

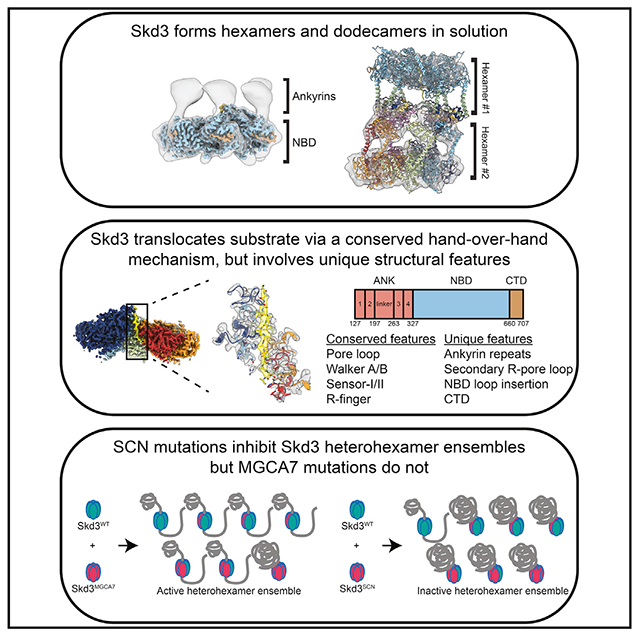
SUMMARY
The AAA+ protein, Skd3 (human CLPB), solubilizes proteins in the mitochondrial intermembrane space, which is critical for human health. Skd3 variants with defective protein-disaggregase activity cause severe congenital neutropenia (SCN) and 3-methylglutaconic aciduria type 7 (MGCA7). How Skd3 disaggregates proteins remains poorly understood. Here, we report a high-resolution structure of a Skd3-substrate complex. Skd3 adopts a spiral hexameric arrangement that engages substrate via pore-loop interactions in the nucleotide-binding domain (NBD). Substrate-bound Skd3 hexamers stack head-to-head via unique, adaptable ankyrin-repeat domain (ANK)-mediated interactions to form dodecamers. Deleting the ANK linker region reduces dodecamerization and disaggregase activity. We elucidate apomorphic features of the Skd3 NBD and C-terminal domain that regulate disaggregase activity. We also define how Skd3 subunits collaborate to disaggregate proteins. Importantly, SCN-linked subunits sharply inhibit disaggregase activity, whereas MGCA7-linked subunits do not. These advances illuminate Skd3 structure and mechanism, explain SCN and MGCA7 inheritance patterns, and suggest therapeutic strategies.
Graphical abstract
In brief
Cupo et al. reveal the structure and mechanism of Skd3, a protein disaggregase found in mitochondria, which is critical for human health. These advances explain the inheritance patterns and suggest therapeutic strategies for debilitating diseases caused by mutations in Skd3.
INTRODUCTION
Protein aggregation and aberrant phase transitions can be deleterious. Thus, specialized protein disaggregases have evolved to safely reverse protein aggregation and restore resolubilized proteins to native form and function (Fare and Shorter, 2021). These include ATP-independent systems, such as DAXX, TRIMs, and nuclear-import receptors, as well as ATP-dependent systems, including specific AAA+ (ATPases associated with diverse cellular activities) proteins, such as Hsp104 and Skd3 (human CLPB)(Cupo and Shorter, 2020b; Fare and Shorter, 2021; Huang et al., 2021; Zhu et al., 2020).
AAA+ proteins couple ATP hydrolysis to mechanical work to drive energetically challenging tasks, including protein disaggregation (Puchades et al., 2020). Thus, Hsp104, a hexameric, double AAA+ ring disaggregase found in all non-metazoan eukaryotes, disassembles stable cross-β structures, disordered aggregates, toxic oligomers, and heat-induced condensates (DeSantis et al., 2012; Shorter and Southworth, 2019; Yoo et al., 2022). To disaggregate proteins, Hsp104 translocates polypeptides into its central channel using tyrosine-bearing pore loops that grip the substrate (Shorter and Southworth, 2019). Curiously, despite conferring neuroprotection when expressed in animals (Cushman-Nick et al., 2013; Lo Bianco et al., 2008), Hsp104 was lost during the transition from protozoa to metazoa, as was its mitochondrial counterpart, Hsp78 (Erives and Fassler, 2015). However, humans express Skd3 (human CLPB), a single AAA+ ring disaggregase found in the mitochondrial intermembrane space (IMS) (Antonicka et al., 2020; Botham et al., 2019; Hung et al., 2014; Rath et al., 2021; Rhee et al., 2013; Thevarajan et al., 2020), which first appears in evolution alongside Hsp104 and Hsp78 in the closest extant protozoan relatives of animals (Erives and Fassler, 2015). Skd3 is related to Hsp104 and Hsp78 via its HCLR clade AAA+ domain, but otherwise shares limited homology (Cupo and Shorter, 2020b; Erives and Fassler, 2015; Erzberger and Berger, 2006; Perier et al., 1995).
Skd3 maintains protein solubility in the IMS to ensure mitochondrial functionality (Chen et al., 2019; Cupo and Shorter, 2020b; Warren et al., 2022). Skd3 exhibits potent protein-disaggregase activity and is critical for human health (Cupo and Shorter, 2020b; Warren et al., 2022). Autosomal dominant mutations in Skd3 that impair disaggregase activity cause severe congenital neutropenia (SCN) (Warren et al., 2022). SCN is a rare bone marrow failure syndrome that presents with impaired neutrophil maturation (Skokowa et al., 2017). Due to low neutrophil counts, SCN patients are prone to life-threatening infections, myelodysplastic syndromes, and acute myeloid leukemia (Skokowa et al., 2017). By contrast, autosomal recessive or distinct biallelic mutations in Skd3 that impair disaggregase activity underlie 3-methylglutaconic aciduria type 7 (MGCA7) (Cupo and Shorter, 2020b; Kanabus et al., 2015; Kiykim et al., 2016; Pronicka et al., 2017; Saunders et al., 2015; Wortmann et al., 2015, 2021). MGCA7 presents with elevated levels of 3-methylglutaconic acid, neurologic deterioration, and neutropenia (Wortmann et al., 2015). In severe cases, patients present with infantile onset of progressive encephalopathy with movement abnormalities and delayed psychomotor development, which can be accompanied by cataracts, seizures, recurrent infections, and death a few weeks after birth (Wortmann et al., 2015). There are no effective therapeutics for severe MGCA7.
Despite the importance of Skd3 for human health, little is known about Skd3 mechanism or structure (Cupo and Shorter, 2020b). Skd3 harbors an N-terminal mitochondrial targeting signal, which is cleaved by mitochondrial processing peptidase upon import into mitochondria (Wortmann et al., 2015). Skd3 then has an autoinhibitory hydrophobic peptide, which is removed by PARL, a rhomboid protease in the mitochondrial inner membrane (Saita et al., 2017; Spinazzi et al., 2019). Removal of this peptide increases Skd3 disaggregase activity by more than 10-fold (Cupo and Shorter, 2020b). Thus, Skd3 is only fully activated upon reaching its final destination in the IMS. After these processing events, the mature form of Skd3 contains an ankyrin-repeat domain (ANK), a nucleotide-binding domain (NBD) from the HCLR clade of the AAA+ family, and a short C-terminal domain (CTD) (Figure 1A).
Figure 1. Structure of PARLSkd3.
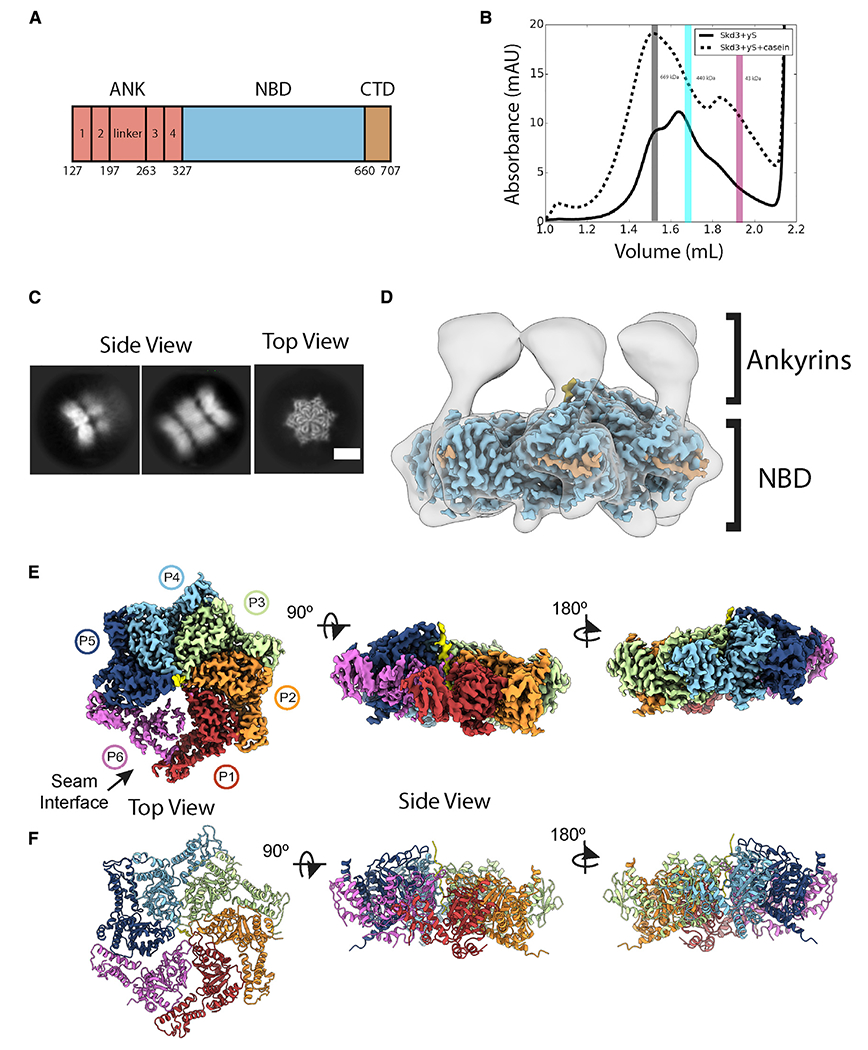
(A) PARLSkd3 domain architecture.
(B) SEC of PARLSkd3 incubated with ATPγS (solid) or ATPγS and FITC-casein (dashed). Vertical bars indicate molecular weight standards: thyroglobulin (669 kDa), ferritin (440 kDa), and ovalbumin (43 kDa), and are representative of PARLSkd3 dodecamer (gray), hexamer (cyan), or monomer (magenta) size. The gray bar also represents the fraction taken for cryo-EM analysis of PARLSkd3:casein.
(C) Cryo-EM 2D class averages of the PARLSkd3:casein:ATPγS complex showing representative side (left) and top (right) views. Scale bar, 100 Å.
(D) Overlay of low-pass filtered hexamer map and high-resolution sharpened map colored by domain as in (A).
(E and F) (E) Final 2.9-Å-resolution sharpened map and (F) molecular model colored by individual protomers (P1–P6), with substrate polypeptide (yellow) in the channel. See also Figures S1 and S2 and Video S1.
The ANK-AAA+ domain combination is an unusual feature of Skd3. Both the ANK and NBD are required for Skd3 disaggregase activity as deletion of either domain ablates activity (Cupo and Shorter, 2020b). How the ANK and NBD collaborate to power disaggregation is unknown. The ANK is comprised of two ankyrin repeats, a linker, and two more ankyrin repeats (Figure 1A). Ankyrin repeats adopt a helix-turn-helix conformation, which can enable specific protein-protein interactions (Kohl et al., 2003; Mosavi et al., 2004; Parra et al., 2015). Intriguingly, ankyrin repeats are a core component of an ATP-independent disaggregase, cpSRP43 (Jaru-Ampornpan et al., 2010, 2013). The Skd3 NBD is homologous to NBD2 of Hsp104 and bacterial ClpB (Cupo and Shorter, 2020b). Like Hsp104, Skd3 couples ATP hydrolysis to protein disaggregation, which requires conserved AAA+ motifs, such as Walker A, Walker B, and pore-loop tyrosines (Cupo and Shorter, 2020b). However, the Skd3 NBD contains an apomorphic insertion at residues L507–I534 that is not observed in any other AAA+ protein (Cupo and Shorter, 2020b). What role this insertion plays in Skd3 activity is unknown. Skd3 has an extended CTD that is patterned with acidic and basic residues (Cupo and Shorter, 2020b). By contrast, S. cerevisiae Hsp104 has an extended, acidic CTD that contributes to hexamerization (Mackay et al., 2008). How the CTD contributes to Skd3 function is unknown.
SCN-linked mutations in Skd3 cluster in the NBD (Warren et al., 2022), whereas biallelic MGCA-7-linked mutations are scattered throughout Skd3 (Wortmann et al., 2015). SCN-linked mutations impair ATPase and disaggregase activity (Warren et al., 2022). MGCA7-linked mutations impair disaggregase activity in a manner that predicts disease severity (Cupo and Shorter, 2020b). However, MGCA7-linked mutations do not always impair ATPase activity (Cupo and Shorter, 2020b). It is not understood why SCN-linked mutations are dominant-negative, whereas MGCA7-linked mutations are recessive.
It is often assumed that Skd3 structure and mechanism closely resemble that of Hsp104/ClpB (Capo-Chichi et al., 2015; Kanabus et al., 2015; Saunders et al., 2015). Yet, there are few studies of Skd3 disaggregase activity (Cupo and Shorter, 2020b; Mroz et al., 2020; Warren et al., 2022; Wortmann et al., 2021). Unlike Hsp104/ClpB, Skd3 does not require Hsp70 or Hsp40 to disaggregate disordered aggregates (Cupo and Shorter, 2020b). Moreover, Skd3 shares only ~20% sequence identity with Hsp104/ClpB and has only one domain, the NBD, in common with Hsp104/ClpB (Cupo and Shorter, 2020b; Erives and Fassler, 2015). Here, we employ cryoelectron microscopy (cryo-EM) and biochemical reconstitution to reveal Skd3 structure and mechanism.
RESULTS
PARLSkd3 structure reveals a substrate-bound AAA+ spiral and flexible ANKs
To capture a substrate-bound state of Skd3, we included a model substrate casein, which binds to wild-type (WT) PARL-protease-activated Skd3 (PARLSkd3; Figure 1A) (Cupo and Shorter, 2020b). PARLSkd3 binding to FITC-labeled casein was assessed under different nucleotide conditions (Figure S1A). Binding was detected under all conditions, but PARLSkd3 bound FITC-casein more effectively in the presence of non-hydrolyzable AMP-PNP (KD ~ 0.1 μM), ATP (KD ~ 0.5 μM), or slowly hydrolyzable ATPγS (KD ~ 0.4 μM) in contrast to ADP or no nucleotide (Figure S1A). Thus, PARLSkd3 differs from Hsp104, where only ATPγS facilitates avid polypeptide binding (Gates et al., 2017; Weaver et al., 2017).
We assessed the oligomeric state of PARLSkd3 by size-exclusion chromatography (SEC) (Figures 1B and S1B–S1D). Following incubation with ATPγS, AMP-PNP, or ADP without FITC-casein substrate, PARLSkd3 exhibits a broad elution profile with peaks likely corresponding to dodecamers (792 kDa), hexamers (396 kDa), smaller oligomers, and monomers (66 kDa; Figure S1B). By contrast, in the presence of FITC-casein, PARLSkd3 shifted toward dodecamers in all nucleotide conditions (Figures 1B and S1C). Thus, substrate binding by PARLSkd3 promotes dodecamerization. Indeed, it has been independently established that PARLSkd3 forms dodecamers (Spaulding et al., 2022; Wu et al., 2022). Moreover, Skd3 forms higher-order structures in cells, consistent with dodecamer formation (Thevarajan et al., 2020).
Previously, we used ATPγS to stabilize substrate-bound states of Hsp104/ClpB (Gates et al., 2017; Rizo et al., 2019). SEC-purified PARLSkd3:casein forms stable complexes in the presence of ATPγS (Figures 1C and S1E). Reference-free 2D class averages show a variety of top and side views with well-resolved features (Figures 1C and S1E). Top views revealed two particle classes: a major class with a hexameric ring containing density in the central channel, and a minor class with a heptameric ring and an empty channel (Figure S1E). Top views of the hexameric ring appeared similar to substrate-bound Hsp104/ClpB (Gates et al., 2017; Rizo et al., 2019), whereas side views revealed a distinct arrangement with two or three bands of density, indicating a stacked-ring arrangement of the PARLSkd3:casein complex (Figures 1C and S1E). Notably, side views exhibit one strong band of density with well-resolved features, whereas the other bands are more diffuse, indicating flexibility or differential occupancy (Figure 1C).
