

Abstract
G-quadruplexes (G4s) are non-canonical nucleic acid structures that form in G-rich regions of the genome and threaten genome stability by interfering with DNA replication. However, the underlying mechanisms are poorly understood. We have recently found that G4s can stall eukaryotic replication forks by blocking the progression of replicative DNA helicase, CMG. In this paper, we detail the methodology of DNA unwinding assays to investigate the impact of G4s on CMG progression. The method details the purification of recombinantly expressed CMG from the budding yeast, Saccharomyces cerevisiae, purification of synthetic oligonucleotides, and covers various aspects of DNA substrate preparation, reaction setup and result interpretation. The use of synthetic oligonucleotides provides the advantage of allowing to control the formation of G4 structures in DNA substrates. The methods discussed here can be adapted for the study of other DNA helicases and provide a general template for the assembly of DNA substrates with distinct G4 structures.
Keywords: CMG, DNA helicase, DNA unwinding, G-quadruplex, DNA replication
1. Introduction
G-quadruplexes (G4 structures/G4s) are stable DNA or RNA secondary structures that are prevalent in characteristic G-rich sequences. Importantly, due to their potential to impede the progression of DNA polymerases, G-quadruplexes in chromosomal DNA have been implicated in genome instability (Lerner and Sale 2019). Moreover, in a recent study we have determined that G-quadruplexes can also cause replication fork stalling by impeding the progression of the eukaryotic replicative DNA helicase, CMG (Cdc45-MCM-GINS), when present on the translocation strand ahead of the CMG (Kumar et al. 2021). Here, we will describe methods to assess the impact of G4s on DNA unwinding by CMG from the budding yeast, Saccharomyces cerevisiae, using oligonucleotide-based DNA templates and recombinant CMG.
G4s are formed by stacking of co-planar guanine tetrads or quartets that are hydrogen bonded through Hoogsteen base pairing (Figure 1) (Spiegel, Adhikari, and Balasubramanian 2020; Bochman, Paeschke, and Zakian 2012). The stacks are stabilized by coordination of monovalent ions to the centrally facing carbonyl oxygens of guanine bases, with K+ ions providing the maximum stabilization (Bhattacharyya, Arachchilage, and Basu 2016). The four G-strands of a G4 can be provided by multiple strands (intermolecular), including hybrid RNA-DNA structures (Wanrooij et al. 2012), or a single nucleic acid strand (intramolecular). G4s can further adopt various topologies (Spiegel, Adhikari, and Balasubramanian 2020; Bochman, Paeschke, and Zakian 2012), classified as parallel, antiparallel or hybrid depending on the orientation of G-strands with respect to each other. Intramolecular G4s in chromosomal DNA that may impact DNA replication or gene expression were thought to predominantly fold from four contiguous G-stretches that are interspersed by 1–7 random nucleotides (G≥3N1–7G≥3N1–7G≥3N1–7G≥3) (Huppert and Balasubramanian 2005; Todd, Johnston, and Neidle 2005). Each G-stretch in the G4 sequence forms a G-strand, while the interspersed sequences form peripheral loops (Figure 1A). However, G4s with longer loops and discontinuous G-stretches have also been described, significantly broadening the G4 potential of genomic regions (Mukundan and Phan 2013; Bedrat, Lacroix, and Mergny 2016).
Figure 1: G-quadruplex structure.
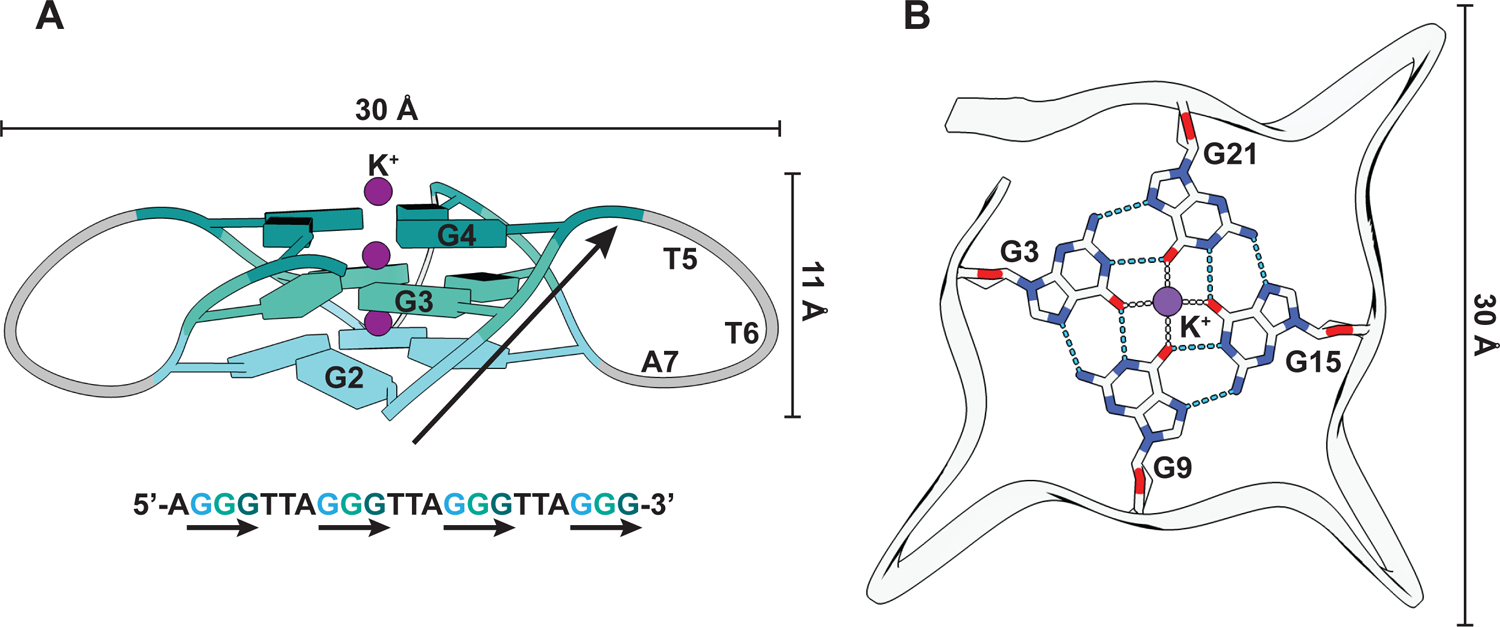
(A) Structural representation of a G4 from human telomeric DNA (PDB: 1KF1) that folds into a parallel G4 structure (Parkinson, Lee, and Neidle 2002). The guanines are represented as slabs and colored differently per quartet. G-strands are marked with black arrows in both the structural model and the corresponding DNA sequence. (B) Interactions between the guanines of one quartet and with K+ ion (purple). Hydrogen bonds and ionic interactions are depicted as blue and gray dashed lines, respectively.
G4-forming sequences are widely distributed across genomes (Chambers et al. 2015; Marsico et al. 2019) and impact several physiological processes, including transcription regulation and telomere maintenance (Spiegel, Adhikari, and Balasubramanian 2020). Irrespective of their physiological significance, G4s can potentially interfere with DNA replication (Sato et al. 2021; Kumar et al. 2021) and must be resolved to provide the template for DNA synthesis; failure in doing so threatens genetic and epigenetic stability (Ribeyre et al. 2009; Sarkies et al. 2010; De and Michor 2011; Paeschke et al. 2013). G4 DNA structures increase during S phase of the cell cycle and subsequently decline upon transition to G2 phase, indicating that G4s are effectively managed during DNA replication (Biffi et al. 2013). Indeed, several monomeric DNA helicases that can unwind G4 DNA structures are important for genetic stability, suggesting that they facilitate replisome progression through G4-containing genomic regions (Vannier et al. 2013; Bosch et al. 2014; Dahan et al. 2018). Examples include helicases belonging to superfamily 1 (DNA2, Pif1) or superfamily 2 (RTEL1, DDX11, FANCJ, RecQ, WRN, BLM, DHX36, DHX9) (Singleton, Dillingham, and Wigley 2007; Lerner and Sale 2019). Structural studies on RecQ and DHX36 provided some mechanistic insights into G4 DNA unwinding (Chen et al. 2018; Voter et al. 2018). However, no general principles for DNA helicase-mediated G4 unwinding could be derived from these studies. RecQ has a guaninespecific pocket (GSP) that flips out a guanine from the 3’ end of the G4, suggesting that sequential sequestration of the terminal guanines could unfold the G4 structure (Voter et al. 2018). DHX36, on the other hand, has a DHX36 specific motif (DSM) which upon binding to G4 transduces structural changes in the helicase core and C-terminal domain that destabilizes the G4 structure even without requiring ATP; ATP hydrolysis is thought to be important for translocation step (Chen et al. 2018). Some of these helicases also exhibit distinct substrate preferences. For example, DHX36 is more active on parallel G4s (Heddi et al. 2015), while Pif1 may prefer non-parallel G4s (Byrd, Bell, and Raney 2018). This diversity in features and mechanisms may hint at a functional specialization of accessory DNA helicases to mediate replisome progression at distinct genomic regions.
