

Abstract
To characterize the dysregulation of chromatin accessibility in Alzheimer’s disease (AD), we generated 636 ATAC-seq libraries in neurons and non-neurons isolated from the superior temporal gyrus and entorhinal cortex of 153 AD cases and 56 controls. By analyzing a total of ~20 billion read pairs, we expanded the repertoire of known open chromatin regions (OCRs) in the human brain and identified cell type-specific enhancer-promoter interactions. We show that inter-individual variability in OCRs can be leveraged to identify cis-regulatory domains (CRDs) that capture the three-dimensional structure of the genome (3D genome). We identified AD-associated effects on chromatin accessibility, the 3D genome and transcription factor regulatory networks. For one of the most AD-perturbed transcription factors, USF2, we validated its regulatory effect on lysosomal genes. Overall, we applied a systematic approach to understand the role of the 3D genome in AD. We provide all data as an online resource for widespread community-based analysis.
Editor summary:
The authors generated the largest epigenome atlas of post-mortem brains with Alzheimer’s disease. They reported regulatory genomic signatures associated with AD, including variability in open chromatin regions, TF networks and cis-regulatory domains.
Alzheimer’s Disease (AD) is a chronic neurodegenerative disorder characterized, clinically, by cognitive decline and, neuropathologically, by accumulation of amyloid beta (Aβ) plaques and intracellular neurofibrillary tangles. Although a growing number of common and rare genetic risk variants have been identified1, the neurobiological causes of the majority of AD cases remain unknown. A number of recent studies have identified epigenomic changes associated with AD2–6. Such abnormalities could result from primary genetic and non-genetic causal factors and epiphenomena, including changes secondary to disease progression. Thus, the epigenome might provide a means to elucidate disease mechanisms, especially in late-onset AD, where there can be a gap of multiple decades before the initial changes in brain function become clinically apparent.
AD related studies of the epigenome have largely been limited to bulk tissue and have only examined one brain region at a time, precluding the identification of brain region and cell-type specific disease signatures. Furthermore, although analyses of bulk tissue can identify apparent changes in transcriptome and epigenome, bulk tissue level changes can reflect alterations in cell composition rather than changes in the function of individual cells. This is particularly important for neurodegenerative diseases, where disease progression involves neuronal loss.
In this study, we expanded the panel of genomic assays in the Mount Sinai Brain Bank AD (MSBB-AD) cohort10. In particular, we generated genome-wide maps of chromatin accessibility using ATAC-seq from neuronal and non-neuronal nuclei isolated from the superior temporal gyrus (STG) and entorhinal cortex (EC) of AD cases and controls. We initially used these data to expand the repertoire of known cell type specific open chromatin regulatory regions and to study their relationship to gene expression. We then examined the shared and distinct molecular mechanisms associated with clinical dementia and neuropathological lesions. Subsequently, we identified regulatory genomic signatures associated with AD, including variability in open chromatin regions (OCRs), transcription factor (TF) regulatory networks and cis-regulatory domains (CRDs). AD-associated signatures showed brain-region and cell-type specificity, implicating non-coding regulatory regions within AD genetic risk loci that participate in a variety of biological pathways as well as changes in transcription factor regulation.
Results
Profiling cell specific chromatin accessibility in brain
We performed ATAC-seq profiling in neuronal (NeuN+) and non-neuronal (NeuN−) nuclei isolated by Fluorescence-Activated Nuclear Sorting (FANS) from the STG and EC in AD cases (n=153) and controls (n=56) (Fig. 1a, Supplementary Fig. 1, Supplementary Table 1). The individuals were selected to represent the full spectrum of clinical and pathological severity (Supplementary Table 2) based on the following phenotypes: (1) case-control status defined using the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria11; (2) Braak AD-staging score for progression of neurofibrillary neuropathology BBScore)12,13; (3) mean density of neuritic plaques (PlaqueMean); and (4) assessment of dementia based on clinical dementia rating scale (CDR)14. These phenotypes were moderately correlated (Supplementary Fig. 2), indicating shared and distinct disease processes15.
Fig. 1 |. Large-scale chromatin accessibility analysis in the human brain.
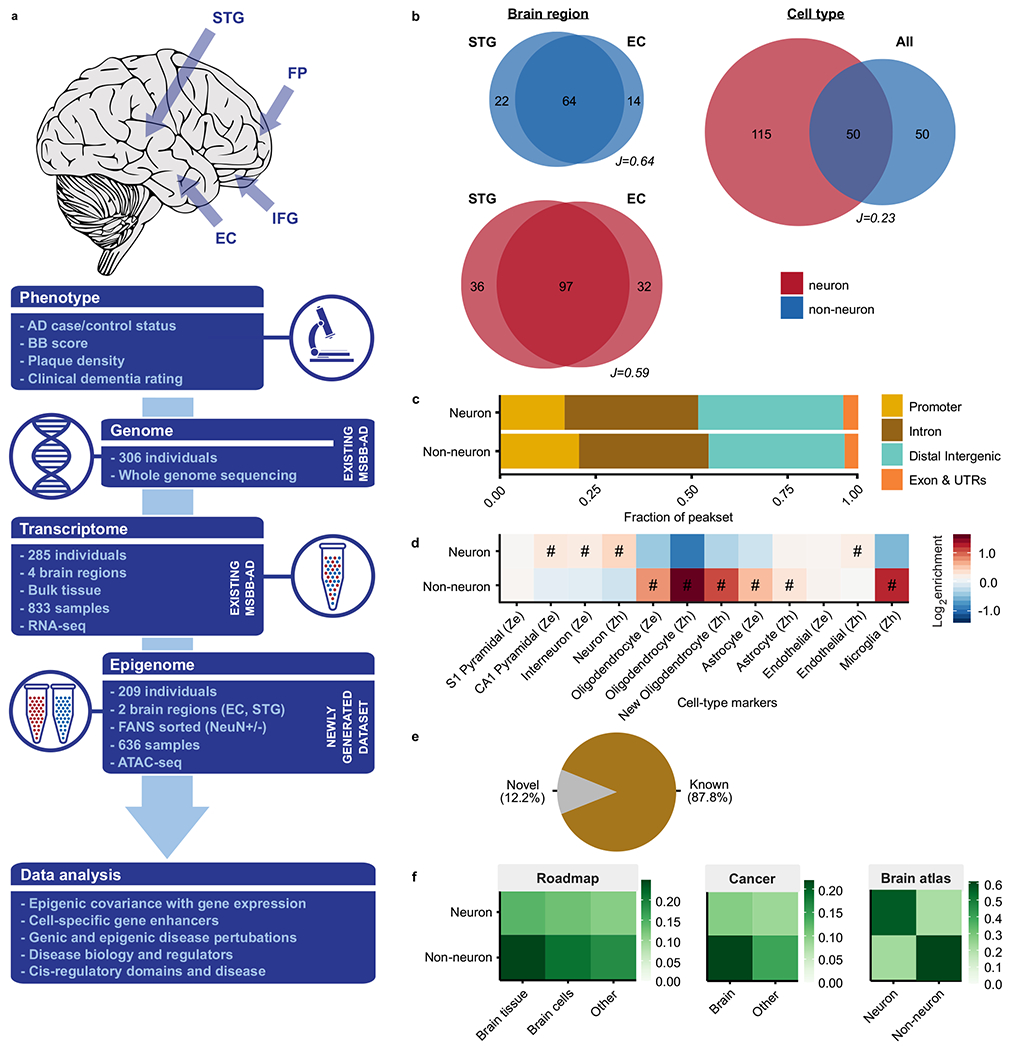
a, Description of the study design. ATAC-seq was performed on neuronal (NeuN+) and non-neuronal (NeuN−) nuclei isolated from two different brain regions (STG and EC) in MSBB-AD samples. Existing RNA-seq profiling in the same cohort was performed on bulk tissue derived from four different brain regions (EC, IFG, FP and STG). All samples have genomic and AD-related phenotypic data. b, Venn diagrams by cell type and brain region summarizing the overlap in megabases of OCRs. “J” indicates the Jaccard index between the respective OCRs. c, Proportions of neuronal and non-neuronal OCRs stratified by genomic context. d, Heatmap showing enrichment of cell-specific neuronal and non-neuronal OCRs with cell type specific markers from “Ze”: Zeisel64 and “Zh”: Zhang65. OCRs were assigned to genes using the GREAT approach (Methods). “#”: Test wide significant at FDR<0.05 “ · ”: Nominally significant at P-value<0.05. e, Novelty of OCRs compared to known OCRs from the union of three broad human tissue OCR resources59,60,66,67 and four brain OCR resources7,16,17,68. f, Overlap of neuronal and non-neuronal OCRs with sets of selected reference studies59,60,68,69 (comparisons with all reference studies in Supplementary Fig. 6).
Quality control of ATAC-seq libraries (Methods) yielded a total of 636 samples constituting 19.6 billion read pairs (Supplementary Fig. 3–5, Supplementary Table 3). Given the large differences in chromatin accessibility profiles between the two cell types (Fig. 1b, Supplementary Fig. 6), neuronal and non-neuronal samples were considered separately for subsequent downstream analysis. A total of 315,630 neuronal and 205,120 non-neuronal open chromatin regions (OCRs) were identified (Fig. 1c–d). While comparison with previous studies identified 50,836 novel OCRs, over 87% of the OCRs overlapped with previously observed regulatory elements detected in the reference repositories (Fig. 1e–f, Supplementary Fig. 7).
