

Abstract
Background and Objectives
Spinal muscular atrophy (SMA) was added to the Recommended Uniform Screening Panel in July 2018 largely on the basis of the availability and efficacy of newly approved disease-modifying therapies. New York State (NYS) started universal newborn screening for SMA in October 2018. The authors report the findings from the first 3 years of screening.
Methods
Statewide neonatal screening was conducted using DNA extracted from dried blood spots using a real-time quantitative PCR assay. Retrospective follow-up data were collected from 9 referral centers across the state on 34 infants.
Results
In the first 3 years since statewide implementation, nearly 650,000 infants have been screened for SMA. Thirty-four babies screened positive and were referred to a neuromuscular specialty care center. The incidence remains lower than previously predicted. The majority (94%), including all infants with 2–3 copies of survival motor neuron (SMN) 2, have received treatment. Among treated infants, the overwhelming majority (94%; 30/32) have received gene replacement. All infants in this cohort with 3 copies of SMN2 are clinically asymptomatic posttreatment based on early clinical follow-up data. Infants with 2 copies of SMN2 are more variable in their outcomes. Electrodiagnostic outcomes data obtained from a subgroup of patients (n = 11) demonstrated either improvement or no change in compound muscle action potential (CMAP) amplitude at last clinical follow-up compared with pretreatment baseline. Most infants were treated before 6 weeks of age (median = 34.5 days of life; range 11–180 days). Delays and barriers to treatment identified by treating clinicians followed 2 broad themes: medical and nonmedical. Medical delays most commonly reported were the presence of AAV9 antibodies and elevated troponin I levels. Nonmedical barriers included delays in obtaining insurance and insurance policies regarding specific treatment modalities.
Discussion
The findings from the NYS cohort of newborn screen-identified infants are consistent with other reports of improved outcomes from early diagnosis and treatment. Additional biomarkers of motor neuron health including EMG can potentially be helpful in detecting preclinical decline.
Spinal muscular atrophy (SMA) is a genetic neuromuscular disorder characterized by progressive degeneration of spinal cord and brainstem motor neurons resulting in progressive muscle weakness and atrophy.1 It is most commonly caused by homozygous loss of exon 7 of the SMN1 gene encoding the survival motor neuron (SMN) protein. The clinical types of SMA are defined by motor milestone achievement: type 1 infants never achieve independent sitting, type 2 infants never achieve independent ambulation, and type 3 infants achieve independent ambulation and invariably lose this skill later on in life.1 Type 0, the most severe form, refers to infants who are symptomatic at birth and have evidence of in utero weakness and immobility, such as arthrogryposis. Type 4 is the mildest form with symptom onset in adulthood. The phenotype is modified, in part, by the number of copies of a paralog gene, SMN2, such that increasing SMN2 copy number correlates with milder phenotypes. For this reason, SMN2 copy number is clinically used as a predictor of disease type.2,3 However, the correlation is not absolute because of a variety of known and yet unknown mechanisms and requires thoughtful application in the era of molecular therapy.2,4 Prior to May 2022, there were 2 Food and Drug Administration -approved therapeutic options for presymptomatic infants with SMA at birth: onasemnogene abeparvovec-xioi (Zolgensma; hereafter referred to as onasemnogene) and nusinersen (Spinraza). A third option, risdiplam (Evrysdi), was previously available for infants older than 2 months but recently received a label extension to include infants under 2 months of age. Nusinersen is an intrathecally administered antisense oligonucleotide that increases production of full-length SMN protein by altering splicing of SMN2 premessenger RNA (mRNA).5 Onasemnogene is a single-dose SMN1 gene replacement therapy given via a peripheral infusion.6 Risdiplam is a small molecule splicing modifier that selectively binds SMN2 pre-mRNA, thereby promoting increased production of SMN protein.7 All 3 treatment modalities have demonstrated superior survival and motor outcomes compared with the well-established natural history of SMA.8-10 Early dosing appears to be critically important, and immediate treatment is recommended for patients with 2–4 copies of SMN2, including presymptomatic infants identified by newborn screening.11,12 Despite these recommendations, insurance policies for coverage are widely variable, particularly with regard to treatment of 4-copy infants.
