

Abstract
PURPOSE OF REVIEW:
This article reviews many of the complex facets of behavioral variant frontotemporal dementia (bvFTD) and frontotemporal lobar degeneration (FTLD). A particular focus is on improving diagnostic accuracy to reduce the arduous diagnostic odyssey that so many patients and families endure. Strategies to promote diagnostic accuracy and approach the management of problematic symptoms are also discussed.
RECENT FINDINGS:
Although the International Consensus Criteria for bvFTD were published more than a decade ago and clinicopathologic studies have confirmed their utility, diagnostic confusion continues. This article presents updated data along with illustrative cases to emphasize the clinical pearls that are most useful for clinicians. Although accurate prediction of the underlying proteinopathy remains a challenge, the ability to differentiate bvFTD from atypical Alzheimer disease, psychiatric disorders, and other mimickers has improved. Knowledge about the genetic underpinnings in a significant minority of individuals with familial FTLD is enabling early and accurate diagnosis. Therapeutic optimism has also increased, particularly in familial FTLD, with a few clinical trials in progress and several more planned, some of which are designed to slow progression or delay the onset of symptoms, or both.
SUMMARY:
The diagnosis and management of bvFTD is challenging for clinicians and particularly for patients and their families. Although much progress has been gained over recent years, several key research questions persist. Treatments that significantly improve symptoms or alter the course of FTLD remain elusive, but optimism is increasing as pathobiology is better understood and novel therapies are being developed.
INTRODUCTION
Frontotemporal dementia (FTD) is a collective term that encompasses a group of clinical syndromes caused by an underlying neurodegenerative disease characterized by progressive changes in behavior, executive function, or language.1 The specific syndromes under the FTD umbrella include behavioral variant FTD (bvFTD) and primary progressive aphasia (PPA). The group of neurodegenerative diseases that manifest clinically as FTD are known as the frontotemporal lobar degeneration (FTLD) spectrum pathologies, and these disorders reflect degeneration in the frontotemporal neural networks. This article focuses on bvFTD and the associated FTLD disorders.
Considerable overlap exists in the clinical and associated features of bvFTD with Alzheimer disease, which causes considerable confusion (CASE 3-1). This article uses the term Alzheimer disease (AD) when referring to the histopathologically defined disease and the term AD dementia when referring to the multidomain dementia syndrome that is commonly associated with, but not specific for, underlying AD.
CASE 3–1.
A 58-year-old attorney was laid off from her legal firm because of performance concerns, as she was unable to complete the tasks relating to her profession. She could not recall details of conversations with clients and with colleagues in meetings. She was anxious and occasionally accused her siblings of trying to coerce her to give them money. Her family was most concerned about her difficulties in making decisions, poor judgment, slowed thought processing, and difficulties expressing her thoughts, yet her comprehension of others appeared intact and geographic orientation was relatively intact. Apathy was prominent. She was not paying some of her bills and not taking some of her medications as prescribed. She had no symptoms of reduced empathy/sympathy, disinhibition, perseveration, or dietary changes. Her family history included a grandparent with late-onset dementia.
She was evaluated by a neurologist, who found no abnormalities on general neurologic examination. She scored 26/30 on the Mini-Mental State Examination (MMSE). Neuropsychological assessment revealed impairment in measures assessing executive function, speeded attention, and verbal fluency, with relatively normal performance on delayed recall and visuospatial tasks. Laboratory studies were unrevealing. MRI of the brain was interpreted as showing frontal atrophy. She was diagnosed with behavioral variant frontotemporal dementia, and her family sought another opinion.
A subsequent neurologic evaluation occurred 3 months later. She scored 25/30 on the MMSE and had obvious psychomotor slowing. She had mild bradykinesia but no rigidity, tremor, or postural instability and no features of amyotrophic lateral sclerosis. MRI and fludeoxyglucose positron emission tomography of her brain were obtained (FIGURE 3-1). The MRI showed frontal more than parietal atrophy but minimal hippocampal atrophy (FIGURES 3-1A and 3-1B), and FDG-PET showed frontal more than temporal, parietal, and posterior cingulate hypometabolism (FIGURE 3-1E). She refused CSF examination. Amyloid PET imaging (not shown) demonstrated prominent uptake in the frontal, parietal, and temporal cortex consistent with amyloid-β deposition. She was diagnosed with a progressive dysexecutive dementia syndrome due to underlying Alzheimer disease and was prescribed a cholinesterase inhibitor.
Serial evaluations over the following 3 years showed progressive dementia and associated behavioral dyscontrol that responded minimally to various psychotropic agents. Bradykinesia increased as well, but no other elements of parkinsonism evolved. Brain MRI at age 62 showed progression of frontal and parietal atrophy but minimal hippocampal atrophy (FIGURES 3-1C and 3-1D). She died at age 66, and autopsy revealed widespread neocortical neuritic plaques and neurofibrillary tangles consistent with Alzheimer disease, without evidence of any other proteinopathy.
FIGURE 3–1.
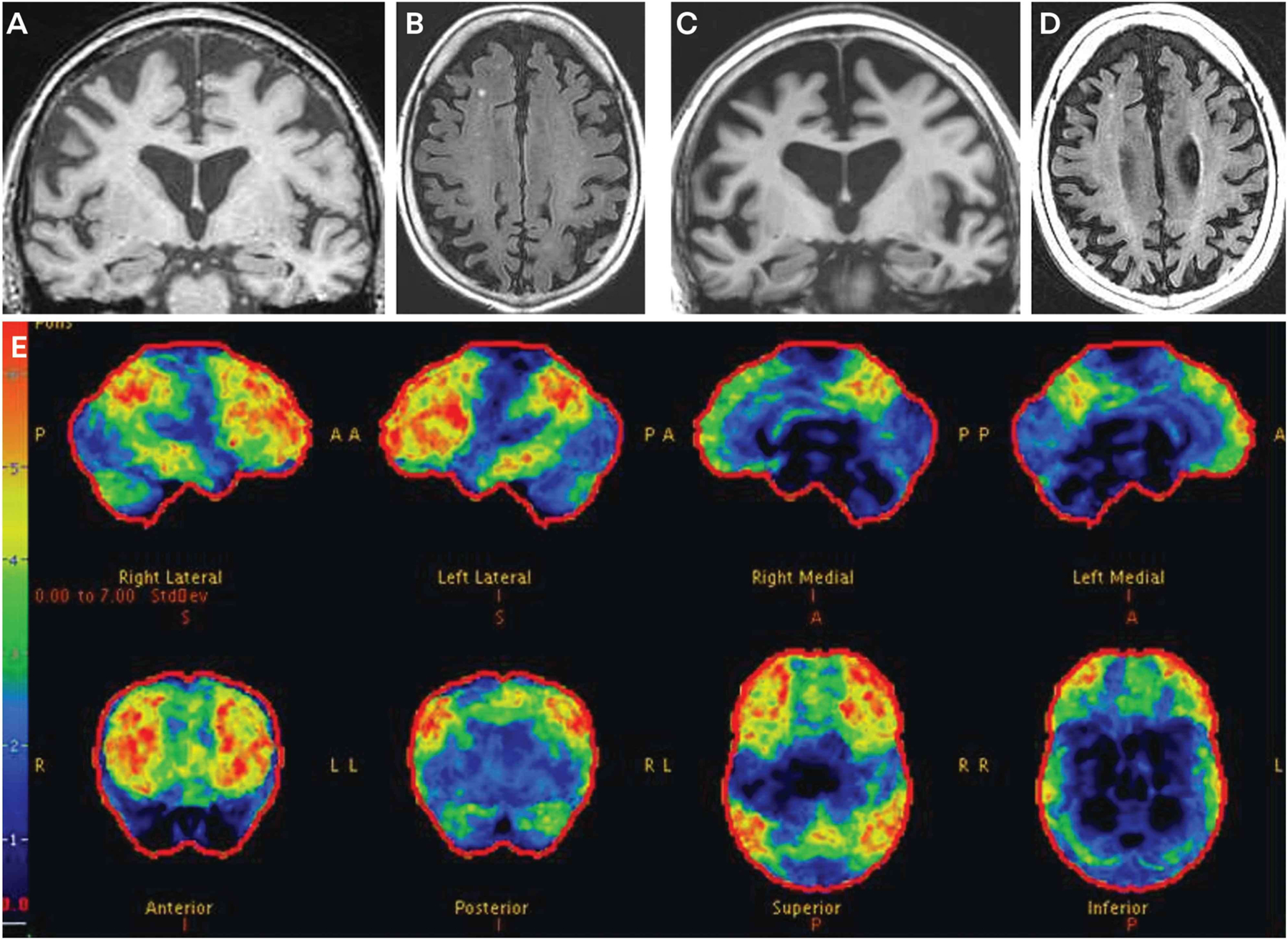
Imaging of the patient in CASE 3-1. Coronal T1-weighted (A) and axial fluid-attenuated inversion recovery (FLAIR) (B) MRI at age 58 show frontal more than parietal atrophy, but minimal hippocampal atrophy. Coronal T1-weighted (C) and axial FLAIR (D) MRI at age 62 show progression of the frontal and parietal atrophy and only minimal hippocampal atrophy. Fludeoxyglucose positron emission tomography (FDG-PET) (E) at age 58 shows marked hypometabolism in the frontal regions, with slightly less prominent hypometabolism in the temporal and parietal regions and the posterior cingulate region. The color bar and z score reference on the far left of the FDG-PET image reflects the degree of FDG hypometabolism, with blue reflecting z scores in the −1 to −2 range and red reflecting a z score of −6. Therefore, the regions with gray or black color reflect normal metabolism, blue color reflects slight to mild hypometabolism, and green to yellow to red color reflects moderate to very severe hypometabolism.
COMMENT
This case exemplifies the diagnostic challenges in differentiating the syndromes of behavioral variant frontotemporal dementia from Alzheimer disease dementia and how biomarkers are useful in predicting the underlying disease.
