


Keywords: cardiac lymphatics, cardiovascular disease, lymphangiogenesis, lymphatics, myocardial edema
Abstract
The heart is imbued with a vast lymphatic network that is responsible for fluid homeostasis and immune cell trafficking. Disturbances in the forces that regulate microvascular fluid movement can result in myocardial edema, which has profibrotic and proinflammatory consequences and contributes to cardiovascular dysfunction. This review explores the complex relationship between cardiac lymphatics, myocardial edema, and cardiac disease. It covers the revised paradigm of microvascular forces and fluid movement around the capillary as well as the arsenal of preclinical tools and animal models used to model myocardial edema and cardiac disease. Clinical studies of myocardial edema and their prognostic significance are examined in parallel to the recent elegant animal studies discerning the pathophysiological role and therapeutic potential of cardiac lymphatics in different cardiovascular disease models. This review highlights the outstanding questions of interest to both basic scientists and clinicians regarding the roles of cardiac lymphatics in health and disease.
CLINICAL HIGHLIGHTS
Cardiovascular diseases are the leading cause of mortality worldwide, and the associated morbidity and disease burden have far-reaching and significant impacts globally. Numerous cardiac diseases are associated with myocardial edema, an excess accumulation of fluid in the interstitium of cardiac tissues. Myocardial edema arises from an imbalance between microvascular fluid filtration and fluid removal by local cardiac lymphatics, leading to a variety of damaging and compounding outcomes such as cardiac dysfunction, electrical imbalances, and tissue fibrosis. Cardiac lymphatic vessels play a key role in regulating interstitial fluid levels in the myocardium and mediating immune responses. These vessels are pathologically altered during cardiovascular diseases. Pharmacological activation of lymphangiogenesis, the growth of new lymphatic vessels, in preclinical models has been shown to improve cardiac function and reduce myocardial fibrosis in the context of myocardial infarction. Therapeutic strategies targeting cardiac lymphatic functions may prove to be a promising strategy to treat myocardial edema and associated cardiovascular diseases. Significant barriers to achieving this goal include establishing mechanisms modulating cardiac edema, developing ways to assess cardiac lymphatic anatomy and function in the clinic, and determining therapeutically tractable lymphatic pathways. This review aims to serve as a survey of the various techniques and models available to assess myocardial edema and lymphatic function in the preclinical and clinical settings, review the clinical cardiovascular pathologies associated with myocardial edema and cardiac lymphatics, and discuss the many outstanding questions regarding the role of cardiac lymphatics in cardiovascular physiology and pathophysiology.
1. INTRODUCTION
Cardiovascular disease is the leading cause of mortality in the United States, accounting for one in every four deaths (1). Hypertension, coronary artery disease, and heart failure are the most common hallmarks of cardiovascular disease. Fluid homeostasis is as a pillar of cardiac function, and it is highly vulnerable to perturbations. Cardiac edema, or myocardial edema, is a deleterious pathology associated with numerous cardiovascular diseases, and it is marked by an accumulation of excess fluid in the interstitium of the heart. Even mild cases of myocardial edema can result in fibrosis, hypoxia, and cardiovascular dysfunction. Resolution of myocardial edema is driven primarily by the drainage action of lymphatic vessels in the heart.
The lymphatic system is a semiopen vascular system that serves to transport fluid, macromolecules, and cells back to blood circulation. This vascular system is composed of lymphatic capillaries or initial lymphatics, precollector and collector lymphatics, and lymph nodes. Initial lymphatics are composed of a single layer of lymphatic endothelial cells (LECs) connected by loose cell-cell junctions, called buttonlike junctions (2), enabling them to take up interstitial fluid, macromolecules, and even cells. Lymphatic muscle cells (LMCs) encapsulating LECs and intraluminal valves are present sporadically in precollectors and become more prevalent in collecting lymphatics. Contractions of vascular muscle cells and the neighboring muscles are the primary driving force of lymphatic flow, and lymphatic valves are responsible for making the flow unidirectional. The lymphatic system is responsible for returning interstitial fluid back into circulation and thereby is essential in the resolution of edema. The lymphatic vasculature is also known to have organ- and tissue bed-specific roles ranging from absorption of dietary lipids in the intestinal lacteals, removing metabolic wastes and immune infiltrate from the liver, to regulating local fluid homeostasis in organs like the heart, helping to maintain an optimal microenvironment for electrical conduction and force generation by cardiomyocytes (3).
Cardiac lymphatics generally conform to the general anatomic and functional role of the lymphatic system described above but are decidedly distinct from other lymphatic beds. Cardiac lymphatics are uniquely adapted to the microenvironment of the heart, and their roles in cardiovascular disease and myocardial edema resolution are just beginning to be unearthed. This review covers the physiology of cardiac lymphatics, the updated paradigm regarding microvascular forces and fluid movement around the capillary, clinical studies of myocardial edema and its prognostic significance, and animal studies that explore the relationship between myocardial edema and cardiac lymphatics in different cardiovascular disease models.
2. OVERVIEW OF CARDIAC LYMPHATIC BIOLOGY
2.1. Anatomy
Scientific interest in vascular anatomy has steadily grown since the earliest identification of the blood vasculature by the ancient Egyptians and more extensive comments by Ayurvedic physicians in ancient India and by ancient Greek texts like Galen’s anatomy. Despite this long history of vascular research, parallel studies on lymphatic anatomy have greatly lagged. Identification of lymphatic vascular beds emerged in the seventeenth century (4). Lymph nodes and vessels containing milky fluid, rather than crimson blood, were noted as early as 500 BC, but it was not until the 1600s that the first appearance of “lymphatics” was noted. In 1653 Olaus Rudbeck dissected subepicardial lymphatics, which he identified as emptying into mediastinal lymph nodes, thereby providing the first identification of cardiac lymphatic vessels (5). Subsequent studies utilized dye injection techniques to identify the anatomy of cardiac lymphatics. In 1939, Patek published a thorough description of cardiac lymphatics in dogs using India ink injections, a study that is still heavily referenced today (6). Of interest, two studies from the 1960s focused on the anatomy of cardiac lymphatics in the human heart and touched on two main questions in the cardiac lymphatic field today. Symbas et al. (7) reported that the lymphatic vessels of the human heart are distributed along the coronary arteries, an important observation about paracrine signaling in lymphatic development that is still being investigated today. Second, Fedyai (8) studied the anatomy of cardiac lymphatics in human hearts from fetuses to adults up to 101 yr of age. Interestingly, he found that the lymphatic vasculature in humans reached maturation around 17 yr of age and that with age the lymphatic system underwent pathological regression (8). Similar postnatal maturation of the cardiac lymphatic vasculature was identified in mice in 2015 (9).
Consistent with the structure of the lymphatic vasculature in other organs, the cardiac lymphatic network consists of highly permeable initial lymphatic vessels that drain into larger valve-containing collecting lymphatics (FIGURE 1). These vessels exhibit structure-specific characteristics, molecule expression, and function similar to the lymphatic networks found in other organs, with the exception that cardiac lymphatic collectors lack lymphatic muscle coverage (10, 11) (FIGURE 1, A–E). An interesting meshlike dense network of interconnected initial and precollecting lymphatics, referred to as a plexus, is often seen on the epicardial surface near the apex of the heart (FIGURE 1, C and F). Given the fairly consistent position of this structure of the epicardium, it is of interest to explore the development and function of these structures, which are to date poorly characterized.
FIGURE 1.
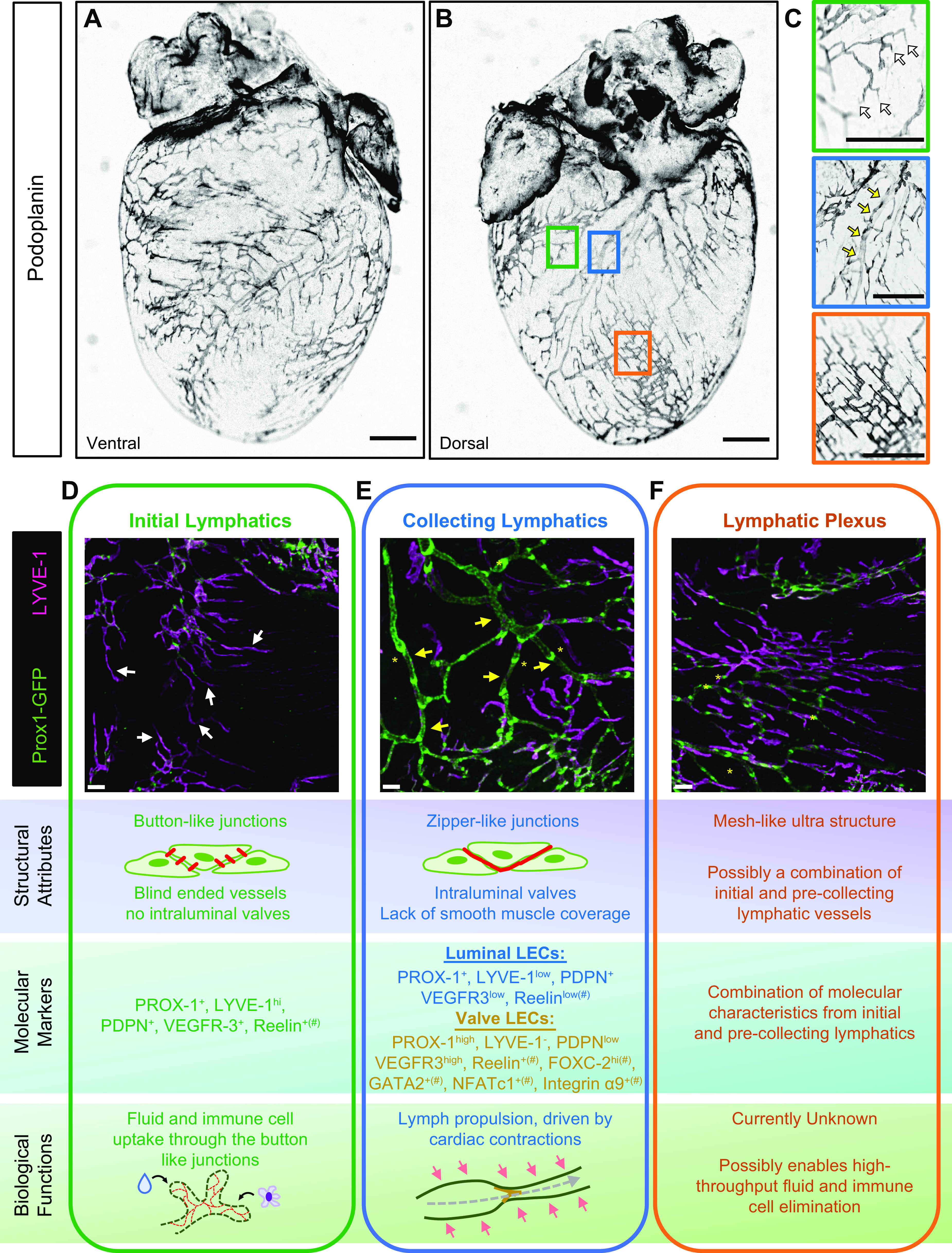
Structural characterization of the cardiac lymphatic vessels. A–C: lymphatic vessels of young adult C57BL/6 heart after whole mount podoplanin staining. Low-magnification representative images are shown of the ventral (A) and dorsal (B) sides of the heart. C: enlarged insets of podoplanin-stained heart demonstrate typical cardiac lymphatic structures, involving initial and collecting lymphatic vessels and lymphatic plexus. Scale bars, 1 mm. D–F: structural and functional characterization of cardiac lymphatic structures. Representative images of green fluorescent protein (GFP)+ and lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE-1+) lymphatic vessels of young adult Prox1GFP hearts are shown. Scale bars, 300 µm. White arrows point at initial lymphatics (C and D), yellow arrows point at collecting lymphatics (C and E), and yellow asterisks mark examples for lymphatic valves (E and F). Structural attributes, molecular characteristics, including classic lymphatic markers, and biological functions are listed for each vessel type. #Presumed expression levels of molecules whose expression patterns have not yet been characterized in detail in the cardiac lymphatics.
Although there is variation in the distribution of cardiac lymphatic vessels between mammalian species, cardiac lymphatics are present in the atrium and ventricles and across layers of the heart including the septum, mitral valve, conduction system, and papillary muscle (5, 6, 10–13). In most species, including humans, lymphatic capillaries are located uniformly in the subendocardial, myocardial, and subepicardial regions, forming a continuous plexus (6). The human heart has a network of lymphatic channels that connect from the endocardium, through the myocardium, and converge on the epicardial surface of the heart, where the densest network of vessels is seen. This anatomical pathway is conserved between humans, dogs, and pigs. However, other species, such as rabbits and mice, are reported to display different anatomical localization of the cardiac lymphatic network (10). Specifically, in mice, the lymphatics are much denser on the epicardial surface of the heart, with less lymphatics penetrating to the myocardium and endocardium (14). Collecting cardiac lymphatic vessels contain intraluminal leaflet valves to maintain unidirectional flow of lymph. In humans, the smaller lymphatic capillaries of the myocardial plexus converge into larger valve-containing collector vessels present in the subepicardium that align along the major coronary artery branches (15). In rodents, these larger collector vessels run parallel to cardiac veins, with one collector situated alongside the left conal vein and the other major collector parallel to the left cardiac vein (16). Regardless, these vessels converge toward the base of the heart, where extracardiac collector vessels empty into mediastinal lymph nodes under the aortic arch and around the trachea (17).
In addition to interspecies differences, sex-based differences in cardiac lymphatic anatomy and physiology have been described (14, 18, 19). Specifically in wild-type C57BL/6 mice, female hearts show increased coverage of cardiac lymphatics on the epicardial surface of the heart compared with male hearts (14). Furthermore, in a model of myocardial edema induced by cauterization of the coronary sinus, female C57BL/6 mice exhibit short-term protection against edema development relative to their male counterparts (18). This is particularly relevant given that a large proportion of cardiovascular diseases show sexual dimorphism regarding disease prevalence and outcomes. Women have less incidence of cardiovascular disease than men but increased mortality and worse prognosis following acute cardiovascular events (20). Sex hormones and now sex chromosomes (21), via X-linked gene dosage, have been shown to modulate cardiac disparities, opening new avenues to study genetic drivers of sex-dependent differences in cardiac lymphatic biology.
Interindividual differences in lymphatic structures have been known for decades. Not surprisingly, anatomical variations of the thoracic duct, the biggest lymphatic structure in the body, point to wide interindividual differences in lymphatic anatomy and function. A recent systematic review compared historical reports on human thoracic duct anatomy and found that the textbook termination of the thoracic duct in the internal jugular vein occurs in approximately only half of the general population, highlighting a high degree of interindividual differences in lymphatic anatomy (22). It was also proposed that the distribution of alternate thoracic duct anatomy may vary among people from different geographic locations, hinting at possible environmental factors influencing development of lymphatic structures. Moreover, advances in lymphangiography techniques have enabled detailed in situ visualization of interindividual differences (23, 24) and have allowed for identification of common structural variations of the larger lymphatic vessels other than the thoracic duct (25). Although current techniques do not allow for complete characterization of all lymphatic structures, evidence of high heterogeneity in large lymphatic structures points toward a possibility of similar interindividual variation in fine lymphatic structures and in organ-specific lymphatic vascular beds. As mentioned above, the developmental program of the cardiac lymphatic vasculature is remarkably complex, with numerous progenitor populations and temporal signaling programs required to develop and maintain the lymphatic plexus. Therefore, ample opportunity exists for minor variation in these processes to occur, leading to potential alterations to cardiac lymphatic patterning and function. Case reports from primary lymphedema patients, whose lymphatic dysfunction results from genetic mutations, have pointed toward rare dysfunctions of cardiac-associated lymphatics (26), like the presence of large pericardial effusions, a supraphysiological accumulation of fluid in the pericardium, associated with faulty lymphatic drainage, discussed in more detail below in this review. The genetic and nongenetic factors that are involved in defining the structure and function of the cardiac lymphatics in an individual present an exciting and yet-unexplored avenue of research.
2.2. Physiology
The principal function of lymphatics and cardiac lymphatics is to shuttle lymph and fluid and molecules collected from the interstitium, actively participating in fluid homeostasis at both a tissue and a systemic level. Lymphatic endothelial cells (LECs) also exert an immunoregulatory response by trafficking immune cells around the body and modulating immune responses through expression of cell adhesion molecules and chemokines, as well as presenting antigens to circulating T cells (2, 27–29). Molecules, macromolecules, and immune cells enter the lymphatics through passive paracellular uptake between permeable intercellular junctions or through active transendothelial processes. Large molecules are preferentially excluded from the blood capillaries and enter lymphatic vessels. Fluid flows into the low-pressure environment of lymphatics relative to the higher-pressure environment of the interstitium and nearby blood vessels. Lymph is propelled continuously from the capillary lymphatic beds to coalescing larger structures, with ultimate return to the thoracic duct and return of fluids and molecules to venous circulation. This movement is aided by contractile units ending in one-way valves, preventing backflow.
Cardiac lymphatics differ from most other organ-specific lymphatic vessels in that there is sparse coverage of lymphatic muscle cells (LMCs) on the collecting lymphatics, which normally propel lymph back to the circulatory system (10, 11). The lymphatic pumping in cardiac lymphatics is unique from other lymphatic beds in that the contraction cycle of the heart is the predominant mechanical factor driving lymph flow (30). This was shown to be true with two studies; the first showed that myocardial lymph flow ceased during cardioplegic arrest, even with coronary perfusion (31, 32). The second study demonstrated that cardiac lymph flow increased when the speed of cardiac contraction was increased with dobutamine infusion (33). Hydrostatic capillary pressure fluctuates dramatically with each heartbeat, and thus fluid exchange probably only occurs during diastole, when interstitial hydrostatic pressure is low (30). During diastole, ventricular pressure drives lymph from subendocardial lymphatics to myocardial lymphatics (34) (FIGURE 2A). As the heart contracts during systole, lymph is propelled from the myocardial lymphatics toward the subepicardial lymphatics (34) (FIGURE 2B). Once at the large collector vessels, lymph is moved from the apex toward the base of the heart, eventually draining to mediastinal lymph nodes (10) (FIGURE 2C). These lymph nodes further transport lymph into the thoracic duct, which empties into the subclavian vein (30) (FIGURE 2D).
FIGURE 2.

Cardiac lymphatic draining routes. A and B: cardiac contractions drive lymphatic propulsion from the deep myocardial layers to the direction of subepicardial lymphatics in systole. A: transverse cross section of the heart. LV, left ventricle; RV, right ventricle. B: zoomed-in view of the major cardiac structures depicting lymph flow from the endocardium through the myocardium and out toward draining epicardial lymphatics. Interstitial fluid and immune cell uptake into the lymphatics take place in diastole. C: subepicardial lymphatic collectors drain lymph from the apical parts of the heart toward the base. Cardiac lymphatic drainage is demonstrated in C57BL/6 wild-type mouse by intramyocardial injection of Qdot605 macromolecule followed by anti-LYVE-1 whole mount immunostaining to visualize lymphatic vessels. Scale bar, 1 mm. Yellow arrows indicate direction of flow. Orange asterisk indicates location of mediastinal lymph node. D: major lymphatic nodes (LNs) and vessels of the thoracic cavity. Cardiac lymphatic vessels drain to the mediastinal lymph nodes predominantly (denoted in orange and with an asterisk).
Cardiac lymphatics, like other lymphatic beds, serve to regulate local fluid homeostasis and contribute to immune regulation. Much of the knowledge base on cardiac lymphatic physiology has been gleaned through the study of cardiac pathologies, which are discussed in depth in this review. Namely, cardiac lymphatics have been shown to resolve myocardial edema and participate in clearance of acute inflammatory responses by draining influxed immune cells away from the heart and to draining mediastinal lymph nodes (14, 18, 35–37).
2.3. Development of Cardiac Lymphatics
The development of cardiac lymphatics has been most heavily studied in murine models, where cardiac lymphatics develop soon after the blood vasculature during embryogenesis. The most extensive description detailing the timeline of cardiac lymphatic development was published by Klotz et al. (9). Lymphatic vessels in mice emerge at embryonic day (E)12.5, sprouting from extracardiac regions near the outflow tract on the ventral side of the heart. At E14.5, lymphatic vessels start populating the dorsal ventricle, originating from a region near the sinus venosus. At E16.5, the lymphatic vessels spread along the dorsal surface, and small vessels start to cover the ventral atria. By E18.5, near the end of embryonic development, an interconnected network of lymphatics blankets the ventral and dorsal sides of the heart, branching toward the apex of the heart. From birth until postnatal day (P)15, the cardiac lymphatic network remodels with additional branching and expansion to blanket the surface of the heart with complete superficial coverage. This network is maintained in quiescence throughout healthy adulthood. The adult network can experience striking remodeling in response to cardiovascular events, both physiological and pathological, and in response to exogenous lymphangiogenic signals, which are discussed in this review. Molecular factors that are key players of systemic lymphatic development have also been identified as participants in the organ-specific developmental program of cardiac lymphatics (TABLE 1).
Table 1.
Key molecular signaling pathways of the systemic lymphatic development that influence the organ-specific developmental program of cardiac lymphatics
| Protein | Function | Role in Embryonic Lymphatic Development | Role in Organ-Specific Development of Cardiac Lymphatics | References |
|---|---|---|---|---|
| Prox1 | Transcription factor | First indicator of lymphatic differentiation from venous endothelial cells during canonical specification | Contributes to the lymphatic cell fate commitment during the development of cardiac lymphatics. | (9, 38–43) |
| Indispensable role in the endothelial lymphatic commitment | Elimination in various cell lines (Tie2+, Vav1+, Isl1+, Cad5+ cells) disrupts the development of cardiac lymphatics. | |||
| VEGF-C | Growth factor | Binds to its lymphatic-specific receptor, VEGFR-3, to promote LEC growth and survival | Mitigation of its binding to its receptor leads to a delay in the developmental program of the cardiac lymphatics. | (39, 44–51) |
| Central role during establishment of the lymphatic vasculature and in adult regenerative lymphangiogenesis | ||||
| AM | Vasoactive hormone | Stabilizes lymphatic barrier function and allows LEC proliferation. | Overexpression leads to cardiac lymphatic hyperplasia. | (14, 52–57) |
| Loss of AM signaling reduces LEC proliferation. |
AM, adrenomedullin; LEC, lymphatic endothelial cell; Prox1, prospero homeobox protein 1; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
Like most lymphatic vessel beds, most cardiac lymphatic vessels are derived from venous endothelial cells from the cardinal vein, such as paraxial mesoderm derived tyrosine-protein kinase receptor 2 (Tie2+) venous endothelial cells (58). However, numerous studies demonstrated a high variety in the origin of cardiac lymphatic endothelial cells (9, 38, 58–60). These have been recently and eloquently reviewed (61). In brief, the hemogenic endothelium, originating from the embryonic yolk sac, has been proposed as a source of nonvenous LEC progenitors (9, 62). Recent lineage tracing-based studies identified three populations of cardiac lymphatic-promoting hemogenic endothelial cells: Pdgfrb+ (platlet-derived growth factor receptor beta), Csf1r+ (macrophage colony-stimulating factor 1 receptor), and Vav1+ (proto-oncogene vav) (9, 63). Two studies have also implicated the secondary heart field as a cardiac lymphatic progenitor source and that cardiac lymphatics on the ventral and dorsal side of the heart arise through different progenitors and molecular mechanisms (38, 60). Isl1+ (insulin gene enhancer protein 1) pharyngeal mesoderm was found to contribute to developing cardiac lymphatics near the outflow tract through a Prospero homeobox protein 1 (Prox1)-dependent mechanism. This Isl1+ secondary heart field progenitor pool was found to contribute preferentially to the development of ventral cardiac lymphatics (38, 60). Interestingly, these explorations have also uncovered a potential epicardial signaling mechanism, via retinoic acid (commonly known as vitamin A), for promoting cardiac lymphangiogenesis (60).
3. INTERSTITIAL FLUID REGULATION
3.1. Classical and Revised Principles of Microvascular Fluid Exchange
The human body contains a vast circulatory network that includes arteries, veins, and capillaries that transport blood to every tissue in the body. This network acts as an exchange system to transport nutrients and gases to surrounding cells and, conversely, to take up and remove waste by-products. The exchange process occurs at the level of the capillary, where plasma from the blood, rich in solutes, will leak out into surrounding tissue. The lymphatic system, a separate but connected vascular network, is vital to this process, as it returns interstitial fluid to the cardiovascular system at the subclavian vein. Traditionally, the movement of fluid and solutes across the capillary bed into the interstitium was thought to be regulated by Starling’s law of fluid filtration, which described the relationship between the hydrostatic and oncotic pressures of these two compartments while taking into consideration vascular permeability of different capillary beds (64). Specifically, Starling’s law stated that the hydrostatic pressure of the capillary (i.e., the physical force of fluid against enclosed barrier) was higher at the arterial side of the capillary versus the venous side because of pressure from upstream arterioles and that both the venous and arterial ends of the capillary saw higher hydrostatic pressure than the interstitium. Additionally, oncotic force (osmotic pressure generated by the presence of protein) was high in the capillary because of albumin and low in the interstitium. This all led to the hypothesis that, based on standard vascular permeability, there was net filtration at the arterial end of the capillary due to high hydrostatic pressure and almost complete resorption (∼90%) of the extravasated fluid back into the venous end of the capillary because of high oncotic forces. The remaining 10% of fluid within the interstitium is absorbed by surrounding lymphatics and returned to blood circulation (65) (FIGURE 3A, classical Starling forces at steady state).
