

Abstract
Purpose:
Phosphatidylinositol 3-kinase inhibitors (PI3Ki) are approved for relapsed chronic lymphocytic leukemia (CLL). While patients may show an initial response to these therapies, development of treatment intolerance or resistance remains clinical challenges. To overcome these, prediction of individual treatment responses based on actionable biomarkers is needed. Here, we characterized the activity and cellular effects of ten PI3Ki and investigated whether functional analyses can identify treatment vulnerabilities in PI3Ki-refractory/intolerant CLL and stratify responders to PI3Ki.
Experimental design:
Peripheral blood mononuclear cell (PBMC) samples (n=51 in total) from treatment naïve and PI3Ki-treated CLL patients were studied. Cells were profiled against ten PI3Ki and the Bcl-2 antagonist venetoclax. Cell signaling and immune phenotypes were analyzed by flow cytometry. Cell viability was monitored by detection of cleaved caspase-3 and the CellTiter-Glo assay.
Results:
pan-PI3Ki were most effective at inhibiting PI3K signaling and cell viability, and showed activity in CLL cells from both treatment-naïve and idelalisib-refractory/intolerant patients. CLL cells from idelalisib-refractory/intolerant patients showed overall reduced protein phosphorylation levels. The pan-PI3Ki copanlisib, but not the p110δ inhibitor idelalisib, inhibited PI3K signaling in CD4+ and CD8+ T cells in addition to CD19+ B cells, but did not significantly affect T cell numbers. Combination treatment with a PI3Ki and venetoclax resulted in synergistic induction of apoptosis. Analysis of drug sensitivities to 73 drug combinations and profiling of 31 proteins stratified responders to idelalisib and umbralisib, respectively.
Conclusions:
Our findings suggest novel treatment vulnerabilities in idelalisib-refractory/intolerant CLL, and indicate that ex vivo functional profiling may stratify PI3Ki responders.
INTRODUCTION
Excessive and autonomous B cell receptor (BCR) signaling is an important pathogenic mechanism in CLL,1 where continuous signaling leads to activation of downstream signaling molecules such as phosphatidylinositol 3-kinase (PI3K).2 Among the four classes of PI3Ks, only class I isoforms are clearly implicated in cancer.3 The catalytic subunit of class I PI3K exists as different isoforms, p110α, β, γ and δ. The α and β isoforms are ubiquitously expressed, while γ and δ are primarily expressed in leukocytes.4 P110δ is highly expressed in CLL cells, which makes it a promising target for CLL treatment.5 So far, two PI3K inhibitors (PI3Ki), idelalisib and duvelisib, have been approved for treatment of CLL, and several next-generation inhibitors are in development.6;7
Even though targeted therapies have revolutionized the management of CLL, treatment intolerance and resistance remain clinical challenges.7–9 To date, no mutations in PI3Ks are known that could explain the resistance mechanism to PI3Ki.10;11 Rather, reports suggest that resistance to PI3Ki may be mediated by upregulation of p110 isoforms other than p110δ12–14.This supports the use of a pan-p110 inhibitor (here referred to as pan-PI3Ki) such as copanlisib, which is approved for indolent lymphoma. Upregulation of alternative signaling pathways may also explain the development of PI3Ki resistance in some cases. Activation of STAT3 or STAT5 by interleukin-6 or ectopic activation of the ERBB signaling were shown to underlie resistance to various PI3Ki in lymphoma,15;16 while activating mutations in MAPK pathway genes and upregulation of IGF1R are reported resistance mechanisms in CLL.17;18 These resistance mechanisms provide a rationale for combination therapies, and we recently showed that MEK inhibitors are effective in idelalisib-resistant CLL.19
Despite the initially encouraging results for PI3Ki in CLL, a more widespread use of approved PI3Ki has been limited by unexpected autoimmune toxicities.5;7;8;20 Current strategies to allow for optimized use of the drug class include exploring next-generation PI3Ki and alternative dosing schedules.7;21 Prospective clinical trials have demonstrated that patients who experience intolerance or resistance to a targeted therapy may still benefit from another drug within the same drug class.22–24 However, there is currently limited knowledge of how the overall sensitivity to PI3Ki evolves in response to idelalisib treatment, and whether idelalisib-refractory/intolerant CLL cells remain sensitive to other PI3Ki. Ex vivo drug testing in patient cells has recently demonstrated clinical utility in guiding functional precision medicine for CLL and other hematological malignancies.25–27 When combined with other functional assays, such an integrated approach can lead to actionable biomarkers that predict patient treatment responses and identify unexpected treatment vulnerabilities such as novel combinations.28
Here, we combined ex vivo profiling of drug sensitivity and cell signaling in response to ten different PI3Ki (buparlisib, compound 7n, copanlisib, duvelisib, idelalisib, nemiralisib, pictilisib, pilaralisib, umbralisib, ZSTK474) in CLL, both alone and in combination with the B-cell lymphoma-2 (Bcl-2) antagonist venetoclax. We found that pan-PI3Ki were most effective in inhibiting cell signaling and viability of CLL cells. Furthermore, pan-PI3Ki single agents and PI3Ki + venetoclax combinations showed activity both in idelalisib-resistant lymphoma cell lines and in CLL cells from idelalisib-refractory/intolerant patients. A systematic analysis of longitudinal samples from idelalisib-treated patients demonstrated significantly higher ex vivo combination sensitivities at baseline in patients who obtained a long-term response to idelalisib than in patients who developed resistance to idelalisib. Furthermore, profiling of 31 proteins in screening samples from patients enrolled in a phase 2 trial of umbralisib (NCT02742090)22 distinguished patients who obtained a stable disease from patients who obtained a partial response to umbralisib therapy.
Our findings indicate PI3Ki drug class activity in idelalisib-refractory/intolerant CLL and suggest that functional precision medicine based on ex vivo testing of drug sensitivity and cell signaling provides an exciting opportunity to predict treatment responses and to identify potential treatment options, both monotherapies and combinations. These results warrant further testing in larger cohorts and in clinical trials.
MATERIALS AND METHODS
Patient material and ethical considerations
Buffy coats from age-matched, anonymized healthy blood donors were received from the Department of Immunology and Transfusion Medicine, Oslo University Hospital, Norway. Blood samples from CLL patients were received from the Department of Haematology, Oslo University Hospital, Norway; the Department of Medicine, Diakonhjemmet Hospital, Norway; Dana-Farber Cancer Institute (DFCI), MA, USA; and TG Therapeutics, New York, NY, USA (NCT02742090). All participants signed a written informed consent prior to sample collection. The study was approved by the Regional Committee for Medical and Health Research Ethics of South-East Norway (2016/947 and 28507). The DFCI tissue bank protocol was approved by the Dana-Farber Harvard Cancer Center Institutional Review Board. The NCT02742090 study was done in compliance with good clinical practice and local and national regulatory guidelines. An institutional review board at each site approved the protocol before any patients were enrolled. Research on blood samples was carried out in agreement with the Declaration of Helsinki.
Reagents and antibodies
Buparlisib, copanlisib, compound 7n, duvelisib, idelalisib, nemiralisib, pictilisib, pilaralisib, umbralisib, venetoclax and ZSTK474 were obtained from Selleckchem (Houston, TX, USA). All compounds and combinations used in this study are listed in Supplementary Table 1 and Supplementary Table 2, respectively. Antibodies against AKT (pS473) (D9E), cleaved caspase-3 (Asp175) (D3PE), NF-κB p65 (pS536) (93H1), p38 MAPK (pT180/Y182) (28B10), p44/42 MAPK (pT202/Y204) (E10), S6-ribosomal protein (pS235/S236) (D57.2.2E) and SYK (pY525/Y526) (C87C1) were from Cell Signaling Technologies (Leiden, The Netherlands). Antibodies against Btk (pY223)/Itk (pY180) (N35–86), Btk (pY551) & p-Itk (pY511) (24a/BTK (Y551), IgG1 Kappa (MOPC-21), MEK1 (pS298) (J114–64), mTOR (pS2448) (O21–404), PLCγ2 (pY759) (K86–689.37), STAT3 (pS727) (49/p-Stat3), ZAP70/Syk (pY319/Y352) (17A/P-ZAP70) were from BD Biosciences (San Jose, CA, USA). These antibodies were conjugated to Alexa Fluor 647. PerCP-Cy5.5 conjugated mouse anti-human CD19 antibody (HIB19) was from eBioscience (San Diego, CA, USA). PE-Cy7 conjugated mouse anti-human CD3 antibody (UCHT1) was from BD Biosciences. Goat F(ab’)2 anti-human IgM was from Southern Biotech (Birmingham, AL, USA). BD Phosflow Fix Buffer I and Perm Buffer III were from BD Biosciences. Alexa Fluor 488, Pacific Blue and Pacific Orange Succinimidyl Esters were from Thermo Fisher Scientific (Waltham, MA, USA).