EPIDEMIOLOGY AND DEMOGRAPHICS
FTD was considered very rare and primarily an early-onset form of dementia years ago, but more recent epidemiologic studies have altered those views. Reported incidence rates range from 1 per 100,000 person-years to 8 per 100,000 person-years,2 and prevalence rates range from 2 per 100,000 to 20 per 100,000.3,4 The incidence and prevalence increase with age and plateau or decrease in the seventies or eighties, depending on the study population; a significant minority of individuals with late-onset dementia have FTD and/or FTLD pathology, or both.5 No convincing sex predilection is seen based on available data. The vast majority of patients with FTD described to date have been White, with far fewer being characterized as Asian or Hispanic.3,6 Very few patients with FTD of African ancestry have been described. Whether these differences in incidence across ethnic and racial groups represent sampling biases, biologic differences, or other factors is not adequately understood.
IMPACT ON PATIENTS/FAMILIES AND SOCIETY
The impact of bvFTD and related disorders is profound. Many potential consequences of altered behavior and cognition during the prediagnosis and postdiagnosis phases of the illness may be seen, including (but not limited to) fractured relationships with family members and friends; divorce; poor job performance and loss of one’s job and the associated loss of income, medical insurance, and pension; sexual indiscretion; gambling or excessive spending; financial devastation because of poor business decisions; and tragic accidents, with major repercussions for patients, their family members, and society. One large survey of caregivers of patients with bvFTD and related disorders found a reduced overall household income after diagnosis, a decline in caregiver health, an increase in caregiver health-related costs, and an increase in patient-related costs (often because of crises).7 These costs are approximately twice as high as the reported costs for AD dementia.7 Remarkably, caregivers rated the quality of life of their loved ones with bvFTD as “worse than dead.”7 These observations underscore the need for improved early diagnosis, better strategies for managing symptoms and associated costs, and, obviously, prevention.
SPORADIC AND FAMILIAL FRONTOTEMPORAL LOBAR DEGENERATION
The majority of patients with bvFTD have an apparently sporadic disorder, with no relevant family history of neurodegenerative disease and no identifiable mutation in any of the known FTLD-associated genes. The mechanisms underlying sporadic bvFTD remain a mystery, and this is a high-priority research issue. At least 20% (and it may be more on the order of 30% to 40%) of patients with bvFTD or similar phenotype have a dominantly inherited familial disorder, commonly termed familial FTLD and abbreviated f-FTLD.8 The most common mutations associated with f-FTLD occur in the microtubule associated protein tau (MAPT) gene,9 the progranulin (GRN) gene,10 and the chromosome 9 open reading frame 72 (C9orf72)11,12 gene; these are sometimes referred to as the big three genes of FTD. These mutations together account for at least 50% of f-FTLD, and penetrance is greater than 95% for MAPT and 70% to 90% for GRN and C9orf72.13 Although many similarities exist in the clinical, neuropsychiatric, and imaging features of sporadic and familial FTLD,6 relatively unique clinical and particularly imaging and pathologic features are also seen in those with f-FTLD with mutations in MAPT, GRN, or C9orf72.14,15 Mutations in more than a dozen other genes have been associated with f-FTLD (each with penetrance at least 70% based on available data), with another new gene seemingly discovered each year. Readers are encouraged to access other excellent sources on f-FTLD and genetics.13,16 Genetic testing is discussed later in this article.
DIAGNOSTIC CRITERIA
The initial criteria for bvFTD were published in 1998,17 and the criteria were updated in 2011 (TABLE 3-1).18 These updated criteria, known as the International Consensus Criteria for bvFTD, have been commonly used in clinical practice and research programs because of their relative simplicity and applicability. One of the main goals of the group establishing these criteria was to achieve adequate sensitivity and specificity for the bvFTD phenotype that is associated with FTLD pathology. Applying the consensus criteria for bvFTD is useful in clinical practice.
TABLE 3–1.
International Consensus Criteria for Behavioral Variant Frontotemporal Dementiaa
|
I Neurodegenerative disease The following symptom must be present to meet criteria for behavioral variant frontotemporal dementia (bvFTD): A Shows progressive deterioration of behavior and/or cognition by observation or history (as provided by a knowledgeable informant) II Possible bvFTD Three of the following behavioral/cognitive symptoms (A-F) must be present to meet criteria; ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events: A Earlyb behavioral disinhibition (one of the following symptoms [A1-A3] must be present): A1 Socially inappropriate behavior A2 Loss of manners or decorum A3 Impulsive, rash, or careless actions B Early apathy or inertia (one of the following symptoms [B1-B2] must be present): B1 Apathy B2 Inertia C Early loss of sympathy or empathy (one of the following symptoms [C1-C2] must be present): C1 Diminished response to other people’s needs and feelings C2 Diminished social interest, interrelatedness, or personal warmth D Early perseverative, stereotyped, or compulsive/ritualistic behavior (one of the following symptoms [D1-D3] must be present): D1 Simple repetitive movements D2 Complex, compulsive, or ritualistic behaviors D3 Stereotypy of speech E Hyperorality and dietary changes (one of the following symptoms [E1-E3] must be present): E1 Altered food preferences E2 Binge eating, increased consumption of alcohol or cigarettes E3 Oral exploration or consumption of inedible objects F Neuropsychological profile: executive/generation deficits with relative sparing of memory and visuospatial functions (all of the following symptoms [F1-F3] must be present): F1 Deficits in executive tasks F2 Relative sparing of episodic memory F3 Relative sparing of visuospatial skills III Probable bvFTD All of the following symptoms (A-C) must be present to meet criteria: A Meets criteria for possible bvFTD B Exhibits significant functional decline (by caregiver report or as evidenced by Clinical Dementia Rating scale or Functional Activities Questionnaire scores) C Imaging results consistent with bvFTD (one of the following [C1-C2] must be present): C1 Frontal and/or anterior temporal atrophy on MRI or CT C2 Frontal and/or anterior temporal hypoperfusion or hypometabolism on PET or SPECT IV bvFTD with definite frontotemporal lobar degeneration (FTLD) pathology Criterion A and either criterion B or C must be present to meet criteria: A Meets criteria for possible or probable bvFTD B Histopathologic evidence of FTLD on biopsy or postmortem C Presence of a known pathogenic mutation V Exclusionary criteria for bvFTD Criteria A and B must be answered negatively for any bvFTD diagnosis; criterion C can be positive for possible bvFTD but must be negative for probable bvFTD: A Pattern of deficits is better accounted for by other nondegenerative nervous system or medical disorders B Behavioral disturbance is better accounted for by a psychiatric diagnosis C Biomarkers strongly indicative of Alzheimer disease or other neurodegenerative process |
CT = computed tomography; MRI = magnetic resonance imaging; PET = positron emission tomography; SPECT = single-photon emission computed tomography.
Modified with permission from Rascovsky K, et al, Brain.18 © 2011 Oxford University Press.
“Early” generally refers to symptom presentation within the first 3 years.
DIAGNOSTIC TOOLS AND APPROACH
In a 2020 study of patients with bvFTD, more than 40% required more than 1 year to establish a diagnosis, more than 60% were evaluated by three or more clinicians, and more than 80% required three or more visits before an FTD diagnosis was established.19 The initial diagnosis was depression or another psychiatric condition in more than 20% of patients.19 These delays and misdiagnoses are part of the so-called diagnostic odyssey of FTD.
Laboratory testing, neuropsychological testing, and structural neuroimaging are recommended for evaluating individuals with mild cognitive impairment and dementia.20 The following discussion presents concepts and data related to the tests most pertinent to bvFTD.
Clinical Features
Any patient with one or more of the core clinical features for possible bvFTD, such as behavioral disinhibition; apathy or inertia; loss of sympathy or empathy; perseverative, stereotyped, or compulsive/ritualistic behavior; or hyperorality and dietary changes (TABLE 3-1), should raise suspicion for bvFTD (CASE 3-2). Interviewing family members separately from the patient may be indicated to more thoroughly elicit features that are often awkward to discuss in the patient’s presence. In some circumstances, clues to a genetic association exist (eg, bizarre delusions and/or hallucinations are rare in bvFTD but, when present, raise suspicion for C9orf72-associated bvFTD) (CASE 3-3).22,23
CASE 3–2.
A 58-year-old minister presented with a 1-year history of progressive personality/behavior changes. He had begun acting sillier and more childish, exhibiting socially inappropriate behavior. He was more apathetic and quieter yet would laugh much more at odd situations. He had become more focused on particular forms of music and had begun gambling, which resulted in the loss of large sums of money. He ate the same meals for breakfast (a particular cereal plus one banana) and lunch (two peanut butter and raspberry jam sandwiches) each day and would get upset if his meal plan was altered in any way. He did not exhibit his usual sense of empathy/sympathy toward family members and members of his congregation. His usual sleep schedule had changed from sleeping 7 hours per night to 4 hours per night. No obvious changes in his cognition were appreciated by his family members, but they and members of his church were greatly concerned about the changes in behavior. He did not understand the concerns expressed by others and repeatedly stated that he felt fine and nothing was wrong. He was otherwise in good general medical health. His mother had developed dementia in her seventies.
The initial evaluation by his primary care provider and then psychiatrist led to the diagnosis of atypical bipolar disorder, and his behavioral features seemed to respond to valproic acid, but the gregariousness and inappropriate comments toward others escalated again 6 months later. He then developed increasing forgetfulness and elements of executive dysfunction. He missed appointments and social engagements. He sometimes forgot details of recent events and was unable to deliver his sermons without referring to notes. He was also unable to manage the financial and organizational complexities of his ministry. His primary care provider referred him for neuropsychological and neurologic evaluation.
Neuropsychological assessment was notable for mild impairment in delayed recall, language, and executive function. His neurologic examination was considered normal, including the mental status examination with a Mini-Mental State Examination (MMSE) score of 25/30, although zero items were recalled on delay. Laboratory studies were unrevealing. Brain MRI was interpreted as showing mild right more than left hippocampal atrophy (not shown). He was diagnosed with clinically probable Alzheimer disease, advised to discontinue working as a minister and discontinue driving, and started on donepezil. His family subsequently believed that the apathy and memory improved following donepezil.
