

ABSTRACT
Antimicrobial resistance (AMR) is widespread within Neisseria gonorrhoeae populations. Recent work has highlighted the importance of commensal Neisseria (cN) as a source of AMR for their pathogenic relatives through horizontal gene transfer (HGT) of AMR alleles, such as mosaic penicillin binding protein 2 (penA), multiple transferable efflux pump (mtr), and DNA gyrase subunit A (gyrA) which impact beta-lactam, azithromycin, and ciprofloxacin susceptibility, respectively. However, nonpathogenic commensal species are rarely characterized. Here, we propose that surveillance of the universally carried commensal Neisseria may play the role of the “canary in the coal mine,” and reveal circulating known and novel antimicrobial resistance determinants transferable to pathogenic Neisseria. We summarize the current understanding of commensal Neisseria as an AMR reservoir, and call to increase research on commensal Neisseria species, through expanding established gonococcal surveillance programs to include the collection, isolation, antimicrobial resistance phenotyping, and whole-genome sequencing (WGS) of commensal isolates. This will help combat AMR in the pathogenic Neisseria by: (i) determining the contemporary AMR profile of commensal Neisseria, (ii) correlating AMR phenotypes with known and novel genetic determinants, (iii) qualifying and quantifying horizontal gene transfer (HGT) for AMR determinants, and (iv) expanding commensal Neisseria genomic databases, perhaps leading to the identification of new drug and vaccine targets. The proposed modification to established Neisseria collection protocols could transform our ability to address AMR N. gonorrhoeae, while requiring minor modifications to current surveillance practices.
KEYWORDS: Neisseria, commensal bacteria, horizontal gene transfer, antibiotic resistance, microbiome, N. gonorrhoeae, Neisseria gonorrhoeae
INTRODUCTION
Antimicrobial resistance (AMR) in Neisseria gonorrhoeae is an urgent threat to public health, with the emergence of resistance in gonococcal populations to all antimicrobials that have been recommended for treatment (1), and infection rates simultaneously on the rise (2). According to the World Health Organization (WHO), an estimated 86.9 million people were infected by the gonococcus worldwide in 2016 (3). In the most recent United States survey by the Gonococcal Isolate Surveillance Project (GISP) from the U.S. Centers for Disease Control and Prevention (CDC), only 44.5% of isolates remained susceptible to all tested antimicrobials (2). Furthermore, treatment failures to dual antimicrobial therapy (ceftriaxone plus azithromycin) have been reported internationally (4–6). This, coupled with a precipitous rise in azithromycin resistance (7), has prompted a change in recommended treatment regimen for uncomplicated gonococcal infection, from dual therapy with ceftriaxone (single intramuscular injection of 250 to 500 mg) and azithromycin (single oral dose of 1 to 2 g), to ceftriaxone monotherapy (single 500 mg intramuscular dose) (8). With only two novel therapeutics currently in development (i.e., zoliflodacin [9] and gepotidacin [10]), new strategies must be implemented to combat the growing threat of AMR in N. gonorrhoeae.
Worldwide, several national and international surveillance programs, such as GISP from the CDC, and the global Gonococcal Antimicrobial Surveillance Program (GASP) and its European version (Euro-GASP) from the WHO, aim to control and prevent the spread of AMR in N. gonorrhoeae. The identification of resistance mechanisms to first-line antibiotics circulating within gonococcal populations has been emphasized in recent years (11–16); however, these efforts often overlook a known source of resistance for gonococci—the commensal Neisseria (4–7). In addition to the two human pathogens within the Neisseria genus (N. gonorrhoeae and N. meningitidis), there are, at minimum, eight closely related commensal Neisseria species carried harmlessly in healthy human adults and children (17–19) that rarely cause disease (20). To note, N. meningitidis is considered an obligate human commensal and opportunistic pathogen, as it is commonly carried in the population asymptomatically but can cause invasive life-threatening disease. Commensal species share alleles with their pathogenic relatives, and genetic mosaicism has been observed genome-wide, whereby loci containing full or partial genes or regulatory sequences within a particular lineage have been acquired from another Neisseria species (4–6, 13, 15, 16, 21, 22). Natural competence, the ability of Neisseria to uptake and integrate DNA at any point during growth and without a necessary external stimulus, as well as high rates of recombination has led many to question the nature of species’ delineations within the genus (11, 23). Likewise, promiscuous allelic exchange has repeatedly been documented to have facilitated rapid adaptive evolution of important phenotypic characteristics such as antimicrobial resistance (4, 21, 24, 25) and body-site colonization niche shifts (16). Thus, without detailed characterization of the resistance alleles in commensals and assessment of their population prevalence, we are blinded to novel mechanisms of resistance in commensal Neisseria that may rapidly and unknowingly disseminate to the pathogenic Neisseria.
A current challenge to inclusion of commensals in genus-level population resistance monitoring is the relative undersampling of commensal populations compared with their pathogenic relatives (20, 26). The diversity of AMR alleles in commensal populations is currently unknown, presenting an issue for complete genotypic-based resistance prediction for any of the Neisseria species, including pathogens. Improved characterization of the Neisseria resistome (i.e., total collection of all resistance alleles available to members of the genus), and subsequent surveillance of commensal populations could serve as a “canary in the coal mine” approach to identifying new or common resistance mechanisms that may emerge from our natural microbial reservoirs. Kenyon et al. have previously championed a call for surveillance of commensal Neisseria as part of their model considering a “pan-Neisseria” approach for choosing appropriate antibiotic therapy for gonorrhea (27), which we expand upon here as an important next step in addressing AMR within the pathogenic Neisseria. Collection and characterization of commensal Neisseria isolates as part of routine surveillance programs by clinical and academic laboratories should provide broad epidemiological surveillance of AMR in commensals, as the carriage rate of commensals (around 100%) is higher than that of their pathogenic relatives (0.01% to 10%) (28–30), and we believe could ultimately serve as an early warning indicator of possible resistance outbreaks in pathogenic Neisseria.
Here, we review the background on commensal Neisseria, horizontal gene transfer within the genus, and the clinically relevant resistance alleles in the pathogenic Neisseria that have been acquired from commensal species. Furthermore, we summarize the current literature on commensal resistance, both the known mechanisms and published surveillance efforts. Finally, we highlight the necessary steps that must be taken to successfully enhance our ability to detect and predict resistance in and from nonpathogenic reservoirs.
THE NEISSERIA GENUS: OUR BIAS TOWARD THE HUMAN PATHOGENS
The genus Neisseria is comprised of several closely related Gram-negative species, which are typically diplococcoid, oxidase-positive and often catalase-positive, and usually isolated from mucosal membranes of humans and animals (20) (only human-associated Neisseria will be reviewed here). Phylogenomic analyses suggest that all human-associated Neisseria evolved from a common ancestor which colonized the oral cavity of an early humanoid (31, 32). Interestingly, metagenomic surveys have demonstrated clear specificity of contemporary Neisseria species to distinct sites within the oro- and nasopharynx, also correlated to clades with different DNA uptake sequence motifs (DUS; see below for further discussion), suggesting distinct ecological niches have resulted in subsequent speciation and genetic divergence (33). Though there are eight broadly cited human-associated commensal species (Neisseria cinerea, N. polysaccharea, N. lactamica, N. mucosa, N. oralis, N. subflava, N. elongata [atypical rod], and N. bacilliformis [atypical rod]), three distinct genetic clusters within the N. polysaccharea species group and seven novel Neisseria species with distinct biochemical and genomic characteristics have recently been identified through phylogenomic analyses (26), suggesting that human commensal Neisseria diversity is likely greater than we currently know and could very likely surpass 20 species (26). Commensal Neisseria are considered part of the “core” oropharyngeal flora, with metagenomic surveys typically identifying Neisseria as the most abundant genus within proteobacteria (~10% of operational taxonomic units [OTUs]) in oral and pharyngeal samples (34, 35). Although correlations between abundance of Neisseria commensals and disease, and systemic infections have been reported, the commensal Neisseria are generally considered nonpathogenic members of the human microbiome (20, 36, 37).
Of the human-associated Neisseria, only N. gonorrhoeae (the gonococcus) and N. meningitidis (the meningococcus) cause disease at notable frequencies. N. meningitidis is an obligate human commensal that typically colonizes the nasopharynx and is carried asymptomatically in 10% of adults; however, it can occasionally become an opportunistic pathogen causing invasive meningococcal disease (meningococcemia) and bacterial meningitis (38, 39). N. gonorrhoeae is the only species within the genus that, in addition to the nasopharyngeal mucosa, also routinely colonizes the urogenital tract and rectum, and causes the sexually transmitted infection gonorrhea (40). N. gonorrhoeae and N. meningitidis are the closest phylogenetic relatives within the genus; and occasional reports of meningococcal urogenital colonizing strains (16, 41) has led to speculation that the two pathogenic Neisseria arose from a common ancestor (likely inhabiting the pharynx), which established in the urogenital niche and developed into the modern gonococcal lineage through adaptive evolution of traits such as the induction of the polymorphonuclear leukocyte (PMN) inflammation response aiding in sexual transmission (42), and nitrite-dependent anaerobic growth which may aid in urethral colonization (16, 43).
Research on commensal Neisseria is in its infancy compared with the pathogenic Neisseria species, N. gonorrhoeae and N. meningitidis. A PubMed query as of June 2022 revealed 25,902 articles in English including the search term “Neisseria” in the title, 12,859 articles including the search terms “Neisseria AND gonorrhoeae NOT commens*” compared with 334 articles with the search terms “Neisseria AND commens* NOT gonorrhoeae,” 11,036 articles including the search terms “Neisseria AND meningitidis NOT commens*,” 230 articles including the search terms “Neisseria AND commens* NOT meningitidis,” and 141 articles with the search terms “Neisseria AND commens* NOT gonorrhoeae NOT meningitidis.” Though restricted in scope, this analysis of the available literature captures the limited research focus, sampling, and sequencing (as pointed later in the text), on the commensals which emphasizes a weakness in our understanding of Neisseria AMR mechanisms that may be, as of yet, undiscovered with the full diversity of the genus undocumented.
DNA PROMISCUITY: THE PREVALENCE OF ALLELIC EXCHANGE
Bacteria replicate asexually through binary fission, in which the chromosome(s) duplicate and subsequently segregate into unique daughter cells. This produces clonal population structures, with relatively low genetic diversity outside the variation created through the accumulation of beneficial or neutral de novo mutations over time (23, 44). This typical clonal structure, however, is disrupted by species, like Neisseria spp, that engage in horizontal gene transfer (HGT) through natural competence. Species capable of natural competence uptake naked extracellular DNA using specialized machinery and integrate homologous tracts into their genomes via RecA-mediated recombination (24), which reassorts alleles across lineages and allows adaptive genetic variation to rapidly spread within and between species (23, 44).
Neisseria are one of the most recombinogenic bacterial genera, with extensive allele sharing observed within and between species in the genus, breaking clonality and resulting in panmictic population structures (23, 44). In part, this is due to the fact that Neisseria are naturally competent for transformation through all stages of growth (45), and constitutively express their competence systems as opposed to many other recombinogenic bacteria like Bacillus subtilis (46), Haemophilus influenzae (47), or Streptococcus pneumoniae (48). The frequency of intragenus genetic exchange has led to the concept of Neisseria as an interconnected consortium of species with “fuzzy” borders (14, 23, 44, 49). Allelic exchange via this mechanism has introduced novel DNA tracts, estimated at 6% genome-wide, into gonococci from other members of the Neisseria genus (14, 22, 50, 51). Examples of intragenus allele sharing have been documented between different species pairs at the loci encoding the IgA proteases (21), Penicillin Binding Protein 2 (encoded by penA, discussed below) (5, 52), the Multiple Transferable Efflux Pump (mtr, discussed below) (13, 15), ribosomal protein genes (discussed below) (51, 53, 54), and ornithine carbamoyltransferase (argF) (6) among others.
Genetic exchange across species’ boundaries has facilitated rapid adaptive evolution of important phenotypic characteristics such as resistance to antibiotics (4, 21, 24, 25) and macrohabitat niche shifts (16). For example, in 2015 in the United States, several cases of men with urethritis caused by Gram-negative diplococci were nucleic acid amplification test (NAAT) negative for N. gonorrhoeae in Ohio and Michigan (16, 41). Further amplification tests revealed the cause to be a clonal group of serogroup C N. meningitidis strains which had acquired the gonococcal aniA/norB cassette. A nitrite reductase (AniA) and a nitric oxide reductase (NorB) are important components of an effective denitrification pathway and are essential for the anaerobic growth typical to the urogenital tract (55–57). N. gonorrhoeae isolates have functional aniA and norB loci, whereas the majority of meningococci which typically experience aerobic conditions in the nasopharynx, have frameshift mutations in aniA and cannot grow anaerobically using nitrite as the electron acceptor (58). However, introduction of a functional in-frame aniA/norB cassette from N. gonorrhoeae has directly led to adaptation to anaerobic conditions and the expansion of urogenital-colonizing N. meningitidis (59), speaking to the incredible power of HGT in facilitating large and rapid evolutionary shifts. Overall, recently incorporated mosaic sequences across Neisseria genomes show signatures consistent with positive selection (e.g., linkage of nearby nonsynonymous mutations, increased intermediate-frequency allele diversity, etc.), suggesting recombination is an important source of beneficial genetic variation through the introgression of coadapted allelic complexes on coinherited DNA tracts from close relatives (14).
