

Abstract
Site-specific protein labeling is important in biomedical research and biotechnology. While many methods allow site-specific protein modification, a straightforward approach for efficient N-terminal protein labeling is not available. We introduce a novel sortase-mediated swapping approach for a one-step site-specific N-terminal labeling with a near-quantitative yield. We show that this method allows rapid and efficient cleavage and simultaneous labeling of the N or C termini of fusion proteins. The method does not require any prior modification beyond the genetic incorporation of the sortase recognition motif. This new approach provides flexibility for protein engineering and site-specific protein modifications.
Graphical Abstract
INTRODUCTION
Site-specific protein labeling is important in modern chemical biology.1–3 Proteins conjugated with fluorophores, polymers, drugs, or antigens have enabled the study of cellular processes and molecular mechanisms. The development of new drugs, vaccines, and other biotechnological tools has relied on such methods.4–7 Site-specific modification of proteins has been accomplished by chemoenzymatic means using enzymes to selectively catalyze the formation or cleavage of covalent bonds (e.g., biotin ligase, sortase, lipoic acid ligase, farnesyl transferase, sialyltransferase, phosphopantetheinyltransferase, transglutaminase, N-myristoyl transferase, and formylglycine-generating enzyme)8–25 or to enable “click” reaction chemistry by the incorporation of non-natural amino acids containing bio-orthogonal reactive groups;26–29 such approaches represent a remarkable technological advance. However, the methods are usually sophisticated, laborious, and technically demanding and may not provide a high yield.
The C termini for many proteins are not accessible or are essential for their function,30–32 highlighting the need for a robust and reproducible N-terminal labeling approach. Several strategies have been developed for N-terminal protein labeling.33,34 Examples include chemical approaches to label the N-terminal α-amine with reactive groups, such as aldehydes including 2-ethynyl benzaldehyde, N-hydroxyphthalimides, 2-pyridinecarboxyaldehydes, and selenobenzaldehydes, among others.35–39 Additionally, chemical modifications to introduce a click handle on the N-terminal α-amine such as an aldehyde, azide, or alkyne, which can subsequently be used to attach functional groups, have been developed as well.40–42 Notwithstanding the encouraging progress, such approaches are usually based on the control of pH, often require extensive optimization, and can result in off-target labeling, which leads to heterogeneous protein mixtures. Additionally, labeling occurs through a chemical reaction other than a native peptide bond, which, in turn, may become a limiting factor when considering clinical translatability. The native chemical ligation is an alternative approach; however, it requires an N-terminal cysteine that would need to be introduced by protein engineering approaches.43,44 Additionally, the introduced cysteine may affect and complicate protein folding. Enzymes such as subtiligase and butelase have been used to perform N-terminal labeling.45–47 Subtiligase mediates the ligation of the N-terminal α-amine of a protein to a C-terminal ester or thioester peptide.47–49 The yield varies depending on the characteristics of the N terminus of the target proteins. Several different subtiligases with different reactivities toward different N-terminal sequences have been developed.50,47 Butilase recognizes an NHV sequence and mediates its ligation with an X1-X2 peptide motif, where the first can be any amino acid except proline, and the N1 terminal residue needs to be Cys, Ile, Leu, or Val.51 The N-terminal sequence needs to be flexible and fully available for the enzyme to function, and thus, engineering a flexible spacer at the N terminus can increase the yield.45 Alternative strategies to site-specifically label the N terminus will provide flexibility when designing proteins, protein-based drugs, and protein-based assays or platforms.
Sortase A is a bacterial transpeptidase mostly used for site-specific C-terminal protein labeling (Figure 1A).8,9,52 Sortase has a short recognition motif “LPXTG,” where “X” denotes several different amino acids such as “E” or “S.”8,9,52 The enzyme cleaves the bond between threonine and glycine, forming a thioester intermediate with the carboxylate of threonine, which is susceptible to a nucleophilic substitution of a multiglycine substrate (n > 2, e.g., “G3-Y,” where “Y” is a molecule intended for conjugation). The final product thus contains the sequence “-LPXT-G3-Y” (Figure 1A). The typical sortase reaction thus relies on an “-LPXTG-” motif and the availability of Gly3-containing substrates, and allows straightforward modification of the C terminus. However, the sortase reaction has found limited applications for N-terminal labeling, as many recombinantly produced protein N termini retain the initiator methionine (M). Proteins expressed in the Escherichia coli cytosol are often demethionylated when the subsequent residue is small; however, demethionylation yields vary for recombinant proteins, potentially due to the saturation of methionine aminopeptidases (MAPs).53–55 Therefore, N-terminal incorporation of a multiglycine residue, which is needed for the sortase reaction, requires additional steps, such as the incorporation of a tobacco etch virus (TEV) cleavage site, as shown before.56–58 A method that allows direct N-terminal labeling without the need for the protease-mediated cleavage step to expose a recognition sequence would be ideal.
Figure 1.
Site-specific N-terminal labeling using the sortase-mediated swapping approach. (A) Traditional sortase reaction for site-specific C-terminal protein labeling. (B) Schematic representation of the direct N-terminal protein labeling using the sortase-swapping approach. (C) The hypothesized mechanism for the direct N-terminal protein labeling. (D) GFP was directly labeled at its N terminus using an azide-functionalized LPETG-containing substrate; LC-MS analyses confirmed the formation of the product. (E) The N-terminally installed click handle can be used to site-specifically attach different molecules to the protein, such as a PEG moiety; SDS-PAGE analysis was used to confirm the formation of PEGylated GFP. Lanes #1: marker, #2: MH6LPETGGGGG-GFP, #3: azide-functionalized GFP, #4: PEGylated-GFP (5 kDa PEG). (F) GFP was directly labeled at its N terminus using an alkyne-functionalized LPETG-containing substrate; LC-MS analyses confirmed the formation of the product. (G) The N-terminally installed alkyne click handle can be used to site-specifically attach different molecules to the protein, such as a PEG moiety; SDS-PAGE analysis was used to confirm the formation of products. Lanes #1: marker, #2: MH6-LPETGGGGG-GFP, #3: alkyne-functionalized GFP, #4: PEGylated-GFP (2 kDa PEG).
Here, we introduce a new one-step approach to use sortase for site-specific labeling of recombinantly produced—and cytosolically expressed—proteins at the N terminus and to synthesize protein fusions. Moreover, it is possible to simultaneously cleave protein fusions and perform site-specific N- or C-terminal labeling. All of this can be accomplished without any prior modification on the proteins other than the genetic provision of the sortase recognition motif.
RESULTS AND DISCUSSION
Sortase-Mediated Swapping: Establishing the Approach.
Site-Specific N-Terminal Labeling without any Prior Modification.
