

Abstract
We report the first well-characterized selective chemical probe for histone deacetylase 10 (HDAC10) with unprecedented selectivity over other HDAC isozymes. HDAC10 deacetylates polyamines and has a distinct substrate specificity, making it unique among the 11 zinc-dependent HDAC hydrolases. Taking inspiration from HDAC10 polyamine substrates, we systematically inserted an amino group (“aza-scan”) into the hexyl linker moiety of the approved drug Vorinostat (SAHA). This one atom replacement (C→N) transformed SAHA from an unselective pan-HDAC inhibitor into a specific HDAC10 inhibitor. Optimization of the aza-SAHA structure yielded the HDAC10 chemical probe DKFZ-748, with potency and selectivity demonstrated by cellular and biochemical target-engagement, as well as thermal-shift, assays. Co-crystal structures of our aza-SAHA derivatives with HDAC10 provide a structural rationale for potency, and chemoproteomic profiling confirmed exquisite cellular HDAC10-selectivity of DKFZ-748 across the target landscape of HDAC drugs. Treatment of cells with DKFZ-748, followed by quantification of selected polyamines, validated for the first time the suspected cellular function of HDAC10 as a polyamine deacetylase. Finally, in a polyamine-limited in vitro tumor model, DKFZ-748 showed dose-dependent growth inhibition of HeLa cells. We expect DKFZ-748 and related probes to enable further studies on the enigmatic biology of HDAC10 and acetylated polyamines in both physiological and pathological settings.
Graphical Abstract
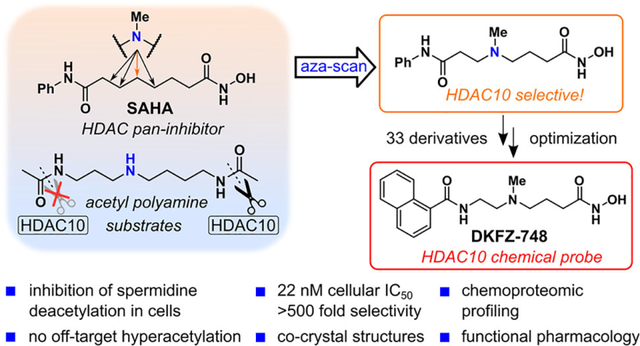
INTRODUCTION
Histone deacetylases (HDACs), a family of 18 hydrolase enzymes that cleave post-translational acyl moieties from lysines, have received a remarkable amount of attention from biologists and chemists over the past decades, and are the primary targets of five approved drugs (1, 2). The entire family of HDAC isozymes are named histone deacetylases analogous to the first identified HDAC, histone deacetylase 1 (HDAC1), which was confirmed to be the molecular target of trapoxin and trichostatin A, two natural products which were known to inhibit histone deacetylation (1).
Histone deacetylase 10 (HDAC10) is arguably the most intriguing member of the eleven zinc-dependent HDACs due to its unique non-epigenetic enzymatic function and still enigmatic physiological role. Isolated and characterized in 2002 by four groups independently (2–5), HDAC10 is categorized as a Class IIB HDAC. together with its closest relative, HDAC6, an α-tubulin deacetylase (6). The Class IIB enzymes are distinct from the Class I isozymes (HDAC1, 2, 3, 8) in that they exhibit poor deacetylase activity on histones and localize primarily in the cytoplasm. Despite decades of intense research on the HDAC family, HDAC10 has received comparatively little attention by the medicinal chemistry community (7, 12–15), resulting in a lack of appropriate pharmacological tools. Indeed, it took 15 years after its isolation before HDAC10 was shown to be a poor lysine deacetylase, which instead recognizes acetylated polyamines as substrates. As such it has been classified as a polyamine deacetylase (PDAC) (11).While the PDAC activity of HDAC10 is established at a biochemical level, to the best of our knowledge no study has directly linked HDAC10 enzymatic activity to polyamine acetylation levels in a cellular context. Blankenship and Fries made seminal contributions with polyamine deacetylase inhibition in the late 1980s, but at the time they could not identify which specific protein(s) were targeted by their inhibitors (16, 9). Since then, our understanding of HDAC10 biology and the (patho)physiological functions of the putative polyamine substrates of HDAC10 within the polyamine metabolome has remained limited (17, 18). HDAC10 knock-out mice are viable and develop normally without signs of disease (19). Still, HDAC10 is reported to be involved in crucial physiological processes, such as autophagy (20, 21), homologous recombination (22), DNA mismatch repair (23), angiogenesis (24), and potentially cell proliferation (25). Furthermore, HDAC10 has been reported to be involved in inflammatory disorders (26) and transplant rejection (19). However, these findings are based on expression analysis, genetic manipulation and non-selective HDAC inhibitors, due to a lack of better tool compounds. Therefore, HDAC10-selective chemical probes are needed to validate HDAC10’s role in physiology and disease and, importantly, to establish a mechanistic link between its activity as a polyamine deacetylase and the higher-level phenotypic observations made after HDAC10 genetic manipulation. Moreover, HDAC10 inhibitors (HDAC10i) have significant potential for therapeutic application in oncology (21)(27)(28) or as immunomodulators (19).
Up to now, no HDAC10i that fulfills the requirements of a selective chemical probe has been described (29). While HDAC10 inhibitors with selectivity over the Class I isozymes are known, the primary challenge lies in achieving selectivity over the other Class IIB member, HDAC6 (7, 13, 30, 12). A good example for this is tubastatin A (1), a well-known HDAC6i, which is an even more potent HDAC10 binder (Figure 1, far left box) (7, 31). We previously showed that an electrostatic interaction between the tertiary amine of the tetrahydro-γ-carboline ring of 1 and the E274 gatekeeper residue in HDAC10 is responsible for its strong binding (7, 15). The gatekeeper residue is unique to HDAC10, and its negative charge provides an attractive interaction with positively charged polyamines (or HDAC10 inhibitors) within HDAC10’s binding site. Ring-opened tubastatin A derivative DKFZ-480 (2) makes a direct hydrogen bond to E274, which results in enhanced binding affinity. While 2 has improved selectivity over HDAC6 (40-fold vs. 7-fold for tubastatin A), this derives only from increased HDAC10 affinity, with no reduction in HDAC6 potency.
Figure 1. HDAC10 inhibitors, substrates, and design concept for a selective HDAC10 inhibitor.
Far left box: Dual HDAC10/6 binders.(7, 8) Middle left box: Selected acetamides, which are good substrates of HDAC10. Middle right box: Selected acetamides, which are poor substrates of HDAC10. Far right box: Concept to merge the structures of SAHA (9) and 10 (9, 10) to create aza-SAHA derivatives, which should produce a potent and selective HDAC10 chemical probe. kcat/KM values are reported for hHDAC10 and taken from Hai et al. (11).
We therefore moved on from the tubastatin A scaffold and took inspiration from the remarkable substrate specificity of HDAC10 (11). Polyamines like N8-acetylspermidine (3), N-acetylputrescine (4), and N-acetylcadaverine (5) are excellent HDAC10 substrates (Figure 1, middle left box). Notably, 3–5 all contain an amine (blue) four to five carbons separated from an N-acetyl group (red). However, not all acetylated polyamines are good substrates as exemplified by N1-acetylspermidine (6), an isomer of 3, which differs only by having a three-carbon amine–acetamide separation (Figure 1, middle right box). Furthermore, typical HDAC substrate peptides, which contain an acetylated lysine (7), and simple acetamides like 8 are poor substrates of HDAC10. Equally informative for our design plans for an HDAC10 chemical probe is the fact that 3–5 are poor substrates of HDAC6. Taken together, these data suggest that HDAC10 has a narrow and unique structural requirement for substrates, which, if appropriately incorporated into an inhibitor structure, could be leveraged to create an HDAC10-selective chemical probe.
SAHA (9), a pan-HDAC inhibitor, was the first clinically approved HDAC inhibitor (Figure 1, right box). The hexyl linker of SAHA (black) can be thought of as a lysine side chain mimic, which joins the hydroxamic acid zinc-binding group (red) to the anilide “cap” group (green). Linear, diamine PDAC inhibitors like 10 have been known for decades (9, 32) and some have recently been crystallized with HDAC10 (33). We hypothesized that merging SAHA (9) and diamine PDAC inhibitors like 10 into aza-SAHA derivatives (11) would render the SAHA linker into a motif only recognized by HDAC10, effectively converting SAHA from a pan-inhibitor into an HDAC10-selective chemical probe. These compounds, containing both an amino group and a hydroxamic acid, would be highly polar, but the anilide cap group would render them more drug-like than 10, and offer a synthetic handle (R2) for optimization of physicochemical properties. Herein, we report the outcome of this study, which has resulted in the discovery of DKFZ-748, a selective HDAC10 chemical probe suitable for studying HDAC10 biology in a cellular context.
RESULTS AND DISCUSSION
Aza-SAHA derivatives have diverse HDAC inhibitory profiles.
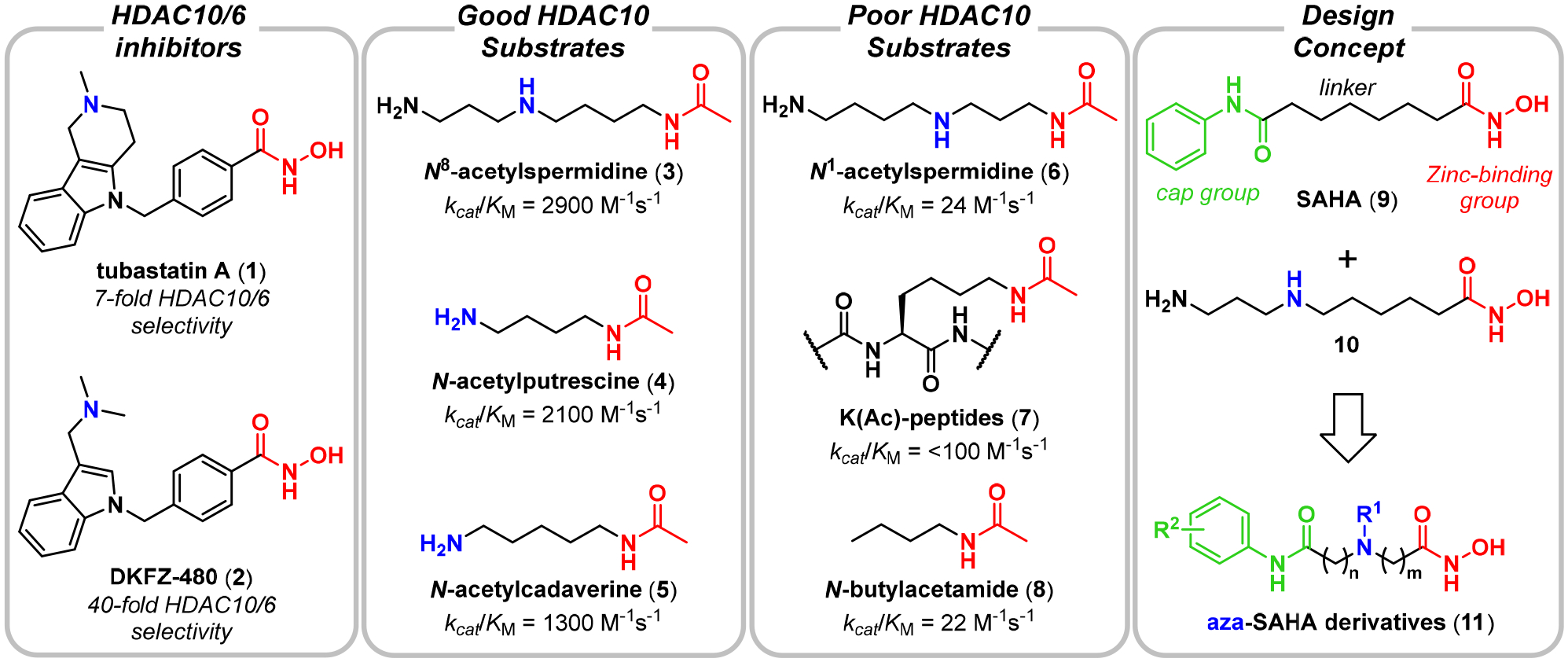
We began by preparing aza-SAHA derivatives, where we “walked” an amino group down the SAHA (9) carbon chain, each time replacing a methylene group and keeping the overall linker length of six atoms the same (Scheme S1). This resulted in the β-, γ-, and δ-amino hydroxamic acids (12–14, Figure 2A). One additional compound, ε-amino hydroxamic acid 15, required extension of the linker by one atom so as not to produce a urea, essentially an “insertion” of the amino group into the SAHA linker. Due to a propensity for lactamization (34), especially for the δ- and γ-aminohydroxamic acids, we prepared all aza-SAHA derivatives as tertiary methylamines.