Taiwan was among the first countries to successfully demonstrate the feasibility of population-based screening for SMA.13 New York State (NYS) completed a pilot study for SMA screening between January 2016 and September 2018.14 One infant was identified during the pilot study and enrolled in a clinical trial for presymptomatic infants treated with antisense oligonucleotide therapy.15 SMA was subsequently added to the Recommended Uniform Screening Panel by the Advisory Committee on Heritable Disorders in Newborns and Children in July 2018. In the United States, 44 states have implemented SMA newborn screening, accounting for 95% of newborns, as of April 1, 2022.16 SMA was added to the NYS newborn screening panel on October 1, 2018. In the first year, 225,093 infants were screened, and 8 screened positive.17 It has now been just over 3 years since the addition of SMA to the NYS screening panel. In this article, we present an update on the incidence, clinical outcomes, and challenges faced from the first 3 years of screening.
Methods
Screening for SMA was accomplished using a modified version of the real-time quantitative PCR (qPCR) assay previously described.14,17 Newborn screening for all babies born in NYS is performed by the NYS Newborn Screening Program at the Wadsworth Center, NYS Department of Health. Briefly, DNA was extracted from one 3-mm dried blood spot punch in a 96-well format. SMA screening was multiplexed with the current newborn screening assay for severe combined immunodeficiency (SCID), modified to include a primer/probe mix targeting the common SMN1 exon 7 deletion (VIC labeled) to the primer/probe mixes targeting T-cell receptor excision circles (TRECs [molecular SCID biomarker]; FAM labeled) and RPPH1 as an internal control (ABY labeled).18 Primer/probe mixes were purchased from Thermo Fisher Scientific (Waltham, MA), and the assay was run in a 384-well format using PerfeCTa Multiplex qPCR ToughMix (QuantaBio, Beverly, MA) on an Applied Biosystems QuantStudio 12 K Flex Real-Time PCR System. Samples with SMN1 Ct ≥ 30 and RPPH1 Ct < 30 were considered screen positive. Heterozygous carriers of the exon 7 deletion and heterozygous individuals with SMA having a deletion on one allele and a pathogenic sequence variation on the other allele are not identifiable by this assay. Confirmation of the SMN1 exon 7 deletion was performed in duplicate using fresh DNA preparations and the originally described qPCR assay with only the SMN1 and RPPH1 primer/probe mixes. SMN2 copy number was determined in infants who screened positive using both the qPCR targeting SMN2 exon 7 and RPPH1 as previously described and a droplet digital PCR kit (ddPCR; Bio-Rad Laboratories, Hercules, CA). Results for SMN2 were provided at the time of referral.
Retrospective data were collected by clinical providers at the individual NYS-designated Neuromuscular Specialty Care Centers (SCCs), and deidentified data were shared with the University of Rochester site for aggregate analyses. Developmental milestones were assessed using parental recall and confirmed in the clinic. Developmental milestone delays were determined based on the upper limits of the World Health Organization (WHO) motor milestone window.19 Among children with motor delays, developmental quotients were calculated as follows: developmental age (upper age limit for the highest level of function achieved) divided by chronological age then multiplied by 100 (22 month old with highest function achieved of walking with assistance: 13.7 months/22 months * 100 = 62%.) We used 4 months as the window for reaching as per the Centers for Disease Control and Prevention guideline.20
