

Abstract
Metabolism is an essential part of life that provides energy for cell growth. During metabolic flux, reactive electrophiles are produced that covalently modify macromolecules, leading to detrimental cellular effects. Methylglyoxal (MG) is an abundant electrophile formed from lipid, protein, and glucose metabolism at intracellular levels of 1–4 μM. MG covalently modifies DNA, RNA, and protein, forming advanced glycation end products (MG-AGEs). MG and MG-AGEs are associated with the onset and progression of many pathologies including diabetes, cancer, and liver and kidney disease. Regulating MG and MG-AGEs is a potential strategy to prevent disease, and they may also have utility as biomarkers to predict disease risk, onset, and progression. Here, we review recent advances and knowledge surrounding MG, including its production and elimination, mechanisms of MG-AGEs formation, the physiological impact of MG and MG-AGEs in disease onset and progression, and the latter in the context of its receptor RAGE. We also discuss methods for measuring MG and MG-AGEs and their clinical application as prognostic biomarkers to allow for early detection and intervention prior to disease onset. Finally, we consider relevant clinical applications and current therapeutic strategies aimed at targeting MG, MG-AGEs, and RAGE to ultimately improve patient outcomes.
Introduction
Metabolism encompasses all of the reactions cells use to convert food into energy and is essential to sustain life. Changes in metabolic flux result from increased food intake, dysregulation of metabolite uptake, and up- and downregulation of proteins involved in metabolic pathways. Altered metabolism is associated with diabetes, cancer, liver, and kidney disease; however, how this drives disease onset and progression is not clear. A proposed mechanism is through changes in cellular physiology caused by reactive electrophiles produced during metabolic flux. Electrophiles are electron-pair-deficient molecules that react with nucleophilic sites within macromolecules to alter the structure and function. An abundant electrophile produced from metabolic flux is methylglyoxal (MG), which is formed intracellularly at levels of 1–4 μM.1 MG covalently modifies nucleophilic sites within nucleic and amino acids, forming advanced glycation end products (MG-AGEs).
Our understanding of the role of MG and MG-AGEs as potential drivers of disease has advanced because of the foundational work by Larry Marnett and other pioneers in the field of chemical toxicology. Marnett described the impact of reactive electrophiles, including malondialdehyde, base propenal, and hydroxynonenal on protein and DNA structure and function.2−8 This work provided the framework to investigate the impact of electrophile stress using chemical tools, analytical methods, biochemistry assays, and models for animal studies. Measuring and targeting electrophiles, their associated byproducts, and receptors has important implications as biomarkers and etiological agents of disease. In this review, we discuss the formation of MG, MG-AGEs, the physiological impact of these molecules on cellular function, and their association with disease.
MG Production
MG (2-oxopropanal or pyruvaldehyde) was discovered in the late 19th century as a byproduct of glucose, protein, and lipid metabolism.9−12 MG is proposed to exert its cellular effect through the formation of MG-AGEs on nucleic acids and protein, leading to changes in macromolecular stability and function.10,13−15 MG is predominantly produced as a byproduct of glycolysis during degradation of the triose phosphate intermediates, dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P) (Figure 1A).16 This occurs through two mechanisms: (1) nonenzymatic breakdown of G3P and DHAP, causing loss of the α-carbonyl group and phosphate, or (2) enzymatic conversion of G3P and DHAP into MG, mediated by enzymes such as MG synthase (MS) and triose phosphate isomerase (TPI) (Figure 1A).16−18 Sources of DHAP include the conversion of glucose to fructose and fructose 1,6-bisphosphate via sorbitol dehydrogenase and phosphofructokinase, respectively, which are then converted to DHAP via aldolase B.19 DHAP is also formed from metabolism of triacylglycerol into glycerol-3-phosphate via glycerol kinase (GK),20 followed by l-glycerol-3-phosphate oxidase (G3PO) or glycerol-3-phosphate dehydrogenase (G3PDH) (Figure 1A).21,22
Figure 1.

MG production from metabolic pathways. (A) During glycolysis, glucose is converted into DHAP or G3P which is metabolized by MS or TPI into MG. (B) In lipid metabolism, triacylglycerol undergoes hydrolysis to form fatty acids which are converted into acetol and acetone and then into MG by AMOO. (C) Ketones such as acetone, β-hydroxybutyric acid, and acetoacetic acid are converted to MG by cytochrome P450, AMOO, or MP. (D) During protein metabolism, amino acids such as glycine or threonine are metabolized by TDH into aminoacetone which is converted into MG by SSAO.
MG is also produced from lipid, ketone, and protein metabolism.19,23,24 Triacylglycerol is hydrolyzed to fatty acids, which form acetol and acetone, giving rise to MG through acetol mono-oxygenase (AMOO) (Figure 1B).25 Ketone metabolism also produces MG through cytochrome P450, AMOO, and myeloperoxidase (MP) (Figure 1C).26,27 Finally, MG is formed from glycine and threonine metabolism to aminoacetone through threonine dehydrogenase (TDH). Aminoacetone is then converted to MG through semicarbazide-sensitive amine oxidase (SSAO) (Figure 1D).28,29
Collectively, this demonstrates that MG and its precursors and cofactors are abundant molecules associated with several metabolic pathways. It is important to note that MG abundance and its rate of formation also largely depends on the state of metabolic flux, the specific organism or tissue being studied, as well as physiological milieu. However, it has been estimated that intracellular MG levels range from 1 to 4 μM.1 However, due to MG’s reactive nature, it has been postulated that MG’s biological half-life is relatively short, and it is therefore likely that the actual amount produced is higher than current estimates.30 As a small molecule, MG is cell permeant and thus is able to diffuse through cell membranes from the extracellular space.31
MG Regulation
Given that MG is reactive and can have detrimental impacts on cellular function, there are multiple mechanisms by which it is detoxified.
MG Detoxification via the Glyoxalase System
One of the most prominent ways cells detoxify MG is through the glyoxalase pathway, a highly evolutionarily conserved system that involves the activity of two enzymes: glyoxalase 1 (GLO1) and glyoxalase 2 (GLO2).32 MG reacts nonenzymatically with glutathione (GSH) to form a hemithioacetal, which is recognized by GLO1 and converted into S-d-lactoylglutathione (Figure 2A).33 GLO2 then converts S-d-lactoylglutathione into d-lactate, regenerating GSH (Figure 2A).33 The rate-limiting step of this pathway is GLO1 recognition of the hemithioacetal.34 Recent work demonstrated a novel role for S-d-lactoylglutathione as a source of protein post-translational modifications, a process inhibited by GLO2.35
Figure 2.

Various pathways for MG detoxification. (A) Glyoxalase system. MG reacts nonenzymatically with GSH to form a hemithioacetal which is recognized by GLO1. GLO1 converts the hemithioacetal to S-d-lactoylglutathione which is recognized by GLO2 to form d-lactate. (B) In addition to GLO1, AR also recognizes the hemithioacetal formed from the reaction of MG with GSH to lactaldehyde through an NADPH-dependent reaction. Lactaldehyde can undergo reduction, forming propanediol. (C) AR also recognizes MG directly through a GSH-independent mechanism, forming acetol which is reduced to form propanediol. GSH-independent reduction of MG by AR. (D) MG is oxidized by ALDH in an NAD-dependent mechanism to form pyruvate. (E) MG is oxidized by 2-ODH in an NADP-dependent reaction to form pyruvate.
GLO1 overexpression protects cells from the accumulation of reactive oxygen species (ROS), glucose-driven apoptosis, and dysfunction arising from angiogenesis and diabetes.36−39 Recently, Alkb homologue 7 (ALKBH7) has been proposed to regulate GLO1 as Alkbh7–/– mice have elevated Glo1 expression and MG-protein adducts.40 In addition, DJ-1, also known as PARK7, is a gene implicated in Parkinson’s disease that detoxifies MG through a GSH-independent glyoxalase mechanism, converting MG to lactic acid.41,42 Clinical trials involving obese patients have implicated combinatory treatment of hesperetin and trans-resveratrol in inducing GLO1 expression and activity, which was found to lower their glucose levels and improve overall vascular function.43
A recent study was among the first to successfully create a viable GLO1 knockout mouse without significant detrimental side effects.44,45 However, the loss of GLO1 has not been fully characterized in mammalian models. A putative Glo1 knockout mouse model was revealed to maintain a normal, healthy phenotype owing to gene duplication prior to gene trapping, preserving the wild-type phenotype.46 It was subsequently reported that aldehyde dehydrogenases (ALDH) and aldose reductases (AR), reviewed below, compensated for and mitigated the loss of GLO1.44 A similar finding implicating compensatory mechanisms for GLO1 has been reported in studies of GLO1 knockout zebrafish (D. rerio), which upregulated ALDH activity, partially compensating for the loss of GLO1.47 The loss of GLO1 in yeast (S. cerevisiae) resulted in hypersensitivity to MG and decreased cell survival and proliferation.48,49 In addition, the loss of GLO1 in fruit flies (D. melanogaster) led to obesity and prolonged lifespan and appeared to recapitulate some diabetic phenotypes, including hyperglycemia.50 In vitro, a GLO1 knockout in HEK293T increased MG-AGEs, specifically MG-hydroimidazolone (MG-H1).51 Further studies in primary human aortic endothelial cells revealed that GLO1 knockdown increased MG levels and subsequent inflammation, apoptosis, and dysfunction that led to vascular damage and impaired function.52 Taken together, this highlights the importance of the glyoxalase system as an indispensable mechanism for detoxifying MG.
MG Detoxification via Oxidation
ALDHs are a class of nicotinamide adenine dinucleotide (NAD) and NAD phosphate (NADP)-dependent enzymes that oxidizes aldehydes to form carboxylic acids.53 ALDHs help detoxify aldehydes, a process that, if left unregulated, can be detrimental. For example, single-nucleotide polymorphisms, particularly ALDH2 rs672 G>A, and ALDH mutations are associated with an enhanced risk of heart disease,54 muscular dystrophy,55 and Alzheimer’s disease.56,57
The E1, E2, and E3 isozymes of ALDH react with MG and oxidize it into pyruvate in an NAD-dependent manner (Figure 2D).58 Likewise, 2-oxoaldehyde dehydrogenase (2-ODH) converts MG to pyruvate but in an NADP-dependent manner (Figure 2E).59 The loss of Aldh in murine models enhanced aldehydic adduct formation, cardiovascular and motor dysfunction, and tissue damage.60Aldh overexpression mitigated the effects of oxidative stress and ROS in various organs, both of which are upregulated following MG accumulation.60,61 Similar to GLO1 knockout cells, glo1 knockout in zebrafish moderately increased MG levels and significantly heightened Aldh activity, supporting the role of ALDH as an additional compensatory mechanism in the event the glyoxalase system is impaired.62
MG Detoxification via Reduction
Aldose reductase (AR) is a 36 kDa enzyme encoded by the human ALR2 gene and is part of the aldo-keto reductase enzyme family. The canonical role of AR is to reduce aldehydes into their respective sugar alcohols via the polyol pathway.63 AR activity is dependent on NADPH and exhibits a higher substrate selectivity and preference than ALDH, particularly for MG, thus making it more efficient at MG breakdown than ALDH.64,65 AR is associated with the development of diabetic complications, such as cardiovascular and renal diseases (reviewed in ref (65)). In addition, AR gene polymorphisms are associated with the risk of developing diabetic complications such as retinopathy,66 nephropathy,67,68 and neuropathy.69 For example, a CA dinucleotide polymorphism located in the 5′ promoter region of the ALR2 gene is correlated with diabetic retinopathy.66 A similar biallelic polymorphism (C-106T) also in the promoter region of ALR2 increased the risk of nephropathy, which was further enhanced if an individual carried both risk alleles.49 Patients with diabetic neuropathy have significantly lower frequency of the Z+2 allele than healthy controls.69 Therefore, AR polymorphisms appear to be closely related to the development of diabetic complications. AR-mediated MG detoxification operates in two distinct pathways: (1) GSH dependent in which the hemithioacetal formed between the nonenzymatic reaction with GSH and MG is converted by AR and NADPH to a lactaldehyde (Figure 2B) and (2) GSH independent in which MG reacts with AR and NADPH to form acetol (Figure 2C).70 Further AR-mediated metabolism of the lactaldehyde and acetol forms propanediol (Figure 2B and 2C). It is important to note that MG reduction by AR is significantly increased in the presence of GSH.70 In Schwann cells with GLO1 knockout, AR inhibition increased intracellular MG levels and elevated sensitivity to MG.71 This suggests that not only is AR-mediated detoxification of MG important for MG elimination, both in the presence and in the absence of GSH, but also it is a compensatory mechanism if the glyoxalase system is impaired.71 However, the effects of AR overexpression have not been fully elucidated in the context of MG detoxification. AR overexpression was found to contribute to drug resistance in cancers,72 neural atrophy,73 and inflammation,74,75 contributing to a slew of disorders such as those impacting the eye73 and nerves.76 Given its ubiquitous nature, pursuing AR overexpression to promote MG breakdown may not be clinically apt.
MG-AGEs on DNA and Protein
The electrophilic properties of MG drive its reaction with nucleophiles within macromolecules, forming covalent adducts. These adducts have been described for DNA and protein, and extensive work has been performed to determine the impact of these modifications on macromolecular function. Here, we will discuss the main adducts formed on DNA and protein, the pathways cells use to remove these adducts, and the impact of adducts on macromolecular function.77−79
MG-AGEs on Nucleic Acids: Impact on Structure and Repair
MG-Nucleic Acid Adducts
The primary target for MG modification in DNA is deoxyguanosine (dG).79 A 20-fold excess of MG with dG resulted in the formation of a cyclic dihydroimidazolone 1,N2-(1,2-dihydroxy-2-methyl)ethano-dG (cMG-dG) (Figure 3A).7 Additional adducts were later characterized including a product with 2-MG equivalents, N2-(1-carboxyethyl)-7-1-hydroxy-2-oxopropyl-dG (MG-CEdG) and N2-(1-carboxyethyl)-2′-dG (CEdG), which was formed at less than stoichiometric amounts of MG (Figure 3A).13,14,80 Although these adducts are formed in vitro, CEdG is the only adduct observed in genomic DNA.13 This is proposed to occur because cMG-dG is unstable and degrades to generate hydrated MG and dG. This hydrated form of MG only modifies dG at the N2 position, driving CEdG production. cMG-dG does not directly convert to CEdG, as was previously hypothesized.13 In addition to DNA, we hypothesize that MG modifies guanosine nucleotides in RNA to form N2-(1-carboxyethyl)-guanosine (CEG) (Figure 3B).81
Figure 3.

Chemical structures of MG-modified nucleic and amino acids. (A) MG modifies dG, forming three main adducts, CEdG, cMG-dG, and MG-CEdG. Stereocenters are indicated by asterisks, and MG addition is shown in red. dR represents the deoxyribose sugar. (B) Proposed structure of MG-modified RNA adduct CEG. R represents the ribose sugar. (C) Arginine, lysine, and cysteine are primary targets for MG modification. Lysine modification forms CEL, and arginine modification forms MG-H1, MG-H2, and MG-H3. MG-H1 and MG-H3 can be hydrolyzed to form CEA. (D) MG can form lysine dimers MOLD and MODIC.
Regulation of MG Adducts—DNA Repair
DNA adducts induce genomic instability, impact transcription, and are mutagenic. To prevent this, cells have multiple DNA repair pathways, including nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR). Each pathway varies in damage recognition and removal.82 The repair pathway activated depends on the chemistry of the lesion and how it perturbs the DNA structure; each repair pathway employs different proteins that have specific interactions with the DNA to trigger the repair pathway. For instance, the NER pathway is the primary pathway cells use to repair bulky DNA adducts that induce helical distortions,83 while BER repairs small lesions that do not significantly distort the helix,84 and MMR removes mispaired bases in the genome.85 The primary pathway for the removal of MG-DNA adducts is not clear, but both BER and NER are proposed to play a role because shuttle vectors modified with MG have persistent DNA adducts when replicated in cells deficient in XPG, a protein involved in both BER and NER.86
DNA repair efficiency is impacted by protein expression and activity and is regulated by complex mechanisms. Hyperglycemia, which increases the levels of MG-DNA adducts, may also play a role in regulating DNA repair. Ciminera et al. recently showed that high glucose decreased the expression of proteins in the NER pathway, leading to accumulation of MG-DNA adducts corresponding to decreased functional repair.87 The authors suggested that high glucose may downregulate NER protein expression through a HIF-1α-dependent mechanism.87
When cells are unable to efficiently repair DNA damage, it leads to persistent lesions that may cause genomic instability and mutations (Figure 4A). CEdG is associated with single-strand DNA breaks and increased mutation frequency.88 In S. cerevisiae D7, MG induced both mitotic gene conversion and reverse point mutations with a dose-dependent response in mutation frequency.89 Forward selection analysis for mutations in the hypoxanthine phosphoribosyl transferase (HPRT) gene also revealed MG to be a mutagen in Chinese hamster lung cells and T-cell lymphocytes.90,91 Mutagenesis in T cells was observed with both a single high-dose MG treatment (1 mM) and multiple low-dose MG treatments (0.1 mM).91
Figure 4.
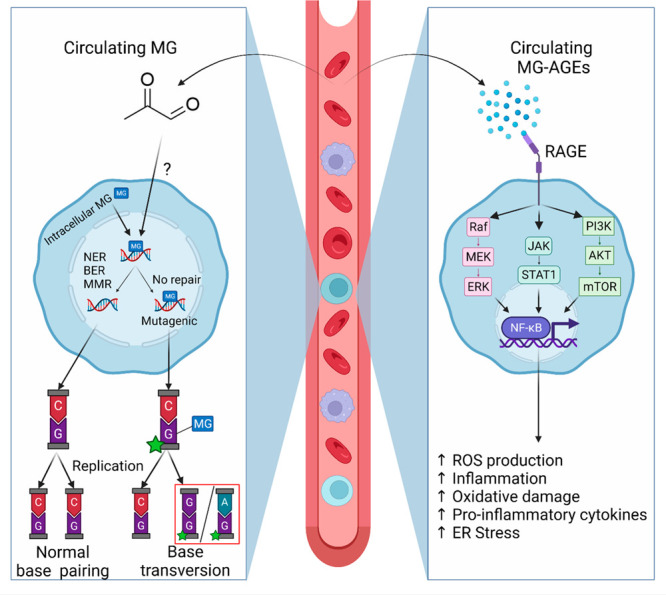
MG forms DNA MG-AGEs. (A) Adducts formed on deoxyguanosine bases can lead to improper base transversions in which guanine incorrectly base pairs with either adenine or guanine. (B) AGEs activate RAGE, and downstream signaling cascades to trigger NF-kB activation, leading to ROS production, inflammation, oxidative stress, etc. Created with BioRender.com.
To define the specific mutations induced by MG, Tamae et al. utilized MG-modified shuttle vectors.86 The MG-adduct density was quantified, and the shuttle vectors were transfected into XPG-proficient or -deficient human fibroblasts, a protein involved in both BER and NER.92 A linear, dose-dependent increase in mutations was observed in the supF tRNA marker gene.86 However, there was a maximal elevated mutation frequency in the XPG-deficient fibroblast cells at varying adduct densities up to 5-fold background levels.86 Specific sites in the supF gene were preferentially modified, suggesting that there may be sites more susceptible to MG modification. These results were recapitulated by Murata-Kamiya et al., who showed that MG-modified shuttle vectors had guanine transversions when replicated in COS-7 fibroblast cells.93 Further characterization of these mutations via sequencing analysis revealed that multibase deletions and base-pair substitutions were predominant in the mutant signature with the latter being primarily G:C → C:G and G:C → T:A transversions in the supF gene of the shuttle vector (Figure 4A).85E. coli deficient in NER also showed increased levels of MG-induced mutations.94 In addition, treatment of human melanoma WM-2664 cells with MG induced the formation of CEdG adducts, which had mutagenic properties in E. coli and were a substrate for DinB DNA polymerase, a known contributor to mutagenesis.95
MG-Protein Adducts: Formation and Impact on Structure and Function
MG-Protein Adduct Formation
In addition to DNA, MG modifies amino acids including lysine, arginine, and cysteine, forming protein adducts that can impact the structure and function (Figure 3C). Initially, Takahashi found that high MG concentrations modified free amine-containing amino acids, specifically lysine and arginine, and hypothesized that thiol-containing amino acids could be modified as well.96 This was later confirmed by Lo et al., who demonstrated that MG modifies cysteine, forming a reversible hemithioacetal, later named carboxyethyl cysteine (CEC).78 Physiological MG concentrations modify proteins, particularly BSA, producing the fluorescent imidazole derivative MG-H1 (Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)ornithine).77 MG-H1 is the predominant adduct, but two other hydroimidazolones also form: MG-H2 (2-amino-5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)pentanoic acid) and the significantly less abundant MG-H3 (2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)pentanoic acid).77 MG-H3 can be hydrolyzed to form carboxyethyl arginine (CEA).97 It was previously hypothesized that MG-H1 is resistant to hydrolysis, but McEwen et al. recently demonstrated that it is also hydrolyzed to form CEA.98 MG also modifies Nα-acetyllysine, forming a glycosylamine, Nε-carboxyethyllysine (CEL) (Figure 3C).78 CEL is found in lens protein and is associated with age.99 While there is significant CEL formation in vitro, MG-H1 is the most abundant amino acid adduct in clinical samples, approximately 10 times greater than CEL.100
Adducted amino acids can undergo a secondary modification, resulting in macromolecule cross-linking. MG forms the protein dimers lysine–lysine (MOLD) and lysine–arginine (MODIC) (Figure 3D).101 Cross-linking also occurs between DNA and polymerases, which is proposed to occur through a dG–lysine bond.102,103
Impact of MG-AGEs on Protein Structure and Function
MG modification of amino acids disrupts the enzymatic activity and protein structure.104,105 Endothelial cells exposed to hyperglycemia had increased cellular MG concentrations and protein MG-AGEs.106 Proteomics analysis revealed that 17% of proteins had low-level MG modification including those involved in protein synthesis, protein folding, kinase signaling, glycolysis, and gluconeogenesis.106 The proteins with the highest number of modifications were pyruvate kinase M, β-actin, α-enolase, and heat shock protein (HSP) 90-β.106 HSPs are chaperone proteins that are elevated in response to stress, and in addition to HSP90-β, MG also modifies HSP6, HSP27, and α-crystallin (protein with HSP domains). This interrupts their interactions with other proteins and impairs chaperone function.107−110 Collagen modified with MG shows a decreased ability to adhere to mesangial cells.111 Human serum albumin is also a target for MG modification.112 There is a hot spot of modification at Arg410, which is located in drug binding site II and in the active site for esterase activity.112 HIF-1α modification by MG decreases heterodimer formation and promoter binding.113 This is a potential mechanism for the decreased DNA repair protein expression in cells grown in high glucose observed by Ciminera et al. as DNA repair proteins are targets for HIF-1α transcription activity.87 Recently, work from Marnett demonstrated that histones are targets for MG modification, potentially regulating gene expression.51 Histone modification by MG has also been associated with changes in chromatin architecture related to disease.114
RAGE Activation and Signaling by MG-AGEs
In addition to changing DNA and protein expression and stability, MG-AGEs are proposed to impact cell function by binding to and activating the receptor for AGEs (RAGE). RAGE is an immunoglobulin transmembrane pattern-recognition receptor that is expressed on a range of cells, including endothelial,115,116 immune,116,117 skeletal muscle,118 and cancer cells.119−121 RAGE is generally present in three primary forms, as a full-length membrane-bound receptor (flRAGE), as a soluble product (sRAGE) created by ADAM10-mediated cleavage of flRAGE, and as a splice variant known as endogenous secretory RAGE (esRAGE).122 Binding of MG-AGEs to RAGE has been investigated both with free modified amino acids and with modified proteins such as albumin. Xie et al. found that CEL does not bind to RAGE, but protein with CEL modifications does.123 However, nuclear magnetic resonance (NMR) studies revealed that CEL-containing peptides bind specifically to a positively charged moiety of the V domain of RAGE.124 Additional NMR studies demonstrated that MG-derived hydroimidazolones (MG-H1–3) bind to a positively charged pocket of the V domain of RAGE, similar to CEL.125 MG-modified albumin (MG-BSA) also binds to RAGE to cause signal transduction; treatment of A549 adenocarcinoma cells with MG-BSA caused an upregulation of JNK phosphorylation in a RAGE-dependent mechanism.125
In addition to AGEs, RAGE is bound by structurally diverse ligands, including phosphatidylserine,126 high-mobility group box-1 (HMGB1) protein,127 S100b,128 lipopolysaccharides,126,129 and nucleic acids.130 RAGE activates JAK/STAT,131 PI3K/AKT/mTOR,132 and MAPK/ERK,133 leading to NFκB activation (Figure 4B).134 This upregulates ER stress, ROS production, inflammation, and oxidative damage (Figure 4B).135,136 The NFκB cascade plays a significant role in mediating cellular responses including inflammation, apoptosis, and cellular survival and proliferation.137 Therefore, AGE-dependent activation of the NFκB pathway via RAGE contributes to numerous disease pathologies.131,138,139
The signaling cascade triggered by RAGE activation is influenced by cell type. For instance, RAGE activation can be detrimental to normal cell growth but advantageous for malignant cell growth. In cancer, increased RAGE expression is correlated with a worse clinical prognosis, which is supported by AGE/RAGE signaling, driving survival,140 proliferation,140 migration,141 angiogenesis,142 and metastasis.142 Therefore, AGE/RAGE signaling is proposed to be a therapeutic target to prevent cancer onset and progression.