Chromatin accessibility explains gene expression variation
Chromatin structure is integral to transcriptional regulation, with chromatin accessibility regulating gene expression by facilitating, or inhibiting, binding of the transcriptional machinery. Given the availability of RNA-seq data from the same individuals10, we sought to quantify the relative contribution of proximal (i.e. promoter) and distal (i.e. enhancer) chromatin accessibility to transcriptional variance. We applied variance decomposition models to each of 20,709 expressed RNA-seq genes, using the covariance of OCRs at transcription start sites (TSSs) and distal regulatory elements as inputs (Supplementary Fig. 8). In this model, more than 70% of expression variance was explained by promoter and enhancer OCRs, confirming that gene expression is broadly associated with chromatin accessibility (Fig. 2a–b, Supplementary Fig. 9–10). The proportion of expression attributed to enhancer OCRs was twice as large in neuronal samples (Fig. 2c, Supplementary Table 4), confirming a larger impact of distal regulatory mechanisms in neuronal cell types7,16–18. Gene set enrichment analysis of the genes with the highest proportion of variance explained by promoter OCRs showed enrichment with known cell type markers (Supplementary Fig. 11a).
Fig. 2 |. Variance component analysis of gene expression.

a-b, The analysis was built upon (a) neuronal and (b) non-neuronal open chromatin datasets. Genes, i.e., columns, are sorted by decreasing proportion of variance explained by epigenome (enhancer and promoter), with the mean variance explained by each component shown in parentheses. c, Comparison of variance explained by each component across all RNA-seq genes. A Wilcoxon two sided signed-rank test was used to test the differences between models using neuronal and non-neuronal ATAC-seq. Horizontal lines in the violin plot indicate median values.
The proportion of expression attributed to inter-individual covariance was similar for neuronal (mean 13.4%) and non-neuronal (mean 13.7%) samples. We hypothesized that the inter-individual covariance was, in part, driven by genetic regulation of gene expression19. Therefore, we estimated, for each gene, the fraction of gene expression variation explained by the cis-genetic component (Methods). Here we observed significant correlations between per-gene inter-individual covariance and gene expression variation explained by the cis-genetic component in neuronal (Spearman ρ=0.22, P-value=1.3×10−54, Supplementary Fig. 11b) and non-neuronal (Spearman ρ=0.16, P-value=3.7×10−30, Supplementary Fig. 11c) OCRs.
Maps of enhancer-promoter regulatory landscape
Next, we sought to determine enhancer-promoter (E-P) interactions. For this purpose, the “activity-by-contact” (ABC) approach20 model was employed, which quantifies the regulatory impact of enhancers quantitatively by assuming proportionality with both E-P contact frequency (inferred from Hi-C) and enhancer activity (inferred from chromatin accessibility and H3K27ac histone modification). To apply this approach using our ATAC-seq data, we generated cell type-specific ChIP-seq and Hi-C data (Methods).
Across the neuronal and non-neuronal datasets, we identified 37,056 and 38,233 E-P interactions, respectively. We determined that at least 63% of the expressed genes (13,135 of 20,709) were linked to one or more distal OCR (OCRABC) (Fig. 3a, Supplementary Fig. 12a, Supplementary Table 5). While most of these enhancers were predicted to interact with a single gene, about one quarter regulated two or more genes (Fig. 3b, Supplementary Fig. 12b). On average, 43-47% of the E-P links were shared between neurons and non-neurons, whereas 83-90% of E-P links were shared across brain regions when comparing within the same cell type, with a high correlation of ABC score (Supplementary Fig. 12f). Among the predicted OCRABC enhancers, only 25% were linked to the nearest gene, clearly demonstrating the shortcomings of distance-based annotation (Fig. 3f, Supplementary Fig. 12c). Still, the frequency of E-P links decreased sharply with distance (Fig. 3c–e and Supplementary Fig. 12d–e)21. Of note, the average distance between E-P links was only 62kb, which is at least 3-5 times shorter than the average for E-P links derived from Hi-C chromatin loops7,18,22.
Fig. 3 |. Linking distal regulatory OCRs (OCRABC) to genes using Activity-By-Contact (ABC) method.

a, Histogram of the number of OCRABC linked per gene. b, Histogram of the number of genes linked per OCRABC. c, Histogram of distance of OCRABC to the TSS of regulated genes. d-e, Scatterplot of genomic distance vs ABC score of enhancer-gene links for (d) neurons and (e) non-neurons. The dashed black line denotes the minimum ABC score for an enhancer-gene link to be reported as a valid association. The solid yellow line indicates LOESS (local regression) fit. f, Histogram of the number of genes “skipped” by an OCRABC to reach their linked genes. g, Correlation between enhancer OCRs and promoter OCRs of linked genes from the ABC method, for links that did not meet the cut-off (low scores, OCRother) versus those that did (high scores, OCRABC). Neuronal dataset: n=4,578 OCRABC-OCRother pairs (P-value=8.7×10−14), non-neuronal dataset: n=4,763 OCRABC-OCRother pairs (P-value=6.3×10−13); P-value of the significance of difference is calculated from Fisher’s Z transformed correlations divided by the standard error of the difference of two z scores. Box plot centered on median, bounds defined between the 25th and 75th percentile with minimum and maximum defined as median ± 1.5 × interquartile range, whiskers extending to the lowest/highest value within this range and potential outliers from this are shown as dots. h, Schematic of CRISPRi system for transcription repression of distal enhancers. i, Expression levels of three genes after KRAB-dCas9-mediated repression of their ABC-predicted distal enhancers as determined with quantitative polymerase chain reaction (qPCR). Each experiment uses two single guide RNAs (sgRNAs), each performed in triplicate (n=6). P-values were determined by the two-tailed t-test (EEF1A2 P-value=9.75×10−9, DBN1: P-value=2.86×10−4, RPS21: P-value=3.87×10−9). Box plots are centered on median, bounds defined between the 25th and 75th percentile with minimum and maximum defined as median ± 1.5 × interquartile range, and whiskers extending to the lowest/highest value within this range.
To corroborate the results of our regulatory analyses, we compared distal OCRs predicted to participate in E-P interactions (OCRABC) with a subset of distal OCRs not predicted to take part in such interactions but matched by distance to nearest TSS and OCR width (OCRother). OCRABC, but not OCRother, were found to be enriched in fine-mapped eQTLs from GTEx23 (OR=1.5-1.8, P-value<10−45, Supplementary Fig. 13a, Supplementary Table 6). When compared based on chromatin states from the Roadmap Epigenomics Project, OCRABC showed a relative depletion in repressed chromatin states as opposed to OCRother (Supplementary Fig. 13b–c). Chromatin accessibility at predicted promoter regions was significantly more correlated with accessibility at OCRABC than accessibility at OCRother (Fig. 3g, Supplementary Fig. 12g). Finally, OCRABC overlapped well with the regions of super-enhancers7, i.e. 83-91% (1.6-2.1 times more than for OCRother) for neuronal and oligodendrocyte super-enhancers (Supplementary Fig. 13d). Lastly, we sought to validate the E-P interactions in vitro, using the CRISPR interference (CRISPRi) platform24 for genetic screens in human neural progenitor cells (NPCs) derived from induced pluripotent stem cells. This approach allowed us to epigenetically silence three OCRABC enhancers and verify their decreased expression using qPCR (Fig. 3h,i). By showing significantly reduced relative expression of the three selected target genes (Methods) after knockout of their distal enhancers by 21%-45% (Fig. 3i), we demonstrated the potential of our data to characterize lineage-specific cis-regulatory interactions.
AD associated changes in chromatin accessibility
We explored AD-associated changes in chromatin accessibility by performing differential analyses across multiple phenotypes considering the brain regions (STG/EC), separately, or in combination, to increase statistical power (Fig. 4a, Supplementary Fig. 14 and 15f–g, Supplementary Table 7–9). EC neurons showed the highest number of associations across all AD phenotypes, with 19,336 OCRs (6.1%) associated with one or more AD phenotype(s) (Fig. 4b). For STG neurons, the corresponding number was 10,490 (3.3%), highlighting the regional specificity of AD-associated epigenetic changes. For non-neurons, the number of significant associations was markedly smaller, with STG and EC non-neurons jointly showing an association in 4,625 OCRs (2.3%) for one or more AD phenotype(s). Despite the substantially different numbers of AD-associated OCRs in STG and EC, disease signatures were remarkably concordant (Supplementary Fig. 15a–b), reflecting the strong interactions previously detected in co-expression network analysis25. Lower magnitudes of epigenomic changes in STG correspond to the established model of AD cortical spread that starts in EC and propagates to connected brain areas in temporal lobe (Supplementary Fig. 15c)26.
Fig. 4 |. Disease-associated chromatin changes.

a, Disease-associated OCRs stratified by cell type, brain regions, and AD-related phenotypes. b, Disease-associated OCRs stratified by brain regions or AD-related phenotypes. c, Genetic context of disease-associated OCRs (risk ratio calculated by unconditional maximum likelihood estimation and small sample adjustment; confidence interval calculated by normal approximation; neuronal OCRs n=61,454, 133,720, 143,925, 14,623; non-neuronal OCRs n=45,177, 75,050, 78,586, 7,888). d, Enrichment of common genetic variants in AD with disease-associated OCRs when assayed by LD-score regression. Only sets of OCRs covering at least 0.05% of the genome were tested. Positive coefficients signify enrichment, error bars indicate ± standard deviation from the mean, and both coefficients, and P-values were obtained directly from LD-score 30. “*”: FDR significant after correction across all tests. Neuronal OCRs: n=4,406 (P-value=0.658), 6,165 (P-value=0.398), 12,282 (P-value=0.658), 33,386 (P-value=0.501), 5,380 (P-value=0.066), 9,345 (P-value=0.064), 28,778 (P-value=0.146), 315,630 (P-value=0.217); non-neuronal OCRs n=2,085 (P-value=0.008), 4,122 (P-value=0.828), 9,514 (P-value=0.386), 205,120 (P-value=0.008). e, Correlation between disease-associated chromatin changes at promoters (ATAC-seq) and disease-associated changes in gene expression (RNA-seq) at the corresponding genes. Pearson correlations were used (95% confidence intervals calculated on Fisher’s Z transformation; P-values determined by two-sided t-test) and only comparisons with over 100 differential OCRs and genes were tested. Neuronal OCRs: n=200 (P-value=1.45×10−1), 727 (P-value=3.77×10−2), 444 (P-value=8.01×10−1), 2,072 (P-value=3.95×10−9), 4,180 (P-value=1.16×10−3), 307 (P-value=1.07×10−6), 691 (P-value=1.49×10−11), 2,434 (P-value=6.85×10−35); non-neuronal OCRs: n=79 (P-value=2.38×10−3), 89 (P-value=1.42×10−3), 404 (P-value=1.59×10−10), 1,212 (P-value=2.79×10−51), 151 (P-value=4.46×10−8), 2,300 (P-value=5.98×10−80). f, Examples of changes in chromatin accessibility and gene expression near the genes encoding NYAP1 and CCKBR.
