

Abstract
Histone deacetylases (HDACs) play critical roles in epigenomic regulation and histone acetylation is dysregulated in many human cancers. While HDAC inhibitors are active in T-cell lymphomas, poor isoform selectivity, narrow therapeutic indices and a deficiency of reliable biomarkers may contribute to the lack of efficacy in solid tumors. In this article, we report the discovery and preclinical development of the novel, orally bioavailable, class I-selective HDAC inhibitor, OKI-179. OKI-179 and its cell active predecessor OKI-005 are thioester prodrugs of the active metabolite OKI-006, a unique congener of the natural product HDAC inhibitor largazole. OKI-006, OKI-005 and subsequently OKI-179, were developed through a lead candidate optimization program designed to enhance physiochemical properties without eroding potency and selectivity relative to largazole. OKI-005 displays anti-proliferative activity in vitro with induction of apoptosis and increased histone acetylation, consistent with target engagement. OKI-179 demonstrated anti-tumor activity in preclinical cancer models with a favorable pharmacokinetic profile and on-target pharmacodynamic effects. Based on its potency, desirable class I HDAC inhibition profile, oral bioavailability, and efficacy against a broad range of solid tumors, OKI-179 is currently being evaluated in a first-in-human phase I clinical trial with plans for continued clinical development in solid tumor and hematologic malignancies.
Keywords: OKI-179, histone deacetylase inhibitor, triple-negative breast cancer, colon cancer
Graphical Abstract
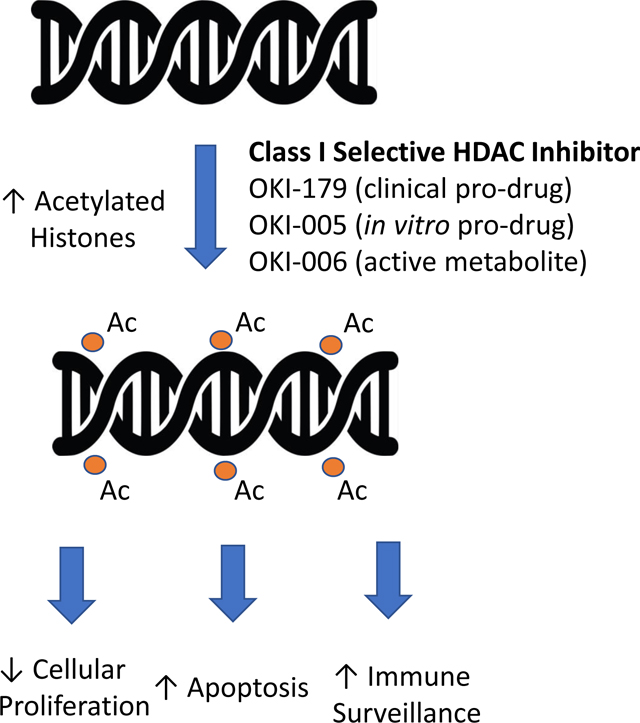
Introduction
Epigenetic modifications result in changes in gene expression without alteration of DNA sequences and play a key role in cancer progression and resistance to cytotoxic and hormonal cancer therapies [1–3]. The activity of epigenetic regulators, such as histone proteins, is manipulated by enzymatic changes, therefore, activity can be altered with pharmacologic inhibitors of specific enzymes [4]. Histone acetylation is one mechanism of epigenetic regulation based on the ability of histones to control chromatin organization and switch DNA structure from un-coiled (available for transcription) to densely packed in nucleosomes (unavailable for transcription) and vice versa [4, 5].
Global dysregulation of histone acetylation correlates with tumorigenesis and cancer progression in many hematologic malignancies and solid tumors [1, 6]. Histone acetylation is managed by histone acetyltransferases and histone deacetylases (HDACs) that enzymatically add or remove acetyl groups from lysine residues on amino-terminal tails of histones, respectively [5]. Acetylation of histones results in increased transcription making HDACs silencers of gene expression. HDAC expression is elevated in many tumor types, including breast and colorectal cancer (CRC) where overexpression is associated with inferior disease-free and overall survival [1, 7–10].
Human HDACs can be grouped as zinc-dependent [Class I (1, 2, 3 and 8), II (4– 7, 9–10), and IV (11)] and NAD-dependent (class III) [11]. Class I HDACs localize to the nucleus where they are ubiquitously expressed in human tissues [1]. Knock-down of class I HDACs leads to cell cycle arrest, decreased cell viability and increased apoptosis in preclinical models of many different solid and hematologic cancers, making class I HDAC inhibitors promising anti-cancer agents [1, 12]. Pharmacologic HDAC inhibitors are active in preclinical models of multiple tumor types where treatment leads to decreases cell proliferation and increased apoptosis [13–15]. HDAC inhibitors also lead to decreased angiogenesis, increased cancer cell differentiation and enhanced cancer cell immunogenicity, including through upregulation of MHC class I and II and natural killer cell activation [16, 17]. Many other nonhistone targets of HDACs have been identified, however, the physiologic ramifications of these modifications are not completely characterized [18].
There are currently four FDA-approved HDAC inhibitors (vorinostat, romidepsin, belinostat and panobinostat) for the treatment of cutaneous T-cell lymphoma (CTCL) and/or peripheral T-cell lymphoma (PTCL) and multiple myeloma (panobinostat) [1, 19, 20]. While active in a subset of hematologic malignancies, the successful clinical development of HDAC inhibitors in solid tumors is perceived to be limited by potency, isoform selectivity, narrow therapeutic indices and/or a non-oral route of administration (romidepsin and belinostat). The natural product romidepsin has attractive potency and selectivity, but to date attempts to engender oral bioavailability have failed. Similar to romidepsin, largazole is a natural product with potent class I-selective HDAC inhibition. While largazole has promising antitumor activity in preclinical solid tumor cancer models, it is not optimized for oral administration [21–23]. Therefore, we initiated a multidisciplinary program with a goal to optimize novel largazole analogs discovered previously [24, 25] with superior drug-like properties and expand clinical applications towards both hematological and non-hematological malignancies.