Physiological Impact of MG and MG-AGEs
MG and MG-AGEs in Disease
MG and MG-AGEs are associated with the pathogenesis of numerous diseases including cancer, diabetes, and cardiac disease.143−146 Here, we discuss the clinical relevance of MG and MG-AGEs in different human diseases and the impact of MG-AGEs in the context of RAGE (Figure 5).
Figure 5.

MG and MG-AGEs have systemic physiological impacts on the body. MG and MG-AGEs have been implicated in the pathogenesis of numerous diseases throughout the body. This includes the brain, heart, skeletomuscular system, liver, pancreas, kidney, and immune system. Created with BioRender.com.
MG and MG-AGEs in Cardiovascular Disease
Patients with heart failure secondary to diabetes have increased MG-AGEs on actin and myosin in heart muscle when compared to nonfailing controls or heart failure in individuals without diabetes.147 These modifications interfere with protein interaction with muscle and disrupt calcium sensitivity, both processes that are critical for proper cardiac function.147 In a study of comorbidity with HIV infection, a positive association between MG in the heart and plasma and an enhanced risk for heart failure was observed.148 Moreover, higher plasma MG levels were consistently associated with fatal cardiovascular events in individuals with type 1 diabetes (T1D).149 A similar finding was also made in individuals with type 2 diabetes (T2D) with elevated plasma MG associated with higher risk for atherosclerosis and high blood pressure, spurring interest in its utility as a predictive biomarker for heart disease.150
In mice, MG-AGEs in the heart were associated with worse outcomes following myocardial infarction, specifically negative cardiac remodeling and cardiac dysfunction.151 In hypertensive rats, elevated aortic and plasma levels of MG and MG-AGEs were associated with oxidative stress, endothelial dysfunction, high blood pressure, and altered vasculature.152 These findings have since been recapitulated in patients with diabetes; elevated plasma and serum MG-AGEs were associated with microvascular complications, higher blood pressure, and markers of atherosclerosis and coronary heart disease.153−157
The AGE/RAGE axis, which describes the interplay between AGE production, binding to RAGE, and RAGE activation, has been associated with coronary artery disease resulting from hyperglycemia due to insulin resistance from T2D.158−160 AGEs are proposed to lead to cardiac dysfunction by cross-linking low-density lipoproteins and extracellular matrix proteins such as collagen and elastin, causing stiffening of the blood vessel lining.159 Blockade of the AGE/RAGE axis via administration of sRAGE in mice demonstrated a dose-dependent drop in markers of atherosclerosis development.161 Taken together, this suggests that MG-AGEs may serve as an early indicator of impending atherosclerosis development and reduced heart function.
MG and MG-AGEs in Neurological Disease
MG and MG-AGEs are associated with the development of neurological disorders such as Alzheimer’s disease (AD), cerebral atrophy, polyneuropathies, and Parkinson’s disease (PD).162−165 In humans, there is a negative association between serum MG levels and memory, overall executive function, and lower gray matter volume.162,166 This suggests that MG is associated with subsequent neurodegeneration and cognitive decline, particularly in older people. In AD, MG and MG-AGEs are posited to accumulate in β-amyloid plaque deposits and neurofibrillarytangles.167,168 In neuroblastoma cells, MG was neurotoxic and associated with increased ROS levels, triggering neuronal damage commonly seen in PD.169 In nerve biopsies of patients with vasculitic polyneuropathy, increased MG-AGEs, RAGE, and NFκB expression were detected in neural mononuclear cells and vessels.170−173
How MG and MG-AGEs drive cognitive decline and neurodegeneration is not known; however, they are proposed to trigger mitochondrial damage and inflammation in a RAGE-dependent manner.174 Older mice have elevated MG-AGEs in their cerebral cortices and hippocampi, leading to mitochondrial dysfunction.175 This observation was recapitulated in rats with streptozotocin-induced AD, which exhibited persistent activation of the MG/AGE/RAGE/NOX-2 pathway.176 Transgenic mouse models mimicking AD have increased expression and activation of RAGE in astrocytes, particularly those in the hippocampus, a key memory controller.177 The brains of patients with AD have nearly 2-fold higher MG levels than control individuals, with MG being 5–7 times higher in cerebrospinal fluid than in plasma.178 Likewise, AD patients have significantly higher hippocampi MG-AGEs, a finding recapitulated in the nigra neurons of PD patients.179 Furthermore, MG-AGEs are elevated in adipose tissue of patients with neuropathy, AD, or neural aging.180−182 MG and MG-AGEs accumulate in glia and astrocytes, which have an increased expression of RAGE positively correlated with age.177,183−185 This suggests a possible link between elevated MG levels, increased MG-AGE production and accumulation, and their dispersal via the cerebrospinal fluid system, driving RAGE activation on cells throughout the central nervous system and driving disease onset and progression.
MG and MG-AGEs in Skeletomuscular Disease
Recent studies have highlighted the impact of MG and MG-AGEs on the skeletomuscular system.186 MG promotes bone degeneration; in both in vivo rat and in vitro models, MG led to osteoclastogenesis, a step in the osteoporosis development.187,188 In the macrophage cell line RAW264.7, MG activated c-Jun N-terminal kinases, suggesting MG propagates improper bone remodeling via the JNK pathway.187 Patients with diabetes are prone to developing pseudoarthrosis in which their ability to heal bone fractures is delayed.189 Diabetic mice exposed to MG and given a bone defect exhibited significantly delayed bone healing and osteoblast differentiation in a dose-dependent manner as opposed to nondiabetic control mice.190 Interestingly, they also had elevated levels of serum and bone MG-AGEs.190 This suggests that MG detoxification may mitigate bone degeneration and loss in patients with diabetes. Limonene, an antioxidative terpene, also mitigates the effects of MG on diabetic osteopathy; pretreatment of murine osteoblast cell line MC3T3-E1 with limonene reduced endoplasmic reticulum stress, ROS release, and cell death, which was recapitulated using spironolactone.191,192 However, the precise mechanism of this protective effect by both limonene and spironolactone is not fully elucidated.
Canonically, RAGE signaling in skeletal muscle is involved in normal skeletomuscular function and maintenance; however, depending on the ligand, RAGE activation also causes wasting, inflammation, and skeletal muscle aging.143,193,194 These effects are proposed to occur by AGE-mediated aging and cross-linking of critical components of the extracellular matrix such as collagen and the basal lamina.195,196 Collectively, this leaves the individual prone to developing overt skeletomuscular atrophy, sarcopenia, and osteoporosis.193,197 In myoblast cells, MG-AGEs increased oxidative stress and reduced myotube formation while upregulating RAGE expression and activation.198 In diabetic mice, skeletal muscle and plasma have significantly higher MG-AGEs compared to controls, suggesting a link between MG-AGE production and accumulation in the skeletal muscle.198 These findings were corroborated in vitro with MG treatment of C2C12 mouse myotube cells, resulting in higher levels of MG-AGEs.198 A mechanism for AGE/RAGE-mediated pathogenesis of myopathy occurred via AMPK-downregulation of the Akt cascade, exacerbating skeletomuscular dysfunction and subsequent loss.199
MG and MG-AGEs in Renal Disease
Kidney disease is associated with hyperglycemia and metabolic dysfunction, suggesting an association with MG and MG-AGEs. Tezuka et al. conducted an observational study with 150 individuals at different stages of chronic kidney disease (CKD) with the goal of measuring plasma MG levels.200 They found plasma MG was positively associated with CKD, thus identifying MG as a potential tool for estimating kidney disease prognosis and stage.200 Rats treated with MG intragastrically had increased expression of mRNA of pro-inflammatory and oxidative pathways in the kidney transcriptome.201 This data was further supported by proteomic and metabolomic analyses which revealed heightened secretion of extracellular matrix components and membrane phospholipids in MG-treated rats, both of which are indispensable for proper kidney function.201,202 Furthermore, Glo1-deficient nondiabetic mice had altered kidney morphology akin to that of diabetic nephropathy, suggesting that MG contributes to the damage that is observed in CKD.203 Similarly, Glo1 overexpression in diabetic mice and rats mitigated MG-AGE production and subsequent oxidative stress, diabetic kidney disease, and retinopathy.203,204 Protein cross-linking by AGEs contributes heavily to tissue damage and is a marker for organ dysfunction.205 Measurement of MG-AGEs has also been used to predict kidney disease prognosis and progression.206−208 Serum MG-AGEs are significantly, positively associated with decreased renal function in humans.209 Individuals with T1D showed higher urinary excretion of MG-AGEs that proved useful as an indicator of early renal failure.210,211 In individuals with end-stage CKD, MG-AGEs were strongly correlated with indicators of endothelial dysfunction and inflammation.212,213
Recently, Lee et al. made a similar finding in humans, demonstrating that in human mesangial cell lines, MG-AGEs trigger nephropathy via upregulating RAGE expression, leading to ROS production and activation of PI3K/AKT and NFκB.214 RAGE suppression via small-molecule inhibition and siRNA diminished oxidative stress and inflammatory response, suggesting the MG-AGE/RAGE axis contributes to nephrotic damage and dysfunction.215 This was also observed in rats with streptozotocin-induced nephropathy, in which treatment with Moutan Cortex mitigated AGE-induced inflammation, resulting in a protective effect.216 MG-AGEs also act through the RAGE/JNK pathway, causing mitochondrial and ER stress dysfunction as well as an upregulation of apoptotic markers such as p53 and Bax.217 Furthermore, gliclazide, a therapeutic for diabetes, conferred a protective effect on renal damage and dysfunction induced by MG-AGEs and hyperglycemia through inhibition of the AGE/RAGE/ROS/NFκB cascade.218 It is important to note that not every AGE/RAGE interaction triggers the same pathways; Baragetti et al. identified −374 T/A polymorphisms in RAGE that increased the risk of progression to CKD.219 This suggests that in renal dysfunction it may be beneficial to target downstream RAGE signaling, as opposed to upstream MG-AGE/RAGE binding.
MG and MG-AGEs in Liver Disease
In rats given carbon tetrachloride (CCl4) to induce early-stage hepatitis, liver MG levels and d-lactate were elevated compared to control untreated rats.220 Similarly, a clinical trial conducted by Michel et al. revealed a positive correlation between elevated levels of serum MG, liver cirrhosis, and widespread inflammation.221 Elevated serum and circulating MG levels were correlated with worsening liver disease prognosis and increased risk of developing additional liver-related complications, such as ascites.221 In HepG2 cells, MG impaired mitochondria, caused cell death via apoptosis, promoted ROS production, and diminished GSH levels, a critical component of the glyoxalase detoxification system.222 In vivo, MG administration led to acute liver toxicity as evidenced by elevated levels of alanine aminotransferase and aspartate aminotransferase, both indicators of liver health. Taken together, this suggests that MG induces liver disease by triggering mitochondrial dysfunction and oxidative stress as a result of excess ROS production.222
To determine the impact of inflammation on MG regulation, rats were treated with CCl4, which led to MG accumulation in the liver, causing decreased Glo1 expression, increased production of MG-AGEs, and RAGE activation, causing inflammation and stress.223 The impact of MG on decreased Glo1 expression is intriguing as it suggests a positive feedback loop in which elevated MG prevents its own detoxification while continuing to cause hepatic dysfunction.223 Excess MG leads to further MG-AGE production, and several studies have identified elevated levels of MG-AGEs in both the plasma and the serum of patients with liver disease and in obese mice.224−226 Serum MG-AGEs and sRAGE in nondiabetic patients are associated with nonalcoholic fatty liver disease.227 In addition, liver steatosis and inflammation led to elevated levels of circulating MG-AGEs.228
Activation of the MG-AGE/RAGE axis increased apoptosis and TGF-β, TNF-α, IL-8, and IFN-γ levels.229,230 Further characterization of the crosstalk between the pro- and the anti-inflammatory cytokines released as a result of the AGE/RAGE axis in liver disease is needed to understand their role in hepatic dysfunction. The upregulation of these cytokines appears to be ablated upon administration of a siRNA-targeting RAGE in primary rat hepatic stellate cells and prevented overt disease progression.231 Subsequent in vivo administration of RAGE siRNA in rats recapitulated these in vitro findings and delayed the development of liver fibrosis via hampered activation of NFκB.232 Interestingly, deletion of Ager (the gene that encodes RAGE) did not prevent development of liver steatosis, which suggests an alternate RAGE-independent mechanism of liver dysfunction mediated by MG-AGEs.226,233
MG and MG-AGEs in Immune Disorders
Recent studies have implicated MG as a potent immunosuppressor. Price et al. were among the first to show that increased MG inhibited T-cell proliferation and triggered a loss of both pro- and anti-inflammatory cytokines, such as IFN-γ in myeloid cells and TNF-α and IL-10 in T cells.234 MG also reduced metabolic activity in myeloid-derived suppressor cells, a group of regulatory immune cells of myeloid origin.235 This suppressive phenotype was also found in CD8+ cytotoxic T cells, which was proposed to occur via MG transfer, thus causing further immunosuppression.235 MG modification of histone H2A increased immunogenicity, providing some evidence that it may be involved in the autoimmune response in cancer and generation of autoantibodies.236
In vitro studies on the effects of MG-AGE accumulation on immune cells revealed that MG-AGEs impaired the activation of inflammasomes in macrophages, thus hampering innate immunosurveillance.237 This effect was independent of MG-AGEs binding to RAGE but rather occurred through the suppression of macrophage M1 polarization, which would otherwise trigger a pro-inflammatory state prone to phagocytosing foreign or unwanted material.237 Jin et al. found conflicting results, noting that MG-AGEs elevated RAGE expression in macrophages, subsequently triggering macrophage polarization into a pro-inflammatory M1 phenotype through NFκB pathway activation.238
RAGE is expressed on T cells and antigen-presenting cells such as macrophages, dendritic cells, and B cells, and its activation drives both innate and adaptive immune responses.239 For example, the RAGE cascade is critical for T-cell priming and proliferation and mediates interactions between dendritic cells and T cells during cross-priming to propagate an adaptive immune response.240,241 However, this can have adverse effects in disease. RAGE is present on macrophages and microglia, which are two major cell groups that infiltrate and attack the central nervous system, leading to multiple sclerosis. In vivo clinical studies in patients with multiple sclerosis demonstrated an increase in MG and MG-AGEs in astrocytes and cerebrospinal fluid.183 This suggests that circulating MG-AGEs may have a role in paracrine signaling in the body by traveling to and activating RAGE-expressing immune cells, thus exacerbating the immune attack on the nerves.183,242 In multiple sclerosis patients, plasma CEL levels were significantly higher than their control counterparts and were also correlated with rate of relapse.243
MG and MG-AGEs in Cancer
Current literature suggests that MG has a hormetic effect in cancer, serving a pro-tumorigenic role in certain conditions and an antitumorigenic effect in others. A selection of studies investigating the pro- or antitumor role of MG and MG-AGEs in various cancers is summarized in Table 1. As reviewed by Leone et al., there is an inverse correlation between MG concentration and cancer growth and metastasis.144 Cancers preferentially use glycolysis and have enhanced glucose uptake; they produce higher levels of MG.244 To counteract this, cancers overexpress GLO1, which may lead to oversaturation of available GLO1, leading to MG accumulation and toxicity.245−249 Nokin et al. demonstrated that preconditioning cells with high glucose (500 mM) or MG (2.5 mM) successfully conditioned yeast to be more tolerant and resistant to higher concentrations of MG (20 mM) and subsequent oxidative damage, an effect found to be independent of GLO1.250 This is posited to be due to a hormetic mechanism by MG in that at low levels MG drives cancer growth but can cause adverse effects at high levels.250
Table 1. Effects of MG and/or MG-AGEs on Cancer.
| cancer type | MG/MG-AGE | pro/anti cancer | model | dose | effect | mechanism | source |
|---|---|---|---|---|---|---|---|
| breast | MG | pro | MDA-MB-231, MDA-MB-468, MCF7 cells | 300 μM | increase growth and metastatic potential | increase Hsp90 glycation, carbonyl stress, and YAP and TAZ accumulation | (291) |
| MG | pro | MDA-MB-231, MDA-MB-468, MCF7 cells | 300–500 μM | increase metastasis and migration | activation of MEK/ERK/SMAD1 pathway; promotes ECM remodeling | (292) | |
| MG | anti | MCF7, T47D, MDA-MB-231 cells | 100–800 μM | decrease viability, colony formation, migration, and invasion; increase apoptosis | increase p-MAPK, p-JNK, and p-ERK; decrease Bcl-2 expression | (293) | |
| MG-AGE | pro | MDA-MB-231 cells | 25–100 μg/mL | increase proliferation, invasion, and migration | upregulated MMP9 and RAGE; p-ERK and p-p70S6K1 | (294) | |
| MG-AGE | pro | primary human TNBC samples | increase tumor aggression and progression | increase MG detoxification; protected against dicarbonyl stress | (295) | ||
| MG and MG-GE | pro | MCF7 cells | 25–200 μg/mL | increase viability, proliferation, migration | AGE-mediated activation of RAGE and p-ERK; increase CREBP | (296) | |
| brain | MG | anti | T98G and U87MG cells | 25 μM | changes in cell cycle, inhibited proliferation; increase apoptosis, senescence | cells arrest in G1/G0; proliferation inhibited | (297) |
| kidney | MG-AGE | pro | 786-O, A498, and HK-2 cells | 100–800 μg/mL | increase proliferation, survival, migration; decrease apoptosis | increase PCNA, MMPs p-AKT, pERK; decrease Bax and Caspase 3 | (298) |
| liver | MG | anti | Huh-7, HepG2, Hep3B cells | 1 μM | decrease migration, invasion, adhesion | proposed to be p53 dependent | (299−301) |
| leukemia | MG | anti | HL-60 cells | 0–1 mM, 0–524 μM | decrease viability/proliferation; increase DNA damage/apoptosis | cells arrested in G1 with nuclear DNA fragments similar to apoptosis | (291,292) |
| prostate | MG | anti | PC-3 cells | 0–5 mM | decrease growth, increase apoptosis | downregulated cyclin expression and degraded PARP-induced G1 arrest, blocked glycolysis | (302) |
| thyroid | MG and MG-AGE | pro | patient samples; B-CPAP, TPC1, 8505C, CAL62 cells | 5 μM | increase cancer aggression, lethality, invasion/migration | differential E-cad, vimentin, MMP-1, TGF-β1 expression, and increase FAK signaling pathway | (303) |
| colon | MG | pro | CT26 mouse models | 50 mg/kg | increase proliferation, migration, inflammation, oxidative stress | increase IL-6 secretion, increase p-ERK, p-p38 MAPK, p-PI3K, and p-mTOR | (304) |
| MG | anti | SW480, SW620, DLD-1, HCT15 cells; CRC mouse models | 400–1600 μM | decrease growth, proliferation, migration, colony formation; increase apoptosis | increase STAT1, p53, Bax; decrease c-Myc and Bcl-2 | (305) | |
| MG | anti | DLD-1 and SW480 cells; CRC mouse models | 25–2000 μM | decrease viability, migration, invasion, proliferation, growth; increase apoptosis | decrease c-Myc; interfered with glycolysis (less ATP and lactate made, less glucose used) | (306) | |
| MG-AGE | pro | primary human samples | EMT progression and increase tumor aggressiveness; cytokine immunomodulation to promote tumor growth | positive correlation between AGEs and production of IL-2, IL-4, IL-6, and IL-1β | (307) | ||
| MG-AGE | pro | primary human samples | increase glycolytic activity and associated with colorectal cancer progression | increase MG-AGEs levels and dicarbonyl stress, decrease GLO1 activity | (308) |
Any pro-tumor effect by MG and MG-AGEs can likely be attributed to a survival mechanism; cancer adapts to withstand detrimental effects of altered metabolic flux and rather uses it to its benefit, while antitumor effects are due to overwhelming dicarbonyl stress exceeding the tumor‘s detoxification capacity (Table 1). We hypothesize this is due to the complex metabolic and physiological milieu as well as biological variation that may cause different responses to these metabolites. Therefore, the precise concentration of MG and MG-AGEs that delineates a pro- or antitumor impact is not yet clear, as there are additional biological factors to consider. Despite this, measuring blood-derived cultures of both healthy and cancer patients revealed an upregulation of MG-AGEs in those with cancer.251 Furthermore, the pro-cancer role of AGE/RAGE activation and its downstream signaling cascades has been well established across multiple types of cancers, showing it inhibits apoptosis252 and promotes autophagy,252 angiogenesis via VEGF,146 growth,253 inflammation,146 and metastasis via pathways such as AP-1, NFκB, STAT3, SMAD4, MAPK, mTOR, and PI3K.119,254−257
MG and MG-AGEs in Diabetes
Given that glucose metabolism is central to MG production, MG is heavily implicated in diseases where glucose levels are elevated, particularly T1D and T2D. In addition to inducing oxidative stress and inflammation via AGE/RAGE signaling, elevated intracellular MG levels impaired cellular responses to insulin, particularly with ERK1/2 and AKT,258 a signaling pathway indispensable in regulating insulin sensitivity and glucose uptake.259−261 Thus, excess MG contributes to insulin resistance, which is characteristic of T2D.258 Both serum MG and MG-AGEs are significantly elevated in individuals with T1D and T2D.208,262−264 When measured in young patients with T1D without complications, serum MG levels were significantly higher than their control nondiabetic counterparts.265 These findings have been recapitulated in newly diagnosed patients with T2D, supporting a link between elevated MG levels and development of either T1D or T2D.266
Individuals with T1D or T2D are at a high risk of developing secondary complications. Because these complications are associated with poor glycemic control, MG and MG-AGEs are proposed to be associated with and potentially drive them through RAGE-dependent mechanisms.267 The association of both MG and MG-AGEs in diabetic complications such as nephropathy, cardiovascular problems, cancer, and skeletomuscular disease have been extensively covered above. In addition to these, retinopathy,268 neuropathy,269 and vascular complications270 are associated with MG-AGEs.271−273
Measuring MG and MG-AGEs
Direct MG quantification is difficult because of its reactivity; therefore, MG-AGEs are proposed to be more accurate indicators of MG production (Figure 6A). Rabbani and Thornalley pioneered a unique technique to detect and quantify MG using stable isotopic dilution liquid chromatography with tandem mass spectrometry (LC-MS/MS).1 Using this method, they achieved a limit of detection of 8 fmol MG and a limit of quantitation of 90 fmol MG.1 This approach has clinical applications by measuring MG in blood and tissue.1,276 Likewise, technologies to measure MG-AGEs have provided diagnostic and prognostic tools. Initial approaches involved the use of skin autofluorescence, a noninvasive technique that measures tissue accumulation of MG-AGEs, a technique that has proven useful in predicting development of cardiovascular disease,277 microvascular complications,278 or kidney transplant rejection.279 In contrast, newer methods use mass spectrometry or colorimetric and fluorometric profiling of MG-AGEs to allow for rapid measurement and analysis of clinically relevant concentrations.278,280−283
Figure 6.

Approaches for measuring and targeting MG, MG-AGEs, and RAGE. (A) MG-AGEs can be quantified in vitro and in biological samples using LC-MS/MS. Example chromatogram is shown of the elution of MG-modified protein and DNA. (B) Molecules such as aminoguanidine and curcumin can be used to scavenge MG. MG and the formation of MG-AGEs can be targeted via scavenger compounds and using thiamine or benfoatiamine. AGEs can be targeted using sRAGE or alagebrium. RAGE activation can be inhibited using DNA aptamers, anti-RAGE antibodies, fusion proteins, peptides, or pharmacological inhibitors. Created with BioRender.com.
Quantification of MG and MG-AGEs via methods such as LC-MS/MS and ELISA support the use of MG and MG-AGEs as biomarkers for diseases such as T2D, Alzheimer’s disease, chronic kidney disease, nonalcoholic fatty liver disease, atherosclerosis, and others.50,150,200,274,275 MG-H1 was measured in biological samples, including aortic tissue and lens protein, with a high correlation to AGE formation.77,284 Taken together, this presents a novel and quantifiable class of metabolites to aid in not only predicting disease but also informing disease state and prognosis.