FIGURE 3.

Classical and revised Starling forces highlight the importance of lymphatics in microvascular fluid exchange. Comparison of classical (A) and revised (B) principles of microvascular fluid exchange. The classic Starling forces depict the endothelium as a continuous semipermeable membrane where fluid movement depends upon a net imbalance between the osmotic pressure of the plasma proteins and hydrostatic pressure of fluid in the capillary. This results in outward fluid filtration on the arterial side of the microcirculation when interstitial hydrostatic pressure is the highest. Net inward filtration occurs on the venous end of the microcirculation, where capillary hydrostatic pressure has declined, and high oncotic pressure drives inward fluid flow from the interstitial space, which has lower oncotic and hydrostatic pressures. Approximately 10% of fluid lost from the arterial end of the circulation is taken up by lymphatic vessels and ultimately returned to circulation. New research has identified the endothelial glycocalyx layer, which sits on top of a protein-devoid space above the capillary endothelial cells, as shown in B. This structure is thought to play the role of the semipermeable membrane. Thus, oncotic pressure difference inside the endothelium rather than across the endothelium drives fluid filtration. In this model, the expected reabsorption of fluid is thought to return to circulation primarily as lymph. The local tissue microenvironment dictates which model most closely matches microvascular fluid exchange. ASL, adsorbed layer; EGL, endothelial glycocalyx layer; Js, solute filtration rate per unit endothelial area; Jv, volume filtration rate per unit endothelial area; Pc, capillary hydrostatic pressure; Pi, interstitial hydrostatic pressure; sub-EGL, subendothelial glycocalyx layer; πc, capillary oncotic pressure; πcollagen, interstitial collagen hydrostatic pressure; πg, glycocalyx oncotic pressure; πGAG, interstitial glycosaminoglycan oncotic pressure; πi, interstitial oncotic pressure; σ, osmotic reflection coefficient.
Indeed, Starling’s law of fluid filtration is still taught as the dogma of microvascular fluid movement. However, many studies have invalidated Starling’s law, calling for an updated paradigm in microvascular fluid movement across the capillary (FIGURE 3B, revised Starling forces at steady state). These studies have already been extensively reviewed (3, 66–73), but the main findings are highlighted below. First, and most importantly, in vivo measurements of capillaries have shown that in steady state there is no resorption of fluid at the venous end of the capillary but rather net filtration across the whole capillary in most capillary beds (exceptions include intestinal mucosa, the kidney, and the heart) (74). However, capillary beds with low blood pressure can absorb fluid transiently before returning to steady state (74, 75). Second, there is now known to be a negatively charged matrix of proteoglycans, glycosaminoglycans (GAGs), and absorbed plasma proteins (termed the glycocalyx layer) on the luminal surface of the endothelium that can repel proteins such as albumin and restrict solute movement across the endothelium (76). Studies using confocal microscopy observed a region beneath the glycocalyx layer that was devoid of protein (77). This led to the hypothesis that oncotic pressure gradient opposing fluid filtration originated across the glycocalyx and had nothing to do with interstitial protein concentration. Put more simply, net capillary filtration may be due to the oncotic pressure difference inside the endothelium rather than across the endothelium (71). Third, studies have shown that albumin can diffuse across the epithelium through large capillary pores (junctions devoid of glycocalyx region) into the interstitium. In fact, as much as half of the body’s albumin content has been measured to be extravascular, with interstitial oncotic pressure measuring 30–60% of plasma oncotic pressure and 10 g of albumin moving from plasma to lymph per hour (66, 72). Additionally, the interstitium itself is now considered a triphasic system composed of free-flowing fluids, a gel phase with GAG molecules, and a collagen matrix. The GAG molecules can bind sodium, which exerts osmotic pressure, whereas the stiffer collagen matrix exerts hydrostatic pressure, which can oppose the filtration force from the capillary (66, 68). Finally, the interstitium was previously thought to have positive interstitial pressure, but measurements have shown that in most tissues interstitial fluid pressure is negative (78). Together, these findings led to a revised hypothesis of microvascular fluid movement in which there is a low rate of filtration and lymph formation in most tissues, mainly due to the protein gradients created from the glycocalyx layer (70). Ultimately, the revised principle elaborates on the original concept that the tissue’s mean interstitial fluid oncotic pressure regulates movement of fluid across a membrane, to highlight the role that small volumes of interstitial fluid within a subcompartment surrounding the microvessels play (79). Contribution of the original Starling forces and updated Starling forces to the microvascular fluid movements may be different among tissues, depending on the local microenvironment. Therefore, future studies measuring Starling forces of different capillaries in different vascular beds, and the accompanying lymph flow, will be essential in understanding the niche role of lymphatics in microvascular fluid movement in different organs.
The microvasculature of the heart is unique compared to other organs, as the heart has a high blood capillary density, with an exchange surface area of ∼500 cm2g−1 compared to only 70 cm2g−1 in skeletal muscle (30). Additionally, the microvascular pore size is larger in the myocardium than in the skeletal muscle (30). Together, this produces a fluid flux that is 10 times greater on a per-gram basis than in other tissues such as lung or skeletal muscles. Moreover, although many organs have negative interstitial fluid pressures, the interstitial fluid pressure in the heart has never been shown to be negative (80). In fact, during the cardiac contraction cycle, the interstitial pressure of the myocardium fluctuates between 15 mmHg and 120 mmHg (81). Because of the constant flux of pressure seen in the myocardium, it is believed that under steady state these capillaries follow the original Starling’s law, where fluid exchange occurs at the venous end of the capillary (82).
3.2. Role of Lymphatics in Regulating Local Fluid Homeostasis
As described above, local pressure characteristics substantially determine fluid exchange between the intravascular and extravascular spaces. In most circumstances, there is net positive filtration across the capillary bed, and the lymphatic system absorbs the totality of this filtrate, highlighting the importance of the lymphatic system in fluid homeostasis. This is supported by studies that have identified lymph flow at the thoracic duct as a measure of how much lymph is being absorbed from the capillary filtrate. In 1938, a study in dogs discovered that the lymphatic system is essential in returning the entire capillary filtrate to the blood circulation via the thoracic duct, as diversion of fluid from the thoracic duct resulted in death due to fluid loss (83). This has been corroborated in humans, where total lymph flow has been measured to be ∼8 L/day. As human plasma volume is only ∼3 L, and the entire plasma volume leaves the circulation approximately once every 9 h, the lymphatic system seems to account for returning the majority of fluid back to the circulatory system (70). However, the lymphatic system is now understood to have a complex role in lymph production due to specialized fluid handling in different organs, which can alter the flow of lymph to the thoracic duct. For example, dermal lymphatic capillaries follow classical lymphatic function, with immune cell surveillance and absorption of interstitial fluid. Deeper dermal collecting lymphatic vessels rely on smooth muscle and large skeletal muscle contraction to propel lymph to distant lymphatic vessels, eventually leading to the thoracic duct (3). Lacteals in the small intestine, on the other hand, absorb fat from digested food, which is packaged into chylomicrons and eventually shuttled to the thoracic duct (84). The liver contains unique capillaries known as sinusoids that have large open pores and lack a basal lamina layer (85). Plasma is filtered out of the sinusoids through its open pores, where it eventually enters interstitial space and is taken up by lymphatic vessels. As the liver filters ∼1.7 L of blood per minute, it is not surprising that the liver produces a large amount of lymph, ∼25–50% of the lymph flowing through the thoracic duct (86). As mentioned above, cardiac lymphatics rely on the contraction of the heart, instead of smooth muscle cells, to propel lymph from the heart to draining mediastinal lymph nodes, meaning that disturbances or fluctuations in the contraction cycle of the heart can influence cardiac lymph flow, which eventually drains to the thoracic duct (30). Not surprisingly, the rate of lymph flow in the thoracic duct can vary widely depending on the volume of fat ingestion, scar tissue in the mediastinum, presence of portal hypertension, cardiac function, as well as other factors (87).
In the heart, the cardiac contraction cycle and myocardial lymph flow are intrinsically linked, such that changes in cardiac function during different cardiovascular diseases or interventions can alter the flow of lymph leaving the heart. Interstitial fluid pressure and intraluminal pressures are both predominantly generated by the contractions of the heart, and interstitial fluid may move according to the original theory of Starling’s forces. This means that drainage of interstitial fluid may be less dependent on the cardiac lymphatic vasculature compared to other organs, as interstitial fluid is also reabsorbed by venous capillaries. Defining the unique local hemodynamic characteristics of the heart will help determine the diverse functions of cardiac lymphatic vessels in cardiac health and disease.
4. MYOCARDIAL EDEMA—FORMATION TO EVALUATION
4.1. Mechanisms Driving Myocardial Edema Formation
Myocardial edema develops when imbalance occurs between the rate of fluid filtration from the coronary vasculature and the rate of interstitial fluid absorption by cardiac lymphatics and epicardial transudation (30). Cardiovascular disease and surgical intervention modulate multiple microvascular fluid exchange forces that result in increased myocardial microvascular filtration rate (Jv) and/or decreased lymphatic drainage (QL), culminating in the formation of myocardial edema. This edema can acutely or chronically contribute to cardiac dysfunction (FIGURE 4). Such edema-favoring alterations include elevated capillary hydrostatic pressure (arterial or venous), increased capillary permeability (via damage to the glycocalyx layer or disruption of the endothelial junctions), lower plasma oncotic pressure, and/or lymphatic obstruction or dysfunction. Interestingly, these modulations are often accompanied by antiedema measures. For example, increased filtration from blood capillaries into the interstitium can increase myocardial lymph flow by increasing lymph driving pressure (due to increased interstitial pressure) and decreasing lymphatic resistance (88). Additionally, protein washdown occurs during edema as increased fluid filtration (Jv) into the interstitium decreases the interstitial protein concentration (πi), thereby opposing edema formation by increasing the colloid osmotic pressure gradient (πi → πc) (81) (FIGURE 3).
FIGURE 4.

Cardiovascular (CV) disease perturbs microvascular fluid exchange forces, leading to myocardial edema formation. Flow chart depicting prevalent CV diseases and the unique and shared microvascular fluid exchange principles impacted in the disease pathophysiology. Singular or multiple insults to microvascular fluid exchange forces can lead to increased myocardial microvascular filtration rates and or decreased lymph flow rate driving cardiac fibrosis and myocardial edema and lead to cardiac dysfunction. Each disease (top) has an associated colored symbol used to identify the force(s) perturbed in that disease’s pathophysiology. MI, myocardial infarction.
The pathological alterations driving edema formation are both unique and shared across prevalent cardiovascular diseases (FIGURE 4). For example, increased Jv driven by an inflammation-induced increase in microvascular permeability is noted in myocarditis, myocardial infarction (MI), and heart failure. However, increased wall tension driving increased capillary hydrostatic pressure also contributes to the increased Jv noted in MI and heart failure. Another point of note is that antiedema measures, in the form of lymphangiogenesis, are also known to occur in cardiovascular diseases such as MI (9). Understanding of the microvascular fluid exchange forces driving myocardial edema is essential in the study of cardiac diseases. These principles guide selection of the appropriate surgical/genetic model of cardiovascular disease and help frame our understanding of the multifaceted role that cardiac lymphatics play in cardiac pathologies and resolution of myocardial edema.
4.2. Clinical and Preclinical Approaches Utilized for the Characterization of the Intersections Between Myocardial Edema and Cardiac Lymphatics
To explore the complex relationship between cardiovascular disease, myocardial edema, and cardiac lymphatics, it is imperative to identify and discuss the array of methods available to assess various aspects of cardiac and lymphatic biology (FIGURE 5). This section discusses the unique and overlapping techniques used in the clinic and at the bench to evaluate myocardial edema and lymphatic structure and function. It discusses advantages and disadvantages of these methods and some of the technical barriers preventing better clinical assessments of cardiac lymphatics in cardiac physiology and pathology.
FIGURE 5.

A quick reference guide comparing the congruency and dichotomy of tools used to assess the relationship between myocardial edema and cardiac lymphatics in the clinic and at the bench. Table depicting common methods utilized in the clinic and in preclinical animal models to evaluate myocardial edema, lymphatic biology, and lymphatic signaling pathways. Knowledge of myocardial edema is aided by shared clinical and preclinical imaging modalities. A wealth of genetic tools, immunohistological analysis, lymphangiographic techniques, and ex vivo molecular analyses has allowed for in-depth characterization of lymphatic anatomy, physiology, and signaling in preclinical animal models. Conversely, the lack of tools in the clinical arsenal highlights the pressing need to develop new methodologies to directly evaluate cardiac lymphatic function and signaling in humans. Together this emphasizes the importance of leveraging animal models to study myocardial edema, its driving forces, and the complex interactions of cardiac lymphatics in cardiovascular (CV) pathophysiology. CMR, cardiac magnetic resonance; CT, computed tomography.
4.2.1. Preclinical assessment of cardiac lymphatics—anatomy and function.
The current knowledge of the anatomy and function of cardiac lymphatic vessels relies highly on preclinical data. Until the development of modern microscopic and imaging techniques, characterization of lymphatic vessels was predominantly dependent on their function rather than gross anatomy. In the eighteenth century, injections of dyes and mercury were used for investigation of the anatomy of the lymphatic system (89, 90). The lymphatic system is not readily amenable to angiographic techniques commonly used to visualize blood vessels, as the lymphatic system is open ended and carries semitransparent lymph fluids, providing little visual contrast. Furthermore, with poor contrast, visualization is often restricted to a particular lymphatic tree, dependent upon the point of tracer injection (91). Lymphangiographic techniques generally rely on peripheral, rather than central, injection of exogenous tracers, which are readily taken up by proximal lymphatics because of their size or other chemical properties (i.e., optimal charge or lipophilicity); then subsequent imaging of the tracer is performed to monitor lymphatic parameters such as structure, flow, and pumping. Current imaging modalities include lymphoscintigraphy, lymphangiography, positron emission tomography coupled with computed tomography (PET/CT), contrast-enhanced ultrasound, noncontrast or contrast magnetic resonance imaging (MRI), and recently near-infrared imaging (NIR) utilizing the fluorescent dye iodocyanine green (ICG) (91, 92). Photoacoustic imaging has emerged as a noninvasive lymphatic imaging tool with high spatial and temporal resolution and has been demonstrated in the clinic to successfully detect sentinel lymph nodes in patients with breast cancer and to observe lymphatic pumping (93–96). In vivo reflectance laser scanning confocal microscopy is emerging as a promising new lymphatic clinical imaging technology to be used in the support of lymphedema diagnosis. It has been recently used in vivo in a rat tail lymphedema model to create three-dimensional lymphatic images with resolution comparable to that of histological examinations (97).
Although these techniques have been used in the clinic to monitor and assess lymphedema, detect metastatic lesions, and locate shallow lymphatic beds, they often bear decisive weaknesses. Namely, many noninvasive techniques, those that do not require peripheral or intranodal injection of a tracer (MRI, ultrasound, CT), are plagued by poor spatial resolution and sensitivity and do not provide functional parameters or visualization of small vessels. Tracer-based imaging provides increased spatial and temporal resolution, allowing for anatomical mapping and functional parameters. These techniques can be expensive, can expose patients and health care providers to radioactivity, and may require invasive surgical administration to access the desired lymphatic bed or lymph node. This is especially problematic in the assessment of cardiac lymphatics. Noninvasive imaging techniques are unable to detect the delicate single layer of lymphatics lying on the epicardial surface of the heart, and tracer-based systems are highly invasive and dangerous in a clinical context. This would require a tracer to be injected directly into the endocardium and allowed to accumulate in cardiac lymphatics (98). Such methods have been developed for animal models. Invasive cardiac lymphangiography relies on an intramyocardial injection of fluorescent quantum dots into the apex of the heart of an anesthetized and ventilated animal (35). The quantum dots are then taken up by lymphatics, and the rhythmic contraction of the heart propels the quantum dot-containing lymph toward draining cardiac lymph nodes at the top of the heart. Fluorescent imaging of this heart allows for visualization of the functional cardiac lymphatic plexus. This technique is restricted to preclinical research, as human application would require a surgically inserted catheterization system to administer tracer or a percutaneous endocardial injection, both of which pose significant risk of injury and cardiotoxicity and monetary costs.
Characterization of the anatomy of cardiac lymphatics has almost exclusively been determined through preclinical assessments. Immunohistochemistry and immunofluorescence for lymphatic markers have provided the best visualization of the cardiac lymphatic plexus. The dense lymphatic network blanketing the surface of the heart has been detected best by whole mount or sectional staining for lymphatic markers such as lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE-1), podoplanin, and vascular endothelial growth factor (VEGF) receptor (VEGFR)-3 (9, 14, 35, 44, 98). Transgenic lymphatic reporter strains, including models in which LacZ (99–102) or fluorescent reporter genes (103–110) driven by LEC-specific promoters, such as Prox-1 (100, 103, 105, 106, 108, 109), VEGFR-3 (99, 101, 104, 107, 110), and LYVE-1 (102), are widely used for visualization of the lymphatic vessels. Novel models that combine LEC-specific Cre-expressing lines with Cre-inducible reporter models (111–113) provide a more sophisticated tool for the detection of lymphatics. It is important to note that only four lymphatic markers are regularly used to detect lymphatics and often a combination of these markers must be used to explore lymphatic biology because of the expression of these markers in other cell types. This problem is especially evident in the heart, as podoplanin is expressed in the developing and injury-reactivated epicardium. Prox1 is expressed in both lymphatic endothelial cells and cardiomyocytes. LYVE-1 is expressed in lymphatics and macrophages, both tissue-resident macrophages and infiltrating macrophages delivered to the heart during injury. Thus, careful consideration must be made when studying the biology of cardiac lymphatics in both cardiac physiology and pathophysiology.
4.2.2. Assessment of myocardial edema.
The most definitive and commonly used assessments of myocardial edema are based upon gravimetric and histological analysis. Gravimetric methods rely on direct measurement of cardiac water content in the cardiac walls, typically transmural pieces of the ventricles. Measurements of cardiac tissue weight before and after extended periods of oven drying provide a wet-to-dry ratio of the heart allowing for a quantitative measurement of water content. Histological assessments rely on visualizing and quantitating the interstitial space between cardiomyocytes along transverse sections of the ventricles. These methods are largely restricted to preclinical analysis and have provided a quantitative and consistent way to measure myocardial edema across species and disease models.
Understanding of the role of myocardial edema in cardiac pathologies has primarily been driven by real-time imaging-based assessments. Clinical imaging serves as a routine aspect of clinical care for cardiovascular disease, aiding in the diagnosis and assessment of cardiovascular pathologies. These multifaceted technologies allow for comprehensive evaluation of myocardial structure and function, tissue viability, ischemia, and cardiac water content (114). These imaging modalities are real time and noninvasive, making them ideal for both clinical and preclinical assessments of cardiovascular disease. Accordingly, techniques such as cardiac magnetic resonance (CMR), CT, and echocardiography are used to evaluate heart function in both humans and animals.
The most robust and studied method to observe myocardial edema clinically is the use of T2-weighted MRI. This method measures relaxation properties of protons after radiofrequency pulses, resulting in tissue contrast. Thus, cardiac MRI signals are primarily derived from water protons, making it the logical choice to study edema. The long relaxation times of water-bound proteins generate a water-specific contrast that results in a high signal intensity of edematous tissue and gives high contrast between blood, fat, normal myocardium, and myocardial edema (115, 116). Older T2-weighted sequences were not as sensitive because of low signal-to-noise ratio and inconsistent image quality, and so older studies investigating the link between myocardial edema and acute allograft rejection following cardiac transplantation had variable results (117). This may be due in part to the inability to distinguish whether the cardiac edema noted was intercellular or extracellular and to poor image resolution that made edema detection more subjective. More recent studies investigating this link have improved the T2-weighted sequences for more sensitive, reproducible, and quantifiable images, and one study found that T2-weighted MRI imaging was a more reliable prognostic marker of transplant rejection than the gold standard, endomyocardial biopsy (118). Cardiac magnetic resonance (CMR) imaging involving T2-weighted sequences to measure myocardial edema has become an integral tool in the diagnosis of some cardiac pathologies and has been extensively applied after MI in both patients and large-animal models. The utilization of CMR imaging to measure myocardial edema could act as a prognostic factor for disease severity and the probability of development into other related cardiovascular diseases. In fact, measurements of myocardial edema by T2-weighted sequences can help define the myocardium at risk in both reperfused and nonreperfused MI and help distinguish between acute and chronic disease (119–122). Other noninvasive imaging technologies such as CT and echocardiography allow for qualitative assessment of myocardial edema. CT has been shown to be effective, and comparable to CMR, in the evaluation of myocardial edema in a porcine model of large acute myocardial infarction (123). Serial echocardiography performed under the context of cardiovascular disease can reveal changes in left ventricular and septal wall thickness as well as indicate changes in systolic and diastolic cardiac function. These changes can be compared to postmortem histological assessments of myocardial edema.
4.3. Animal Models of Cardiovascular Diseases and Myocardial Edema
4.3.1. Overview of preclinical surgical models used to induce myocardial edema.
Myocardial edema in animals and humans primarily occurs as a secondary response to direct cardiac injury or acute hemodynamic imbalance. Reproducible surgical and/or genetic animal models have been successfully employed to evaluate the complex role of cardiac lymphatic vessels in the pathophysiology of myocardial edema. These various surgical methods allow for regional or global cardiac injury, induce myocardial edema, and provide temporal modeling of cardiovascular disease, thereby supplying researchers with representative models of clinical cardiac pathologies (FIGURE 6).
FIGURE 6.
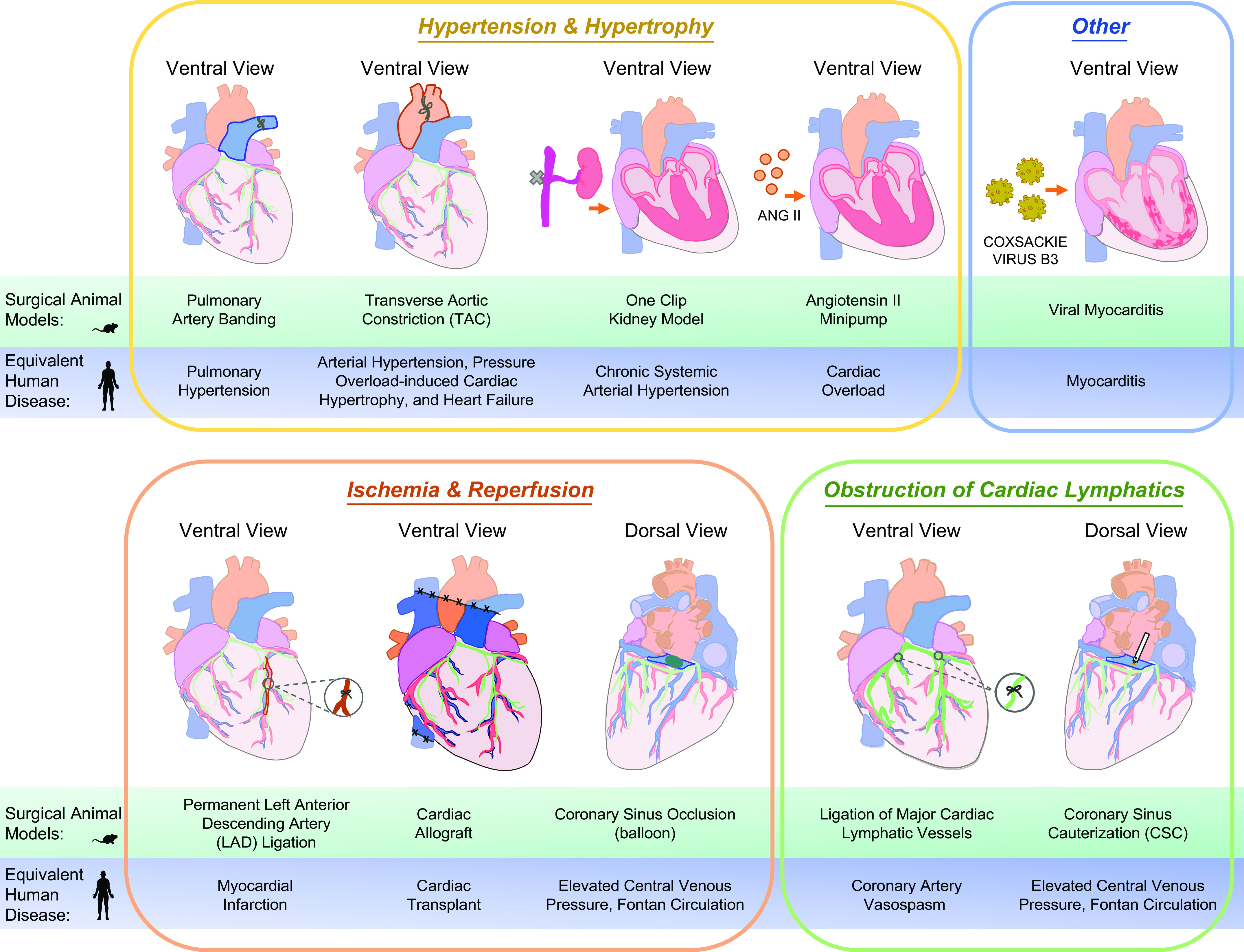
Preclinical surgical modeling of myocardial edema and cardiovascular disease. Various techniques have been employed in both small and large animals to mimic cardiovascular disease and myocardial edema observed in humans. These surgical models, in addition to transgenic/disease prone mouse models, have greatly aided in the exploration of cardiac lymphatic vessels in the pathophysiology of myocardial edema. Displayed are visual representations of common surgical procedures performed directly on the heart or in the periphery to mimic a particular cardiovascular disease such as focal ischemia caused by an infarct. The surgical model is listed above the correlating clinical disease/pathology.
