

Summary
Transcriptomics data have been integrated with genome-wide association studies (GWASs) to help understand disease/trait molecular mechanisms. The utility of metabolomics, integrated with transcriptomics and disease GWASs, to understand molecular mechanisms for metabolite levels or diseases has not been thoroughly evaluated. We performed probabilistic transcriptome-wide association and locus-level colocalization analyses to integrate transcriptomics results for 49 tissues in 706 individuals from the GTEx project, metabolomics results for 1,391 plasma metabolites in 6,136 Finnish men from the METSIM study, and GWAS results for 2,861 disease traits in 260,405 Finnish individuals from the FinnGen study. We found that genetic variants that regulate metabolite levels were more likely to influence gene expression and disease risk compared to the ones that do not. Integrating transcriptomics with metabolomics results prioritized 397 genes for 521 metabolites, including 496 previously identified gene-metabolite pairs with strong functional connections and suggested 33.3% of such gene-metabolite pairs shared the same causal variants with genetic associations of gene expression. Integrating transcriptomics and metabolomics individually with FinnGen GWAS results identified 1,597 genes for 790 disease traits. Integrating transcriptomics and metabolomics jointly with FinnGen GWAS results helped pinpoint metabolic pathways from genes to diseases. We identified putative causal effects of UGT1A1/UGT1A4 expression on gallbladder disorders through regulating plasma (E,E)-bilirubin levels, of SLC22A5 expression on nasal polyps and plasma carnitine levels through distinct pathways, and of LIPC expression on age-related macular degeneration through glycerophospholipid metabolic pathways. Our study highlights the power of integrating multiple sets of molecular traits and GWAS results to deepen understanding of disease pathophysiology.
Keywords: genome-wide association study, metabolomics, transcriptomics, transcriptome-wide association study, colocalizataion
Graphical abstract
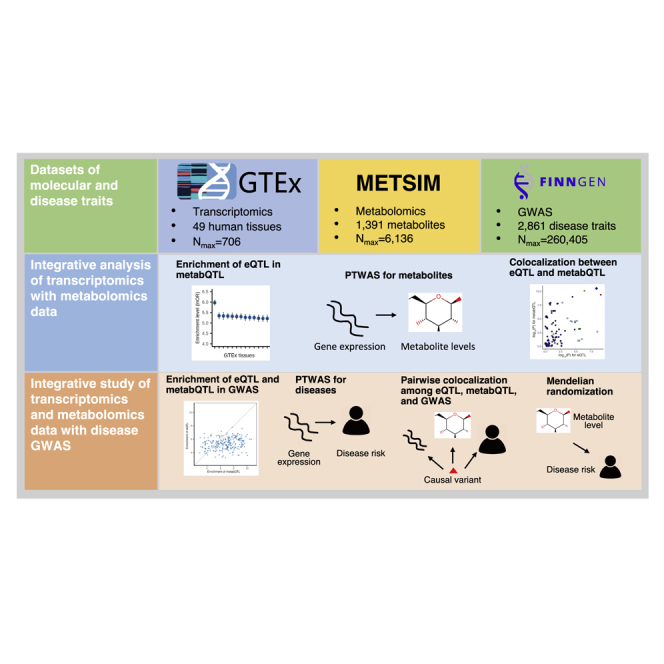
We integrated transcriptomics data for 49 human tissues, metabolomics data for 1,391 metabolites, and genome-wide association studies for 2,861 diseases. We identified 397 genes for 521 metabolites and 1,597 genes for 790 diseases. The results help understand the metabolic pathways underlying disease genetic associations.
Introduction
Genome-wide association studies (GWASs) have identified hundreds of thousands of genetic variants robustly associated with human diseases and traits.1 However, the underlying molecular mechanisms for most associations remain unclear. Many recent studies have investigated the molecular mechanisms through integrative analysis of GWAS and omics data.2 These integrative studies have helped prioritize genes3 and putative causal variants4 and tissues and cell types of action5 for GWAS associations. Most of these integrative studies have used only transcriptomics data and typically have provided few clues into disease metabolic pathways. The recent advent of high-throughput technology makes large-scale metabolomics profiling feasible and affordable,6 opening an avenue for integrating metabolomics data with disease GWAS and transcriptomics data.
Metabolomics analysis comprehensively profiles small molecules (i.e., metabolites) in biosamples. Many metabolites are associated with human diseases.7 Studies have used metabolomics data to identify disease biomarkers and to investigate disease metabolic pathways.8 Disease GWASs are usually carried out in much larger cohorts with little or no transcriptomics or metabolomics data. To help understand molecular mechanisms for disease GWAS associations, current studies often integrate disease GWASs with external transcriptomics or metabolomics data that are measured in individuals different from the GWAS samples,3,9 but seldom have all three data types been integrated jointly. Compared with transcriptomics, large-scale metabolomics data have become widely available only more recently and have been integrated with disease GWASs in only a few studies.9, 10, 11, 12 The extent to which metabolomics data in large cohorts improve our understanding of disease molecular mechanisms beyond the integration of transcriptomics and GWAS results has not been systematically investigated.
Metabolite levels are biological readouts of age, environment, lifestyle, and the genome.13 Understanding genetic mechanisms underlying the variation in metabolite levels benefits the investigation of disease molecular mechanisms when integrating metabolomics with disease GWASs. GWASs have discovered thousands of genetic associations (metabQTLs) with many metabolite levels.14 However, the underlying mechanisms for many metabQTLs are unknown. Most metabQTLs fall in non-coding genomic regions but the extent to which gene expression is responsible for these metabQTLs has not been fully characterized. Transcriptomics data have been integrated with metabQTLs, but integration was limited to transcriptomics data of lymphoblastoid cell lines or to only 64 blood metabolites.15,16
Transcriptome-wide association studies (TWASs) integrate GWAS and transcriptomics datasets to identify gene-trait associations.17, 18, 19, 20, 21 Colocalization analysis evaluates the probability that two or more traits share the same causal variant(s).22, 23, 24 Additionally, enrichment analysis can evaluate the overall over-representation of colocalized sites by assessing whether genetic variants associated with one trait are more likely to be associated with another trait.25, 26, 27 TWASs and colocalization analysis are statistical methods used to integrate omics and GWAS data. Combining TWASs and colocalization analysis increases the positive predictive value for gene association discovery compared to TWASs alone.15 Probabilistic TWASs (PTWASs), which implement TWASs in an instrument variable framework, facilitate the testing and estimation of causal effects for genes.28 Compared with variant-level colocalization analysis, locus-level colocalization increases statistical power to identify colocalization.27
We profiled plasma metabolites in 6,136 Finnish men from the Metabolic Syndrome in Men (METSIM) study by using the Metabolon mass spectrometry platform. We recently performed GWASs for 1,391 metabolites, identifying 2,030 independent metabolite genetic associations (metabQTLs) at experiment-wide significance.29 Through statistical fine-mapping analysis and linking metabolite biochemical features with functions for genes near the metabQTLs, we nominated 290 genes underlying 1,427 of the 2,030 metabQTLs, comprising 1,495 gene-metabolite pairs between the 290 genes and 631 metabolites.
Here, we apply state-of-the-art PTWASs and locus-level colocalization to systematically integrate Genotype-Tissue Expression (GTEx) transcriptomics and METSIM metabolomics data to improve our mechanistic understanding of metabQTLs and disease GWAS associations. We investigated three interrelated scientific questions. (1) To what extent is the genetic basis shared between gene expression and metabolite levels? (2) Does integrating two types of omics data (here, metabolomics and transcriptomics) with disease GWASs identify gene-disease associations that were not identified in studies integrating only one type of omics data (here metabolomics or transcriptomics alone)? (3) Does integrating transcriptomics and metabolomics data simultaneously with disease GWASs identify disease molecular mechanisms that were not identified in studies integrating a single type of omics data? Our study provides a strategy for integrating two types of omics data simultaneously with GWAS results. The findings shed metabolic insights into disease molecular mechanisms.
Material and Methods
GTEx
The GTEx project aims to build a catalog of genetic regulatory sites and their effects on gene expression across a variety of human tissues and to elucidate the molecular mechanisms of disease genetic associations.30 GTEx determined genotypes at 46.5M variants through genome and exome sequencing and assayed gene expression and splicing levels in 54 tissues by using RNA sequencing. Genetic regulatory effects on gene expression and splicing were investigated in expression (eQTL) and splicing (sQTL) quantitative trait locus analysis.