Targeting MG and MG-AGEs and Preventing Their formation
The prevalence of MG and MG-AGEs during both normal and disease states makes regulating their levels an attractive target to mitigate disease severity and progression. Aside from the canonical pathways involved in MG detoxification, recent advances have generated considerable interest in approaches to selectively target MG and MG-AGEs to offset their associated damage and effects (Figure 6B, Table 2).145 While there are pharmacological approaches to preventing MG and MG-AGE formation and accumulation, their potential for clinical application requires additional investigation. Alternatively, nonpharmacological approaches such as diet and exercise may have utility in decreasing MG and MG-AGE formation by regulating metabolic flux.145
Table 2. Approaches to Targeting MG and/or AGEs.
| target | name | modality | model | effect | proposed mechanism | source |
|---|---|---|---|---|---|---|
| MG | aminoguanidine | scavenger | diabetic rats, human endothelial cells, rat mesangial cells | prevents AGE formation, attenuates diabetic complications in vivo | binds to carbonyl groups and converts them to nontoxic byproducts (3-amino-6-methyl-1,2,4-triazine and 3-amino-5-methyl-1,2,4-triazine) | (309−317) |
| MG | curcumin | scavenger | mouse blastocysts and embryonic stem cells, human mononuclear and endothelial cells, diabetic rats | decreases apoptosis and oxidative stress, mitigates MG-induced DNA damage, anti-inflammatory and antioxidant | scavenges MG by forming adducts at the 10th carbon between keto carbon groups; synergizes with aminoguanidine for increased benefit | (313,318−322) |
| MG | aucubin | scavenger | in vitro models and in vivo MG-injected rats | inhibits AGE formation and prevents their accumulation | (323) | |
| MG | genistein | traps MG | in vitro | inhibits AGE formation | forms mono-MG and di-MG adducts of genistein | (324) |
| MG | quercetin | traps MG | in vitro | inhibits AGE formation in dose-dependent manner; traps MG and glyoxal | forms MG adducts to make mono-MGO and di-MGO adducts | (325) |
| MG | Eucommia ulmoides | promotes MG detox | in vitro and diabetic mice | inhibits AGE formation and accumulation, decreases RAGE expression, and reduces oxidative stress | upregulates Glo1 and Nrf2 pathway to increase GLO1 production and oxidative protection | (326) |
| AGEs | sRAGE | scavenger | human endothelial cells, diabetic and nondiabetic apoE-null mice | significant reduction of atherosclerotic lesions and inflammation, ameliorates vascular permeability | (115,161,327) | |
| AGEs | thiamine | vitamin | human endothelial cells, bovine retinal endothelial cells | inhibits AGE formation and mitigates oxidative stress | promotes metabolism of glycolysis metabolites | (328) |
| AGEs | benfoatiamine | vitamin | diabetic rats, T1D and T2D patients | restores nerve conduction velocity, inhibits AGE formation, prevents diabetes-induced glycoxidation products, prevents micro- and macrovascular endothelial dysfunction, mitigates oxidative stress | (329−333) | |
| AGEs | alagebrium | cross-link breaker | old primates and humans, diabetic rats | improves cardiac function and output, and endothelial function | removes new AGEs by separating α-dicarbonyl carbon–carbon bonds formed in cross-links | (334−338) |
Targeting RAGE
Pharmacological targeting of RAGE has been explored using antibody and small-molecule-based methods. Blocking RAGE activation by treating endothelial cells with anti-RAGE antibodies decreased the oxidative damage caused by RAGE activation, highlighting its potential as a therapeutic approach.285 Since then, RAGE inhibition using compounds such as FPS-ZM1 or azeliragon in a murine model of acute lung injury reduced RAGE activation and expression and decreased inflammation and damage.286 Similar findings have been reported in murine models of Alzheimer’s disease as well, finding that azeliragon was nontoxic in rats and helped reduced Alzheimer’s injury and progression.287 Additional human clinical trial evidence has supported the use of azeliragon in ameliorating the deleterious effects associated with RAGE activation, yielding promising results and demonstrating its safety profile and tolerance in humans.288,289 Although the precise mechanism by which azeliragon acts has not been fully elucidated, it is proposed to be a RAGE antagonist.287 Similar findings have been reported in studies of GM-1111, a semisynthetic glycosaminoglycan ether, which was found to inhibit interactions between RAGE and its ligands, such as S100B, HMGB-1, and CML-BSA, an AGE.290 In addition, other inhibitors and approaches to targeting RAGE have been developed. They are shown in Figure 6B and summarized in Table 3. However, due to RAGE’s ubiquitous nature, there is a risk of off-target toxicity and unwanted side effects that could result from indiscriminate RAGE targeting.
Table 3. Approaches to Targeting RAGE.
| type | name/nature | model | effect | source |
|---|---|---|---|---|
| antibody | rat antimouse monoclonal | septic mouse model | prolonged survival | (339) |
| humanized monoclonal | pneumonic mouse model | prolonged survival | (340) | |
| rabbit polyclonal | rat liver injury model | decreased necrosis, inflammation, and fibrosis; protected from further liver injury | (341) | |
| goat polyclonal | Duchenne muscular dystrophy mouse model | reduced necrosis and inflammation | (342) | |
| rabbit polyclonal | systemic inflammation mouse model | reduced inflammation and activation of ERK1/2, p65, and IkB | (343) | |
| mouse monoclonal | uremic mouse model | reduces atherosclerosis | (344) | |
| antihuman/mouse monoclonal | neuropathic pain mouse model | attenuation of inflammation and neuropathic pain | (345) | |
| antimouse/rat monoclonal | lung cancer mouse model | suppressed metastasis | (346) | |
| peptide | S100P-derived RAGE antagonist | glioma and pancreatic cancer mouse model | blocked ligands’ ability to bind and stimulate RAGE and NFkB; reduced growth and metastasis of tumors | (347) |
| inhibitory peptides for RAGE signaling | SH-SY5Y and U-87MG cells | reduced neuronal cell death, inhibit invasion and migration of glioma cells | (348) | |
| fusion protein | TAT-SAM blocks interaction between RAGE and SLP76, a critical adaptor protein for RAGE function | septic mouse model | decreases tissue damage and RAGE cytokine release and downstream signaling; prolonged survival of mice | (349) |
| aptamer | short DNA sequence created using SELEX | mesangial cells and diabetic nephropathy rat model | suppression of AGE-induced oxidative stress, inflammation, fibrosis, albuminuria, and podocyte damage | (350) |
| HCT116 cells and colorectal cancer mouse model | suppression of RAGE/NFkB signaling, inhibition of tumor growth, decreases cell proliferation and migration | (351) | ||
| melanoma mouse model | inhibit tumor growth, decrease AGE and RAGE expression, lowered oxidative stress and angiogenesis | (352) | ||
| pharmacological inhibitor | 4,6-bis(4-chlorphenyl)pyrimidine analogue | Alzheimer’s disease mouse model | inhibition of amyloid-beta plaque accumulation, improvement of cognitive function | (353) |
| pyrazole-5-carboxamide | Alzheimer’s disease mouse model | inhibition of amyloid-beta plaque accumulation | (354) | |
| FPS-ZM1 | breast cancer mouse model | impaired tumor growth and angiogenesis, decreased inflammation, inhibition of metastasis | (355) | |
| Alzheimer’s disease mouse model | decreased amyloid-beta plaque accumulation in brain, suppressed inflammation, improved cognitive performance | (356) | ||
| acute lung injury mouse model | decreased RAGE expression and inflammation, restored cell contacts and epithelium integrity | (286) | ||
| azeliragon | acute lung injury mouse model | decreased RAGE expression and inflammation, restored cell contacts and epithelium integrity | (286) | |
| Alzheimer’s disease rat model | reduced AD injury, reversed neuronal damage, increased neurological function | (287) | ||
| semisynthetic glycosaminoglycan ethers (SAGE) | in vitro models and rosacea mouse model | inhibits ligands from binding to RAGE, reduces inflammation | (290) | |
| secondhand smoke mouse model | decreased inflammation, RAGE signaling; increased AXL and Gas6 protein expression | (357,358) |
Conclusion
All living organisms perform metabolism, a process vital for life. Despite its necessity in sustaining life, metabolic perturbations underlie the pathology of many diseases. The mechanisms leading this are not clear, but MG, MG-AGEs, and RAGE are shown to play a critical role in many metabolism-driven diseases. Elucidation of their role in promoting disease onset and progression has been increasingly appreciated and studied as an invaluable asset to aid in our understanding of how and why certain diseases develop.
Despite many significant clinical advances in the treatment of human disease, there remains a gap in knowledge allowing us to predict and intervene to prevent or treat diseases early rather than after irreversible damage has occurred. Furthermore, there is a shortage of clinically relevant and quantifiable biomarkers of disease that would aid in the process. Current ways of predicting disease, such as the measurement of HbA1c in diabetes, only allow physicians to see a snapshot in time. On the other hand, metabolism is a process that can be continually monitored, and metabolism flux can be studied as it occurs. Therefore, we believe that the study of metabolites and their role in disease as progressors and predictors may yield a slew of novel viable biomarker candidates for clinical application. As such, further characterization of the biochemistry and interactions of MG, MG-AGEs, and RAGE is critical to study how their role in the body can be exploited for therapeutic benefit.
Acknowledgments
This work was supported from funding from the NIDDK under award number R21DK12785 to S.C.S.
Glossary
Abbreviations
- 2-ODH
2-oxoaldehyde dehydrogenase
- AD
Alzheimer’s disease
- ALDH
aldehyde dehydrogenase
- AGEs
advanced glycation end products
- AKT
protein kinase B
- ALKBH7
Alkb homologue 7
- AMOO
acetol mono-oxygenase
- AR
aldose reductase
- Bcl-2
B-cell lymphoma-2
- BER
base excision repair
- CEdG
N2-(1-carboxyethyl)-2′-deoxyguanosine
- CEG
N2-(1-carboxyethyl)-guanosine
- CEA
N7-carboxyethyl arginine
- CEC
carboxyethyl cysteine
- CEL
Nε-carboxyethyllysine
- CKD
chronic kidney disease
- cMG-dG
1,N2-(1,2-dihydroxy-2-methyl)ethano-deoxyguanosine
- dG
deoxyguanosine
- DHAP
dihydroxyacetone phosphate
- ERK
extracellular signal-regulated kinase
- esRAGE
endogenous secretory receptor for advanced glycation end products
- G3P
glyceraldehyde-3-phosphate
- G3PDH
glycerol-3-phosphate dehydrogenase
- G3PO
l-glycerol-3-phosphate oxidase
- GK
glycerol kinase
- GLO1 (human), Glo1 (murine)
glyoxalase 1
- GLO2
glyoxalase 2
- GSH
glutathione
- HMGB1
high-mobility group box-1
- HPLC
high-performance liquid chromatography
- HPRT
hypoxanthine phosphorylribosyltransferase
- HSP
heat shock protein
- JAK
Janus kinase
- JNK
c-Jun N-terminal kinase
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- MAPK
mitogen-activated protein kinases
- MEK
mitogen-activated ERK kinase
- MG
methylglyoxal
- MG-AGEs
methylglyoxal-derived advanced glycation end products
- MG-BSA
methylglyoxal bovine serum albumin
- MG-CEdG
N2-(1-carboxyethyl)-7-1-hydroxy-2-oxopropyl-dG
- MG-H1
Nδ -(5-hydro-5-methyl-4-imidazolon-2-yl)ornithine
- MG-H2
2-amino-5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)pentanoic acid
- MG-H3
2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)pentanoic acid
- MMP
matrix metalloproteinase
- MMR
mismatch repair
- MODIC
lysine–arginine protein dimer induced by MG
- MOLD
lysine–lysine protein dimer induced by MG
- MP
myeloperoxidase
- MS
methylglyoxal synthase
- mTOR
mammalian target of rapamycin
- NAD
nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- NER
nucleotide excision repair
- NFκB
nuclear factor kappa B
- PD
Parkinson’s disease
- PI3K
phosphatidylinositol 3-kinase
- RAGE
receptor for advanced glycation end products
- ROS
reactive oxygen species
- SMAD1
SMAD family member 1
- sRAGE
soluble receptor for advanced glycation end products
- SSAO
semicarbazide-sensitive amine oxidase
- STAT
signal transducer and activator of transcription
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TAZ
transcriptional coactivator with PDZ-binding motif
- TDH
threonine dehydrogenase
- TGF-β1
transforming growth factor beta 1
- TNBC
triple-negative breast cancer
- TPI
triose phosphate isomerase
Biographies
Seigmund Wai Tsuen Lai is a Ph.D. candidate at the Irell and Manella Graduate School of Biological Sciences at City of Hope Comprehensive Cancer Center. He earned his B.Sc. degree in Neurobiology, Physiology, and Behavior at University of California, Davis. He currently works in the lab of Sarah Shuck in the Department of Diabetes and Cancer Metabolism at the Arthur Riggs Diabetes and Metabolism Research Institute.
Edwin De Jesus Lopez Gonzalez is a research associate working in the lab of Sarah Shuck in the Department of Diabetes and Cancer Metabolism at the Arthur Riggs Diabetes and Metabolism Research Institute at City of Hope. He earned his B.A. degree in Neuroscience and Chemistry at Pomona College.
Tala Zoukari is the lab manager of the lab of Sarah Shuck, where she also works as a research associate, in the Department of Diabetes and Cancer Metabolism at the Arthur Riggs Diabetes and Metabolism Research Institute at City of Hope. She completed her B.S. degree in Biology at the University of California, Riverside and recently earned her MPH degree at George Washington University’s Milken Institute School of Public Health. She will be attending Loma Linda University in Fall 2022 to begin her studies towards a Doctorate in Public Health.
Priscilla Ki works as a research associate in the lab of Sarah Shuck in the Department of Diabetes and Cancer Metabolism at the Arthur Riggs Diabetes and Metabolism Research Institute at the City of Hope. She completed her B.A. degree in Neuroscience at Pomona College.
Sarah Shuck is an assistant professor in the Department of Diabetes and Cancer Metabolism at the Arthur Riggs Diabetes and Metabolism Research Institute at City of Hope. She completed her Ph.D. degree in Biochemistry at Indiana University School of Medicine with John Turchi, studying small-molecule inhibitors of the nucleotide excision repair pathway. She performed her postdoctoral work with Larry Marnett at Vanderbilt University School of Medicine, where she was an NIH Ruth Kirschstein postdoctoral scholar. She focused on electrophile chemistry, mass spectrometry, and DNA adducts. She next worked as a Research Assistant Professor with John Termini in the Department of Molecular Medicine of City of Hope, where she identified novel biomarkers for predicting metabolic disease. In March of 2021, she began her independent laboratory, where she is exploring the clinical utility of methylglyoxal adducts to predict stage type 1 diabetes and related complications. She is also exploring the role of these adducts on genomic stability and as potential drivers of disease. She is a member of the American Chemical Society Toxicology Division, where she serves on the executive committee.
Author Contributions
S.W.T.L., E.D.J.L.G., T.Z., P.K., and S.C.S. researched data for the article. S.W.T.L., E.D.J.L.G., and S.C.S. prepared the figures. S.C.S. supervised the laboratory. All authors contributed to the writing and revision of the article and approved of the final version.
The authors declare no competing financial interest.
References
- Rabbani N.; Thornalley P. J. Measurement of Methylglyoxal by Stable Isotopic Dilution Analysis LC-MS/MS with Corroborative Prediction in Physiological Samples. Nat. Protoc. 2014, 9 (8), 1969–1979. 10.1038/nprot.2014.129. [DOI] [PubMed] [Google Scholar]
- Shuck S. C.; Wauchope O. R.; Rose K. L.; Kingsley P. J.; Rouzer C. A.; Shell S. M.; Sugitani N.; Chazin W. J.; Zagol-Ikapitte I.; Boutaud O.; Oates J. A.; Galligan J. J.; Beavers W. N.; Marnett L. J. Protein Modification by Adenine Propenal. Chem. Res. Toxicol. 2014, 27 (10), 1732–1742. 10.1021/tx500218g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddukuri L.; Shuck S. C.; Eoff R. L.; Zhao L.; Rizzo C. J.; Guengerich F. P.; Marnett L. J. Replication, Repair, and Translesion Polymerase Bypass of N6-Oxopropenyl-2′-Deoxyadenosine. Biochemistry 2013, 52 (48), 8766–8776. 10.1021/bi401103k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavers W. N.; Rose K. L.; Galligan J. J.; Mitchener M. M.; Rouzer C. A.; Tallman K. A.; Lamberson C. R.; Wang X.; Hill S.; Ivanova P. T.; Brown H. A.; Zhang B.; Porter N. A.; Marnett L. J. Protein Modification by Endogenously Generated Lipid Electrophiles: Mitochondria as the Source and Target. ACS Chem. Biol. 2017, 12 (8), 2062–2069. 10.1021/acschembio.7b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarillo J. M.; Rose K. L.; Galligan J. J.; Xu S.; Marnett L. J. Covalent Modification of CDK2 by 4-Hydroxynonenal as a Mechanism of Inhibition of Cell Cycle Progression. Chem. Res. Toxicol. 2016, 29 (3), 323–332. 10.1021/acs.chemrestox.5b00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluise C. D.; Rose K.; Boiani M.; Reyzer M. L.; Manna J. D.; Tallman K.; Porter N. A.; Marnett L. J. Peptidyl-Prolyl Cis/Trans-Isomerase A1 (Pin1) Is a Target for Modification by Lipid Electrophiles. Chem. Res. Toxicol. 2013, 26 (2), 270–279. 10.1021/tx300449g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath C. E.; Tallman K. A.; Porter N. A.; Marnett L. J. Structure-Activity Analysis of Diffusible Lipid Electrophiles Associated with Phospholipid Peroxidation: 4-Hydroxynonenal and 4-Oxononenal Analogues. Chem. Res. Toxicol. 2011, 24 (3), 357–370. 10.1021/tx100323m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila A.; Tallman K. A.; Jacobs A. T.; Liebler D. C.; Porter N. A.; Marnett L. J. Identification of Protein Targets of 4-Hydroxynonenal Using Click Chemistry for Ex Vivo Biotinylation of Azido and Alkynyl Derivatives. Chem. Res. Toxicol. 2008, 21 (2), 432–444. 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S.; Karmakar K.; Chakravortty D. Cells Producing Their Own Nemesis: Understanding Methylglyoxal Metabolism. IUBMB Life 2014, 66 (10), 667–678. 10.1002/iub.1324. [DOI] [PubMed] [Google Scholar]
- Thornalley P. J.; Langborg A.; Minhas H. S. Formation of Glyoxal, Methylglyoxal and 3-Deoxyglucosone in the Glycation of Proteins by Glucose. Biochem. J. 1999, 344 (1), 109–116. 10.1042/bj3440109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willetts A. J.; Turner J. M. Threonine Metabolism in a Strain of Bacillus Subtilis. Biochem. J. 1970, 117 (2), 27P–28P. 10.1042/bj1170027Pb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard G. A. J.; Skutches C. L.; Hoeldtke R. D.; Owen O. E. Acetone Metabolism in Humans during Diabetic Ketoacidosis. Diabetes 1986, 35 (6), 668–674. 10.2337/diab.35.6.668. [DOI] [PubMed] [Google Scholar]
- Shuck S. C.; Wuenschell G. E.; Termini J. S. Product Studies and Mechanistic Analysis of the Reaction of Methylglyoxal with Deoxyguanosine. Chem. Res. Toxicol. 2018, 31 (2), 105–115. 10.1021/acs.chemrestox.7b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmann M.; Bidmon C.; Angerer J.; Pischetsrieder M. Identification of DNA Adducts of Methylglyoxal. Chem. Res. Toxicol. 2005, 18 (10), 1586–1592. 10.1021/tx0501278. [DOI] [PubMed] [Google Scholar]
- Shapiro R.; Cohen B. I.; Shiuey S. J.; Maurer H. On the Reaction of Guanine with Glyoxal, Pyruvaldehyde, and Kethoxal, and the Structure of the Acylguanines. A New Synthesis of N2-Alkylguanines. Biochemistry 1969, 8 (1), 238–245. 10.1021/bi00829a034. [DOI] [PubMed] [Google Scholar]
- Phillips S. A.; Thornalley P. J. The Formation of Methylglyoxal from Triose Phosphates. Investigation Using a Specific Assay for Methylglyoxal. Eur. J. Biochem. 1993, 212 (1), 101–105. 10.1111/j.1432-1033.1993.tb17638.x. [DOI] [PubMed] [Google Scholar]
- Richard J. P. Mechanism for the Formation of Methylglyoxal from Triosephosphates. Biochem. Soc. Trans. 1993, 21 (2), 549–553. 10.1042/bst0210549. [DOI] [PubMed] [Google Scholar]