Following 3D classification with four classes, we identified three distinct oligomeric arrangements: a hexameric double-ring complex that contains density in the channel (class 1), a hexameric three-ring complex that contains density in the central channel for one well-resolved ring (class 2), and a heptameric form containing two rings and an empty central channel (class 3) (Figure S1F). Given the low abundance of the heptameric ring and the absence of density for substrate, class 3 was not pursued further. Class 1 had the highest percentage of particles (41%) and a well-defined AAA+ ring. Thus, refinement was performed with class 1, resulting in a final resolution of 2.9Å for the PARLSkd3:casein complex (Figures S2A–2H; Table S1; Video S1). A molecular model for the hexameric ring comprised of the NBD, which refined to the highest resolution (~2.5 Å) in the map (Figure S2E), was determined using homology models generated by SWISS-Model (Waterhouse et al., 2018).
At an increased threshold, lower-resolution density extends from the N-terminal end of the NBDs and forms a second ring of separated and flexible globular structures (Figure 1D). These separated regions contrast with the extensive contact interfaces of the NBDs in the AAA+ ring (Figure 1D). Based on the molecular model of the NBDs and the position of the N-terminal AAA+ residues, we conclude that these separated regions are the N-terminal ANKs (Figures 1A and 1D). Given this architecture for a hexameric arrangement, we propose that the three-ring structures identified in the 2D class averages and in class 2 in the 3D classification are dodecamers comprised of two PARLSkd3 hexamers that interact via the ANKs, which form the middle ring of density (Figures 1C and S1F). Considering the flexibility of the ANKs and the second AAA+ ring, we postulate that, in addition to the hexamer form, class 1 likely contains dodecamers in which the second ring is poorly aligned and not visible in the reconstruction due to its flexibility. Indeed, when 2D classification of the class 1 particle is performed, weak density for a second AAA+ ring is identified in many side-view class averages, indicating a dodecamer (Figure S2A). Furthermore, SEC in tandem with multi-angle light scattering confirms PARLSkd3 dodecamers in solution (Figure S1G). We suggest that the active, substrate-bound form of PARLSkd3 likely exists in a dynamic equilibrium between hexamers and dodecamers.
The PARLSkd3 NBDs adopt a right-handed spiral, wherein five protomers directly contact substrate polypeptide along a 40-Å-length of the channel (Figures 1E and 1F;, Video S1). These well-resolved protomers (P1–P5) are positioned in a helical arrangement, each with a rise of ~6 Å and rotation of ~60° along the substrate. Protomer P6 is at the seam interface between the lowest (P1) and highest (P5) substrate contact sites but is disconnected and has lower resolution, resulting in an asymmetric position within the spiral (Figures 1E and 1F). This architecture is akin to other substrate-bound AAA+ structures, including Hsp104/ClpB (Gates et al., 2017; Rizo et al., 2019). Additional density is identified in protomers P3–P5 that extend from the small sub-domain of the NBD toward the adjacent clockwise protomer, and is likely the CTD (Figure 1D).
Substrate contacts and NBD occupancy support a conserved, stepwise translocation model
Pore loop-substrate interactions and nucleotide states in the PARLSkd3 hexamer structure suggest a translocation mechanism. An extended polypeptide is well resolved in the PARLSkd3 channel and modeled as a 14-residue polyA peptide (Figure 2A). Based on previous work (Gates et al., 2017; Lopez et al., 2020; Rizo et al., 2019) and binding data (Figures S1A and S1D), we conclude that this extended polypeptide is a nonspecific portion of FITC-casein. The canonical pore loops (residues 429–432) for protomers P1–P5 directly bind the substrate backbone via a conserved YV motif (Y430 and V431; Figures 2A and 2B). These pore loops form a spiral staircase of contacts and comprise the primary substrate-binding sites identified in the structure, supporting their established role in translocase function (Shorter and Southworth, 2019). Indeed, mutation of the conserved tyrosine to alanine (Y430A) reduces ATPase activity and abolishes disaggregase activity (Cupo and Shorter, 2020b). We now find that V431G also reduces PARLSkd3 ATPase activity and abolishes disaggregase activity (Figures 2C and 2D). These results are consistent with our PARLSkd3 structure where Y430 and V431 directly engage substrate.
Figure 2. Spiral of pore loop-substrate contacts and nucleotide states of PARLSkd3.
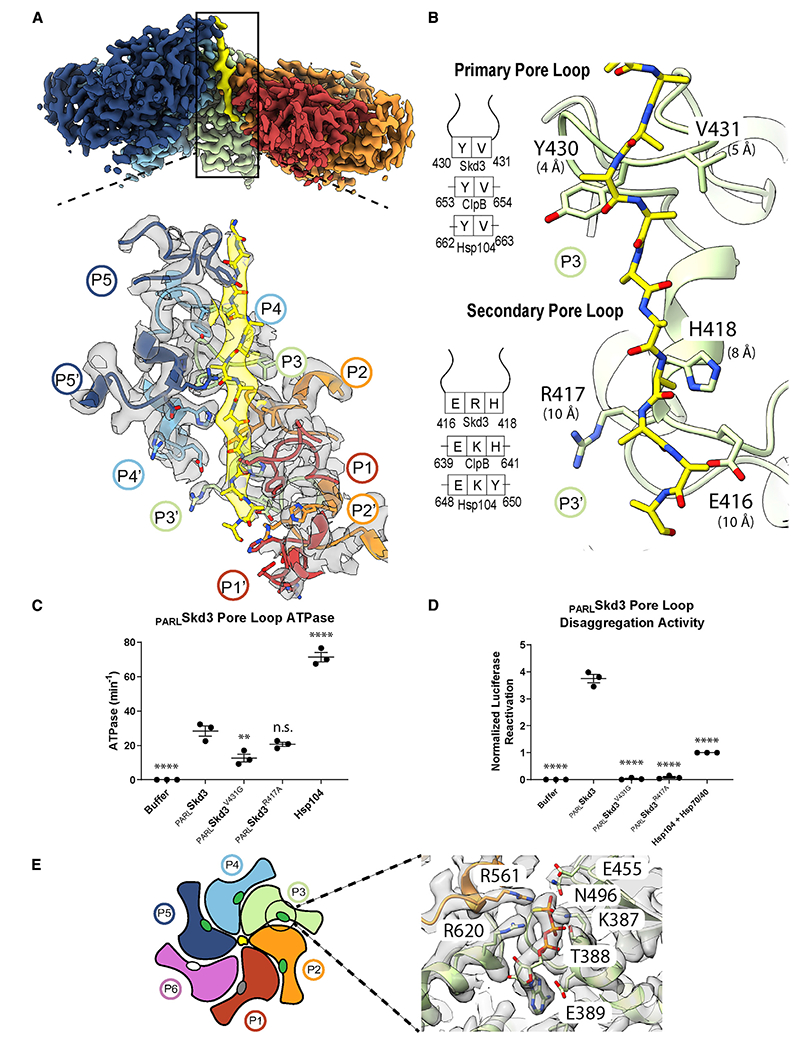
(A) Cryo-EM density map (top) of protomers (P1–P5) and substrate (yellow) and expanded view (bottom) of the channel, including the primary (P1–P5) and secondary (P1′–P5′) loops interacting in a spiral along the 14-residue substrate strand. P1 is the canonical pore loop from protomer 1 and P1′ is the secondary pore loop from protomer 1.
(B) The primary and secondary pore loop-substrate contacts for P4, including distances to substrate (measured between α-carbons) and a schematic indicating conservation with Hsp104/ClpB.
(C) ATPase activity of PARLSkd3, PARLSkd3V431G, PARLSkd3R417A, and Hsp104. ATPase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n = 3, individual data points shown as dots, bars show mean ± SEM, **p < 0.01, ****p < 0.0001).
(D) Luciferase disaggregase activity of PARLSkd3, PARLSkd3V431G, PARLSkd3R417A, and Hsp104 plus Hsp70 and Hsp40. Luciferase activity was normalized to Hsp104 plus Hsp70 and Hsp40. Disaggregase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n = 3, individual data points shown as dots, bars show mean ± SEM, ****p < 0.0001).
(E) Schematic indicating nucleotide states (ovals) for each protomer (ATP, green; ADP, gray; apo, white) and expanded view of P4 nucleotide-binding pocket showing density for ATP and conserved interacting residues, including Arg finger (R561), sensor-1 (N496), sensor-2 (R620), Walker A (K387), and Walker B (E455). See also Figure S2 and Video S1.
An additional spiral of substrate interactions at the channel exit is formed by secondary pore-loop motifs from protomers P2–P5 (Figures 2A and 2B). In PARLSkd3, residues E416, R417, and H418 comprise this secondary pore loop, which is positioned in line with canonical YV loops above, but slightly further away (~9 Å) from the substrate backbone (Figures 2A and 2B). To define the role of the secondary pore loop, we generated PARLSkd3R417A, which exhibited similar ATPase activity to PARLSkd3 (Figure 2C), but diminished disaggregase activity (Figure 2D). This loss of function is much more severe than that caused by equivalent mutations in Hsp104/ClpB (Howard et al., 2020; Rizo et al., 2019). Thus, the secondary pore loops play a more critical role in PARLSkd3 disaggregase activity than in Hsp104/ClpB.
The nucleotide-binding pockets in substrate-bound PARLSkd3 are positioned at inter-protomer interfaces with conserved AAA+ residues contacting the nucleotide (Figures 2E and S2I). For protomers P2–P5, these pockets are well resolved, revealing a bound ATP that is contacted by canonical Walker A (K387), Walker B (E455), sensor-1 (N496), and sensor-2 (R620) residues (Figures 2E and S2I). The Arg-finger (R561) is provided by the neighboring clockwise protomer, and positioned one step lower along the substrate, contacting the γ-phosphate of ATP in protomers P3–P5 (Figures 2E and S2I). For protomer P2, ATP is present, but the Arg-finger from P1 is positioned further away and not in contact, indicating an intermediate state (Figure S2I). Notably, ATP-bound states are only found for substrate-engaged protomers (Figures 2A and S2I). Density for the nucleotide is more poorly resolved in protomers P1 and P6 at the spiral seam (Figures 2A and S2I). The nucleotide is absent from P6, indicating an apo state, whereas P1 is likely ADP bound. Thus, post-hydrolysis states coincide with substrate release at the seam. Thus, PARLSkd3 may employ a conserved hydrolysis cycle similar to other AAA+ translocases (Gates et al., 2017; Puchades et al., 2017; Rizo et al., 2019). Based on this model, ATP hydrolysis and substrate release occur at the lower contact sites in the spiral (P1), whereas ATP binding promotes substrate re-binding to the top position (P5) along the substrate, enabling a rotary mechanism involving two residue steps along the substrate during processive translocation (Shorter and Southworth, 2019). However, other kinetic paths or non-processive events may also occur (Durie et al., 2019; Fei et al., 2020; Lin et al., 2022).
ANKs mediate PARLSkd3 dodecamerization and enable disaggregase activity
The ANK is an unusual feature of Skd3 that is required for disaggregase activity (Cupo and Shorter, 2020b). Cryo-EM of SEC-purified PARLSkd3NBD, which lacks the ANK domain, reveals well-resolved single hexamers but no dodecamers (Figure S3A) (Wu et al., 2022). Thus, the ANK is not required for hexamerization, but is important for stabilizing the dodecamer. SEC indicates that the dodecamer predominates in the presence of substrate (Figures 1B and S1C). However, the dodecamer is less well represented following 2D and 3D cryo-EM analysis, with ~15% of particles possessing the three-ring architecture of class 2 (Figure S1F). Moreover, flexibility of the ANKs and the tilted arrangement of the AAA+ rings likely limit structure determination of the full dodecamer complex from class 2. Nonetheless, two full Skd3 hexamer models could be docked into the low-resolution class 2 map, revealing that the ANKs mediate contacts between the two hexamers (Figure S3B; Table S1). Resolution of the more poorly resolved NBD ring was insufficient to identify substrate in the channel, whereas density is present in the second NBD ring, which is likely casein contacted by pore loops from five protomers (Figure S3B). Conversely, in addition to the high-resolution AAA+ ring, the final map of the hexamer class (class 1) contains strong globular density extending from the N-terminal face that is likely the helical bundles of ankyrin repeats (Figure 1D). Thus, analysis of the full hexamer with the ANKs was further pursued with the class 1 map.
Structural information for the Skd3 ANK is unavailable. Thus, we used the Alpha-fold to model the ANK (Jumper et al., 2021). This secondary structural model is predicted with high confidence based on the pLDDT score and low predicted align error values (Figures S3C and S3D). Confidence was highest in the ANK and NBD domains (Figure S3D). Based on the Alpha-fold model, the ANK adopts four two-helix bundle structures that match canonical ANKs (Figure 3A). Starting at the N terminus, this structure consists of two ankyrin repeats (1 and 2), a 66-residue linker (L) that is mostly disordered, and two further ankyrin repeats (3 and 4) (Figure 3A). The linker is the exact length of two ankyrin repeats and has cryptic elements of ankyrin repeats within its primary sequence (Figure S3F). Alpha-fold predicts some helical regions within the linker, and these regions partially align to the other repeats (Figures 3A, S3C, and S3D). Thus, the linker may impart some ankyrin-like functions. Notably, repeat 4 forms an extended helix that transitions directly into the N-terminal region of the NBD without a separate linker between domains (Figure 3A). This continuous helix likely stabilizes the position of the ANKs given that interprotomer contacts are not present in the ANK ring. The four ankyrin repeats bundle together in the Alpha-fold model and dock well into the globular density adjacent to the NBD (Figure 3B; Video S1). The density for the ANK is more prominent for protomers P2–P5, which are bound to the substrate and better resolved compared with the spiral seam (Figure 3B). To further resolve the ANK, focus classification was performed on the P3 ANK. Resulting classes reveal that the ANK adopts different positions, indicating the flexibility of the ankyrin-repeat 4/NBD connecting helix (Figures 3C, 3D, and S3E). Notably, class 1 contains additional density that projects from the globular ANKs toward the central channel and is likely the linker (Figure S3E).
Figure 3. The ANK mediates head-to-head contacts required for dodecamerization.
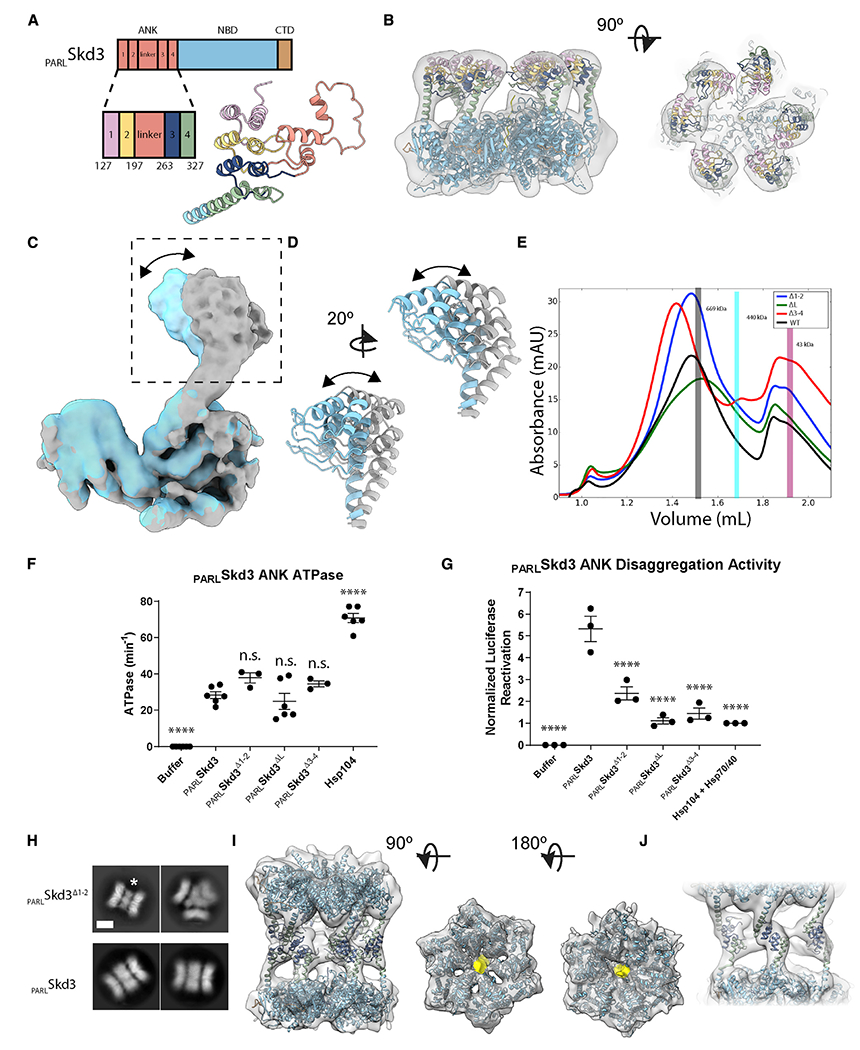
(A) Schematic and model of PARLSkd3 ANK, colored-based repeat number and linker.