Historically, some of the most informative and pivotal studies interrogating the role of cardiac lymphatics in myocardial edema have been conducted in large animals, primarily because of the large size of the heart relative to that of rodents. Additionally, large animals, like dogs and pigs, possess cardiac architecture, heart rates, oxygen consumption, contractility, protein expression, and stem cell populations similar to human hearts (124). Furthermore, cardiac lymphatic anatomy and lymph drainage pathways are like humans, as mentioned above in this review. These models, however, lack the genetic power of rodent models to specifically interrogate signaling pathways and candidate drivers of myocardial edema through generating gene knockout or overexpression models. With the advancement of surgical tools and expertise, new and existing surgical models have been adapted for small mammals. Therefore, leveraging large- and small-animal surgical models in complement to transgenic mouse models is critical for investigating CVD drivers and potential therapies.
Pulmonary hypertension and other conditions of elevated central venous pressure comprise a large proportion of myocardial edema cases in humans. Consequently, numerous surgical techniques have been developed to induce elevated central venous pressure in the primary context of stressed lymphatic drainage or in a secondary context resulting from focal complications of ischemic cardiac events, such as myocardial infarction. Elegant studies in dogs in the 1990s used chronic pulmonary artery banding [resulting in elevated central venous pressure (↑CVP)], acute coronary lymphatic ligation (CLL) and thoracic duct ligation (TDL), or acute ligation of the coronary sinus (CSL, a large conduit vessel that drains coronary venous blood into the right atria) to induce myocardial edema in the absence of ischemia (125–127). Recently the authors of the present review have performed and validated CSL in mice and have also published and validated a new coronary sinus cauterization (CSC) technique in mice to induce ↑CVP (18). Chronic balloon obstruction of the coronary sinus in sheep has also been used to generate a model of ↑CVP (128). A host of animal models including pigs, guinea pigs, rats, rabbits, mice, and dogs have been used in ischemic surgical models to study secondary myocardial edema (35, 129–133). Common among these surgical methods is the generation of myocardial edema, which is principally measured by gravimetric analyses focused on analyzing the water content of the ventricle or pieces of myocardium. A retrospective analysis of these historical and contemporary surgical studies reveals that 1) chronic surgeries/lingering cardiovascular pathologies (on the scale of days to weeks) cause greater myocardial edema than acute, hourly challenges and that 2) the absolute magnitude of myocardial edema induced is conserved across techniques (regardless of ischemia) and across species, with nearly all surgical techniques producing <10% increase in myocardial water content (FIGURE 7, A and B). The broad diversity of animal models and techniques available provides a wide repertoire of approaches to study myocardial edema in various conditions, thereby exploring the cardiac lymphatic response in each of these diseases. Additionally, this hints at a finite tolerance of myocardial edema and perhaps conserved antiedema mechanisms deployed by the heart during various cardiovascular pathologies.
FIGURE 7.

Absolute magnitude of edema is conserved across species and surgical models of myocardial edema. Review and analysis of seminal and contemporary studies evaluating myocardial edema in the context of ischemic injury, lymphatic obstruction, and elevated central venous pressure reveal that myocardial edema is highly conserved across mammals and across myocardial edema-inducing pathologies. Myocardial edema data gathered as gravimetric analysis of cardiac wet:dry weight (A) or as water content (%) in the left ventricle (B) from literature review or through never-before published primary data from the authors of the present review (coronary sinus ligation, mouse). The experimental animals in each surgical model presented with increased myocardial edema, which persisted over time, compared with sham-treated control animals. C: serial cardiac magnetic resonance imaging (CMR) analysis of infarcted myocardium of humans and pigs reveals a similar bimodal pattern of myocardial edema. Myocardial edema was assessed by identifying the area of myocardium at risk and calculating this as % of total area of the left ventricle (LV). This literature-based cross-species and cross-surgical comparison reveals a highly similar % change in edema, highlighting the ability to use any of these models to study the interplay of cardiac lymphatics and myocardial edema. Legend depicts color-coded species; color shades refer to sham or experimental groups. CLL, coronary lymphatic ligation; CSC, coronary sinus cauterization; CSL, coronary sinus ligation; CVP, central venous pressure; d, day; Expt., experimental; MI, myocardial infarction; Sham, sham treated; TDL, thoracic duct ligation.
4.3.2. Foundational knowledge gained from preclinical animal models of myocardial edema.
The first major explorations into the pathophysiology of myocardial edema centered on obstructing cardiac lymphatic flow in dogs and pigs by means of ligating the major cardiac lymphatic vessels. In most cases, this surgical technique involves injecting the myocardium with Evans blue dye to visualize the draining lymphatic system and then resecting cardiac lymph nodes, ligating the major lymphatic vessels coronary lymphatic ligation (CLL) and sometimes also ligating the thoracic duct (TDL). These studies generated foundational knowledge of the acute and chronic consequences of myocardial edema and of lymphatic anatomy. These studies provided detailed descriptions of interstitial edema, dilation of lymphatic capillaries, interstitial fibrosis, subendocardial hemorrhage, electrocardiogram abnormalities consistent with hypoxia, thickening valve leaflets, and decreased cardiac contractility (126, 134–139). Interestingly, one study focused on the effect of cardiac lymphatic obstruction on the coronary arterial wall and coronary circulation. Surprisingly, they found evidence of subendothelial edema, interstitial and intracellular edema in the tunica media, and dilated lymph vessels and fibrosis in the adventitial space (140). Additionally, coronary circulatory reserve was found to decrease with lymphatic obstruction, in conjunction with development of numerous coronary arteriovenous microshunts (140).
Increasing pressure within the coronary sinus is a popular surgical method used to induce acute myocardial edema. This technique elevates capillary hydrostatic pressure within the venous coronary vasculature system, preventing absorption of interstitial fluid back into the capillary, leading to interstitial edema. Most of these preclinical studies were focused on mere hours after surgery on the acute consequences of myocardial edema. As expected, increasing coronary sinus pressure increased lymphatic drainage and caused significant myocardial edema compared with control animals (125, 140, 141). This acute incidence of myocardial edema was enough to reduce contractility within 3 h of surgery (125, 127, 128, 140, 142, 143). In fact, 3 h after coronary sinus pressure was reduced to baseline values and myocardial edema resolved, diastolic stiffness remained significantly elevated and cardiac function compromised (142). A more recent study was the first to implement this technique in mice, by cauterizing the coronary sinus, and found, as previously reported, a significant increase in myocardial water content 3 h after surgery compared with control mice (18). There were remarkable differences in the resolution of myocardial edema between sexes, in which females had a short period of cardioprotection before developing myocardial edema compared with males. This emphasizes the importance of studying sex differences in cardiovascular and cardiac lymphatic biology. Interestingly, ejection fraction and fractional shortening were significantly reduced 24 h after surgery, at a time when myocardial edema had resolved. This study was the only one to measure long-term effects of elevated coronary sinus pressure, which included increased lymphangiogenesis and fibrosis 28 days after surgery (18). Further in-depth experiments are needed to understand the relationship between cardiac lymphatics, myocardial edema, and cardiac contractile dysfunction.
To better understand the connection between myocardial edema and cardiac dysfunction, researchers have used a combination of the methods described above to produce chronic myocardial edema with an additional acute myocardial edema insult. Laine and Allen (125) found that reducing myocardial lymph flow in combination with elevated coronary sinus pressure produced more myocardial edema than just elevated coronary sinus pressure alone. Additionally, arterial hypertensive animals subjected to acute coronary sinus pressure increase had significantly more myocardial edema than both normotensive and hypertensive control animals (144). Desai et al. (145) used a model of chronic pulmonary artery banding to simulate chronic myocardial edema and acutely increased myocardial edema even more through elevating pressure in the coronary sinus. Their results show that generating acute myocardial edema in control animals resulted in an increase in interstitial fluid pressure that was significantly higher than that resulting from acute myocardial edema in animals with chronic edema, which they suggest could be due to enhanced venous outflow from the thebesian veins or shifting fluid movement across the epicardium (transudation) (145).
Other groups have studied the effect of increased myocardial edema following myocardial infarction (MI) in large-animal models. Ischemia-reperfusion studies in pigs and dogs have revealed that during early stages of ischemic injury myocardial edema occurs as intercardiomyocyte swelling (146). Upon reperfusion, a sharp increase in interstitial edema is observed, secondary to rupturing of cardiomyocytes, fluid loss from damaged capillaries, and reactive hyperemia (147, 148). CMR studies examining ischemic myocardium, in both humans and pigs, have revealed that myocardial edema is unstable and follows a bimodal pattern (129, 149) (FIGURE 7C). An initial wave of edema peaks abruptly after reperfusion and sharply reduces by 24 h, which is followed by a deferred progressive wave of edema that peaks between 4 and 7 days after injury (129). Kline et al. (150) studied the effect of cardiac lymphatic obstruction following MI compared with MI alone and found that lymphatic obstruction produced larger infarct size, increased fibrosis and calcium salt deposits, and more inflammation compared with MI alone. Diab et al. (151) subjected animals to coronary sinus ligation after MI and found a significant decrease in left ventricular systolic function, increase in necrosis, and presence of dilated congested thebesian sinusoids compared with MI alone. These studies highlight the importance of fluid homeostasis within the heart and provide evidence that myocardial edema has a variety of pathological consequences involving all aspects of the heart including the myocardium, endocardium, lymphatics, conduction system, and coronary circulation.
5. EXAMINING THE INTERSECTION BETWEEN MYOCARDIAL EDEMA AND CARDIAC LYMPHATICS IN CARDIOVASCULAR PATHOLOGIES
The interdependence of the cardiac cycle and myocardial lymph flow has been recognized for some time, as ventricular fibrillation and cardioplegic arrest have been associated with stalled lymphatic drainage (31, 32, 152, 153). A multitude of cardiac conditions that are associated with poor myocardial contraction are also associated with myocardial edema. It is well recognized that myocardial edema impairs both systolic and diastolic cardiac function by decreasing cardiac output, decreasing ventricular compliance, and increasing isovolumic ventricular relaxation (125, 154–156). Moreover, increased interstitial volume in the myocardium can lead to poor tissue oxygenation via increasing oxygen diffusion distances to cardiomyocytes, pathological alterations of the interstitial matrix, local inflammation and fibrosis, and disruption of electrical conduction, all of which contribute to ventricular stiffening and/or suboptimal cardiac function (125, 127, 142, 154, 157). Cardiac lymphatics play a critical role in maintaining local fluid balance in the heart, and structural and functional alterations in these vessels have been shown in both acute and chronic cardiac diseases. Antiedema measures, in the form of lymphangiogenesis, have been shown to occur in cardiovascular diseases such as MI. This has been thought to increase lymphatic flow rates and temper excessive formation of myocardial edema. The clinical evaluation of myocardial edema, or cardiac lymphatics, has not yet emerged as a common diagnostic tool or criterion because of the limited availability to accurately measure myocardial edema and cardiac lymph flow via available imaging modalities, as discussed above. Thus, surgical animal models of cardiovascular disease are employed to characterize and define the causes and consequences of myocardial edema and its relationship to cardiac lymphatics. This section reviews the clinical and preclinical studies focused on elucidating the complex interplay between myocardial edema and cardiac lymphatics, which are shared and unique among cardiovascular diseases.
5.1. Coronary Bypass Surgery
Coronary bypass surgery involves grafting blood vessels to coronary arteries to bypass blocked or narrowed arteries to improve blood flow to the myocardium. Traditionally, this surgery required cardioplegic arrest and a cardiopulmonary bypass machine, which takes the place of the heart to pump blood throughout the body, called “on-pump” surgery. With the knowledge of microvasculature fluid dynamics in the heart, it is no surprise that on-pump coronary bypass surgery creates myocardial edema through a multitude of different mechanisms. First, as cardiac lymph flow requires the cardiac contraction cycle to move lymph away from the heart, stopping the heart in diastole to perform the surgery creates edema by halting lymph flow. Second, the physical surgery itself produces multiple cytokines and inflammatory signals that increase microvascular permeability and thus fluid filtration into the interstitium. Finally, the solutions used to create cardioplegic arrest result in low capillary colloid osmotic pressure, which results in less absorption of fluid at the venular end of the capillary (30). Indeed, this procedure has been known to create such extreme myocardial edema that in some patients surgeons must wait for the edema to resolve after surgery before closing the patients’ chest to ensure that their heart will fit within their chest cavity (81). Additionally, this method can cause local loss of blood flow, known as ischemia. One elegant animal study indicated that ischemia-reperfusion injury following coronary bypass surgery was the main cause of hemodynamic dysfunction and myocardial edema (158).
In recent years, more sophisticated technology has allowed for coronary bypass surgery to be performed without cardioplegic arrest, termed “off-pump” surgery. However, this technique requires stabilization platforms or suctioning devices for cardiac positioning and local compression or vacuum suction of the epicardium for cardiac stabilization (159). Both of these methods can compromise the hemodynamics of the heart by altering cardiac output, stroke volume, and mean arterial pressure (159). Additionally, these methods can cause transient regional myocardial ischemia, which may further contribute to hemodynamic instability. However, one study has shown that a small degree of cardiac contraction during coronary bypass surgery is sufficient to maintain lymph flow, and thus minimize myocardial edema (160). Thus, it would seem that the off-pump technique may be beneficial compared with the on-pump technique in minimizing myocardial edema. However, the advantages to either technique in regard to minimizing edema have yet to be directly studied.
Finally, there is a new hybrid method, on-pump beating-heart coronary artery bypass, which maintains coronary blood flow to reduce myocardial injury and stabilize hemodynamics and avoids complications caused by cardioplegic arrest (161). Although this method seems to limit hemodynamic instability and therefore potentially decrease formation of myocardial edema, more studies need to be done to verify this hypothesis. Additionally, the advantages of these three techniques for coronary bypass surgery are still being debated, as some studies claim a benefit for one over the other, and other studies find no long-term differences between techniques (161–165). Future studies comparing these techniques should focus on the development of myocardial edema as a consideration for long-term outcomes.
5.2. Cardiac Transplantation
Cardiac transplantation involves placing patients on a heart-lung bypass machine, removing the diseased heart, and attaching the donor heart. When the patient’s major blood vessels are attached to the donor heart, the restoration of blood flow often initializes cardiac contractions (166). This restoration of blood flow results in ischemia-reperfusion injury that can produce cytokines and alter capillary permeability; substantial edema has been shown to develop following reperfusion after transplantation (167). However, donor cardiac lymphatics are not anastomosed to receipt lymphatics during this surgery, leaving reactionary lymphangiogenesis as the only mechanism to reestablish myocardial lymphatic flow (168). This has pathological consequences due to the fact that cardiac lymphatics have two main functions: first, to traffic immune cells to lymph nodes, which can initiate an immune response, and second, to regulate myocardial fluid homeostasis resulting in edema prevention. Despite the use of potent immunosuppressive drugs, one of the most devastating complications following transplantation is acute cardiac allograft rejection.
Two studies investigated the role of lymphangiogenesis in allograft rejection and found increased graft expression of the lymphangiocrine factor VEGF-C and its receptor VEGFR-3 in mice (169). When researchers pharmacologically mimicked this effect with VEGF-C/VEGFR-3 stimulation, they found early lymphatic activation that correlated with an increase in allograft inflammation (170). Moreover, when they blocked the VEGF-C/VEGFR-3 pathway, the lymphatic vessel response was decreased, as of acute and chronic rejection (170). These studies have broad clinical implications for targeting the VEGF-C/VEGFR-3 pathway to decrease allograft rejection that should be followed up with clinical trials.
Additionally, 10–12% of cardiac transplantation patients develop a condition called cardiac allograft vasculopathy (CAV) each year, making CAV the leading cause of death in patients >5 yr after transplantation (167). CAV occurs when the coronary arteries of the allograft narrow or become occluded, and it has been hypothesized that this results from lack of lymphatic flow in the transplanted heart (167, 171). One study tested this hypothesis with computed tomography lymphoscintigraphy in a murine model of chronic rejection following cardiac transplantation. Surprisingly, their results demonstrated that chronic rejection resulted in increased lymphatic flow to the draining lymph nodes, which they hypothesized could promote immune cell trafficking, alloimmunity, and CAV (167). Thus, blunting the lymphangiogenic response following surgery may decrease the chance of rejection and CAV development in these patients. Recent studies have shown a direct link between myocardial edema and acute rejection (117, 118, 172, 173). However, more studies are needed to fully understand the role of cardiac lymphatics in the recovery of patients after cardiac transplantation and their relationship with myocardial edema, allograft rejection, and CAV.
5.3. Myocarditis
There are many causes of myocarditis, or inflammation of the heart, but acute cases are most commonly due to viral infection. The infection causes direct cardiomyocyte injury, triggering an inflammatory response that results in edema, necrosis, and possible contractile dysfunction (174). Prolonged autoimmune response or virus presence can cause chronic inflammation, which is considered a frequent cause of dilated cardiomyopathy (DCM) (175). The pathogenesis of myocarditis in humans is not completely understood, but murine models have provided a template for the disease progression of myocarditis into cardiomyopathy. After the acute inflammatory phase, there is a second autoimmune phase, which can last from weeks to months, during which activated T lymphocytes target viral proteins and some cardiac proteins (176). This inflammatory response can further aggravate myocardial damage and cause contractile dysfunction. Although this immune response may decline with elimination of the virus, chronic inflammation may develop, leading to left ventricular remodeling and development of dilated cardiomyopathy (175).
The etiology of edema seen with myocarditis can be explained by inflammatory cytokines and the inflammatory response, which alters capillary permeability and can increase fluid movement into the myocardium interstitium. This has been supported by findings of another study, which found that, by improving the glycocalyx function, the secreted glycoprotein SPARC protects against myocardial inflammation during viral myocarditis, thus strengthening endothelial barrier function (177). The role of cardiac lymphatics in myocarditis has not been heavily studied, but an increase in cardiac lymphatic density has been found in humans suffering from myocarditis (178). Additionally, one study found that exposure of isolated rat cardiac lymphatic smooth muscle cells to inflammatory mediators like tumor necrosis factor (TNF)-α, commonly seen during inflammatory conditions such as myocarditis, significantly decreased the contractility of these cells (179). Further investigation into the response of cardiac lymphatics and mediastinal lymph nodes to myocarditis could help future researchers understand the relationship between inflammation, myocardial edema, and contractile dysfunction.
5.4. Myocardial Infarction
Myocardial infarction (MI) results from a sudden blockage of coronary blood flow resulting in damage to the downstream heart muscle. If blood flow is not restored, cardiomyocyte death occurs, triggering an immune response and pathogenic remodeling of the tissue into a fibrous collagen scar. Dissolution of the clot, either spontaneously or through pharmacological or surgical interventions, has been shown to significantly increase survival (180). This restores blood flow to the damaged myocardium, which causes a phenomenon known as reperfusion injury, a pathological consequence of restoring blood flow to ischemic myocardium characterized by myocardial, electrophysiological, and vascular dysfunction, including myocardial edema (181, 182). Reperfusion injury has been shown to contribute to infarct size, as researchers have shown that both reducing perfusion pressure and performing brief episodes of coronary reocclusion/reflow at the time of perfusion can reduce myocardial edema and infarct size (183, 184). This is important, as infarct size may be predictive for adverse outcomes following MI, including heart failure (185).
The pathogenesis of myocardial edema following ischemia-reperfusion is relatively well understood, mainly because of research performed in dogs. In the early stages of ischemia before cardiomyocyte necrosis, myocardial edema occurs with the swelling of cardiomyocytes (146). This coincides with a significant reduction in lymphatic draining in the ischemic zone, which was hypothesized to allow accumulation of toxic products and exacerbate myocardial damage (186). Longer periods of ischemia result in a large increase in fluid accumulation in the interstitium due to cardiomyocyte death, reactive hyperemia, and leakage from damaged capillaries (119, 187). After reperfusion, the restoration of capillary hydrostatic pressure dramatically increases fluid movement into the interstitium, compounding edema. Additionally, the large amount of fluid in the interstitium can cause microvascular obstruction by external compression (188). This sudden increase in capillary hydrostatic pressure can also result in microvascular destruction and cause intramyocardial hemorrhage. Both microvascular obstruction and intramyocardial hemorrhage are linked to large MI size and worse clinical outcomes (188). In patients, myocardial edema manifests within hours after MI and can remain present up to 7 mo (149, 189). However, there is controversy regarding the pattern of myocardial edema following MI in patients (bimodal vs. transient) and whether T2-weighted imaging, a method to quantify edema, can be used to quantify the area at risk in the myocardium (187, 190). Although there is ongoing debate in the field on the prognostic value of myocardial edema imaged via CMR, there is no argument that cardioprotective therapies that reduce infarct size and ischemia also reduce edema (187).
The timeline between when myocardial edema resolves and when lymphangiogenesis occurs has not been defined in humans. Unlike blood vessels, which quickly undergo angiogenesis and then regress as the scar matures, increased lymphatic density is maintained in the scarred tissue (191). Studies using clinical samples to evaluate the density of lymphatics in post-MI patients have found lymphangiogenesis in the peri-infarct region; researchers hypothesized that lymphangiogenesis had a significant role in scar maturation by removing excess fluid and immune cells (178, 191). Conversely, the role of cardiac lymphatics after MI in rodent models has been much more comprehensively studied, providing a template for future clinical studies. Whole mount and immunofluorescent staining of murine hearts after left anterior descending artery (LAD) ligation have confirmed that after MI there is a dramatic increase in the number of lymphatic vessels in the heart, especially near the infarct, but the endogenous response consists mostly of small capillary-like vessels (9, 35, 192). Henri et al. (35) were the first to quantify cardiac lymphangiogenesis, extent of myocardial edema (with both T2 MRI mapping and gravimetric analysis), and function of cardiac lymphatics after MI simultaneously. Surprisingly, they concluded that the endogenous lymphangiogenic response following MI resulted in dysfunctional cardiac lymphatics that hampered the drainage of excess fluid, an effect observed with sustained myocardial edema (35).
5.5. Arterial Hypertension
Arterial hypertension is a disease in which blood pressure in the arteries is persistently elevated. Chronic arterial hypertension is associated with an increased risk of coronary artery disease, stroke, and heart failure. The cause of arterial hypertension is multifactorial, including lifestyle and genetic factors. Remarkably, there have been no clinical studies measuring myocardial edema in patients with chronic arterial hypertension. The utilization of CMR imaging to measure myocardial edema could act as a prognostic factor for disease severity and the probability of development into other cardiovascular diseases. However, there have been two critical studies in dogs measuring microvascular fluid dynamics in a model of chronic arterial hypertension. In the first study, Laine (143) found significant myocardial edema in the hypertensive group compared to control animals, which was found to be due to increased microvascular permeability. This edema was present even with the finding that increased microvascular permeability led to increased fluid accumulation in the interstitium, increasing interstitial hydrostatic pressure, which increased myocardial lymph flow (143, 144). In a follow-up study, Laine and Allen (125) confirmed the presence of myocardial edema in a chronic arterial hypertensive dog model. One interesting caveat to their first study was that it was not determined what caused the increase in vascular permeability in chronic arterial hypertension model. Unrelated studies have found that angiotensin II can disrupt the blood-brain barrier in hypertensive rats and that aldosterone can diminish endothelial nitric oxide synthase activity, both of which can affect endothelial permeability (193). Studying myocardial edema and cardiac lymphatics in patients with chronic arterial hypertension becomes even more important with the knowledge that patients with arterial hypertension have a worse prognosis after MI compared to patients with normal blood pressure (194).
5.6. Pulmonary Hypertension
Pulmonary hypertension is a disease marked by increased arterial pressure in the lung due to pathology in pulmonary vasculature. This results in increased right ventricular pressure. Pulmonary hypertension has also been shown to cause left ventricular diastolic function (195, 196). To date, there have been no clinical studies to quantitatively measure the presence of myocardial edema in these patients. However, preclinical models that imitate pulmonary hypertension have found the presence of myocardial edema and fibrosis (88, 125, 145, 197). One study found that myocardial edema preceded an increase in collagen deposition, leading to fibrosis, and proposed that edema itself may trigger fibroblasts to increase collagen production (88). The pathogenesis of myocardial edema in patients with pulmonary hypertension results from the fact that central venous pressure is elevated because of increased right heart pressure. Because cardiac lymphatics drain into the thoracic duct at the level of the subclavian vein, increased central venous pressure decreases cardiac lymph drainage (30). Additionally, increased central venous pressure increases pressure in the coronary sinus, increasing pressure along the venous end of coronary venous capillaries, decreasing fluid resorption from the interstitium (30, 197). How this affects the morphology and density of cardiac lymphatics long term has yet to be studied.