Here, we report the structures of largazole-derivatives OKI-006, OKI-005 and OKI-179. OKI-005 and OKI-179 are prodrugs metabolized in cells and in vivo, respectively, to OKI-006 which selectively inhibits class I HDACs with no significant inhibition of class IIa HDACs [26]. We present the pharmacokinetic and pharmacodynamic profiles of these drugs in vitro and in vivo, and efficacy studies in preclinical models of CRC and breast cancer. Based on the results presented herein, OKI-179 was selected as a clinical development candidate and is currently being evaluated in a phase I dose-escalation clinical trial (NCT03931681, Graphical Abstract).
Material and Methods
Drugs
OKI-005 was dissolved in dimethyl sulfoxide solution (DMSO) to make a 10 mM stock solution in media for in vitro experiments. OKI-005 was dissolved in corn oil, OKI-179 was dissolved in 0.1M citric acid and entinostat was dissolved in 0.5% carboxymethylcellulose sodium (CMC-Na) for in vivo experiments. 4-hydroxytamoxifen was dissolved in DMSO to make a 30 mM stock solution for in vitro experiments and tamoxifen was dissolved in peanut oil for in vivo experiments.
Murine in vivo pharmacokinetic and colorectal and breast cancer xenograft studies
For pharmacokinetic studies, male BALB/c nude mice (6–8 weeks) were inoculated subcutaneously in the right flank with HCT-116 cells (5 × 106) in 0.1 ml phosphate-buffered saline (PBS). When average tumor size reached 200 mm3, mice were treated with a single dose of OKI-005 10, 30, 100 mg/kg by oral gavage (PO) or 5 mg/kg intraperitoneal (IP) and plasma samples were obtained at 0.5, 1, 2, 4 and 8 hours post-dose. Animals were euthanized and tumor samples were obtained at the same timepoints and 100 mg of tumor tissue was flash frozen for pharmacokinetic (PK) analysis. Plasma and tumor concentrations of the metabolite OKI-006 were determined by liquid chromatography mass spectrometry in at least 3 individual mice/tumors and concentration-time curves were generated.
For OKI-005 efficacy studies, when average tumor size reached 200 mm3, BALB/c nude mice were randomized to treatment groups vehicle control (corn oil), OKI-005 10, 30, or 100 mg/kg PO, OKI-005 5 mg/kg IP or entinostat 20 mg/kg PO daily × 14 days. Body weight was monitored daily and dose was reduced in the entire treatment group by 50% for > 8% body weight loss and stopped for > 15%. To determine the effect of OKI-005 on tumor metastasis, luciferase-labeled MDA-MB-231 cells (1×106/injection) were engrafted into the mammary fat pad and BALB/c mice were randomized to vehicle or OKI-005 10 mg/kg IP three times per week beginning 5 weeks post-engraftment. Bioluminescent imaging was used to quantify pulmonary metastasis in the two groups. Briefly, mice were injected with D-luciferin, anesthetized with isofluorane, and imaged 5 min after injection using the In Vivo Imaging System (IVIS) Spectrum (Perkin Elmer). For OKI-179 efficacy studies, when average tumor size reached 2003 mm, mice were randomized to vehicle control (0.1 M citric acid), OKI-179 40 or 80 mg/kg PO daily or 120 mg PO every other day. Tumor size was measured twice weekly using calipers and tumor volume was calculated (V = (length × width2) × 0.52). Tumor samples were collected for pharmacodynamic analysis from nude mice with MDA-MB-231 implanted tumors treated with OKI-179 40 mg/kg, 60 mg/kg or 80 mg/kg for 3–10 days. A one-way ANOVA was performed to compare tumor volume and tumor weight among groups and comparisons between groups were carried out with Games-Howell test using SPSS 17.0.
Cancer cell lines and proliferation assays
Human TNBC cell lines MDA-MB-231, MDA-MB-436, MDA-MB-157, HCC1806, HCC1937, Hs578T, HCC38, BT549, HCC1187, SW527, HCC1143 and HCC1395 were obtained from American Type Culture Collection (ATCC, Manassas, VA). CAL-51, CAL-85–1, CAL-120, CAL-148 and HDQ-P1 were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Braunschweig, Germany). MDA-MB-468, BT20, and HCC70 were obtained from the University of Colorado Cancer Center (UCCC) Tissue Culture Core laboratory. Human colorectal cancer cell lines were obtained from ATCC (Manassas, VA, USA), DSMZ Cell Line Bank (Braunschweig, Germany), ECACC (Sigma, St. Louis, MO) and the Korean Cell Line Bank (KCLB) (Seoul, South Korea). The GEO cell line was a generous gift from Dr. Fortunato Ciardiello (Cattedra di Oncologia Medica, Dipartimento Medico-Chirurgico di Internistica Clinica e Sperimentale “F Magrassi e A Lanzara,” Seconda Università degli Studi di Napoli, Naples, Italy). KM20 and KM12C were a generous gift from Dr. Scott Kopetz from MD Anderson Cancer Center, Houston, TX, USA. The 55 human colorectal cancer cell lines used in this study were: CL-11(DSMZ), CL-34(DSMZ), COLO205 (ATCC), COLO 741 (ECACC), DLD1 (ATCC), GP5D (SIGMA), HCT116 (ATCC), HCT15 (ATCC), HT15 (SIGMA), HT55 (SIGMA), LOVO (ATCC), LS180 (ATCC), LS513 (ATCC), MDST8 (SIGMA), Mip101 (ECACC), NCI-H716 (ATCC), NCI-H747 (ATCC), RKO (ATCC), SKCO1 (ATCC), SNU-1235 (KCLB), SNU-977 (KCLB), SNU-C1 (KCLB), SNU1460 (KCLB), SW1116 (ATCC), SW1417 (ATCC), SW48 (ATCC), SW480 (ATCC), T84 (ATCC), WiDr (ATCC). All cell lines were cultured in RPMI media supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin and 1% MEM nonessential amino acids and routinely screened monthly for the presence of mycoplasma (MycoAlert, Cambrex Bio Science, Baltimore, MD, USA). Cell lines were maintained at 37 °C with 5% CO2. Cell lines were authenticated using STR DNA Profiling Globalfiler PCR Amplification Kit (Thermo Fisher) prior to use and cells were passaged < 20 times for all experiments.