The site-specific N-terminal modification of proteins remains a challenge. Enzymes such as sortase and OaAEP1 have been used to site-specifically label proteins at the N termini.56,57,59,60 These methods call for specific N-terminal sequences, Gly3 and GV, requiring additional steps to introduce recognition elements at the N terminus, for example through exposure of cryptic Gly3 or GV sequences by cleavage with a TEV protease.57,59
We hypothesized that incorporation of an “LPETGn” motif (n > 2) at the protein N terminus, upon the reaction of the protein with sortase, should result in the in situ formation of a Glyn-protein. Therefore, when reacted with sortase, the in situ formed Glyn-protein should act as a nucleophile, allowing labeling with an “LPETG”-containing probe (Figure 1B,C). Sortase will form a thioester intermediate with the LPETG-containing probe (probe-LPET-thioester-sortase) and will release a G-X in the solution as well (Figure 1C). However, the in situ formed oligo-glycine-protein should act as a much more efficient nucleophile compared to the G-X released from the probe,61,62 and therefore, an excess of the “probe-LPETG-X” should drive equilibrium toward the formation of the “probe-LPET-Gn-protein” construct. To test this, we engineered a green fluorescent protein (GFP) to contain an LPETG5 sequence at the N terminus (MH6-LPETG5-GFP). The protein was expressed cytosolically in E. coli with a high expression yield (~40 mg per liter of culture) and purified via a Ni-NTA affinity column. We next performed a sortase reaction using an azide-functionalized LPETG peptide (with the azide group attached to the N-terminal amine of the leucine; see the structure in the SI) to introduce a click-handle azide moiety and determine whether this approach allows direct labeling of the GFP at its N terminus. Interestingly, the reaction yielded near-quantitative formation of the azide-labeled GFP (Figure 1D,E) (reaction included ~100 μM GFP, 5 μM sortase, 4 mM azide substrate, and 10 mM CaCl2). The product was characterized with liquid chromatography–mass spectrometry (LC-MS) and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 1D,E). To confirm the reactivity of the introduced azide group, we performed a “click” reaction between the azide-modified protein and a 5 kDa polyethylene glycol (PEG) moiety functionalized with a dibenzocyclooctyne (DBCO) group. The PEGylation reaction proceeded with a near-quantitative yield (Figure 1E) (reaction condition: azide-GFP (25 μM), DBCO-PEG5 (100 μM), room temperature, 2 h). A similar reaction was performed to install an alkyne-functionalized LPETG substrate at the N terminus of the GFP protein, providing quantitative alkyne-labeled GFP (Figure 1F,G) (reaction included ~100 μM GFP, 5 μM sortase, 4 mM alkyne substrate, and 10 mM CaCl2). PEGylation using a 2 kDa azide-PEG provided near-quantitative formation of N-terminally PEGylated protein (Figure 1G) (reaction condition: alkyne-GFP (25 μM), azide-PEG2 (100 μM), CuSO4 1 mM, BTTAA 5 mM, ascorbic acid 2.5 mM, room temperature, 2 h). Therefore, we concluded that the new sortase-mediated swapping approach can be used to site-specifically label proteins at the N terminus in a single-step reaction to a near-quantitative yield. The 5 Gs were included in the GFP protein design to ensure maximum flexibility and availability of the recognition motif and to ensure that, after cleavage, it will act as a strong nucleophile to resolve the probe-LPET-thioester intermediate. The incorporation of 3 Gs (LPETGGG) would have likely behaved similarly. Of note, the sortase reaction is reversible, and addition of the excess amount of substrate can drive the protein labeling to completion.
Synthesis of C-to-N Protein Fusions without any Prior Modifications.
Considering the initial success of the N-terminal labeling approach, we tested the method in forming direct C-to-N protein fusions by mixing two proteins (A and B) and sortase together (Figure 2A). Protein A, in this case a nanobody (single-domain antibody or VHH, ~14 kDa in size), was engineered to contain an LPETG sequence at the C terminus, and protein B, N-terminally modified with LPETGGGGG to fulfill the requirement of an “LPETGn” (n > 2) motif at the N terminus (Figure 2B). Strikingly, the addition of sortase to the mixture of proteins A and B resulted in the formation of the nanobody–GFP fusion with high specificity and yield (>60% in 2 h) (Figure 2C) (reaction condition: GFP (25 μM), VHH (120 μM), 10 mM CaCl2; the reaction was initiated with the addition of immobilized sortase beads (50% slurry) were added to a final 1/10th of the reaction volume; 2 h at 4 °C). The product formation was confirmed by LC-MS and SDS-PAGE (Figure 2B,C). The nanobody (VHH4) used for this protein fusion targets human class II MHC molecules.63 We characterized the VHH4-GFP fusion by performing flow cytometric studies on Nalm-6 cells, a human B cell precursor leukemia cell line, confirming the fusion protein binds to the cells when used in low nM concentration range, suggesting that the approach does not compromise protein characteristics (Figure 2D). As discussed earlier, the sortase reaction is reversible, and thus, the addition of starting substrates VHH4, GFP, or both can increase the yield of the product. Of note, the intermediate thioester is prone to hydrolysis, as a water molecule can act as a nucleophile yet with a very slow rate. However, the hydrolysis is irreversible as it results in the formation of an “X-LPET”, which is no more recognized as a substrate by the enzyme. Therefore, in any sortase-mediated reaction, including the approach developed here, the product will eventually be hydrolyzed, and thus, the reaction must be quenched by deactivating or removing the sortase, for example by addition of EDTA (as sortase activity is calcium-dependent) or purification of the product via size exclusion chromatography.
Figure 2.
One-step synthesis of C to N protein fusions using the sortase-mediated swapping approach. (A) Schematic representation of synthesizing a C-to-N protein fusion via the sortase-swapping approach. (B) A nanobody equipped with an LPETG motif at its C terminus was fused to the GFP equipped with an LPETGGGGG at its N terminus via the sortase-swapping approach. LC-MS analyses confirmed the formation of the product. (C) SDS-PAGE was used to further confirm the formation of products after 2 h of reaction at 4 °C using immobilized sortase beads. (D) Flow cytometry analysis confirmed the functionality of the anti-human class II MHC nanobody after the synthesis of the nanobody–GFP fusion. Jurkat cells are class II MHC-negative, and Nalm6 cells are class II MHC-positive.
One-Step Cleavage and Site-Specific N or C Termini Labeling of Fusion Proteins.
Many proteins are difficult to express recombinantly without the presence of a fusion tag, both in terms of yield and folding status.64–73 Characteristics of fusion tags are their high solubility and stability, high expression yield, rapid and highly efficient folding upon translation, and decreased proteolytic degradation.69,70,74 Examples of fusion tags include glutathione-S-transferase (GST),64 maltose-binding protein (MBP),75,76 and small ubiquitin-like modifier (SUMO).65,66 To obtain the free protein, tags must be removed. Therefore, many cleavage sites have been engineered at the junction of the fusion tags and the desired protein. The corresponding enzymes required for cleavage are commercially available.77 Examples include thrombin (LVPR↓GS), TEV protease (ENLYFQ↓S (G,A)), Factor Xa (I(E or D)GR↓X), and human rhinovirus (HRV) 3C protease (LEVLFQ↓GP).77
This cleavage reaction can pose a challenge of protein fusion technology.78 Common hurdles include labor-intensive optimization of cleavage conditions and precipitation of the protein after cleavage. We hypothesized that the incorporation of a sortase recognition tag in the linker between the two fused proteins can be used not only as a cleavage site79 but also as a landing pad for further site-specific modifications80 (Figure 3A). As a proof-of-concept, we first tried this approach with the LPETG5 N-terminal-engineered GFP. Addition of the sortase and the commercially available Gly3 molecule resulted in rapid and quantitative cleavage of the MH6-LPET motif from the GFP N terminus in less than 30 min (100 μM GFP, 2 μM sortase, 10 mM Gly3 substrate) (Figure 3B,C). The reaction product was confirmed via SDS-PAGE and LC-MS analyses (Figure 3B,C). Of note, and in general, sortase can be used in a wide concentration range relative to the substrate. A typical range is ~0.01 to 0.1 equimolar relative to the “LPETG” substrate. The pentamutant sortase used in this study9 has a Km,LPETG of ~230 μM, a kcat of ~5.4 s−1 (kcat/Km ~23,000 M−1 s−1), and Km,GGG of ~1617 μM.9 For comparison, the kcat/Km for thrombin,81,82 TEV,83 HRV 3C84,85 and Factor Xa86 are ~94,600, ~2620, ~920 and ~39,000 M−1 s−1, respectively.