For the purposes of this article, we defined presymptomatic as the absence of clinical examination findings of SMA, including evidence of hypotonia out of proportion with age, or reduced/absent reflexes in a patient prior to receiving treatment. The term asymptomatic is used in posttreatment babies without clinical examination evidence of SMA and normal acquisition of expected developmental milestones for age.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was reviewed by the University of Rochester Research Subjects Review Board (STUDY00006923) and determined to meet federal and University criteria for exemption. Waiver of consent was included in the protocol for exempt status.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Screening
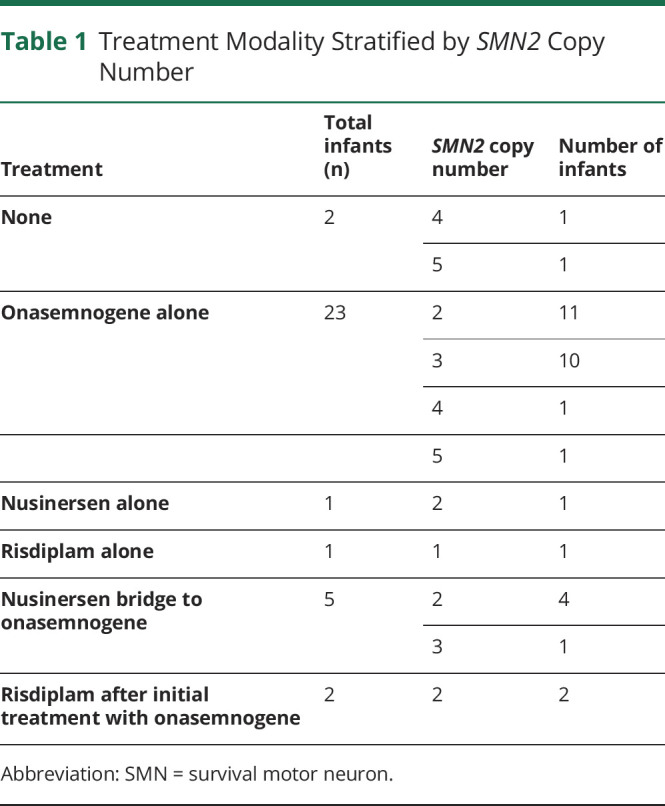
NYS screened approximately 650,000 infants from October 1, 2018, through September 30, 2021, and 34 screen-positive infants were referred by the newborn screening program for SMA. The referred cohort included 1 infant with 1 SMN2 copy, 18 infants with 2 SMN2 copies, 11 infants with 3 SMN2 copies, and 4 infants with ≥ 4 SMN2 copies (Table 1). Positive results were reported by NYS on median day of life (DOL) 7 (range 4–12 days). All infants with positive newborn screening results had confirmatory genetic testing, and there have been no false-positive results. All infants were referred to NYS Neuromuscular SCCs for facilitation of diagnostic confirmation, clinical evaluation, and treatment. Infants were seen for their first evaluation at an SCC at median DOL 9 (range 1–58 days). All positive screens were confirmed via diagnostic testing. Three infants were evaluated and seen at an SCC before NYS NBS results were available because of prenatally known risk from parental carrier testing or a family history of an affected sibling. One infant was formally evaluated at an SCC at 58 DOL but had confirmatory clinical testing by DOL 8 and was seen at an out-of-state clinic at DOL 9.
Table 1.
Treatment Modality Stratified by SMN2 Copy Number
Treatment
In our cohort, 94% (32/34) of infants diagnosed with SMA via newborn screening received treatment. Two infants have not been treated. A total of 23 infants (71%) were treated with onasemnogene alone, 1 infant (3%) is receiving nusinersen alone, 1 infant (3%) is receiving risdiplam alone, 5 infants (17%) were treated with nusinersen before onasemnogene, and 2 infants (6%) initially treated with onasemnogene were additionally started on risdiplam (Table 2). All infants who received nusinersen followed by onasemnogene have discontinued nusinersen. Two infants initially treated with onasemnogene were additionally started on risdiplam.
Table 2.
Clinical Summary of 34 Infants Identified in the First 3 Years of Spinal Muscular Atrophy Newborn Screening in New York State

Motor and Developmental Outcomes
One-Copy SMN2 Infants (n = 1)
This infant was noted to be severely affected at birth, including respiratory failure, dysphagia, immobility, diffuse contractures, and cardiac malformation. At parental request, risdiplam was started in the neonatal intensive care unit at 2.5 months of age. He remains ventilator dependent, is unable to feed orally, and has severe global weakness with minimal finger movement.
Two-Copy SMN2 Infants (n = 18)
There were 18 infants with 2 copies of SMN2. All received treatment at a median age of 34 DOL (range 12–89 days). Ten were presymptomatic at the time of treatment, and 8 were symptomatic at the time of treatment. Of the 10 children who were noted to be presymptomatic at the time of treatment, 4 were noted to have symptoms at last follow-up. Symptoms reported for these infants include mild hypotonia and areflexia, but all 4 infants have achieved motor milestones respective to their age. The median initial CHOP-INTEND score for those reported (n = 8) was 47 (range = 37–54).