Breast cancer cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) and CRC cell lines were cultured in Roswell Park Memorial Institute Medium (RPMI); both supplemented with 10% FBS, 1% penicillin–streptomycin, and 1% MEM nonessential amino acids (Supplementary Methods). Cells were grown in an incubator at 37°C which contained 5% CO2 and lines were routinely authenticated and screened for mycoplasma.
The effect of OKI-005 on cell proliferation was evaluated using the Sulforhodamine B (SRB) assay as previously described [27]. In brief, 3,000 – 5000 cells were plated in 96-well plates and allowed to adhere for 24 hours before being exposed to increasing concentrations of OKI-005 (0–5 uM) for 72 hours. The number of cells remained consistent for each cell line through each experiment and experimental arm. The number of cells plated for different cells lines was varied due to differences in doubling times in order to normalize confluency at the time of dosing. Media was then discarded, cells were fixed, washed and stained with 0.4% SRB (Fisher Scientific) for 30 minutes. Cells were washed with 1% acetic acid and treated with 10 mM Tris. Plates were read at an absorbance wavelength of 565 nm using a Biotek Synergy 2 96-well plate reader. Raw absorbance data was used to determine IC50 (half-maximal inhibitory concentration) and proliferation curves were generated using GraphPad Prism.
Caspase −3/−7 and cell cycle analysis
Apoptosis was assessed using the Caspase −3/−7 apoptosis assay as previously described [27]. In brief, 1500–5000 cells were plated in black-walled 96 well plates and allowed to adhere overnight. Cells were then exposed to increasing concentrations of OKI-005 (0–5 uM) for 24 or 48 hours and caspase-3 and −7 activity was assessed using the luminometric Caspase-Glo-3/7 assay (Promega).
For cell cycle analysis, cells were plated in 6-well plates, incubated overnight and then exposed to increasing concentrations of OKI-005 (0–200 nM) for 24 hours. Culture media was removed, cells were washed 1X with PBS and harvested by trypsinization. Cells were centrifuged at 1500 rpm and resuspended in Krishan’s stain. Cells were incubated at 4°C for 24 hours and analyzed at the University of Colorado Cancer Center Flow Cytometry Core Facility using a Gallios flow cytometer [22, 23].
Immunoblotting
Cells were seeded in 6 well plates and allowed to adhere for 24 hours. Cells were then exposed to increasing concentrations of OKI-005 (0, 50, 200 or 500 nM) for 24 hours. Cells were harvested by trypsinization and then lysed in Cell Lysis Buffer (Cell Signaling Technology). Fifteen to 50 μg of total protein was loaded onto a 4% to 20% BIS-TRIS 1.5 mm gradient gel (NuPage, Novex Invitrogen), electrophoresed, and transferred to nitrocellulose using the invitrogen Xcell II blotting apparatus (BioRad). Membranes were blocked in blocking buffer for 1 hour, washed × 3 for 10 min with tris-buffered saline (TBS)-Tween (0.1%) and incubated overnight at 4°C with primary antibodies: acetylated histone H3 (Bethyl Lab), acetylated α-tubulin (Cell Signaling), p21 (Cell Signaling) and α-tubulin (Abcam) (Supplementary Table 1). Following primary antibody incubation, membranes were washed in TBS-Tween (0.1%) × 3 and incubated with secondary anti-rabbit or anti-mouse IgG1 horseradish peroxidase-linked antibody at 1:15,000 (Jackson Immuno Research, West Grove, PA, USA) for 1 hour at room temperature (RT). Blots were developed using the enhanced chemiluminescence detection system 9 pierce, (Rockford, IL) or the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Experiments were performed in at least triplicate.
Tissue samples were snap-frozen and stored in liquid nitrogen until lysate preparation for immunoblotting. Fifteen to 50 μg of total protein was loaded onto a 4–12% bis-tris gel (NuPage, Novex Invitrogen), electrophoresed, and transferred to nitrocellulose using the iPierce G2 Fast Blotter, and analyzed as above.
Pharmacokinetic studies of OKI-179 in Balb/c mice, rats and beagle dogs
Male and female Balb/c mice were treated with a single dose PO of 25, 50 or 100 mg/kg OKI-179. Serum samples were obtained at 0.5, 1, 2 and 4 hours post dose. Additional samples were obtained at 8 hours post dose for animals treated at 50 mg/kg and 12 hours for animals treated with 100 mg/kg. Male and female rats were treated with a single 10, 30 or 100 mg/kg dose of OKI-179 PO and plasma samples were obtained from 0.5–24 hours. Male and female Beagle dogs were treated with a single dose of 1, 3 or 10 mg/kg OKI-179 PO and plasma samples were obtained from 0.5–24 hours. Quantification of the metabolite OKI-006 and analysis of PK parameters was performed as described above.