For each cis-eQTL (≤1 megabase [Mb]), GTEx analyzed individual-level expression and genotype data by using deterministic approximation of posteriors (DAP-g)26 to fine map causal variants and distinguish them from their linkage disequilibrium (LD) proxies.30 DAP-g seeks to identify all independent association signals for each cis-eQTL. For each such signal, DAP-g computes (1) a posterior inclusion probability (SPIP) to quantify the probability of at least one causal variant and (2) a variant posterior inclusion probability (VPIP) to quantify the probability that the variant is causal for the signal.
GTEx data processing, eQTL, and DAP-g fine-mapping analyses were previously described.30 For this integrative study, we used cis-eQTL and DAP-g fine-mapping results in all 49 tissues with 73 to 706 individuals from GTEx version 8 (hereafter transcriptomics results). We complied with the GTEx data use agreement.
METSIM metabolomics study
METSIM is a single-site study of 10,197 Finnish men aged 45 to 74 years at baseline from Kuopio, Finland, designed to investigate risk factors for type 2 diabetes and cardiovascular diseases.31 All METSIM participants provided written informed consent. We performed non-targeted metabolomics profiling in 6,136 randomly selected non-diabetics by using the Metabolon DiscoveryHD4 mass spectrometry platform (Durham, North Carolina, USA) on EDTA-plasma samples obtained after ≥10 h overnight fast during baseline visits from 2005 to 2010. The ethics committee at the University of Eastern Finland and the institutional review board at the University of Michigan approved the METSIM metabolomics study.
We previously performed single-variant GWASs for 1,391 metabolites, identifying 2,030 independent associations (metabQTLs).29 To identify the causal variants for these 2,030 associations, we created 1,501 genomic regions of ∼2 Mb centered on the index variants and performed fine-mapping analysis in each region by using DAP-g.26 Within the 1,501 regions, DAP-g analysis identified 1,952 signals with SPIP ≥ 0.95.29 For this integrative study, we used GWAS summary statistics at 16.2M genetic variants for the 1,391 metabolites and the DAP-g fine-mapping results (hereafter metabolomics results).
FinnGen study
The FinnGen study is designed to collect and analyze genome and digital healthcare data from 500,000 Finnish participants to identify new therapeutic and diagnostic targets for diseases.32 Participants in FinnGen provided informed consent for biobank research on the basis of the Finnish Biobank Act. Alternatively, separate research cohorts, collected prior to when the Finnish Biobank Act came into effect (in September 2013) and the start of FinnGen (August 2017), were collected on the basis of study-specific consents and later transferred to the Finnish biobanks after approval by Fimea, the National Supervisory Authority for Welfare and Health. Recruitment protocols followed biobank protocols approved by Fimea. The Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa (HUS) approved the FinnGen study protocol Nr HUS/990/2017. The FinnGen study is approved by Finnish Institute for Health and Welfare (permit numbers: THL/2031/6.02.00/2017, THL/1101/5.05.00/2017, THL/341/6.02.00/2018, THL/2222/6.02.00/2018, THL/283/6.02.00/2019, THL/1721/5.05.00/2019, THL/1524/5.05.00/2020, and THL/2364/14.02/2020), Digital and Population Data Service Agency (permit numbers: VRK43431/2017-3, VRK/6909/2018-3, and VRK/4415/2019-3), Social Insurance Institution (permit numbers: KELA 58/522/2017, KELA 131/522/2018, KELA 70/522/2019, KELA 98/522/2019, KELA 138/522/2019, KELA 2/522/2020, and KELA 16/522/2020), and Statistics Finland (permit numbers: TK-53-1041-17 and TK-53-90-20).
FinnGen identified disease status (hereafter disease trait) with the following Finnish national registries: Drug Purchase and Drug Reimbursement and Digital and Population Data Services Agency; Digital and Population Data Services Agency; Statistics Finland; Register of Primary Health Care Visits (AVOHILMO); Care Register for Health Care (HILMO); and Finnish Cancer Registry. For each participant, these registries recorded disease-relevant codes of the International Classification of Diseases (ICD) revisions 8, 9, and 10, cancer-specific ICD-O-3, Nordic Medico-Statistical Committee (NOMESCO) procedure, Finnish-specific Social Insurance Institute (KELA) drug reimbursement, and Anatomical Therapeutic Chemical (ATC). FinnGen genotyped each participant with an Illumina or Affymetrix array, followed by genotype imputation with the Finnish-specific Sequencing Initiative Suomi (SISu) v3 reference panel.33
For each disease trait, a GWAS was carried out via mixed model logistic regression in SAIGE.34 For each disease trait with an association at p < 10−6, FinnGen created a 3 Mb region (±1.5 Mb) around each lead variant and merged overlapping regions. They performed statistical fine-mapping analysis within each region by using FINEMAP v1.4_051035 and SuSiE 0.8.1.0545.36
For this integrative study, we used the GWAS summary statistics at 16.96M genetic variants (imputation quality info score > 0.9) and FINEMAP fine-mapping analysis results for all 2,861 disease traits in up to 260,405 individuals from FinnGen release 6 (hereafter FinnGen disease trait GWAS results).
Enrichment of eQTLs in metabQTLs and enrichment of molecular QTLs in FinnGen disease trait GWASs
We applied enrichment analysis to test for greater overlap than expected by chance between causal genetic associations for two traits by using the statistical procedure implemented in fastENLOC v2.0.27 FastENLOC explicitly models the latent causal associations (denoted by Ɣ and d) for the two traits and accounts for the uncertainty of inferring unobserved Ɣ and d values in genetic association analysis. The enrichment level is quantified by
where each probability represents the frequency of a possible causal association scenario. Note that log odds ratio (OR) = 0 indicates no enrichment and Pr(, d) = Pr()×Pr(d).
To characterize the extent to which metabQTL sites are more likely to confer eQTL effects, we estimated the enrichment of eQTLs in METSIM metabQTLs for each of the 49 GTEx tissues. We estimate the enrichment level by using the natural logarithm odds ratio of being an eQTL in a specific tissue for a metabQTL versus a non-metabQTL. We used a Bonferroni significance threshold of p < 0.05/49 = 1.0 × 10−3 to account for 49 GTEx tissues.
Similarly, to evaluate the extent to which eQTLs and metabQTLs influence the risk of human diseases, we estimated the enrichment levels of molecular QTLs (i.e., eQTLs and metabQTLs) in causal GWAS hits for the 2,861 FinnGen disease traits. We used a Bonferroni significance threshold of p < 0.05/2,861 = 1.7 × 10−5 to account for 2,861 disease traits.
PTWAS
To take advantage of the availability of GTEx eQTL DAP-g fine-mapping results and identify genes associated with metabolites and disease traits, we implemented a PTWAS for each METSIM metabolite and FinnGen disease trait.28 PTWASs employ an instrument variable framework to infer causal relationships and estimate putative causal effects of gene expression on outcome traits (e.g., metabolites or disease traits). PTWASs use the signals identified in GTEx eQTL DAP-g fine-mapping analysis as instrument variables. By using eQTL DAP-g probabilistic annotations, PTWASs take advantage of widespread allelic heterogeneity and accounts for LD.28 We used the PTWASs to estimate gene expression’s putative causal effects on metabolites and disease traits for each of the corresponding eQTL DAP-g signals with SPIP ≥ 0.5. We quantified heterogeneity of gene expression’s putative causal effect sizes by I2.37 We performed PTWASs in each of the 49 GTEx tissues and limited our analysis to up to 19,988 protein-coding genes.38 Given widespread eQTL sharing across tissues39 and pervasive phenotypic correlation between metabolites29 and FinnGen disease traits,32 we followed the common practice of TWASs18 and applied false discovery rate (FDR) < 5%, which can achieve a better trade-off between type I and II errors,40 to claim significant gene associations. When an eQTL had ≥2 causal signals with SPIP ≥ 0.5 in the DAP-g fine-mapping analysis, we required heterogeneity I2 < 0.15 to focus on genes with low heterogeneity of putative causal effects across DAP-g signals.