Epigenetically perturbed regulatory regions in AD were located near transcripts as well as in intergenic regions, suggesting that a combination of proximal and distal regulatory elements contribute to AD (Fig. 4c, Supplementary Fig. 15d–e, Supplementary Table 7). We then confirmed the robustness of our differential chromatin accessibility analysis by checking the concordance with three epigenetic studies of AD in human postmortem brains3,27 and iPSC-derived neurons3,28. We found significant concordance with an average Pearson coefficient of 0.35, while higher correlations were observed in neuronal compared to non-neuronal analysis (mean Pearson correlation of 0.41 vs 0.28, Supplementary Fig. 16).
While it is well-established that AD genetic risk variants are enriched in OCRs in microglia and astrocyte genes29, the relationship with quantitative regulation of chromatin accessibility in AD is less clear. Here, we quantified the relationship between genetic risk variation and AD-associated OCRs using LD-score partitioned heritability30. We found the set of all non-neuronal OCRs and the PlaqueMean-associated non-neuronal OCRs to be significantly enriched in AD genetic variants, even when accounting for the general genetic context of the OCRs (Fig. 4d). Finally, we saw moderate enrichment when we overlapped OCRs with common variants of AD co-heritable traits, such as neuroticism, insomnia or bipolar disorder29 (Supplementary Fig. 17, Supplementary Table 10).
Concordance of the epigenome and transcriptome changes in AD
To investigate changes in gene expression, we reprocessed 833 homogenate RNA-seq samples from four brain regions in a highly overlapping set of individuals (Supplementary Fig. 18). To perform differential analysis, a deconvolution parameter was added to account for differences in cell type composition in the bulk tissue, driven by the neuronal loss associated with disease progression (Supplementary Fig. 19–20). A higher proportion of genes was differentially expressed in EC and frontal pole (FP) (24.4% and 21.5% of expressed genes, respectively), followed by STG and inferior frontal gyrus (IFG) (8.1% and 3.0% of expressed genes, respectively) (Supplementary Fig. 21, Supplementary Table 11).
Comparing the epigenomic changes at TSSs with the changes in gene expression revealed strong correlations (Fig. 4e, Supplementary Fig. 22). Two illustrative examples demonstrating decreased chromatin accessibility and gene expression for AD cases through differentially regulated OCRs are shown in Fig. 4f. NYAP1 (Neuronal Tyrosine Phosphorylated Phosphoinositide-3-Kinase Adaptor 1), encodes a late-onset AD GWAS-linked candidate gene31 that plays a role in the regulation of PI3K signaling pathway and controls cytoskeletal remodeling in outgrowing neurites32. Loss of expression of CCKBR (Cholecystokinin B Receptor) negatively affects spatial reference memory33 and has repeatedly been identified among the top hits in differential gene expression analyses in AD34–36.
Identification of perturbed processes and pathways in AD
To elucidate biological processes implicated in AD, we performed gene set enrichment analyses for our two ATAC-seq cell types, bulk RNA-seq, as well as the aforementioned GWAS study29. We did this separately for each phenotype (Supplementary Fig. 23–24) and aggregated these by taking the most significant association across all phenotypes (Fig. 5, Supplementary Fig. 25).
Fig. 5 |. Gene set enrichment analysis using general gene sets.
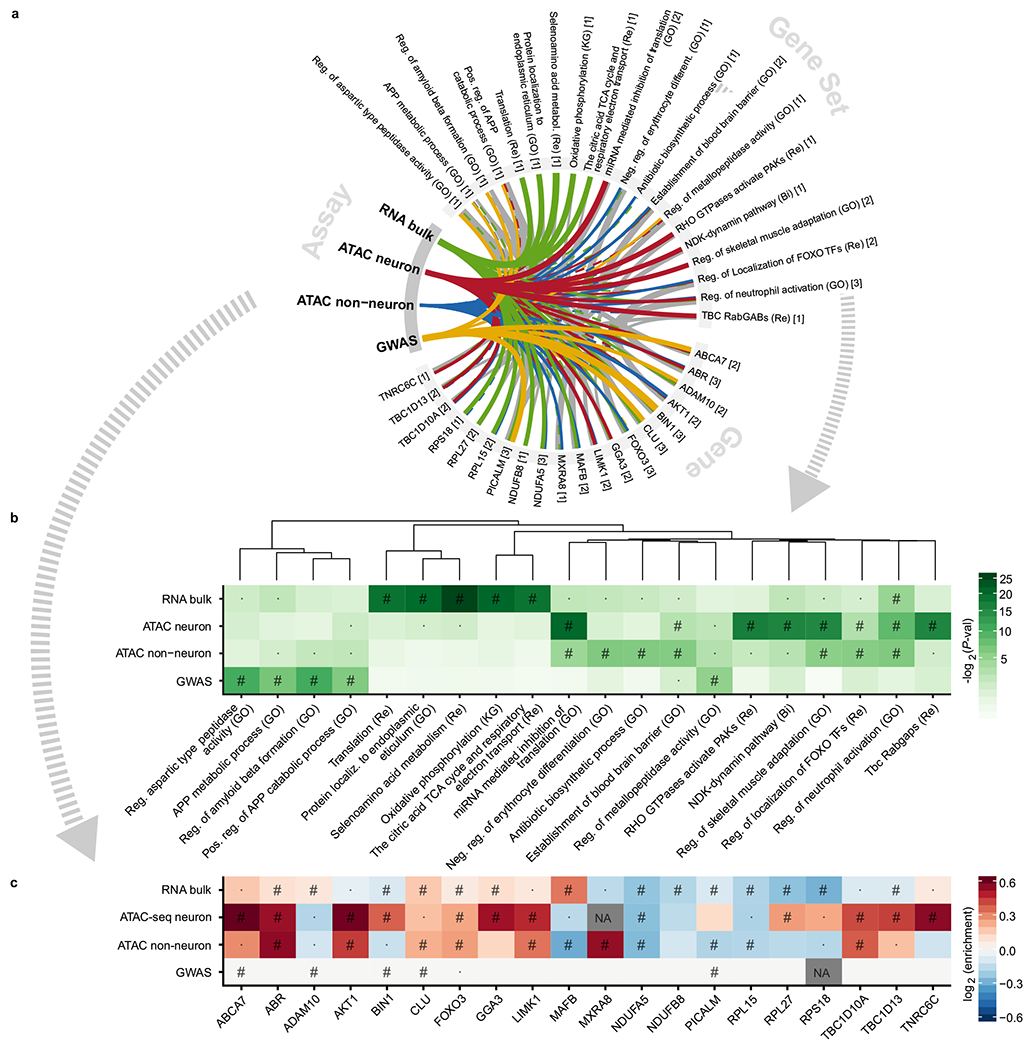
a-b, Top five gene sets of the four overall assays and the top genes within each. Thickness indicates the strength of association. A full line indicates significance at FDR<5%, whereas a dashed line indicates nominal significance (P-values were directly outputted from CameraPR70). Grey lines between genes and gene sets indicate gene set membership and the thickness is inversely proportional to the size of the gene set. The numbers in square brackets indicate the number of assays in which the gene or gene set was found significant at FDR<5%. The gene sets are clustered based on the constituent member genes. FDR throughout is calculated for all tests within one assay (e.g. all contrasts analyzed in ATAC Neuron times the number of gene sets tested). c, Heatmap of fold changes in chromatin accessibility/gene expression and significance of change. The GWAS associations are genetic variants, which might increase, decrease or otherwise alter gene function. Thus no colors are applied to these. P-values were directly outputted from dream71 and for GWAS from MAGMA72. “#” Significance at FDR<5%. “ · ”: Nominal significance. “NA”: not available. “Bi”: Biocarta. “GO”: Gene Ontology. “KG”: Kegg. “Re”: Reactome. The ApoE- and MHC-loci were excluded from the GWAS analysis, as is customary. Not all genes are directly linked to an OCR in neuron and non-neuron ATAC-seq.
The AD-associated changes in gene expression seen in RNA-seq implicated very general molecular pathways, such as “Oxidative Phosphorylation” and “Translation”. However, chromatin accessibility pointed to more specific molecular pathways. For example, using neuronal ATAC-seq we identified “the endocytotic role of NDK, phosphins and dynamin” and “RHO GTPase activation of PAKs”, among the top associations. Interestingly, PAKs (p21-activated kinases) are abundant in the brain and associated with cell death and survival signaling37. The non-neuronal ATAC-seq samples implicated “establishment of the blood-brain barrier”, which was also nominally significant with neuronal ATAC-seq, RNA-seq, and the AD GWAS. The blood-brain barrier has a purported role in the initiation and maintenance of chronic inflammation during AD38. Other pathways associated with multiple assays included “regulation of neutrophil activation” and “regulation of localization of FOXO transcription factors”. Of note, the latter is involved in cell death and longevity39,40, and one member of the gene set, FOXO341, showed at least a nominal association across all assays. Within these top gene sets, the top genes (e.g. ABCA7, BIN1, and PICALM) often showed an association across multiple assays (Fig. 5, Supplementary Fig. 26, Supplementary Table 12).