OKI-179 in combination with tamoxifen
Five- to six-week-old female athymic nude (nu/nu) mice (Envigo) were housed in groups of five per cage and allowed to acclimate for a week before handling. Sterilized food and water were provided ad libitum and animals were kept on a 12-hour light/dark cycle. Estrogen pellets were prepared in autoclaved medical beeswax packed with estrogen hormone in a biosafety cabinet (BSC) and transported in sterile sealed containers. The estrogen pellets were implanted a week before the MCF7 cell line was injected to assist in MCF7 tumor growth. Once the estrogen pellet was implanted to the base of the neck by a 10-gauge needle, the neck lesion was sealed by Vetbond Tissue Adhesive to ensure the pellet stayed in place. MCF-7 cells were harvested during logarithmic growth and resuspended in a 1:1 mixture of DMEM and Matrigel (BD Biosciences). The cells were injected bilaterally into the flank using 5 × 106 cells/injection in a volume of 100 μL. Mice were weighed, and tumor measurements were collected twice a week with digital calipers and recorded with the Study Director software package (Studylog Systems). When tumors reached an average volume of 200 mm3, mice were randomized to treatment with vehicle, OKI-179 60 mg/kg PO daily 5 days on 2 days off, tamoxifen 25 mg/kg SQ 3 times a week, or the combination of OKI-179 and tamoxifen. At the end of the study, mice were euthanized, and tumor samples were collected.
Statistical Analysis.
Results from the proliferation, apoptosis, and cell cycle experiments were analyzed for statistical significance using GraphPad Prism 8.1 (San Diego, CA) using an unpaired t-test. In vivo models were analyzed with one-way ANOVA with Dunnett’s multiple comparison test. Log-rank (Mantel-Cox) test was used to determine significance for in vivo survival. Differences were determined to be statistically significant with P-values ≤ 0.05.
All xenograft studies were performed in accordance with the NIH guidelines for the care and use of laboratory animals in a facility accredited by the American Association for Accreditation of Laboratory Animal Care with approval by the Institutional Animal Care and Use Committee prior to initiation of experiments.
Data availability statement.
The data generated in this study are available within the article and its supplementary data files.
Results
HDAC inhibitors OKI-006, OKI-005 and OKI-179
Largazole is a potent class I-selective HDAC inhibitor isolated from a marine cyanobacterium, but it is not optimized for clinical drug development. A series of analogs of the active component of largazole were undertaken, resulting in an optimized molecule, OKI-006 [24, 25] (Figure 1). The biochemical potency and selectivity of OKI-006 for HDACs using a cell-free biochemical assay has been previously published [26]. OKI-006 potently inhibits class I HDACs, including HDAC-1 (IC50 1.2 nM), HDAC-2 (IC50 2.4 nM), HDAC-3 (IC50 2.0 nM) and HDAC-8 (IC50 47 nM); class IIb HDACs, including HDAC-6 (IC50 47 nM) and HDAC-10 (IC50 2.8 nM); and the class IV HDAC-11 (IC50 2.3 nM). OKI-006 does not significantly inhibit class IIa HDACs (HDAC-4,−5,−7 and −9 IC50’s >1000 nM) [26]. While OKI-006 is an optimized HDAC inhibitor, protection of the free sulfhydryl group was felt to be a superior strategy to improve drug delivery. With that objective in mind, we proceeded to make two prodrugs. OKI-005 was optimized for use in cell culture, while OKI-179 was optimized for properties for oral deliver in both preclinical models and in humans.
Figure 1.

Structure of largazole-derivative OKI-006 and prodrugs OKI-005 and OKI-179. Largazole thiol was used a lead structure for medicinal chemistry optimization. Following identification of OKI-006, a first generation prodrug, OKI-005 was prepared and used for in vitro pharmacology studies. Subsequent prodrug optimization afforded OKI-179.
OKI-005 has antiproliferative and pro-apoptotic activity in CRC and TNBC cancer cell lines in vitro
Based on documented higher expression of class I HDACs correlating with clinical outcomes in both CRC and TNBC, these two disease types were selected for our initial evaluation of the preclinical activity of OKI-005 [7, 10]. We tested OKI-005 in vitro against a broad panel of CRC and TNBC cell lines and found OKI-005 to have potent antiproliferative activity against both TNBC and CRC cell lines (Figure 2A, Supplementary Figure 1-2). The majority of cell lines were sensitive to OKI-005 with IC50’s < 500 nM and the most sensitive cell lines exhibited IC50’s < 100 nM.
Figure 2.

Effects of OKI-005 in vitro in TNBC and CRC cell lines. A) Anti-proliferative effects were assessed using the SRB assay. Cells were treated with OKI-005 at doses of 0–5 uM and proliferation was assessed at 72 hours. Absolute IC50 was calculated using GraphPad Prism software. B) Apoptotic effects were evaluated in 2 sensitive (CAL-120 and MDA-MB-231) and 2 resistant (HCC1395 and Hs578T) TNBC cell lines at 24 and 48 hours using the Caspase-Glo 3/7 assay. Cells were treated with OKI-005 doses 0–500 nM. C) Cell cycle analysis was performed in sensitive TNBC cell lines at 24 hours using Krishans staining followed by flow cytometry. * P < 0.05, ** P < 0.01 compared to ND. D) Effects of OKI-005 on protein acetylation and p21 in sensitive and resistant TNBC cell lines treated with OKI-005 for 24 hours. Total protein was extracted and assessed for expression of indicated proteins by immunoblotting.
We selected 2 sensitive and 2 resistant TNBC cell lines for further evaluation including OKI-005-induced apoptosis, changes in cell cycle and effect on histone acetylation. In the CAL-120 and MDA-MB-231 cell lines sensitive to OKI-005 (IC50 < 100 nM), exposure to OKI-005 resulted in a concentration-dependent increase in apoptosis as measured by caspase −3 and −7 activity with up to 7–9-fold increase compared to no drug control (Figure 2B). In the resistant HCC1395 and Hs578T cell lines (IC50 to OKI-005 >500 nM), the degree of apoptosis was much less. As expected, exposure to OKI-005 resulted in a trend towards an increase in G1 and a decrease in the cell fraction in S phase in CAL-120 cells and a statistically significant difference in the MDA-MB-231 cell line (Figure 2B).
Exposure to OKI-005 in vitro resulted in a concentration-dependent increase in acetylated histone-H3 and p21 consistent with class I HDAC inhibition in sensitive and resistant TNBC cell lines (Figure 2D). Interestingly, a small increase in acetylated α-tubulin was observed at the highest dose levels which may be related to Class IIb HDAC inhibition.
