

ABSTRACT
Listeria monocytogenes is a Gram-positive facultative anaerobe and an excellent model pathogen for investigating regulatory changes that occur during infection of a mammalian host. SpxA1 is a widely conserved transcriptional regulator that induces expression of peroxide-detoxifying genes in L. monocytogenes and is thus required for aerobic growth. SpxA1 is also required for L. monocytogenes virulence, although the SpxA1-dependent genes important in this context remain to be identified. Here, we sought to investigate the role of SpxA1 in a tissue culture model of infection and made the surprising discovery that ΔspxA1 cells are dramatically elongated during growth in the host cytosol. Quantitative microscopy revealed that ΔspxA1 cells also form elongated filaments extracellularly during early exponential phase in rich medium. Scanning and transmission electron microscopy analysis found that the likely cause of this morphological phenotype is aberrantly placed division septa localized outside cell midpoints. Quantitative mass spectrometry of whole-cell lysates identified SpxA1-dependent changes in protein abundance, including a significant number of motility and flagellar proteins that were depleted in the ΔspxA1 mutant. Accordingly, we found that both the filamentation and the lack of motility contributed to decreased phagocytosis of ΔspxA1 cells by macrophages. Overall, we identify a novel role for SpxA1 in regulating cell elongation and motility, both of which impact L. monocytogenes virulence.
KEYWORDS: filamentation, Gram-positive bacteria, pathogenesis, redox signaling, transcriptional regulation
INTRODUCTION
Listeria monocytogenes is the causative agent of the foodborne disease listeriosis and is a well-described model intracellular pathogen (1). L. monocytogenes lives as an environmental saprophyte until consumed by a susceptible mammalian host. Once ingested, L. monocytogenes is phagocytosed by professional phagocytes or induces its own uptake into nonphagocytic cells (2). Quickly after internalization, L. monocytogenes escapes the vacuole and replicates within the host cytosol (3). Here, L. monocytogenes produces the surface-associated protein ActA, which recruits host actin to mediate intracellular motility and intercellular spread via characteristic actin comet tails (4). The asymmetric distribution of ActA along the bacterial cell surface is tightly coupled to the bacterial cell cycle and critical for motility initiation, comet tail formation, and bacterial movement within the host cytosol (5). Although the complex set of biophysical steps necessary for efficient actin-dependent cytosolic motility of L. monocytogenes is not fully understood, the rod shape of this bacterium is thought to be essential to this process (6, 7).
Alterations to cell shape can have detrimental effects for many bacterial pathogens, since cell shape dictates motility, surface protein localization, innate immune activation, adherence, and invasion into host cells. For example, the helical shape of the foodborne pathogens Helicobacter pylori and Campylobacter jejuni is critical for flagellar motility in mucus and host colonization (8–10). Similarly, the curvature of the comma-shaped pathogen Vibrio cholerae enhances colonization of the small intestine (11). In L. monocytogenes, mutants that form chains due to septation defects are deficient for invasion and virulence (12, 13). Further, the shape and geometry of L. monocytogenes influence actin tail formation and function, while the mechanical stress of actin-based motility affects cell elongation and division (5, 13, 14).
Alternate morphologies can also provide survival advantages to bacterial pathogens. For example, many pathogenic organisms form filaments in vivo that are resistant to both phagocytosis and antimicrobials (15). Filamentation describes the process of continual longitudinal growth without formation of septa between newly replicated chromosomes. Increasing cell envelope volume absent separation results in cells that are much longer than their bacillary counterparts (16). Filamentous morphologies are induced in rod-shaped bacteria by a variety of stressors, such as metabolic changes, DNA damage (the SOS response), or alterations to the stoichiometry of cell division components (15). In addition to these stressors, L. monocytogenes filamentation is induced extracellularly in response to high- or low-pH, salt, cold, or heat stress (17–19). Despite these extracellular observations, the regulation of L. monocytogenes morphology in vivo and the relevance to pathogenesis are not well understood.
One L. monocytogenes regulator that is important for both extracellular and intracellular growth is the redox-responsive transcriptional regulator SpxA1 (20). SpxA1 belongs to the ArsC-like Spx family of proteins that is highly conserved in low G+C Firmicutes (21). L. monocytogenes SpxA1 is essential for aerobic growth in rich broth, as well as virulence in a murine model of infection (22, 23). Similarly, Staphylococcus aureus Spx is essential for growth in rich media, and Spx homologues are required for virulence in Streptococcus spp. and Enterococcus faecalis (24–28). Our previous work found that L. monocytogenes lacking spxA1 are unable to replicate aerobically due to toxic levels of endogenously produced hydrogen peroxide, resulting from a combination of dysregulated cytochrome oxidase and insufficient catalase production (29). However, the SpxA1-regulated genes necessary for aerobic growth are dispensable for infection, so the role of SpxA1 in pathogenesis remains to be defined.
We sought to investigate the role of SpxA1 in L. monocytogenes virulence and made the surprising discovery that ΔspxA1 mutant cells exhibit variable and elongated morphology during growth in the host cytosol. Quantitative microscopy further revealed ΔspxA1 cells undergo filamentation extracellularly in anaerobic rich medium. A whole-cell proteomics approach identified SpxA1-dependent changes in protein abundance, including motility and flagellar proteins that were depleted in the ΔspxA1 mutant. Finally, we show that filamentation and decreased motility of ΔspxA1 cells contribute to the virulence defect of ΔspxA1 cells in tissue culture models of infection. Together, these results significantly advance our understanding of the role of SpxA1 in L. monocytogenes replication and pathogenesis.
RESULTS
The L. monocytogenes ΔspxA1 mutant exhibits an elongated morphology.
The L. monocytogenes ΔspxA1 strain is significantly impaired for intracellular replication and intercellular spread, although the SpxA1-regulated genes important for these processes have yet to be identified (23, 29). To further examine the role of SpxA1 in L. monocytogenes virulence, we took a microscopy-based approach and infected monolayers of murine fibroblasts grown on glass coverslips. Monolayers were fixed 10 h postinfection, and L. monocytogenes, DNA, and host actin were fluorescently labeled. As expected, wild-type (wt) L. monocytogenes appeared as short rods in the host cytosol. Many bacteria colocalized with increased densities of host actin (red) and some appeared to be forming canonical actin comet tails (Fig. 1A). In contrast, intracellular ΔspxA1 cells were more heterogeneous in shape, with some appearing as short rods and many appearing as chains of cells or elongated single cells. Furthermore, elongated ΔspxA1 cells appeared to colocalize less frequently with highly dense areas of actin. Cells that did colocalize with host actin displayed disorganized or nonpolar actin recruitment and were rarely associated with actin comet tails (Fig. 1B). These results led us to question whether the morphological defect of the ΔspxA1 strain was specific to growth in the mammalian cytosol.
FIG 1.

Microscopy of L. monocytogenes ΔspxA1 during intracellular and extracellular growth. (A) L2 murine fibroblasts were infected for 10 h with wt or ΔspxA1 cells and fluorescently labeled to visualize DNA (blue), L. monocytogenes (green), and host actin (red). The yellow arrow indicates a canonical actin comet tail. Images were taken using a 100× objective. (B) Additional examples of cells infected with the ΔspxA1 mutant. White arrows indicate elongated bacteria with disorganized or nonpolar actin recruitment. (C) Phase-contrast images of wt and ΔspxA1 cells grown to early exponential phase in rich anaerobic broth. Bacteria were imaged with a 40× objective. All scale bars represent 10 μm, and all images represent three biological replicates and at least 10 fields of view.
To determine whether the observed ΔspxA1 morphological heterogeneity was specific to intracellular growth, bacteria were grown to early exponential phase in a rich medium (i.e., brain heart infusion [BHI]) in an anaerobic chamber and spotted onto phosphate-buffered saline (PBS) agar pads for phase-contrast microscopy. Under these conditions, wt L. monocytogenes was almost exclusively rod-shaped and of uniform length, whereas the ΔspxA1 mutant exhibited varied and elongated morphologies (Fig. 1C). Together, these images suggested that SpxA1 regulation of cell shape could play a significant role in L. monocytogenes growth in both intracellular and extracellular environments.
Quantification of ΔspxA1 elongation.