Among symptomatic infants at the time of treatment (n = 8), all are noted to be symptomatic at last follow-up. The median initial CHOP-INTEND score for those reported (n = 6) was 35.5 (range = 14–47). Of these patients, 1 is independently walking, 1 is standing with minimal assistance, and 3 are sitting with support. Five infants are requiring respiratory support with noninvasive ventilatory measures like bilevel positive airway pressure. Three infants have required some degree of feeding assistance, but none have required gastrostomy tube placement. Of those with serial CHOP-INTEND scores, 1 infant has experienced a decline and has not achieved adequate head control or ability to roll or sit at 6 months of age (Figure, A).
Figure. Ulnar CMAP Amplitudes (A) and CHOP-INTEND Scores (B) Over Time.

Orange: 3 SMN2 copies and asymptomatic at last follow-up; green: 2 SMN2 copies and asymptomatic at last follow-up; blue: 2 SMN2 copies and symptomatic at last follow-up. Age at treatment for each patient is indicated in the graph legends. SMN = survival motor neuron.
Among the 2 SMN2 copy infants old enough to assess motor development (had either achieved a motor milestone early or was over 6 months of age, n = 12), 7 infants showed delayed motor development. Developmental quotients ranged from 30% to 95%. The mean developmental quotient among 2 SMN2 copy children treated symptomatically was 50.6% (n = 5, range 30%–77%) and 86.5% (range 78%–95%) among those treated presymptomatically.
Three-Copy SMN2 Infants (n = 11)
There were 11 infants with 3 copies of SMN2. All received treatment with a median age of 34 DOL (range = 11–94 days). All patients were presymptomatic at the time of treatment. Initial CHOP-INTEND scores were available for 5 infants and demonstrated a median of 49 (range = 40–60). All are reported to be clinically asymptomatic, meeting age-appropriate developmental milestones with normal neurologic examinations, at last follow-up. Seven are walking independently. The other 4 infants were less than 12 months old and reported to be meeting all age-relevant milestones. None in this group have required respiratory or feeding support.
≥Four-Copy SMN2 Infants (n =4)
Two infants were confirmed to have 4 copies of SMN2, and 2 confirmed to have 5 copies of SMN2. Two of these infants, 1 with 4 SMN2 copies and 1 with 5 SMN2 copies, were treated with onasemnogene. Two infants have not yet received treatment. One untreated infant with 5 copies of SMN2 is being followed clinically. The patient achieved independent ambulation at 13 months and was presymptomatic at last clinical follow-up at 18 months of age. One untreated infant with 4 copies was last seen at 8 months of age and achieved sitting and crawling at 6 months of age.
Electrodiagnostic Outcomes
Electrodiagnostic outcomes data were analyzed from a subgroup of patients for whom pretreatment and posttreatment data were collected (n = 11). This included 7 infants with 2 copies of SMN2 and 4 infants with 3 copies of SMN2. Lower nadir compound muscle action potential (CMAP) amplitudes were seen in infants with 2 copies of SMN2 with a mean ulnar CMAP value of 1.4 mV (median 0.9 mV, range 0.3–4.5 mV). Higher CMAP values were recorded in 3-copy infants with a mean initial ulnar CMAP value of 5.8 mV (median 5.4 mV, range 4.9–7.4 mV). All infants had either improvement or no change in CMAP amplitude at last clinical follow-up compared with pretreatment baseline (Figure, A).
Of note, 2 infants with 2 copies of SMN2 were born prematurely (34 6/7 weeks, 34 1/7 weeks gestational age) and had relatively high ulnar CMAP amplitudes at first assessment (5.1 and 6.2 mV, respectively). The other five 2-copy infants who were born full term had initial mean CMAP amplitudes of 1.4 mV (median 0.9 mV, range 0.3–2.9 mV). One of these infants was noted to be significantly symptomatic at birth and had initial CMAP amplitude of 0.3 mV, which rose to 0.7 mV at 15 months of age with combinatorial onasemnogene and risdiplam treatment. One prematurely born infant was noted to have a precipitous drop in CMAP to 1.2 mV at the time of treatment (40 weeks GA), with posttreatment CMAP nadir at 0.7 mV, and subsequent slow improvement to CMAP of 1.5 mV at 11 months posttreatment. The other infant also had a drop in CMAP to 4.5 mV at the time of treatment (3 weeks of age) and has had improvement in CMAP to 5.7 mV at 2.4 months posttreatment.