The likely site for the bulk of cross-species gene exchange is in the oro- and nasopharynx, where most Neisseria species typically reside in proximal yet distinct ecological niches (33). Here, exogenous DNA uptake is mediated by functional type IV pili (Tfp). Tfp expression greatly enhances transformation efficiency in N. gonorrhoeae, with piliated strains exhibiting transformation frequencies in excess of 20% in the presence of high DNA concentrations (60), whereas piliation loss decreases these frequencies closer to 1 × 10−7 (61). Pili are embedded in the outer membrane and protrude into the extracellular milieu through the multimeric PilQ pore complex (62, 63), and are formed by pilin subunits (PilE and ComP) which assemble into a helical structure with a conserved core, central layer, and hypervariable outer region (64–66). Competence is facilitated by a protein exposed on the surface of the pilus, ComP, which has high affinity for Neisseria-specific DNA uptake sequences (DUS) (67–69). These conserved clade-specific 12-bp sequences (e.g., AT-DUS: 5′-AT-GCCGTCTGAA-3′), located every ~1,100 bp throughout Neisseria genomes (70), greatly increase transformation efficiency in Neisseria species with the same sequence motif “dialects.” The cytoplasmic NTPase motors PilF (also known as PilB), PilT, and PilU are involved in regulating pilus assembly and retraction (71–73). Retraction of pili is necessary for competence and is thought to facilitate transport of ComP-bound DNA through the outer membrane and peptidoglycan layer (74). Single-stranded DNA is then imported through the inner membrane and into the cytoplasm through a pore formed by ComA (75). Finally, two RecA-mediated pathways (RecBCD and RecF) integrate the DNA into the chromosome through homologous recombination (76–78).
Although interspecies recombination can provide rapid access to new advantageous alleles, it can also introduce variants that are deleterious in divergent genomic backgrounds through gene disruption or the creation of maladapted gene combinations (i.e., locus A + locus B = advantageous combination; locus C + locus D = advantageous combination; however, locus A + locus D = deleterious combination). Thus, there are some barriers to allelic exchange that do exist within the genus that limit transfer from more evolutionarily distant relatives. For example, coevolution of DUS motifs and their binding protein ComP (79) has contributed to the emergence of “reproductive isolation” between clades of Neisseria species. Eight sequence variations of DUSs coupled with alternative forms of the ComP protein create divergent “dialects,” which directly increase the frequency of DNA binding, uptake, and transformation within groups compared with between groups (70). A second barrier to recombination is divergence in the number and identity of methyltransferases between pathogenic and commensal Neisseria species. Compared with N. gonorrhoeae which harbors 14 to 19 DNA methyltransferases (80–82), the commensal N. elongata encodes only 7 to 10. Divergence in methyltransferase activity across these species differentially modifies CpG and GpC sequence motifs producing species-specific methylation signatures. Mismatched methylation has been demonstrated to kill Neisseria which have recently integrated novel DNA into their chromosomes through restriction enzyme(s) cleavage and the formation of double-strand breaks (83, 84). However, despite the aforementioned barriers, observable genome-wide recombination (14, 22, 85) and reports of specific alleles acquired across species’ borders (86, 87) directly support the importance of commensals as reservoirs of readily available adaptive genetic variation for the pathogens within the genus.
COMMENSALS AS A KEY RESERVOIR OF RESISTANCE
Full or partial gene exchange from commensals has historically played a large role in rendering antimicrobial therapies ineffective in the pathogenic Neisseria (see below and Fig. 1). In gonococci, reduced susceptibility to extended-spectrum cephalosporins (ESC) is predominantly associated with the acquisition of multiple commensal haplotypes at the penA locus, which have been reported worldwide (22, 88–91). Mosaic penA alleles also contribute to reduced penicillin susceptibility in gonococci and meningococci (86, 92). Furthermore, a large proportion of macrolide resistance in gonococci has been acquired from other Neisseria species through the transfer of mosaic multiple transferable efflux pump (mtr) alleles (22, 93–97). Mutations in the ribosomal protein genes rplB, rplD, rpsY, and rpsJ have likely been acquired from commensal Neisseria and correlate to azithromycin and tetracycline resistance (53, 98). Finally, meningococcal quinolone resistance has recently been reported to have been acquired through transfer of gyrA alleles from commensal Neisseria (99). These multiple documented HGT events between commensals and pathogens underscore the importance of commensals as a proximal reservoir of resistance (see Fig. 1). In Fig. 1, we also depict other well-documented mechanisms of resistance in N. gonorrhoeae that have not yet been studied in commensal Neisseria. These include changes of the cell surface permeability by the decoration of lipooligosaccharides (LOS) with phosphoethanolamine (PEA) (100) (Fig. 1A), and bypassing antibiotic-targeted biochemical pathways through the expression of insensitive enzymes (101) (Fig. 1A). In addition to AMR acquisition from commensal Neisseria (Fig. 1B), we depict the potential for ceftriaxone resistance through acquisition of novel mutations from other proximal human-associated bacterial species (Fig. 1C). Together, these examples of both intra- and intergenus AMR acquisition (discussed below) and unexplored mechanisms of resistance in commensal species highlight the importance of further characterization of commensals to fully understand the alleles at risk of rapid dissemination across species’ boundaries.
FIG 1.
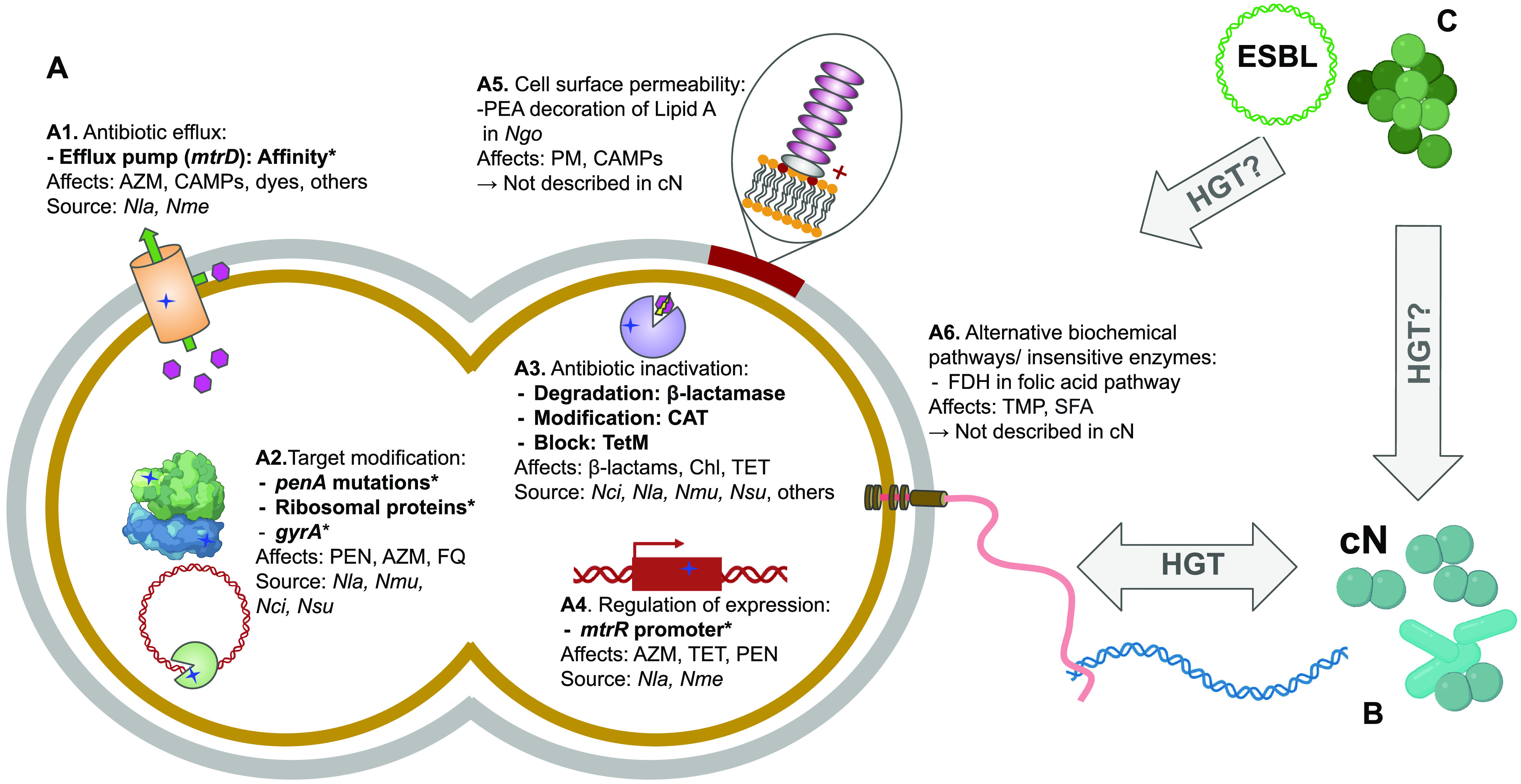
(A) Schematic representation of some of the known mechanisms of resistance in N. gonorrhoeae, including those that have been acquired from (bold text and *) or have been documented to be present in the commensal Neisseria (bold text). Known resistance encompasses multiple mechanisms, including: (A1) mutations enhancing the affinity of antibiotics for the Mtr efflux pump, increasing their efflux from the cell; (A2) mutations altering the structures of the targets of antibiotics reducing binding affinity (e.g., β-lactams [PEN] and PBP2, azithromycin [AZM] and ribosomal proteins, and fluoroquinolones [FQ] and DNA Gyrase Subunit A); (A3) genes encoding enzymes that modify antibiotics through degradation or blocking their target site; (A4) altered promoter motifs which increase or decrease the expression of genes; (A5) changes in cell surface permeability through mechanisms such as the decoration of Lipid A with phosphoethanolamine (PEA) (not yet described in commensal Neisseria); and (A6) development of alternative pathways to antibiotic targets (not yet described in commensal Neisseria). (B) Of the six described mechanisms, four have been shared from or documented to be present within Neisseria commensal communities and thus available for horizontal gene transfer (HGT) to pathogenic Neisseria through their natural competence and pilus-mediated DNA uptake machinery; the other two (A5 and A6) are yet to be described in commensal Neisseria. (C) Of great concern is the possibility that commensal Neisseria could acquire extended spectrum β-lactamase (ESBL) genes from other bacteria with which they live proximally to in the oral and nasopharynx, rendering them resistant to ceftriaxone, our last line of defense against gonorrhea. To note, chromosomal mutations encoding ceftriaxone resistance are already present in gonococcal populations (164); however, additional acquired β-lactam resistance mechanisms could hasten the wide-spread failure of this drug as an effective anti-gonococcal therapy. cN, commensal Neisseria; Nci, N. cinerea; Ngo, N. gonorrhoae; Nla, N. lactamica; Nme, N. meningitidis; Nmu, N. mucosa; Nsu, N. subflava; CAT, chloramphenicol acetyltransferase; CAMP, cationic antimicrobial peptides; SFA, sulfonamides; TMP, trimethoprim; TET, tetracycline; Chl, chloramphenicol; FDH, folate dehydrogenase; PM, polymyxins.
Hybrid penicillin binding protein 2 sequences.
The first documentation of resistance transferred from commensal to pathogenic Neisseria was reported in 1988 by Brian G. Spratt, as part of an investigation of a gonococcal isolate collected from an individual in Boston in 1984 (86). The isolate had an elevated penicillin MIC, which prompted the comparison and discovery of a sequence with low similarity to native gonococcal alleles in the carboxy-terminal region of the transpeptidase domain of Penicillin-Binding Protein 2 (PBP2, encoded by penA). Shortly thereafter, the genetic basis of penicillin resistance in a N. meningitidis strain (isolated in 1978 from the United Kingdom) was also identified as being conferred by a “hybrid” (now referred to as “mosaic”) penA allele (92). This allele was 22% divergent from the native meningococcal sequence, containing 44 amino acid substitutions and one insertion, and was most similar to the penA alleles present in N. flavescens (now reclassified as N. subflava) (32). Intragenus transfer of penA alleles has also been reported in penicillin-resistant N. lactamica strains (52, 87, 102). In gonococci, thirty-six “flavors” or haplotypes of penA alleles have been described and are classified using the roman numeric system as alleles I-XXXVI, based on substitutions at 82 amino acid positions (88). Mosaic alleles in gonococci include fragments acquired from N. cinerea and N. perflava (now also N. subflava), or N. perflava (N. subflava) alone (5). These mutations act by both (i) lowering the affinity of the beta-lactam antibiotics (e.g., penicillin, ceftriaxone, and cefixime) for PBP2, and (ii) restricting the motions of PBP2 which are important for acylation by beta-lactams (103).