The eukaryotic replisome is a large multi-subunit complex structurally organized around the replicative DNA helicase, CMG. (Attali, Botchan, and Berger 2021). CMG is composed of 11 subunits comprising a hetero-hexamer of catalytic subunits, Mcm2-7 (MCM), and two essential non-catalytic subunits, Cdc45 and the hetero-tetrameric GINS (Go, Ichi, Ni, San, an acronym for Sld5 and subunits Psf1-3) complex (Attali, Botchan, and Berger 2021). The C-terminal AAA+ ATPase domains of the MCM hexamer power the CMG helicase, while the non-catalytic subunits promote CMG processivity (Ilves et al. 2010). CMG translocates in 3’−5’ direction on the leading strand template with the MCM N-terminal face leading at the fork junction (Yardimci et al. 2010; Douglas et al. 2018; Fu et al. 2011; Georgescu et al. 2017). Other replisome components that directly contact the CMG helicase include the trimeric complex of Csm3, Tof1 and Mrc1, known as fork protection complex (FPC), which enhances fork rates and maintains normal replisome progression under replication stress (Bando et al. 2009; Szyjka, Viggiani, and Aparicio 2005; Tourriere et al. 2005; Yeeles et al. 2017). Of note, Csm3-Tof1 form a heterodimer that associates with the CMG through interactions with the N-terminal domains of Mcm2, −4, −6 and −7, contacting duplex DNA ahead of the fork junction (Baretic et al. 2020). This places Csm3-Tof1 at the fork apex and thus in prime position to detect G4s ahead of the CMG. Consistent with this notion, the vertebrate homologue of Tof1, Timeless, has been shown to possess a C-terminal DNA binding domain (CTD) with enhanced specificity for G4 DNA that is important for the maintenance of epigenetic stability around G4 sequences (Lerner et al. 2020). In addition, the CTD of Timeless interacts with Ddx11 (Cali et al. 2016). It is, therefore, thought that Timeless mediates replisome progression through G4 DNA by recruiting Ddx11 (Cali et al. 2016; Lerner et al. 2020).
We found that replisome stalling occurs at G4s during budding yeast DNA replication in vitro independent of the FPC, suggesting that G4s impact CMG directly (Kumar et al. 2021). Moreover, we found that a G4 specifically in the template strand can impede DNA unwinding by purified CMG (Kumar et al. 2021). This is consistent with CMG being generally capable of bypassing bulky groups on the non-template (lagging) strand, since the lagging strand is sterically excluded from the central CMG channel (Fu et al. 2011). However, the specific structure of a lagging strand adduct may affect its potential to block CMG progression (Langston et al. 2017; Kose et al. 2019). Thus, it remains a possibility that certain G4 structures on the lagging also impact CMG progression. G4s on the translocation strand could conceivably block the central CMG channel. Although, the central channel is flexible enough to accommodate ~ 20 Å wide double stranded DNA (Langston and O’Donnell 2017), it might struggle to accommodate G4s that are generally 23 – 28 Å in diameter (Do et al. 2011) (Figure 1). Moreover, G4s display a variability in structure and size (Spiegel, Adhikari, and Balasubramanian 2020), illustrating the importance of determining the effect of distinct G4s on CMG activity.
Here we present the experimental procedure to perform CMG helicase assays to investigate the impact of G4s on CMG progression. For this we have focused on the sequence d(GGGTGGGTGGGTGGG), which folds into a highly thermostable parallel G4 in vitro (Do et al. 2011) and is known to interfere with DNA replication in yeast cells (Piazza et al. 2015). Recombinant Saccharomyces cerevisae CMG is purified from budding yeast cells co-overexpressing all eleven CMG subunits. While CMG is normally formed in a complex and highly cell cycle-regulated sequence of events involving many accessory factors at cellular replication origins (Parker, Botchan, and Berger 2017), co-overexpression of the eleven CMG subunits in yeast or baculovirus expression systems has been demonstrated previously to allow reconstitution of functional CMG complexes in the absence of replication origin activity (Georgescu et al. 2014; Ilves et al. 2010; Zhou et al. 2017). The assay monitors the unwinding of oligo-nucleotide-based DNA substrates in the presence of CMG and can be readily modified to test the impact of different G4s and accessory factors.
2. CMG Purification
Recombinant CMG is purified from a diploid budding yeast strain that conditionally overexpresses all eleven Saccharomyces cerevisiae CMG subunits. For this, respective open reading frames (ORFs) are cloned downstream of the GAL1–10 promoter in expression vectors that are integrated at auxotrophic marker loci. Analogously to a previously published approach (Frigola et al. 2013; Posse, Johansson, and Diffley 2021), to maximize expression levels, ORFs have been codonoptimized for expression in budding yeast by converting the endogenous codon usage to that of the twenty most abundant proteins in the budding yeast proteome (Ghaemmaghami et al. 2003). In addition, each ORF is flanked by PGK1 5’ and 3’ UTR sequences. The strain also harbors a galactose-inducible expression cassette for the Gal4 transcriptional activator to boost GAL1–10 promoter activity. Expression is induced in asynchronous cultures by the addition of galactose to the medium. Mcm5 is N-terminally fused to a 3xFLAG tag and Sld5 is N-terminally fused to a calmodulin binding peptide (CBP)-tag, allowing for enrichment of the CMG holo-complex by tandem affinity purification. CMG is subsequently purified by anion exchange chromatography using an FPLC system designed for micro-preparative columns, which helps to concentrate the sample.
2.1. Equipment
Sorvall RC 3C Plus centrifuge with Rotor HBB6
Thermo Scientific Sorvall Lynx 6000 centrifuge with Rotor F12–6×500
Eppendorf 5810 R centrifuge with Rotor A-4–81
Freezer/Mill 6875D (Spex)
Thermo Scientific Sorvall WX+ ultracentrifuge with Rotor T-647.5
Bio-Rad Econo Pump and Columns 1.0 × 10 cm and 0.7 × 5 cm
Bio-Rad Mini-PROTEAN Tetra Cell – casting and electrophoresis systems
MiniQ PC 3.2/3 (GE)
AKTA micro FPLC system
2.2. Buffers and reagents
YPD: 1 % (w/v) yeast extract and 2 % (w/v) peptone in ddH2O. Autoclave and add 2 % (w/v) glucose from a sterile stock of 20 % (w/v) glucose before use.
YPD-Agar: 1 % (w/v) yeast extract, 2 % (w/v) and 2.5 % (w/v) agar in ddH2O. Autoclave and add 2 % (w/v) glucose from a sterile stock of 20 % (w/v) glucose before pouring the plates.
YPLG: For 12 L of YPLG, 120 g yeast extract and 240 g peptone are dissolved in 11.4 l of ddH2O. 950 mL of this YP media is distributed to each of twelve 2 L flasks and autoclaved. After the media has cooled down, 40 mL of 50 % (v/v) glycerol, 23 mL of lactic acid (88 % concentrated solution) and 15 mL of 50 % (w/v) NaOH are added in that order.
Buffer M(+): 40 mM HEPES-KOH pH 7.6/ 10 % (v/v) glycerol / 300 mM potassium chloride / 2 mM magnesium acetate / 0.005 % TWEEN 20 / 1 mM DTT
Buffer M(−): 40 mM HEPES-KOH pH 7.6 / 10 % (v/v) glycerol / 300 mM potassium chloride / 0.005 % TWEEN 20 / 1 mM DTT
Buffer K(−): 40 mM HEPES-KOH pH 7.6 / 10 % (v/v) glycerol / 2 mM magnesium acetate / 0.005 % (v/v) TWEEN 20 / 1 mM DTT
Buffer T: 25 mM Tris-HCl pH 7.2 / 10 % (v/v) glycerol/ 2 mM magnesium acetate / 0.005 % (v/v) TWEEN 20 / 1 mM DTT
CMG storage buffer: 25 mM HEPES-KOH pH 7.6 / 10 % (v/v) glycerol / 200 mM potassium acetate/ 2 mM magnesium acetate/ 1 mM DTT
Pierce protease inhibitor tablets – EDTA free (1 tablet per 50 mL of buffer)
Anti-FLAG M2 affinity resin (Sigma)
3x FLAG peptide (Sigma)
GelCode blue stain (Thermo Scientific)
4–20 % TGX gels (Bio-Rad)
Calmodulin affinity resin (Agilent)
2.3. Strain genotype
YSD35: MATa/a ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 pep4::kanMX bar::hphNAT1 his3::Gal-Gal4 (HIS3) trp1) ura3::GAL- MCM2/FLAG-MCM3 (URA3) leu2::GAL-MCM7/MCM6 (LEU2) trp1::GAL-CDC45 (TRP1) ura3::GAL-PSF2/PSF3 (URA3) leu2::GAL-PSF1/CBP-SDL5 (LEU2)
2.4. Protein expression and cell extract preparation
Streak out strain YSD35 from glycerol stock on YPD-agar plate and grow at 30°C for 3 days.
Inoculate 3–4 colonies in 60 mL of YPD media in a 250 mL conical flask and culture at 30 °C and 200 rpm for 20 hours. The cell density of this pre-culture should reach ~5 × 107 cells / mL.
Inoculate 12 L of YPLG media with 60 mL of pre-culture by adding 5 mL of inoculum to each 2 L Erlenmeyer flask containing 1 L of YPLG. Incubate the cultures at 30 °C and 200 rpm for 20 – 22 hours until they reach a cell density of ~ 5 × 107 cells / mL.
Induce expression by adding 20 g of galactose to each flask and continue incubation at 30 °C and 200 rpm for another 6 hours.
After induction, harvest cells by centrifugation in 1 L bottles at 4,500 rpm for 10 min at 4 °C, using a Sorvall RC 3C Plus centrifuge or equivalent.
Resuspend cell pellets in 40–50 mL of 25 mM HEPES-KOH pH 7.6 / 1 M sorbitol per centrifuge bottle.
