

Abstract
Psychostimulant drugs, such as cocaine, inhibit dopamine reuptake via blockade of the dopamine transporter (DAT), which is the primary mechanism underpinning their abuse. Atypical DAT inhibitors are dissimilar to cocaine and can block cocaine or methamphetamine induced behaviors, supporting their development as part of a treatment regimen for psychostimulant use disorders. When developing these atypical DAT inhibitors as medications, it is necessary to avoid off-target binding that can produce unwanted side effects or toxicities. In particular, the blockade of a potassium channel, human ether-a-go-go (hERG), can lead to the potentially lethal ventricular tachycardia. In this study, we established a counter screening platform for DAT and against hERG binding by combining machine learning-based quantitative structure-activity relationship (QSAR) modeling, experimental validation, and molecular modeling and simulations. Our results show the available data are adequate to establish robust QSAR models, as validated by chemical synthesis and pharmacological evaluation of a validation set of DAT inhibitors. Further, the QSAR models based on subsets of the data according to experimental approaches used have predictive power as well, which opens the door to target specific functional states of a protein. Complementarily, our molecular modeling and simulations identified the structural elements responsible for a pair of DAT inhibitors having opposite binding affinity trends at DAT and hERG, which can be leveraged for rational optimization of lead atypical DAT inhibitors with desired pharmacological properties.
Graphical Abstract
INTRODUCTION
When drugs interact with undesired targets (off-targets), adverse side effects can arise;1, 2 if such effects are severe enough to cause toxicity, clinical trials fail or post-marketing drugs can be recalled.3 On the other hand, some other drugs may require interactions with more than one target to exert their therapeutic effects in a polypharmacological way.4 Thus, in drug development, it is critical to comprehensively identify the potential targets for a given lead molecule, and to rationally optimize it toward the desired pharmacological profile, either selectively or synergistically, for appropriate target(s).
The dopamine transporter (DAT) serves to terminate dopamine neurotransmission by transporting released dopamine back into the presynaptic neuron. DAT is the primary target for medications used to treat ADHD (e.g., Ritalin) and sleep disorders (e.g., modafinil), but also for abused psychostimulants such as cocaine and methamphetamine, the abuse of which can lead to psychostimulant use disorders (PSUD). Interestingly, although cocaine is a DAT inhibitor, a large number of studies over the past ~25 years have shown that several other structural classes of DAT inhibitors, based on benztropine, modafinil, and rimcazole, have limited or no rewarding effects in numerous animal models across species.5–9 Indeed, based on these data, several lead candidates have been proposed as potential medications to treat PSUD.10–13
In the development of these classes of DAT inhibitors, as in many other drug development campaigns, however, a frequently encountered challenge is how to avoid hitting other targets that may cause toxic side effects; in particular, the human ether-a-go-go (hERG) potassium channel (Kv11.1). hERG enables the delayed rectifying potassium current in cardiomyocytes. Compounds that block the hERG channel interfere with this current, resulting in a prolonged QT interval on an EKG and can lead to Torsades de Pointes (TdP), a potentially lethal ventricular tachycardia.14–16 hERG is extremely promiscuous in binding small compounds, especially those with protonatable amines and aromatic groups,17, 18 a hallmark of many neurotransmitter transport inhibitors, as well as G-protein coupled receptor ligands, and thus a common challenge for drug design and development.19, 20 It was estimated that 40–70% of new drug candidates have measurable affinities to hERG.21, 22 Indeed, one of the main reasons for drug withdrawals in the 1990s and early 2000s was drug-induced cardiotoxicity, for which hERG blockade was identified as one of the major causes.23, 24 At present, due to revised FDA regulations in early 2000,25 drug candidates have to be evaluated for TdP liability in vitro by patch-clamp electrophysiology and in vivo in phase 1 clinical trials, both of which are expensive and time consuming.16, 22
Thus, when targeting DAT, accurate computational predictions of both DAT and hERG affinities would facilitate medicinal chemistry campaigns in discovering new medication candidates with desired properties, i.e., high-affinity DAT inhibitors possessing low hERG affinity. Such virtual counter screening can be applied in an early stage of drug discovery and help guide drug design and ultimately chemical synthesis. As a result of increased concerns of the risks that hERG binding poses, there has been a significant increase in the amount of hERG binding affinity data in public databases and the literature. These data have been used to generate machine-learning based, quantitative structure-activity relationship (QSAR) models to correlate chemical structures with binding affinities at hERG.26–31 On the other hand, while there is abundance of data in the literature and databases, machine learning-based QSAR models for DAT or its homologs have only begun to emerge.32
Compared to protein structure-based QSAR, ligand-based QSAR is a much faster way to predict the correlation between the small-molecule chemical structures and activities, such as binding affinities.18, 33 However, the early linear-regression based QSAR models suffered from inadequate data and intrinsically non-linear QSAR.26 Unlike linear-regression modeling, there is more freedom in the machine learning method to search relevant variables in descriptors space, in order to develop a good QSAR model. During the learning process, some descriptors can be forward included or backward excluded to better interpret the model. Studies have shown that machine-learning based QSAR models outperform traditional linear-regression based QSAR models.26, 34, 35
In this study, we established a platform to predict and validate the binding affinities of compounds at both DAT and hERG by combining machine-learning based QSAR models (Figure S1), experimental binding affinity measurements, and molecular modeling approaches.
RESULTS
The hERG datasets divided by experimental methods result in QSAR models with comparable qualities
We queried and retrieved all the available hERG data from ChEMBL (version 25)36 (see Methods). Compared to previous work,26, 27 the availability of a larger dataset allowed us to further divide and characterize the dataset according to how the binding affinities were represented (IC50 versus Ki) and by experimental methods (patch-clamp electrophysiology versus radioligand binding assays), and to evaluate the impact of more homogeneous data on the quality and predictive power of QSAR models.
First, we found there are three-fold more IC50 than Ki data for hERG (Table S1). While Ki conceptually better represents the binding affinity than IC50 (half maximal inhibitory concentration), as the latter may be sensitive to the approaches used in the measurements according to the Cheng-Prusoff equation,37 the Ki value should always be smaller than the IC50 from which it derives, thus the trends of Ki and IC50 should be the same if similar approaches were used.37 The abundance of IC50 measurements in the databases is not surprising, as it streams from the apparent advances in cellular electrophysiology and the availability of robust cell-lines over-expressing hERG1 channel.38, 39 The deployment of the automated patch-clamp platforms enabled a broad coverage of hERG inhibitory potency for various drug scaffolds.40 An IC50 in the low micro-molar range is strongly associated with QT prolongation, a surrogate marker for torsadogenicity of the compound,40, 41 and often used for binary classification, e.g., blocker vs. non-blocker. Practically, in order to adequately train machine-learning based QSAR models, we would like to have as many non-redundant homogenous data points as possible. Thus, based on these considerations, we chose to use IC50 data to train our hERG QSAR models (see below), without mixing Ki and IC50 data together.
We then developed filters to sort the data based on how they were acquired, i.e., from either patch-clamp electrophysiology (referred to as “clamp” below) or radioligand binding assays (“binding”). We note that there are a number of inaccurate or ambiguous annotations in ChEMBL regarding the types of assays being carried out and had to curate lists of key words to comprehensively retrieve all the relevant entries for each dataset (Table S2). The resulting datasets, after removal of (nearly) identical compounds (Tanimoto similarly > 0.999), are ~2000 IC50 data points for the clamp dataset and ~1400 IC50 data points for the binding dataset. Interestingly, when we plot the distributions of these two hERG datasets, we found that both of them have single sharp peaks of pIC50 (i.e., −logIC50) centering around 5.2 (Figure 1A). Compared to the IC50 datasets, the binding Ki dataset shows the slightly higher peak value (5.8) of the pKi distribution curve, suggesting that homogeneous approaches were used in acquiring hERG binding data and, therefore, the IC50 trend likely reliably represents that of Ki. In contrast, the drastically larger peak value (8.5) of the clamp pKi curve, based on only 44 data points, suggests this Ki dataset does not adequately cover chemical space.
Figure 1. Distribution of the pIC50 and pKi values of hERG ligands retrieved from ChEMBL.
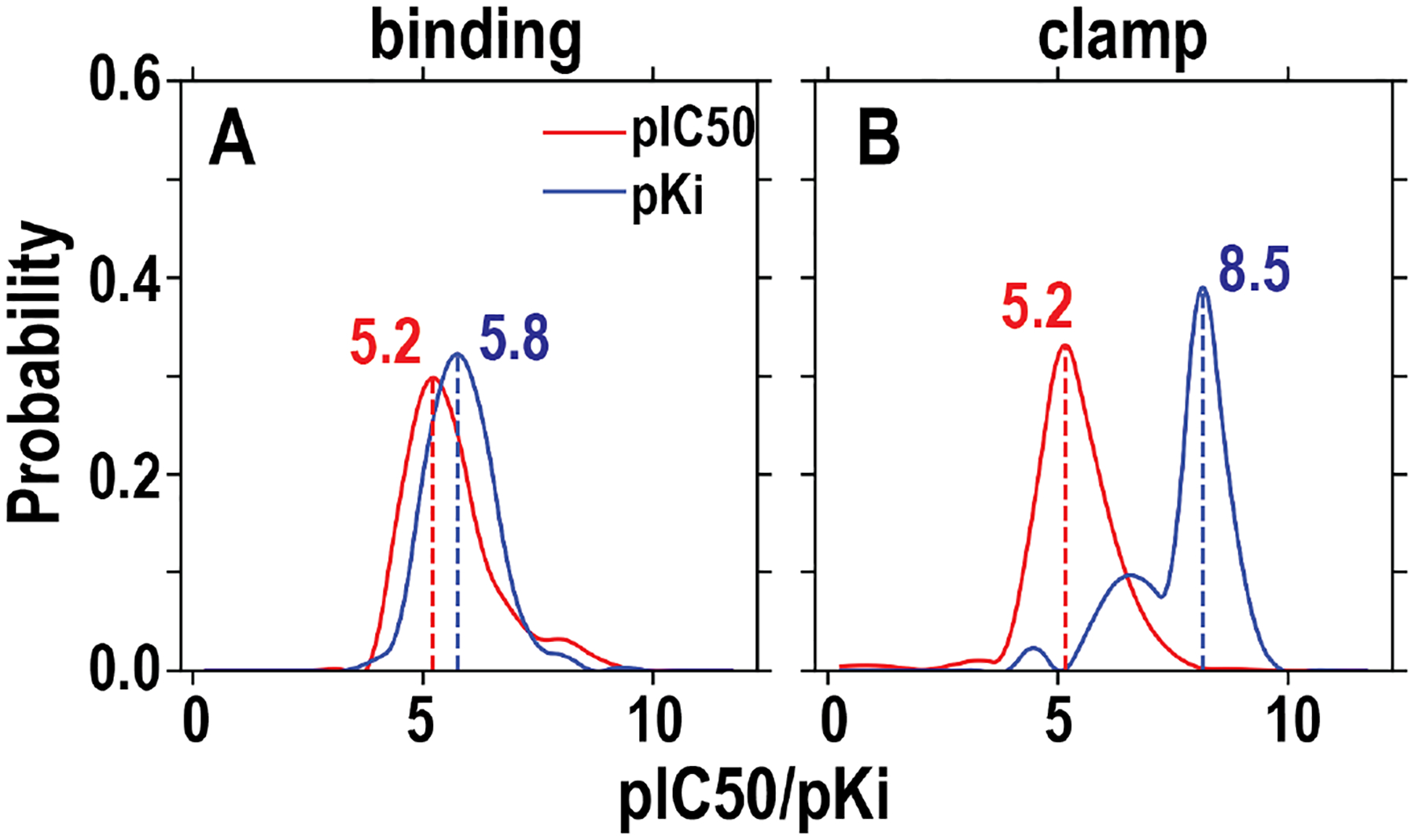
We divided the initially retrieved dataset into “binding” and “clamp” datasets according to the experimental methods used (see text and Table S2) and further filtered them (Table S1). The included data for these two panels are the final datasets to be used for training. The peak values for each dataset are indicated.