To quantitatively evaluate ΔspxA1 morphology, phase-contrast images of wt and ΔspxA1 cells grown anaerobically in BHI were analyzed with the cell segmentation software Celltool (30). The cell perimeter was used as the primary metric to describe cell size and account for the irregular shape and curvature of some ΔspxA1 cells. Despite demonstrating no significant difference in growth rate compared to wt cells (see Fig. S1 in the supplemental material), ΔspxA1 cells showed increased mean, median, range, and variance values in cell perimeters (Fig. 2A). The cell perimeters of wt and ΔspxA1 cells differed most at the early exponential phase, with the mean for wt cells measuring 8.5 μm compared to 16.9 μm for ΔspxA1 cells. In addition, the maximum cell perimeter observed for the ΔspxA1 mutant was 146 μm, which was nearly triple the size of the largest wt cell. While the average cell perimeters of both wt and ΔspxA1 cells decreased over time, ΔspxA1 cells were not restored to wt size at any point during the 8-h time course. In order to accurately assess the biological relevance of the observed elongation, we calculated the percentages of elongated bacteria. Elongated cells were defined as having a perimeter >1 standard deviation (SD) above the mean for wt cells at each time point. Approximately 10% of wt cells were elongated at each time point examined (Fig. 2B). Throughout growth, ΔspxA1 cultures contained much higher percentages of elongated cells, with elongated cells comprising 54% of the population at 2 h. This phenotype was rescued via genetic complementation by providing a copy of spxA1 with its native promoter at an ectopic site on the chromosome using the integrative plasmid pPL2 (ΔspxA1 pPL2.spxA1) (31). These data demonstrated that L. monocytogenes lacking spxA1 forms significantly longer cells, particularly at the early exponential phase.
FIG 2.

Quantification of L. monocytogenes ΔspxA1 elongation and cell viability during broth growth. (A) Bacteria were grown anaerobically in rich broth, and cell perimeters were measured from phase-contrast images using Celltool. The lines indicate the means. Distributions were compared using Kolmogorov-Smirnov tests. ***, P < 0.001. (B) The percentages of elongated cells were measured for at least 10 fields of view or 1,000 cells. Elongated cells were defined as perimeter lengths greater than one SD above the wt mean at each time point. Each symbol represents a biological replicate, the bars indicate the mean, and the error bars denote the SEM. P values were calculated using an unpaired Student t test compared to wt. *, P < 0.05; ***, P < 0.001. (C) The percentages of propidium iodide (PI)-positive cells were calculated from at least 1,000 cells or 10 fields of view. The data are not significantly different, as determined by an unpaired Student t test (n.s. P > 0.3). (D) The cell perimeters of bacteria from PI-negative (–) and PI-positive (+) populations were measured. Graphs represent cell perimeters measured from three biological replicates where at least 1,000 cells or 10 fields of view were quantified per replicate. The lines indicate the means.
Due to the observed morphological irregularities, we hypothesized that highly elongated ΔspxA1 cells may have irregular or damaged membranes and therefore decreased viability. To determine the viability and membrane integrity of the ΔspxA1 mutant, early-log-phase cultures were stained with propidium iodide (PI), a live-dead stain that cannot penetrate intact lipid bilayers. We observed an ~2-fold increase in the percentage of PI-positive ΔspxA1 cells compared to wt cells, although this difference was not statistically significant (Fig. 2C). Neither strain exhibited significant PI staining, with the percentage of PI-positive cells never reaching 2% for wt cells or 3% for ΔspxA1 cells. To evaluate the viability of elongated cells specifically, the cell perimeters of the PI-positive and PI-negative populations were measured for each strain. The distributions of PI-positive and PI-negative ΔspxA1 cells appeared to be nearly identical, indicating that the elongated ΔspxA1 cells were not more likely to be PI positive (Fig. 2D). Taken together, these results demonstrated that ΔspxA1 cells are morphologically heterogenous and that a significant portion of the cells are dramatically elongated. However, this elongation does not correlate to a critical defect in viability or membrane integrity.
Fluorescence microscopy of membranes.
Phase-contrast microscopy revealed that the majority of ΔspxA1 cells were significantly elongated compared to wt cells, but the quantitative analysis of these images could not distinguish between incompletely separated chains of cells or filamentous single cells. To examine the specific nature of elongated ΔspxA1 cells, bacteria were labeled with the fluorescent membrane dye TMA-DPH, which intercalates into lipid bilayers containing fatty acyl chains (32). L. monocytogenes wt cells appeared as typical rods at various stages of division with sites of septation localized to the cell midpoint (Fig. 3A). In contrast, ΔspxA1 cells exhibited a range of elongated filamentous morphologies that rarely contained septa at cell midpoints and often exhibited a complete lack of septa. These results suggested that the significant elongation of the ΔspxA1 mutant was likely the result of filamentation rather than chaining.
FIG 3.
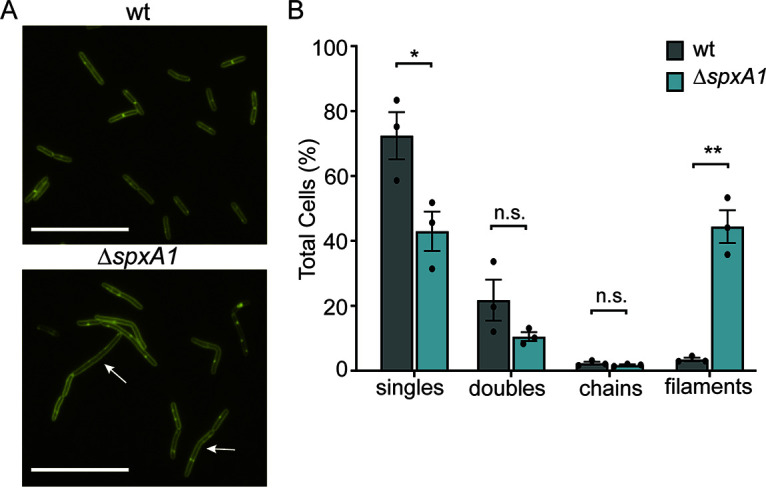
Fluorescent labeling of membranes indicates the prevalence of ΔspxA1 filamentation. (A) L. monocytogenes wt and ΔspxA1 cells grown to early exponential phase were labeled with the fluorescent membrane stain TMA-DPH. Arrows indicate elongated cells without regular septa, and scale bars represent 10 μm. (B) Quantification of elongated cells observed in panel A. Data displayed are the means and the SEM of three biological replicates of 1,000 cells each. P values were calculated using an unpaired Student t test compared to wt cells. *, P < 0.05; **, P < 0.01; n.s., not significant.
To quantify the frequency of ΔspxA1 filamentation, we visually evaluated the TMA-DPH images and categorized >1,000 cells per replicate for each strain. Single cells were defined as cells lacking fully formed septa that were approximately the length of the wt mean (2 to 5 μm). Doublets and chains were defined as two or more linked bacteria, respectively, composed of 2- to 5-μm cells attached by evenly placed septa. Filaments were defined as cells longer than 2 to 5 μm that either lack septa completely, have irregular numbers of septa relative to the length of the cell, or contain septa placed outside the midpoint of the cell. As expected, approximately 94% of wt bacteria were either single cells or doublets, with a small minority of chains and filaments (Fig. 3B). In contrast, >44% of ΔspxA1 cells were elongated filaments with aberrant septum localization. These results confirmed that the majority of elongated ΔspxA1 cells are filamentous single cells.
Scanning transmission electron microscopy.
After determining that the majority of ΔspxA1 cells were filaments, we next sought to investigate the observed differences in membrane architecture at higher resolution. To that end, we performed scanning transmission electron microscopy (STEM) on wt, ΔspxA1, and complemented ΔspxA1 strains. Early-exponential-phase cultures were negatively stained with uranyl acetate, which incorporates into organic materials with a high affinity for phosphorylated molecules. This enables resolution of dense and highly phosphorylated components such as membranes, nucleic acids, and some proteins (33). In many respects, the ΔspxA1 cell envelope appeared indistinguishable from that of wt and the complemented strain (Fig. 4). For example, all three strains had a similarly thick outer layer of peptidoglycan covering a thin layer of highly contrasting phospholipid membrane, with no obvious differences in phospholipid or peptidoglycan abundance (Fig. 4A). In addition, the division septa appeared to have similar architecture and composition in all three strains. However, several differences in the spatial organization of the ΔspxA1 envelope were observed. Consistent with our phase-contrast and fluorescence microscopy images, ΔspxA1 cells appeared elongated compared to wt cells (Fig. 4B to D). Multiple septa were observed at various stages of development within ΔspxA1 filaments, and these were unevenly distributed along the cell (Fig. 4C). We observed ΔspxA1 cells in the process of initiating septum formation in aberrant locations outside the cell midpoint, sometimes irregularly spaced and atypically close together (Fig. 4B and C). The formation of septa was not only initiated, but successfully completed, outside cell midpoints (Fig. 4D). Finally, ΔspxA1 cells could initiate full separation of segmented cells at sites of mislocalized septation (Fig. 4D). Taken together, these images suggested that ΔspxA1 filamentation is due to both improper localization and frequency of septum formation.
FIG 4.