- Seo G.-Y.; Lee H.-S.; Kim H.; Cho S.; Na J.-G.; Yeon Y. J.; Lee J. A Novel Hyperthermophilic Methylglyoxal Synthase: Molecular Dynamic Analysis on the Regional Fluctuations. Sci. Rep. 2021, 11 (1), 2538. 10.1038/s41598-021-82078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery J.; Jörnvall H. Enzyme Relationships in a Sorbitol Pathway That Bypasses Glycolysis and Pentose Phosphates in Glucose Metabolism. Proc. Natl. Acad. Sci. U. S. A. 1983, 80 (4), 901–905. 10.1073/pnas.80.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao L.; Su F.; Yang Y.; Liu Y.; Wang L.; Zhan Y.; Yin R.; Yu M.; Li C.; Yang X.; Ge C. Glycerol Kinase Enhances Hepatic Lipid Metabolism by Repressing Nuclear Receptor Subfamily 4 Group A1 in the Nucleus. Biochem. Cell Biol. Biochim. Biol. Cell. 2020, 98 (3), 370–377. 10.1139/bcb-2019-0317. [DOI] [PubMed] [Google Scholar]
- Maenpuen S.; Watthaisong P.; Supon P.; Sucharitakul J.; Parsonage D.; Karplus P. A.; Claiborne A.; Chaiyen P. Kinetic Mechanism of L-α-Glycerophosphate Oxidase from Mycoplasma Pneumoniae. FEBS J. 2015, 282 (16), 3043–3059. 10.1111/febs.13247. [DOI] [PubMed] [Google Scholar]
- Hartley C. J.; French N. G.; Scoble J. A.; Williams C. C.; Churches Q. I.; Frazer A. R.; Taylor M. C.; Coia G.; Simpson G.; Turner N. J.; Scott C. Sugar Analog Synthesis by in Vitro Biocatalytic Cascade: A Comparison of Alternative Enzyme Complements for Dihydroxyacetone Phosphate Production as a Precursor to Rare Chiral Sugar Synthesis. PloS One 2017, 12 (11), e0184183. 10.1371/journal.pone.0184183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstad R. I.; McKinley-McKee J. S. Methylglyoxal and the Polyol Pathway. Three-Carbon Compounds Are Substrates for Sheep Liver Sorbitol Dehydrogenase. FEBS Lett. 1993, 330 (1), 31–35. 10.1016/0014-5793(93)80913-F. [DOI] [PubMed] [Google Scholar]
- Beisswenger B. G. K.; Delucia E. M.; Lapoint N.; Sanford R. J.; Beisswenger P. J. Ketosis Leads to Increased Methylglyoxal Production on the Atkins Diet. Ann. N.Y. Acad. Sci. 2005, 1043, 201–210. 10.1196/annals.1333.025. [DOI] [PubMed] [Google Scholar]
- Casazza J. P.; Felver M. E.; Veech R. L. The Metabolism of Acetone in Rat. J. Biol. Chem. 1984, 259 (1), 231–236. 10.1016/S0021-9258(17)43646-5. [DOI] [PubMed] [Google Scholar]
- Bondoc F. Y.; Bao Z.; Hu W. Y.; Gonzalez F. J.; Wang Y.; Yang C. S.; Hong J. Y. Acetone Catabolism by Cytochrome P450 2E1: Studies with CYP2E1-Null Mice. Biochem. Pharmacol. 1999, 58 (3), 461–463. 10.1016/S0006-2952(99)00111-2. [DOI] [PubMed] [Google Scholar]
- Zhang M. M.; Ong C. Y.; Walker M. J.; McEwan A. G. Defence against Methylglyoxal in Group A Streptococcus: A Role for Glyoxylase I in Bacterial Virulence and Survival in Neutrophils?. Pathog. Dis. 2016, 74 (2), ftv122. 10.1093/femspd/ftv122. [DOI] [PubMed] [Google Scholar]
- Tressel T.; Thompson R.; Zieske L. R.; Menendez M. I.; Davis L. Interaction between L-Threonine Dehydrogenase and Aminoacetone Synthetase and Mechanism of Aminoacetone Production. J. Biol. Chem. 1986, 261 (35), 16428–16437. 10.1016/S0021-9258(18)66584-6. [DOI] [PubMed] [Google Scholar]
- Bird M. I.; Nunn P. B.; Lord L. A. Formation of Glycine and Aminoacetone from L-Threonine by Rat Liver Mitochondria. Biochim. Biophys. Acta 1984, 802 (2), 229–236. 10.1016/0304-4165(84)90166-1. [DOI] [PubMed] [Google Scholar]
- Kalapos M. P. The Tandem of Free Radicals and Methylglyoxal. Chem. Biol. Interact. 2008, 171 (3), 251–271. 10.1016/j.cbi.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Allaman I.; Bélanger M.; Magistretti P. J. Methylglyoxal, the Dark Side of Glycolysis. Front. Neurosci. 2015, 9, 23. 10.3389/fnins.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragonès G.; Rowan S.; Francisco S. G.; Whitcomb E. A.; Yang W.; Perini-Villanueva G.; Schalkwijk C. G.; Taylor A.; Bejarano E. The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy. Cells 2021, 10 (8), 1852. 10.3390/cells10081852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley P. J. Pharmacology of Methylglyoxal: Formation, Modification of Proteins and Nucleic Acids, and Enzymatic Detoxification--a Role in Pathogenesis and Antiproliferative Chemotherapy. Gen. Pharmacol. 1996, 27 (4), 565–573. 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- Shinohara M.; Thornalley P. J.; Giardino I.; Beisswenger P.; Thorpe S. R.; Onorato J.; Brownlee M. Overexpression of Glyoxalase-I in Bovine Endothelial Cells Inhibits Intracellular Advanced Glycation Endproduct Formation and Prevents Hyperglycemia-Induced Increases in Macromolecular Endocytosis. J. Clin. Invest. 1998, 101 (5), 1142–1147. 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings E. Q.; Ray J. D.; Zerio C. J.; Trujillo M. N.; McDonald D. M.; Chapman E.; Spiegel D. A.; Galligan J. J. Sirtuin 2 Regulates Protein LactoylLys Modifications. Chembiochem Eur. J. Chem. Biol. 2021, 22 (12), 2102–2106. 10.1002/cbic.202000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z.; Yang X.; Qin J.; Ye K.; Wang X.; Shi H.; Jiang M.; Liu X.; Lu X. Glyoxalase-1 Overexpression Reverses Defective Proangiogenic Function of Diabetic Adipose-Derived Stem Cells in Streptozotocin-Induced Diabetic Mice Model of Critical Limb Ischemia. Stem Cells Transl. Med. 2017, 6 (1), 261–271. 10.5966/sctm.2015-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D.; Brownlee M. Hyperglycemia-Induced Reactive Oxygen Species Increase Expression of the Receptor for Advanced Glycation End Products (RAGE) and RAGE Ligands. Diabetes 2010, 59 (1), 249–255. 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed U.; Dobler D.; Larkin S. J.; Rabbani N.; Thornalley P. J. Reversal of Hyperglycemia-Induced Angiogenesis Deficit of Human Endothelial Cells by Overexpression of Glyoxalase 1 in Vitro. Ann. N.Y. Acad. Sci. 2008, 1126, 262–264. 10.1196/annals.1433.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers O.; Niessen P. M.; Haenen G.; Miyata T.; Brownlee M.; Stehouwer C. D.; De Mey J. G.; Schalkwijk C. G. Hyperglycaemia-Induced Impairment of Endothelium-Dependent Vasorelaxation in Rat Mesenteric Arteries Is Mediated by Intracellular Methylglyoxal Levels in a Pathway Dependent on Oxidative Stress. Diabetologia 2010, 53 (5), 989–1000. 10.1007/s00125-010-1677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni C. A.; Nadtochiy S. M.; Kennedy L.; Zhang J.; Chhim S.; Alwaseem H.; Murphy E.; Fu D.; Brookes P. S. ALKBH7Mediates Necrosis via Rewiring of Glyoxal Metabolism. eLife 2020, 9, e58573. 10.7554/eLife.58573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A.; Bekkhozhin Z.; Omertassova N.; Baizhumanov T.; Yeltay G.; Akhmetali M.; Toibazar D.; Utepbergenov D. The Apparent Deglycase Activity of DJ-1 Results from the Conversion of Free Methylglyoxal Present in Fast Equilibrium with Hemithioacetals and Hemiaminals. J. Biol. Chem. 2019, 294 (49), 18863–18872. 10.1074/jbc.RA119.011237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N.; Kimura M.; Queliconi B. B.; Kojima W.; Mishima M.; Takagi K.; Koyano F.; Yamano K.; Mizushima T.; Ito Y.; Tanaka K. Parkinson’s Disease-Related DJ-1 Functions in Thiol Quality Control against Aldehyde Attack in Vitro. Sci. Rep. 2017, 7 (1), 12816. 10.1038/s41598-017-13146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M.; Weickert M. O.; Qureshi S.; Kandala N.-B.; Anwar A.; Waldron M.; Shafie A.; Messenger D.; Fowler M.; Jenkins G.; Rabbani N.; Thornalley P. J. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65 (8), 2282–2294. 10.2337/db16-0153. [DOI] [PubMed] [Google Scholar]
- Schumacher D.; Morgenstern J.; Oguchi Y.; Volk N.; Kopf S.; Groener J. B.; Nawroth P. P.; Fleming T.; Freichel M. Compensatory Mechanisms for Methylglyoxal Detoxification in Experimental & Clinical Diabetes. Mol. Metab. 2018, 18, 143–152. 10.1016/j.molmet.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S.; Kwon D. M.; Kwon K.; Park C. Generation and Characterization of Mouse Knockout for Glyoxalase 1. Biochem. Biophys. Res. Commun. 2017, 490 (2), 460–465. 10.1016/j.bbrc.2017.06.063. [DOI] [PubMed] [Google Scholar]
- Shafie A.; Xue M.; Barker G.; Zehnder D.; Thornalley P. J.; Rabbani N. Reappraisal of Putative Glyoxalase 1-Deficient Mouse and Dicarbonyl Stress on Embryonic Stem Cells in Vitro. Biochem. J. 2016, 473 (22), 4255–4270. 10.1042/BCJ20160691. [DOI] [PubMed] [Google Scholar]
- Lodd E.; Wiggenhauser L. M.; Morgenstern J.; Fleming T. H.; Poschet G.; Büttner M.; Tabler C. T.; Wohlfart D. P.; Nawroth P. P.; Kroll J. The Combination of Loss of Glyoxalase1 and Obesity Results in Hyperglycemia. JCI Insight 2019, 4 (12), 126154. 10.1172/jci.insight.126154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemva J.; Fink C. A.; Fleming T. H.; Schmidt L.; Loft A.; Herzig S.; Knieß R. A.; Mayer M.; Bukau B.; Nawroth P. P.; Tyedmers J. Hormesis Enables Cells to Handle Accumulating Toxic Metabolites during Increased Energy Flux. Redox Biol. 2017, 13, 674–686. 10.1016/j.redox.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y.; Choi B. Y.; Murata K.; Kimura A. Sexual Response of Saccharomyces Cerevisiae: Phosphorylation of Yeast Glyoxalase I by a Cell Extract of Mating Factor-Treated Cells. J. Biochem. (Tokyo) 1990, 108 (1), 4–6. 10.1093/oxfordjournals.jbchem.a123159. [DOI] [PubMed] [Google Scholar]
- Moraru A.; Wiederstein J.; Pfaff D.; Fleming T.; Miller A. K.; Nawroth P.; Teleman A. A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27 (4), 926. 10.1016/j.cmet.2018.02.003. [DOI] [PubMed] [Google Scholar]
- Galligan J. J.; Wepy J. A.; Streeter M. D.; Kingsley P. J.; Mitchener M. M.; Wauchope O. R.; Beavers W. N.; Rose K. L.; Wang T.; Spiegel D. A.; Marnett L. J. Methylglyoxal-Derived Posttranslational Arginine Modifications Are Abundant Histone Marks. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (37), 9228–9233. 10.1073/pnas.1802901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratmann B.; Engelbrecht B.; Espelage B. C.; Klusmeier N.; Tiemann J.; Gawlowski T.; Mattern Y.; Eisenacher M.; Meyer H. E.; Rabbani N.; Thornalley P. J.; Tschoepe D.; Poschmann G.; Stühler K. Glyoxalase 1-Knockdown in Human Aortic Endothelial Cells - Effect on the Proteome and Endothelial Function Estimates. Sci. Rep. 2016, 6, 37737. 10.1038/srep37737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortall K.; Djeghader A.; Magner E.; Soulimane T. Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective. Front. Mol. Biosci. 2021, 8, 659550. 10.3389/fmolb.2021.659550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.-Y.; Wang Y.-B.; Han B.; Yang B.; Qiang Y.-W.; Zhang Y.; Wang Z.; Huang X.; Liu J.; Chen Y.-D.; Ren J.; Cao F.; Xu Y. Activation of Aldehyde Dehydrogenase 2 Slows down the Progression of Atherosclerosis via Attenuation of ER Stress and Apoptosis in Smooth Muscle Cells. Acta Pharmacol. Sin. 2018, 39 (1), 48–58. 10.1038/aps.2017.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne J.; Joanne P.; Catelain C.; Riveron S.; Bayer A. C.; Lafable J.; Punzon I.; Blot S.; Agbulut O.; Vilquin J.-T. Aldehyde Dehydrogenases Contribute to Skeletal Muscle Homeostasis in Healthy, Aging, and Duchenne Muscular Dystrophy Patients. J. Cachexia Sarcopenia Muscle 2020, 11 (4), 1047–1069. 10.1002/jcsm.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A. U.; Van Wassenhove L. D.; Logas K. R.; Minhas P. S.; Andreasson K. I.; Weinberg K. I.; Chen C.-H.; Mochly-Rosen D. Aldehyde Dehydrogenase 2 Activity and Aldehydic Load Contribute to Neuroinflammation and Alzheimer’s Disease Related Pathology. Acta Neuropathol. Commun. 2019, 7 (1), 190. 10.1186/s40478-019-0839-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Huang W.; Cheng C.-H.; Zhou L.; Jiang G.-B.; Hu Y.-Y. Association Between Aldehyde Dehydrogenase-2 Polymorphisms and Risk of Alzheimer’s Disease and Parkinson’s Disease: A Meta-Analysis Based on 5,315 Individuals. Front. Neurol. 2019, 10, 290. 10.3389/fneur.2019.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaguirre G.; Kikonyogo A.; Pietruszko R. Methylglyoxal as Substrate and Inhibitor of Human Aldehyde Dehydrogenase: Comparison of Kinetic Properties among the Three Isozymes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 119 (4), 747–754. 10.1016/S0305-0491(98)00051-0. [DOI] [PubMed] [Google Scholar]
- Vander Jagt D. L.; Hunsaker L. A. Methylglyoxal Metabolism and Diabetic Complications: Roles of Aldose Reductase, Glyoxalase-I, Betaine Aldehyde Dehydrogenase and 2-Oxoaldehyde Dehydrogenase. Chem. Biol. Interact. 2003, 143–144, 341–351. 10.1016/S0009-2797(02)00212-0. [DOI] [PubMed] [Google Scholar]
- Chen C.-H.; Ferreira J. C. B.; Gross E. R.; Mochly-Rosen D. Targeting Aldehyde Dehydrogenase 2: New Therapeutic Opportunities. Physiol. Rev. 2014, 94 (1), 1–34. 10.1152/physrev.00017.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai K. M.; Chang T.; Wang H.; Banigesh A.; Dhar A.; Liu J.; Untereiner A.; Wu L. Oxidative Stress and Aging: Is Methylglyoxal the Hidden Enemy?. Can. J. Physiol. Pharmacol. 2010, 88 (3), 273–284. 10.1139/Y10-001. [DOI] [PubMed] [Google Scholar]
- Lou B.; Boger M.; Bennewitz K.; Sticht C.; Kopf S.; Morgenstern J.; Fleming T.; Hell R.; Yuan Z.; Nawroth P. P.; Kroll J. Elevated 4-Hydroxynonenal Induces Hyperglycaemia via Aldh3a1 Loss in Zebrafish and Associates with Diabetes Progression in Humans. Redox Biol. 2020, 37, 101723. 10.1016/j.redox.2020.101723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S. K.; Ramana K. V.; Bhatnagar A. Role of Aldose Reductase and Oxidative Damage in Diabetes and the Consequent Potential for Therapeutic Options. Endocr. Rev. 2005, 26 (3), 380–392. 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- Vander Jagt D. L.; Robinson B.; Taylor K. K.; Hunsaker L. A. Reduction of Trioses by NADPH-Dependent Aldo-Keto Reductases. Aldose Reductase, Methylglyoxal, and Diabetic Complications. J. Biol. Chem. 1992, 267 (7), 4364–4369. 10.1016/S0021-9258(18)42844-X. [DOI] [PubMed] [Google Scholar]
- Jannapureddy S.; Sharma M.; Yepuri G.; Schmidt A. M.; Ramasamy R. Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 2021, 12, 636267. 10.3389/fendo.2021.636267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaine A.; Cross D.; Millward A. Polymorphisms of the Aldose Reductase Gene and Susceptibility to Retinopathy in Type 1 Diabetes Mellitus. Invest. Ophthalmol. Vis. Sci. 2000, 41 (13), 4064–4068. [PubMed] [Google Scholar]
- Moczulski D. K.; Scott L.; Antonellis A.; Rogus J. J.; Rich S. S.; Warram J. H.; Krolewski A. S. Aldose Reductase Gene Polymorphisms and Susceptibility to Diabetic Nephropathy in Type 1 Diabetes Mellitus. Diabet. Med. J. Br. Diabet. Assoc. 2000, 17 (2), 111–118. 10.1046/j.1464-5491.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- Shah V. O.; Scavini M.; Nikolic J.; Sun Y.; Vai S.; Griffith J. K.; Dorin R. I.; Stidley C.; Yacoub M.; Vander Jagt D. L.; Eaton R. P.; Zager P. G. Z-2 Microsatellite Allele Is Linked to Increased Expression of the Aldose Reductase Gene in Diabetic Nephropathy. J. Clin. Endocrinol. Metab. 1998, 83 (8), 2886–2891. 10.1210/jcem.83.8.5028. [DOI] [PubMed] [Google Scholar]
- Heesom A. E.; Millward A.; Demaine A. G. Susceptibility to Diabetic Neuropathy in Patients with Insulin Dependent Diabetes Mellitus Is Associated with a Polymorphism at the 5′ End of the Aldose Reductase Gene. J. Neurol. Neurosurg. Psychiatry 1998, 64 (2), 213–216. 10.1136/jnnp.64.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Jagt D. L.; Hassebrook R. K.; Hunsaker L. A.; Brown W. M.; Royer R. E. Metabolism of the 2-Oxoaldehyde Methylglyoxal by Aldose Reductase and by Glyoxalase-I: Roles for Glutathione in Both Enzymes and Implications for Diabetic Complications. Chem. Biol. Interact. 2001, 130–132 (1–3), 549–562. 10.1016/S0009-2797(00)00298-2. [DOI] [PubMed] [Google Scholar]
- Morgenstern J.; Fleming T.; Schumacher D.; Eckstein V.; Freichel M.; Herzig S.; Nawroth P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292 (8), 3224–3238. 10.1074/jbc.M116.760132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. W.; Ko B. C.; Jiang Z.; Cao D.; Chung S. S. Overexpression of Aldose Reductase in Liver Cancers May Contribute to Drug Resistance. Anticancer. Drugs 2001, 12 (2), 129–132. 10.1097/00001813-200102000-00005. [DOI] [PubMed] [Google Scholar]
- Chang K.-C.; Shieh B.; Petrash J. M. Aldose Reductase Mediates Retinal Microglia Activation. Biochem. Biophys. Res. Commun. 2016, 473 (2), 565–571. 10.1016/j.bbrc.2016.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S. K.; Yadav U. C. S.; Reddy A. B. M.; Saxena A.; Tammali R.; Shoeb M.; Ansari N. H.; Bhatnagar A.; Petrash M. J.; Srivastava S.; Ramana K. V. Aldose Reductase Inhibition Suppresses Oxidative Stress-Induced Inflammatory Disorders. Chem. Biol. Interact. 2011, 191 (1–3), 330–338. 10.1016/j.cbi.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikramadithyan R. K.; Hu Y.; Noh H.-L.; Liang C.-P.; Hallam K.; Tall A. R.; Ramasamy R.; Goldberg I. J. Human Aldose Reductase Expression Accelerates Diabetic Atherosclerosis in Transgenic Mice. J. Clin. Invest. 2005, 115 (9), 2434–2443. 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagihashi S.; Yamagishi S. I.; Wada Ri R.; Baba M.; Hohman T. C.; Yabe-Nishimura C.; Kokai Y. Neuropathy in Diabetic Mice Overexpressing Human Aldose Reductase and Effects of Aldose Reductase Inhibitor. Brain J. Neurol. 2001, 124 (12), 2448–2458. 10.1093/brain/124.12.2448. [DOI] [PubMed] [Google Scholar]
- Ahmed N.; Thornalley P. J.; Dawczynski J.; Franke S.; Strobel J.; Stein G.; Haik G. M. Methylglyoxal-Derived Hydroimidazolone Advanced Glycation End-Products of Human Lens Proteins. Invest. Ophthalmol. Vis. Sci. 2003, 44 (12), 5287–5292. 10.1167/iovs.03-0573. [DOI] [PubMed] [Google Scholar]
- Lo T. W.; Westwood M. E.; McLellan A. C.; Selwood T.; Thornalley P. J. Binding and Modification of Proteins by Methylglyoxal under Physiological Conditions. A Kinetic and Mechanistic Study with N Alpha-Acetylarginine, N Alpha-Acetylcysteine, and N Alpha-Acetyllysine, and Bovine Serum Albumin. J. Biol. Chem. 1994, 269 (51), 32299–32305. 10.1016/S0021-9258(18)31635-1. [DOI] [PubMed] [Google Scholar]
- Li Y.; Cohenford M. A.; Dutta U.; Dain J. A. The Structural Modification of DNA Nucleosides by Nonenzymatic Glycation: An in Vitro Study Based on the Reactions of Glyoxal and Methylglyoxal with 2′-Deoxyguanosine. Anal. Bioanal. Chem. 2008, 390 (2), 679–688. 10.1007/s00216-007-1682-4. [DOI] [PubMed] [Google Scholar]
- Schneider M.; Quistad G. B.; Casida J. E. N2,7-Bis(1-Hydroxy-2-Oxopropyl)-2′-Deoxyguanosine: Identical Noncyclic Adducts with 1,3-Dichloropropene Epoxides and Methylglyoxal. Chem. Res. Toxicol. 1998, 11 (12), 1536–1542. 10.1021/tx9801256. [DOI] [PubMed] [Google Scholar]
- al-Abed Y.; Schleicher E.; Voelter W.; Liebich H.; Papoulis A.; Bucala R. Identification of N2-(1-Carboxymethyl)Guanine (CMG) as a Guanine Advanced Glycation End Product. Bioorg. Med. Chem. Lett. 1998, 8 (16), 2109–2110. 10.1016/S0960-894X(98)00398-9. [DOI] [PubMed] [Google Scholar]
- Schärer O. D. Chemistry and Biology of DNA Repair. Angew. Chem., Int. Ed. Engl. 2003, 42 (26), 2946–2974. 10.1002/anie.200200523. [DOI] [PubMed] [Google Scholar]
- Schärer O. D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5 (10), a012609. 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard S.; Schurman S. H.; Harboe C.; de Souza-Pinto N. C.; Bohr V. A. Base Excision Repair of Oxidative DNA Damage and Association with Cancer and Aging. Carcinogenesis 2008, 30 (1), 2–10. 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18 (1), 85–98. 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Tamae D.; Lim P.; Wuenschell G. E.; Termini J. Mutagenesis and Repair Induced by the DNA Advanced Glycation End Product N2–1-(Carboxyethyl)-2′-Deoxyguanosine in Human Cells. Biochemistry 2011, 50 (12), 2321–2329. 10.1021/bi101933p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciminera A. K.; Shuck S. C.; Termini J. Elevated Glucose Increases Genomic Instability by Inhibiting Nucleotide Excision Repair. Life Sci. Alliance 2021, 4 (10), e202101159. 10.26508/lsa.202101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischetsrieder M.; Seidel W.; Münch G.; Schinzel R. N(2)-(1-Carboxyethyl)Deoxyguanosine, a Nonenzymatic Glycation Adduct of DNA, Induces Single-Strand Breaks and Increases Mutation Frequencies. Biochem. Biophys. Res. Commun. 1999, 264 (2), 544–549. 10.1006/bbrc.1999.1528. [DOI] [PubMed] [Google Scholar]
- Bronzetti G.; Corsi C.; Chiaro D. D.; Boccardo P.; Vellosi R.; Rossi F.; Paolini M.; Forti G. C. Methylglyoxal: Genotoxicity Studies and Its Effect in Vivo on the Hepatic Microsomal Mono-Oxygenase System of the Mouse. Mutagenesis 1987, 2 (4), 275–277. 10.1093/mutage/2.4.275. [DOI] [PubMed] [Google Scholar]
- Cajelli E.; Canonero R.; Martelli A.; Brambilla G. Methylglyoxal-Induced Mutation to 6-Thioguanine Resistance in V79 Cells. Mutat. Res. 1987, 190 (1), 47–50. 10.1016/0165-7992(87)90081-9. [DOI] [PubMed] [Google Scholar]
- Hou S. M.; Nori P.; Fang J. L.; Vaca C. E. Methylglyoxal Induces Hprt Mutation and DNA Adducts in Human T-Lymphocytes in Vitro. Environ. Mol. Mutagen. 1995, 26 (4), 286–291. 10.1002/em.2850260404. [DOI] [PubMed] [Google Scholar]