Finally, we performed a targeted gene set enrichment analysis using the synaptic gene ontology resource42 which revealed both pre- and post-synaptic dysfunction (Supplementary Fig. 24–25). For the AD GWAS, synaptic dysfunction was also implicated, which, to the best of our knowledge, has not previously been reported. Of particular interest, “synaptic vesicle cycle” was significant in GWAS, RNA-seq and neuronal ATAC-seq, highlighting the shared enrichment of fundamental neuronal functions between the genetic drivers and subsequent chromatin accessibility and gene expression profiling in post-mortem brain.
Perturbed transcription factor regulatory networks in AD
Using footprinting analysis43 we systematically examined transcription factor (TF) activity patterns underlying cell type differences and AD-related chromatin changes. We utilized 431 TF motifs, which, due to sharing of binding motifs, represented 798 TFs. TFs clustered into two cell type-specific groups with predominantly neuronal and non-neuronal regulatory patterns (Fig. 6a–b, Supplementary Fig. 27a, Supplementary Table 13). The TF cell specificity identified here was broadly concordant with existing literature (Supplementary Table 14). Corroborating our findings, the TF cell type specificities identified here were highly concordant (Fig. 6a, Spearman ρ = 0.87, P-value < 10−15) with TF footprinting analyses from the BOCA study (Brain Open Chromatin Atlas16). Additionally, the genes that were controlled exclusively by cell-specific TFs (Supplementary Table 15) showed enrichment in known cell type markers of the appropriate cells (Supplementary Fig. 27b).
Fig. 6 |. Mapping of transcription factors to cell types and AD-related phenotypes.
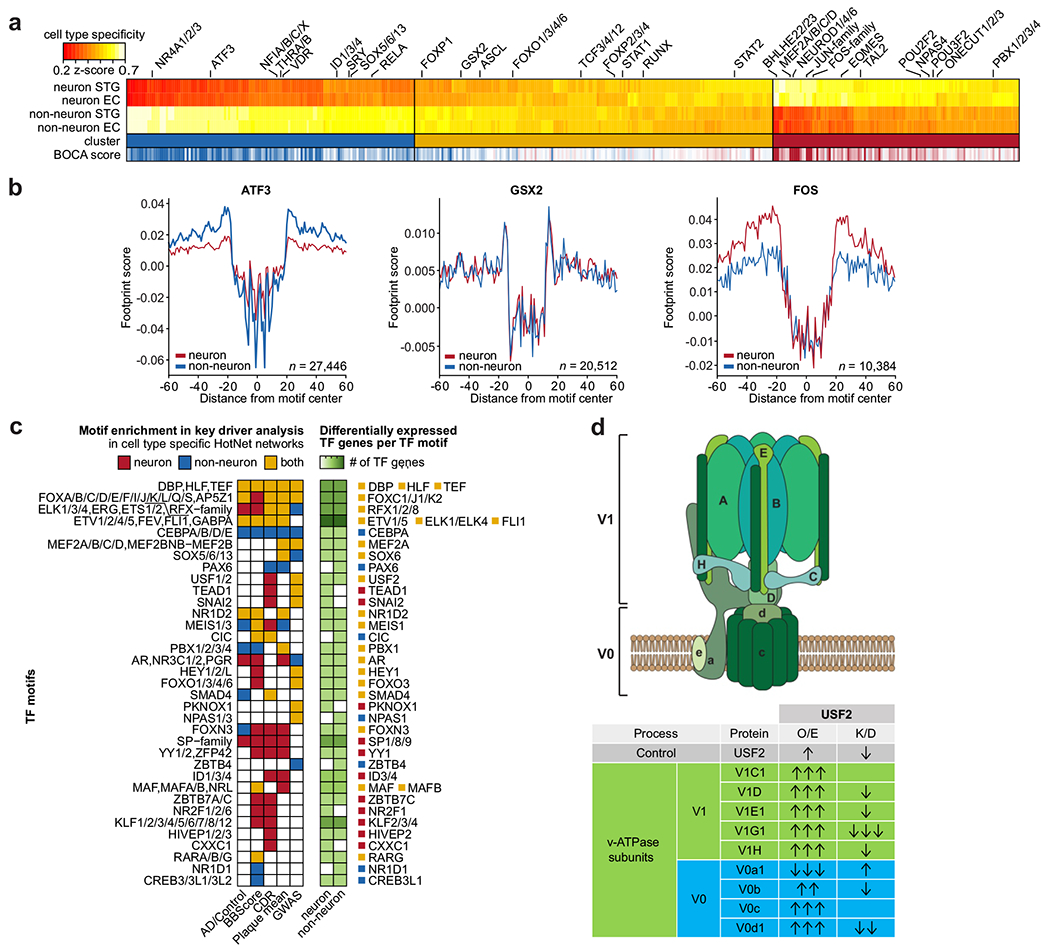
a, Hierarchical clustering of TF motifs based on a cell type specificity score across neuronal and non-neuronal OCRs. High specificity scores indicate high specificity to the given cell type. The concordance with the Brain Open Chromatin Atlas is indicated as “BOCA score“ ranging from blue (strong non-neuronal signal) to red (strong neuronal signal). Select motifs are further described in Supplementary Table 14. b, Aggregated footprint scores across all potential TF binding sites for three motifs that represent the three major clusters of TFs according to cell type specificity. c, Overview of prioritized TF genes. The prioritized TF genes (i) belong to TF motifs whose neuronal and/or non-neuronal TFRNs are enriched in AD genes, (ii) show a significant correlation of their gene expression with their regulatory targets, and (iii) show significant AD-related dysregulation of their gene expression in at least one AD phenotype comparison. d, Top: Schematic of the V0 and V1 subunits of the v-ATPase complex responsible for maintaining lysosomal pH. Bottom: Summary of western blot data in SH-SY5Y human neuroblastoma cells stably overexpressing USF2 or transfected with siUSF2. Arrow direction indicates protein level increase or decrease, while the number of arrows represents statistical significance by Student’s two-tailed unpaired t-test, i.e. one arrow: P-value<0.05; two arrows: P-value<0.005; three arrows: P-value<0.0005. O/E: over-expressed; K/D: knocked-down.
Having broadly characterized TF activity in the two cell types, we then sought to examine TF dynamics in AD. We constructed TF regulatory networks (TFRNs) for each cell type by analyzing actively bound TF motifs within both proximal and distal regulatory regions of genes representing the aforementioned 798 TFs. Using the neuronal and non-neuronal TFRN topology of directed TF-to-TF interactions and TF risk scores based on RNA-seq (across four AD-related phenotypes) and GWAS29 data, we identified high-scoring subnetworks associated with AD (P-value<0.05) (Fig. 6c). For each cell type, we subsequently combined the resulting subnetworks derived from the five TF risk scores into consensus subnetworks. Overall, we identified 95 and 176 TF motifs enriched in AD risk genes for the neuronal and non-neuronal TFRNs, respectively. To whittle down a list of candidate TF genes represented by the TF motifs prioritized by the TFRN analysis, we filtered out genes that were not differentially expressed between AD cases and controls (Fig. 6c, Supplementary Fig. 28). The resulting set of 51 TF genes shows a notable overlap with three studies that sought to identify AD regulatory hubs44–46. We found the TF USF2 to be highlighted in all three studies, as well as in our data, where AD perturbation on TFRN was supported by both GWAS and RNA-seq evidence. Genes regulated by USF2 are enriched primarily in lysosomal-dependent protein degradation pathways, which are known to be affected in AD47. While the literature on USF2 is limited, there is evidence that USF2 plays a role in regulating lysosomal gene expression48. To further support the link between USF2 and lysosomal dysfunction, we evaluated the effects of USF2 knockdown and over-expression on key components of the lysosomal pathway essential for maintaining lysosomal pH49 and activity in a human neuronal cell line, SH-SY5Y (Fig. 6d, Supplementary Table 16). We found a reduction in the abundance of the members of the V0 (V0b, V0d1) and V1 (V1D, V1E1, V1G1, V1H) subunits of v-ATPase, which are responsible for maintaining intra-lysosomal pH (Supplementary Fig. 29a–d), indicating impairments in lysosomal enzyme function. Convincingly, we observed changes in protein abundance in the opposite direction when USF2 was overexpressed (Supplementary Fig. 29e–h). v-ATPase hydrolyzes ATP via its V1 domain and uses the energy released to transport protons across membranes via its V0 domain. This activity is critical for pH homeostasis and generation of a membrane potential that drives cellular metabolism50. Accordingly, reduced levels of V1 sector v-ATPase subunits on lysosomes isolated from USF2 knockdown cells lowered the rate of ATP hydrolysis of v-ATPase by ~60% (Supplementary Fig. 29i). The ATPase activity decline is expected to block proton translocation via the V0 sector51 and impair acidification of lysosomes, as we observed (Supplementary Fig. 29j–k). The increased level of V0a1 may be a compensatory, but incomplete, response to the loss of V1 subunits. Taken together, these findings suggest that the TF USF2 plays a role in maintaining pH dependent lysosomal function via v-ATPase activity.
