

Summary
Proteins containing the FERM (four-point-one, ezrin, radixin, and moesin) domain link the plasma membrane with cytoskeletal structures at specific cellular locations and have been implicated in the localization of cell-membrane-associated proteins and/or phosphoinositides. FERM domain-containing protein 5 (FRMD5) localizes at cell adherens junctions and stabilizes cell-cell contacts. To date, variants in FRMD5 have not been associated with a Mendelian disease in OMIM. Here, we describe eight probands with rare heterozygous missense variants in FRMD5 who present with developmental delay, intellectual disability, ataxia, seizures, and abnormalities of eye movement. The variants are de novo in all for whom parental testing was available (six out of eight probands), and human genetic datasets suggest that FRMD5 is intolerant to loss of function (LoF). We found that the fly ortholog of FRMD5, CG5022 (dFrmd), is expressed in the larval and adult central nervous systems where it is present in neurons but not in glia. dFrmd LoF mutant flies are viable but are extremely sensitive to heat shock, which induces severe seizures. The mutants also exhibit defective responses to light. The human FRMD5 reference (Ref) cDNA rescues the fly dFrmd LoF phenotypes. In contrast, all the FRMD5 variants tested in this study (c.340T>C, c.1051A>G, c.1053C>G, c.1054T>C, c.1045A>C, and c.1637A>G) behave as partial LoF variants. In addition, our results indicate that two variants that were tested have dominant-negative effects. In summary, the evidence supports that the observed variants in FRMD5 cause neurological symptoms in humans.
Keywords: FRMD5, Drosophila, CG5022, dFrmd, developmental delay, intellectual disability, ataxia, seizures, nystagmus, opsoclonus
Graphical abstract
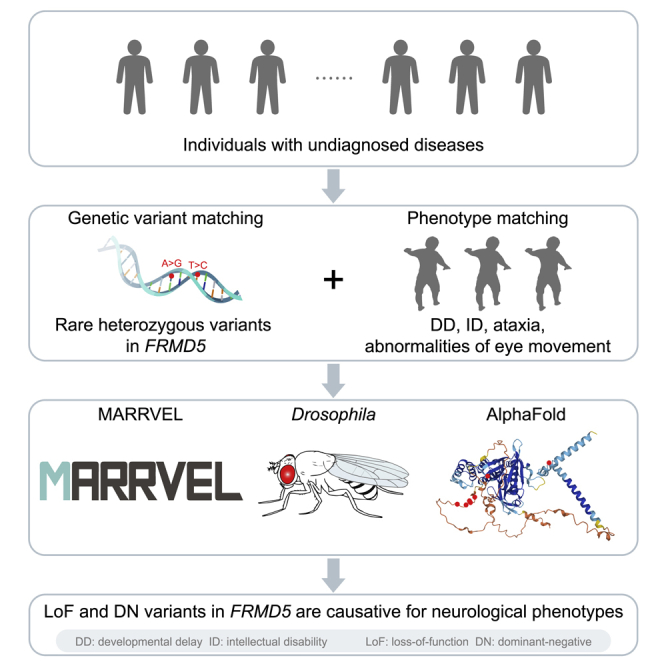
We report eight individuals with rare heterozygous variants in FRMD5 who present with developmental delay, intellectual disability, ataxia and abnormalities of eye movement. Experimental evidence based on Drosophila studies and protein structure predictions indicate that these variants cause loss-of-function as well as dominant-negative effects.
Main text
The FERM (four-point-one, ezrin, radixin, and moesin) domain is often located at the N terminus of FERM domain-containing proteins (FDCPs), linking the cytoskeletal network to the plasma membrane.1 The FDCPs play important roles in cellular movements and migration by binding to a variety of proteins and lipids.2 They contribute to membrane dynamics to mediate migration of the cell when responding to directional cues.3,4 There are about 50 FDCPs in the human genome, and they participate in a variety of biological processes, such as wound healing and immune responses in health as well as cancer metastasis.2 Fewer than 20 FDCPs have been reported to be associated with human diseases,5 and the functions of the majority of the FDCPs remain to be discovered.