- Bessho T. Nucleotide Excision Repair 3′ Endonuclease XPG Stimulates the Activity of Base Excision Repairenzyme Thymine Glycol DNA Glycosylase. Nucleic Acids Res. 1999, 27 (4), 979–983. 10.1093/nar/27.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata-Kamiya N.; Kamiya H.; Kaji H.; Kasai H. Methylglyoxal Induces G:C to C:G and G:C to T:A Transversions in the SupF Gene on a Shuttle Vector Plasmid Replicated in Mammalian Cells. Mutat. Res. 2000, 468 (2), 173–182. 10.1016/S1383-5718(00)00044-9. [DOI] [PubMed] [Google Scholar]
- Murata-Kamiya N.; Kaji H.; Kasai H. Deficient Nucleotide Excision Repair Increases Base-Pair Substitutions but Decreases TGGC Frameshifts Induced by Methylglyoxal in Escherichia Coli. Mutat. Res. 1999, 442 (1), 19–28. 10.1016/S1383-5718(99)00054-6. [DOI] [PubMed] [Google Scholar]
- Yuan B.; Cao H.; Jiang Y.; Hong H.; Wang Y. Efficient and Accurate Bypass of N2-(1-Carboxyethyl)-2′-Deoxyguanosine by DinB DNA Polymerase in Vitro and in Vivo. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (25), 8679–8684. 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K. The Reaction of Phenylglyoxal with Arginine Residues in Proteins. J. Biol. Chem. 1968, 243 (23), 6171–6179. 10.1016/S0021-9258(18)94475-3. [DOI] [PubMed] [Google Scholar]
- Klöpfer A.; Spanneberg R.; Glomb M. A. Formation of Arginine Modifications in a Model System of Nα-Tert-Butoxycarbonyl (Boc)-Arginine with Methylglyoxal. J. Agric. Food Chem. 2011, 59 (1), 394–401. 10.1021/jf103116c. [DOI] [PubMed] [Google Scholar]
- McEwen J. M.; Fraser S.; Guir A. L. S.; Dave J.; Scheck R. A. Synergistic Sequence Contributions Bias Glycation Outcomes. Nat. Commun. 2021, 12 (1), 3316. 10.1038/s41467-021-23625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. U.; Brinkmann Frye E.; Degenhardt T. P.; Thorpe S. R.; Baynes J. W. N-Epsilon-(Carboxyethyl)Lysine, a Product of the Chemical Modification of Proteins by Methylglyoxal, Increases with Age in Human Lens Proteins. Biochem. J. 1997, 324 (2), 565–570. 10.1042/bj3240565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley P. J.; Battah S.; Ahmed N.; Karachalias N.; Agalou S.; Babaei-Jadidi R.; Dawnay A. Quantitative Screening of Advanced Glycation Endproducts in Cellular and Extracellular Proteins by Tandem Mass Spectrometry. Biochem. J. 2003, 375 (3), 581–592. 10.1042/bj20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade S. J.; Miller A. G.; Gerrard J. A. The Role of Dicarbonyl Compounds in Non-Enzymatic Crosslinking: A Structure-Activity Study. Bioorg. Med. Chem. 2003, 11 (6), 853–862. 10.1016/S0968-0896(02)00564-3. [DOI] [PubMed] [Google Scholar]
- Murata-Kamiya N.; Kamiya H. Methylglyoxal, an Endogenous Aldehyde, Crosslinks DNA Polymerase and the Substrate DNA. Nucleic Acids Res. 2001, 29 (16), 3433–3438. 10.1093/nar/29.16.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova K. V.; Millsap A. D.; Stec D. F.; Rizzo C. J. Characterization of the Deoxyguanosine-Lysine Cross-Link of Methylglyoxal. Chem. Res. Toxicol. 2014, 27 (6), 1019–1029. 10.1021/tx500068v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoncini G.; Maresca M.; Ronchi S.; Bonsignore A. Studies on the Inactivation of Glyceraldehyde-3-Phosphate Dehydrogenase by Methylglyoxal. Experientia 1981, 37 (5), 443–444. 10.1007/BF01986121. [DOI] [PubMed] [Google Scholar]
- Mira M. L.; Martinho F.; Azevedo M. S.; Manso C. F. Oxidative Inhibition of Red Blood Cell ATPases by Glyceraldehyde. Biochim. Biophys. Acta 1991, 1060 (3), 257–261. 10.1016/S0005-2728(05)80315-9. [DOI] [PubMed] [Google Scholar]
- Irshad Z.; Xue M.; Ashour A.; Larkin J. R.; Thornalley P. J.; Rabbani N. Activation of the Unfolded Protein Response in High Glucose Treated Endothelial Cells Is Mediated by Methylglyoxal. Sci. Rep. 2019, 9 (1), 7889. 10.1038/s41598-019-44358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranova L. K.; Perfilov M. M.; Serebryakova M. V.; Gusev N. B. Effect of Methylglyoxal Modification on the Structure and Properties of Human Small Heat Shock Protein HspB6 (Hsp20). Cell Stress Chaperones 2016, 21 (4), 617–629. 10.1007/s12192-016-0686-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R. H.; Oya-Ito T.; Padayatti P. S.; Kumar R.; Mehta S.; West K.; Levison B.; Sun J.; Crabb J. W.; Padival A. K. Enhancement of Chaperone Function of Alpha-Crystallin by Methylglyoxal Modification. Biochemistry 2003, 42 (36), 10746–10755. 10.1021/bi034541n. [DOI] [PubMed] [Google Scholar]
- Padival A. K.; Crabb J. W.; Nagaraj R. H. Methylglyoxal Modifies Heat Shock Protein 27 in Glomerular Mesangial Cells. FEBS Lett. 2003, 551 (1–3), 113–118. 10.1016/S0014-5793(03)00874-3. [DOI] [PubMed] [Google Scholar]
- Sakamoto H.; Mashima T.; Yamamoto K.; Tsuruo T. Modulation of Heat-Shock Protein 27 (Hsp27) Anti-Apoptotic Activity by Methylglyoxal Modification. J. Biol. Chem. 2002, 277 (48), 45770–45775. 10.1074/jbc.M207485200. [DOI] [PubMed] [Google Scholar]
- Pozzi A.; Zent R.; Chetyrkin S.; Borza C.; Bulus N.; Chuang P.; Chen D.; Hudson B.; Voziyan P. Modification of Collagen IV by Glucose or Methylglyoxal Alters Distinct Mesangial Cell Functions. J. Am. Soc. Nephrol. JASN 2009, 20 (10), 2119–2125. 10.1681/ASN.2008080900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N.; Dobler D.; Dean M.; Thornalley P. J. Peptide Mapping Identifies Hotspot Site of Modification in Human Serum Albumin by Methylglyoxal Involved in Ligand Binding and Esterase Activity. J. Biol. Chem. 2005, 280 (7), 5724–5732. 10.1074/jbc.M410973200. [DOI] [PubMed] [Google Scholar]
- Ceradini D. J.; Yao D.; Grogan R. H.; Callaghan M. J.; Edelstein D.; Brownlee M.; Gurtner G. C. Decreasing Intracellular Superoxide Corrects Defective Ischemia-Induced New Vessel Formation in Diabetic Mice. J. Biol. Chem. 2008, 283 (16), 10930–10938. 10.1074/jbc.M707451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.; Omans N. D.; Leicher R.; Osunsade A.; Agustinus A. S.; Finkin-Groner E.; D’Ambrosio H.; Liu B.; Chandarlapaty S.; Liu S.; David Y. Reversible Histone Glycation Is Associated with Disease-Related Changes in Chromatin Architecture. Nat. Commun. 2019, 10 (1), 1289. 10.1038/s41467-019-09192-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J.; Lee J.; Lim J.; Cho S.; An S.; Lee M.; Yoon N.; Seo M.; Lim S.; Park S. Soluble RAGE Attenuates AngII-Induced Endothelial Hyperpermeability by Disrupting HMGB1-Mediated Crosstalk between AT1R and RAGE. Exp. Mol. Med. 2019, 51 (9), 1–15. 10.1038/s12276-019-0312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehl A.; Németh J.; Angel P.; Hess J. The Receptor RAGE: Bridging Inflammation and Cancer. Cell Commun. Signal. CCS 2009, 7, 12. 10.1186/1478-811X-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K.; Fritz G. RAGE Regulation and Signaling in Inflammation and Beyond. J. Leukoc. Biol. 2013, 94 (1), 55–68. 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- Hansen L.; Joseph G.; Valdivia A.; Taylor W. R. Satellite Cell Expression of RAGE (Receptor for Advanced Glycation End Products) Is Important for Collateral Vessel Formation. J. Am. Heart Assoc. 2021, 10 (21), e022127. 10.1161/JAHA.120.022127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.-C.; Chen K.-C.; Chang G.-C.; Lin H.; Wu C.-C.; Kao W.-H.; Teng C.-L. J.; Hsu S.-L.; Yang T.-Y. RAGE Acts as an Oncogenic Role and Promotes the Metastasis of Human Lung Cancer. Cell Death Dis. 2020, 11 (4), 265. 10.1038/s41419-020-2432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R.; Loux T.; Tang D.; Schapiro N. E.; Vernon P.; Livesey K. M.; Krasinskas A.; Lotze M. T.; Zeh H. J. 3rd. The Expression of the Receptor for Advanced Glycation Endproducts (RAGE) Is Permissive for Early Pancreatic Neoplasia. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (18), 7031–7036. 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusterla T.; Nèmeth J.; Stein I.; Wiechert L.; Knigin D.; Marhenke S.; Longerich T.; Kumar V.; Arnold B.; Vogel A.; Bierhaus A.; Pikarsky E.; Hess J.; Angel P. Receptor for Advanced Glycation Endproducts (RAGE) Is a Key Regulator of Oval Cell Activation and Inflammation-Associated Liver Carcinogenesis in Mice. Hepatol. Baltim. Md 2013, 58 (1), 363–373. 10.1002/hep.26395. [DOI] [PubMed] [Google Scholar]
- Sparvero L. J.; Asafu-Adjei D.; Kang R.; Tang D.; Amin N.; Im J.; Rutledge R.; Lin B.; Amoscato A. A.; Zeh H. J.; Lotze M. T. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and Their Role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Reverdatto S.; Frolov A.; Hoffmann R.; Burz D. S.; Shekhtman A. Structural Basis for Pattern Recognition by the Receptor for Advanced Glycation End Products (RAGE. J. Biol. Chem. 2008, 283 (40), 27255–27269. 10.1074/jbc.M801622200. [DOI] [PubMed] [Google Scholar]
- Xue J.; Rai V.; Singer D.; Chabierski S.; Xie J.; Reverdatto S.; Burz D. S.; Schmidt A. M.; Hoffmann R.; Shekhtman A. Advanced Glycation End Product Recognition by the Receptor for AGEs. Structure 2011, 19 (5), 722–732. 10.1016/j.str.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J.; Ray R.; Singer D.; Böhme D.; Burz D. S.; Rai V.; Hoffmann R.; Shekhtman A. The Receptor for Advanced Glycation End Products (RAGE) Specifically Recognizes Methylglyoxal-Derived AGEs. Biochemistry 2014, 53 (20), 3327–3335. 10.1021/bi500046t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.; Kubo H.; Morimoto K.; Fujino N.; Suzuki T.; Takahasi T.; Yamada M.; Yamaya M.; Maekawa T.; Yamamoto Y.; Yamamoto H. Receptor for Advanced Glycation End Products Binds to Phosphatidylserine and Assists in the Clearance of Apoptotic Cells. EMBO Rep. 2011, 12 (4), 358–364. 10.1038/embor.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J.; Avalos A. M.; Mao S.-Y.; Chen B.; Senthil K.; Wu H.; Parroche P.; Drabic S.; Golenbock D.; Sirois C.; Hua J.; An L. L.; Audoly L.; La Rosa G.; Bierhaus A.; Naworth P.; Marshak-Rothstein A.; Crow M. K.; Fitzgerald K. A.; Latz E.; Kiener P. A.; Coyle A. J. Toll-like Receptor 9-Dependent Activation by DNA-Containing Immune Complexes Is Mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8 (5), 487–496. 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- Huttunen H. J.; Kuja-Panula J.; Sorci G.; Agneletti A. L.; Donato R.; Rauvala H. Coregulation of Neurite Outgrowth and Cell Survival by Amphoterin and S100 Proteins through Receptor for Advanced Glycation End Products (RAGE) Activation. J. Biol. Chem. 2000, 275 (51), 40096–40105. 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y.; Harashima A.; Saito H.; Tsuneyama K.; Munesue S.; Motoyoshi S.; Han D.; Watanabe T.; Asano M.; Takasawa S.; Okamoto H.; Shimura S.; Karasawa T.; Yonekura H.; Yamamoto H. Septic Shock Is Associated with Receptor for Advanced Glycation End Products Ligation of LPS. J. Immunol. 2011, 186 (5), 3248–3257. 10.4049/jimmunol.1002253. [DOI] [PubMed] [Google Scholar]
- Sirois C. M.; Jin T.; Miller A. L.; Bertheloot D.; Nakamura H.; Horvath G. L.; Mian A.; Jiang J.; Schrum J.; Bossaller L.; Pelka K.; Garbi N.; Brewah Y.; Tian J.; Chang C.; Chowdhury P. S.; Sims G. P.; Kolbeck R.; Coyle A. J.; Humbles A. A.; Xiao T. S.; Latz E. RAGE Is a Nucleic Acid Receptor That Promotes Inflammatory Responses to DNA. J. Exp. Med. 2013, 210 (11), 2447–2463. 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. S.; Guh J. Y.; Chen H. C.; Hung W. C.; Lai Y. H.; Chuang L. Y. Role of Receptor for Advanced Glycation End-Product (RAGE) and the JAK/STAT-Signaling Pathway in AGE-Induced Collagen Production in NRK-49F Cells. J. Cell. Biochem. 2001, 81 (1), 102–113. . [DOI] [PubMed] [Google Scholar]
- Qin Q.; Niu J.; Wang Z.; Xu W.; Qiao Z.; Gu Y. Heparanase Induced by Advanced Glycation End Products (AGEs) Promotes Macrophage Migration Involving RAGE and PI3K/AKT Pathway. Cardiovasc. Diabetol. 2013, 12, 37. 10.1186/1475-2840-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P.; Ren M.; Yang C.; Hu Y.-X.; Ran J.-M.; Yan L. Involvement of RAGE, MAPK and NF-KB Pathways in AGEs-Induced MMP-9 Activation in HaCaT Keratinocytes. Exp. Dermatol 2012, 21 (2), 123–129. 10.1111/j.1600-0625.2011.01408.x. [DOI] [PubMed] [Google Scholar]
- Tóbon-Velasco J. C.; Cuevas E.; Torres-Ramos M. A. Receptor for AGEs (RAGE) as Mediator of NF-KB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord. Drug Targets 2014, 13 (9), 1615–1626. 10.2174/1871527313666140806144831. [DOI] [PubMed] [Google Scholar]
- Chen Y.-H.; Chen Z.-W.; Li H.-M.; Yan X.-F.; Feng B. AGE/RAGE-Induced EMP Release via the NOX-Derived ROS Pathway. J. Diabetes Res. 2018, 2018, 6823058. 10.1155/2018/6823058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Park S.; Lakatta E. G. RAGE Signaling in Inflammation and Arterial Aging. Front. Biosci. (Landmark Ed.) 2009, 14 (4), 1403–1413. 10.2741/3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K.; Karin M. NF-KB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18 (5), 309–324. 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- Lappas M.; Permezel M.; Rice G. E. Advanced Glycation Endproducts Mediate Pro-Inflammatory Actions in Human Gestational Tissues via Nuclear Factor-KappaB and Extracellular Signal-Regulated Kinase 1/2. J. Endocrinol 2007, 193 (2), 269–277. 10.1677/JOE-06-0081. [DOI] [PubMed] [Google Scholar]
- Park S.; Yoon S.-J.; Tae H.-J.; Shim C. Y. RAGE and Cardiovascular Disease. Front. Biosci. (Landmark Ed.) 2011, 16 (2), 486–497. 10.2741/3700. [DOI] [PubMed] [Google Scholar]
- Bao J.-M.; He M.-Y.; Liu Y.-W.; Lu Y.-J.; Hong Y.-Q.; Luo H.-H.; Ren Z.-L.; Zhao S.-C.; Jiang Y. AGE/RAGE/Akt Pathway Contributes to Prostate Cancer Cell Proliferation by Promoting Rb Phosphorylation and Degradation. Am. J. Cancer Res. 2015, 5 (5), 1741–1750. [PMC free article] [PubMed] [Google Scholar]
- Ko S.-Y.; Ko H.-A.; Shieh T.-M.; Chang W.-C.; Chen H.-I.; Chang S.-S.; Lin I.-H. Cell Migration Is Regulated by AGE-RAGE Interaction in Human Oral Cancer Cells in Vitro. PloS One 2014, 9 (10), e110542. 10.1371/journal.pone.0110542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthyalaiah Y. S.; Jonnalagadda B.; John C. M.; Arockiasamy S. Impact of Advanced Glycation End Products (AGEs) and Its Receptor (RAGE) on Cancer Metabolic Signaling Pathways and Its Progression. Glycoconj. J. 2021, 38 (6), 717–734. 10.1007/s10719-021-10031-x. [DOI] [PubMed] [Google Scholar]
- Schalkwijk C. G.; Stehouwer C. D. A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100 (1), 407–461. 10.1152/physrev.00001.2019. [DOI] [PubMed] [Google Scholar]
- Leone A.; Nigro C.; Nicolò A.; Prevenzano I.; Formisano P.; Beguinot F.; Miele C. The Dual-Role of Methylglyoxal in Tumor Progression - Novel Therapeutic Approaches. Front. Oncol. 2021, 11, 645686. 10.3389/fonc.2021.645686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Castillo C.; Shuck S. C. Diet and Obesity-Induced Methylglyoxal Production and Links to Metabolic Disease. Chem. Res. Toxicol. 2021, 34 (12), 2424–2440. 10.1021/acs.chemrestox.1c00221. [DOI] [PubMed] [Google Scholar]
- Yamagishi S.; Matsui T.; Fukami K. Role of Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Cancer Risk. Rejuvenation Res. 2015, 18 (1), 48–56. 10.1089/rej.2014.1625. [DOI] [PubMed] [Google Scholar]
- Papadaki M.; Holewinski R. J.; Previs S. B.; Martin T. G.; Stachowski M. J.; Li A.; Blair C. A.; Moravec C. S.; Van Eyk J. E.; Campbell K. S.; Warshaw D. M.; Kirk J. A. Diabetes with Heart Failure Increases Methylglyoxal Modifications in the Sarcomere, Which Inhibit Function. JCI Insight 2018, 3 (20), 121264. 10.1172/jci.insight.121264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P. K.; Alomar F. A.; Cox J. L.; McMillan J.; Hackfort B. T.; Makarov E.; Morsey B.; Fox H. S.; Gendelman H. E.; Gorantla S.; Bidasee K. R. A Link Between Methylglyoxal and Heart Failure During HIV-1 Infection. Front. Cardiovasc. Med. 2021, 8, 792180. 10.3389/fcvm.2021.792180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen N. M. J.; Scheijen J. L. J. M.; Jorsal A.; Parving H.-H.; Tarnow L.; Rossing P.; Stehouwer C. D. A.; Schalkwijk C. G. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease in Individuals With Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes 2017, 66 (8), 2278–2283. 10.2337/db16-1578. [DOI] [PubMed] [Google Scholar]
- Ogawa S.; Nakayama K.; Nakayama M.; Mori T.; Matsushima M.; Okamura M.; Senda M.; Nako K.; Miyata T.; Ito S. Methylglyoxal Is a Predictor in Type 2 Diabetic Patients of Intima-Media Thickening and Elevation of Blood Pressure. Hypertension 2010, 56 (3), 471–476. 10.1161/HYPERTENSIONAHA.110.156786. [DOI] [PubMed] [Google Scholar]
- Blackburn N. J. R.; Vulesevic B.; McNeill B.; Cimenci C. E.; Ahmadi A.; Gonzalez-Gomez M.; Ostojic A.; Zhong Z.; Brownlee M.; Beisswenger P. J.; Milne R. W.; Suuronen E. J. Methylglyoxal-Derived Advanced Glycation End Products Contribute to Negative Cardiac Remodeling and Dysfunction Post-Myocardial Infarction. Basic Res. Cardiol. 2017, 112 (5), 57. 10.1007/s00395-017-0646-x. [DOI] [PubMed] [Google Scholar]
- Wang X.; Desai K.; Chang T.; Wu L. Vascular Methylglyoxal Metabolism and the Development of Hypertension. J. Hypertens 2005, 23 (8), 1565–1573. 10.1097/01.hjh.0000173778.85233.1b. [DOI] [PubMed] [Google Scholar]
- Hanssen N. M. J.; Beulens J. W. J.; van Dieren S.; Scheijen J. L. J. M.; van der A D. L.; Spijkerman A. M. W.; van der Schouw Y. T.; Stehouwer C. D. A.; Schalkwijk C. G. Plasma Advanced Glycation End Products Are Associated with Incident Cardiovascular Events in Individuals with Type 2 Diabetes: A Case-Cohort Study with a Median Follow-up of 10 Years (EPIC-NL). Diabetes 2015, 64 (1), 257–265. 10.2337/db13-1864. [DOI] [PubMed] [Google Scholar]
- Aso Y.; Inukai T.; Tayama K.; Takemura Y. Serum Concentrations of Advanced Glycation Endproducts Are Associated with the Development of Atherosclerosis as Well as Diabetic Microangiopathy in Patients with Type 2 Diabetes. Acta Diabetol 2000, 37 (2), 87–92. 10.1007/s005920070025. [DOI] [PubMed] [Google Scholar]
- Kilhovd B. K.; Juutilainen A.; Lehto S.; Rönnemaa T.; Torjesen P. A.; Birkeland K. I.; Berg T. J.; Hanssen K. F.; Laakso M. High Serum Levels of Advanced Glycation End Products Predict Increased Coronary Heart Disease Mortality in Nondiabetic Women but Not in Nondiabetic Men: A Population-Based 18-Year Follow-up Study. Arterioscler. Thromb. Vasc. Biol. 2005, 25 (4), 815–820. 10.1161/01.ATV.0000158380.44231.fe. [DOI] [PubMed] [Google Scholar]
- Chiarelli F.; de Martino M.; Mezzetti A.; Catino M.; Morgese G.; Cuccurullo F.; Verrotti A. Advanced Glycation End Products in Children and Adolescents with Diabetes: Relation to Glycemic Control and Early Microvascular Complications. J. Pediatr. 1999, 134 (4), 486–491. 10.1016/S0022-3476(99)70208-8. [DOI] [PubMed] [Google Scholar]
- Miura J.; Yamagishi S.-i.; Uchigata Y.; Takeuchi M.; Yamamoto H.; Makita Z.; Iwamoto Y. Serum Levels of Non-Carboxymethyllysine Advanced Glycation Endproducts Are Correlated to Severity of Microvascular Complications in Patients with Type 1 Diabetes. J. Diabetes Its Complications 2003, 17 (1), 16–21. 10.1016/S1056-8727(02)00183-6. [DOI] [PubMed] [Google Scholar]
- Ramasamy R.; Yan S. F.; Schmidt A. M. Receptor for AGE (RAGE): Signaling Mechanisms in the Pathogenesis of Diabetes and Its Complications. Ann. N.Y. Acad. Sci. 2011, 1243, 88–102. 10.1111/j.1749-6632.2011.06320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman S. L.; Sonmez H.; Basman C.; Singh V.; Poretsky L. The Role of Advanced Glycation End-Products in the Development of Coronary Artery Disease in Patients with and without Diabetes Mellitus: A Review. Mol. Med. 2018, 24 (1), 59. 10.1186/s10020-018-0060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegab Z.; Gibbons S.; Neyses L.; Mamas M. A. Role of Advanced Glycation End Products in Cardiovascular Disease. World J. Cardiol. 2012, 4 (4), 90–102. 10.4330/wjc.v4.i4.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L.; Raman K. G.; Lee K. J.; Lu Y.; Ferran L. J. J.; Chow W. S.; Stern D.; Schmidt A. M. Suppression of Accelerated Diabetic Atherosclerosis by the Soluble Receptor for Advanced Glycation Endproducts. Nat. Med. 1998, 4 (9), 1025–1031. 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- Srikanth V.; Westcott B.; Forbes J.; Phan T. G.; Beare R.; Venn A.; Pearson S.; Greenaway T.; Parameswaran V.; Münch G. Methylglyoxal, Cognitive Function and Cerebral Atrophy in Older People. J. Gerontol. A. Biol. Sci. Med. Sci. 2013, 68 (1), 68–73. 10.1093/gerona/gls100. [DOI] [PubMed] [Google Scholar]