Figure 3.
Rapid and efficient fusion tag cleavage using sortase. (A) Schematic representation of using sortase as a cleavage enzyme. Incorporation of an LPETG motif between the two proteins would allow sortase to cut the protein between the T and G. (B) Addition of the Gly3 substrate and sortase resulted in rapid and quantitative cleavage of the MH6-LPET- sequence from the GFP’s N terminus; LC-MS analyses confirmed the formation of the product. (C) SDS-PAGE confirmed the formation of the product. (D to H) SUMO-BCMA, GST-BCMA, SUMO-IL2, and SUMO-anti-CD4 scFv fusion proteins were rapidly and quantitatively cleaved via the addition of sortase and Gly3 substrate. SDS-PAGE and LC-MS analyses confirmed the formation of the products.
To further explore the capabilities of this sortase cleavage approach, B cell maturation antigen (BCMA) was selected as the target, a protein often challenging to express. BCMA, also known as tumor necrosis factor receptor superfamily member 17 (TNFRSF17), is a cell surface receptor that recognizes the B cell-activating factor.87,88 Preferentially expressed in mature B lymphocytes, BCMA is thought to play a role in B cell development and autoimmune response.87,89 BCMA chimeric antigen receptor (CAR) T cell therapy (idecabtagene vicleuce) is now FDA-approved and has shown success in treating patients with relapsed or refractory multiple myeloma.90–92 Fluorophore-labeled BCMA (commercially available as a BCMA-Fc fusion) is routinely used to characterize BCMA CAR T cells.
Based on the crystal structure of the BCMA ectodomain,93 we designed and expressed GST-BCMA with a sortase recognition LPETG motif located between the two proteins. Incubating the resulting fusion protein with sortase and Gly3 substrate (~100 μM GST-BCMA, 2 μM sortase, and 10 mM Gly3 substrate) resulted in rapid and quantitative cleavage of BCMA from the GST tag. SDS-PAGE analysis showed near-complete cleavage in less than 60 min (Figure 3F). Three more fusion proteins were synthesized and tested using the same protocol: SUMO-BCMA, SUMO-interleukin-2 (IL2), and SUMO-anti-CD4 single-chain variable fragment (scFv). The addition of sortase and Gly3 substrate resulted in rapid and quantitative cleavage for all protein fusions (Figure 3E,G,H). SDS-PAGE and LC-MS analyses confirmed the formation of the products (Figure 3E,G,H and Figure S1A–C). The cleavage process can also be performed using immobilized sortase on agarose beads at 4 °C as well, making the approach further straightforward (Figure 3F).
It should be possible to use sortase not only to release the protein of interest from the fusion tag but also site-specifically label its N or C terminus (Figure 4). For instance, an additional LPETG motif incorporated at the C terminus of the protein should allow the concurrent sortase-mediated release of the fusion and site-specific C-terminal labeling of the protein of interest (Figure 4A). By incorporating an “LPETGn” (n > 2) sequence in the spacer, the approach can be used for simultaneous cleavage and N-terminal labeling as well (Figure 4B). Thus, this approach not only cleaves the protein of interest from the fusion tag but also site-specifically labels it at its N or C terminus (Figure 4A–B).
Figure 4.
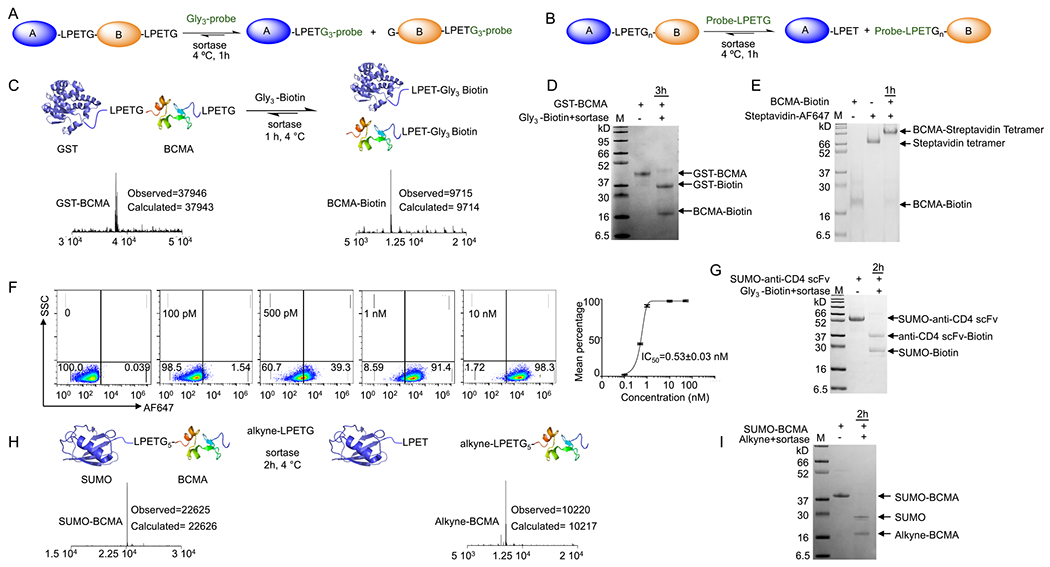
Simultaneous cleavage and site-specific C- or N-terminal protein labeling using sortase. (A,B) Schematic representation of simultaneous cleavage and site-specific C- or N-terminal protein labeling using sortase. (C) GST-BCMA fusion protein was engineered to contain a sortase recognition sequence between the two proteins and at the C terminus; addition of sortase and Gly3-biotin substrate resulted in simultaneous and near-quantitative cleavage and formation of C-terminally biotin-labeled BCMA; LC-MS analyses confirmed the formation of the products. (D) SDS-PAGE confirmed the formation of the products. (E,F) BCMA-biotin was tetramerized using streptavidin. Flow cytometry analyses on BCMA-CAR Jurkat T cells confirmed retention of BCMA functionality (EC50 = 0.53 nM). (G) SUMO-anti-CD4 scFv fusion protein was engineered to contain a sortase recognition sequence between the two proteins and at the C terminus; addition of sortase and Gly3-biotin substrate resulted in simultaneous and near-quantitative cleavage and formation of the C-terminally biotin-labeled anti-CD4 scFv; SDS-PAGE confirmed the formation of the products. (H) SUMO-BCMA fusion with an LPETG4 sequence between the SUMO and BCMA was produced by periplasmic expression in E. coli. Addition of alkyne-functionalized LPETG substrate and sortase yielded near-quantitative cleavage and facilitated N-terminally alkyne-labeled BCMA. (I) SDS-PAGE confirmed the formation of the products.
