked intellectual disability affects synaptic vesicle life cycle, clustering and mobility. Hum Mol Genet 26: 4699–4714, 2017. doi: 10.1093/hmg/ddx352. [DOI] [PubMed] [Google Scholar]
- 504. Accogli A, Wiegand G, Scala M, Cerminara C, Iacomino M, Riva A, Carlini B, Camerota L, Belcastro V, Prontera P, Fernández-Jaén A, Bebek N, Scudieri P, Baldassari S, Salpietro V, Novelli G, De Luca C, von Stülpnagel C, Kluger F, Kluger GJ, Wohlrab GC, Ramantani G, Lewis-Smith D, Thomas RH, Lai M, Verrotti A, Striano S, Depienne C, Minetti C, Benfenati F, Brancati F, Zara F, Striano P. Clinical and genetic features in patients with reflex bathing epilepsy. Neurology 97: e577–e586, 2021. doi: 10.1212/WNL.0000000000012298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Rogawski MA. A new SV2A ligand for epilepsy. Cell 167: 587, 2016. doi: 10.1016/j.cell.2016.09.057. [DOI] [PubMed] [Google Scholar]
- 506. Benke TA, Park K, Krey I, Camp CR, Song R, Ramsey AJ, Yuan H, Traynelis SF, Lemke J. Clinical and therapeutic significance of genetic variation in the GRIN gene family encoding NMDARs. Neuropharmacology 199: 108805, 2021. doi: 10.1016/j.neuropharm.2021.108805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet 45: 1067–1072, 2013. doi: 10.1038/ng.2728. [DOI] [PubMed] [Google Scholar]
- 508. Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J, Wright S, Tsintsadze V, Michel A, Doummar D, Lascelles K, Strug L, Waters P, de Bellescize J, Vrielynck P, de Saint Martin A, Ville D, Ryvlin P, Arzimanoglou A, Hirsch E, Vincent A, Pal D, Burnashev N, Sanlaville D, Szepetowski P. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet 45: 1061–1066, 2013. doi: 10.1038/ng.2726. [DOI] [PubMed] [Google Scholar]
- 509. Platzer K, Yuan H, Schütz H, Winschel A, Chen W, Hu C, et al. GRIN2B encephalopathy: novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet 54: 460–470, 2017. doi: 10.1136/jmedgenet-2016-104509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Bertocchi I, Eltokhi A, Rozov A, Chi VN, Jensen V, Bus T, Pawlak V, Serafino M, Sonntag H, Yang B, Burnashev N, Li SB, Obenhaus HA, Both M, Niewoehner B, Single FN, Briese M, Boerner T, Gass P, Rawlins JN, Köhr G, Bannerman DM, Sprengel R. Voltage-independent GluN2A-type NMDA receptor Ca2+ signaling promotes audiogenic seizures, attentional and cognitive deficits in mice. Commun Biol 4: 59, 2021. doi: 10.1038/s42003-020-01538-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Shin W, Kim K, Serraz B, Cho YS, Kim D, Kang M, Lee EJ, Lee H, Bae YC, Paoletti P, Kim E. Early correction of synaptic long-term depression improves abnormal anxiety-like behavior in adult GluN2B-C456Y-mutant mice. PLOS Biol 18: e3000717, 2020. doi: 10.1371/journal.pbio.3000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Amador A, Bostick CD, Olson H, Peters J, Camp CR, Krizay D, Chen W, Han W, Tang W, Kanber A, Kim S, Teoh J, Sah M, Petri S, Paek H, Kim A, Lutz CM, Yang M, Myers SJ, Bhattacharya S, Yuan H, Goldstein DB, Poduri A, Boland MJ, Traynelis SF, Frankel WN. Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain 143: 2039–2057, 2020. doi: 10.1093/brain/awaa147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Mayer ML. Glutamate receptor ion channels. Curr Opin Neurobiol 15: 282–288, 2005. doi: 10.1016/j.conb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 514. Greger IH, Ziff EB, Penn AC. Molecular determinants of AMPA receptor subunit assembly. Trends Neurosci 30: 407–416, 2007. doi: 10.1016/j.tins.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 515. Salpietro V, Dixon CL, Guo H, Bello OD, Vandrovcova J, Efthymiou S, et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun 10: 3094, 2019. doi: 10.1038/s41467-019-10910-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945–956, 1996. doi: 10.1016/S0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- 517. Wu Y, Arai AC, Rumbaugh G, Srivastava AK, Turner G, Hayashi T, Suzuki E, Jiang Y, Zhang L, Rodriguez J, Boyle J, Tarpey P, Raymond FL, Nevelsteen J, Froyen G, Stratton M, Futreal A, Gecz J, Stevenson R, Schwartz CE, Valle D, Huganir RL, Wang T. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc Natl Acad Sci USA 104: 18163–18168, 2007. doi: 10.1073/pnas.0708699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Trivisano M, Santarone ME, Micalizzi A, Ferretti A, Dentici ML, Novelli A, Vigevano F, Specchio N. GRIA3 missense mutation is cause of an x-linked developmental and epileptic encephalopathy. Seizure 82: 1–6, 2020. doi: 10.1016/j.seizure.2020.08.032. [DOI] [PubMed] [Google Scholar]