- Hipkiss A. R. On the Relationship between Energy Metabolism, Proteostasis, Aging and Parkinson’s Disease: Possible Causative Role of Methylglyoxal and Alleviative Potential of Carnosine. Aging Dis. 2017, 8 (3), 334–345. 10.14336/AD.2016.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes R.; Sousa Silva M.; Quintas A.; Cordeiro C.; Freire A.; Pereira P.; Martins A.; Monteiro E.; Barroso E.; Ponces Freire A. Argpyrimidine, a Methylglyoxal-Derived Advanced Glycation End-Product in Familial Amyloidotic Polyneuropathy. Biochem. J. 2005, 385 (2), 339–345. 10.1042/BJ20040833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Liu D.; Sun L.; Lu Y.; Zhang Z. Advanced Glycation End Products and Neurodegenerative Diseases: Mechanisms and Perspective. J. Neurol. Sci. 2012, 317 (1–2), 1–5. 10.1016/j.jns.2012.02.018. [DOI] [PubMed] [Google Scholar]
- Beeri M. S.; Moshier E.; Schmeidler J.; Godbold J.; Uribarri J.; Reddy S.; Sano M.; Grossman H. T.; Cai W.; Vlassara H.; Silverman J. M. Serum Concentration of an Inflammatory Glycotoxin, Methylglyoxal, Is Associated with Increased Cognitive Decline in Elderly Individuals. Mech. Ageing Dev. 2011, 132 (11–12), 583–587. 10.1016/j.mad.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautwald M.; Münch G. Advanced Glycation End Products as Biomarkers and Gerontotoxins - A Basis to Explore Methylglyoxal-Lowering Agents for Alzheimer’s Disease?. Exp. Gerontol. 2010, 45 (10), 744–751. 10.1016/j.exger.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Kimura T.; Takamatsu J.; Araki N.; Goto M.; Kondo A.; Miyakawa T.; Horiuchi S. Are Advanced Glycation End-Products Associated with Amyloidosis in Alzheimer’s Disease?. Neuroreport 1995, 6 (6), 866–868. 10.1097/00001756-199504190-00010. [DOI] [PubMed] [Google Scholar]
- Xie B.; Lin F.; Peng L.; Ullah K.; Wu H.; Qing H.; Deng Y. Methylglyoxal Increases Dopamine Level and Leads to Oxidative Stress in SH-SY5Y Cells. Acta Biochim. Biophys. Sin. 2014, 46 (11), 950–956. 10.1093/abbs/gmu094. [DOI] [PubMed] [Google Scholar]
- Haslbeck K.-M.; Bierhaus A.; Erwin S.; Kirchner A.; Nawroth P.; Schlötzer U.; Neundörfer B.; Heuss D. Receptor for Advanced Glycation Endproduct (RAGE)-Mediated Nuclear Factor-KappaB Activation in Vasculitic Neuropathy. Muscle Nerve 2004, 29 (6), 853–860. 10.1002/mus.20039. [DOI] [PubMed] [Google Scholar]
- Haslbeck K. M.; Schleicher E. D.; Friess U.; Kirchner A.; Neundörfer B.; Heuss D. N(Epsilon)-Carboxymethyllysine in Diabetic and Non-Diabetic Polyneuropathies. Acta Neuropathol. (Berl.) 2002, 104 (1), 45–52. 10.1007/s00401-002-0518-8. [DOI] [PubMed] [Google Scholar]
- Haslbeck K. M.; Neundörfer B.; Schlötzer-Schrehardtt U.; Bierhaus A.; Schleicher E.; Pauli E.; Haslbeck M.; Hecht M.; Nawroth P.; Heuss D. Activation of the RAGE Pathway: A General Mechanism in the Pathogenesis of Polyneuropathies?. Neurol. Res. 2007, 29 (1), 103–110. 10.1179/174313206X152564. [DOI] [PubMed] [Google Scholar]
- Haslbeck K.-M.; Schleicher E.; Bierhaus A.; Nawroth P.; Haslbeck M.; Neundörfer B.; Heuss D. The AGE/RAGE/NF-(Kappa)B Pathway May Contribute to the Pathogenesis of Polyneuropathy in Impaired Glucose Tolerance (IGT). Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2005, 113 (5), 288–291. 10.1055/s-2005-865600. [DOI] [PubMed] [Google Scholar]
- Derk J.; MacLean M.; Juranek J.; Schmidt A. M. The Receptor for Advanced Glycation Endproducts (RAGE) and Mediation of Inflammatory Neurodegeneration. J. Alzheimers Dis. Parkinsonism 2018, 8 (1), 421. 10.4172/2161-0460.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter F.; Chen D.; Akhter A.; Yan S. F.; Yan S. S. Age-Dependent Accumulation of Dicarbonyls and Advanced Glycation Endproducts (AGEs) Associates with Mitochondrial Stress. Free Radic. Biol. Med. 2021, 164, 429–438. 10.1016/j.freeradbiomed.2020.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira A. P.; Vizuete A. F. K.; Zin L. E. F.; de Marques C. O.; Pacheco R. F.; Leal M. B.; Gonçalves C.-A. The Methylglyoxal/RAGE/NOX-2 Pathway Is Persistently Activated in the Hippocampus of Rats with STZ-Induced Sporadic Alzheimer’s Disease. Neurotox. Res. 2022, 40 (2), 395–409. 10.1007/s12640-022-00476-9. [DOI] [PubMed] [Google Scholar]
- Choi B.-R.; Cho W.-H.; Kim J.; Lee H. J.; Chung C.; Jeon W. K.; Han J.-S. Increased Expression of the Receptor for Advanced Glycation End Products in Neurons and Astrocytes in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Exp. Mol. Med. 2014, 46 (2), e75. 10.1038/emm.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhla B.; Lüth H.-J.; Haferburg D.; Boeck K.; Arendt T.; Münch G. Methylglyoxal, Glyoxal, and Their Detoxification in Alzheimer’s Disease. Ann. N.Y. Acad. Sci. 2005, 1043, 211–216. 10.1196/annals.1333.026. [DOI] [PubMed] [Google Scholar]
- Fan X.; Sell D. R.; Hao C.; Liu S.; Wang B.; Wesson D. W.; Siedlak S.; Zhu X.; Kavanagh T. J.; Harrison F. E.; Monnier V. M. Vitamin C Is a Source of Oxoaldehyde and Glycative Stress in Age-Related Cataract and Neurodegenerative Diseases. Aging Cell 2020, 19 (7), e13176. 10.1111/acel.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jono T.; Kimura T.; Takamatsu J.; Nagai R.; Miyazaki K.; Yuzuriha T.; Kitamura T.; Horiuchi S. Accumulation of Imidazolone, Pentosidine and N(Epsilon)-(Carboxymethyl)Lysine in Hippocampal CA4 Pyramidal Neurons of Aged Human Brain. Pathol. Int. 2002, 52 (9), 563–571. 10.1046/j.1320-5463.2002.01390.x. [DOI] [PubMed] [Google Scholar]
- Aubert C. E.; Michel P.-L.; Gillery P.; Jaisson S.; Fonfrede M.; Morel F.; Hartemann A.; Bourron O. Association of Peripheral Neuropathy with Circulating Advanced Glycation End Products, Soluble Receptor for Advanced Glycation End Products and Other Risk Factors in Patients with Type 2 Diabetes. Diabetes Metab. Res. Rev. 2014, 30 (8), 679–685. 10.1002/dmrr.2529. [DOI] [PubMed] [Google Scholar]
- Angeloni C.; Zambonin L.; Hrelia S. Role of Methylglyoxal in Alzheimer’s Disease. BioMed. Res. Int. 2014, 2014, 238485. 10.1155/2014/238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzels S.; Vanmierlo T.; Scheijen J. L. J. M.; van Horssen J.; Amor S.; Somers V.; Schalkwijk C. G.; Hendriks J. J. A.; Wouters K. Methylglyoxal-Derived Advanced Glycation Endproducts Accumulate in Multiple Sclerosis Lesions. Front. Immunol. 2019, 10, 855. 10.3389/fimmu.2019.00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N.; Hirano A.; Hedley-Whyte E. T.; Dal Canto M. C.; Nagai R.; Uchida K.; Horiuchi S.; Kawaguchi M.; Yamamoto T.; Kobayashi M. Selective Formation of Certain Advanced Glycation End Products in Spinal Cord Astrocytes of Humans and Mice with Superoxide Dismutase-1 Mutation. Acta Neuropathol. (Berl.) 2002, 104 (2), 171–178. 10.1007/s00401-002-0537-5. [DOI] [PubMed] [Google Scholar]
- Kim J.; Waldvogel H. J.; Faull R. L. M.; Curtis M. A.; Nicholson L. F. B. The RAGE Receptor and Its Ligands Are Highly Expressed in Astrocytes in a Grade-Dependant Manner in the Striatum and Subependymal Layer in Huntington’s Disease. J. Neurochem. 2015, 134 (5), 927–942. 10.1111/jnc.13178. [DOI] [PubMed] [Google Scholar]
- Suzuki A.; Yabu A.; Nakamura H. Advanced Glycation End Products in Musculoskeletal System and Disorders. Methods 2020, 203, 179–186. 10.1016/j.ymeth.2020.09.012. [DOI] [PubMed] [Google Scholar]
- Lee K. M.; Lee C. Y.; Zhang G.; Lyu A.; Yue K. K. M. Methylglyoxal Activates Osteoclasts through JNK Pathway Leading to Osteoporosis. Chem. Biol. Interact. 2019, 308, 147–154. 10.1016/j.cbi.2019.05.026. [DOI] [PubMed] [Google Scholar]
- Bi H.; Chen X.; Gao S.; Yu X.; Xiao J.; Zhang B.; Liu X.; Dai M. Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 2017, 4, 234. 10.3389/fmed.2017.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H.; Xiao E.; Graves D. T. Diabetes and Its Effect on Bone and Fracture Healing. Curr. Osteoporos. Rep. 2015, 13 (5), 327–335. 10.1007/s11914-015-0286-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa T.; Matsubara H.; Ugaji S.; Shirakawa J.; Nagai R.; Munesue S.; Harashima A.; Yamamoto Y.; Tsuchiya H. Contribution of Methylglyoxal to Delayed Healing of Bone Injury in Diabetes. Mol. Med. Rep. 2017, 16 (1), 403–409. 10.3892/mmr.2017.6589. [DOI] [PubMed] [Google Scholar]
- Suh K. S.; Chon S.; Choi E. M. Limonene Attenuates Methylglyoxal-Induced Dysfunction in MC3T3-E1 Osteoblastic Cells. Food Agric. Immunol. 2017, 28 (6), 1256–1268. 10.1080/09540105.2017.1337082. [DOI] [Google Scholar]
- Park S. Y.; Suh K. S.; Jung W.-W.; Chin S. O. Spironolactone Attenuates Methylglyoxal-Induced Cellular Dysfunction in MC3T3-E1 Osteoblastic Cells. J. Korean Med. Sci. 2021, 36 (38), e265. 10.3346/jkms.2021.36.e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riuzzi F.; Sorci G.; Sagheddu R.; Chiappalupi S.; Salvadori L.; Donato R. RAGE in the Pathophysiology of Skeletal Muscle. J. Cachexia Sarcopenia Muscle 2018, 9 (7), 1213–1234. 10.1002/jcsm.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mey J. T.; Haus J. M. Dicarbonyl Stress and Glyoxalase-1 in Skeletal Muscle: Implications for Insulin Resistance and Type 2 Diabetes. Front. Cardiovasc. Med. 2018, 5, 117. 10.3389/fcvm.2018.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson R. B.; Smith S. T.; Moyer P. J.; Magnusson S. P. Effects of Maturation and Advanced Glycation on Tensile Mechanics of Collagen Fibrils from Rat Tail and Achilles Tendons. Acta Biomater. 2018, 70, 270–280. 10.1016/j.actbio.2018.02.005. [DOI] [PubMed] [Google Scholar]
- Pastino A. K.; Greco T. M.; Mathias R. A.; Cristea I. M.; Schwarzbauer J. E. Stimulatory Effects of Advanced Glycation Endproducts (AGEs) on Fibronectin Matrix Assembly. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 59, 39–53. 10.1016/j.matbio.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson L. C.; Redden J. T.; Schwartz Z.; Cohen D. J.; McClure M. J. Advanced Glycation End-Products in Skeletal Muscle Aging. Bioengineering (Basel) 2021, 8 (11), 168. 10.3390/bioengineering8110168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig M. H.; Jan A. T.; Rabbani G.; Ahmad K.; Ashraf J. M.; Kim T.; Min H. S.; Lee Y. H.; Cho W.-K.; Ma J. Y.; Lee E. J.; Choi I. Methylglyoxal and Advanced Glycation End Products: Insight of the Regulatory Machinery Affecting the Myogenic Program and of Its Modulation by Natural Compounds. Sci. Rep. 2017, 7 (1), 5916. 10.1038/s41598-017-06067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C.-Y.; Yang R.-S.; Sheu M.-L.; Chan D.-C.; Yang T.-H.; Tsai K.-S.; Chiang C.-K.; Liu S.-H. Advanced Glycation End-Products Induce Skeletal Muscle Atrophy and Dysfunction in Diabetic Mice via a RAGE-Mediated, AMPK-down-Regulated, Akt Pathway. J. Pathol. 2016, 238 (3), 470–482. 10.1002/path.4674. [DOI] [PubMed] [Google Scholar]
- Tezuka Y.; Nakaya I.; Nakayama K.; Nakayama M.; Yahata M.; Soma J. Methylglyoxal as a Prognostic Factor in Patients with Chronic Kidney Disease. Nephrology (Carlton) 2019, 24 (9), 943–950. 10.1111/nep.13526. [DOI] [PubMed] [Google Scholar]
- Markova I.; Hüttl M.; Oliyarnyk O.; Kacerova T.; Haluzik M.; Kacer P.; Seda O.; Malinska H. The Effect of Dicarbonyl Stress on the Development of Kidney Dysfunction in Metabolic Syndrome - a Transcriptomic and Proteomic Approach. Nutr. Metab. 2019, 16, 51. 10.1186/s12986-019-0376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y.; Gupta R. K. Alterations in Heart and Kidney Membrane Phospholipids in Hypertension as Observed by 31P Nuclear Magnetic Resonance. Lipids 1998, 33 (10), 1023–1030. 10.1007/s11745-998-0301-z. [DOI] [PubMed] [Google Scholar]
- Giacco F.; Du X.; D’Agati V. D.; Milne R.; Sui G.; Geoffrion M.; Brownlee M. Knockdown of Glyoxalase 1 Mimics Diabetic Nephropathy in Nondiabetic Mice. Diabetes 2014, 63 (1), 291–299. 10.2337/db13-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner A. K.; Brouwers O.; Pringle R.; Klaassen I.; Colhoun L.; McVicar C.; Brockbank S.; Curry J. W.; Miyata T.; Brownlee M.; Schlingemann R. O.; Schalkwijk C.; Stitt A. W. Protection against Methylglyoxal-Derived AGEs by Regulation of Glyoxalase 1 Prevents Retinal Neuroglial and Vasodegenerative Pathology. Diabetologia 2012, 55 (3), 845–854. 10.1007/s00125-011-2393-0. [DOI] [PubMed] [Google Scholar]
- Goldin A.; Beckman J. A.; Schmidt A. M.; Creager M. A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114 (6), 597–605. 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- Beisswenger P. J.; Howell S. K.; Russell G. B.; Miller M. E.; Rich S. S.; Mauer M. Early Progression of Diabetic Nephropathy Correlates with Methylglyoxal-Derived Advanced Glycation End Products. Diabetes Care 2013, 36 (10), 3234–3239. 10.2337/dc12-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisswenger P. J.; Drummond K. S.; Nelson R. G.; Howell S. K.; Szwergold B. S.; Mauer M. Susceptibility to Diabetic Nephropathy Is Related to Dicarbonyl and Oxidative Stress. Diabetes 2005, 54 (11), 3274–3281. 10.2337/diabetes.54.11.3274. [DOI] [PubMed] [Google Scholar]
- Waris S.; Winklhofer-Roob B. M.; Roob J. M.; Fuchs S.; Sourij H.; Rabbani N.; Thornalley P. J. Increased DNA Dicarbonyl Glycation and Oxidation Markers in Patients with Type 2 Diabetes and Link to Diabetic Nephropathy. J. Diabetes Res. 2015, 2015, 915486. 10.1155/2015/915486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K.; Sakata N.; Nagai R.; Shirakawa J.-I.; Watanabe M.; Mimata A.; Abe Y.; Yasuno T.; Sasatomi Y.; Miyake K.; Ueki N.; Hamauchi A.; Nakashima H. High Serum Level of Methylglyoxal-Derived AGE, Nδ-(5-Hydro-5-Methyl-4-Imidazolone-2-Yl)-Ornithine, Independently Relates to Renal Dysfunction. Clin. Exp. Nephrol. 2017, 21 (3), 398–406. 10.1007/s10157-016-1301-9. [DOI] [PubMed] [Google Scholar]
- Forbes J. M.; Le Bagge S.; Righi S.; Fotheringham A. K.; Gallo L. A.; McCarthy D. A.; Leung S.; Baskerville T.; Nisbett J.; Morton A.; Teasdale S.; D’Silva N.; Barrett H.; Jones T.; Couper J.; Donaghue K.; Isbel N.; Johnson D. W.; Donnellan L.; Deo P.; Akison L. K.; Moritz K. M.; O’Moore-Sullivan T. Advanced Glycation End Products as Predictors of Renal Function in Youth with Type 1 Diabetes. Sci. Rep. 2021, 11 (1), 9422. 10.1038/s41598-021-88786-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins B. A.; Rabbani N.; Weston A.; Adaikalakoteswari A.; Lee J. A.; Lovblom L. E.; Cardinez N.; Thornalley P. J. High Fractional Excretion of Glycation Adducts Is Associated with Subsequent Early Decline in Renal Function in Type 1 Diabetes. Sci. Rep. 2020, 10 (1), 12709. 10.1038/s41598-020-69350-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens R. J. H.; Broers N. J. H.; Canaud B.; Christiaans M. H. L.; Cornelis T.; Gauly A.; Hermans M. M. H.; Konings C. J. A. M.; van der Sande F. M.; Scheijen J. L. J. M.; Stifft F.; Wirtz J. J. J. M.; Kooman J. P.; Schalkwijk C. G. Relations of Advanced Glycation Endproducts and Dicarbonyls with Endothelial Dysfunction and Low-Grade Inflammation in Individuals with End-Stage Renal Disease in the Transition to Renal Replacement Therapy: A Cross-Sectional Observational Study. PloS One 2019, 14 (8), e0221058. 10.1371/journal.pone.0221058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens R. J. H.; Broers N. J. H.; Canaud B.; Christiaans M. H. L.; Cornelis T.; Gauly A.; Hermans M. M. H.; Konings C. J. A. M.; van der Sande F. M.; Scheijen J. L. J. M.; Stifft F.; Kooman J. P.; Schalkwijk C. G. Advanced Glycation Endproducts and Dicarbonyls in End-Stage Renal Disease: Associations with Uraemia and Courses Following Renal Replacement Therapy. Clin. Kidney J. 2020, 13 (5), 855–866. 10.1093/ckj/sfz099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-W.; Gu M. J.; Lee J.-Y.; Lee S.; Kim Y.; Ha S. K. Methylglyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Inflammation via Interaction with RAGE in Mesangial Cells. Mol. Nutr. Food Res. 2021, 65 (13), 2000799. 10.1002/mnfr.202000799. [DOI] [PubMed] [Google Scholar]
- Lee H.-W.; Gu M. J.; Kim Y.; Lee J.-Y.; Lee S.; Choi I.-W.; Ha S. K. Glyoxal-Lysine Dimer, an Advanced Glycation End Product, Induces Oxidative Damage and Inflammatory Response by Interacting with RAGE. Antioxidants (Basel) 2021, 10 (9), 1486. 10.3390/antiox10091486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Feng L.; Zhu M.; Gu J.; Jiang J.; Cheng X.; Ding S.; Wu C.; Jia X. The Anti-Inflammation Effect of Moutan Cortex on Advanced Glycation End Products-Induced Rat Mesangial Cells Dysfunction and High-Glucose-Fat Diet and Streptozotocin-Induced Diabetic Nephropathy Rats. J. Ethnopharmacol. 2014, 151 (1), 591–600. 10.1016/j.jep.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Jeong S.-R.; Lee K.-W. Methylglyoxal-Derived Advanced Glycation End Product (AGE4)-Induced Apoptosis Leads to Mitochondrial Dysfunction and Endoplasmic Reticulum Stress through the RAGE/JNK Pathway in Kidney Cells. Int. J. Mol. Sci. 2021, 22 (12), 6530. 10.3390/ijms22126530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P.-Y.; Li P.-C.; Feng B. Protective Effects of Gliclazide on High Glucose and AGEs-Induced Damage of Glomerular Mesangial Cells and Renal Tubular Epithelial Cells via Inhibiting RAGE-P22phox-NF-KB Pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23 (20), 9099–9107. 10.26355/eurrev_201910_19313. [DOI] [PubMed] [Google Scholar]
- Baragetti I.; Norata G. D.; Sarcina C.; Baragetti A.; Rastelli F.; Buzzi L.; Grigore L.; Garlaschelli K.; Pozzi C.; Catapano A. L. 374 T/A RAGE Polymorphism Is Associated with Chronic Kidney Disease Progression in Subjects Affected by Nephrocardiovascular Disease. PloS One 2013, 8 (4), e60089. 10.1371/journal.pone.0060089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.-C.; Chou C.-K.; Chuang M.-C.; Li Y.-C.; Lee J.-A. Elevated Levels of Liver Methylglyoxal and D-Lactate in Early-Stage Hepatitis in Rats. Biomed. Chromatogr. 2018, 32 (2), 4039. 10.1002/bmc.4039. [DOI] [PubMed] [Google Scholar]
- Michel M.; Hess C.; Kaps L.; Kremer W. M.; Hilscher M.; Galle P. R.; Moehler M.; Schattenberg J. M.; Wörns M.-A.; Labenz C.; Nagel M. Elevated Serum Levels of Methylglyoxal Are Associated with Impaired Liver Function in Patients with Liver Cirrhosis. Sci. Rep. 2021, 11 (1), 20506. 10.1038/s41598-021-00119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo K.; Ki S. H.; Shin S. M. Methylglyoxal Induces Mitochondrial Dysfunction and Cell Death in Liver. Toxicol. Res. 2014, 30 (3), 193–198. 10.5487/TR.2014.30.3.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbach M.; Thonig A.; Pohl S.; Ripoll C.; Michel M.; Zipprich A. Expression of Glyoxalase-I Is Reduced in Cirrhotic Livers: A Possible Mechanism in the Development of Cirrhosis. PloS One 2017, 12 (2), e0171260. 10.1371/journal.pone.0171260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N.; Thornalley P. J.; Lüthen R.; Häussinger D.; Sebekova K.; Schinzel R.; Voelker W.; Heidland A. Processing of Protein Glycation, Oxidation and Nitrosation Adducts in the Liver and the Effect of Cirrhosis. J. Hepatol. 2004, 41 (6), 913–919. 10.1016/j.jhep.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Spanos C.; Maldonado E. M.; Fisher C. P.; Leenutaphong P.; Oviedo-Orta E.; Windridge D.; Salguero F. J.; Bermudez-Fajardo A.; Weeks M. E.; Evans C.; Corfe B. M.; Rabbani N.; Thornalley P. J.; Miller M. H.; Wang H.; Dillon J. F.; Quaglia A.; Dhawan A.; Fitzpatrick E.; Bernadette Moore J. Proteomic Identification and Characterization of Hepatic Glyoxalase 1 Dysregulation in Non-Alcoholic Fatty Liver Disease. Proteome Sci. 2018, 16, 4. 10.1186/s12953-018-0131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters K.; Cento A. S.; Gaens K. H.; Teunissen M.; Scheijen J. L. J. M.; Barutta F.; Chiazza F.; Collotta D.; Aragno M.; Gruden G.; Collino M.; Schalkwijk C. G.; Mastrocola R. Deletion of RAGE Fails to Prevent Hepatosteatosis in Obese Mice Due to Impairment of Other AGEs Receptors and Detoxifying Systems. Sci. Rep. 2021, 11 (1), 17373. 10.1038/s41598-021-96859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma-Duran S. A.; Kontogianni M. D.; Vlassopoulos A.; Zhao S.; Margariti A.; Georgoulis M.; Papatheodoridis G.; Combet E. Serum Levels of Advanced Glycation End-Products (AGEs) and the Decoy Soluble Receptor for AGEs (SRAGE) Can Identify Non-Alcoholic Fatty Liver Disease in Age-, Sex- and BMI-Matched Normo-Glycemic Adults. Metabolism. 2018, 83, 120–127. 10.1016/j.metabol.2018.01.023. [DOI] [PubMed] [Google Scholar]