STEM images of L. monocytogenes. (A) L. monocytogenes strains grown to early exponential phase were fixed and imaged by STEM. (B to D) Representative examples of ΔspxA1 filaments. Boxed portions of each image are shown magnified below. Black arrows indicate septa forming outside the cell midpoint, irregularly spaced, and atypically close together. Scale bars are indicated in each panel.
Whole-cell proteomics.
To identify SpxA1-dependent factors influencing cell morphology, we performed whole-cell proteomics on early-exponential-phase bacterial cultures. Previous work demonstrated that SpxA1 impacts the transcription of 217 genes, activating 145 and repressing 72, including several proteins involved in protein degradation (29). Thus, a proteomics approach has the advantage of identifying the proteins that are changed directly via SpxA1 transcriptional regulation and indirectly via SpxA1 regulation of protein turnover. Bacteria were grown in triplicate to early exponential phase, and proteins were isolated and quantified using unlabeled mass spectrometry coupled to liquid chromatography. We identified 1,692 proteins from 23,219 peptides, accounting for 60.1% of all protein coding regions. Proteins that differed at least 2-fold from wt abundance (P < 0.05) or were absent in one strain and abundant (more than 5 peptides detected) in the other were considered for further analysis. Using these criteria, we identified 122 proteins that were significantly decreased in abundance in ΔspxA1 cells compared to wt cells (see Table S1) and 46 proteins that were significantly increased in abundance in ΔspxA1 cells (see Table S2).
The largest category of proteins depleted in ΔspxA1 cells compared to wt cells were proteins involved in redox homeostasis and respiration. Specifically, the largest decrease observed was for the protein catalase (50-fold), which detoxifies hydrogen peroxide and is produced during aerobic growth in an SpxA1-dependent manner (29). Further, proteomics detected thiol peroxidase (Tpx) and heme biosynthesis proteins (HemE and HemH) in wt cells at high abundance, but these proteins were below the limit of detection in ΔspxA1 cells (see Table S1). Other redox-related proteins depleted in ΔspxA1 cells compared to wt cells included ChdC (heme peroxidase), Lmo0983 (glutathione peroxidase), Lmo1609 (thioredoxin), Rex (redox-responsive regulator), and the SpxA1 paralogue SpxA2 (Lmo2426) (23). To test whether the depletion of these proteins contributed to the morphology of ΔspxA1 cells, we measured the cell perimeters of deletion mutants grown to early exponential phase in anaerobic BHI. L. monocytogenes cells lacking kat, tpx, hemEH, chdC, lmo0983, lmo1609, rex, or spxA2 were not significantly elongated compared to wt cells (see Fig. S2). Moreover, a mutant lacking both spxA1 and spxA2 exhibited filamentation at levels indistinguishable from ΔspxA1 alone. These results indicated that the genes critical for ΔspxA1 aerobic growth and redox homeostasis are not required for proper cell elongation and division.
We next evaluated protein turnover, since proteins in this category were the most dramatically enriched in ΔspxA1 cells compared to wt cells (see Table S2). Specifically, the protease adaptor proteins MecA and ClpE were the most increased in ΔspxA1 cells (19- and 10-fold, respectively). To explore the relevance of these proteins to L. monocytogenes morphology, mutants lacking mecA or clpE were generated via allelic exchange. Quantitative microscopy of L. monocytogenes ΔmecA and ΔclpE cells revealed no change in the frequency of cell elongation compared to wt cells (Fig. 5). Similarly, deletion of mecA and clpE in the ΔspxA1 background did not rescue the elongation of ΔspxA1 cells. Interestingly, the ΔspxA1 ΔclpE double mutant was significantly more elongated than the ΔspxA1 parental strain. To extend our analysis of protein turnover, mutants lacking yjbH and clpX were generated in both wt and ΔspxA1 backgrounds. SpxA1 abundance is regulated by YjbH- and ClpX-dependent degradation such that mutants deficient for yjbH or clpX have increased levels of SpxA1 (20, 34). L. monocytogenes ΔyjbH and clpX::Tn mutants formed cells that were significantly smaller than wt cells (Fig. 5). Furthermore, double mutants that lack spxA1 and yjbH or clpX did not differ in elongation compared to the ΔspxA1 parental strain. Together, these data suggested that cell size maintenance requires precise regulation of SpxA1, since mutants with increased SpxA1 abundance form cells smaller than wt and spxA1-deficient cells are significantly elongated.
FIG 5.
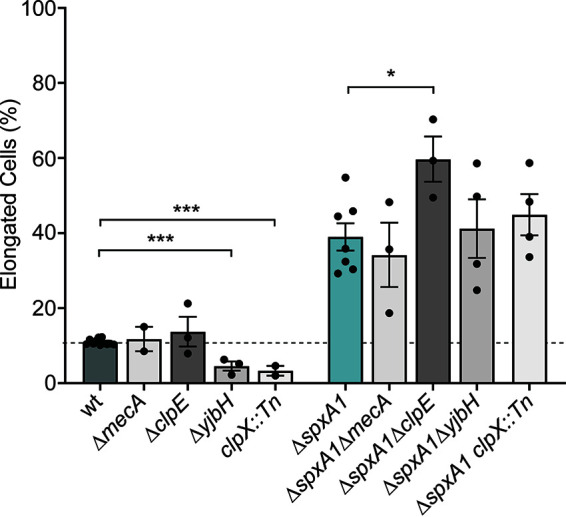
Proteolysis influences morphology of L. monocytogenes. The percentages of elongated cells were measured for at least 10 fields of view or 1,000 cells. Elongated cells were defined as perimeter lengths greater than one SD above the wt mean. Each symbol represents a biological replicate, the bars indicate the mean, and the error bars denote the SEM. P values were calculated using an unpaired Student t test compared to the parental strain, as indicated. *, P < 0.05; ***, P < 0.001.
We next examined whether spxA1 elongation was mediated by well-described inducers of filamentation in other Firmicutes. Filamentation can arise due to changes in the abundance of proteins involved in cell envelope biosynthesis and modification, induction of the DNA damage response (the SOS response), or alterations to the stoichiometry of cell division proteins (15). Interestingly, none of the canonical factors critical for these processes were significantly changed in ΔspxA1 cells compared to wt cells. Specifically, proteins involved in the synthesis and modification of wall teichoic acid (DltACD and TagABDH), lipoteichoic acid (LtaPS, LafAB, and GtlAB), and peptidoglycan (MurBCDEFGIZ, PBPs, Ami, and PgdA) were equally abundant in wt and ΔspxA1 cells (see Table S3). Furthermore, immunoblot detection of lipoteichoic acid revealed no changes in the size or composition of lipoteichoic acid between wt and ΔspxA1 strains (data not shown). The SOS response induces filamentation in response to DNA damage through RecA-mediated transcriptional changes. RecA binds to single-stranded DNA and induces the autocleavage of the major SOS regulator LexA, leading to inhibition of septation (35). Whole-cell proteomics identified no difference in the abundance of RecA or LexA in ΔspxA1 cells compared to wt cells, and several other canonical SOS proteins were also unchanged (RecF, RecQ, RuvAB, and UvrABC). Lastly, critical cell shape determinants (MreBC, RodA1, and RodZ), proteins necessary for the formation of the divisome (FtsAEHKYZ, SepF, and ZapA), and proteins required for septum localization to the cell midpoint (DivIB, EngB, EzrA, MinCDJ, and Noc) were unchanged in the ΔspxA1 mutant compared to wt cells. In considering these results together, we did not find evidence of cell envelope alterations, induction of the SOS response, or aberrant production of divisome proteins in ΔspxA1 filamentous cells.
Finally, we examined whether ΔspxA1 filamentation was the result of a deficiency in secreted factors such as autolysins, small peptides, or signaling molecules that would not be readily measured by whole-cell proteomics. If this was the case, we hypothesized that coculture with wt cells would rescue ΔspxA1 filamentation in trans. Indeed, L. monocytogenes mutants lacking the secreted peptidoglycan hydrolases NamA or p60 form chains that are unable to divide unless exposed to wt culture supernatants (12, 13). However, anaerobic coculture of wt cells constitutively expressing green fluorescent protein (GFP) with ΔspxA1 cells expressing mCherry did not rescue the elongated phenotype of ΔspxA1 cells (see Fig. S3). Overall, our proteomic and microscopic analyses determined that SpxA1-dependent factors regulate cell size, since proteolysis-deficient strains with increased SpxA1 abundance (ΔyjbH and ΔclpX) are significantly smaller than wt strains, and L. monocytogenes lacking spxA1 are significantly elongated. Moreover, we determined that SpxA1-dependent filamentation is not due to SOS response induction, altered synthesis or modification of teichoic acids or peptidoglycan, changes in cell division protein abundance, or defects in secreted factors such as autolysins or small molecules.