- 519. Martin S, Chamberlin A, Shinde DN, Hempel M, Strom TM, Schreiber A, Johannsen J, Ousager LB, Larsen MJ, Hansen LK, Fatemi A, Cohen JS, Lemke J, Sørensen KP, Helbig KL, Lessel D, Abou Jamra R. De novo variants in GRIA4 lead to intellectual disability with or without seizures and gait abnormalities. Am J Hum Genet 101: 1013–1020, 2017. doi: 10.1016/j.ajhg.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Maljevic S, Møller RS, Reid CA, Pérez-Palma E, Lal D, May P, Lerche H. Spectrum of GABAA receptor variants in epilepsy. Curr Opin Neurol 32: 183–190, 2019. doi: 10.1097/WCO.0000000000000657. [DOI] [PubMed] [Google Scholar]
- 521. Butler KM, Moody OA, Schuler E, Coryell J, Alexander JJ, Jenkins A, Escayg A. De novo variants in GABRA2 and GABRA5 alter receptor function and contribute to early-onset epilepsy. Brain 141: 2392–2405, 2018. doi: 10.1093/brain/awy171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522. Maljevic S, Keren B, Aung YH, Forster IC, Mignot C, Buratti J, Lafitte A, Freihuber C, Rodan LH, Bergin A, Hubert L, Poirier K, Munnich A, Besmond C, Hauser N, Miller R, McWalter K, Nabbout R, Héron D, Leguern E, Depienne C, Petrou S, Nava C. Novel GABRA2 variants in epileptic encephalopathy and intellectual disability with seizures. Brain 142: e15, 2019. doi: 10.1093/brain/awz079. [DOI] [PubMed] [Google Scholar]
- 523. Hernandez CC, XiangWei W, Hu N, Shen D, Shen W, Lagrange AH, Zhang Y, Dai L, Ding C, Sun Z, Hu J, Zhu H, Jiang Y, Macdonald RL. Altered inhibitory synapses in de novo GABRA5 and GABRA1 mutations associated with early onset epileptic encephalopathies. Brain 142: 1938–1954, 2019. doi: 10.1093/brain/awz123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Møller RS, Hjalgrim H, Cook J, Geraghty E, O’Roak BJ, Petrou S, Clarke A, Gill D, Sadleir LG, Muhle H, von Spiczak S, Nikanorova M, Hodgson BL, Gazina EV, Suls A, Shendure J, Dibbens LM, De Jonghe P, Helbig I, Berkovic SF, Scheffer IE, Mefford HC. GABRA1 and STXBP1: Novel genetic causes of Dravet syndrome. Neurology 82: 1245–1253, 2014. doi: 10.1212/WNL.0000000000000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Janve VS, Hernandez CC, Verdier KM, Hu N, Macdonald RL. Epileptic encephalopathy de novo GABRB mutations impair γ-aminobutyric acid type A receptor function. Ann Neurol 79: 806–825, 2016. doi: 10.1002/ana.24631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. Ishii A, Kang JQ, Schornak CC, Hernandez CC, Shen W, Watkins JC, Macdonald RL, Hirose S. A de novo missense mutation of GABRB2 causes early myoclonic encephalopathy. J Med Genet 54: 202–211, 2017. doi: 10.1136/jmedgenet-2016-104083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. Shen D, Hernandez CC, Shen W, Hu N, Poduri A, Shiedley B, Rotenberg A, Datta AN, Leiz S, Patzer S, Boor R, Ramsey K, Goldberg E, Helbig I, Ortiz-Gonzalez XR, Lemke JR, Marsh ED, Macdonald RL. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 140: 49–67, 2017. doi: 10.1093/brain/aww272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Ahring PK, Liao VW, Gardella E, Johannesen KM, Krey I, Selmer KK, et al. Gain-of-function variants in GABRD reveal a novel pathway for neurodevelopmental disorders and epilepsy. Brain 145: 1299–1309, 2022. doi: 10.1093/brain/awab391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Mermer F, Poliquin S, Rigsby K, Rastogi A, Shen W, Romero-Morales A, Nwosu G, McGrath P, Demerast S, Aoto J, Bilousova G, Lal D, Gama V, Kang JQ. Common molecular mechanisms of SLC6A1 variant-mediated neurodevelopmental disorders in astrocytes and neurons. Brain 144: 2499–2512, 2021. doi: 10.1093/brain/awab207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Arain FM, Boyd KL, Gallagher MJ. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor α1 subunit. Epilepsia 53: e161–e165, 2012. doi: 10.1111/j.1528-1167.2012.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Yeung RK, Xiang ZH, Tsang SY, Li R, Ho TY, Li Q, Hui CK, Sham PC, Qiao MQ, Xue H. Gabrb2-knockout mice displayed schizophrenia-like and comorbid phenotypes with interneuron–astrocyte–microglia dysregulation. Transl Psychiatry 8: 128, 2018. doi: 10.1038/s41398-018-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Mäkelä R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW. Mice devoid of gamma-aminobutyrate type A receptor 3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci USA 94: 4143–4148, 1997. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the β3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 18: 8505–8514, 1998. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534. Schofield CM, Kleiman-Weiner M, Rudolph U, Huguenard JR. A gain in GABAA receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures. Proc Natl Acad Sci USA 106: 7630–7635, 2009. doi: 10.1073/pnas.0811326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535. Warner TA, Shen W, Huang X, Liu Z, Macdonald RL, Kang JQ. Differential molecular and behavioural alterations in mouse models of GABRG2 haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum Mol Genet 25: 3192–3207, 2016. doi: 10.1093/hmg/ddw168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536. Yoo Y, Jung J, Lee YN, Lee Y, Cho H, Na E, et al. GABBR2 mutations determine phenotype in Rett syndrome and epileptic encephalopathy. Ann Neurol 82: 466–478, 2017. doi: 10.1002/ana.25032. [DOI] [PubMed] [Google Scholar]
- 537. Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15: 637–654, 2014. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Di Cristo G, Awad PN, Hamidi S, Avoli M. KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol 162: 1–16, 2018. doi: 10.1016/j.pneurobio.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 539. Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, Poduri A. The KCC2 cotransporter and human epilepsy. Neuroscientist 22: 555–562, 2016. doi: 10.1177/1073858416645087. [DOI] [PubMed] [Google Scholar]
- 540. Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep 15: 723–729, 2014. doi: 10.1002/embr.201438749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Jentsch TJ, Pusch M. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev 98: 1493–1590, 2018. doi: 10.1152/physrev.00047.2017. [DOI] [PubMed] [Google Scholar]
- 542. He H, Guzman RE, Cao D, Sierra-Marquez J, Yin F, Fahlke C, Peng J, Stauber T. The molecular and phenotypic spectrum of CLCN4 ‐related epilepsy. Epilepsia 62: 1401–1415, 2021. doi: 10.1111/epi.16906. [DOI] [PubMed] [Google Scholar]
- 543. Palmer EE, Stuhlmann T, Weinert S, Haan E, Van Esch H, Holvoet M, et al. De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol Psychiatry 23: 222–230, 2018. doi: 10.1038/mp.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544. Stergachis AB, Pujol-Giménez J, Gyimesi G, Fuster D, Albano G, Troxler M, Picker J, Rosenberg PA, Bergin A, Peters J, El Achkar CM, Harini C, Manzi S, Rotenberg A, Hediger MA, Rodan LH. Recurrent SLC1A2 variants cause epilepsy via a dominant negative mechanism. Ann Neurol 85: 921–926, 2019. doi: 10.1002/ana.25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699–1702, 1997. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 546. Goodspeed K, Pérez-Palma E, Iqbal S, Cooper D, Scimemi A, Johannesen KM, Stefanski A, Demarest S, Helbig KL, Kang J, Shaffo FC, Prentice B, Brownstein CA, Lim B, Helbig I, De Los Reyes E, McKnight D, Crunelli V, Campbell AJ, Møller RS, Freed A, Lal D. Current knowledge of SLC6A1-related neurodevelopmental disorders. Brain Commun 2: fcaa170, 2020. doi: 10.1093/braincomms/fcaa170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Bröer S, Gether U. The solute carrier 6 family of transporters. Br J Pharmacol 167: 256–278, 2012. doi: 10.1111/j.1476-5381.2012.01975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45: 825–830, 2013. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549. Menuz K, Nicoll RA. Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J Neurosci 28: 10599–10603, 2008. doi: 10.1523/JNEUROSCI.2732-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Harrison V, Connell L, Hayesmoore J, McParland J, Pike MG, Blair E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A 155A: 2826–2831, 2011. doi: 10.1002/ajmg.a.34255. [DOI] [PubMed] [Google Scholar]
- 551. Dinopoulos A, Stefanou MI, Attilakos A, Tsirouda M, Papaevangelou V. A case of startle epilepsy associated with IL1RAPL1 gene deletion. Pediatr Neurol 51: 271–274, 2014. doi: 10.1016/j.pediatrneurol.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 552. Rodenas-Cuadrado P, Pietrafusa N, Francavilla T, La Neve A, Striano P, Vernes SC. Characterisation of CASPR2 deficiency disorder—a syndrome involving autism, epilepsy and language impairment. BMC Med Genet 17: 8, 2016. doi: 10.1186/s12881-016-0272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 30: 335–341, 2002. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Lovero KL, Fukata Y, Granger AJ, Fukata M, Nicoll RA. The LGI1-ADAM22 protein complex directs synapse maturation through regulation of PSD-95 function. Proc Natl Acad Sci USA 112: E4129–E4137, 2015. doi: 10.1073/pnas.1511910112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555. Schirwani S, McConnell V, Willoughby J, Balasubramanian M; DDD Study. Exploring the association between SRPX2 variants and neurodevelopment: how causal is it? Gene 685: 50–54, 2019. doi: 10.1016/j.gene.2018.10.067. [DOI] [PubMed] [Google Scholar]
- 556. Dazzo E, Fanciulli M, Serioli E, Minervini G, Pulitano P, Binelli S, Di Bonaventura C, Luisi C, Pasini E, Striano S, Striano P, Coppola G, Chiavegato A, Radovic S, Spadotto A, Uzzau S, La Neve A, Giallonardo AT, Mecarelli O, Tosatto SC, Ottman R, Michelucci R, Nobile C. Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am J Hum Genet 96: 992–1000, 2015. doi: 10.1016/j.ajhg.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Michelucci R, Pulitano P, Di Bonaventura C, Binelli S, Luisi C, Pasini E, Striano S, Striano P, Coppola G, La Neve A, Giallonardo AT, Mecarelli O, Serioli E, Dazzo E, Fanciulli M, Nobile C. The clinical phenotype of autosomal dominant lateral temporal lobe epilepsy related to reelin mutations. Epilepsy Behav 68: 103–107, 2017. doi: 10.1016/j.yebeh.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Dazzo E, Nobile C. Epilepsy-causing Reelin mutations result in impaired secretion and intracellular degradation of mutant proteins. Hum Mol Genet 31: 665–673, 2022. doi: 10.1093/hmg/ddab271. [DOI] [PubMed] [Google Scholar]
- 559. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364, 2018. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 560. Fassio A, Falace A, Esposito A, Aprile D, Guerrini R, Benfenati F. Emerging role of the autophagy/lysosomal degradative pathway in neurodevelopmental disorders with epilepsy. Front Cell Neurosci 14: 39, 2020. doi: 10.3389/fncel.2020.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561. Rocchi A, Carminati E, De Fusco A, Kowalska JA, Floss T, Benfenati F. REST/NRSF deficiency impairs autophagy and leads to cellular senescence in neurons. Aging Cell 20: e13471, 2021. doi: 10.1111/acel.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of mTOR. Neuroscience 341: 112–153, 2017. doi: 10.1016/j.neuroscience.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 563. Nguyen LH, Bordey A. Corrigendum: convergent and divergent mechanisms of epileptogenesis in mTORopathies. Front Neuroanat 15: 715363, 2021. doi: 10.3389/fnana.2021.715363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Baulac S. mTOR signaling pathway genes in focal epilepsies. Prog Brain Res 226: 61–79, 2016. doi: 10.1016/bs.pbr.