Anti-tumor activity and pharmacokinetic profile of OKI-005 in xenograft models
We selected the MDA-MB-231 (TNBC) and HCT-116 (CRC) cell lines for xenograft studies to confirm activity of OKI-005 in vivo due to their sensitivity to OKI-005 in vitro and consistent growth patterns in vivo. First, we treated HCT-116 xenografts with OKI-005 at escalating doses PO or IP and observed a dose-dependent and statistically significant inhibition of tumor growth at doses of OKI-005 higher than 30 mg/kg, with TGI 28.1% (P = 0.025) for 30 mg/kg, 75.6% ( P < 0.001) for 100 mg/kg compared to vehicle control (Figure 3A). Mice treated with OKI-005 at 100 mg/kg experienced weight loss related to treatment requiring dose reduction at approximately 1 week which may explain the initial tumor shrinkage observed in this group that was followed by tumor growth (Supplemental Figure 3). Treatment with OKI-005 resulted in a dose-dependent increase in acetyl-histone H3 and acetyl H3K9 in OKI-005 treatment groups at 1 hour post-dose (Supplementary Figure 4A). OKI-005 dosed at 100 mg/kg exerted a time dependent increase of acetyl-Histone H3 and acetyl H3K9 with highest levels occurring 4 hours post-treatment and returning to baseline 24 hours post-dose (Supplemental Figure 4B-C).
Figure 3.

Effect of OKI-005 in CRC and TNBC models and OKI-005 plasma and tumor pharmacokinetics. A) HCT116 (CRC) xenograft model in BALb/c nude mice treated with vehicle, OKI-005 (PO or IP) or etinostat × 12 days. Arrow indicates time of dose reduction in OKI-005 100 mg/kg PO group due to weight loss. * P < 0.05, *** P < 0.001 compared to vehicle group. B) Mean plasma concentration-time profiles of OKI-006 following a single dose of OKI-005 (PO or IP) in BALb/c nude mice. C) Mean tumor concentration-time profiles of OKI-006 following single dose of OKI-005 (PO or IP) in BALb/c nude mice. D) Luciferase-labeled MDA-MB-231 TNBC were engrafted onto the mammary fat pads of 6-week-old female BALB/c nude mice, which were left untreated (vehicle) or treated with OKI-005 IP (10 mg/kg, 3X/week IP). Data are the mean (± SEM) tumor volumes measured at the indicated time points after engraftment (*P < 0.05). E) MDA-MB-231 tumors excised at day 35 following treatment with either vehicle or OKI-005. F & G) Mice treated with OKI-005 as in panel E displayed significantly decreased pulmonary metastasis at day 35 post-engraftment. Data are expressed as the mean (± SEM). A t-test was used to compare groups.
Next, we evaluated the pharmacokinetic profile of OKI-005 at escalating doses PO and a single dose IP in BALB/c HCT-116 tumor-bearing mice. Treatment with a single PO or IP dose of OKI-005 resulted in a dose-proportional increase in the plasma concentration of its active metabolite OKI-006 with highest exposure at 100 mg/kg PO and 5 mg/kg IP (Figure 3B). The half-life in plasma was short with PO dosing (< 2 hours), however, levels were more sustained with IP dosing. Interestingly, tumor concentrations of OKI-006 exceeded plasma levels at all time points and crossed the threshold for antiproliferative activity observed in vitro consistently with OKI-005 dosed at 100 mg/kg PO (Figure 3C). This finding was consistent with the early tumor shrinkage observed in the HCT-116 xenograft model before dose reduction for weight loss occurred (Figure 3A).
Finally, we evaluated the ability of OKI-005 to prevent metastasis in addition to inhibit primary tumor growth using a MDA-MB-231 luciferase-labeled metastasis model. Based on sustained plasma exposure with IP dosing, this route of administration was selected and the dose was increased to 10 mg/kg to increase exposure. In this model, treatment with OKI-005 10 mg/kg IP 3 times a week for 30 days resulted in statistically significant inhibition of primary tumor growth and metastasis as measured by the number of observed thoracic metastasis (Figure 3D-G).
Pharmacokinetic profile of OKI-179 in BALB/c nude mice, rats and beagle dogs
While OKI-005 demonstrated antitumor activity and favorable exposures in blood and tumor in mouse models, prodrug optimization provided OKI-179 which was used for further preclinical in vivo studies and subsequently human clinical trials. Following a single dose of OKI-179 in Balb/c mice and rats, we observed a dose-proportional increase in blood concentration of the active metabolite OKI-006 with no significant difference between male and female animals (Figure 4A). The maximal concentration (Cmax) exceeded 1 uM following a dose of OKI-179 100 mg/kg PO in both models. Exposure was somewhat higher in mice compared to rats. In beagle dogs, lower doses were evaluated based on predicted differences in drug absorption and metabolism across species. The Cmax exceeded 1 uM at doses ≥ 1 mg/kg in male and female dogs (Figure 4A).
Figure 4.