In order to build machine-learning based QSAR models, we further filtered the datasets as described in Methods. In particular, 55 compounds that appeared in a high-quality patch-clamp electrophysiology study41 were first filtered out from the clamp set, then the results of those 55 compounds from that study were added to the dataset after all other filters were applied. Similar to a previous study,27 in the training datasets for the classification models, we further excluded the data points with pIC50s between 5 and 6, to define a clearer boundary between binders and non-binders.
Using these training datasets, we first built and evaluated regression QSAR models, which would predict numeric values of the binding affinities. We compared two machine learning algorithms, Random Forest (RF) and eXtreme Gradient Boosting (XGBoost), and systematically scanned and optimized their parameters (see Methods). The benchmarks of the regression models, i.e., the coefficient of determination (R2) and root mean square error (RMSE), indicate that both algorithms can result in reasonably good models, while the XGBoost-trained models outperform the RF-trained models, and the binding data-based models have slightly better benchmarks than those based on the clamp data (Table 1). We noticed there is a strong stochastic element in training of the models when splitting the dataset into training and testing sets (see Methods), i.e., if we build the model twice with the same algorithm but with different splittings, it will result in somewhat different benchmarks. Our statistics show that more models would reduce the stochastic uncertainty (Figure S2, see Methods). Thus, to eliminate the stochastic effect in evaluating the quality of the models, we use multiple models constructed with different random numbers.
Table 1.
Benchmarks of the hERG regression and classification models.
| Models | Metrics | Dataset | XGBoost | Random Forest | ||||
|---|---|---|---|---|---|---|---|---|
| Ave. | S.D. | Best | Ave. | S.D. | Best | |||
| Regression | R 2 | binding | 0.66 | 0.04 | 0.76 | 0.59 | 0.04 | 0.70 |
| clamp | 0.65 | 0.08 | 0.80 | 0.54 | 0.08 | 0.67 | ||
| RMSE | binding | 0.58 | 0.03 | -- | 0.64 | 0.03 | -- | |
| clamp | 0.59 | 0.06 | -- | 0.62 | 0.06 | -- | ||
| Classification | Accuracy | binding | 0.89 | 0.03 | -- | 0.90 | 0.02 | -- |
| clamp | 0.87 | 0.03 | -- | 0.89 | 0.03 | -- | ||
| Sensitivity | binding | 0.89 | 0.04 | -- | 0.89 | 0.04 | -- | |
| clamp | 0.71 | 0.06 | -- | 0.75 | 0.07 | -- | ||
| Specificity | binding | 0.88 | 0.03 | -- | 0.90 | 0.03 | -- | |
| clamp | 0.96 | 0.02 | -- | 0.97 | 0.02 | -- | ||
| F Score | binding | 0.88 | 0.03 | -- | 0.89 | 0.03 | -- | |
| clamp | 0.80 | 0.05 | -- | 0.83 | 0.05 | -- | ||
We then used correspondingly prepared training datasets (Table S1) to build the classification QSAR models, which would predict whether a compound is a binder or a non-binder. We evaluated their qualities with the benchmarks, accuracy, sensitivity, and specificity (see Methods for definitions). Both clamp and binding datasets result in high accuracy (≥0.87) models. However, while the binding data-based models have relatively good and balanced sensitivity, specificity, and F-score, the clamp data-based models have relatively low sensitivity and high specificity, regardless of the machine learning algorithm used, which is likely due to the smaller number of binders in this dataset (see Discussion).
Adequate DAT Ki data result in reasonably good QSAR models
Among the available DAT data in ChEMBL (including all species, see Methods and below), there are comparable amount of IC50 and Ki data (Table S1). For DAT, when radioligand binding assays were carried out with various selective DAT inhibitor probes, the resulting binding affinities represent the situation in which the transporter is trapped in outward-facing conformations. In comparison, the inhibition of dopamine uptake using [3H]dopamine as the probe may reflect a mixed scenario, wherein the protein is in an equilibrium of outward- and inward-facing conformations.42, 43 Thus, we developed filters to sort the data according to their assay types, from either the binding assay using radiolabeled inhibitors (“binding”) or inhibition of dopamine uptake (“uptake”).
After applying additional filters, like those developed for hERG datasets (Table S2), both the binding and uptake DAT datasets have broader distributions than the hERG datasets and show more than one peak (Figure 2A and B). In addition, while the pIC50 curve of DAT binding data peaks at 6.7, ~1.5 unit larger than that of hERG, the peak of DAT pKi curve at 5.9 is comparable to that of hERG binding dataset (Figure 1A). The higher peak value of the pIC50 curve suggest that the DAT binding data were likely collected with heterogeneous approaches and that IC50 data using different approaches may not be directly comparable. Thus, based on these considerations, we chose to use the Ki datasets to train our DAT QSAR models.
Figure 2. Distribution of the pIC50 and pKi values of DAT ligands retrieved from ChEMBL.
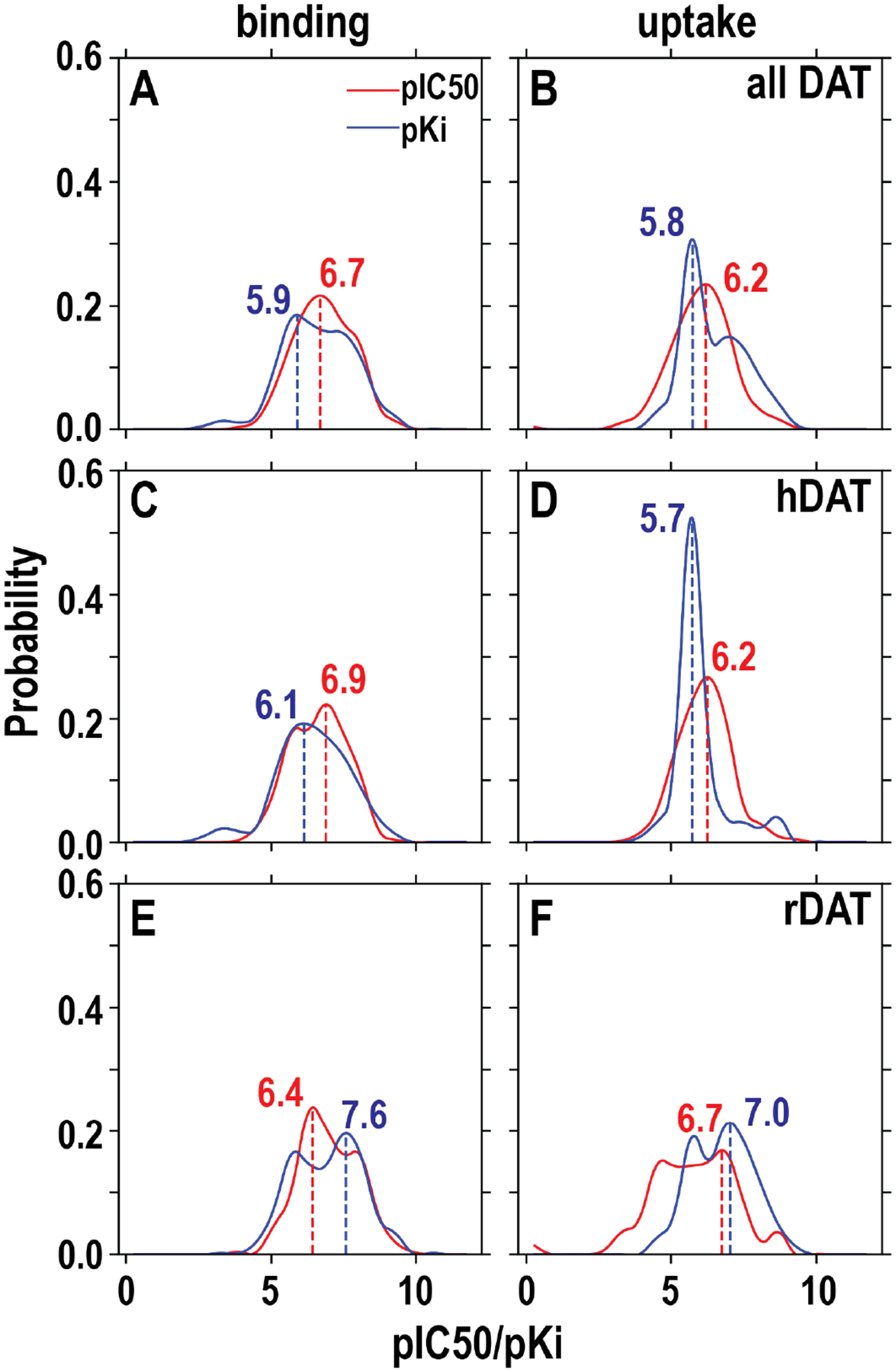
Similar to the hERG datasets, we divided the initially retrieved dataset into “binding” and “uptake” datasets according to the experimental methods used (see text and Table S2); in addition, we evaluated the situations for species specific datasets, i.e., hDAT and rDAT. We further filtered them similarly as the hERG dataset (Table S1). The included data for these panels are the final datasets to be used for training. The peak values for each dataset are indicated.
We used either the binding or the uptake dataset to train DAT regression models and compared the XGBoost and RF algorithms. While most of the resulting models have reasonably good benchmarks, the R2 of XGBoost models are in most cases better than those of RF ones (Table 2). Interestingly, the models based on the binding data are slightly better than those based on uptake data. Using the correspondingly prepared datasets (same as the regression datasets except that the data points with pKi of 5 to 6 were excluded), our DAT classification models have high accuracy, sensitivity, and F-score, but poor specificity, which may be related to relatively smaller numbers of nonbinders in the training datasets (see Discussion).
Table 2.