Pairwise colocalization between GTEx eQTLs, METSIM metabQTLs, and FinnGen disease trait GWAS loci
To identify whether pairs of eQTLs, metabQTLs, and disease trait GWAS loci shared causal variants, we carried out pairwise Bayesian colocalization analysis by using fastENLOC v2.0.24,27 We avoided using existing colocalization tools that allow simultaneous consideration of >2 data types41,42 because certain assumptions (e.g., at most a single causal variant in the region of interest) and the required prior information cannot easily be justified. FinnGen release 6 includes FINEMAP-based35 fine-mapping analysis results of genetic variant posterior inclusion probabilities for GWAS loci of 2,797 disease traits with at least one association at p < 10−6. FastENLOC v2.0 allows multiple causal variants in colocalization analysis and computes two probabilities by using these FinnGen fine-mapping results and the DAP-g-based fine-mapping results for GTEx eQTLs and METSIM metabQTLs.24,27 The locus-level colocalization posterior probability (LCP) is the probability that the same variant within the locus is causal for a pair of traits. The variant-level colocalization posterior probability (SCP) is the probability that a specific variant is causal for both traits. We presented colocalizations with LCP ≥ 0.5. We used the enrichment estimate (see enrichment of eQTLs in metabQTLs and enrichment of molecular QTLs in FinnGen disease trait GWASs) as the prior for Bayesian analysis in fastENLOC v2.0.24,26,27
Identification of gene-metabolite-disease trait combinations
To leverage both transcriptomics and metabolomics results and investigate molecular mechanisms for disease traits, we jointly analyzed the results for the three pairwise integrative analyses among transcriptomics, metabolomics, and disease trait GWAS results. In step 1, we identified gene-disease trait pairs with PTWAS FDR < 5% and colocalization LCP ≥ 0.5 in the integrative analysis of GTEx transcriptomics results and FinnGen disease trait GWASs. In step 2, we identified metabolite-disease trait pairs with colocalization LCP ≥ 0.5 in the integrative analysis of METSIM metabolomics results and FinnGen disease trait GWASs. In step 3, we built gene-metabolite-disease trait combinations by matching the gene-disease trait pairs identified in step 1 and the metabolite-disease trait pairs identified in step 2 by disease trait. Last, we required the resulting gene-metabolite-disease trait combinations to have PTWAS for metabolites FDR < 5% and eQTL-metabQTL colocalization LCP ≥ 0.5.
Mendelian randomization
We used PTWASs to infer the putative causal effects of gene expression on both metabolites and disease traits (see PTWAS). To complete the inference of causal relationships between gene expression, metabolite levels, and disease traits, we also examined the putative causal effects of METSIM plasma metabolite levels on FinnGen disease traits or vice versa by applying four two-sample Mendelian randomization methods: inverse variance weighted,43 weighted median,44 Egger regression,45 and MR-PRESSO.46 These methods make different assumptions and use different strategies to account for horizontal pleiotropy to control false positive inference of causality. For each exposure, we identified nearly independent genetic instrument variables (LD r2 < 0.1, distance ≥ 500 kilobases [kb]) with single-variant association p < 10−6. We considered findings significant if they had consistent effect direction and p < 0.05 for all four Mendelian randomization methods. We present MR-PRESSO effect sizes and p values in the main text.
To account for the possible confounding effects of HDL-C, LDL-C, and triglycerides on the putative causal effects of ten plasma metabolites relevant to glycerophospholipid metabolic pathways—i.e., 1-palmitoyl-GPE (16:0), 1-stearoyl-2-oleoyl-GPE (18:0/18:1), 1-palmitoyl-2-dihomo-linolenoyl-GPE (16:0/20:3), 1-stearoyl-2-docosahexaenoyl-GPE (18:0/22:6), 1-oleoyl-2-arachidonoyl-GPE (18:1/20:4), 1-palmitoyl-2-oleoyl-GPE (16:0/18:1), 1-stearoyl-2-arachidonoyl-GPE (18:0/20:4), 1-palmitoyl-2-arachidonoyl-GPE (16:0/20:4), 1-oleoyl-2-docosahexaenoyl-GPE (18:1/22:6), and 1-palmitoyl-2-docosahexaenoyl-GPE (16:0/22:6)—on age-related macular degeneration, we repeated GWASs for each metabolite after controlling for the effects of HDL-C, LDL-C, and triglyceride levels in METSIM and reran the Mendelian randomization analysis by using the resulting metabolite GWAS summary statistics.
Results
We sought to elucidate disease molecular mechanisms by integrating transcriptomics and metabolomics results with disease GWAS results. To improve the mechanistic understanding of metabQTLs before integrating METSIM metabolomics with FinnGen disease trait GWAS results, we integrated GTEx transcriptomics with METSIM metabolomics results in PTWASs and colocalization analysis (Figure 1A). To identify gene-disease associations, we integrated GTEx transcriptomics and METSIM metabolomics separately with FinnGen disease trait GWAS results in PTWASs and colocalization analysis (Figure 1B). We showed that jointly analyzing the results for the aforementioned three sets of pairwise integrative analyses improved the understanding of disease molecular mechanisms (Figure 1C).
Figure 1.

Study design
(A) Integrative analysis of GTEx transcriptomics and METSIM metabolomics results helps identify the underlying molecular mechanisms for metabQTLs by using PTWASs and colocalization analyses.
(B) Integrative analyses of one type of omics (transcriptomics or metabolomics) and FinnGen disease trait GWAS results helps identify gene-disease associations by using PTWASs and colocalization analyses.
(C) Joint integrative analysis of transcriptomics, metabolomics, and disease trait GWAS results by intersecting three sets of pairwise integrative analysis results improves understanding of disease molecular mechanisms compared with integrative analysis of a single type of omics results. LCP, locus-level colocalization posterior probability.
Relationship between eQTLs and metabQTLs
MetabQTL sites also likely confer eQTL effects
To evaluate the extent to which metabQTL sites also confer eQTL effects, we estimated the enrichment of GTEx eQTLs from 49 tissues in METSIM metabQTLs relative to non-metabQTLs as quantified by log odds ratios (see material and methods). We found eQTLs in all 49 tissues were significantly enriched in plasma metabQTLs (enrichment median = 5.12; p < 0.05/49 = 1.0 × 10−3 after Bonferroni correction for the 49 tissues; Figure 2A and S1). This most likely reflects eQTL sharing39 and the nature of plasma metabolite levels as metabolite aggregate concentrations across tissues.47 Of the 49 tissues, liver showed the strongest enrichment (enrichment = 5.98; 95% confidence interval: 5.74–6.21; p = 2.6 × 10−532), consistent with the liver's central role in regulating plasma metabolite levels.
Figure 2.

Enrichment levels of GTEx eQTLs in METSIM metabQTLs across 49 tissues and eQTLs and metabQTLs in GWASs for FinnGen disease traits
(A) Enrichment levels (natural logarithm of odds ratio ± standard error) of GTEx eQTLs in METSIM metabQTLs across 49 tissues. The sample size for eQTL analysis in each of the 49 tissues is included in parentheses.
(B) Enrichment levels of GTEx eQTLs (yellow) and METSIM metabQTLs (blue) in GWASs for all 2,861 FinnGen disease traits with overlap (teal).
(C) Enrichment levels of GTEx eQTLs and METSIM metabQTLs in GWASs for the 246 FinnGen disease traits with significant enrichment of both eQTLs and metabQTLs. lnOR, natural logarithm of odds ratio.
Impact of gene expression on plasma metabolite levels
To identify the impact of gene expression on plasma metabolite levels, we performed PTWASs28 for the 1,391 metabolites assayed in METSIM.29 For gene-metabolite pairs identified in PTWASs, we subsequently performed a Bayesian locus-level colocalization analysis27 to evaluate whether the same causal variants were shared between their genetic associations.
PTWASs identified 3,914 genes associated with 1,274 of the 1,391 metabolites (FDR < 0.05) and estimated the putative causal effects of gene expression on plasma metabolite levels (Figure S2 and Table S1). PTWASs identified 1 to 83 genes per metabolite (mean = 9.9, median = 7.0), for a total of 12,575 gene-metabolite pairs. For 1,354 of the 12,575 gene-metabolite pairs (between 397 genes and 521 metabolites), colocalization analysis suggested that the causal variants for gene expression and metabolite level are the same (LCP ≥ 0.5; Table S2).