Kinome scan
The activities of compound 7n, copanlisib, nemiralisib, and pilaralisib were profiled at 1 μM across a panel of 468 human kinases, including atypical, mutant, lipid, and pathogen kinases, using the DiscoveRx competition binding assay (https://www.discoverx.com/services/drug-discovery-development-services/kinase-profiling/kinomescan, Eurofins Scientific, Luxembourg). The measured percent of inhibition data are shown in Supplementary Table 3. Similar kinase activity data for buparlisib, duvelisib, idelalisib, pictilisib, umbralisib and ZSTK474, profiled following the same kinome scan protocol, but with a panel of 442 or 468 kinases, were available from previous studies.29;30 The activity data were visualized using the web-based tools TREEspot (Eurofins Scientific) (Figure 1) and iTOL31 (Supplementary Figure 1).
Figure 1. Target specificity and activity profile for ten PI3Ki.
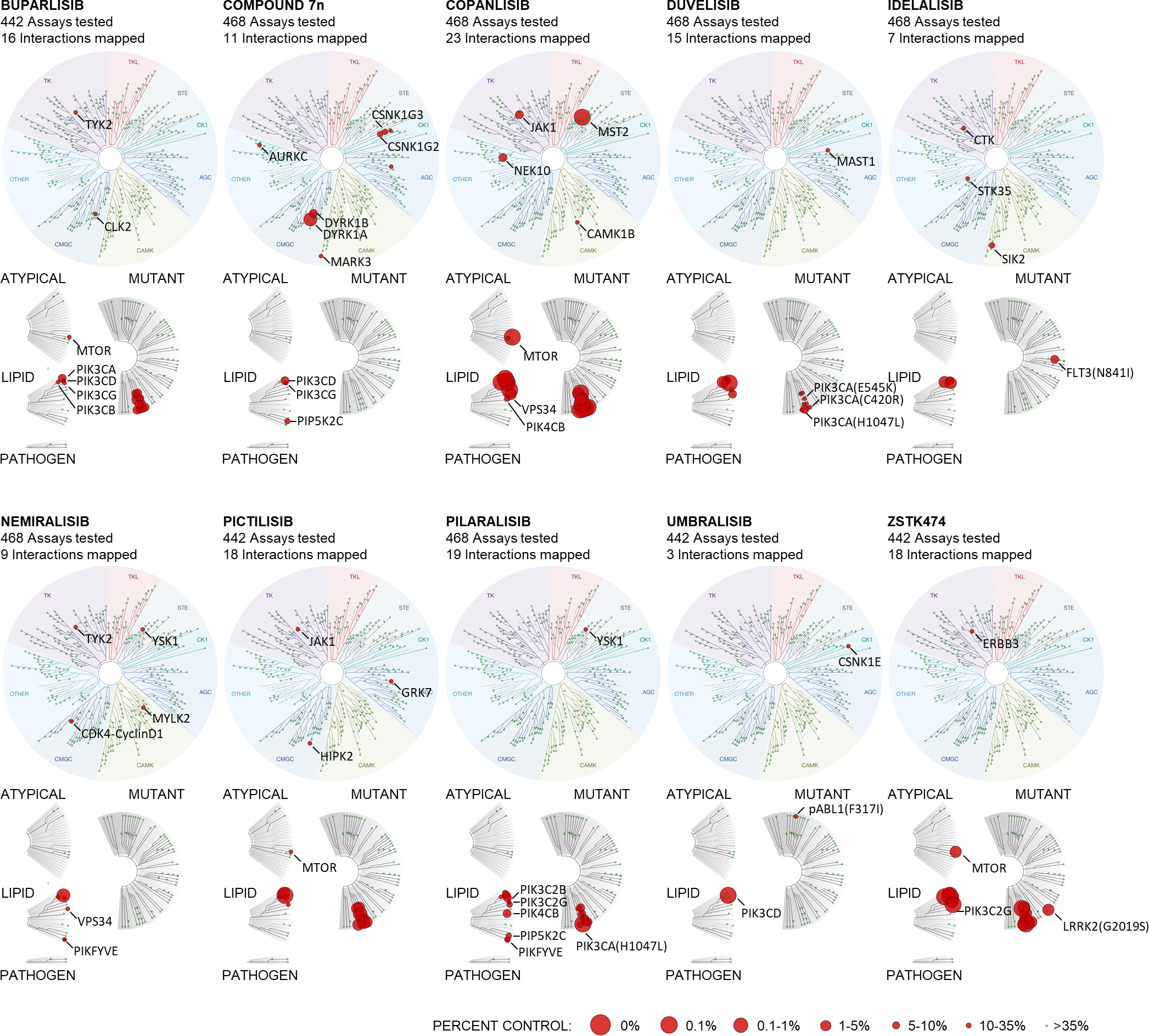
The activity of buparlisib, compound 7n, copanlisib, duvelisib, idelalisib, nemiralisib, pictilisib, pilaralisib, umbralisib, and ZSTK474 were profiled at 1 μM over a panel of up to 468 human kinases, including atypical, mutant, lipid, and pathogen kinases (lower dendrograms), using the DiscoveRx kinase assays. The upper dendrograms show (moving clockwise from the upper, purple section) tyrosine kinases (TK), tyrosine kinase-like kinases (TKL), STE protein kinases (STE), casein kinase 1 family (CK1), protein kinase A, G, and C families (AGC), Ca2+/calmodulin-dependent kinases (CAMK), CMGC kinase group (CMGC), and other. The size of the circles represents the percentage of target inhibition, with larger circles indicating a stronger inhibition compared with control, as defined in the scale.
Isolation of peripheral blood mononuclear cells (PBMCs)
Isolation of PBMCs from CLL patient samples was performed as previously described.32 Isolated cells were cryopreserved in liquid nitrogen.33 See Supplementary Table 4 for patient characteristics.
Phospho flow with fluorescent cell barcoding
Experiments were performed as previously described.34;35 For experiments shown in Figure 2 and Figure 4 (panels a-c in both figures), PBMCs from CLL153, CLL159, CLL160 and CLL216 (Supplementary Table 4) were treated with the indicated compound and concentration for 30 min, followed by 5 min stimulation with anti-IgM (10 μg/mL; Southern Biotech, Birmingham, AL, USA) to activate the BCR. For experiments shown in Figure 2d–i, PBMCs from CLL002D, CLL216 and CLL248 (Supplementary Table 4) were simultaneously co-cultured with irradiated (50 Gy/125 Gy/125 Gy, respectively) CD40L+/APRIL+, BAFF+ and APRIL+ 3T3 fibroblasts (ratio 1:1:1) to prevent induction of spontaneous apoptosis,36 and treated with the indicated compound and concentration for 24h. The treated CLL cells were then fixed, barcoded and permeabilized as previously described.34 The samples were stained with the indicated antibodies and analyzed with a BD LSR Fortessa cytometer (BD Biosciences) equipped with 488 nm, 561 nm, 640 nm and 407 nm lasers. The data were analyzed in Cytobank (https://cellmass.cytobank.org/cytobank/) as previously described.35
Figure 2. Drug-induced changes in cell signaling and cell viability are PI3Ki specific.

a-b) Peripheral blood mononuclear cells (PBMCs) from CLL patient samples (n=4) were treated with ten PI3Ki at the indicated concentrations for 30 min, followed by 5 min anti-IgM stimulation. The cells were then fixed, permeabilized and stained with the indicated antibodies. Signals were analyzed by flow cytometry. Results are shown for CD19+ B cells. Raw data were transformed to an arcsinh ratio relative to the signal in DMSO treated control cells, which was set to zero. Curves show the mean of the four experiments. Each curve represents the response to one inhibitor. Error bars indicate standard error of the mean (SEM).
c) Results are shown for experiments described in (a-b). The mean inhibitory effects of the ten PI3Ki on phosphorylation of AKT (pS473) and S6-ribosomal protein (pS235/S236) are plotted at the 1000 nM concentration. Error bars indicate SEM.
d) CLL cells were simultaneously co-cultured with APRIL/BAFF/CD40L+ fibroblasts and treated with the indicated concentration of a PI3Ki or DMSO (0.1%) for 24h. The CLL cells were then separated from the fibroblast layer, fixed, permeabilized and stained with the indicated antibodies. Signals were analyzed by flow cytometry. The cells were gated on CD19. The numbers indicate the fraction (%) of cells in the respective gate. Results are shown for one representative experiment
e-g) Experiments were performed as described in (d) on CLL cells from n=3 patients. The bars show the mean fraction (%) of positive cells ± standard error of the mean (SEM). The dotted line indicates the fraction of positive cells in the DMSO control. The blue bars indicate pan-PI3Ki.
h-i) Pearson’s correlation analyses were performed on the indicated protein levels detected in (d). Each point represents one treatment.
j) PBMCs from n=6 treatment naïve CLL patients were co-cultured with APRIL/BAFF/CD40L+ fibroblasts and DMSO (0.1%), copanlisib (100 nM), or idelalisib (1 μM) for 24h. The PBMCs were then separated from the fibroblast layer, fixed, barcoded, permeabilized and stained with surface markers and anti-S6-ribosomal protein (pS235/S236) antibody. Experiments were analyzed with a BD FACSymphony A5 cytometer (BD Biosciences) and further processed in Cytobank (https://cellmass.cytobank.org/cytobank/). FlowSOM clustering algorithm was applied to identify cell populations, which were validated by manual gating. The UMAP dimensionality reduction algorithm was used to visualize the data. The UMAP for one representative patient sample is shown.
k) Experiments were performed as described in (j). CD19+/CD185lo B cells were quantified as percent of CD3− lymphocytes.
l) Experiments were performed as described in (j). Signals are shown for CD19+ B cells, CD4+ T cells, and CD8+ T cells. Raw data were transformed to an arcsinh ratio relative to the signal in DMSO treated control cells, which was set to zero. Statistical testing was done with 2-way ANOVA. **p<0.01, n.s; not significant.