We first tested sortase-mediated cleavage and C-terminal protein labeling by modifying the GST-BCMA fusion protein by incorporating two LPETG motifs at the N and C terminus of the BCMA. Addition of sortase and a Gly3-biotin substrate to the fusion protein yielded near-quantitative cleavage between the GST and BCMA and resulted in the concomitant formation of C-terminally biotinylated BCMA, as confirmed by SDS-PAGE and LC-MS analyses (~50 μM GST-BCMA, 2 μM sortase, and 2 mM Gly3 substrate) (Figure 44DC,). To test the functionality of the biotinylated BCMA, we reacted BCMA-biotin with streptavidin tetramers to form a tetrameric BCMA. The resulting tetramer stained BCMA Jurkat CAR T cells with an EC50 of ~0.53 nM (calculated for BCMA tetramer), proving conservation of BCMA functionality (Figure 4E,F). We repeated the reaction, following the same protocol, using a SUMO-anti-CD4 scFv with two LPETG motifs at the N and C termini of the scFv. Addition of sortase and Gly3-biotin substrate resulted in near-quantitative cleavage and biotinylation of the anti-CD4 scFv, as confirmed by SDS-PAGE and LC-MS analyses (Figure 4G).
Considering this result, we explored simultaneous cleavage and N-terminal labeling of a protein through design and expression of a SUMO-BCMA fusion with an LPETG4 sequence between the SUMO and BCMA. Addition of the alkyne-functionalized LPETG substrate and sortase to the SUMO-BCMA fusion protein resulted in near-quantitative cleavage and facilitated the production of N-terminally alkyne-labeled BCMA (reaction condition: ~100 μM SUMO-BCMA, 5 μM sortase, 4 mM alkyne substrate, 10 mM CaCl2, 2 h at 4 °C). Product formation was confirmed by SDS-PAGE and LC-MS analyses (Figure 4H,I).
The sortase-mediated concurrent cleavage and site-specific labeling is rapid with excellent yield and does not require extensive optimization. Sortase expression is an easily adopted laboratory technique, where 1 L of the bacterial culture can yield ~20–40 mg of the highly stable enzyme, which can be stored at −20 °C for several years. Of note, sortase can be immobilized on N-hydroxysuccinimide (NHS)-activated agarose beads and remains functional for >1 month at 4 °C.8,56,94 Sortase beads can be used for labeling or cleavage, and addition of more beads can be used as an approach to increase or optimize the rate of the cleavage or labeling. Moreover, the sortase beads can be recovered, washed, and reused for several times. The triglycine (Gly3) nucleophile molecule, needed for cleavage, is commercially available. Furthermore, this approach would only leave one glycine at the N terminus of the protein of interest as the sortase cuts between the T and G in the LPETG recognition motif, whereas many of the widely used cleavage enzymes leave more amino acids at the N terminus after the cut; for example, HRV 3C would leave a GP, and thrombin would leave a GS at the newly exposed N terminus.
CONCLUSIONS
The new sortase-mediated swapping approach introduced here allows the installation of biomolecules at the N terminus in near-quantitative yield and without modifications other than the genetic incorporation of the sortase recognition tag. As sortase is a transpeptidase, the linker between the N terminus and the probe remains to be a native peptide bond. Additionally, it allows efficient and rapid simultaneous cleavage and N- or C-terminal labeling on fusion proteins, a unique feature of the approach. Using orthogonal sortases,9,95 one can use this approach to label both C and N termini of proteins as well, as shown before.9,95 Therefore, this approach can be considered an attractive alternative for N-terminal protein labeling and provide a possibility for simultaneous protein cleavage and N- or C-terminal site-specific labeling.
EXPERIMENTAL PROCEDURES
Cloning and Expression.
The bacterially codon-optimized double-stranded DNA (from either SynBio or IDT companies) for SUMO-IL2, SUMO-BCMA, and SUMO-anti-CD4 scFv, containing about 30 bp overlaps (from each side) with their backbones were synthesized and cloned downstream of the pelB signal sequence of the linearized pHEN6 plasmid by Gibson assembly61 for periplasmic expression. For GFP protein and GST-BCMA fusion protein, similarly, the bacterially codon-optimized double-stranded DNA was synthesized and directly cloned into a linearized pET28a plasmid for bacterial cytosolic expression. Any other modification on these plasmids to add sortase recognition tags (LPETG or LPETGn) at the N and C termini were performed using site-directed mutation. Insertion of fragments and modification of plasmids were verified by DNA Sanger sequencing (at GeneWiz Company). Proteins were designed to have a multihistidine tag for metal affinity purification (sequences available in the SI).
The sequence-verified pHEN6 and pET28a plasmids were transformed into E. coli WK6 and E. coli BL21 (DE3), respectively. For the E. coli BL21 (DE3) expression (GFP and GST-BCMA), transformed colonies were grown at 37 °C in Terrific broth, induced with 0.5 mM IPTG at OD600 of 0.6 for protein expression, and harvested at 6000g for 10 min 4 h after IPTG induction. This was followed by cell lysis and purification to obtain the soluble protein. Briefly, bacterial cell pellets were resuspended in lysis buffer (20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 20 mM imidazole, and 1% Triton X-100 with 1 mM freshly prepared PMSF) followed by lysis using sonication (5 min, 10 s pulses with 10 s in between pulses; 80% amplitude). The lysate was centrifuged at 15,000g for 10 min at 4 °C. The supernatant was harvested and purified by immobilized metal affinity chromatography (IMAC) using a 5 mL HisTrap column preloaded with NiSO4.
For the E. coli WK6 expression (SUMO-IL2, SUMO-BCMA, and SUMO-anti-CD4 scFv), after reaching OD600 of ~0.6–0.8, cells were induced with 0.5 mM IPTG and grown overnight at 30 °C for protein expression. Cells were then harvested by centrifugation (6000g for 10 min), the pellet was resuspended in ice-cold TES buffer (200 mM Tris, 600 μM EDTA, 500 mM sucrose, pH 8) and incubated at 4 °C for 2 h with gentle shaking. This was followed by osmotic shock where the solution was diluted by adding Tris buffer (50 mM Tris, pH 8) (4-fold dilution) and an overnight incubation at 4 °C with gentle shaking. The cell suspension was centrifuged at 6000g for 10 min, and the process was repeated as necessary to obtain a clear supernatant; the supernatant was then collected and further purified by immobilized metal affinity chromatography (IMAC) using a 5 mL HisTrap column preloaded with NiSO4.
Sortase-Mediated Labeling Reactions.
The Penta mutant sortase A with an improved kcat was used for all experiments.62 The reaction mixtures contained 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, ~2–10 μM sortase, and the substrates. When sortase was used as a cleavage enzyme, >5 mM triglycine (Gly3) molecule was used to accelerate the cleavage and achieve quantitative yield. For N-terminal labeling reactions, ~1–2 mM LPETG-containing probe and ~ 50–200 μM LPETGn-containing substrate were used. After incubation at 4 °C with agitation for ~60 min, reaction products were analyzed by LC-MS with yields of generally >80%. When the yield was below 80%, the reaction was allowed to proceed for an additional hour, with the addition of extra 2 μM sortase and a 1 mM LPETG-containing probe; this process can be repeated until a satisfactory yield is obtained, and we could achieve >90% yield for all the reactions discussed in this work. The reactions were quenched by addition of EDTA (25 mM), and the labeled proteins were purified by size exclusion chromatography.