Sensitivity and precision of metabQTL gene nominations through integrating transcriptomics results with metabQTLs
For 1,427 of the 2,030 metabQTLs in METSIM, we previously identified 1,495 gene-metabolite pairs (between 290 genes and 631 metabolites) by matching metabolite biochemical activities and nearby gene functions of metabQTLs.29 To evaluate the ability of our PTWAS and colocalization results to identify genes underlying metabQTLs, we used these 1,495 gene-metabolite pairs as ground truths. Of these 1,495 pairs, 956 (63.9%; between 216 genes and 535 metabolites) achieved significant association in PTWASs, consistent with a previous study showing that TWASs have 67% sensitivity for identifying true genes for metabQTLs.15 Of the 956 pairs, 496 (between 125 genes and 355 metabolites) also showed significant colocalization (LCP ≥ 0.5; Figure S3). Combining PTWASs and colocalization analysis increased the precision for assigning genes for metabQTLs to 36.6% = 496/1,354 from 7.6% = 956/12,575 when using TWASs alone.
Our results suggest strong connections between gene expression and metabolite abundance. Many metabQTLs most likely share causal variants with eQTLs. For the remaining 539 (1,495 − 956) gene-metabolite pairs we previously nominated29 but PTWASs failed to identify, possible explanations include insufficient statistical power in PTWASs or a mechanism besides gene regulation underlying the corresponding metabQTL. For example, our GWAS identified an association at SLC22A16 deleterious missense variant p.Met409Thr (rs12210538 [c.1226A>G]; β = −0.44, p = 9.3 × 10−59) for arachidonoylcarnitine level in METSIM and nominated SLC22A16 as the underlying gene.29 The PTWAS failed to identify the causal role of SLC22A16 expression for arachidonoylcarnitine. GTEx showed p.Met409Thr affects SLC22A16 alternative splicing (β = −0.39, p = 2.7 × 10−5) rather than total expression,30 which might explain the nonsignificance of SLC22A16 in PTWASs that did not consider sQTLs.
Complementary metabQTL gene nominations through integrating transcriptomics results with metabQTLs
Integrating transcriptomics results with metabQTLs helps nominate genes underlying metabQTLs and facilitates gene effect estimation for metabolite levels. In contrast to the 1,427 metabQTLs for which we previously nominated 290 genes through linking metabolite biochemical activities with metabQTLs nearby genes’ functions or statistical fine-mapping,29 PTWASs and colocalization analysis together nominated genes for 1,059 metabQTLs including 283 without prior gene nominations (Figure S4).
Our METSIM metabolomics GWAS previously identified an association with pentose acid at rs705379 (β = 0.13, p = 2.9 × 10−11).29 Because pentose acid, as measured in the Metabolon panel, reflects the combined levels of multiple related metabolites, our knowledge-based approach matching metabolite biochemical characteristics and functions of nearby genes failed to nominate any genes for this association.29 The PTWAS suggested a causal role of PON1 expression for this pentose association and that decreased PON1 expression in the small intestine increased plasma pentose acid level (β = −0.021, p = 1.2 × 10−9). Colocalization analysis suggested that genetic associations for PON1 expression and pentose acid level most likely shared putative causal variant rs705379 (SCP = 0.40, LCP = 0.79; Figure 3A), which resides in gene regulatory elements in the small intestine.48 PON1 encodes a paraoxonase enzyme, which affects the component levels in the pentose phosphate pathway that generates pentoses.49
Figure 3.

Colocalizations between eQTL and metabQTL
(A and B) eQTL and metabQTL colocalizations between eQTLs for PON1 in the small intestine and metabQTLs for plasma pentose acid level (A) and between eQTLs for AGA in the stomach and metabQTLs for plasma aspartate level (B).
Improved metabQTL fine-mapping via colocalizing metabQTLs with eQTLs
Colocalizing metabQTLs with eQTLs improved metabQTL fine-mapping resolution (VPIP median: 0.20 versus 0.11; two-sided paired t-test p = 1.8 × 10−94; Figure S5). This colocalization analysis identified 173 genetic variants with VPIP ≥ 0.8 for metabQTLs.
AGA encodes a member of the N-terminal nucleophile hydrolase cleaving asparagine from N-acetyl glucosamine.50 Our metabolomics GWAS previously identified independent association signals with aspartate at two AGA variants.29 We had suggested AGA underlies the first signal and prioritized p.Leu105Ile (rs76491548 [c.313C>T]) as the putative causal variant for that signal (VPIP = 0.56). However, we were unable to estimate AGA’s effect on regulating plasma aspartate levels or to identify the putative causal variant for the second signal (highest VPIP = 0.16).29 The PTWAS validated the potential causal role of AGA expression and suggested that elevated AGA expression results in increased plasma aspartate levels (β = 0.0070, p = 3.2 × 10−9). Colocalization analysis for AGA expression and plasma aspartate level prioritized the lead variant rs11131799 as the putative causal variant for the second aspartate association signal (SCP = LCP = 0.89; Figure 3B). Lead variant rs11131799 overlaps AGA gene enhancers and promoters,48 making it a promising candidate causal variant.
Metabolomics results complement transcriptomics results to nominate gene-disease associations
Stronger enrichment for metabQTLs than eQTLs in disease trait GWAS-associated variants
Compared with gene expression, metabolites represent intermediate molecular phenotypes more proximal to human diseases.51,52 To examine and compare the extent to which eQTLs and metabQTLs influence the risk of human diseases, we estimated enrichment levels of GTEx eQTLs and METSIM metabQTLs among GWAS associations for 2,861 disease traits (Table S3) in up to 260,405 FinnGen participants.
Across the 2,861 disease traits, the enrichment levels for metabQTLs showed a wider range compared with the ones for eQTLs (Figure 2B). We detected significant GTEx eQTL enrichment for 216 to 553 disease traits in the 49 tissues (mean = 407; median = 414; p < 0.05/2,861 = 1.7 × 10−5), significant METSIM metabQTL enrichment for 328 disease traits, and significant enrichment of both eQTLs and metabQTLs for 246 disease traits. For these 246 disease traits, the metabQTL enrichment level was generally greater than the greatest eQTL enrichment level across the 49 GTEx tissues (mean = 6.8 versus 5.3; two-sided paired t-test p = 5.1 × 10−28; Figure 2C), consistent with a more proximal impact of metabolite levels than gene expression on human diseases.51,52
Integrating transcriptomics or metabolomics results with disease trait GWASs individually
To identify gene-disease associations, we used PTWASs and colocalization analysis to integrate (1) GTEx transcriptomics30 or (2) METSIM metabolomics29 with GWAS results for the 2,861 FinnGen disease traits (Figure 1B).
Integrating GTEx transcriptomics with FinnGen GWAS results via PTWASs identified 63,591 gene-disease trait pairs between 9,443 genes and 2,754 disease traits (FDR < 0.05; Figure S6 and Table S4) and estimated the putative causal effects of gene expression on disease risk. For 4,990 of the 63,591 gene-disease trait pairs between 1,539 genes and 721 disease traits, colocalization analysis suggested shared causal variants (LCP ≥ 0.5; Table S5). We identified 1 to 78 genes per disease trait (mean = 6.9, median = 3.0).
Integrating METSIM metabolomics with FinnGen GWAS results via colocalization analysis suggested shared causal variants for 2,857 metabolite-disease trait pairs between 388 metabolites and 242 disease traits (LCP ≥ 0.5; Table S6). For colocalization between FinnGen disease trait GWASs and metabQTLs with gene nominations from the PTWASs and colocalization analyses between transcriptomics and metabolomics results (see above results section), we suggested 92 genes for 145 disease traits, which comprised 388 gene-disease trait pairs (Table S6). We identified 1 to 20 genes per disease trait (mean = 2.7, median = 2.0).
Together, these parallel pairwise integrative analyses identified 5,378 pairs of 1,597 genes for 790 disease traits. Of the 5,378 pairs, 188 (3.5%) of 34 genes and 76 disease traits were identified in both analyses; 5,002 = 4,990 − 188 + 388 − 188 (93.0%) achieved significance only in the integrative analysis of a single molecular type.
Jointly analyzing transcriptomics, metabolomics, and disease trait GWAS results facilitates discovery of disease molecular mechanisms
Integrating transcriptomics, metabolomics, and disease trait GWAS results together
To investigate disease molecular mechanisms by integrating transcriptomics and metabolomics results with disease GWASs simultaneously, we intersected the results for all three pairwise integrative analyses (Figure 1C). For the 188 gene-disease trait pairs that were identified in both of the integrative analyses of a single type of omics results (i.e., transcriptomics or metabolomics) and the FinnGen disease trait GWASs, we matched the two sets of integrative results for the same disease trait (see above) and intersected the resulting matches with the integrative analysis of transcriptomics and metabolomics results (see above). This strategy identified 2,610 gene-metabolite-disease trait combinations (based on 29 genes, 169 metabolites, and 72 disease traits). In each combination, the gene achieved significance for both metabolite and disease trait in PTWASs (FDR < 5%) and all three pairwise colocalizations between gene expression, metabolite level, and disease trait GWAS showed significance (LCP ≥ 0.5).