(B) Side (left) and top (right) views of the filtered class 1 map and docked model, colored as in (A), identifying ANK position.
(C and D) Overlay of two different classes resolved from focus classification around P3 identifying rotation of the ANK region (arrow).
(E) SEC of PARLSkd3 (black), PARLSkd3Δ1-2 (blue), PARLSkd3ΔL (green), and PARLSkd3Δ3-4 (red) incubated with ATPγS and casein. Vertical bars indicate molecular weight standards: thyroglobulin (669 kDa), ferritin (440 kDa), and ovalbumin (43 kDa), and are representative of PARLSkd3 dodecamer (gray), hexamer (cyan), or monomer (magenta) size.
(F) ATPase activity of PARLSkd3, PARLSkd3Δ1-2, PARLSkd3ΔL, PARLSkd3Δ3-4, and Hsp104. ATPase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n = 3–6, individual data points shown as dots, bars are mean ± SEM, ****p < 0.0001).
(G) Luciferase disaggregase activity of PARLSkd3, PARLSkd3Δ1-2, PARLSkd3ΔL, PARLSkd3Δ3-4, and Hsp104 plus Hsp70 and Hsp40. Luciferase activity was normalized to Hsp104 plus Hsp70 and Hsp40. Disaggregase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n = 3, individual data points shown as dots, bars show mean ± SEM, ****p < 0.0001).
(H) 2D class averages of PARLSkd3Δ1-2 (top) and PARLSkd3 (bottom) with the middle band of ANK density indicated (*). Note that the triple-hexamer arrangement (top right) is only found for PARLSkd3Δ1-2. Scale bar, 100 Å.
(I) Dodecamer map and model of PARLSkd3Δ1-2 colored by individual domains (left). Color-zoned map of substrate (yellow) present in the central channel of each AAA+ ring with PARLSkd3Δ1-2 model docked in (middle, right).
(J) Dodecamer map and model of PARLSkd3Δ1-2 showing ankyrin-repeat interactions across hexamers. See also Figure S3 and Video S2.
To assess how specific ANK regions enable Skd3 function, we generated PARLSkd3 variants with ankyrin repeats 1 and 2 deleted (ΔY127-G196, PARLSkd3Δ1-2), the linker deleted (ΔD197-A262, PARLSkd3ΔL), or ankyrin repeats 3 and 4 deleted (ΔS263-K325, PARLSkd3Δ3-4; Figure S3G). In the presence of casein and ATPγS, PARLSkd3Δ1-2 and PARLSkd3Δ3-4 formed predominantly dodecamers like PARLSkd3 (Figures 1B and 3E). By contrast, PARLSkd3ΔL exhibited reduced dodecamer formation, and was shifted more toward hexamers (Figure 3E). Unlike PARLSkd3NBD, which exhibits reduced ATPase activity (Cupo and Shorter, 2020b), PARLSkd3Δ1-2, PARLSkd3ΔL, and PARLSkd3Δ3-4 had similar ATPase activity to PARLSkd3 (Figure 3F). Thus, a portion of the N-terminal ANK is required to maintain PARLSkd3 ATPase activity. Nonetheless, PARLSkd3Δ1-2, PARLSkd3ΔL, and PARLSkd3Δ3-4 exhibited reduced disaggregase activity (Figure 3G), indicating that the ANK enables PARLSkd3 to efficiently couple ATP hydrolysis to protein disaggregation. Deletion of the linker had the largest effect (Figure 3G). PARLSkd3ΔL is impaired in dodecamer formation (Figure 3E). Thus, dodecamerization may promote disaggregase activity. We suggest that the ANKs play multiple roles in protein disaggregation, including dodecamerization, potentially supported by the linker, and substrate engagement mediated by ankyrin repeats 1–4 (Wu et al., 2022).
EM indicated that PARLSkd3Δ1-2 forms more stable dodecamers than PARLSkd3ΔL and PARLSkd3Δ3-4. Thus, we investigated PARLSkd3Δ1-2 further by cryo-EM. 2D averages of PARLSkd3Δ1-2 show a well-resolved middle ring of ANKs that is smaller in diameter than PARLSkd3 (Figure 3H). 3D classification of PARLSkd3Δ1-2 identified three distinct forms. Class 1 and class 2 are dodecamers with different relative positions of the AAA+ rings, whereas class 3 is a trimer of hexamers (Figures S3H and S3I). Refinement of class 1 was pursued due to the more homogeneous arrangement of the ANK ring and improved density for the second AAA+ ring compared with the PARLSkd3 complex (Figures 3I, 3J, and S3J–S3M). Notably, we identify density in the two AAA+ channels that corresponds to the substrate, indicating that both AAA+ rings can bind substrate in the dodecamer (Figure 3I), Although the resolution was low (~9 Å), a dodecameric model with ankyrin repeats 3 and 4 fit well into the density, and revealed head-to-head ANK contacts around the central ring that mediate dodecamer formation (Figures 3I and 3J; Table S1; Video S2). Given that PARLSkd3Δ3-4 can also form dodecamers, as can PARLSkd3ΔL to a lesser extent (Figure 3E), our findings indicate plasticity in how the ANK mediates cross-contacts to form dodecamers. We suggest that deleting specific ankyrin repeats or the linker reduces this interactive plasticity and disaggregase activity (Figure 3G).
A unique insertion within the PARLSkd3 NBD regulates the AAA+ motor
Skd3 contains an insertion (residues L507-I534) within the NBD that is conserved across Skd3 homologs but is not found in Hsp104 or other HCLR class AAA+ proteins (Figures 4A and S4A) (Cupo and Shorter, 2020b). We modeled part of the insertion, but 17 residues (517–533) were missing (Figures 4B and S4B). The insertion extends past the loop that is found in Hsp104 (Figure S4C) and protrudes from the hexamer exterior (Figure 4B). Purified PARLSkd3ΔL507-I534 formed large oligomers that are not observed for PARLSkd3 (Figure S4D). However, upon addition of casein, PARLSkd3ΔL507-I534 forms primarily dodecamers (Figure S4D). PARLSkd3ΔL507-I534 had elevated ATPase and disaggregase activity compared with PARLSkd3 (Figures 4C and 4D). Thus, the L507-I534 insertion is a regulatory element, which slows PARLSkd3 ATPase activity and tunes disaggregase activity. The location of the L507-I534 insertion on the hexamer exterior could provide a site for regulatory factors to bind or post-translationally modify Skd3.
Figure 4. An NBD insertion and the CTD regulate PARLSkd3.
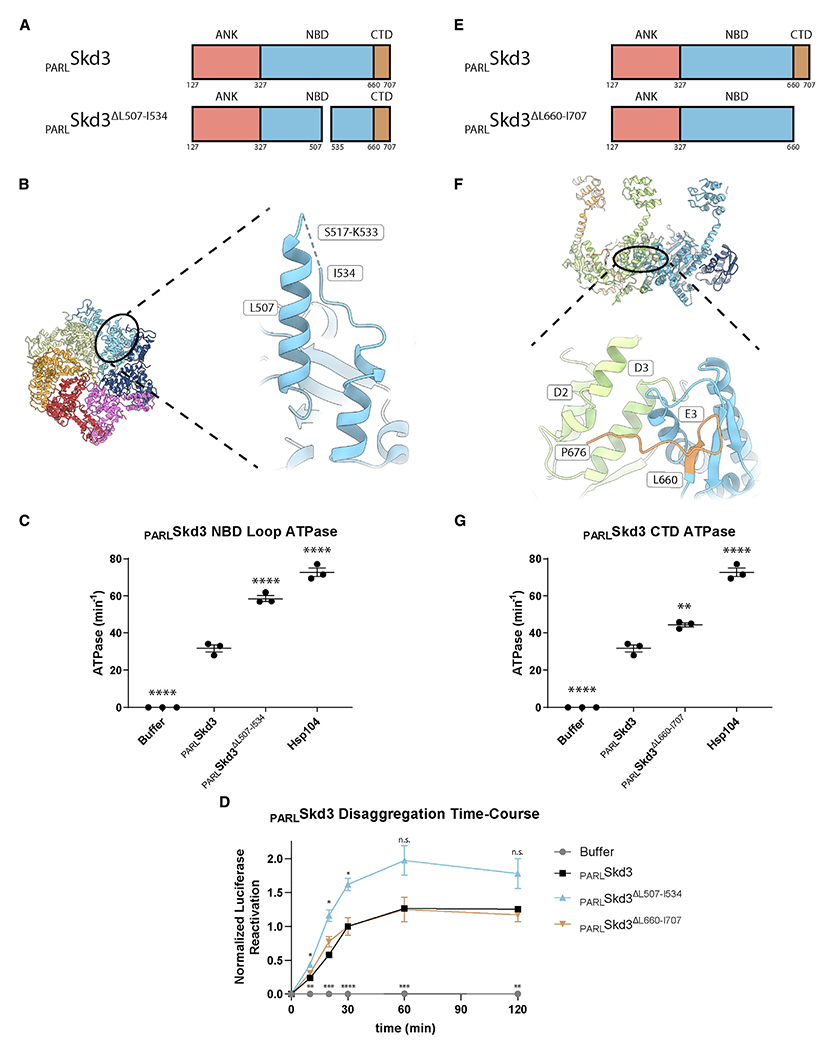
(A) Domain architecture of PARLSkd3 and PARLSkd3ΔL507-I534.
(B) Top view of PARLSkd3 hexamer and expanded view of NBD-insertion region residues 449–552 (residues 517–533 are not resolved [dashed line]) in P4.
(C) ATPase activity of PARLSkd3, PARLSkd3ΔL507-I534, and Hsp104. Data are from the same experiments as Figure 3F. ATPase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n= 3, individual data points shown as dots, bars are mean ± SEM, ****p < 0.0001).
(D) Luciferase disaggregation kinetics showing that PARLSkd3ΔL507-I534 has enhanced disaggregase activity at early times, whereas PARLSkd3ΔL660-I707 does not. Luciferase activity was normalized to the PARLSkd3 30 min time point. Luciferase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n = 3, values are means ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
(E) Domain architecture of PARLSkd3 and PARLSkd3ΔL660-I707.
(F) Side view of PARLSkd3 hexamer and expanded view of P3–P4 with the P4 CTD model (brown) shown adjacent to P3 with potential interacting helices indicated.
(G) ATPase activity of PARLSkd3 and PARLSkd3ΔL660-I707. Data are from the same experiments as Figure 3F. ATPase activity was compared with PARLSkd3 using one-way ANOVA and a Dunnett’s multiple comparisons test (n = 3, individual data points shown as dots, bars show mean ± SEM, **p < 0.01, ****p < 0.0001). See also Figure S4.
Deletion of the PARLSkd3 CTD mildly stimulates ATPase activity
Deletion of the extended, acidic CTD of S. cerevisiae Hsp104 results in hexamerization defects (Mackay et al., 2008). Like Hsp104 and in contrast to other HCLR clade AAA+ proteins, Skd3 has an extended CTD from residues 660 to 707 (Figure 4E) (Cupo and Shorter, 2020b). However, the Skd3 CTD is patterned with acidic and basic residues, whereas the Hsp104 CTD is acidic (Cupo and Shorter, 2020b). In the PARLSkd3 reconstruction, 14 residues of the CTD were evident in protomers P3–P5 (Figure 1D). These CTD residues fit along the side of the adjacent protomer and are near helices D2 and D3 (Figures 4F and S2F). Residues within ~4 Å of the CTD include E340 and Q341 in D2, and R362 in D3. Additional contacts may occur with helix E3 of the same protomer (Figures 4F and S2F). We purified PARLSkd3 lacking the CTD (PARLSkd3ΔL660-I707), which formed hexamers and dodecamers similar to PARLSkd3 (Figures 1A and S4E). PARLSkd3ΔL660-I707 exhibited mildly increased ATPase activity (Figure 4G), whereas disaggregase activity was similar to PARLSkd3 (Figure 4D). Thus, the CTD enables efficient coupling of PARLSkd3 ATPase and disaggregase activity. We suggest that the PARLSkd3 CTD plays a different role than the Hsp104 CTD.
PARLSkd3 is functional at low ATP concentrations
Mitochondria maintain lower ratios of ATP:ADP and lower ATP concentrations than the cytoplasm (Gellerich et al., 2002; Heldt et al., 1972; Imamura et al., 2009). To determine how PARLSkd3 might operate under various nucleotide conditions, we established that PARLSkd3 ATPase activity has a Vmax of ~24 min−1 and a KM of ~65 μM (Figure 5A). By contrast, the KM of Hsp104 is ~5–11 mM (Grimminger et al., 2004; Schirmer et al., 1998). PARLSkd3 maintains disaggregase activity at low ATP concentrations, including ~50% disaggregase activity at the lowest ATP concentration tested (0.434 mM) (Figure 5B). Thus, PARLSkd3 is likely adapted to operate effectively at lower ATP concentrations than Hsp104.
Figure 5. PARLSkd3 is functional at low ATP concentrations.
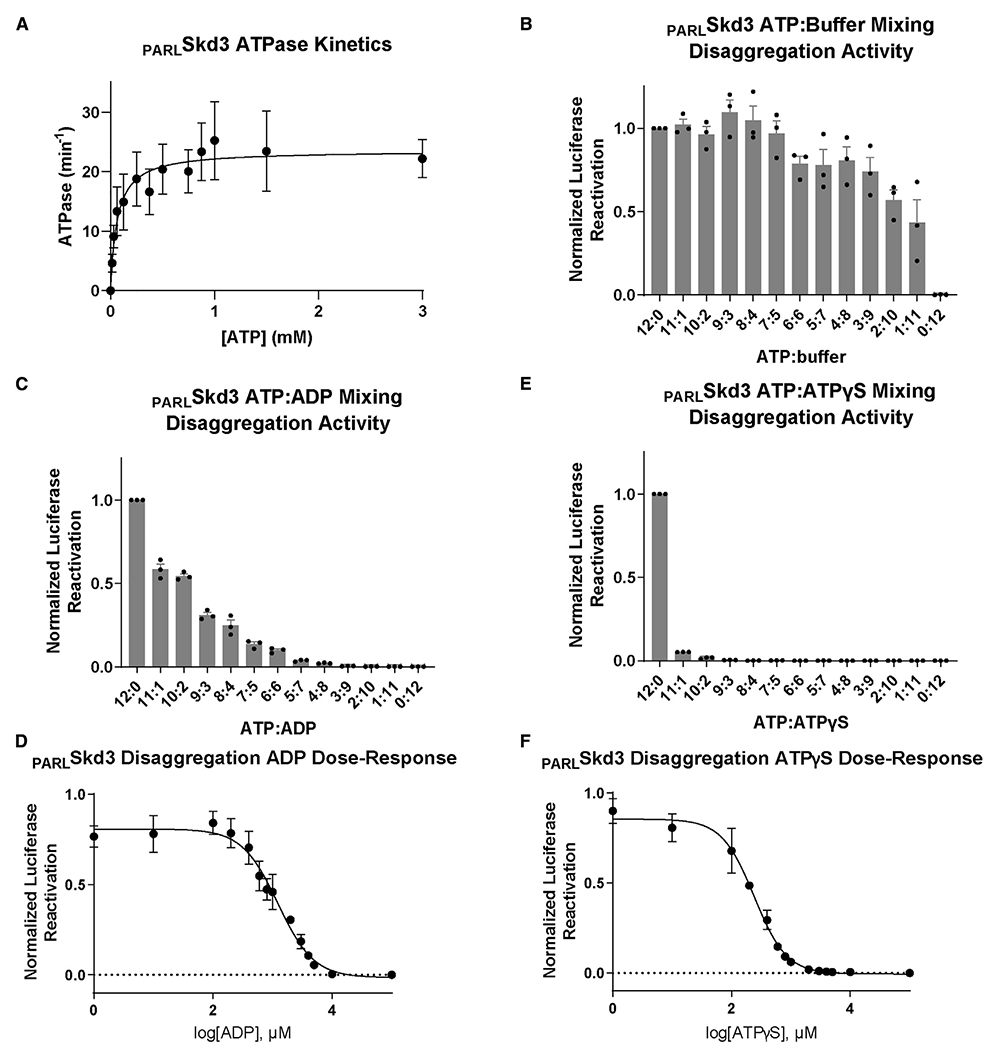
(A) Michaelis-Menten plot of PARLSkd3 ATPase activity. Vmax ~ 23.6 min−1 and KM ~ 64.6 μM (n = 3, values are means ± SEM).