Pool cell suspensions in 500 mL centrifuge bottle and centrifuge at 5,000 rpm for 5 min at 4 °C in Sorvall Lynx 6000 centrifuge or equivalent.
Resuspend cell pellet from the sorbitol wash in 50 mL of Buffer M(+). Divide the suspension into four 50 mL centrifuge tubes and centrifuge in a swing out rotor at 4,000 rpm for 10 min at 4 °C using an Eppendorf 5810 R centrifuge or equivalent.
Note the volumes of the cell pellets, add 0.5 × volume of Buffer M(+) (supplemented with protease inhibitor), and resuspend the pellets by stirring with a glass rod to form a thick slurry.
Freeze the cell slurry dropwise into liquid nitrogen using a 10 mL serological pipette. The frozen cell pellets are referred as popcorn and can be stored at −80 °C until further processing.
Crush the popcorn in a cryogenic mill with a run time of 2 min at 15 CPS for 10 cycles. Transfer the crushed powder to a glass beaker and allow to thaw on ice.
After the powder liquifies, add an equal volume of the Buffer M(+) (supplemented with protease inhibitor) and mix thoroughly on a magnetic stirrer for 10 min at 4 °C.
Transfer the cell extracts to six 60 mL ultra-centrifuge bottles and centrifuge at 41,000 rpm for 1 hour at 4 °C.
Transfer the clarified soluble phase to fresh tubes and snap-freeze in liquid nitrogen. The frozen soluble phase can be stored at −80 °C until further processing.
2.5. CMG affinity purification
All steps are performed at 4 °C.
FLAG pulldown
Transfer 3 mL of anti-FLAG M2 affinity resin (Sigma) to a 1.0 × 10 cm Econo-column and equilibrate with 5 column volumes (CV) of Buffer M(+).
Pass the soluble phase over the anti-FLAG resin at a flow rate of 1.5 mL / min using a peristaltic pump (e.g. Bio-Rad Econo pump).
Wash the resin with 5 CV of Buffer M(+) to remove unbound proteins.
Wash the resin with 25 mL of Buffer M(+) / 1 mM ATP, followed by one wash with 5 CV of Buffer M(+).
Elute bound proteins by gravity flow in 5 fractions of 1 CV each. Elute fraction 1 with 3 mL Buffer M(+) / 2 mM CaCl2 / 0.2 mg / mL 3x FLAG peptide, fractions 2 and 3 with 3 mL of Buffer M(+) / 2 mM CaCl2 / 0.1 mg / mL 3x FLAG peptide each, and fractions 4 and 5 with 3 mL of Buffer M(+) / 2 mM CaCl2 each. Snap-freeze eluates in liquid nitrogen and store at −80 °C.
Analyze 5 μL of each elution fraction by SDS-PAGE on 4–20 % precast TGX gels (Bio-Rad). Stain the gels with GelCode blue.
Repeat steps 1 – 6 until CMG is depleted from the soluble phase.
CBP pulldown
Equilibrate 1 mL calmodulin affinity resin with 5 CV of Buffer M(−) / 2 mM CaCl2 in a 0.7 × 5 cm Econo-column.
Pool the eluates from the FLAG pulldown and pass over the calmodulin affinity resin using a peristaltic pump at a flow rate of 1 mL / min.
Wash the resin with 15 CV of Buffer M(−) / 2 mM CaCl2.
Elute bound proteins by gravity flow with 8 × 1 CV of Buffer M(−) / 2 mM EGTA / 2mM EDTA.
Supplement the eluates with 5 mM magnesium acetate, snap-freeze in liquid nitrogen and store at −80 °C.
Analyze 5 μl of each fraction by SDS-PAGE.
Repeat steps 1 – 5 until the CMG is depleted from FLAG elution fractions.
Anion exchange chromatography
Pool the eluates from the CBP pulldown and dilute by adding an equal volume of Buffer K(−) to reduce the KCl concentration to 150 mM.
Connect the MiniQ column to the FPLC system.
Equilibrate the MiniQ column in Buffer T / 150 mM KCl.
Pass the CBP eluate over the MiniQ column at a flow rate of 0.12 mL / min.
Wash the column with 10 CV of Buffer T/ 150 mM KCl.
Elute bound proteins with a KCl gradient of 150 mM – 1000 mM over 30 CV in Buffer T.
Collect 0.12 mL fractions and analyze 2 μl from each peak fraction by SDS-PAGE (Figure 2).
Pool peak fractions containing stoichiometric amounts of each CMG subunit and dialyze for 4 hours against 1 L of CMG storage buffer. Aliquot dialyzed fractions, snap-freeze in liquid nitrogen and store at −80 °C.
Determine the concentration of CMG by SDS-PAGE alongside protein standard (e.g. BSA). For this, scan the gel and determine band intensities with ImageJ software.
Figure 2: Purification of CMG.
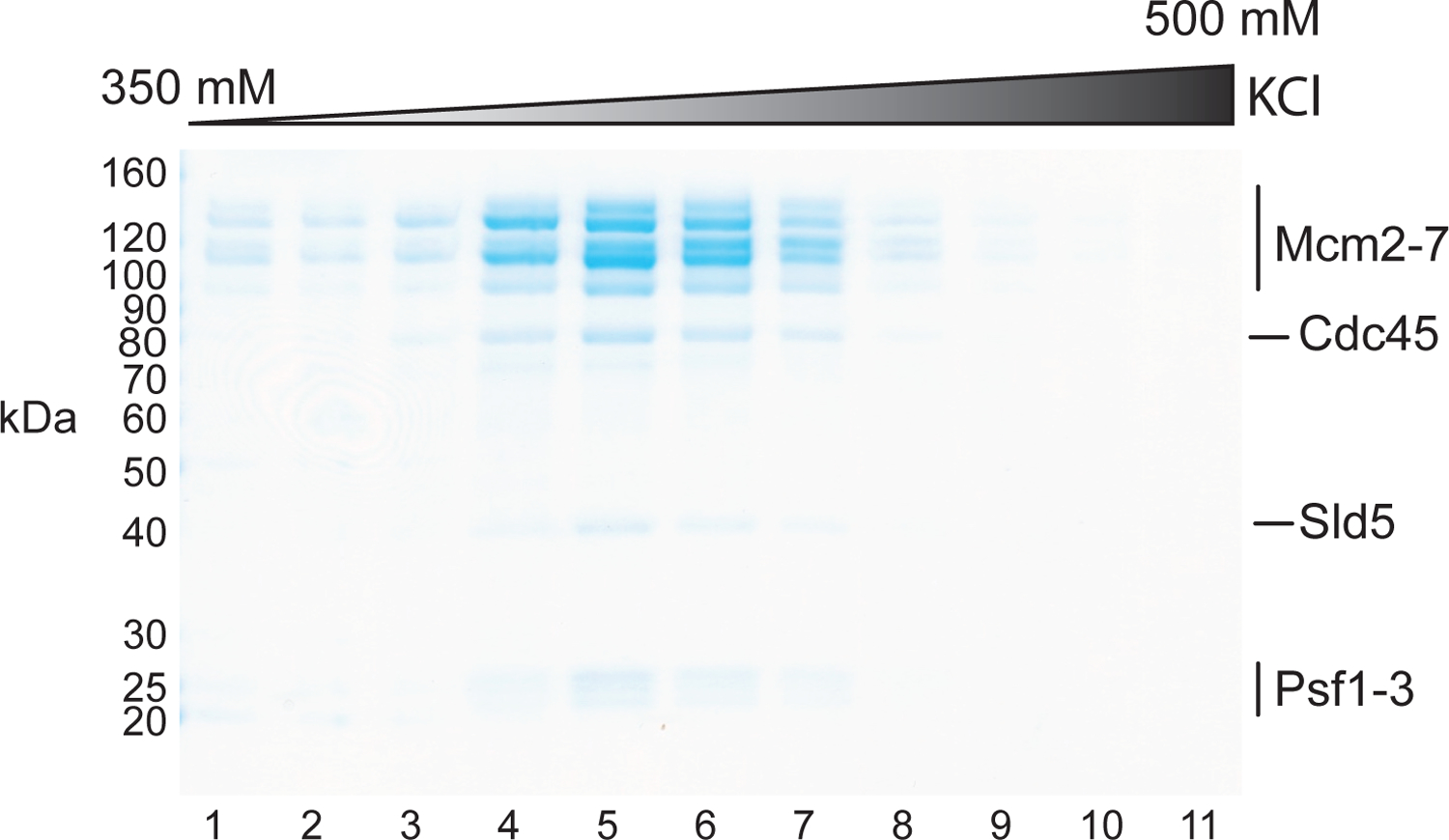
Coomassie-stained SDS-PAGE analysis of MiniQ fractions.
3. DNA substrates
Our forked DNA substrates contain a 3’ (dT)40 tail which allows the loading of CMG on the translocating strand. The non-template strand features a secondary structure-forming (GGCA)40 sequence on the 5’ end to disfavor CMG loading and aid the steric exclusion of this strand by CMG (Figure 3A–F) (Georgescu et al. 2017; Kose et al. 2019; Petojevic et al. 2015). A G4-forming sequence [d(TTGGGTGGGTGGGTGGGTT)] is inserted either in the template or non-template strand of the DNA duplex region. To stabilize the G4 structure, the corresponding region in the complementary strand features a poly(dT) stretch that prevents reannealing of the G4 sequence (Figure 3A + C). In control substrates, one of the G-stretches is mutated to CCC to interfere with the folding of the G4 structure (Figure 3B + D). In additional control substrates, the G4 sequence is not pre-formed into a G4 structure and remains in B-form DNA (Figure 3E).