For these 2,610 combinations, we further tested for possible causal relationships between metabolites and disease traits through Mendelian randomization (Table S8). We illustrate how integrating transcriptomics and metabolomics results jointly with disease GWASs can clarify disease molecular mechanisms through three examples: (1) a metabolite mediating gene effects on disease (vertical pleiotropy); (2) a gene influencing metabolite level and disease risk through distinct pathways (horizontal pleiotropy); and (3) a group of metabolites together facilitating the discovery of unknown disease metabolic pathways.
UGT1A1/UGT1A4 affects gallbladder disorders through regulation of plasma (E,E)-bilirubin levels
FinnGen identified a genome-wide significant association at rs1976391 for disorders of the gallbladder, biliary tract, and pancreas (gallbladder disorders; OR = 1.08, p = 1.5 × 10−15). The identified genomic region contains ten paralogous genes encoding UDP-glucuronosyltransferases, complicating the identification of the underlying gene(s) and genetic mechanism. The PTWAS integrating liver eQTL results with this disease association suggested a causal role of gene expression for two of the paralogs: UGT1A1 and UGT1A4. However, the pathways mediating gene expression’s putative causal effect remained unclear.
In the same region, we identified a significant association for (E,E)-bilirubin and identified UGT1A1 and UGT1A4 underlying this metabQTL through linking metabolite biochemical activities with the functions of nearby genes.29 Colocalization analysis suggested a single shared causal variant between genetic associations for UGT1A1/UGT1A4 expression in the liver, plasma (E,E)-bilirubin level, and the risk of gallbladder disorders (all pairwise LCP > 0.86; Figure 4A). PTWASs showed that higher UGT1A1 and lower UGT1A4 expression in the liver reduced plasma (E,E)-bilirubin level (β = −0.12 and 0.17, p < 3.0 × 10−302) and risk of gallbladder disorders (β = −0.14 and 0.020, p < 6.0 × 10−13). Mendelian randomization suggested elevated plasma (E,E)-bilirubin level increases risk of gallbladder disorders (65 instrument variables, β = 0.10, p = 3.2 × 10−17) (Figure 4B and 4C).
Figure 4.

Estimated putative causal effects of UGT1A1/UGT1A4 expression on the risk of disorders of the gallbladder, biliary tract, and pancreas through regulation of plasma (E,E)-bilirubin levels
(A) Colocalization of genetic associations for UGT1A1/UGT1A4 expression in the liver, plasma (E,E)-bilirubin, and disorders of the gallbladder, biliary tract, and pancreas.
(B) Comparison of effect sizes of 65 instrument variables in the genome used in the Mendelian randomization analysis between (E,E)-bilirubin and gallbladder disorders. The slope of the blue dashed line depicts the estimated putative causal effect of (E,E)-bilirubin on gallbladder disorders in Mendelian randomization analysis.
(C) Effects (β) on gallbladder disorders of 65 instrument variables used in the Mendelian randomization analysis. Each point in (B) and (C) represents an instrument variable. The vertical and horizontal dashed lines depict β = 0 on disorders of the gallbladder, biliary tract, and pancreas and plasma (E,E)-bilirubin. The error bars in (B) indicate ± standard error of the association effect of the instrument variable on (E,E)-bilirubin and disorders of the gallbladder, biliary tract, and pancreas. The error bar in (C) shows 95% confidence interval of the association effect of the instrument variable on disorders of the gallbladder, biliary tract, and pancreas.
These results together suggested the causal variant in this region stimulated UGT1A1 expression and repressed UGT1A4 expression in the liver, resulting in decreased plasma (E,E)-bilirubin level and decreased risk of gallbladder disorders. UDP-glucuronosyltransferases transform small lipophilic molecules into water-soluble and excretable metabolites.53 Bilirubin is their preferred substrate. Bilirubin level has been shown as a causal factor for gallbladder disorders.54
SLC22A5 affects plasma carnitine levels and risk of nasal polyps through distinct pathways
FinnGen discovered a genome-wide significant association with risk of nasal polyps at rs56399423 (OR = 0.86, p = 3.1 × 10−9). In the same region, we identified an association with plasma carnitine level at rs2073643 (LD r2 = 0.60 with rs56399423; β = −0.14, p = 2.4 × 10−13) and nominated SLC22A5 as the underlying gene.29 PTWASs suggested a causal role of SLC22A5 expression for both associations and showed elevated SLC22A5 expression increased plasma carnitine level (β = 0.030, p = 2.5 × 10−7) and risk of nasal polyps (β = 0.035, p = 1.2 × 10−9). Pairwise colocalization analysis among eQTLs for SLC22A5 in the coronary artery, metabQTLs for plasma carnitine, and the GWAS for nasal polyps suggested a shared causal variant in this region (all pairwise LCP > 0.51; Figure 5). However, Mendelian randomization did not support a causal relationship between plasma carnitine level and the risk of nasal polyps (46 and 44 instrument variables for carnitine to nasal polyps and vice versa; p = 0.52 and 0.51; Figure S7), suggesting SLC22A5 might affect plasma carnitine level and nasal polyps risk through distinct pathways. SLC22A5 encodes a carnitine transporter that contributes to cellular uptake of carnitine and elimination of environmental toxins. Mucosal inflammation caused by allergies and infections results in nasal polyps.55 SLC22A5, which has been implicated in risk of asthma,56 might contribute to nasal polyps through transporting allergens or modulating microbial interactions57 (Figure 6A).
Figure 5.

Colocalization of genetic associations for SLC22A5 expression in the coronary artery, plasma carnitine levels, and risk of nasal polyps
Figure 6.

Genetic causal pathways
(A) Two potentially distinct metabolic pathways mediate the effect of SLC22A5 expression on the risk of nasal polyps and the level of plasma carnitine level; (B) glycerophospholipid metabolic pathways mediate the effect of LIPC expression on the risk of age-related macular degeneration (AMD). Blue circles represent the shared putative causal genes between metabolites and diseases. Yellow circles and rectangles represent metabolites. Gray circles represent disease outcomes. Dashed rectangles and gray dashed lines depict potential environmental risk factors that mediate the effect of SLC22A5 expression levels on carnitine levels and nasal polyps risk. The black dashed arrows depict the estimated effects of gene expression on metabolite levels or disease risk in PTWASs. The black solid arrow denotes the putative causal effects of the ten glycerophospholipids on the risk of AMD, which are estimated in the Mendelian randomization analysis. The flash symbol indicates that Mendelian randomization was unable to detect significant causal relationship between nasal polyps risk and plasma carnitine level.
A group of metabolites helps clarify glycerophospholipid metabolic pathways for age-related macular degeneration
Gene-metabolite-disease trait combinations without significant pairwise colocalization can also help elucidate disease mechanisms. GWASs provide strong association evidence for LIPC with age-related macular degeneration (AMD).58, 59, 60 PTWASs identified LIPC expression associated with the risk of AMD and ten plasma glycerophospholipid levels. Increased LIPC expression in the pancreas decreased metabolite levels (β < −0.043, p < 2.0 × 10−20) and increased AMD risk (β = 0.021, p = 5.4 × 10−6; Figure 6B). These ten metabolites are highly correlated and relevant to the glycerophospholipid metabolic pathways (Figure S8). Mendelian randomization showed that higher levels of each of the ten metabolites protect against AMD (β < −0.21, p < 6.0 × 10−7) (Figure S9). LIPC, hepatic triglyceride lipase, is a key enzyme for HDL metabolism and catalyzes hydrolysis of phospholipids, mono-, di-, and triglycerides, and acyl-CoA thioesters.61 In METSIM, controlling for blood HDL, LDL, and triglyceride levels did not alter substantially the estimated putative causal effects or significance of the ten metabolites on AMD risk in Mendelian randomization analysis (Figure S10). A recent study identified LIPC polymorphisms associated with phosphatidylethanolamine metabolites in AMD case-control populations, suggesting that glycerophospholipid metabolic pathways play a role in AMD.62 Our findings suggest LIPC expression exerts its putative causal effect on AMD through glycerophospholipid metabolic pathways, and this putative causal effect is independent of HDL, LDL, and triglyceride levels.