Figure 4. PI3Ki act in synergy with venetoclax.
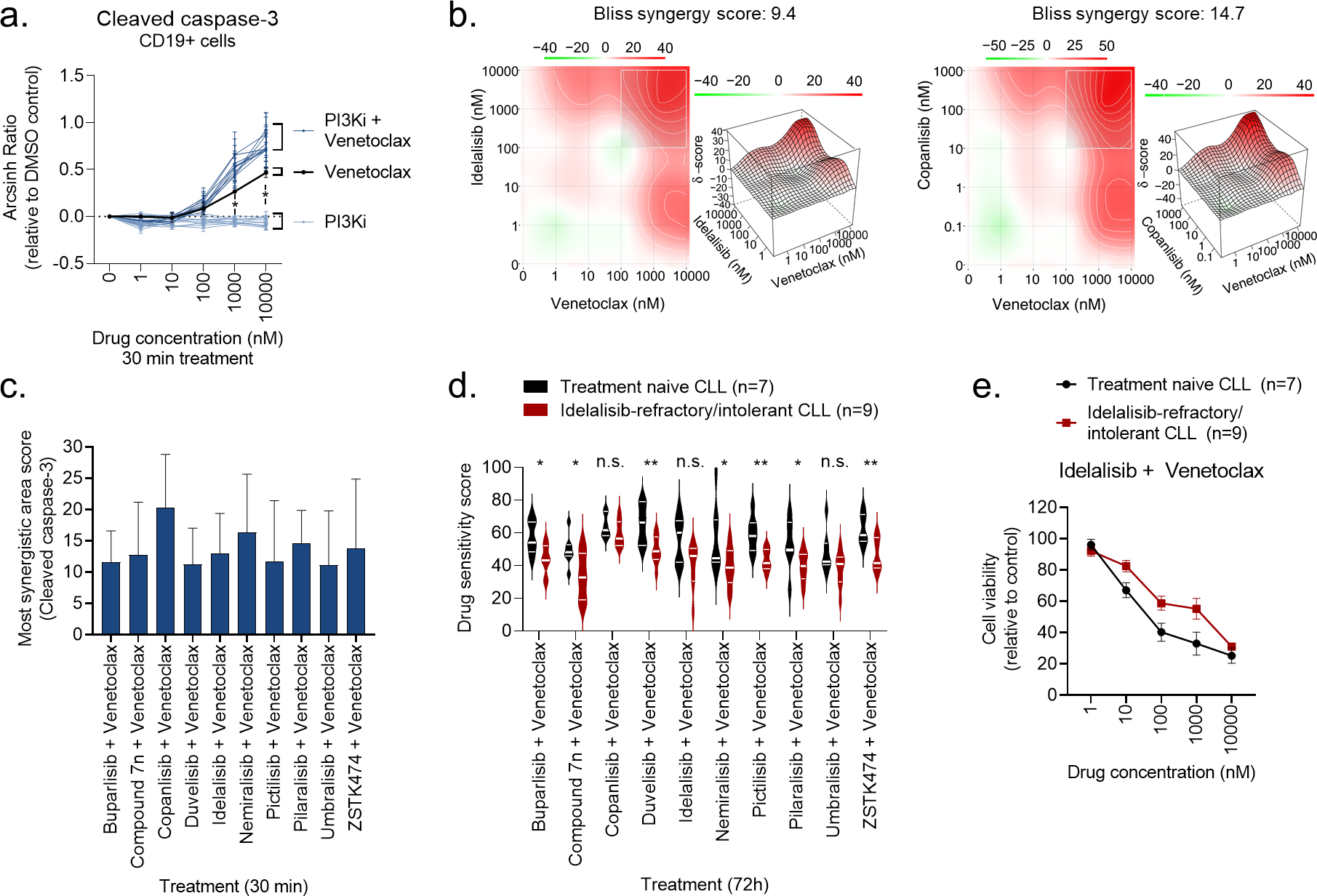
a) Peripheral blood mononuclear cells (PBMCs) from CLL patient samples (n=4) were treated with a PI3Ki, venetoclax, or PI3Ki + venetoclax combinations, at the indicated concentrations for 30 min, followed by 5 min anti-IgM stimulation. The cells were then fixed, permeabilized and stained with anti-cleaved caspase-3. Signals were analyzed by flow cytometry. Results are shown for CD19+ B cells. Raw data were transformed to an arcsinh ratio relative to the signal in DMSO treated control cells, which was set to zero. Curves show the mean ± standard error of the mean (SEM). Statistical testing was done with a one-way ANOVA with Holm-Sidak’s multiple comparisons test. *p<0.05.
b) Normalized data from experiments described in (a) were used in DECREASE (https://decrease.fimm.fi/) to predict the full drug combination dose-response matrices, which were analyzed using SynergyFinder (https://synergyfinder.fimm.fi/) to score the synergy of the drug combinations. Bliss synergy over the full matrix is indicated. A representative plot is shown for the idelalisib + venetoclax and copanlisib + venetoclax combinations.
c) Results are shown for analyses described in (b). The most synergistic area score was calculated by SynergyFinder (https://synergyfinder.fimm.fi/) for the indicated combination treatments. Bars show mean (n=4) ± standard error of the mean (SEM).
d) Peripheral blood mononuclear cells (PBMCs) from treatment naïve CLL patients (n=7) or idelalisib-refractory/intolerant patients (n=9) were co-cultured with APRIL/BAFF/CD40L+ fibroblasts for 24h. The CLL cells were then separated from the fibroblast layer and treated with the indicated drug combinations for 72h. Cell viability was assessed with the CellTiter-Glo assay. The drug sensitivity score was calculated for each treatment based on the area under the dose-response curve. High score indicates high sensitivity to the treatment. Violin plots show min to max response with lines at quartiles and median. Statistical testing was done with an unpaired t test. *p<0.05, **p<0.01, n.s; not significant.
e) Peripheral blood mononuclear cells (PBMCs) from treatment naïve CLL patients (n=7) or idelalisib-refractory/intolerant patients (n=9) were co-cultured with APRIL/BAFF/CD40L+ fibroblasts for 24h. The CLL cells were then separated from the fibroblast layer and treated with idelalisib + venetoclax combination at the indicated concentrations for 72h. Cell viability was assessed with the CellTiter-Glo assay. The graph shows mean relative cell viability ± standard error of the mean (SEM).
Immune phenotyping
PBMCs from each CLL donor were fixed, barcoded,34 and stained for 30 min with the antibody panel. The following antibodies were used: CD3-BUV395, CD4-BUV563, HLA-DR-BUV615, CD16-BUV737, CXCR5 (CD185)-BUV805, CCR7 (CD197)-BV605, CCR6 (CD196)-BV711, CD56-BV750, CD127-BV786, PD-1 (CD279)-BB700, CD14-PE, CD25-PE-CF594, CCR3 (CD183)-PE-Cy5, CD45RA-PE-Cy7, CD69-APC-R700, CD8/CD19-APC-Cy7 (BD Biosciences). Experiments were analyzed with a BD FACSymphony A5 cytometer (BD Biosciences) and further processed in Cytobank (https://cellmass.cytobank.org/cytobank/). The FlowSOM clustering algorithm was applied to identify cell populations, which were validated by manual gating. The UMAP dimensionality reduction algorithm was used to visualize the data.
Cell lines
Idelalisib-resistant lines from VL5137 and KARPAS171838 in vitro models were developed by prolonged exposure to the IC90 concentration of idelalisib, as previously described.16;39 The cell lines were confirmed mycoplasma negative using the MycoAlert Detection Kit (Lonza, Basel, Switzerland).