Although mosaic alleles were originally found to render gonococci and other Neisseria resistant to penicillin, more concerningly mosaic alleles are also correlated with decreased susceptibility to ESCs, including ceftriaxone, the last remaining option for first-line empirical antimicrobial monotherapy for gonorrhea. In a U.S. population survey from 2000 to 2013, the predominant mosaic allele (penA XXXIV) was found in 91% of isolates with reduced susceptibility to ceftriaxone (MIC ≥0.125 μg/mL), and 98% of isolates with reduced susceptibility to another ESC cefixime (MIC ≥0.25 μg/mL) (22). Additionally, one of the penA XXXIV derivatives, designated penA41 and described in the H041 isolate collected from Japan in 2009, confers high-level resistance to ceftriaxone (MIC = 2 μg/mL) (88, 104). This allele has four unique substitutions, of which the amino acid changes A311V, V316P, and T483S are key to producing high-level ceftriaxone and cefixime MICs (105). Additional mutations in mosaic alleles important for intermediate-level ESC resistance include I312M, V316T, and G545S (106, 107). A A501V substitution has also been associated with decreased ESC susceptibility (106, 108), but has only been reported in nonmosaic isolates (107). Population prevalence of mosaic alleles is alarmingly increasing in some countries. In South Korea, prevalence increased from 1.1% to 23.9% from 2012 through 2017 (89); and in Vietnam, it increased from 0% in 2011% to 27% in 2015 to 2016 (90). Therefore, though fitness costs do exist for mosaic penA alleles (109), sustained spread (22, 50, 89–91), repeated acquisition (22, 50), and the evolution of compensatory mutations (i.e., AcnB Q371K) in natural populations in cooccurrence (110) with these alleles points to the continued importance of commensals in transmission of clinically relevant resistance to the pathogenic Neisseria.
The mosaic multiple transferable efflux pump (mtr) locus.
In 2012, a novel mtrR promoter sequence was described in a small cluster of gonococcal isolates from Australia (n = 10 of 397 total) with reduced susceptibility to azithromycin (≥2 μg/mL) (96). Sequences from these isolates failed to amplify on the Sequenom MassARRAY iPLEX platform, which identifies known antimicrobial resistance single nucleotide polymorphisms (SNP). Speculating mispriming due to divergent primer binding sites, the authors further characterized the locus in these isolates and found it to be more meningococcal-like than typical gonococcal promoter sequences. Since then, genomic surveys have discovered multiple “flavors” of mosaic mtr alleles in gonococcal populations, acquired from N. lactamica, a commensal species, and the meningococcus. Isolates with mosaic mtr alleles have a global distribution, and have been described in gonococci collected from the United States (22, 93), Canada (94), Australia (95, 96), and Germany (97). In a U.S. population data set, the prevalence of mosaic mtr sequences has increased precipitously since the recommendation to add azithromycin as part of the treatment for gonorrhea in 2012, from 1% before (2000 to 2011) to 6% (2012 to 2013) after the addition of the drug (22). Additionally, a recent 2017 New South Wales (Australia) survey estimated that 76% of reduced susceptibility to azithromycin had been derived from inheritance of mosaic mtr alleles (95), suggesting the persistence and expansion of these alleles in gonococcal populations over the last 2 decades globally.
The mechanistic basis of reduced azithromycin susceptibility in mtr mosaics stems from the interaction of multiple mutations across the mtr operon. The operon includes three coding domains: mtrC (which encodes a periplasmic membrane fusion protein), mtrD (encoding an inner membrane transporter of the resistance/nodulation/cell division [RND] family), and mtrE (encoding the outer membrane channel). The resulting tripartite efflux pump system spans the inner and outer membranes and exports diverse hydrophobic antimicrobial agents such as antibiotics, nonionic detergents, antibacterial peptides, bile salts, and gonadal steroid hormones from the cell (111–115). Using directed transformations of segments of mosaic mtr pump components, both Wadsworth et al. (13) and Rouquette-Loughlin et al. (15) found that two loci acting together were required to raise azithromycin MIC values above the reduced susceptibility threshold. These were: (i) a cis-acting single nucleotide change within the −35 A-nucleotide hexamer of the mtrCDE promoter which increases expression of the pump and efflux of azithromycin, and (ii) gain-of-function amino acid changes at the N- and C-terminal regions of MtrD which interacts epistatically with the promoter mutation.
Commensally acquired mtr sequences appear to have been acquired from N. lactamica and N. meningitidis as evidenced by high sequence similarity to these taxa (13, 15, 96, 116). Genomic signatures around the mtr locus were also found to be consistent with recent horizontal gene transfer and introgression of commensal alleles in the gonococcal genomic background: (i) elevated genetic diversity at loci recently acquired from other species, as these alleles would have diverged due to selection or drift in commensal strain backgrounds; (ii) increased linkage, as recombination would not have sufficient time to break down associations between loci inherited on the same recombination tract from divergent species; and (iii) admixed phylogenies between Neisseria species (13). These signatures have also been used to describe other loci transferred across species boundaries genome-wide (14).
Horizontal gene transfer of ribosomal protein genes.
Antibiotics such as tetracyclines, macrolides, aminoglycosides, or chloramphenicols, target ribosomal proteins thereby reducing protein synthesis. Hence, mutations within ribosomal proteins can reduce the affinity of these antibiotics to their targets and decrease the susceptibility of the bacterium. Manoharan-Basil et al. determined whether mutations within genes encoding ribosomal proteins acquired through HGT were associated with increased azithromycin resistance in N. gonorrhoeae (98). Azithromycin, a macrolide with a bacteriostatic effect, binds to the large subunit of the ribosome (50S) and prevents the translocation of the peptidyl-tRNA, releasing incomplete and nonfunctional proteins. The 50S ribosomal subunit contains 36 ribosomal proteins. Curating the Pathogenwatch (117) and NCBI databases (including 11,644 isolates of N. gonorrhoeae and 15 commensal Neisseria isolates), the team identified 23 alleles out of 36 as highly conserved ribosomal genes, and performed an extensive comparative analysis to assess the likelihood of the alleles having been acquired through horizontal gene transfer (98). The authors determined that alleles of the ribosomal genes rplB, rplD, and rplY coding for ribosomal proteins L2, L4 and L25, respectively, were likely to have been acquired by the gonococcus from commensals through HGT. Mutations from commensal Neisseria within these genes were associated with MICs 4 to 16 times greater than basal susceptible azithromycin MICs of 0.125 to 0.25 μg/mL. The authors noted that these mutations, however, were not sufficient to increase MICs alone, and may be compensatory mutations impacting fitness, or epistatic, requiring additional interacting mutations to exert their effects. While the direction of acquisition of the mutations is unknown, mutations are shared mostly with N. cinerea, N. mucosa, and N. lactamica. This study concludes that HGT among Neisseria species is widespread in time and space. Indeed, the observations of HGT were present in the majority (46/68) of countries investigated, for isolates across time, from preantibiotic (isolates predating 1950s), golden age (between 1950 and the 1970s), and postmodern (between 1980 and the present) eras (98).
Additional resistance-encoding SNPs in loci encoding ribosomal proteins have been identified in both pathogenic and commensal Neisseria, suggesting their importance in different genomic backgrounds for resistance emergence, and/or as latent reserves of resistance for other species. For example, Hu et al. identified a single point mutation in the rpsJ1 gene (previously called tet-2) of N. gonorrhoeae that leads to increased resistance to tetracycline. The single point mutation G → A in rpsJ1, leading to the amino-acid residue change V57M in the 30S ribosomal protein S10, observed in clinical isolates, appeared to increase resistance 4-fold compared with several parental laboratory strains (53). This mutation was also observed in commensal Neisseria species (118). However, a definitive correlation between tetracycline resistance and the V57M substitution was not observed, suggesting that additional mutations in mtrR and penB may also be needed to develop high levels of resistance to tetracycline (53). Although this example does not directly identify HGT of this particular SNP between commensal and pathogenic Neisseria, it highlights conserved mechanisms of resistance broadly distributed across the genus and suggests their immediate availability for rapid resistance transfer.
Transfer of quinolone resistance to N. meningitidis from commensals.
Reports of ciprofloxacin resistance in meningococcal populations are increasing and have been documented globally (119–121). In part, these cases are due to HGT from the commensal Neisseria. The first records of commensally acquired quinolone resistance in meningococci are from the United States in 2007 and 2008. Three cases of meningococci from serogroup B were identified as ciprofloxacin-resistant (with MICs of 0.25 μg/mL) in two states (North Dakota and Minnesota). These isolates all had acquired a T91I substitution in the gene encoding DNA Gyrase Subunit A (gyrA) from N. lactamica, which rendered them resistant. A more recent survey in China found a high prevalence of quinolone resistant meningococci (>70%) with multiple divergent gyrA and parC (encoding topoisomerase IV) haplotypes (99). Isolates with elevated MICs (≥0.25 μg/mL) had all acquired the same T91I mutation as the North American isolates mentioned above, with 53.4% of resistant alleles being acquired from the commensals: N. lactamica, N. cinerea, and N. subflava. Chen et al. also found that of 293 commensal Neisseria sampled, 99% were quinolone resistant, suggesting that quinolone resistance may have an especially high likelihood of interspecies transfer from commensal reservoirs to pathogens (99).
ANTIBIOTIC SELECTION ON COMMENSALS: FREQUENT BYSTANDERS TAKE UP ARMS
Antibiotic resistance in many bacterial species increases with the consumption of antibiotics in the community (122–126), and in gonococci several studies have suggested a link between antibiotic use and the emergence and spread of AMR (29, 30). For example, a survey across 24 European countries found that cephalosporin and fluoroquinolone use significantly increased ceftriaxone/cefixime and ciprofloxacin MICs, respectively (127). Increased azithromycin MICs have also been reported after azithromycin usage in patients at a Dutch sexual health clinic (128) and at a U.S. clinic in Portland (129). Additionally, seasonal variability in azithromycin use in the United States (increased in the winter compared with other months), has been correlated with seasonal fluctuations in azithromycin MICs in N. gonorrhoeae populations (higher in the winter and spring compared with summer and fall [130]). Finally, more generally, resistance has emerged to all new antimicrobials used to treat gonorrhea in only a few years or decades after their initial recommendation (131). These examples are unsurprising, and simply a function of the selective pressure of antibiotic use killing off susceptible bacteria, followed by the survivors (the more resistant members of those populations) propagating in ecological niches that become vacant postantibiotic treatment.
Importantly, most of the aforementioned studies investigating a link between antibiotic use and resistance support bystander selection as a main driver in the evolution of AMR. Bystander selection is the unintentional selection of resistance in bacteria not targeted by the prescribed antimicrobial therapy, and is higher in bacteria with high carriage rates, as they more frequently experience exposure to consumed drugs (132). Extrapolating this finding to Neisseria suggests that commensals (carriage rate ~100%) should experience higher bystander selection from antibiotic use compared with their pathogenic relatives (carriage rate between 0.01 and 10%) (29, 30, 133), and should be more likely to “take up arms” and display resistance to clinically important antimicrobials. Reduced drug susceptibility in commensal Neisseria populations has been shown to be directly selected for after antibiotic usage in a study of Vietnamese men, in which recent antibiotic use (within 1 month) was strongly associated with increased MICs to cefixime, ceftriaxone, and cefpodoxime in commensal Neisseria compared with a control population with no antibiotic use between 1 and 6 months prior to the study (28). This work highlights the impact of bystander selection on commensals, their persistent threat as evolutionary engines for resistance donation, and the underlying importance of characterizing the resistance phenotypes and genotypes of commensals to fully describe the Neisseria resistome.
THE CURRENT STATE OF WGS SURVEILLANCE IN COMMENSAL NEISSERIA
Currently, the characterization of resistance in commensal reservoirs has only been conducted by individual academic laboratory groups that have often focused on a small sample of strains, rather than a concerted effort, nationally supported, on a large population of clinical or general population isolates (27, 99, 134–136). Hence, there is a disproportionate amount of genomic data for N. gonorrhoeae and N. meningitidis isolates, compared with the number of deposits for commensal Neisseria species. A search of WGS deposits to the SRA (June 2022) revealed that pathogenic species, N. gonorrhoeae and N. meningitidis, are 100 to 1,000 times more represented compared with commensal species (N. gonorrhoeae: 42,799, N. meningitidis: 34,050, N. lactamica: 1,166, and less than 100 for each of the other human commensal Neisseria species, as of June 21, 2022). This undoubtedly leads to a molecular bias in comparative genomics analyses impacting findings and conclusions on Neisseria historic and contemporary evolution, including interspecific AMR gene transfer events (e.g., their frequency, presence, direction, etc.). A broad WGS database associated with AMR profiles of commensal Neisseria species is necessary for the scientific community to further anticipate the evolution of AMR in pathogenic species. We are at a critical inflection point for the maintenance of effective antimicrobial treatment of N. gonorrhoeae, and only with a better understanding of the spread of AMR across Neisseria species will we be able to develop better antimicrobial stewardship strategies against gonorrhea.