Figure 2. Aza-SAHA derivative DKFZ-711 is a potent and selective HDAC10 binder.
A. Molecules obtained from an “aza-scan” of the SAHA linker; B. Differential scanning fluorimetry thermal shift assay of aza-SAHA derivatives (500 μM) performed in duplicate with recombinant HDAC10 (4 μM); C. HDAC isozyme selectivity heatmap and pIC50 values of aza-SAHA derivatives reveal diverse inhibitory profiles depending on the nitrogen position. pIC50 values are the mean of two replicates, each determined by four-parameter non-linear regression analysis with eight dose levels in triplicate with HDAC Glo assaya or FRET assayb. c Mean of three individual experiments. d Curve fit was not convergent at 100 μM. For 95% confidence intervals, see Table S1.
All four compounds, with SAHA (9) as a benchmark comparison, were screened for HDAC10 binding in a differential scanning fluorimetry thermal shift assay (Figure 2B). SAHA stabilized HDAC10 by 5.0 °C relative to the vehicle control, while β-amino derivative 12 gave a smaller thermal shift of 2.2 °C, indicative of weaker binding. Compared to SAHA, the γ- and δ-amines (DKFZ-711 (13) and 14) induced higher thermal shifts of 8.2 °C and 7.1 °C, respectively. ε-Aminohydroxamic acid 15, which contains a linker length like 10, gave the largest thermal shift of 11.7 °C, suggesting that it may be a potent HDAC10 binder.
In order to establish qualitative selectivity profiles with respect to other HDAC enzymes, we tested SAHA and its aza-derivatives against HDACs 1, 2, 3, 6 and 8 with the enzymatic HDAC Glo assay system. As previously described (7, 11, 35), HDAC10 does not recognize peptidic substrates, and we therefore used a FRET ligand displacement assay to measure HDAC10 binding affinity (7, 36). Our data (Figure 2C, Table S1) confirmed SAHA to be a pan-HDAC inhibitor, with moderately higher potency for HDAC1 (pIC50 = 7.30) and HDAC6 (pIC50 = 7.72) than for HDAC8 (pIC50 = 6.16) and HDAC10 (pIC50 = 6.93). The data for β-aminohydroxamic acid 12 show a nearly complete loss of activity against all isozymes with a pIC50 > 5 only for HDAC10. A three-carbon spacer (DKFZ-711), gives a 3.5-fold increase in HDAC10 activity (pIC50 = 7.48) relative to SAHA. Pleasingly, it also strongly reduces activity against all other tested enzymes, resulting in HDAC10/6 and HDAC10/1 selectivities of 108- and 624-fold, respectively. The δ-amino hydroxamic acid 14 shows a noticeable decrease in affinity compared to SAHA for all measured isozymes, except for HDAC10, resulting in a moderate (<10-fold) selectivity for HDAC10 over HDAC 1 and 6. Lastly, 15 has an over 60-fold higher affinity (pIC50 = 8.73) for HDAC10 than SAHA, consistent with the thermal shift data. Notably, 15 shows potency similar to SAHA against HDAC 1, 2, 6 and 8, resulting overall in moderate HDAC10/6 and HDAC10/1 selectivities of 26- and 44-fold, respectively. This selectivity profile is similar to that of tubastatin A derivative DKFZ-480 (2).
Structure–Activity–Relationship (SAR) of the Linker Region.
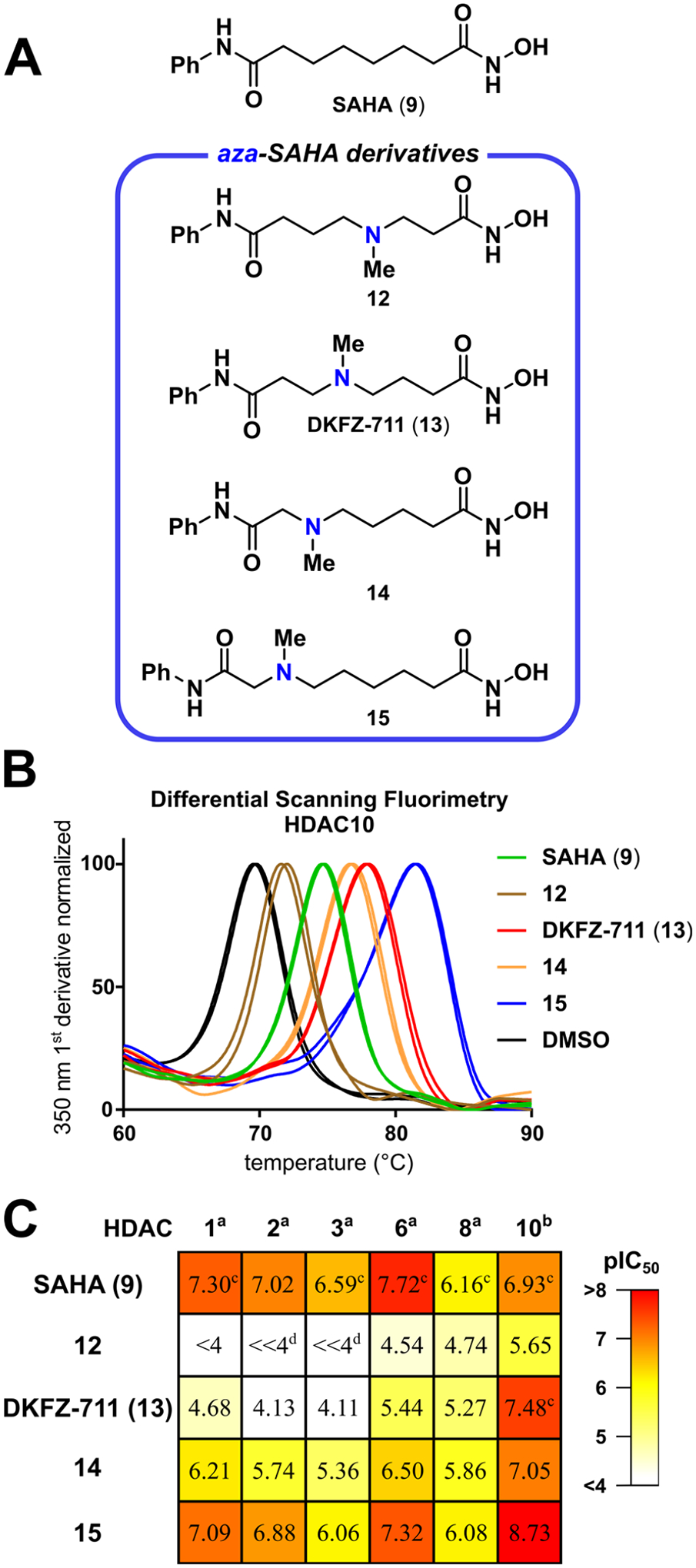
Although 15 is the most potent compound of the small aza-SAHA series, γ-aminohydroxamate DKFZ-711 has the best selectivity profile (37). This confirms the high potency of the previously reported ε-aminohydroxamic acid motif of 10, but reveals that a two carbon shorter amine–hydroxamate separation is superior for selective HDAC10 inhibition. We therefore decided to explore SAR around DKFZ-711. Examination of substituents other than methyl for the tertiary nitrogen (Table 1, orange box, see Scheme S2 for synthesis routes) revealed the small methyl substituent in DKFZ-711 to be the best in terms of potency and selectivity. An inverse correlation between potency against HDAC10 and steric bulk was observed (16–21), consistent with the inhibitors residing in the relatively narrow binding tunnel of HDAC10. The basicity of the nitrogen atom in the linker was also probed: 2,2,2-trifluoroethyl derivative 22 (pIC50 = 5.84) showed over 10-fold reduced HDAC10 activity as compared to ethyl derivative 16 (pIC50 = 6.93). At the same time, HDAC6 and Class I activity is changed little by the electron withdrawing substituent, highlighting that a basic linker is important only for HDAC10 binding.
Table 1.
pIC50 valuesa of γ-amino hydroxamic acids.
|
pIC50 values are the mean of two replicates, each determined by four-parameter non-linear regression analysis with eight dose levels in triplicate with HDAC Glo assayb or FRET assayc.
Mean of at least three individual experiments.
Curve fit analysis not convergent at 100 μM. SF (selectivity factor) = 10(pIC50(HDAC10) − pIC50(HDAC6 or 1)) rounded to two significant figures. Activity profile for the best HDAC10 inhibitors are highlighted in bold. For an extended version of this table with 95% confidence intervals, see Table S2.
We also constrained the three carbon-linker between the amino group and zinc-binder hydroxamic acid group as trans- and cis-cyclobutanes 23 and 24, respectively, as azetidine 25, and as piperidine 26 (Table 1, green box, see Scheme S3 for synthesis routes). Four-membered rings all result in losses of potency and selectivity, with only piperidine 26 showing selectivity similar to DKFZ-711, but with reduced HDAC10 activity (pIC50 = 7.06). Removal of a carbon between the amino and cap groups (27, purple box, see Scheme S4 for synthesis routes) has little effect on HDAC10 activity (pIC50 = 7.39), but increases HDAC6 inhibition (pIC50 = 6.05), resulting in a loss of selectivity. On the other hand, extension of the DKFZ-711 linker on the cap group side to propyl (28), significantly increases HDAC10 activity (pIC50 = 8.08). At the same time, activity against all other tested isozymes decreases, resulting in about 2700- and 6200-fold HDAC10/6 and HDAC10/1 selectivities, respectively.
In addition to altering the linker length, we investigated the potential influence of the amide bridging the linker with the capping region (Table 1, gray box, see Scheme S5 for synthesis routes). Inverting the anilide in DKFZ-711 to a benzamide gave DKFZ-728 (29), which has improved HDAC10 activity (pIC50 = 7.97) and decreased off-target activities with about 890- and 2900-fold HDAC10/6 and HDAC10/1 selectivities, respectively. Interestingly, 30, where the benzamide of DKFZ-728 is replaced with a benzenesulfonamide, has a similar HDAC10 activity profile to that of DKFZ-711. Also noteworthy is the over 10-fold reduction in HDAC10 potency of methyl amide 31, indicating the importance of the amide NH (vide infra). We therefore employed benzimidazoles in 32 and 33 in order to provide a potentially better hydrogen bond donor. Ethyl-linked 32 exhibits a significant gain in potency (pIC50 = 8.08) compared to the parent compound DKFZ-711, and the HDAC10/6 selectivity is improved to 220-fold. Propyl-linked 33 has similar HDAC10 binding (pIC50 = 8.09) and slightly reduced selectivity compared to 28.
SAR of the Capping Region. We next optimized the cap group of DKFZ-728 (see Scheme S5 for synthesis routes). Compounds 28 and 33 have the highest HDAC10 potency and largest selectivity factors from
Table 1; however, DKFZ-728 also has an excellent biochemical selectivity profile as well as superior cellular selectivity (vide infra). We reasoned that keeping the linker as short as possible was favorable and that cap group diversification of DKFZ-728 by amide bond formation would be simpler than for benzimidazole 33.
Structures of cap SAR compounds are shown in Figure 3, together with a graphical summary of data (red dots) as a plot of potency vs. HDAC10/6 selectivity (see Table S2 for complete assay data). Selected compounds from Figure 2 and Table 1 are also included for reference (other colors). Many derivatives had decreased selectivity, especially 2-methyl indole compounds 34 and 35. Potency, on the other hand, could be well modulated with differently sized cap groups. Fused bi- and tricyclic (hetero)aromatic ring systems provide a gain in HDAC10 potency, but often at the price of slightly reduced selectivity over HDAC6, as seen with the indazoles 36 and 37, anthracene 38, and hydroxynaphthalene 39. The highest gain in potency with retention of selectivity could be achieved by the naphthamide cap groups in 40 and 41 (DKFZ-748), while biphenyl 43 resulted in a loss of selectivity and potency despite the increased hydrophobic surface area. Among the naphthamides, DKFZ-748 had the best HDAC10-selectivity, superior to its regioisomer 42.