Genome wide cis-regulatory domains of open chromatin
Previous work on identifying correlation structures using histone modification at the population level has yielded high-resolution maps of cis-regulatory domains (CRDs) in lymphoblastoid cell lines52. We employed this approach to explore the coordinated activity and chromatin organization in the adult human brain and its perturbations in AD (Fig. 7a, Supplementary Fig. 30–32). Initially, we explored the correlation of chromatin accessibility between pairs of OCRs and found that they decrease in frequency with genomic distance (Supplementary Fig. 33a). Subsequently, we used hierarchical clustering53,54 to identify 13,671 STG neuronal, 13,334 EC neuronal, 8,861 STG non-neuronal, and 8,688 EC non-neuronal CRDs, which included 37-39% of all OCRs (Fig. 7b–c, Supplementary Table 17). Each CRD contained an average of 8 OCRs (Fig. 7c, Supplementary Fig. 33b–d). Next, we compared CRDs to cell type-specific Hi-C-derived Topologically Associated Domains (TADs) to investigate the efficiency with which CRDs captured the 3D genome. CCCTC-binding factor (CTCF) binding sites at TAD boundaries enhance contact insulation between regulatory elements of adjacent TADs55. Both CRD and TAD boundaries were enriched in CTCF binding sites, with the former showing the highest density (Fig. 7d). Furthermore, there was a marked overlap between CRD and TAD boundaries (Supplementary Fig. 33e–g). While TADs capture the spatial organization of the genome at the scale of hundreds of kb (mean of 409-413kb in neurons and non-neurons), CRDs represent finer-scale regulatory clusters on the order of tens of kb (mean of 90kb in neurons; 120kb in non-neurons) (Fig. 7e). We leveraged Hi-C to divide the genome into A and B compartments, which are known to be associated with active and inactive chromatin, respectively56, and confirmed that CRDs were enriched in type A compartments (Supplementary Fig. 33h). To explore the regulatory interactions within each CRD, we leveraged the outcome of the ABC analysis and found enrichment of E-P interactions that are within the same CRDs (Fig. 7f). Interestingly, the correlation was higher for OCR interactions that have support for Hi-C loops and diminished with distance (Supplementary Fig. 33i).
Fig. 7 |. Definition of neuronal and non-neuronal cis-regulatory domains.

a, An example of cis-regulatory domains in STG neurons identified by estimating the interindividual correlation structure between nearby OCRs. From top to bottom: genes that are present in the locus; a single Hi-C TAD (dark gray horizontal bar) and Hi-C loops (red arcs); OCRs (gray vertical bars); CRDs, where each colorbar represents a different regulatory domain; correlation matrix of OCRs including colored triangles that highlight CRDs. b, Number of CRDs stratified by cell type and brain region. The percentage is the fraction of OCRs within CRDs to the total number of OCRs. c, Venn diagrams by cell type and brain region summarizing the overlap of CRDs. “J” indicates the Jaccard index between the respective CRDs. d, CTCF density at and around CRD and TAD boundaries. e, Size distribution of cell type and region specific CRDs and cell type specific TADs. f, Associations of enhancer-promoter regions (ABC method) that are within the same CRDs. The odds ratios with their 95% confidence intervals are plotted as a function of the distance between enhancer and gene TSSs. P-values are estimated based on a two-sided Fisher’s exact test. CRD: cis-regulatory domain; TAD: topologically associating domain; kb: kilobase; and Mb: megabase.
Differential analysis of cis-regulatory domains in AD
We performed differential analyses of CRDs across all AD-related phenotypes (Supplementary Fig. 30). The AD epigenomic changes in CRDs varied markedly by cell type, brain region, and phenotype (Fig. 8a, Supplementary Table 18), and showed similar patterns to the changes observed in the single OCR analysis (Fig. 4a). The highest number of significant CRDs (n=2,603) was observed for the CDR phenotype in EC neurons and involved 26,365 OCRs (21.6%) that were also dysregulated (Fig. 8a). On average, across every phenotype, brain region, and cell type, dysregulation of the 3D genome in AD spanned 92.5 Mb or 3% of the genome (Supplementary Fig. 34a–b). For the PlaqueMean phenotype, perturbations of CRDs in EC neurons spanned 362.7 Mbp, or 12.1% of the genome, suggesting that restructuring of the 3D genome is widespread in these regions of the AD brain.
Fig. 8 |. Disease-associated perturbations in CRDs.
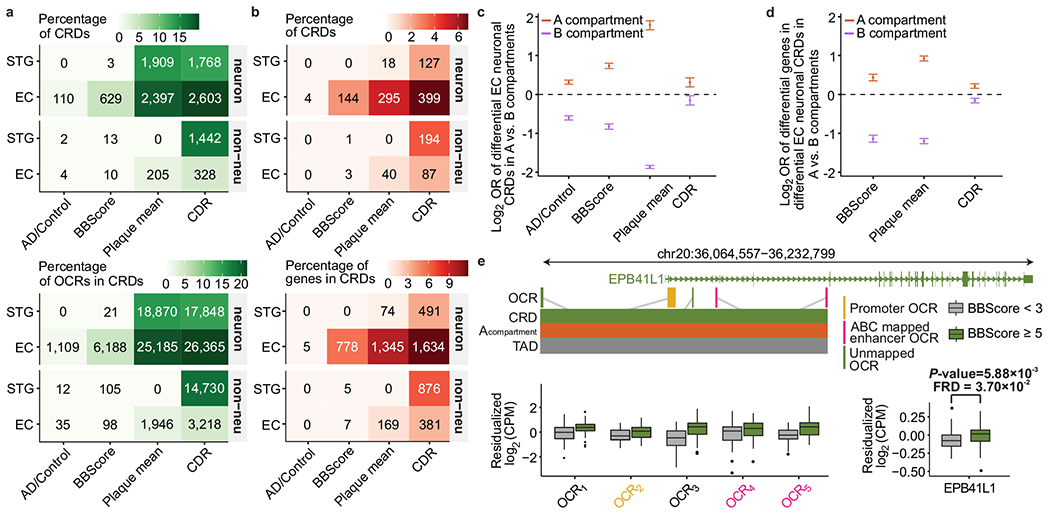
a, Counts of CRDs and OCRs within CRDs that are associated with AD-related phenotypes stratified by cell type and brain regions. b, Counts of CRDs and genes mapped to CRDs using the ABC model, that showed association with AD-related phenotypes in both epigenomic and transcriptomic levels using differential CRD test, stratified by brain region and cell type. c, Odds ratios of disease associated EC neuronal CRDs to be in A compartments vs. B compartments, measured for AD related phenotype: AD/Control: n=110 CRDs, P-value=5.25×10−32 (3.75×10−111); BBscore: n=629 CRDs, P-value=2.35×10−106 (1.15×10−128); Plaque mean: n=2,397 CRDs, P-value=2.37×10−199 (<10−200); CDR: n=2,603 CRDs, P-value=5.18×10−7 (1.30×10−2) in A(B) compartments. d, Odds ratios of disease associated genes in EC neuronal CRDs to be in A compartments vs. B compartments, measured for each AD related phenotype: 1) BBscore: n=778, P-value=6.17×10−24 (5.0×10−142), 2) Plaque mean: n=1,345, P-value=1.3×10−181(1.3×10−282) and 3) CDR: n=1,634, P-value=6.58×10−13(9.93×10−7) in A(B) compartments. Odds ratios and P-values in c-d are estimated using two-sided Fisher’s exact test and error bars show upper and lower bounds of odds ratio at 95% confidence interval. e, Examples of changes in OCR expression (OCR1: P-value=4.20×10−4, FDR=5.67×10−2; OCR2: P-value=2.16×10−2, FDR=2.56×10−1; OCR3: P-value=5.26e×10−5, FDR=2.83×10−2; OCR4: P-value=3.08×10−1 FDR=7.05e-1; OCR5: P-value= 2.59×10−4, FDR=4.8×10−2) and gene expression near the EPB41L1 gene (P-value=5.88×10−3, FDR=3.7×10−2) in BBScore associated CRD in EC neurons. P-values and FDR values were directly outputted from dream71. Box plot centered on median, bounds defined between the 25th and 75th percentile with minimum and maximum defined as median ± 1.5 × interquartile range, whiskers extending to the lowest/highest value within this range and potential outliers are shown as dots.
Next, we explored whether AD-associated structural epigenomic perturbations cause transcriptional changes in nearby genes. For this, we mapped OCRs within CRDs to genes using the E-P interactions of our ABC analysis (Supplementary Table 5). Then, we correlated changes in OCRs with changes in gene expression and found our ABC-mapped CRD-derived OCRs to show a similar degree of correlation to genes when compared to the single OCR analysis (Supplementary Fig. 34c–d). Using CRDs as the functional unit, we identified dysregulation of the transcriptome in AD (Fig. 8b, Supplementary Table 18), which show similar cell type, brain region, and phenotype specificity as the epigenetically defined perturbations (Fig. 8a). Transcriptome-defined CRD perturbations were highly reproducible (range of π1 estimates between 0.39-0.88) in an independent gene expression dataset57 (Supplementary Fig. 35a–b). Gene set enrichment analysis for the transcriptome-defined CRD perturbations identified significant associations for multiple canonical pathways, including AD, cell adhesion molecules from the immunoglobulin family, chemokine receptors CXCR3/CXCR4, SHC adaptor proteins and glutamate metabolic processes (Supplementary Fig. 35c–d).
A previous study reported a larger effect for tau-related changes in H3K9ac peaks located in Hi-C defined type A (active) compared to B (inactive) compartments3. Consistent with the referred study3, differentially upregulated CRDs in AD and the genes they contain were enriched for type A compartments (Fig. 8c,d); in contrast, downregulated CRDs and their constituent genes were enriched in B compartments. Spatial organization of differential CRDs was further corroborated by a negative correlation between the magnitude of AD-related phenotypic effects of CRDs that had no overlap with nuclear lamina compared to those that had an overlap58 (Supplementary Fig. 35e). An illustrative example of a differentially upregulated CRD in EC neurons associated with BBScore is shown in Fig. 8e. The spatial organization of this CRD was observed in type A compartments in neurons and it involved proximal and distal OCRs regulating EPB41L1, which were all upregulated in more severe tau pathology (based on BBScores) (Supplementary Fig. 35e).