(B) Luciferase disaggregase activity of PARLSkd3 at various ATP:buffer ratios. ATP concentrations used were 12:0 (5 mM), 11:1 (4.58 mM), 10:2 (4.17 mM), 9:3 (3.75 mM), 8:4 (3.33 mM), 7:5 (2.92 mM), 6:6 (2.5 mM), 5:7 (2.08 mM), 4:8 (1.67 mM), 3:9 (1.25 mM), 2:10 (0.83 mM), 1:11 (0.42 mM), and 0:12 (0 mM). Disaggregase activity was normalized to PARLSkd3 plus ATP (n = 3, individual data points shown as dots, bars show mean ± SEM).
(C) Luciferase disaggregase activity of PARLSkd3 at various ATP:ADP ratios where the total nucleotide concentration was 5 mM. Disaggregase activity was normalized to PARLSkd3 plus ATP (n= 3, individual data points shown as dots, bars show mean ± SEM).
(D) Luciferase disaggregase activity of PARLSkd3 at a constant concentration of ATP (5 mM) and increasing ADP concentrations. Disaggregase activity was normalized to PARLSkd3 plus ATP (n = 3, values are means ± SEM).
(E) Luciferase disaggregase activity of PARLSkd3 at various ATP:ATPγS ratios where the total nucleotide concentration was 5 mM. Disaggregase activity was normalized to PARLSkd3 plus ATP (n = 3, individual data points shown as dots, bars show mean ± SEM).
(F) Luciferase disaggregase activity of PARLSkd3 at a constant concentration of ATP (5 mM) and increasing ATPγS concentrations. Disaggregase activity was normalized to PARLSkd3 plus ATP (n = 3, data shown as mean ± SEM).
PARLSkd3 disaggregase activity is inhibited by ADP
Hsp104 is sharply inhibited by mixing ADP with ATP (Grimminger et al., 2004; Hattendorf and Lindquist, 2002; Klosowska et al., 2016). Even a 5:1 ATP:ADP ratio diminishes Hsp104 activity (Klosowska et al., 2016). To test the effect of ADP on PARLSkd3, we assessed PARLSkd3 disaggregase activity under different ATP:ADP ratios while keeping the total nucleotide concentration constant. PARLSkd3 is inhibited by ADP, but maintains ~50% activity at a 5:1 ATP:ADP ratio (Figure 5C), which inactivates Hsp104 (Klosowska et al., 2016). The half-maximal inhibitory concentration (IC50) of ADP at a constant concentration of ATP (5 mM) was ~1.2 mM (Figure 5D). Thus, PARLSkd3 is less sensitive than Hsp104 to inhibition by ADP and is likely adapted to function at the lower ATP:ADP ratios found in mitochondria.
PARLSkd3 disaggregase activity is sharply inhibited by ATPγS
Next, we assessed how PARLSkd3 disaggregase activity is affected by the slowly hydrolyzable ATP analog, ATPγS. Like Hsp104, PARLSkd3 is inactive in the presence of ATPγS as the sole nucleotide (Cupo and Shorter, 2020b; DeSantis et al., 2012). However, Hsp104 disaggregase activity against disordered aggregates can be stimulated at specific ratios of ATP:ATPγS (~3:1–1:5), whereas Hsp104 disaggregase activity against amyloid is inhibited by ATPγS in the presence of ATP (DeSantis et al., 2012). Thus, Hsp104 employs distinct mechanisms of subunit collaboration to disaggregate disordered aggregates versus amyloid (DeSantis et al., 2012). To assess the effect of ATPγS on PARLSkd3, we measured PARLSkd3 disaggregase activity under different ATP:ATPγS ratios while keeping the total nucleotide concentration constant. PARLSkd3 is sharply inhibited by ATPγS (Figure 5E). Even an 11:1 ATP:ATPγS ratio inhibits PARLSkd3 (Figure 5E). The IC50 of ATPγS at a constant concentration of ATP (5 mM) was ~242 μM (Figure 5F). Thus, in contrast to Hsp104 (DeSantis et al., 2012), PARLSkd3 disaggregase activity against disordered aggregates is not stimulated by ATP:ATPγS mixtures. The distinctive responses of PARLSkd3 to ADP and ATPγS reveal differences in how PARLSkd3 and Hsp104 disaggregase activity are regulated. The sharp inhibition of PARLSkd3 disaggregase activity by ATPγS indicates that PARLSkd3 is sensitive to individual subunits that hydrolyze ATP slowly.
PARLSkd3 is a subglobally cooperative protein disaggregase
Next, to define mechanochemical coupling mechanisms of PARLSkd3, we used a mutant subunit doping strategy to assess how individual PARLSkd3 subunits contribute to ATPase and disaggregase activity. We modeled Skd3 as a hexamer, which forms the functional AAA+ cassette (Figure 2E). In this strategy, mutant PARLSkd3 subunits defective in ATP hydrolysis or substrate binding are mixed with WT PARLSkd3 subunits to generate heterohexameric ensembles according to a binomial distribution dictated by the WT:mutant ratio (Figure 6A). As mutant PARLSkd3 concentration in the mixture increases, the probability of mutant PARLSkd3 incorporation into a PARLSkd3 hexamer increases (Figure 6A). This approach has revealed mechanochemical coupling mechanisms of other NTPases, including Hsp104/ClpB (DeSantis et al., 2012; Moreau et al., 2007; Shivhare et al., 2019; Werbeck et al., 2008).
Figure 6. Skd3 is a subglobally cooperative disaggregase.
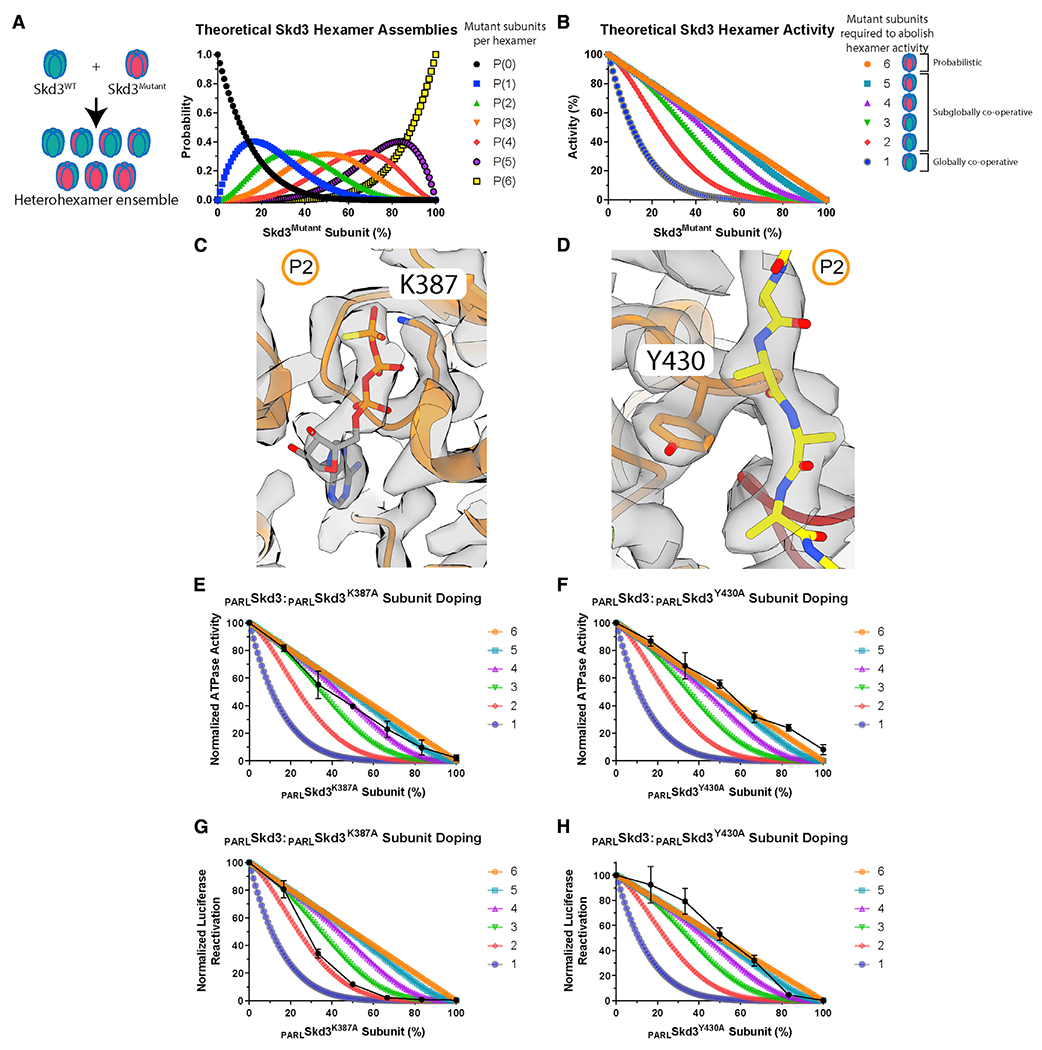
(A) Theoretical PARLSkd3 hexamer ensembles containing zero (black), one (blue), two (green), three (orange), four (red), five (purple), and six (yellow) mutant subunits as a function of the fraction of mutant subunit.
(B) Theoretical PARLSkd3 activity curves where one or more (blue), two or more (red), three or more (green), four or more (purple), five or more (light blue), or six (orange) mutant subunits ablate hexamer activity. In a probabilistic model, six mutant subunits per hexamer ablate activity. In a subglobally cooperative model, two to five mutant subunits per hexamer ablate activity. In a globally cooperative model, one mutant subunit per hexamer ablates activity.
(C and D) Protomer P2 with nucleotide and K387 shown in (C) or substrate and Y430 shown in (D).
(E and F) ATPase activity of PARLSkd3 mixed with various ratios of PARLSkd3K387A (E) or PARLSkd3Y430A (F). ATPase activity was normalized to PARLSkd3 (n = 3, black dots are means ± SEM).
(G and H) Luciferase disaggregase activity of PARLSkd3 mixed with various ratios of PARLSkd3K387A(G) or PARLSkd3Y430A (H). Disaggregase activity was normalized to PARLSkd3 (n = 3, black dots are means ± SEM). See also Figure S5.
This strategy depends on robust formation of randomized heterohexamer ensembles, which requires exchange of subunits between WT and mutant PARLSkd3 hexamers such that mutant subunits mix equally well into heterohexamers as WT (Figure 6A). To assess subunit mixing, we labeled PARLSkd3 with Alexa 488 or Alexa 594, which can form a Förster resonance energy transfer (FRET) pair. Labeled PARLSkd3 retained ATPase and disaggregase activity (Figures S5A and S5B). We mixed Alexa 488-labeled PARLSkd3 and Alexa 594-labeled PARLSkd3 in the absence of substrate, where the hexamer is more populated (Figures S1B and S1C). Hence, FRET likely reflects subunit mixing within the hexamer. For WT PARLSkd3, a robust FRET signal was observed within a few minutes, indicating subunit mixing on the minute timescale similar to Hsp104 (Figure S5D) (DeSantis et al., 2012). Thus, PARLSkd3 forms dynamic hexamers that exchange subunits on the minute timescale.
Importantly, mutant PARLSkd3 subunits were effectively incorporated into WT PARLSkd3 hexamers (Figure S5D). PARLSkd3K387A (Walker A) subunits likely incorporate into WT PARLSkd3 hexamers as effectively as WT PARLSkd3 subunits, whereas PARLSkd3Y430A (pore loop) subunits incorporated into WT PARLSkd3 hexamers ~16% less effectively than WT PARLSkd3 subunits (Figure S5D). Thus, specific mutant PARLSkd3 subunits incorporate effectively into WT PARLSkd3 hexamers, indicating that the PARLSkd3 mechanism can be probed via mutant doping studies.
This rapid subunit exchange enables formation of PARLSkd3 heterohexamer ensembles comprised of WT and mutant subunits according to a binomial distribution (Figure 6A). Using this distribution, we can predict how PARLSkd3 activity would be inhibited at various WT:mutant ratios if a specific number of PARLSkd3 mutant subunits inactivate the hexamer (Figure 6B). For example, if all six PARLSkd3 subunits must work together, then one mutant subunit would abolish hexamer activity (Figure 6B, dark blue curve). By contrast, if the activity of a single PARLSkd3 subunit within the hexamer is sufficient, then some activity would still be observed with five mutant subunits per hexamer, and only six mutant subunits would abolish activity (Figure 6B, orange line). By comparing experimental data with theoretical plots, we can determine whether subunit collaboration within PARLSkd3 hexamers is probabilistic (six mutant subunits abolish activity), subglobally cooperative (two to five mutant subunits abolish activity), or globally cooperative (one mutant subunit abolishes activity).
We titrated PARLSkd3 with buffer over the concentration range of the subunit doping ATPase assay and found a linear decline in ATPase activity (Figure S5E). Thus, when titrating mutant PARLSkd3, a sharper than linear decline in ATPase activity indicates inhibitory effects of mutant subunits incorporated into hexamers. We also titrated PARLSkd3 with buffer over a range of concentrations to assess disaggregase activity (Figure S5F). We selected saturating PARLSkd3 concentrations to ensure that any observed effects on disaggregase activity upon mixing WT and mutant are not caused by a mere decrease in the concentration of WT PARLSkd3 (DeSantis et al., 2012; Werbeck et al., 2008).
PARLSkd3K387A (Walker A) and PARLSkd3Y430A (pore loop) are inactive for ATPase and disaggregase activity (Cupo and Shorter, 2020b). The Walker A residue, K387, contacts the β- and γ-phosphate of ATP and a K387A mutation is predicted to reduce ATP binding and hydrolysis (Figure 6C) (Puchades et al., 2020). The pore-loop tyrosine, Y430, engages substrate, and a Y430A mutation is predicted to reduce substrate binding (Figures 2B and 6D) (Cupo and Shorter, 2020b). We assembled heterohexamer ensembles of PARLSkd3 with PARLSkd3K387A (Walker A) or PARLSkd3Y430A (pore loop). PARLSkd3K387A (Walker A) subunits inhibited PARLSkd3 ATPase activity in a manner that suggested the incorporation of approximately three to five mutant subunits inactivates the hexamer (Figure 6E). Thus, basal Skd3 ATPase activity appears to be subglobally cooperative. By contrast, titrating PARLSkd3Y430A (pore loop) subunits did not affect PARLSkd3 ATPase activity any more than dilution in buffer (Figure 6F). Hence, the ATPase activity of the PARLSkd3 hexamer is more resistant to pore-loop mutant subunits than Walker A mutant subunits. Our findings contrast with observations made with Hsp104 (DeSantis et al., 2012), where Walker A mutant subunits do not affect the ATPase activity of the hexamer more than dilution in buffer, and pore-loop mutant subunits have no effect (DeSantis et al., 2012). Thus, Hsp104 and PARLSkd3 display distinct subunit cooperativity with respect to ATP hydrolysis.
We next examined how PARLSkd3K387A (Walker A) and PARLSkd3Y430A (pore loop) subunits affected PARLSkd3 disaggregase activity. Incorporation of two PARLSkd3K387A (Walker A) subunits is sufficient to inactivate the PARLSkd3 hexamer (Figure 6G). Thus, PARLSkd3 hexamers are sensitive to individual subunits that are unable to bind or hydrolyze ATP due to a defective Walker A motif. This finding reinforces our previous observation that PARLSkd3 disaggregase activity is sharply inhibited by the slowly hydrolyzable ATP analog, ATPγS (Figures 5E and 5F). By contrast, five PARLSkd3Y430A (pore loop) subunits are needed to inactivate the PARLSkd3 hexamer (Figure 6H). Even though PARLSkd3Y430A has reduced ATPase activity (Cupo and Shorter, 2020b), this ATPase defect is more readily buffered by WT subunits upon incorporation into PARLSkd3 hexamers (Figure 6F). Thus, insufficient substrate binding by the pore loops must contribute to inhibition by PARLSkd3Y430A subunits. We suggest that PARLSkd3 utilizes a subglobally cooperative mechanism to disaggregate disordered luciferase aggregates. Specifically, at least five subunits must have a functional Walker A motif to bind and hydrolyze ATP and at least two subunits must be able to engage substrate tightly via Y430 for productive luciferase disaggregase activity. Our findings illustrate that PARLSkd3 hexamers display robustness and can buffer a defined number of specific mutant subunits. For example, PARLSkd3 hexamers can tolerate one subunit with a defective Walker A motif, and four subunits with a defective pore loop, and still drive luciferase disaggregation. This variable robustness has implications for SCN and MGCA7 etiology.