Three months later, he drove the family car without his family’s knowledge and crashed it into a tree. He had no apparent head injury.
The family sought another neurologic opinion 6 months after the initial neurologic evaluation. This examination revealed a gregarious and socially inappropriate man with no significant language impairment and mild impairment on the MMSE, with a score of 23/30. Other than sparse fasciculations in both gastrocnemius muscles, the general neurologic examination was normal. Updated brain MRI showed atrophy in the right amygdala and mesial frontal regions (FIGURE 3-2). EMG showed no evidence of motor neuron disease.
The patient was diagnosed with behavioral variant frontotemporal dementia (bvFTD), and he and his family were counseled on management, prognostic, and psychosocial issues. Escitalopram was prescribed in an attempt to decrease the problematic behavioral issues, which was dramatically successful, and donepezil was subsequently discontinued.
He was followed annually in person for the next 8 years (serial MRI scans at ages 61, 64, and 66 are shown in FIGURE 3-2) and then by phone for the subsequent 7 years until his death at age 73. He developed a profound affinity for sweets and gained more than 100 pounds early in his course; pharmacotherapy did not impact this issue, and his family had to resort to chaining and locking the refrigerator and locking cupboards. Behavioral dyscontrol was very challenging to manage for several years despite multiple pharmacologic manipulations. He developed significant parkinsonism by age 71, which complicated his caregiver’s ability to assist with ambulation, dressing, and transfers; carbidopa/levodopa provided a modest but noticeable improvement. He was bedridden for the final months of his life.
Neuropathologic examination revealed marked frontotemporal atrophy with Pick bodies, ballooned neurons, and glial inclusions in the neocortex, medial temporal structures, and midbrain.21 Evidence of brainstem-predominant Lewy body disease was also seen, with Lewy bodies in the substantia nigra and moderate associated neuronal cell loss. No Alzheimer disease neuropathologic changes were seen.
FIGURE 3–2.
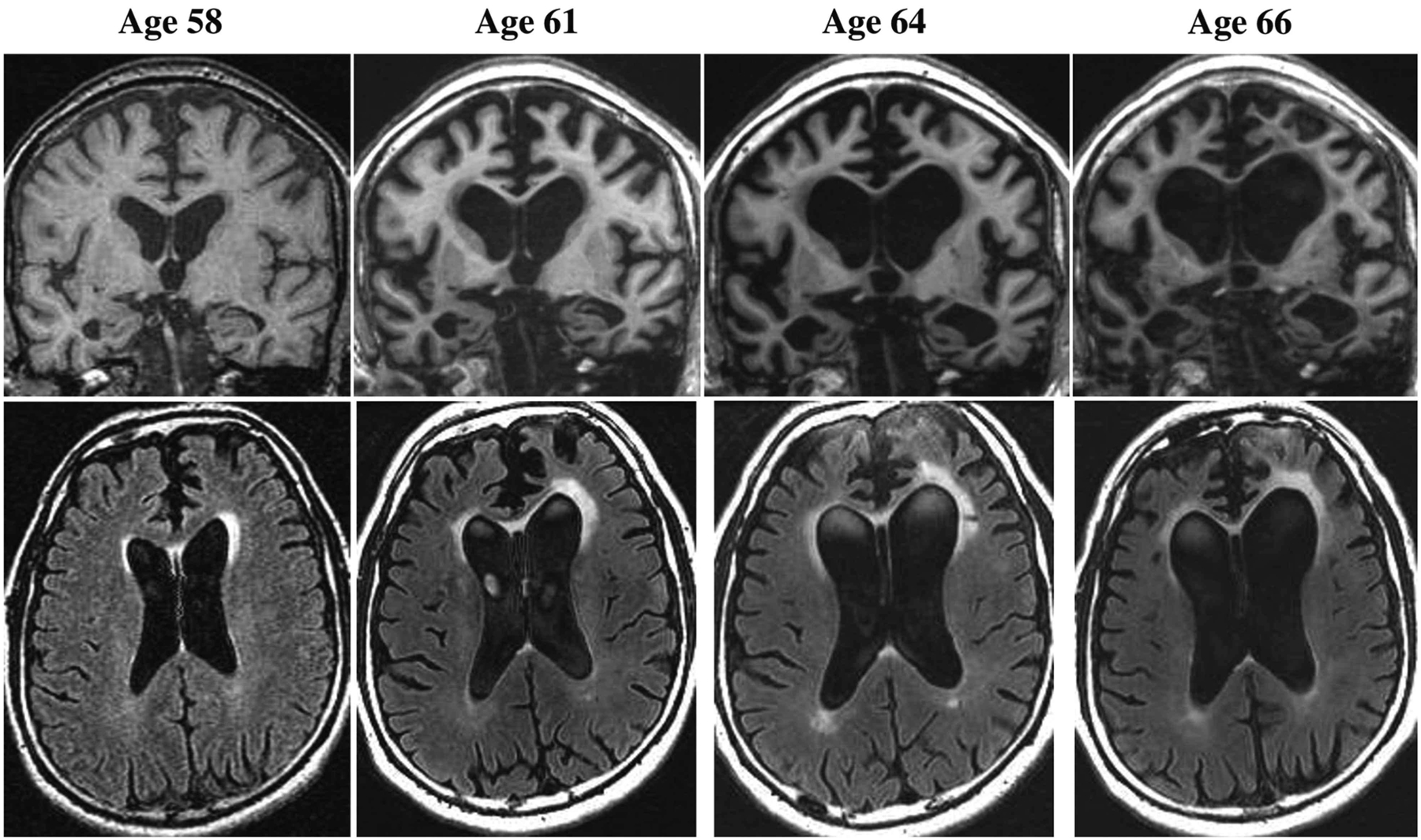
Imaging of the patient in CASE 3-2. Representative coronal T1-weighted (top row) and axial fluid-attenuated inversion recovery (FLAIR) (bottom row) images at ages 58, 61, 64, and 66. The MRI at age 58 shows atrophy in the right amygdala and mesial frontal regions, along with mildly increased signal on FLAIR in the left more than right anterior periventricular regions, including the anterior corpus collosum. Over time, progressive atrophy in the frontal and temporal regions and associated ventricular dilatation are apparent, along with expansion of the increased signal particularly in the left anterior periventricular region.
COMMENT
This case illustrates many of the typical clinical and MRI features of bvFTD as well as the challenges families and clinicians face when attempting to manage behavioral problems. Pick disease has long been considered as the prototypical histopathology associated with the syndrome of bvFTD, but autopsy studies indicate that among the frontotemporal lobar degeneration spectrum of diseases, Pick disease is relatively rare.
CASE 3–3.
A 60-year-old woman who lived alone repeatedly expressed concern to her daughter that she was unable to find certain items in her home, such as a particular item of clothing, jewelry, kitchen utensils, and money. She was convinced that someone was burglarizing her home. She repeatedly called local law enforcement for a suspected intruder, but no evidence of a break-in was ever found, leading the officers to suggest to the daughter that a medical evaluation may be warranted. Months earlier, she had also told her family that she was approved by her employer for early retirement. She adamantly stated to her family that she had ample financial resources and would soon be receiving a pension, having worked at her previous employer for more than 30 years.
Initial evaluation by her primary care provider did not elicit any suggestion of cognitive decline. She had been paying her bills and shopping independently and had not had any car accidents. She showed no symptoms of depression. A psychiatric consultation was arranged, which elicited a much more elaborate description of delusions and paranoia. The psychiatrist noted that the patient’s father had been diagnosed with amyotrophic lateral sclerosis in his sixties. She scored 25/30 on the Montreal Cognitive Assessment (MoCA). Other than the delusions, no other elements of psychosis were present, and her mood and affect appeared within normal limits. A neurologic consultation was arranged.
The neurologist interviewed the patient and the daughter separately, and the patient felt that everything was normal for her other than the intruders who were stealing items from her home. Her daughter reported other behavioral changes, such as a tendency to be a bit more outgoing toward strangers, a reduced interest in socializing with family and friends, and a few inappropriate comments made to others in stores and restaurants. No convincing changes in cognition were voiced by the daughter. The general neurologic examination was within normal limits. The neurologist was suspicious for evolving behavioral variant frontotemporal dementia considering the behavioral features. Considering her family history, familial frontotemporal lobar degeneration (FTLD) was also a concern, possibly associated with the C9orf72 hexanucleotide expansion. Additional diagnostic studies were sought. The patient refused to undergo a neuropsychological assessment. Brain MRI was obtained (FIGURES 3-3A through 3-3D), which revealed mild atrophy along the falx anteriorly (FIGURES 3-3A, and 3-3B). Fludeoxyglucose positron emission tomography (FDG-PET) showed convincing evidence of bilateral frontal hypometabolism (FIGURE 3-3E). She was diagnosed with mild cognitive/behavioral impairment due to underlying FTLD. Genetic counseling and genetic testing were then arranged, leading to the identification of >500 GGGGCC repeats in one allele of the C9orf72 gene.
Her daughter subsequently learned that her mother’s medical insurance had been discontinued upon her leaving her job. She no longer had any source of income, and, to her family’s surprise, the balances in her checking and savings accounts were low. The patient admitted to giving large donations to people she barely knew, and she had been taken in by telephone and mail scams.
Her former employer was approached about disability coverage, and her daughter learned that the patient had not been meeting productivity expectations in the workplace and was repeatedly reprimanded, but she had never been terminated. Since she had decided to quit her job, the employer argued that she could not receive any pension. After repeated attempts by her family and the clinicians involved in her care to support the premise that evolving familial FTLD was underlying her performance issues at work and led to her decision to quit, the employer agreed to support long-term disability.