Figure 3: Design of forked DNA substrates for CMG helicase assays.
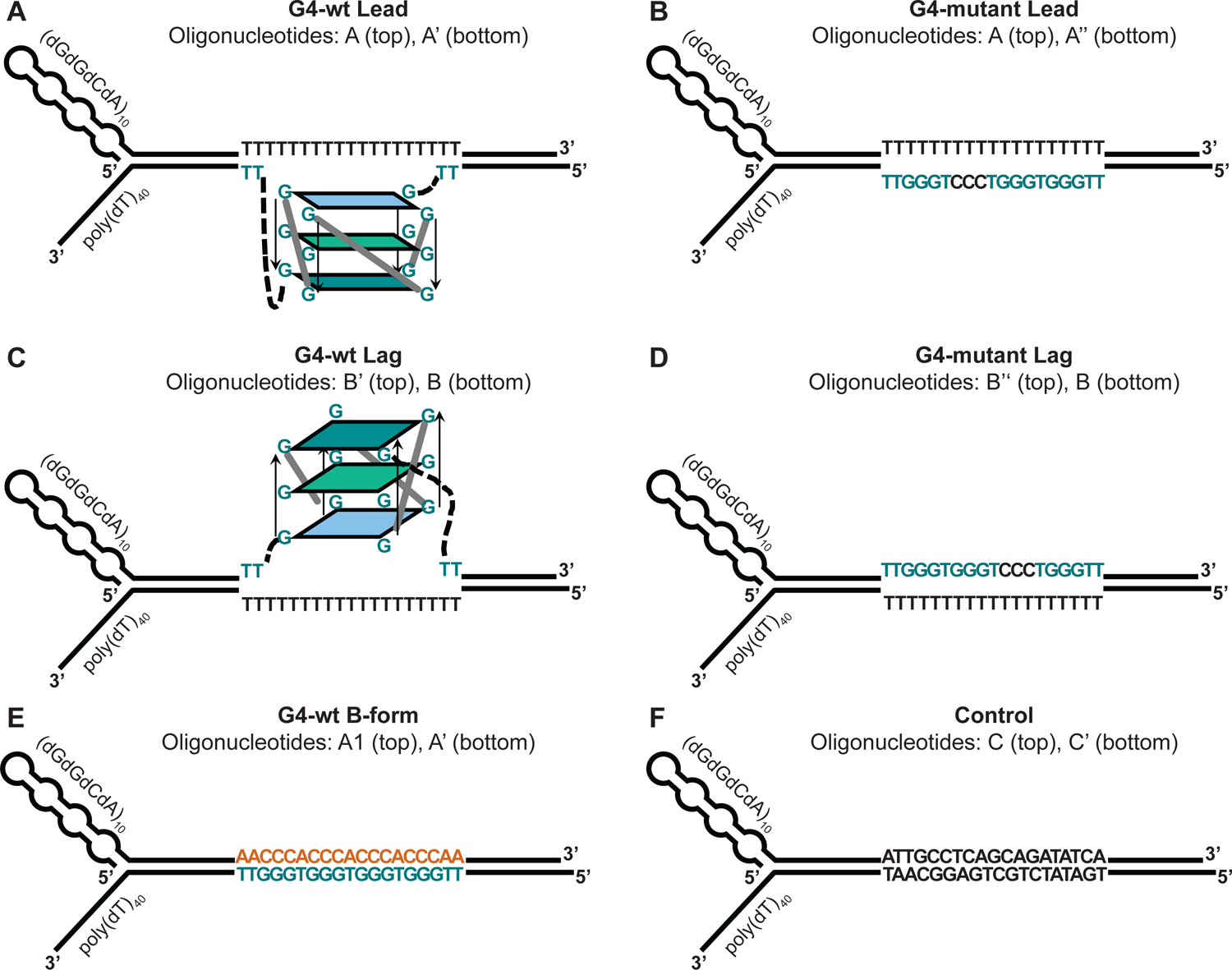
A poly(dT) stretch on the translocation strand serves as the loading site for CMG. Secondary structure on the 5’ end of the non-template strand prevents aberrant CMG loading and promotes the strand exclusion. The duplex region contains the following features: (A) G4 on the template strand. (B) Mutant G4 sequence on the template strand. (C) G4 on the non-template strand. (D) Mutant G4 sequence on the non-template strand. (E) Template strand G4 sequence in B-form DNA. (F) Non-G4 duplex DNA.
To prepare the DNA substrates, oligonucleotides are annealed by boiling and subsequent slow cooling. The oligonucleotides are commercially synthesized as standard desalted oligonucleotides (Table 1). Oligonucleotides produced in this way generally have significant impurities and truncations that should be eliminated prior to substrate assembly. We, therefore, purify oligonucleotides by denaturing PAGE. Bands corresponding to full-length oligonucleotide are excised from the gel and the DNA is extracted and further purified by ethanol precipitation. PAGE-purified oligonucleotides are also available directly from the manufacturers. However, considering the yield, quality and turnaround times, we prefer in-house oligonucleotide purification. One of the oligonucleotides will be radiolabeled with γ−32P-ATP and T4 polynucleotide kinase (T4 PNK) prior to annealing to the unlabeled complementary oligonucleotide. Alternatively, annealed DNA substrates may also be labeled following annealing. For this, the translocation strand features a 5’ overhang of two basepairs to promote phosphorylation by T4 PNK. Annealed products are gel-purified after native PAGE to eliminate un-annealed oligonucleotides or higher-order inter-strand G4 structures.
Table1:
List of oligonucleotides
| Name | Sequence (5’−3’) |
|---|---|
| A | GGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGTTGGCGAATTCCCTTTTTTTTTTTTTTTTTTTCCTCAGCACGACGTTGTAAAACGAG |
| A’ | GGCTCGTTTTACAACGTCGTGCTGAGGTTGGGTGGGTGGGTGGGTTGGGAATTCGCCAACCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT |
| A” | GGCTCGTTTTACAACGTCGTGCTGAGGTTGGGTGGGTCCCTGGGTTGGGAATTCGCCAACCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT |
| A1 | GGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGTTGGCGAATTCCCAACCCACCCACCCACCCAACCTCAGCACGACGTTGTAAAACGAG |
| B | GGCTCGTTTTACAACGTCGTGCTGAGGTTTTTTTTTTTTTTTTTTTGGGAATTCGCCAACCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT |
| B’ | GGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGTTGGCGAATTCCCTTGGGTGGGTGGGTGGGTTCCTCAGCACGACGTTGTAAAACGAG |
| B” | GGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGTTGGCGAATTCCCTTGGGTGGGTCCCTGGGTTCCTCAGCACGACGTTGTAAAACGAG |
| C | GGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGCAGGTTGGCGAATTCCCATTGCCTCAGCAGATATCACCTCAGCACGACGTTGTAAAACGAG |
| C’ | GGCTCGTTTTACAACGTCGTGCTGAGGTGATATCTGCTGAGGCAATGGGAATTCGCCAACCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT |
3.1. Equipment
Bio-Rad Mini-PROTEAN Tetra Cell – casting and electrophoresis systems
Dark Reader (Clare Chemical Research)
Eppendorf ThermoMixer F1.5
Eppendorf 5424 centrifuge with Rotor FA-45-24-11
Econospin mini spin columns for gel filtration resin (Epoch)
Eppendorf 5417R centrifuge with Rotor F45-30-11
Nanodrop 2000c (Thermo Scientific)
Bio-Rad Universal hood II GelDoc.
Bio-Rad C1000 thermocycler
Konica SRX-101A Film processor
3.2. Buffers and reagents
1x TBE: 90 mM Tris-borate / 2 mM EDTA
2x denaturing buffer: 95 % (v/v) Formamide / 5 mM EDTA / 0.025 % (w/v) Bromophenol Blue / 0.025 % (w/v) Xylene cyanol
1x TE: 10 mM Tris-HCl pH 8.0 / 1mM EDTA
Acrylamide elution buffer: 500 mM ammonium acetate/ 10 mM magnesium acetate / 1 mM EDTA
3 M sodium acetate pH 5.2
1 M potassium acetate pH 7.6
Sequagel urea gel system (National Diagnostics)
Ethidium bromide solution (Thermo Scientific)
Blue light transilluminator (Clare Chemical Research)
γ−32P-ATP (Perkin Elmer)
Polynucleotide kinase (NEB)
6x purple gel loading dye (NEB)
3.3. Oligonucleotide purification
Gel-isolation of oligonucleotides
Prepare a 12 mL solution of desired strength, using the Sequagel urea gel system. Typically, we cast gels with 8 % monomer strength, which we find suitable for the purification of 70 – 120 nt long oligonucleotides.
Assemble the Bio-Rad mini-PROTEAN gel casting apparatus with 1.5 mm spacer plates as per the manufacturer’s instructions; pour the monomer solution, insert 5-well comb and set aside for polymerization.
Pre-run the gels in 1x TBE at 200 V for 30 min. After the pre-run, rinse the wells with reservoir buffer to flush out excess urea.
Mix 50 μL of each 100 μM oligonucleotide stock solution with an equal volume of 2x denaturing buffer, heat at 95 °C for 5 min and snap-chill on ice for 2 min.