Prematurity (n = 7)
Seven infants in this cohort were born prematurely, defined as less than 37 weeks GA. Gestational age ranged from 34w1d to 36w5d. A summary of their treatment timeline and clinical symptoms is extracted and included in Table 3. One infant born at 34w6d was noted to be presymptomatic with a CHOP-INTEND score of 40/64 at 10 DOL. His examination on the day of treatment (40w1d) was notable for areflexia, hypotonia, weakness, and drop in the CHOP-INTEND score to 26/64. The child was last seen at 11.5 months old and can sit independently for brief periods. The repeat CHOP-INTEND score was 59/64 at that time. The 6 other infants are reported to be presymptomatic at the time of treatment and remain clinically asymptomatic at their last clinical follow-up. No adverse side effects following onasemnogene were reported for the 2 infants treated at 37w1d GA.
Table 3.
Summary of Infants Born Prematurely (<37w GA) Including Age at Treatment

Treatment Timing
Most infants were treated before 6 weeks of age (median = 34.5 DOL; range 11–197 days). The significant outlier was an infant with 4 copies of SMN2 treated at 180 DOL. Of the infants with 2–3 copies of SMN2, treatment occurred between 11 and 94 days. Delays and barriers to treatment identified by treating clinicians followed 2 broad themes: medical and nonmedical. Medical delays most commonly reported were the presence of AAV9 antibodies and elevated troponin I levels. Nonmedical delays reported included, most commonly, delays in attaining insurance and insurance approval of requested treatment and, less commonly, delays in obtaining birth certificate during the COVID-19 pandemic and lack of contractual agreement between the patient's insurer and the specialty pharmacy.
Discussion
After the first year of screening, the reported incidence was 1 in 28,137.17 The incidence after data accumulation from 2 additional years of screening is approximately 1 in 19,000. However, the incidence remains lower than previously expected based on published estimates of 1 in 6,000–11,000.21 The current estimate of 1 in 19,000 is likely closer to the true NYS incidence because of a larger sample size and more precise estimate. The authors maintain that the increase in prenatal counseling and informed reproductive decision due to advances in genetic testing capabilities contribute to this effect. All referrals were made before 2 weeks of life, and the majority of infants were seen within a few days of a referral notification at a specialty care center demonstrating the practical feasibility of urgently establishing specialized care and treatment plans.
Of the 34 states offering population-wide screening for SMA as of June 2021, New York was 1 of 8 states simultaneously reporting SMN2 copy number.22 SMN2 copy number must also be confirmed with repeat clinical testing, but it is helpful for risk stratification of infants at the highest risk of earlier clinical symptom onset.
This NYS cohort redemonstrates the consistently observed theme that early identification and treatment of newborns with SMA leads to improved motor outcomes. In our group, all infants with 3 copies of SMN2 were treated before symptom onset and remained asymptomatic at follow-up. All infants in this group who are now older than 1 year are independently ambulating. We noted more variability in the outcomes of 2 SMN2 copy infants, with some demonstrating rapid clinical decline over the first weeks to months of life, stressing the urgency of treatment initiation in this group. The youngest 2 SMN2 copy infant in our cohort noted to be symptomatic at the time of treatment was 19 days old.
Serial CHOP-INTEND scores were available from 1 center on 10 infants (Figure, B). All but 1 infant had improvement in CHOP-INTEND over time, although the rate of rise in score appears to be stratified by SMN2 copy number.
In this cohort, all 2 and 3 SMN2 copy infants received treatment concordant with expert recommendations that treatment initiation occurs as soon as possible after diagnosis.11 Although the consensus statement was revised to include 4 SMN2 copy infants, there is still practice variability among providers. Of interest, Germany's NBS program identified 15 children with 4 SMN2 copies who were, by protocol, not treated presymptomatically. This included 1 infant who developed symptoms at 8 months of age. This patient was retested and found to have 3 SMN2 copies, reminding us the importance of confirming SMN2 copy number using orthogonal methods for a more accurate, albeit imperfect, prediction of disease type.23
Among the treated infants, the overwhelming majority (94%; 30/32) have received onasemnogene, either as initial therapy or following nusinersen bridge therapy. The only infant on nusinersen alone was noted to have persistently elevated AAV9 antibodies, precluding treatment with onasemnogene. This observation is likely reflective of both provider and parent choice. Recent studies have demonstrated that physician counseling, frequency of treatment, and method of delivery are important factors to caregivers for treatment decisions.24,25
All infants who received nusinersen followed by onasemnogene have discontinued nusinersen. Two symptomatic infants, who had initially received onasemnogene, received adjunct therapy with risdiplam at 6 months of age because of a perceived plateau. No short-term adverse effects have been reported by the clinical teams following these infants. Still too little is known about the potential benefits and risks of combination therapy at this time.