Discussion
Here we provide an extensive characterization of the cell type and regional chromatin regulatory landscape in human brain tissue derived from AD cases and controls. We initially utilized 19.6 billion read pairs from 636 individual ATAC-seq libraries to identify hundreds of thousands of cell type specific regulatory regions, expanding the annotation of the chromatin accessibility landscape in the brain from previous large-scale efforts16,59,60.
Studying the epigenome at the population level allowed for broad inferences about gene regulation in the human brain. In fact, chromatin accessibility explained, globally, over 70% of the variance in gene expression. Building on this, we generated additional epigenome datasets and performed integrative analysis to define cell-specific enhancer-promoter interactions. This analysis identified putative links for more than half of the protein-coding genes, expanding previous efforts to catalogue E-P interactions in the human brain7 and further increasing our understanding of the cell type-specific regulome in brain tissue. This valuable resource can be leveraged in future studies to better understand how genetic variation can affect regulatory regions, and to link these risk variants to specific genes.
In addition, the functional relevance of this dataset of high-confidence putative enhancers was demonstrated by experimental validation in neural progenitor cells. We used CRISPRi-mediated repression of enhancer activity to observe a significant loss of expression of regulated genes in all three interrogated E-P interactions. We noticed in our analysis that OCRs and genes participating in interrogated E-P interactions were dysregulated in AD. These findings are broadly concordant with existing literature that suggests the association of all three genes with AD and neurodegenerative processes61–63 and further provides a regulatory mechanism affecting those changes.
Disease-associated changes to chromatin accessibility reported in this study were extensive, involving thousands of regulatory sequences, many of which displayed specificity for a given cell-type and/or brain region. In addition to overlapping with AD common risk variation and gene expression perturbations revealed by bulk tissue analysis, these epigenetic changes also revealed additional cell-type specific AD associated molecular perturbations. Of note, common genetic variants related to the non-neuronal epigenome showed the most pronounced changes in AD, particularly so for non-neurons of the entorhinal cortex.
By applying footprinting analysis43, we generated TFRNs and identified AD perturbations related to TF activity patterns that captured changes in gene expression and genetic variation in AD. This approach has the potential to elucidate biology as an aggregate of the multitudinous disease signals telling the story of gene dysregulation and dysfunction. As an example, we highlighted a putative role for USF2 in Alzheimer’s disease. This TF was predicted, in silico, to affect lysosomal genes, an observation that was supported by subsequent validation experiments using a human neuronal cell line.
Further, we leveraged inter-individual variation in OCRs to infer cis-regulatory domains, where OCRs work in concert to regulate nearby or more distant genes. These domains also recaptured chromosomal conformation information as evidenced by their overlap with Hi-C derived TADs. Using these domains to investigate changes in gene co-regulation structures turned out to be a powerful tool for interrogating epigenetic changes in AD. Of note, perturbed domains were enriched in active “A” compartments of the genome and depleted in inactive “B” compartments. On a more general level, we saw a disease-associated decrease in both chromatin accessibility and gene expression for the lamina-associated domains. This is in accordance with the previous reports3, but we do not yet understand the biological significance of this finding.
Collectively, this study augments our knowledge of AD pathogenesis, but also represents a valuable omics resource. With this cell specific map of chromatin accessibility, the MSBB-AD cohort contains genetics, epigenomics and gene expression data, which can be applied to downstream studies both genome-wide and at the single gene level, and, in time, should contribute to improved diagnosis and/or treatment of the disease.
Methods
Study cohort
Frozen brain tissue samples derived from STG (Superior Temporal Gyrus / Brodmann area 22) and EC (Entorhinal cortex / Brodmann area 36) were obtained from the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB–Mount Sinai NIH Neurobiobank), which holds over 2,000 human brains. Since we wanted to leverage existing transcriptomics data, we narrowed our initial selection of brain donors to those with available high-quality RNA-seq data for both targeted brain regions (STG and EC). All neuropsychological, diagnostic, and autopsy protocols were approved by the Mount Sinai and JJ Peters VA Medical Center Institutional Review Boards. AD cases and controls were selected to include donors with either no discernable neuropathology or cognitive complaints (controls) or those with only AD-associated neuropathology (cases; excluding donors with comorbid lesions such as significant cerebrovascular disease or Lewy bodies, etc.). Neuropathological assessments, cognitive, medical status, and neurological status were performed according to established procedures73. Here neuropathological assessments were performed according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) protocol74 and included assessment by hematoxylin and eosin, modified Bielschowski, modified thioflavin S, and anti-β amyloid (4G8), anti-tau (AD2) and anti-ubiquitin (Dakoa Corp.). Further, a Braak AD-staging score for progression of neurofibrillary neuropathology was assigned to each sample12,13. Additionally, the mean density of neuritic plaques in the middle frontal gyrus, orbital frontal cortex, STG, inferior parietal cortex and calcarine cortex (PlaqueMean) was calculated73. For assessing cognitive function, the clinical dementia rating (CDR) was applied14. Supplementary Table 1 summarizes the demographic information of the present study population, including sex, age at the time of death, mean plaque density, CDR, and Braak & Braak score, stratified by cell type, brain region, and AD diagnosis. AD diagnosis was based on CERAD with cases consisting of ”definite”, ”probable”, and ”possible” cases. The complete demographic information of the present MSBB AD study population is provided at https://dx.doi.org/10.7303/syn3159438 and https://dx.doi.org/10.7303/syn7392158. The description of FANS, generation of ATAC-seq, ChIP-seq, and Hi-C libraries as well as computational preprocessing of sequencing data is further described in Supplementary Methods.
ATAC-seq data generation and processing
ATAC-seq libraries were generated from neuronal and non-neuronal nuclei isolated by FANS from frozen post-mortem human brain tissue dissections using an established protocol75 with modifications described in Supplementary Methods. All libraries were processed by our computational pipeline (Supplementary Fig. 3) that performs read mapping using (STAR76), peak calling (MACS77), genotype calling (GATK78 and KING79), and quality control checking. Extensive quality control of ATAC-seq libraries based on cell-type, sex, and genotype concordance, as well as sample quality metrics and sequencing depth, yielded a total of 636 samples constituting a total of 19.6 billion read pairs with an average of 30.8 million non-duplicated read pairs per library (Supplementary Fig. 4 and 5, Supplementary Table 3).
Hi-C data generation and processing
Hi-C data libraries were generated from neuronal and non-neuronal nuclei isolated by FANS from frozen post-mortem human brain tissue dissections using the in situ Hi-C protocol55 described in Supplementary Methods. Hi-C libraries were pooled and deep sequenced on the Illumina NovaSeq S4 platform obtaining 100 bp paired-end reads, to generate approximately 250 million reads per sample. In total, we generated Hi-C on 17 neuronal samples and 17 non-neuronal samples from 7 individuals. After pooling unique and valid pairwise contacts from multiple technical and biological replicates, we obtained 2.7 billion pairs from neurons and 3.2 billion pairs from non-neurons, producing ultra-deep cell type specific 3D genome maps in primary human brain tissue across development (Supplementary Table 19). Hi-C data were aligned using the HiC-Pro strategy80 to human genome hg38 using bowtie281, chromatin loops were called with HICCUPS82, topological associated domains (TADs) were identified with Topdom83. The detailed setting of all tools is specified in Supplementary Materials.
ChIP-seq data generation and processing
ChIP-seq data were generated from neuronal and non-neuronal nuclei isolated by FANS from frozen post-mortem human brain tissue dissections using the protocol described in Supplementary Methods. In total, we generated 20 neuronal and 20 non-neuronal samples from STG and EC brain regions of 10 control individuals from our study cohort. Approximately 40 million paired-end reads were generated per sample and subsequently aligned on hg38.
Annotation of ATAC-seq open chromatin regions
The Ensembl 95 genes were used for all analyses in this paper. Further, ChIPSeeker84 (v.1.18.0) was used to assign genomic context and the closest gene for all ATAC-seq OCRs. For ChIPSeeker, a transcript database was created using GenomicFeatures85 (v.1.14.8) and the Ensembl genes. The genomic contexts were defined as promoter (+/− 3kb of any TSS), 5’-UTR, 3’-UTR, exon, intron, and distal intergenic. Besides annotation based on genomic context, we also quantified overlap with previously published epigenomic annotations (REMC59,69, Cancer Atlas60, and the Brain Open Chromatin Atlas) as well as with published cell-marker genes64,86 as detailed in Supplementary Materials.
Gene set enrichment analyses
For gene sets, we used the MSigDB 7.0 gene sets87 of sizes 10 to 1000 genes. For ATAC-seq data, we used the GREAT approach to assign OCRs to genes17,88. Then, we used those genes as an input to CameraPR gene set analysis89. For RNA-seq, the CameraPR function was used directly. For enrichment analyses on the GWAS data, we used MAGMA72. The setting of this analysis is detailed in Supplementary Materials.
Variance component modeling of gene expression
A variance component analysis was used to examine how much gene expression variability could be correlated to patterns of chromatin covariance (Fig. 2). To implement such a model, we followed an implementation suggested by a previous report90. First, we modeled negative binomial distribution of RNA-seq count data by a variance stabilizing transformation91 (vst; varistran R package v.1.0.492). Then, for each gene represented by vst-normalized vector g, we considered the following variance component model:
where P and E are sample-sample covariance matrices of chromatin accessibility in promoter (OCRs overlapping region within 1kb from the transcription start site) and enhancer regions (OCRs overlapping region within 1-100kb from the transcription start site), captures per-individual covariance matrix and is the noise term. The values of , and , were estimated by the average information restricted likelihood estimation (AIREML; gaston R package v.1.5.5; https://cran.r-project.org/web/packages/gaston). We used residualized count matrices of OCRs where the effect of technical covariates was regressed out. For clarification, this approach does not model the relationship of each gene to its own promoter/enhancer OCRs but to the overall status of all enhancers/promoter OCRs. The principle of this analysis is also summarized in Supplementary Fig. 8.