The patient underwent annual neurologic evaluations, in which clinical progression was rapidly progressive. Her neuropsychiatric features were managed with various psychotropic agents, with limited apparent benefit. Follow-up MRI at age 63 showed progressive frontotemporal cortical atrophy (FIGURES 3-3C and 3-3D). She became progressively less verbal and was moved into a skilled nursing care facility. No features of parkinsonism or amyotrophic lateral sclerosis evolved. She died at age 64, and neuropathologic examination revealed FTLD with transactive response DNA-binding protein 43 (TDP-43) pathology in the brain.
FIGURE 3–3.
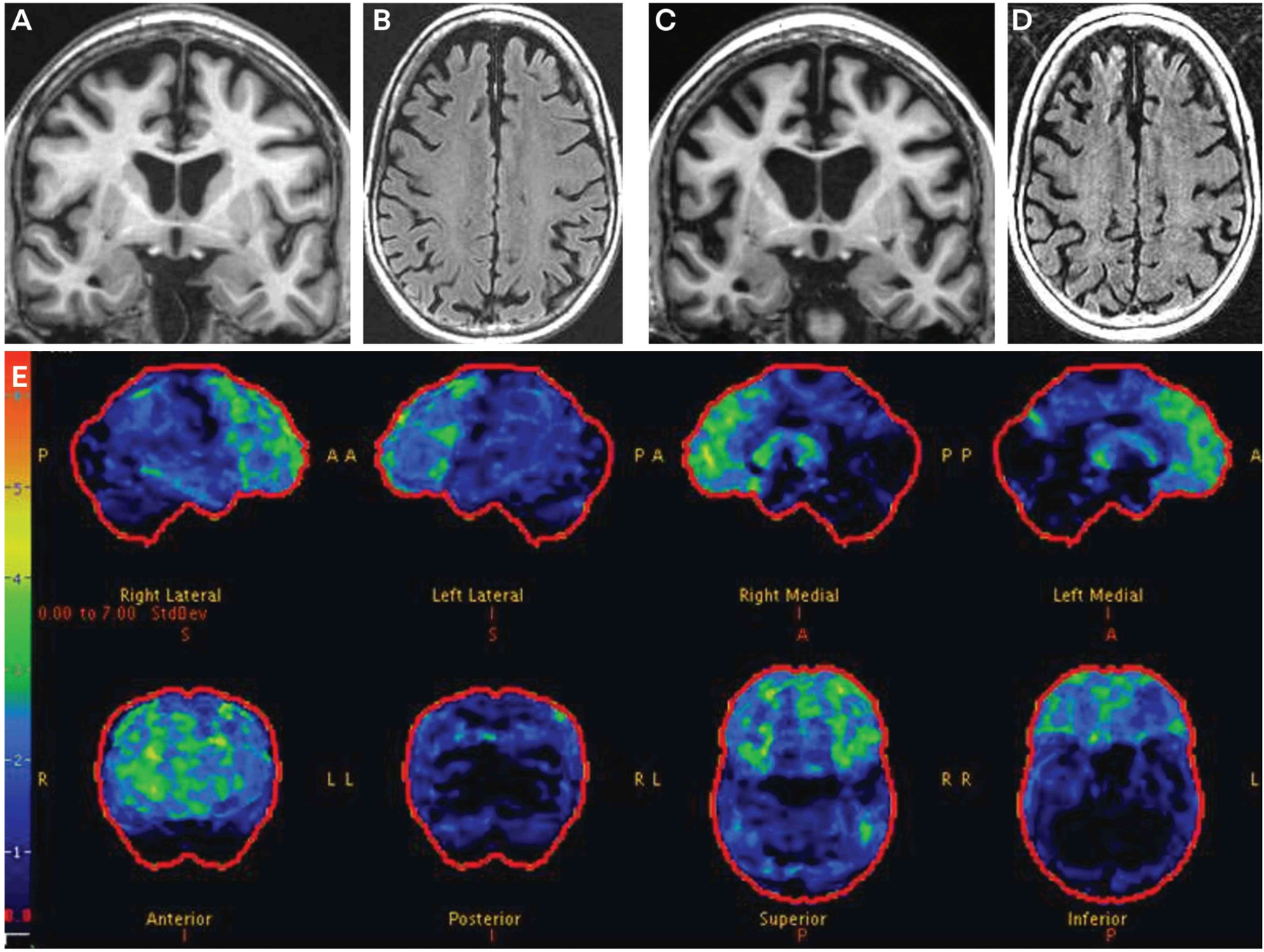
Imaging of the patient in CASE 3-3. Coronal T1-weighted (A) and axial fluid-attenuated inversion recovery (FLAIR) MRI (B) at age 61 show obvious frontal atrophy and more subtle amygdala atrophy. Coronal T1-weighted (C) and axial FLAIR MRI (D) at age 63 show progression of the frontal atrophy, with more obvious temporal atrophy. The lateral ventricles have also increased in size. Fludeoxyglucose positron emission tomography (FDG-PET) (E) at age 61 shows mild to moderate hypometabolism in the frontal regions. The color bar and z score reference on the far left of the FDG-PET image reflects the degree of FDG hypometabolism, with blue reflecting z scores in the −1 to −2 range and red reflecting a z score of −6. Therefore, the regions with gray or black color reflect normal metabolism, blue color reflects slight to mild hypometabolism, and green to yellow to red color reflects moderate to very severe hypometabolism.
COMMENT
Several key points are illustrated in this case. A primarily “psychiatric” presentation can occur early in the course of FTLD, and the impact on interpersonal relationships, employment, and financial status can be devastating. The findings on FDG-PET are often unquestionably supportive of a frontotemporal dementia diagnosis, and the presence of a family history of amyotrophic lateral sclerosis raises suspicion of a genetically mediated process, particularly an expansion in the C9orf72 gene.
Another scenario in bvFTD is the onset of behavioral/cognitive changes at a very young age (CASE 3-4). Although this is rather rare in bvFTD, it often makes an accurate diagnosis even more challenging to establish.
CASE 3–4.
A 27-year-old woman began exhibiting cognitive/behavioral changes following the birth of her first and only child. She had been treated for depression during high school but had not experienced mood symptoms or been treated with psychoactive agents for several years before her pregnancy. A few months after the birth of her child, she told family members that she was hearing voices telling her that people that she knew had died or were dying and frequently cursing at her. She soon developed other features, including obsessions, the urge to count aloud repeatedly, concrete thinking, reduced hygiene, reduced judgment, reduced responses, apathy, childlike behavior, laughing inappropriately, some degree of religious rituals, and a tendency to repeat statements. She often punched others without apparent appreciation of the pain she was inflicting. Her food selection became focused on high-calorie items, and she gained more than 9 kg (20 lb). She was hospitalized locally and treated in an inpatient psychiatric service for suspected postpartum psychosis versus schizophrenia. She was prescribed numerous antipsychotic medications and became catatonic with parkinsonian features. She underwent numerous diagnostic studies, including MRI, CSF examination, and multiple autoimmune/infectious/paraneoplastic laboratory studies, all of which were interpreted as normal except for bilateral caudate head atrophy on her MRI. A limited neuropsychological assessment showed cognitive slowing. Schizophrenia and neuroleptic-induced parkinsonism were the working diagnoses at this time. She underwent electroconvulsive therapy, and her tremor and parkinsonian features improved, but profound obsessive-compulsive tendencies and emotional withdrawal continued.
After she was discharged from the hospital, she lived with her family and attempted to return to some of her usual activities, but concerning behaviors continued. She repeatedly went to the grocery stores forgetting to bring a method to pay for groceries, yet still attempted to leave with the groceries. She became less interested in caring for her child, which alarmed her family greatly. She was treated with various psychotropic agents over the following 2 years, but her behavioral, cognitive, and motor changes continued to worsen. Her family history included two distant relatives with late-onset dementia.
Her family sought a second opinion, which occurred at 29 years of age. On examination, she sat rigidly, stared forward with rare eye blinks, and had no spontaneous verbal output. She was fully oriented, and constructional praxis was intact. She had good recall of current events. A more extensive mental status examination could not be completed because of limited cooperation. Mild bradykinesia was present, but she had no rigidity, no extraocular movement abnormalities, and no features of motor neuron disease.
Blood and CSF laboratory studies for infectious, autoimmune/inflammatory, and paraneoplastic etiologies were negative. CSF amyloid-β, total tau, and phosphorylated tau values were not consistent with Alzheimer disease. MRI and fludeoxyglucose positron emission tomography (FDG-PET) scans are shown in FIGURE 3-4. The MRI showed frontal and temporal atrophy as well as thinning of the caudate heads, and the FDG-PET showed marked hypometabolism in the frontal and temporal regions. Genetic testing did not reveal mutations in any of the known frontotemporal lobar degeneration–associated genes. She was diagnosed with behavioral variant frontotemporal dementia, and her family was counseled on frontotemporal dementia. Over 2 years of follow-up, behavioral dyscontrol was managed with varying doses of psychotropic agents and behavioral measures. At age 31, she was incontinent and dependent on her family for most activities of daily living.
FIGURE 3–4.