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 565. Moloney PB, Cavalleri GL, Delanty N. Epilepsy in the mTORopathies: opportunities for precision medicine. Brain Commun 3: fcab222, 2021. doi: 10.1093/braincomms/fcab222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Lasarge CL, Danzer SC. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci 7: 18, 2014. doi: 10.3389/fnmol.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. LaSarge CL, Pun RY, Gu Z, Riccetti MR, Namboodiri DV, Tiwari D, Gross C, Danzer SC. mTOR-driven neural circuit changes initiate an epileptogenic cascade. Prog Neurobiol 200: 101974, 2021. doi: 10.1016/j.pneurobio.2020.101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, Muraca G, Roussel D, Navarro V, Leguern E, Miles R, Baulac S. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest 128: 2452–2458, 2018. doi: 10.1172/JCI99384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569. De Fusco A, Cerullo MS, Marte A, Michetti C, Romei A, Castroflorio E, Baulac S, Benfenati F. Acute knockdown of Depdc5 leads to synaptic defects in mTOR-related epileptogenesis. Neurobiol Dis 139: 104822, 2020. doi: 10.1016/j.nbd.2020.104822. [DOI] [PubMed] [Google Scholar]
- 570. Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, Rubinsztein DC. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93: 1015–1034, 2017. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 571. Byrne S, Jansen L, U-King-Im JM, Siddiqui A, Lidov HG, Bodi I, et al. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain 139: 765–781, 2016. doi: 10.1093/brain/awv393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Thomas AC, Williams H, Setó-Salvia N, Bacchelli C, Jenkins D, et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet 95: 611–621, 2014. doi: 10.1016/j.ajhg.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8: 917–929, 2007. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 574. Cotter K, Stransky L, McGuire C, Forgac M. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem Sci 40: 611–622, 2015. doi: 10.1016/j.tibs.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Morel N, Poëa-Guyon S. The membrane domain of vacuolar H+ATPase: a crucial player in neurotransmitter exocytotic release. Cell Mol life Sci 72: 2561–2573, 2015. doi: 10.1007/s00018-015-1886-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576. Van Damme T, Gardeitchik T, Mohamed M, Guerrero-Castillo S, Freisinger P, Guillemyn B, et al. Mutations in ATP6V1E1 or ATP6V1A cause autosomal-recessive cutis laxa. Am J Hum Genet 100: 216–227, 2017. doi: 10.1016/j.ajhg.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577. Wang M, Li A, Sekiya M, Beckmann ND, Quan X, Schrode N, et al. Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer’s disease. Neuron 109: 257–272.e14, 2021. doi: 10.1016/j.neuron.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Hirose T, Cabrera-Socorro A, Chitayat D, Lemonnier T, Féraud O, Cifuentes-Diaz C, Gervasi N, Mombereau C, Ghosh T, Stoica L, Bacha JD, Yamada H, Lauterbach MA, Guillon M, Kaneko K, Norris JW, Siriwardena K, Blasér S, Teillon J, Mendoza-Londono R, Russeau M, Hadoux J, Ito S, Corvol P, Matheus MG, Holden KR, Takei K, Emiliani V, Bennaceur-Griscelli A, Schwartz CE. ATP6AP2 variant impairs CNS development and neuronal survival to cause fulminant neurodegeneration. J Clin Invest 129: 2145–2162, 2019. doi: 10.1172/JCI79990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Nagano F, Kawabe H, Nakanishi H, Shinohara M, Deguchi-Tawarada M, Takeuchi M, Sasaki T, Takai Y. Rabconnectin-3, a novel protein that binds both GDP/GTP exchange protein and GTPase-activating protein for Rab3 small G protein family. J Biol Chem 277: 9629–9632, 2002. doi: 10.1074/jbc.C100730200. [DOI] [PubMed] [Google Scholar]
- 580. Kawabe H, Sakisaka T, Yasumi M, Shingai T, Izumi G, Nagano F, Deguchi-Tawarada M, Takeuchi M, Nakanishi H, Takai Y. A novel rabconnectin-3-binding protein that directly binds a GDP/GTP exchange protein for Rab3A small G protein implicated in Ca2+-dependent exocytosis of neurotransmitter. Genes Cells 8: 537–546, 2003. doi: 10.1046/j.1365-2443.2003.00655.x. [DOI] [PubMed] [Google Scholar]