The FERM domain-containing protein 5 (FRMD5 [MIM: 616309]) is localized to adherens junctions.6 Previous studies have documented that knockdown of FRMD5 promotes lung cancer cell migration and invasion.6,7 FRMD5 inhibits migration through binding to integrin subunit beta 5 (ITGB5) and Rho-associated coiled-coil-containing protein kinase 1 (ROCK1).7 However, other scientists showed that knockdown of FRMD5 suppresses hepatocellular carcinoma cell (HCC) proliferation and tumorigenesis and that FRMD5 is elevated by Wnt/β-catenin activation in human HCCs.8 In addition, the transcriptional activity of FRMD5 is regulated by β-catenin in colorectal cancer cells.9 These data indicate different functional outcomes of loss of FRMD5 in different contexts. However, variants in FRMD5 have not been associated with a disease in the Online Mendelian Inheritance in Man (OMIM) database.10
We identified eight individuals with rare heterozygous missense FRMD5 variants who present with neurodevelopmental disorders. Proper informed consent was obtained from legal guardians of the individuals. The variants are de novo in all the cases except for probands 7 and 8, for whom the variants were not detected in maternal samples, but the paternal samples are unavailable. A summary of the clinical information, including nucleotide changes, of these probands can be found in Table 1. All probands exhibit developmental delay including motor delay. All probands present with intellectual disability, except proband 1, who is too young to be diagnosed. Seven probands have ataxia. They all exhibit abnormalities of eye movement. Among them, probands 2, 4, 5, and 7 have nystagmus, whereas probands 3, 6, and 7 have opsoclonus. Proband 1 has strabismus, and proband 8 has intermittent esotropia. Nystagmus and opsoclonus are abnormal involuntary eye movements, whereas strabismus is an abnormal conjugate eye movement. Five individuals have seizures, and proband 8 has an abnormal EEG. Some individuals have refractory seizures. Three of the eight individuals have abnormal brain MRIs (probands 2, 7, and 8). Proband 2 exhibited pachygyria in bilateral temporal lobes at the age of 6 (Figures 1A and 1B). For more detailed information, other symptoms, and other potential variants not within FRMD5, please see the case reports in the supplemental information.
Table 1.
Clinical features of affected individuals with FRMD5 variants
| Proband | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Exome sequencing (ES) | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| FRMD5 variant (GenBank: NM_032892.5) | c.340T>C | c.1051A>G | c.1053C>G | c.1054T>C | c.1054T>C | c.1060T>C | c.1045A>C | c.1637A>G |
| Protein change | p.Phe114Leu | p.Ser351Gly | p.Ser351Arg | p.Cys352Arg | p.Cys352Arg | p.Ser354Pro | p.Ser349Arg | p.Tyr546Cys |
| CADD | 29.1 | 23.3 | 22.6 | 23.6 | 23.6 | 27.8 | 23.4 | 26.3 |
| Current age (years) | 3 | 8 | 27 | 17 | 18 | 9 | 16 | 15.5 |
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | no paternal sample | no paternal sample |
| Age of onset | 6 months | 3 months | neonatal period | neonatal period | neonatal period | neonatal period | neonatal period | 8 months |
| Sex | M | M | F | M | M | F | M | M |
| Motor delay | + | +++ | + | + | ++ | + | + | ++ |
| Developmental delay | + | +++ | + | + | ++ | + | + | ++ |
| Intellectual disability | N/A | +++ | + (borderline) | + | ++ | + | + | + |
| Seizures | ++ | +++ | + | + | – | – | + | abnormal EEG |
| Ataxia | + | + | + | ++ | + | +++ | + | – |
| Hypotonia | – | + | – | + | + | + | N/A | + |
| Spasticity | + | + | – | + | – | – | N/A | + |
| Abnormality of the eye | strabismus | nystagmus | opsoclonus | ocular vertical nystagmus/flutter | nystagmus, intermittent flutter with poor fixation, mild myopia, delayed visual maturation | opsoclonus, hypermetropia, visually impaired | nystagmus and opsoclonus | intermittent esotropia |