5.7. Heart Failure
Heart failure is a chronic progressive condition in which the heart becomes unable to sufficiently pump enough blood to meet the body’s needs for blood and oxygen. The most common cause of heart failure is coronary artery disease, but it can also arise as an end-stage or chronic outcome stemming from another cardiovascular disease including dilated cardiomyopathy, MI, heart valve disease, and hypertension. Myocardial edema has been found to be associated with heart failure in both humans and animal models of heart failure. One study in patients diagnosed with heart failure identified the presence of myocardial edema in these patients (198). They also found a reduction in myocardial edema after successful heart failure treatment due to improvement in hemodynamic forces (198). Heart failure causes the accumulation of interstitial fluid, most likely due to decreased systolic function driving insufficient cardiac lymphatic drainage, increased capillary permeability caused by inflammatory cytokines, and increased venous pressure that impedes cardiac lymphatic drainage (198). Biopsy samples from patients with terminal heart failure showed a significantly higher number of open lymphatic capillaries compared with control samples, indicating that cardiac lymphatics morphologically change during terminal heart failure, perhaps as a compensatory mechanism to drain excess interstitial fluid (199). One study found evidence of myocardial edema in transverse aortic constriction (TAC) mice with heart failure but did not investigate how cardiac lymphatics were affected (200). Another study found that TAC-induced pressure overload increased proinflammatory cytokines, lymphatic markers such as LYVE-1 and VEGFR-3, and macrophages compared with controls, suggesting cardiac inflammation as a potential therapeutic target for heart failure (201).
A recent study utilizing TAC to model pressure overload-induced cardiac hypertrophy found a striking sex- and strain-dependent cardiac hypertrophic response linked to lymphatic expansion (202). The authors performed TAC-induced cardiac hypertrophy in male and female C57BL/6 and Balb/c mice to see whether an increase in lymphangiogenesis would temporally correlate with the need for increased fluid drainage capacity accompanied by the expanding ventricular mass of the heart. Surprisingly, they found in males no significant increase in lymphatic density or lymphatic gene expression 3 wk after TAC, which did not become significant until 8 wk after TAC in C57BL/6 males and was found to be driven by vascular endothelial growth factor-D (VEGF-D). Consistent with prior studies showing increased lymphatics in female C57BL/6 mice (14), C57BL/6 females showed a transient increase in lymphatic density and lymphatic markers at 3 wk after TAC, which subsided at the 8 wk post-TAC evaluation. Balb/c mice showed striking vascular endothelial growth factor-C (VEGF-C)-driven lymphangiogenesis after TAC, but this increase in cardiac lymphatics did not resolve myocardial edema. Blockade of the lymphangiogenic response in C57BL/6 males by anti-VEGFR-3 antibody treatment led to increased markers of cardiac inflammation, greater hypertrophy, and perivascular fibrosis, accelerating cardiac dysfunction. This study highlights the sex-dependent genetic and molecular signaling complexity of the lymphatic response to heart failure.
Microvascular dysfunction and chronic but low-grade inflammation have recently been indicated as a mechanism contributing to clinical heart failure with preserved ejection fraction (HFpEF). New experimental evidence has shown that metabolic impairments associated with HFpEF can drive lymphatic dysfunction, including dilation of lymphatic capillaries and leaky molecule transport (203). These findings have been echoed in the clinic, as peripheral lymphatic structural alterations and reduced lymphatic reserved have been identified in HFpEF patients (204).
New technologies are beginning to be tested in the clinic that target lymphatic function to treat the excessive edema and congestion associated with advanced decompensated heart failure (ADHF) (205). This condition can be marked by significant peripheral edema and can be accompanied by pulmonary edema leading to life-threatening difficulty in breathing and the need for hospitalization to perform decongestion measures. The most common decongestive treatments for ADHF are the use of vasodilators, to alter microvascular fluid dynamics in favor of fluid reabsorption, and loop diuretics, to pharmacologically induce natriuresis and diuresis by lowering the levels of sodium and chloride reabsorption in the kidney (206). This treatment focuses on removal of excess fluid through the blood vascular system and does not generally affect interstitial fluid levels, which are also greatly increased in ADHF. However, some patients do not respond well to diuretic therapy. High and prolonged doses of diuretics can lead to hypotension or worsened kidney function or create ion and metabolic imbalances that can increase risk of arrhythmia (206–208). A new medical device, undergoing early safety and feasibility studies, aims to provide both vascular and interstitial decongestion while sparing renal function (205, 209). The device involves the implantation of an impeller pump catheterization system into veins proximal to the thoracic duct, thereby creating a localized low-pressure zone around the duct to enhance lymphatic drainage of interstitial fluid. This device highlights the promising potential of lymphatic-directed therapies to treat cardiac disease.
5.8. Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is a disease marked by the presence of ventricular dilation with impaired systolic function. The most common etiology of DCM is idiopathic without any discernible causes. Secondary DCM can occur from a variety of factors including alcohol and infections or as the final stage of extensive myocardial damage, for example after myocardial infarction (210, 211). Not unexpectedly, myocardial edema has been detected in patients presenting with DCM (212, 213). Myocardial edema has been shown to increase with disease severity, providing a useful technique for the diagnosis and quantification of myocardial edema in patients with DCM (213). The etiology behind myocardial edema seen in patients with DCM can partly be attributed to insufficient contractility, which would reduce cardiac lymph flow, resulting in edema. However, patients with DCM have been shown to have prolonged inflammatory responses, even in the absence of systemic infection, which could contribute to increased capillary permeability and increased fluid extravasation to the interstitium (212). Additionally, the presence of increased lymphatics and lymphatic markers in patients presenting with DCM has been found (178, 199, 214, 215). Contemporary surgical mouse studies have correlated clinically increased lymphatic density and lymphatic signaling and are beginning to define the mechanistic relationship between the chronic inflammatory response and cardiac lymphatics (202, 216). These studies will set the framework to explore lymphangiogenic-targeted therapies to resolve cardiac inflammation and improve cardiac outcomes in DCM.
5.9. Pericardial Effusion
The pericardium is a fluid-filled membranous sac surrounding the heart that serves multiple functions, including separation of the heart from surrounding thoracic structures like the lungs, and mechanically constrains the heart during the contraction cycle to prevent overdilation and overfilling (217). The pericardial fluid, which fills the pericardial sac, provides lubrication for the heart as it beats and stocks bioactive substances such as ions, cytokines, and cardiac hormones such as adrenomedullin proximal to the epicardium (218–220). This fluid is the net product of plasma ultrafiltration from epicardial capillaries and fluid transudation from both the myocardium and epicardium.
Classical Starling forces (FIGURE 3A) drive the transudation of fluid from the epicardial capillaries to the pericardial cavity. The net difference in hydrostatic pressure (▵P) between the epicardial capillaries and the pericardial cavity, ∼2–10 mmHg, favors movement of fluid from the epicardium to the pericardial cavity (221). Interestingly, the hydrostatic pressure of the pericardial cavity is sensitive to the cardiac contraction cycle, with ▵P increasing during diastole and decreasing during systole (222, 223). However, this difference does not affect favored movement of fluid toward the pericardial cavity. Pericardial fluid volume is typically held at 20–60 mL in humans and is reflective of a tightly regulated balance between rates of formation and removal (221).
Pathologies that alter the rate of pericardial fluid accumulation and/or drainage can result in supraphysiological fluid levels, referred to in the clinic as pericardial effusion. Similar to myocardial edema, the causes of pericardial effusion vary widely and can be idiopathic in nature or can occur secondary to other disease. Cardiac conditions like cardiovascular surgery/trauma, acute myocardial infarction, heart failure, arterial and venous hypertension, atherosclerosis, metabolic disease, infection (pericarditis and myocarditis), inflammation, and renal disease can all lead to the development of pericardial effusion (224–226). The volume of pericardial effusion varies widely; however, sudden and/or excessive accumulation can constrict the ability of the heart to properly fill and pump, leading to decreased cardiac output and shock (225). This critical condition is known as cardiac tamponade, and it requires immediate clinical intervention, typically by pericardiocentesis, to drain excess pericardial fluid to improve hemodynamics. Pericardial effusion and cardiac tamponade are typically diagnosed by echocardiography (225, 227).
Lymphatics have been shown to participate in the regulation of pericardial fluid homeostasis in sheep, dogs, and humans (228–232). A complex network of lymphatics drain fluid away from the pericardium to mediastinal, pericardial, and tracheobronchial lymph nodes (229). It has been found in sheep that drainage of pericardial fluid increases proportionally to the increase in fluid volume or pressure in the pericardial cavity (232). Animal models of lymphatic obstruction (FIGURE 6) have demonstrated that impaired lymphatic drainage can lead to the development of pericardial effusions (228, 231, 232). In rare cases, patients with primary lymphedema have been reported to develop pericardial effusions, providing further evidence that proper lymphatic function is integral to pericardial fluid homeostasis (26, 233–235).
A recent report has drawn attention to the connection between lymphatics, myocardial edema, and pericardial fluid (226). It is noted that increased pulmonary arterial pressure drives increased pressure in the atria and ventricles that ultimately results in increased microvascular filtration, which leads to the formation of myocardial edema. In turn, this myocardial edema creates an increase in hydrostatic pressure between the epicardium and pericardial space, leading to increased fluid transudation into the pericardial cavity and formation of pericardial effusion. The increase in both myocardial edema and pericardial fluid results in increased lymph flow. However, the drainage of pericardial fluid is diminished by increased systemic venous pressure. Thus, this creates a cycle in favor of further myocardial and pericardial fluid accumulation. Further exploration of the connection between myocardial edema, pericardial effusion, and cardiac lymphatics in the context of various cardiovascular diseases will provide valuable information on the complex change in hemodynamics seen in these diseases.
6. CARDIOVASCULAR DISEASE TRIGGERS MODULATION OF LYMPHANGIOCRINE SIGNALING BY THE REACTIVATION OF EMBRYONIC EPICARDIAL PATHWAYS
Investigations of cardiac repair pathways have been a major focus of cardiovascular disease research over the last decade, and numerous therapeutic strategies have been tested. These include introduction of cell-based therapeutics such as delivery of stem cells and stem cell-derived cardiomyocytes, cell cycle-modulating agents to induce cardiomyocyte proliferation, and delivery of regenerative factors by viral vectors or specialized cardiac patches (236–238). A number of these studies focused on reactivation of the epicardium, a single-cell layer covering the outermost surface of the myocardium, upon which cardiac lymphatics sit. This tissue serves a critical role in the development of the heart by serving as a source of multipotent stem cells and releasing a variety of paracrine signals that contribute to the growth and patterning of the heart (239). This cellular layer becomes dormant in the adult heart and functions primarily as a physical barrier between the myocardium and the pericardial space. However, during cardiac injury the epicardium is thought to undergo a reactivation of its fetal gene programming and serve as a source of paracrine signals with regenerative potential (240). Of note, delivery of mesenchymal stem cells (MSCs) after surgical MI triggered endogenous cardiac repair and improvement of cardiac function through activation of the epicardium that resulted in differentiation of the MSCs into a variety of cell types including smooth muscle cells, cardiomyocytes, and endothelial cells (241). A significant increase in endothelial cell density, including cardiac lymphatics, was found in this study, suggesting a correlation between epicardial reactivation following cardiac injury and lymphangiogenesis.
This potential link between epicardial reactivation and lymphangiogenesis may lie in the secretory function of the epicardium. During development, the embryonic epicardium secretes several growth factors and chemokines responsible for cardiovascular growth, many of which act in an autocrine or paracrine fashion (242, 243). Several of these factors are known to promote angiogenesis, myocardial regeneration, and cardiac fibroblast proliferation (52). Recent work has identified that injury-activated adult epicardium reverts to embryonic-like epicardial gene programs and releases some of its embryonic secretome factors after cardiac injury (239, 240, 244). Some of these reactivated genes and secreted factors overlap with pathways influencing lymphangiogenesis such as Podoplanin and Adrenomedullin. Interestingly, direct stimulation of lymphangiogenesis by intracardial injection of LECs and VEGF-C showed an expected increase in lymphangiogenesis accompanied by a surprising increase in angiogenesis and myocardial regeneration. This suggests a complex cross talk between the epicardium, cardiac lymphatics, and the myocardium, likely regulated by secretion of paracrine factors from these three tissue beds triggering key signaling pathways (FIGURE 8). This section summarizes the significance of elevated levels of the secreted factors VEGF-C, Adrenomedullin, Apelin, and Reelin, and expression of Podoplanin, in epicardial reactivation and our current understanding of their role in the interactions between the cardiac lymphatics and the cardiac tissues.
FIGURE 8.

Molecular interactions between the cardiac and lymphatic endothelial cells in cardiac disease. Upon cardiac injury, epicardial cells promote expansion of the cardiac lymphatic vasculature by secreting adrenomedullin and vascular endothelial growth factor (VEGF)-C, a phenomenon induced by reactivation of embryonic epicardial pathways typically quiescent in healthy adult hearts. Apelin, expressed by myocardial cells and lymphatic cells, also promotes lymphatic response under hypoxic conditions, independently of the VEGF-C-VEGF receptor (VEGFR)-3 axis. Lymphatic-derived reelin promotes cardiomyocyte proliferation and tissue regeneration after injury, in addition to its role during heart development heart development. Adrenomedullin (AM) also has a proproliferative effect on cardiomyocytes, further promoting tissue regeneration. APJ, apelin receptor; CLR, calcitonin gene-related receptor; LEC, lymphatic endothelial cell; RAMP, receptor activity-modifying protein.
6.1. VEGF-C
6.1.1. Sources of VEGF-C in the heart.
Adult lymphangiogenesis has been described in response to tissue injury and tumor growth (245, 246). During this processes, smooth muscle cells (247), infiltrating macrophages (248), and tumor-associated macrophages (249) have all been shown to promote lymphatic expansion by secreting VEGF-C. It is appreciated that epicardial cells secrete VEGF-C during embryonic development before the initiation of the development of cardiac lymphatics (250). In healthy adult hearts, VEGF-C is not expressed in the epicardium (37). Four days after LAD ligation, however, VEGF-C-expressing epicardial cells and macrophages have been observed (37), which is in line with increased VEGF-C and VEGFR-3 expression reported by others (9, 44, 133). The presence of VEGF-C-expressing epicardial cells upon ischemic heart injury suggests that a reactivation of embryonic epicardial pathways is at least in part responsible for the lymphatic expansion after injury. Additionally, noncardiomyocyte cells such as macrophages produce VEGF-C in the infarct zone after MI, likely contributing to cardiac lymphangiogenesis (36).
6.1.2. Role of VEGF-C signaling in cardiac diseases.
Recent research has suggested an importance for both the lymphatic vasculature and VEGF-C signaling in multiple cardiac diseases. These studies focus on inhibition or stimulation of VEGF-C signaling, through means of genetic disruption or therapeutic stimulation. Most of these studies have been performed in the context of ischemic injury resulting from myocardial infarction. Vuorio et al. (44) used transgenic mice expressing a soluble decoy VEGFR-3 (sVEGFR-3) as well as Chy mice (Flt4Chy), which possess a natural inactivating VEGFR-3 mutation, to examine the necessity of VEGF-C signaling in healthy and infarcted hearts. Loss of VEGF-C signaling produced basal disruption of the cardiac lymphatics, specifically dilation and disorganization of the network. After MI, loss of VEGF-C signaling was associated with significantly higher mortality, modifications to fibrotic composition in the infarct, and vascular hyperpermeability as evidenced by increased leukocyte infiltration and accumulation of α-smooth muscle actin-positive myofibroblasts. Another study utilizing sVEGFR-3 mice revealed decreased cardiac lymphatic expansion after MI in comparison to wild-type control mice, suggesting that post-MI lymphatic expansion is at least partially dependent upon VEGF-C/VEGFR-3 signaling (251). A recent study utilized three genetic approaches to VEGF-C signaling blockade to investigate the role of lymphatic vessel growth after MI: pan-endothelial (Flt4fl/fl;Cdh5-CreERT2) or lymphatic-specific (Flt4fl/fl;Prox1-CreERT2) loss of VEGFR-3 or global loss of VEGF-C and VEGF-D (VEGF-C−/−/VEGF-D−/−) (252). All three models indicated increased mortality and a >80% reduction of cardiac lymphatic growth in the infarct zone. Interestingly, the severely blunted lymphangiogenesis did not result in any significant changes to cardiac function in comparison to wild-type controls. This finding was also echoed in post-MI sVEGFR-3 mice (251).
A recent and noteworthy study by the authors of the present review demonstrated that vascular endothelial (VE)-cadherin serves as a scaffolding protein to maintain prolymphangiogenic VEGFR-3 signaling nodes at the plasma membrane in lymphatic endothelial cells, which is required for cardiac lymphatic maintenance and signaling in healthy and injured hearts (253). Temporal loss of lymphatic VE-cadherin, achieved with Cdh5fl/fl;Prox1-CreERT2 mice, resulted in complete disassembly of the cardiac lymphatic network during postnatal development or a significant and progressive regression of the network in adulthood. Loss of VE-cadherin, in cultured LECs, was demonstrated to prevent cell surface presentation of VEGFR-3 and canonical activation of VEGFR-3 by VEGF-C. Thus, this mouse model serves as an indirect approach to inhibit VEGF-C signaling. Like the four genetic models of direct VEGF-C signaling disruption, lymphatic VE-cadherin loss was associated with severe lymphatic dysfunction but spared basal cardiac function and did not result in significant cardiac function changes after MI in comparison to controls. This study did note a significant increase in myocardial collagen content, a finding similar to MI studies done in sVEGFR-3 mice.
Studies utilizing exogenous methods to induce VEGF-C signaling blockade have produced similar but contrasting results to the studies above using genetic mouse models of VEGF-C signaling downregulation. Shimizu and colleagues (133) observed a rarefaction of subendocardial lymphatic vessel density 7 days after reperfusion in mice treated with a hydrogel containing the VEGFR-3 inhibitor MAZ-51 or a VEGFR-3-neutralizing antibody attached to the infarcted myocardium, compared with vehicle-treated mice. These structural changes of the cardiac lymphatic system were correlated to impaired cardiac lymphatic function, increased cardiac water content, B cell accumulation, and increased inflammatory response 7 days after reperfusion. Mice with attenuated VEGFR-3 signaling exhibited increased infarct scar size, increased ventricular hypertrophy, increased ventricular dilation, and decreased ventricular function 4 wk after reperfusion, compared with controls (133). Another research group applied an adeno-associated virus (AAV)-based delivery of a soluble VEGF-C trap and reported a reducing number of cardiac lymphatics in the infarct zone after LAD ligation, which unexpectedly resulted in an improved cardiac function, perhaps due to a decreased T-cell infiltration into the infarcted area (36).
A recent prospective study on 2,418 patients with known coronary artery disease revealed that VEGF-C level is both inversely and independently associated with mortality (254). Whereas previous studies have shown that high VEGF-D levels are associated with heart failure and stroke, this study showed that low VEGF-C levels were also associated with these pathologies (254, 255). This may indicate an increased lymphangiogenic demand for VEGF-C in coronary artery diseases, which would correlate with the demonstrated lymphangiogenic response seen in these diseases and MI.
Taken together, these findings suggest that lymphangiogenesis in the heart is at least partly driven by VEGF-C/VEGFR-3 pathway, and although it is debated whether attenuated VEGF-C signaling influences cardiac function after MI, it may play an important role in determining the cardiac edema formation, leukocyte infiltration, inflammation, and modulating fibrotic tissue formation, therefore suggesting VEGF-C as a potential therapeutic target in MI.
6.2. Adrenomedullin
6.2.1. Plasma adrenomedullin levels are elevated in patients after ischemic cardiac injury.
Adrenomedullin (AM) is a peptide hormone with potent vasodilatory effects and is essential for cardiovascular and lymphatic development. Circulating levels of AM have also been implicated as prognostic factors in cardiovascular disease. A study by Jougasaki et al. (256) identified that plasma AM concentrations were significantly higher in patients with chronic congestive heart failure (CHF) compared to healthy patients without ventricular dysfunction. These findings were later validated in dogs, identifying an ∼2.6-fold increase in plasma AM concentrations in the experimental CHF animals versus the control dogs (257). Elevated AM levels in CHF correlated with decreased cardiac output, decreased left ventricular ejection fraction, and increased ventricular mass index, a predictor of heart failure (257). A recent clinical study by Kiris et al. (258) analyzed left ventricle (LV) function and plasma AM levels in patients, drawing comparisons between those who had experienced anterior myocardial infarction (MI), posterior/inferior MI, or no previous MI before coronary artery bypass grafting (CABG). Plasma AM levels were reported to be elevated in both MI groups, 3 wk or longer after MI, compared with patients who had never experienced MI (258). Furthermore, plasma AM levels were higher in the anterior MI group, which was associated with a more significant LV function reduction (258). Increased plasma levels of AM have also been found in patients with arteriosclerosis or hypertension, relative to control subjects (259, 260). It is thought that these increased AM levels serve to combat blood pressure elevation associated with these conditions.
AM has been demonstrated to promote myocyte survival and apoptosis and inhibit fibrotic response (261). Reactivation of the vessels during cardiac stress results in increased AM levels and is thought to provide a cardioprotective effect by modulating factors that improve cardiac function (14). A study conducted by Kataoka et al. (262) found that patients who were treated with intravenous AM after acute MI showed significant cardiovascular improvement. Trincot et al. (14) utilized a mouse model in which AM was genetically overexpressed, leading to increased cardiac lymphatic coverage. After MI, this increase in cardiac lymphatic coverage resulted in less edema, reduced remodeling, and improved heart function compared with controls. Given that epicardium is a significant source of AM during heart development (52), elevated AM levels in cardiac disease may also support paradigms associated with disease-induced epicardial reactivation upon cardiac injury. Trincot et al. demonstrated that genetic upregulation of AM resulted in more vessels along the epicardial border zone and increased vessel diameter and dilation after MI. Therefore, not only is AM a biomarker of cardiovascular disease, but it may also serve as a robust therapeutic target.
6.2.2. Sex differences and adrenomedullin levels linked to disparities in cardiac disease and lymphatic function.
Numerous cardiovascular diseases exhibit sexual dimorphism regarding disease prevalence and outcomes. As discussed above, lymphatic coverage is also influenced by sex. As a potent lymphangiogenic peptide and biomarker of cardiac disease, it is no surprise that AM has been shown to be estrogen responsive. AM as well as receptor activity-modifying protein 3 (RAMP3; Ramp3), a protein known to modulate the activity of the AM receptor calcitonin gene-related receptor (CLR; Calcrl), have been shown to be induced upon exogenous estrogen administration (263). Additionally, Adm expression is also regulated via estrogen-induced miRNAs (264). This has led to speculation about estrogen’s role in AM regulation, presupposing that cardioprotection is granted for females during the reproductive period. One study found that overexpression of the estrogen receptor in cardiomyocytes in mice resulted in enhanced angiogenesis and lymphangiogenesis after MI in both sexes, which reduced fibrosis in females only (265).
Genetic overexpression of AM has been shown to increase basal cardiac lymphatic coverage and improve post-MI cardiac outcomes and lymphangiogenesis (14). Furthermore, downstream targets of AM signaling have also shown sex-dependent differences in expression and activity. One such downstream target is connexin-43 (Cx43), a gap junction protein that assists in propagation of electrical activity and contraction between cardiomyocytes. This is the most abundant connexin in the heart and is necessary for normal cardiac conductivity. Additionally, loss of lymphatic Cx43 results in dysregulated lymphatic valve formation and capillary patterning and function (14). Interestingly, assessment of basal Cx43 expression in wild-type rats revealed increased expression of Cx43 in the ventricular myocardium relative to age-matched males (266). Furthermore, a study conducted by Knezl et al. (267) in spontaneously hypertensive (SHR) rats identified an inverse correlation between ventricular fibrillation susceptibility and Cx43 expression, with female rats expressing significantly higher levels of Cx43 in the ventricles. It is believed that adrenomedullin increases expression of Cx43 in lymphatics, thereby improving lymphatic function, reducing myocardial edema, and improving heart function after MI (14).
6.3. Other Signaling Mechanisms
6.3.1. Apelin.
Apelin, an adipokine that signals through the G protein-coupled receptor apelin receptor (APJ), has been implicated in numerous human cardiovascular diseases including atherosclerosis, hypertension, coronary artery disease, heart failure, and myocardial ischemia (268). A significant increase in Apelin expression was found in patients with coronary heart disease and in early heart failure (269, 270). In line with these observations, apelin expression is induced in cultured rat cardiomyocytes under hypoxic conditions (271), and apelin-deficient mice have been shown to exhibit increased infarct size, elevated levels of inflammatory cytokines, and decreased cardiac function after permanent LAD ligation compared with control animals (272).
Apelin exerts a wide range of physiological effects including vasodilation, myocardial contractility, fluid homeostasis, and angiogenesis (268). Apelin has also been demonstrated to be involved in embryonic lymphatic development in zebrafish (273). Although lack of apelin signaling does not affect lymphatic vessel density in mice, a dilation of lymphatics has been reported (274, 275), suggesting a role in regulating lymphatic function. Interestingly, apelin is not expressed in quiescent cardiac lymphatics but has been found to be significantly upregulated in cardiac lymphatics after MI (275). This study also uncovered a sudden, transient upregulation of apelin in the infarct zone 2 days after MI and a prolonged increase in the expression of APJ in the LECs of the newly formed lymphatic vessels in the infarct area. Apelin signaling, which has been previously shown to promote lymphangiogenesis independently of the VEGF-C-VEGFR-3 axis (273), was found to promote the stability of lymphatic barrier function, as apelin-deficient mice possess dilated and leaky cardiac lymphatic vessels (275). Importantly, VEGF-C expression was significantly increased in apelin-deficient mice compared with wild-type control mice 2 days after MI, reflective of a compensatory mechanism. Long-term overexpression of apelin did not improve cardiac function after MI but significantly reduced the fibrotic tissue area, which may be related to its action in regulating the expression of atrial natriuretic peptide (ANP), proinflammatory mediators, matrix metalloproteases, and sphingosine kinase 1, promoting fibrosis (275). LECs are an in vivo source of sphingosine-1-phosphate (S1P), allowing for lymphocyte egress from lymph nodes, further highlighting the role that Apelin may have in lymphatic function after MI (111). Together these studies highlight Apelin signaling as a novel therapeutic approach to treat cardiovascular disease and/or modulate the lymphatic response after cardiac injury.