Most available studies linking MIC data to genomic sequences focus on small panels of commensals that are publicly available, or isolated by independent research groups. For example, Fiore et al. described a panel of 26 Neisseria, of which 14 isolates are typically considered human commensals, which are publicly available through the CDC and Food and Drug Administration’s (FDA) Antibiotic Resistance (AR) Isolate Bank within the “Neisseria species MALDI-TOF Verification panel” (118). Of these isolates, 10 had reduced susceptibility to at least one of the antibiotics tested (penicillin, ceftriaxone, cefixime, tetracycline, azithromycin, or ciprofloxacin); and the single N. bacilliformis was resistant to all tested drugs. Notably, all the N. subflava isolates (n = 5) were resistant to azithromycin, and some also displayed reduced susceptibility to tetracycline, penicillin, and cefixime. The authors coupled MIC test results with genome sequence to identify putative resistance-encoding mutations and found several known loci or alleles previously described in gonococci, including: gyrA (S91F), tetM, and TEM-type β-lactamases. The authors also noted that not all resistance-contributing mutations were able to be described in the study, evidenced by isolates with the same resistance haplotypes displaying variation in MIC. For example, two N. subflava isolates had MICs to penicillin of 3 and 1 μg/mL, despite having the same mutations in penA and porB. The field will remain severely limited in the genetic diversity of commensals represented given the extreme scarcity of commensal samples stored at public strain repositories; this highlights the importance of increasing the support for commensal collection through additional mechanisms (described below).
A thorough and comprehensive evaluation of the availability of published commensal genomic sequences with associated antimicrobial resistance data has recently been conducted by Vanbaelen et al. (30). In their systematic review, the authors found that despite over 295 published studies on commensals, only 17 contained sufficient data to link WGS with MIC values. Reasons for exclusion of the remaining studies included: No MIC data (n = 179), only a single isolate reported (n = 9), MIC data present but for nonrelevant antimicrobials (n = 6), animal-associated Neisseria reported (n = 3), missing data (n = 84), repeated characterization of an isolate (n = 1), and inferred MIC values (n = 1). Of the 15 retained studies (two studies were not accessible), the number of isolates characterized ranged from 4 to 491, with a mean of 115.3 ± 31.82 isolates per study. However, those collecting larger numbers of isolates within this list contained N. gonorrhoeae and N. meningitidis isolates as the primary focal species sampled. Limiting the list to those studies that only focused on collection of commensals (n = 10) the upper limit of isolates drops from 4 to 286, and the mean shrinks to 88 ± 30.0 isolates per study. Despite the small number of studies identified, key epidemiological and biological information was obtained such as: (i) commensal species do not appear to be intrinsically resistant, (ii) commensal species have increasing MIC values over time in multiple countries, and (iii) AMR prevalence in commensal species is a clear threat to AMR in pathogenic Neisseria. The paucity of commensal data sets, and their limited and biased geographic sampling (11 from Europe, five from Asia, one from the United States), identified by this comprehensive analysis of available data strongly supports the need for increased characterization and sequencing of commensal species through a broader national or international strategy. As we propose below, we believe that these data could be generated through established surveillance programs, and this expansion could provide the necessary pool of commensal Neisseria for a broad sampling of diversity within the genus, for the analysis of AMR profiles and sequence correlation to AMR.
CANARY IN THE COAL MINE: THE BENEFITS OF SURVEILLANCE IN COMMENSALS
The importance of commensals as a reservoir for resistance donation has led Kenyon et al. to propose a “pan-Neisseria” approach to selecting drugs for treatment of pathogenic Neisseria infections which should limit the selection of resistant commensal species, and prevent the spread of resistance determinants to pathogenic species (27). Building upon those goals, we call for the expansion of collection protocols to include commensal Neisseria to aid in the fight against AMR in pathogenic Neisseria. Specifically, we recommend that a point be added to the eGISP-optional analysis section (137), so that the collection of Neisseria from extragenital sites, from male and female patients with possible N. gonorrhoeae exposure (n = 25 per sentinel site per month), includes additional human-borne Neisseria species and is not restricted to pathogenic Neisseria. (Fig. 2). To achieve this, we recommend prioritizing oropharyngeal samples, which are more likely to harbor commensals than rectal samples, through oral rinse-and-gargle to collect diverse Neisseria species from distinct ecological sites in the mouth and nasopharynx, as described by Laumen et al. (138). The oral rinse-and-gargle method has been demonstrated to capture increased Neisseria species diversity as opposed to a posterior oropharyngeal/tonsillar swab, is less invasive than a swab, and is more readily implementable as a standardized tool across clinics (138).
FIG 2.

Flowchart of the current protocol for the Gonococcal Isolate Surveillance Project (GISP, run by the CDC). For each clinical collection site, the current GISP (orange) and eGISP (blue) programs sample 25 individuals from genital and 25 from nongenital sites per month, respectively. Currently, all samples are submitted for morphological testing and nucleic acid amplification testing (NAAT) and retained if bacterial isolates are Gram-negative, diplococci, oxidase-positive, have a tan/transparent colony morphology on modified Thayer-Martin media, and NAAT-positive for N. gonorrhoeae (Ngo). aSentinel sites not participating in eGISP discard their non-Ngo samples. Retained colonies are then subjected to AMR profiling and WGS. We propose a minor modification to the eGISP program (bold and red), in which all pharyngeal samples collected via oral rinse-and-gargle (138) are conserved if they display features inconsistent with N. gonorrhoeae, yet consistent with other members of the Neisseria genus (Gram−, diplococci or rods, and oxidase+) (bold and red). We also suggest that pharyngeal samples are inoculated onto LBVT.SNR media, which is selective for commensal Neisseria. Subsequent development of a pipeline for complete analysis of each resultant non-N. gonorrhoeae strain (AMR profiling, WGS, etc.) will allow for detailed characterization of Neisseria species and strain diversity, allowing for a comprehensive evaluation of the Neisseria resistome and other comparative genomics analyses of interest.
In addition, the current eGISP protocol isolates pathogenic Neisseria from patient samples through the use of a selective medium (modified Thayer-Martin media [MTM]) containing vancomycin (inhibits Gram-positive bacteria), nystatin (inhibits fungi), and colistin and trimethoprim (which inhibit many Gram-negative bacteria, but not pathogenic Neisseria) (137). However, commensal Neisseria display variable sensitivity to colistin which limits the utility of MTM media for sampling commensals (139, 140). Therefore, we suggest the use of an additional plate of complementary medium, specifically designed to isolate commensal Neisseria (LB containing vancomycin, trimethoprim, sucrose, neutral red, LBVT.SNR) (19, 138). While labor-intensive, this new strategy will undoubtedly create a wider and more diverse collection of commensal Neisseria than is currently available. Downstream morphology-, biochemical-, and genomic-based approaches (currently undertaken as part of eGSIP) should be used to further characterize the commensal Neisseria species. The integration of genus-wide Neisseria NAATs (e.g., sequencing of rplF is sufficient to differentiate pathogenic and commensal Neisseria species) will further aid in selecting commensal Neisseria for downstream analytical pipelines (141).
Once isolates are obtained, AMR profiling and WGS bioinformatics workflows used with N. gonorrhoeae can be expanded to newly isolated commensal Neisseria (142, 143). Alternatively, low-cost semiquantitative AMR profiling, by adding antibiotics of interest at an established concentration to plates, could be used to further select for AMR Neisseria (138). WGS and AMR profiling data can be used as the input for established bioinformatics pipelines. For example, the combined PubMLST (https://pubmlst.org/neisseria) and Bacterial Isolate Genome Sequence Database (BIGSdb) platforms (144, 145), which store both (i) information on gene-by-gene allelic variation across genera and species, and (ii) an isolate database with associated metadata and sequencing data, already have workflows for identification of AMR loci across Neisseria spp. The utility of these platforms has recently been demonstrated in describing associations between AMR, the presence of accessory genes, and population structure within N. gonorrhoeae (146). We believe that expanding this type of approach to commensal species could enhance epidemiological surveillance and alert us to early warning indicators of new resistance phenotypes and genotypes that could sweep through pathogenic Neisseria populations and possibly cause resistance outbreaks.
Therefore, the surveillance of commensal Neisseria species would require minor changes in the methodology of the clinical microbiology lab, and would not modify patients’ experiences nor sample collection at large (138). We acknowledge that our proposed modification to eGISP (adding the collection, characterization, and storage of commensal isolates) will be both a costly endeavor and require significantly increased effort for the personnel involved in surveillance programs; however, we also believe that our current blindness of known AMR reservoirs for Neisseria pathogens is a key gap in addressing AMR within the genus. Implementing this approach as a pilot within a single program (e.g., GISP) for a limited time, may thus prove a necessary first step to assess the overall impact of scaling modified eGISP to worldwide surveillance efforts (e.g., GASP and Euro-GASP). Ultimately, this expansion could uncover the global diversity of Neisseria species, the AMR phenotypes and genotypes harbored, and the prevalence of allelic exchange across species’ boundaries (see 14).
Complementary lab-based approaches to broad field-work surveillance of AMR in Neisseria include transformation of susceptible strains of different species with DNA from a resistant donor strain, and experimental evolution. Transformation experiments have proven useful and effective in uncovering molecular mechanisms of antimicrobial resistance (5, 61, 147). These experiments can be conducted intraspecies, and interspecies but with lower efficiency across species due to the barriers described previously, and the likely dependence of AMR expression on additive or epistatic interactions in divergent genomic backgrounds. Additionally, in vitro evolution experiments can reveal the spontaneous mutations caused by DNA replication and repair errors which increase mean fitness in new selective environments (148), and are quick and easy to implement in the research laboratory due to the short generation times of Neisseria (~60 min). Using this approach, multiple tandem duplications in the locus encoding the 50S ribosomal L34 protein (rpmH) and the intergenic region proximal to the 30S ribosomal S3 protein (rpsC) were identified to increase resistance (≥2 μg/mL) to the macrolide antibiotic azithromycin in the commensal N. elongata in 20 days (or approximately 480 generations) (149). A similar experiment also described an identical 7LKRTYQ12 sequence duplication in rpmH as a transient steppingstone to high level azithromycin resistance in N. gonorrhoeae (150). Multiple mutations, in particular in penA (encoding a A501V substitution) and ftsX (encoding a T31P substitution), have also been uncovered as causal to reduced ceftriaxone susceptibility in N. gonorrhoeae through in vitro selection (108). Thus, expansion of these experiments to additional drug and species combinations could help characterize the potential Neisseria resistome, improving genotype-based surveillance efforts (WGS or probe-based) which depend on a known database of resistance determinants in both pathogenic and commensal species within the genus. A key caveat of such evolutionary approaches is that mutations evolving in vitro may not be representative of those that can persist in natural populations. Despite this limitation, lab-based evolution experiments can still provide valuable information on potential resistance genes and should be viewed as complementary to the expansion of the surveillance program mentioned above.
One additional concern highlighting the immense importance of global surveillance across the Neisseria is the growing prevalence of transferable plasmids containing extended spectrum β-lactamase (ESBL) genes, which are present in ~30 human-associated bacterial species (151). ESBLs confer resistance to broad-spectrum cephalosporins with oxyimino side chains (e.g., ceftriaxone) and other β-lactam antibiotics (152), and were designated a serious and increasing threat within the CDC’s 2019 Antibiotic Resistance Threat Report (153). As commensal Neisseria species coevolve in the nasopharynx with respiratory tract pathogens which harbor plasmids containing ESBL genes (Fig. 1C), there is a serious threat that a commensal Neisseria could acquire these ESBL loci rendering them resistant to ceftriaxone, our last line of defense against gonorrhea. From there, either full or partial transfer of ESBL encoding sequences from carrying commensal Neisseria to N. gonorrhoeae may lead to a disastrous outcome for gonococcal treatment. Indeed, acquisition of β-lactamase expressing plasmids in the 1970s by N. gonorrhoeae from H. influenzae (154), which have persisted in contemporary gonococcal populations (22), imparted resistance to penicillin (typically > 8 μg/mL), and was a main driver for the discontinued use of penicillin as a treatment for gonorrhea. Considering the frequent genetic exchange across Neisseria species (11, 14, 23), known examples of gene transfer from other bacteria to Neisseria (154–156), and broad mobilization and dissemination of ESBL-containing plasmids across bacteria species and genera (157–159), we must consider the above nightmare scenario which underscores the importance of surveillance of commensals, coresidents of the same ecological niche as ESBL-carrying respiratory tract pathogens.
In closing, we highly encourage the Neisseria research community to support and focus on increasing the collection of commensal Neisseria by both clinical and academic laboratory groups, and sequence these isolates to broaden the database of genomic data for commensal Neisseria species. Not only will these isolates and corresponding sequence data sets aid in the fight against resistance by characterizing Neisseria alleles available for intragenus transfer, they may also aid the Neisseria research community by providing a pool of data to study other common/divergent genomic features and Neisseria biology at large, such as the presence and importance of prophages (160), the diversity of anti-phage defense systems (161), the identification of toxin/antitoxin systems (146), patterns of killing through DNA methylation (83), and identifying immunogenic antigens as candidate vaccines (162).