Load 100 μl sample per well and electrophorese at 120 V. The length of the run will vary according to the length of the oligonucleotides and should be estimated based on the migration of tracking dyes. Typically, we run our gels for 85 – 100 minutes.
Transfer the gel to a staining tray containing 1 mg / mL ethidium bromide in 1x TBE buffer and stain for 10 minutes.
Visualize DNA bands on a blue-light transilluminator. Each sample should feature a prominent band corresponding to the full-length oligonucleotide. Depending on the quality of oligonucleotide synthesis, a smear of faster migrating truncated products and some higher molecular weight non-specific products may also be observed (Figure 4A). The gel slices corresponding to full-length product are excised using razor blades.
Figure 4: DNA substrate preparation.
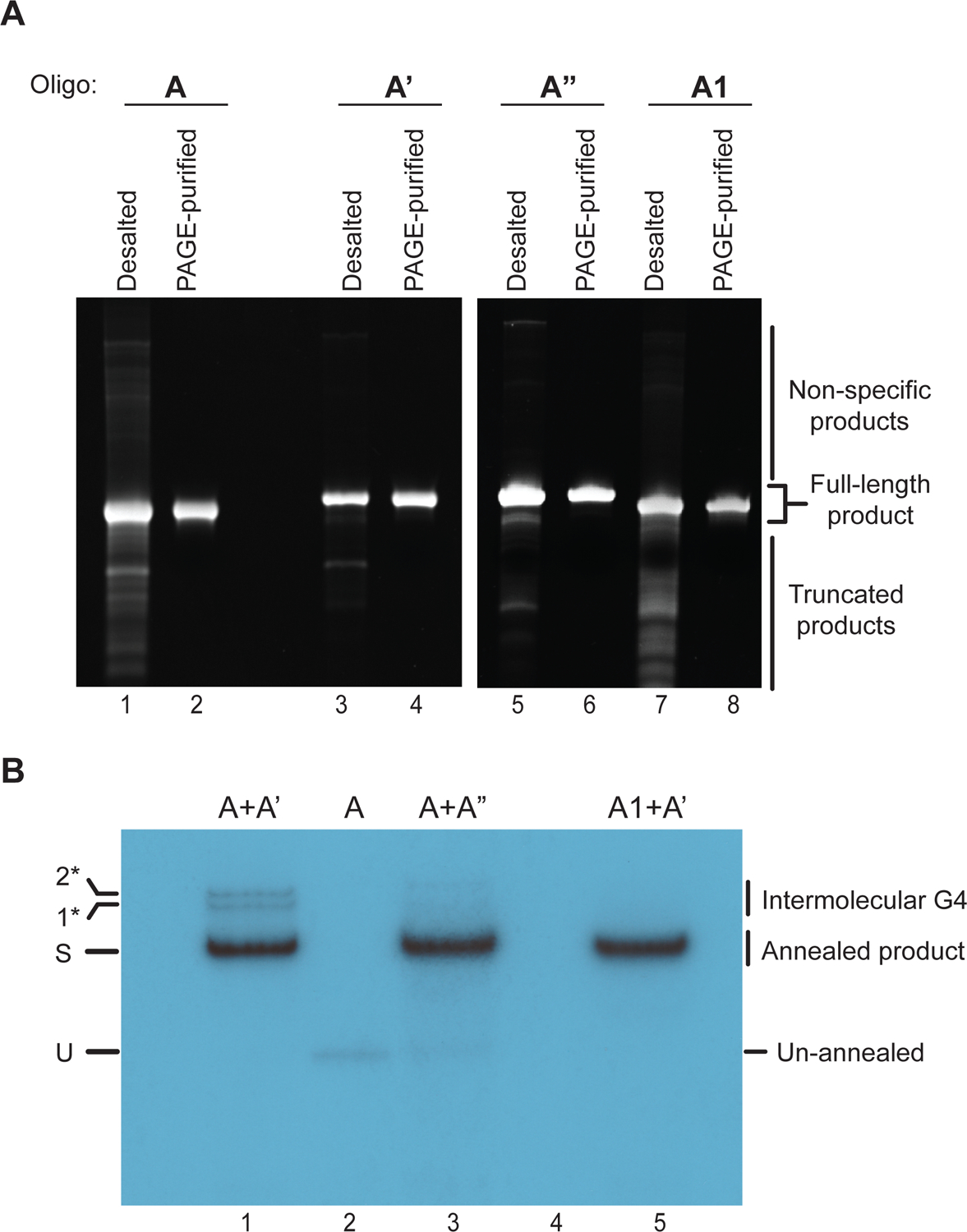
(A) Denaturing polyacrylamide gel-analysis of oligonucleotides before (desalted) and after PAGE purification (PAGE-purified). The gels are stained with ethidium bromide. (B) Autoradiogram of annealing reaction products after native PAGE. U: unannealed oligonucleotide; S: Substrate; 1* and 2*: Higher molecular weight products likely corresponding to intermolecular G4s.
Extraction of oligonucleotides
Transfer the excised slices to 1.5 mL centrifuge tubes and crush them with micro-pipette tips that have been melted close at the tip using an open flame.
Add 100 μL of 1x TE to the crushed gel, vortex and freeze at −80 °C for 30 min. Subsequently, heat the samples for 5 minutes at 95 °C.
Add 400 μl of acrylamide elution buffer to each tube and shake in a thermo-mixer at 37 °C and 1,400 rpm overnight.
Next day, transfer the gel suspension to an empty spin column and spin at 13,000 rpm for 1 minute in an Eppendorf centrifuge to remove gel chunks. Collect the flow-through and transfer to a fresh 1.5 mL centrifuge tube.
Add 1 mL of ice-cold ethanol to each flow-through fraction, mix and incubate at −20 °C for 30 minutes to allow precipitation of oligonucleotides.
Centrifuge tubes at 4 °C and 13,000 rpm for 10 minutes. Discard the supernatant, taking care not to disturb the pellet.
Dissolve the pellet in 200 μL of 1x TE and add 25 μL of 3 M sodium acetate. Precipitate oligonucleotides by adding 500 μL of ice-cold ethanol and incubate at −20 °C for 30 minutes.
Centrifuge samples at 4 °C and 13,000 rpm for 10 minutes, discard supernatant.
Wash pellets with 500 μL of 70 % ethanol (v/v) and spin again at 4 °C and 13,000 rpm for 10 minutes.
Discard supernatant and air-dry pellets in tubes.
Resuspend pellets in 40 μL of 1x TE buffer. Determine the concentration of oligonucleotide using a Nanodrop and adjust to a final concentration of 10 μM.
Check a small aliquot on a denaturing urea-polyacrylamide gel. For this, we generally cast 10-well gels of 0.75 mm thickness which requires 6 mL of monomer solution per gel. The sample is electrophoresed and stained as above (Figure 4A). The gel can be imaged using a gel documentation system.
3.4. Substrate formation and extraction
Radiolabeling of oligonucleotides
- Perform a 5’-radiolabeling reaction with one of the oligonucleotides from a complementary pair (Table 2, Figure 3) using PNK. Set up the reaction in a PCR tube as shown below:
Component Volume 10x PNK Buffer 3 μl ddH2O 20.25 μl Oligonucleotide A/C (10 μM) 0.75 μl γ-P32-ATP 4.5 μl PNK 1.5 μl Total 30 μl Perform the reaction in a thermocycler which is programmed to maintain 37 °C for 1 hour. Heat-inactivate the enzyme by raising the temperature to 80 °C for 20 minutes followed by 90 °C for 10 minutes.
Table 2:
List of DNA substrates
| Substrate | Oligonucleotide pair |
|---|---|
| G4-wt lead | A + A’ |
| G4-mutant lead | A + A” |
| G4-wt lag | B + B’ |
| G4-mutant lag | B + B” |
| G4-wt B-form | A1 + A’ |
| Control | C + C’ |
Red color denotes radiolabeled oligonucleotides
Annealing of complementary oligonucleotides
Withdraw a 1 μL aliquot from the labeling reaction and keep aside. This will serve as a control during PAGE analysis alongside the annealed products.
Add 1.5 μL of 1 M KOAc to the remaining reaction to adjust the K+ concentration to ~ 50 mM. Add 1 μL of complementary oligonucleotide (from 10 μM stock) to the reaction (Table 2).
Mix all the components thoroughly by pipetting and proceed with the annealing reaction in a thermocycler, programmed to heat the samples at 95 °C for 5 minutes and then slow-cool at a rate of 1 °C / minute until the temperature reaches 10 °C.
Gel-isolation of annealed products
Prepare 6 mL acrylamide solution of 10 % monomer strength in TBE to cast a native gel with acrylamide to bis-acrylamide ratio of 29:1.
Assemble the Bio-Rad mini-PROTEAN gel casting apparatus with 0.75 mm spacer plates as per the manufacturer’s instructions; pour the above prepared monomer solution, insert a 5-well comb and set aside for polymerization.
Mix each of the annealed reaction products with 6.5 μL of 6x purple gel loading dye (NEB) and electrophorese through 10 % native gel in 0.5x TBE at 150 V for 45 minutes. Run 1 μL of the labeled unannealed oligonucleotide alongside the reaction products.
In the meantime, develop an unexposed autoradiography film. This film will be used as a transparent gel support for the excision of DNA bands containing the desired DNA substrates.
Place two phosphorescent stickers on the transparent support film and place the gel on the film. Wrap this setup in cling film to seal the gel and fix it in position.