6.3.2. Reelin.
Reelin is a large secreted extracellular matrix glycoprotein that has important roles in lymphatic and cardiovascular development. This glycoprotein serves as an important lymphangiocrine molecule allowing LECs to communicate with surrounding cells such as smooth muscle cells (SMCs) and cardiomyocytes (39, 276). This mechanism is best described for SMC recruitment to lymphatic collecting vessels: Reelin is produced and expressed by LECs and upon SMC contact is cleaved and secreted into the extracellular matrix, where secondary signals serve to recruit additional SMCs to the lymphatic vasculature (276). A recent study found that LEC-secreted Reelin glycoprotein promotes the proliferation and survival of cardiomyocytes in an integrin β1-dependent manner (39). Furthermore, they demonstrated that reelin expression is upregulated in newly formed cardiac lymphatic vessels after MI, and application of a reelin-releasing collagen patch onto the heart significantly reduced fibrotic scar area at 6 wk after MI and improved cardiac function 3 wk after MI compared with control-treated animals (39). These findings suggest that reelin acts in a lymphangiocrine manner to directly influence cardiomyocyte proliferation and modulate the fibrotic response and cardiovascular function after ischemic heart injury. Interestingly, upregulation of Reelin is not universally beneficial across cardiac disease. Two studies have highlighted that Reelin depletion protects against atherosclerosis through reducing the accumulation of macrophages and adhesion of leukocytes to endothelial cells (277, 278). This indicates that therapeutic targeting of Reelin signaling to treat cardiovascular disease will be highly dependent on disease context and perhaps at the expense of cardiac lymphatic function.
6.3.3. Podoplanin.
Podoplanin (PDPN) is a membrane-integrated protein that plays a central role in the separation of the blood and lymphatic circulatory systems (279). Podoplanin is expressed in numerous cell types, but this expression is temporally regulated, with the protein being most widely expressed during development (280). The roles of Podoplanin in cardiac physiology have been recently and eloquently reviewed (281). Importantly, during early embryonic development, podoplanin is expressed by both epicardial and myocardial cells. It participates in the epithelial-to-mesenchymal transition of proepicardial cells, by regulating E-cadherin (cadherin-1) and Ras homolog family member A (RhoA), which are required for the adhesion and spreading of the epicardium and development of sinus venosus myocardium (282, 283). In the fully developed healthy heart, podoplanin expression is restricted to LECs, type I pneumocytes, and glomerular podocytes, making it a popular lymphatic marker in adult tissues (284).
As mentioned above, the quiescent epicardium has been shown to undergo a program of embryonic-like gene reactivation and begins to reexpress PDPN (239, 240, 244). One study demonstrated that after MI PDPN is expressed by nonlymphatic lymphangiogenic, fibrogenic, and mesenchymal progenitor cells (285). Antibody neutralization of PDPN results in reduced infarct scar size and improved cardiac function and promoted anti-inflammatory mechanisms after LAD ligation, indicating that the reactivation of PDPN is likely pathological and not beneficial to the healing process (286). The underlying mechanisms involved in the injury-induced reprograming of PDPN and other developmentally expressed gene programs are yet to be understood.
7. EXPLORING THE THERAPEUTIC POTENTIAL OF CARDIAC LYMPHATICS
There have surprisingly been few successful innovative therapies to minimize cardiac injury or improve cardiac function after MI (287). Most patients are treated with a combination of antithrombotics, invasive surgery, and statins to try to mitigate cardiovascular damage and remodeling (287). As detailed above, cardiac lymphatics and their physiological functions are modulated in multiple cardiac diseases. Accordingly, the therapeutic potential of cardiac lymphatics has been explored, namely though stimulation of lymphangiogenesis achieved by activating prolymphangiogenic signaling pathways, administering lymphatic progenitor cells, or modulating endothelial function. Despite successful preclinical evaluation of potential cardiac lymphatic-directed therapies, monumental barriers are currently barring translation of these therapies to the clinic.
7.1. Stimulation of Prolymphangiogenic Pathways
Klotz and colleagues (9) observed that treatment of mice with VEGFR-3-selective recombinant VEGF-CC156S led to a more pronounced lymphatic response and improved cardiac function after MI. These findings were echoed in a similar study utilizing a surgical rat MI and administration of VEGF-CC156S-loaded albumin-alginate microparticles. This study pioneeringly quantified post-MI lymphatic coverage in the heart, utilizing cardiac lymphangiography, and examined myocardial edema (35). They found significantly increased lymphatic capillary density in the subepicardium and moderate increases in midmyocardial lymphatic density. Importantly, in mice treated with recombinant VEGF-C no significant difference between the groups was observed in the cardiac lymphatic structure 8 wk after LAD ligation. In relation to these findings, VEGF-C treatment accelerated the reduction of MI-induced myocardial edema and inflammatory response in the chronic phase of MI, but by week 8 after LAD ligation no significant difference was observed in cardiac water content between groups. Importantly, recombinant VEGF-CC156S treatment prevented cardiac fibrosis and mitigated cardiac dysfunction, although it did not affect infarct size (35). It has been also demonstrated that VEGF-CC156S treatment enhances cardiac lymphangiogenic response 7 days after permanent LAD ligation and promotes immune cell clearance into the mediastinal lymph nodes (37). Similarly, application of recombinant VEGF-CC156S-containing hydrogel on the infarcted myocardium led to increased lymphatic expansion and improved lymphatic function 7 days after reperfusion compared with vehicle-treated animals. Cardiac water content, leukocyte infiltration, and inflammatory response decreased in comparison to control animals. Furthermore, infarct scar size, ventricular hypertrophy, ventricular dilation were diminished and ventricular function was increased 4 wk after reperfusion (133). A more clinically applicable administration of single-dose intraperitoneal injection of adeno-associated virus (AAV)-hVEGF-CC156S enabled a more efficient, systemic increase in VEGF-C levels (36).
7.2. Therapeutic Administration of Lymphatic Progenitor Cells
In an effort to increase cardiac lymphatic coverage and lymphangiogenesis after MI, multiple studies have injected populations of endothelial progenitor cells into the myocardium after MI. Zhang et al. (288) administered endothelial progenitor cells in conjunction with VEGF-C in infarcted rats. These rats expectedly exhibited increased lymphangiogenesis, reduced myocardial edema, improved drainage of inflammatory cells, and improved cardiac function (288). Interestingly, they also found increased angiogenesis and myocardial regeneration subsequent to reduction of myocardial edema, highlighting the important role of cardiac lymphatics in both fluid homeostasis and cardiac repair after injury (288). Another study by Park et al. (289) injected bone marrow-derived endothelial progenitor cells (EPCs) into the infarct and border zone of mice after MI. Surprisingly, they found the downregulation of multiple lymphatic markers, including LYVE-1, PROX1, and VEGF-C, compared to control mice. This correlated with decreased lymphangiogenesis, decreased adverse ventricular remodeling, and increased cardiac function, suggesting a possible deleterious role for lymphangiogenesis in post-MI cardiac remodeling (289). Further research into cell-based therapies should help determine whether these interventions are beneficial or not.
Beside these approaches, other investigators have focused on preclinical testing of cell-based therapies to increase lymphangiogenesis in a postinfarct heart. Wang et al. (241) tested gelatin patches loaded with mesenchymal stem cells (MSCs) on activating endogenous cardiac repair after MI. Mice grafted with the patch had increased cardiac function hypothesized to be from MSCs activating the epicardium and differentiating into a variety of cell types, including endothelial cells, smooth muscle cells, and cardiomyocytes (241). They also found increased density of endothelial cells, including cardiac lymphatics, although this was not the major focus of their paper (241). Still, this study suggests correlation between epicardial reactivation following MI and enhanced lymphangiogenesis, an intriguing avenue for future research.
7.3. Modulating Vascular Permeability
Another study aimed to investigate the role of extracellular RNA (eRNA) released from injured cells and its role in vascular permeability and myocardial edema following MI in mice (290). There was a significant increase in plasma eRNA 30 min after ligation. Intravenously treating mice with eRNA increased vascular permeability, leading to significant myocardial edema (290). To address the therapeutic potential of this finding, mice were treated with RNase 1 to degrade eRNA after MI. This increased perfusion of collateral arteries, reducing infarct size and improving heart function, and reduced myocardial edema (290). Although there was no mention of the effect of this treatment on cardiac lymphatic density or function, this study provides an exciting example of innovative preclinical studies aimed at developing new therapeutics to improve patient outcomes after MI.
7.4. Immunological Consequences of Manipulating Cardiac Lymphatics
Inflammation can serve as both an initiator and a consequence of multiple cardiovascular pathologies such as myocarditis and myocardial infarction, respectively. Clinical studies indicate a correlation between inflammatory parameters and cardiac dysfunction in patients with heart failure, making inflammation-targeted therapies of great interest (291–294). In addition to its fluid homeostatic role, a critical function of lymphatics is to traffic immune cells and antigens to draining lymph nodes (295). Thus, lymphatics participate in immune surveillance and modulation of immune responses. Cardiac lymphatic vessels may play similar roles in determining immune responses in cardiac disease and influence disease outcome.
Studies have demonstrated bidirectional communication between cardiac lymphatics and the immune system. It has been shown that cardiac lymphatic coverage is inversely correlated between macrophage counts in the heart (35), and another study reported that during MI macrophages produce VEGF-C in the infarct zone, likely contributing to post-MI lymphangiogenesis in this region (36). Interestingly, studies investigating the effects of impaired lymphatic responses in cardiac injury report contradictory consequences on immune signaling. Select studies did not observe differences in immune responses in VEGF-C signaling-impaired mice that underwent heart injury (36, 44, 252), whereas other reports indicate that disruption of the VEGF-C-VEGFR-3 axis or apelin signaling results in proinflammatory cytokine levels and leukocyte accumulation (39, 133, 275). Despite these conflicting reports, the majority of studies to date support that stimulation of prolymphangiogenic pathways improves leukocyte clearance and reduces proinflammatory cytokine levels after cardiac injury (35–37, 133, 288, 296), indicating that modulation of lymphatic response not only helps edema resolution but also promotes cardiac healing through promoting immune cell trafficking and regulating inflammatory responses.
7.5. Barriers to Cardiac Lymphatic-Directed Therapies
Most preclinical models have shown that therapeutically stimulating cardiac lymphangiogenesis can improve immune cell trafficking, reduce edema, and improve cardiac function after MI. Conversely, knowledge gained from cardiac allograft studies indicates that inhibition of lymphangiogenesis may be beneficial in preventing inflammation and reducing immune cell surveillance, which leads to organ rejection. These considerations will be important in moving forward with clinical applications and determining the best treatment options for patients. Ultimately, the field has yet to conclude whether mitigated lymphatic function worsens edema or fibrosis or hinders cardiac function and whether lymphangiogenesis improves these conditions after cardiac injury. In a situation where lymphangiogenesis would be beneficial, understanding the interplay of lymphatic vessel function in different organs and in organ-specific contexts is important in driving decisions around delivery methods for the administration of prolymphangiogenic molecules and raises major concerns about the systemic effects of lymphatic-directed therapies. Although these findings provide promising potential therapeutic approaches to treat patients with cardiac disease, numerous studies are needed to address the current theoretical and methodological questions.
An important and yet-unresolved concern is that the primary delivery mode of prolymphangiogenic molecules lacks site specificity because of peripheral administration. More direct means of therapeutic delivery to the heart have only been tested in animals, where therapies have been administered in a highly invasive surgical setting in which therapy-loaded patches have been placed directly on the epicardium or injected intramyocardially. In the clinic, therapies need to be administered through intracoronary routes to achieve direct cardiac application. Such opportunities exist during surgical and nonsurgical interventional cardiac procedures. For example, a prolymphangiogenic therapy could be administered during a percutaneous coronary intervention to treat patients with ST segment-elevation myocardial infarction with ongoing ischemia. This would achieve local cardiac administration of the therapy and allow for theoretical lymphangiogenesis in the post-MI window of recovery, hopefully leading to improvements in cardiac function as seen with the animal models mentioned above. However, engaging in direct application of cardiac lymphatic-targeted therapies without any other cardiovascular surgical need poses significant risks that may not outweigh the therapeutic benefit. A necessary step forward for cardiac lymphatic therapies is the development of a technique that allows for cardiac accumulation of a peripherally administered therapy, thereby providing a noninvasive method with minimal side effects and maximal efficacy.
Another major challenge to evaluating potential cardiac lymphatic therapies is the current inability to assess the efficacy of the therapies in real time in the clinic. As described above, one of the central challenges of understanding clinical cardiac lymphatic anatomy, physiology, and pathophysiology is the current limitations of clinical imaging modalities to visualize the cardiac lymphatics. Many of the noninvasive imaging techniques, (MRI, ultrasound, and CT) are plagued by poor spatial resolution and sensitivity and do not provide functional parameters or visualization of small vessels. Imaging techniques that provide increased spatial and temporal resolution allow for anatomical mapping and assessment of functional parameters but require direct injection of tracers into regional vascular beds. In the case of cardiac lymphatics, this requires invasive surgical administration to access the desired lymphatic bed or lymph node, which is again a very risky and costly procedure. Without a reliable means to visualize the cardiac lymphatics or their fluid trafficking functions, it is nearly impossible to evaluate the clinical efficacy of cardiac lymphatic-targeted therapies.
8. CONCLUDING REMARKS
This review has provided a comprehensive dive into the study of fluid homeostasis around the capillary, the role of myocardial edema in numerous cardiovascular diseases. Surgical animal models and clinical findings have been leveraged to illuminate the role of cardiac lymphatics in disease pathogenesis. Foundational genetic animal models have illuminated a suite of lymphangiocrine molecules and pathways that are modulated in cardiac disease and have served to guide the development of lymphatic-targeted therapies. These therapeutic strategies have shown that increasing lymphangiogenesis in the context of acute cardiac injury can lead to an improvement in overall cardiac function by means of reducing myocardial edema, inflammation, and fibrosis. Surprising new evidence has emerged indicating that cardiac lymphatics may be dispensable for preserving basal cardiac function before and after injury. This is an important finding for the consideration of antiangiogenic therapies, where benefit can be drawn from dampening undesired inflammation.
Despite recent advances in understanding the physiological and pathophysiological role of cardiac lymphatic vessels, several outstanding questions remain in the field (FIGURE 9). Open questions remain regarding the response and function of the cardiac lymphatics in acute versus chronic cardiac disease, as well as which lymphatic function contributes most to the disease pathology. Given that females are predominantly affected by disorders of lymphatic vascular function and that sexual dimorphism exists in cardiac disease prevalence and outcomes, sex is an important variable affecting lymphatic biology and pathophysiology, yet we lack a comprehensive understanding of the effects of sex and sex hormones on lymphatic growth, function, and dysfunction. Continued exploration of basic lymphatic biology as well as its role in cardiovascular disease is needed on both the clinical and preclinical fronts to develop safe and effective cardiac lymphatic-targeted therapies to treat cardiovascular pathologies.
FIGURE 9.
Outstanding questions in the quest to understand the physiological and pathophysiological roles of lymphatics in cardiovascular (CV) disease and myocardial edema. Despite recent advances in understanding the physiological and pathophysiological roles of cardiac lymphatic vessels, several outstanding questions remain in the field. Here, we feature some of the most compelling and critical questions to be answered.
GRANTS
This work was supported by National Institutes of Health (NIH) grants from the National Heart, Lung, and Blood Institute (NHLBI) (HL1290986) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK119145) and an American Heart Association (AHA) Innovator Award to K.M.C., NHLBI T32HL069768 and AHA Pre-doctoral Fellowship no. 834898 to N.R.H., NHLBI F31 HL143836 to N.R.N., and a Howard Hughes Medical Institute Gilliam Fellowship for Advanced Study to H.G.M.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.R.H., L.B., D.M.D., N.R.N., H.G.M., A.A., and K.M.C. conceived and designed research; N.R.H., L.B., D.M.D., and H.G.M. prepared figures; N.R.H., L.B., D.M.D., N.R.N., H.G.M., and K.M.C. drafted manuscript; N.R.H., L.B., A.A., and K.M.C. edited and revised manuscript; N.R.H., L.B., D.M.D., N.R.N., H.G.M., A.A., and K.M.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Brian Jensen (UNC Department of Medicine, Division of Cardiology) and Dr. Audrey Garneau (UNC Department of Medicine, Department of Obstetrics and Gynecology) for valuable input during the preparation of this review.
REFERENCES
- 1. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics—2019 update: a report from the American Heart Association. Circulation 139: e56–e528, 2019. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2. Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 204: 2349–2362, 2007. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Breslin JW, Yang Y, Scallan JP, Sweat RS, Adderley SP, Murfee WL. Lymphatic vessel network structure and physiology. Compr Physiol 9: 207–299, 2018. doi: 10.1002/cphy.c180015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Natale G, Bocci G, Ribatti D. Scholars and scientists in the history of the lymphatic system. J Anat 231: 417–429, 2017. doi: 10.1111/joa.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bradham RR, Parker EF. The cardiac lymphatics. Ann Thorac Surg 15: 526–535, 1973. doi: 10.1016/s0003-4975(10)65339-8. [DOI] [PubMed] [Google Scholar]
- 6. Patek P. The morphology of the lymphatics of the mammalian heart. Am J Anat 64: 203–249, 1939. doi: 10.1002/aja.1000640202. [DOI] [Google Scholar]
- 7. Symbas PN, Cooper T, Gantner GA Jr, Willman VL. Lymphatic drainage of the heart: effects of experimental interruption of lymphatics. Surg Forum 14: 254–256, 1963. [PubMed] [Google Scholar]
- 8. Fedyai VV. Age changes in the intrinsic lymphatics of the heart. Fed Proc Transl Suppl 25: 177–180, 1966. [PubMed] [Google Scholar]
- 9. Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dubé KN, Bollini S, Matsuzaki F, Carr CA, Riley PR. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 522: 62–67, 2015. doi: 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ratajska A, Gula G, Flaht-Zabost A, Czarnowska E, Ciszek B, Jankowska-Steifer E, Niderla-Bielinska J, Radomska-Lesniewska D. Comparative and developmental anatomy of cardiac lymphatics. ScientificWorldJournal 2014: 183170, 2014. doi: 10.1155/2014/183170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sacchi G, Weber E, Aglianò M, Cavina N, Comparini L. Lymphatic vessels of the human heart: precollectors and collecting vessels. A morpho-structural study. J Submicrosc Cytol Pathol 31: 515–525, 1999. [PubMed] [Google Scholar]
- 12. Loukas M, Abel N, Tubbs RS, Grabska J, Birungi J, Anderson RH. The cardiac lymphatic system. Clin Anat 24: 684–691, 2011. doi: 10.1002/ca.21104. [DOI] [PubMed] [Google Scholar]
- 13. Shimada T, Noguchi T, Takita K, Kitamura H, Nakamura M. Morphology of lymphatics of the mammalian heart with special reference to the architecture and distribution of the subepicardial lymphatic system. Acta Anat (Basel) 136: 16–20, 1989. doi: 10.1159/000146791. [DOI] [PubMed] [Google Scholar]
- 14. Trincot CE, Xu W, Zhang H, Kulikauskas MR, Caranasos TG, Jensen BC, Sabine A, Petrova TV, Caron KM. Adrenomedullin induces cardiac lymphangiogenesis after myocardial infarction and regulates cardiac edema via connexin 43. Circ Res 124: 101–113, 2019. doi: 10.1161/CIRCRESAHA.118.313835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimada T, Morita T, Oya M, Kitamura H. Morphological studies of the cardiac lymphatic system. Arch Histol Cytol 53, Suppl: 115–126, 1990. doi: 10.1679/aohc.53.suppl_115. [DOI] [PubMed] [Google Scholar]
- 16. Norman S, Riley PR. Anatomy and development of the cardiac lymphatic vasculature: its role in injury and disease. Clin Anat 29: 305–315, 2016. doi: 10.1002/ca.22638. [DOI] [PubMed] [Google Scholar]
- 17. Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nat Rev Cardiol 16: 56–68, 2019. doi: 10.1038/s41569-018-0087-8. [DOI] [PubMed] [Google Scholar]
- 18. Nielsen NR, Rangarajan KV, Mao L, Rockman HA, Caron KM. A murine model of increased coronary sinus pressure induces myocardial edema with cardiac lymphatic dilation and fibrosis. Am J Physiol Heart Circ Physiol 318: H895–H907, 2020. doi: 10.1152/ajpheart.00436.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trincot CE, Caron KM. Lymphatic function and dysfunction in the context of sex differences. ACS Pharmacol Transl Sci 2: 311–324, 2019. doi: 10.1021/acsptsci.9b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao Z, Chen Z, Sun A, Deng X. Gender differences in cardiovascular disease. Med Nov Technol Devices 4: 100025, 2019. doi: 10.1016/j.medntd.2019.100025. [DOI] [Google Scholar]
- 21. Shi W, Sheng X, Dorr KM, Hutton JE, Emerson JI, Davies HA, Andrade TD, Wasson LK, Greco TM, Hashimoto Y, Federspiel JD, Robbe ZL, Chen X, Arnold AP, Cristea IM, Conlon FL. Cardiac proteomics reveals sex chromosome-dependent differences between males and females that arise prior to gonad formation. Dev Cell 56: 3019–3034.e7, 2021. doi: 10.1016/j.devcel.2021.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bellier A, Pardo Vargas JS, Cassiba J, Desbrest P, Guigui A, Chaffanjon P. Anatomical variations in distal portion of the thoracic duct—a systematic review. Clin Anat 33: 99–107, 2020. doi: 10.1002/ca.23476. [DOI] [PubMed] [Google Scholar]