Roles of motility and morphology during infection.
Upon examining our whole-cell proteomics for protein changes that could impact virulence, we discovered that proteins associated with motility and chemotaxis were significantly decreased in abundance in ΔspxA1 cells compared to wt cells (Table 1). Specifically, the ΔspxA1 strain was significantly deficient for 16 motility and chemotaxis proteins, including a 7-fold decrease in the flagellin monomer FlaA. While previous work demonstrated that L. monocytogenes strain 10403S significantly downregulates flaA at 37°C, flagellum production and motility is not completely abrogated at this temperature (36). Indeed, we observed that wt L. monocytogenes was approximately 40% less motile at 37°C compared to room temperature when incubated anaerobically (see Fig. S4). Compared to the wt strain, the ΔspxA1 mutant exhibited reduced motility at room temperature and was nonmotile at 37°C.
TABLE 1.
Motility proteins decreased in ΔspxA1 cells
| 10403S | EGD-e | Protein | Protein function | Average LFQ |
Fold decrease (wt/ΔspxA1) | P | |
|---|---|---|---|---|---|---|---|
| wt | ΔspxA1 mutant | ||||||
| LMRG_00386 | lmo0697 | FlgE | Flagellar hook protein | 6.9E+07 | 3.0E+06 | 23.3 | 1.4E–03 |
| LMRG_00378 | lmo0690 | FlaA | Flagellin | 2.5E+09 | 3.6E+08 | 7.1 | 4.1E–04 |
| LMRG_00412 | lmo0723 | Methyl-accepting chemotaxis protein | 6.1E+08 | 9.2E+07 | 6.7 | 2.9E–03 | |
| LMRG_00402 | lmo0713 | FliF | Flagellar M-ring protein | 9.0E+07 | 1.5E+07 | 6.2 | 2.3E–05 |
| LMRG_00395 | lmo0706 | FlgL | Flagellar hook-associated protein | 4.9E+07 | 8.1E+06 | 6.1 | 2.1E–02 |
| LMRG_02773 | lmo1699 | Methyl-accepting chemotaxis protein | 2.3E+08 | 4.0E+07 | 5.9 | 3.3E–03 | |
| LMRG_00394 | lmo0705 | FlgK | Flagellar hook-associated protein | 1.3E+08 | 2.2E+07 | 5.7 | 2.4E–03 |
| LMRG_00380 | lmo0692 | CheA | Signal transduction histidine kinase | 2.4E+08 | 5.0E+07 | 4.7 | 3.5E–04 |
| LMRG_00396 | lmo0707 | FliD | Flagellar hook-associated protein | 7.0E+07 | 1.5E+07 | 4.6 | 1.8E–02 |
| LMRG_00379 | lmo0691 | CheY | Chemotaxis regulator | 2.5E+08 | 6.1E+07 | 4.2 | 3.9E–05 |
| LMRG_00403 | lmo0714 | FliG | Flagellar motor switch protein | 1.2E+08 | 3.1E+07 | 3.8 | 6.5E–04 |
| LMRG_00373 | lmo0685 | MotA | Flagellar motor rotation protein | 5.6E+07 | 1.8E+07 | 3.2 | 7.5E–03 |
| LMRG_00377 | lmo0689 | CheV | Chemotaxis | 4.1E+08 | 1.3E+08 | 3.0 | 1.9E–04 |
| LMRG_00376 | lmo0688 | GmaR | Flagellin glycosyltransferase | 2.3E+08 | 8.4E+07 | 2.8 | 2.7E–04 |
| LMRG_00388 | lmo0699 | FliM | Flagellar motor switch protein | 2.8E+07 | 0 | 6.2E–04 | |
| LMRG_00389 | lmo0700 | CheC | Chemotaxis | 2.7E+07 | 0 | 2.9E–04 | |
We hypothesized that decreased motility contributes to the virulence defect of ΔspxA1 cells in tissue culture infection models, since previous research demonstrated that nonmotile mutants are unable to swim toward host cell monolayers and thus cannot efficiently invade (37, 38). To address this hypothesis, gentamicin protection assays were performed on infected immortalized murine bone marrow-derived macrophages (iBMMs), and the uptake of ΔspxA1 cells was compared to that of wt cells or a ΔflaA mutant that lacks flagella (38). After a 30-min infection with L. monocytogenes, cells were washed and incubated with media containing gentamicin for 30 min to kill extracellular bacteria. Host cells were then lysed and plated to enumerate the CFU, and intracellular bacteria were calculated as a percentage of the total inoculum. The nonmotile ΔflaA strain exhibited an ~3-fold reduction in intracellular bacteria compared to wt L. monocytogenes (Fig. 6A). The ΔspxA1 mutants exhibited a similar ~4-fold reduction in intracellular bacteria, which was fully restored in the ΔspxA1 pPL2.spxA1 strain. To test whether the ΔspxA1 defect was due to impaired motility, centrifugation was applied immediately after infection to force the bacteria to contact the host cell monolayer. After centrifugation, the ΔflaA and ΔspxA1 mutants were taken up by iBMMs with the same efficiency as wt L. monocytogenes (Fig. 6B). These results suggested that the observed defect in phagocytosis of the ΔspxA1 strain can be attributed to decreased motility that limits interactions with the host cells.
FIG 6.

The roles of bacterial motility and morphology in macrophage phagocytosis of the ΔspxA1 mutant. (A) Gentamicin protection assay measuring intracellular bacteria at 1 h postinfection of iBMMs. (B) Gentamicin protection assay after bacteria were centrifuged onto the host cells immediately after infection. The means and SEM of three biological replicates are shown for panels A and B. P values were calculated using an unpaired Student t test compared to wt cells. *, P < 0.05; n.s., P > 0.05. (C) iBMMs were infected with centrifugation for 15 min and stained to enable differentiation of extracellular (E) and intracellular (I) bacteria. The perimeters of cells in each compartment were measured using Celltool. Cell perimeters were measured from two biological replicates where at least 1,000 bacteria or 10 fields of view were quantified. The lines represent the means. Distributions were compared using Kolmogorov-Smirnov tests. *, P < 0.05.
While motility is clearly important for ΔspxA1 cells to access the host cell monolayer, it is also well known that filamentous bacteria and fungi are more resistant to phagocytosis than bacilli (15). To test whether filamentation contributes to decreased host cell uptake of ΔspxA1, we performed quantitative immunofluorescence microscopy of infected iBMMs. L. monocytogenes wt or ΔspxA1 strains constitutively expressing mCherry were added to cells with centrifugation and allowed to infect for 15 min. The cells were then washed, fixed, immunostained with a L. monocytogenes-specific antibody, and labeled with a green fluorophore. Since the iBMMs were not permeabilized prior to immunostaining, extracellular bacteria could be identified by the colocalization of red and green fluorescence, while intracellular bacteria could be differentiated by the absence of green fluorescence. Celltool was then used to measure the cell perimeters of intracellular and extracellular bacterial populations. We observed a significant decrease in the mean perimeter of intracellular ΔspxA1 cells compared to extracellular cells, and all filamentous ΔspxA1 cells with a perimeter of 26 μm or larger remained extracellular (Fig. 6C). Together, these results suggested that macrophage uptake of the ΔspxA1 mutant is impaired due to both decreased motility and impaired phagocytosis of ΔspxA1 filaments.
DISCUSSION
This study set out to define the role of the transcriptional regulator SpxA1 in L. monocytogenes pathogenesis. Surprisingly, we observed that the ΔspxA1 strain exhibits striking morphological heterogeneity during intracellular and extracellular growth, forming single rod-shaped bacteria, chains of cells, and filamentous single cells. Quantitative microscopic analysis of this morphology phenotype in vitro revealed that L. monocytogenes lacking spxA1 primarily form filamentous cells with an average cell perimeter twice that of wt cells. These filamentous cells did not have compromised cell membranes and appeared to result from irregular frequency and localization of division septum formation. Using a whole-cell proteomics approach, we determined that SpxA1 does not influence the abundance of proteins critical to cell division, the SOS response, or the synthesis or modification of cell envelope components. However, proteins important for motility and chemotaxis were significantly depleted in ΔspxA1 cells compared to wt L. monocytogenes and that bacterial motility was necessary for the infection of macrophages in tissue culture. Furthermore, highly filamentous ΔspxA1 cells were more resistant to phagocytosis than their bacillary counterparts, supporting a multifaceted role of SpxA1 in virulence regulation. Together, our results provide novel insight into the function of SpxA1 in L. monocytogenes and its roles in regulating cell division, motility, and virulence.