Figure 3. Biochemical potency versus selectivity plot of HDAC10 inhibitors.
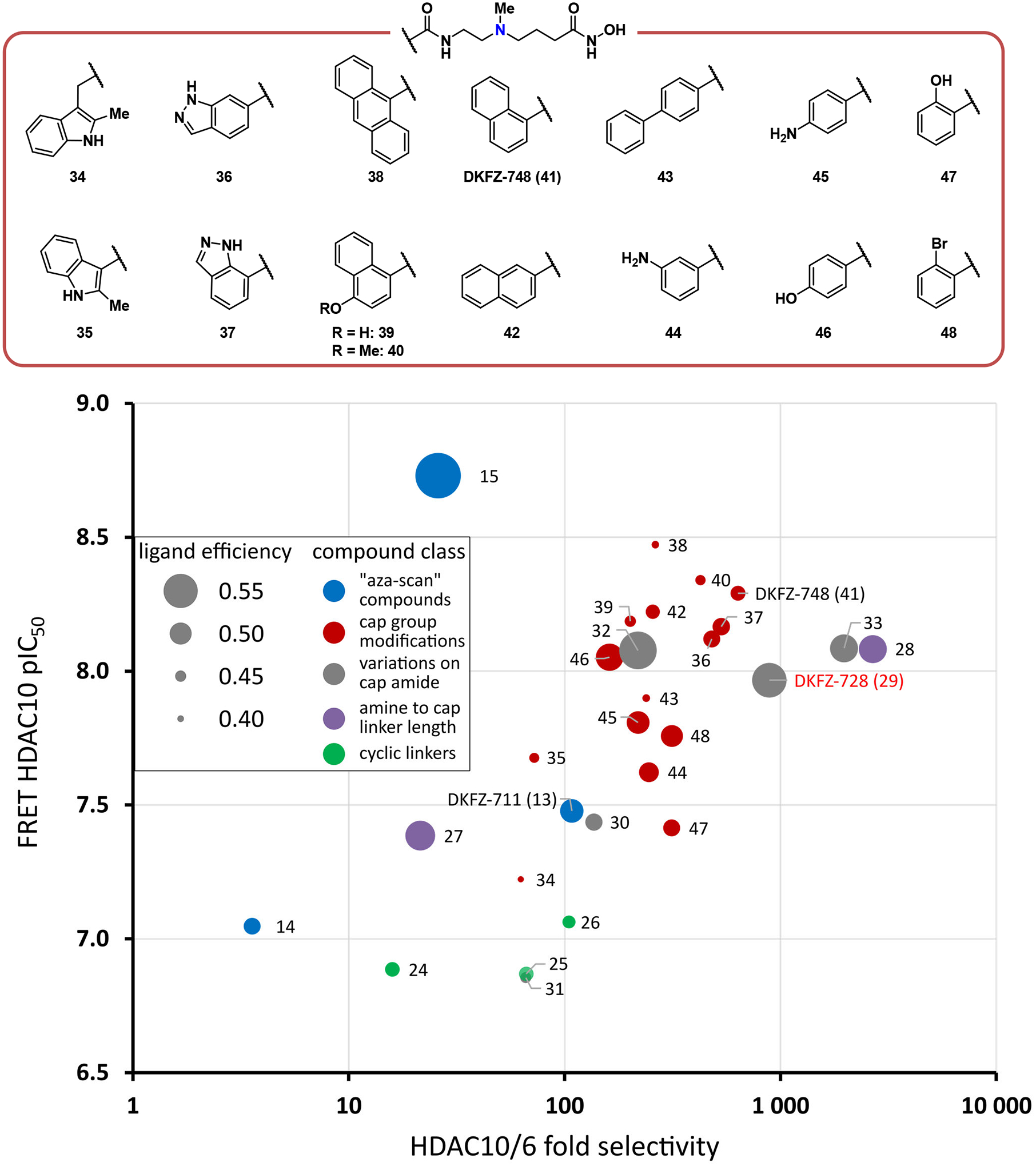
Cap group modifications of DKFZ-728 modulate HDAC10 potency, but do not achieve an increase in selectivity beyond that of DKFZ-728, 28, and 33. Dot size represents ligand efficiency (LE = 1.37 × pIC50(HDAC10) / number of heavy atoms) (38). Colors represent from which “box” from Table 1 and Figures 2 and 3 the compounds originate: Blue dots: “aza-scan” compounds (Figure 2); Red dots: cap modifications based on DKFZ-728 (Figure 3); Grey dots: cap attachment amide modifications (Table 1); Purple dots: amine to cap amide linker length variations of DKFZ-728 scaffold (Table 1); Green dots: cyclic γ-amine linkers (Table 1). For a tabulated and extended version of this plot with 95% confidence intervals, see Table S2.
We also probed the three aromatic substitution vectors of benzamide DKFZ-728 with aniline (44 and 45) and phenol functionalities (46 and 47), but no beneficial vector for additional H-bond donor/acceptors was identified. Constraining the cap group phenyl ring either in plane with the amide (47) or out of plane (48) did not show increased HDAC10 binding or selectivity over HDAC6. Overall, the cap group modifications did not increase HDAC10/6 selectivity beyond that of DKFZ-728, 28, and 33. Still, the SAR study suggests that HDAC10 is remarkably tolerant towards differently sized and substituted (hetero)aromatic cap groups and demonstrates that physicochemical properties and potency can be modulated with different cap groups, while maintaining excellent HDAC10 selectivity, as long as the γ-amino linker is employed.
Crystal Structures of Linear HDAC10i Complexed with Humanized Danio rerio HDAC10.
In order to gain insight into the structural features of HDAC10 binding, we solved crystal structures of SAHA, 14, DKFZ-711, and DKFZ-728 bound to zebrafish (D. rerio) HDAC10, which has been “humanized” by the introduction of two mutations near the binding site (39). The hydroxamate group of each inhibitor coordinates to the catalytic Zn2+ ion with bidentate geometry, except for DKFZ-711 which tends toward monodentate coordination due to a relatively long C=O--- Zn2+ separation of 2.8 Å. Additionally, each Zn2+-bound hydroxamate N–O− group accepts a hydrogen bond from H136, each hydroxamate C=O group accepts a hydrogen bond from Y307, and each hydroxamate NH group donates a hydrogen bond to H137.
The 2.10 Å-resolution crystal structure of the HDAC10–SAHA complex (Figure 4A) reveals that the NH of the anilide capping group donates a hydrogen bond to a water molecule that in turn donates a hydrogen bond to the active site gatekeeper, E274. The Zn2+ ligand H176 donates a hydrogen bond to E274 as well, as also observed in the HDAC10–tubastatin A complex (15), thereby forming a hydrogen bond network between H176 and the anilide NH group. Water-mediated hydrogen bond networks with H176 are also observed in other HDAC10–inhibitor complexes (15, 13, 12, 40).
Figure 4. X-ray crystal structures of inhibitors bound to HDAC10.
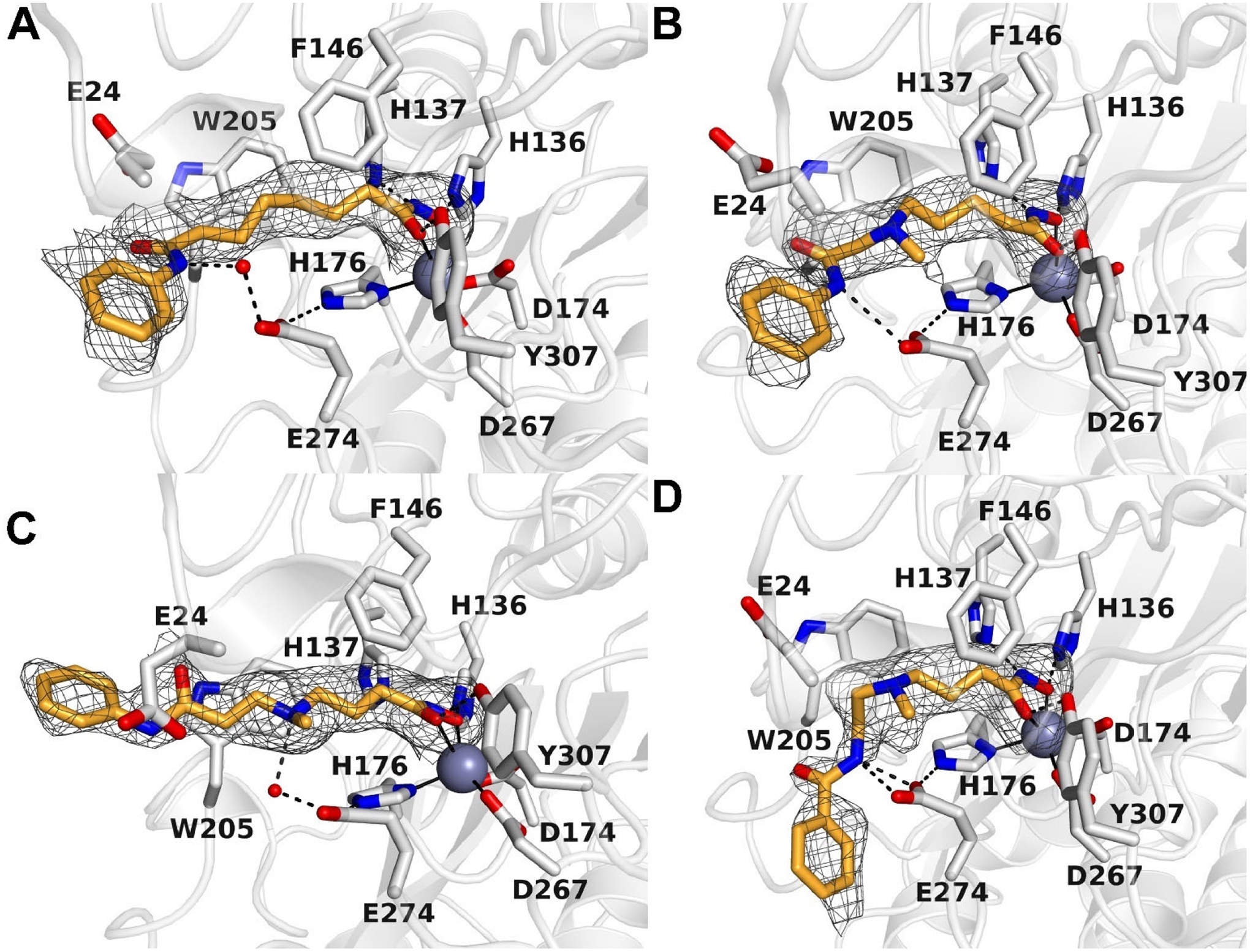
Simulated annealing omit maps of inhibitors in the active site of HDAC10: A. SAHA (contoured at 1.7σ); B. Inhibitor 14 (contoured at 2.3σ); C. DKFZ-711 (contoured at 2.4σ); D. DKFZ-728 (contoured at 2.2σ). Atoms are color-coded as follows: C = light gray (HDAC10) or orange (inhibitors), N = blue, O = red, and Zn2+ = gray sphere. Metal coordination interactions are shown as solid black lines and hydrogen bonds are represented as dashed black lines.
The 2.15 Å-resolution crystal structure of the HDAC10–14 complex (Figure 4B) reveals that the tertiary amine installed at the δ–position of the inhibitor, which is protonated and hence positively charged at physiological pH, makes a weak cation–π interaction with W205 and a long-range electrostatic interaction with gatekeeper residue E274 (the N---O separation of 4.2 Å is too long for hydrogen bonding). As in the SAHA and tubastatin A structures, E274 forms a hydrogen bond with H176. Whereas the anilide NH group of SAHA makes a water-mediated hydrogen bond with E274, the anilide NH group of 14 makes a direct hydrogen bond interaction with E274. Additionally, the sidechain of E24 interacts with the anilide C=O group, so it is possible that E24 is protonated to accommodate this interaction.
The crystal structure of the HDAC10–DKFZ-711 complex (Figure 4C) was determined at 2.25 Å resolution. In contrast to 14, DKFZ-711 contains a tertiary ammonium group not at the δ-, but at the γ-position of the inhibitor. Even so, the tertiary ammonium group is also stabilized by a weak cation–π interaction with W205 and does not make a direct hydrogen bonding interaction with E274. However, it donates a hydrogen bond to water molecule #194 which in turn donates a hydrogen bond to E274. The gatekeeper residue (E274) also forms a hydrogen bond with the Zn2+ ligand H176. The anilide capping group makes no direct hydrogen bonds with protein residues.