| Brain MRI | normal | pachygyria in bilateral temporal lobes | normal | normal | normal | normal | cystic foci in the periventricular white matter | delays in myelination |
| Additional features | feeding difficulties | feeding difficulties, severe constipation | myoclonus and dystonia; difficulty with fine motor skills; dyslexia; migraines | N/A | learning disability, behavioral problems, dystonia, dyskinetic and spasm-like movements and postures | moderate learning disability, urinary incontinence, behavior and self-regulation concerns, poor sleep, anxiety | learning problems, fatigue, headaches, interrupted sleep | ASD, renal anomalies |
We report eight individuals with rare heterozygous variants in FRMD5 who present with developmental delay, intellectual disability, ataxia, and abnormalities of eye movement. Experimental evidence based on Drosophila studies and protein structure predictions indicate that these variants cause loss-of-function as well as dominant-negative effects. CADD, combined annotation-dependent depletion; M, male; F, female; N/A, not available; EEG, electroencephalography; MRI, magnetic resonance imaging; ASD, autism spectrum disorder; –, +, ++, and +++, none, mild, moderate, and severe.
Figure 1.

Brain MRI of proband 2
(A and B) Axial T1-weighted image from proband 2 at 6 years shows pachygyria in bilateral temporal lobes (A, white arrows), when compared to the unaffected control (B) of the same age and sex.
To gather information on human FRMD5 and the potential impact of the variants, we used the Model organism Aggregated Resources for Rare Variant ExpLoration (MARRVEL) tool,11 which gathers information from multiple sources including Genome Aggregation Database (gnomAD12), OMIM, Database of Genomic Variants (DGV13), etc. FRMD5 has a probability of loss-of-function (LoF) intolerance (pLI) score of 1.00 based on gnomAD,12 suggesting that FRMD5 may be a haploinsufficient gene and that loss of a single copy of the gene may cause the observed phenotypes. FRMD5 has a missense Z score of 1.98, suggesting that FRMD5 missense variants may not be tolerable.14 However, there are few heterozygous LoF variants, and individuals with deletions that uncover FRMD5 locus are observed in control12,13 and disease datasets.15,16 Together, these human population genetic data suggest that haploinsufficient or dominant-negative variants of FRMD5 may create phenotypes.
Seven different missense variants in FRMD5 (GenBank: NM_032892.5) were identified among the eight probands (c.1054T>C is shared by probands 4 and 5), and none of the variants are found in gnomAD. All the variants are predicted to be deleterious based on combined annotation dependent depletion (CADD) scores above 20 (Table 1).17 Interestingly, five of the seven FRMD5 variants (p.Ser349Arg, p.Ser351Gly, p.Ser351Arg, p.Cys352Arg, and p.Ser354Pro) are clustered within very few amino acids (aa 349–354) in the FERM-adjacent (FA) domain, suggesting a hotspot region for FRMD5. Since DECIPHER15 has not annotated any hotspot region for FRMD5, we queried MutScore,18 a pathogenicity predictor, for region-specific constraint/missense variant analysis. There is no significant clustering for pathogenic or benign variants detected by MutScore (Figure S1). The MutLand plot from MutScore for FRMD5 domains shows relatively higher scores for the FERM domain region (aa 21–354), suggesting that missense variants in this region are more likely to be pathogenic (Figure S1). The aa 530–555 region in the C terminus of the protein displays intermediate scores, while the aa 349–354 region does not show higher scores than the FERM domain region (Figure S1). In summary, little information is available about the aa 349–354 region of the FA domain.