SCN-linked subunits inhibit PARLSkd3 activity more severely than MGCA7-linked PARLSkd3 subunits
We surveyed the location of disease-linked mutations in the Skd3 structure. Biallelic MGCA7-linked mutations are dispersed throughout Skd3 (Figure 7A) (Wortmann et al., 2015). MGCA7-linked mutations are found in the MTS, ANK, NBD, and CTD (Figure 7A). T268M, A269T, and Y272C cluster in ankyrin repeat 3 and are conserved residues of the ankyrin-repeat motif (Figure 7A). Specifically, residues T268 and A269 lie near the N-terminal portion of the first α helix in ankyrin repeat 3 (Figure S3F). Mutations at T268 and A269 might destabilize the α helix and disrupt ankyrin repeat 3 (Mosavi et al., 2002). MGCA7-linked mutations are also found in the large (e.g., M411I, R460P, C486R, and E501K) and small (e.g., A591V, R628C, and R650P) NBD subdomains (Figure 7A). Some MGCA7-linked NBD mutations, such as R475Q and R408G, are in residues that make interprotomer contacts (Figure 7B).
Figure 7. SCN-linked subunits inhibit PARLSkd3 activity more severely than MGCA7-linked PARLSkd3 subunits.
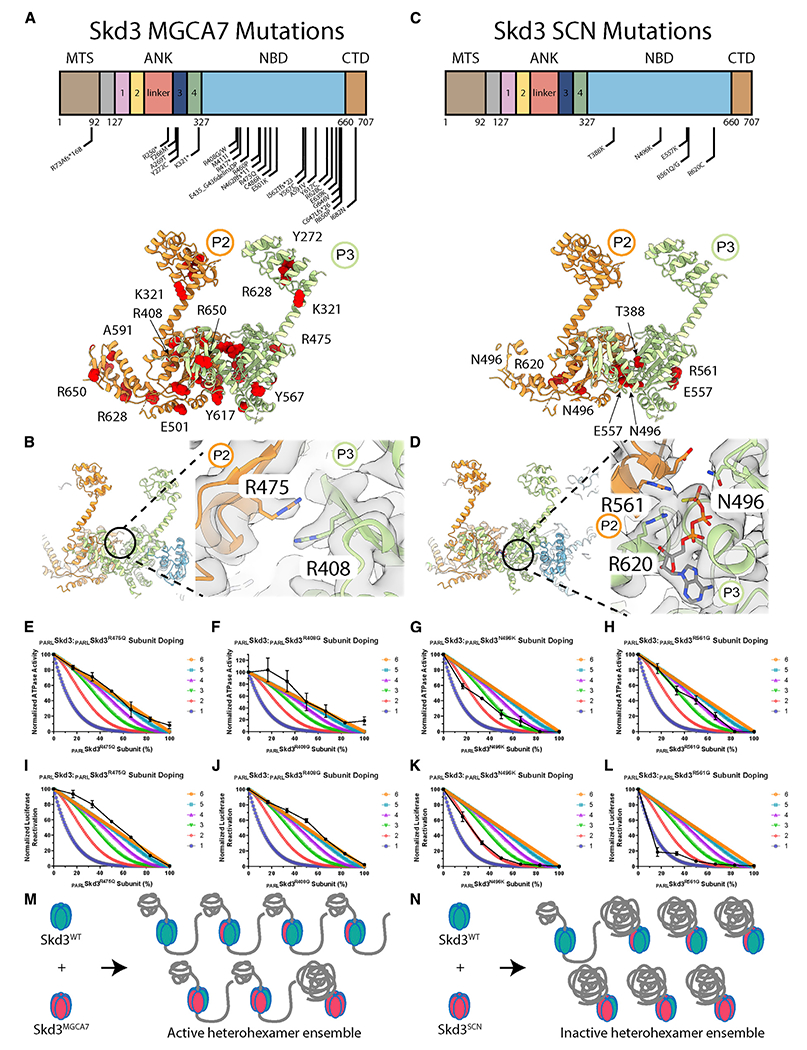
(A) Location of MGCA7-linked biallelic mutations in Skd3 (top). Model of protomers P2 and P3 with MGCA7-linked mutations colored in red (bottom).
(B) Model of back protomers colored by individual protomers (left). Interaction interface of residue R475 from protomer P2 and residue R408 from protomer P3.
(C) Location of SCN-linked mutations in Skd3 (top). Model of protomers P2 and P3 with SCN-linked mutations colored in red.
(D) Model of back protomers colored by individual protomers (left). Interaction interface of residues E557 and R561 of protomer P2 and residues N496 and R620 from protomer P3 within the nucleotide-binding pocket of protomer P3.
(E–H) ATPase activity of PARLSkd3 mixed with various ratios of PARLSkd3R475Q (E), PARLSkd3R408G (F), PARLSkd3N496K (G), or PARLSkd3R561G (H). ATPase activity was normalized to PARLSkd3 (n = 3, black dots are means ± SEM).
(I–L) Luciferase disaggregase activity of PARLSkd3 mixed with various ratios of PARLSkd3R475Q (I), PARLSkd3R408G (J), PARLSkd3N496K (K), or PARLSkd3R561G (L). Disaggregase activity was normalized to PARLSkd3 (n = 3, black dots are means ± SEM).
(M) PARLSkd3 hexamers containing a mixture of WT and MGCA7-linked subunits are typically active disaggregases.
(N) PARLSkd3 hexamers containing a mixture of WT and SCN-linked subunits are typically less active disaggregases. See also Figure S6.
By contrast, SCN-linked mutations are found exclusively in the NBD and cluster within the nucleotide-binding pocket (Figures 7C and 7D) (Warren et al., 2022). Most of the SCN-linked mutations are in canonical AAA+ motifs. N496K mutates the sensor-1 motif, which coordinates the attacking water molecule relative to the γ-phosphate of ATP (Figures 2E, S2I, and 7D) (Puchades et al., 2020). R561G mutates the Arg finger, which contacts the γ-phosphate of ATP in the nucleotide-binding pocket of the adjacent protomer and is key for ATP hydrolysis (Figures 2E, S2I, and 7D) (Puchades et al., 2020). R620C mutates the sensor-2 motif, which contacts the β- and γ-phosphates of ATP (Figures 2E, S2I, and 7D) (Puchades et al., 2020). T388K is directly adjacent to the Walker A motif, is highly conserved among other HCLR clade AAA+ proteins, and faces into the nucleotide-binding pocket (Figures 2E and 7C). Finally, E557K lies within a conserved stretch of residues near the Arg finger and contacts the nucleotide (Figures 7C and 7D).
MGCA7-linked mutations impair disaggregase activity in a way that predicts disease severity (Cupo and Shorter, 2020b). However, MGCA7-linked mutations do not always impair ATPase activity (Cupo and Shorter, 2020b). By contrast, SCN-linked mutations invariably impair ATPase and disaggregase activity (Warren et al., 2022). It is not understood why SCN-linked mutations are dominant negative, whereas MGCA7-linked mutations are recessive. To assess how severely MGCA7-linked and SCN-linked variants affect WT PARLSkd3 activity, we selected three MGCA-7 linked variants (R408G, R475Q, and A591V) and three SCN-linked variants (N496K, R561G, and R620C) for subunit doping studies (Figures 7A–7D) (Pronicka et al., 2017; Warren et al., 2022). All of these mutations severely impair disaggregase activity (Cupo and Shorter, 2020b; Warren et al., 2022). Likewise, these disease-linked variants have diminished ATPase activity, with the exception of the MGCA7-linked variant PARLSkd3R408G, which retains ~20% ATPase activity (Cupo and Shorter, 2020b; Warren et al., 2022).
We assessed the ability of the disease-linked PARLSkd3 variants to form hexamers and dodecamers in the presence of ATPγS and absence of substrate. Unlike PARLSkd3, which was shifted toward the hexameric form, two of the MGCA7-linked variants, PARLSkd3R408G and PARLSkd3R475Q, were shifted toward dodecamers (Figure S6A). By contrast, MGCA7-linked PARLSkd3A591V was shifted to lower-molecular-weight oligomers (Figure S6A). PARLSkd3A591V formed some hexamers, but dodecamers were reduced (Figure S6A). The SCN-linked variant, PARLSkd3N496K, was shifted toward hexamers like PARLSkd3 (Figure S6B). The remaining SCN-linked variants, PARLSkd3R561G and PARLSkd3R620C, were shifted toward dodecamers (Figure S6B).
To test how severely disease-linked variants affected WT PARLSkd3 activity, we mixed each disease-linked variant and WT PARLSkd3 and assessed ATPase activity. FRET studies showed that disease-linked PARLSkd3 subunits were effectively incorporated into WT PARLSkd3 hexamers (Figures S6C and S6D). Addition of the MGCA7-linked variant PARLSkd3R475Q to PARLSkd3 revealed that six PARLSkd3R475Q subunits are required to reduce ATPase activity to the same level as PARLSkd3R475Q (Figure 7E). PARLSkd3R408G retains ~20% of WT ATPase activity and five PARLSkd3R408G subunits per hexamer reduced ATPase activity to this level (Figure 7F). Likewise, five PARLSkd3A591V subunits per hexamer are required to eliminate ATPase activity (Figure S6E). Thus, MGCA7-linked subunits have mild inhibitory effects on the ATPase activity of WT subunits.
The inhibitory effect of SCN-linked subunits on ATPase activity was more severe. Incorporation of two to four PARLSkd3N496K subunits inactivated the hexamer (Figure 7G). Moreover, three to four SCN-linked PARLSkd3R561G subunits or four PARLSkd3R620C subunits inactivated the hexamer (Figures 7H and S6G). Thus, SCN-linked subunits more sharply inhibit the ATPase activity of WT subunits than MGCA7-linked subunits.
We assessed how MGCA7-linked subunits affected PARLSkd3 disaggregase activity in mixing experiments. Six MGCA7-linked PARLSkd3R408G, PARLSkd3R475Q, or PARLSkd3A591V subunits were needed to eliminate PARLSkd3 disaggregase activity (Figures 7I, 7J, and S6F). Strikingly, PARLSkd3A591V subunits barely affected disaggregase activity even when three mutant subunits were incorporated into the hexamer (Figure S6F). Thus, even one WT PARLSkd3 subunit in an otherwise PARLSkd3R408G, PARLSkd3R475Q, or PARLSkd3A591V hexamer enables disaggregase activity. Indeed, MGCA7-linked mutant subunits have only minor effects on the disaggregase activity of WT subunits within the hexamer. The strong buffering activity of WT PARLSkd3 subunits explains why MGCA7-linked mutations are biallelic and recessive.
Finally, we assessed how SCN-linked subunits affected PARLSkd3 disaggregase activity in mixing experiments (Figures 7K, 7L, and S6H). Incorporation of only two SCN-linked PARLSkd3N496K subunits inactivated the hexamer (Figure 7K). Only one or two PARLSkd3R561G subunits were required to inactivate the hexamer (Figure 7L). Finally, incorporation of three SCN-linked PARLSkd3R620C subunits inactivated the hexamer (Figure S6H). Generally, SCN-linked subunits have a sharper inhibitory effect on WT PARLSkd3 than MGCA7-linked subunits (Figures 7E–7L and S6E–S6J). These results explain why SCN-linked mutations are dominant negative and typically monoallelic.
DISCUSSION
Here, we describe PARLSkd3 (human CLPB) structure and define mechanisms by which PARLSkd3 disaggregates proteins. PARLSkd3 forms a hexamer with an asymmetric seam between protomers P1 and P6, analogous to other AAA+ proteins, such as Hsp104/ClpB and ClpA (Gates et al., 2017; Lopez et al., 2020; Rizo et al., 2019). PARLSkd3 hexamers engage substrate in their central channel via pore-loop interactions in the NBD. Indeed, PARLSkd3 may employ a conserved translocation mechanism of other AAA+ translocases (Gates et al., 2017; Puchades et al., 2017; Rizo et al., 2019). Mutation of conserved primary pore-loop residues that engage the substrate (e.g., Y430 and V431) reduce disaggregase activity (Cupo and Shorter, 2020b). Interestingly, mutations at V431 are found in humans (V431D, V431A, and V431I) with low frequency, although none of the known carriers are homozygous (Karczewski et al., 2020). We predict that specific biallelic mutations to V431 would be highly pathogenic.
One of the most prominent features of the PARLSkd3 structure is the dodecamer, which is created by two hexamers making head-to-head contacts via the ANK domain. While our paper was under review, it has been independently established that PARLSkd3 forms dodecamers (Spaulding et al., 2022; Wu et al., 2022). Moreover, Skd3 forms higher-order structures in cells, consistent with dodecamerization (Thevarajan et al., 2020). The type of dodecameric arrangement formed by PARLSkd3 is unusual for a AAA+ protein, but the AAA+ Lon protease can form dodecamers, which exhibit reduced activity for some substrates (Vieux et al., 2013). By contrast, our findings suggest that dodecamer formation may enhance PARLSkd3 disaggregase activity. PARLSkd3 hexamers and dodecamers are in dynamic equilibrium, but the dodecamer predominates upon polypeptide binding. The head-to-head ANK contacts could concentrate PARLSkd3 disaggregases on the aggregate surface and enable stronger pulling forces by maximizing the number of hexamers, simultaneously processing the substrate. Indeed, PARLSkd3ΔL, which lacks the ANK linker region, exhibited reduced dodecamerization and reduced disaggregase activity, whereas ATPase activity was unaffected. Moreover, PARLSkd3ΔL507-I534, which lacks the insertion in the NBD, exhibits increased dodecamerization, disaggregase activity, and ATPase activity. Thus, dodecamerization might enhance PARLSkd3 disaggregase activity.
The ankyrin repeats are an unusual feature of PARLSkd3. The ANK and NBD are required for Skd3 disaggregase activity (Cupo and Shorter, 2020b). Alpha-fold predicts that N-terminal ankyrin repeats 1 and 2 stack on the C-terminal ankyrin repeats 3 and 4, with the largely disordered linker excluded (Figure 3A). Deletion of ankyrin repeats 1 and 2, the linker, or ankyrin repeats 3 and 4 from the ANK reduces disaggregase activity, but not ATPase activity. However, each of these deletion variants retained ~20%-45% PARLSkd3 disaggregase activity, indicating that the remaining ankyrin repeats and linker can support some activity. The linker region helps to promote dodecamer formation, whereas deletion of ankyrin repeats 1 and 2 or 3 and 4 does not perturb dodecamerization. The ankyrin repeats likely also play a role in substrate engagement (Wu et al., 2022) or disaggregase plasticity analogous to the Hsp104 N-terminal domain (Sweeny et al., 2015).
Interestingly, several Skd3 transcript variants are found in humans, which differ only in the ANK (The UniProt Consortium, 2021). Residues R152-N180, corresponding to part of ankyrin-repeats 1 and 2, are absent in transcript variant 3. Residues D216-G245, corresponding to the middle section of the linker, are absent in transcript variants 2 and 3. The functional consequences of these transcript variants are not clear, but both deletions correspond to the length of almost exactly one ankyrin repeat (Figure 1A). Thus, cells may tune the level of Skd3 disaggregase activity via translation of these distinct Skd3 transcripts.