The crystal structure of the HDAC10–DKFZ-728 complex (Figure 4D) was determined at 2.20 Å resolution. Like DKFZ-711, DKFZ-728 contains a tertiary ammonium group at the γ-position of the inhibitor. Interactions are similar in that it appears to make a weak cation–π interaction with W205, but in contrast with DKFZ-711, DKFZ-728 makes a long-range electrostatic interaction with gatekeeper E274. Here, too, E274 forms a hydrogen bond with H176. DKFZ-728 differs from DKFZ-711 in that the amide bond has been “inverted” from an anilide to a benzamide. This benzamide NH group donates a bifurcated hydrogen bond directly to E274, which is not observed for the anilide NH in DKFZ-711. Moreover, the benzamide capping group adopts a different conformation in comparison with that of DKFZ-711 as well as 14 (Figure 5). Finally, the benzamide carbonyl of DKFZ-728 replaces a water molecule within a surface water network that is found in both 14 and DKFZ-711.
Figure 5. Overlay of HDAC10–inhibitor structures.
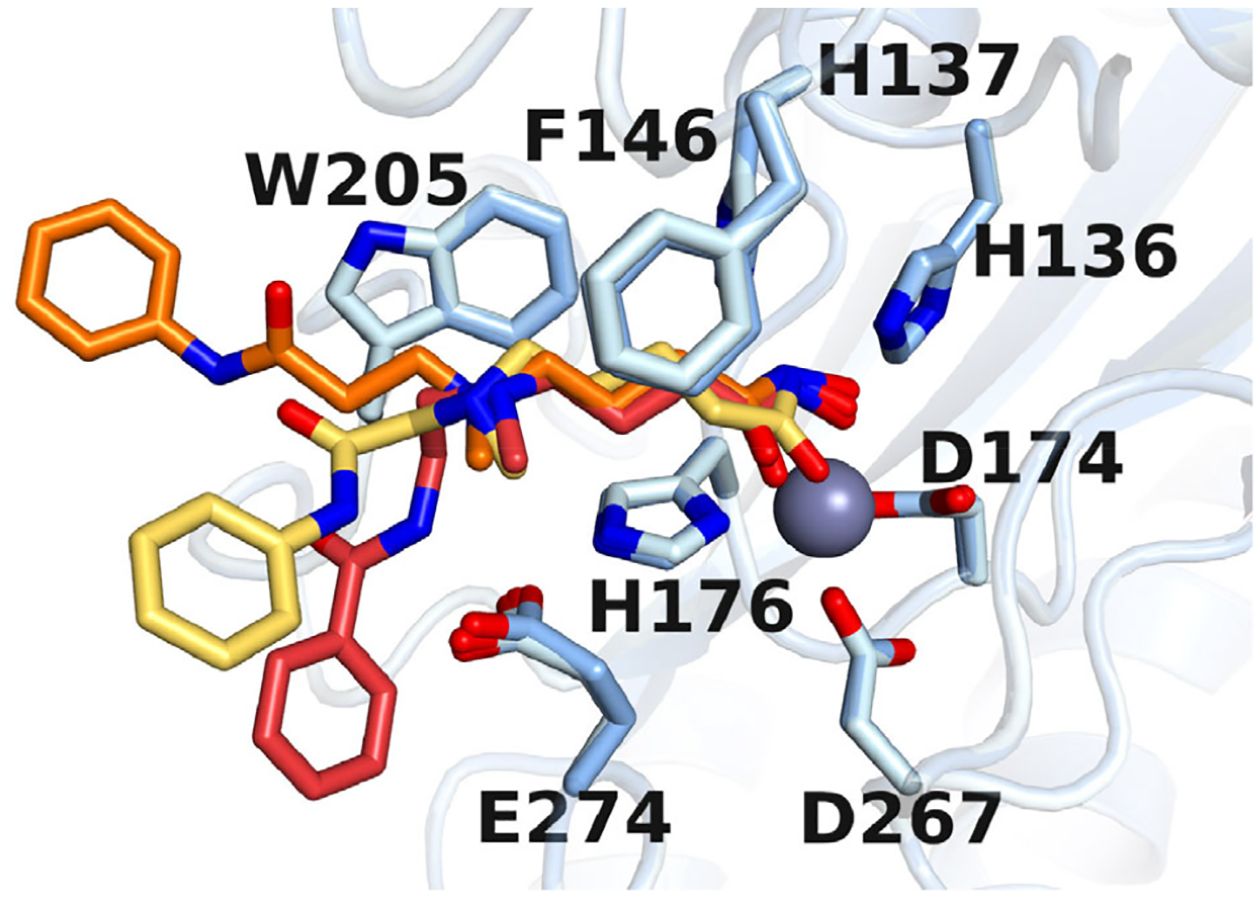
Blue: HDAC10 residues; Yellow: inhibitor 14; Orange: DKFZ-711; Red: DKFZ-728.
We incorporated a tertiary ammonium cation into the SAHA scaffold to mimic N8-acetylspermidine, an excellent substrate of HDAC10. On the basis of previous crystal structures of HDAC10 bound by polyamine 10 (33) and tubastatin A (15), we expected that the ammonium cations of potent binders would directly interact with E274, the gatekeeper residue. To our surprise, the distinguishing feature of the three newly solved aza-SAHA structures is that each ammonium establishes a cation-π interaction with W205. Despite the ammonium cations being located at γ– or δ–positions relative to the hydroxamic acid, they are in approximately the same location relative to W205 in each complex, suggesting that the cation-π interaction acts as an anchor around which the molecules orient themselves (Figure 5). In the crystal structure of 10 bound to HDAC10 (PDB 6UHV), (33) the secondary ammonium of 10 is at the ε position and makes a bifurcated hydrogen bond with E274. Presumably, compound 15, with an ε-amino linkage like 10, also makes a direct H-bond interaction between the ammonium and E274. As seen with tubastatin A derivative 2, such a polarized H-bond to the gatekeeper residue provides excellent potency toward HDAC10; however, binding to the gatekeeper residue is apparently not sufficient for diminishing potency against other HDAC isozymes, as 15 shows little difference to SAHA with respect to HDACs 1, 2, 3, 6, and 8. Instead, the cation-π interaction with W205 appears to be the critical feature for determining selectivity. Class I HDAC isozymes have a phenylalanine at the position of W205 and presumably DKFZ-711, DKFZ-728, and their derivatives have poorly matched electrostatics with the relatively non-polar surfaces of other HDAC isozymes (11).
These crystal structure data help to rationalize the SAR data surrounding the linker amine and cap group amide. In the case of β-amino derivative 12, the linker length is such that it is geometrically impossible for the hydroxamic acid to bind the zinc at the bottom of the binding tunnel simultaneous to the ammonium cation making a cation-π interaction with W205, resulting in a very weak binder. It is reasonable to assume that conformationally restricted γ-amino hydroxamic acid inhibitors with amino cyclobutane (23 and 24) or azetidine linkers (25) do not allow for optimal location of the ammonium cation to engage in the cation–π interaction with W205. The important contribution of hydrogen bonding from the cap group amide NH to the gatekeeper E274 is reflected in the distinct gain of HDAC10 affinity by inversion of the linker amide from the anilide in DKFZ-711 to the benzamide orientation in DKFZ-728. Contribution of the NH hydrogen bond donor was confirmed by the nearly 10-fold loss of HDAC10 activity by methylation of the DKFZ-728 amide in 31.
In contrast to 14 and DKFZ-728, the amide NH group of DKFZ-711 does not form a hydrogen bond with HDAC10. Even so, DKFZ-711 has higher affinity and selectivity for HDAC10 than 14. Possibly, water #194 contributes to increased affinity. Structures of inactivated HDAC10 complexed with substrates reveal that 1–2 water molecules engage the secondary ammonium group of N8-acetylspermidine and the primary ammonium group of N-acetylputrescine in hydrogen bond interactions that dictate substrate specificity (40). The implicated protons on these ammonium cations point directly to water molecules that are also stabilized by hydrogen bonds with E274, much like the proton on the tertiary ammonium cation of DKFZ-711 that donates a hydrogen bond to water #194.
Docking studies with HDAC6 and HDAC10.
In order to rationalize the decrease in HDAC6 activity of DKFZ-711 compared to its close analogue SAHA, we built HDAC6 docking models (Figure 6A and B) based on the crystal structure of human HDAC6 in complex with the linear inhibitor trichostatin A (PDB 5EDU). Furthermore, we modeled the hydrogen positions into the HDAC10 crystal structures of DKFZ-711 and SAHA (Figure 6C and D) to highlight differences in the water network associated with the introduction of an amine to the SAHA linker region.
Figure 6. Comparison of HDAC6 docking models with HDAC10 crystal structures.
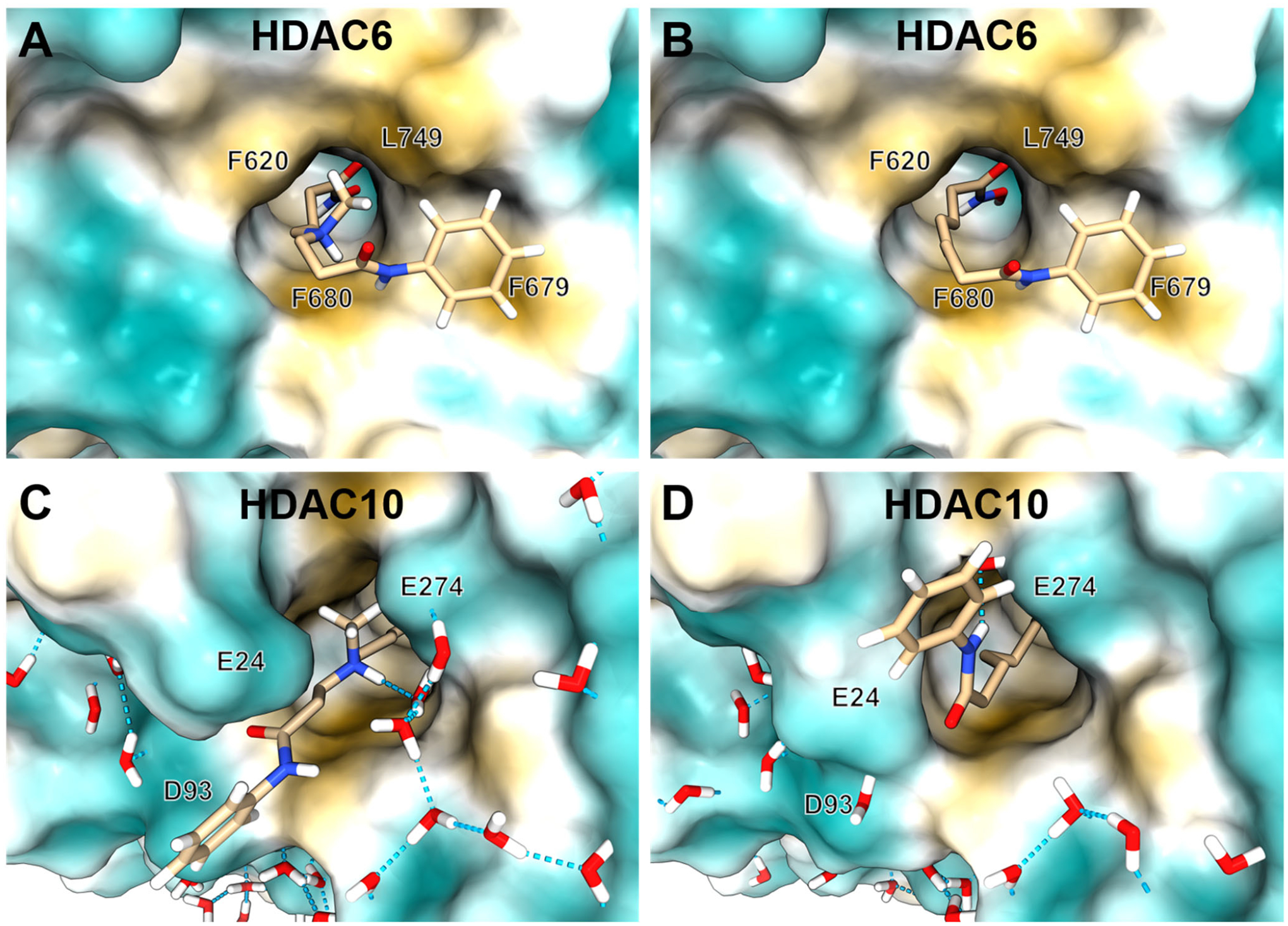
Selectivity-driving interactions are revealed by comparison of HDAC6 docking models with HDAC10–DKFZ-711 and HDAC10–SAHA crystal structures. HDAC6 docking models of DKFZ-711 (A) or SAHA (B) based on PDB 5EDU. Crystal structures of HDAC10 with DKFZ-711 (C) or SAHA (D) with hydrogen atoms added by modeling to emphasize surface water networks. Surface colors: ochre: hydrophobic, turquois: hydrophilic. Methylene hydrogens are omitted for clarity. Hydrogen bonds depicted as dashed blue lines.