Immobilizing Sortase.
In order to immobilize sortase on beads, we used dried NHS-activated agarose resin (Thermo), which reacts with primary amines to form covalent bonds. Approximately 20 mg of sortase in ~3 mL of HEPES buffer (50 mM, pH 7.5) was added to 150 mg of resin in a 15 mL tube and incubated at 4 °C overnight on a Rotisserie tube rotator. The tube was centrifuged at 1000g for 1 min. The beads were then washed twice with 10 mL of HEPES (50 mM pH 7.5) buffer followed by 30 min incubation in quenching buffer (1 M Tris, pH 7.4) at room temperature. The tube was then centrifuged, and beads were washed as previously mentioned. The washed sortase-immobilized beads were stored upright at 4 °C in HEPES buffer (50 mM pH 7.5; containing at least 0.5 mL of buffer above the resin). The flowthrough and washes can be used to determine coupling efficiency by measuring protein absorption and SDS-PAGE analyses. The immobilized sortase beads remain functional for at least several weeks.
Liquid Chromatography–Mass Spectroscopy (LC-MS).
LC-MS analyses were performed using a Waters LC-MS system consisting of an ACQUITY Arc Sample Manager FTNR coupled with a Waters 2489 UV–Vis Detector and QDa mass detector. Reverse-phase chromatography was performed using an XBridge Protein BEH C4 column, 300A, 3.5 μm 2.1 × 50 mm, 10–500 K. The LC method used was a gradient of water (0.1% formic acid)-acetonitrile (0.1% formic acid) 95–5 to 20–80 over 3.5 min, followed by 1.5 min of 95–5. Concentrations of samples ranged from 30–100 μg/mL of protein in HEPES buffer (50 mM HEPES, with 150 mM NaCl, pH 7.5); 25 μL of each sample was injected into the instrument. The mass spectroscopy was performed in positive mode using electrospray ionization (ESI), using a 600–1250 m/z detection range. The data were acquired and analyzed using MassLynx Software. The total ion current peaks corresponding to the proteins were selected for MS analyses. The mass spectra were then deconvoluted to obtain the parent protein mass. The deconvoluted mass spectra were then plotted using GraphPad Prism 8.
Flow Cytometry.
The binding capacity of VHH4-GFP and BCMA-biotin-streptavidin-Alexa Fluor 647 fusion proteins to Nalm6 cells and anti-BCMA Jurkat CAR T cells, respectively, was tested using flow cytometry analysis. The cultured cells were washed with PBS + 5% BSA solution twice by centrifuging at 350g for 5 min to remove the culture media. Cells were then resuspended in blocking solution (PBS + 5% BSA), and cell count and viability were determined by mixing with Trypan blue and analyzing using a Countess II cell counter. Approximately 1 million live cells were used for each staining. Fluorophore-conjugated proteins were diluted to concentration as mentioned in the figures in blocking solution and incubated with cells for 30 min on ice. Cells were then washed with PBS twice and resuspended in 200 μL PBS. An Accuri (BD) flow cytometer system and FlowJo software (TreeStar) were used for cytometer analysis. A total of 30,000 cells were collected for each sample to enable data normalization.
Supplementary Material
ACKNOWLEDGMENTS
Sortase 5M and VHH4 plasmids were gifts from the Dr. Hidde Ploegh laboratory of the Boston Children’s Hospital. We thank Christopher Podracky for helpful discussions. We thank Omar Abousaway for technical assistance throughout the study. BCMA Jurkat Cells was provided by the Dr. Eric Smith laboratory of the Dana-Farber Cancer Institute. This work was supported by an Innovation Research Fund Basic Research Award from the Dana-Farber Cancer Institute (M.R.) and NIH-K22 award NIH-K22CA226040 (M.R.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.1c00442.
More details on proteins and peptide substrates characterization and sequences of recombinant proteins produced in this study (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.1c00442
The authors declare no competing financial interest.
Contributor Information
Min Cong, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Soheil Tavakolpour, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Lea Berland, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States; Université Côte d’Azur, CNRS, INSERM, IRCAN, 06100 Nice, France.
Hannah Glöckner, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Bohdan Andreiuk, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Taha Rakhshandehroo, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Safak Uslu, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States; Medical Scientist Training Program, Hacettepe University Faculty of Medicine, Ankara 06230, Turkey.
Shruti Mishra, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Louise Clark, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Mohammad Rashidian, Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States; Department of Radiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02215, United States.
REFERENCES
- (1).Bertozzi CR A Decade of Bioorthogonal Chemistry. Acc. Chem. Res 2011, 44, 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- (3).Rashidian M; Dozier JK; Distefano MD Enzymatic Labeling of Proteins: Techniques and Approaches. Bioconjugate Chem. 2013, 24, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Junutula JR; Raab H; Clark S; Bhakta S; Leipold DD; Weir S; Chen Y; Simpson M; Tsai SP; Dennis MS; Lu Y; Meng YG; Ng C; Yang J; Lee CC; Duenas E; Gorrell J; Katta V; Kim A; McDorman K; Flagella K; Venook R; Ross S; Spencer SD; Lee Wong W; Lowman HB; Vandlen R; Sliwkowski MX; Scheller RH; Polakis P; Mallet W Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol 2008, 26, 925–932. [DOI] [PubMed] [Google Scholar]
- (5).Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (6).Woodham AW; Cheloha RW; Ling J; Rashidian M; Kolifrath SC; Mesyngier M; Duarte JN; Bader JM; Skeate JG; Da Silva DM; Kast WM; Ploegh HL Nanobody-Antigen Conjugates Elicit HPV-Specific Antitumor Immune Responses. Cancer Immunol. Res 2018, 6, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hinner MJ; Johnsson K How to Obtain Labeled Proteins and What to Do with Them. Curr. Opin. Biotechnol 2010, 21, 766–776. [DOI] [PubMed] [Google Scholar]