Mutant subunit-doping studies suggest that PARLSkd3 uses a subglobally cooperative mechanism (i.e., two to five subunits collaborate) to disaggregate disordered luciferase aggregates. PARLSkd3 disaggregase activity was very sensitive to mutant subunits that were defective in ATP hydrolysis. Thus, one or two Arg-finger mutant (R561G) subunits, two Walker A mutant (K387A) or sensor-1 mutant (N496K) subunits, or three sensor-2 mutant (R620C) subunits per PARLSkd3 hexamer ablated activity. PARLSkd3 hexamers exhibit some robustness and can buffer the incorporation of a specific number of mutant subunits, i.e., one subunit with a defective Walker-A motif or sensor-1 motif and two subunits with a defective sensor-2 motif. These results reveal that some AAA+ motifs are more important for subunit cooperativity within the hexamer than others. For example, the Arg finger appears to be more critical than the sensor-2 motif. PARLSkd3 hexamers also exhibited robustness against subunits with a defective primary pore loop (Y430A). Thus, PARLSkd3 could tolerate four subunits with the Y430A mutation, indicating that two functional pore loops are required for PARLSkd3 to maintain a grip on substrate during disaggregation. PARLSkd3 differs from Hsp104, which uses a probabilistic mechanism to disaggregate disordered luciferase aggregates (DeSantis et al., 2012).
Skd3 is a therapeutic target to inhibit in prostate cancer and Venetoclax-resistant acute myeloid leukemia (Chen et al., 2019; Pudova et al., 2020). Our structures of PARLSkd3 will enable design of small-molecule inhibitors. They are also useful for interpreting disease-linked mutations. SCN-linked mutations cluster within the nucleotide-binding pocket, whereas MGCA7-linked mutations are scattered throughout Skd3. We establish that SCN-linked mutant subunits more sharply inhibit PARLSkd3 ATPase and disaggregase activity than MGCA7-linked subunits. The robustness of PARLSkd3 against inhibition by MGCA7-linked subunits explains why MGCA7-linked mutations are recessive and must be biallelic to cause disease. Moreover, the sharp inhibition by SCN-linked mutant subunits explains why SCN-linked mutations are dominant negative.
MGCA7 and SCN are caused by loss-of-function Skd3 mutations (Cupo and Shorter, 2020b; Warren et al., 2022). In principle, both diseases could be treated by increasing Skd3 activity. However, due to the mechanistic differences between how MGCA7-linked and SCN-linked subunits affect activity of WT PARLSkd3, different treatment modalities will likely be beneficial for each disease. For treating biallelic MGCA7 mutations, expression of WT Skd3 via adeno-associated viruses (AAVs) is a therapeutic option. Indeed, expression of WT genes via AAVs has yielded therapies for congenital blindness and spinal muscular atrophy (Al-Zaidy et al., 2019; Mendell et al., 2017). Here, the robustness of PARLSkd3 hexamers against MGCA7-linked subunits will enable restoration of PARLSkd3 activity (Figure S6I). By contrast, SCN-linked mutations are dominant negative, and SCN-linked subunits sharply inhibit WT PARLSkd3. Consequently, an AAV strategy to deliver the WT Skd3 gene is likely to be less effective (Figure S6J). We suggest that a therapeutic strategy that reduces or eliminates expression of the mutant Skd3 allele is likely to be more beneficial for SCN. Here, gene editing, specific antisense oligonucleotides, or AAV-delivered siRNA to specifically reduce mutant allele expression could be therapeutic strategies to enable restoration of Skd3 activity.
Limitations of the study
We were unable to resolve a high-resolution structure for the ANK and thus an Alpha -fold model was used. Future studies should solve the atomic structure and dynamics of the ANK. We resolve a PARLSkd3 dodecamer that is formed from the head-to-head contact of two hexamers. We establish that the dodecamer exists in solution, but future studies with endogenous substrates and cell models will be needed to further probe the importance of PARLSkd3 dodecamers in mitochondria.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James Shorter (jshorter@pennmedicine.upenn.edu).
Materials availability
Plasmids newly generated in this study will be made readily available to the scientific community. We will honor requests in a timely fashion. Material transfers will be made with no more restrictive terms than in the Simple Letter Agreement or the Uniform Biological Materials Transfer Agreement and without reach through requirements.
Data and code availability
PARLSkd3:casein:ATPγS cryo-EM maps and atomic coordinates have been deposited in the EMDB and PDB with accession codes EMDB: 26121 (State 1), EMDB: 26122 (State 1 filtered), PDB: 7TTR (State 1, AAA + only), and PDB: 7TTS (State 1, Full Model). The original/source data in the paper are available from the lead contact upon reasonable request.
This paper did not generate code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli BL21-CodonPlus (DE3)-RIL competent cells (Genotype: B F– ompT hsdS(rB – mB –) dcm + Tetr gal l (DE3) endA Hte [argU ileY leuW Camr) (Agilent, Cat#230245) were used to express recombinant proteins for purification.
METHOD DETAILS
Multiple sequence alignments
NBD sequences were acquired via UniProtKB for Homo sapiens Skd3, Escherichia coli ClpA, Escherichia coli ClpB, Staphylococcus aureus ClpC, Escherichia coli ClpX, Saccharomyces cerevisiae Hsp78, Arabidopsis thaliana Hsp101, Saccharomyces cerevisiae Hsp104, Escherichia coli RuvB, and Pseudomonas aeruginosa ClpG. Ankyrin-repeat sequences were acquired via UniProtKB for Homo sapiens Skd3. Consensus ankyrin repeat was derived from Mosavi, et al. (Mosavi et al., 2002). Compiled sequences were aligned via Clustal Omega (Madeira et al., 2019). The linker region of the ankyrin repeats was aligned manually to the Clustal Omega alignment. Alignment image was generated via BoxShade tool as described previously (Cupo and Shorter, 2020b).
Purification of PARLSkd3
PARLSkd3 and variants were purified as previously described (Cupo and Shorter, 2020a, b). In short, PARLSkd3 and variants were expressed with an N-terminal MBP-tag in BL21 (DE3) RIL cells (Agilent). Cells were lysed via sonication in lysis buffer (40 mM HEPES-KOH pH = 7.4, 500 mM KCl, 20% [w/v] glycerol, 5 mM ATP, 10 mM MgCl2, 2 mM β-mercaptoethanol, 2.5 μM PepstatinA, and complete Protease Inhibitor Cocktail [one tablet/250 mL, Millipore Sigma]). Lysates were cleared via centrifugation at 30,597xg and 4°C for 20 min and the supernatant was applied to amylose resin (NEB). The column was washed with 15 column volumes (CV) of wash buffer (WB: 40 mM HEPES-KOH pH = 7.4, 500 mM KCl, 20% [w/v] glycerol, 5 mM ATP, 10 mM MgCl2, 2 mM β-mercaptoethanol, 2.5 μM PepstatinA, and cOmplete Protease Inhibitor Cocktail [1 full size tablet/50mL, Millipore Sigma]) at 4°C, 3 CV of WB supplemented with 20 mM ATP at 25°C for 30 min, and an additional 15 CV of WB at 4°C. The protein was then washed with ~8 CV of elution buffer (EB: 50 mM Tris-HCl pH = 8.0, 300 mM KCl, 10% glycerol, 5 mM ATP, 10 mM MgCl2, and 2 mM β-mercaptoethanol) and eluted via TEV protease cleavage at 34°C. The protein was run over a size exclusion column (GE Healthcare HiPrep 26/60 Sephacryl S-300 HR) in sizing buffer (50 mM Tris-HCl pH = 8.0, 500 mM KCl, 10% glycerol, 1 mM ATP, 10 mM MgCl2, and 1 mM DTT). Peak fractions were collected, concentrated to ~5 mg/mL, supplemented with 5 mM ATP, and snap frozen. Protein purity was determined to be >95% by SDS-PAGE and Coomassie staining.
Purification of Hsp104
Hsp104 was purified as previously described (DeSantis et al., 2012). In short, Hsp104 was expressed in BL21 (DE3) RIL cells, lysed via sonication in lysis buffer (50 mM Tris-HCl pH = 8.0, 10 mM MgCl2, 2.5% glycerol, 2 mM β-mercaptoethanol, 2.5 μM PepstatinA, and cOmplete Protease Inhibitor Cocktail [one mini EDTA-free tablet/50 mL, Millipore Sigma]), centrifuged at 30,597xg and 4°C for 20 min, and purified on Affi-Gel Blue Gel (Bio-Rad). Hsp104 was eluted in elution buffer (50 mM Tris-HCl pH = 8.0, 1M KCl, 10 mM MgCl2, 2.5% glycerol, and 2 mM β-mercaptoethanol) and then exchanged into storage buffer (40 mM HEPES-KOH pH = 7.4, 500 mM KCl, 20 mM MgCl2, 10% glycerol, 1 mM DTT). The protein was diluted to 10% in buffer Q (20 mM Tris-HCl pH = 8.0, 50 mM NaCl, 5 mM MgCl2, and 0.5 mM EDTA) and loaded onto a 5 mL RESOURCE Q anion exchange chromatography (GE Healthcare). Hsp104 was eluted via linear gradient of buffer Q+ (20 mM Tris pH = 8.0, 1M NaCl, 5 mM MgCl2, and 0.5 mM EDTA). The protein was exchanged into storage buffer and snap frozen. Protein purity was determined to be >95% by SDS-PAGE and Coomassie staining.
Purification of Hsc70 and Hdj1
Hsc70 and Hdj1 were purified as previously described (Michalska et al., 2019). Hsc70 and Hdj1 were expressed in BL21 (DE3) RIL cells with an N-terminal His-SUMO tag. Cells were lysed via sonication into lysis buffer (50 mM HEPES-KOH pH = 7.5, 750 mM KCl, 5 mM MgCl2, 10% glycerol, 20 mM imidazole, 2 mM β-mercaptoethanol, 5 μM pepstatin A, and cOmplete Protease Inhibitor Cocktail [one mini EDTA-free tablet/50 mL, Millipore Sigma]). Lysates were cleared via centrifugation at 30,597xg and 4°C for 20 min. The supernatant was bound to Ni-NTA Agarose resin (Qiagen), washed with 10 CV of wash buffer (50 mM HEPES-KOH pH = 7.5, 750 mM KCl, 10 mM MgCl2, 10% glycerol, 20 mM imidazole, 1 mM ATP, and 2 mM β-mercaptoethanol), and eluted with 2 CV of elution buffer (wash buffer supplemented with 300 mM imidazole). The tag was removed via Ulp1 (1:100 Ulp1:Protein molar ratio) cleavage during dialysis into wash buffer. The protein was further purified via loading onto a 5 mL HisTrap HP column (GE Healthcare) and pooling the untagged elution. The protein was pooled and concentrated, and then purified further via Resource Q ion-exchange chromatography. The elution was pooled, concentrated, and snap frozen. Protein purity was determined to be >95% via SDS-PAGE and Coomassie staining.
Size-exclusion chromatography
All size-exclusion chromatography experiments were run on a Superose 6 Increase 3.2/300 (Cytiva) column pre-equilibrated in buffer containing: 40 mM HEPES (pH = 8.0), 40 mM KCl, 10 MgCl2, and 1 mM DTT. To form a substrate-bound complex, PARLSkd3 (20 μM) was incubated with FITC-casein (55 μM) (#C0528; Sigma) in the presence of nucleotide (ATPγS, ATP, ADP, or AMP-PNP) (5 mM) for 15 min at room temperature. For experiments without FITC-casein, PARLSkd3 (20 μM) and nucleotide (ATPγS, ATP, ADP, or AMP-PNP) (5 mM) were incubated for 15 min at room temperature. After the incubation period, the samples were spin-filtered before injecting on column.
Size-exclusion chromatography with multiangle light scattering (SEC-MALS)
Experiments were conducted at the Johnson Foundation Structural Biology and Biophysics Core at the Perelman School of Medicine (Philadelphia, PA). The SEC experiments were performed using 100 μL injections (125μM PARLSkd3) with a Superose 6 Increase 10/300 GL column (GE Life Sciences) at 0.5 mL/min at room temperature. The column was equilibrated with 20 mM Tris-HCl (pH = 8.00), 150 mM KCl, and 10 mM MgCl2 and 1 mM DTT at 25°C. Absolute molecular weights were determined by MALS. The scattered light intensity of the column eluent was recorded at 18 different angles using a DAWN-HELEOS MALS detector (Wyatt Technology Corporation) operating at 658 nm after calibration with the monomer fraction of Type V BSA (Sigma). The protein concentration of the eluent was determined using an in-line Optilab T-rex interferometric refractometer (Wyatt Technology Corporation). The weight-averaged molecular weight of species within defined chromatographic peaks was calculated using the software ASTRA, version 8.0 (Wyatt Technology Corporation), by construction of Debye plots [(KC/Rθ versus sin2(θ/2)] at 1/S data intervals. The weight-averaged molecular weight was then calculated at each point of the chromatographic trace from the Debye plot intercept, and an overall average molecular weight was calculated by averaging across the peak.
Cryo-EM data collection and processing for PARLSkd3:casein:ATPγS Complex
To form a substrate-bound complex, PARLSkd3 (55 μM) was incubated with FITC-casein (55 μM) (#C0528; Sigma) in the presence of ATPγS (5 mM) in buffer containing: 40 mM HEPES (pH = 8.0), 40 mM KCl, 10 MgCl2, 1 mM DTT. After incubating for 15 min at room temperature, the sample was applied to a Superose 6 Increase 3.2/300 column (GE Healthcare) for size exclusion chromatography (SEC) analysis. The fraction corresponding to the largest molecular weight complex from SEC of PARLSkd3 and FITC-casein (Figure 1B) was isolated and incubated with 1 mM ATPγS. Before freezing, proper dilutions were made to a final concentration of ~.7 mg/mL and a 3.0 μL drop was applied to glow discharged holey carbon (R 1.2/1.3; Quantifoil), then blotted for 3 s at 4°C and 100% humidity with a blot force of 1, followed by an additional 3.0μL drop. The sample was then blotted again for 2 s with a blot force of 0 with Whatman No. 1 filter paper before being plunge frozen in liquid ethane using a Vitrobot (Thermo Fischer Scientific).
The sample was then imaged on a Titan Krios TEM (Thermo Fischer Scientific) operated at 300 keV and equipped with a Gatan BioQuantum imaging energy filter using a 20eV zero loss energy slit (Gatan Inc). Movies were acquired in super-resolution mode on a K3 direct electron detector (Gatan Inc.) at a nominal magnification of 105,000X corresponding to a physical pixel size of 0.417 Å/pixel. A defocus range of 0.8–1.2 μm was used with a total exposure time of 2 s fractionated into 0.2s subframes for a total dose of 68 e−/Å2 at a dose rate of 25 e−/pixel/s. Movies were subsequently corrected for drift using MotionCor2 (Zheng et al., 2017) and were Fourier cropped by a factor of 2 to a final pixel size of 0.834 Å/pixel.
A total of ~30,000 micrographs were collected over multiple datasets. Micrograph quality was assessed and poor micrographs, including those above the resolution cutoff of ~5Å, were discarded. The individual datasets were processed separately to ensure data quality before combining them all together for further processing. Data processing was performed in cryoSPARC v3.2 (Punjani et al., 2017). For particle picking, blob picker was set to 180 Å-200 Å for minimum and maximum particle diameter and the particles picked were inspected before extracting particles. 2D classification was performed to remove contamination and junk particles and good classes were selected which left ~900,000 remaining particles. Four different ab-initio models were reconstructed which were then used in 3D classification.
Heterogeneous refinement was performed with 4 different classes, which resulted in 3 distinct classes: hexamer, Class 1 (41%, ~358K particles); heptamer, Class 3 (19%, ~167K particles); and dodecamer, Class 2 (15%, ~130K particles); Other, Class 4 (24%, ~210K particles) (Figure S1F). Each of the classes underwent homogeneous refinement which resulted in resolutions of 9Å for the dodecamer, 7 Å for the heptamer and 3.2Å for the hexamer. To improve the resolution, non-uniform refinement was completed on both the dodecamer and hexamer to improve the resolutions to 7.2Å and 2.9Å respectively (Figure S2B). Both classes underwent local CTF refinement which did not result in an improvement in resolution.
Cryo-EM data collection and processing for PARLSkd3Δ1-2:casein:ATPγS Complex
To form a substrate-bound complex, PARLSkd3Δ1-2 (40 μM) was incubated with FITC-casein (40 μM) (#C0528; Sigma) in the presence of ATPγS (5 mM) in buffer containing: 40 mM HEPES (pH = 8.0), 40 mM KCl, 10 MgCl2, 1 mM DTT. After incubating for 15 min at room temperature, grids were prepared. For grid freezing, a 3.0 μL drop was applied to glow discharged holey carbon (R 1.2/1.3; Quantifoil), then blotted for 3 s at 4°C and 100% humidity with a blot force of 1 followed by an additional 3.0μL drop. The sample was then blotted again for 2 s with a blot force of 0 with Whatman No. 1 filter paper before being plunge frozen in liquid ethane using a Vitrobot (Thermo Fischer Scientific).