Comparison of the HDAC6 docking models with the respective HDAC10 crystal structures shows that while DKFZ-711 benefits from a polar water network and hydrophilic surface in HDAC10 (Figure 6C), in HDAC6 (Figure 6A) the protonated amine is surrounded by a hydrophobic tunnel composed of F620, F680 and especially L749, which replaces the HDAC10 gatekeeper E274. Consequently, no comparably favorable binding pose for DKFZ-711 in HDAC6 was found. For SAHA on the other hand, the HDAC6 binding tunnel matches the hydrophobic alkyl linker (Figure 6B). In HDAC10 (Figure 6D), only the anilide NH group of SAHA can interact with the gatekeeper E247 via a water mediated hydrogen bond, but the aliphatic linker cannot benefit from the glutamate residues E24 and E274 forming the tunnel entrance in HDAC10.
We also built a docking model of DKFZ-748 bound to HDAC10 based on the HDAC10–DKFZ-728 complex (Figure S1). The model suggests a very similar binding pose for DKFZ-748 as observed in the DKFZ-728–complex, including the hydrogen bond from the amide NH to E274. We conclude that the improved binding is mainly driven by unspecific hydrophobic interactions with the protein surface, while unfavorable conformations of the linker region due to steric limitations of the capping group are avoided.
Confirmation of Intracellular HDAC10 Engagement.
With an array of potent and selective compounds in hand, we tested selected substances for target engagement and selectivity over HDAC6 in a cell-based bioluminescence resonance energy transfer (BRET) assay (41). BRET data (plotted in Figure 7 and listed in Table S3) confirms the excellent HDAC10 selectivity of the γ-amino hydroxamic acid motif, verifies the biochemical SAR data, and proves cellular target engagement. Some compounds like 28 showed reduced potency and selectivity in the cellular assay format, while inhibitors with large, hydrophobic cap groups like DKFZ-748 and 38 perform well in both the biochemical and cellular HDAC10 assay.
Figure 7. Cellular potency versus selectivity plot of selected best HDAC10 inhibitors.
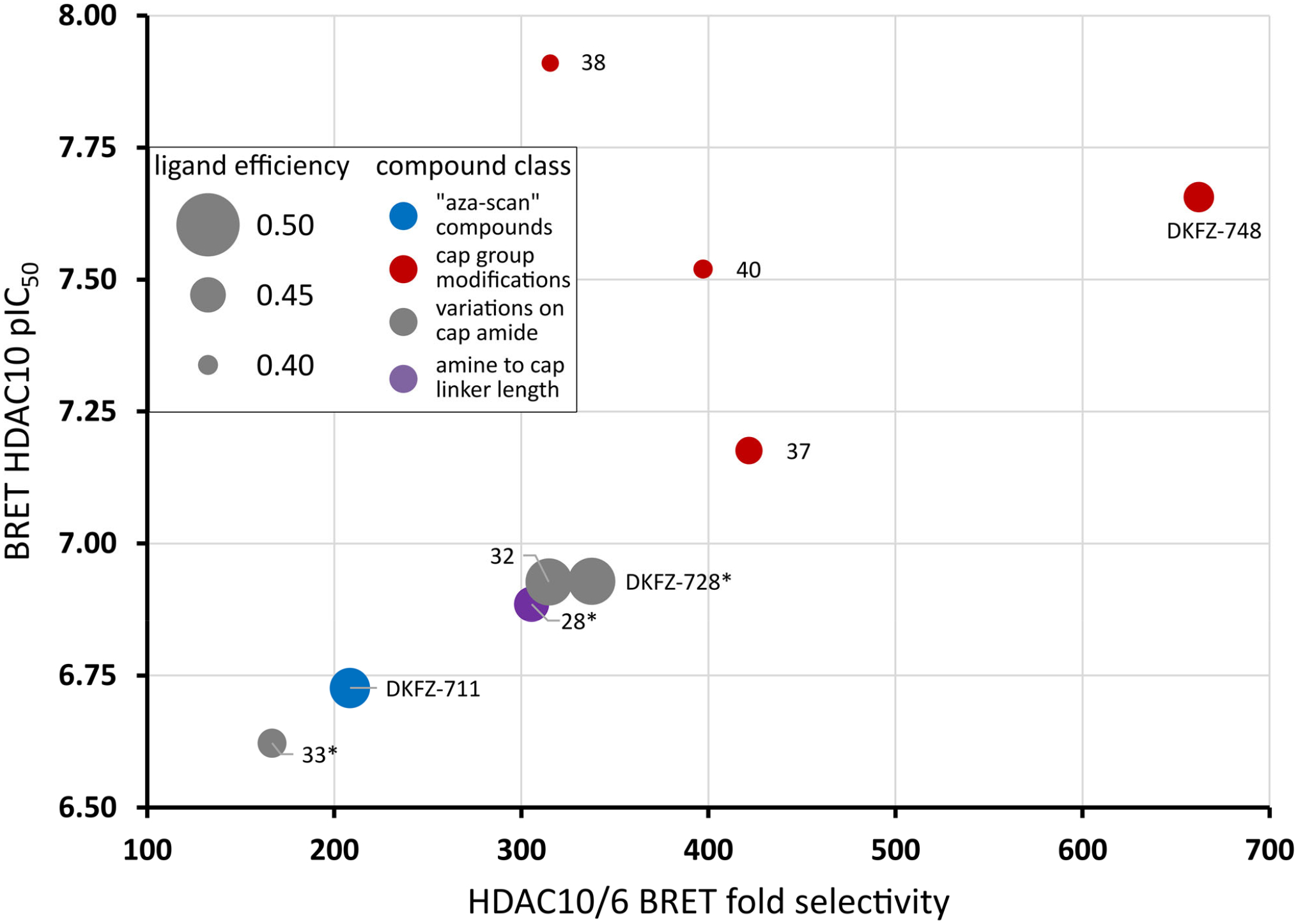
DKFZ-748 achieves the best cellular target engagement and HDAC10 selectivity. Dot size represents ligand efficiency (LE = 1.37 × pIC50(HDAC10) / number of heavy atoms) (38). Colors represent from which “box” from Table 1 and Figures 2 and 3 the compounds originate: Blue dots: “aza-scan” compounds (Figure 2); Red dots: cap modifications based on DKFZ-728 (Figure 3); Grey dots: cap attachment amide modifications (Table 1); Purple dots: amine to cap amide linker length variations of DKFZ-728 scaffold (Table 1). *: HDAC6 pIC50 could not be accurately determined due to lack of activity. Selectivity was calculated using the HDAC6 assay limit (pIC50 of 4.4), actual selectivity may be higher. For a tabulated and extended version of this plot with 95% confidence intervals, see Table S3.
Most amino hydroxamic acids show less than 30% of their biochemical activity in the cellular BRET assay. This can be attributed to the high polarity, and resulting moderate cell permeability of the compounds (Table S4). Less polar HDAC10 binders like SAHA, hindered amines or tubastatin A retain typically 30–60% of their biochemical potency in the cellular assay system (Table S3).
Chemoproteomic Profiling Confirms Selectivity Over Common HDACi Off-Targets.
Having established HDAC10-selectivity in biochemical assay systems with purified proteins and overexpressed target constructs in cells, we aimed for an unbiased selectivity determination within the whole proteome. We therefore employed a recently developed chemoproteomic profiling technology, that uses an HDAC affinity matrix composed of three structurally diverse immobilized hydroxamic acid probes and a bottom-up proteomics readout (31). This approach is capable of assaying up to 9 of the 11 HDACs, plus common HDACi off-targets like GATD3A, ISOC1/2 and ALDH1/2, which were notably hit by SAHA. We chose to profile DKFZ-748 in the human neuroblastoma BE(2)-C cell line because of HDAC10’s role in chemotherapy-resistant neuroblastomas (21). As controls, SAHA and CHDI00465983 were also profiled; SAHA was selected due to its structural similarity to DKFZ-748 and as an HDAC Class I, IIB and non-HDAC off-target reference, while CHDI00465983 served as a control for Class IIA, HDAC8, and MBLAC2. Competition of the inhibitors against the HDAC affinity matrix followed by mass-spectrometry measurement demonstrated dose-dependent binding of the inhibitors to HDAC1, 2, 5, 6, 8, and 10, as well as six non-HDAC off-targets (Table 2, Figure S2). SAHA was found to have apparent pKD values (pKDapp) between 5.7 and 6.4 for HDAC1, 2, 6 and 10, with the highest pKDapp for HDAC6, confirming the biochemical HDAC-Glo selectivity profile (Figure 2C). As reported previously with mixed lysates of MV4–11 and SW620 cells (31), a higher affinity for ISOC1 and ISOC2 than for HDAC6 was also observed for SAHA-treated BE(2)-C lysates. ALDH2 was confirmed as an off-target and is accompanied in BE(2)-C cells by its close relative and novel off-target ALDH1B1.
Table 2.
Apparent pKD valuesa of DKFZ-748 and reference compounds by chemical proteomics profiling.
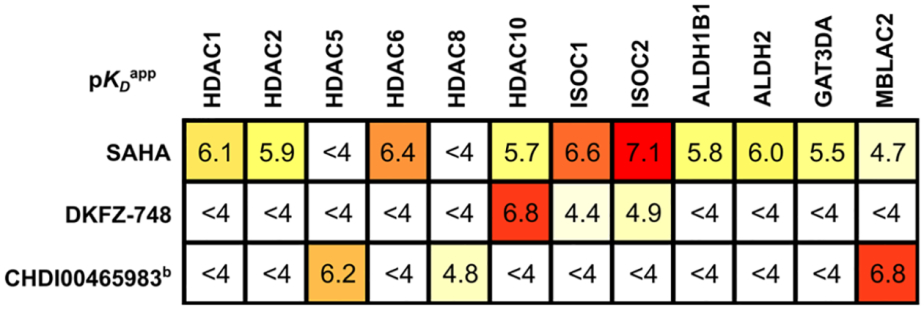
|
Determined by LC-MS/MS after dose-dependent competition pulldown assay from three or twob separate experiments, each performed with eleven dose levels. For structure of CHDI00465983 and dose-response curves with individual datapoints, see Figure S2. Apparent pKD values with standard deviations are listed in Table S6.
On the other hand, DKFZ-748 does not bind any of the non-HDAC metalloenzyme off-targets with a pKDapp ≥ 5.0 and exhibits high HDAC10-potency (pKDapp = 6.8). Moreover, all of the enriched HDAC isozymes have pKDapp < 4, indicating greater than 600-fold selectivity for HDAC10. This data adds a proof of selectivity over HDAC Class IIA with HDAC5 as a representative, which can be competed off the affinity matrix by the Class IIA-selective inhibitor CHDI00465983. Other Class IIA enzymes and HDAC11 could not be robustly enriched from BE(2)-C lysates, likely due to their general low abundance and/or tissue-specific expression. As SAHA is known to be inactive against all Class IIA enzymes as well as HDAC11, we consider it unlikely that DKFZ-748 binds with appreciable potency to those targets.
Selective HDAC10 Inhibitors have no Effect on Histone and Tubulin Acetylation, and are not Cytotoxic.