- 581. Tata B, Huijbregts L, Jacquier S, Csaba Z, Genin E, Meyer V, Leka S, Dupont J, Charles P, Chevenne D, Carel JC, Léger J, de Roux N. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol 12: e1001952, 2014. doi: 10.1371/journal.pbio.1001952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, Roos M, McGillewie L, Bär S, Minocha S, Chevalier C, Po C, Chelly J, Mandel JL, Borgatti R, Piton A, Kinnear C, Loos B, Adams DJ, Hérault Y, Collins SC, Friant S, Godin JD, Yalcin B; Sanger Mouse Genetics Project. WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci USA 114: E9308–E9317, 2017. doi: 10.1073/pnas.1713625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Ebrahimi-Fakhari D, Saffari A, Wahlster L, Lu J, Byrne S, Hoffmann GF, Jungbluth H, Sahin M. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain 139: 317–337, 2016. doi: 10.1093/brain/awv371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. Löscher W. Single-target versus multi-target drugs versus combinations of drugs with multiple targets: preclinical and clinical evidence for the treatment or prevention of epilepsy. Front Pharmacol 12: 730257, 2021. doi: 10.3389/fphar.2021.730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Yum MS, Lee M, Ko TS, Velíšek L. A potential effect of ganaxolone in an animal model of infantile spasms. Epilepsy Res 108: 1492–1500, 2014. doi: 10.1016/j.eplepsyres.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 586. Chern CR, Chern CJ, Velíšková J, Velíšek L. ACTON PROLONGATUM® suppresses spasms head to head with Acthar® Gel in the model of infantile spasms. Epilepsy Behav 105: 106950, 2020. doi: 10.1016/j.yebeh.2020.106950. [DOI] [PubMed] [Google Scholar]
- 587. Janicot R, Stafstrom CE, Shao LR. 2-Deoxyglucose terminates pilocarpine-induced status epilepticus in neonatal rats. Epilepsia 61: 1528–1537, 2020. doi: 10.1111/epi.16583. [DOI] [PubMed] [Google Scholar]
- 588. Hawkins NA, Jurado M, Thaxton TT, Duarte SE, Barse L, Tatsukawa T, Yamakawa K, Nishi T, Kondo S, Miyamoto M, Abrahams BS, During MJ, Kearney JA. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia 62: 2845–2857, 2021. doi: 10.1111/epi.17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Jancovski N, Baldwin T, Orford M, Li M, Jones GD, Burbano LE, Rutherford T, Reid C, Heales S, Eaton S, Petrou S. Protective effects of medium chain triglyceride diet in a mouse model of Dravet syndrome. Epilepsia 62: 3131–3142, 2021. doi: 10.1111/epi.17101. [DOI] [PubMed] [Google Scholar]
- 590. Hawkins NA, Nomura T, Duarte S, Barse L, Williams RW, Homanics GE, Mulligan MK, Contractor A, Kearney JA. Gabra2 is a genetic modifier of Dravet syndrome in mice. Mamm Genome 32: 350–363, 2021. doi: 10.1007/s00335-021-09877-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Satpute Janve V, Anderson LL, Bahceci D, Hawkins NA, Kearney JA, Arnold JC. The heat sensing Trpv1 receptor is not a viable anticonvulsant drug target in the Scn1a +/- mouse model of Dravet syndrome. Front Pharmacol 12: 675128, 2021. doi: 10.3389/fphar.2021.675128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592. Anderson LL, Heblinski M, Absalom NL, Hawkins NA, Bowen MT, Benson MJ, Zhang F, Bahceci D, Doohan PT, Chebib M, McGregor IS, Kearney JA, Arnold JC. Cannabigerolic acid, a major biosynthetic precursor molecule in cannabis, exhibits divergent effects on seizures in mouse models of epilepsy. Br J Pharmacol 178: 4826–4841, 2021. doi: 10.1111/bph.15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Anderson LL, Ametovski A, Lin Luo J, Everett-Morgan D, McGregor IS, Banister SD, Arnold JC. Cannabichromene, related phytocannabinoids, and 5-fluoro-cannabichromene have anticonvulsant properties in a mouse model of Dravet syndrome. ACS Chem Neurosci 12: 330–339, 2021. doi: 10.1021/acschemneuro.0c00677. [DOI] [PubMed] [Google Scholar]