- 23. Itkin M, Nadolski GJ. Modern techniques of lymphangiography and interventions: current status and future development. Cardiovasc Intervent Radiol 41: 366–376, 2018. doi: 10.1007/s00270-017-1863-2. [DOI] [PubMed] [Google Scholar]
- 24. Okuda I, Udagawa H, Hirata K, Nakajima Y. Depiction of the thoracic duct by magnetic resonance imaging: comparison between magnetic resonance imaging and the anatomical literature. Jpn J Radiol 29: 39–45, 2011. doi: 10.1007/s11604-010-0515-0. [DOI] [PubMed] [Google Scholar]
- 25. Hsu MC, Itkin M. Lymphatic anatomy. Tech Vasc Interv Radiol 19: 247–254, 2016. doi: 10.1053/j.tvir.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 26. Feldman L, Kleiner-Baumgarten A, Maislos M. Large pericardial and pleural effusions associated with familial lymphedema. Isr Med Assoc J 3: 769–770, 2001. [PubMed] [Google Scholar]
- 27. Jackson DG. Leucocyte trafficking via the lymphatic vasculature—mechanisms and consequences. Front Immunol 10: 471, 2019. doi: 10.3389/fimmu.2019.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21st century: novel functional roles in homeostasis and disease. Cell 182: 270–296, 2020. doi: 10.1016/j.cell.2020.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP. The lymphatic system: integral roles in immunity. Annu Rev Immunol 35: 31–52, 2017. doi: 10.1146/annurev-immunol-041015-055354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mehlhorn U, Geissler HJ, Laine GA, Allen SJ. Myocardial fluid balance. Eur J Cardiothorac Surg 20: 1220–1230, 2001. doi: 10.1016/s1010-7940(01)01031-4. [DOI] [PubMed] [Google Scholar]
- 31. Mehlhorn U, Davis KL, Burke EJ, Adams D, Laine GA, Allen SJ. Impact of cardiopulmonary bypass and cardioplegic arrest on myocardial lymphatic function. Am J Physiol Heart Circ Physiol 268: H178–H183, 1995. doi: 10.1152/ajpheart.1995.268.1.H178. [DOI] [PubMed] [Google Scholar]
- 32. Ullal SR, Kluge TH, Gerbode F. Studies on cardiac lymph during extracorporeal circulation. Chest 61: 662–667, 1972. doi: 10.1378/chest.61.7.662. [DOI] [PubMed] [Google Scholar]
- 33. Allen SJ, Geissler HJ, Davis KL, Gogola GR, Warters RD, de Vivie ER, Mehlhorn U. Augmenting cardiac contractility hastens myocardial edema resolution after cardiopulmonary bypass and cardioplegic arrest. Anesth Analg 85: 987–992, 1997. doi: 10.1097/00000539-199711000-00006. [DOI] [PubMed] [Google Scholar]
- 34. Cui Y. Impact of lymphatic vessels on the heart. Thorac Cardiovasc Surg 58: 1–7, 2010. doi: 10.1055/s-0029-1240553. [DOI] [PubMed] [Google Scholar]
- 35. Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Lévy F, Henry JP, Dumesnil A, Boukhalfa I, Banquet S, Schapman D, Thuillez C, Richard V, Mulder P, Brakenhielm E. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation 133: 1484–1497; discussion 1497, 2016. doi: 10.1161/CIRCULATIONAHA.115.020143. [DOI] [PubMed] [Google Scholar]
- 36. Houssari M, Dumesnil A, Tardif V, Kivelä R, Pizzinat N, Boukhalfa I, Godefroy D, Schapman D, Hemanthakumar KA, Bizou M, Henry JP, Renet S, Riou G, Rondeaux J, Anouar Y, Adriouch S, Fraineau S, Alitalo K, Richard V, Mulder P, Brakenhielm E. Lymphatic and immune cell cross-talk regulates cardiac recovery after experimental myocardial infarction. Arterioscler Thromb Vasc Biol 40: 1722–1737, 2020. doi: 10.1161/ATVBAHA.120.314370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vieira JM, Norman S, Villa Del Campo C, Cahill TJ, Barnette DN, Gunadasa-Rohling M, Johnson LA, Greaves DR, Carr CA, Jackson DG, Riley PR. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest 128: 3402–3412, 2018. doi: 10.1172/JCI97192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maruyama K, Miyagawa-Tomita S, Mizukami K, Matsuzaki F, Kurihara H. Isl1-expressing non-venous cell lineage contributes to cardiac lymphatic vessel development. Dev Biol 452: 134–143, 2019. doi: 10.1016/j.ydbio.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 39. Liu X, De la Cruz E, Gu X, Balint L, Oxendine-Burns M, Terrones T, Ma W, Kuo HH, Lantz C, Bansal T, Thorp E, Burridge P, Jakus Z, Herz J, Cleaver O, Torres M, Oliver G. Lymphoangiocrine signals promote cardiac growth and repair. Nature 588: 705–711, 2020. doi: 10.1038/s41586-020-2998-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong YK, Harvey N, Noh YH, Schacht V, Hirakawa S, Detmar M, Oliver G. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev Dyn 225: 351–357, 2002. doi: 10.1002/dvdy.10163. [DOI] [PubMed] [Google Scholar]
- 41. Petrova TV, Mäkinen T, Mäkelä TP, Saarela J, Virtanen I, Ferrell RE, Finegold DN, Kerjaschki D, Ylä-Herttuala S, Alitalo K. Lymphatic endothelial reprogramming of vascular endothelial cells by the Prox-1 homeobox transcription factor. EMBO J 21: 4593–4599, 2002. doi: 10.1093/emboj/cdf470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, Jackson DG, Oliver G. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J 21: 1505–1513, 2002. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell 98: 769–778, 1999. doi: 10.1016/s0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- 44. Vuorio T, Ylä-Herttuala E, Laakkonen JP, Laidinen S, Liimatainen T, Ylä-Herttuala S. Downregulation of VEGFR3 signaling alters cardiac lymphatic vessel organization and leads to a higher mortality after acute myocardial infarction. Sci Rep 8: 16709, 2018. doi: 10.1038/s41598-018-34770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Ylä-Herttuala S, Jackson DG, Alitalo K, McDonald DM. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest 115: 247–257, 2005. doi: 10.1172/JCI22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goldman J, Le TX, Skobe M, Swartz MA. Overexpression of VEGF-C causes transient lymphatic hyperplasia but not increased lymphangiogenesis in regenerating skin. Circ Res 96: 1193–1199, 2005. doi: 10.1161/01.RES.0000168918.27576.78. [DOI] [PubMed] [Google Scholar]
- 47. Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, Swartz M, Fukumura D, Jain RK, Alitalo K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 276: 1423–1425, 1997. doi: 10.1126/science.276.5317.1423. [DOI] [PubMed] [Google Scholar]
- 48. Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, Breitman M, Alitalo K. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA 92: 3566–3570, 1995. doi: 10.1073/pnas.92.8.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol 5: 74–80, 2004. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- 50. Mäkinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG, Alitalo K. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J 20: 4762–4773, 2001. doi: 10.1093/emboj/20.17.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rauniyar K, Jha SK, Jeltsch M. Biology of vascular endothelial growth factor C in the morphogenesis of lymphatic vessels. Front Bioeng Biotechnol 6: 7, 2018. doi: 10.3389/fbioe.2018.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wetzel-Strong SE, Li M, Klein KR, Nishikimi T, Caron KM. Epicardial-derived adrenomedullin drives cardiac hyperplasia during embryogenesis. Dev Dyn 243: 243–256, 2014. doi: 10.1002/dvdy.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci USA 98: 615–619, 2001. doi: 10.1073/pnas.98.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dunworth WP, Fritz-Six KL, Caron KM. Adrenomedullin stabilizes the lymphatic endothelial barrier in vitro and in vivo. Peptides 29: 2243–2249, 2008. doi: 10.1016/j.peptides.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fritz-Six KL, Dunworth WP, Li M, Caron KM. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J Clin Invest 118: 40–50, 2008. doi: 10.1172/JCI33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klein KR, Caron KM. Adrenomedullin in lymphangiogenesis: from development to disease. Cell Mol Life Sci 72: 3115–3126, 2015. doi: 10.1007/s00018-015-1921-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu W, Wittchen ES, Hoopes SL, Stefanini L, Burridge K, Caron KM. Small GTPase Rap1A/B is required for lymphatic development and adrenomedullin-induced stabilization of lymphatic endothelial junctions. Arterioscler Thromb Vasc Biol 38: 2410–2422, 2018. doi: 10.1161/ATVBAHA.118.311645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stone OA, Stainier DY. Paraxial mesoderm is the major source of lymphatic endothelium. Dev Cell 50: 247–255.e3, 2019. doi: 10.1016/j.devcel.2019.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gancz D, Raftrey BC, Perlmoter G, Marín-Juez R, Semo J, Matsuoka RL, Karra R, Raviv H, Moshe N, Addadi Y, Golani O, Poss KD, Red-Horse K, Stainier DY, Yaniv K. Distinct origins and molecular mechanisms contribute to lymphatic formation during cardiac growth and regeneration. Elife 8: e44153, 2019. doi: 10.7554/eLife.44153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lioux G, Liu X, Temiño S, Oxendine M, Ayala E, Ortega S, Kelly RG, Oliver G, Torres M. A second heart field-derived vasculogenic niche contributes to cardiac lymphatics. Dev Cell 52: 350–363.e6, 2020. doi: 10.1016/j.devcel.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Klaourakis K, Vieira JM, Riley PR. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat Rev Cardiol 18: 368–379, 2021. doi: 10.1038/s41569-020-00489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ulvmar MH, Mäkinen T. Heterogeneity in the lymphatic vascular system and its origin. Cardiovasc Res 111: 310–321, 2016. doi: 10.1093/cvr/cvw175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ulvmar MH, Martinez-Corral I, Stanczuk L, Mäkinen T. Pdgfrb-Cre targets lymphatic endothelial cells of both venous and non-venous origins. Genesis 54: 350–358, 2016. doi: 10.1002/dvg.22939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol 19: 312–326, 1896. doi: 10.1113/jphysiol.1896.sp000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Taylor AE. Capillary fluid filtration. Starling forces and lymph flow. Circ Res 49: 557–575, 1981. doi: 10.1161/01.res.49.3.557. [DOI] [PubMed] [Google Scholar]
- 66. Bhave G, Neilson EG. Body fluid dynamics: back to the future. J Am Soc Nephrol 22: 2166–2181, 2011. doi: 10.1681/ASN.2011080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huxley VH, Scallan J. Lymphatic fluid: exchange mechanisms and regulation. J Physiol 589: 2935–2943, 2011. doi: 10.1113/jphysiol.2011.208298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lent-Schochet D, Jialal I. Physiology, edema. In: StatPearls. Treasure Island, FL: StatPearls, 2020. [PubMed] [Google Scholar]
- 69. Levick JR. Revision of the Starling principle: new views of tissue fluid balance. J Physiol 557: 704, 2004. doi: 10.1113/jphysiol.2004.066118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res 87: 198–210, 2010. doi: 10.1093/cvr/cvq062. [DOI] [PubMed] [Google Scholar]
- 71. Pillinger NL, Kam P. Endothelial glycocalyx: basic science and clinical implications. Anaesth Intensive Care 45: 295–307, 2017. doi: 10.1177/0310057X1704500305. [DOI] [PubMed] [Google Scholar]
- 72. Renkin EM. Some consequences of capillary permeability to macromolecules: Starling’s hypothesis reconsidered. Am J Physiol Heart Circ Physiol 250: H706–H710, 1986. doi: 10.1152/ajpheart.1986.250.5.H706. [DOI] [PubMed] [Google Scholar]
- 73. Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth 108: 384–394, 2012. doi: 10.1093/bja/aer515. [DOI] [PubMed] [Google Scholar]
- 74. Michel CC, Phillips ME. Steady-state fluid filtration at different capillary pressures in perfused frog mesenteric capillaries. J Physiol 388: 421–435, 1987. doi: 10.1113/jphysiol.1987.sp016622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang X, Adamson RH, Curry FE, Weinbaum S. Transient regulation of transport by pericytes in venular microvessels via trapped microdomains. Proc Natl Acad Sci USA 105: 1374–1379, 2008. doi: 10.1073/pnas.0710986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng 9: 121–167, 2007. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 77. Adamson RH, Lenz JF, Zhang X, Adamson GN, Weinbaum S, Curry FE. Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J Physiol 557: 889–907, 2004. doi: 10.1113/jphysiol.2003.058255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guyton AC, Coleman TG. Regulation on interstitial fluid volume and pressure. Ann NY Acad Sci 150: 537–547, 1968. doi: 10.1111/j.1749-6632.1968.tb14705.x. [DOI] [PubMed] [Google Scholar]
- 79. Michel CC, Woodcock TE, Curry FE. Understanding and extending the Starling principle. Acta Anaesthesiol Scand 64: 1032–1037, 2020. doi: 10.1111/aas.13603. [DOI] [PubMed] [Google Scholar]
- 80. Stewart RH, Rohn DA, Mehlhorn U, Davis KL, Allen SJ, Laine GA. Regulation of microvascular filtration in the myocardium by interstitial fluid pressure. Am J Physiol Regul Integr Comp Physiol 271: R1465–R1469, 1996. doi: 10.1152/ajpregu.1996.271.6.R1465. [DOI] [PubMed] [Google Scholar]
- 81. Dongaonkar RM, Stewart RH, Geissler HJ, Laine GA. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc Res 87: 331–339, 2010. doi: 10.1093/cvr/cvq145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wearn JT; Technical Assistance of Zschiesche LJ. The extent of the capillary bed of the heart. J Exp Med 47: 273–290, 1928. doi: 10.1084/jem.47.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Drinker CK. The functional significance of the lymphatic system: Harvey Lecture, December 16, 1937. Bull NY Acad Med 14: 231–251, 1938. [PMC free article] [PubMed] [Google Scholar]
- 84. Cifarelli V, Eichmann A. The Intestinal lymphatic system: functions and metabolic implications. Cell Mol Gastroenterol Hepatol 7: 503–513, 2019. doi: 10.1016/j.jcmgh.2018.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol 1: 1, 2002. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chung C, Iwakiri Y. The lymphatic vascular system in liver diseases: its role in ascites formation. Clin Mol Hepatol 19: 99–104, 2013. doi: 10.3350/cmh.2013.19.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Puligandla PS, Laberge JM. Infections and diseases of the lungs, pleura, and mediastinum. In: Pediatric Surgery (7th ed.). Philadelphia PA: Elsevier, 2012, p. 855–880. [Google Scholar]
- 88. Davis KL, Laine GA, Geissler HJ, Mehlhorn U, Brennan M, Allen SJ. Effects of myocardial edema on the development of myocardial interstitial fibrosis. Microcirculation 7: 269–280, 2000. doi: 10.1111/j.1549-8719.2000.tb00127.x. [DOI] [PubMed] [Google Scholar]
- 89. Lord RS. The white veins: conceptual difficulties in the history of the lymphatics. Med Hist 12: 174–184, 1968. doi: 10.1017/s0025727300013053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hewson W, Hunter W. An account of the lymphatic system in fish. By the same. Philosophical Transactions (1683–1775) 59: 204–215, 1769. [Google Scholar]
- 91. Munn LL, Padera TP. Imaging the lymphatic system. Microvasc Res 96: 55–63, 2014. doi: 10.1016/j.mvr.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rasmussen JC, Tan IC, Marshall MV, Fife CE, Sevick-Muraca EM. Lymphatic imaging in humans with near-infrared fluorescence. Curr Opin Biotechnol 20: 74–82, 2009. doi: 10.1016/j.copbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Forbrich A, Heinmiller A, Zemp RJ. Photoacoustic imaging of lymphatic pumping. J Biomed Opt 22: 1–6, 2017. doi: 10.1117/1.JBO.22.10.106003. [DOI] [PubMed] [Google Scholar]
- 94. Martel C, Yao J, Huang CH, Zou J, Randolph GJ, Wang LV. Photoacoustic lymphatic imaging with high spatial-temporal resolution. J Biomed Opt 19: 116009, 2014. doi: 10.1117/1.JBO.19.11.116009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Song KH, Stein EW, Margenthaler JA, Wang LV. Noninvasive photoacoustic identification of sentinel lymph nodes containing methylene blue in vivo in a rat model. J Biomed Opt 13: 054033, 2008. doi: 10.1117/1.2976427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Suzuki Y, Kajita H, Imanishi N, Aiso S, Kishi K. Observation of a lymphatic pump in a human by using photoacoustic imaging. Plast Reconstr Surg Glob Open 8: e2914, 2020. doi: 10.1097/GOX.0000000000002914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jin S, Zhang C, Gao M, Wang T, Li L, Yang G, Ou Y, Li Y, Li S. Validation of laser scanning confocal microscopy as a diagnostic method for lymphedema using a rat model. Lasers Med Sci 36: 811–819, 2021. doi: 10.1007/s10103-020-03106-y. [DOI] [PubMed] [Google Scholar]
- 98. Jankowska-Steifer E, Ratajska A, Czarnowska E, Badurek I, Matryba P, Niderla-Bielińska J, Ciszek B, Brakenhielm E. Assessing functional status of cardiac lymphatics: from macroscopic imaging to molecular profiling. Trends Cardiovasc Med 31: 333–338, 2021. doi: 10.1016/j.tcm.2020.06.006. [DOI] [PubMed] [Google Scholar]
- 99. Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282: 946–949, 1998. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- 100. Wigle JT, Chowdhury K, Gruss P, Oliver G. Prox1 function is crucial for mouse lens-fibre elongation. Nat Genet 21: 318–322, 1999. doi: 10.1038/6844. [DOI] [PubMed] [Google Scholar]
- 101. Iljin K, Karkkainen MJ, Lawrence EC, Kimak MA, Uutela M, Taipale J, Pajusola K, Alhonen L, Halmekytö M, Finegold DN, Ferrell RE, Alitalo K. VEGFR3 gene structure, regulatory region, and sequence polymorphisms. FASEB J 15: 1028–1036, 2001. doi: 10.1096/fj.00-0383com. [DOI] [PubMed] [Google Scholar]
- 102. Gale NW, Prevo R, Espinosa J, Ferguson DJ, Dominguez MG, Yancopoulos GD, Thurston G, Jackson DG. Normal lymphatic development and function in mice deficient for the lymphatic hyaluronan receptor LYVE-1. Mol Cell Biol 27: 595–604, 2007. doi: 10.1128/MCB.01503-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B, Hong YK. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood 117: 362–365, 2011. doi: 10.1182/blood-2010-07-298562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Calvo CF, Fontaine RH, Soueid J, Tammela T, Makinen T, Alfaro-Cervello C, Bonnaud F, Miguez A, Benhaim L, Xu Y, Barallobre MJ, Moutkine I, Lyytikkaä J, Tatlisumak T, Pytowski B, Zalc B, Richardson W, Kessaris N, Garcia-Verdugo JM, Alitalo K, Eichmann A, Thomas JL. Vascular endothelial growth factor receptor 3 directly regulates murine neurogenesis. Genes Dev 25: 831–844, 2011. doi: 10.1101/gad.615311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hägerling R, Pollmann C, Kremer L, Andresen V, Kiefer F. Intravital two-photon microscopy of lymphatic vessel development and function using a transgenic Prox1 promoter-directed mOrange2 reporter mouse. Biochem Soc Trans 39: 1674–1681, 2011. doi: 10.1042/BST20110722. [DOI] [PubMed] [Google Scholar]
- 106. Truman LA, Bentley KL, Smith EC, Massaro SA, Gonzalez DG, Haberman AM, Hill M, Jones D, Min W, Krause DS, Ruddle NH. ProxTom lymphatic vessel reporter mice reveal Prox1 expression in the adrenal medulla, megakaryocytes, and platelets. Am J Pathol 180: 1715–1725, 2012. doi: 10.1016/j.ajpath.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Martínez-Corral I, Olmeda D, Diéguez-Hurtado R, Tammela T, Alitalo K, Ortega S. In vivo imaging of lymphatic vessels in development, wound healing, inflammation, and tumor metastasis. Proc Natl Acad Sci USA 109: 6223–6228, 2012. doi: 10.1073/pnas.1115542109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hong M, Jung E, Yang S, Jung W, Seong YJ, Park E, Bramos A, Kim KE, Lee S, Daghlian G, Seo JI, Choi I, Choi IS, Koh CJ, Kobielak A, Ying QL, Johnson M, Gardner D, Wong AK, Choi D, Hong YK. Efficient assessment of developmental, surgical and pathological lymphangiogenesis using a lymphatic reporter mouse and its embryonic stem cells. PLoS One 11: e0157126, 2016. doi: 10.1371/journal.pone.0157126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jung E, Gardner D, Choi D, Park E, Jin Seong Y, Yang S, Castorena-Gonzalez J, Louveau A, Zhou Z, Lee GK, Perrault DP, Lee S, Johnson M, Daghlian G, Lee M, Jin Hong Y, Kato Y, Kipnis J, Davis MJ, Wong AK, Hong YK. Development and characterization of a novel Prox1-EGFP lymphatic and Schlemm’s canal reporter rat. Sci Rep 7: 5577, 2017. doi: 10.1038/s41598-017-06031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Redder E, Kirschnick N, Bobe S, Hägerling R, Hansmeier NR, Kiefer F. Vegfr3-tdTomato, a reporter mouse for microscopic visualization of lymphatic vessel by multiple modalities. PLoS One 16: e0249256, 2021. doi: 10.1371/journal.pone.0249256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, Coughlin SR, McDonald DM, Schwab SR, Cyster JG. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med 207: 17–27, 2010. doi: 10.1084/jem.20091619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bianchi R, Teijeira A, Proulx ST, Christiansen AJ, Seidel CD, Rülicke T, Mäkinen T, Hägerling R, Halin C, Detmar M. A transgenic Prox1-Cre-tdTomato reporter mouse for lymphatic vessel research. PLoS One 10: e0122976, 2015. doi: 10.1371/journal.pone.0122976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Connor AL, Kelley PM, Tempero RM. Lymphatic endothelial lineage assemblage during corneal lymphangiogenesis. Lab Invest 96: 270–282, 2016. doi: 10.1038/labinvest.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mavrogeni S, Dimitroulas T, Gabriel S, Sfikakis PP, Pohost GM, Kitas GD. Why currently used diagnostic techniques for heart failure in rheumatoid arthritis are not enough: the challenge of cardiovascular magnetic resonance imaging. Rev Cardiovasc Med 15: 320–331, 2014. doi: 10.3909/ricm0724. [DOI] [PubMed] [Google Scholar]
- 115. Amano Y, Tachi M, Tani H, Mizuno K, Kobayashi Y, Kumita S. T2-weighted cardiac magnetic resonance imaging of edema in myocardial diseases. ScientificWorldJournal 2012: 194069, 2012., doi: 10.1100/2012/194069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Eitel I, Friedrich MG. T2-weighted cardiovascular magnetic resonance in acute cardiac disease. J Cardiovasc Magn Reson 13: 13, 2011. doi: 10.1186/1532-429X-13-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Usman AA, Taimen K, Wasielewski M, McDonald J, Shah S, Giri S, Cotts W, McGee E, Gordon R, Collins JD, Markl M, Carr JC. Cardiac magnetic resonance T2 mapping in the monitoring and follow-up of acute cardiac transplant rejection: a pilot study. Circ Cardiovasc Imaging 5: 782–790, 2012. doi: 10.1161/CIRCIMAGING.111.971101. [DOI] [PubMed] [Google Scholar]
- 118. Butler CR, Savu A, Bakal JA, Toma M, Thompson R, Chow K, Wang H, Kim DH, Mengel M, Haykowsky M, Pearson GJ, Kaul P, Paterson I. Correlation of cardiovascular magnetic resonance imaging findings and endomyocardial biopsy results in patients undergoing screening for heart transplant rejection. J Heart Lung Transplant 34: 643–650, 2015. doi: 10.1016/j.healun.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 119. Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I. Myocardial edema: a translational view. J Mol Cell Cardiol 52: 931–939, 2012. doi: 10.1016/j.yjmcc.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 120. García-Dorado D, Oliveras J, Gili J, Sanz E, Pérez-Villa F, Barrabés J, Carreras MJ, Solares J, Soler-Soler J. Analysis of myocardial oedema by magnetic resonance imaging early after coronary artery occlusion with or without reperfusion. Cardiovasc Res 27: 1462–1469, 1993. doi: 10.1093/cvr/27.8.1462. [DOI] [PubMed] [Google Scholar]
- 121. Tilak GS, Hsu LY, Hoyt RF Jr, Arai AE, Aletras AH. In vivo T2-weighted magnetic resonance imaging can accurately determine the ischemic area at risk for 2-day-old nonreperfused myocardial infarction. Invest Radiol 43: 7–15, 2008. doi: 10.1097/RLI.0b013e3181558822. [DOI] [PubMed] [Google Scholar]
- 122. Wince WB, Suranyi P, Schoepf UJ. Contemporary cardiovascular imaging methods for the assessment of at-risk myocardium. J Am Heart Assoc 3: e000473, 2013. doi: 10.1161/JAHA.113.000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mahnken AH, Bruners P, Bornikoel CM, Krämer N, Guenther RW. Assessment of myocardial edema by computed tomography in myocardial infarction. JACC Cardiovasc Imaging 2: 1167–1174, 2009. doi: 10.1016/j.jcmg.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 124. Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, Blanco-Colio L, Lavin B, Mallavia B, Tarin C, Mas S, Ortiz A, Egido J. Animal models of cardiovascular diseases. J Biomed Biotechnol 2011: 497841, 2011. doi: 10.1155/2011/497841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Laine GA, Allen SJ. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res 68: 1713–1721, 1991. doi: 10.1161/01.res.68.6.1713. [DOI] [PubMed] [Google Scholar]