We selected DKFZ-728, DKFZ-748 and 37 for further biological testing due to their high selectivity, high cellular target engagement, and good ligand efficiency. To confirm HDAC10 selectivity in a functional, cellular context, we investigated the effects of our inhibitors on the acetylation status of potential off-target substrates by Western blot in BE(2)-C cells (Figure 8A and S3). No increase in acetylation of the HDAC1 substrate histone H3 is detected even at 100 μM DKFZ-728, about 40-fold above the concentration required for complete cellular HDAC10 target occupancy (~2.5 μM). We also observe no effects at 10 μM with the more potent inhibitors DKFZ-748 and 37. Similar results were obtained for the HDAC6 target α-tubulin: while tubastatin A and SAHA treatment both result in hyperacetylation of tubulin at low micromolar concentrations, aza-SAHA inhibitor doses sufficient for full cellular HDAC10 inhibition do not affect tubulin acetylation. Only the highest tested concentration of DKFZ-748 (100 μM) induced significant acetylation out of the three aza-SAHA derivatives.
Figure 8. Cellular drug-effect markers and cytotoxicity data.
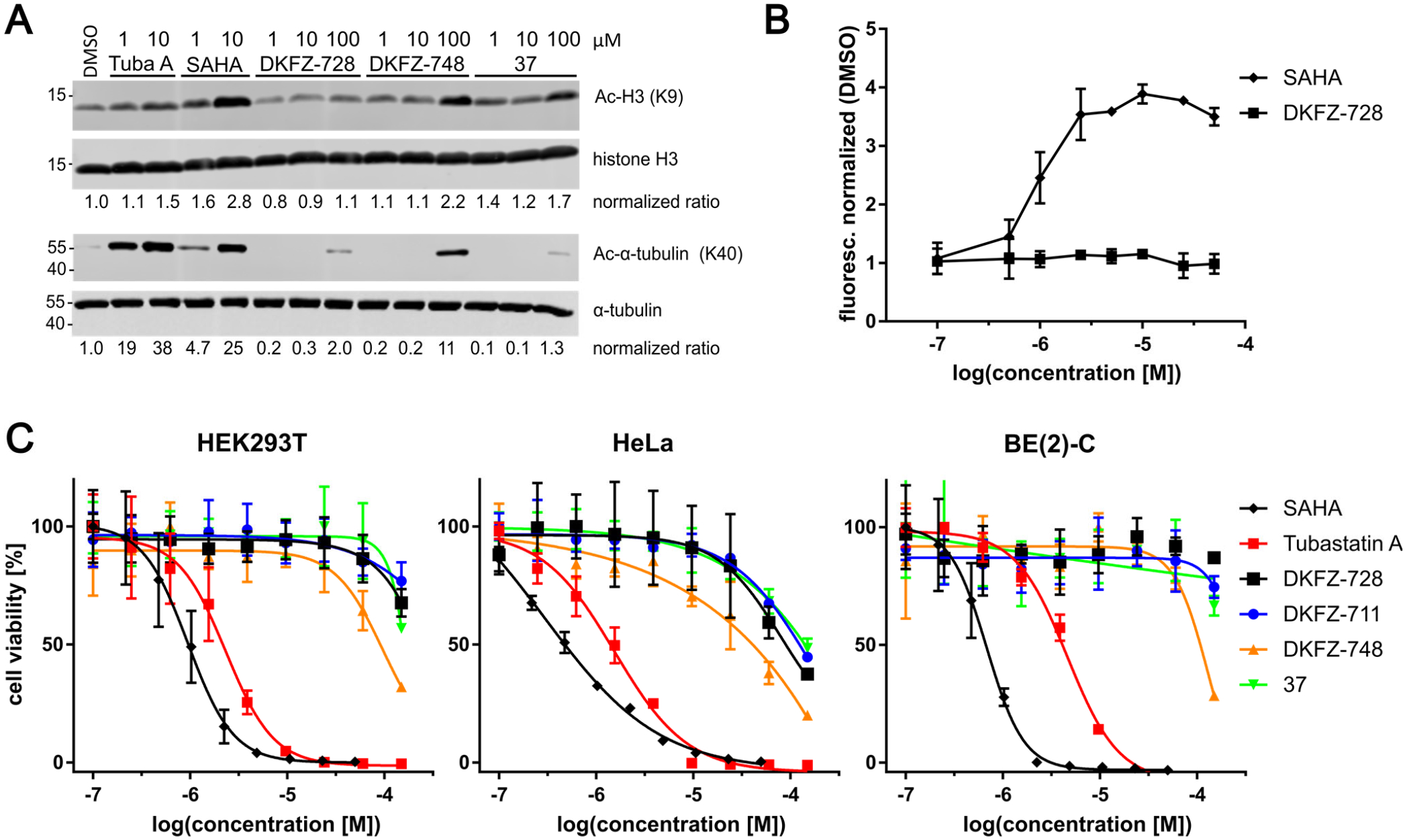
A: Acetylation Western blots of histone H3 and α-tubulin from BE(2)-C cells demonstrates no off-target hyperacetylation at concentrations required for full inhibition of HDAC10. Whole blots of two independent biological replicates are provided in Figure S3. B: HDAC10-selective DKFZ-728 does not lead to acidification of the lysosomal compartment, a previously used measure of HDAC10 interference. Shown by increased fluorescence from the LysoTracker DND-99 probe in BE(2)-C cells, represented as mean with range of two independent experiments. C: No cytotoxicity (Cell-Titer-Blue) associated with HDAC10 inhibition in three representative cell lines is observed after 72 h treatment, in contrast to SAHA (pan-inhibitor) and tubastatin A (HDAC6/10 selective).
Previously, we measured acidification of the lysosomal compartment as a marker for cellular HDAC10 interference due to the involvement of HDAC10 in autophagy (7, 42). Surprisingly, the potent and highly selective HDAC10i DKFZ-728 did not lead to lysosomal acidification (Figure 8B), a phenotype observed after HDAC10 knockdown and with comparably potent, but less selective HDAC10 binders like many tubastatin A derivatives (7) or the pan inhibitor SAHA. Interestingly, Western blots against p62 and LC3-I/II of BE(2)-C cells treated with DKFZ-748 suggest autophagy may be reduced (Figure S4).
Selective HDAC10 inhibitors show a lack of growth inhibition or cytotoxic effects in HEK293T, HeLa, and BE(2)-C cells (Figure 8C). Although DKFZ-728 is stable in cell culture with BE(2)-C cells for at least 72 h (Figure S5), we observed no reduction in cell viability linked to HDAC10 inhibition. This is remarkable, as our selective HDAC10 inhibitors differ from the cytotoxic drug SAHA only by an appropriately placed amino functionality, and still show comparable intracellular target engagement as well as acceptable ADME profiles (Table S4). To expand beyond these three cell lines, one-dose NCI-60 screening was performed with DKFZ-711 at 10 μM, which resulted in an average viability of 99 ± 8 % over all 60 cell lines with the lowest 10th percentile covering 72–91 % viability (for complete data see SI NCI60 Screen Data xlsx table).
We also observed no cooperative toxicity in BE(2)-C cells by co-treatment of 5 μM 37 with the Helmholtz Drug Repurposing Library, a collection of over 5600 pharmaceuticals and bioactive compounds, covering a wide range of biological targets. After manual curation of hits, we tested 12 drugs with synergistic effects in the screen, but none of these hits could be reproduced and validated (see SI Sensitization screen with repurposing library data xlsx table).
Selective Inhibition of HDAC10 Leads to Spermidine N8-Hyperacetylation in Cells.
The fact that our selective HDAC10 binders induce no proliferative phenotype in cells, in spite of demonstrated cellular target engagement, prompted us to question whether they inhibit HDAC10 enzymatic activity. To our knowledge, there is no report on the enzymatic inhibition of native cellular HDAC10 since its classification as the cytosolic polyamine deacetylase. Although polyamines are abundant in cells, endogenous levels of acetylated polyamines are very low. We therefore established a targeted metabolomics LC-MS/MS approach to measure cellular acetylated spermidine species: N1-acetyl-, N8-acetyl- and N1,8-diacetylspermidine were quantified in BE(2)-C cells treated with hydroxamic acid DKFZ-748, its corresponding ester (49) as a negative control (see Table S2), or vehicle after 24 h (Figure 9A and Figure S6). We observed significant accumulation of N8-acetyl- and N1,8-diacetylspermidine in a dose-dependent manner with DKFZ-748, but no accumulation of N1-acetylspermidine. Effective dose-levels align with the pIC50 for cellular target engagement (BRET pIC50 of DKFZ-748 = 7.66), and the inactive amino ester 49 has no effect on acetylspermidine levels. HDAC10 knockdown resulted in a similar metabolomic profile (Figure 9B) with significant accumulation of only N8-acetyl- and N1,8-diacetylspermidine. In contrast to pharmacological inhibition, HDAC10 knockdown decreased N1-acetylspermidine relative to the control, which might be a result of regulatory processes of polyamine metabolism involving the HDAC10 protein. Notably, these data confirm for the first time in cells the previously reported (11) polyamine substrate specificity of recombinant HDAC10.
Figure 9. HDAC10 inhibition and knockdown affect polyamine acetylation.
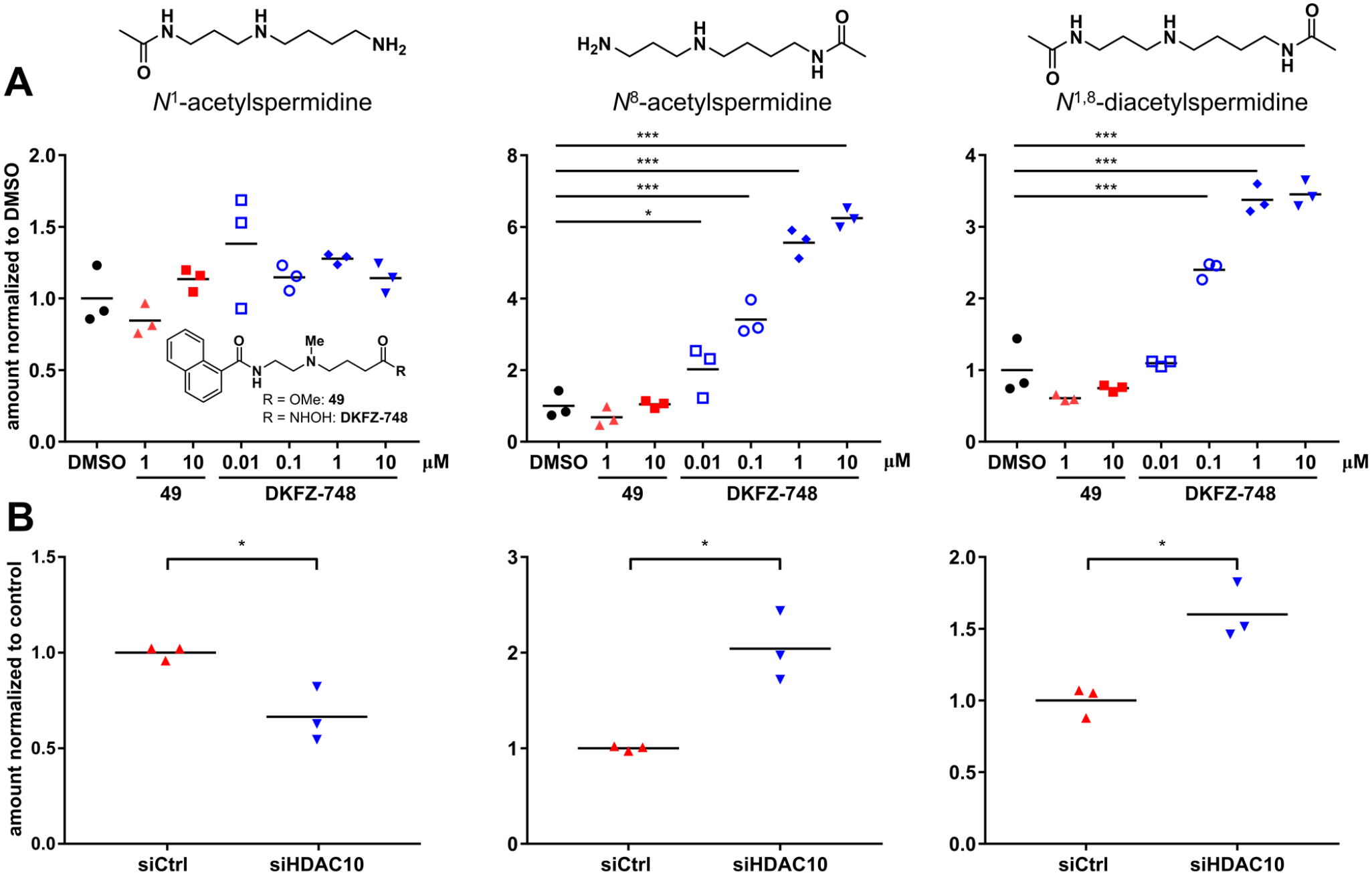
Targeted metabolomics quantification by LC-MS/MS of N1-acetylspermidine, N8-acetylspermidine and N1,8-diacetylspermidine in BE(2)-C cells after 24 h treatment with DKFZ-748 or 49 (A) and after knockdown (B). Results in (A) were reproduced in a second, independent experiment (Figure S6). Significance of inhibitor treatment was calculated between DMSO controls and samples by one-way ANOVA, following the Dunnett test for multiple comparison. Significance of knockdown vs. control was determined by unpaired t-test with Welch’s correction. *: p-value < 0.05, ***: <0.001.