- 594. Crotts MS, Kim Y, Bravo E, Richerson GB, Teran FA. A ketogenic diet protects DBA/1 and Scn1aR1407X/+ mice against seizure-induced respiratory arrest independent of ketosis. Epilepsy Behav 124: 108334, 2021. doi: 10.1016/j.yebeh.2021.108334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Mora-Jimenez L, Valencia M, Sanchez-Carpintero R, Tønnesen J, Fadila S, Rubinstein M, Gonzalez-Aparicio M, Bunuales M, Fernandez-Pierola E, Nicolas MJ, Puerta E, Miguelez C, Minguez PG, Lumbreras S, Gonzalez-Aseguinolaza G, Ricobaraza A, Hernandez-Alcoceba R. Transfer of SCN1A to the brain of adolescent mouse model of Dravet syndrome improves epileptic, motor, and behavioral manifestations. Mol Ther Nucleic Acids 25: 585–602, 2021. doi: 10.1016/j.omtn.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Sturgeon ML, Langton R, Sharma S, Cornell RA, Glykys J, Bassuk AG. The opioid antagonist naltrexone decreases seizure-like activity in genetic and chemically induced epilepsy models. Epilepsia Open 6: 528–538, 2021. doi: 10.1002/epi4.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Dinday MT, Baraban SC. Large-scale phenotype-based antiepileptic drug screening in a zebrafish model of Dravet syndrome. Eneuro 2: ENEURO.0068-15.2015, 2015. doi: 10.1523/ENEURO.0068-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Griffin A, Hamling KR, Knupp K, Hong S, Lee LP, Baraban SC. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 140: 669–683, 2017. doi: 10.1093/brain/aww342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599. Banerji R, Huynh C, Figueroa F, Dinday MT, Baraban SC, Patel M. Enhancing glucose metabolism via gluconeogenesis is therapeutic in a zebrafish model of Dravet syndrome. Brain Commun 3: fcab004, 2021. doi: 10.1093/braincomms/fcab004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600. Hatini PG, Commons KG. A 5-HT1D-receptor agonist protects Dravet syndrome mice from seizure and early death. Eur J Neurosci 52: 4370–4374, 2020. doi: 10.1111/ejn.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601. Yamagata T, Raveau M, Kobayashi K, Miyamoto H, Tatsukawa T, Ogiwara I, Itohara S, Hensch TK, Yamakawa K. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis 141: 104954, 2020. doi: 10.1016/j.nbd.2020.104954. [DOI] [PubMed] [Google Scholar]
- 602. Weuring WJ, Singh S, Volkers L, Rook MB, van ’t Slot RH, Bosma M, Inserra M, Vetter I, Verhoeven-Duif NM, Braun KP, Rivara M, Koeleman BP. NaV1.1 and NaV1.6 selective compounds reduce the behavior phenotype and epileptiform activity in a novel zebrafish model for Dravet Syndrome. PLoS One 15: e0219106, 2020. doi: 10.1371/journal.pone.0219106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603. Doumlele K, Conway E, Hedlund J, Tolete P, Devinsky O. A case report on the efficacy of vigabatrin analogue (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) in a patient with infantile spasms. Epilepsy Behav Case Rep 6: 67–69, 2016. doi: 10.1016/j.ebcr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. Samueli S, Dressler A, Gröppel G, Scholl T, Feucht M. Everolimus in infants with tuberous sclerosis complex–related West syndrome: first results from a single-center prospective observational study. Epilepsia 59: e142–e146, 2018. doi: 10.1111/epi.14529. [DOI] [PubMed] [Google Scholar]
- 605. Cross JH, Galer BS, Gil-Nagel A, Devinsky O, Ceulemans B, Lagae L, Schoonjans AS, Donner E, Wirrell E, Kothare S, Agarwal A, Lock M, Gammaitoni AR. Impact of fenfluramine on the expected SUDEP mortality rates in patients with Dravet syndrome. Seizure 93: 154–159, 2021. doi: 10.1016/j.seizure.2021.10.024. [DOI] [PubMed] [Google Scholar]
- 606. Sullivan J, Specchio N, Devinsky O, Auvin S, Perry MS, Strzelczyk A, Gil‐Nagel A, Dai D, Galer BS, Gammaitoni AR. Fenfluramine significantly reduces day-to-day seizure burden by increasing number of seizure-free days and time between seizures in patients with Dravet syndrome: a time-to-event analysis. Epilepsia 63: 130–138, 2022. doi: 10.1111/epi.17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Ceulemans B, Boel M, Leyssens K, Van Rossem C, Neels P, Jorens PG, Lagae L. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia 53: 1131–1139, 2012. doi: 10.1111/j.1528-1167.2012.03495.x. [DOI] [PubMed] [Google Scholar]