CellTiter-Glo luminescent cell viability assay
Dose-response experiments were performed as previously described.19;36 Compounds (Supplementary Table 1) were pre-printed in 384-well cell culture microplates using the Echo 550 liquid handler (Labcyte Inc., San Jose, CA, USA). Each compound was tested at five different concentrations in ten-fold increments ranging from 1 nM - 10000 nM (0.1 nM - 1000 nM for copanlisib). Combinations were designed using a fixed molar concentration series identical to those used for single agents. PBMCs from CLL patient samples were co-cultured with irradiated (50 Gy/125 Gy/125 Gy, respectively) CD40L+/APRIL+, BAFF+ and APRIL+ 3T3 fibroblasts (ratio 1:1:1) for 24 h prior to initiation of the experiment to mimic the tumor microenvironment and to prevent spontaneous apoptosis. The CLL cells were then separated from the adherent fibroblast layer by carefully re-suspending the culturing medium and transferring it to a separate tube. Experiments on “JB” samples (Supplementary Table 4) were performed using a slightly modified protocol, in which non-irradiated fibroblasts were removed from the CLL cells by positive selection with a PE-conjugated anti-CD47 antibody and anti-PE microbeads, as previously described.19 Single-cell suspension (10000 cells/well in 25 μl) was distributed to each well of the 384-well assay plate using the CERTUS Flex liquid dispenser (Fritz Gyger, Thun, Switzerland). Dose-response experiments on cell lines were performed on freshly thawed cells to reduce variation between experiments introduced by culturing conditions. The cells were incubated with the compounds at 37°C for 72h. Cell viability was measured using the CellTiter-Glo luminescent assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Luminescence was recorded with an EnVision 2102 Multilabel Reader (PerkinElmer, Waltham, MA, USA). The response readout was normalized to the negative (0.1% DMSO) and positive (100 μM benzethonium chloride) controls. The measured dose–response data were processed with the KNIME software (KNIME AG, Zurich, Switzerland).
Data analyses
Measured data from phospho flow experiments and drug sensitivity screens were processed in GraphPad Prism 8 (San Diego, CA, USA). Applied statistical tests are indicated in the figure legends. The normality of the data distribution was tested using the Kolmogorov-Smirnov test in GraphPad Prism 8. To quantify the compound responses, a modified drug sensitivity score (DSS) was calculated for each sample and compound separately.40 Area under the dose-response curve was calculated using an activity window from 100% to 10%, and a dose-window from the minimum concentration tested to the concentration where the viability reached 10%. DSS3 metric was used, without the division by the logarithm of the upper asymptote of the logistic curve. Higher levels of DSS indicate higher sensitivity to the compound. The DECREASE tool was used to predict the full dose-response matrices based on the diagonal (dose ratio 1:1) combination experiments based on normalized phospho flow data.36;41 Combination synergy was scored using the Bliss model using the SynergyFinder web-tool.42;43
To predict the umbralisib responders, the support vector machine (SVM) models were trained with the R-package e1071.44 Due to the small cohort size, we treated this as a binary classification between the patients who obtained a partial response (PR) or stable disease (SD) in response to umbarlisib treatment. Balanced cross-validation (CV) guaranteed that each CV fold must include at least one patient with PR and one patient with SD.
Recursive Feature Elimination (RFE) was performed using the R package caret.45;46 Since each RFE run may select different proteins having most predictive contribution in the particular CV fold, we repeated the 3-fold CV 500 times to investigate the robustness of the protein selection and model accuracy. Finally, we ranked the proteins according to their frequency across the RFE runs (i.e., each protein can be selected a maximum of 1500 times).
Data availability
The data generated in this study are available upon reasonable request to the corresponding author (sigrid.skanland@ous-research.no).
RESULTS
Target specificity and activity profiles of PI3K inhibitors
The target activity of ten PI3Ki was profiled at 1 μM over a panel of up to 468 human kinases using the kinome scan assay (Figure 1), and includes earlier reported data.29;30 The inhibitors showed expected differences in their p110 isoform specificity and off-target activity profiles (Figure 1 and Supplementary Table 3). Among the PI3Ki, compound 7n, nemiralisib and umbralisib showed p110δ isoform-specific activity with relatively few off-target activities, while copanlisib and ZSTK474 showed broad activity against all four p110 isoforms and several off-target activities (Figure 1 and Supplementary Figure 1).
Drug-induced changes in cell signaling are PI3Ki specific
To study how the different compounds inhibit PI3K signaling, CLL cells were treated with the individual PI3Ki at five concentrations (1 nM - 10000 nM) for 30 min. Copanlisib was tested at 10-fold lower concentrations (0.1 nM – 1000 nM) due to its higher potency.47 The cells were then stimulated with anti-IgM for 5 min to activate the BCR. As shown in Figure 2a–b, each PI3Ki inhibited the phosphorylation of downstream effectors AKT (pS473) and S6-ribosomal protein (pS235/S236), but with varying potency. Overall, the pan-PI3Ki buparlisib, copanlisib, pictilisib and ZSTK474 were most effective at inhibiting PI3K signaling (Figure 2c). To study whether this trend could be reproduced with longer incubation times, CLL cells were incubated with each PI3Ki for 24h. To prevent induction of spontaneous apoptosis which would influence the read-out, the CLL cells were simultaneously co-cultured with irradiated fibroblasts expressing APRIL/BAFF/CD40L. As shown in Figure 2d–e, 24h incubation with a PI3Ki inhibited phosphorylation of S6-ribosomal protein (pS235/S236), and the pan-PI3Ki were again most effective. Furthermore, 24h incubation with a PI3Ki induced apoptosis, as indicated by increased expression of cleaved caspase-3 (Figure 2d, f). Similarly, expression of the anti-apoptotic protein Mcl-1 was reduced in response to PI3Ki exposure. These phenotypes were most pronounced in response to a pan-PI3Ki (Figure 2f–g). The drug-induced changes in S6-ribosomal protein (pS236/S235) and Mcl-1 levels significantly correlated with cleavage of caspase-3 (Figure 2h–i), indicating that alteration of cell signaling or protein expression can serve as substitute markers for induction of apoptosis.
Effects of copanlisib and idelalisib on different immune cell compartments
To study the effect of PI3K inhibition on different immune cell compartments, PBMCs from treatment naïve CLL patients were simultaneously cultured with fibroblasts expressing APRIL/BAFF/CD40L and treated either with DMSO (control), copanlisib (100 nM), or idelalisib (1 μM) for 24h (Figure 2j). The CD185lo (CXCR5) B cell population was shown to consistently expand in response to copanlisib treatment (Figure 2k). This effect was not as pronounced upon idelalisib treatment, although two of the six patient samples showed a similar trend (Figure 2k). CD185 is a mediator of CLL tissue homing, and it has been shown to decrease also in response to ibrutinib + rituximab treatment.48 Other analyzed immune subsets were not significantly modulated by the treatments. This is in agreement with a previous study showing specificity of idelalisib and copanlisib towards CLL B cells.47 However, we observed that copanlisib, but not idelalisib, reduced S6-ribosomal protein phosphorylation also in CD4+ and CD8+ T cells (Figure 2l).
Pan-PI3Ki show activity in idelalisib-resistant lymphoma cells and idelalisib-refractory/intolerant CLL cells
To study the ability of the different PI3Ki to inhibit cell viability, 72h drug sensitivity assays were performed on the lymphoma cell lines VL51 and KARPAS1718 (Figure 3a–b). Each PI3Ki was tested at five different concentrations (1 nM - 10000 nM). Due to its higher potency, copanlisib was tested at 10-fold lower concentrations (0.1 nM - 1000 nM). In agreement with the effects observed on cell signaling, the pan-PI3Ki buparlisib, copanlisib, pictilisib and ZSTK474 were most effective at inhibiting cell viability in these cell lines (Figure 3a–b, black bars).
Figure 3. Pan-PI3Ki are active in idelalisib-refractory/intolerant CLL cells.