- Bijnen M.; van Greevenbroek M. M. J.; van der Kallen C. J. H.; Scheijen J. L.; van de Waarenburg M. P. H.; Stehouwer C. D. A.; Wouters K.; Schalkwijk C. G. Hepatic Fat Content and Liver Enzymes Are Associated with Circulating Free and Protein-Bound Advanced Glycation End Products, Which Are Associated with Low-Grade Inflammation: The CODAM Study. J. Diabetes Res. 2019, 2019, 6289831. 10.1155/2019/6289831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serban A. I.; Stanca L.; Geicu O. I.; Munteanu M. C.; Dinischiotu A. RAGE and TGF-B1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PloS One 2016, 11 (3), e0152376. 10.1371/journal.pone.0152376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Kim O. S.; Kim C.-S.; Sohn E.; Jo K.; Kim J. S. Accumulation of Argpyrimidine, a Methylglyoxal-Derived Advanced Glycation End Product, Increases Apoptosis of Lens Epithelial Cells Both in Vitro and in Vivo. Exp. Mol. Med. 2012, 44 (2), 167–175. 10.3858/emm.2012.44.2.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.-R.; Chen T.-T.; Li W.-D.; Lu F.-L.; Liu J.; Cai X.-G.; Lu Q.; Yang C.-P. Inhibitory Effect of Receptor for Advanced Glycation End Product-specific Small Interfering RNAs on the Development of Hepatic Fibrosis in Primary Rat Hepatic Stellate Cells. Mol. Med. Rep. 2015, 12 (1), 569–574. 10.3892/mmr.2015.3342. [DOI] [PubMed] [Google Scholar]
- Cai X.-G.; Xia J.-R.; Li W.-D.; Lu F.-L.; Liu J.; Lu Q.; Zhi H. Anti-Fibrotic Effects of Specific-SiRNA Targeting of the Receptor for Advanced Glycation End Products in a Rat Model of Experimental Hepatic Fibrosis. Mol. Med. Rep. 2014, 10 (1), 306–314. 10.3892/mmr.2014.2207. [DOI] [PubMed] [Google Scholar]
- Bijnen M.; Beelen N.; Wetzels S.; Gaar J. v. d.; Vroomen M.; Wijnands E.; Scheijen J. L.; van de Waarenburg M. P. H; Gijbels M. J.; Cleutjens J. P.; Biessen E. A. L.; Stehouwer C. D. A.; Schalkwijk C. G.; Wouters K. RAGE Deficiency Does Not Affect Non-Alcoholic Steatohepatitis and Atherosclerosis in Western Type Diet-Fed Ldlr(−/−) Mice. Sci. Rep. 2018, 8 (1), 15256. 10.1038/s41598-018-33661-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price C. L.; Hassi H. O. S. A.; English N. R.; Blakemore A. I. F.; Stagg A. J.; Knight S. C. Methylglyoxal Modulates Immune Responses: Relevance to Diabetes. J. Cell. Mol. Med. 2010, 14 (6B), 1806–1815. 10.1111/j.1582-4934.2009.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann T.; Dunkel A.; Schmid C.; Schmitt S.; Hiltensperger M.; Lohr K.; Laketa V.; Donakonda S.; Ahting U.; Lorenz-Depiereux B.; Heil J. E.; Schredelseker J.; Simeoni L.; Fecher C.; Körber N.; Bauer T.; Hüser N.; Hartmann D.; Laschinger M.; Eyerich K.; Eyerich S.; Anton M.; Streeter M.; Wang T.; Schraven B.; Spiegel D.; Assaad F.; Misgeld T.; Zischka H.; Murray P. J.; Heine A.; Heikenwälder M.; Korn T.; Dawid C.; Hofmann T.; Knolle P. A.; Höchst B. Regulatory Myeloid Cells Paralyze T Cells through Cell-Cell Transfer of the Metabolite Methylglyoxal. Nat. Immunol. 2020, 21 (5), 555–566. 10.1038/s41590-020-0666-9. [DOI] [PubMed] [Google Scholar]
- Mir A. R.; Moinuddin; Habib S.; Khan F.; Alam K.; Ali A. Structural Changes in Histone H2A by Methylglyoxal Generate Highly Immunogenic Amorphous Aggregates with Implications in Auto-Immune Response in Cancer. Glycobiology 2016, 26 (2), 129–141. 10.1093/glycob/cwv082. [DOI] [PubMed] [Google Scholar]
- Son S.; Hwang I.; Han S. H.; Shin J.-S.; Shin O. S.; Yu J.-W. Advanced Glycation End Products Impair NLRP3 Inflammasome-Mediated Innate Immune Responses in Macrophages. J. Biol. Chem. 2017, 292 (50), 20437–20448. 10.1074/jbc.M117.806307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X.; Yao T.; Zhou Z.; Zhu J.; Zhang S.; Hu W.; Shen C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-KB Pathway. BioMed. Res. Int. 2015, 2015, 732450. 10.1155/2015/732450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S. F.; Ramasamy R.; Schmidt A. M. The RAGE Axis: A Fundamental Mechanism Signaling Danger to the Vulnerable Vasculature. Circ. Res. 2010, 106 (5), 842–853. 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser B.; Desai D. D.; Downie M. P.; Chen Y.; Yan S. F.; Herold K.; Schmidt A. M.; Clynes R. Receptor for Advanced Glycation End Products Expression on T Cells Contributes to Antigen-Specific Cellular Expansion in Vivo. J. Immunol. 2007, 179 (12), 8051–8058. 10.4049/jimmunol.179.12.8051. [DOI] [PubMed] [Google Scholar]
- Akirav E. M.; Preston-Hurlburt P.; Garyu J.; Henegariu O.; Clynes R.; Schmidt A. M.; Herold K. C. RAGE Expression in Human T Cells: A Link between Environmental Factors and Adaptive Immune Responses. PloS One 2012, 7 (4), e34698. 10.1371/journal.pone.0034698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzels S.; Wouters K.; Schalkwijk C. G.; Vanmierlo T.; Hendriks J. J. A. Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18 (2), 421. 10.3390/ijms18020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg Z.; Hennies C.; Sternberg D.; Wang P.; Kinkel P.; Hojnacki D.; Weinstock-Guttman B.; Munschauer F. Diagnostic Potential of Plasma Carboxymethyllysine and Carboxyethyllysine in Multiple Sclerosis. J. Neuroinflammation 2010, 7, 72. 10.1186/1742-2094-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Chen X.; Sun X.; Wang L.; Chen S. The Glycolytic Switch in Tumors: How Many Players Are Involved?. J. Cancer 2017, 8 (17), 3430–3440. 10.7150/jca.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda F.; Arai Y.; Okada N.; Shimizu H.; Miyamoto M.; Kitagawa N.; Katai H.; Taniguchi H.; Yanagihara K.; Imoto I.; Inazawa J.; Ohki M.; Shibata T. Integrated Genomic and Functional Analyses Reveal Glyoxalase I as a Novel Metabolic Oncogene in Human Gastric Cancer. Oncogene 2015, 34 (9), 1196–1206. 10.1038/onc.2014.57. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kuramitsu Y.; Ueno T.; Suzuki N.; Yoshino S.; Iizuka N.; Akada J.; Kitagawa T.; Oka M.; Nakamura K. Glyoxalase I (GLO1) Is up-Regulated in Pancreatic Cancerous Tissues Compared with Related Non-Cancerous Tissues. Anticancer Res. 2012, 32 (8), 3219–3222. [PubMed] [Google Scholar]
- Bair W. B. 3rd; Cabello C. M.; Uchida K.; Bause A. S.; Wondrak G. T. GLO1 Overexpression in Human Malignant Melanoma. Melanoma Res. 2010, 20 (2), 85–96. 10.1097/CMR.0b013e3283364903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamori S.; Nozaki Y.; Motomura H.; Nakane H.; Katayama R.; Onaga C.; Kikuchi E.; Shimada N.; Suzuki Y.; Noike M.; Hara Y.; Sato K.; Sato T.; Yamamoto K.; Hanawa T.; Imai M.; Abe R.; Yoshimori A.; Takasawa R.; Tanuma S.-I.; Akimoto K. Glyoxalase 1 Gene Is Highly Expressed in Basal-like Human Breast Cancers and Contributes to Survival of ALDH1-Positive Breast Cancer Stem Cells. Oncotarget 2018, 9 (92), 36515–36529. 10.18632/oncotarget.26369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abordo E. A.; Minhas H. S.; Thornalley P. J. Accumulation of Alpha-Oxoaldehydes during Oxidative Stress: A Role in Cytotoxicity. Biochem. Pharmacol. 1999, 58 (4), 641–648. 10.1016/S0006-2952(99)00132-X. [DOI] [PubMed] [Google Scholar]
- Nokin M.-J.; Durieux F.; Bellier J.; Peulen O.; Uchida K.; Spiegel D. A.; Cochrane J. R.; Hutton C. A.; Castronovo V.; Bellahcène A. Hormetic Potential of Methylglyoxal, a Side-Product of Glycolysis, in Switching Tumours from Growth to Death. Sci. Rep. 2017, 7 (1), 11722. 10.1038/s41598-017-12119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio M. L.; Gentile F.; Presta I.; Donato G.; Coppedè N.; Valprapuram I.; Mignogna C.; Lavecchia A.; Figuccia F.; Garo V. M.; Fabrizio E. D.; Candeloro P.; Viglietto G.; Malara N. Tailoring Chemometric Models on Blood-Derived Cultures Secretome to Assess Personalized Cancer Risk Score. Cancers 2020, 12 (6), 1362. 10.3390/cancers12061362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R.; Tang D.; Schapiro N. E.; Livesey K. M.; Farkas A.; Loughran P.; Bierhaus A.; Lotze M. T.; Zeh H. J. The Receptor for Advanced Glycation End Products (RAGE) Sustains Autophagy and Limits Apoptosis, Promoting Pancreatic Tumor Cell Survival. Cell Death Differ. 2010, 17 (4), 666–676. 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki S.; Kasayama S.; Miki Y.; Nakamura Y.; Yamamoto M.; Sato B.; Kishimoto T. Expression of Receptors for Advanced Glycosylation End Products on Renal Cell Carcinoma Cells in Vitro. Biochem. Biophys. Res. Commun. 1993, 196 (2), 984–989. 10.1006/bbrc.1993.2346. [DOI] [PubMed] [Google Scholar]
- Sakellariou S.; Fragkou P.; Levidou G.; Gargalionis A. N.; Piperi C.; Dalagiorgou G.; Adamopoulos C.; Saetta A.; Agrogiannis G.; Theohari I.; Sougioultzis S.; Tsioli P.; Karavokyros I.; Tsavaris N.; Kostakis I. D.; Zizi-Serbetzoglou A.; Vandoros G. P.; Patsouris E.; Korkolopoulou P. Clinical Significance of AGE-RAGE Axis in Colorectal Cancer: Associations with Glyoxalase-I, Adiponectin Receptor Expression and Prognosis. BMC Cancer 2016, 16, 174. 10.1186/s12885-016-2213-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghela B. N.; Vaidya F. U.; Ranjan K.; Chhipa A. S.; Tiwari B. S.; Pathak C. AGE-RAGE Synergy Influences Programmed Cell Death Signaling to Promote Cancer. Mol. Cell. Biochem. 2021, 476 (2), 585–598. 10.1007/s11010-020-03928-y. [DOI] [PubMed] [Google Scholar]
- Logsdon C. D.; Fuentes M. K.; Huang E. H.; Arumugam T. RAGE and RAGE Ligands in Cancer. Curr. Mol. Med. 2007, 7 (8), 777–789. 10.2174/156652407783220697. [DOI] [PubMed] [Google Scholar]
- Azizian-Farsani F.; Abedpoor N.; Hasan Sheikhha M.; Gure A. O.; Nasr-Esfahani M. H.; Ghaedi K. Receptor for Advanced Glycation End Products Acts as a Fuel to Colorectal Cancer Development. Front. Oncol. 2020, 10, 552283. 10.3389/fonc.2020.552283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riboulet-Chavey A.; Pierron A.; Durand I.; Murdaca J.; Giudicelli J.; Van Obberghen E. Methylglyoxal Impairs the Insulin Signaling Pathways Independently of the Formation of Intracellular Reactive Oxygen Species. Diabetes 2006, 55 (5), 1289–1299. 10.2337/db05-0857. [DOI] [PubMed] [Google Scholar]
- Ng Y.; Ramm G.; Lopez J. A.; James D. E. Rapid Activation of Akt2 Is Sufficient to Stimulate GLUT4 Translocation in 3T3-L1 Adipocytes. Cell Metab. 2008, 7 (4), 348–356. 10.1016/j.cmet.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Thompson B. J.; Hietakangas V.; Cohen S. M. MAPK/ERK Signaling Regulates Insulin Sensitivity to Control Glucose Metabolism in Drosophila. PLoS Genet. 2011, 7 (12), e1002429. 10.1371/journal.pgen.1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko A. J.; Miller R. A.; Kelman A.; Frauwirth K. A. Induction of Glucose Metabolism in Stimulated T Lymphocytes Is Regulated by Mitogen-Activated Protein Kinase Signaling. PloS One 2010, 5 (11), e15425. 10.1371/journal.pone.0015425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N.; Babaei-Jadidi R.; Howell S. K.; Beisswenger P. J.; Thornalley P. J. Degradation Products of Proteins Damaged by Glycation, Oxidation and Nitration in Clinical Type 1 Diabetes. Diabetologia 2005, 48 (8), 1590–1603. 10.1007/s00125-005-1810-7. [DOI] [PubMed] [Google Scholar]
- Duran-Jimenez B.; Dobler D.; Moffatt S.; Rabbani N.; Streuli C. H.; Thornalley P. J.; Tomlinson D. R.; Gardiner N. J. Advanced Glycation End Products in Extracellular Matrix Proteins Contribute to the Failure of Sensory Nerve Regeneration in Diabetes. Diabetes 2009, 58 (12), 2893–2903. 10.2337/db09-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilker S. C.; Chellan P.; Arnold B. M.; Nagaraj R. H. Chromatographic Quantification of Argpyrimidine, a Methylglyoxal-Derived Product in Tissue Proteins: Comparison with Pentosidine. Anal. Biochem. 2001, 290 (2), 353–358. 10.1006/abio.2001.4992. [DOI] [PubMed] [Google Scholar]
- Han Y.; Randell E.; Vasdev S.; Gill V.; Gadag V.; Newhook L. A.; Grant M.; Hagerty D. Plasma Methylglyoxal and Glyoxal Are Elevated and Related to Early Membrane Alteration in Young, Complication-Free Patients with Type 1 Diabetes. Mol. Cell. Biochem. 2007, 305 (1–2), 123–131. 10.1007/s11010-007-9535-1. [DOI] [PubMed] [Google Scholar]
- Kong X.; Ma M.; Huang K.; Qin L.; Zhang H.; Yang Z.; Li X.; Su Q. Increased Plasma Levels of the Methylglyoxal in Patients with Newly Diagnosed Type 2 Diabetes 2. J. Diabetes 2014, 6 (6), 535–540. 10.1111/1753-0407.12160. [DOI] [PubMed] [Google Scholar]
- Bellier J.; Nokin M.-J.; Lardé E.; Karoyan P.; Peulen O.; Castronovo V.; Bellahcène A. Methylglyoxal, a Potent Inducer of AGEs, Connects between Diabetes and Cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. 10.1016/j.diabres.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Stitt A. W.; Bhaduri T.; McMullen C. B.; Gardiner T. A.; Archer D. B. Advanced Glycation End Products Induce Blood-Retinal Barrier Dysfunction in Normoglycemic Rats. Mol. Cell Biol. Res. Commun. MCBRC 2000, 3 (6), 380–388. 10.1006/mcbr.2000.0243. [DOI] [PubMed] [Google Scholar]
- Griggs R. B.; Laird D. E.; Donahue R. R.; Fu W.; Taylor B. K. Methylglyoxal Requires AC1 and TRPA1 to Produce Pain and Spinal Neuron Activation. Front. Neurosci. 2017, 11, 679. 10.3389/fnins.2017.00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobler D.; Ahmed N.; Song L.; Eboigbodin K. E.; Thornalley P. J. Increased Dicarbonyl Metabolism in Endothelial Cells in Hyperglycemia Induces Anoikis and Impairs Angiogenesis by RGD and GFOGER Motif Modification. Diabetes 2006, 55 (7), 1961–1969. 10.2337/db05-1634. [DOI] [PubMed] [Google Scholar]
- Xu J.; Chen L.-J.; Yu J.; Wang H.-J.; Zhang F.; Liu Q.; Wu J. Involvement of Advanced Glycation End Products in the Pathogenesis of Diabetic Retinopathy. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol 2018, 48 (2), 705–717. 10.1159/000491897. [DOI] [PubMed] [Google Scholar]
- Sugimoto K.; Yasujima M.; Yagihashi S. Role of Advanced Glycation End Products in Diabetic Neuropathy. Curr. Pharm. Des. 2008, 14 (10), 953–961. 10.2174/138161208784139774. [DOI] [PubMed] [Google Scholar]
- Bierhaus A.; Hofmann M. A.; Ziegler R.; Nawroth P. P. AGEs and Their Interaction with AGE-Receptors in Vascular Disease and Diabetes Mellitus. I. The AGE Concept. Cardiovasc. Res. 1998, 37 (3), 586–600. 10.1016/S0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- Haddad M.; Perrotte M.; Khedher M. R. B.; Demongin C.; Lepage A.; Fülöp T.; Ramassamy C. Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20 (19), 4906. 10.3390/ijms20194906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knani I.; Bouzidi H.; Zrour S.; Bergaoui N.; Hammami M.; Kerkeni M. Methylglyoxal: A Relevant Marker of Disease Activity in Patients with Rheumatoid Arthritis. Dis. Markers 2018, 2018, 8735926. 10.1155/2018/8735926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley P. J.; Rabbani N. Detection of Oxidized and Glycated Proteins in Clinical Samples Using Mass Spectrometry--a User’s Perspective. Biochim. Biophys. Acta 2014, 1840 (2), 818–829. 10.1016/j.bbagen.2013.03.025. [DOI] [PubMed] [Google Scholar]
- Conway B.; Edmundowicz D.; Matter N.; Maynard J.; Orchard T. Skin Fluorescence Correlates Strongly with Coronary Artery Calcification Severity in Type 1 Diabetes. Diabetes Technol. Ther. 2010, 12 (5), 339–345. 10.1089/dia.2009.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits E. G.; Lutgers H. L.; Kleefstra N.; Graaff R.; Groenier K. H.; Smit A. J.; Gans R. O.; Bilo H. J. Skin Autofluorescence: A Tool to Identify Type 2 Diabetic Patients at Risk for Developing Microvascular Complications. Diabetes Care 2008, 31 (3), 517–521. 10.2337/dc07-1755. [DOI] [PubMed] [Google Scholar]
- Hartog J. W. L.; Gross S.; Oterdoom L. H.; van Ree R. M.; de Vries A. P. J.; Smit A. J.; Schouten J. P.; Nawroth P. P.; Gans R. O. B.; van Son W. J.; Bierhaus A.; Bakker S. J. L. Skin-Autofluorescence Is an Independent Predictor of Graft Loss in Renal Transplant Recipients. Transplantation 2009, 87 (7), 1069–1077. 10.1097/TP.0b013e31819d3173. [DOI] [PubMed] [Google Scholar]
- Ammanath G.; Delachi C. G.; Karabacak S.; Ali Y.; Boehm B. O.; Yildiz U. H.; Alagappan P.; Liedberg B. Colorimetric and Fluorometric Profiling of Advanced Glycation End Products. ACS Appl. Mater. Interfaces 2022, 14 (1), 94–103. 10.1021/acsami.1c16261. [DOI] [PubMed] [Google Scholar]
- Münch G.; Keis R.; Wessels A.; Riederer P.; Bahner U.; Heidland A.; Niwa T.; Lemke H. D.; Schinzel R. Determination of Advanced Glycation End Products in Serum by Fluorescence Spectroscopy and Competitive ELISA. Eur. J. Clin. Chem. Clin. Biochem. 1997, 35 (9), 669–677. 10.1515/cclm.1997.35.9.669. [DOI] [PubMed] [Google Scholar]
- de Vos L. C.; Lefrandt J. D.; Dullaart R. P. F.; Zeebregts C. J.; Smit A. J. Advanced Glycation End Products: An Emerging Biomarker for Adverse Outcome in Patients with Peripheral Artery Disease. Atherosclerosis 2016, 254, 291–299. 10.1016/j.atherosclerosis.2016.10.012. [DOI] [PubMed] [Google Scholar]
- Ahmed N.; Argirov O. K.; Minhas H. S.; Cordeiro C. A. A.; Thornalley P. J. Assay of Advanced Glycation Endproducts (AGEs): Surveying AGEs by Chromatographic Assay with Derivatization by 6-Aminoquinolyl-N-Hydroxysuccinimidyl-Carbamate and Application to Nepsilon-Carboxymethyl-Lysine- and Nepsilon-(1-Carboxyethyl)Lysine-Modified Albumin. Biochem. J. 2002, 364 (1), 1–14. 10.1042/bj3640001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K.; Khor O. T.; Oya T.; Osawa T.; Yasuda Y.; Miyata T. Protein Modification by a Maillard Reaction Intermediate Methylglyoxal. Immunochemical Detection of Fluorescent 5-Methylimidazolone Derivatives in Vivo. FEBS Lett. 1997, 410 (2–3), 313–318. 10.1016/S0014-5793(97)00610-8. [DOI] [PubMed] [Google Scholar]
- Yan S. D.; Schmidt A. M.; Anderson G. M.; Zhang J.; Brett J.; Zou Y. S.; Pinsky D.; Stern D. Enhanced Cellular Oxidant Stress by the Interaction of Advanced Glycation End Products with Their Receptors/Binding Proteins. J. Biol. Chem. 1994, 269 (13), 9889–9897. 10.1016/S0021-9258(17)36966-1. [DOI] [PubMed] [Google Scholar]
- Li J.; Wang K.; Huang B.; Li R.; Wang X.; Zhang H.; Tang H.; Chen X. The Receptor for Advanced Glycation End Products Mediates Dysfunction of Airway Epithelial Barrier in a Lipopolysaccharides-Induced Murine Acute Lung Injury Model. Int. Immunopharmacol. 2021, 93, 107419. 10.1016/j.intimp.2021.107419. [DOI] [PubMed] [Google Scholar]
- Yang L.; Liu Y.; Wang Y.; Li J.; Liu N. Azeliragon Ameliorates Alzheimer’s Disease via the Janus Tyrosine Kinase and Signal Transducer and Activator of Transcription Signaling Pathway. Clin. Sao Paulo Braz. 2021, 76, e2348. 10.6061/clinics/2021/e2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasko D.; Bell J.; Mancuso J. Y.; Kupiec J. W.; Sabbagh M. N.; van Dyck C.; Thomas R. G.; Aisen P. S. Clinical Trial of an Inhibitor of RAGE-Aβ Interactions in Alzheimer Disease. Neurology 2014, 82 (17), 1536–1542. 10.1212/WNL.0000000000000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbagh M. N.; Agro A.; Bell J.; Aisen P. S.; Schweizer E.; Galasko D. PF-04494700, an Oral Inhibitor of Receptor for Advanced Glycation End Products (RAGE), in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2011, 25 (3), 206–212. 10.1097/WAD.0b013e318204b550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Xu X.; Rao N. V.; Argyle B.; McCoard L.; Rusho W. J.; Kennedy T. P.; Prestwich G. D.; Krueger G. Novel Sulfated Polysaccharides Disrupt Cathelicidins, Inhibit RAGE and Reduce Cutaneous Inflammation in a Mouse Model of Rosacea. PloS One 2011, 6 (2), e16658. 10.1371/journal.pone.0016658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nokin M.-J.; Durieux F.; Peixoto P.; Chiavarina B.; Peulen O.; Blomme A.; Turtoi A.; Costanza B.; Smargiasso N.; Baiwir D.; Scheijen J. L.; Schalkwijk C. G.; Leenders J.; De Tullio P.; Bianchi E.; Thiry M.; Uchida K.; Spiegel D. A.; Cochrane J. R.; Hutton C. A.; De Pauw E.; Delvenne P.; Belpomme D.; Castronovo V.; Bellahcène A. Methylglyoxal, a Glycolysis Side-Product, Induces Hsp90 Glycation and YAP-Mediated Tumor Growth and Metastasis. eLife 2016, 5, e19375. 10.7554/eLife.19375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nokin M.-J.; Bellier J.; Durieux F.; Peulen O.; Rademaker G.; Gabriel M.; Monseur C.; Charloteaux B.; Verbeke L.; van Laere S.; Roncarati P.; Herfs M.; Lambert C.; Scheijen J.; Schalkwijk C.; Colige A.; Caers J.; Delvenne P.; Turtoi A.; Castronovo V.; Bellahcène A. Methylglyoxal, a Glycolysis Metabolite, Triggers Metastasis through MEK/ERK/SMAD1 Pathway Activation in Breast Cancer. Breast Cancer Res. BCR 2019, 21 (1), 11. 10.1186/s13058-018-1095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Zhang Y.; Yang X.; Lu P.; Yan X.; Xiao F.; Zhou H.; Wen C.; Shi M.; Lu J.; Meng Q. H. Effects of Methylglyoxal and Glyoxalase I Inhibition on Breast Cancer Cells Proliferation, Invasion, and Apoptosis through Modulation of MAPKs, MMP9, and Bcl-2. Cancer Biol. Ther. 2016, 17 (2), 169–180. 10.1080/15384047.2015.1121346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharaf H.; Matou-Nasri S.; Wang Q.; Rabhan Z.; Al-Eidi H.; Al Abdulrahman A.; Ahmed N. Advanced Glycation Endproducts Increase Proliferation, Migration and Invasion of the Breast Cancer Cell Line MDA-MB-231. Biochim. Biophys. Acta 2015, 1852 (3), 429–441. 10.1016/j.bbadis.2014.12.009. [DOI] [PubMed] [Google Scholar]