Benchmarks of the DAT regression and classification models.
| Models | Metrics | Dataset | XGBoost | Random Forest | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ave. | S.D. | Best | Ave. | S.D. | Best | ||||
| Regression | R 2 | all-DAT | binding | 0.71 | 0.04 | 0.80 | 0.66 | 0.05 | 0.75 |
| uptake | 0.66 | 0.11 | 0.83 | 0.64 | 0.11 | 0.78 | |||
| hDAT | binding | 0.70 | 0.05 | 0.79 | 0.68 | 0.05 | 0.76 | ||
| uptake | 0.35 | 0.37 | 0.76 | 0.35 | 0.33 | 0.80 | |||
| rDAT | binding | 0.69 | 0.10 | 0.85 | 0.67 | 0.08 | 0.80 | ||
| uptake | 0.62 | 0.14 | 0.84 | 0.60 | 0.14 | 0.84 | |||
| RMSE | all-DAT | binding | 0.65 | 0.04 | -- | 0.70 | 0.04 | -- | |
| uptake | 0.60 | 0.08 | -- | 0.63 | 0.08 | -- | |||
| hDAT | binding | 0.68 | 0.06 | -- | 0.71 | 0.06 | -- | ||
| uptake | 0.58 | 0.15 | -- | 0.58 | 0.14 | -- | |||
| rDAT | binding | 0.61 | 0.10 | -- | 0.68 | 0.10 | -- | ||
| uptake | 0.63 | 0.09 | -- | 0.65 | 0.09 | -- | |||
| Classification | Accuracy | all-DAT | binding | 0.96 | 0.02 | -- | 0.96 | 0.02 | -- |
| uptake | 0.95 | 0.03 | -- | 0.95 | 0.03 | -- | |||
| hDAT | binding | 0.95 | 0.02 | -- | 0.96 | 0.02 | -- | ||
| uptake | 0.79 | 0.17 | -- | 0.77 | 0.17 | -- | |||
| rDAT | binding | 0.97 | 0.02 | -- | 0.96 | 0.03 | -- | ||
| uptake | 0.99 | 0.02 | -- | 0.99 | 0.02 | -- | |||
| Sensitivity | all-DAT | binding | 0.99 | 0.01 | -- | 0.99 | 0.01 | -- | |
| uptake | 0.99 | 0.02 | -- | 0.99 | 0.02 | -- | |||
| hDAT | binding | 0.98 | 0.02 | -- | 0.99 | 0.01 | -- | ||
| uptake | 0.96 | 0.12 | -- | 0.94 | 0.14 | -- | |||
| rDAT | binding | 0.99 | 0.01 | -- | 0.99 | 0.01 | -- | ||
| uptake | 1.00 | 0.01 | -- | 1.00 | 0.01 | -- | |||
| Specificity | all-DAT | binding | 0.63 | 0.13 | -- | 0.61 | 0.11 | -- | |
| uptake | 0.52 | 0.32 | -- | 0.56 | 0.27 | -- | |||
| hDAT | binding | 0.68 | 0.16 | -- | 0.70 | 0.15 | -- | ||
| uptake | 0.00 | 0.00 | -- | 0.00 | 0.00 | -- | |||
| rDAT | binding | 0.52 | 0.31 | -- | 0.40 | 0.33 | -- | ||
| uptake | 0.84 | 0.30 | -- | 0.87 | 0.30 | -- | |||
| F Score | all-DAT | binding | 0.98 | 0.01 | -- | 0.98 | 0.01 | -- | |
| uptake | 0.97 | 0.02 | -- | 0.97 | 0.02 | -- | |||
| hDAT | binding | 0.97 | 0.01 | -- | 0.98 | 0.01 | -- | ||
| uptake | 0.87 | 0.11 | -- | 0.86 | 0.11 | -- | |||
| rDAT | binding | 0.98 | 0.01 | -- | 0.98 | 0.01 | -- | ||
| uptake | 0.99 | 0.01 | -- | 1.00 | 0.01 | -- | |||
Models based on human or rat DAT dataset alone have predictive powers but these datasets can be combined
In the DAT dataset, as expected, the majority of the data points are of human DAT (hDAT) and rat DAT (rDAT), because the majority of the experiments were carried out with hDAT heterologously expressed in in vitro cell lines or with rat brain tissues. When we specifically filtered the DAT dataset by species, after removing redundancy, hDAT has 684 Ki values in the binding dataset, while rDAT has 541 binding Ki values. The uptake datasets are significantly smaller, only 126 hDAT Ki values and 229 rDAT Ki values. To evaluate whether the datasets of these sizes can result in QSAR models with predictive power, we built both regression and classification models using individual hDAT or rDAT binding or uptake datasets.
While hDAT binding, rDAT binding, and rDAT uptake datasets all result in regression models with good benchmarks, the regression models based on only 126 hDAT uptake data points, which does not cover sufficient chemical space, have poor benchmarks. For classification modeling, hDAT binding, rDAT binding, and rDAT uptake datasets result in models with high accuracy and sensitivity, but with poor specificity. Similar to the situation of the hDAT uptake regression model, the small hDAT uptake dataset (only 45 data points after excluding pKi 5–6) generated classification models with very poor benchmarks, regardless of the machine learning algorithm used.
Curiously, the rDAT binding dataset appears to have more distribution at higher pKi values than that of hDAT, i.e., the rDAT pKi curve has two peaks, one of them is at 7.6, 1.5 unit higher than that of the hDAT (Figure 2C and E). To investigate whether this trend represents rDAT having higher affinities than hDAT, we identified that there are 18 common compounds that have been tested for binding at both hDAT and rDAT (Figure S3A), and our analysis showed that ~85% of these compounds have a ΔpKi (hDAT pKi - rDAT pKi) less than 1 (Figure S3B). Therefore, the binding sites of hDAT and rDAT have very similar binding affinities for the same compounds. Indeed, the central binding site residues of hDAT and rDAT are identical.44 We further compare how different the compounds in the hDAT and rDAT binding datasets are by their chemical similarity (see Methods). The results show that these two datasets have only a smaller percentage of highly similar compounds, i.e., with a similarity score > 0.85 (Figure S3C). Thus, we concluded that the compounds in the rDAT and hDAT binding datasets do not have significant overlap.
Taken together, due to their highly similar binding sites, we can combine hDAT and rDAT datasets as the training data for the QSAR model building, as described in the previous section. Such a combination is also necessary to cover adequate chemical space as the hDAT uptake dataset appears not large enough to build robust models.
Experimental validation of the QSAR models on a selected set of DAT inhibitors
To establish a counter screening platform to discover small-molecule reagents for DAT and against hERG, the affinity prediction at these two targets is the initial, but critical, step for efficient lead discovery. Reliable predictions would minimize the efforts in chemical synthesis and pharmacological measurements. Thus, it is important to evaluate whether our QSAR models have adequate predictive power at both DAT and hERG. To this end, we selected a set of modafinil-derivatized DAT inhibitors, and carried out both computational predictions and experimental measurements of their affinities, at both rDAT and hERG.
Computationally, we built QSAR models using the same protocol described above but 100% data retrieved and filtered from ChEMBL (see Figure S1 and Methods), and used them to predict both the affinity values and the binary classifications of binder versus non-binder for each compound. Experimentally, we measured their binding affinities at DAT, with [3H]WIN35,428 as the radioligand, and at hERG, with patch-clamp pipette experiments (see Methods). We then performed the correlation analysis between the computational predicted and experimental measured affinities of these compounds (Figure 3). For hERG, the affinity predictions with the regression models based on the clamp data, are moderately correlated with the experimental measurements; the models trained with the XGBoost algorithm performed better than those trained with RF (R2 = 0.70 and 0.54, respectively, Figure 3A). These clamp data-based models slightly outperformed the binding data-based models (R2 = 0.62 and 0.47 for XGBoost- and RF-trained models, respectively). For DAT, the binding data based regression models have better performance than those based on the uptake data in their predicted affinities, in terms of the correlation with the experimentally measured affinities, regardless of either XGBoost- or RF-trained models (Figure 3B). When we evaluated the models based on species-specific DAT datasets (Figure S4), the best performed model was rDAT binding data (R2 = 0.76 and 0.67 for XGBoost- and RF-trained models, respectively), the same experimental approach used in collecting the DAT affinity data in the current study. Interestingly, the models based rDAT uptake data have no predictive power (Figure S4G,H). Taken together, in addition to their good benchmarks described above, these correlations suggest our regression models have significant predictive powers, while the XGBoost algorithm may better fit these datasets than RF. When comparing the correlations of the predictions to experimental data using the Pearson correlation coefficient (R), XGBoost models outperform RF models as well for both hERG and DAT (Figure 3).
Figure 3. Correlations between the predicted and experimentally measured hERG and DAT affinities.
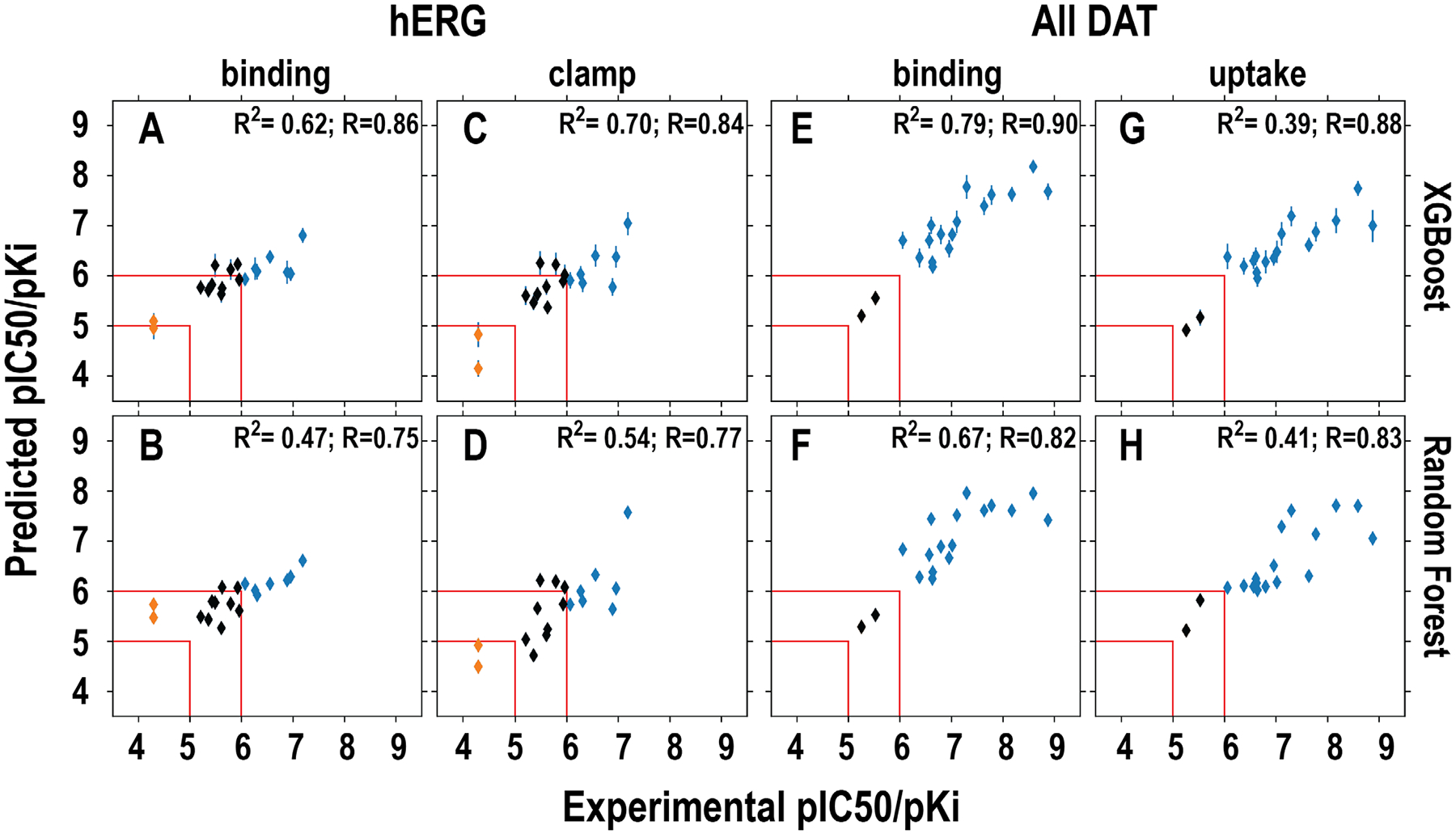
The models in the upper row (A, C, E, G) used XGBoost and those in bottom row (B, D, F, H) used RF in training. The specific datasets used for training, i.e., binding and clamp for hERG and binding and uptake for DAT, are indicated on top of each column. The results of classification prediction are color-coded. The blue and red represent binder and non-binder, respectively. The compounds that have been experimentally measured to have pIC50 or pKi between 5 and 6 are colored in black. The red lines indicate the regions that <5 and >6 are defined as non-binder and binder, respectively. Note that among 18 compounds in this set, ten of them have been published previously13, 45, 46.