To examine the morphological heterogeneity of ΔspxA1 cells, we took several complementary microscopy approaches. Phase-contrast images revealed that ~5-fold more ΔspxA1 cells are elongated in early exponential phase than wt cells, but these elongated cells did not exhibit a significant loss in viability or membrane integrity. Membrane staining identified that ΔspxA1 cultures are dominated by filamentous cells, the physiology of which we investigated more closely using STEM. Previous research successfully applied STEM to identify changes in the thickness and composition of peptidoglycan in the L. monocytogenes envelope (39). However, our STEM images suggested that the composition of the ΔspxA1 cell envelope did not differ significantly from wt cells. Instead, we observed notable irregularities in division septum formation in ΔspxA1 filamentous cells. Overall, these microscopic analyses suggested that the altered frequency and localization of division septa are likely responsible for ΔspxA1 filamentation.
To elucidate the mechanism of ΔspxA1 filamentation, quantitative label-free mass spectrometry of whole-cell lysates identified SpxA1-dependent changes in protein abundance. These whole-cell proteomics data validated conclusions from previous transcriptomic results and demonstrated that SpxA1 regulates the abundance of several proteins responsible for peroxide stress resistance, including those involved in heme biosynthesis and catalase (29). In addition, we identified that 48 proteins decreased and 6 proteins increased in ΔspxA1 cells that were similarly changed in transcript abundance (29). While the commonalities shared by these two data sets provide confirmation of the proteomic results, the differences can likely be attributed to the altered conditions under which bacterial cultures were grown. Here, proteomic analyses were performed under strict anaerobiosis, with cultures grown in degassed media in an anaerobic chamber. Furthermore, samples for proteomic analysis were harvested at early exponential phase when the most filamentation was observed for the ΔspxA1 mutant, rather than late exponential phase. Despite these differences, the proteomic and transcriptomic results overlapped considerably.
Results from this global proteomic analysis disproved many of our initial hypotheses about the potential mechanism of ΔspxA1 filamentation. The largest category of proteins influenced by SpxA1 were those involved in redox homeostasis and respiration. However, the SpxA1-regulated redox genes essential for aerobic growth (kat and hemEH) were dispensable for SpxA1 regulation of morphology. Furthermore, the experiments described here were performed in strictly anaerobic conditions, eliminating redox stress as a potential cause of ΔspxA1 filamentation. Our whole-cell proteomics enforced observations from previous transcriptional analysis that SpxA1 plays a distinct role in regulating components of protein turnover. Although the proteins most increased in ΔspxA1 cells compared to wt cells were MecA and ClpE, both related to protein turnover, deleting these proteins did not rescue ΔspxA1 filamentation. In fact, the increased filamentation of ΔspxA1ΔclpE cells compared to ΔspxA1 cells suggests that ClpE overproduction in ΔspxA1 cells may be a direct response to the disrupted stoichiometries of SpxA1-regulated factors. Our data therefore support a novel model for SpxA1 influence over morphology that is independent of the other previously described roles for this protein.
Whole-cell proteomics results excluded the influence of several canonical filamentation pathways on ΔspxA1 morphology, including the SOS response, alterations to cell division machinery, and cell envelope modifications. Filamentation of bacteria is often associated with induction of the SOS response to DNA damage (15), but key SOS response proteins were unchanged in the ΔspxA1 mutant. The production and modification of cell envelope components can also influence both morphology and pathogenesis in L. monocytogenes (40–42). However, proteins required for biosynthesis and modification of teichoic acids and peptidoglycan were not altered in the ΔspxA1 mutant. Given the septation defects of the ΔspxA1 mutant, it was most surprising that proteins important for cell division were unchanged in ΔspxA1 cells compared to wt cells. The Bacillus subtilis homologue Spx directly regulates the divisome component ZapA, which interacts with the cell division Z-ring protein FtsZ to promote septum formation (43). In L. monocytogenes, neither FtsZ nor ZapA were changed in abundance in the ΔspxA1 mutant compared to wt cells. Overall, we found that ΔspxA1 filamentation was occurring independently of the redox response, SOS response, cell division, and cell envelope biosynthesis. Current research is aimed at elucidating the SpxA1-dependent mechanism of filamentation.
Our data suggest that a combination of motility and morphology defects contribute to reduced phagocytosis of the ΔspxA1 mutant. Whole-cell proteomics revealed a significant depletion of proteins involved in motility and chemotaxis in ΔspxA1 cells compared to wt cells. Indeed, we observed that when incubated at 37°C, the ΔspxA1 mutant exhibited the same lack of motility as a mutant unable to produce flagella. We further demonstrate that flagellar motility is important for L. monocytogenes to interact with the host monolayer during tissue culture models of infection, since forcing ΔspxA1 to contact host cells via centrifugation rescued the defect in uptake by macrophages. Immunofluorescence microscopy of the L. monocytogenes-host cell surface interaction revealed that the longest filaments were exclusively extracellular. Together, these results demonstrated roles for both motility and bacterial cell size in macrophage phagocytosis.
It is not yet clear which SpxA1-dependent factors are necessary during murine infection, during which the ΔspxA1 mutant is attenuated >500-fold in the spleen and 5 logs in the liver (23). Flagellar motility is important for adhesion and invasion of host cells in vitro but dispensable for infection in both oral and intravenous models of murine infection (37, 44). While impaired phagocytosis of filamentous ΔspxA1 cells may contribute to its virulence defect, entry into host cells is not the only barrier to virulence of the ΔspxA1 mutant. Once in the host cytosol, the ΔspxA1 mutant has a doubling time of 109 min compared to 47 min for wt L. monocytogenes and is severely defective for intercellular spread (23). The rod shape of L. monocytogenes is required in the host cytosol for establishing the asymmetric actin cloud necessary for canonical comet tail formation and motility (6, 7). Thus, the elongated shape and improper actin polarization observed for ΔspxA1 filaments may limit cytosolic motility and intercellular spread, which would significantly impair host colonization (3, 45, 46). The impact of disorganized septum formation and filamentation on cytosolic growth, intercellular spread, and systemic infection is an area of ongoing investigation.
This study aimed to identify SpxA1-dependent factors important for L. monocytogenes pathogenesis. We discovered that ΔspxA1 cells form elongated filaments that are impaired for motility and resistant to phagocytosis. While the mechanism of filamentation is not yet known, it is intriguing that the SpxA1-regulated genes necessary for aerobic growth in vitro are dispensable during infection and are not involved in cell size regulation or motility. SpxA1-dependent regulation thus independently impacts many areas of L. monocytogenes physiology. We predict that the complexity of SpxA1 regulation likely results from selective pressure to respond to a variety of signals in diverse environments. Future research will elucidate the cues that influence SpxA1 activity and the genes it regulates during both extracellular growth and pathogenesis.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used are listed in Table S4. All L. monocytogenes strains were derived from the 10403S background and cultured in degassed BHI at 37°C in an anaerobic workstation (Don Whitley Scientific), unless otherwise described. All chemicals were purchased from Sigma-Aldrich unless otherwise stated. The following concentrations of antibiotics were used: streptomycin, 200 μg/mL; chloramphenicol, 10 μg/mL (E. coli) or 5 μg/mL (L. monocytogenes); tetracycline, 2 μg/mL; and carbenicillin, 100 μg/mL.
Growth curves were carried out in an anaerobic chamber (10% carbon dioxide, 10% hydrogen, balanced with nitrogen) at 37°C in degassed BHI media. First, colonies were inoculated into broth and grown overnight. Fresh BHI was inoculated with overnight cultures to an optical density at 600 nm (OD600) of 0.02 in 25 mL in 100-mL flasks and grown statically for 8 h. Every 2 h, cultures were serially diluted with sterile degassed PBS and plated on BHI-streptomycin agar to enumerate CFU.
For coculture experiments, wt constitutively expressing GFP was mixed with ΔspxA1 cells constitutively expressing mCherry at 1:1 or 2:1 wt/ΔspxA1 ratios in fresh degassed BHI. Cultures were grown to early exponential phase, incubated aerobically for 10 min to activate fluorophores, and spotted onto PBS-agar pads for visualization in phase-contrast or fluorescence channels using a 40× high-resolution objective.
Strain construction.
Generally, plasmids were introduced into chemically competent E. coli strains via transformation. Using E. coli SM10 (47), plasmids were transferred into L. monocytogenes by conjugation. Alternatively, transducing lysates were generated from L. monocytogenes containing desired constructs and used to infect recipient strains. Preparation of lysates was accomplished by mixing a donor strain with U153 phage, followed by incubation overnight in soft agar at 30°C, as described previously (48). Phage were subsequently eluted from agar and filter sterilized, and the resulting lysate was mixed with recipient L. monocytogenes for 30 min at room temperature. Transductants were then selected on antibiotic-containing agar at 37°C.