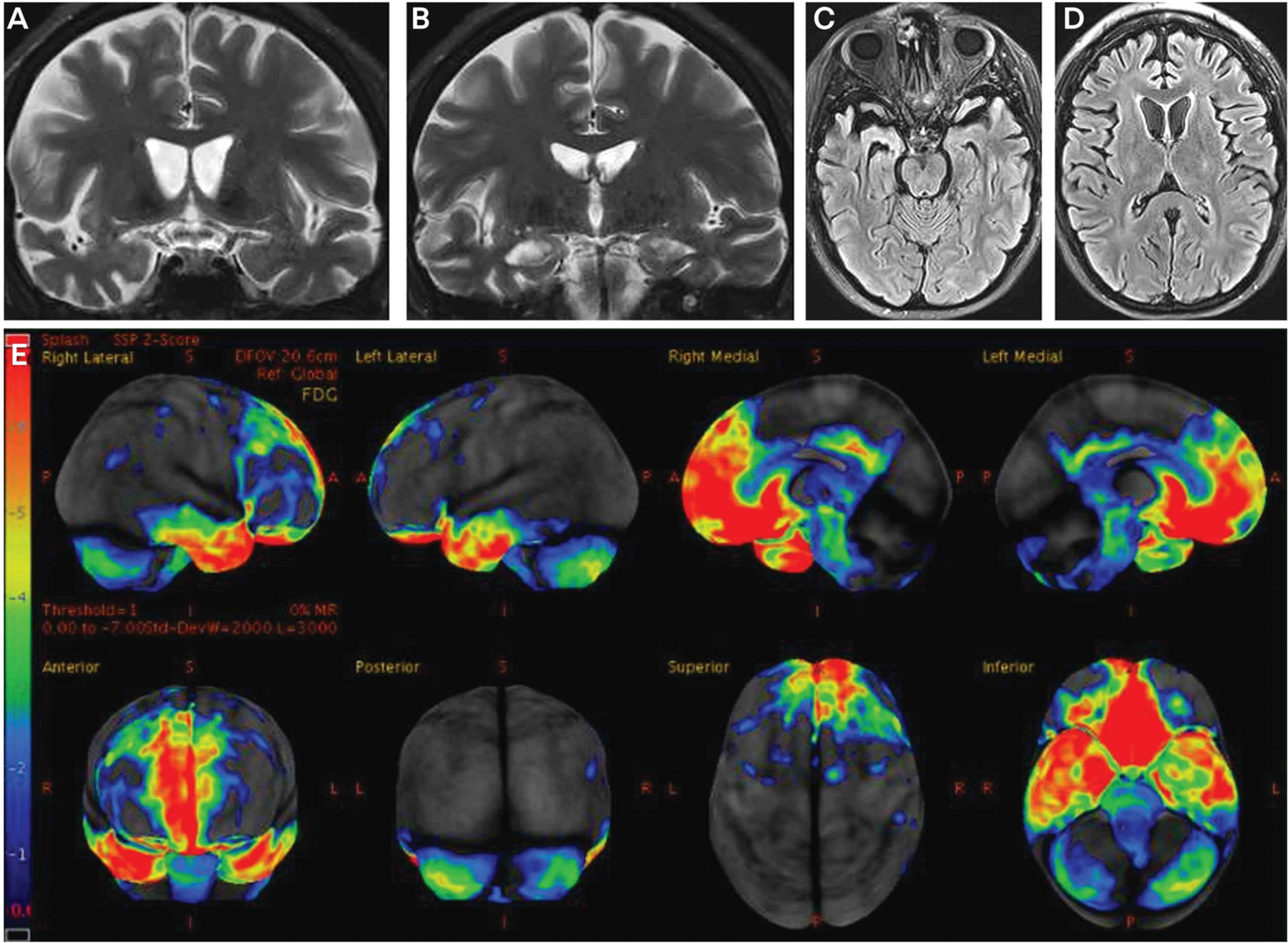
Imaging of the patient in CASE 3-4. Coronal (A, B) T2-weighted and axial (C, D) fluid-attenuated inversion recovery (FLAIR) MRI slices show frontal and temporal atrophy as well as thinning of the caudate heads. Fludeoxyglucose positron emission tomography (FDG-PET) (E) shows hypometabolism in the frontal and temporal regions that is most pronounced in the mesial frontal, orbitofrontal, and anterior temporal regions. The color bar andz score reference on the far left of the FDG-PET image reflect the degree of FDG hypometabolism, with blue reflecting z scores in the −1 to −2 range and red reflecting a z score of −6. Therefore, the regions with gray or black color reflect normal metabolism, blue color reflects slight to mild hypometabolism, and green to yellow to red color reflects moderate to very severe hypometabolism.
COMMENT
This case illustrates the “diagnostic odyssey of FTD,” with features that overlap with a primary psychiatric disorder and challenges in managing behaviors using nonpharmacologic and pharmacologic means.
Blood/Urine
No specific findings on laboratory testing of blood or urine have yet been identified as characteristic of FTLD pathology. However, growing evidence indicates that plasma biomarkers are sensitive and specific for AD,24,25 and plasma phosphorylated tau at position 181 (p-tau181)25 and at position 217 (p-tau217)25,26 differentiate clinically diagnosed and autopsy-confirmed AD from FTLD. If these findings are replicated and costs associated with their use in clinical practice are justified, one could envision that these biomarkers will be increasingly used in supporting or refuting underlying AD and FTLD. This would be a major advancement in simplifying diagnostic clarification and reducing the need for expensive diagnostic tests (eg, CSF amyloid and tau quantification, positron emission tomography [PET]) that are not easily obtained in many practice settings. Furthermore, neurofilament light chain (Nfl) is a sensitive marker of neurodegeneration, and this may become useful in differentiating between a neurodegenerative disease and a primary psychiatric disorder in patients with a bvFTD-like presentation.27,28
CSF Analysis
Although developing markers for FTLD-tau and FTLD–transactive response DNA-binding protein 43 (TDP-43) is an active area of research, no currently identified CSF markers are sensitive or specific for FTLD. Phosphorylated tau levels in CSF are useful for differentiating AD from FTLD as a cause of bvFTD.25,29 NfL in the CSF is elevated in FTD, but levels do not differentiate FTLD from non-FTLD pathology, nor do they distinguish the different proteinopathies in FTLD.25,30,31 Therefore, for at least the next few years, the role of CSF testing in a bvFTD-like presentation will be most useful in ruling out AD and in investigating the very rare infectious, autoimmune/inflammatory, and paraneoplastic processes.
Neuropsychological Assessment
Neuropsychological assessment in clinical practice typically involves a battery of tests that measure performance across the primary cognitive domains of attention/concentration, executive functioning, learning and memory, language functioning, and visuospatial functioning. In patients with bvFTD, the typical profile of impairment is on measures of attention/concentration, executive functioning, and language.6,32,33 Usually, performance on measures of memory and visuospatial functioning is relatively preserved compared to performance in attention/concentration, executive functioning, and language.
Several important caveats should be mentioned. The syndrome of bvFTD is first and foremost a disorder of social cognition (ie, impaired emotion recognition, empathy, mentalizing).34 Although many relatively new measures and questionnaires have been shown to be sensitive to impaired social cognition in patients with bvFTD,35,36 these measures are rarely, if ever, included in the neuropsychological batteries that are used in most clinical practice settings. Particularly in patients early in the course of bvFTD, performance on standard neuropsychological measures may be within normal limits. Also, many examples exist of patients with sporadic or familial bvFTD who have evidence of memory and/or visuospatial impairment. Finally, impaired performance in attention/concentration, executive functioning, and language can be seen in non-FTLD disorders such as atypical AD or Lewy body disease; hence, a “typical bvFTD profile” of impairment on neuropsychological testing is not specific for bvFTD.
Structural Neuroimaging
The presence and topography of atrophy on CT or MRI may provide diagnostic information. Atrophy in the frontal and/or temporal lobes, which can be symmetric or very focal/asymmetric, supports the clinical suspicion of bvFTD. However, the absence of obvious atrophy does not preclude an underlying neurodegenerative disorder, and other disorders (such as atypical AD) can present as a dysexecutive syndrome with associated frontal and/or temporal atrophy.37 Rare cases also exist of a bvFTD syndrome associated with a tumor in the frontal and/or temporal lobes as well as a bvFTD syndrome associated with intracranial hypotension causing the curious frontotemporal brain sagging syndrome.38
Functional Neuroimaging
Fludeoxyglucose positron emission tomography (FDG-PET) is often used to support the clinical suspicion of bvFTD.39–41 FDG-PET is therefore reasonable in many patients with suspected bvFTD, particularly if the MRI findings are normal or only questionably consistent with FTD. However, the progressive dysexecutive syndrome can appear clinically somewhat like bvFTD, but hypometabolism in this syndrome is more typical of underlying AD.37 Therefore, although the clinical and research utility of FDG-PET has been demonstrated in suspected bvFTD,39–41 the findings are not 100% sensitive or specific for an FTLD-spectrum disorder.
Molecular Positron Emission Tomography Brain Imaging
Amyloid PET imaging identifies individuals with amyloid deposition in the brain and can be used to differentiate underlying AD from non-AD disorders in patients with a bvFTD-like presentation.42 Data have not yet supported the use of tau PET imaging for differentiating a tauopathy from a non-tauopathy among those with an underlying FTLD spectrum disorder, despite early enthusiasm.43 Clinicians must wait for other ligands for tau and TDP-43 to be developed. If and when such ligands are shown to be highly sensitive and specific for differentiating tau, TDP-43, and others in the setting of bvFTD, this differentiation advancement will have major implications for diagnosis, tracking progression, aiding prognosis, and supporting target engagement of specific therapies.
Genetic Testing
Clinical genetic testing for the known genetic mutations associated with FTLD is reasonable in those with bvFTD and a positive family history of dementia, parkinsonism, or amyotrophic lateral sclerosis, particularly in those with an autosomal dominant pattern of inheritance. In previous years, targeted genetic testing for single or a few genes was the norm, which proved to be cumbersome and expensive if the initial results were negative. Many Clinical Laboratory Improvement Amendments (CLIA)–approved genetic panels are now available that will test several of the FTLD-associated genes, and the costs are not exorbitant. It is advised that adequate pretest and posttest counseling is performed and sufficient psychological support is ensured before and after testing for any patient/family who seeks genetic testing.
A more challenging situation is when a clinician encounters a patient with bvFTD who does not have a compelling family history for an autosomal dominant disorder. In some rare instances, a mutation is identified in a seemingly sporadic case of bvFTD.16 The decision on whether to recommend genetic testing in patients with sporadic bvFTD should depend on individual circumstances, recognizing that the yield is low.