Pharmacokinetic profile of OKI-179 and effects in vivo in CRC and TNBC xenograft models. A) Serum/plasma concentration-time profiles of OKI-006 following a single dose of OKI-179 PO in male and female BALB/c nude mice, rats and beagle dogs. B) Effect of OKI-179 in HCT116 and MDA-MB-231 xenograft models in BALB/c nude mice. Treatment groups were compared to vehicle control for end of treatment statistical analysis. C) Effect of OKI-179 treatment PO daily × 3 or 10 days in MDA-MB-231 xenograft model. Tumors were excised, protein extracted and expression of acetylated proteins assessed by immunoblotting. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Activity of OKI-179 in CRC and TNBC xenograft models
Given favorable exposure in vivo with OKI-179 when compared to OKI-005 we evaluated the anti-tumor activity of OKI-179 in HCT-116 and MDA-MB-231 xenograft mouse models. Doses of OKI-179 of 40 – 80 mg/kg PO daily and 120 mg PO every other day were evaluated. Figure 4B depicts the tumor growth curves where we observed in the HCT-116 model, OKI-179 treatment at all doses and schedules evaluated resulted in statistically significant decreased tumor growth at day 24 which compared favorably to the anti-tumor activity of the same dose of OKI-005 in this model (Figure 3A). In the MDA-MB-231 model, OKI-179 treatment resulted in statistically significant tumor growth inhibition at higher doses (Figure 4B). Treatment with OKI-179 in the MDA-MB-231 xenograft model resulted in an increase in acetylated histone H3 consistent with target engagement that was greatest after 10 days of dosing compared to 3 days of dosing and greater with 60–80 mg/kg compared to 40 mg/kg (Figure 4C). We did not observe consistent changes in acetylated α-tubulin following treatment, although a small increase was observed at some dose levels.
Combining tamoxifen and OKI-179 in ER+ breast cancer xenograft
In preclinical models of estrogen receptor (ER)-positive HER2-negative breast cancer, HDAC inhibitors potentiate the activity of tamoxifen likely resulting from epigenetic modulation of ER signaling [28, 29]. OKI-179 was administered on an intermittent dosing schedule 5 days on 2 days off based on emerging data from the ongoing first-in-human phase I clinical trial on OKI-179 in patients with advanced solid tumors demonstrating dose-limiting thrombocytopenia with continuous dosing [30]. Intermittent dosing schedules are generally required for HDAC inhibitors to allow for recovery of thrombocytopenia. As depicted in Figure 5A, we observed a trend towards improved tumor growth inhibition in an MCF-7 xenograft model with the combination of OKI-179 and tamoxifen compared to either single agent alone, however, both compounds were active in this model. Treatment with the combination resulted in a statistically significant increase in survival with the combination compared to OKI-179 (P = 0.02) and a trend towards increased survival compared with tamoxifen (P = NS) (Figure 5B).
Figure 5.

Effect of OKI-179 or tamoxifen alone or in combination in MCF7 xenograft BALB/c nude mouse model. A) Tumor volumes for mice treated with OKI-179 PO 5 days on 2 days off, tamoxifen SQ 3 times a week or the combination. B) Probability of survival for MCF7 xenograft mice treated with OKI-179, tamoxifen or the combination.
Discussion
Histone acetylation is dysregulated in a wide array of solid and hematologic cancers, however, to-date the development of selective, potent and active HDAC inhibitors in solid tumors has remained elusive [1]. In this manuscript, we report the creation of prodrug OKI-179 that is metabolized in vivo to the largazole-derivative OKI-006. We demonstrated that OKI-005, an alternative prodrug of OKI-006, is optimized for in vitro experimentation as evidenced by potent anti-proliferative activity against TNBC and CRC cell lines with IC50’s in the nanomolar range, robust induction of apoptosis in sensitive cell lines and proof of target engagement with concentration-proportional increases in histone acetylation. While OKI-005 was active in preclinical in vivo models, OKI-179 was optimized for in vivo experimentation in animal models and humans resulting in a superior PK profile in vivo. Based on these results, OKI-179 was selected to move forward into clinical development.
OKI-179 represents a second-generation prodrug design that leverages sulfhydryl group derivatization of OKI-006 and salt form optimization to aid in absorption with oral dosing. Interestingly, with oral or IP administration of OKI-005, tumor concentrations of the active metabolite OKI-006 exceeded plasma concentrations at most doses. Further work will be needed moving forward to investigate the pattern of metabolism in human tissues including in the gastrointestinal tract, liver and target tumor tissue. The PK profile of OKI-179 in mouse, rat and beagle dogs was linear with a dose-proportional increase in exposure with oral dosing. OKI-179 is converted to the active metabolite OKI-006 rapidly in preclinical models which may be an asset clinically. OKI-006 has a short half-life with plasma concentrations > 100 nM following a single dose persisting for ~4–12 hours depending on the dose and model (ie mouse vs. rat vs. dog). The short half-life should be taken into consideration in determining the ideal dosing strategy in the clinic.
Various dosing strategies were employed in our preclinical evaluation of the efficacy of OKI-005 and OKI-179 in xenograft models. In our initial study in the MDA-MD-231 xenograft model, we evaluated OKI-005 administered either IP or orally once daily (10–100 mg/kg). Our data demonstrate similar efficacy and intra-tumor drug concentrations for the IP and oral formulations. At the higher dose of 100 mg/kg oral daily, animals experienced dose-limiting treatment-related toxicity (evidenced by weight loss) and the dose was reduced. This limits our ability to make conclusions regarding the dose response for OKI-005 in this model. For the second generation compound, OKI-179, we administered doses of 40–80 mg/kg orally daily or 120 mg/kg oral every other day in the HCT-116 and MDA-MB-231 xenograft models. The higher dose of 120 mg/kg with every other day dosing was included based on the weight loss observed in animals treated with OKI-179 100 mg/kg oral daily and our desire to increase Cmax while maintaining tolerability. This dosing schedule was better tolerated in the animals. For our final in vivo study, we incorporated a dosing schedule currently being evaluated in the ongoing phase I clinical trial which was an intermittent dosing schedule with 5 days on and 2 days off. This schedule was selected clinically to allow for recovery from drug-related thrombocytopenia. Ultimately, the recommended phase 2 dose of OKI-179 for future clinical trials will likely be based on clinical toxicity, however, we made efforts in our preclinical experimental design to evaluate dosing strategies anticipated to move forward in the clinic.