In-frame deletions were constructed using PheS* counterselection of the conjugation-proficient suicide vector pLIM1 (a gift from Arne Rietsch, Case Western Reserve University). First, upstream and downstream regions of the gene of interest were amplified, fused via SOE-PCR, restriction digested, and ligated into pLIM1. The sequences of all pLIM1-derived vectors were confirmed by Sanger DNA sequencing. Deletion vectors were conjugated into recipient L. monocytogenes and integrated into the chromosome. After antibiotic selection, integrants were cured of pLIM1, as previously described (49). Allelic exchange was confirmed by PCR, antibiotic sensitivity, and sensitivity to oxygen. The wt and ΔspxA1 genomes were fully sequenced, and it was determined that these strains differed only at the spxA1 locus.
Genes were either natively expressed or overexpressed from an ectopic locus using the integrative plasmids pPL1 and pPL2 (31). For complementation, an insert containing a gene and corresponding promoter was PCR amplified, restriction digested, and ligated into the appropriate vector. Constitutive expression of fluorophores was accomplished by incorporating the desired gene downstream of the pHyper promoter in pPL2, as previously described (48).
Tissue culture.
L2 murine fibroblasts were generated from L929 cells described previously (22). iBMMs were a generous gift from Joshua Woodward (50). Cell lines were grown at 37°C and 5% CO2 in high-glucose Dulbecco modified Eagle medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM sodium pyruvate, and 1 mM l-glutamine. During passaging, 100 U/mL of Pen-Strep was added to cell culture media. The day before infection, cells were plated in antibiotic-free media.
Immunofluorescence microscopy.
To visualize intracellular bacteria, L2 fibroblasts were seeded at 1.5 × 106 cells per well in 6-well tissue culture-treated plates containing collagen-coated glass coverslips (Thermo Fisher Scientific) and inoculated with L. monocytogenes the next day at a multiplicity of infection (MOI) of 50. Bacterial cultures were grown overnight in anaerobic BHI at 37°C and washed twice in PBS prior to inoculation. At 1 h postinfection, monolayers were washed twice with PBS, and medium containing 30 μg/mL gentamicin was added to kill extracellular bacteria. At 10 h postinfection, coverslips were washed twice with PBS, fixed for 10 min in 4% formaldehyde (Pierce), washed in TBS-Tx (0.1% Triton, 50 mM Tris [pH 7.6], 150 mM NaCl), and incubated in antibody buffer (TBS-Tx, 1% bovine serum albumin [BSA]) at 4°C overnight. Coverslips were then incubated for 30 min with rabbit anti-Listeria O antigen antiserum (Difco) at a concentration of 1:100 in antibody buffer and washed in TBS-Tx, followed by a secondary incubation with a 1:200 solution of goat anti-rabbit-Alexa488 (Life Tech) in antibody buffer and Alexa555-phalloidin (Life Tech) at a concentration of 1:1,000. After immunolabeling and washing in TBS-Tx a final time, coverslips were attached to glass slides with ProLong Anti-fade Diamond with DAPI (4′,6′-diamidino-2-phenylindole; Life Tech), left to cure at room temperature overnight, and imaged with a 100× objective.
For fluorescence microscopy of extracellular bacteria, overnight cultures of selected strains were diluted in BHI to an OD600 of 0.02 and grown for 2 h to early exponential phase (OD600 ~0.2) in an anaerobic chamber at 37°C. Prior to staining, 1 mL of culture was pelleted at 10,000 × g for 1 min in a tabletop centrifuge and resuspended in PBS, which halts further bacterial growth and cell death (23). To label cell membranes, cultures were incubated with 20 μg/mL membrane intercalating agent TMA-DPH for 10 min in the dark and then washed twice in PBS prior to spotting on PBS-agar pads. To visualize membrane integrity and viability of bacterial cells, cultures were mixed with a 5-μg/mL propidium iodide solution, incubated in the dark for 15 min, and then spotted immediately onto agar pads.
All imaging was performed using a Keyence BZ-X710 All-in-One fluorescence microscope with either a 40× objective or a 100× oil immersion objective and corresponding filter cubes (Keyence). Phase-contrast images were taken using bright-field settings. Propidium iodide and mCherry were detected using BZ-X Filter Texas Red. TMA-DPH was detected using BZ-X Filter DAPI. GFP and Alexa488 were detected using BZ-X Filter GFP.
Quantitative analysis of bacterial morphology using Celltool.
Phase contrast and fluorescence microscopy was performed on L. monocytogenes grown anaerobically shaking in BHI and then resuspended in PBS. Culture resuspensions were immediately spotted on PBS–2% agarose pads, sealed under a glass coverslip, and imaged. All quantified images were taken using a 40× objective. Fluorescence images were generated in the appropriate channel for each fluorescent label as specified per experiment. At least 10 fields of view were imaged such that at least 1,000 cells could be analyzed per biological replicate per sample. Binary masks were uniformly applied to each field of view using the MATLAB imbinarize function. Quantitative analysis of binarized images was performed using the Celltool software package (30). Image contours were extracted using the extract_contours functions and smoothed via interpolation to polygons with 100 evenly placed vertices. The fluorescence intensities of labeled bacteria were measured using the extract_images function which specifically extracts intensity per area of fluorescence microscopy images based on contours previously generated from phase-contrast images. The measure_contours function was used for the final generation of data output. Perimeter length of contours was used as the primary metric for analysis in order to account for curved and irregular morphologies. Unpaired t tests were used to measure the differences in perimeter length distributions between populations of bacteria. Percent of elongated cells was calculated by measuring the number of cell perimeters in a population which were >1 standard deviation (SD) above the wt mean for each corresponding replicate and time point. Means and standard errors of the mean (SEM) are displayed for three biological replicates of percent elongation analysis.
Scanning transmission electron microscopy.
The appropriate strains were grown to early exponential phase anaerobically in 400 mL of BHI. Cultures were centrifuged for 10 min at 4,500 rpm, and pellets were washed once in 1 M PBS. Pellets (~100 μL) were mixed with equal volumes of fixative buffer consisting of 4% mass spectrometry-grade paraformaldehyde and 0.1 M sodium cacodylate. After 30 min, cells were pelleted and mixed with equal volumes fresh fixative buffer for 1 h at room temperature. Samples were then rinsed with 0.1 M sodium cacodylate buffer, postfixed in osmium tetroxide buffered with 0.1 M sodium cacodylate for 2 h, and then washed to remove osmium tetroxide. Samples were then progressively dehydrated with increasing concentrations of ethanol (50, 70, 90, and 100%) and twice with 100% acetone. Bacteria were then embedded in 50:50 acetone-resin (Epon) and incubated rotating for 4 h. Incubation was next performed using 20:80 acetone-resin. After drying for 2 h, the samples were placed in a 100% resin gelatin capsule under light vacuum for 2 h. Finally, samples were placed in fresh resin molds and incubated at 60°C overnight. Resin blocks were sectioned at 80 nm using a Leica Ultracut 6 Microtome and stained using uranyl acetate and Reynolds lead citrate stains. Samples were imaged with an FEI Tecnai G2 F20 Twin transmission electron microscope at 200 kV.
Whole-cell proteomics.
Protein isolation was performed as previously described (34). L. monocytogenes were grown anaerobically in BHI for 2 h. Bacteria were pelleted, washed once in PBS, and immediately flash-frozen. Bacterial pellets were resuspended in lysis buffer containing 8 M urea and lysed by sonification. Proteins in lysates were reduced via incubation with dithiothreitol and then alkylated via incubation with iodoacetamide. After quantification of protein concentration via BCA, 300 μg of protein per sample were treated with Trypsin Gold (Promega) overnight. The pH was adjusted to pH 2 using trifluoroacetic acid, and then proteins were purified using C18-300 peptide purification columns per manufacturer instructions (Nest Group). After elution, the peptides were dried and resuspended in 0.1% formic acid. Autosampler vials were loaded with 100-μL samples at a concentration of 0.5 μg/mL. Mass spectrometry was performed on each sample using an Orbitrap Eclipse mass spectrometer (Thermo Scientific) and liquid chromatography was performed using the Easy-nLC 1000 liquid chromatograph (Thermo Scientific). Raw spectral data were processed using the MaxQuant suite of software with default settings, and peptide sequences were searched against the L. monocytogenes 10403S reference proteome (UniProt).