Discussion
Here, we systematically integrated transcriptomics results for 49 tissues and metabolomics results for 1,391 plasma metabolites with GWAS results for 2,861 disease traits. We identified 397 genes for 521 metabolites and 1,597 genes for 790 disease traits. Notably, we estimated that gene expression impacts levels of 1,274 of the 1,391 (91.6%) measured plasma metabolites and that 63.9% of metabQTLs regulate metabolite levels by influencing gene expression. We showed that enrichment of metabQTLs is generally greater than enrichment of eQTLs in disease trait GWAS associations. We demonstrated that parallel and joint analyses of transcriptomics and metabolomics results facilitate discovery of unknown disease molecular mechanisms. These findings provide insights into disease pathophysiology and highlight the value of combining transcriptomics and metabolomics results to deepen our understanding of disease molecular mechanisms.
The relationship between gene expression and metabolite levels has been examined in humans,47 model organisms,63 and cell lines64 through simultaneous profiling of transcriptomics and metabolomics data in the same set of individuals. Two recent studies integrated separate metabolomics and transcriptomics datasets to test the associations between metabolites and predicted gene expression but were limited to only 64 blood metabolites15 or a single type of cell-line-based transcriptomics data.16 Here, we systematically integrated transcriptomics results on 49 human tissues with metabolomics results on 1,391 plasma metabolites. Our results suggested wide impact of the genome on regulating metabolite levels.
GWASs have identified thousands of metabQTLs for plasma metabolite levels,12,14 but the underlying genes for many metabQTLs remain unclear. Our PTWAS analysis showed that integrating transcriptomics results with metabQTLs for the 1,391 metabolites recovered 63.9% of prior gene findings with strong biochemical evidence,29 consistent with a previous study that showed TWASs had 67% sensitivity for metabQTL gene nominations.15 We also estimated that 33.2% of metabQTLs, for which there exists strong biochemical evidence linking target metabolites to corresponding genes, shared causal variants with eQTLs. Our results confirmed that combining TWASs and colocalization analysis improved the precision of metabQTL gene nominations.15
Omics data are often integrated with GWAS results to help identify underlying disease mechanisms, but usually only a single type of omics data are used.2,3 For example, transcriptomics data are routinely integrated with disease GWASs through TWASs or colocalization analysis.3 Here, we integrated transcriptomics results of 49 human tissues and metabolomics results of 1,391 plasma metabolites together with GWAS associations for 2,861 disease traits. Few approaches exist for integrating multiple molecular traits with GWASs.41,42 They are either computationally challenging or sensitive to prior settings. Instead, we applied a straightforward approach to integrate transcriptomics and metabolomics with GWAS results simultaneously through matching the results for the three sets of pairwise integrative analyses. Our strategy can be easily extended to up to ∼10 traits by integrating results from all pairwise analyses. For even larger numbers of traits, alternative strategies will most likely be needed. We showed that pairwise integration of transcriptomics, metabolomics, and GWAS results facilitates uncovering unknown or more specific disease mechanisms compared to integration of a single type of molecular data. Metabolomics data are becoming available in increasingly large samples, for example the metabolomics profiling of UK Biobank participants.65 We anticipate that our study will encourage the integration of transcriptomics, metabolomics, and disease GWASs in the future both within and across studies.
Gallbladder disorders are common, having a combined prevalence of 12.5% in US adults.66 A previous study identified a causal role of plasma bilirubin on symptomatic gallstone disease.54 GWASs for bilirubin levels have uncovered an association of the UGT1A1 locus,67,68 at which a genetic variant was associated with increased risk of gallstone disease.54 However, the effect of UGT1A1 expression on plasma bilirubin levels in gallstone disease has previously been unclear. Here, we identified a putative causal effect of UGT1A1/UGT1A4 expression on elevated plasma (E,E)-bilirubin levels. We demonstrated that the elevated plasma (E,E)-bilirubin levels increase risk of gallbladder disorders.
AMD is the major common cause of blindness in developed countries.69 The association of LIPC with AMD is well established.58, 59, 60 A recent study identified LIPC polymorphisms associated with phosphatidylethanolamine metabolites in AMD case-control populations.62 Our study provides the evidence to support a putative causal role of LIPC expression on AMD through glycerophospholipid metabolic pathways.
TWASs and colocalization are complementary approaches for integrating omics data with GWAS results.27 We applied state-of-the-art PTWASs,28 enabling simultaneous gene association screening and causal effect estimation. We used the recently developed locus-level colocalization method.27 Compared with variant-level colocalization that was previously used,22,23,26 locus-level colocalization27 provides greater sensitivity and power at the cost of lower genomic resolution. We evaluated the probability of sharing the same causal variant among eQTLs, metabQTLs, and disease GWASs through running all three pairwise colocalizations. We also ran moloc,61 a multi-trait colocalization tool, with default priors and marginal GWAS results in comparison to pairwise fastENLOC, which estimated priors in the data and used Bayesian statistical fine-mapping results. For the colocalization among genetic associations for UGT1A1/UGT1A4 expression in the liver, plasma (E,E)-bilirubin levels, and the risk of gallbladder disorder, moloc identified significant colocalization (posterior probability [PPA] = 0.98 and 0.88).61 In contrast, for the colocalization among genetic associations for SLC22A5 expression in the coronary artery, plasma carnitine levels, and the risk of nasal polyps, moloc did not find colocalization (PPA = 0.00). The lead variants of the genetic associations for SLC22A5 expression in the coronary artery, plasma carnitine levels, and the risk of nasal polyps are in high LD (r2 ≥ 0.599 for all three pairs), suggesting these three genetic associations share a single causal variant. Our previous GWAS identified two independent association signals for plasma carnitine level in this region.29 That might explain the failure of moloc to identify the colocalization. These results highlight the weakness of the moloc method and the need for careful consideration and method development in multi-trait colocalization analysis. Our study identified gene associations for both metabolites and diseases outside loci without conventional GWAS significance, which highlights the value of PTWASs and colocalization analysis that use multi-SNP fine-mapping results for new discovery. These gene associations might provide clues for future investigations.
Notably, fine-mapping results used in both PTWASs and colocalization analysis take allelic heterogeneity into account and provide improved statistical power for both analyses.27 We acknowledge that fine mapping was only applied in genomic regions with prior GWAS associations. Genetic variants affect disease risk likely in a tissue/cell-type-specific manner. We ignored the exploration of tissue-specific gene effects on disease mainly because of incomparable statistical power in eQTLs across 49 GTEx tissues. We might miss rare variants in the integrative analysis because of low statistical power for rare variants in the original eQTLs and GWASs.
In summary, we performed an integrative study of transcriptomics for 49 human tissues, metabolomics for 1,391 plasma metabolites, and GWAS results for 2,861 disease traits. We found strong connections between gene expression and metabolite abundance. We demonstrated that integrating transcriptomics with metabolomics results reveals regulatory mechanisms underlying metabQTLs. Integrating transcriptomics and metabolomics results with disease GWASs individually complements the identification of genes underlying disease GWAS associations. Our results highlight that integrating transcriptomics and metabolomics results together can help deepen our understanding of disease molecular mechanisms.
Acknowledgments
We thank all the participants and investigators in the GTEx, METSIM, and FinnGen studies. This work was supported by National Institutes of Health (NIH) awards U01 DK062370 (M.B.), R35 GM138121 (X.Q.W.), R01 DK119380 (X.Q.W.), P01 HL151328 (N.O.S.), R01 HL131961 (N.O.S.), UM1 HG008853 (I.H., N.O.S., L.G., A.E.L.), R01 DK093757 (K.L.M.), and U01 DK105561 (K.L.M.); an American Diabetes Association Postdoctoral Fellowship (1-19-PDF-061, X.Y.); a University of Michigan Precision Health Scholarship (X.Y.); Academy of Finland grant 321428 (M.L.); the Sigrid Jusélius Foundation (M.L., S.R.); Academy of Finland Center of Excellence in Complex Disease Genetics grants 312062 and 336820 (S.R.) and grants 312074 and 336824 (A.P.); the Finnish Foundation for Cardiovascular Research (S.R.); University of Helsinki HiLIFE Fellow and Grand Challenge grants (S.R.); and Horizon 2020 Research and Innovation Programme (grant 101016775 “INTERVENE”, S.R.). F.S.C., M.R.E., and L.L.B. were supported by the NIH Intramural Research Program of the National Human Genome Research Institute (ZIA HG000024).