CONCLUSION
AMR in commensal Neisseria species serves as a reservoir for AMR in pathogenic Neisseria species, in particular N. gonorrhoeae. N. gonorrhoeae has evolved resistance mechanisms which reduce susceptibility to all antimicrobials used as first-line therapies for the treatment of uncomplicated gonorrhea, and as this review demonstrates, they are often acquired through horizontal gene transfer from closely related commensals. Commensal Neisseria, which are carried in nearly 100% of the human population, undergo unintentional antimicrobial treatment and selection, making these “bystanders” major participants in the acquisition, maintenance, and transfer of AMR determinants. Surveillance of gonorrhea is critical for sexually transmitted infection prevention and addressing AMR (163). As such, we propose that expanding surveillance to all Neisseria species will greatly strengthen our capacity to monitor AMR in N. gonorrhoeae and predict and react to antimicrobial resistance outbreaks across the globe by locating early warning indicators (i.e., AMR determinants) that could be on the verge of rapid dissemination into pathogenic Neisseria populations. Finally, surveillance, isolation, collection, sequencing, and analysis of commensal Neisseria genomes could broadly expand our understanding of the biology and AMR determinants within the Neisseria genus at large, and may provide novel insights into new strategies to treat and prevent pathogenic Neisseria infections. Ultimately, commensal Neisseria are key drivers of AMR emergence in important human pathogens, and thus it is critical for the Neisseria research community to consider supporting, developing, and funding the timely integrations of commensal Neisseria into established gonococcal clinical surveillance programs worldwide. We believe that public access to the resultant omics and phenotypic data sets will be critical to generating new insights for addressing AMR in the pathogenic Neisseria, species of global concern to public health.
ACKNOWLEDGMENTS
We thank William M. Shafer, of Emory University–School of Medicine, for his insightful advice and feedback on this manuscript. We thank Joshua S. Weitz, of the Georgia Institute of Technology, for feedback and editing of the manuscript.
Figures were created, in part, with BioRender.com.
Research by C.B.W. is supported by the College of Science and Thomas H. Gosnell School of Life Sciences at RIT. Research by M.G. is supported by the Department of Biology at Spelman College, the Learning Planet Institute (LPI) in Paris, France, and NSF Research Initiation Award (RIA #1800691).
Contributor Information
Maira Goytia, Email: mgoytia@spelman.edu.
Crista B. Wadsworth, Email: cbwsbi@rit.edu.
Nathan John Weyand, Ohio University.
Jacob Yount, Ohio State University.
REFERENCES
- 1.Mortimer TD, Grad YH. 2019. Applications of genomics to slow the spread of multidrug-resistant Neisseria gonorrhoeae. Ann N Y Acad Sci 1435:93–109. doi: 10.1111/nyas.13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Workowski KA, Bachmann LH, Chan PA, Johnston CM, Muzny CA, Park I, Reno H, Zenilman JM, Bolan GA. 2021. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep 70:1–187. doi: 10.15585/mmwr.rr7004a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowley J, Vander Hoorn S, Korenromp E, Low N, Unemo M, Abu-Raddad LJ, Chico RM, Smolak A, Newman L, Gottlieb S, Thwin SS, Broutet N, Taylor MM. 2019. Chlamydia, gonorrhoea, trichomoniasis and syphilis: global prevalence and incidence estimates, 2016. Bull World Health Organ 97:548–562P. doi: 10.2471/BLT.18.228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feavers IM, Heath AB, Bygraves JA, Maiden MCJ. 1992. Role of horizontal genetic exchange in the antigenic variation of the class 1 outer membrane protein of Neisseria meningitidis. Mol Microbiol 6:489–495. doi: 10.1111/j.1365-2958.1992.tb01493.x. [DOI] [PubMed] [Google Scholar]
- 5.Spratt BG, Bowler LD, Zhang QY, Zhou J, Smith JM. 1992. Role of interspecies transfer of chromosomal genes in the evolution of penicillin resistance in pathogenic and commensal Neisseria species. J Mol Evol 34:115–125. doi: 10.1007/BF00182388. [DOI] [PubMed] [Google Scholar]
- 6.Zhou J, Spratt BG. 1992. Sequence diversity within the argF, fbp and recA genes of natural isolates of Neisseria meningitidis: interspecies recombination within the argF gene. Mol Microbiol 6:2135–2146. doi: 10.1111/j.1365-2958.1992.tb01387.x. [DOI] [PubMed] [Google Scholar]
- 7.Derbie A, Mekonnen D, Woldeamanuel Y, Abebe T. 2020. Azithromycin resistant gonococci: a literature review. Antimicrob Resist Infect Control 9:138. doi: 10.1186/s13756-020-00805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St Cyr SB. 2020. Update to CDC’s treatment guidelines for gonococcal infection, 2020. MMWR Morb Mortal Wkly Rep 69. doi: 10.15585/mmwr.mm6950a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradford PA, Miller AA, O'Donnell J, Mueller JP. 2020. Zoliflodacin: an oral spiropyrimidinetrione antibiotic for the treatment of Neisseria gonorrhoeae, including multi-drug-resistant isolates. ACS Infect Dis 6:1332–1345. doi: 10.1021/acsinfecdis.0c00021. [DOI] [PubMed] [Google Scholar]
- 10.Taylor SN, Morris DH, Avery AK, Workowski KA, Batteiger BE, Tiffany CA, Perry CR, Raychaudhuri A, Scangarella-Oman NE, Hossain M, Dumont EF. 2018. Gepotidacin for the treatment of uncomplicated urogenital gonorrhea: a phase 2, randomized, dose-ranging, single-oral dose evaluation. Clin Infect Dis 67:504–512. doi: 10.1093/cid/ciy145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corander J, Connor TR, O'Dwyer CA, Kroll JS, Hanage WP. 2012. Population structure in the Neisseria, and the biological significance of fuzzy species. J R Soc Interface 9:1208–1215. doi: 10.1098/rsif.2011.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Wintersdorff CJH, Penders J, van Niekerk JM, Mills ND, Majumder S, van Alphen LB, Savelkoul PHM, Wolffs PFG. 2016. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front Microbiol 7. doi: 10.3389/fmicb.2016.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wadsworth CB, Arnold BJ, Sater MRA, Grad YH. 2018. Azithromycin resistance through interspecific acquisition of an epistasis-dependent efflux pump component and transcriptional regulator in Neisseria gonorrhoeae. mBio 9:e01419-18. doi: 10.1128/mBio.01419-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold B, Sohail M, Wadsworth C, Corander J, Hanage WP, Sunyaev S, Grad YH. 2020. Fine-scale haplotype structure reveals strong signatures of positive selection in a recombining bacterial pathogen. Mol Biol Evol 37:417–428. doi: 10.1093/molbev/msz225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouquette-Loughlin CE, Reimche JL, Balthazar JT, Dhulipala V, Gernert KM, Kersh EN, Pham CD, Pettus K, Abrams AJ, Trees DL, St Cyr S, Shafer WM. 2018. Mechanistic basis for decreased antimicrobial susceptibility in a clinical isolate of Neisseria gonorrhoeae possessing a mosaic-like mtr efflux pump locus. mBio 9:e02281-18. doi: 10.1128/mBio.02281-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Retchless AC, Kretz CB, Chang H-Y, Bazan JA, Abrams AJ, Norris Turner A, Jenkins LT, Trees DL, Tzeng Y-L, Stephens DS, MacNeil JR, Wang X. 2018. Expansion of a urethritis-associated Neisseria meningitidis clade in the United States with concurrent acquisition of N. gonorrhoeae alleles. BMC Genomics 19:176. doi: 10.1186/s12864-018-4560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamorro G, Ibarz-Pavon AB, Kawabata A, León ME, Orrego V, Nagai M, Gabastou JM. 2019. Carriage of Neisseria meningitidis and other Neisseria species among children and young adults in Paraguay. J Med Microbiol 68:1793–1801. [DOI] [PubMed] [Google Scholar]
- 18.Diallo K, Trotter C, Timbine Y, Tamboura B, Sow SO, Issaka B, Dano ID, Collard J-M, Dieng M, Diallo A, Mihret A, Ali OA, Aseffa A, Quaye SL, Bugri A, Osei I, Gamougam K, Mbainadji L, Daugla DM, Gadzama G, Sambo ZB, Omotara BA, Bennett JS, Rebbetts LS, Watkins ER, Nascimento M, Woukeu A, Manigart O, Borrow R, Stuart JM, Greenwood BM, Maiden MCJ. 2016. Pharyngeal carriage of Neisseria species in the African meningitis belt. J Infect 72:667–677. doi: 10.1016/j.jinf.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knapp JS, Hook EW. 1988. Prevalence and persistence of Neisseria cinerea and other Neisseria spp. in adults. J Clin Microbiol 26:896–900. doi: 10.1128/jcm.26.5.896-900.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu G, Tang CM, Exley RM. 2015. Non-pathogenic Neisseria: members of an abundant, multi-habitat, diverse genus. Microbiology (Reading) 161:1297–1312. doi: 10.1099/mic.0.000086. [DOI] [PubMed] [Google Scholar]
- 21.Halter R, Pohlner J, Meyer TF. 1989. Mosaic-like organization of IgA protease genes in Neisseria gonorrhoeae generated by horizontal genetic exchange in vivo. EMBO J 8:2737–2744. doi: 10.1002/j.1460-2075.1989.tb08415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grad YH, Harris SR, Kirkcaldy RD, Green AG, Marks DS, Bentley SD, Trees D, Lipsitch M. 2016. Genomic epidemiology of gonococcal resistance to extended-spectrum cephalosporins, macrolides, and fluoroquinolones in the United States, 2000–2013. J Infect Dis 214:1579–1587. doi: 10.1093/infdis/jiw420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanage WP, Fraser C, Spratt BG. 2005. Fuzzy species among recombinogenic bacteria. BMC Biol 3:6. doi: 10.1186/1741-7007-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton HL, Dillard JP. 2006. Natural transformation of Neisseria gonorrhoeae: from DNA donation to homologous recombination. Mol Microbiol 59:376–385. doi: 10.1111/j.1365-2958.2005.04964.x. [DOI] [PubMed] [Google Scholar]
- 25.Fussenegger M, Rudel T, Barten R, Ryll R, Meyer TF. 1997. Transformation competence and type-4 pilus biogenesis in Neisseria gonorrhoeae–a review. Gene 192:125–134. doi: 10.1016/s0378-1119(97)00038-3. [DOI] [PubMed] [Google Scholar]
- 26.Diallo K, MacLennan J, Harrison OB, Msefula C, Sow SO, Daugla DM, Johnson E, Trotter C, MacLennan CA, Parkhill J, Borrow R, Greenwood BM, Maiden MCJ. 2019. Genomic characterization of novel Neisseria species. Sci Rep 9:13742. doi: 10.1038/s41598-019-50203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenyon C, Laumen J, Manoharan-Basil S. 2021. Choosing new therapies for gonorrhoea: we need to consider the impact on the pan-Neisseria genome. a viewpoint. Antibiotics 10:515. doi: 10.3390/antibiotics10050515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong HV, Pham LQ, Nguyen HT, Nguyen MXB, Nguyen TV, May F, Le GM, Klausner JD. 2020. Decreased cephalosporin susceptibility of oropharyngeal Neisseria species in antibiotic-using men who have sex with men in Hanoi, Vietnam. Clin Infect Dis 70:1169–1175. doi: 10.1093/cid/ciz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenyon CR, Schwartz IS. 2018. Effects of sexual network connectivity and antimicrobial drug use on antimicrobial resistance in Neisseria gonorrhoeae. Emerg Infect Dis J 24:1195–1203. doi: 10.3201/eid2407.172104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanbaelen T, Van Dijck C, Laumen J, Gonzalez N, De Baetselier I, Manoharan-Basil SS, De Block T, Kenyon C. 2022. Global epidemiology of antimicrobial resistance in commensal Neisseria species: a systematic review. Int J Med Microbiol 312. doi: 10.1016/j.ijmm.2022.151551. [DOI] [PubMed] [Google Scholar]
- 31.Weyrich LS, Duchene S, Soubrier J, Arriola L, Llamas B, Breen J, Morris AG, Alt KW, Caramelli D, Dresely V, Farrell M, Farrer AG, Francken M, Gully N, Haak W, Hardy K, Harvati K, Held P, Holmes EC, Kaidonis J, Lalueza-Fox C, de la Rasilla M, Rosas A, Semal P, Soltysiak A, Townsend G, Usai D, Wahl J, Huson DH, Dobney K, Cooper A. 2017. Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544:357–361. doi: 10.1038/nature21674. [DOI] [PubMed] [Google Scholar]
- 32.Bennett JS, Jolley KA, Earle SG, Corton C, Bentley SD, Parkhill J, Maiden MCJ. 2012. A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology (Reading) 158:1570–1580. doi: 10.1099/mic.0.056077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donati C, Zolfo M, Albanese D, Tin Truong D, Asnicar F, Iebba V, Cavalieri D, Jousson O, De Filippo C, Huttenhower C, Segata N. 2016. Uncovering oral Neisseria tropism and persistence using metagenomic sequencing. Nat Microbiol 1:16070. doi: 10.1038/nmicrobiol.2016.70. [DOI] [PubMed] [Google Scholar]