Effect of genetic regulation on gene expression
To determine whether the inter-individual covariance was, at least partially, driven by genetic regulation of gene expression, we calculated the fraction of variation of gene expression explained by cis-genetic component (for each gene). This fraction was proxied by the R2 obtained by performing nested 5-fold cross validation (R2CV) when training transcriptomic imputation models with the EpiXcan method65 in an independent dataset with genotype (SNP array) and gene expression information (bulk RNA-seq) composed of human postmortem brains (prefrontal cortex) from the PsychENCODE Consortium93. Specifically, the R2CV corresponds to the fraction of gene expression variation explained by common (mean allele frequency ≥ 0.01) SNPs located ±1 Mbp from the transcription start site of the gene and is a good proxy (despite it being a somewhat conservative estimate since it doesn’t account for rare variants and very distal elements) for how much of the expression variance is driven by cis-genetic variation94.
Prediction of enhancer-gene interactions
We used Activity-by-contact model (ABC v.0.2)20 to construct a comprehensive regulatory map of enhancer-promoter (E-P) interactions in neuronal and non-neuronal cell types of the two investigated brain regions (STG and EC). This model requires (1) contact frequency between putative enhancers and promoters of regulated genes and (2) enhancer activity data. Contact frequency matrices were generated from neuronal and non-neuronal Hi-C datasets composed of seven post-mortem human brains. Here different neocortical regions (dorsolateral prefrontal cortex, orbital frontal cortex, and anterior prefrontal cortex) were profiled across multiple donors aged 34-103 years. Enhancer activity data were represented by the cell type and brain region specific ATAC-seq signal (current study) and the H3K27ac ChIP-seq signal. ChIP-seq data were generated in a subset of ten controls (STG and EC; age of donors ranged between 61-103 years). In accordance with the authors’ instructions, we filtered out predictions for genes on chromosome Y and lowly expressed genes (genes that did not meet inclusion criteria in our RNA-seq dataset). We used the default threshold of ABC score (a minimum score of 0.02) and the default screening window (5MB around the TSS of each gene). Additional information on computational validation of E-P predictions using (i) overlap with chromatin states from Epigenomics Roadmap Project59,66 and (ii) enrichment in sets of SNPs for eGenes in GTEx brain samples (95% credible set interval)23 are described in Supplementary Materials.
Experimental validation of putative enhancers using CRISPRi
The functional relevance of predicted enhancer-promoter (E-P) links was demonstrated via epigenetic silencing of three high-confidence enhancers in neural progenitor cells derived from human induced stem cells using CRISPR interference (CRISPRi) platform24 for genetic screens. In CRISPRi, catalytically dead Cas9 nuclease (dCas9) is fused to a Kruppel-associated box (KRAB) effector domain to mediate transcriptional repression by spreading repressive histone modifications24. To consider enhancer-promoter (E-P) link for experimental validation, the following criteria needed to be met: (i) E-P distance is between 10kb and 500kb, (ii) gene needs to be at least moderately expressed (CPM>2) in external RNA-seq data from iPSC-derived NPC as well as post-mortem brains, (iii) both the enhancer and promoter of the supposedly regulated gene need to be at least moderately accessible (CPM>2) in our ATAC-seq data from post-mortem brains as well as in iPSC-derived NGN (NIH GEO GSE203082), (iv) both enhancer and gene need to be differentially accessible / expressed in at least one AD-related comparison (AD case/control, BBscore, CDR, PlaqueMean), (v) Spearman correlation between enhancer and promoter (ATAC-seq vs ATAC-seq) as well as enhancer and gene (ATAC-seq vs RNA-seq) need to be higher than 0.3 for data from post-mortem brains as well as data from iPSC-derived NPCs and NGNs. Out of 65 shortlisted E-P links, we chose 3 of them that kept CNS function. More specifically, we tested E-P for the following genes: (1) translation elongation factor (EEF1A2) that is a key modulator of structural plasticity in the dendritic spine95, (2) developmentally regulated brain protein (DBN1) that acts as actin-binding protein modulating synaptic morphology and long-term memory96 and, (3) ribosomal protein S21 (RPS21), a brain-specific component of ribosomal biogenesis whose aberrant function perturbs synapse development and functionality97. All steps of experimental work performed on iPSC line NSB553-S1-16098 are described in Supplementary Materials.
Differential chromatin accessibility and gene expression analysis
To identify OCRs showing differential accessibility and genes showing differential expression in AD-related phenotypes, we evaluated them statistically. First, we filtered out OCRs/genes that were lowly accessible/expressed in most of the samples. Then, we applied an approach based on the Bayesian information criterion to select covariates. Because we keep multiple samples per specimen, we used dream19,71 to properly model correlation structure and, thus, keep the false discovery rate low. Multiple hypothesis testing was adjusted using FDR<5%. A detailed description of all steps is specified in Supplementary Materials.
Disease heritability analysis
To examine the role that OCRs identified in this paper might play in various diseases and traits, we tested if the OCRs were enriched in common trait-associated genetic variants from a selection of GWAS studies. For this, LD-score partitioned heritability30 was used. The settings for this analysis are detailed in Supplementary Materials.
Transcription factor analysis
Transcription factor binding motifs collected from CIS-BP 1.02 meta-database99 were filtered to keep only one motif per TF using TomTom100 for quantification of pairwise similarities. To assess genome-wide putative chromatin occupancy by transcription factors, we performed footprinting analysis using TOBIAS43. This way, we were able to determine cell type specificity (neuronal/non-neuronal) for each TF. Finally, we used information about TF binding to create TF regulatory networks that were analyzed by the HotNet algorithm101 to find altered subnetworks containing TF motifs that are highly dysregulated based on transcriptomics or on the GWAS level, and are topologically close on an interaction network. Please see Supplementary Methods for a detailed description of all steps of this computational analysis as well as experimental validation of USF2 that was performed on Human neuroblastoma cell line SH-SY5Y (purchased from the ATCC, Cat# CRL-2266).
Three-dimensional chromatin interactions from population-level maps of chromatin accessibility
To determine three-dimensional (3D) chromatin interactions and evaluate differential accessibility in 3D interactions associated with AD-related phenotypes, we integrated the decorate pipeline54 and applied additional statistical tests (Supplementary Fig. 30). Supplementary Methods contain a comprehensive description of all steps of this analysis, i.e. (i) data preprocessing and cis-regulatory domain (CRD) calling, (ii) CRD filtering and merging, (iii) CRD biological validation, (iv) differential CRD analysis and (v) differential gene-CRD analysis.
Supplementary Material
Supplementary Methods
Supplementary Figures 1-35
Supplementary Note
Supplementary Tables 1-19
Acknowledgements:
We thank the patients and families who donated material for these studies. We thank the members of the Roussos laboratory for thoughtful advice and critique and the computational resources and staff expertise provided by the Scientific Computing at the Icahn School of Medicine at Mount Sinai. This study was supported by grants from the National Institute on Aging, NIH grants R01-AG067025 (to P.R. and V.H.), R01-AG065582 (to P.R. and V.H.) and R01-AG050986 (to P.R.) and by grants from the National Institute of Mental Health, NIH grants, R56-MH101454 (to K.J.B), R01-MH106056 (to P.R. and K.J.B.), R01-MH109897 (to P.R. and K.J.B.) and R01-MH121074 (K.J.B.). J.B. was supported in part by Alzheimer’s Association Research Fellowship AARF-21-722200. K.G. was supported in part by Alzheimer’s Association Research Fellowship AARF-21-722582. G.E.H. was supported in part by NARSAD Young Investigator Grant 26313 from the Brain & Behavior Research Foundation, P.D. was supported in part by NARSAD Young Investigator Grant 29683 from the Brain & Behavior Research Foundation. S.P.K. is a recipient of an NIH LRP award. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award numbers S10OD018522 and S10OD026880. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests: The authors declare no competing interests.
Code availability: The code used to perform the analysis described in this study is available at https://doi.org/10.7303/syn34034120.
Data availability:
Raw (FASTQ files) and processed data (BigWig files, peaks, and raw / normalized count matrices) are available via the AD Knowledge Portal (https://adknowledgeportal.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). For access to the content described in this manuscript see: https://doi.org/10.7303/syn21513145. Browsable UCSC genome browser tracks of processed data are available at: https://labs.icahn.mssm.edu/roussos-lab/atacad/.
External validation sets: MSBB RNA-seq of post-mortem brains (Synapse ID: syn3157743), ATAC-seq on FANS-sorted NeuN+/− from post-mortem brains (synID syn20755767), H3K9ac ChIP-seq of post-mortem brains (Synapse ID: syn4896408). ATAC-seq iPSC-derived neurons overexpressing MAPT gene (GEO GSE97409), ROSMAP RNA-seq of post-mortem brains (Synapse ID: syn3388564), fine-mapped eQTLs (https://alkesgroup.broadinstitute.org/LDSCORE/LDSC_QTL/, version “FE_META_TISSUE_GTEx_Brain_MaxCPP”), CTCF ChIP-seq peaks on human neural cell (GEO GSE127577). Open chromatin regions (peaks) from The Cancer Genome Atlas (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG), BOCA/BOCA2 - brain epigenome atlas (https://icahn.mssm.edu/boca, https://icahn.mssm.edu/boca2), Dong. et al. 2021 (Synapse ID: syn25716684), Nott et al. 2019 (dbGaP ID: phs001373), and Meuleman et al. 2020 (ENCODE ID: ENCSR857UZV), fine-mapped eQTLs (https://alkesgroup.broadinstitute.org/LDSCORE/LDSC_QTL/, version “FE_META_TISSUE_GTEx_Brain_MaxCPP”), CTCF ChIP-seq on human neural cell (GEO GSE127577), The Cancer Genome Atlas (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG), REMC (http://www.roadmapepigenomics.org), mSigDB 7.0 (http://www.gsea-msigdb.org/), dbSNP v.151 (https://www.ncbi.nlm.nih.gov/snp/), PsychENCODE SNP-array: Capstone collection (https://psychencode.synapse.org/).