In our evaluation of the hERG classification models, the 9 compounds with experimentally measured pIC50 values between 5 and 6 were not included. For the rest of 9 compounds, our models are 100% accurate in differentiating the 7 binders (pIC50 > 6) from the two non-binders (pIC50 <5) (Figure 3A–D). For the same 18 compounds, 16 of them were DAT binders, while the pKi of the other two were between 5 and 6. Similar high accuracy was observed for the predictions of DAT binders with our classification models based on either all-DAT or hDAT data (Figure 3E–H, and Figure S4A–D). However, while most of the models based on rDAT data had good predictions as well, the one based on the uptake data incorrectly classified the most potent binder as a non-binder (Figure S4E–H).
Impact of dataset size and activity distribution on the predictive power
To further evaluate the impact of the size and activity distribution of the dataset on the outcome of our approach, we applied the same protocol of model building on an in-house DAT binding dataset, which includes 277 compounds synthesized by Newman group throughout the years.6, 7 The strength of this dataset is that the binding affinities have been measured using same or similar approaches. For the regression modeling, the benchmarking results show moderate R2 values of 0.48 for the XGBoost models and 0.46 for the RF models (Table S3). For the classification modeling, both XGBoost and RF models show high accuracy, sensitivity, and F-score, but poor specificity in benchmarking. Similar to the ChEMBL datasets, the unbalanced numbers of binders and nonbinder compounds in this in-house dataset is likely the major cause for the poor specificity (see Discussion).
The regression prediction for the validation dataset (Figure S5) shows that the models based on the in-house DAT binding dataset perform worse than the models based on the “all-DAT” binding dataset from ChEMBL. We found that the insufficient high-affinity compounds in the in-house dataset renders poor predictions of high-affinity compounds in the validation dataset. In the all-DAT binding dataset, there are 69 compounds (5.8%) with pKi ≥ 8.5, while in the in-house DAT binding dataset, there are only 6 compounds (2.2%) with pKi ≥ 8.5. The Pearson R value noticeably increases without the high affinity compounds (pKi ≥ 8.5) in the validation dataset (Figure S5). For the classification prediction of the validation dataset, the results are as good as those using the all-DAT binding models (Figure S5).
Thus, a dataset with a few hundred compounds is potentially sufficient to build QSAR models with predictive powers; however, adequate training data covering the entire activity range, especially for the high-affinity range, is also critical for accurate predictions.
Molecular modeling reveals structural features responsible for the opposite trends of DAT and hERG affinities of two DAT inhibitors
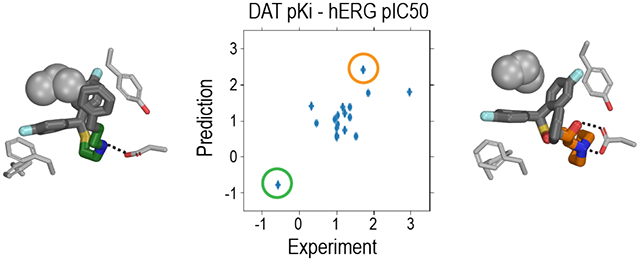
From this validation set of DAT inhibitors, we noticed that a pair of analogs with similar chemical structures, JJC8-01646 and JJC8-08813 (Tanimoto similarity = 0.62, Figure S6), have opposite trends of affinities at DAT and hERG. JJC8-088 has ~90-fold higher affinity than JJC8-016 at DAT (Ki = 2.6 and 234.4 nM, respectively), but has ~2-fold lower affinity than JJC8-016 at hERG (IC50 = 0.13 and 0.06 μM, respectively). We searched compound pairs in the DAT and hERG datasets retrieved from ChEMBL with similar criteria (>90 fold better in DAT, and >2 fold better in hERG), and found that 6 pairs have Tanimoto similarity >0.6 (Figure S7). However, none of these pairs have the DAT or hERG binding data collected from the same study; thus, because the binding affinities of our pair (JJC8-088 and JJC8-016) of DAT and hERG are measured by the same laboratories, their affinity differences are potentially more reliable. Hence, we used this pair of compounds to explore the protein structure-based clues in DAT and hERG responsible for these opposite trends by carrying out molecular modeling and simulation studies, using both of our hDAT and hERG models (see Methods).13, 16, 47, 48
The central ligand binding (S1) site of neurotransmitter transporters to which DAT belongs can be divided into three subsites, A, B, and C.49 For the mono-amine transporters, a conserved Asp in subsite A is responsible for forming a salt-bridge with the tertiary amine moieties of their cognate endogenous neurotransmitter substrates,44 while the subsites B and C of S1 have aromatic and hydrophobic residues to accommodate the aromatic moieties of the ligands.50, 51 As expected in our molecular dynamics (MD) simulations, the bisphenyl moieties of both JJC8-016 and JJC8-088 are similarly accommodated by subsites B and C, and the two phenyl rings closely interact with Ile152 located in between these two subsites, while their charged nitrogen atoms form stable salt bridges with Asp79 of subsite A. Our analysis indicates that JJC8-088 forms an additional stable H-bond between its -OH and Asp79, which very likely contributes to its higher affinity compared to JJC8-016 at DAT (Figures 4A–C and S5). The moieties on the N-termini of these two DAT inhibitors: phenylpropyl for JJC8-016 and phenylpropanol for JJC8-088 (referred to as “tail”, Figure S6), protrude into the extracellular vestibule of DAT and make aromatic interactions with Phe155, Tyr156, and/or Phe320.
Figure 4. JJC8-016 and JJC8-088 forms distinct interactions with DAT but not with hERG.
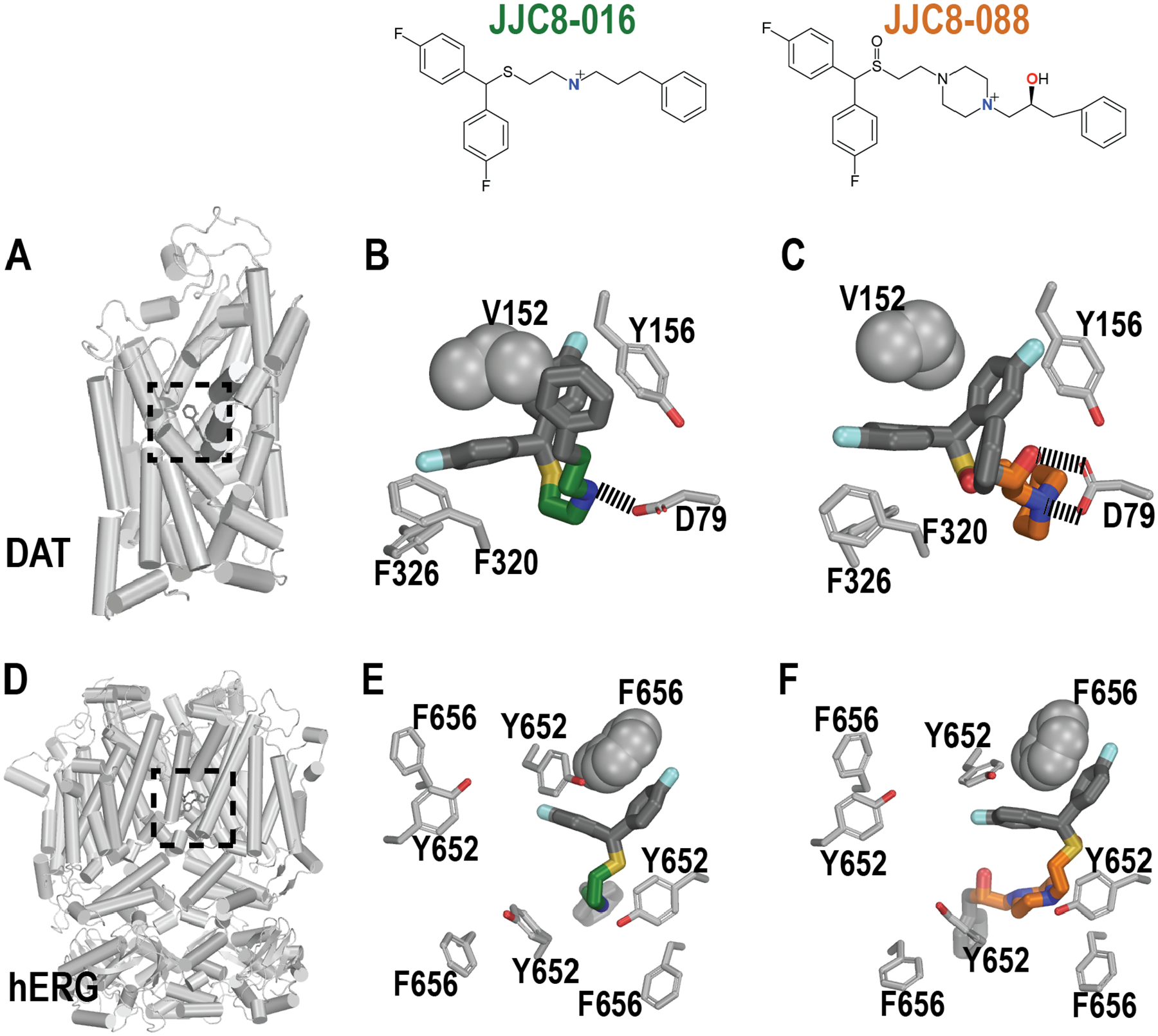
Panels A and D show the overviews of the DAT and hERG models viewed parallel to the membrane plane, and the locations of their binding sites (dotted boxes). Panels B-F show the representative binding poses of bound JJC8-016 and JJC8-088 in DAT (B and C) and hERG (E and F) resulting from the MD simulations. The critical residues in the binding sites are shown, including Val152 in DAT and one of Phe656 in hERG that are in close contact with the bisphenol moieties of the ligands.