- 34.Keijser BJF, Zaura E, Huse SM, van der Vossen JMBM, Schuren FHJ, Montijn RC, ten Cate JM, Crielaard W. 2008. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res 87:1016–1020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- 35.Burcham ZM, Garneau NL, Comstock SS, Tucker RM, Knight R, Metcalf JL, Genetics of Taste Lab Citizen Scientists . 2020. Patterns of oral microbiota diversity in adults and children: a crowdsourced population study. Sci Rep 10:2133. doi: 10.1038/s41598-020-59016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu J, Iragavarapu S, Nadkarni GN, Huang R, Erazo M, Bao X, Verghese D, Coca S, Ahmed MK, Peter I. 2018. Location-specific oral microbiome possesses features associated with CKD. Kidney Int Rep 3:193–204. doi: 10.1016/j.ekir.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeigler CC, Persson GR, Wondimu B, Marcus C, Sobko T, Modéer T. 2012. Microbiota in the oral subgingival biofilm is associated with obesity in adolescence. Obesity 20:157–164. doi: 10.1038/oby.2011.305. [DOI] [PubMed] [Google Scholar]
- 38.Christensen H, May M, Bowen L, Hickman M, Trotter CL. 2010. Meningococcal carriage by age: a systematic review and meta-analysis. Lancet Infect Dis 10:853–861. doi: 10.1016/S1473-3099(10)70251-6. [DOI] [PubMed] [Google Scholar]
- 39.Unemo M, Seifert HS, Hook EW, Hawkes S, Ndowa F, Dillon J-AR. 2019. Gonorrhoea. Nat Rev Dis Primer 5:1–23. [DOI] [PubMed] [Google Scholar]
- 40.Hill SA, Masters TL, Wachter J. 2016. Gonorrhea – an evolving disease of the new millennium. Microb Cell 3:371–389. doi: 10.15698/mic2016.09.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bazan JA, Turner AN, Kirkcaldy RD, Retchless AC, Kretz CB, Briere E, Tzeng Y-L, Stephens DS, Maierhofer C, Del Rio C, Abrams AJ, Trees DL, Ervin M, Licon DB, Fields KS, Roberts MW, Dennison A, Wang X. 2017. Large cluster of Neisseria meningitidis urethritis in Columbus, Ohio, 2015. Clin Infect Dis 65:92–99. doi: 10.1093/cid/cix215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seifert HS. 2019. Location, location, location—commensalism, damage and evolution of the pathogenic Neisseria. J Mol Biol 431:3010–3014. doi: 10.1016/j.jmb.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Taha M-K, Claus H, Lappann M, Veyrier FJ, Otto A, Becher D, Deghmane A-E, Frosch M, Hellenbrand W, Hong E, du Châtelet IP, Prior K, Harmsen D, Vogel U. 2016. Evolutionary events associated with an outbreak of meningococcal disease in men who have sex with men. PLoS One 11:e0154047. doi: 10.1371/journal.pone.0154047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith JM, Smith NH, O'Rourke M, Spratt BG. 1993. How clonal are bacteria? Proc Natl Acad Sci USA 90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biswas GD, Sox T, Blackman E, Sparling PF. 1977. Factors affecting genetic transformation of Neisseria gonorrhoeae. J Bacteriol 129:983–992. doi: 10.1128/jb.129.2.983-992.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamoen LW, Venema G, Kuipers OP. 2003. Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology (Reading) 149:9–17. doi: 10.1099/mic.0.26003-0. [DOI] [PubMed] [Google Scholar]
- 47.Goodgal SH. 1982. DNA uptake in Haemophilus transformation. Annu Rev Genet 16:169–192. doi: 10.1146/annurev.ge.16.120182.001125. [DOI] [PubMed] [Google Scholar]
- 48.Steinmoen H, Knutsen E, Håvarstein LS. 2002. Induction of natural competence in Streptococcus pneumoniae triggers lysis and DNA release from a subfraction of the cell population. Proc Natl Acad Sci USA 99:7681–7686. doi: 10.1073/pnas.112464599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Rourke M, Stevens E. 1993. Genetic structure of Neisseria gonorrhoeae populations: a non-clonal pathogen. J Gen Microbiol 139:2603–2611. doi: 10.1099/00221287-139-11-2603. [DOI] [PubMed] [Google Scholar]
- 50.Grad YH, Lipsitch M. 2014. Epidemiologic data and pathogen genome sequences: a powerful synergy for public health. Genome Biol 15:538. doi: 10.1186/s13059-014-0538-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma KC, Mortimer TD, Duckett MA, Hicks AL, Wheeler NE, Sánchez-Busó L, Grad YH. 2020. Increased power from conditional bacterial genome-wide association identifies macrolide resistance mutations in Neisseria gonorrhoeae. Nat Commun 11:5374. doi: 10.1038/s41467-020-19250-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowler L, Zhang Q-Y, Riou JY, Spratt BG. 1994. Interspecies recombination between the penA genes of Neisseria meningitidis and commensal Neisseria species during the emergence of penicillin resistance in N. meningitidis: natural events and laboratory simulation. J Bacteriol 176:333–337. doi: 10.1128/jb.176.2.333-337.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu M, Nandi S, Davies C, Nicholas RA. 2005. High-level chromosomally mediated tetracycline resistance in Neisseria gonorrhoeae results from a point mutation in the rpsJ gene encoding ribosomal protein s10 in combination with the mtrR and penB resistance determinants. Antimicrob Agents Chemother 49:4327–4334. doi: 10.1128/AAC.49.10.4327-4334.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ilina E, Malakhova M, Bodoev I, Filimonova A, Oparina N, Govorun V. 2013. Mutation in ribosomal protein S5 leads to spectinomycin resistance in Neisseria gonorrhoeae. Front Microbiol 4. doi: 10.3389/fmicb.2013.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isabella V, Wright LF, Barth K, Spence JM, Grogan S, Genco CA, Clark VL. 2008. cis- and trans-acting elements involved in regulation of norB (norZ), the gene encoding nitric oxide reductase in Neisseria gonorrhoeae. Microbiology (Reading) 154:226–239. doi: 10.1099/mic.0.2007/010470-0. [DOI] [PubMed] [Google Scholar]
- 56.Barth KR, Isabella VM, Clark VL. 2009. Biochemical and genomic analysis of the denitrification pathway within the genus Neisseria. Microbiology (Reading) 155:4093–4103. doi: 10.1099/mic.0.032961-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Potter AJ, Kidd SP, Edwards JL, Falsetta ML, Apicella MA, Jennings MP, McEwan AG. 2009. Thioredoxin reductase is essential for protection of Neisseria gonorrhoeae against killing by nitric oxide and for bacterial growth during interaction with cervical epithelial cells. J Infect Dis 199:227–235. doi: 10.1086/595737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ku SC, Schulz BL, Power PM, Jennings MP. 2009. The pilin O-glycosylation pathway of pathogenic Neisseria is a general system that glycosylates AniA, an outer membrane nitrite reductase. Biochem Biophys Res Commun 378:84–89. doi: 10.1016/j.bbrc.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 59.Tzeng Y-L, Bazan JA, Turner AN, Wang X, Retchless AC, Read TD, Toh E, Nelson DE, Del Rio C, Stephens DS. 2017. Emergence of a new Neisseria meningitidis clonal complex 11 lineage 11.2 clade as an effective urogenital pathogen. Proc Natl Acad Sci USA 114:4237–4242. doi: 10.1073/pnas.1620971114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gunn JS, Stein DC. 1996. Use of a non-selective transformation technique to construct a multiply restriction/modification-deficient mutant of Neisseria gonorrhoeae. Mol Gen Genet 251:509–517. doi: 10.1007/BF02173639. [DOI] [PubMed] [Google Scholar]
- 61.Sparling PF. 1966. Genetic transformation of Neisseria gonorrhoeae to streptomycin resistance. J Bacteriol 92:1364–1371. doi: 10.1128/jb.92.5.1364-1371.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jain S, Mościcka KB, Bos MP, Pachulec E, Stuart MCA, Keegstra W, Boekema EJ, van der Does C. 2011. Structural characterization of outer membrane components of the type IV pili system in pathogenic Neisseria. PLoS One 6:e16624. doi: 10.1371/journal.pone.0016624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nandi S, Swanson S, Tomberg J, Nicholas RA. 2015. Diffusion of antibiotics through the PilQ secretin in Neisseria gonorrhoeae occurs through the immature, sodium dodecyl sulfate-labile form. J Bacteriol 197:1308–1321. doi: 10.1128/JB.02628-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swanson J. 1973. Studies on gonococcus infection. IV. Pili: their role in attachment of gonococci to tissue culture cells. J Exp Med 137:571–589. doi: 10.1084/jem.137.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schoolnik GK, Fernandez R, Tai JY, Rothbard J, Gotschlich EC. 1984. Gonococcal pili. Primary structure and receptor binding domain. J Exp Med 159:1351–1370. doi: 10.1084/jem.159.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.So M, Billyard E, Deal C, Getzoff E, Hagblom P, Meyer TF, Segal E, Tainer J. 1985. Gonococcal pilus: genetics and structure. Curr Top Microbiol Immunol 118. [DOI] [PubMed] [Google Scholar]
- 67.Chen I, Dubnau D. 2004. DNA uptake during bacterial transformation. Nat Rev Microbiol 2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- 68.Findlay WA, Redfield RJ. 2009. Coevolution of DNA uptake sequences and bacterial proteomes. Genome Biol Evol 1:45–55. doi: 10.1093/gbe/evp005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolfgang M, van Putten JP, Hayes SF, Koomey M. 1999. The comP locus of Neisseria gonorrhoeae encodes a type IV prepilin that is dispensable for pilus biogenesis but essential for natural transformation. Mol Microbiol 31:1345–1357. doi: 10.1046/j.1365-2958.1999.01269.x. [DOI] [PubMed] [Google Scholar]
- 70.Frye SA, Nilsen M, Tønjum T, Ambur OH. 2013. Dialects of the DNA uptake sequence in Neisseriaceae. PLoS Genet 9:e1003458. doi: 10.1371/journal.pgen.1003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freitag NE, Seifert HS, Koomey M. 1995. Characterization of the pilF-pilD pilus-assembly locus of Neisseria gonorrhoeae. Mol Microbiol 16:575–586. doi: 10.1111/j.1365-2958.1995.tb02420.x. [DOI] [PubMed] [Google Scholar]
- 72.Brossay L, Paradis G, Fox R, Koomey M, Hébert J. 1994. Identification, localization, and distribution of the PilT protein in Neisseria gonorrhoeae. Infect Immun 62:2302–2308. doi: 10.1128/iai.62.6.2302-2308.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park H-SM, Wolfgang M, Koomey M. 2002. Modification of type IV pilus-associated epithelial cell adherence and multicellular behavior by the PilU protein of Neisseria gonorrhoeae. Infect Immun 70:3891–3903. doi: 10.1128/IAI.70.7.3891-3903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piepenbrink KH. 2019. DNA uptake by type IV filaments. Front Mol Biosci 6:1. doi: 10.3389/fmolb.2019.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Facius D, Meyer TF. 1993. A novel determinant (comA) essential for natural transformation competence in Neisseria gonorrhoeae and the effect of a comA defect on pilin variation. Mol Microbiol 10:699–712. doi: 10.1111/j.1365-2958.1993.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 76.Koomey JM, Falkow S. 1987. Cloning of the recA gene of Neisseria gonorrhoeae and construction of gonococcal recA mutants. J Bacteriol 169:790–795. doi: 10.1128/jb.169.2.790-795.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mehr IJ, Seifert HS. 1998. Differential roles of homologous recombination pathways in Neisseria gonorrhoeae pilin antigenic variation, DNA transformation and DNA repair. Mol Microbiol 30:697–710. doi: 10.1046/j.1365-2958.1998.01089.x. [DOI] [PubMed] [Google Scholar]
- 78.Stohl EA, Seifert HS. 2001. The recX gene potentiates homologous recombination in Neisseria gonorrhoeae. Mol Microbiol 40:1301–1310. doi: 10.1046/j.1365-2958.2001.02463.x. [DOI] [PubMed] [Google Scholar]
- 79.Cehovin A, Simpson PJ, McDowell MA, Brown DR, Noschese R, Pallett M, Brady J, Baldwin GS, Lea SM, Matthews SJ, Pelicic V. 2013. Specific DNA recognition mediated by a type IV pilin. Proc Natl Acad Sci USA 110:3065–3070. doi: 10.1073/pnas.1218832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stein DC, Gunn JS, Radlinska M, Piekarowicz A. 1995. Restriction and modification systems of Neisseria gonorrhoeae. Gene 157:19–22. doi: 10.1016/0378-1119(94)00649-D. [DOI] [PubMed] [Google Scholar]