- (8).Guimaraes CP; Witte MD; Theile CS; Bozkurt G; Kundrat L; Blom AEM; Ploegh HL Site-Specific C-Terminal and Internal Loop Labeling of Proteins Using Sortase-Mediated Reactions. Nat. Protoc 2013, 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dorr BM; Ham HO; An C; Chaikof EL; Liu DR Reprogramming the Specificity of Sortase Enzymes. Proc. Natl. Acad. Sci 2014, 111, 13343–13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Duckworth BP; Zhang Z; Hosokawa A; Distefano MD Selective Labeling of Proteins by Using Protein Farnesyltransferase. ChemBioChem 2007, 8, 98–105. [DOI] [PubMed] [Google Scholar]
- (11).Rashidian M; Song JM; Pricer RE; Distefano MD Chemoenzymatic Reversible Immobilization and Labeling of Proteins without Prior Purification. J. Am. Chem. Soc 2012, 134, 8455–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rush JS; Bertozzi CR New Aldehyde Tag Sequences Identified by Screening Formylglycine Generating Enzymes in Vitro and in Vivo. J. Am. Chem. Soc 2008, 130, 12240–12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen I; Howarth M; Lin W; Ting AY Site-Specific Labeling of Cell Surface Proteins with Biophysical Probes Using Biotin Ligase. Nat. Methods 2005, 2, 99–104. [DOI] [PubMed] [Google Scholar]
- (14).Cohen JD; Zou P; Ting AY Site-Specific Protein Modification Using Lipoic Acid Ligase and Bis-Aryl Hydrazone Formation. ChemBioChem 2012, 13, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Abe H; Goto M; Kamiya N Protein Lipidation Catalyzed by Microbial Transglutaminase. Chem. – Eur. J 2011, 17, 14004–14008. [DOI] [PubMed] [Google Scholar]
- (16).Fontana A; Spolaore B; Mero A; Veronese FM Site-Specific Modification and PEGylation of Pharmaceutical Proteins Mediated by Transglutaminase. Adv. Drug Delivery Rev 2008, 60, 13–28. [DOI] [PubMed] [Google Scholar]
- (17).Tanaka T; Kamiya N; Nagamune T N-Terminal Glycine-Specific Protein Conjugation Catalyzed by Microbial Transglutaminase. FEBS Lett. 2005, 579, 2092–2096. [DOI] [PubMed] [Google Scholar]
- (18).Lin C-W; Ting AY Transglutaminase-Catalyzed Site-Specific Conjugation of Small-Molecule Probes to Proteins in Vitro and on the Surface of Living Cells. J. Am. Chem. Soc 2006, 128, 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wong LS; Thirlway J; Micklefield J Direct Site-Selective Covalent Protein Immobilization Catalyzed by a Phosphopantetheinyl Transferase. J. Am. Chem. Soc 2008, 130, 12456–12464. [DOI] [PubMed] [Google Scholar]
- (20).Zhou Z; Cironi P; Lin AJ; Xu Y; Hrvatin S; Golan DE; Silver PA; Walsh CT; Yin J Genetically Encoded Short Peptide Tags for Orthogonal Protein Labeling by Sfp and AcpS Phosphopantetheinyl Transferases. ACS Chem. Biol 2007, 2, 337–346. [DOI] [PubMed] [Google Scholar]
- (21).Yin J; Straight PD; McLoughlin SM; Zhou Z; Lin AJ; Golan DE; Kelleher NL; Kolter R; Walsh CT Genetically Encoded Short Peptide Tag for Versatile Protein Labeling by Sfp Phosphopantetheinyl Transferase. Proc. Natl. Acad. Sci 2005, 102, 15815–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yu H; Huang S; Chokhawala H; Sun M; Zheng H; Chen X Highly Efficient Chemoenzymatic Synthesis of Naturally Occurring and Non-Natural α-2,6-Linked Sialosides: AP. Damsela α-2,6-Sialyltransferase with Extremely Flexible Donor–Substrate Specificity. Angew. Chem., Int. Ed 2006, 45, 3938–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wu ZL; Person AD; Burton AJ; Singh R; Burroughs B; Fryxell D; Tatge TJ; Manning T; Wu G; Swift KAD; Kalabokis V Direct Fluorescent Glycan Labeling with Recombinant Sialyltransferases. Glycobiology 2019, 29, 750–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Steffen W; Ko FC; Patel J; Lyamichev V; Albert TJ; Benz J; Rudolph MG; Bergmann F; Streidl T; Kratzsch P; Boenitz-Dulat M; Oelschlaegel T; Schraeml M Discovery of a Microbial Transglutaminase Enabling Highly Site-Specific Labeling of Proteins. J. Biol. Chem 2017, 292, 15622–15635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Heal WP; Wickramasinghe SR; Bowyer PW; Holder AA; Smith DF; Leatherbarrow RJ; Tate EW Site-Specific N-Terminal Labelling of Proteins in Vitro and in Vivo Using N-Myristoyl Transferase and Bioorthogonal Ligation Chemistry. Chem. Commun 2008, 4, 480. [DOI] [PubMed] [Google Scholar]
- (26).Chen PR; Groff D; Guo J; Ou W; Cellitti S; Geierstanger BH; Schultz PG A Facile System for Encoding Unnatural Amino Acids in Mammalian Cells. Angew. Chem., Int. Ed 2009, 48, 4052–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fleissner MR; Brustad EM; Kalai T; Altenbach C; Cascio D; Peters FB; Hideg K; Peuker S; Schultz PG; Hubbell WL Site-Directed Spin Labeling of a Genetically Encoded Unnatural Amino Acid. Proc. Natl. Acad. Sci 2009, 106, 21637–21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Guo J; Melançon CE III; Lee HS; Groff D; Schultz PG Evolution of Amber Suppressor TRNAs for Efficient Bacterial Production of Proteins Containing Nonnatural Amino Acids. Angew. Chem., Int. Ed 2009, 48, 9148–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Seitchik JL; Peeler JC; Taylor MT; Blackman ML; Rhoads TW; Cooley RB; Refakis C; Fox JM; Mehl RA Genetically Encoded Tetrazine Amino Acid Directs Rapid Site-Specific in Vivo Bioorthogonal Ligation with Trans -Cyclooctenes. J. Am. Chem. Soc 2012, 134, 2898–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Reyes-Turcu FE; Horton JR; Mullally JE; Heroux A; Cheng X; Wilkinson KD The Ubiquitin Binding Domain ZnF UBP Recognizes the C-Terminal Diglycine Motif of Unanchored Ubiquitin. Cell 2006, 124, 1197–1208. [DOI] [PubMed] [Google Scholar]
- (31).Schwede A; Jones N; Engstler M; Carrington M The VSG C-Terminal Domain Is Inaccessible to Antibodies on Live Trypanosomes. Mol. Biochem. Parasitol 2011, 175, 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sharma S; Schiller MR The Carboxy-Terminus, a Key Regulator of Protein Function. Crit. Rev. Biochem. Mol. Biol 2019, 54, 85–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rosen CB; Francis MB Targeting the N Terminus for Site-Selective Protein Modification. Nat. Chem. Biol 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- (34).De Rosa L; Di Stasi R; Romanelli A; D’Andrea LD Exploiting Protein N-Terminus for Site-Specific Bioconjugation. Molecules 2021, 26, 3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Purushottam L; Adusumalli SR; Singh U; Unnikrishnan VB; Rawale DG; Gujrati M; Mishra RK; Rai V Single-Site Glycine-Specific Labeling of Proteins. Nat. Commun 2019, 10, 2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen D; Disotuar MM; Xiong X; Wang Y; Chou DH-C Selective N-Terminal Functionalization of Native Peptides and Proteins. Chem. Sci 2017, 8, 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Deng J-R; Lai NC-H; Kung KK-Y; Yang B; Chung S-F; Leung AS-L; Choi M-C; Leung Y-C; Wong M-K N-Terminal Selective Modification of Peptides and Proteins Using 2-Ethynylbenzaldehydes. Commun. Chem 2020, 3, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Raj M; Wu H; Blosser SL; Vittoria MA; Arora PS Aldehyde Capture Ligation for Synthesis of Native Peptide Bonds. J. Am. Chem. Soc 2015, 137, 6932–6940. [DOI] [PubMed] [Google Scholar]