Expose the gel to a second autoradiography film in a Hypercassette up to 1 minute and develop the film (Figure 4B). Place this film underneath the gel and align with the signals from the phosphorescent stickers to indicate the positions of the DNA bands in the gel.
An example autoradiogram depicting annealing products is shown in Figure 4B. Lane 1 contains the annealed product of oligonucleotides A and A’, which results in the formation of a fork substrate that has a G4 on the translocation strand (Figure 3A). Lane 3 contains the DNA substrate with the G4-mutant sequence, formed through annealing of oligonucleotides A and A” (Figure 3B). Comparison of the gel-mobilities of the annealed products with that of unannealed oligonucleotide A in Lane 2 reveals a prominent band shift of the annealed product. Lane 5 contains substrate with the G4 sequence in B-form DNA (Figure 3E). We also observe higher molecular weight bands in Lane 1 (labeled 1* and 2*) that we attribute to intermolecular G4 species (Figure 4B). Consistent with this interpretation, these species are not observed in substrates devoid of G4 structure.
Excise gel slices corresponding to the annealed substrates with a scalpel and transfer to a 1.5 mL centrifuge tube.
Crush the gel slices with a pipette tip that was sealed at the tip with a flame and add 100 μL of 1x TE supplemented with 50 mM potassium acetate to each tube.
Shake the slurry in a thermo-mixer at 22 °C and 1,400 rpm overnight to extract the DNA.
Next day, transfer the gel suspension to an empty spin column and spin at 13,000 rpm for 1 minute to remove gel chunks. Collect the flow-through and transfer to a fresh 1.5 mL centrifuge tube. Stored at −20 °C.
Calculating substrate concentration
To determine the concentration of our final DNA substrate, we take the following considerations. We start with a known quantity of radiolabeled oligonucleotide which after annealing and gel-electrophoresis separates into distinct product bands: un-annealed oligonucleotide, fully annealed DNA substrate and non-specific annealing products. The total of all these species is equal to the starting material. Therefore, we quantify the total intensity of all bands in a lane and calculate the fraction contained in the band corresponding to the fully annealed DNA substrate. Furthermore, by measuring the radioactivity in the eluate and left behind in the separated gel chunks after the gel-extraction step we typically observe a gel extraction efficiency of 85 %.
Given below is an example of how to calculate the concentration of a DNA substrate, using the substrate shown in Figure 4B, lane 1. We radiolabeled 7.5 pmoles of oligonucleotide A in a 30 μL reaction and removed 1 μL prior to the annealing reaction. Therefore, our starting material is:
After the annealing reaction, products are separated by native PAGE. We observe multiple bands (Figure 4B, lane 1): 1) Un-annealed oligonucleotide A (U), 2) Substrate (S) and 3) Inter-strand G4 structures (1*, 2*). The intensity of each band is determined using ImageJ:
| Band | Intensity |
|---|---|
| U | not detected |
| S | 19461.6 |
| 1* | 2098.1 |
| 2* | 1717.0 |
| Total | 23276.7 |
The fraction of radiolabeled oligo in the correctly annealed DNA substrate therefore is:
The annealed DNA substrate is extracted into 100 μL of buffer. At an efficiency of extraction of 85 %, the final concentration of the substrate is:
4. Helicase assay
For CMG helicase assays, CMG is initially loaded onto the DNA substrate in the presence of non-hydrolyzable ATP analogue. Addition of an excess of ATP subsequently initiates CMG translocation and concomitant DNA unwinding. Aliquots are removed at designated time points and the reaction is quenched by adding an excess of EDTA to chelate Mg2+ ions required for ATP hydrolysis. The stopped reaction aliquots are analyzed by native PAGE to separate forked DNA substrate from product single strands.
4.1. Equipment
Slab gel dryer GD2000 (Hoefer)
Imaging plate BAS MS 2040 (FUJIFILM)
Typhoon FLA 7000 (GE)
Eppendorf ThermoMixer F1.5
Bio-Rad C1000 thermocycler
Bio-Rad Mini-PROTEAN Tetra Cell – casting and electrophoresis systems
4.2. Buffers and reagents
1x TAE: 40 mM Tris-acetate / 1 mM EDTA
10x Helicase Buffer: 250 mM HEPES-KOH pH 7.6 / 1 M potassium acetate / 100 mM magnesium acetate
Adenosine 5’-O-(3-thiotriphosphate) (ATPγS) [Roche]
ATP (100 mM) (Thermo Scientific)
4.3. DNA unwinding assay
-
1
Cast 10-well native polyacrylamide gels of 10 % monomer strength in 1x TAE / 5 % glycerol.
-
2Prepare the following reaction mix:
Component Volume 10x Helicase Buffer 3.25 μL DTT (100 mM) 0.81 μL BSA (1 mg/mL) 3.25 μL ATPγS (5 mM) 0.65 μL Substrate X μL for 4 nM ddH2O ad 32.5 μL Total 32.5 μL -
3
Remove an aliquot of 3 μL from the above reaction mix and keep aside as no-protein control.
-
4
Add 20 nM of CMG or a corresponding volume of CMG storage buffer (buffer control) to the remaining mix.
-
5
Mix the reactions thoroughly by pipetting and incubate in thermomixer at 30 °C for 1 hour without shaking.
-
6
In the meantime, prepare the following solutions:
2x ATP mix
| Component | Volume |
|---|---|
| 10x Helicase Buffer | 3.5 μL |
| DTT (100 mM) | 0.9 μL |
| BSA (1 mg/mL) | 3.5 μL |
| ATP (100 mM) | 3.5 μL |
| ddH2O | 23.6 μL |
| Total | 35 ml |
No-ATP mix
As 2x ATP mix, but without ATP. Aliquot 3 μL into a 1.5 mL tube for each substrate.
2x Stop buffer
Dilute 6x Purple gel loading dye (NEB) two-fold with water and supplement with 40 mM EDTA and 0.24 % SDS (w/v).
| Component | Volume |
|---|---|
| 6x Purple gel loading dye | 100 μL |
| (NEB) | |
| SDS (10 % w/v) | 2.4 μL |
| EDTA (0.5 M) | 12 μL |
| ddH2O | 185.6 μL |
| Total | 300 μL |
-
7
Add 6 μL aliquots each of 2x Stop buffer to 7 tubes that will receive reaction aliquots at the following time points [minutes]: 0.5, 1, 2, 3, 5, 15 and 60.
-
8
After the pre-incubation step, withdraw a 3 μL aliquot from the pre-incubation mix and add to the tube with 3 μL of no-ATP mix. This is a control reaction to ensure that DNA unwinding is ATP-dependent.
-
9
Add 3 μL of 2x ATP mix to the no-protein control tube. Place both the ATP- and no-protein control tubes back in the thermomixer for 1 hour.
-
10
Mix the remaining ~ 27 μL pre-incubation reaction mix with 2x ATP mix and place in thermomixer to initiate DNA unwinding.
-
11
Remove aliquots of 6 μL at designated times and add to prepared tube containing 6 μL of 2x Stop buffer to stop the reaction. Store tubes on ice until they are ready to be loaded on the gel.
-
12
Dilute an equivalent amount of substrate – as used in the reaction – in water (leaving out the volume required for 10x Helicase buffer) and denature in thermocycler at 95 °C for 5 min, snap-cool on ice for 2 minutes. Add 10x Helicase buffer and 2x Stop buffer to the denatured substrate. This boiled control is a marker for unwound product.
-
13
At the 1 hour mark, remove final reaction aliquot along with no-protein and no-ATP control reactions.
-
14
Assemble native polyacrylamide gel in the Bio-Rad mini-PROTEAN electrophoretic system and pre-run in 0.5x TAE at 110 V for 30 minutes. Rinse the wells with reservoir buffer and load 6 μL from each of the stopped reaction mixes. Perform gel-electrophoresis at 75 V for 2 hours.
-
15
After electrophoresis, transfer the gels onto Blotting paper (VWR) and cover with cling film. Dry the gel on a gel dryer at 80 °C for 5 minutes.
-
16
Expose the dried gel to storage phosphor screen. Suitable exposure times and scanning parameters that avoid over-exposure and pixel saturation will depend on the specific activity of the DNA substrate and need to be determined empirically.
-
17
Scan exposed screen on Typhoon FLA 7000 laser scanner.
-
18
Analyze the scanned images by ImageJ to quantify band intensities: Use the rectangular selection tool to draw rectangle around individual lanes, measure the intensity profile across each lane and plot profiles using the plot lanes tool. Each lane will feature two distinct peaks, corresponding to the substrate and unwound product bands (Figure 5). The intensity profiles are baseline corrected and the area under each of the bell-shaped intensity curves is determined. The values are then used to calculate the fraction of unwound product relative to the sum of substrate unwound product for each lane. Plot the values using GraphPad Prism software.
Figure 5: CMG helicase assay.