The relative abundance of proteins was determined using the label-free quantification (LFQ) intensity metric generated by analysis with the MaxLFQ algorithm, a generic label-free quantification technology available in MaxQuant (51, 52). The LFQ intensity metric was chosen since it does not depend on normalization with housekeeping proteins for accurate abundance prediction sample to sample, relying instead on the assumption that most proteins are minimally changed between experimental conditions. Furthermore, LFQ intensities are the result of calculations using the maximum possible information extracted from samples and retain the absolute scale of original peptide intensities, giving a highly accurate proxy for absolute protein abundance (51). Proteins changed in abundance between wt and ΔspxA1 strains were considered for further analysis if they met the following criteria: (i) the proteins differed in average abundance between wt and ΔspxA1 cells by at least 2-fold; (ii) an unpaired t test comparing the LFQ intensities of three wt and ΔspxA1 biological replicates yielded a P value of <0.05; and (iii) to increase the stringency of our hit identification and reduce possible false positives, protein abundance calculations were based on the identification of at least five peptides per protein in the sample with higher abundance.
Measuring phagocytosis via gentamicin protection assay and microscopy.
Gentamicin protection assays were performed as previously described (49). Immortalized murine bone marrow-derived macrophages (iBMMs) were seeded in 24-well plates at a density of 6 × 105 cells per well. The next day, overnight anaerobic cultures were washed twice with sterile PBS. Macrophages were infected at an MOI of 10, and the inocula were serially diluted and plated on BHI-strep agar to enumerate the CFU. To force bacteria to the cell surface, select six-well plates were then centrifuged at 300 × g for 2 min after inoculation. After 30 min, monolayers were washed twice in sterile PBS. New media with 30 μg/mL gentamicin was added for 30 min to eliminate any extracellular bacteria, and then the monolayers were washed twice and lysed in 250 μL of cold 0.1% Triton X-100 in PBS. Lysates were serially diluted and plated on BHI-strep agar to enumerate the CFU. The percent uptake was calculated by dividing the number of bacteria recovered from each well by the starting inoculum CFU.
To image uptake of bacteria, iBMMs were seeded in 24-well plates containing ethanol-sterilized glass coverslips at a density of 6 × 105 cells per well. Overnight anaerobic cultures were washed twice with sterile PBS. Macrophages were infected with mCherry-expressing bacteria at an MOI of 10 and centrifuged for 2 min at 300 rpm to normalize strain contact with the monolayer. Infections were incubated for 15 min and then washed twice in sterile PBS. Coverslips were removed and fixed for 10 min in 4% formaldehyde (Pierce), washed in PBS, and blocked in PBS with 1% BSA at 4°C overnight. PBS replaced TBST-Tx for all buffers used to prevent the permeabilization and immunolabel infiltration of host cells. Thus, only extracellular bacteria were labeled. Coverslips where then incubated successively with the described immunostaining agents diluted in PBS with 1% BSA, with PBS washes between incubations, as follows: 1:20 Fc Block (BD), 1:100 rabbit anti-Listeria O antigen antiserum (Difco), and 1:200 goat anti-rabbit-Alexa488 (Life Tech). After a washing step with PBS a final time, coverslips were attached to glass slides with ProLong Anti-fade Diamond (Life Tech), left to cure at room temperature overnight, and imaged with a 100× objective. Celltool was used to extract contours from mCherry images (30). Cells were determined to be extracellular based on the intensity of fluorescence extracted from GFP images within the same area as the Celltool-derived contours.
Motility assay.
Degassed motility plates consisting of BHI and 0.4% agar were poured fresh within a week of use and inoculated via stab with pipette tip dipped in a bacterial colony. Plates were then incubated anaerobically at 37°C or room temperature for 4 days. After incubation, plates were scanned, and the diameter of the radial bacterial spread was measured using ImageJ software.
ACKNOWLEDGMENTS
We thank members of the Woodward (University of Washington) and Reniere labs for helpful discussions and critical reading of the manuscript. We also thank the lab of Joseph Mougous (University of Washington) for access to reagents and equipment and, specifically, Yaxi Wang and Andi Liu for assistance with the processing of whole-cell proteomics data. STEM imaging was conducted at the Molecular Analysis Facility (MAF) with the help of Ellen Lavoie.
The MAF is supported in part by funds from the Molecular Engineering and Sciences Institute, the Clean Energy Institute, and the National Science Foundation (NNCI-2025489 and NNCI-1542101). Research in the Reniere lab is funded by NIH R01 A132356. M.R.C. was funded by the Howard Hughes Medical Institute through the James H. Gilliam Fellowship for Advanced Study program (GT11030). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material is available online only.
Contributor Information
Michelle L. Reniere, Email: reniere@uw.edu.
Victor J. Torres, New York University School of Medicine
REFERENCES
- 1.Gray MJ, Freitag NE, Boor KJ. 2006. How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect Immun 74:2505–2512. 10.1128/IAI.74.5.2505-2512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ireton K. 2007. Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell Microbiol 9:1365–1375. 10.1111/j.1462-5822.2007.00933.x. [DOI] [PubMed] [Google Scholar]
- 3.Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109:1597–1608. 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pillich H, Puri M, Chakraborty T. 2017. ActA of Listeria monocytogenes and its manifold activities as an important listerial virulence factor, p 113–132. In Mannherz HG (ed), The actin cytoskeleton and bacterial infection. Springer International Publishing, Cham, Switzerland. [DOI] [PubMed] [Google Scholar]
- 5.Rafelski SM, Theriot JA. 2006. Mechanism of polarization of Listeria monocytogenes surface protein ActA. Mol Microbiol 59:1262–1279. 10.1111/j.1365-2958.2006.05025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernheim-Groswasser A, Wiesner S, Golsteyn RM, Carlier M-F, Sykes C. 2002. The dynamics of actin-based motility depend on surface parameters. Nature 417:308–311. 10.1038/417308a. [DOI] [PubMed] [Google Scholar]
- 7.Cameron LA, Robbins JR, Footer MJ, Theriot JA. 2004. Biophysical parameters influence actin-based movement, trajectory, and initiation in a cell-free system. Mol Biol Cell 15:2312–2323. 10.1091/mbc.e03-12-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frirdich E, Biboy J, Adams C, Lee J, Ellermeier J, Gielda LD, DiRita VJ, Girardin SE, Vollmer W, Gaynor EC. 2012. Peptidoglycan-modifying enzyme Pgp1 is required for helical cell shape and pathogenicity traits in Campylobacter jejuni. PLoS Pathog 8:e1002602. 10.1371/journal.ppat.1002602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sycuro LK, Pincus Z, Gutierrez KD, Biboy J, Stern CA, Vollmer W, Salama NR. 2010. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell 141:822–833. 10.1016/j.cell.2010.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor JA, Bratton BP, Sichel SR, Blair KM, Jacobs HM, DeMeester KE, Kuru E, Gray J, Biboy J, VanNieuwenhze MS, Vollmer W, Grimes CL, Shaevitz JW, Salama NR. 2020. Distinct cytoskeletal proteins define zones of enhanced cell wall synthesis in Helicobacter pylori. Elife 9:e52482. 10.7554/eLife.52482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartlett TM, Bratton BP, Duvshani A, Miguel A, Sheng Y, Martin NR, Nguyen JP, Persat A, Desmarais SM, VanNieuwenhze MS, Huang KC, Zhu J, Shaevitz JW, Gitai Z. 2017. A periplasmic polymer curves Vibrio cholerae and promotes pathogenesis. Cell 168:172–185. 10.1016/j.cell.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn M, Goebel W. 1989. Identification of an extracellular protein of Listeria monocytogenes possibly involved in intracellular uptake by mammalian cells. Infect Immun 57:55–61. 10.1128/iai.57.1.55-61.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonzo F, McMullen PD, Freitag NE. 2011. Actin polymerization drives septation of Listeria monocytogenes namA hydrolase mutants, demonstrating host correction of a bacterial defect. Infect Immun 79:1458–1470. 10.1128/IAI.01140-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegrist MS, Aditham AK, Espaillat A, Cameron TA, Whiteside SA, Cava F, Portnoy DA, Bertozzi CR. 2015. Host actin polymerization tunes the cell division cycle of an intracellular pathogen. Cell Rep 11:499–507. 10.1016/j.celrep.2015.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Justice SS, Hunstad DA, Cegelski L, Hultgren SJ. 2008. Morphological plasticity as a bacterial survival strategy. Nat Rev Microbiol 6:162–168. 10.1038/nrmicro1820. [DOI] [PubMed] [Google Scholar]