Several clinical trials in progress or under development are designed to alter the pathophysiology in some of the genes associated with f-FTLD.44 Furthermore, several natural history studies that are enrolling family members with f-FTLD are also in progress across many countries (refer to the section on research programs below for more details). Enrollment of interested family members in such research programs is critical to ensure sufficient numbers of known potential participants for current and future clinical trials.
NEUROPATHOLOGY
As described above, FTLD is the overarching term referring to the spectrum of histopathologic entities that can manifest as bvFTD and related syndromes. The primary proteinopathies associated with the FTLD spectrum disorders include tau (often termed the tauopathies), TDP-43 proteinopathies, and the FET (FUS, EWSR1, TAF15)-related proteins (TABLE 3-2).45 The challenge for clinicians is that in the current practice environment with nonspecific biomarkers (except for genetic test results), it is not possible to predict with accuracy the underlying proteinopathy in anyone with bvFTD.
TABLE 3–2.
Primary Proteinopathies Associated With Frontotemporal Lobar Degeneration Spectrum Disordersa
|
Tauopathies (approximately 40–45% of cases) ♦ Pick disease ♦ Progressive supranuclear palsy (PSP) ♦ Corticobasal degeneration (CBD) ♦ Argyrophilic grain disease (AGD) ♦ Globular glial tauopathy (GGT) ♦ Age-related tau astrogliopathy (ARTAG) ♦ Primary age-related tauopathy (PART) Transactive response DNA-binding protein 43 (TDP-43) proteinopathies (approximately 40–45% of cases) ♦ Subtypes (TDP types A-D +/− U) are based on the relative abundance of different types of TDP+ neuronal inclusions and their laminar distribution within the cerebral neocortex ♦ Limbic-predominant age-related TDP-43 encephalopathy (LATE) FET protein familyb and other disorders (approximately 5–10% of cases) ♦ Basophilic inclusion body disease (BIBD) ♦ Neuronal intermediate filament inclusion body disease (NIFID) ♦ Atypical frontotemporal lobar degeneration with ubiquitin-positive inclusions (aFTLD-U) ♦ Frontotemporal lobar degeneration with inclusions labeled with markers of the ubiquitin/proteasome system ♦ Hereditary diffuse leukoencephalopathy with spheroids (HDLS) ♦ Others |
Data from Neumann M, Mackenzie IRA, Neuropathol Appl Neurobiol.45
FUS, EWSR1, TAF15.
MANAGEMENT
Management of symptoms is challenging in bvFTD. Clinicians should consider both nonpharmacologic and pharmacologic modes of management, realizing the evidence supporting any of these approaches is very limited.
Nonpharmacologic Modalities for Managing Symptoms
Family members should be counseled on the many psychosocial aspects of bvFTD. The Association for Frontotemporal Degeneration (theaftd.org) is an excellent resource. Patients with bvFTD often lack the capacity to avoid danger because of disinhibition, apathy, and poor understanding of the internal state of others (ie, they have impaired theory of mind).46 Although violent behaviors are rarely exhibited, patients with FTD are at risk of occupational, financial, or physical victimization because of their impairments in social cognition. Discontinuation of driving, whether voluntary or through other measures (eg, disconnecting spark plugs, hiding car keys), is sometimes needed to reduce the risk to patients and other drivers. Firearms should be removed from the home. Readers are encouraged to review other nonpharmacologic approaches for managing problematic FTD features,47–49 including information from The Association for Frontotemporal Degeneration (theaftd.org/living-with-ftd/managing-ftd).
Pharmacologic Modalities for Managing Symptoms
Several small series and trials of therapies to manage symptoms related to FTD have been conducted, with few encouraging results.36,44,50 No pharmacologic agents have been approved by the US Food and Drug Administration (FDA) for managing symptoms related to bvFTD. Cholinesterase inhibitors or memantine for treatment of cognition have not been shown to be effective.51–54 For behavioral dyscontrol and agitation, the serotonergic agents55–57 and atypical antipsychotic agents50 have been studied, with the best evidence for trazodone and selective serotonin reuptake inhibitors (SSRIs). Psychostimulants may help disinhibition.58 Antiseizure medications have also been reported to decrease problematic behaviors, but these reports are largely anecdotal.59–61 Oxytocin is currently being studied after encouraging early findings.62
With little guidance from the literature, what should clinicians do? Most take the reasonable approach of discussing with the patient and family which target symptoms are most problematic and then deciding on which nonpharmacologic and pharmacologic options are most appropriate for each patient. This is admittedly a trial-and-error approach, with some notable improvements in select patients.
FUTURE DIRECTIONS
Two major efforts in progress include characterizing prodromal bvFTD and enrolling in putative disease-modifying treatment trials.
Prodromal Behavioral Variant Frontotemporal Dementia
Whereas the clinical diagnosis of bvFTD can be based on established criteria, no such criteria exist for prodromal bvFTD. One would predict that mild changes in behavior, personality, or comportment without major changes in social/occupational functioning would occur before the development of more overt behavioral and cognitive changes and the associated functional decline.
The concepts of mild cognitive impairment (MCI)63 and mild behavioral impairment64 are pertinent here. These terms are intended to represent the intermediate clinical stage between normal aging and an overt neurodegenerative phenotype, most commonly evolving AD. Considerable work is in progress in key natural history studies to characterize prodromal FTLD further, particularly in the ARTFL (Advancement in Research and Treatment in Frontotemporal Lobar Degeneration)6 and LEFFTDS (Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects)65 studies (now combined into the ALLFTD [ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration] Consortium), the GENFI (Genetic Frontotemporal Dementia Initiative) Consortium,66 the FPI (Frontotemporal Dementia Prevention Initiative),13 and other cohorts. Based on a large cohort of asymptomatic and symptomatic members of kindreds with f-FTLD, the ALLFTD Consortium recently proposed a set of criteria for prodromal bvFTD in which the term mild behavioral and/or cognitive impairment in bvFTD (MBCI-FTD) was characterized.67 The proposed MBCI-FTD criteria correctly classified 74% to 95% of two groups of prodromal f-FTLD mutation carriers, with a false-positive rate of less than 10% in healthy controls and 11% to 16% of individuals with prodromal AD.67 Future research will need to refine the sensitivity and specificity of these criteria.
An obvious challenge to applying these criteria is exemplified by three of the four case examples in this article, in which even in the midst of overt bvFTD, the diagnosis was delayed. However, the patient in CASE 3-3 was evaluated relatively early in her course; if these prodromal criteria are applied to this patient, one can see that she satisfies the criteria. In clinical practice, it is unlikely that prodromal bvFTD will be suspected in any sporadic case unless the clinician is familiar with the term and criteria. It is more likely to be applied when patients in families with known f-FTLD develop symptoms and seek an evaluation or when family members convince a person with mild cognitive or behavioral changes to seek medical attention. These criteria can also be applied in asymptomatic at-risk members of kindreds with f-FTLD who are being followed in research programs. As clinical trial platforms continue to be refined, the utility of these criteria will be most applicable for inclusion in trials designed to slow the rate of progression and as an end point (ie, the onset of phenoconversion) for disease-modifying therapies designed to delay the onset of evolving bvFTD in those who are genetically at risk.
Trials of Disease-modifying Therapies
Most current therapeutic programs target autosomal dominant forms of FTLD, including C9orf72 repeat expansions and GRN or MAPT mutations. The strategies include using antisense oligonucleotides, adeno-associated virus vector therapies, antibodies to block peripheral receptors to elevate central progranulin protein, and antibodies to presumably alter cell-to-cell transmission of misfolded proteins, among others. Readers are encouraged to access other excellent resources on the direction of future FTLD therapies.36,44
RESEARCH PROGRAMS
Several multicenter natural history research programs are focused on sporadic and familial bvFTD and associated disorders, and clinicians are encouraged to consider referring patients for possible enrollment. These programs are designed to conduct comprehensive longitudinal clinical and biomarker measures to help inform clinical trial methodology as new potential interventions are developed. Clinicians and their patients/families are also encouraged to consider participation in the FTD Disorders Registry (ftdregistry.org), which is designed to educate patients and families as well as foster research advances in bvFTD and related disorders.
CONCLUSION
The prototypical patient with bvFTD presents in their forties to seventies with personality/behavior changes, and the patient usually does not perceive anything is amiss because of the limited insight that is inherent to the disorder. The personality/behavior changes are often more pronounced than cognitive changes early in the course, and since such changes can mimic other more common psychiatric disorders (eg, major depression, bipolar disorder, schizophrenia), diagnostic confusion often exists for family members and clinicians. As the illness progresses, more obvious changes in cognition or progression in personality/behavior changes may trigger consideration of an underlying neurologic disease; even then, diagnostic confusion is common because of the similarities of bvFTD to AD dementia. Although many challenges remain regarding establishing an early diagnosis of bvFTD, determining the underlying proteinopathy causing the clinical syndrome, and optimizing management using nonpharmacologic and pharmacologic methods, the scientific advances in bvFTD and FTLD have escalated markedly in recent years. This article discussed concepts and data to help guide the diagnosis and management of patients with bvFTD. The optimism for therapeutic advancements is high considering the large network of research programs working toward a common goal, increased funding for FTLD research, strong interest among industry partners working on therapeutic pipelines, and continued advances in our understanding of the mechanisms underlying FTLD pathobiology.
KEY POINT.
The altered behavior and cognition during the prediagnosis and postdiagnosis phases of behavioral variant frontotemporal dementia (bvFTD) have many potential consequences, with profound repercussions for patients, their family members, and society.
KEY POINTS.
At least 20% of patients with bvFTD or similar phenotype have a dominantly inherited familial disorder, with mutations being most common in the genes encoding microtubule associated protein tau (MAPT), progranulin (GRN), and chromosome 9 open reading frame 72 (C9orf72).
Applying the consensus criteria for bvFTD is useful in clinical practice.
The diagnostic odyssey of frontotemporal dementia (FTD) that many patitents and families endure is often convoluted and prolonged.