- 126. Ludwig LL, Schertel ER, Pratt JW, McClure DE, Ying AJ, Heck CF, Myerowitz PD. Impairment of left ventricular function by acute cardiac lymphatic obstruction. Cardiovasc Res 33: 164–171, 1997. doi: 10.1016/s0008-6363(96)00177-0. [DOI] [PubMed] [Google Scholar]
- 127. Pratt JW, Schertel ER, Schaefer SL, Esham KE, McClure DE, Heck CF, Myerowitz PD. Acute transient coronary sinus hypertension impairs left ventricular function and induces myocardial edema. Am J Physiol Heart Circ Physiol 271: H834–H841, 1996. doi: 10.1152/ajpheart.1996.271.3.H834. [DOI] [PubMed] [Google Scholar]
- 128. Charan NB, Ripley R, Carvalho P. Effect of increased coronary venous pressure on left ventricular function in sheep. Respir Physiol 112: 227–235, 1998. doi: 10.1016/S0034-5687(98)00012-7. [DOI] [PubMed] [Google Scholar]
- 129. Fernández-Jiménez R, Sánchez-González J, Agüero J, García-Prieto J, López-Martín GJ, García-Ruiz JM, Molina-Iracheta A, Rosselló X, Fernández-Friera L, Pizarro G, García-Álvarez A, Dall’Armellina E, Macaya C, Choudhury RP, Fuster V, Ibáñez B. Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: imaging and histological tissue characterization. J Am Coll Cardiol 65: 315–323, 2015. doi: 10.1016/j.jacc.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 130. Jacob M, Paul O, Mehringer L, Chappell D, Rehm M, Welsch U, Kaczmarek I, Conzen P, Becker BF. Albumin augmentation improves condition of guinea pig hearts after 4 hr of cold ischemia. Transplantation 87: 956–965, 2009. doi: 10.1097/TP.0b013e31819c83b5. [DOI] [PubMed] [Google Scholar]
- 131. Powell WJ Jr, Wittenberg J, Maturi RA, Dinsmore RE, Miller SW. Detection of edema associated with myocardial ischemia by computerized tomography in isolated, arrested canine hearts. Circulation 55: 99–108, 1977. doi: 10.1161/01.cir.55.1.99. [DOI] [PubMed] [Google Scholar]
- 132. Qiu Y, Hearse DJ. Comparison of ischemic vulnerability and responsiveness to cardioplegic protection in crystalloid-perfused versus blood-perfused hearts. J Thorac Cardiovasc Surg 103: 960–968, 1992. [PubMed] [Google Scholar]
- 133. Shimizu Y, Polavarapu R, Eskla KL, Pantner Y, Nicholson CK, Ishii M, Brunnhoelzl D, Mauria R, Husain A, Naqvi N, Murohara T, Calvert JW. Impact of lymphangiogenesis on cardiac remodeling after ischemia and reperfusion injury. J Am Heart Assoc 7: e009565, 2018. doi: 10.1161/JAHA.118.009565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gloviczki P, Solti F, Szlavy L, Jellinek H. Ultrastructural and electrophysiologic changes of experimental acute cardiac lymphostasis. Lymphology 16: 185–192, 1983. [PubMed] [Google Scholar]
- 135. Kluge T, Ullal SR. Pathology of the heart following chronic cardiac lymphatic obstruction. An experimental study in dogs. Acta Pathol Microbiol Scand A 80: 150–158, 1972. doi: 10.1111/j.1699-0463.1972.tb02159.x. [DOI] [PubMed] [Google Scholar]
- 136. Kong D, Kong X, Wang L. Effect of cardiac lymph flow obstruction on cardiac collagen synthesis and interstitial fibrosis. Physiol Res 55: 253–258, 2006. doi: 10.33549/physiolres.930727. [DOI] [PubMed] [Google Scholar]
- 137. Miller AJ, Pick R, Katz LN. Ventricular endomyocardial pathology produced by chronic cardiac lymphatic obstruction in the dog. Circ Res 8: 941–947, 1960. doi: 10.1161/01.res.8.5.941. [DOI] [PubMed] [Google Scholar]
- 138. Sun SC, Lie JT. Cardiac lymphatic obstruction: ultrastructure of acute-phase myocardial injury in dogs. Mayo Clin Proc 52: 785–792, 1977. [PubMed] [Google Scholar]
- 139. Wang YL, Wang XH, Liu YL, Kong XQ, Wang LX. Cardiac lymphatic obstruction impairs left ventricular function and increases plasma endothelin-1 and angiotensin II in rabbits. Lymphology 42: 182–187, 2009. [PubMed] [Google Scholar]
- 140. Solti F, Lengyel E, Jellinek H, Schneider F, Juhász-Nagy A, Kékesi V. Coronary arteriopathy after lymphatic blockade: an experimental study in dogs. Lymphology 27: 173–180, 1994. [PubMed] [Google Scholar]
- 141. Leeds SE, Uhley HN, Sampson JJ, Friedman M. The cardiac lymphatics after ligation of the coronary sinus. Proc Soc Exp Biol Med 135: 59–62, 1970. doi: 10.3181/00379727-135-34987. [DOI] [PubMed] [Google Scholar]
- 142. Dongaonkar RM, Stewart RH, Quick CM, Uray KL, Cox CS Jr, Laine GA. Award article: Microcirculatory Society Award for Excellence in Lymphatic Research: time course of myocardial interstitial edema resolution and associated left ventricular dysfunction. Microcirculation 19: 714–722, 2012. doi: 10.1111/j.1549-8719.2012.00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Laine GA. Microvascular changes in the heart during chronic arterial hypertension. Circ Res 62: 953–960, 1988. doi: 10.1161/01.res.62.5.953. [DOI] [PubMed] [Google Scholar]
- 144. Mehlhorn U, Davis KL, Laine GA, Geissler HJ, Allen SJ. Myocardial fluid balance in acute hypertension. Microcirculation 3: 371–378, 1996. doi: 10.3109/10739689609148309. [DOI] [PubMed] [Google Scholar]
- 145. Desai KV, Laine GA, Stewart RH, Cox CS Jr, Quick CM, Allen SJ, Fischer UM. Mechanics of the left ventricular myocardial interstitium: effects of acute and chronic myocardial edema. Am J Physiol Heart Circ Physiol 294: H2428–H2434, 2008. doi: 10.1152/ajpheart.00860.2007. [DOI] [PubMed] [Google Scholar]
- 146. Whalen DA Jr, Hamilton DG, Ganote CE, Jennings RB. Effect of a transient period of ischemia on myocardial cells. I. Effects on cell volume regulation. Am J Pathol 74: 381–397, 1974. [PMC free article] [PubMed] [Google Scholar]
- 147. Kloner RA, Ganote CE, Whalen DA Jr, Jennings RB. Effect of a transient period of ischemia on myocardial cells. II. Fine structure during the first few minutes of reflow. Am J Pathol 74: 399–422, 1974. [PMC free article] [PubMed] [Google Scholar]
- 148. Mewton N, Rapacchi S, Augeul L, Ferrera R, Loufouat J, Boussel L, Micolich A, Rioufol G, Revel D, Ovize M, Croisille P. Determination of the myocardial area at risk with pre- versus post-reperfusion imaging techniques in the pig model. Basic Res Cardiol 106: 1247–1257, 2011. doi: 10.1007/s00395-011-0214-8. [DOI] [PubMed] [Google Scholar]
- 149. Fernández-Jiménez R, Barreiro-Pérez M, Martin-García A, Sanchez-Gonzalez J, Aguero J, Galan-Arriola C, Garcia-Prieto J, Diaz-Pelaez E, Vara P, Martinez I, Zamarro I, Garde B, Sanz J, Fuster V, Sanchez PL, Ibanez B. Dynamic edematous response of the human heart to myocardial infarction: implications for assessing myocardial area at risk and salvage. Circulation 136: 1288–1300, 2017. doi: 10.1161/CIRCULATIONAHA.116.025582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kline IK, Miller AJ, Pick R, Katz LN. The effects of chronic impairment of cardiac lymph flow on myocardial reactions after coronary artery ligation in dogs. Am Heart J 68: 515–523, 1964. doi: 10.1016/0002-8703(64)90153-x. [DOI] [PubMed] [Google Scholar]
- 151. Diab OA, Amer MS, Salah El-Din RA. Effect of experimental coronary sinus ligation on myocardial structure and function in the presence or absence of structural heart disease: an insight for the interventional electrophysiologist. Europace 18: 1897–1904, 2016. doi: 10.1093/europace/euv431. [DOI] [PubMed] [Google Scholar]
- 152. Mehlhorn U, Allen SJ, Adams DL, Davis KL, Gogola GR, de Vivie ER, Laine GA. Normothermic continuous antegrade blood cardioplegia does not prevent myocardial edema and cardiac dysfunction. Circulation 92: 1940–1946, 1995. doi: 10.1161/01.CIR.92.7.1940. [DOI] [PubMed] [Google Scholar]
- 153. Taira A, Yamashita M, Arikawa K, Hamada Y, Toyohira H, Akita H. Cardia lymph in electrical ventricular fibrillation: an experimental study. Ann Thorac Surg 27: 144–147, 1979. doi: 10.1016/s0003-4975(10)63256-0. [DOI] [PubMed] [Google Scholar]
- 154. Miyamoto M, McClure DE, Schertel ER, Andrews PJ, Jones GA, Pratt JW, Ross P, Myerowitz PD. Effects of hypoproteinemia-induced myocardial edema on left ventricular function. Am J Physiol Heart Circ Physiol 274: H937–H944, 1998. doi: 10.1152/ajpheart.1998.274.3.H937. [DOI] [PubMed] [Google Scholar]
- 155. Rubboli A, Sobotka PA, Euler DE. Effect of acute edema on left ventricular function and coronary vascular resistance in the isolated rat heart. Am J Physiol Heart Circ Physiol 267: H1054–H1061, 1994. doi: 10.1152/ajpheart.1994.267.3.H1054. [DOI] [PubMed] [Google Scholar]
- 156. Spotnitz HM, Hsu DT. Myocardial edema: importance in the study of left ventricular function. Adv Card Surg 5: 1–25, 1994. [PubMed] [Google Scholar]
- 157. Weng ZC, Nicolosi AC, Detwiler PW, Hsu DT, Schierman SW, Goldstein AH, Spotnitz HM. Effects of crystalloid, blood, and University of Wisconsin perfusates on weight, water content, and left ventricular compliance in an edema-prone, isolated porcine heart model. J Thorac Cardiovasc Surg 103: 504–513, 1992. doi: 10.1016/S0022-5223(19)34992-X. [DOI] [PubMed] [Google Scholar]
- 158. Egan JR, Butler TL, Cole AD, Aharonyan A, Baines D, Street N, Navaratnam M, Biecker O, Zazulak C, Au CG, Tan YM, North KN, Winlaw DS. Myocardial ischemia is more important than the effects of cardiopulmonary bypass on myocardial water handling and postoperative dysfunction: a pediatric animal model. J Thorac Cardiovasc Surg 136: 1265–1273.e2, 2008. doi: 10.1016/j.jtcvs.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 159. Verma S, Fedak PW, Weisel RD, Szmitko PE, Badiwala MV, Bonneau D, Latter D, Errett L, LeClerc Y. Off-pump coronary artery bypass surgery: fundamentals for the clinical cardiologist. Circulation 109: 1206–1211, 2004. doi: 10.1161/01.CIR.0000120292.65143.F5. [DOI] [PubMed] [Google Scholar]
- 160. Mehlhorn U, Allen SJ, Adams DL, Davis KL, Gogola GR, Warters RD. Cardiac surgical conditions induced by beta-blockade: effect on myocardial fluid balance. Ann Thorac Surg 62: 143–150, 1996. doi: 10.1016/0003-4975(96)00221-4. [DOI] [PubMed] [Google Scholar]
- 161. Wang W, Wang Y, Piao H, Li B, Wang T, Li D, Zhu Z, Xu R, Liu K. Early and medium outcomes of on-pump beating-heart versus off-pump CABG in patients with moderate left ventricular dysfunction. Braz J Cardiovasc Surg 34: 62–69, 2019.doi: 10.21470/1678-9741-2018-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Hueb W, Lopes NH, Pereira AC, Hueb AC, Soares PR, Favarato D, Vieira RD, Lima EG, Garzillo CL, da Silva Paulitch F, César LA, Gersh BJ, Ramires JA. Five-year follow-up of a randomized comparison between off-pump and on-pump stable multivessel coronary artery bypass grafting. The MASS III Trial. Circulation 122: S48–S52, 2010. [DOI] [PubMed] [Google Scholar]
- 163. Kim HJ, Oh YN, Ju MH, Kim JB, Jung SH, Chung CH, Lee JW, Choo SJ. On-pump beating heart versus conventional coronary artery bypass grafting: comparative study on early and long-term clinical outcomes. J Thorac Dis 10: 2656–2665, 2018. doi: 10.21037/jtd.2018.05.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Miyahara K, Matsuura A, Takemura H, Saito S, Sawaki S, Yoshioka T, Ito H. On-pump beating-heart coronary artery bypass grafting after acute myocardial infarction has lower mortality and morbidity. J Thorac Cardiovasc Surg 135: 521–526, 2008. doi: 10.1016/j.jtcvs.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 165. Zhou P, Zhu P, Xiao Z, Lin X, Xu R, Zheng S. Meta-analysis of repeat revascularization of off-pump and on-pump coronary artery bypass surgery. Ann Thorac Surg 106: 526–531, 2018. doi: 10.1016/j.athoracsur.2018.02.068. [DOI] [PubMed] [Google Scholar]
- 166.Mayo Clinic. Heart transplant. 2020. https://www.mayoclinic.org/tests-procedures/heart-transplant/about/pac-20384750.
- 167. Edwards LA, Nowocin AK, Jafari NV, Meader LL, Brown K, Sarde A, Lam C, Murray A, Wong W. Chronic rejection of cardiac allografts is associated with increased lymphatic flow and cellular trafficking. Circulation 137: 488–503, 2018. doi: 10.1161/CIRCULATIONAHA.117.028533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Soong TR, Pathak AP, Asano H, Fox-Talbot K, Baldwin WM 3rd.. Lymphatic injury and regeneration in cardiac allografts. Transplantation 89: 500–508, 2010. doi: 10.1097/TP.0b013e3181c73c34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Nykänen AI, Sandelin H, Krebs R, Keränen MA, Tuuminen R, Karpanen T, Wu Y, Pytowski B, Koskinen PK, Yla-Herttuala S, Alitalo K, Lemstrom KB. Targeting lymphatic vessel activation and CCL21 production by vascular endothelial growth factor receptor-3 inhibition has novel immunomodulatory and antiarteriosclerotic effects in cardiac allografts. Circulation 121: 1413–1422, 2010. doi: 10.1161/CIRCULATIONAHA.109.910703. [DOI] [PubMed] [Google Scholar]
- 170. Dashkevich A, Raissadati A, Syrjala SO, Zarkada G, Keränen MA, Tuuminen R, Krebs R, Anisimov A, Jeltsch M, Leppänen VM, Alitalo K, Nykänen AI, Lemstrom KB. Ischemia-reperfusion injury enhances lymphatic endothelial VEGFR3 and rejection in cardiac allografts. Am J Transplant 16: 1160–1172, 2016. doi: 10.1111/ajt.13564. [DOI] [PubMed] [Google Scholar]
- 171. Bhagra SK, Pettit S, Parameshwar J. Cardiac transplantation: indications, eligibility and current outcomes. Heart 105: 252–260, 2019. doi: 10.1136/heartjnl-2018-313103. [DOI] [PubMed] [Google Scholar]
- 172. Marie PY, Carteaux JP, Escanye JM, Claudon O, David N, Mattei S, Hassan N, Danchin N, Karcher G, Bertrand A, Villemot JP. Detection and prediction of acute heart transplant rejection with the myocardial T2 determination provided by a black-blood magnetic resonance imaging sequence. J Heart Lung Transplant 20: 193–194, 2001. doi: 10.1016/s1053-2498(00)00406-x. [DOI] [PubMed] [Google Scholar]
- 173. Vermes E, Pantaléon C, Auvet A, Cazeneuve N, Machet MC, Delhommais A, Bourguignon T, Aupart M, Brunereau L. Cardiovascular magnetic resonance in heart transplant patients: diagnostic value of quantitative tissue markers: T2 mapping and extracellular volume fraction, for acute rejection diagnosis. J Cardiovasc Magn Reson 20: 59, 2018. doi: 10.1186/s12968-018-0480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Friedrich MG, Marcotte F. Cardiac magnetic resonance assessment of myocarditis. Circ Cardiovasc Imaging 6: 833–839, 2013. doi: 10.1161/CIRCIMAGING.113.000416. [DOI] [PubMed] [Google Scholar]
- 175. Schultz JC, Hilliard AA, Cooper LT Jr, Rihal CS. Diagnosis and treatment of viral myocarditis. Mayo Clin Proc 84: 1001–1009, 2009. doi: 10.1016/S0025-6196(11)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M, Yilmaz A, Klingel K, Kandolf R, Sechtem U, Cooper LT, Böhm M. Update on myocarditis. J Am Coll Cardiol 59: 779–792, 2012. doi: 10.1016/j.jacc.2011.09.074. [DOI] [PubMed] [Google Scholar]
- 177. Rienks M, Carai P, van Teeffelen J, Eskens B, Verhesen W, Hemmeryckx B, Johnson DM, van Leeuwen R, Jones EA, Heymans S, Papageorgiou AP. SPARC preserves endothelial glycocalyx integrity, and protects against adverse cardiac inflammation and injury during viral myocarditis. Matrix Biol 74: 21–34, 2018. doi: 10.1016/j.matbio.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 178. Kholová I, Dragneva G, Cermáková P, Laidinen S, Kaskenpää N, Hazes T, Cermáková E, Steiner I, Ylä-Herttuala S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest 41: 487–497, 2011. doi: 10.1111/j.1365-2362.2010.02431.x. [DOI] [PubMed] [Google Scholar]
- 179. Al-Kofahi M, Omura S, Tsunoda I, Sato F, Becker F, Gavins FN, Woolard MD, Pattillo C, Zawieja D, Muthuchamy M, Gashev A, Shihab I, Ghoweba M, Von der Weid PY, Wang Y, Alexander JS. IL-1beta reduces cardiac lymphatic muscle contraction via COX-2 and PGE2 induction: Potential role in myocarditis. Biomed Pharmacother 107: 1591–1600, 2018. doi: 10.1016/j.biopha.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 180. Eapen ZJ, Tang WH, Felker GM, Hernandez AF, Mahaffey KW, Lincoff AM, Roe MT. Defining heart failure end points in ST-segment elevation myocardial infarction trials: integrating past experiences to chart a path forward. Circ Cardiovasc Qual Outcomes 5: 594–600, 2012. doi: 10.1161/CIRCOUTCOMES.112.966150. [DOI] [PubMed] [Google Scholar]
- 181. Andrés-Villarreal M, Barba I, Poncelas M, Inserte J, Rodriguez-Palomares J, Pineda V, Garcia-Dorado D. Measuring water distribution in the heart: preventing edema reduces ischemia-reperfusion injury. J Am Heart Assoc 5: e003843, 2016. doi: 10.1161/JAHA.116.003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ibáñez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65: 1454–1471, 2015. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 183. Okamoto F, Allen BS, Buckberg GD, Bugyi H, Leaf J. Reperfusion conditions: importance of ensuring gentle versus sudden reperfusion during relief of coronary occlusion. J Thorac Cardiovasc Surg 92: 613–620, 1986. [PubMed] [Google Scholar]
- 184. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 285: H579–H588, 2003. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 185. Stone GW, Selker HP, Thiele H, Patel MR, Udelson JE, Ohman EM, Maehara A, Eitel I, Granger CB, Jenkins PL, Nichols M, Ben-Yehuda O. Relationship between infarct size and outcomes following primary PCI: patient-level analysis from 10 randomized trials. J Am Coll Cardiol 67: 1674–1683, 2016. doi: 10.1016/j.jacc.2016.01.069. [DOI] [PubMed] [Google Scholar]
- 186. Szlavy L, Koster K, de Courten A, Hollenberg NK. Early disappearance of lymphatics draining ischemic myocardium in the dog. Angiology 38: 73–84, 1987. doi: 10.1177/000331978703800111. [DOI] [PubMed] [Google Scholar]
- 187. Ibanez B, Aletras AH, Arai AE, Arheden H, Bax J, Berry C, Bucciarelli-Ducci C, Croisille P, Dall’Armellina E, Dharmakumar R, Eitel I, Fernandez-Jimenez R, Friedrich MG, Garcia-Dorado D, Hausenloy DJ, Kim RJ, Kozerke S, Kramer CM, Salerno M, Sanchez-Gonzalez J, Sanz J, Fuster V. Cardiac MRI endpoints in myocardial infarction experimental and clinical trials: JACC Scientific Expert Panel. J Am Coll Cardiol 74: 238–256, 2019. doi: 10.1016/j.jacc.2019.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Hamirani YS, Wong A, Kramer CM, Salerno M. Effect of microvascular obstruction and intramyocardial hemorrhage by CMR on LV remodeling and outcomes after myocardial infarction: a systematic review and meta-analysis. JACC Cardiovasc Imaging 7: 940–952, 2014. doi: 10.1016/j.jcmg.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Krauss XH, van der Wall EE, van der Laarse A, Doornbos J, de Roos A, Matheijssen NA, van Dijkman PR, van Voorthuisen AE, Bruschke AV. Follow-up of regional myocardial T2 relaxation times in patients with myocardial infarction evaluated with magnetic resonance imaging. Eur J Radiol 11: 110–119, 1990. doi: 10.1016/0720-048x(90)90159-9. [DOI] [PubMed] [Google Scholar]
- 190. Bulluck H, Dharmakumar R, Arai AE, Berry C, Hausenloy DJ. Cardiovascular magnetic resonance in acute ST-segment-elevation myocardial infarction: recent advances, controversies, and future directions. Circulation 137: 1949–1964, 2018. doi: 10.1161/CIRCULATIONAHA.117.030693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Ishikawa Y, Akishima-Fukasawa Y, Ito K, Akasaka Y, Tanaka M, Shimokawa R, Kimura-Matsumoto M, Morita H, Sato S, Kamata I, Ishii T. Lymphangiogenesis in myocardial remodelling after infarction. Histopathology 51: 345–353, 2007. doi: 10.1111/j.1365-2559.2007.02785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Sun QN, Wang YF, Guo ZK. Reconstitution of myocardial lymphatic vessels after acute infarction of rat heart. Lymphology 45: 80–86, 2012. [PubMed] [Google Scholar]
- 193. Martinez-Quinones P, McCarthy CG, Watts SW, Klee NS, Komic A, Calmasini FB, Priviero F, Warner A, Chenghao Y, Wenceslau CF. Hypertension induced morphological and physiological changes in cells of the arterial wall. Am J Hypertens 31: 1067–1078, 2018. doi: 10.1093/ajh/hpy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Carrick D, Haig C, Maznyczka AM, Carberry J, Mangion K, Ahmed N, Yue May VT, McEntegart M, Petrie MC, Eteiba H, Lindsay M, Hood S, Watkins S, Davie A, Mahrous A, Mordi I, Ford I, Radjenovic A, Welsh P, Sattar N, Wetherall K, Oldroyd KG, Berry C. Hypertension, microvascular pathology, and prognosis after an acute myocardial infarction. Hypertension 72: 720–730, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Mahmud E, Raisinghani A, Hassankhani A, Sadeghi HM, Strachan GM, Auger W, DeMaria AN, Blanchard DG. Correlation of left ventricular diastolic filling characteristics with right ventricular overload and pulmonary artery pressure in chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol 40: 318–324, 2002. doi: 10.1016/S0735-1097(02)01959-9. [DOI] [PubMed] [Google Scholar]
- 196. Moustapha A, Kaushik V, Diaz S, Kang SH, Barasch E. Echocardiographic evaluation of left-ventricular diastolic function in patients with chronic pulmonary hypertension. Cardiology 95: 96–100, 2001. doi: 10.1159/000047353. [DOI] [PubMed] [Google Scholar]
- 197. Davis KL, Mehlhorn U, Laine GA, Allen SJ. Myocardial edema, left ventricular function, and pulmonary hypertension. J Appl Physiol (1985) 78: 132–137, 1995. doi: 10.1152/jappl.1995.78.1.132. [DOI] [PubMed] [Google Scholar]
- 198. Verbrugge FH, Bertrand PB, Willems E, Gielen E, Mullens W, Giri S, Tang WH, Raman SV, Verhaert D. Global myocardial oedema in advanced decompensated heart failure. Eur Heart J Cardiovasc Imaging 18: 787–794, 2017. doi: 10.1093/ehjci/jew131. [DOI] [PubMed] [Google Scholar]
- 199. Dashkevich A, Bloch W, Antonyan A, Fries JU, Geissler HJ. Morphological and quantitative changes of the initial myocardial lymphatics in terminal heart failure. Lymphat Res Biol 7: 21–27, 2009.doi: 10.1089/lrb.2008.1010. [DOI] [PubMed] [Google Scholar]
- 200. Song D, Yang Y, He N, Tian X, Sang DS, Li YJ. The involvement of AQP1 in myocardial edema induced by pressure overload in mice. Eur Rev Med Pharmacol Sci 22: 4969–4974, 2018. [DOI] [PubMed] [Google Scholar]
- 201. Ge Q, Zhao L, Ren XM, Ye P, Hu ZY. LCZ696, an angiotensin receptor-neprilysin inhibitor, ameliorates diabetic cardiomyopathy by inhibiting inflammation, oxidative stress and apoptosis. Exp Biol Med (Maywood) 244: 1028–1039, 2019. doi: 10.1177/1535370219861283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Heron C, Dumesnil A, Houssari M, Renet S, Lemarcis T, Lebon A, Godefroy D, Schapman D, Henri O, Riou G, Nicol L, Henry JP, Valet M, Pieronne-Deperrois M, Ouvrard-Pascaud A, Hägerling R, Chiavelli H, Michel JB, Mulder P, Fraineau S, Richard V, Tardif V, Brakenhielm E. Regulation and impact of cardiac lymphangiogenesis in pressure-overload-induced heart failure. Cardiovasc Res 2022: cvac086, 2022. doi: 10.1093/cvr/cvac086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Cuijpers I, Simmonds SJ, van Bilsen M, Czarnowska E, González Miqueo A, Heymans S, Kuhn AR, Mulder P, Ratajska A, Jones EA, Brakenhielm E. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res Cardiol 115: 39–39, 2020. doi: 10.1007/s00395-020-0798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Rossitto G, Mary S, McAllister C, Neves KB, Haddow L, Rocchiccioli JP, Lang NN, Murphy CL, Touyz RM, Petrie MC, Delles C. Reduced lymphatic reserve in heart failure with preserved ejection fraction. J Am Coll Cardiol 76: 2817–2829, 2020. doi: 10.1016/j.jacc.2020.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.National Library of Medicine. Early feasibility study to evaluate safety and performance of the WhiteSwell System in the treatment of fluid overload in hospitalized patients with acutely decompensated heart failure. ClinicalTrials.gov Identifier NCT02863796, 2021.
- 206. Felker GM, Mentz RJ. Diuretics and ultrafiltration in acute decompensated heart failure. J Am Coll Cardiol 59: 2145–2153, 2012. doi: 10.1016/j.jacc.2011.10.910. [DOI] [PubMed] [Google Scholar]
- 207. Cooper HA, Dries DL, Davis CE, Shen YL, Domanski MJ. Diuretics and risk of arrhythmic death in patients with left ventricular dysfunction. Circulation 100: 1311–1315, 1999. doi: 10.1161/01.CIR.100.12.1311. [DOI] [PubMed] [Google Scholar]
- 208. Muñoz D, Felker GM. Approaches to decongestion in patients with acute decompensated heart failure. Curr Cardiol Rep 15: 335–335, 2013. doi: 10.1007/s11886-012-0335-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.WhiteSwell. Leverging the lymphatic system to treat ADHF. https://whiteswell.com/.2022.