- 16.Yang DC, Blair KM, Salama NR. 2016. Staying in shape: the impact of cell shape on bacterial survival in diverse environments. Microbiol Mol Biol Rev 80:187–203. 10.1128/MMBR.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isom LL, Khambatta ZS, Moluf JL, Akers DF, Martin SE. 1995. Filament formation in Listeria monocytogenes. J Food Prot 58:1031–1033. 10.4315/0362-028X-58.9.1031. [DOI] [PubMed] [Google Scholar]
- 18.Vail KM, McMullen LM, Jones TH. 2012. Growth and filamentation of cold-adapted, log-phase Listeria monocytogenes exposed to salt, acid, or alkali stress at 3°C. J Food Prot 75:2142–2150. 10.4315/0362-028X.JFP-12-199. [DOI] [PubMed] [Google Scholar]
- 19.Rowan NJ, Anderson JG. 1998. Effects of above-optimum growth temperature and cell morphology on thermotolerance of Listeria monocytogenes cells suspended in bovine milk. Appl Environ Microbiol 64:2065–2071. 10.1128/AEM.64.6.2065-2071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruhland BR, Reniere ML. 2019. Sense and sensor ability: redox-responsive regulators in Listeria monocytogenes. Curr Opin Microbiol 47:20–25. 10.1016/j.mib.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuber P. 2004. Spx-RNA polymerase interaction and global transcriptional control during oxidative stress. J Bacteriol 186:1911–1918. 10.1128/JB.186.7.1911-1918.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reniere ML, Whiteley AT, Portnoy DA. 2016. An in vivo selection identifies Listeria monocytogenes genes required to sense the intracellular environment and activate virulence factor expression. PLoS Pathog 12:e1005741. 10.1371/journal.ppat.1005741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiteley AT, Ruhland BR, Edrozo MB, Reniere ML. 2017. A redox-responsive transcription factor is critical for pathogenesis and aerobic growth of Listeria monocytogenes. Infect Immun 85:e00978-16. 10.1128/IAI.00978-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villanueva M, Jousselin A, Baek KT, Prados J, Andrey DO, Renzoni A, Ingmer H, Frees D, Kelley WL. 2016. Rifampin resistance rpoB alleles or multicopy thioredoxin/thioredoxin reductase suppresses the lethality of disruption of the global stress regulator spx in Staphylococcus aureus. J Bacteriol 198:2719–2731. 10.1128/JB.00261-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kajfasz JK, Rivera-Ramos I, Abranches J, Martinez AR, Rosalen PL, Derr AM, Quivey RG, Lemos JA. 2010. Two Spx proteins modulate stress tolerance, survival, and virulence in Streptococcus mutans. J Bacteriol 192:2546–2556. 10.1128/JB.00028-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kajfasz JK, Mendoza JE, Gaca AO, Miller JH, Koselny KA, Giambiagi-Demarval M, Wellington M, Abranches J, Lemos JA. 2012. The Spx regulator modulates stress responses and virulence in Enterococcus faecalis. Infect Immun 80:2265–2275. 10.1128/IAI.00026-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Ge X, Wang X, Patel JR, Xu P. 2012. SpxA1 involved in hydrogen peroxide production, stress tolerance and endocarditis virulence in Streptococcus sanguinis. PLoS One 7:e40034. 10.1371/journal.pone.0040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Port GC, Cusumano ZT, Tumminello PR, Caparon MG. 2017. SpxA1 and SpxA2 act coordinately to fine-tune stress responses and virulence in Streptococcus pyogenes. mBio 8:e00288-17. 10.1128/mBio.00288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cesinger MR, Thomason MK, Edrozo MB, Halsey CR, Reniere ML. 2020. Listeria monocytogenes SpxA1 is a global regulator required to activate genes encoding catalase and heme biosynthesis enzymes for aerobic growth. Mol Microbiol 114:230–243. 10.1111/mmi.14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pincus Z, Theriot JA. 2007. Comparison of quantitative methods for cell-shape analysis. J Microsc 227:140–156. 10.1111/j.1365-2818.2007.01799.x. [DOI] [PubMed] [Google Scholar]
- 31.Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol 184:4177–4186. 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prendergast FG, Haugland RP, Callahan PJ. 1981. 1-[4-(Trimethylamino)phenyl]-6-phenylhexa-1,3,5-triene: synthesis, fluorescence properties, and use as a fluorescence probe of lipid bilayers. Biochemistry 20:7333–7338. 10.1021/bi00529a002. [DOI] [PubMed] [Google Scholar]
- 33.Terzakis JA. 1968. Uranyl acetate, a stain and a fixative. J Ultrastruct Res 22:168–184. 10.1016/S0022-5320(68)90055-5. [DOI] [PubMed] [Google Scholar]
- 34.Ruhland BR, Reniere ML. 2020. YjbH requires its thioredoxin active motif for the nitrosative stress response, cell-to-cell spread, and protein-protein interactions in Listeria monocytogenes. J Bacteriol 202:e00099-20. 10.1128/JB.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawai Y, Moriya S, Ogasawara N. 2003. Identification of a protein, YneA, responsible for cell division suppression during the SOS response in Bacillus subtilis. Mol Microbiol 47:1113–1122. 10.1046/j.1365-2958.2003.03360.x. [DOI] [PubMed] [Google Scholar]
- 36.Gründling A, Burrack LS, Bouwer HGA, Higgins DE. 2004. Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. Proc Natl Acad Sci USA 101:12318–12323. 10.1073/pnas.0404924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bigot A, Pagniez H, Botton E, Fréhel C, Dubail I, Jacquet C, Charbit A, Raynaud C. 2005. Role of FliF and FliI of Listeria monocytogenes in flagellar assembly and pathogenicity. Infect Immun 73:5530–5539. 10.1128/IAI.73.9.5530-5539.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Neil HS, Marquis H. 2006. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect Immun 74:6675–6681. 10.1128/IAI.00886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan Zeevi M, Shafir NS, Shaham S, Friedman S, Sigal N, Nir Paz R, Boneca IG, Herskovits AA. 2013. Listeria monocytogenes multidrug resistance transporters and cyclic di-AMP, which contribute to type I interferon induction, play a role in cell wall stress. J Bacteriol 195:5250–5261. 10.1128/JB.00794-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abachin E, Poyart C, Pellegrini E, Milohanic E, Fiedler F, Berche P, Trieu-Cuot P. 2002. Formation of d-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol Microbiol 43:1–14. 10.1046/j.1365-2958.2002.02723.x. [DOI] [PubMed] [Google Scholar]
- 41.Rismondo J, Schulz LM, Yacoub M, Wadhawan A, Hoppert M, Dionne MS, Gründling A. 2021. EslB is required for cell wall biosynthesis and modification in Listeria monocytogenes. J Bacteriol 203:e00553-20. 10.1128/JB.00553-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webb AJ, Karatsa-Dodgson M, Gründling A. 2009. Two-enzyme systems for glycolipid and polyglycerolphosphate lipoteichoic acid synthesis in Listeria monocytogenes. Mol Microbiol 74:299–314. 10.1111/j.1365-2958.2009.06829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Y, Dempwolff F, Oshiro RT, Gueiros-Filho FJ, Jacobson SC, Kearns DB. 2021. The division defect of a Bacillus subtilis minD noc double mutant can be suppressed by Spx-dependent and Spx-independent mechanisms. J Bacteriol 203:e00249-21. 10.1128/JB.00249-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Way SS, Thompson LJ, Lopes JE, Hajjar AM, Kollmann TR, Freitag NE, Wilson CB. 2004. Characterization of flagellin expression and its role in Listeria monocytogenes infection and immunity. Cell Microbiol 6:235–242. 10.1046/j.1462-5822.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- 45.Smith GA, Portnoy DA, Theriot JA. 1995. Asymmetric distribution of the Listeria monocytogenes ActA protein is required and sufficient to direct actin-based motility. Mol Microbiol 17:945–951. 10.1111/j.1365-2958.1995.mmi_17050945.x. [DOI] [PubMed] [Google Scholar]
- 46.Rafelski SM, Theriot JA. 2005. Bacterial shape and ActA distribution affect initiation of Listeria monocytogenes actin-based motility. Biophys J 89:2146–2158. 10.1529/biophysj.105.061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat Biotechnol 1:784–791. 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 48.Cesinger MR, Schwardt NH, Halsey CR, Thomason MK, Reniere ML. 2021. Investigating the roles of Listeria monocytogenes peroxidases in growth and virulence. Microbiol Spectr 9:e0044021. 10.1128/Spectrum.00440-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halsey CR, Glover RC, Thomason MK, Reniere ML. 2021. The redox-responsive transcriptional regulator Rex represses fermentative metabolism and is required for Listeria monocytogenes pathogenesis. PLoS Pathog 17:e1009379. 10.1371/journal.ppat.1009379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McFarland AP, Burke TP, Carletti AA, Glover RC, Tabakh H, Welch MD, Woodward JJ. 2018. RECON-dependent inflammation in hepatocytes enhances Listeria monocytogenes cell-to-cell spread. mBio 9:e00526-18. 10.1128/mBio.00526-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13:2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyanova S, Temu T, Cox J. 2016. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4 and Tables S1 to S4. Download iai.00211-22-s0001.pdf, PDF file, 4.2 MB (4.2MB, pdf)