The functional unit of hERG is a homotetramer. Each monomer contains six transmembrane segments. The hERG binding site is formed at the center and interface of the homotetramer, with identical binding residues contributed from all four monomers, and is a large and highly hydrophobic cavity located on the intracellular side of the transmembrane domains.52 The ligand binding site at hERG is highly promiscuous for small molecules with a wide range of chemical scaffolds defined by a combination of amphipathic residues capable of hydrogen-bonding from the pore helix, two aromatic residues Tyr652 and Phe656 from S6 helix, and in some cases Phe557 from S5.16, 17, 53–56 Therefore, the water-filled intracavitary site of hERG1 channel provides a diverse range of chemical moieties to enable stable binding and blockade by a large cohort of drug-like molecules.19, 20, 57 Similar to the situation in our hDAT simulations, the results of our MD simulations of hERG show that the binding modes of the bisphenyl moieties of JJC8-016 and JJC8-088 are highly similar, and they closely interact with Phe656 in the binding site. In comparison, their N-termini (tails) do not have very defined binding modes, and are flexible in the large binding cavity that opens to the intracellular water phase (Figure 4D–F). Thus, the extra -OH in JJC8-088 could not contribute any additional favored interactions, like in hDAT, which is consistent with JJC8-088 and JJC8-016 having very small affinity differences at hERG.
Taken together, our results indicate that functionality extending from the tertiary amines of these DAT inhibitors can be further tuned without affecting the affinities at hERG, which opens the door for structure-based optimization of DAT inhibitors, without simultaneously improving hERG affinity.
DISCUSSION
To facilitate the development of high-affinity DAT inhibitors possessing low hERG affinity, we built machine-learning based QSAR models to predict the binding affinities of compounds at both DAT and hERG, and identified the structural clues responsible for different affinity trends in these two proteins with molecular modeling and simulations. Our computational predictions were validated with experimental binding affinity measurements.
In classification models, we found that the sensitivity and specificity may have been driven by the balance of binders and nonbinders used in training (Tables 1 and S1). Many more binders may lead the classification models to have high sensitivity and low specificity. To test this hypothesis, we randomly selected binders to match the number of nonbinders from the all-DAT binding dataset (resulting in a training dataset with 89 binders and 89 non-binders) to build classification models with XGBoost, using the same training and validation procedure as for the classification models described above. The benchmarks of the models trained with equal numbers of binders and non-binders show that the accuracy became slightly worse than using all available binders, though the specificity and sensitivity were balanced (Table S4). The lower accuracy is a consequence of much smaller number of binders (from 798 to 89), which result in ~89% information of binders not being used. While the trend of benchmarks is consistent with our hypothesis, practically, we may not want to use only part of the data just for the purpose of benchmarks, which would sacrifice the accuracy of predictions by limiting the chemical space that the models would be able to cover. However, such an issue in building the classification models may be partially addressed by adjusting the definition of binder or nonbinder, specifically in our modeling building process, the pKi or pIC50 range to be excluded. For example, we can shift this range to a higher range, which may not affect the purpose to discover compounds that bind with nanomolar affinity to DAT.
Overall, the benchmarks and prediction performances of our DAT regression QSAR models based on binding data are better than those based on uptake data. However, the models based on either rDAT or hDAT binding datasets alone are either not better or significantly worse in some cases than the models based on the all-DAT binding dataset. These trends may reflect the corresponding coverages of chemical space. Interestingly, using our rDAT regression models to predict the hDAT binding data, the models appear to have no predictive power, with an R2 of only 0.20. Conversely, the prediction using our hDAT regression models on the rDAT data results in an R2 of 0.40 (Figure S8). We found that the rDAT models were not well trained with compounds with high pKi values, and therefore making poor predictions on the high pKi compounds in hDAT dataset. Thus, the hDAT and rDAT binding datasets may be differentially enriched in certain chemical scaffolds, e.g., rDAT dataset has more distribution at higher pKi values. In addition, our analysis showed that the rDAT and hDAT datasets do not have significant overlap and, importantly, that the binding affinities of a given compound at rDAT and hDAT are very similar (Figure S3), which justifies our combination of these two datasets in building the final models. Similar impact of high affinity training data on the predictive power was also observed for the models trained with our in-house DAT binding dataset.
One potential application of our DAT and hERG QSAR models is to identify new lead DAT inhibitors with novel scaffolds by counter screening. If we plot DAT affinities against hERG affinities, the ideal region is the upper left region in Figure 5, i.e., high-affinity DAT inhibitors possessing low hERG affinity. In our validation set of compounds, one compound appears to be promising, with ~1000-fold affinity difference in favor of DAT over hERG, according to our experimental measurements (Figure 5B). Encouragingly, our QSAR model predictions of this compound also showed ~63-fold affinity difference in the same direction (Figure 5A,C). In order to further evaluate the potential of our models to identify the ideal candidates, we then applied these models to screening other databases, such as the NCI open database compounds (Release 4; 260,277 compounds), and the results are shown in Figure S9. By filtering predicted DAT binders that were likely to have low affinity for hERG, i.e., satisfying either of the following two conditions, i) pKiDAT ≥ 7 and pIC50hERG ≤ 5.5, or ii) pKiDAT > 7 and pKiDAT - pIC50hERG ≥ 2, we found 161 hits, which account for 0.06% of the screened compounds. Thus, when a chemical library is large enough to cover adequate chemical space, such virtual counter screening is promising in identifying novel compound scaffolds with desired affinity profiles (high DAT and low hERG). However, iterations of experimental measurements and refinement of models are required to eventually validate hits.
Figure 5. The predicted differences between DAT pKi and hERG pIC50 for the validation compound set show a similar trend as those based on experimental measurements.
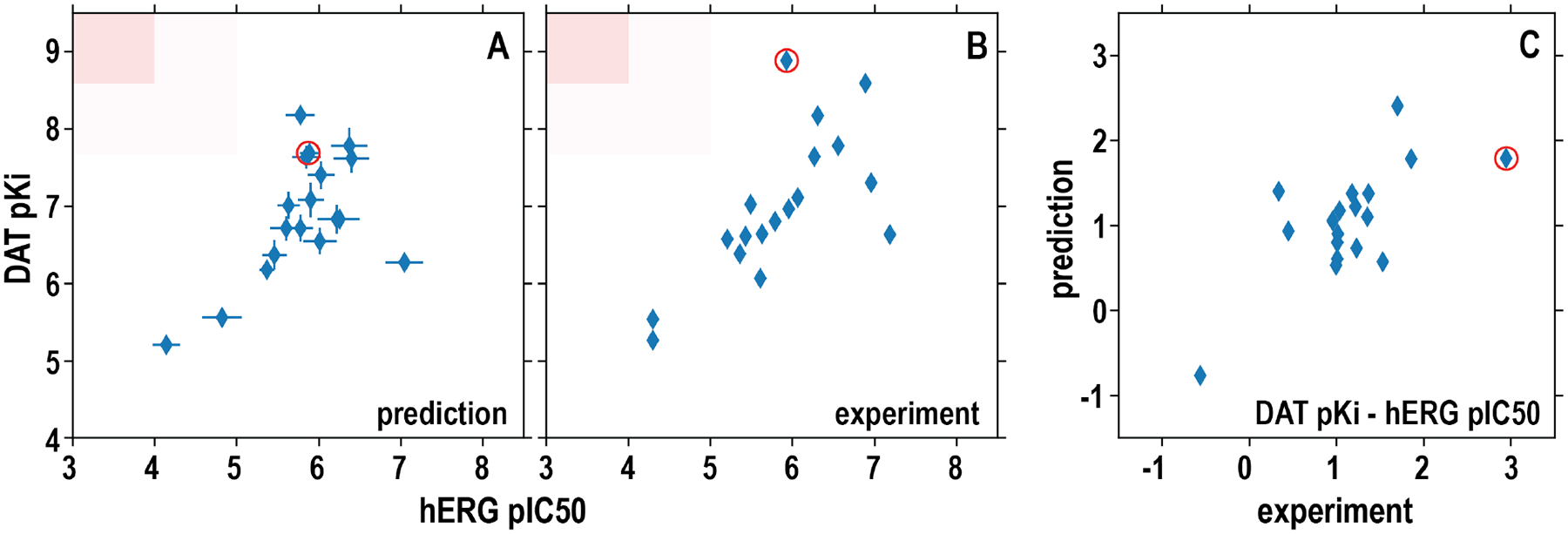
(A) the computationally predicted DAT pKi are plotted against hERG pIC50 for each compound in the validation set. The results from XGBoost-trained all-DAT (binding) and hERG (clamp) models were used. (B) The experimentally measured DAT pKi are plotted against the hERG pIC50 for each compound. The red shades are desired regions as hERG nonbinder and DAT binder. (C) The differences of DAT pKi and hERG pIC50 for each compound are plotted against the corresponding differences of the experimentally measured values. The red circle marks the compound with the highest affinity difference between DAT and hERG based on experimentally measured values. Pearson correlation coefficient R is 0.58 for the DAT versus hERG predicted affinities (A) and 0.69 for their experimentally measured affinities (B).
The quality and predictive power of QSAR prediction is associated with the data quality, coverage, and size.58 While the upper boundary of data size that can be utilized in model building is limited by computer resources, it is not necessarily true that the more compounds, the better. On the other hand, at least 40 compounds have been proposed as the lower limit of the dataset to build QSAR model with an optimum data size balance of 150 to 300 compounds.59, 60 With improved computational powers, the quality and the chemical space coverage have significant impact on the final outcome. In this study, we used eight datasets of various sizes to build 16 different sets of models (see Tables 1 and 2). Applicability domain for each model, which is the chemical space on which the training set of the model has been developed, cover all the entries in testing or validation datasets (Figure S10). Among the comparable datasets, the size of the rDAT binding dataset is smaller than that of the hDAT binding dataset (Table S1), but the prediction made with the rDAT binding models is better than that with the hDAT binding models (Figure S4), suggesting that the chemical space that rDAT binding dataset covers may fit the validation dataset better. However, combining hDAT binding with the rDAT binding data facilitates more adequate chemical space to be covered, resulting in slightly better prediction using XGBoost models (R2 = 0.79, Figure 3E). While only the hDAT uptake dataset has less than 150 compounds (126 compounds), the R2 of the prediction with the corresponding models on the validation dataset are 0.58 and 0.32, using the XGBoost and RF methods, respectively, are better than those of the rDAT uptake data based models, which do not have any predictive power on the validation dataset (Figure S4).
To evaluate whether any chemical descriptors may be differentially correlated with DAT and hERG affinities, we identified the most correlated descriptors for both the DAT binding and hERG clamp datasets used in our model building. Interestingly, when comparing 10 most positively correlated descriptors for each dataset, a few ring-related descriptors, “NumAliphaticHeterocycles”, “NumSaturatedHeterocycles”, “RingCount”, and “NumSaturatedRings” have Pearson correlation coefficient R > 0.4 for DAT binding affinity but not for hERG binding (Table S5). These differences likely represent some characteristics of a more defined binding site in DAT, while the generally low R values of the most correlated descriptors for hERG binding are likely related to its promiscuous binding of small compounds. For the validation datasets, some descriptors, such as “NumRotatableBonds”, can have high R for both the DAT and hERG affinities, reflecting the correlated DAT and hERG binding affinities of this compound set (Figure 5), and therefore the challenge in further optimizing the related scaffolds for DAT binding without increasing hERG binding (Table S5). In addition, the lack of connections between the most correlated descriptors for training and validation datasets, demonstrates the difficulty in QSAR modeling with linear regression. Thus, machine learning-based approach is necessary to integrate the information encoded in these features in building the QSAR models.