Selective Inhibition of HDAC10 Suppresses Tumor Cell Growth under Polyamine-Limiting Conditions.
Polyamines are essential for tumor growth and progression, but polyamine-targeting anti-cancer strategies are challenging due to a variety of compensatory mechanisms within the highly regulated polyamine metabolic pathway (18). Therefore, we utilized DKFZ-748 as a pharmacological tool to investigate the role of HDAC10 as a source of spermidine under polyamine-limiting conditions.
HeLa cells expressing HDAC10 (wildtype) or lacking HDAC10 due to CRISPR/Cas9-mediated knockout were treated with α-difluoromethylornithine (DFMO), an irreversible inhibitor of ornithine decarboxylase, a critical enzyme in polyamine biosynthesis. Exposure of wildtype HeLa cells to DFMO caused growth inhibition that could be rescued by exogenous supply with N8-acetylspermidine (N8-AcSPD) (Figure 10A, blue curve). However, rescue by N8-AcSPD was greatly reduced in HeLa cells genetically lacking HDAC10 (Figure 10A, green curve). Similarly, treatment with DKFZ-748 caused a dose-dependent inhibition of rescue by N8-AcSPD.
Figure 10. HDAC10 inhibition suppresses rescue by N8-AcSPD under polyamine-limiting conditions.
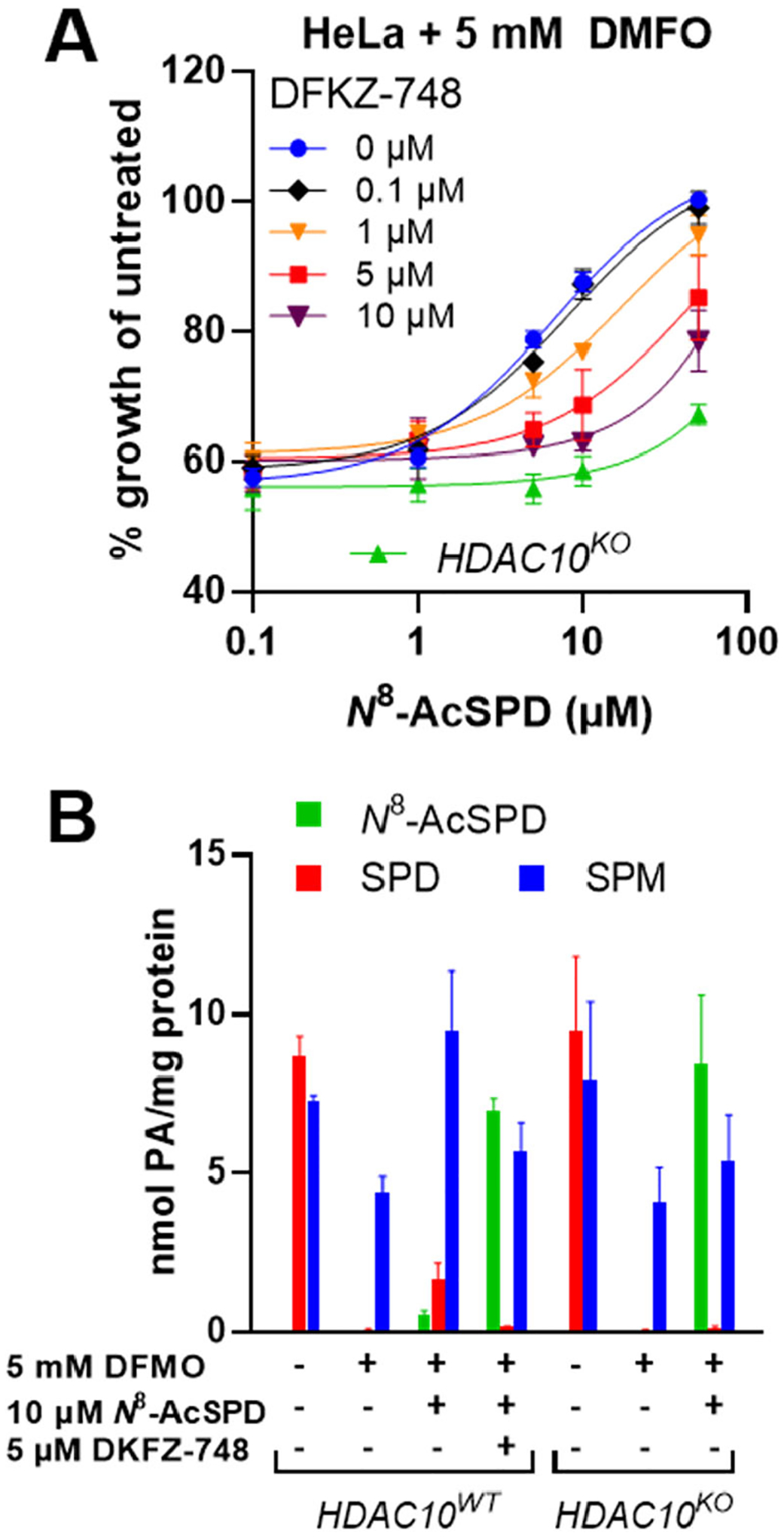
A: DKFZ-748 prevents growth rescue by N8-AcSPD from DMFO treatment in HeLa S3 cells dose-dependently. B: HPLC quantification of polyamines in treated HeLa S3 cells. Cells were pre-treated with DKFZ-748 for 24 h, followed by co-treatment with DMFO and N8-AcSPD for 96 h. Data is pooled from at least two biological replicates, each performed in technical triplicates, and represented as mean with standard error.
HPLC-based quantification of intracellular polyamine pools under these conditions further confirmed HDAC10 cellular function as a PDAC (Figure 10B). Treatment of HeLa cells with DFMO depleted spermidine (SPD) and reduced spermine levels (SPM) in association with growth arrest. Interestingly, supplementation with extracellular N8-AcSPD restored the intracellular spermine level and increased the spermidine concentration, with very little accumulation of N8-AcSPD. This is consistent with HDAC10-catalyzed N8-AcSPD deacetylation. In contrast, HeLa cells with genetic knockout of HDAC10 or wildtype cells treated with DKFZ-748 had no significant restoration of spermidine or spermine, and N8-AcSPD concentrations were substantially increased. These results indicate, that HDAC10 is responsible for N8-AcSPD rescue in cells depleted of polyamines by DFMO treatment. Similarly, HDAC10 inhibition with DKFZ-748 also prevents rescue dose-dependently and elicits a phenotypic response that recapitulates that of genetic HDAC10 loss.
CONCLUSIONS
We report the rational design of HDAC10 inhibitors with unprecedented selectivity over HDAC6, Class I isozymes, and other metalloenzyme off-targets. Compounds with an amino functionality inserted into the linear linker region of the pan-inhibitor SAHA have distinct HDAC isozyme selectivity profiles depending on the location of the amino group. While linker variations resulted in a dispersed selectivity profile, cap group modifications based on the HDAC10-selective linker resulted in a mostly clustered SAR. We identified the flexible, slender γ-amino hydroxamic acid scaffold as the driver for HDAC10 selectivity, which is enhanced by an appropriately located hydrogen bond-donating NH group, which bridges the linker to a flat, bicyclic hydrophobic capping region.
Structural insights from HDAC10–inhibitor complexes, together with the SAR study paint a clear picture of the HDAC10 active site and provide a good understanding of the structural features for potent and selective HDAC10 inhibition. Docking models with HDAC6 provide a structural rational for HDAC10 over 6 selectivity, which is driven by the hydrophilic nature of the added amino group within the SAHA linker.
We demonstrated potent HDAC10 binding with a ligand-displacement FRET assay (best inhibitors with pIC50 > 8) and by thermal shift of recombinant HDAC10. Intracellular target engagement was shown with a BRET assay (best inhibitors with pIC50 > 7.5), and cellular inhibition of HDAC10 PDAC activity was confirmed by targeted metabolomics of acetylated spermidines. Over 500-fold HDAC10/6 selectivity for the best inhibitors was observed in cell-free and cellular target engagement assays, while selectivity over Class I enzymes was measured to be several 1000-fold in a biochemical assay format. Western blotting of HDAC substrates confirmed functional cellular selectivity over Class I enzymes and HDAC6. Finally, chemoproteomic profiling confirmed HDAC10-selectivity within the whole target landscape of HDAC drugs. This includes selectivity over Class IIA enzyme HDAC5 and non-HDAC off-targets of SAHA.
We therefore conclude that γ-amino hydroxamic compounds, like DKFZ-748, fulfill the requirements of a selective chemical probe (43) for HDAC10 and are valuable tool compounds to investigate HDAC10 biology and the role of acetylated polyamines. As a proof of principle, we applied DKFZ-748 in a polyamine-limited in vitro tumor model, where pharmacological inhibition of HDAC10 phenocopied its genetic knockout by dose-dependent suppression of N8-AcSPD growth rescue. We anticipate that our chemical probe will enable further studies on the role of acetylated polyamines in oncology and beyond.
Selective HDAC10i exhibit surprisingly low cytotoxicity in cancer cell lines and HEK293T compared to the HDAC6/10 inhibitor tubastatin A. This may be beneficial for future therapeutic applications of selective HDAC10i as immunomodulators, as previously suggested (19) by the long-term allograft survival of HDAC10−/− mice receiving fully MHC-mismatched cardiac transplants. That inhibition of HDAC10 enzymatic function is not cytotoxic, and does not sensitize neuroblastoma (BE(2)-C) cells toward drug treatment, suggests that a so far undescribed scaffolding or adaptor function of HDAC10 may exist. The development of selective HDAC10 degraders (44) and rescue experiments using mutant, enzymatically inactive HDAC10 could help to clarify these currently unanswered questions.
Supplementary Material
ACKNOWLEDGMENT
A.K.M. acknowledges the Helmholtz Drug Initiative and the German Cancer Research Center (DKFZ) for financial support. I.O. acknowledges the H.W. & J. Hector Foundation for grant M91. D.W.C. thanks the NIH for grant GM49758. R.A.C and T.M.S. thank the University of Pennsylvania Orphan Disease Center Million Dollar Bike Ride (MDBR-20-135-SRS) and the Chan Zuckerberg Initiative. R.A.C. acknowledges the National Institutes of Health National Cancer Institute for grants CA204345 and CA235863. S.L. and B.K. acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG) (SFB 1309, project 401883058). We thank Dr. Karel Klika and Gabriele Schwebel for NMR spectroscopy support. We thank Johanna Hummel-Eisenbeiß for performing Western blot analysis. We thank Dr. Karine Lapouge for performing thermal shift experiments and Kerstin Putzker for performing the co-treatment screening with the Helmholtz Drug Repurposing Library. We thank Dr. Elisa Nuti for MMP activity measurements, and Hannah Steffke for the synthesis of 26. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Supplementary tables and figures, synthesis schemes, detailed experimental procedures, compound characterization, and 1H and 13C NMR spectra of newly synthesized substances. (PDF) Molecular docking of DKFZ-748 with HDAC10 (PDB)
Molecular docking of SAHA with HDAC6 (PDB)
Molecular docking of DKFZ-711 with HDAC6 (PDB)
NCI60 Screen Data (XLSX)
Sensitization screen with repurposing library data (XLSX)
ACCESSION CODES
The atomic coordinates and crystallographic structure factors of enzyme-inhibitor complexes have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes as follows: HDAC10-SAHA complex, 7SGG; HDAC10-compound 14 complex, 7SGI; HDAC10-DKFZ-711 complex, 7SGJ; HDAC10-DKFZ-728 complex, 7SGK.