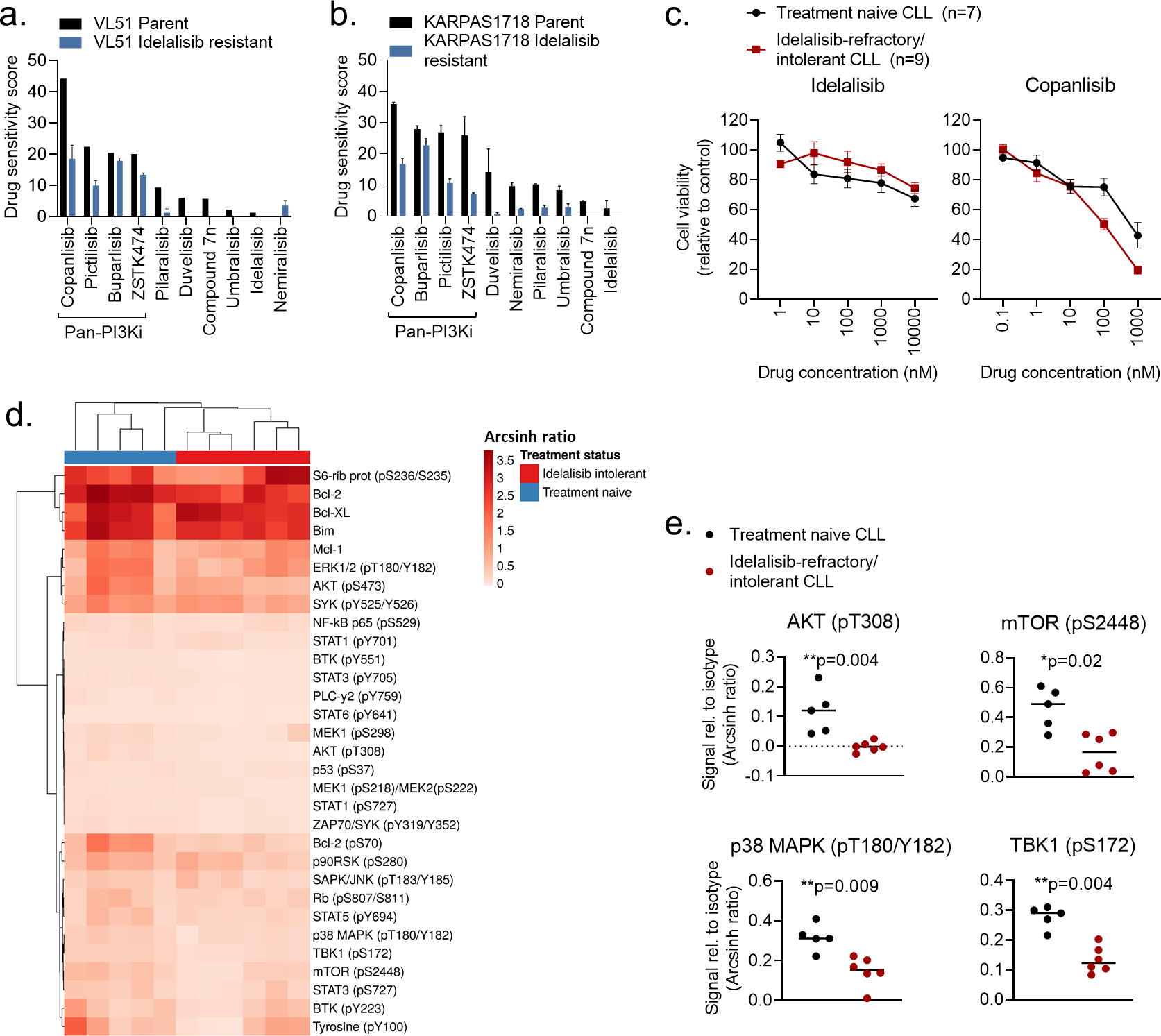
a-b) VL51 and KARPAS1718 (parent and idelalisib resistant) cell lines were treated with the indicated PI3Ki at 5 concentrations (0.1 nM – 1000 nM for copanlisib, 1 nM – 10000 nM for the others) for 72h. Cell viability was assessed with the CellTiter-Glo assay. The drug sensitivity score was calculated for each treatment based on the area under the dose-response curve. High score indicates high sensitivity to the treatment. The experiment were performed twice or once (VL51 parent). The bars show average with standard deviation.
c) PBMCs from treatment naïve CLL patients (n=7) or idelalisib refractory patients (n=9) were co-cultured with APRIL/BAFF/CD40L+ fibroblasts for 24h. The CLL cells were then separated from the fibroblast layer and treated with the indicated compound and concentrations for 72h. Cell viability was assessed with the CellTiter-Glo assay. The graphs show mean relative cell viability ± standard error of the mean (SEM).
d) Freshly thawed PBMCs from treatment naïve CLL patients (n=5) or idelalisib refractory patients (n=6) were fixed, permeabilized and stained with antibodies against the indicated proteins (rows). Signals were detected in CD19+ B cells by flow cytometry. Raw data were transformed to an arcsinh ratio relative to the signal of an isotype control (color key), which was set to zero. The heatmap was created using ClustVis (https://biit.cs.us.ee/clustvis/). Rows were clustered using Manhattan distance and Ward linkage. Columns were clustered using correlation distance and average linkage.
e) Phosphorylation levels of the indicated proteins are shown for experiments described in (d). The horizontal line indicates median. Statistical testing was done with the Mann-Whitney test.
To identify potential alternative therapies for patients who experience intolerance or develop resistance to idelalisib, we investigated whether idelalisib-resistant lymphoma cell lines remained sensitive to other PI3Ki. As expected, PI3Ki showed reduced efficacy in idelalisib-resistant cells, however, pan-PI3Ki still maintained a relatively high activity (Figure 3a–b, blue bars). In the idelalisib resistant VL51 cell line, the activity of the four indicated pan-PI3Ki remained between 42% (copanlisib) and 87.5% (buparlisib) relative to the activity in the parent cell line. For KARPAS1718, the corresponding range was between 27.8% (ZSTK474) and 81.4% (buparlisib). The sensitivity to idelalisib was reduced in CLL cells from idelalisib-refractory/intolerant patients as well (Figure 3c, left). However, the sensitivity to copanlisib was maintained at similar or even higher levels than in CLL cells from treatment naïve patients (Figure 3c, right). This trend was also observed for the pan-PI3Ki buparlisib, pictilisib, and ZSTK474 (Supplementary Figure 2). These findings demonstrate that pan-PI3Ki remain active in idelalisib-refractory/intolerant cells.
Idelalisib-refractory/intolerant CLL cells show reduced protein phosphorylation levels
To better understand the mechanisms underlying idelalisib-refractory/intolerant disease, we performed single-cell protein profiling of CLL cells from patients who were either treatment naïve or refractory/intolerant to idelalisib (Figure 3d). We found that expression or phosphorylation of several proteins was downregulated in CLL cells from idelalisib-refractory/intolerant patients relative to treatment naïve patients, including Bcl-2 (p = 0.02 with an unpaired t test), Bcl-2 (pS70) (p = 0.009), BTK (pY551) (p = 0.004), MEK1 (pS218)/MEK2 (pS222) (p = 0.004), p53 (pS37) (p = 0.004), PLCγ2 (pY759) (p = 0.02), STAT1 (pS727) (p = 0.01), STAT3 (pY705) (p = 0.02), STAT6 (pY641) (p = 0.004), and ZAP70/SYK (pY319/Y352) (p = 0.004). Of note, phosphorylation of proteins downstream of PI3K, including AKT (pT308), mTOR (pS2448) and TBK1 (pS172), were all significantly reduced in idelalisib-refractory/intolerant CLL cells (Figure 3e). These findings support previous reports that have demonstrated that higher response rates to copanlisib are associated with high expression of PI3K/BCR signaling pathway genes.49;50 We also found that the activity of p38 MAPK (pT180/Y182) was reduced in idelalisib-refractory/intolerant cells (Figure 3e). Interestingly, we previously showed that low phosphorylation levels of p38 MAPK correlates with poor response to venetoclax,19 suggesting that cell signaling profiles may provide response markers for different classes of targeted therapies.
PI3Ki act in synergy with venetoclax to induce apoptosis in CLL cells
Targeted therapies are increasingly studied in combinations, and venetoclax has been reported to be a good combination partner for copanlisib in B cell lymphoma (NCT03886649).51 We therefore investigated the potential benefit of combining a PI3Ki with venetoclax. CLL cells were treated with five different concentrations of each PI3Ki, venetoclax, or their combinations, for 30 min. The cleavage of caspase-3 was then analyzed by flow cytometry. None of the PI3Ki as a single agent induced apoptosis after this short incubation time, while treatment with venetoclax did (Figure 4a). Interestingly, each PI3Ki + venetoclax combination induced higher levels of cleaved caspase-3 than the venetoclax treatment alone (Figure 4a), suggesting a synergistic effect of the combination. Dose-response combination assays confirmed synergy among all ten combinations (Figure 4b–c).
Next, we tested whether PI3Ki + venetoclax combinations were effective in co-inhibiting the viability of CLL cells from idelalisib-refractory/intolerant patients over 72h. Overall, the sensitivities to most of these combinations were reduced in idelalisib-refractory/intolerant CLL relative to treatment naïve CLL (Figure 4d). However, the combinations did reduce the cell viability in a concentration-dependent manner consistently in both patient groups, as shown for idelalisib + venetoclax (Figure 4e). Taken together, these findings show that PI3Ki act in synergy with venetoclax to induce apoptosis, and that PI3Ki + venetoclax combinations are active in both treatment naïve and idelalisib-refractory/intolerant CLL.
Ex vivo drug sensitivity and protein profiles stratify responders to idelalisib and umbralisib therapy
To assess the predictive value of ex vivo drug sensitivity profiles, we analyzed drug responses to 73 combinations (Supplementary Table 2) on CLL cells from patients treated with idelalisib (“JB” samples, Supplementary Table 4). CLL cells collected at baseline from patients who obtained a long-term response to idelalisib showed significantly higher drug sensitivity scores than CLL cells from patients who obtained a short-term response (i.e., patients who developed resistance to idelalisib) (Figure 5a). Annotated drug responses are shown in Supplementary Figure 3. After correcting for multiple comparisons, we identified significant differences in responses to three PI3Ki + venetoclax combinations (Figure 5b). Interestingly, cells collected at the time the patients were responding to idelalisib showed lower levels of AKT (pS473) in long-term responders than in short-term responders (Figure 5c), but this difference was not statistically significant, possibly due to the low number of samples in each group (n=3). However, the two groups showed completely non-overlapping AKT (pS473) levels, which gives a good indication that this protein may be an accurate biomarker with high sensitivity and specificity.