- 210. Francone M. Role of cardiac magnetic resonance in the evaluation of dilated cardiomyopathy: diagnostic contribution and prognostic significance. ISRN Radiol 2014: 365404, 2014. doi: 10.1155/2014/365404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Hazebroek M, Dennert R, Heymans S. Idiopathic dilated cardiomyopathy: possible triggers and treatment strategies. Neth Heart J 20: 332–335, 2012. doi: 10.1007/s12471-012-0285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Jeserich M, Föll D, Olschewski M, Kimmel S, Friedrich MG, Bode C, Geibel A. Evidence of myocardial edema in patients with nonischemic dilated cardiomyopathy. Clin Cardiol 35: 371–376, 2012. doi: 10.1002/clc.21979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Nishii T, Kono AK, Shigeru M, Takamine S, Fujiwara S, Kyotani K, Aoyama N, Sugimura K. Cardiovascular magnetic resonance T2 mapping can detect myocardial edema in idiopathic dilated cardiomyopathy. Int J Cardiovasc Imaging 30: 65–72, 2014. doi: 10.1007/s10554-014-0414-z. [DOI] [PubMed] [Google Scholar]
- 214. Abraham D, Hofbauer R, Schäfer R, Blumer R, Paulus P, Miksovsky A, Traxler H, Kocher A, Aharinejad S. Selective downregulation of VEGF-A165, VEGF-R1, and decreased capillary density in patients with dilative but not ischemic cardiomyopathy. Circ Res 87: 644–647, 2000. doi: 10.1161/01.RES.87.8.644. [DOI] [PubMed] [Google Scholar]
- 215. Dashkevich A, Bloch W, Antonyan A, Goebel H, Fries JU, Schlensak C, Beyersdorf F, Geissler HJ. Immunohistochemical study of remodeling of myocardial lymphatic and blood microvascular structures in terminal heart failure: differences between ischemic and dilated cardiomyopathy. Lymphology 43: 110–117, 2010. [PubMed] [Google Scholar]
- 216. Lin QY, Zhang YL, Bai J, Liu JQ, Li HH. VEGF-C/VEGFR-3 axis protects against pressure-overload induced cardiac dysfunction through regulation of lymphangiogenesis. Clin Transl Med 11: e374, 2021. doi: 10.1002/ctm2.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Rehman I, Nassereddin A, Rehman A. Anatomy, thorax, pericardium. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC, 2022. [PubMed] [Google Scholar]
- 218. Ben-Horin S, Shinfeld A, Kachel E, Chetrit A, Livneh A. The composition of normal pericardial fluid and its implications for diagnosing pericardial effusions. Am J Med 118: 636–640, 2005. doi: 10.1016/j.amjmed.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 219. Nishikimi T, Asakawa H, Iida H, Matsushita Y, Shibasaki I, Tadokoro K, Mori Y, Mori H, Mochizuki Y, Okamura Y, Miyoshi S, Kangawa K, Matsuoka H. Different secretion patterns of two molecular forms of cardiac adrenomedullin in pressure- and volume-overloaded human heart failure. J Card Fail 10: 321–327, 2004. doi: 10.1016/j.cardfail.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 220. Trindade F, Vitorino R, Leite-Moreira A, Falcão-Pires I. Pericardial fluid: an underrated molecular library of heart conditions and a potential vehicle for cardiac therapy. Basic Res Cardiol 114: 10, 2019. doi: 10.1007/s00395-019-0716-3. [DOI] [PubMed] [Google Scholar]
- 221. Vogiatzidis K, Zarogiannis SG, Aidonidis I, Solenov EI, Molyvdas PA, Gourgoulianis KI, Hatzoglou C. Physiology of pericardial fluid production and drainage. Front Physiol 6: 62–62, 2015. doi: 10.3389/fphys.2015.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Hamilton DR, Sas R, Tyberg JV. Atrioventricular nonuniformity of pericardial constraint. Am J Physiol Heart Circ Physiol 287: H1700–H1704, 2004. doi: 10.1152/ajpheart.00117.2004. [DOI] [PubMed] [Google Scholar]
- 223. Smiseth OA, Frais MA, Kingma I, Smith ER, Tyberg JV. Assessment of pericardial constraint in dogs. Circulation 71: 158–164, 1985. doi: 10.1161/01.cir.71.1.158. [DOI] [PubMed] [Google Scholar]
- 224. Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. Lymphatic system in cardiovascular medicine. Circ Res 118: 515–530, 2016. doi: 10.1161/CIRCRESAHA.115.306544. [DOI] [PubMed] [Google Scholar]
- 225. Sagristà-Sauleda J, Mercé AS, Soler-Soler J. Diagnosis and management of pericardial effusion. World J Cardiol 3: 135–143, 2011. doi: 10.4330/wjc.v3.i5.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Stewart RH, Cox CS Jr, Allen SJ, Laine GA. Myocardial edema provides a link between pulmonary arterial hypertension and pericardial effusion. Circulation 145: 793–795, 2022. doi: 10.1161/CIRCULATIONAHA.121.057666. [DOI] [PubMed] [Google Scholar]
- 227. Pérez-Casares A, Cesar S, Brunet-Garcia L, Sanchez-de-Toledo J. Echocardiographic evaluation of pericardial effusion and cardiac tamponade. Front Pediatr 5: 79–79, 2017. doi: 10.3389/fped.2017.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Boulanger B, Yuan Z, Flessner M, Hay J, Johnston M. Pericardial fluid absorption into lymphatic vessels in sheep. Microvasc Res 57: 174–186, 1999. doi: 10.1006/mvre.1998.2127. [DOI] [PubMed] [Google Scholar]
- 229. Eliskova M, Eliska O, Miller AJ. The lymphatic drainage of the parietal pericardium in man. Lymphology 28: 208–217, 1995. [PubMed] [Google Scholar]
- 230. Pang LM, Mellins RB, Rodriguez-Martinez F. Effect of acute lymphatic obstruction on fluid accumulation in the chest in dogs. J Appl Physiol 39: 985–989, 1975. doi: 10.1152/jappl.1975.39.6.985. [DOI] [PubMed] [Google Scholar]
- 231. Stewart RH, Rohn DA, Allen SJ, Laine GA. Basic determinants of epicardial transudation. Am J Physiol Heart Circ Physiol 273: H1408–H1414, 1997. doi: 10.1152/ajpheart.1997.273.3.H1408. [DOI] [PubMed] [Google Scholar]
- 232. Yuan Z, Boulanger B, Flessner M, Johnston M. Relationship between pericardial pressure and lymphatic pericardial fluid transport in sheep. Microvasc Res 60: 28–36, 2000. doi: 10.1006/mvre.2000.2239. [DOI] [PubMed] [Google Scholar]
- 233. Beer DJ, Pereira W Jr, Snider GL. Pleural effusion associated with primary lymphedema: a perspective on the yellow nail syndrome. Am Rev Respir Dis 117: 595–599, 1978. [DOI] [PubMed] [Google Scholar]
- 234. Jiang L, Tao T, Zheng J, Jia Z, Xu H, Ni Y. Case report of refractory pericardial effusion associated with lymphatic fistula due to surgical injury during sternotomy. Medicine (Baltimore) 97: e9892, 2018. doi: 10.1097/MD.0000000000009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Patel R. Cardiac Manifestation of Lymphedema (Clinical Vignette), Proceedings Department of Medicine. Los Angeles, CA: UCLA, 2016. [Google Scholar]
- 236. Forte E, Furtado MB, Rosenthal N. The interstitium in cardiac repair: role of the immune-stromal cell interplay. Nat Rev Cardiol 15: 601–616, 2018. doi: 10.1038/s41569-018-0077-x. [DOI] [PubMed] [Google Scholar]
- 237. Heallen TR, Kadow ZA, Kim JH, Wang J, Martin JF. Stimulating cardiogenesis as a treatment for heart failure. Circ Res 124: 1647–1657, 2019. doi: 10.1161/CIRCRESAHA.118.313573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Vagnozzi RJ, Molkentin JD, Houser SR. New myocyte formation in the adult heart: endogenous sources and therapeutic implications. Circ Res 123: 159–176, 2018. doi: 10.1161/CIRCRESAHA.118.311208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Quijada P, Trembley MA, Small EM. The role of the epicardium during heart development and repair. Circ Res 126: 377–394, 2020. doi: 10.1161/CIRCRESAHA.119.315857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ. Cardiac regeneration from activated epicardium. PLoS One 7: e44692, 2012. doi: 10.1371/journal.pone.0044692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Wang QL, Wang HJ, Li ZH, Wang YL, Wu XP, Tan YZ. Mesenchymal stem cell-loaded cardiac patch promotes epicardial activation and repair of the infarcted myocardium. J Cell Mol Med 21: 1751–1766, 2017. doi: 10.1111/jcmm.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Olivey HE, Svensson EC. Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res 106: 818–832, 2010. doi: 10.1161/CIRCRESAHA.109.209197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Redpath AN, Smart N. Recapturing embryonic potential in the adult epicardium: prospects for cardiac repair. Stem Cells Transl Med 10: 511–521, 2021. doi: 10.1002/sctm.20-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C, Lin RZ, Melero-Martin JM, Dolmatova E, Duffy HS, Gise A, Zhou P, Hu YW, Wang G, Zhang B, Wang L, Hall JL, Moses MA, McGowan FX, Pu WT. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest 121: 1894–1904, 2011. doi: 10.1172/JCI45529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol 193: 607–618, 2011. doi: 10.1083/jcb.201012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Wang S, Yamakawa M, Santosa SM, Chawla N, Guo K, Montana M, Hallak JA, Han KY, Ema M, Rosenblatt MI, Chang JH, Azar DT. Quantification of angiogenesis and lymphangiogenesis in the dual ex vivo aortic and thoracic duct assay. Protein Pept Lett 27: 30–40, 2020. doi: 10.2174/0929866526666190925145842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Nurmi H, Saharinen P, Zarkada G, Zheng W, Robciuc MR, Alitalo K. VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol Med 7: 1418–1425, 2015. doi: 10.15252/emmm.201505731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE, Han SH, Alitalo K, Koh GY. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood 113: 5650–5659, 2009. doi: 10.1182/blood-2008-09-176776. [DOI] [PubMed] [Google Scholar]
- 249. Schoppmann SF, Fenzl A, Nagy K, Unger S, Bayer G, Geleff S, Gnant M, Horvat R, Jakesz R, Birner P. VEGF-C expressing tumor-associated macrophages in lymph node positive breast cancer: impact on lymphangiogenesis and survival. Surgery 139: 839–846, 2006. doi: 10.1016/j.surg.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 250. Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivelä R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, Jacobs AH, Epstein JA, Stankunas K, Alitalo K, Red-Horse K. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 141: 4500–4512, 2014. doi: 10.1242/dev.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Mäkinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, Kauppinen R, Jackson DG, Kubo H, Nishikawa S, Ylä-Herttuala S, Alitalo K. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med 7: 199–205, 2001. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 252. Keller TC 4th, Lim L, Shewale SV, McDaid K, Martí-Pàmies I, Tang AT, Wittig C, Guerrero AA, Sterling S, Leu NA, Scherrer-Crosbie M, Gimotty PA, Kahn ML. Genetic blockade of lymphangiogenesis does not impair cardiac function after myocardial infarction. J Clin Invest 131: e147070, 2021. doi: 10.1172/JCI147070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Harris NR, Nielsen NR, Pawlak JB, Aghajanian A, Rangarajan K, Serafin DS, Farber G, Dy DM, Nelson-Maney NP, Xu W, Ratra D, Hurr SH, Qian L, Scallan JP, Caron KM. VE-Cadherin Is Required for Cardiac Lymphatic Maintenance and Signaling. Circ Res 130: 5–23, 2022. doi: 10.1161/CIRCRESAHA.121.318852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Wada H, Suzuki M, Matsuda M, Ajiro Y, Shinozaki T, Sakagami S, et al. Distinct characteristics of VEGF-D and VEGF-C to predict mortality in patients with suspected or known coronary artery disease. J Am Heart Assoc 9: e015761, 2020. doi: 10.1161/JAHA.119.015761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Wada H, Suzuki M, Matsuda M, Ajiro Y, Shinozaki T, Sakagami S, Yonezawa K, Shimizu M, Funada J, Takenaka T, Morita Y, Nakamura T, Fujimoto K, Matsubara H, Kato T, Unoki T, Takagi D, Ura S, Wada K, Iguchi M, Masunaga N, Ishii M, Yamakage H, Shimatsu A, Kotani K, Satoh-Asahara N, Abe M, Akao M, Hasegawa K; ANOX Study Investigators. VEGF-C and mortality in patients with suspected or known coronary artery disease. J Am Heart Assoc 7: e010355, 2018. doi: 10.1161/JAHA.118.010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Jougasaki M, Wei CM, McKinley LJ, Burnett JC Jr.. Elevation of circulating and ventricular adrenomedullin in human congestive heart failure. Circulation 92: 286–289, 1995. doi: 10.1161/01.CIR.92.3.286. [DOI] [PubMed] [Google Scholar]
- 257. Jougasaki M, Stevens TL, Borgeson DD, Luchner A, Redfield MM, Burnett JC Jr.. Adrenomedullin in experimental congestive heart failure: cardiorenal activation. Am J Physiol Regul Integr Comp Physiol 273: R1392–R1399, 1997. doi: 10.1152/ajpregu.1997.273.4.R1392. [DOI] [PubMed] [Google Scholar]
- 258. Kiris I, Kapan S, Narin C, Ozaydın M, Cure MC, Sutcu R, Okutan H. Relationship between site of myocardial infarction, left ventricular function and cytokine levels in patients undergoing coronary artery surgery. Cardiovasc J Afr 27: 299–306, 2016. doi: 10.5830/CVJA-2016-027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Ishimitsu T, Nishikimi T, Saito Y, Kitamura K, Eto T, Kangawa K, Matsuo H, Omae T, Matsuoka H. Plasma levels of adrenomedullin, a newly identified hypotensive peptide, in patients with hypertension and renal failure. J Clin Invest 94: 2158–2161, 1994. doi: 10.1172/JCI117573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Suzuki Y, Horio T, Hayashi T, Nonogi H, Kitamura K, Eto T, Kangawa K, Kawano Y. Plasma adrenomedullin concentration is increased in patients with peripheral arterial occlusive disease associated with vascular inflammation. Regul Pept 118: 99–104, 2004. doi: 10.1016/j.regpep.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 261. Yanagawa B, Nagaya N. Adrenomedullin: molecular mechanisms and its role in cardiac disease. Amino Acids 32: 157–164, 2007. doi: 10.1007/s00726-005-0279-5. [DOI] [PubMed] [Google Scholar]
- 262. Kataoka Y, Miyazaki S, Yasuda S, Nagaya N, Noguchi T, Yamada N, Morii I, Kawamura A, Doi K, Miyatake K, Tomoike H, Kangawa K. The first clinical pilot study of intravenous adrenomedullin administration in patients with acute myocardial infarction. J Cardiovasc Pharmacol 56: 413–419, 2010. doi: 10.1097/FJC.0b013e3181f15b45. [DOI] [PubMed] [Google Scholar]
- 263. Watanabe H, Takahashi E, Kobayashi M, Goto M, Krust A, Chambon P, Iguchi T. The estrogen-responsive adrenomedullin and receptor-modifying protein 3 gene identified by DNA microarray analysis are directly regulated by estrogen receptor. J Mol Endocrinol 36: 81–89, 2006. doi: 10.1677/jme.1.01825. [DOI] [PubMed] [Google Scholar]
- 264. Wetzel-Strong SE, Li M, Espenschied ST, Caron KM. Cohort of estrogen-induced microRNAs regulate adrenomedullin expression. Am J Physiol Regul Integr Comp Physiol 310: R209–R216, 2016. doi: 10.1152/ajpregu.00305.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Mahmoodzadeh S, Leber J, Zhang X, Jaisser F, Messaoudi S, Morano I, Furth PA, Dworatzek E, Regitz-Zagrosek V. Cardiomyocyte-specific estrogen receptor alpha increases angiogenesis, lymphangiogenesis and reduces fibrosis in the female mouse heart post-myocardial infarction. J Cell Sci Ther 5: 153, 2014. doi: 10.4172/2157-7013.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Rosenkranz-Weiss P, Tomek RJ, Mathew J, Eghbali M. Gender-specific differences in expression of mRNAs for functional and structural proteins in rat ventricular myocardium. J Mol Cell Cardiol 26: 261–270, 1994. doi: 10.1006/jmcc.1994.1029. [DOI] [PubMed] [Google Scholar]
- 267. Knezl V, Bacova B, Kolenova L, Mitasikova M, Weismann P, Drimal J, Tribulova N. Distinct lethal arrhythmias susceptibility is associated with sex-related difference in myocardial connexin-43 expression. Neuro Endocrinol Lett 29: 798–801, 2008. [PubMed] [Google Scholar]
- 268. Wysocka MB, Pietraszek-Gremplewicz K, Nowak D. The role of apelin in cardiovascular diseases. Front Physiol 9: 557, 2018. doi: 10.3389/fphys.2018.00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Chen MM, Ashley EA, Deng DX, Tsalenko A, Deng A, Tabibiazar R, Ben-Dor A, Fenster B, Yang E, King JY, Fowler M, Robbins R, Johnson FL, Bruhn L, McDonagh T, Dargie H, Yakhini Z, Tsao PS, Quertermous T. Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction. Circulation 108: 1432–1439, 2003. doi: 10.1161/01.CIR.0000091235.94914.75. [DOI] [PubMed] [Google Scholar]
- 270. Földes G, Horkay F, Szokodi I, Vuolteenaho O, Ilves M, Lindstedt KA, Mäyränpää M, Sármán B, Seres L, Skoumal R, Lakó-Futó Z, deChâtel R, Ruskoaho H, Tóth M. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure. Biochem Biophys Res Commun 308: 480–485, 2003. doi: 10.1016/S0006-291X(03)01424-4. [DOI] [PubMed] [Google Scholar]
- 271. Ronkainen VP, Ronkainen JJ, Hänninen SL, Leskinen H, Ruas JL, Pereira T, Poellinger L, Vuolteenaho O, Tavi P. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. FASEB J 21: 1821–1830, 2007. doi: 10.1096/fj.06-7294com. [DOI] [PubMed] [Google Scholar]
- 272. Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, Penninger JM, Kassiri Z, Vederas JC, Murray AG, Oudit GY. Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic Apelin analogues. J Am Heart Assoc 2: e000249, 2013. doi: 10.1161/JAHA.113.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Kim JD, Kang Y, Kim J, Papangeli I, Kang H, Wu J, Park H, Nadelmann E, Rockson SG, Chun HJ, Jin SW. Essential role of Apelin signaling during lymphatic development in zebrafish. Arterioscler Thromb Vasc Biol 34: 338–345, 2014. doi: 10.1161/ATVBAHA.113.302785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Kidoya H, Naito H, Muramatsu F, Yamakawa D, Jia W, Ikawa M, Sonobe T, Tsuchimochi H, Shirai M, Adams RH, Fukamizu A, Takakura N. APJ regulates parallel alignment of arteries and veins in the skin. Dev Cell 33: 247–259, 2015. doi: 10.1016/j.devcel.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 275. Tatin F, Renaud-Gabardos E, Godet AC, Hantelys F, Pujol F, Morfoisse F, Calise D, Viars F, Valet P, Masri B, Prats AC, Garmy-Susini B. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight 2: e93887, 2017. doi: 10.1172/jci.insight.93887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Lutter S, Xie S, Tatin F, Makinen T. Smooth muscle-endothelial cell communication activates Reelin signaling and regulates lymphatic vessel formation. J Cell Biol 197: 837–849, 2012. doi: 10.1083/jcb.201110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Calvier L, Xian X, Lee RG, Sacharidou A, Mineo C, Shaul PW, Kounnas MZ, Tsai S, Herz J. Reelin depletion protects against atherosclerosis by decreasing vascular adhesion of leukocytes. Arterioscler Thromb Vasc Biol 41: 1309–1318, 2021. doi: 10.1161/ATVBAHA.121.316000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Ding Y, Huang L, Xian X, Yuhanna IS, Wasser CR, Frotscher M, Mineo C, Shaul PW, Herz J. Loss of Reelin protects against atherosclerosis by reducing leukocyte-endothelial cell adhesion and lesion macrophage accumulation. Sci Signal 9: ra29, 2016. doi: 10.1126/scisignal.aad5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Schacht V, Ramirez MI, Hong YK, Hirakawa S, Feng D, Harvey N, Williams M, Dvorak AM, Dvorak HF, Oliver G, Detmar M. T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J 22: 3546–3556, 2003. doi: 10.1093/emboj/cdg342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Astarita JL, Acton SE, Turley SJ. Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol 3: 283, 2012. doi: 10.3389/fimmu.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Cimini M, Kishore R. Role of podoplanin-positive cells in cardiac fibrosis and angiogenesis after ischemia. Front Physiol 12: 667278, 2021. doi: 10.3389/fphys.2021.667278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Mahtab EA, Vicente-Steijn R, Hahurij ND, Jongbloed MR, Wisse LJ, DeRuiter MC, Uhrin P, Zaujec J, Binder BR, Schalij MJ, Poelmann RE, Gittenberger-de Groot AC. Podoplanin deficient mice show a RhoA-related hypoplasia of the sinus venosus myocardium including the sinoatrial node. Dev Dyn 238: 183–193, 2009. doi: 10.1002/dvdy.21819. [DOI] [PubMed] [Google Scholar]
- 283. Mahtab EA, Wijffels MC, Van Den Akker NM, Hahurij ND, Lie-Venema H, Wisse LJ, Deruiter MC, Uhrin P, Zaujec J, Binder BR, Schalij MJ, Poelmann RE, Gittenberger-De Groot AC. Cardiac malformations and myocardial abnormalities in podoplanin knockout mouse embryos: Correlation with abnormal epicardial development. Dev Dyn 237: 847–857, 2008. doi: 10.1002/dvdy.21463. [DOI] [PubMed] [Google Scholar]
- 284. Ugorski M, Dziegiel P, Suchanski J. Podoplanin—a small glycoprotein with many faces. Am J Cancer Res 6: 370–386, 2016. [PMC free article] [PubMed] [Google Scholar]
- 285. Cimini M, Cannatá A, Pasquinelli G, Rota M, Goichberg P. Phenotypically heterogeneous podoplanin-expressing cell populations are associated with the lymphatic vessel growth and fibrogenic responses in the acutely and chronically infarcted myocardium. PLoS One 12: e0173927, 2017. doi: 10.1371/journal.pone.0173927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Cimini M, Garikipati VN, de Lucia C, Cheng Z, Wang C, Truongcao MM, Lucchese AM, Roy R, Benedict C, Goukassian DA, Koch WJ, Kishore R. Podoplanin neutralization improves cardiac remodeling and function after acute myocardial infarction. JCI Insight 5: e126967, 2019. doi: 10.1172/jci.insight.126967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Spath NB, Mills NL, Cruden NL. Novel cardioprotective and regenerative therapies in acute myocardial infarction: a review of recent and ongoing clinical trials. Future Cardiol 12: 655–672, 2016. doi: 10.2217/fca-2016-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Zhang HF, Wang YL, Tan YZ, Wang HJ, Tao P, Zhou P. Enhancement of cardiac lymphangiogenesis by transplantation of CD34+VEGFR-3+ endothelial progenitor cells and sustained release of VEGF-C. Basic Res Cardiol 114: 43, 2019. doi: 10.1007/s00395-019-0752-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Park JH, Yoon JY, Ko SM, Jin SA, Kim JH, Cho CH, Kim JM, Lee JH, Choi SW, Seong IW, Jeong JO. Endothelial progenitor cell transplantation decreases lymphangiogenesis and adverse myocardial remodeling in a mouse model of acute myocardial infarction. Exp Mol Med 43: 479–485, 2011. doi: 10.3858/emm.2011.43.8.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Stieger P, Daniel JM, Thölen C, Dutzmann J, Knöpp K, Gündüz D, Aslam M, Kampschulte M, Langheinrich A, Fischer S, Cabrera-Fuentes H, Wang Y, Wollert KC, Bauersachs J, Braun-Dullaeus R, Preissner KT, Sedding DG. Targeting of extracellular RNA reduces edema formation and infarct size and improves survival after myocardial infarction in mice. J Am Heart Assoc 6: e004541, 2017. doi: 10.1161/JAHA.116.004541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Fukunaga T, Soejima H, Irie A, Sugamura K, Oe Y, Tanaka T, Kojima S, Sakamoto T, Yoshimura M, Nishimura Y, Ogawa H. Expression of interferon-gamma and interleukin-4 production in CD4+ T cells in patients with chronic heart failure. Heart Vessels 22: 178–183, 2007. doi: 10.1007/s00380-006-0955-8. [DOI] [PubMed] [Google Scholar]
- 292. Perticone M, Zito R, Miceli S, Pinto A, Suraci E, Greco M, Gigliotti S, Hribal ML, Corrao S, Sesti G, Perticone F. Immunity, inflammation and heart failure: their role on cardiac function and iron status. Front Immunol 10: 2315, 2019. doi: 10.3389/fimmu.2019.02315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Pistulli R, Hammer N, Rohm I, Kretzschmar D, Jung C, Figulla HR, Yilmaz A. Decrease of circulating myeloid dendritic cells in patients with chronic heart failure. Acta Cardiol 71: 165–172, 2016. doi: 10.1080/AC.71.2.3141846. [DOI] [PubMed] [Google Scholar]
- 294. Yndestad A, Holm AM, Müller F, Simonsen S, Frøland SS, Gullestad L, Aukrust P. Enhanced expression of inflammatory cytokines and activation markers in T-cells from patients with chronic heart failure. Cardiovasc Res 60: 141–146, 2003. doi: 10.1016/s0008-6363(03)00362-6. [DOI] [PubMed] [Google Scholar]
- 295. Hampton HR, Chtanova T. Lymphatic migration of immune cells. Front Immunol 10: 1168, 2019. doi: 10.3389/fimmu.2019.01168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Song L, Chen X, Swanson TA, LaViolette B, Pang J, Cunio T, Nagle MW, Asano S, Hales K, Shipstone A, Sobon H, Al-Harthy SD, Ahn Y, Kreuser S, Robertson A, Ritenour C, Voigt F, Boucher M, Sun F, Sessa WC, Roth Flach RJ. Lymphangiogenic therapy prevents cardiac dysfunction by ameliorating inflammation and hypertension. Elife 9: e58376, 2020. doi: 10.7554/eLife.58376. [DOI] [PMC free article] [PubMed] [Google Scholar]