The sample was then imaged on a Glacios TEM (Thermo Fischer Scientific) operated at 200 keV (Gatan Inc). Movies were acquired in super-resolution mode on a K2 direct electron detector (Gatan Inc.) at a nominal magnification of ,45,000X corresponding to a physical pixel size of 0.486 Å/pixel. A defocus range of 1.0–2.0 μm was used with a total exposure time of 6 s fractionated into 0.06s subframes for a total dose of 55.8 e−/Å2 at a dose rate of 8 e−/pixel/s. Movies were subsequently corrected for drift using MotionCor2 (Zheng et al., 2017) and were Fourier cropped by a factor of 2 to a final pixel size of 0.972 Å/pixel.
A total of ~15,000 micrographs were collected over multiple datasets. The individual datasets were processed separately to ensure data quality before combining them all together for further processing. Micrograph quality was assessed and poor micrographs, including those above the resolution cutoff of ~5Å, were discarded. Data processing was performed in cryoSPARC v3.2 (Punjani et al., 2017). For particle picking, blob picker was set to 180 Å-200 Å for minimum and maximum particle diameter and the particles picked were inspected before extracting particles. 2D classification was performed in two rounds to remove contamination and junk particles and good classes were selected which left ~700,000 remaining particles. The results from a previous 3D classification were used as the starting models for 3D classification.
Heterogeneous refinement was performed with 4 different classes, which resulted in 3 distinct classes: dodecamer, Class 1 (24%, ~165K particles); bent dodecamer, Class 2 (30%, ~203K particles); trimer, Class 3 (32%, ~213K particles); and other, Class 4 (14%, ~95K particles) (Figure S3I). Each of the classes underwent homogeneous refinement which resulted in resolutions of 8Å for the dodecamer, 7Å for the bent dodecamer and 8Å for the trimer (Figure S3J).
Molecular modeling
An initial model for PARLSkd3was generated in SWISS-MODEL (Waterhouse et al., 2018) and was docked into the EM map using the UCSF chimera’s function fit in map (Pettersen et al., 2004). The initial model lacked the ANK so the SWISS-MODEL generated was combined with the Alpha-fold prediction of the ANK taken from the AlphaFold Protein Structure Database. Initial refinement was performed using Rosetta_Relax in cartesian space to generate 30 different models. The map/model quality for each model generated was examined in Chimera (Pettersen et al., 2004) and the lowest energy minimized model was used moving forward. Various outliers and poorly fit density were manually fixed using ISOLDE (Croll, 2018) in ChimeraX (Pettersen et al., 2021). To fix most of the outliers another round of Rosetta_Relax in cartesian space was performed followed by iterative rounds of refinement in Phenix Real Space Refine (Liebschner et al., 2019). The model from Phenix Real Space refinement was taken and used in a final round of Rosetta FastRelax in torsion space to remove the various clashes that were introduced during Phenix refinement.
ATPase assays
Hsp104, PARLSkd3, and PARLSkd3 variants (0.25 μM monomer) were incubated with ATP (1 mM) (Innova Biosciences) at 37°C for 5 min in luciferase reactivation buffer (LRB; 25 mM HEPES-KOH [pH = 8.0], 150 mM KAOc, 10 mM MgAOc, 10 mM DTT). ATPase activity was assessed via inorganic phosphate release with a malachite green detection assay (Expedeon) and measured in Nunc 96 Well Optical plates on a Tecan Infinite M1000 plate reader. Background hydrolysis was measured at time zero and subtracted (Cupo and Shorter, 2020b; DeSantis et al., 2012). ATPase kinetics for PARLSkd3 were calculated using GraphPad Prism with a Michaelis-Menten least squares fit which was subsequently used to derive KM and Vmax.
Luciferase disaggregation and reactivation assays
Firefly luciferase was aggregated by incubating luciferase (50 μM) in LRB (pH = 7.4) with 8M urea at 30°C for 30 min. The denatured luciferase was then rapidly diluted 100-fold into ice-cold LRB, snap frozen, and stored at −80°C until use. Hsp104 (1μM monomer) was incubated with 50 nM aggregated firefly luciferase in the presence or absence of Hsc70 and Hdj2 (0.167 μM each) in LRB plus 5 mM ATP plus an ATP regeneration system (ARS; 1 mM creatine phosphate and 0.25 μM creatine kinase) at 37°C for 90 min (unless otherwise indicated). PARLSkd3 and variants (1 μM monomer, unless otherwise indicated) were incubated with 50 nM aggregated firefly luciferase in LRB plus 5 mM ATP plus ARS at 37°C for 90 min (unless otherwise indicated). Nucleotide-inhibitor assays for PARLSkd3 disaggregation activity were tested in the presence of ATP (Sigma), ATPγS (Roche), or ADP (MP Biomedicals) for 30 min at 37°C without ARS. IC50 curves for ADP and ATPγS were fitted using GraphPad Prism with a variable slope (four parameters) least squares fit. Recovered luminescence was monitored in Nunc 96 Well Optical plates using a Tecan Infinite M1000 plate reader (Cupo and Shorter, 2020b; DeSantis et al., 2012; Glover and Lindquist, 1998). Typically, Hsp104, Hsc70, and Hdj2 recovered ~10% of native luciferase activity, whereas PARLSkd3 recovered ~45% native luciferase activity (Cupo and Shorter, 2020b). Under our conditions, neither ADP nor ATPγS had an inhibitory effect on native luciferase (Figure 5).
FITC-casein binding assays
Fluorescence polarization was performed essentially as described previously (Rizo et al., 2019). PARLSkd3 was exchanged into 40 mM HEPES-KOH pH 8.0, 20 mM MgCl2, 150 mM KCl, 10% Glycerol (v/v), 2 mM β-mercaptoethanol. To assess FITC-casein binding, FITC-casein (60 nM, Sigma) was incubated with increasing concentrations (0-2.5μM hexameric) of PARLSkd3 with 2 mM of the indicated nucleotide for 10 min at 25°C. For the ATP condition, an ATP regeneration system (5 mM creatine phosphate and 0.125 μM creatine kinase) was included to maintain 2 mM ATP. Fluorescence polarization was measured (excitation 470 nm, emission 520 nm) using a Tecan Infinite M1000 plate reader. The binding isotherms were analyzed using Prism.
Modeling heterohexamer ensemble activity
The binomial distribution was used to model the activity of various heterohexamer ensembles (DeSantis et al., 2012; Werbeck et al., 2008):
Where: P(k) is the probability of a hexamer containing k mutant subunits, n is total number of subunits (which for a hexamer, n = 6), and p is the probability that a mutant subunit is incorporated. FRET subunit mixing experiments demonstrated that mutant and WT subunits have a similar probability of being incorporated into a hexamer (Figures S5D, 6A and 6B). Thus, p is calculated as the molar ratio of mutant and WT protein present:
Therefore, for any specified concentration of mutant protein, the probability distribution of PARLSkd3 hexamers containing zero, one, two, three, four, five, or six mutant subunits can be derived (Figure 6A). Activity versus p plots (Figure 6B) can then be generated assuming each WT subunit makes an equal contribution to the total activity (one-sixth per subunit). Consequently, if subunits within the hexamer operate independently then activity should decline linearly upon incorporation of mutant subunits. Conversely, if subunit activity is coupled then the incorporation of a specific number of subunits will be sufficient to abolish activity. In our model, zero activity is assigned to hexamers that exceed the specific threshold number of mutant subunits. In this way, we generate activity versus p plots by assuming that 1 or more, 2 or more, 3 or more, 4 or more, or 5 or more mutant subunits are required to eliminate activity. This formula can be expressed as:
Where: P(k) is the probability of hexamer containing k mutant subunits derived above and A(k) is the relative assigned contribution to activity of a hexamer containing k mutant subunits.
Alexa fluor labeling of PARLSkd3
For Förster resonance energy transfer (FRET) studies, we labeled separate pools of PARLSkd3 with Alexa Fluor 488 (Alexa 488, ThermoFisher Scientific CAT# A20000) as the FRET donor and Alexa Fluor 594 (Alexa 594, ThermoFisher Scientific CAT# A20004) as the FRET acceptor. In brief, PARLSkd3 (WT and mutants) was extensively exchanged into labeling buffer (LB; 50 mM HEPES-KOH [pH = 8.0], 150 mM KCl, 20 mM MgCl2, 10% glycerol, 10mM BME) at room temperature using Micro Bio-Spin 6 columns (Bio-Rad CAT# 7326200). PARLSkd3 concentration was measured via A280 and the molar extinction coefficient and PARLSkd3 concentration was adjusted to 30 μM. The primary amine (R-NH2) reactive dye, Alexa Fluor 488 NHS Ester (Succinimidyl Ester) (Alexa 488) or Alexa Fluor 594 NHS Ester (Succinimidyl Ester) (Alexa 594), was then added to PARLSkd3 samples to achieve a 10-fold molar excess over PARLSkd3. Samples were then incubated at 25°C in the dark. After 75min, the labeling reaction was quenched by rapidly and extensively exchanging into labeling buffer +10 mM DTT. To ensure that all unreacted dye is removed, the buffer exchange step was repeated at least twice. PARLSkd3 concentration and labeling efficiency were determined by UV/Vis spectrometry according to the manufacturer’s instructions (Invitrogen). Typically, we achieved ~50% labeling efficiency.
Fluorescence resonance energy transfer and subunit mixing
We employed Förster resonance energy transfer (FRET) to measure subunit mixing (Figures S5D, 6A and 6B). Donor (Alexa Fluor 488) labeled PARLSkd3 (Alexa 488- PARLSkd3), Acceptor (Alexa Fluor 594) labeled PARLSkd3 (Alexa 594-PARLSkd3), or free dye were mixed in equal stoichiometric parts to a final total dye concentration of 1μM in labeling buffer with ATP (5 mM). Because these dyes function as an FRET pair with a Förster radius of 60 Å, primary amine labeling in PARLSkd3 is expected to yield intermolecular FRET once mixed oligomers are formed. Given the R0 value of 60Å for the Alexa 488-Alexa594 FRET pair, it is possible to observe both intrahexameric FRET (e.g. solvent exposed residues K538 from P3 and K658 from P4 are 8.5 Å apart) and interhexameric FRET within a dodecamer (closest two lysines are K134 to K265 at 22.4 Å apart, but it is unclear if they are solvent exposed). However, the hexameric state is the predominant species in the absence of substrate and thus our FRET assay likely reports on the hexameric state of PARLSkd3 rather than the dodecamer (Figure 1B). A similar strategy has been employed to demonstrate the formation of mixed hexamers by bacterial ClpB (Werbeck et al., 2008), Hsp104 (DeSantis et al., 2012), and MCM helicase, another AAA + ATPase, from Sulfolobus solfataricus (McGeoch et al., 2005; Moreau et al., 2007). Prior to any measurements, samples were allowed to equilibrate for 10 min at room temperature. Mixed and equilibrated samples were excited at the donor excitation wavelength of 480nm. Donor fluorescence was measured at 519nm with a bandwidth of 5nm. Acceptor fluorescence was measured at 630nm with a bandwidth of 5nm. Apparent FRET efficiency (Figures S5D, S6C and S6D) was calculated from Alexa 488-PARLSkd3 emission (488nm) and Alexa 594-PARLSkd3 emission (519nm) as Fa/(Fd + Fa), where Fd is the measured Alexa 488-PARLSkd3 (donor) fluorescence and Fa is the Alexa 594-PARLSkd3 (acceptor) fluorescence in the presence of Alexa 488-Skd3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification is as described in the figure legends. Statistical analyses were performed using the GraphPad Prism (GraphPad Software, Inc,; La Jolla, CA, USA) as described in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli BL21-CodonPlus (DE3)-RIL competent cells | Agilent | Cat#230245 |
| Chemicals, peptides, and recombinant proteins | ||
| Creatine phosphate | Roche | Cat#10621722001 |
| Adenosine-5′-Diphosphate Monosodium Salt (ADP) | MP Biomedicals | Cat#150260 |
| Adenylyl-imidodiphosphate (AMP-PNP) | Roche | Cat#10102547001 |
| Adenosine 5′-triphosphate disodium salt hydrate (ATP) | Sigma-Aldrich | Cat#A3377 |
| Adenosine-5′-o-(3-thio-triphosphate) (ATPγS) | Roche | Cat#10102342001 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#4693159001 |
| Pepstatin A, microbial, ≥90% (HPLC) | Sigma-Aldrich | Cat#P5318 |
| Alexa Fluor™ 488 NHS Ester (Succinimidyl Ester) | ThermoFisher Scientific | Cat#A20000 |
| Alexa Fluor™ 594 NHS Ester (Succinimidyl Ester) | ThermoFisher Scientific | Cat#A20004 |
| Casein fluorescein isothiocyanate from bovine milk (FITC-Casein) | Sigma-Aldrich | Cat#C0528 |
| Creatine kinase | Roche | Cat#10127566001 |
| Firefly luciferase | Sigma-Aldrich | Cat#L9506 |
| Lysozyme | Sigma-Aldrich | Cat#L6876 |
| His-TEV protease | (Cupo and Shorter, 2020a, b) | N/A |
| Hdj1 | (Cupo and Shorter, 2020b; Michalska et al., 2019) | N/A |
| Hsc70 | (Cupo and Shorter, 2020b; Michalska et al., 2019) | N/A |
| Hsp104 | (DeSantis et al., 2012; Jackrel et al., 2014) | N/A |
| PARLSkd3 | (Cupo and Shorter, 2020a, b) | N/A |
| PARLSkd3V431G | This paper | N/A |
| PARLSkd3R417A | This paper | N/A |
| PARLSkd3NBD | (Cupo and Shorter, 2020b) | N/A |
| PARLSkd3Δ1-2 | This paper | N/A |
| PARLSkd3ΔL | This paper | N/A |
| PARLSkd3Δ3-4 | This paper | N/A |
| PARLSkd3ΔL507-I534 | This paper | N/A |
| PARLSkd3ΔL660-I707 | This paper | N/A |
| PARLSkd3K387A | (Cupo and Shorter, 2020b) | N/A |
| PARLSkd3Y430A | (Cupo and Shorter, 2020b) | N/A |
| PARLSkd3R408G | (Warren et al., 2022) | N/A |
| PARLSkd3R475Q | (Cupo and Shorter, 2020b) | N/A |
| PARLSkd3A591V | (Cupo and Shorter, 2020b) | N/A |
| PARLSkd3N496K | (Warren et al., 2022) | N/A |
| PARLSkd3R561G | (Warren et al., 2022) | N/A |
| PARLSkd3R62°C | (Warren et al., 2022) | N/A |
| Critical commercial assays | ||
| ATPase Activity Kit (Colorimetric) | Innova Biosciencens | Cat#601–0120 |
| Luciferase Assay Reagent | Promega | Cat#E1483 |
| Deposited data | ||
| EMDB: 26121 | This paper | N/A |
| EMDB: 26122 | This paper | N/A |
| PDB: 7TTR | This paper | N/A |
| PDB: 7TTS | This paper | N/A |
| Recombinant DNA | ||
| pHis-TEV | (Cupo and Shorter, 2020a, b) | N/A |
| pE-SUMO-Hdj1 | (Cupo and Shorter, 2020b; Michalska et al., 2019) | N/A |
| pE-SUMO-Hsc70 | (Cupo and Shorter, 2020b; Michalska et al., 2019) | N/A |
| pNOTAG-Hsp104 | (DeSantis et al., 2012; Jackrel et al., 2014) | N/A |
| pMAL-C2-TEV-PARLSkd3 | (Cupo and Shorter, 2020a, b) | N/A |
| pMAL-C2-TEV-PARLSkd3V431G | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3R417A | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3NBD | (Cupo and Shorter, 2020b) | N/A |
| pMAL-C2-TEV-PARLSkd3Δ1-2 | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3ΔL | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3Δ3-4 | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3ΔL507-I534 | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3ΔL660-I707 | This paper | N/A |
| pMAL-C2-TEV-PARLSkd3K387A | (Cupo and Shorter, 2020b) | N/A |
| pMAL-C2-TEV-PARLSkd3Y430A | (Cupo and Shorter, 2020b) | N/A |
| pMAL-C2-TEV-PARLSkd3R408G | (Warren et al., 2022) | N/A |
| pMAL-C2-TEV-PARLSkd3R475Q | (Cupo and Shorter, 2020b) | N/A |
| pMAL-C2-TEV-PARLSkd3A591V | (Cupo and Shorter, 2020b) | N/A |
| pMAL-C2-TEV-PARLSkd3N496K | (Warren et al., 2022) | N/A |
| pMAL-C2-TEV-PARLSkd3R561G | (Warren et al., 2022) | N/A |
| pMAL-C2-TEV-PARLSkd3R62°C | (Warren et al., 2022) | N/A |
| Software and algorithms | ||
| BOXSHADE | Github | https://github.com/mdbaron42/pyBoxshade |
| Clustal Omega | (Madeira et al., 2019) | N/A |
| Prism 8 | GraphPad | N/A |
| ASTRA, version 8.0 | Wyatt Technology | N/A |
| cryoSPARC version 2.0–3.3.2 | Structura Biotechnology Inc. | N/A |
| Chimera | (Pettersen et al., 2004) | N/A |
| ChimeraX version 1.2.5–1.3 | (Pettersen et al., 2021) | N/A |
| ISOLDE | (Croll, 2018) | N/A |
| cisTEM | (Grant et al., 2018) | N/A |
| Phenix (Phenix Real Space Refine) | (Liebschner et al., 2019) | N/A |
| Coot | (Emsley et al., 2010) | N/A |
| Rosetta (2020.08 Release) | (Wang et al., 2016) | N/A |
| AlphaFold | (Jumper et al., 2021; Varadi et al., 2022) | N/A |
Highlights.