One of the promising features of HDAC inhibitors as anti-cancer agents is the ability to induce apoptosis and convert growth arrest to apoptosis when used in combination with other agents. The apoptotic potential of HDAC inhibitors likely depends on the intrinsic cellular threshold for apoptosis that may be tumor cell-dependent and the mechanism by which death can be triggered by HDAC inhibition in the cell. Given the multiple potential effects of HDACs in cancer cells, there may be differential effects depending on the cellular milieu. In our study, OKI-005 exposure resulted in a robust 4- to 9-fold increase in apoptosis in TNBC cell lines sensitive to the antiproliferative activity of OKI-005 compared to a modest 2- to 3-fold increase in resistant cell lines. The pharmacodynamic effect of OKI-005 on histone acetylation was similar in sensitive and resistant cell lines, pointing to another factor in cellular downstream signaling as mediating a pro-apoptotic response in some cells but not others. Biomarkers predictive of response to HDAC inhibitors are yet to be fully identified, however enhanced JAK/STAT signaling has been reported as associated with resistance to HDAC inhibitor-induced cell death and resistance to vorinostat in patents with CTCL [31]. Biomarkers predictive of a pro-apoptotic response to OKI-005 and OKI-179 should be actively investigated as they may aid in selecting a responding patient population in future trials in advanced solid tumors.
OKI-179 is a promising agent for the treatment of TNBC based on preclinical activity and the ongoing clinical need for therapies that may potentiate the activity of programmed cell death 1 (PD-1) and programmed death-ligand 1(PD-L1)-inhibitors in TNBC. While both atezolizumab and pembrolizumab are FDA-approved in combination with chemotherapy in patients with metastatic PD-L1-positive TNBC, the rates of 1-year progression-free survival remain in the 30–40% range, highlighting room for improvement in achieving long-term disease control for these patients with immunotherapy [32, 33]. HDAC inhibitors potentiate the activity of immunotherapy agents in many preclinical models of solid tumors with evidence of increased tumor immunogenicity as a potential mechanism [13, 34, 35]. Supporting these findings, the addition of OKI-179 to nivolumab in a humanized mouse MDA-MB-231 xenograft, resulted in improved tumor growth inhibition and increased T-cell activation in the tumor microenvironment [36]. Similar findings were observed for OKI-179 in combination with anti-PD-1 inhibition in preclinical models of B-cell lymphomas resistant to anti-PD-1 where tumor-derived MHC class I was required and MHC class II expression correlated with response [26]. Other emerging roles for histone deacetylases include an expanding landscape of the removal of acyl lysine groups from histones [37, 38]. This may expand the potential impact of HDAC inhibitors and should be investigated further, as well as the impact on nonhistone proteins which remains largely uncharacterized. It is possible that these underexplored effects might contribute to the modulation of immune response and potentiation of immunotherapy.
While there are many opportunities for clinical investigation of OKI-179 as a potent, class I-selective HDAC inhibitor in solid tumors, we chose to explore the combination of OKI-179 with tamoxifen preclinically. Resistance to endocrine therapy in hormone receptor-positive, HER2- breast cancer remains a clinical challenge and HDAC inhibitors are promising agents for reversing endocrine resistance [3, 39, 40]. In preclinical models, HDAC inhibitors downregulate ER expression and can reverse tamoxifen-induced ER stabilization leading to increased apoptosis [28]. Many clinical trials have evaluated HDAC inhibitors in combination with tamoxifen or aromatase inhibitors with some promising results. The combination of vorinostat and tamoxifen was evaluated in a phase II clinical trial in patients with previously treated ER+ HER2- metastatic breast cancer and demonstrated an objective response rate of 19% with a clinical benefit rate of 40%, which is higher than expected clinical activity of tamoxifen alone based on historical controls [41]. In a phase III clinical trial, the combination of the subtype-selective HDACi chidamide and exemestane improved PFS from 3.8 mos with exemestane + placebo to 7.4 months with exemestane + chidamide, however, a substantial portion of patients required dose interruption of chidamide for toxicity [42]. The isoform selectivity of OKI-179 may lead to improved clinical tolerability, and this is part of the on-going investigation in a Phase 1 clinical trial. There are many potential clinical opportunities for OKI-179 in hematologic and solid tumors, including potential combination with hormonal therapy in ER-positive HER2-negative breast cancer.
Supplementary Material
Acknowledgements:
This work was supported by the National Institutes of Health (NIH) and the National Cancer Institute (NCI) through 5P30CA046934-25 (University of Colorado Cancer Center Support Grant), 1K23CA172691-01A1 (J.R. Diamond), R01GM113141 (X. Liu), W81XWH-10-1-0989 (X. Liu, A. Phillips, W. Schiemann) and CPRIT Scholar Award #RR160093 (S. G. Eckhardt). Support was also received from OnKure, Inc. The funding bodies played a role in study design, data collection, analysis, and interpretation, and manuscript preparation.
Potential Conflict of Interest:
Research funding from OnKure, Inc. to JRD, SGE and TMP. JAD, JW, and ADP are employees of OnKure, Inc. JRD receives consulting funds from OnKure, Inc. and owns equity in the company. XL is a co-founder and member of the scientific advisory board of OnKure, Inc. and owns equity in the company. XL served as a paid consultant, CSO and member of the board of directors during the time of this study. SGE serves on the Scientific Advisory Board of OnKure, Inc. The University of Colorado Boulder holds the patent of OKI-005 and licensed it to OnKure, Inc.