Taken together, we have established a combined platform for targeted compound discovery by combining machine learning based QSAR modeling, experimental validation, and molecular modeling and simulations. By dividing the training data to subsets that correspond to different functional state(s) of the target, we still obtained QSAR models with adequate predictive powers. These encouraging results open the door for future work to better connect the ligand-based QSAR modeling to protein structure-based molecular modeling and simulations in the compound discovery focusing on specific functional states of a target. In addition, this platform considers more than one target - while we conducted the counter screening of DAT against hERG, the current platform can be easily expanded and adapted for other targets of interest, as well as other applications such as synergy screening.
METHODS
Data preparation
Our training datasets were assembled from entries in a locally installed instance of ChEMBL (version 25) (March 2019).36 Initially, the target_dictionary, assays, activities, compound_structures, docs, source, relationship_type, molecule_hierarchy, and molecule_dictionary tables were joined on overlapping primary keys (tid, assay_id, molregno, doc_id, src_id, and relationship_type) and the resulting table was queried for entries related to the interested target (hERG or DAT). For the evaluations described in text, we also prepared specifies-specific filtered the hDAT and rDAT datasets.
The query results were then further cleaned up and filtered using a Python script. First, entries having a confidence value of less than 9 and those not arising from binding or functional assays were eliminated. Next, the remaining entries were split into IC50 and Ki datasets accordingly, followed by removing the entries where the standard_relationship type was not ‘=’. At this point, target-specific assay description filters (Table S2) were applied to sort the data according to specific experimental conditions. Specifically, the hERG dataset was split into binding and clamp dataset, while each of the DAT datasets were divided into binding and uptake datasets. Subsequently, all entries were excluded that did not come from assays with at least four distinct compounds.
In the hERG clamp dataset, 55 compounds present in a set of high-quality measurements of hERG affinity by gigaseal patch clamp electrophysiology41 were removed (the affinity data from that study were then added back to the dataset after applying the filters in Table S2). At this point, all salt forms present in the datasets were converted to neutral form, and entries with compounds containing unusual elements (defined as boron, deuterium, and silicon) were excluded. All entries with a compound having a molecular weight of over 650 were then excluded. For any entry without a pchembl (defined as: −Log(molar IC50, XC50, EC50, AC50, Ki, Kd or Potency)) value, but a numerical value with a compatible unit in standard_value and standard_units fields, we calculated the pchembl value accordingly.
Finally, all entries were clustered by the Tanimoto distances between the 2048-bit, 2-radius Morgan fingerprints of their respective chemical structures. Entries with a Tanimoto similarity of greater than 0.99 were considered as having identical structures; for each group of identical structures, the entry with the lowest pchembl value was selected to be part of the training set. As the chemical structure and pIC50 values for the hERG training set were written out, structures and values for the gigaseal patch clamp electrophysiology41 data set, described above, were added to the final training dataset.
After cleaning up the dataset with the filters above, the final dataset was used for the QSAR regression model construction. The numbers of compounds after each filter applied can be found in Table S1.
To prepare the training dataset for the classification models, the final datasets for regression were then divided into binary classification. The nonbinders were defined when pIC50 or pKi value is smaller than or equal to 5 (i.e., nonbinder). The binders were the log value bigger than or equal to 6 (i.e., binder). Molecules with pIC50 or pKi value between 5 and 6 were excluded.
Building QSAR models with machine learning approach
To predict hERG and DAT binding affinities, machine learning methods of eXtreme gradient boosting (XGBoost) and random forest (RF) in scikit-learn61 were used for constructing regression and classification QSAR models. Using the RDkit Python package,62 we first used all the available 200 descriptors defined in _descList of rdkit.Chem.Descriptors to calculate various properties for each compound, as well as the Gobbi 2D pharmacophore to calculate pharmacophore descriptors. In building the machine learning based model, we first reserve 15% data as the testing dataset for evaluating a model and 85% data as the training dataset to construct a model. It is a stochastic process to split the dataset and affect the model parameters.
To calculate the applicability domain (Figure S9), we first calculated the Morgan similarities of all pairs of compounds in a training dataset. The similarity was then converted into distance (1-similarity). The average and standard deviation of the distance distributions from all calculation were then used to determine the radius of applicability domain of the training dataset. For a testing dataset, the minimum distance of each testing molecule to any entry in the training dataset was calculated to confirm the radius of applicability domain is large enough to cover all testing molecules.
During the model training with machine learning approaches, the hyper-parameters space was grid-searched with sci-learning kit. 10-fold cross-validation was also used to determine the best performance with unique descriptors and optimized parameters. The initial parameters of using XGBoost for this grid search included colsample_bytree: [0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1]; subsample: [0.2, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1]; max_depth: [2, 3, 4, 5, 6, 7, 8]; and learning_rate: [0.001, 0.01, 0.02, 0.08, 0.1]. Similarly, the initial parameters of using RF was the following: n_estimators: [30, 100, 300, 1000, 3000, 10000, 30000, 100000], while we set the criterion to be gini (i.e., Gini importance sums over the number of tree splittings for each feature, the higher the better) for classification model and mae (i.e., mean absolute error) for regression model. Those initial parameters were the same in building regression and classification models. To evaluate how many models during training were adequate to reduce the stochastic uncertainty from randomly splitting the dataset (85% training and 15% testing), we first constructed a set of 35 independent models for each dataset. For each set of models, we randomly select certain number (n=1, …, 35) of models by a bootstrapping sampling with 100 repeats, and calculated the averages and standard deviations of R2 values for n models (Figure S2, left panels). We then calculated the slope of the trending curve for the standard deviations (Figure S2, right panels). Our results show that 15 models would be adequate to eliminate the stochastic uncertainty in most of cases, except for the models based on the hDAT uptake data. Therefore, we included 35 models with different random splittings for benchmarking the regression models.
The random_state in both XGBoost and RF is a stochastic element as well. Our benchmarks show that the random_state has some impact in model building, but far less than that in randomly splitting the dataset into training and testing datasets. Thus, we used fixed random_state in optimizing the parameters to build the final models. In building the final model set using 100% data for a condition, we build 10 models each with different random_state.
Metrics for model evaluation
The coefficient of determination (R2) and root mean square error (RMSE) are the metrics for evaluating the regression model performance. We used the toolkits implemented in scikit-learn to calculate R2 and RMSE between the model predicted and experimentally measured pIC50/pKi values.
The performance of a classification model can be described by a confusion matrix containing true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN), which are used to assess accuracy, sensitivity, and specificity. Accuracy is the most common model evaluation metric in classification validation.
Sensitivity (true positive rate or recall) is the ratio of correct positive prediction to the total positive prediction.
Specificity (true negative rate) is the ratio of correct negative prediction to the total negative prediction.
F-Score (F1 score) is the harmonic mean of precision and recall, where precision is defined as .
Molecular docking and modeling of hDAT
The representative human DAT (hDAT) model was selected from our previous hDAT models47 with outward-open conformations. Based on our established structure-activity relationship of DAT inhibitors and our previous modeling and simulation studies with hDAT models,47 from the resulting docking poses of JJC8-016 using AutoDock Vina,63 we chose the pose with the bisphenyl moiety occupying the S1 site and the charged nitrogen forming an ionic interaction with Asp79 of hDAT. The model of hDAT/JJC8-088 complex has been reported in our previous study.13 The pKa predictions of both JJC8-088 and JJC8-016 were performed using both Jaguar and Epik programs in Schrodinger suite (2019–2) and force field parameterization from force field builder in Schrodinger suite (2019–2) was carried out for JJC8-016. The protein/ligand complexes were then embedded with explicit 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipid bilayer (POPC) with the same orientation of dDAT/nortriptyline structure (PDB ID 4M48 from the Orientation of Proteins in Membranes database).64 The systems were then solvated using simple point charge (SPC) water model65 and neutralized with 0.15 M NaCl. The final model system contained ~131,000 atoms.
Molecular docking and modeling of hERG
The molecular dynamics flexible fitting (MDFF)-refined cryo-EM structure of hERG (PDB ID 5VA2) was used for molecular docking, as described below. The details of the structure refinement can be found in Khan et al.48 The missing loops built using ROSETTA loop modelling, were minimized and briefly equilibrated to avoid steric clashes, and did not contain the Per-Arnt-Sim (PAS) domain.66 The selected hERG models above were then used as the model for docking. The docking of JJC8-016 and JJC8-088 to hERG model was performed using Glide package in Schrodinger suite. The Glide-XP (extra-precision)67 from the Maestro suite in Schrödinger was used for all docking calculations with a ligand vdW scale factors set to 0.80 and a RMSD cut-off of 2.0 Å. JJC8-016 and JJC8-088 were docked to the primary binding pocket identified in a number of studies: the internal central cavity. The position of the docking grid was set to 0,0,Z, where Z corresponds to the position of a center of mass for residue F656 in distal S6.53, 56, 68 As hERG1 biological assembly is tetrameric, all four subunits were considered to build receptor grids. The receptor grid dimensions were then sub-divided into inner and outer cubes. The length of the inner cube box edge was set to 10 Å and represents the space explored by Glide as acceptable positions for the geometrical center of the drugs. The outer box edge, representing the total space explored by all atoms comprising ligand was set to 26 Å. Only poses with energy score (Gscore) of ≤ −3 kcal/mol were used in further analysis. The absence of the membrane phase in docking studies represents a natural challenge to the direct interpretations of the binding scores, and therefore we only use these scores to assess a relative likelihood (relative to the most favorable binding score) of different binding poses. Hence, coordinates of hERG-ligand from molecular docking were used to seed MD simulations with explicit lipid bilayer. After docking poses were selected, we immersed the hERG/ligand complexes in explicit POPC lipid bilayer and water environment. The orientation of the complexes in the membrane was guided by that calculated for the cryo-EM structure of hERG (PDB ID 5VA152) in the Orientation of Proteins in Membranes database.64 Similar to hDAT, the model systems of hERG complexes were then solvated using SPC water model65 and neutralized with 0.15 M KCl. The final simulated system contained ~247,000 atoms.
MD simulations and analysis
MD simulations of hDAT/JJC8-016, hDAT/JJC8-088, hERG/JJC8-016 and hERG/JJC8-088 complexes were performed using Desmond MD engine (D. E. Shaw Research, New York, NY) and OPLS3e force field.69 Langevin dynamics was performed with NPγT ensemble at constant temperature (310 K) and 1 atm constant pressure with the hybrid Nose-Hoover Langevin piston method70 on an anisotropic flexible periodic cell, and a constant surface tension (x-y plane). The systems were first minimized and equilibrated with restraints on the ligand heavy atoms and protein backbone atoms. The restraints were removed during the production phase of the simulations. Seven independent trajectories of hDAT/JJC8-016 and five of hDAT/JJC8-088 complexes were collected with the aggregated simulation time of 15.5 and 11.1 μs, respectively (Table S6). For hERG, we collected six hERG/JJC8-016 and three hERG/JJC8-088 trajectories with accumulated lengths of 4.86 and 3.6 μs, respectively (Table S6). The analysis was performed using MDAnalysis71, VMD72 and in-house Python scripts.