High-resolution structure of a PARLSkd3-substrate complex
Substrate-bound PARLSkd3 hexamers stack head-to-head to form dodecamers
Mechanisms by which PARLSkd3 subunits collaborate to disaggregate proteins
Mechanisms by which Skd3 mutations cause dominant and recessive forms of disease
ACKNOWLEDGMENTS
We thank J. Lin, L. Miller, C. Fare, K. Copley, and Z. Tabassum for feedback and the Johnson Foundation Structural Biology and Biophysics Core for SEC-MALS. Our work was funded by a Blavatnik Family Foundation Fellowship (to R.R.C.), The G Harold and Leila Y Mathers Foundation (to J.S.), and NIH grants T32GM008275 (to R.R.C.), F31AG060672 (to R.R.C.), R01GM099836 (to J.S.), R21AG061784 (to J.S.), and R01GM138690 (to D.R.S).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111408.
DECLARATIONS OF INTERESTS
J.S. is a consultant for Dewpoint Therapeutics, Confluence Therapeutics, Neumora, and ADRx.
SUPPORTING CITATIONS
The following reference appears in the supplemental information: Lee et al. (2003).
REFERENCES
- Al-Zaidy SA, Kolb SJ, Lowes L, Alfano LN, Shell R, Church KR, Nagendran S, Sproule DM, Feltner DE, Wells C, et al. (2019). AVXS-101 (onasemnogene abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J. Neuromuscul. Dis 6, 307–317. [DOI] [PubMed] [Google Scholar]
- Antonicka H, Lin ZY, Janer A, Aaltonen MJ, Weraarpachai W, Gingras AC, and Shoubridge EA (2020). A high-density human mitochondrial proximity interaction network. Cell Metabol. 32, 479–497.e479. [DOI] [PubMed] [Google Scholar]
- Botham A, Coyaud E, Nirmalanandhan VS, Gronda M, Hurren R, Maclean N, St-Germain J, Mirali S, Laurent E, Raught B, et al. (2019). Global interactome mapping of mitochondrial intermembrane space proteases identifies a novel function for HTRA2. Proteomics 19, e1900139. [DOI] [PubMed] [Google Scholar]
- Capo-Chichi JM, Boissel S, Brustein E, Pickles S, Fallet-Bianco C, Nassif C, Patry L, Dobrzeniecka S, Liao M, Labuda D, et al. (2015). Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J. Med. Genet 52, 303–311. [DOI] [PubMed] [Google Scholar]
- Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, Sakellaropoulos T, Gong Y, Kloetgen A, Yap YS, et al. (2019). Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment. Cancer Discov. 9, 890–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI (2018). ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol 74, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupo RR, and Shorter J (2020a). Expression and purification of recombinant Skd3 (human ClpB) protein and tobacco Etch virus (TEV) protease from Escherichia coli. Bio Protoc. 10, e3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupo RR, and Shorter J (2020b). Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman-Nick M, Bonini NM, and Shorter J (2013). Hsp104 suppresses polyglutamine-induced degeneration post onset in a Drosophila MJD/SCA3 model. PLoS Genet. 9, e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN, and Shorter J (2012). Operational plasticity enables Hsp104 to disaggregate diverse amyloid and non-amyloid clients. Cell 151, 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durie CL, Lin J, Scull NW, Mack KL, Jackrel ME, Sweeny EA, Castellano LM, Shorter J, and Lucius AL (2019). Hsp104 and potentiated variants can operate as distinct nonprocessive translocases. Biophys. J 116, 1856–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erives A, and Fassler J (2015). Metabolic and chaperone gene loss marks the origin of animals: evidence for hsp104 and hsp78 chaperones sharing mitochondrial enzymes as clients. PLoS One 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, and Berger JM (2006). Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct 35, 93–114. [DOI] [PubMed] [Google Scholar]
- Fare CM, and Shorter J (2021). Dis)Solving the problem of aberrant protein states. Dis. Model. Mech 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei X, Bell TA, Jenni S, Stinson BM, Baker TA, Harrison SC, and Sauer RT (2020). Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, Buell CE, Sweeny EA, Mack KL, Chuang E, et al. (2017). Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellerich FN, Laterveer FD, Zierz S, and Nicolay K (2002). The quantitation of ADP diffusion gradients across the outer membrane of heart mitochondria in the presence of macromolecules. Biochim. Biophys. Acta 1554, 48–56. [DOI] [PubMed] [Google Scholar]
- Glover JR, and Lindquist S (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7, e35383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimminger V, Richter K, Imhof A, Buchner J, and Walter S (2004). The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J. Biol. Chem 279, 7378–7383. [DOI] [PubMed] [Google Scholar]
- Hattendorf DA, and Lindquist SL (2002). Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J. 21, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt HW, Klingenberg M, and Milovancev M (1972). Differences between the ATP-ADP ratios in the mitochondrial matrix and in the extramitochondrial space. Eur. J. Biochem 30, 434–440. [DOI] [PubMed] [Google Scholar]
- Howard MK, Sohn BS, von Borcke J, Xu A, and Jackrel ME (2020). Functional analysis of proposed substrate-binding residues of Hsp104. PLoS One 15, e0230198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Agrawal T, Zhu G, Yu S, Tao L, Lin J, Marmorstein R, Shorter J, and Yang X (2021). DAXX represents a new type of protein-folding enabler. Nature 597, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, Carr SA, Mootha VK, and Ting AY (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Nhat KP, Togawa H, Saito K, lino R, Kato-Yamada Y, Nagai T, and Noji H (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 106, 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaru-Ampornpan P, Shen K, Lam VQ, Ali M, Doniach S, Jia TZ, and Shan SO (2010). ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol 17, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, and Shorter J (2014). Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell. 156, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaru-Ampornpan P, Liang FC, Nisthal A, Nguyen TX, Wang P, Shen K, Mayo SL, and Shan SO (2013). Mechanism of an ATP-independent protein disaggregase: II. distinct molecular interactions drive multiple steps during aggregate disassembly. J. Biol. Chem 288, 13431–13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanabus M, Shahni R, Saldanha JW, Murphy E, Plagnol V, Hoff WV, Heales S, and Rahman S (2015). Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation. J. Inherit. Metab. Dis 38, 211–219. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiykim A, Garncarz W, Karakoc-Aydiner E, Ozen A, Kiykim E, Yesil G, Boztug K, and Baris S (2016). Novel CLPB mutation in a patient with 3-methylglutaconic aciduria causing severe neurological involvement and congenital neutropenia. Clin. Immunol 165, 1–3. [DOI] [PubMed] [Google Scholar]
- Klosowska A, Chamera T, and Liberek K (2016). Adenosine diphosphate restricts the protein remodeling activity of the Hsp104 chaperone to Hsp70 assisted disaggregation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl A, Binz HK, Forrer P, Stumpp MT, Pluckthun A, and Grutter MG (2003). Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. USA 100, 1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sowa ME, Watanabe Y, Sigler PB, Chiu W, Yoshida M, and Tsai FTF (2003). The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 115, 229–240. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Shorter J, and Lucius AL (2022). AAA+ proteins: one motor, multiple ways to work. Biochem. Soc. Trans 50, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Shorter J, Regulier E, Lashuel H, Iwatsubo T, Lindquist S, and Aebischer P (2008). Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Invest 118, 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez KE, Rizo AN, Tse E, Lin J, Scull NW, Thwin AC, Lucius AL, Shorter J, and Southworth DR (2020). Conformational plasticity of the ClpAP AAA+ protease couples protein unfolding and proteolysis. Nat. Struct. Mol. Biol 27, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay RG, Helsen CW, Tkach JM, and Glover JR (2008). The C-terminal extension of Saccharomyces cerevisiae Hsp104 plays a role in oligomer assembly. Biochemistry 47, 1918–1927. [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch AT, Trakselis MA, Laskey RA, and Bell SD (2005). Organization of the archaeal MCM complex on DNA and implications for the helicase mechanism. Nat. Struct. Mol. Biol 72, 756–762. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, et al. (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Michalska K, Zhang K, March ZM, Hatzos-Skintges C, Pintilie G, Bigelow L, Castellano LM, Miles LJ, Jackrel ME, Chuang E, et al. (2019). Structure of calcarisporiella thermophila Hsp104 disaggregase that antagonizes diverse proteotoxic misfolding events. Structure 27, 449–463.e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau MJ, McGeoch AT, Lowe AR, Itzhaki LS, and Bell SD (2007). ATPase site architecture and helicase mechanism of an archaeal MCM. Mol. Cell 28, 304–314. [DOI] [PubMed] [Google Scholar]
- Mosavi LK, Cammett TJ, Desrosiers DC, and Peng ZY (2004). The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 73, 1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosavi LK, Minor DL Jr., and Peng ZY (2002). Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl. Acad. Sci. USA 99, 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz D, Wyszkowski H, Szablewski T, Zawieracz K, Dutkiewicz R, Bury K, Wortmann SB, Wevers RA, and Zietkiewicz S (2020). CLPB (caseinolytic peptidase B homolog), the first mitochondrial protein refoldase associated with human disease. Biochim. Biophys. Acta Gen. Subj 1864, 129512. [DOI] [PubMed] [Google Scholar]
- Parra RG, Espada R, Verstraete N, and Ferreiro DU (2015). Structural and energetic characterization of the ankyrin repeat protein family. PLoS Comput. Biol 11, e1004659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier F, Radeke CM, Raab-Graham KF, and Vandenberg CA (1995). Expression of a putative ATPase suppresses the growth defect of a yeast potassium transport mutant: identification of a mammalian member of the Clp/HSP104 family. Gene 152, 157–163. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronicka E, Ropacka-Lesiak M, Trubicka J, Pajdowska M, Linke M, Ostergaard E, Saunders C, Horsch S, van Karnebeek C, Yaplito-Lee J, et al. (2017). A scoring system predicting the clinical course of CLPB defect based on the foetal and neonatal presentation of 31 patients. J. Inherit. Metab. Dis 40, 853–860. [DOI] [PubMed] [Google Scholar]
- Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, Glynn SE, and Lander GC (2017). Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchades C, Sandate CR, and Lander GC (2020). The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat. Rev. Mol. Cell Biol 21, 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudova EA, Krasnov GS, Kobelyatskaya AA, Savvateeva MV, Fedorova MS, Pavlov VS, Nyushko KM, Kaprin AD, Alekseev BY, Trofimov DY, et al. (2020). Gene expression changes and associated pathways involved in the progression of prostate cancer advanced stages. Front. Genet 11, 613162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, et al. (2021). MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, and Ting AY (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo AN, Lin J, Gates SN, Tse E, Bart SM, Castellano LM, DiMaio F, Shorter J, and Southworth DR (2019). Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat. Commun 10, 2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saita S, Nolte H, Fiedler KU, Kashkar H, Venne AS, Zahedi RP, Kruger M, and Langer T (2017). PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat. Cell Biol 19, 318–328. [DOI] [PubMed] [Google Scholar]
- Saunders C, Smith L, Wibrand F, Ravn K, Bross P, Thiffault I, Christensen M, Atherton A, Farrow E, Miller N, et al. (2015). CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am. J. Hum. Genet 96, 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, Queitsch C, Kowal AS, Parsell DA, and Lindquist S (1998). The ATPase activity of Hsp104, effects of environmental conditions and mutations. J. Biol. Chem 273, 15546–15552. [DOI] [PubMed] [Google Scholar]
- Shivhare D, Ng J, Tsai YC, and Mueller-Cajar O (2019). Probing the rice Rubisco-Rubisco activase interaction via subunit heterooligomerization. Proc. Natl. Acad. Sci. USA 116, 24041–24048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, and Southworth DR (2019). Spiraling in control: structures and mechanisms of the Hsp104 disaggregase. Cold Spring Harb. Perspect. Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skokowa J, Dale DC, Touw IP, Zeidler C, and Welte K (2017). Severe congenital neutropenias. Nat. Rev. Dis Primers 3, 17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding Z, Thevarajan I, Schrag LG, Zubcevic L, Zolkiewska A, and Zolkiewski M (2022). Human mitochondrial AAA+ ATPase SKD3/CLPB assembles into nucleotide-stabilized dodecamers. Biochem. Biophys. Res. Commun 602, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzi M, Radaelli E, Horre K, Arranz AM, Gounko NV, Agostinis P, Maia TM, Impens F, Morais VA, Lopez-Lluch G, et al. (2019). PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc. Natl. Acad. Sci. USA 116, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, Gupta K, and Shorter J (2015). The Hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Mol. Cell 57, 836–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevarajan I, Zolkiewski M, and Zolkiewska A (2020). Human CLPB forms ATP-dependent complexes in the mitochondrial intermembrane space. Int. J. Biochem. Cell Biol 127, 105841. [DOI] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux EF, Wohlever ML, Chen JZ, Sauer RT, and Baker TA (2013). Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc. Natl. Acad. Sci. USA 110, E2002–E2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY, Song Y, Barad BA, Cheng Y, Fraser JS, and DiMaio F (2016). Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 5, e17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JT, Cupo RR, Wattanasirakul P, Spencer DH, Locke AE, Makaryan V, Bolyard AA, Kelley ML, Kingston NL, Shorter J, et al. (2022). Heterozygous variants of CLPB are a cause of severe congenital neutropenia. Blood 139, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CL, Duran EC, Mack KL, Lin J, Jackrel ME, Sweeny EA, Shorter J, and Lucius AL (2017). Avidity for polypeptide binding by nucleotide-bound Hsp104 structures. Biochemistry 56, 2071–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbeck ND, Schlee S, and Reinstein J (2008). Coupling and dynamics of subunits in the hexameric AAA+ chaperone ClpB. J. Mol. Biol 378, 178–190. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Zietkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, Muntau AC, Rakovic A, Renkema GH, Rodenburg RJ, et al. (2015). CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am. J. Hum. Genet 96, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortmann SB, Zietkiewicz S, Guerrero-Castillo S, Feichtinger RG, Wagner M, Russell J, Ellaway C, Mroz D, Wyszkowski H, Weis D, et al. (2021). Neutropenia and intellectual disability are hallmarks of biallelic and de novo CLPB deficiency. Genet. Med 23, 1705–1714. [DOI] [PubMed] [Google Scholar]
- Wu D, Liu Y, Dai Y, Wang G, Lu G, Chen Y, Li N, Lin J, and Gao N (2022). Structure and mechanism of a mitochondrial AAA+ disaggregase CLBP. Preprint at bioRxiv. 10.1101/2022.03.10.483744. [DOI] [Google Scholar]
- Yoo H, Bard JAM, Pilipenko EV, and Drummond DA (2022). Chaperones directly and efficiently disperse stress-triggered biomolecular condensates. Mol. Cell 82, 741–755.e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Harischandra DS, Ghaisas S, Zhang P, Prall W, Huang L, Maghames C, Guo L, Luna E, Mack KL, et al. (2020). TRIM11 prevents and reverses protein aggregation and rescues a mouse model of Parkinson’s disease. Cell Rep. 33, 108418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PARLSkd3:casein:ATPγS cryo-EM maps and atomic coordinates have been deposited in the EMDB and PDB with accession codes EMDB: 26121 (State 1), EMDB: 26122 (State 1 filtered), PDB: 7TTR (State 1, AAA + only), and PDB: 7TTS (State 1, Full Model). The original/source data in the paper are available from the lead contact upon reasonable request.
This paper did not generate code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.