- 81.Roberts RJ, Vincze T, Posfai J, Macelis D. 2015. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43:D298–299. doi: 10.1093/nar/gku1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blow MJ, Clark TA, Daum CG, Deutschbauer AM, Fomenkov A, Fries R, Froula J, Kang DD, Malmstrom RR, Morgan RD, Posfai J, Singh K, Visel A, Wetmore K, Zhao Z, Rubin EM, Korlach J, Pennacchio LA, Roberts RJ. 2016. The epigenomic landscape of prokaryotes. PLoS Genet 12:e1005854. doi: 10.1371/journal.pgen.1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim WJ, Higashi D, Goytia M, Rendón MA, Pilligua-Lucas M, Bronnimann M, McLean JA, Duncan J, Trees D, Jerse AE, So M. 2019. Commensal Neisseria kill Neisseria gonorrhoeae through a DNA-dependent mechanism. Cell Host Microbe 26:228–239.e8. doi: 10.1016/j.chom.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.So M, Rendón MA. 2019. Tribal warfare: commensal Neisseria kill pathogen Neisseria gonorrhoeae using its DNA. Microb Cell 6:544–546. doi: 10.15698/mic2019.12.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mortimer TD, Zhang JJ, Ma KC, Grad YH. 2022. Loci for prediction of penicillin and tetracycline susceptibility in Neisseria gonorrhoeae: a genome-wide association study. Lancet Microbe 3:e376–e381. doi: 10.1016/S2666-5247(22)00034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Spratt BG. 1988. Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature 332:173–176. doi: 10.1038/332173a0. [DOI] [PubMed] [Google Scholar]
- 87.Ameyama S, Onodera S, Takahata M, Minami S, Maki N, Endo K, Goto H, Suzuki H, Oishi Y. 2002. Mosaic-like structure of penicillin-binding protein 2 gene (penA) in clinical isolates of Neisseria gonorrhoeae with reduced susceptibility to cefixime. Antimicrob Agents Chemother 46:3744–3749. doi: 10.1128/AAC.46.12.3744-3749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ohnishi M, Golparian D, Shimuta K, Saika T, Hoshina S, Iwasaku K, Nakayama S, Kitawaki J, Unemo M. 2011. Is Neisseria gonorrhoeae initiating a future era of untreatable gonorrhea?: detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrob Agents Chemother 55:3538–3545. doi: 10.1128/AAC.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee H, Suh YH, Lee S, Kim Y-K, Han M-S, Bae HG, Unemo M, Lee K. 2019. Emergence and spread of cephalosporin-resistant Neisseria gonorrhoeae with mosaic penA alleles, South Korea, 2012-2017. Emerg Infect Dis 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lan PT, Golparian D, Ringlander J, Van Hung L, Van Thuong N, Unemo M. 2020. Genomic analysis and antimicrobial resistance of Neisseria gonorrhoeae isolates from Vietnam in 2011 and 2015–16. J Antimicrob Chemother 75:1432–1438. doi: 10.1093/jac/dkaa040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Demczuk W, Lynch T, Martin I, Van Domselaar G, Graham M, Bharat A, Allen V, Hoang L, Lefebvre B, Tyrrell G, Horsman G, Haldane D, Garceau R, Wylie J, Wong T, Mulvey MR. 2015. Whole-genome phylogenomic heterogeneity of Neisseria gonorrhoeae isolates with decreased cephalosporin susceptibility collected in Canada between 1989 and 2013. J Clin Microbiol 53:191–200. doi: 10.1128/JCM.02589-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spratt BG, Zhang Q-Y, Jones DM, Hutchison A, Brannigan JA, Dowson CG. 1989. Recruitment of a penicillin-binding protein gene from Neisseria flavescens during the emergence of penicillin resistance in Neisseria meningitidis. Proc Natl Acad Sci USA 86:8988–8992. doi: 10.1073/pnas.86.22.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gernert KM, Seby S, Schmerer MW, Thomas JC, Pham CD, St Cyr S, Schlanger K, Weinstock H, Shafer WM, Raphael BH, Kersh EN, Hun S, Hua C, Ruiz R, Soge OO, Dominguez C, Patel A, Loomis J, Leavitt J, Zhang J, Baldwin T, Wang C, Moore C, Whelen C, O'Brien P, Harvey A, Antimicrobial-Resistant Neisseria Gonorrhoeae Working Group . 2020. Azithromycin susceptibility of Neisseria gonorrhoeae in the USA in 2017: a genomic analysis of surveillance data. Lancet Microbe 1:e154–e164. doi: 10.1016/S2666-5247(20)30059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Demczuk W, Martin I, Peterson S, Bharat A, Van Domselaar G, Graham M, Lefebvre B, Allen V, Hoang L, Tyrrell G, Horsman G, Wylie J, Haldane D, Archibald C, Wong T, Unemo M, Mulvey MR. 2016. Genomic epidemiology and molecular resistance mechanisms of azithromycin-resistant Neisseria gonorrhoeae in Canada from 1997 to 2014. J Clin Microbiol 54:1304–1313. doi: 10.1128/JCM.03195-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Whiley DM, Kundu RL, Jennison AV, Buckley C, Limnios A, Hogan T, Enriquez R, El Nasser J, George CR, Lahra MM. 2018. Azithromycin-resistant Neisseria gonorrhoeae spreading amongst men who have sex with men (MSM) and heterosexuals in New South Wales, Australia, 2017. J Antimicrob Chemother 73:1242–1246. doi: 10.1093/jac/dky017. [DOI] [PubMed] [Google Scholar]
- 96.Trembizki E, Doyle C, Jennison A, Smith H, Bates J, Lahra M, Whiley D, on behalf of the GRAND study investigators . 2014. A Neisseria gonorrhoeae strain with a meningococcal mtrR sequence. J Med Microbiol 63:1113–1115. doi: 10.1099/jmm.0.074286-0. [DOI] [PubMed] [Google Scholar]
- 97.Banhart S, Selb R, Oehlmann S, Bender J, Buder S, Jansen K, Heuer D. 2021. The mosaic mtr locus as major genetic determinant of azithromycin resistance of Neisseria gonorrhoeae-Germany, 2018. J Infect Dis 224:1398–1404. doi: 10.1093/infdis/jiab091. [DOI] [PubMed] [Google Scholar]
- 98.Manoharan-Basil SS, Laumen JGE, Van Dijck C, De Block T, De Baetselier I, Kenyon C. 2021. Evidence of horizontal gene transfer of 50S ribosomal genes rplB, rplD, and rplY in Neisseria gonorrhoeae. Front Microbiol 12. doi: 10.3389/fmicb.2021.683901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen M, Zhang C, Zhang X, Chen M. 2020. Meningococcal quinolone resistance originated from several commensal Neisseria species. Antimicrob Agents Chemother 64:e01494-19. doi: 10.1128/AAC.01494-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lewis LA, Choudhury B, Balthazar JT, Martin LE, Ram S, Rice PA, Stephens DS, Carlson R, Shafer WM. 2009. Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect Immun 77:1112–1120. doi: 10.1128/IAI.01280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Averett DR, Roth B, Burchall JJ, Baccanari DP. 1979. Dihydrofolate reductase from Neisseria sp. Antimicrob Agents Chemother 15:428–435. doi: 10.1128/AAC.15.3.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Demczuk W, Sidhu S, Unemo M, Whiley DM, Allen VG, Dillon JR, Cole M, Seah C, Trembizki E, Trees DL, Kersh EN, Abrams AJ, de Vries HJC, van Dam AP, Medina I, Bharat A, Mulvey MR, Van Domselaar G, Martin I. 2017. Neisseria gonorrhoeae sequence typing for antimicrobial resistance, a novel antimicrobial resistance multilocus typing scheme for tracking global dissemination of N. gonorrhoeae strains. J Clin Microbiol 55:1454–1468. doi: 10.1128/JCM.00100-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Singh A, Turner JM, Tomberg J, Fedarovich A, Unemo M, Nicholas RA, Davies C. 2020. Mutations in penicillin-binding protein 2 from cephalosporin-resistant Neisseria gonorrhoeae hinder ceftriaxone acylation by restricting protein dynamics. J Biol Chem 295:7529–7543. doi: 10.1074/jbc.RA120.012617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohnishi M, Saika T, Hoshina S, Iwasaku K, Nakayama S, Watanabe H, Kitawaki J. 2011. Ceftriaxone-resistant Neisseria gonorrhoeae, Japan. Emerg Infect Dis 17:148–149. doi: 10.3201/eid1701.100397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tomberg J, Unemo M, Ohnishi M, Davies C, Nicholas RA. 2013. Identification of amino acids conferring high-level resistance to expanded-spectrum cephalosporins in the penA gene from Neisseria gonorrhoeae strain H041. Antimicrob Agents Chemother 57:3029–3036. doi: 10.1128/AAC.00093-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takahata S, Senju N, Osaki Y, Yoshida T, Ida T. 2006. Amino acid substitutions in mosaic penicillin-binding protein 2 associated with reduced susceptibility to cefixime in clinical isolates of Neisseria gonorrhoeae. Antimicrob Agents Chemother 50:3638–3645. doi: 10.1128/AAC.00626-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomberg J, Unemo M, Davies C, Nicholas RA. 2010. Molecular and structural analysis of mosaic variants of penicillin-binding protein 2 conferring decreased susceptibility to expanded-spectrum cephalosporins in Neisseria gonorrhoeae: role of epistatic mutations. Biochemistry 49:8062–8070. doi: 10.1021/bi101167x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gong Z, Lai W, Liu M, Hua Z, Sun Y, Xu Q, Xia Y, Zhao Y, Xie X. 2016. Novel genes related to ceftriaxone resistance found among ceftriaxone-resistant Neisseria gonorrhoeae strains selected in vitro. Antimicrob Agents Chemother 60:2043–2051. doi: 10.1128/AAC.00149-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vincent LR, Kerr SR, Tan Y, Tomberg J, Raterman EL, Dunning Hotopp JC, Unemo M, Nicholas RA, Jerse AE. 2018. In vivo-selected compensatory mutations restore the fitness cost of mosaic penA alleles that confer ceftriaxone resistance in Neisseria gonorrhoeae. mBio 9:e01905-17. doi: 10.1128/mBio.01905-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thomas JC, Seby S, Abrams AJ, Cartee J, Lucking S, Vidyaprakash E, Schmerer M, Pham CD, Hong J, Torrone E, Cyr SS, Shafer WM, Bernstein K, Kersh EN, Gernert KM, Antimicrobial-Resistant Neisseria Gonorrhoeae Working Group . 2019. Evidence of recent genomic evolution in gonococcal strains with decreased susceptibility to cephalosporins or azithromycin in the United States, 2014–2016. J Infect Dis 220:294–305. doi: 10.1093/infdis/jiz079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Delahay RM, Robertson BD, Balthazar JT, Shafer WM, Ison CA. 1997. Involvement of the gonococcal MtrE protein in the resistance of Neisseria gonorrhoeae to toxic hydrophobic agents. Microbiol Read Engl 143:2127–2133. doi: 10.1099/00221287-143-7-2127. [DOI] [PubMed] [Google Scholar]
- 112.Hagman KE, Shafer WM. 1995. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J Bacteriol 177:4162–4165. doi: 10.1128/jb.177.14.4162-4165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hagman KE, Lucas CE, Balthazar JT, Snyder L, Nilles M, Judd RC, Shafer WM. 1997. The MtrD protein of Neisseria gonorrhoeae is a member of the resistance/nodulation/division protein family constituting part of an efflux system. Microbiol Read Engl 143:2117–2125. doi: 10.1099/00221287-143-7-2117. [DOI] [PubMed] [Google Scholar]
- 114.Lucas CE, Balthazar JT, Hagman KE, Shafer WM. 1997. The MtrR repressor binds the DNA sequence between the mtrR and mtrC genes of Neisseria gonorrhoeae. J Bacteriol 179:4123–4128. doi: 10.1128/jb.179.13.4123-4128.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pan W, Spratt BG. 1994. Regulation of the permeability of the gonococcal cell envelope by the mtr system. Mol Microbiol 11:769–775. doi: 10.1111/j.1365-2958.1994.tb00354.x. [DOI] [PubMed] [Google Scholar]
- 116.Shafer WM. 2018. Mosaic drug efflux gene sequences from commensal Neisseria can lead to low-level azithromycin resistance expressed by Neisseria gonorrhoeae clinical isolates. mBio 9:e01747-18. doi: 10.1128/mBio.01747-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sánchez-Busó L, Yeats CA, Taylor B, Goater RJ, Underwood A, Abudahab K, Argimón S, Ma KC, Mortimer TD, Golparian D, Cole MJ, Grad YH, Martin I, Raphael BH, Shafer WM, Town K, Wi T, Harris SR, Unemo M, Aanensen DM. 2021. A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of Neisseria gonorrhoeae at Pathogenwatch. Genome Med 13:61. doi: 10.1186/s13073-021-00858-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fiore MA, Raisman JC, Wong NH, Hudson AO, Wadsworth CB. 2020. Exploration of the Neisseria resistome reveals resistance mechanisms in commensals that may be acquired by N. gonorrhoeae through horizontal gene transfer. Antibiotics 9:656. doi: 10.3390/antibiotics9100656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kawasaki Y, Matsubara K, Takahashi H, Morita M, Ohnishi M, Hori M, Isome K, Iwata A, Nigami H, Ikemachi M, Yamamoto G, Ohkusu K. 2018. Invasive meningococcal disease due to ciprofloxacin-resistant Neisseria meningitidis sequence type 4821: the first case in Japan. J Infect Chemother 24:305–308. doi: 10.1016/j.jiac.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 120.Gorla MC, Cassiolato AP, Pinhata JMW, de Moraes C, Corso A, Gagetti P, Lemos AP. 2018. Emergence of resistance to ciprofloxacin in Neisseria meningitidis in Brazil. J Med Microbiol 67:286–288. doi: 10.1099/jmm.0.000685. [DOI] [PubMed] [Google Scholar]