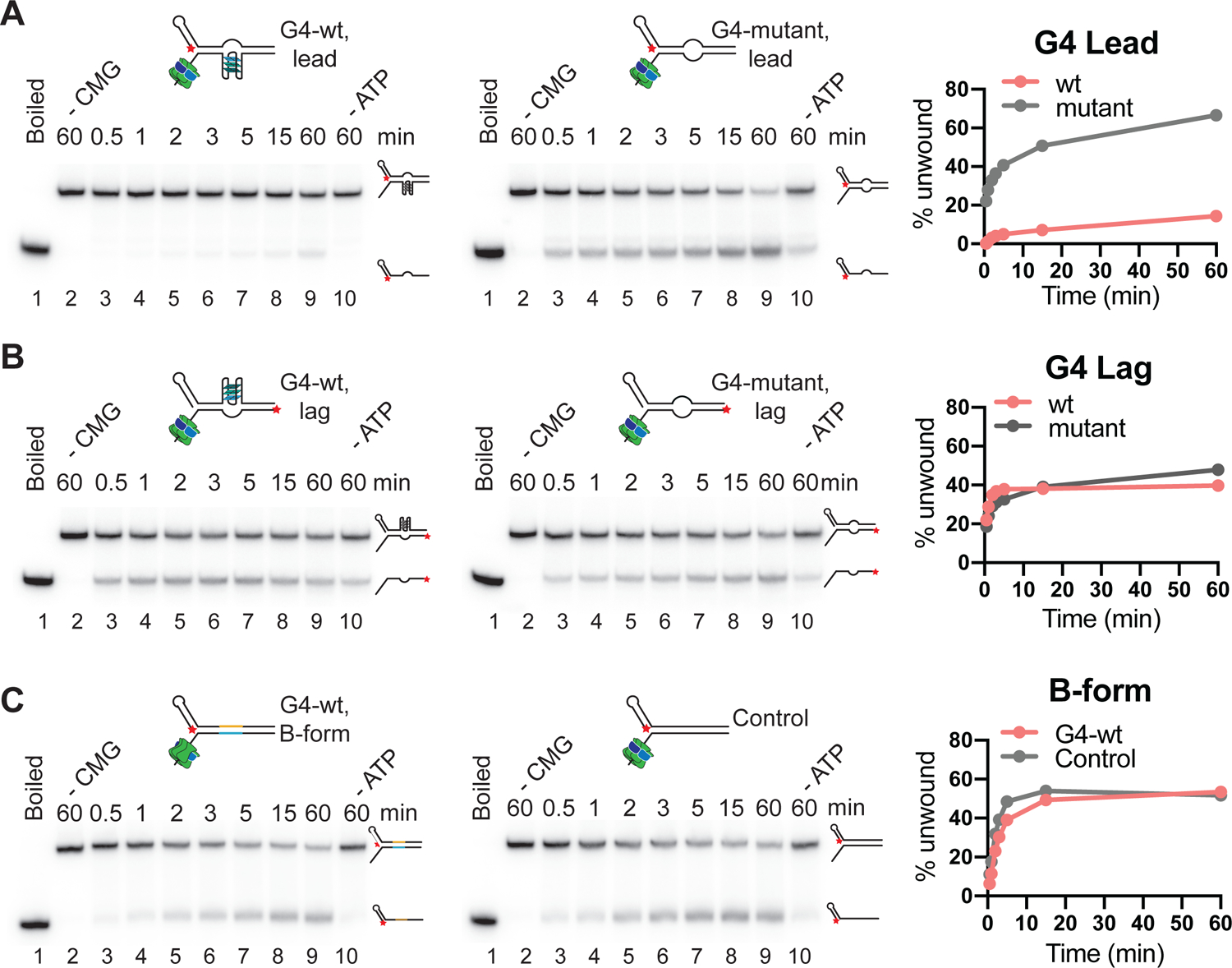
(A) Wildtype (left) or mutant (right) G4 on translocation strand; (B) Wildtype (left) or mutant (right) G4 on non-template strand. (C) Wildtype G4 sequence (left) or random DNA sequence (right) in B-form duplex DNA. Red star indicates the position of radiolabel. Reaction products were separated by native PAGE and imaged on a phosphor screen. Product formation is quantified using ImageJ and plotted using Prism 9 software.
Interpretation of results
Here, we have performed the unwinding assays on three types of substrates: 1) Wild-type or mutant G4 on the translocating strand (Figure 5A), 2) Wild-type or mutant G4 on the non-template strand (Figure 5B) and 3) B-form duplex DNA with or without the G4 sequence (Figure 5C). The ‘boiled’ control (lane 1 in respective gels) indicates the position of the unwound product, while the ‘no CMG’ controls in lane 2 of the respective gels indicate the position of the DNA substrate. When CMG is incubated with duplex DNA fork substrate (Figure 3E–F and Figure 5C), the unwound product accumulates over time, indicating substrate unwinding by CMG. The DNA unwinding activity of CMG is dependent on ATP, as can be inferred from ‘no ATP’ control in lane 10 (compare to lane 9). The DNA unwinding activity of CMG is unaltered when the G4 sequence is incorporated in the duplex DNA. However, if the G4 structure is present on the translocation strand (Figure 3A–B), unwinding is strongly inhibited (Figure 5A). Disruption of the G4 structure by mutation of the G4 sequence relieves the inhibition of DNA unwinding by CMG, demonstrating that the inhibition is specific for the G4 structure. In contrast, the same G4 structure does not pose an impediment to CMG if present on the non-template strand (Figure 5B).
Limitations
Bulk assays are limited in their ability to determine rates of helicase progression, as the observed DNA unwinding rates are a cumulative product of helicase progression rates and intermittent helicase pause or stall events. In addition, helicase loading onto the DNA substrate may be rate-limiting for product formation in this assay. To bypass the latter, we perform a CMG preloading step in the presence of the non-hydrolyzable ATP analogue, ATPγS. It has recently been noted that adenylyl-imidodiphosphate (AMP-PNP) may be more suitable for the preloading step as CMG has been observed to exhibit limited DNA unwinding activity also in the presence of ATPγS (Yao et al. 2022).
In the absence of competitor oligo-nucleotides that may trap free CMG after the initiation of DNA unwinding it is unclear if the extent of DNA unwinding observed over the course of the experiment is due to a single round of CMG loading onto the DNA substrate or whether it is the result of repeated initiation events. Under the conditions described here we have not noted a reduction in product formation in the presence of trapping oligo-nucleotides, indicating that repeated DNA unwinding events do not significantly contribute to the DNA unwinding activity observed here. However, depending on the exact reaction conditions (e.g. CMG:DNA ratio) trapping oligonucleotides may be desirable to eliminate the potential for repeated CMG loading onto the DNA substrate for a more quantitative read-out of activity.
5. Summary and conclusions
In this chapter we have presented a procedure to assess the DNA helicase activity of budding yeast CMG and to determine the impact of G-quadruplexes on this activity. For this, we have tested the activity of CMG on DNA substrates harboring a G4 on either the template or non-template strand (Figure 5). We observe that a G4 on the excluded non-tempate strand does not affect the CMG progression, which concurs with previous observations that bulky groups linked to the lagging strand can be readily bypassed by CMG (Fu et al. 2011; Kose et al. 2019). In contrast, a G4 on the template strand severely impedes DNA unwinding by CMG, suggesting that CMG is unable to resolve or bypass the G4 structure. Mutating a G-strand in the G4 forming sequence or pairing it with complementary sequence in B-form DNA relievies the inhibition of DNA unwinding by CMG, demonstrating that the inhibition of CMG activity is specific to the G4 structure and not the G4 sequence per se. The central channel of CMG has a variable width but can be ~ 25 Å wide (Yuan et al. 2016). This width is similar to the width of the G4 structure formed in the DNA substrates here (Do et al. 2011). The specific G4 structure formed here is symmetrical and can be approximated to a square cuboid with the plane parallel to the quartets forming a square (Figure 1). A quartet which is 25 Å wide will thus have a diagonal of √2×25 Å = ~ 35 Å. Therefore, depending on the orientation in which the G4 enters the inner channel and depending on its conformational flexibility, even a minimal G4 may overwhelm the CMG, thus warranting the need for other helicases to resolve this obstruction (Sato et al. 2021). Future studies involving assays as described here but with distinct G4 structures should help elucidate the structural determinants that govern the impact of G4s on CMG activity.
Acknowledgement
This work was supported by NIGMS grants R01-GM127428 and R01-GM107239 (DR) and NIH/NCI Cancer Center Support Grant P30 CA008748.