Radioligand binding assay of rDAT
Frozen striatal membranes, dissected from male Sprague−Dawley rat brains (supplied on ice by Bioreclamation, Hicksville, NY), were homogenized in 20 volumes (w/v) of ice cold modified sucrose phosphate buffer (0.32 M sucrose, 7.74 mM Na2HPO4, and 2.26 mM NaH2PO4, pH adjusted to 7.4) using a Brinkman Polytron (Setting 6 for 20 s) and centrifuged at 48,400 × g for 10 min at 4 °C. The resulting pellet was resuspended in buffer, recentrifuged, and suspended in ice cold buffer again to a concentration of 20 mg/mL, original wet weight (OWW). Experiments were conducted in 96-well polypropylene plates containing 50 μL of various concentrations of the inhibitor, diluted using 30% DMSO vehicle, 300 μL of sucrose phosphate buffer, 50 μL of [3H]WIN 35,428 (final concentration 1.5 nM; PerkinElmer Life Sciences, Waltham, MA), and 100 μL of tissue (2.0 mg/well OWW). All compound dilutions were tested in triplicate and the competition reactions started with the addition of tissue, and the plates were incubated for 120 min at 0–4 °C. Nonspecific binding was determined using 10 μM indatraline.
Electrophysiology of hERG blockade
The extracellular solution contained (in mM) NaCl 140, KCl 5.4, CaCl2 1, MgCl2 1, HEPES 5, glucose 5.5, and was kept at pH 7.4 with NaOH. Micropipettes were pulled from borosilicate glass capillary tubes on a programmable horizontal puller (Sutter Instruments, Novato, CA). The pipette solution contained the following: 10 mM KCl, 110 mM K-aspartate, 5 mM MgCl2, 5mM Na2ATP, 10 mM EGTA - ethylene glycol-bis(-aminoethyl ether)-N,N,N,N tetra-acetic acid, 5 mM HEPES, and 1mM CaCl2. The solution was adjusted to pH 7.2 with KOH. Standard patch-clamp methods were used to measure the whole cell currents of hERG mutants expressed in HEK 293 cells using the AXOPATCH 200B amplifier (Axon Instruments). The holding potential was −80 mV. The amplitudes of tail currents were measured when the voltage was returned to −100 mV after + 50 mV 1-second depolarization. The compounds were dissolved in Tyrode solution immediately before the experiments and the solutions were used for the next two hours during the experiments.
Statistical analysis of electrophysiology data
The data are presented as the mean +/− S.D. One-way ANOVA test was used to analyze the data. P < 0.05 was designated as being significant.
Supplementary Material
Acknowledgements
Support for this research was provided by the National Institute on Drug Abuse - Intramural Research Program, Z1A DA000389 (A.H.N) and Z1A DA000606 (L.S.). Work in Calgary (JG, SW, MK and SYN) was supported with the Canadian Institutes for Health Research Project Grant (FRN-CIHR: 156236). MK was supported by Queen Elizabeth II graduate scholarship and Jake Duerksen Memorial scholarships. JG was supported with IBM CAS Innovation Eco-System as part of “Cardiotoxicity Modelling Project”.
Footnotes
Supporting Information Available
The workflow of model building; the number of models needed for each set of XGBoost regression models; comparison of the rDAT and hDAT binding datasets; correlations between the predicted and experimentally measured affinities for the models trained with the hDAT, rDAT and in-house DAT datasets; hydrogen bonding analyses for hDAT MD simulations; the compound pairs found in the ChEMBL datasets showing opposite affinity trends at hERG and DAT; cross predictions between hDAT and rDAT binding XGBoost models; counter screening of the NCI open database; the applicability domain coverage for the testing and validation datasets; the filters and the numbers of datapoints after applying each filter; keywords used in assay description filter; benchmarks of the models trained with the in-house DAT dataset; benchmarks of the XGBoost classification models trained with equal numbers of binders and non-binders from the all-DAT binding dataset; most correlated descriptors for DAT and hERG ligands; summary of MD simulations (PDF)
Data and software availability
The code for machine-learning based QSAR modeling is freely available on GitHub at https://github.com/NIDA-IRP-CCMB/QSAR_DAT-hERG. The QSAR models described herein are available upon request.
REFERENCE
- 1.Lounkine E; Keiser MJ; Whitebread S; Mikhailov D; Hamon J; Jenkins JL; Lavan P; Weber E; Doak AK; Cote S; Shoichet BK; Urban L, Large-Scale Prediction and Testing of Drug Activity on Side-Effect Targets. Nature 2012, 486, 361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campillos M; Kuhn M; Gavin AC; Jensen LJ; Bork P, Drug Target Identification Using Side-Effect Similarity. Science 2008, 321, 263–6. [DOI] [PubMed] [Google Scholar]
- 3.Schuster D; Laggner C; Langer T, Why Drugs Fail--a Study on Side Effects in New Chemical Entities. Curr Pharm Des 2005, 11, 3545–59. [DOI] [PubMed] [Google Scholar]
- 4.Roth BL; Sheffler DJ; Kroeze WK, Magic Shotguns Versus Magic Bullets: Selectively Non-Selective Drugs for Mood Disorders and Schizophrenia. Nat Rev Drug Discov 2004, 3, 353–9. [DOI] [PubMed] [Google Scholar]
- 5.Tanda G; Newman AH; Katz JL, Discovery of Drugs to Treat Cocaine Dependence: Behavioral and Neurochemical Effects of Atypical Dopamine Transport Inhibitors. Adv Pharmacol 2009, 57, 253–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanda G; Hersey M; Hempel B; Xi ZX; Newman AH, Modafinil and its Structural Analogs as Atypical Dopamine Uptake Inhibitors and Potential Medications for Psychostimulant Use Disorder. Curr Opin Pharmacol 2020, 56, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman AH; Ku T; Jordan CJ; Bonifazi A; Xi ZX, New Drugs, Old Targets: Tweaking the Dopamine System to Treat Psychostimulant Use Disorders. Annu Rev Pharmacol Toxicol 2021, 61, 609–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman AH; Kulkarni S, Probes for the Dopamine Transporter: New Leads toward a Cocaine-Abuse Therapeutic--a Focus on Analogues of Benztropine and Rimcazole. Med Res Rev 2002, 22, 429–64. [DOI] [PubMed] [Google Scholar]
- 9.Reith ME; Blough BE; Hong WC; Jones KT; Schmitt KC; Baumann MH; Partilla JS; Rothman RB; Katz JL, Behavioral, Biological, and Chemical Perspectives on Atypical Agents Targeting the Dopamine Transporter. Drug Alcohol Depend 2015, 147, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanda G; Newman AH; Ebbs AL; Tronci V; Green JL; Tallarida RJ; Katz JL, Combinations of Cocaine with Other Dopamine Uptake Inhibitors: Assessment of Additivity. J Pharmacol Exp Ther 2009, 330, 802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desai RI; Kopajtic TA; Koffarnus M; Newman AH; Katz JL, Identification of a Dopamine Transporter Ligand That Blocks the Stimulant Effects of Cocaine. J Neurosci 2005, 25, 1889–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiranita T; Soto PL; Newman AH; Katz JL, Assessment of Reinforcing Effects of Benztropine Analogs and Their Effects on Cocaine Self-Administration in Rats: Comparisons with Monoamine Uptake Inhibitors. J Pharmacol Exp Ther 2009, 329, 677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman AH; Cao J; Keighron JD; Jordan CJ; Bi GH; Liang Y; Abramyan AM; Avelar AJ; Tschumi CW; Beckstead MJ; Shi L; Tanda G; Xi ZX, Translating the Atypical Dopamine Uptake Inhibitor Hypothesis toward Therapeutics for Treatment of Psychostimulant Use Disorders. Neuropsychopharmacology 2019, 44, 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanguinetti MC; Tristani-Firouzi M, hERG Potassium Channels and Cardiac Arrhythmia. Nature 2006, 440, 463–9. [DOI] [PubMed] [Google Scholar]
- 15.Hancox JC; McPate MJ; El Harchi A; Zhang YH, The hERG Potassium Channel and hERG Screening for Drug-Induced Torsades de Pointes. Pharmacol Ther 2008, 119, 118–32. [DOI] [PubMed] [Google Scholar]
- 16.Yang PC; DeMarco KR; Aghasafari P; Jeng MT; Dawson JRD; Bekker S; Noskov SY; Yarov-Yarovoy V; Vorobyov I; Clancy CE, A Computational Pipeline to Predict Cardiotoxicity: From the Atom to the Rhythm. Circ Res 2020, 126, 947–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez D; Ghanta A; Kauffman GW; Sanguinetti MC, Physicochemical Features of the hERG Channel Drug Binding Site. J Biol Chem 2004, 279, 10120–7. [DOI] [PubMed] [Google Scholar]
- 18.Cavalluzzi MM; Imbrici P; Gualdani R; Stefanachi A; Mangiatordi GF; Lentini G; Nicolotti O, Human Ether-a-go-go-Related Potassium Channel: Exploring SAR to Improve Drug Design. Drug Discov Today 2020, 25, 344–366. [DOI] [PubMed] [Google Scholar]
- 19.Gintant G; Sager PT; Stockbridge N, Evolution of Strategies to Improve Preclinical Cardiac Safety Testing. Nat Rev Drug Discov 2016, 15, 457–71. [DOI] [PubMed] [Google Scholar]
- 20.Kalyaanamoorthy S; Barakat KH, Binding Modes of hERG Blockers: An Unsolved Mystery in the Drug Design Arena. Expert Opin Drug Discov 2018, 13, 207–210. [DOI] [PubMed] [Google Scholar]
- 21.Vandenberg JI; Perry MD; Perrin MJ; Mann SA; Ke Y; Hill AP, hERG K(+) Channels: Structure, Function, and Clinical Significance. Physiol Rev 2012, 92, 1393–478. [DOI] [PubMed] [Google Scholar]
- 22.Wallis R; Benson C; Darpo B; Gintant G; Kanda Y; Prasad K; Strauss DG; Valentin JP, CiPA Challenges and Opportunities from a Non-Clinical, Clinical and Regulatory Perspectives. An Overview of the Safety Pharmacology Scientific Discussion. J Pharmacol Toxicol Methods 2018, 93, 15–25. [DOI] [PubMed] [Google Scholar]
- 23.Shah RR, Can Pharmacogenetics Help Rescue Drugs Withdrawn from the Market? Pharmacogenomics 2006, 7, 889–908. [DOI] [PubMed] [Google Scholar]
- 24.Onakpoya IJ; Heneghan CJ; Aronson JK, Post-Marketing Withdrawal of 462 Medicinal Products Because of Adverse Drug Reactions: A Systematic Review of the World Literature. BMC Med 2016, 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Food; Drug Administration, H. H. S., International Conference on Harmonisation; Guidance on S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals; Availability. Notice. Fed Regist 2005, 70, 61133–4. [PubMed] [Google Scholar]
- 26.Wacker S; Noskov SY, Performance of Machine Learning Algorithms for Qualitative and Quantitative Prediction Drug Blockade of hERG1 Channel. Comput Toxicol 2018, 6, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siramshetty VB; Chen Q; Devarakonda P; Preissner R, The Catch-22 of Predicting hERG Blockade Using Publicly Accessible Bioactivity Data. J Chem Inf Model 2018, 58, 1224–1233. [DOI] [PubMed] [Google Scholar]