References
- 1.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014; 124: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457–463. [DOI] [PubMed] [Google Scholar]
- 3.Sukocheva OA, Lukina E, Friedemann M et al. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: Current findings and future perspectives. Semin Cancer Biol 2020. [DOI] [PubMed] [Google Scholar]
- 4.Biel M, Wascholowski V, Giannis A. Epigenetics--an epicenter of gene regulation: histones and histone-modifying enzymes. Angew Chem Int Ed Engl 2005; 44: 3186–3216. [DOI] [PubMed] [Google Scholar]
- 5.Dickinson M, Johnstone RW, Prince HM. Histone deacetylase inhibitors: potential targets responsible for their anti-cancer effect. Invest New Drugs 2010; 28 Suppl 1: S3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraga MF, Ballestar E, Villar-Garea A et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 2005; 37: 391–400. [DOI] [PubMed] [Google Scholar]
- 7.Weichert W, Roske A, Niesporek S et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res 2008; 14: 1669–1677. [DOI] [PubMed] [Google Scholar]
- 8.Krusche CA, Wulfing P, Kersting C et al. Histone deacetylase-1 and −3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat 2005; 90: 15–23. [DOI] [PubMed] [Google Scholar]
- 9.Guo P, Chen W, Li H et al. The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications. Pathol Oncol Res 2018; 24: 807–813. [DOI] [PubMed] [Google Scholar]
- 10.Garmpis N, Damaskos C, Garmpi A et al. Histone Deacetylases as New Therapeutic Targets in Triple-negative Breast Cancer: Progress and Promises. Cancer Genomics Proteomics 2017; 14: 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 2004; 338: 17–31. [DOI] [PubMed] [Google Scholar]
- 12.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol 2012; 6: 637–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology 2014; 3: e27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skov S, Pedersen MT, Andresen L et al. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res 2005; 65: 11136–11145. [DOI] [PubMed] [Google Scholar]
- 15.Magner WJ, Kazim AL, Stewart C et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol 2000; 165: 7017–7024. [DOI] [PubMed] [Google Scholar]
- 16.Lindemann RK, Newbold A, Whitecross KF et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A 2007; 104: 8071–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newbold A, Lindemann RK, Cluse LA et al. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Ther 2008; 7: 1066–1079. [DOI] [PubMed] [Google Scholar]
- 18.Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41: 185–198. [DOI] [PubMed] [Google Scholar]
- 19.Yoon S, Eom GH. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med J 2016; 52: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly WK, Marks P, Richon VM. CCR 20th Anniversary Commentary: Vorinostat-Gateway to Epigenetic Therapy. Clin Cancer Res 2015; 21: 2198–2200. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Salvador LA, Byeon S et al. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J Pharmacol Exp Ther 2010; 335: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ying Y, Taori K, Kim H et al. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J Am Chem Soc 2008; 130: 8455–8459. [DOI] [PubMed] [Google Scholar]
- 23.Benelkebir H, Marie S, Hayden AL et al. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg Med Chem 2011; 19: 3650–3658. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Phillips AJ, Ungermannova D, Nasveschuk CG, Zhang G. Macrocyclic compounds useful as inhibitors of histone deacetylases. In States U (ed). United States: 2016. [Google Scholar]
- 25.Ungermannova D. P27 as a molecular target for cancer therapeutics: Discovering small molecule inhibitors of p27 proteolysis and structure-activity relationship and mechanistic studies of largazole, a potent inhibitor of histone deacetylase. In. University of Colorado-Boulder, Boulder, CO, USA: 2010. [Google Scholar]
- 26.Wang X, Waschke BC, Woolaver RA et al. Histone Deacetylase Inhibition Sensitizes PD1 Blockade-Resistant B-cell Lymphomas. Cancer Immunol Res 2019; 7: 1318–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diamond J. A novel multi-targeted Aurora A and VEGFR2 kinase inhibitor, ENMD-2076, demonstrates synergistic antiproliferative and proapoptotic effects in combination with chemotherapy and trastuzumab in breast cancer cell lines. . In AACR/NCI/EORTC Molecular Targets and Cancer Therapeutics poster presentation. Boston, MA: 2009. [Google Scholar]
- 28.Hodges-Gallagher L, Valentine CD, Bader SE, Kushner PJ. Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res Treat 2007; 105: 297–309. [DOI] [PubMed] [Google Scholar]
- 29.Jang ER, Lim SJ, Lee ES et al. The histone deacetylase inhibitor trichostatin A sensitizes estrogen receptor alpha-negative breast cancer cells to tamoxifen. Oncogene 2004; 23: 1724–1736. [DOI] [PubMed] [Google Scholar]
- 30.Jodi A Kagihara BRC, Jose Pacheco, Davis S. Lindsey, Lieu Christopher, Kim Sunnie, Jimeno Antonio, Heim Amy M, DeMattei John A, Gordon Gilad, Triplett Todd A., Eckhardt S. Gail, Winkler James, Piscopio Anthony D, Diamond Jennifer R. Phase 1 study of OKI-179, an oral class 1-selective depsipeptide HDAC inhibitor, in patients with advanced solid tumors: Final results. In ASCO. 2021. [Google Scholar]
- 31.Fantin VR, Loboda A, Paweletz CP et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res 2008; 68: 3785–3794. [DOI] [PubMed] [Google Scholar]
- 32.Cortes J, Cescon DW, Rugo HS et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020; 396: 1817–1828. [DOI] [PubMed] [Google Scholar]
- 33.Schmid P, Adams S, Rugo HS et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018; 379: 2108–2121. [DOI] [PubMed] [Google Scholar]
- 34.Zheng H, Zhao W, Yan C et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res 2016; 22: 4119–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gameiro SR, Malamas AS, Tsang KY et al. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget 2016; 7: 7390–7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capasso A, Lang J, Pitts TM et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J Immunother Cancer 2019; 7: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsen CA. Expansion of the lysine acylation landscape. Angew Chem Int Ed Engl 2012; 51: 3755–3756. [DOI] [PubMed] [Google Scholar]
- 38.Tan M, Luo H, Lee S et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011; 146: 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Normanno N, Di Maio M, De Maio E et al. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer 2005; 12: 721–747. [DOI] [PubMed] [Google Scholar]
- 40.Zucchetti B, Shimada AK, Katz A, Curigliano G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 2019; 43: 130–134. [DOI] [PubMed] [Google Scholar]
- 41.Munster PN, Thurn KT, Thomas S et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 2011; 104: 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang Z, Li W, Hu X, et al. Phase III trial of chidamide, a subtype-selective histone deacetylase (HDAC) inhibitor, in combination with exemestane in patients with hormone receptor-positive advanced breast cancer. Annals of Oncology 2018; 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.