- 121.Tsang RSW, Law DKS, Deng S, Hoang L. 2017. Ciprofloxacin-resistant Neisseria meningitidis in Canada: likely imported strains. Can J Microbiol 63:265–268. doi: 10.1139/cjm-2016-0716. [DOI] [PubMed] [Google Scholar]
- 122.Bronzwaer SLAM, Cars O, Buchholz U, Mölstad S, Goettsch W, Veldhuijzen IK, Kool JL, Sprenger MJW, Degener JE, European Antimicrobial Resistance Surveillance System . 2002. A European study on the relationship between antimicrobial use and antimicrobial resistance. Emerg Infect Dis 8:278–282. doi: 10.3201/eid0803.010192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Riedel S, Beekmann SE, Heilmann KP, Richter SS, Garcia-de-Lomas J, Ferech M, Goosens H, Doern GV. 2007. Antimicrobial use in Europe and antimicrobial resistance in Streptococcus pneumoniae. Eur J Clin Microbiol Infect Dis 26:485–490. doi: 10.1007/s10096-007-0321-5. [DOI] [PubMed] [Google Scholar]
- 124.Goossens H, Ferech M, Vander Stichele R, Elseviers M, ESAC Project Group . 2005. Outpatient antibiotic use in Europe and association with resistance: a cross-national database study. Lancet Lond Engl 365:579–587. doi: 10.1016/S0140-6736(05)17907-0. [DOI] [PubMed] [Google Scholar]
- 125.Bruyndonckx R, Hens N, Aerts M, Goossens H, Cortiñas Abrahantes J, Coenen S. 2015. Exploring the association between resistance and outpatient antibiotic use expressed as DDDs or packages. J Antimicrob Chemother 70:1241–1244. doi: 10.1093/jac/dku525. [DOI] [PubMed] [Google Scholar]
- 126.Olesen SW, Barnett ML, MacFadden DR, Brownstein JS, Hernández-Díaz S, Lipsitch M, Grad YH. 2018. The distribution of antibiotic use and its association with antibiotic resistance. Elife 7:e39435. doi: 10.7554/eLife.39435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kenyon C, Buyze J, Spiteri G, Cole MJ, Unemo M. 2020. Population-level antimicrobial consumption is associated with decreased antimicrobial susceptibility in Neisseria gonorrhoeae in 24 European countries: an ecological analysis. J Infect Dis 221:1107–1116. [DOI] [PubMed] [Google Scholar]
- 128.Wind CM, de Vries E, Schim van der Loeff MF, van Rooijen MS, van Dam AP, Demczuk WHB, Martin I, de Vries HJC. 2017. Decreased azithromycin susceptibility of Neisseria gonorrhoeae isolates in patients recently treated with azithromycin. Clin Infect Dis 65:37–45. doi: 10.1093/cid/cix249. [DOI] [PubMed] [Google Scholar]
- 129.Soge OO, Harger D, Schafer S, Toevs K, Raisler KA, Venator K, Holmes KK, Kirkcaldy RD. 2012. Emergence of increased azithromycin resistance during unsuccessful treatment of Neisseria gonorrhoeae infection with azithromycin (Portland, OR, 2011). Sex Transm Dis 39:877–879. doi: 10.1097/OLQ.0b013e3182685d2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Olesen SW, Torrone EA, Papp JR, Kirkcaldy RD, Lipsitch M, Grad YH. 2019. Azithromycin susceptibility among Neisseria gonorrhoeae isolates and seasonal macrolide use. J Infect Dis 219:619–623. doi: 10.1093/infdis/jiy551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Unemo M, Shafer WM. 2014. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev 27:587–613. doi: 10.1128/CMR.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tedijanto C, Olesen SW, Grad YH, Lipsitch M. 2018. Estimating the proportion of bystander selection for antibiotic resistance among potentially pathogenic bacterial flora. Proc Natl Acad Sci USA 115:E11988–E11995. doi: 10.1073/pnas.1810840115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dong HV, Klausner JD. 2019. Neisseria gonorrhoeae resistance driven by antibiotic use. Nat Rev Urol 16:509–510. doi: 10.1038/s41585-019-0206-2. [DOI] [PubMed] [Google Scholar]
- 134.De Block T, Laumen JGE, Van Dijck C, Abdellati S, De Baetselier I, Manoharan-Basil SS, Van den Bossche D, Kenyon C. 2021. WGS of commensal Neisseria reveals acquisition of a new ribosomal protection protein (MsrD) as a possible explanation for high level azithromycin resistance in Belgium. Pathogens 10:384. doi: 10.3390/pathogens10030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Goytia M, Thompson ST, Jordan SVL, King KA. 2021. Antimicrobial resistance profiles of human commensal Neisseria species. Antibiotics 10:538. doi: 10.3390/antibiotics10050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Laumen JGE, Van Dijck C, Abdellati S, De Baetselier I, Serrano G, Manoharan-Basil SS, Bottieau E, Martiny D, Kenyon C. 2022. Antimicrobial susceptibility of commensal Neisseria in a general population and men who have sex with men in Belgium. Sci Rep 12:9. doi: 10.1038/s41598-021-03995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.St Cyr SB, Kriesel K, Pham CD, Raphael BH. 2021. Gonococcal Isolate Surveillance Project (GISP) and Enhanced GISP (eGISP) Protocol. CDC. [Google Scholar]
- 138.Laumen JGE, Abdellati S, Van Dijck C, Martiny D, De Baetselier I, Manoharan-Basil SS, Van den Bossche D, Kenyon C. 2022. A novel method to assess antimicrobial susceptibility in commensal oropharyngeal Neisseria—a pilot study. Antibiotics 11:100. doi: 10.3390/antibiotics11010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Clark SA, Gray S, Finn A, Borrow R. 2021. Colistin sensitivity and factor H-binding protein expression among commensal Neisseria species. mSphere 6:e0017521. doi: 10.1128/mSphere.00175-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Anand CM, Ashton F, Shaw H, Gordon R. 1991. Variability in growth of Neisseria polysaccharea on colistin-containing selective media for Neisseria spp. J Clin Microbiol 29:2434–2437. doi: 10.1128/jcm.29.11.2434-2437.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bennett JS, Watkins ER, Jolley KA, Harrison OB, Maiden MCJ. 2014. Identifying Neisseria species by use of the 50S ribosomal protein L6 (rplF) gene. J Clin Microbiol 52:1375–1381. doi: 10.1128/JCM.03529-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yahara K, Nakayama S-I, Shimuta K, Lee K-I, Morita M, Kawahata T, Kuroki T, Watanabe Y, Ohya H, Yasuda M, Deguchi T, Didelot X, Ohnishi M. 2018. Genomic surveillance of Neisseria gonorrhoeae to investigate the distribution and evolution of antimicrobial-resistance determinants and lineages. Microb Genom 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Singh R, Dillon J-AR, Demczuk W, Kusalik A. 2019. Gen2Epi: an automated whole-genome sequencing pipeline for linking full genomes to antimicrobial susceptibility and molecular epidemiological data in Neisseria gonorrhoeae. BMC Genomics 20:165. doi: 10.1186/s12864-019-5542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jolley KA, Maiden MCJ. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jolley KA, Bray JE, Maiden MCJ. 2018. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124. doi: 10.12688/wellcomeopenres.14826.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de Korne-Elenbaas J, Bruisten SM, van Dam AP, Maiden MCJ, Harrison OB. 2022. The Neisseria gonorrhoeae accessory genome and its association with the core genome and antimicrobial resistance. Microbiol Spectr 10:e0265421. doi: 10.1128/spectrum.02654-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zarantonelli L, Borthagaray G, Lee E-H, Shafer WM. 1999. Decreased azithromycin susceptibility of Neisseria gonorrhoeae due to mtrR mutations. Antimicrob Agents Chemother 43:2468–2472. doi: 10.1128/AAC.43.10.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jansen G, Barbosa C, Schulenburg H. 2013. Experimental evolution as an efficient tool to dissect adaptive paths to antibiotic resistance. Drug Resist Updat 16:96–107. doi: 10.1016/j.drup.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 149.Raisman JC, Fiore MA, Tomin L, Adjei JKO, Aswad VX, Chu J, Domondon CJ, Donahue BA, Masciotti CA, McGrath CG, Melita J, Podbielski PA, Schreiner MR, Trumpore LJ, Wengert PC, Wrightstone EA, Hudson AO, Wadsworth CB. 2022. Evolutionary paths to macrolide resistance in a Neisseria commensal converge on ribosomal genes through short sequence duplications. PLoS One 17:e0262370. doi: 10.1371/journal.pone.0262370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Laumen JGE, Manoharan-Basil SS, Verhoeven E, Abdellati S, De Baetselier I, Crucitti T, Xavier BB, Chapelle S, Lammens C, Van Dijck C, Malhotra-Kumar S, Kenyon C. 2021. Molecular pathways to high-level azithromycin resistance in Neisseria gonorrhoeae. J Antimicrob Chemother 76:1752–1758. doi: 10.1093/jac/dkab084. [DOI] [PubMed] [Google Scholar]
- 151.Zhao W-H, Hu Z-Q. 2013. Epidemiology and genetics of CTX-M extended-spectrum β-lactamases in Gram-negative bacteria. Crit Rev Microbiol 39:79–101. doi: 10.3109/1040841X.2012.691460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bradford PA. 2001. Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev 14:933–951. doi: 10.1128/CMR.14.4.933-951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Centers for Disease Control and Prevention (U.S.). 2019. Antibiotic resistance threats in the United States, 2019. Centers for Disease Control and Prevention (U.S.). [Google Scholar]
- 154.Elwell LP, Roberts M, Mayer LW, Falkow S. 1977. Plasmid-mediated beta-lactamase production in Neisseria gonorrhoeae. Antimicrob Agents Chemother 11:528–533. doi: 10.1128/AAC.11.3.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McNicol PJ, Albritton WL, Ronald AR. 1986. Transfer of plasmid-mediated ampicillin resistance from Haemophilus to Neisseria gonorrhoeae requires an intervening organism. Sex Transm Dis 13:145–150. doi: 10.1097/00007435-198607000-00006. [DOI] [PubMed] [Google Scholar]
- 156.Morse SA, Johnson SR, Biddle JW, Roberts MC. 1986. High-level tetracycline resistance in Neisseria gonorrhoeae is result of acquisition of streptococcal tetM determinant. Antimicrob Agents Chemother 30:664–670. doi: 10.1128/AAC.30.5.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lerminiaux NA, Cameron ADS. 2019. Horizontal transfer of antibiotic resistance genes in clinical environments. Can J Microbiol 65:34–44. doi: 10.1139/cjm-2018-0275. [DOI] [PubMed] [Google Scholar]
- 158.Kocer K, Boutin S, Dalpke AH, Heeg K, Mutters NT, Nurjadi D. 2020. Comparative genomic analysis reveals a high prevalence of inter-species in vivo transfer of carbapenem-resistance plasmids in patients with haematological malignancies. Clin Microbiol Infect 26:780.e1-780–e8. doi: 10.1016/j.cmi.2019.10.014. [DOI] [PubMed] [Google Scholar]
- 159.Conlan S, Thomas PJ, Deming C, Park M, Lau AF, Dekker JP, Snitkin ES, Clark TA, Luong K, Song Y, Tsai Y-C, Boitano M, Dayal J, Brooks SY, Schmidt B, Young AC, Thomas JW, Bouffard GG, Blakesley RW, Mullikin JC, Korlach J, Henderson DK, Frank KM, Palmore TN, Segre JA, NISC Comparative Sequencing Program . 2014. Single-molecule sequencing to track plasmid diversity of hospital-associated carbapenemase-producing Enterobacteriaceae. Sci Transl Med 6:254ra126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Orazi G, Collins AJ, Whitaker RJ. 2022. Prediction of prophages and their host ranges in pathogenic and commensal Neisseria species. mSystems 7:e0008322. doi: 10.1128/msystems.00083-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tesson F, Hervé A, Mordret E, Touchon M, d'Humières C, Cury J, Bernheim A. 2022. Systematic and quantitative view of the antiviral arsenal of prokaryotes. Nat Commun 13:2561. doi: 10.1038/s41467-022-30269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Baarda BI, Zielke RA, Holm AK, Sikora AE. 2021. Comprehensive bioinformatic assessments of the variability of Neisseria gonorrhoeae vaccine candidates. mSphere 6:e00977-20. doi: 10.1128/mSphere.00977-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.World Health Organization. 2016. Global health sector strategy on sexually transmitted infections 2016–2021. Towards ending STIs. WHO/RHR/16.09. World Health Organization. Department of Reproductive Health and Research. [Google Scholar]
- 164.Zhao S, Duncan M, Tomberg J, Davies C, Unemo M, Nicholas RA. 2009. Genetics of chromosomally mediated intermediate resistance to ceftriaxone and cefixime in Neisseria gonorrhoeae. Antimicrob Agents Chemother 53:3744–3751. doi: 10.1128/AAC.00304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]