Declaration of interests
A.E.L. is an employee and stockholder of Regeneron Pharmaceuticals. N.O.S. has received research funding from Regeneron Pharmaceuticals unrelated to this work. E.B.F. is an employee and stockholder of Pfizer.
Published: September 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.08.007.
Contributor Information
Markku Laakso, Email: markku.laakso@uef.fi.
Xiaoquan Wen, Email: xwen@umich.edu.
Web resources
DAP-g, https://github.com/xqwen/dap
fastENLOC, https://github.com/xqwen/fastenloc
FinnGen, https://www.finngen.fi
FinnGen documentation, https://finngen.gitbook.io/documentation
GTEx portal, https://gtexportal.org/home/
METSIM metabolomics PheWeb, https://pheweb.org/metsim-metab
PTWAS, https://github.com/xqwen/ptwas
TwoSampleMR, https://github.com/MRCIEU/TwoSampleMR
Supplemental information
Data and code availability
GTEx genotype and gene expression data can be accessed at dbGaP with accession number dbGaP: phs000424.v8.p2. FinnGen genome-wide summary statistics and Bayesian statistical fine-mapping results are available at https://r6.finngen.fi. Full summary statistics from the genome-wide association studies of the 1,391 plasma metabolites are available at https://pheweb.org/metsim-metab/.
Each use of software tools has been clearly identified in the material and methods section. Integrative analysis code and scripts are available upon request from Dr. Xiaoquan Wen.
References
- 1.Loos R.J.F. 15 years of genome-wide association studies and no signs of slowing down. Nat. Commun. 2020;11:5900. doi: 10.1038/s41467-020-19653-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mortezaei Z., Tavallaei M. Recent innovations and in-depth aspects of post-genome wide association study (Post-GWAS) to understand the genetic basis of complex phenotypes. Heredity. 2021;127:485–497. doi: 10.1038/s41437-021-00479-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li B., Ritchie M.D. From GWAS to gene: transcriptome-wide association studies and other methods to functionally understand GWAS discoveries. Front. Genet. 2021;12:713230. doi: 10.3389/fgene.2021.713230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cano-Gamez E., Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 2020;11:424. doi: 10.3389/fgene.2020.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres J.M., Abdalla M., Payne A., Fernandez-Tajes J., Thurner M., Nylander V., Gloyn A.L., Mahajan A., McCarthy M.I. A multi-omic integrative scheme characterizes tissues of action at loci associated with type 2 diabetes. Am. J. Hum. Genet. 2020;107:1011–1028. doi: 10.1016/j.ajhg.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasin Y., Seldin M., Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:83. doi: 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mamas M., Dunn W.B., Neyses L., Goodacre R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch. Toxicol. 2011;85:5–17. doi: 10.1007/s00204-010-0609-6. [DOI] [PubMed] [Google Scholar]
- 8.Zhang A., Sun H., Yan G., Wang P., Wang X. Mass spectrometry-based metabolomics: applications to biomarker and metabolic pathway research. Biomed. Chromatogr. 2016;30:7–12. doi: 10.1002/bmc.3453. [DOI] [PubMed] [Google Scholar]
- 9.Tanha H.M., Sathyanarayanan A., International Headache Genetics Consortium Genetic overlap and causality between blood metabolites and migraine. Am. J. Hum. Genet. 2021;108:2086–2098. doi: 10.1016/j.ajhg.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feofanova E.V., Chen H., Dai Y., Jia P., Grove M.L., Morrison A.C., Qi Q., Daviglus M., Cai J., North K.E., et al. A genome-wide association study discovers 46 loci of the human metabolome in the hispanic community health study/study of latinos. Am. J. Hum. Genet. 2020;107:849–863. doi: 10.1016/j.ajhg.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu X., Jaeger M., Beumer J., Bakker O.B., Aguirre-Gamboa R., Oosting M., Smeekens S.P., Moorlag S., Mourits V.P., Koeken V.A.C.M., et al. Integration of metabolomics, genomics, and immune phenotypes reveals the causal roles of metabolites in disease. Genome Biol. 2021;22:198. doi: 10.1186/s13059-021-02413-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lotta L.A., Pietzner M., Stewart I.D., Wittemans L.B.L., Li C., Bonelli R., Raffler J., Biggs E.K., Oliver-Williams C., Auyeung V.P.W., et al. A cross-platform approach identifies genetic regulators of human metabolism and health. Nat. Genet. 2021;53:54–64. doi: 10.1038/s41588-020-00751-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollywood K., Brison D.R., Goodacre R. Metabolomics: current technologies and future trends. Proteomics. 2006;6:4716–4723. doi: 10.1002/pmic.200600106. [DOI] [PubMed] [Google Scholar]
- 14.Kastenmüller G., Raffler J., Gieger C., Suhre K. Genetics of human metabolism: an update. Hum. Mol. Genet. 2015;24:r93–r101. doi: 10.1093/hmg/ddv263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ndungu A., Payne A., Torres J.M., van de Bunt M., McCarthy M.I. A multi-tissue transcriptome analysis of human metabolites guides interpretability of associations based on multi-snp models for gene expression. Am. J. Hum. Genet. 2020;106:188–201. doi: 10.1016/j.ajhg.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sönmez Flitman R., Khalili B., Kutalik Z., Rueedi R., Brümmer A., Bergmann S. Untargeted metabolome- and transcriptome-wide association study suggests causal genes modulating metabolite concentrations in urine. J. Proteome Res. 2021;20:5103–5114. doi: 10.1021/acs.jproteome.1c00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., GTEx Consortium. Cox N.J., Im H.K. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W.J.H., Jansen R., de Geus E.J.C., Boomsma D.I., Wright F.A., et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M., Yang J. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 20.Wainberg M., Sinnott-Armstrong N., Mancuso N., Barbeira A.N., Knowles D.A., Golan D., Ermel R., Ruusalepp A., Quertermous T., Hao K., et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019;51:592–599. doi: 10.1038/s41588-019-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu A., Matoba N., Wilson E.P., Tapia A.L., Li Y., Ibrahim J.G., Stein J.L., Love M.I. MRLocus: identifying causal genes mediating a trait through Bayesian estimation of allelic heterogeneity. PLoS Genet. 2021;17:e1009455. doi: 10.1371/journal.pgen.1009455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hormozdiari F., van de Bunt M., Segrè A.V., Li X., Joo J.W.J., Bilow M., Sul J.H., Sankararaman S., Pasaniuc B., Eskin E. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 2016;99:1245–1260. doi: 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hukku A., Pividori M., Luca F., Pique-Regi R., Im H.K., Wen X. Probabilistic colocalization of genetic variants from complex and molecular traits: promise and limitations. Am. J. Hum. Genet. 2021;108:25–35. doi: 10.1016/j.ajhg.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen X., Pique-Regi R., Luca F. Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genet. 2017;13:e1006646. doi: 10.1371/journal.pgen.1006646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hukku A., Sampson M.G., Luca F., Pique-Regi R., Wen X. Analyzing and reconciling colocalization and transcriptome-wide association studies from the perspective of inferential reproducibility. Am. J. Hum. Genet. 2022;109:825–837. doi: 10.1016/j.ajhg.2022.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Quick C., Yu K., Barbeira A., GTEx Consortium. Pique-Regi R., Kyung Im H., Wen X. PTWAS: investigating tissue-relevant causal molecular mechanisms of complex traits using probabilistic TWAS analysis. Genome Biol. 2020;21:232. doi: 10.1186/s13059-020-02026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin X., Chan L.S., Bose D., Jackson A.U., VandeHaar P., Locke A.E., Fuchsberger C., Stringham H.M., Welch R., Yu K., et al. Genome-wide association studies of metabolites in Finnish men identify disease-relevant loci. Nat. Commun. 2022;13:1644. doi: 10.1038/s41467-022-29143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laakso M., Kuusisto J., Stančáková A., Kuulasmaa T., Pajukanta P., Lusis A.J., Collins F.S., Mohlke K.L., Boehnke M. The Metabolic Syndrome in Men study: a resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 2017;58:481–493. doi: 10.1194/jlr.O072629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurki M.I., Karjalainen J., Palta P., Sipilä T.P., Kristiansson K., Donner K., Reeve M.P., Laivuori H., Aavikko M., Kaunisto M.A., et al. FinnGen: Unique genetic insights from combining isolated population and national health register data. medRxiv. 2022 doi: 10.1101/2022.03.03.22271360. Preprint at. [DOI] [Google Scholar]