- (39).MacDonald JI; Munch HK; Moore T; Francis MB One-Step Site-Specific Modification of Native Proteins with 2-Pyridinecarboxyaldehydes. Nat. Chem. Biol 2015, 11, 326–331. [DOI] [PubMed] [Google Scholar]
- (40).Chan AO-Y; Ho C-M; Chong H-C; Leung Y-C; Huang J-S; Wong M-K; Che C-M Modification of N-Terminal α-Amino Groups of Peptides and Proteins Using Ketenes. J. Am. Chem. Soc 2012, 134, 2589–2598. [DOI] [PubMed] [Google Scholar]
- (41).Schoffelen S; van Eldijk MB; Rooijakkers B; Raijmakers R; Heck AJR; van Hest JCM Metal-Free and PH-Controlled Introduction of Azides in Proteins. Chem. Sci 2011, 2, 701. [Google Scholar]
- (42).De Rosa L; Di Stasi R; Longhitano L; D’Andrea LD Labeling of VEGFR1D2 through Oxime Ligation. Bioorg. Chem 2019, 91, 103160. [DOI] [PubMed] [Google Scholar]
- (43).Dawson P; Muir T; Clark-Lewis I; Kent S Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- (44).Johnson ECB; Kent SBH Insights into the Mechanism and Catalysis of the Native Chemical Ligation Reaction. J. Am. Chem. Soc 2006, 128, 6640–6646. [DOI] [PubMed] [Google Scholar]
- (45).Nguyen GKT; Cao Y; Wang W; Liu CF; Tam JP Site-Specific N-Terminal Labeling of Peptides and Proteins using Butelase|1 and Thiodepsipeptide. Angew. Chem., Int. Ed 2015, 54, 15694–15698. [DOI] [PubMed] [Google Scholar]
- (46).Harmand TJ; Bousbaine D; Chan A; Zhang X; Liu DR; Tam JP; Ploegh HL One-Pot Dual Labeling of IgG 1 and Preparation of C-to-C Fusion Proteins Through a Combination of Sortase A and Butelase 1. Bioconjugate Chem. 2018, 29, 3245–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Weeks AM; Wells JA N-Terminal Modification of Proteins with Subtiligase Specificity Variants. Curr. Protoc. Chem. Biol 2020, 12, e79. [DOI] [PubMed] [Google Scholar]
- (48).Weeks AM; Wells JA Subtiligase-Catalyzed Peptide Ligation. Chem. Rev 2020, 120, 3127–3160. [DOI] [PubMed] [Google Scholar]
- (49).Yoshihara HAI; Mahrus S; Wells JA Tags for Labeling Protein N-Termini with Subtiligase for Proteomics. Bioorg. Med. Chem. Lett 2008, 18, 6000–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Weeks AM; Wells JA Engineering Peptide Ligase Specificity by Proteomic Identification of Ligation Sites. Nat. Chem. Biol 2018, 14, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nguyen GKT; Wang S; Qiu Y; Hemu X; Lian Y; Tam JP Butelase 1 Is an Asx-Specific Ligase Enabling Peptide Macrocyclization and Synthesis. Nat. Chem. Biol 2014, 10, 732–738. [DOI] [PubMed] [Google Scholar]
- (52).Mazmanian SK; Liu G; Ton-That H; Schneewind O Staphylococcus Aureus Sortase, an Enzyme That Anchors Surface Proteins to the Cell Wall. Science 1999, 285, 760–763. [DOI] [PubMed] [Google Scholar]
- (53).Hirel PH; Schmitter MJ; Dessen P; Fayat G; Blanquet S Extent of N-Terminal Methionine Excision from Escherichia Coli Proteins Is Governed by the Side-Chain Length of the Penultimate Amino Acid. Proc Natl. Acad. Sci 1989, 86, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Vassileva-Atanassova A; Mironova R; Nacheva G; Ivanov I N-Terminal Methionine in Recombinant Proteins Expressed in Two Different Escherichia Coli Strains. J. Biotechnol 1999, 69, 63–67. [DOI] [PubMed] [Google Scholar]
- (55).Wingfield PT N-Terminal Methionine Processing. Curr. Protoc. Protein Sci 2017, 88, 6.14.1–6.14.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Theile CS; Witte MD; Blom AEM; Kundrat L; Ploegh HL; Guimaraes CP Site-Specific N-Terminal Labeling of Proteins Using Sortase-Mediated Reactions. Nat. Protoc 2013, 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sarpong K; Bose R Efficient Sortase-Mediated N-Terminal Labeling of TEV Protease Cleaved Recombinant Proteins. Anal. Biochem 2017, 521, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Williamson DJ; Fascione MA; Webb ME; Turnbull WB Efficient N-Terminal Labeling of Proteins by Use of Sortase. Angew. Chem., Int. Ed 2012, 51, 9377–9380. [DOI] [PubMed] [Google Scholar]
- (59).Rehm FBH; Harmand TJ; Yap K; Durek T; Craik DJ; Ploegh HL Site-Specific Sequential Protein Labeling Catalyzed by a Single Recombinant Ligase. J. Am. Chem. Soc 2019, 141, 17388–17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Harris KS; Guarino RF; Dissanayake RS; Quimbar P; McCorkelle OC; Poon S; Kaas Q; Durek T; Gilding EK; Jackson MA; Craik DJ; van der Weerden NL; Anders RF; Anderson MA A Suite of Kinetically Superior AEP Ligases Can Cyclise an Intrinsically Disordered Protein. Sci. Rep 2019, 9, 10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Huang X; Aulabaugh A; Ding W; Kapoor B; Alksne L; Tabei K; Ellestad G Kinetic Mechanism of Staphylococcus Aureus Sortase SrtA. Biochemistry 2003, 42, 11307–11315. [DOI] [PubMed] [Google Scholar]
- (62).Chen I; Dorr BM; Liu DR A General Strategy for the Evolution of Bond-Forming Enzymes Using Yeast Display. Proc. Natl. Acad. Sci 2011, 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Van Elssen CHMJ; Rashidian M; Vrbanac V; Wucherpfennig KW; Habre ZE; Sticht J; Freund C; Jacobsen JT; Cragnolini J; Ingram J; Plaisier L; Spierings E; Tager AM; Ploegh HL Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J. Nucl. Med 2017, 58, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Harper S; Speicher DW Expression and Purification of GST Fusion Proteins. In Current Protocols in Protein Science; Coligan JE; Dunn BM; Speicher DW; Wingfield PT Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 6.6.1–6.6.26, DOI: 10.1002/0471140864.ps0606s52. [DOI] [Google Scholar]
- (65).Butt TR; Edavettal SC; Hall JP; Mattern MR SUMO Fusion Technology for Difficult-to-Express Proteins. Protein Expression Purif. 2005, 43, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Marblestone JG Comparison of SUMO Fusion Technology with Traditional Gene Fusion Systems: Enhanced Expression and Solubility with SUMO. Protein Sci. 2006, 15, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chatterjee DK; Esposito D Enhanced Soluble Protein Expression Using Two New Fusion Tags. Protein Expression Purif. 2006, 46, 122–129. [DOI] [PubMed] [Google Scholar]