Two infants in this cohort were treated within the first 2 weeks of life. In both cases, diagnosis was known before newborn screen results. One infant was screened prenatally via noninvasive prenatal testing for SMA because of a previous pregnancy with fetal demise. The other infant had clinical testing sent on DOL 1 because of known parental carrier status. Carrier screening for spinal muscular atrophy has been recommended by the American College of Medical Genetics and Genomics since 2008, but the practice of offering routine carrier screening for SMA is variable.26 The American College of Obstetricians and Gynecologists recently reiterated their recommendation that all pregnant women be offered carrier screening for SMA.27 Although the primary objective of carrier screening has historically been to inform risk and reproductive decisions, an increase in carrier screening rates may result in earlier diagnosis and treatment. As the treatment and care of SMA evolves and the new natural history of treated SMA emerges, pretest and posttest counseling will continue to have a critical role in this process.
When there was a treatment delay, recurrently encountered reasons leading to delay in treatment with onasemnogene included elevated troponin I levels and presence of AAV9 antibodies. It can be difficult to navigate the decisions about how long to wait when a medical barrier to treatment is encountered given the risk of rapid clinical symptom onset, particularly in infants with 2 copies of SMN2. Additional biomarkers of motor neuron health including CMAP can potentially be helpful in detecting preclinical decline.
Delays in treatment were also seen because of delays in obtaining insurance and insurance policies regarding specific treatment modalities. Of note, this led to treatment delay in a 2 SMN2 copy preterm infant for whom insurance required the infant reach 40 weeks GA before treatment.
The premature infants in this cohort were born moderate to late preterm (32–37 weeks GA). No major neonatal complications were reported. No infants were treated before reaching term gestation (37 weeks as defined by the WHO). Early treatment with onasemnogene at 37w GA occurred in 2 infants, demonstrating feasibility. Both infants are reported to have tolerated treatment without significant short-term events. Long-term safety and neurodevelopmental outcomes will be important to follow.
Measurement of CMAP amplitude in SMA has been studied as a marker of motor neuron health and disease burden. The degree of innervation as reflected by maximum ulnar CMAP amplitudes has previously been shown to correlate with disease severity in children with SMA.28-30 Early observations of prenatally identified infants with SMA type 1 also demonstrated early and rapid decline in CMAP amplitudes that corresponded to development of symptoms.31 Infants with SMA type 2 were observed to have slower decline in CMAP amplitudes.29 Additional studies have shown that CMAP correlates with motor function on a variety of motor function assessments, with children obtaining higher motor function scores with higher CMAP amplitudes.28,32 Since the implementation of newborn screening for SMA, new data reflecting CMAP changes with SMA treatment have been reported from the Taiwan and Germany newborn screening programs. A rapid decrease in CMAP amplitudes was observed in infants predicted to have SMA type 1 compared with infants predicted to have SMA types 2 or 3.33 Infants who were already symptomatic at the time of treatment had ulnar CMAP amplitudes of less than 1 mV.34 Both programs hypothesized that CMAP amplitude could be used as an early detector of the transition from being presymptomatic to symptomatic based on observations that CMAP amplitude decreased significantly just before development of weakness in some patients.33,34 Both programs also noted improvement in CMAP after treatment.
Our data are consistent with previous studies that demonstrated correlation between CMAP amplitudes and predicted disease severity. This highlights the importance of early treatment, particularly in infants predicted to have SMA type 1 because symptom onset can be rapid. Based on observations comparing our premature and term infants with 2 copies of SMN2, we also postulate that motor neuron function may be more likely to be preserved before 40 weeks gestation. This notion is supported by high CMAP values seen in the 2 premature infants (mean initial CMAP amplitude = 5.6 mV) compared with term infants (mean initial CMAP amplitude = 1.6 mV). Additional long-term data will be needed to see whether the CMAP and clinical outcome differences persist with time. Overall, CMAP amplitude appears to be a useful marker of motor neuron health. In the era of SMA treatment, it can also potentially be used to stratify treatment response and help guide the use of adjunctive therapies.