- 608. Schoonjans A, Paelinck BP, Marchau F, Gunning B, Gammaitoni A, Galer BS, Lagae L, Ceulemans B. Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: a prospective study of a new cohort of patients. Eur J Neurol 24: 309–314, 2017. doi: 10.1111/ene.13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, Devinsky O, Cross JH, Guerrini R, Talwar D, Miller I, Farfel G, Galer BS, Gammaitoni A, Mistry A, Morrison G, Lock M, Agarwal A, Lai WW, Ceulemans B; FAIRE DS Study Group. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet 394: 2243–2254, 2019. doi: 10.1016/S0140-6736(19)32500-0. [DOI] [PubMed] [Google Scholar]
- 610. Guerrini R, Balestrini S, Wirrell EC, Walker MC. Monogenic epilepsies: disease mechanisms, clinical phenotypes, and targeted therapies. Neurology 97: 817–831, 2021. doi: 10.1212/WNL.0000000000012744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611. Liu D, Zhu M, Zhang Y, Diao Y. Crossing the blood-brain barrier with AAV vectors. Metab Brain Dis 36: 45–52, 2021. doi: 10.1007/s11011-020-00630-2. [DOI] [PubMed] [Google Scholar]
- 612. Turner TJ, Zourray C, Schorge S, Lignani G. Recent advances in gene therapy for neurodevelopmental disorders with epilepsy. J Neurochem 157: 229–262, 2021. doi: 10.1111/jnc.15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613. Morris G, O’Brien D, Henshall DC. Opportunities and challenges for microRNA-targeting therapeutics for epilepsy. Trends Pharmacol Sci 42: 605–616, 2021. doi: 10.1016/j.tips.2021.04.007. [DOI] [PubMed] [Google Scholar]
- 614. Mooney C, Becker BA, Raoof R, Henshall DC. EpimiRBase: a comprehensive database of microRNA-epilepsy associations. Bioinformatics 32: 1436–1438, 2016. doi: 10.1093/bioinformatics/btw008. [DOI] [PubMed] [Google Scholar]
- 615. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157: 1262–1278, 2014. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616. Redman M, King A, Watson C, King D. What is CRISPR/Cas9? Arch Dis Child Educ Pract Ed 101: 213–215, 2016. doi: 10.1136/archdischild-2016-310459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617. Guerrini R. Epilepsy in children. Lancet 367: 499–524, 2006. doi: 10.1016/S0140-6736(06)68182-8. [DOI] [PubMed] [Google Scholar]
- 618. Shewmon DA, Erwin RJ. The effect of focal interictal spikes on perception and reaction time. II. Neuroanatomic specificity. Electroencephalogr Clin Neurophysiol 69: 338–352, 1988. doi: 10.1016/0013-4694(88)90005-3. [DOI] [PubMed] [Google Scholar]
- 619. Shewmon DA, Erwin RJ. Focal spike-induced cerebral dysfunction is related to the after-coming slow wave. Ann Neurol 23: 131–137, 1988. doi: 10.1002/ana.410230205. [DOI] [PubMed] [Google Scholar]
- 620. Brancati C, Barba C, Metitieri T, Melani F, Pellacani S, Viggiano MP, Guerrini R. Impaired object identification in idiopathic childhood occipital epilepsy. Epilepsia 53: 686–694, 2012. doi: 10.1111/j.1528-1167.2012.03410.x. [DOI] [PubMed] [Google Scholar]
- 621. Chilosi AM, Brovedani P, Ferrari AR, Ziegler AL, Guerrini R, Deonna T. Language regression associated with autistic regression and electroencephalographic (EEG) abnormalities. J Child Neurol 29: 855–859, 2014. doi: 10.1177/0883073813482767. [DOI] [PubMed] [Google Scholar]
- 622. Gordon K, Bawden H, Camfield P, Mann S, Orlik P. Valproic acid treatment of learning disorder and severely epileptiform EEG without clinical seizures. J Child Neurol 11: 41–43, 1996. doi: 10.1177/088307389601100110. [DOI] [PubMed] [Google Scholar]
- 623. Guzzetta F, Frisone MF, Ricci D, Randò T, Guzzetta A. Development of visual attention in West syndrome. Epilepsia 43: 757–763, 2002. doi: 10.1046/j.1528-1157.2002.34601.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.19666521.v2.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.17694728.v2.