- Lee K. J.; Yoo J. W.; Kim Y. K.; Choi J. H.; Ha T.-Y.; Gil M. Advanced Glycation End Products Promote Triple Negative Breast Cancer Cells via ERK and NF-KB Pathway. Biochem. Biophys. Res. Commun. 2018, 495 (3), 2195–2201. 10.1016/j.bbrc.2017.11.182. [DOI] [PubMed] [Google Scholar]
- Khan M. S.; Tabrez S.; Al-Okail M. S.; Shaik G. M.; Bhat S. A.; Rehman T. M.; Husain F. M.; AlAjmi M. F. Non-Enzymatic Glycation of Protein Induces Cancer Cell Proliferation and Its Inhibition by Quercetin: Spectroscopic, Cytotoxicity and Molecular Docking Studies. J. Biomol. Struct. Dyn. 2021, 39 (3), 777–786. 10.1080/07391102.2020.1715838. [DOI] [PubMed] [Google Scholar]
- Paul-Samojedny M.; Łasut B.; Pudełko A.; Fila-Daniłow A.; Kowalczyk M.; Suchanek-Raif R.; Zieliński M.; Borkowska P.; Kowalski J. Methylglyoxal (MGO) Inhibits Proliferation and Induces Cell Death of Human Glioblastoma Multiforme T98G and U87MG Cells. Biomed. Pharmacother. Biomedecine Pharmacother. 2016, 80, 236–243. 10.1016/j.biopha.2016.03.021. [DOI] [PubMed] [Google Scholar]
- Nam H.-K.; Jeong S.-R.; Pyo M. C.; Ha S.-K.; Nam M.-H.; Lee K.-W. Methylglyoxal-Derived Advanced Glycation End Products (AGE4) Promote Cell Proliferation and Survival in Renal Cell Carcinoma Cells through the RAGE/Akt/ERK Signaling Pathways. Biol. Pharm. Bull. 2021, 44 (11), 1697–1706. 10.1248/bpb.b21-00382. [DOI] [PubMed] [Google Scholar]
- Loarca L.; Sassi-Gaha S.; Artlett C. M. Two α-Dicarbonyls Downregulate Migration, Invasion, and Adhesion of Liver Cancer Cells in a P53-Dependent Manner. Dig. Liver Dis. 2013, 45 (11), 938–946. 10.1016/j.dld.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Ayoub F. M.; Allen R. E.; Thornalley P. J. Inhibition of Proliferation of Human Leukaemia 60 Cells by Methylglyoxal in Vitro. Leuk. Res. 1993, 17 (5), 397–401. 10.1016/0145-2126(93)90094-2. [DOI] [PubMed] [Google Scholar]
- Kang Y.; Edwards L. G.; Thornalley P. J. Effect of Methylglyoxal on Human Leukaemia 60 Cell Growth: Modification of DNA G1 Growth Arrest and Induction of Apoptosis. Leuk. Res. 1996, 20 (5), 397–405. 10.1016/0145-2126(95)00162-X. [DOI] [PubMed] [Google Scholar]
- Milanesa D. M.; Choudhury M. S.; Mallouh C.; Tazaki H.; Konno S. Methylglyoxal-Induced Apoptosis in Human Prostate Carcinoma: Potential Modality for Prostate Cancer Treatment. Eur. Urol. 2000, 37 (6), 728–734. 10.1159/000020226. [DOI] [PubMed] [Google Scholar]
- Antognelli C.; Moretti S.; Frosini R.; Puxeddu E.; Sidoni A.; Talesa V. N. Methylglyoxal Acts as a Tumor-Promoting Factor in Anaplastic Thyroid Cancer. Cells 2019, 8 (6), 547. 10.3390/cells8060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-A.; Wu C.-H.; Yen G.-C. Methylglyoxal Displays Colorectal Cancer-Promoting Properties in the Murine Models of Azoxymethane and CT26 Isografts. Free Radic. Biol. Med. 2018, 115, 436–446. 10.1016/j.freeradbiomed.2017.12.020. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Fang L.; Li G.; Zhang J.; Li C.; Ma M.; Guan C.; Bai F.; Lyu J.; Meng Q. H. Synergistic Inhibition of Colon Cancer Growth by the Combination of Methylglyoxal and Silencing of Glyoxalase I Mediated by the STAT1 Pathway. Oncotarget 2017, 8 (33), 54838–54857. 10.18632/oncotarget.18601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T.; Zhou H.; Li C.; Chen Y.; Chen X.; Li C.; Mao J.; Lyu J.; Meng Q. H. Methylglyoxal Suppresses Human Colon Cancer Cell Lines and Tumor Growth in a Mouse Model by Impairing Glycolytic Metabolism of Cancer Cells Associated with Down-Regulation of c-Myc Expression. Cancer Biol. Ther. 2016, 17 (9), 955–965. 10.1080/15384047.2016.1210736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio M. L.; Presta I.; Greco M.; Gervasi R.; La Torre D.; Renne M.; Voci C. P.; Lunelli L.; Donato G.; Malara N. Microenvironment Molecular Profile Combining Glycation Adducts and Cytokines Patterns on Secretome of Short-Term Blood-Derived Cultures during Tumour Progression. Int. J. Mol. Sci. 2020, 21 (13), 4711. 10.3390/ijms21134711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavarina B.; Nokin M.-J.; Bellier J.; Durieux F.; Bletard N.; Sherer F.; Lovinfosse P.; Peulen O.; Verset L.; Dehon R.; Demetter P.; Turtoi A.; Uchida K.; Goldman S.; Hustinx R.; Delvenne P.; Castronovo V.; Bellahcène A. Methylglyoxal-Mediated Stress Correlates with High Metabolic Activity and Promotes Tumor Growth in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18 (1), 213. 10.3390/ijms18010213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva M. A.; Borges C. M.; Florêncio M. H. Reactions of Aminoguanidine with α-Dicarbonyl Compounds Studied by Electrospray Ionization Mass Spectrometry. Eur. J. Mass Spectrom. Chichester Engl. 2012, 18 (4), 385–397. 10.1255/ejms.1191. [DOI] [PubMed] [Google Scholar]
- Thornalley P. J. Use of Aminoguanidine (Pimagedine) to Prevent the Formation of Advanced Glycation Endproducts. Arch. Biochem. Biophys. 2003, 419 (1), 31–40. 10.1016/j.abb.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Thornalley P. J.; Yurek-George A.; Argirov O. K. Kinetics and Mechanism of the Reaction of Aminoguanidine with the Alpha-Oxoaldehydes Glyoxal, Methylglyoxal, and 3-Deoxyglucosone under Physiological Conditions. Biochem. Pharmacol. 2000, 60 (1), 55–65. 10.1016/S0006-2952(00)00287-2. [DOI] [PubMed] [Google Scholar]
- Solís-Calero C.; Ortega-Castro J.; Hernández-Laguna A.; Muñoz F. DFT Study of the Mechanism of the Reaction of Aminoguanidine with Methylglyoxal. J. Mol. Model. 2014, 20 (4), 2202. 10.1007/s00894-014-2202-z. [DOI] [PubMed] [Google Scholar]
- Lima T. F. O.; Costa M. C.; Figueiredo I. D.; Inácio M. D.; Rodrigues M. R.; Assis R. P.; Baviera A. M.; Brunetti I. L. Curcumin, Alone or in Combination with Aminoguanidine, Increases Antioxidant Defenses and Glycation Product Detoxification in Streptozotocin-Diabetic Rats: A Therapeutic Strategy to Mitigate Glycoxidative Stress. Oxid. Med. Cell. Longevity 2020, 2020, 1036360. 10.1155/2020/1036360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Hu Q.; Ren X.; He P.; Xu H.; Dai H.; Chen Z. Aminoguanidine suppresses methylglyoxal-mediated oxygen-glucose deprivation injury in human brain microvascular endothelial cells. J. Zhejiang Univ. Med. Sci. 2013, 42 (3), 261–266. [PubMed] [Google Scholar]
- Liu B.-F.; Miyata S.; Hirota Y.; Higo S.; Miyazaki H.; Fukunaga M.; Hamada Y.; Ueyama S.; Muramoto O.; Uriuhara A.; Kasuga M. Methylglyoxal Induces Apoptosis through Activation of P38 Mitogen-Activated Protein Kinase in Rat Mesangial Cells. Kidney Int. 2003, 63 (3), 947–957. 10.1046/j.1523-1755.2003.00829.x. [DOI] [PubMed] [Google Scholar]
- Magdaleno F.; Blajszczak C. C.; Charles-Niño C. L.; Guadrón-Llanos A. M.; Vázquez-Álvarez A. O.; Miranda-Díaz A. G.; Nieto N.; Islas-Carbajal M. C.; Rincón-Sánchez A. R. Aminoguanidine Reduces Diabetes-Associated Cardiac Fibrosis. Exp. Ther. Med. 2019, 18 (4), 3125–3138. 10.3892/etm.2019.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo T. W.; Selwood T.; Thornalley P. J. The Reaction of Methylglyoxal with Aminoguanidine under Physiological Conditions and Prevention of Methylglyoxal Binding to Plasma Proteins. Biochem. Pharmacol. 1994, 48 (10), 1865–1870. 10.1016/0006-2952(94)90584-3. [DOI] [PubMed] [Google Scholar]
- Hsuuw Y.-D.; Chang C.-K.; Chan W.-H.; Yu J.-S. Curcumin Prevents Methylglyoxal-Induced Oxidative Stress and Apoptosis in Mouse Embryonic Stem Cells and Blastocysts. J. Cell. Physiol. 2005, 205 (3), 379–386. 10.1002/jcp.20408. [DOI] [PubMed] [Google Scholar]
- Chan W.; Wu H. Protective Effects of Curcumin on Methylglyoxal-Induced Oxidative DNA Damage and Cell Injury in Human Mononuclear Cells. Acta Pharmacol. Sin. 2006, 27 (9), 1192–1198. 10.1111/j.1745-7254.2006.00374.x. [DOI] [PubMed] [Google Scholar]
- Sun Y. P.; Gu J. F.; Tan X. B.; Wang C. F.; Jia X. B.; Feng L.; Liu J. P. Curcumin Inhibits Advanced Glycation End Product-Induced Oxidative Stress and Inflammatory Responses in Endothelial Cell Damage via Trapping Methylglyoxal. Mol. Med. Rep. 2016, 13 (2), 1475–1486. 10.3892/mmr.2015.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T.-Y.; Liu C.-L.; Chyau C.-C.; Hu M.-L. Trapping of Methylglyoxal by Curcumin in Cell-Free Systems and in Human Umbilical Vein Endothelial Cells. J. Agric. Food Chem. 2012, 60 (33), 8190–8196. 10.1021/jf302188a. [DOI] [PubMed] [Google Scholar]
- Roxo D. F.; Arcaro C. A.; Gutierres V. O.; Costa M. C.; Oliveira J. O.; Lima T. F. O.; Assis R. P.; Brunetti I. L.; Baviera A. M. Curcumin Combined with Metformin Decreases Glycemia and Dyslipidemia, and Increases Paraoxonase Activity in Diabetic Rats. Diabetol. Metab. Syndr. 2019, 11, 33. 10.1186/s13098-019-0431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung E.; Park S.-B.; Jung W. K.; Kim H. R.; Kim J. Antiglycation Activity of Aucubin In Vitro and in Exogenous Methylglyoxal Injected Rats. Molecules 2019, 24 (20), 3653. 10.3390/molecules24203653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L.; Shao X.; Chen H.; Ho C.-T.; Sang S. Genistein Inhibits Advanced Glycation End Product Formation by Trapping Methylglyoxal. Chem. Res. Toxicol. 2011, 24 (4), 579–586. 10.1021/tx100457h. [DOI] [PubMed] [Google Scholar]
- Li X.; Zheng T.; Sang S.; Lv L. Quercetin Inhibits Advanced Glycation End Product Formation by Trapping Methylglyoxal and Glyoxal. J. Agric. Food Chem. 2014, 62 (50), 12152–12158. 10.1021/jf504132x. [DOI] [PubMed] [Google Scholar]
- Do M. H.; Hur J.; Choi J.; Kim M.; Kim M. J.; Kim Y.; Ha S. K. Eucommia Ulmoides Ameliorates Glucotoxicity by Suppressing Advanced Glycation End-Products in Diabetic Mice Kidney. Nutrients 2018, 10 (3), 265. 10.3390/nu10030265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciarelli L. G.; Wendt T.; Qu W.; Lu Y.; Lalla E.; Rong L. L.; Goova M. T.; Moser B.; Kislinger T.; Lee D. C.; Kashyap Y.; Stern D. M.; Schmidt A. M. RAGE Blockade Stabilizes Established Atherosclerosis in Diabetic Apolipoprotein E-Null Mice. Circulation 2002, 106 (22), 2827–2835. 10.1161/01.CIR.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- La Selva M.; Beltramo E.; Pagnozzi F.; Bena E.; Molinatti P. A.; Molinatti G. M.; Porta M. Thiamine Corrects Delayed Replication and Decreases Production of Lactate and Advanced Glycation End-Products in Bovine Retinal and Human Umbilical Vein Endothelial Cells Cultured under High Glucose Conditions. Diabetologia 1996, 39 (11), 1263–1268. 10.1007/s001250050568. [DOI] [PubMed] [Google Scholar]
- Stracke H.; Hammes H. P.; Werkmann D.; Mavrakis K.; Bitsch I.; Netzel M.; Geyer J.; Köpcke W.; Sauerland C.; Bretzel R. G.; Federlin K. F. Efficacy of Benfotiamine versus Thiamine on Function and Glycation Products of Peripheral Nerves in Diabetic Rats. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2001, 109 (6), 330–336. 10.1055/s-2001-17399. [DOI] [PubMed] [Google Scholar]
- Alkhalaf A.; Klooster A.; van Oeveren W.; Achenbach U.; Kleefstra N.; Slingerland R. J.; Mijnhout G. S.; Bilo H. J. G.; Gans R. O. B.; Navis G. J.; Bakker S. J. L. A Double-Blind, Randomized, Placebo-Controlled Clinical Trial on Benfotiamine Treatment in Patients with Diabetic Nephropathy. Diabetes Care 2010, 33 (7), 1598–1601. 10.2337/dc09-2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirban A.; Negrean M.; Stratmann B.; Gawlowski T.; Horstmann T.; Götting C.; Kleesiek K.; Mueller-Roesel M.; Koschinsky T.; Uribarri J.; Vlassara H.; Tschoepe D. Benfotiamine Prevents Macro- and Microvascular Endothelial Dysfunction and Oxidative Stress Following a Meal Rich in Advanced Glycation End Products in Individuals with Type 2 Diabetes. Diabetes Care 2006, 29 (9), 2064–2071. 10.2337/dc06-0531. [DOI] [PubMed] [Google Scholar]
- Du X.; Edelstein D.; Brownlee M. Oral Benfotiamine plus Alpha-Lipoic Acid Normalises Complication-Causing Pathways in Type 1 Diabetes. Diabetologia 2008, 51 (10), 1930–1932. 10.1007/s00125-008-1100-2. [DOI] [PubMed] [Google Scholar]
- Contreras C. L.; Guzman-Rosiles I.; Del Castillo D.; Gomez-Ojeda A.; Garay-Sevilla Ma. E. Advanced Glycation End Products (AGEs) and SRAGE Levels after Benfotiamine Treatment in Diabetes Mellitus Type 2. FASEB J. 2017, 31 (S1), 646.32. 10.1096/fasebj.31.1_supplement.646.32. [DOI] [Google Scholar]
- Bakris G. L.; Bank A. J.; Kass D. A.; Neutel J. M.; Preston R. A.; Oparil S. Advanced Glycation End-Product Cross-Link Breakers. A Novel Approach to Cardiovascular Pathologies Related to the Aging Process. Am. J. Hypertens. 2004, 17 (12), 23S–30S. 10.1016/j.amjhyper.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Toprak C.; Yigitaslan S. Alagebrium and Complications of Diabetes Mellitus. Eurasian J. Med. 2019, 51 (3), 285–292. 10.5152/eurasianjmed.2019.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaitkevicius P. V.; Lane M.; Spurgeon H.; Ingram D. K.; Roth G. S.; Egan J. J.; Vasan S.; Wagle D. R.; Ulrich P.; Brines M.; Wuerth J. P.; Cerami A.; Lakatta E. G. A Cross-Link Breaker Has Sustained Effects on Arterial and Ventricular Properties in Older Rhesus Monkeys. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (3), 1171–1175. 10.1073/pnas.98.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieman S. J.; Melenovsky V.; Clattenburg L.; Corretti M. C.; Capriotti A.; Gerstenblith G.; Kass D. A. Advanced Glycation Endproduct Crosslink Breaker (Alagebrium) Improves Endothelial Function in Patients with Isolated Systolic Hypertension. J. Hypertens. 2007, 25 (3), 577–583. 10.1097/HJH.0b013e328013e7dd. [DOI] [PubMed] [Google Scholar]
- Kass D. A.; Shapiro E. P.; Kawaguchi M.; Capriotti A. R.; Scuteri A.; deGroof R. C.; Lakatta E. G. Improved Arterial Compliance by a Novel Advanced Glycation End-Product Crosslink Breaker. Circulation 2001, 104 (13), 1464–1470. 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- Lutterloh E. C.; Opal S. M.; Pittman D. D.; Keith J. C. J.; Tan X.-Y.; Clancy B. M.; Palmer H.; Milarski K.; Sun Y.; Palardy J. E.; Parejo N. A.; Kessimian N. Inhibition of the RAGE Products Increases Survival in Experimental Models of Severe Sepsis and Systemic Infection. Crit. Care London Engl. 2007, 11 (6), R122. 10.1186/cc6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christaki E.; Opal S. M.; Keith J. C. J.; Kessimian N.; Palardy J. E.; Parejo N. A.; Tan X. Y.; Piche-Nicholas N.; Tchistiakova L.; Vlasuk G. P.; Shields K. M.; Feldman J. L.; Lavallie E. R.; Arai M.; Mounts W.; Pittman D. D. A Monoclonal Antibody against RAGE Alters Gene Expression and Is Protective in Experimental Models of Sepsis and Pneumococcal Pneumonia. Shock Augusta Ga 2011, 35 (5), 492–498. 10.1097/SHK.0b013e31820b2e1c. [DOI] [PubMed] [Google Scholar]
- Xia P.; Deng Q.; Gao J.; Yu X.; Zhang Y.; Li J.; Guan W.; Hu J.; Tan Q.; Zhou L.; Han W.; Yuan Y.; Yu Y. Therapeutic Effects of Antigen Affinity-Purified Polyclonal Anti-Receptor of Advanced Glycation End-Product (RAGE) Antibodies on Cholestasis-Induced Liver Injury in Rats. Eur. J. Pharmacol. 2016, 779, 102–110. 10.1016/j.ejphar.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Sagheddu R.; Chiappalupi S.; Salvadori L.; Riuzzi F.; Donato R.; Sorci G. Targeting RAGE as a Potential Therapeutic Approach to Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2018, 27 (21), 3734–3746. 10.1093/hmg/ddy288. [DOI] [PubMed] [Google Scholar]
- Gasparotto J.; Ribeiro C. T.; Bortolin R. C.; Somensi N.; Fernandes H. S.; Teixeira A. A.; Guasselli M. O. R.; Agani C. A. J. O.; Souza N. C.; Grings M.; Leipnitz G.; Gomes H. M.; de Bittencourt Pasquali M. A.; Dunkley P. R.; Dickson P. W.; Moreira J. C. F.; Gelain D. P. Anti-RAGE Antibody Selectively Blocks Acute Systemic Inflammatory Responses to LPS in Serum, Liver, CSF and Striatum. Brain. Behav. Immun. 2017, 62, 124–136. 10.1016/j.bbi.2017.01.008. [DOI] [PubMed] [Google Scholar]
- Bro S.; Flyvbjerg A.; Binder C. J.; Bang C. A.; Denner L.; Olgaard K.; Nielsen L. B. A Neutralizing Antibody against Receptor for Advanced Glycation End Products (RAGE) Reduces Atherosclerosis in Uremic Mice. Atherosclerosis 2008, 201 (2), 274–280. 10.1016/j.atherosclerosis.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Brederson J.-D.; Strakhova M.; Mills C.; Barlow E.; Meyer A.; Nimmrich V.; Leddy M.; Simler G.; Schmidt M.; Jarvis M.; Lacy S. A Monoclonal Antibody against the Receptor for Advanced Glycation End Products Attenuates Inflammatory and Neuropathic Pain in the Mouse. Eur. J. Pain London Engl. 2016, 20 (4), 607–614. 10.1002/ejp.775. [DOI] [PubMed] [Google Scholar]
- Mizumoto S.; Takahashi J.; Sugahara K. Receptor for Advanced Glycation End Products (RAGE) Functions as Receptor for Specific Sulfated Glycosaminoglycans, and Anti-RAGE Antibody or Sulfated Glycosaminoglycans Delivered in Vivo Inhibit Pulmonary Metastasis of Tumor Cells. J. Biol. Chem. 2012, 287 (23), 18985–18994. 10.1074/jbc.M111.313437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam T.; Ramachandran V.; Gomez S. B.; Schmidt A. M.; Logsdon C. D. S100P-Derived RAGE Antagonistic Peptide Reduces Tumor Growth and Metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18 (16), 4356–4364. 10.1158/1078-0432.CCR-12-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putranto E. W.; Murata H.; Yamamoto K.-I.; Kataoka K.; Yamada H.; Futami J.-I.; Sakaguchi M.; Huh N.-H. Inhibition of RAGE Signaling through the Intracellular Delivery of Inhibitor Peptides by PEI Cationization. Int. J. Mol. Med. 2013, 32 (4), 938–944. 10.3892/ijmm.2013.1467. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Luo H.; Xie B.; Tian T.; Li S.; Chen Z.; Liu J.; Zhao X.; Zhang L.; Deng Y.; Billiar T. R.; Jiang Y. Targeting Adaptor Protein SLP76 of RAGE as a Therapeutic Approach for Lethal Sepsis. Nat. Commun. 2021, 12 (1), 308. 10.1038/s41467-020-20577-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T.; Higashimoto Y.; Nishino Y.; Nakamura N.; Fukami K.; Yamagishi S.-I. RAGE-Aptamer Blocks the Development and Progression of Experimental Diabetic Nephropathy. Diabetes 2017, 66 (6), 1683–1695. 10.2337/db16-1281. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Zhu W.; He F.; Li Z.; Cai N.; Wang H.-H. An Aptamer-Based Antagonist against the Receptor for Advanced Glycation End-Products (RAGE) Blocks Development of Colorectal Cancer. Mediators Inflammation 2021, 2021, 9958051. 10.1155/2021/9958051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N.; Matsui T.; Nishino Y.; Sotokawauchi A.; Higashimoto Y.; Yamagishi S.-I. Long-Term Local Injection of RAGE-Aptamer Suppresses the Growth of Malignant Melanoma in Nude Mice. J. Oncol. 2019, 2019, 7387601. 10.1155/2019/7387601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y. T.; Choi G.-I.; Son D.; Kim N.-J.; Yun H.; Lee S.; Chang D. J.; Hong H.-S.; Kim H.; Ha H.-J.; Kim Y.-H.; Park H.-J.; Lee J.; Suh Y.-G. Ligand-Based Design, Synthesis, and Biological Evaluation of 2-Aminopyrimidines, a Novel Series of Receptor for Advanced Glycation End Products (RAGE) Inhibitors. J. Med. Chem. 2012, 55 (21), 9120–9135. 10.1021/jm300172z. [DOI] [PubMed] [Google Scholar]
- Han Y. T.; Kim K.; Choi G.-I.; An H.; Son D.; Kim H.; Ha H.-J.; Son J.-H.; Chung S.-J.; Park H.-J.; Lee J.; Suh Y.-G. Pyrazole-5-Carboxamides, Novel Inhibitors of Receptor for Advanced Glycation End Products (RAGE. Eur. J. Med. Chem. 2014, 79, 128–142. 10.1016/j.ejmech.2014.03.072. [DOI] [PubMed] [Google Scholar]
- Kwak T.; Drews-Elger K.; Ergonul A.; Miller P. C.; Braley A.; Hwang G. H.; Zhao D.; Besser A.; Yamamoto Y.; Yamamoto H.; El-Ashry D.; Slingerland J. M.; Lippman M. E.; Hudson B. I. Targeting of RAGE-Ligand Signaling Impairs Breast Cancer Cell Invasion and Metastasis. Oncogene 2017, 36 (11), 1559–1572. 10.1038/onc.2016.324. [DOI] [PubMed] [Google Scholar]
- Deane R.; Singh I.; Sagare A. P.; Bell R. D.; Ross N. T.; LaRue B.; Love R.; Perry S.; Paquette N.; Deane R. J.; Thiyagarajan M.; Zarcone T.; Fritz G.; Friedman A. E.; Miller B. L.; Zlokovic B. V. A Multimodal RAGE-Specific Inhibitor Reduces Amyloid β-Mediated Brain Disorder in a Mouse Model of Alzheimer Disease. J. Clin. Invest. 2012, 122 (4), 1377–1392. 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergar A.; Egbert K.; Clark C.; Hall P.; Davis T.; Hirschi K. M.; Arroyo J. A.; Reynolds P. R. Semi-Synthetic Glycosaminoglycan Ethers Decrease Receptors for Advanced Glycation End-Products and Increase AXL Receptor in the Lung from Secondhand Smoke Treated Mice. FASEB J. 2018, 32 (S1), 143.5. 10.1096/fasebj.2018.32.1_supplement.143.5.28904019 [DOI] [Google Scholar]
- Hirschi K. M.; Lewis J. B.; Hall P. D.; Wright T. J.; Egbert K. M.; Ogden K. C.; Nelson S. M.; Clark J. C.; Milner D. C.; Arroyo J. A.; Reynolds P. R. Abrogation of RAGE Signaling Using Semi-Synthetic Glycosaminoglycan Ethers (SAGEs) Ameliorates Inflammation in Mice Exposed to Secondhand Tobacco Smoke. FASEB J. 2017, 31 (S1), 656.1. 10.1096/fasebj.31.1_supplement.656.1. [DOI] [Google Scholar]