Figure 5. Ex vivo drug sensitivity and protein profiles stratify responders to idelalisib and umbralisib therapy.
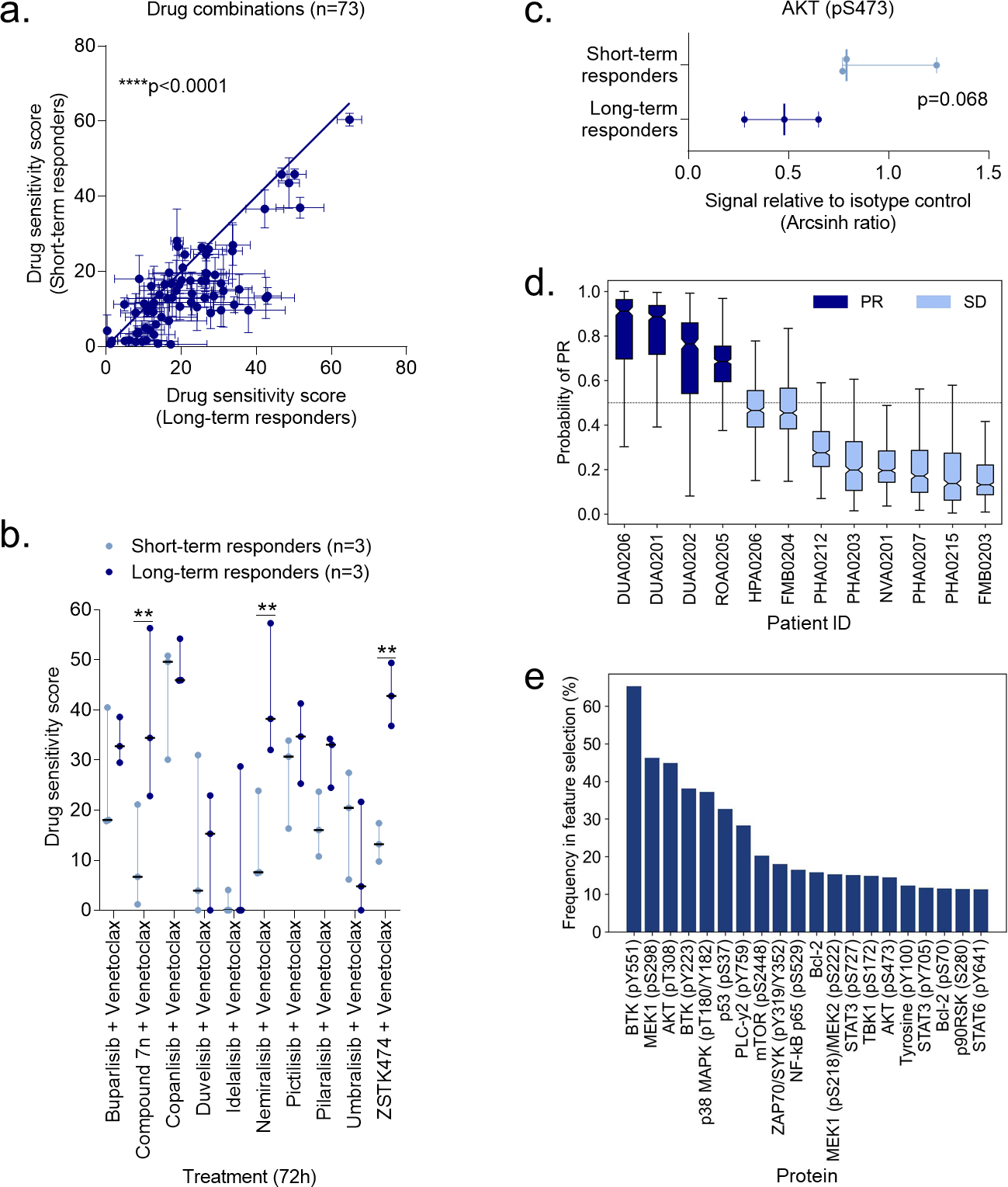
a) Drug sensitivity screens were performed with 73 drug combinations on PBMCs collected from CLL patients before the patients started treatment with idelalisib (n=3 short-term responders, i.e. developed resistance to idelalisib, and n=3 long-term responders). Each data point indicates the mean drug sensitivity score to one drug combination. Error bars show standard error of the mean (SEM) over the patients (n=3). The diagonal line indicates equal sensitivity in long- and short-term responders. Statistical testing was done with a paired t test comparing short-term responders to long-term responders. ****p<0.0001.
b) As in (a), but with the indicated PI3Ki + venetoclax combinations. The dot plot shows median with range. Statistical testing was done with a 2-way ANOVA with Sidak’s multiple comparisons within treatment groups for the 73 drug combinations. **p<0.01.
c) Peripheral blood mononuclear cells (PBMCs) collected from short-term and long-term responders at the time of response to idelalisib were fixed, permeabilized and stained with anti-AKT (pS473). Signals were detected in CD19+ B cells by flow cytometry. Raw data were transformed to an arcsinh ratio relative to the signal of an isotype control, which was set to zero. The scatter dot plot shows median with range. Statistical testing was done with an unpaired t test.
d) Peripheral blood mononuclear cells (PBMCs) collected at screening from CLL patients enrolled in a phase 2 trial of umbralisib (NCT02742090) were fixed, permeabilized and stained with antibodies against 31 proteins. The plot shows the predicted probability of partial response (PR) to umbralisib across the 12 CLL patients. A support vector machine (SVM) model was trained using the 31 protein levels as input features with a repeated cross-validation (CV) to avoid over-fitting. Three-fold CV was repeated 500 times to study the stability of the prediction model and its features. The default probability cut-off of 0.5 (the dotted horizontal line) distinguishes patients who obtained a PR from patients who obtained a stable disease (SD) as best response to umbralisib therapy. The boxes show the interquartile ranges, and the solid horizontal lines in each box indicate the median. Error bars show the minimum and maximum probabilities of the SVM predictions across 3-fold cross-validations repeated 500 times.
e) The experiments are described in (d). Recursive Feature Elimination (RFE) was performed using the R package caret. Three-fold cross-validation (CV) was repeated 500 times to investigate the robustness of the protein selection and model accuracy. Proteins were ranked according to their frequency across the RFE runs (i.e., each protein can be selected a maximum of 1500 times). The plot shows the frequency of the top 20 proteins selected by RFE, and the percentage indicates the proportion of CV folds. The higher the frequency the more stable and predictive is the protein for classification of patients between partial response (PR) and stable disease (SD) classes.
To further test the predictive value of protein profiles for PI3Ki treatment outcome, we analyzed samples from 12 CLL patients enrolled in a phase 2 clinical trial with umbralisib (NCT02742090) (see Supplementary Table 4 for the patient characteristics).22 The samples were collected at screening stage and profiled for expression or phosphorylation status of 31 proteins. We trained a support vector machine (SVM), a non-linear classification algorithm, using the profiles of the 31 proteins as input features with a repeated cross-validation (CV) to avoid over-fitting. Three-fold CV was repeated 500 times to study the stability of the prediction model and its predictive features. The SVM model was able to robustly distinguish between patients who obtained a partial response (PR) or stable disease (SD) in response to umbralisib therapy (Figure 5d, median prediction probabilities). However, due to the small cohort size, there was rather large variability across the CV folds (Figure 5d, error bars). Despite the low number of patient samples, the prediction model obtained surprisingly high sensitivity, specificity, and accuracy across the 12 patients (Supplementary Figure 4a–c).
Next, we used Recursive Feature Elimination (RFE) to identify the proteins with most contribution to the classification accuracy. This resulted in a ranked list of the 31 proteins in terms of their importance for umbralisib response prediction (Figure 5e). The top three predictors were BTK (pY551), MEK1 (pS298), and AKT (pT308), which are effectors of the BCR and PI3K (Figure 5e). The classification accuracy remained relatively high also when the models were built with the proteins selected by RFE (Supplementary Figure 4d–f).
We further investigated whether adding the ex vivo drug response readouts of the patient cells to various monotherapies and combinations could improve the prediction accuracy. Due to the moderate cohort size and significant cross-correlations between the drug sensitivity score (DSS) and protein levels in the samples, the ex vivo drug sensitivities did not lead to improved accuracy compared to the protein profile model in this patient cohort and for umbralisib treatment.
Taken together, these results demonstrate that functional profiling data may identify responders to PI3Ki, and warrant further studies in larger cohorts and clinical trials.