Any patient with one or more of the following core clinical features should be considered as possibly having bvFTD: behavioral disinhibition; apathy or inertia; loss of sympathy or empathy; perseverative, stereotyped, or compulsive/ritualistic behavior; or hyperorality and dietary changes.
In the setting of suspected bvFTD, interviewing family members separately from the patient may be indicated to more thoroughly query about the core features.
Although no specific findings on laboratory testing of blood, urine, or CSF have yet been identified as characteristic of frontotemporal lobar degeneration (FTLD) pathology, the plasma and CSF biomarkers that are sensitive and specific for Alzheimer disease may aid in differentiating between Alzheimer disease and FTLD.
KEY POINTS.
Neuropsychological test results in patients with suspected bvFTD are usually informative and support the initial clinical impression.
A normal profile or an atypical profile of impairment on neuropsychological testing should not dissuade the clinician from suspecting bvFTD if the history is very compelling.
A “typical bvFTD profile” of impairment on neuropsychological testing should not supersede other clinical features and biomarker findings when making a diagnosis of bvFTD.
Atrophy in the frontal and/or temporal lobes on brain CT or MRI, which can be symmetric or very focal/asymmetric, supports the diagnosis of bvFTD in the appropriate clinical setting. However, the absence of obvious atrophy in the frontal and/or temporal lobes on brain CT or MRI does not rule out bvFTD.
Atrophy in the frontal and/or temporal lobes on brain CT or MRI can be seen in other disorders, including in atypical AD.
Fludeoxyglucose positron emission tomography is reasonable in many patients with suspected bvFTD, particularly if the MRI findings are normal or only questionably consistent with FTD.
KEY POINTS.
The clinical utility of molecular positron emission tomography (PET) imaging with amyloid and tau ligands is not fully defined; amyloid PET imaging may have a role in differentiating underlying Alzheimer disease from non–Alzheimer disease disorders in patients with a bvFTD-like presentation.
Clinical genetic counseling and testing for the known genetic mutations associated with FTLD is reasonable in those with bvFTD and a positive family history of dementia, parkinsonism, or amyotrophic lateral sclerosis, particularly in those with an autosomal dominant pattern of inheritance.
Clinical genetic counseling and testing for the known genetic mutations associated with FTLD may be reasonable in other select circumstances, such as in patients with very-early-onset bvFTD who do not have any known family history of a neurodegenerative disorder.
In the current practice environment with nonspecific biomarkers (except for genetic test results), it is not possible to predict with accuracy the underlying proteinopathy in anyone with bvFTD.
Management of symptoms is challenging in bvFTD, and clinicians should consider several nonpharmacologic and pharmacologic modes of management.
USEFUL WEBSITES.
THE ASSOCIATION FOR FRONTOTEMPORAL DEGENERATION
The Association for Frontotemporal Degeneration website provides information about frontotemporal degeneration, including genetics, where to find help, and how to participate in clinical trials.
FTD DISORDERS REGISTRY
The FTD Disorders Registry website provides information about clinical trials and resources for patients and families.
CLINICALTRIALS.GOV TRIALS FOR FRONTOTEMPORAL DEMENTIA
This page on the ClinicalTrials.gov website provides a list of clinical trials related to frontotemporal dementia.
NATIONAL INSTITUTE OF NEUROLOGICAL DISORDERS AND STROKE FRONTOTEMPORAL DEMENTIA INFORMATION PAGE
This page provides a definition of frontotemporal dementia and its diagnosis and treatment as well as information on clinical trials in the United States and internationally.
ninds.nih.gov/Disorders/All-Disorders/Frontotemporal-Dementia-Information-Page
THE ALLFTD STUDY
This website offers research study information on the Advancement in Research and Treatment in Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) studies, now combined into the ARTFL LEFFTDS Longitudinal Frontotemporal Lobar Degeneration (ALLFTD) program. This study is being conducted in the United States and Canada.
GENETIC FTD INITIATIVE (GENFI)
The website provides research study information on the GENFI program taking place in Europe and Eastern Canada.
LATIN AMERICAN AND CARIBBEAN CONSORTIUM ON DEMENTIA
This website provides research study information on the Research Dementia Latin America (ReDLAT) Program being conducted in Latin America.
FTD PREVENTION INITIATIVE (FPI) PROGRAM
This website provides information on a variety of research studies and other projects, including some of the programs mentioned above, with a focus on familial frontotemporal lobar degeneration.
NATIONAL INSTITUTE ON AGING – ALZHEIMER’S DISEASE RESEARCH CENTERS (ADRC) PROGRAM
This website provides information on research centers located at major medical institutions across the United States. The ADRC program works in close collaboration with the ALLFTD program.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health (AG063911). The author expresses his gratitude to his many collaborators at Mayo Clinic as well as colleagues focused on FTLD clinical practice and research across the globe. He particularly thanks the many patients and families for their interest and participation in aging and neurodegenerative disease research.
RELATIONSHIP DISCLOSURE:
Dr Boeve has received personal compensation in the range of $10,000 to $49,999 for serving as an officer or member of the scientific advisory board of the Rainwater Charitable Foundation. Dr Boeve has received publishing royalties from a publication relating to health care. The institution of Dr Boeve has received research/grant support from Alector, Inc; Biogen; EIP Pharma, Inc;. GE Healthcare; and the National Institutes of Health.
Footnotes
UNLABELED USE OF PRODUCTS/INVESTIGATIONAL USE DISCLOSURE:
Dr Boeve discusses the unlabeled/investigational use of antiseizure medications, atypical antipsychotics, cholinesterase inhibitors, memantine, oxytocin, psychostimulants, selective serotonin reuptake inhibitors/serotonin norepinephrine reuptake inhibitors, and trazodone for treating symptoms related to frontotemporal dementia, none of which are approved by the US Food and Drug Administration.
REFERENCES
- 1.Bang J, Spina S, Miller B. Frontotemporal dementia. Lancet 2015;386(10004):1672–1682. doi: 10.1016/S0140-6736(15)00461-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turcano P, Stang CD, Mielke MM, et al. Incidence of frontotemporal disorders in Olmsted County: a population-based study. Alzheimers Dement 2020;16(3):482–490. doi: 10.1016/j.jalz.2019.08.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry 2013; 25(2):130–137. doi: 10.3109/09540261.2013.776523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hendriks S, Peetoom K, Bakker C, et al. Global prevalence of young-onset dementia: a systematic review and meta-analysis. JAMA Neurol 2021;78(9): 1080–1090. doi: 10.1001/jamaneurol.2021.2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsson C, Landqvist Waldö M, Nilsson K, et al. Age-related incidence and family history in frontotemporal dementia: data from the Swedish Dementia Registry. PLoS One 2014;9(4). doi: 10.1371/journal.pone.0094901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heuer HW, Wang P, Rascovsky K, et al. Comparison of sporadic and familial behavioral variant frontotemporal dementia (FTD) in a North American cohort. Alzheimers Dement 2020;16(1): 60–70. doi: 10.1002/alz.12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galvin JE, Howard DH, Denny SS, et al. The social and economic burden of frontotemporal degeneration. Neurology 2017;89(20):2049–2056. doi: 10.1212/WNL.0000000000004614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen HJ, Boeve BF, Boxer AL. Tracking disease progression in familial and sporadic frontotemporal lobar degeneration: recent findings from ARTFL and LEFFTDS. Alzheimers Dement 2020;16(1):71–78. doi: 10.1002/alz.12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393(6686):702–705. doi: 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 10.Baker M, Mackenzie I, Pickering-Brown S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442(7105):916–919. doi: 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- 11.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol 2020; 19(2):145–156. doi: 10.1016/S1474-4422(19)30394-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boeve BF, Rosen H. Clinical and neuroimaging aspects of familial frontotemporal lobar degeneration associated with MAPT and GRN mutations. Adv Exp Med Biol 2021;1281:77–92. doi: 10.1007/978-3-030-51140-1_6 [DOI] [PubMed] [Google Scholar]
- 15.Saracino D, Le Ber I. Clinical update on C9orf72: frontotemporal dementia, amyotrophic lateral sclerosis, and beyond. Adv Exp Med Biol 2021; 1281:67–76. doi: 10.1007/978-3-030-51140-1_5 [DOI] [PubMed] [Google Scholar]
- 16.Ramos EM, Dokuru DR, Van Berlo V, et al. Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimers Dement 2020;16(1):118–130. doi: 10.1002/alz.12011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neary D, Snowden J, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51(6):1546–1554. doi: 10.1212/wnl.51.6.1546 [DOI] [PubMed] [Google Scholar]
- 18.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(9):2456–2477. doi: 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Besser LM, Galvin JE. Diagnostic experience reported by caregivers of patients with frontotemporal degeneration. Neurol Clin Pract 2020;10(4):298–306. doi: 10.1212/CPJ.0000000000000738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knopman D, Boeve B, Petersen R. Essentials of the proper diagnoses of mild cognitive impairment, dementia, and major subtypes of dementia. Mayo Clin Proc 2003;78(10):1290–1308. doi: 10.4065/78.10.1290 [DOI] [PubMed] [Google Scholar]
- 21.Choudhury P, Scharf EL, Paolini MA, et al. Pick’s disease: clinicopathologic characterization of 21 cases. J Neurol 2020;267(9):2697–2704. doi: 10.1007/s00415-020-09927-9 [DOI] [PubMed] [Google Scholar]