References
- Attali I, Botchan MR, and Berger JM. 2021. ‘Structural Mechanisms for Replicating DNA in Eukaryotes’, Annu Rev Biochem, 90: 77–106. [DOI] [PubMed] [Google Scholar]
- Bando M, Katou Y, Komata M, Tanaka H, Itoh T, Sutani T, and Shirahige K. 2009. ‘Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks’, Journal of Biological Chemistry, 284: 34355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretic D, Jenkyn-Bedford M, Aria V, Cannone G, Skehel M, and Yeeles JTP. 2020. ‘Cryo-EM Structure of the Fork Protection Complex Bound to CMG at a Replication Fork’, Molecular Cell, 78: 926–40 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedrat A, Lacroix L, and Mergny JL. 2016. ‘Re-evaluation of G-quadruplex propensity with G4Hunter’, Nucleic Acids Research, 44: 1746–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya D, Arachchilage GM, and Basu S. 2016. ‘Metal Cations in G-Quadruplex Folding and Stability’, Frontiers in Chemistry, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Tannahill D, McCafferty J, and Balasubramanian S. 2013. ‘Quantitative visualization of DNA G-quadruplex structures in human cells’, Nature Chemistry, 5: 182–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman ML, Paeschke K, and Zakian VA. 2012. ‘DNA secondary structures: stability and function of G-quadruplex structures’, Nat Rev Genet, 13: 770–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch PC, Segura-Bayona S, Koole W, van Heteren JT, Dewar JM, Tijsterman M, and Knipscheer P. 2014. ‘FANCJ promotes DNA synthesis through G-quadruplex structures’, Embo Journal, 33: 2521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AK, Bell MR, and Raney KD. 2018. ‘Pif1 helicase unfolding of G-quadruplex DNA is highly dependent on sequence and reaction conditions’, Journal of Biological Chemistry, 293: 17792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali F, Bharti SK, Di Perna R, Brosh RM Jr., and Pisani FM. 2016. ‘Tim/Timeless, a member of the replication fork protection complex, operates with the Warsaw breakage syndrome DNA helicase DDX11 in the same fork recovery pathway’, Nucleic Acids Research, 44: 705–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, and Balasubramanian S. 2015. ‘High-throughput sequencing of DNA G-quadruplex structures in the human genome’, Nature Biotechnology, 33: 877–+. [DOI] [PubMed] [Google Scholar]
- Chen MC, Tippana R, Demeshkina NA, Murat P, Balasubramanian S, Myong S, and Ferre-D’Amare AR. 2018. ‘Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36’, Nature, 558: 465–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan D, Tsirkas I, Dovrat D, Sparks MA, Singh SP, Galletto R, and Aharoni A. 2018. ‘Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures’, Nucleic Acids Research, 46: 11847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, and Michor F. 2011. ‘DNA secondary structures and epigenetic determinants of cancer genome evolution’, Nature Structural & Molecular Biology, 18: 950–U115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do NQ, Lim KW, Teo MH, Heddi B, and Phan AT. 2011. ‘Stacking of G-quadruplexes: NMR structure of a G-rich oligonucleotide with potential anti-HIV and anticancer activity’, Nucleic Acids Research, 39: 9448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas ME, Ali FA, Costa A, and Diffley JFX. 2018. 'The mechanism of eukaryotic CMG helicase activation', Nature, 555: 265–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J, Remus D, Mehanna A, and Diffley JF. 2013. ‘ATPase-dependent quality control of DNA replication origin licensing’, Nature, 495: 339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, and Walter JC. 2011. ‘Selective Bypass of a Lagging Strand Roadblock by the Eukaryotic Replicative DNA Helicase’, Cell, 146: 930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, and O’Donnell ME. 2014. ‘Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork’, Nature Structural & Molecular Biology, 21: 664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R, Yuan ZN, Bai L, Santos RDA, Sun JC, Zhang D, Yurieva O, Li HL, and O’Donnell ME. 2017. ‘Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation’, Proceedings of the National Academy of Sciences of the United States of America, 114: E697–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, and Weissman JS. 2003. ‘Global analysis of protein expression in yeast’, Nature, 425: 737–41. [DOI] [PubMed] [Google Scholar]
- Heddi B, Cheong VV, Martadinata H, and Phan AT. 2015. ‘Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: Solution structure of a peptide-quadruplex complex’, Proceedings of the National Academy of Sciences of the United States of America, 112: 9608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL, and Balasubramanian S. 2005. ‘Prevalence of quadruplexes in the human genome’, Nucleic Acids Research, 33: 2908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves I, Petojevic T, Pesavento JJ, and Botchan MR. 2010. ‘Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins’, Molecular Cell, 37: 247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kose HB, Larsen NB, Duxin JP, and Yardimci H. 2019. ‘Dynamics of the Eukaryotic Replicative Helicase at Lagging-Strand Protein Barriers Support the Steric Exclusion Model’, Cell Reports, 26: 2113–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar C, Batra S, Griffith JD, and Remus D. 2021. ‘The interplay of RNA:DNA hybrid structure and G-quadruplexes determines the outcome of R-loop-replisome collisions’, Elife, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston LD, Mayle R, Schauer GD, Yurieva O, Zhang D, Yao NY, Georgescu RE, and O’Donnell ME. 2017. ‘Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks’, Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston L, and O’Donnell M. 2017. ‘Action of CMG with strand-specific DNA blocks supports an internal unwinding mode for the eukaryotic replicative helicase’, Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner LK, Holzer S, Kilkenny ML, Svikovic S, Murat P, Schiavone D, Eldridge CB, Bittleston A, Maman JD, Branzei D, Stott K, Pellegrini L, and Sale JE. 2020. ‘Timeless couples G-quadruplex detection with processing by DDX11 helicase during DNA replication’, Embo Journal, 39: e104185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner LK, and Sale JE. 2019. ‘Replication of G Quadruplex DNA’, Genes, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsico G, Chambers VS, Sahakyan AB, McCauley P, Boutell JM, Di Antonio M, and Balasubramanian S. 2019. ‘Whole genome experimental maps of DNA G-quadruplexes in multiple species’, Nucleic Acids Research, 47: 3862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukundan VT, and Phan AT. 2013. ‘Bulges in G-Quadruplexes: Broadening the Definition of G-Quadruplex-Forming Sequences’, Journal of the American Chemical Society, 135: 5017–28. [DOI] [PubMed] [Google Scholar]
- Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, and Zakian VA. 2013. ‘Pif1 family helicases suppress genome instability at G-quadruplex motifs’, Nature, 497: 458–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Botchan MR, and Berger JM. 2017. ‘Mechanisms and regulation of DNA replication initiation in eukaryotes’, Crit Rev Biochem Mol Biol, 52: 107–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson GN, Lee MPH, and Neidle S. 2002. ‘Crystal structure of parallel quadruplexes from human telomeric DNA’, Nature, 417: 876–80. [DOI] [PubMed] [Google Scholar]
- Petojevic T, Pesavento JJ, Costa A, Liang JD, Wang ZJ, Berger JM, and Botchan MR. 2015. ‘Cdc45 (cell division cycle protein 45) guards the gate of the Eukaryote Replisome helicase stabilizing leading strand engagement’, Proceedings of the National Academy of Sciences of the United States of America, 112: E249–E58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Adrian M, Samazan F, Heddi B, Hamon F, Serero A, Lopes J, Teulade-Fichou MP, Phan AT, and Nicolas A. 2015. ‘Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites’, Embo Journal, 34: 1718–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posse V, Johansson E, and Diffley JFX. 2021. ‘Eukaryotic DNA replication with purified budding yeast proteins’, Methods Enzymol, 661: 1–33. [DOI] [PubMed] [Google Scholar]
- Ribeyre C, Lopes J, Boule JB, Piazza A, Guedin A, Zakian VA, Mergny JL, and Nicolas A. 2009. ‘The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo’, PLoS Genet, 5: e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Reams C, Simpson LJ, and Sale JE. 2010. ‘Epigenetic instability due to defective replication of structured DNA’, Molecular Cell, 40: 703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Martin-Pintado N, Post H, Altelaar M, and Knipscheer P. 2021. ‘Multistep mechanism of G-quadruplex resolution during DNA replication’, Science Advances, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, and Wigley DB. 2007. ‘Structure and mechanism of helicases and nucleic acid translocases’, Annu Rev Biochem, 76: 23–50. [DOI] [PubMed] [Google Scholar]
- Spiegel J, Adhikari S, and Balasubramanian S. 2020. ‘The Structure and Function of DNA G-Quadruplexes’, Trends Chem, 2: 123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyjka SJ, Viggiani CJ, and Aparicio OM. 2005. ‘Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae’, Molecular Cell, 19: 691–7. [DOI] [PubMed] [Google Scholar]
- Todd AK, Johnston M, and Neidle S. 2005. ‘Highly prevalent putative quadruplex sequence motifs in human DNA’, Nucleic Acids Research, 33: 2901–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourriere H, Versini G, Cordon-Preciado V, Alabert C, and Pasero P. 2005. ‘Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53’, Molecular Cell, 19: 699–706. [DOI] [PubMed] [Google Scholar]
- Vannier JB, Sandhu S, Petalcorin MIR, Wu XL, Nabi Z, Ding H, and Boulton SJ. 2013. ‘RTEL1 Is a Replisome-Associated Helicase That Promotes Telomere and Genome-Wide Replication’, Science, 342: 239–42. [DOI] [PubMed] [Google Scholar]
- Voter AF, Qiu YP, Tippana R, Myong S, and Keck JL. 2018. ‘A guanine-flipping and sequestration mechanism for G-quadruplex unwinding by RecQ helicases’, Nature Communications, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij PH, Uhler JP, Shi Y, Westerlund F, Falkenberg M, and Gustafsson CM. 2012. ‘A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop’, Nucleic Acids Research, 40: 10334–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao NY, Zhang D, Yurieva O, and O’Donnell ME. 2022. ‘CMG helicase can use ATPgammaS to unwind DNA: Implications for the rate-limiting step in the reaction mechanism’, Proc Natl Acad Sci U S A, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardimci H, Loveland AB, Habuchi S, van Oijen AM, and Walter JC. 2010. ‘Uncoupling of Sister Replisomes during Eukaryotic DNA Replication’, Molecular Cell, 40: 834–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Janska A, Early A, and Diffley JFX. 2017. ‘How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication’, Molecular Cell, 65: 105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZN, Bai L, Sun JC, Georgescu R, Liu J, O’Donnell ME, and Li HL. 2016. ‘Structure of the eukaryotic replicative CMG helicase suggests a pumpjack motion for translocation’, Nature Structural & Molecular Biology, 23: 217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JC, Janska A, Goswami P, Renault L, Abid Ali F, Kotecha A, Diffley JFX, and Costa A. 2017. ‘CMG-Pol epsilon dynamics suggests a mechanism for the establishment of leading-strand synthesis in the eukaryotic replisome’, Proc Natl Acad Sci U S A, 114: 4141–46. [DOI] [PMC free article] [PubMed] [Google Scholar]