DISCUSSION
Targeted therapies have considerably improved patient outcome in CLL, but development of treatment intolerance and resistance remains clinical challenges. One strategy to prevent resistance to monotherapy is to combine therapies.9 Here, we studied the efficacy and synergy of ten PI3Ki, both as single agents and in combination with the Bcl-2 antagonist venetoclax. Venetoclax targets the intrinsic apoptotic pathway and is therefore an attractive partner for BCR inhibitors. The combination of venetoclax with the BTK inhibitor ibrutinib has shown promising results in CLL.52–54 Numerous additional studies on CLL are currently investigating the effect of venetoclax in combination with other targeted therapies, including various PI3Ki (NCT03534323, NCT03886649, NCT05209308). Resistance to both PI3Ki and venetoclax may be prevented or delayed with this strategy, as a suggested mechanism of resistance to venetoclax is upregulation of the PI3K/AKT/mTOR pathway.55
Here, we showed that pan-PI3Ki were more effective than p110δ selective inhibitors at reducing cell viability of CLL cells, both as single agents and in combination with venetoclax. Studies of copanlisib in relapsed or refractory lymphoma have demonstrated significant efficacy and a manageable safety profile,49;56;57 suggesting that copanlisib is a relevant treatment option for lymphoproliferative diseases. Copanlisib plus venetoclax combination showed the highest efficacy in our ex vivo assays. Synergy between these compounds has been demonstrated in B- and T-cell lymphoma models,51 and the combination is currently being studied in relapsed/refractory B-cell lymphomas (NCT03886649). The results will provide further information on its safety and efficacy.
Other strategies to prevent treatment resistance include improved patient stratification or precision medicine.9 CLL is a highly heterogeneous disease, and better model systems and biomarkers to guide clinical decision-making are likely to be useful. Functional assays can be valuable to this end.2;28 Ex vivo drug sensitivity has successfully predicted clinical activity in hematological malignancies.25–27 For instance, the EXALT trial (NCT03096821) investigated the feasibility and clinical impact of image-based ex vivo drug sensitivity-guided treatment decisions in patients with aggressive refractory hematological malignancies, and showed that integration of sensitivity testing in clinical decisions led to improved treatment outcomes.25
Here, we showed that ex vivo drug sensitivities and protein profiles could stratify patients based on clinical treatment responses, suggesting that such functional read-outs may serve as predictive biomarkers for treatment outcome. Our analyses showed that CLL cells from patients who are resistant to idelalisib remain sensitive to pan-PI3Ki. This finding is in agreement with studies showing that CLL patients who fail on a targeted therapy may still benefit from a second therapy in the same drug class,22–24 and warrants further studies on how to maximize the clinical value of PI3Ki.
Taken together, our findings indicate PI3Ki drug class activity in idelalisib-refractory/intolerant CLL and suggest that functional tests may guide precision medicine and predict treatment responses.
Supplementary Material
TRANSLATIONAL RELEVANCE STATEMENT.
The phosphatidylinositol 3-kinase inhibitors (PI3Ki) idelalisib and duvelisib are approved for relapsed chronic lymphocytic leukemia (CLL), but their use has been limited by severe toxicity and acquired resistance. Identification of biomarkers that predict individual treatment responses, as well as alternative treatment vulnerabilities in PI3Ki refractory/intolerant patients, is needed to optimally tailor CLL therapy. We performed functional analyses of baseline and drug-induced cell signaling and viability in CLL cells from treatment naïve and PI3Ki treated CLL patients to identify clinically actionable biomarkers. We show that CLL cells from idelalisib-refractory/intolerant patients remain sensitive to pan-PI3Ki and PI3Ki plus venetoclax combinations. A systematic analysis of drug sensitivities to 73 drug combinations stratified responders to idelalisib, while profiling of 31 proteins stratified responders to umbralisib. Our study suggests that functional testing may be used to guide precision medicine in relapsed CLL.
ACKNOWLEDGMENTS
The authors are thankful to all patients who contributed to this study. We are grateful to Silje Hjellbrekke, Hallvard Zapffe, Mentowa Fürst Bright and Martine Schrøder for technical assistance. We thank the High-Throughput Chemical Biology Screening Platform at Centre for Molecular Medicine Norway (NCMM), University of Oslo, and the High Throughput Biomedicine Unit at Institute for Molecular Medicine Finland (FIMM), University of Finland, for assistance with drug sensitivity screens. We thank the Flow Cytometry Core Facilities at Oslo University Hospital for assistance and access to instrumentation. This work was supported by the Research Council of Norway under the frames of ERA PerMed (to S. S. Skånland, project number 322898) and Digital Life Norway (to K. Taskén, project number 294916), the Norwegian Cancer Society (to K. Taskén), the Regional Health Authority for South-Eastern Norway (to K. Taskén), Stiftelsen Kristian Gerhard Jebsen (to K. Taskén and G. E. Tjønnfjord, Grant 19), Lilly Constance og Karl Ingolf Larssons stiftelse (to S. S. Skånland), and the Medical Student Research Program at the University of Oslo (to L. Karlsen and S. S. Skånland). F. Bertoni was supported by Swiss National Science Foundation (SNSF 31003A_163232/1). J. R. Brown was supported by NIH R01 CA 213442. T. Aittokallio was supported by the Norwegian Cancer Society (grant 216104), Helse Sør-Øst (2020026), Radium Hospital Foundation, Finnish Cancer Foundation, the Academy of Finland (grants 310507, 313267, 326238, 340141, 345803 and 344698), and the European Union’s Horizon 2020 Research and Innovation Programme (ERA PerMed CLL-CLUE project).
Footnotes
CONFLICT OF INTEREST STATEMENT
A.J.A. has received travel grants from AstraZeneca.
F.B. has received institutional research funds from Acerta, ADC Therapeutics, Bayer AG, Cellestia, CTI Life Sciences, EMD Serono, Helsinn, ImmunoGen, Menarini Ricerche, NEOMED Therapeutics 1, Nordic Nanovector ASA, Oncology Therapeutic Development, PIQUR Therapeutics AG; consultancy fee from Helsinn, Menarini; expert statements provided to HTG; travel grants from Amgen, AstraZeneca, Jazz Pharmaceuticals, PIQUR Therapeutics AG.
A.R.M. has received grants, personal fees, DSMB membership, and other funds from TG Therapeutics; grants and personal fees from AbbVie, Adaptive, AstraZeneca, Genentech, Janssen, Pharmacyclics; grants and other funds from Celgene; grants from DTRM Biopharm, Loxo, Regeneron, Sunesis; and personal fees from BeiGene.
E.N. reports employment and ownership of stock with TG Therapeutics.
J.R.B. has served as a consultant for AbbVie, Acerta/AstraZeneca, BeiGene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Genentech/Roche, Eli Lilly, Janssen, MEI Pharma, Morphosys AG, Nextcea, Novartis, Pfizer, Rigel; received research funding from Gilead, Loxo/Lilly, Verastem/SecuraBio, Sun, TG Therapeutics; and served on the data safety monitoring committee for Invectys.
G.E.T. has received advisory board honoraria from Alexion Pharmaceuticals and Janssen-Cilag, study support from Mundipharma, and lecture honoraria from Novartis, Janssen-Cilag, Alexion Pharmaceuticals, and Mundipharma.
S.S.S. has received honoraria from AbbVie and AstraZeneca, and research support from BeiGene and TG Therapeutics.
The other authors declare no competing financial interests.