The clinical outcomes from the NYS cohort of newborn screen-identified infants are consistent with previous studies and reports. Risk stratification by SMN2 copy number, albeit imperfect, appears to be clinically helpful in identifying infants at the highest risk for early decline and argues for the addition of SMN2 copy number reporting along with initial screening results. Our cohort illustrates the rapidity at which neonatal screening to initial specialty evaluation can feasibly occur. However, the presence of real-life clinical and system barriers and the complexities of treatment decisions must be recognized and addressed. Additional biomarkers of motor neuron health including electrodiagnostic measures should be considered for detection of preclinical decline and could be a useful posttreatment outcome marker. Additional long-term follow-up data will be critically important in the era of the new treated SMA phenotypes. Standardization of recommended clinical follow-up measures will help inform continued improvement of clinical SMA care and management.
The authors recognize the limitations of a retrospective, descriptive study and the small sample size. Some data points were unavailable because of limited access to medical charts and some visits occurring via telemedicine during the height of the COVID-19 pandemic. Additional long-term follow-up data will be informative with regard to sustainability of motor benefit and incidence of long-term side effects or complications of therapy.
Acknowledgment
The authors thank the laboratory and follow-up staff of the New York State Newborn Screening Program, especially April Parker, Allison Brown-Madole, Robert Sicko, and Virginia Sack. The authors thank additional members of the Neuromuscular Disease Specialty Care Center staff, especially Dawn Dawson, Debra Guntrum, Erin Collins, and Joan Mountain.
Glossary
- DOL
day of life
- mRNA
messenger RNA
- NYS
New York State
- qPCR
quantitative PCR
- SCC
Specialty Care Center
- SCID
severe combined immunodeficiency
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
- WHO
World Health Organization
Appendix. Authors
Study Funding
Funding for universal SMA newborn screening was provided by the New York State Department of Health with additional funding from the Centers for Disease Control and Prevention (CDC) (NU88EH001319).
Disclosure
B.H. Lee has received funding from AveXis, AMO Pharma, Sarepta, and Sanofi Genzyme; she received personal compensation for serving on advisor boards for Roche. C.A. Chiriboga has received funding from AveXis, Biogen, and Roche; she is a consultant (advisory board) for AveXis/Novartis, Genentech, Roche, and PTC Therapeutics; she has served as an educational speaker for Biogen and Roche. D.M Kay's work has been funded by the CDC and Biogen Idec. M. Caggana's work has been funded by the CDC and Biogen Idec. C.F. Stevens's work has been funded by the CDC and Biogen Idec. E. Ciafaloni has received personal compensation for serving on advisory boards and/or as a consultant for AveXis, Biogen, Medscape, Pfizer, PTC Therapeutics, Sarepta Therapeutics, Ra Pharma, Wave, and Strongbridge Biopharma; she has received personal compensation for serving on a speaker's bureau for Biogen and has received research and/or grant support from the CDC, CureSMA, Muscular Dystrophy Association, National Institutes of Health, the Patient-Centered Outcomes Research Institute, Parent Project Muscular Dystrophy, PTC Therapeutics, Santhera, Sarepta Therapeutics, Orphazyme, and the US Food and Drug Administration; she has received royalties from Oxford University Press and compensation from MedLink for editorial duties. The other authors have no relevant disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83(9):810-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cusco I, Barcelo MJ, Rojas-Garcia R, et al. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol. 2006;253(1):21-25. [DOI] [PubMed] [Google Scholar]
- 3.Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu Rev Genomics Hum Genet. 2020;21:231-261. [DOI] [PubMed] [Google Scholar]
- 5.Kemper AR, Brosco J, Comeau AM, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. 2017;19(1):121-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vos B, Senterre C, Lagasse R; SurdiScreen Group, Leveque A. Newborn hearing screening programme in Belgium: a consensus recommendation on risk factors. BMC Pediatr. 2015;15:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levenson D. Newborn screening for Duchenne muscular dystrophy gains support: researchers to push for federal recommendation to have states add DMD test to newborn panel. Am J Med Genet A. 2012;158A(12):viii-ix. [DOI] [PubMed] [Google Scholar]