- 28.Cai C; Guo P; Zhou Y; Zhou J; Wang Q; Zhang F; Fang J; Cheng F, Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J Chem Inf Model 2019, 59, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryu JY; Lee MY; Lee JH; Lee BH; Oh KS, DeepHIT: A Deep Learning Framework for Prediction of hERG-Induced Cardiotoxicity. Bioinformatics 2020, 36, 3049–3055. [DOI] [PubMed] [Google Scholar]
- 30.Kim H; Nam H, hERG-Att: Self-Attention-Based Deep Neural Network for Predicting hERG Blockers. Comput Biol Chem 2020, 87, 107286. [DOI] [PubMed] [Google Scholar]
- 31.Siramshetty VB; Nguyen DT; Martinez NJ; Southall NT; Simeonov A; Zakharov AV, Critical Assessment of Artificial Intelligence Methods for Prediction of hERG Channel Inhibition in the “Big Data” Era. J Chem Inf Model 2020, 60, 6007–6019. [DOI] [PubMed] [Google Scholar]
- 32.Kong W; Wang W; An J, Prediction of 5-Hydroxytryptamine Transporter Inhibitors Based on Machine Learning. Comput Biol Chem 2020, 87, 107303. [DOI] [PubMed] [Google Scholar]
- 33.Abel R; Manas ES; Friesner RA; Farid RS; Wang L, Modeling the Value of Predictive Affinity Scoring in Preclinical Drug Discovery. Curr Opin Struct Biol 2018, 52, 103–110. [DOI] [PubMed] [Google Scholar]
- 34.Davis AM 3.15 - Quantitative Structure–Activity Relationships. In Comprehensive Medicinal Chemistry III, Chackalamannil S; Rotella D; Ward SE, Eds.; Elsevier: Oxford, 2017, pp 379–392. [Google Scholar]
- 35.Gertrudes JC; Maltarollo VG; Silva RA; Oliveira PR; Honorio KM; da Silva AB, Machine Learning Techniques and Drug Design. Curr Med Chem 2012, 19, 4289–97. [DOI] [PubMed] [Google Scholar]
- 36.Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP, ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res 2012, 40, D1100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng Y; Prusoff WH, Relationship between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem Pharmacol 1973, 22, 3099–108. [DOI] [PubMed] [Google Scholar]
- 38.Strauss DG; Gintant G; Li Z; Wu W; Blinova K; Vicente J; Turner JR; Sager PT, Comprehensive In Vitro Proarrhythmia Assay (CiPA) Update from a Cardiac Safety Research Consortium / Health and Environmental Sciences Institute / FDA Meeting. Ther Innov Regul Sci 2019, 53, 519–525. [DOI] [PubMed] [Google Scholar]
- 39.Anwar-Mohamed A; Barakat KH; Bhat R; Noskov SY; Tyrrell DL; Tuszynski JA; Houghton M, A Human Ether-a-go-go-Related (hERG) Ion Channel Atomistic Model Generated by Long Supercomputer Molecular Dynamics Simulations and Its Use in Predicting Drug Cardiotoxicity. Toxicol Lett 2014, 230, 382–92. [DOI] [PubMed] [Google Scholar]
- 40.Vicente J; Zusterzeel R; Johannesen L; Mason J; Sager P; Patel V; Matta MK; Li Z; Liu J; Garnett C; Stockbridge N; Zineh I; Strauss DG, Mechanistic Model-Informed Proarrhythmic Risk Assessment of Drugs: Review of the “CiPA” Initiative and Design of a Prospective Clinical Validation Study. Clin Pharmacol Ther 2018, 103, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kramer J; Obejero-Paz CA; Myatt G; Kuryshev YA; Bruening-Wright A; Verducci JS; Brown AM, MICE Models: Superior to the hERG Model in Predicting Torsade de Pointes. Sci Rep 2013, 3, 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi L 9.7 Molecular Modeling and Simulations of Transporter Proteins – the Transmembrane Allosteric Machinery. In Comprehensive Biophysics, Egelman EH, Ed.; Elsevier: Amsterdam, 2012; Chapter 9.7, pp 105–122. [Google Scholar]
- 43.Shan J; Javitch JA; Shi L; Weinstein H, The Substrate-Driven Transition to an Inward-Facing Conformation in the Functional Mechanism of the Dopamine Transporter. PLoS One 2011, 6, e16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beuming T; Shi L; Javitch JA; Weinstein H, A Comprehensive Structure-Based Alignment of Prokaryotic and Eukaryotic Neurotransmitter/Na+ Symporters (NSS) Aids in the Use of the LeuT Structure to Probe NSS Structure and Function. Mol Pharmacol 2006, 70, 1630–42. [DOI] [PubMed] [Google Scholar]
- 45.Keighron JD; Giancola JB; Shaffer RJ; DeMarco EM; Coggiano MA; Slack RD; Newman AH; Tanda G, Distinct Effects of (R)-Modafinil and Its (R)- and (S)-Fluoro-Analogs on Mesolimbic Extracellular Dopamine Assessed by Voltammetry and Microdialysis in Rats. Eur J Neurosci 2019, 50, 2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang HY; Bi GH; Yang HJ; He Y; Xue G; Cao J; Tanda G; Gardner EL; Newman AH; Xi ZX, The Novel Modafinil Analog, JJC8-016, as a Potential Cocaine Abuse Pharmacotherapeutic. Neuropsychopharmacology 2017, 42, 1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abramyan AM; Stolzenberg S; Li Z; Loland CJ; Noe F; Shi L, The Isomeric Preference of an Atypical Dopamine Transporter Inhibitor Contributes to Its Selection of the Transporter Conformation. ACS Chem Neurosci 2017, 8, 1735–1746. [DOI] [PubMed] [Google Scholar]
- 48.Khan HM; Guo J; Duff HJ; Tieleman DP; Noskov SY, Refinement of a Cryo-EM Structure of hERG: Bridging Structure and Function. Biophys J 2021, 120, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorensen L; Andersen J; Thomsen M; Hansen SM; Zhao X; Sandelin A; Stromgaard K; Kristensen AS, Interaction of Antidepressants with the Serotonin and Norepinephrine Transporters: Mutational Studies of the S1 Substrate Binding Pocket. J Biol Chem 2012, 287, 43694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abramyan AM; Slack RD; Meena S; Davis BA; Newman AH; Singh SK; Shi L, Computation-Guided Analysis of Paroxetine Binding to hSERT Reveals Functionally Important Structural Elements and Dynamics. Neuropharmacology 2019, 161, 107411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slack RD; Abramyan AM; Tang H; Meena S; Davis BA; Bonifazi A; Giancola JB; Deschamps JR; Naing S; Yano H; Singh SK; Newman AH; Shi L, A Novel Bromine-Containing Paroxetine Analogue Provides Mechanistic Clues for Binding Ambiguity at the Central Primary Binding Site of the Serotonin Transporter. ACS Chem Neurosci 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang W; MacKinnon R, Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 2017, 169, 422–430 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kudaibergenova M; Guo J; Khan HM; Zahid F; Lees-Miller J; Noskov SY; Duff HJ, Allosteric Coupling between Drug Binding and the Aromatic Cassette in the Pore Domain of the hERG1 Channel: Implications for a State-Dependent Blockade. Front Pharmacol 2020, 11, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mousaei M; Kudaibergenova M; MacKerell AD Jr.; Noskov S, Assessing hERG1 Blockade from Bayesian Machine-Learning-Optimized Site Identification by Ligand Competitive Saturation Simulations. J Chem Inf Model 2020, 60, 6489–6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen J; Seebohm G; Sanguinetti MC, Position of Aromatic Residues in the S6 Domain, Not Inactivation, Dictates Cisapride Sensitivity of hERG and eag Potassium Channels. Proc Natl Acad Sci U S A 2002, 99, 12461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saxena P; Zangerl-Plessl EM; Linder T; Windisch A; Hohaus A; Timin E; Hering S; Stary-Weinzinger A, New Potential Binding Determinant for hERG Channel Inhibitors. Sci Rep 2016, 6, 24182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perrin MJ; Kuchel PW; Campbell TJ; Vandenberg JI, Drug Binding to the Inactivated State Is Necessary but Not Sufficient for High-Affinity Binding to Human Ether-a-go-go-Related Gene Channels. Mol Pharmacol 2008, 74, 1443–52. [DOI] [PubMed] [Google Scholar]
- 58.Fourches D; Muratov E; Tropsha A, Trust, but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J Chem Inf Model 2010, 50, 1189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tropsha A, Best Practices for QSAR Model Development, Validation, and Exploitation. Mol Inform 2010, 29, 476–88. [DOI] [PubMed] [Google Scholar]
- 60.Lima AN; Philot EA; Trossini GH; Scott LP; Maltarollo VG; Honorio KM, Use of Machine Learning Approaches for Novel Drug Discovery. Expert Opin Drug Discov 2016, 11, 225–39. [DOI] [PubMed] [Google Scholar]
- 61.Pedregosa F; Varoquaux G. l.; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay d., Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res 2011, 12, 2825–2830. [Google Scholar]
- 62.Landrum G RDKit: Open-Source Cheminformatics.
- 63.Trott O; Olson AJ, AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem 2010, 31, 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lomize MA; Lomize AL; Pogozheva ID; Mosberg HI, OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22, 623–5. [DOI] [PubMed] [Google Scholar]
- 65.Berendsen HJC; Postma JPM; van Gunsteren WF; Hermans J Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, April 13–16, 1981, Pullman B, Ed.; Springer; Netherlands: Dordrecht, 1981, pp 331–342. [Google Scholar]
- 66.Perissinotti LL; De Biase PM; Guo J; Yang PC; Lee MC; Clancy CE; Duff HJ; Noskov SY, Determinants of Isoform-Specific Gating Kinetics of hERG1 Channel: Combined Experimental and Simulation Study. Front Physiol 2018, 9, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J Med Chem 2006, 49, 6177–96. [DOI] [PubMed] [Google Scholar]
- 68.Zangerl-Plessl EM; Berger M; Drescher M; Chen Y; Wu W; Maulide N; Sanguinetti M; Stary-Weinzinger A, Toward a Structural View of hERG Activation by the Small-Molecule Activator ICA-105574. J Chem Inf Model 2020, 60, 360–371. [DOI] [PubMed] [Google Scholar]
- 69.Roos K; Wu C; Damm W; Reboul M; Stevenson JM; Lu C; Dahlgren MK; Mondal S; Chen W; Wang L; Abel R; Friesner RA; Harder ED, OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J Chem Theory Comput 2019, 15, 1863–1874. [DOI] [PubMed] [Google Scholar]
- 70.Feller SE; Zhang Y; Pastor RW; Brooks BR, Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J Chem Phys 1995, 103, 4613–4621. [Google Scholar]
- 71.Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O, MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J Comput Chem 2011, 32, 2319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humphrey W; Dalke A; Schulten K, VMD: Visual Molecular Dynamics. J Mol Graph 1996, 14, 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code for machine-learning based QSAR modeling is freely available on GitHub at https://github.com/NIDA-IRP-CCMB/QSAR_DAT-hERG. The QSAR models described herein are available upon request.