REFERENCES
- 1.Duhren-von Minden M, Ubelhart R, Schneider D et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012;489:309–312. [DOI] [PubMed] [Google Scholar]
- 2.Skånland SS, Karlsen L, Taskén K. B cell signaling pathways - new targets for precision medicine in CLL. Scand.J.Immunol. 2020e12931. [DOI] [PubMed] [Google Scholar]
- 3.Ortiz-Maldonado V, Garcia-Morillo M, Delgado J. The biology behind PI3K inhibition in chronic lymphocytic leukaemia. Ther.Adv.Hematol. 2015;6:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrer G, Montserrat E. Critical molecular pathways in CLL therapy. Mol.Med. 2018;24:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JR. Phosphatidylinositol 3 Kinase delta Inhibitors: Present and Future. Cancer J. 2019;25:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, Perry MWD, Brown JR, André F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nat.Rev.Drug Discov. 2021;20:741–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skånland SS, Brown JR. PI3K inhibitors in chronic lymphocytic leukemia: where do we go from here? Haematologica 2022;Epub ahead of print: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanlon A, Brander DM. Managing toxicities of phosphatidylinositol-3-kinase (PI3K) inhibitors. Hematology.Am.Soc.Hematol.Educ.Program. 2020;2020:346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skånland SS, Mato AR. Overcoming resistance to targeted therapies in chronic lymphocytic leukemia. Blood Adv. 2021;5:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghia P, Ljungstrom V, Tausch E et al. Whole-Exome Sequencing Revealed No Recurrent Mutations within the PI3K Pathway in Relapsed Chronic Lymphocytic Leukemia Patients Progressing Under Idelalisib Treatment. Blood 2016;128:2770.27697770 [Google Scholar]
- 11.Scheffold A, Jebaraj BMC, Tausch E et al. In Vivo modeling of Resistance to PI3Kδ Inhibitor Treatment Using EμTCL1-Tg Tumor Transfer Model. Blood 2016;128:190. [Google Scholar]
- 12.Iyengar S, Clear A, Bödör C et al. P110α-mediated Constitutive PI3K Signaling Limits the Efficacy of p110δ-selective Inhibition in Mantle Cell Lymphoma, Particularly With Multiple Relapse. Blood 2013;121:2274–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huw LY, O’Brien C, Pandita A et al. Acquired PIK3CA amplification causes resistance to selective phosphoinositide 3-kinase inhibitors in breast cancer. Oncogenesis. 2013;2:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juric D, Castel P, Griffith M et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015;518:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JH, Kim WS, Park C. Interleukin-6 mediates resistance to PI3K-pathway-targeted therapy in lymphoma. BMC.Cancer 2019;19:936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arribas AJ, Napoli S, Gaudio E et al. Secreted Factors Determine Resistance to Idelalisib in Marginal Zone Lymphoma Models of Resistance [abstract]. Blood 2022;135 (Supplement_1):2569. [Google Scholar]
- 17.Murali I, Kasar S, Naeem A et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia (cll). Blood 2021;138:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tausch E, Ljungström V, Agathangelidis A et al. Secondary resistance to idelalisib is characterized by upregulation of IGF1R rather than MAPK/ERK pathway mutations. Blood 2022;139:3340–3344. [DOI] [PubMed] [Google Scholar]
- 19.Melvold K, Giliberto M, Karlsen L et al. Mcl-1 and Bcl-xL levels predict responsiveness to dual MEK/Bcl-2 inhibition in B-cell malignancies. Mol.Oncol. 2022;16:1153–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richardson NC, Kasamon Y, Pazdur R, Gormley N. The saga of PI3K inhibitors in haematological malignancies: survival is the ultimate safety endpoint. Lancet Oncol. 2022 [DOI] [PubMed] [Google Scholar]
- 21.Roeker LE, Thompson MC, Mato AR. Searching for a home: phosphoinositide 3-kinase inhibitors for chronic lymphocytic leukaemia in modern clinical practice. Br.J.Haematol. 2021;194:9–10. [DOI] [PubMed] [Google Scholar]
- 22.Mato AR, Ghosh N, Schuster SJ et al. Phase 2 Study of the Safety and Efficacy of Umbralisib in Patients with CLL Who Are Intolerant to BTK or PI3Kδ Inhibitor Therapy. Blood 2021;137:2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers KA, Thompson PA, Allan JN et al. Phase 2 study of acalabrutinib in ibrutinib-intolerant patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica 2021;106:2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mato AR, Shah NN, Jurczak W et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet 2021;397:892–901. [DOI] [PubMed] [Google Scholar]
- 25.Kornauth C, Pemovska T, Vladimer GI et al. Functional Precision Medicine Provides Clinical Benefit in Advanced Aggressive Hematologic Cancers and Identifies Exceptional Responders. Cancer Discov. 2022;12:372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malani D, Kumar A, Brück O et al. Implementing a Functional Precision Medicine Tumor Board for Acute Myeloid Leukemia. Cancer Discov. 2022;12:388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skånland SS, Inngjerdingen M, Bendiksen H et al. Functional testing of relapsed chronic lymphocytic leukemia guides precision medicine and maps response and resistance mechanisms. An index case. Haematologica 2022;107:1994–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Letai A, Bhola P, Welm AL. Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell 2022;40:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rewcastle GW, Kolekar S, Buchanan CM et al. Biological characterization of SN32976, a selective inhibitor of PI3K and mTOR with preferential activity to PI3Kα, in comparison to established pan PI3K inhibitors. Oncotarget. 2017;8:47725–47740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burris HA III, Flinn IW, Patel MR et al. Umbralisib, a novel PI3Kdelta and casein kinase-1epsilon inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: an open-label, phase 1, dose-escalation, first-in-human study. Lancet Oncol. 2018;19:486–496. [DOI] [PubMed] [Google Scholar]
- 31.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skånland SS, Cremaschi A, Bendiksen H et al. An in vitro assay for biomarker discovery and dose prediction applied to ibrutinib plus venetoclax treatment of CLL. Leukemia 2020;34:478–487. [DOI] [PubMed] [Google Scholar]
- 33.Hermansen JU, Tjønnfjord GE, Munthe LA, Taskén K, Skånland SS. Cryopreservation of primary B cells minimally influences their signaling responses. Sci.Rep. 2018;8:17651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skånland SS. Phospho Flow Cytometry with Fluorescent Cell Barcoding for Single Cell Signaling Analysis and Biomarker Discovery. J.Vis.Exp. 2018;140:e58386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myhrvold IK, Cremaschi A, Hermansen JU et al. Single cell profiling of phospho-protein levels in chronic lymphocytic leukemia. Oncotarget. 2018;9:9273–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Athanasiadis P, Ianevski A, Skånland SS, Aittokallio T. Computational Pipeline for Rational Drug Combination Screening in Patient-Derived Cells. Methods Mol.Biol. 2022;2449:327–348. [DOI] [PubMed] [Google Scholar]
- 37.Inokuchi K, Abo J, Takahashi H et al. Establishment and characterization of a villous lymphoma cell line from splenic B-cell lymphoma. Leuk Res. 1995;19:817–822. [DOI] [PubMed] [Google Scholar]
- 38.Martinez-Climent JA, Sanchez-Izquierdo D, Sarsotti E et al. Genomic abnormalities acquired in the blastic transformation of splenic marginal zone B-cell lymphoma. Leuk Lymphoma 2003;44:459–464. [DOI] [PubMed] [Google Scholar]
- 39.Arribas AJ, Napoli S, Cascione L et al. Resistance to PI3Kδ inhibitors in marginal zone lymphoma can be reverted by targeting the IL-6/PDGFRA axis. Haematologica 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yadav B, Pemovska T, Szwajda A et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci.Rep. 2014;4:5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ianevski A, Giri AK, Gautam P et al. Prediction of drug combination effects with a minimal set of experiments. Nat.Mach.Intell. 2019;1:568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bliss C The toxicity of poisons applied jointly. Ann.Appl.Biol. 1939;26:585–615. [Google Scholar]
- 43.Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017;33:2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer D, Dimitriadou E, Hornik K, Weingessel A, Leisch F, Chang C-C, and Lin C-C Package ‘e1071’: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien. R Package version 1.7–3, 1–63. 2022. 26–11-2019. Ref Type: Online Source [Google Scholar]
- 45.Guyon I, Weston J, Barnhill S, Vapnik V. Gene Selection for Cancer Classification using Support Vector Machines. Machine Learning 2002;46:389–422. [Google Scholar]
- 46.Kuhn M Building predictive models in R using the caret package. J Stat Softw 2008;28:1–26.27774042 [Google Scholar]
- 47.Göckeritz E, Kerwien S, Baumann M et al. Efficacy of phosphatidylinositol-3 kinase inhibitors with diverse isoform selectivity profiles for inhibiting the survival of chronic lymphocytic leukemia cells. Int.J.Cancer 2015;137:2234–2242. [DOI] [PubMed] [Google Scholar]
- 48.Peragine N, De Propris MS, Intoppa S et al. Modulated expression of adhesion, migration and activation molecules may predict the degree of response in chronic lymphocytic leukemia patients treated with ibrutinib plus rituximab. Haematologica 2020;106:1500–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dreyling M, Santoro A, Mollica L et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J.Clin Oncol. 2017;35:3898–3905. [DOI] [PubMed] [Google Scholar]
- 50.Dreyling M, Morschhauser F, Bouabdallah K et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann.Oncol. 2017;28:2169–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarantelli C, Lange M, Gaudio E et al. Copanlisib synergizes with conventional and targeted agents including venetoclax in B- and T-cell lymphoma models. Blood Adv. 2020;4:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hillmen P, Rawstron AC, Brock K et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J.Clin.Oncol. 2019;37:2722–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jain N, Keating M, Thompson P et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N.Engl.J.Med. 2019;380:2095–2103. [DOI] [PubMed] [Google Scholar]
- 54.Kater AP, Levin MD, Dubois J et al. Minimal residual disease-guided stop and start of venetoclax plus ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia (HOVON141/VISION): primary analysis of an open-label, randomised, phase 2 trial. Lancet Oncol. 2022;23:818–828. [DOI] [PubMed] [Google Scholar]
- 55.Choudhary GS, Al-Harbi S, Mazumder S et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death.Dis. 2015;6:e1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lenz G, Hawkes E, Verhoef G et al. Single-agent activity of phosphatidylinositol 3-kinase inhibition with copanlisib in patients with molecularly defined relapsed or refractory diffuse large B-cell lymphoma. Leukemia 2020;34:2184–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panayiotidis P, Follows GA, Mollica L et al. Efficacy and safety of copanlisib in patients with relapsed or refractory marginal zone lymphoma. Blood Adv. 2021;5:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon reasonable request to the corresponding author (sigrid.skanland@ous-research.no).