References
- 1.Andrews SJ, Fulton-Howard B & Goate A Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 19, 326–335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marzi SJ et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci 21, 1618–1627 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Klein H-U et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci 22, 37–46 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gasparoni G et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 11, 41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li P et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun 10, 2246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj T et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet 50, 1584–1592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nott A et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Y, Matevossian A, Huang H-S, Straubhaar J & Akbarian S Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 9, 42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozlenkov A et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 44, 2593–2612 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci. Data 5, 180185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fillenbaum GG et al. Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): the first twenty years. Alzeimers Dement 4, 96–109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braak H & Braak E Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H & Del Tredici K Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris JC The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43, 2412–2414 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Nelson PT et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol 71, 362–381 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fullard JF et al. An atlas of chromatin accessibility in the adult human brain. Genome Res. 28, 1243–1252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauberg ME et al. Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons. Nat. Commun 11, 5581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu B et al. Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun 12, 3968 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman GE & Schadt EE variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17, 483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fulco CP et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mifsud B et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet 47, 598–606 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Corces MR et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hormozdiari F et al. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat. Genet 50, 1041–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickar-Oliver A & Gersbach CA The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol 20, 490–507 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 8, 104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khan UA et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci 17, 304–311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrera J et al. Sex dependent glial-specific changes in the chromatin accessibility landscape in late-onset Alzheimer’s disease brains. Mol. Neurodegener 16, 58 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowles KR et al. 17q21.31 sub-haplotypes underlying H1-associated risk for Parkinson’s disease and progressive supranuclear palsy converge on altered glial regulation. BioRxiv (2019) doi: 10.1101/860668. [DOI] [Google Scholar]
- 29.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokoyama K et al. NYAP: a phosphoprotein family that links PI3K to WAVE1 signalling in neurons. EMBO J. 30, 4739–4754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X et al. Cholecystokinin release triggered by NMDA receptors produces LTP and sound-sound associative memory. Proc Natl Acad Sci USA 116, 6397–6406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Long J, He T, Belshaw R & Scott J Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci. Rep 5, 12393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castillo E et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep 7, 17762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hokama M et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan PM & Manser E PAKs in human disease. Prog. Mol. Biol. Transl. Sci 106, 171–187 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Bell RD & Zlokovic BV Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 118, 103–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan Z et al. Regulation of neuronal cell death by MST1-FOXO1 signaling. J. Biol. Chem 284, 11285–11292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greer EL & Brunet A FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24, 7410–7425 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Webb AE, Kundaje A & Brunet A Characterization of the direct targets of FOXO transcription factors throughout evolution. Aging Cell 15, 673–685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koopmans F et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 103, 217–234.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bentsen M et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun 11, 4267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rahman MR et al. Network-based approach to identify molecular signatures and therapeutic agents in Alzheimer’s disease. Comput. Biol. Chem 78, 431–439 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Acquaah-Mensah GK & Taylor RC Brain in situ hybridization maps as a source for reverse-engineering transcriptional regulatory networks: Alzheimer’s disease insights. Gene 586, 77–86 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Qin L et al. Ethnicity-specific and overlapping alterations of brain hydroxymethylome in Alzheimer’s disease. Hum. Mol. Genet 29, 149–158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malik BR, Maddison DC, Smith GA & Peters OM Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 12, 100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamanaka T et al. Genome-wide analyses in neuronal cells reveal that upstream transcription factors regulate lysosomal gene expression. FEBS J. 283, 1077–1087 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Johnson DE, Ostrowski P, Jaumouillé V & Grinstein S The position of lysosomes within the cell determines their luminal pH. J. Cell Biol 212, 677–692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayek SR, Rane HS & Parra KJ Reciprocal Regulation of V-ATPase and Glycolytic Pathway Elements in Health and Disease. Front. Physiol 10, 127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Couoh-Cardel S, Milgrom E & Wilkens S Affinity purification and structural features of the yeast vacuolar atpase vo membrane sector. J. Biol. Chem 290, 27959–27971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delaneau O et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science 364, (2019). [DOI] [PubMed] [Google Scholar]
- 53.Ambroise C, Dehman A, Neuvial P, Rigaill G & Vialaneix N Adjacency-constrained hierarchical clustering of a band similarity matrix with application to genomics. Algorithms Mol. Biol 14, 22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffman GE, Bendl J, Girdhar K & Roussos P decorate: differential epigenetic correlation test. Bioinformatics 36, 2856–2861 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao SSP et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Jager PL et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 5, 180142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meuleman W et al. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 23, 270–280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corces MR et al. The chromatin accessibility landscape of primary human cancers. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishizuka Y & Hanamura K Drebrin in alzheimer’s disease. Adv. Exp. Med. Biol 1006, 203–223 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Turi Z, Lacey M, Mistrik M & Moudry P Impaired ribosome biogenesis: mechanisms and relevance to cancer and aging. Aging (Albany NY) 11, 2512–2540 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chambers DM, Peters J & Abbott CM The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1alpha, encoded by the Eef1a2 gene. Proc Natl Acad Sci USA 95, 4463–4468 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zeisel A et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Zhang W et al. Integrative transcriptome imputation reveals tissue-specific and shared biological mechanisms mediating susceptibility to complex traits. Nat. Commun 10, 3834 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ernst J & Kellis M Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc 12, 2478–2492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meuleman W et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong P et al. Population-level variation of enhancer expression identifies novel disease mechanisms in the human brain. Nat. Genet 54, 1493–1503 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ernst J & Kellis M Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol 33, 364–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoffman GE & Roussos P Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics 37, 192–201 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 73.Haroutunian V, Katsel P & Schmeidler J Transcriptional vulnerability of brain regions in Alzheimer’s disease and dementia. Neurobiol. Aging 30, 561–573 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris JC et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 39, 1159–1165 (1989). [DOI] [PubMed] [Google Scholar]
- 75.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Servant N et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Durand NC et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shin H et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 44, e70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu G, Wang L-G & He Q-Y ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Lawrence M et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Law CW et al. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. [version 3; peer review: 3 approved]. F1000Res. 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoshida H et al. The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anscombe FJ The Transformation of Poisson, Binomial and Negative-Binomial Data. Biometrika 35, 246–254 (1948). [Google Scholar]
- 92.Francis Harrison P Varistran: Anscombe’s variance stabilizing transformation for RNA-seq gene expression data. JOSS 2, 257 (2017). [Google Scholar]
- 93.Gandal MJ et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nagpal S et al. TIGAR: an improved bayesian tool for transcriptomic data imputation enhances gene mapping of complex traits. Am. J. Hum. Genet 105, 258–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Becker M, Kuhse J & Kirsch J Effects of two elongation factor 1A isoforms on the formation of gephyrin clusters at inhibitory synapses in hippocampal neurons. Histochem. Cell Biol 140, 603–609 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Shirao T et al. The role of drebrin in neurons. J. Neurochem 141, 819–834 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Hetman M & Slomnicki LP Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem 148, 325–347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schrode N et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet 51, 1475–1485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gupta S, Stamatoyannopoulos JA, Bailey TL & Noble WS Quantifying similarity between motifs. Genome Biol. 8, R24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Reyna MA, Leiserson MDM & Raphael BJ Hierarchical HotNet: identifying hierarchies of altered subnetworks. Bioinformatics 34, i972–i980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Supplementary Figures 1-35
Supplementary Note
Supplementary Tables 1-19
Data Availability Statement
Raw (FASTQ files) and processed data (BigWig files, peaks, and raw / normalized count matrices) are available via the AD Knowledge Portal (https://adknowledgeportal.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). For access to the content described in this manuscript see: https://doi.org/10.7303/syn21513145. Browsable UCSC genome browser tracks of processed data are available at: https://labs.icahn.mssm.edu/roussos-lab/atacad/.
External validation sets: MSBB RNA-seq of post-mortem brains (Synapse ID: syn3157743), ATAC-seq on FANS-sorted NeuN+/− from post-mortem brains (synID syn20755767), H3K9ac ChIP-seq of post-mortem brains (Synapse ID: syn4896408). ATAC-seq iPSC-derived neurons overexpressing MAPT gene (GEO GSE97409), ROSMAP RNA-seq of post-mortem brains (Synapse ID: syn3388564), fine-mapped eQTLs (https://alkesgroup.broadinstitute.org/LDSCORE/LDSC_QTL/, version “FE_META_TISSUE_GTEx_Brain_MaxCPP”), CTCF ChIP-seq peaks on human neural cell (GEO GSE127577). Open chromatin regions (peaks) from The Cancer Genome Atlas (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG), BOCA/BOCA2 - brain epigenome atlas (https://icahn.mssm.edu/boca, https://icahn.mssm.edu/boca2), Dong. et al. 2021 (Synapse ID: syn25716684), Nott et al. 2019 (dbGaP ID: phs001373), and Meuleman et al. 2020 (ENCODE ID: ENCSR857UZV), fine-mapped eQTLs (https://alkesgroup.broadinstitute.org/LDSCORE/LDSC_QTL/, version “FE_META_TISSUE_GTEx_Brain_MaxCPP”), CTCF ChIP-seq on human neural cell (GEO GSE127577), The Cancer Genome Atlas (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG), REMC (http://www.roadmapepigenomics.org), mSigDB 7.0 (http://www.gsea-msigdb.org/), dbSNP v.151 (https://www.ncbi.nlm.nih.gov/snp/), PsychENCODE SNP-array: Capstone collection (https://psychencode.synapse.org/).