B.K. is cofounder and shareholder of OmicScouts and msAId. He has no operational role in either company. The other authors declare no competing financial interests.
References
- 1.Taunton J; Hassig CA; Schreiber SL A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. DOI: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 2.Fischer DD; Cai R; Bhatia U; Asselbergs FA; Song CZ; Terry R; Trogani N; Widmer R; Atadja P; Cohen D Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem 2002, 277, 6656–6666. DOI: 10.1074/jbc.M108055200. [DOI] [PubMed] [Google Scholar]
- 3.Guardiola AR; Yao TP Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem 2002, 277, 3350–3356. DOI: 10.1074/jbc.M109861200. [DOI] [PubMed] [Google Scholar]
- 4.Kao HY; Lee CH; Komarov A; Han CC; Evans RM Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem 2002, 277, 187–193. DOI: 10.1074/jbc.M108931200. [DOI] [PubMed] [Google Scholar]
- 5.Tong JJ; Liu JH; Bertos NR; Yang XJ Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res 2002, 30, 1114–1123. DOI: 10.1093/nar/30.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hubbert C; Guardiola A; Shao R; Kawaguchi Y; Ito A; Nixon A; Yoshida M; Wang X-F; Yao T-P HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. DOI: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 7.Géraldy M; Morgen M; Sehr P; Steimbach RR; Moi D; Ridinger J; Oehme I; Witt O; Malz M; Nogueira MS; Koch O; Gunkel N; Miller AK Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J. Med. Chem 2019, 62, 4426–4443. DOI: 10.1021/acs.jmedchem.8b01936. [DOI] [PubMed] [Google Scholar]
- 8.Butler KV; Kalin J; Brochier C; Vistoli G; Langley B; Kozikowski AP Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc 2010, 132, 10842–10846. DOI: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang TL; Dredar SA; Manneh VA; Blankenship JW; Fries DS Inhibition of N8-acetylspermidine deacetylase by active-site-directed metal coordinating inhibitors. J. Med. Chem 1992, 35, 2414–2418. DOI: 10.1021/jm00091a009. [DOI] [PubMed] [Google Scholar]
- 10.Decroos C; Bowman CM; Christianson DW Synthesis and evaluation of N8-acetylspermidine analogues as inhibitors of bacterial acetylpolyamine amidohydrolase. Bioorg. Med. Chem 2013, 21, 4530–4540. DOI: 10.1016/j.bmc.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hai Y; Shinsky SA; Porter NJ; Christianson DW Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun 2017, 8, 15368. DOI: 10.1038/ncomms15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herp D; Ridinger J; Robaa D; Shinsky SA; Schmidtkunz K; Yesiloglu TZ; Bayer T; Steimbach RR; Herbst-Gervasoni CJ; Merz A; Romier C; Sehr P; Gunkel N; Miller AK; Christianson DW; Oehme I; Sippl W; Jung M First fluorescent acetylspermidine deacetylation assay for HDAC10 identifies selective inhibitors with cellular target engagement. ChemBioChem 2022, e202200180. DOI: 10.1002/cbic.202200180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeyen P; Zeyn Y; Herp D; Mahmoudi F; Yesiloglu TZ; Erdmann F; Schmidt M; Robaa D; Romier C; Ridinger J; Herbst-Gervasoni CJ; Christianson DW; Oehme I; Jung M; Krämer OH; Sippl W Identification of histone deacetylase 10 (HDAC10) inhibitors that modulate autophagy in transformed cells. Eur. J. Med. Chem 2022, 234, 114272. DOI: 10.1016/j.ejmech.2022.114272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melesina J; Simoben CV; Praetorius L; Bülbül EF; Robaa D; Sippl W Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. DOI: 10.1002/cmdc.202000934. [DOI] [PubMed] [Google Scholar]
- 15.Herbst-Gervasoni CJ; Steimbach RR; Morgen M; Miller AK; Christianson DW Structural Basis for the Selective Inhibition of HDAC10, the Cytosolic Polyamine Deacetylase. ACS Chem. Biol 2020, 15, 2154–2163. DOI: 10.1021/acschembio.0c00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchant P; Dredar S; Manneh V; Alshabanah O; Matthews H; Fries D; Blankenship J A selective inhibitor of N8-acetylspermidine deacetylation in mice and hela cells without effects on histone deacetylation. Arch. Biochem. Biophys 1989, 273, 128–136. DOI: 10.1016/0003-9861(89)90170-7. [DOI] [PubMed] [Google Scholar]
- 17.Miller-Fleming L; Olin-Sandoval V; Campbell K; Ralser M Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol 2015, 427, 3389–3406. DOI: 10.1016/j.jmb.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 18.Casero RA; Murray Stewart T; Pegg AE Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. DOI: 10.1038/s41568-018-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahiya S; Beier UH; Wang L; Han R; Jiao J; Akimova T; Angelin A; Wallace DC; Hancock WW HDAC10 deletion promotes Foxp3+ T-regulatory cell function. Sci. Rep 2020, 10, 424. DOI: 10.1038/s41598-019-57294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Obayashi H; Nagano Y; Takahashi T; Seki T; Tanaka S; Sakai N; Matsumoto M; Maruyama H Histone deacetylase 10 knockout activates chaperone-mediated autophagy and accelerates the decomposition of its substrate. Biochem. Biophys. Res. Commun 2020, 523, 246–252. DOI: 10.1016/j.bbrc.2019.12.048. [DOI] [PubMed] [Google Scholar]
- 21.Oehme I; Linke J-P; Böck BC; Milde T; Lodrini M; Hartenstein B; Wiegand I; Eckert C; Roth W; Kool M; Kaden S; Gröne H-J; Schulte JH; Lindner S; Hamacher-Brady A; Brady NR; Deubzer HE; Witt O Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Nat. Acad. Sci 2013, 110, E2592–2601. DOI: 10.1073/pnas.1300113110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotian S; Liyanarachchi S; Zelent A; Parvin JD Histone Deacetylases 9 and 10 Are Required for Homologous Recombination. J. Biol. Chem 2011, 286, 7722–7726. DOI: 10.1074/jbc.C110.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radhakrishnan R; Li Y; Xiang S; Yuan F; Yuan Z; Telles E; Fang J; Coppola D; Shibata D; Lane WS; Zhang Y; Zhang X; Seto E Histone Deacetylase 10 Regulates DNA Mismatch Repair and May Involve the Deacetylation of MutS Homolog 2. J. Biol. Chem 2015, 290, 22795–22804. DOI: 10.1074/jbc.M114.612945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duan B; Ye D; Zhu S; Jia W; Lu C; Wang G; Guo X; Yu Y; Wu C; Kang J HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ERK axis. Oncotarget 2017, 8, 61338–61349. DOI: 10.18632/oncotarget.18130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z; Fries D; Blankenship J Effect of N8-acetylspermidine deacetylase inhibition on the growth of L1210 cells. Biochem. Pharmacol 1999, 57, 1095–1103. DOI: 10.1016/S0006-2952(99)00028-3. [DOI] [PubMed] [Google Scholar]
- 26.Liao W; Sun J; Liu W; Li W; Jia J; Ou F; Su K; Zheng Y; Zhang Z; Sun Y HDAC10 up-regulation contributes to interleukin 1β-mediated inflammatory activation of synovium-derived mesenchymal stem cells in temporomandibular joint. J. Cell. Physiol 2019, 234, 12646–12662. DOI: 10.1002/jcp.27873. [DOI] [PubMed] [Google Scholar]
- 27.Islam MM; Banerjee T; Packard CZ; Kotian S; Selvendiran K; Cohn DE; Parvin JD HDAC10 as a potential therapeutic target in ovarian cancer. Gynecol. Oncol 2017, 144, 613–620. DOI: 10.1016/j.ygyno.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y; Zhang X; Zhu S; Dejene EA; Peng W; Sepulveda A; Seto E HDAC10 Regulates Cancer Stem-like Cell Properties in KRAS-driven Lung Adenocarcinoma. Cancer Res 2020, 80, 3265–3278. DOI: 10.1158/0008-5472.CAN-19-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Chemical Probes Portal search query: “HDAC10” https://new.chemicalprobes.org/?q=HDAC10 (accessed 2022-05-10).
- 30.Ho TCS; Chan AHY; Ganesan A Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem 2020, 63, 12460–12484. DOI: 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- 31.Lechner S; Malgapo MIP; Grätz C; Steimbach RR; Baron A; Rüther P; Nadal S; Stumpf C; Loos C; Ku X; Prokofeva P; Lautenbacher L; Heimburg T; Würf V; Meng C; Wilhelm M; Sippl W; Kleigrewe K; Pauling JK; Kramer K; Miller AK; Pfaffl MW; Linder ME; Kuster B; Médard G Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common off-target. Nat. Chem. Biol 2022. DOI: 10.1038/s41589-022-01015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dredar SA; Blankenship JW; Marchant PE; Manneh V; Fries DS Design and synthesis of inhibitors of N8-acetylspermidine deacetylase. J. Med. Chem 1989, 32, 984–989. DOI: 10.1021/jm00125a010. [DOI] [PubMed] [Google Scholar]
- 33.Herbst-Gervasoni CJ; Christianson DW Binding of N8-Acetylspermidine Analogues to Histone Deacetylase 10 Reveals Molecular Strategies for Blocking Polyamine Deacetylation. Biochemistry 2019, 58, 4957–4969. DOI: 10.1021/acs.biochem.9b00906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. The desmethyl secondary amine TFA-salt analog of 13 slowly cyclized to the corresponding pyrrolidinone in DMSO solution, which was evident from activity-loss in between assay replicates, despite DMSO stock solutions being stored at −20 °C. Pyrrolidinone formation was confirmed by LC-MS.
- 35.Bradner JE; West N; Grachan ML; Greenberg EF; Haggarty SJ; Warnow T; Mazitschek R Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol 2010, 6, 238–243. DOI: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marks BD; Fakhoury SA; Frazee WJ; Eliason HC; Riddle SM A substrate-independent TR-FRET histone deacetylase inhibitor assay. J. Biomol. Screen 2011, 16, 1247–1253. DOI: 10.1177/1087057111422102. [DOI] [PubMed] [Google Scholar]
- 37. DKFZ-711 was also tested against matrix metalloproteinases (MMPs) and found to inhibit MMP-9 and MMP-13 less than 10% at 100 μM. For details see Table S5.
- 38.Hopkins AL; Keserü GM; Leeson PD; Rees DC; Reynolds CH The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. DOI: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- 39.Morgen M; Steimbach RR; Géraldy M; Hellweg L; Sehr P; Ridinger J; Witt O; Oehme I; Herbst-Gervasoni CJ; Osko JD; Porter NJ; Christianson DW; Gunkel N; Miller AK Design and Synthesis of Dihydroxamic Acids as HDAC6/8/10 Inhibitors. ChemMedChem 2020, 15, 1163–1174. DOI: 10.1002/cmdc.202000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herbst-Gervasoni CJ; Christianson DW X-ray Crystallographic Snapshots of Substrate Binding in the Active Site of Histone Deacetylase 10. Biochemistry 2021, 60, 303–313. DOI: 10.1021/acs.biochem.0c00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robers MB; Dart ML; Woodroofe CC; Zimprich CA; Kirkland TA; Machleidt T; Kupcho KR; Levin S; Hartnett JR; Zimmerman K; Niles AL; Ohana RF; Daniels DL; Slater M; Wood MG; Cong M; Cheng Y-Q; Wood KV Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun 2015, 6, 10091. DOI: 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ridinger J; Koeneke E; Kolbinger FR; Koerholz K; Mahboobi S; Hellweg L; Gunkel N; Miller AK; Peterziel H; Schmezer P; Hamacher-Brady A; Witt O; Oehme I Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep 2018, 8, 10039. DOI: 10.1038/s41598-018-28265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bunnage ME; Chekler ELP; Jones LH Target validation using chemical probes. Nat. Chem. Biol 2013, 9, 195–199. DOI: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y; Donovan KA; Eleuteri NA; Kirmani N; Yue H; Razov A; Krupnick NM; Nowak RP; Fischer ES Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem. Biol 2021, 28, 1514–1527. DOI: 10.1016/j.chembiol.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.