- 8.Finkel RS, Mercuri E, Darras BT, et al. ; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-1732. [DOI] [PubMed] [Google Scholar]
- 9.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-1722. [DOI] [PubMed] [Google Scholar]
- 10.Baranello G, Darras BT, Day JW, et al. ; FIREFISH Working Group. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med. 2021;384(10):915-923. [DOI] [PubMed] [Google Scholar]
- 11.Glascock J, Sampson J, Haidet-Phillips A, et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glascock J, Sampson J, Connolly AM, et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chien YH, Chiang SC, Weng WC, et al. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. J Pediatr. 2017;190:124-129 e1. [DOI] [PubMed] [Google Scholar]
- 14.Kraszewski JN, Kay DM, Stevens CF, et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet Med. 2018;20(6):608-613. [DOI] [PubMed] [Google Scholar]
- 15.De Vivo DC, Bertini E, Swoboda KJ, et al. ; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newborn Screening Status for All Disorders. 2021. Accessed April 25, 2022. newsteps.org/resources/data-visualizations/newborn-screening-status-all-disorders?q=resources/newborn-screening-status-all-disorders. [Google Scholar]
- 17.Kay DM, Stevens CF, Parker A, et al. Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genet Med. 2020;22(8):1296-1302. [DOI] [PubMed] [Google Scholar]
- 18.Vogel BH, Bonagura V, Weinberg GA, et al. Newborn screening for SCID in New York State: experience from the first two years. J Clin Immunol. 2014;34(3):289-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.WHO Multicentre Growth Reference Study Group. WHO Motor Development Study: windows of achievement for six gross motor development milestones. Acta Paediatr Suppl. 2006;450:86-95. [DOI] [PubMed] [Google Scholar]
- 20.Prevention CfDCa. Important Milestones: Your Baby by Four Months.
- 21.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1(1):19-29. [DOI] [PubMed] [Google Scholar]
- 22.Hale K, Ojodu J, Singh S. Landscape of spinal muscular atrophy newborn screening in the United States: 2018-2021. Int J Neonatal Screen. 2021;7(3):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vill K, Schwartz O, Blaschek A, et al. Newborn screening for spinal muscular atrophy in Germany: clinical results after 2 years. Orphanet J Rare Dis. 2021;16(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng S, Lee BH, Ciafaloni E. Parent perceptions in choosing treatment for infants with spinal muscular atrophy diagnosed through newborn screening. J Child Neurol. 2022;37(1):43-49. [DOI] [PubMed] [Google Scholar]
- 25.Monnette A, Chen E, Hong D, et al. Treatment preference among patients with spinal muscular atrophy (SMA): a discrete choice experiment. Orphanet J Rare Dis. 2021;16(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregg AR, Aarabi M, Klugman S, et al. ; ACMG Professional Practice and Guidelines Committee. Correction to: screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(10):2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hopkins MK, Dugoff L, Kuller JA. Spinal muscular atrophy: inheritance, screening, and counseling for the obstetric provider. Obstet Gynecol Surv. 2021;76(3):166-169. [DOI] [PubMed] [Google Scholar]
- 28.Bishop KM, Montes J, Finkel RS. Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological Exam-Part 2: experience from a nusinersen clinical study. Muscle Nerve. 2018;57(1):142-146. [DOI] [PubMed] [Google Scholar]
- 29.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yonekawa T, Komaki H, Saito Y, Sugai K, Sasaki M. Peripheral nerve abnormalities in pediatric patients with spinal muscular atrophy. Brain Dev. 2013;35(2):165-171. [DOI] [PubMed] [Google Scholar]
- 31.Finkel RS. Electrophysiological and motor function scale association in a pre-symptomatic infant with spinal muscular atrophy type I. Neuromuscul Disord. 2013;23(2):112-115. [DOI] [PubMed] [Google Scholar]
- 32.Lewelt A, Krosschell KJ, Scott C, et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve. 2010;42(5):703-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weng W-C, Hsu YK, Chang FM, et al. CMAP changes upon symptom onset and during treatment in spinal muscular atrophy patients: lessons learned from newborn screening. Genet Med. 2021;23(2):415-420. [DOI] [PubMed] [Google Scholar]
- 34.Vill K, Kolbel H, Schwartz O, et al. One year of newborn screening for SMA - results of a German pilot Project. J Neuromuscul Dis. 2019;6(4):503-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.