- 33.Lim E.T., Würtz P., Havulinna A.S., Palta P., Tukiainen T., Rehnström K., Esko T., Mägi R., Inouye M., Lappalainen T., et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benner C., Spencer C.C.A., Havulinna A.S., Salomaa V., Ripatti S., Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. B. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higgins J.P.T., Thompson S.G. Quantifying heterogeneity in a meta-analysis. Stat. Med. 2002;21:1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 38.Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J.E., Mudge J.M., Sisu C., Wright J.C., Armstrong J., Barnes I., et al. GENCODE 2021. Nucleic Acids Res. 2021;49:d916–d923. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.GTEx Consortium. Laboratory Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18:275–294. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giambartolomei C., Zhenli Liu J., Zhang W., Hauberg M., Shi H., Boocock J., Pickrell J., Jaffe A.E., CommonMind Consortium. Roussos P. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics. 2018;34:2538–2545. doi: 10.1093/bioinformatics/bty147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foley C.N., Staley J.R., Breen P.G., Sun B.B., Kirk P.D.W., Burgess S., Howson J.M.M. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 2021;12:764. doi: 10.1038/s41467-020-20885-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burgess S., Butterworth A., Thompson S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowden J., Davey Smith G., Haycock P.C., Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowden J., Davey Smith G., Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verbanck M., Chen C.Y., Neale B., Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartel J., Krumsiek J., Schramm K., Adamski J., Gieger C., Herder C., Carstensen M., Peters A., Rathmann W., Roden M., et al. The human blood metabolome-transcriptome interface. PLoS Genet. 2015;11:e1005274. doi: 10.1371/journal.pgen.1005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roadmap Epigenomics Consortium. Meuleman W., Bilenky M., Heravi-Moussavi A., Zhang Z., Ziller M.J., Whitaker J.W., Ward L.D., Quon G., Eaton M.L., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meneses M.J., Silvestre R., Sousa-Lima I., Macedo M.P. Paraoxonase-1 as a regulator of glucose and lipid homeostasis: impact on the onset and progression of metabolic disorders. Int. J. Mol. Sci. 2019;20:E4049. doi: 10.3390/ijms20164049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rip J.W., Coulter-Mackie M.B., Rupar C.A., Gordon B.A. Purification and structure of human liver aspartylglucosaminidase. Biochem. J. 1992;288:1005–1010. doi: 10.1042/bj2881005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zelezniak A., Sheridan S., Patil K.R. Contribution of network connectivity in determining the relationship between gene expression and metabolite concentration changes. PLoS Comput. Biol. 2014;10:e1003572. doi: 10.1371/journal.pcbi.1003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ivanisevic J., Elias D., Deguchi H., Averell P.M., Kurczy M., Johnson C.H., Tautenhahn R., Zhu Z., Watrous J., Jain M., et al. Arteriovenous blood metabolomics: a readout of intra-tissue metabostasis. Sci. Rep. 2015;5:12757. doi: 10.1038/srep12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King C.D., Rios G.R., Green M.D., Tephly T.R. UDP-glucuronosyltransferases. Curr. Drug Metab. 2000;1:143–161. doi: 10.2174/1389200003339171. [DOI] [PubMed] [Google Scholar]
- 54.Stender S., Frikke-Schmidt R., Nordestgaard B.G., Tybjærg-Hansen A. Extreme bilirubin levels as a causal risk factor for symptomatic gallstone disease. JAMA Intern. Med. 2013;173:1222–1228. doi: 10.1001/jamainternmed.2013.6465. [DOI] [PubMed] [Google Scholar]
- 55.Hulse K.E., Stevens W.W., Tan B.K., Schleimer R.P. Pathogenesis of nasal polyposis. Clin. Exp. Allergy. 2015;45:328–346. doi: 10.1111/cea.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valette K., Li Z., Bon-Baret V., Chignon A., Bérubé J.C., Eslami A., Lamothe J., Gaudreault N., Joubert P., Obeidat M., et al. Prioritization of candidate causal genes for asthma in susceptibility loci derived from UK Biobank. Commun. Biol. 2021;4:700. doi: 10.1038/s42003-021-02227-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moffatt M.F., Gut I.G., Demenais F., Strachan D.P., Bouzigon E., Heath S., von Mutius E., Farrall M., Lathrop M., Cookson W.O.C.M., GABRIEL Consortium A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010;363:1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neale B.M., Fagerness J., Reynolds R., Sobrin L., Parker M., Raychaudhuri S., Tan P.L., Oh E.C., Merriam J.E., Souied E., et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC) Proc. Natl. Acad. Sci. USA. 2010;107:7395–7400. doi: 10.1073/pnas.0912019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fritsche L.G., Igl W., Bailey J.N.C., Grassmann F., Sengupta S., Bragg-Gresham J.L., Burdon K.P., Hebbring S.J., Wen C., Gorski M., et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016;48:134–143. doi: 10.1038/ng.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han X., Gharahkhani P., Mitchell P., Liew G., Hewitt A.W., MacGregor S. Genome-wide meta-analysis identifies novel loci associated with age-related macular degeneration. J. Hum. Genet. 2020;65:657–665. doi: 10.1038/s10038-020-0750-x. [DOI] [PubMed] [Google Scholar]
- 61.Hasham S.N., Pillarisetti S. Vascular lipases, inflammation and atherosclerosis. Clin. Chim. Acta. 2006;372:179–183. doi: 10.1016/j.cca.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 62.Lains I., Zhu S., Han X., Chung W., Yuan Q., Kelly R.S., Gil J.Q., Katz R., Nigalye A., Kim I.K., et al. Genomic-metabolomic associations support the role of lipc and glycerophospholipids in age-related macular degeneration. Ophthalmol. Sci. 2021;1:100017. doi: 10.1016/j.xops.2021.100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lempp M., Farke N., Kuntz M., Freibert S.A., Lill R., Link H. Systematic identification of metabolites controlling gene expression in E. coli. Nat. Commun. 2019;10:4463. doi: 10.1038/s41467-019-12474-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H., Barbour J.A., Zhu X., Wong J.W.H. Gene expression is a poor predictor of steady-state metabolite abundance in cancer cells. FASEB. J. 2022;36:e22296. doi: 10.1096/fj.202101921RR. [DOI] [PubMed] [Google Scholar]
- 65.Julkunen H., Cichońska A., Slagboom P.E., Würtz P., Nightingale Health UK Biobank Initiative Metabolic biomarker profiling for identification of susceptibility to severe pneumonia and COVID-19 in the general population. Elife. 2021;10:e63033. doi: 10.7554/eLife.63033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Everhart J.E., Khare M., Hill M., Maurer K.R. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology. 1999;117:632–639. doi: 10.1016/s0016-5085(99)70456-7. [DOI] [PubMed] [Google Scholar]
- 67.Johnson A.D., Kavousi M., Smith A.V., Chen M.H., Dehghan A., Aspelund T., Lin J.P., van Duijn C.M., Harris T.B., Cupples L.A., et al. Genome-wide association meta-analysis for total serum bilirubin levels. Hum. Mol. Genet. 2009;18:2700–2710. doi: 10.1093/hmg/ddp202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen G., Adeyemo A., Zhou J., Doumatey A.P., Bentley A.R., Ekoru K., Shriner D., Rotimi C.N. A UGT1A1 variant is associated with serum total bilirubin levels, which are causal for hypertension in African-ancestry individuals. NPJ Genom. Med. 2021;6:44. doi: 10.1038/s41525-021-00208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong W.L., Su X., Li X., Cheung C.M.G., Klein R., Cheng C.Y., Wong T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet. Glob. Health. 2014;2 doi: 10.1016/S2214-109X(13)70145-1. e106–116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GTEx genotype and gene expression data can be accessed at dbGaP with accession number dbGaP: phs000424.v8.p2. FinnGen genome-wide summary statistics and Bayesian statistical fine-mapping results are available at https://r6.finngen.fi. Full summary statistics from the genome-wide association studies of the 1,391 plasma metabolites are available at https://pheweb.org/metsim-metab/.
Each use of software tools has been clearly identified in the material and methods section. Integrative analysis code and scripts are available upon request from Dr. Xiaoquan Wen.