- 22.Boeve B, Graff-Radford N. Cognitive and behavioral features of c9FTD/ALS. Alzheimers Res Ther 2012;4(4). doi: 10.1186/alzrt132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012;135(pt 3):693–708. doi: 10.1093/brain/awr355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.West T, Kirmess KM, Meyer MR, et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegener 2021;16(1). doi: 10.1186/s13024-021-00451-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med 2020;26(3):387–397. doi: 10.1038/s41591-020-0762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thijssen EH, La Joie R, Strom A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol 2021; 20(9):739–752. doi: 10.1016/S1474-4422(21)00214-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eratne D, Loi SM, Walia N, et al. A pilot study of the utility of cerebrospinal fluid neurofilament light chain in differentiating neurodegenerative from psychiatric disorders: a “C-reactive protein” for psychiatrists and neurologists? Aust N Z J Psychiatry 2020;54(1):57–67. doi: 10.1177/0004867419857811 [DOI] [PubMed] [Google Scholar]
- 28.Katisko K, Cajanus A, Jaaskelainen O, et al. Serum neurofilament light chain is a discriminative biomarker between frontotemporal lobar degeneration and primary psychiatric disorders. J Neurol 2020;267(1):162–167. doi: 10.1007/s00415-019-09567-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 2020;324(8): 772–781. doi: 10.1001/jama.2020.12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016;3(8):623–636. doi: 10.1002/acn3.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Illán-Gala I, Lleo A, Karydas A, et al. Plasma tau and neurofilament light in frontotemporal lobar degeneration and Alzheimer disease. Neurology 2021;96(5). doi: 10.1212/WNL.0000000000011226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olney NT, Ong E, Goh SM, et al. Clinical and volumetric changes with increasing functional impairment in familial frontotemporal lobar degeneration. Alzheimers Dement 2020;16(1): 49–59. doi: 10.1016/j.jalz.2019.08.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staffaroni AM, Bajorek L, Casaletto KB, et al. Assessment of executive function declines in presymptomatic and mildly symptomatic familial frontotemporal dementia: NIH-EXAMINER as a potential clinical trial endpoint. Alzheimers Dement 2020;16(1):11–21. doi: 10.1016/j.jalz.2019.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumfor F, Honan C, McDonald S, et al. Assessing the “social brain” in dementia: applying TASIT-S. Cortex 2017;93:166–177. doi: 10.1016/j.cortex.2017.05.022 [DOI] [PubMed] [Google Scholar]
- 35.Rankin KP. Measuring behavior and social cognition in FTLD. Adv Exp Med Biol 2021;1281: 51–65. doi: 10.1007/978-3-030-51140-1_4 [DOI] [PubMed] [Google Scholar]
- 36.Boeve B, Boxer A, Kumfor F, et al. Advances and controversies in frontotemporal dementia: characterization, diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol 2022; 21(3):258–272. doi: 10.1016/S1474-4422(21)00341-0 [DOI] [PubMed] [Google Scholar]
- 37.Townley RA, Graff-Radford J, Mantyh WG, et al. Progressive dysexecutive syndrome due to Alzheimer’s disease: a description of 55 cases and comparison to other phenotypes. Brain Commun 2020;2(1). doi: 10.1093/braincomms/fcaa068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wicklund MR, Mokri B, Drubach DA, et al. Frontotemporal brain sagging syndrome: an SIH-like presentation mimicking FTD. Neurology 2011;76(16):1377–1382. doi: 10.1212/WNL.0b013e3182166e42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster NL. Validating FDG-PET as a biomarker for frontotemporal dementia. Exp Neurol 2003; 184(suppl 1):S2–S8. doi: 10.1016/s0014-4886(03)00360-1 [DOI] [PubMed] [Google Scholar]
- 40.Foster NL, Heidebrink JL, Clark CM, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain 2007;130(pt 10):2616–2635. doi: 10.1093/brain/awm177 [DOI] [PubMed] [Google Scholar]
- 41.Bejanin A, Tammewar G, Marx G, et al. Longitudinal structural and metabolic changes in frontotemporal dementia. Neurology 2020;95(2). doi: 10.1212/WNL.0000000000009760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghirelli A, Tosakulwong N, Weigand SD, et al. Sensitivity-specificity of tau and amyloid β positron emission tomography in frontotemporal lobar degeneration. Ann Neurol 2020;88(5):1009–1022. doi: 10.1002/ana.25893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai RM, Bejanin A, Lesman-Segev O, et al. 18F-flortaucipir (AV-1451) tau PET in frontotemporal dementia syndromes. Alzheimers Res Ther 2019;11(1). doi: 10.1186/s13195-019-0470-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ljubenkov PA, Boxer AL. FTLD treatment: current practice and future possibilities. Adv Exp Med Biol 2021;1281:297–310. doi: 10.1007/978-3-030-51140-1_18 [DOI] [PubMed] [Google Scholar]
- 45.Neumann M, Mackenzie IRA. Review: neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 2019; 45(1):19–40. doi: 10.1111/nan.12526 [DOI] [PubMed] [Google Scholar]
- 46.Gregory C, Lough S, Stone V, et al. Theory of mind in patients with frontal variant frontotemporal dementia and Alzheimer’s disease: theoretical and practical implications. Brain 2002;125(pt 4): 752–764. doi: 10.1093/brain/awf079 [DOI] [PubMed] [Google Scholar]
- 47.Shnall A, Agate A, Grinberg A, et al. Development of supportive services for frontotemporal dementias through community engagement. Int Rev Psychiatry 2013;25(2):246–252. doi: 10.3109/09540261.2013.767780 [DOI] [PubMed] [Google Scholar]
- 48.Wylie MA, Shnall A, Onyike CU, Huey ED. Management of frontotemporal dementia in mental health and multidisciplinary settings. Int Rev Psychiatry 2013;25(2):230–236. doi: 10.3109/09540261.2013.776949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tookey SA, Greaves CV, Rohrer JD, Stott J. Specific support needs and experiences of carers of people with frontotemporal dementia: a systematic review. Dementia (London) 2021; 20(8):3032–3054. doi: 10.1177/14713012211022982 [DOI] [PubMed] [Google Scholar]
- 50.Boxer AL, Boeve BF. Frontotemporal dementia treatment: current symptomatic therapies and implications of recent genetic, biochemical, and neuroimaging studies. Alzheimer Dis Assoc Disord 2007;21(4). doi: 10.1097/WAD.0b013e31815c345e [DOI] [PubMed] [Google Scholar]
- 51.Kertesz A, Morlog D, Light M, et al. Galantamine in frontotemporal dementia and primary progressive aphasia. Dement Geriatr Cogn Disord 2008;25(2):178–185. doi: 10.1159/000113034 [DOI] [PubMed] [Google Scholar]
- 52.Moretti R, Torre P, Antonello R, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931–937. doi: 10.2165/00002512-200421140-00003 [DOI] [PubMed] [Google Scholar]
- 53.Kimura T, Takamatsu J. Pilot study of pharmacological treatment for frontotemporal dementia: risk of donepezil treatment for behavioral and psychological symptoms. Geriatr Gerontol Int 2013;13(2):506–507. doi: 10.1111/j.1447-0594.2012.00956.x [DOI] [PubMed] [Google Scholar]
- 54.Boxer AL, Knopman DS, Kaufer DI, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2013;12(2):149–156. doi: 10.1016/S1474-4422(12)70320-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huey ED, Putnam KT, Grafman J. A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia. Neurology 2006;66(1):17–22. doi: 10.1212/01.wnl.0000191304.55196.4d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herrmann N, Black SE, Chow T, et al. Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am J Geriatr Psychiatry 2012;20(9): 789–797. doi: 10.1097/JGP.0b013e31823033f3 [DOI] [PubMed] [Google Scholar]
- 57.Lebert F, Stekke W, Hasenbroekx C, Pasquier F. Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement Geriatr Cogn Disord 2004;17(4):355–359. doi: 10.1159/000077171 [DOI] [PubMed] [Google Scholar]
- 58.Huey ED, Garcia C, Wassermann EM, et al. Stimulant treatment of frontotemporal dementia in 8 patients. J Clin Psychiatry 2008;69(12): 1981–1982. doi: 10.4088/JCP.v69n1219a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galvez-Andres A, Blasco-Fontecilla H, Gonzalez-Parra S, et al. Secondary bipolar disorder and Diogenes syndrome in frontotemporal dementia: behavioral improvement with quetiapine and sodium valproate. J Clin Psychopharmacol 2007; 27(6):722–723. doi: 10.1097/JCP.0b013e31815a57c1 [DOI] [PubMed] [Google Scholar]
- 60.Poetter CE, Stewart JT. Treatment of indiscriminate, inappropriate sexual behavior in frontotemporal dementia with carbamazepine. J Clin Psychopharmacol 2012;32(1):137–138. doi: 10.1097/JCP.0b013e31823f91b9 [DOI] [PubMed] [Google Scholar]
- 61.Shinagawa S, Tsuno N, Nakayama K. Managing abnormal eating behaviours in frontotemporal lobar degeneration patients with topiramate. Psychogeriatrics 2013;13(1):58–61. doi: 10.1111/j.1479-8301.2012.00429.x [DOI] [PubMed] [Google Scholar]
- 62.Finger EC, MacKinley J, Blair M, et al. Oxytocin for frontotemporal dementia: a randomized dose-finding study of safety and tolerability. Neurology 2015;84(2):174–181. doi: 10.1212/WNL.0000000000001133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petersen R, Lopez O, Armstrong MJ, et al. Practice guideline update summary: mild cognitive impairment: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018;90(3): 126–135. doi: 10.1212/WNL.0000000000004826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ismail Z, Smith EE, Geda Y, et al. Neuropsychiatric symptoms as early manifestations of emergent dementia: provisional diagnostic criteria for mild behavioral impairment. Alzheimers Dement 2016; 12(2):195–202. doi: 10.1016/j.jalz.2015.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boeve B, Bove J, Brannelly P, et al. The longitudinal evaluation of familial frontotemporal dementia subjects protocol: framework and methodology. Alzheimers Dement 2020;16(1): 22–36. doi: 10.1016/j.jalz.2019.06.4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 2015;14(3):253–262. doi: 10.1016/S1474-4422(14)70324-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barker MD, Gottesman RT, Manoochehri M, et al. Proposed research criteria for prodromal behavioural variant frontotemporal dementia. Brain 2022;awab365. doi: 10.1093/brain/awab365 [DOI] [PMC free article] [PubMed] [Google Scholar]