- (68).Costa S; Almeida A; Castro A; Domingues L Fusion Tags for Protein Solubility, Purification and Immunogenicity in Escherichia Coli: The Novel Fh8 System. Front. Microbiol 2014, 5, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Ki M-R; Pack SP Fusion Tags to Enhance Heterologous Protein Expression. Appl Microbiol. Biotechnol 2020, 104, 2411–2425. [DOI] [PubMed] [Google Scholar]
- (70).Kosobokova EN; Skrypnik KA; Kosorukov VS Overview of Fusion Tags for Recombinant Proteins. Biochemistry (Moscow) 2016, 81, 187–200. [DOI] [PubMed] [Google Scholar]
- (71).Zhao X; Li G; Liang S Several Affinity Tags Commonly Used in Chromatographic Purification. J. Anal. Methods Chem 2013, 2013, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Uhlén M; Forsberg G; Moks T; Hartmanis M; Nilsson B Fusion Proteins in Biotechnology. Curr. Opin. Biotechnol 1992, 3, 363–369. [DOI] [PubMed] [Google Scholar]
- (73).Kamihira M; Ono K; Esaka K; Nishijima K; Kigaku R; Komatsu H; Yamashita T; Kyogoku K; Iijima S High-Level Expression of Single-Chain Fv-Fc Fusion Protein in Serum and Egg White of Genetically Manipulated Chickens by Using a Retroviral Vector. J. Virol 2005, 79, 10864–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Esposito D; Chatterjee DK Enhancement of Soluble Protein Expression through the Use of Fusion Tags. Curr. Opin. Biotechnol 2006, 17, 353–358. [DOI] [PubMed] [Google Scholar]
- (75).Pryor KD; Leiting B High-Level Expression of Soluble Protein InEscherichia ColiUsing a His6-Tag and Maltose-Binding-Protein Double-Affinity Fusion System. Protein Expression Purif. 1997, 10, 309–319. [DOI] [PubMed] [Google Scholar]
- (76).Fox JD Single Amino Acid Substitutions on the Surface of Escherichia Coli Maltose-Binding Protein Can Have a Profound Impact on the Solubility of Fusion Proteins. Protein Sci. 2001, 10, 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Waugh DS An Overview of Enzymatic Reagents for the Removal of Affinity Tags. Protein Expression Purif. 2011, 80, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Pina AS; Lowe CR; Roque ACA Challenges and Opportunities in the Purification of Recombinant Tagged Proteins. Biotechnol. Adv 2014, 32, 366–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Mao H A Self-Cleavable Sortase Fusion for One-Step Purification of Free Recombinant Proteins. Protein Expression Purif. 2004, 37, 253–263. [DOI] [PubMed] [Google Scholar]
- (80).Warden-Rothman R; Caturegli I; Popik V; Tsourkas A Sortase-Tag Expressed Protein Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step. Anal. Chem 2013, 85, 11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Chang J-Y Thrombin Specificity. Requirement for Apolar Amino Acids Adjacent to the Thrombin Cleavage Site of Polypeptide Substrate. Eur. J. Biochem 1985, 151, 217–224. [DOI] [PubMed] [Google Scholar]
- (82).Pozsgay M; Cs Szabó G; Elödi P; Gáspár R; Bajusz S; Simonsson R Study of the Specificity of Thrombin with Tripeptidyl-p-Nitroanilide Substrates. Eur. J. Biochem 1981, 115, 491–495. [DOI] [PubMed] [Google Scholar]
- (83).Stols L; Gu M; Dieckman L; Raffen R; Collart FR; Donnelly MI A New Vector for High-Throughput, Ligation-Independent Cloning Encoding a Tobacco Etch Virus Protease Cleavage Site. Protein Expression Purif. 2002, 25, 8–15. [DOI] [PubMed] [Google Scholar]
- (84).Walker PA; Leong LE-C; Ng PWP; Tan SH; Waller S; Murphy D; Porter AG Efficient and Rapid Affinity Purification of Proteins Using Recombinant Fusion Proteases. Biotechnology 1994, 12, 601–605. [DOI] [PubMed] [Google Scholar]
- (85).Wang QM; Johnson RB; Cox GA; Villarreal EC; Loncharich RJ A Continuous Colorimetric Assay for Rhinovirus-14 3C Protease Using Peptide p-Nitroanilides as Substrates. Anal. Biochem 1997, 252, 238–245. [DOI] [PubMed] [Google Scholar]
- (86).Cho K; Tanaka T; Cook RR; Kisiel W; Fujikawa K; Kurachi K; Powers JC Active-Site Mapping of Bovine and Human Blood Coagulation Serine Proteases Using Synthetic Peptide 4-Nitroanilide and Thio Ester Substrates. Biochemistry 1984, 23, 644–650. [DOI] [PubMed] [Google Scholar]
- (87).Bossen C; Schneider P BAFF, APRIL and Their Receptors: Structure, Function and Signaling. Semin. Immunol 2006, 18, 263–275. [DOI] [PubMed] [Google Scholar]
- (88).Schiemann B; Gommerman JL; Vora K; Cachero TG; Shulga-Morskaya S; Dobles M; Frew E; Scott ML An Essential Role for BAFF in the Normal Development of B Cells Through a BCMA-Independent Pathway. Science 2001, 293, 2111–2114. [DOI] [PubMed] [Google Scholar]
- (89).Gross JA; Johnston J; Mudri S; Enselman R; Dillon SR; Madden K; Xu W; Parrish-Novak J; Foster D; Lofton-Day C; Moore M; Littau A; Grossman A; Haugen H; Foley K; Blumberg H; Harrison K; Kindsvogel W; Clegg CH TACI and BCMA Are Receptors for a TNF Homologue Implicated in B-Cell Autoimmune Disease. Nature 2000, 404, 995–999. [DOI] [PubMed] [Google Scholar]
- (90).Markham A Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [DOI] [PubMed] [Google Scholar]
- (91).Raje N; Berdeja J; Lin Y; Siegel D; Jagannath S; Madduri D; Liedtke M; Rosenblatt J; Maus MV; Turka A; Lam L-P; Morgan RA; Friedman K; Massaro M; Wang J; Russotti G; Yang Z; Campbell T; Hege K; Petrocca F; Quigley MT; Munshi N; Kochenderfer JN Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med 2019, 380, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Mailankody S; Jakubowiak AJ; Htut M; Costa LJ; Lee K; Ganguly S; Kaufman JL; Siegel DSD; Bensinger W; Cota M; Doerr T; DeVries T; Wong SWK Orvacabtagene Autoleucel (Orva-Cel), a B-Cell Maturation Antigen (BCMA)-Directed CAR T Cell Therapy for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Update of the Phase 1/2 EVOLVE Study (NCT03430011). JCO 2020, 38, 8504–8504. [Google Scholar]
- (93).Liu Y; Hong X; Kappler J; Jiang L; Zhang R; Xu L; Pan C-H; Martin WE; Murphy RC; Shu H-B; Dai S; Zhang G Ligand–Receptor Binding Revealed by the TNF Family Member TALL-1. Nature 2003, 423, 49–56. [DOI] [PubMed] [Google Scholar]
- (94).Witte MD; Theile CS; Wu T; Guimaraes CP; Blom AEM; Ploegh HL Production of Unnaturally Linked Chimeric Proteins Using a Combination of Sortase-Catalyzed Transpeptidation and Click Chemistry. Nat. Protoc 2013, 8, 1808–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Podracky CJ; An C; DeSousa A; Dorr BM; Walsh DM; Liu DR Laboratory Evolution of a Sortase Enzyme That Modifies Amyloid-β Protein. Nat. Chem. Biol 2021, 17, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.