Besides, there are two variants that map outside the aa 349–354 region: p.Phe114Leu maps to the FERM-middle (M) domain, whereas p.Tyr546Cys maps to the C terminus of the protein. There are two variants that are not confirmed to be de novo: p.Ser349Arg (maps to aa 349–354) and p.Tyr546Cys. Although p.Tyr546Cys maps to an uncharacterized region of the protein, the in silico data suggest that p.Tyr546Cys is deleterious, whereas the other variants observed in gnomAD with high frequencies are mostly predicted to be benign/tolerated (Table S1).
Drosophila dFrmd is an ortholog of FRMD5
To investigate the function of FRMD5 in vivo, we utilized Drosophila as the model organism.19 The Drosophila RNAi Screening Center (DRSC) Integrative Ortholog Prediction Tool (DIOPT)20 predicts one fly gene, CG5022 (hereafter referred to as dFrmd), as the ortholog of both human FRMD5 and FRMD3. The DIOPT score between FRMD5 and dFrmd is 12 out of 16, suggesting a high level of homology between the two genes. The overall similarity and identity between FRMD5 and dFrmd are 47% and 33%, respectively (Figures 2A and S2), and the two proteins show similar domain topology, including the well-conserved FERM domain (Figure 2B). Taken together, these data indicate that fly dFrmd is orthologous to FRMD5 in humans.
Figure 2.

CG5022 (dFrmd) is the FRMD5 ortholog in fly
(A) FRMD5 and FRMD3 share the same fly ortholog, dFrmd. Data were obtained from DIOPT (DRSC Integrative Ortholog Prediction Tool).
(B) Protein domains are conserved between FRMD5 and dFrmd.
(C) Genomic structure of dFrmd locus and reagents used in this study. Real-time PCR primers to detect the dFrmd mRNA levels are also labeled.
(D) Strategy to study FRMD5 in flies. Using dFrmdCRIMIC-TG4, we determined the expression pattern and the loss-of-function (LoF) phenotypes and performed rescue assays. We also ectopically expressed FRMD5 reference (Ref) and variants using different GAL4 drivers to assess their effects in vivo.
(E) Real-time PCR data show that dFrmdCRIMIC-TG4 is a severe LoF or null mutant. Relative dFrmd mRNA expression levels in dFrmdCRIMIC-TG4 mutant larvae decrease to <1% when compared to controls (yw/yw). Each dot represents an independent sample that contains 3–5 larvae. Data are represented as mean + SEM. Unpaired t tests. ∗p < 0.05.
To study FRMD5 in flies, we generated the fly reagents listed in Figure 2C and Table S2. These include a CRISPR-Mediated Integration Cassette (CRIMIC) allele of the dFrmd (dFrmdCRIMIC-TG4),21 which has a Splice Acceptor (SA)-T2A-GAL4-polyA cassette inserted in the first intron of the gene (Figure 2C). The dFrmdCRIMIC-TG4 is likely a null allele, as it creates a truncated dFrmd mRNA (Figure 2D), and our real-time PCR data show that the dFrmdCRIMIC-TG4 reduced the dFrmd mRNA levels to less than 1% (Figure 2E). This dFrmdCRIMIC-TG4 allele also leads to the expression of GAL4 under the endogenous gene-regulatory elements (Figure 2D) and allows us to assess the expression pattern of dFrmd, to explore LoF phenotypes, and to test the rescue ability of fly and human cDNAs.21, 22, 23
dFrmd is expressed primarily in neurons of the fly CNS
We first determined the expression pattern of dFrmd by crossing the dFrmdCRIMIC-TG4 allele to UAS-mCherry.NLS (nuclear-localized mCherry fluorescent protein). The mCherry expression is obviously enriched in the larval central nervous system (CNS) (Figures 3A–3C). In both larval CNS and adult brain, mCherry (dFrmd) co-localizes with some Elav (pan-neuronal nuclear marker)-positive cells, but no obvious overlap was observed between mCherry and Repo (pan-glial nuclear marker) (Figures 3C and 3D), indicating that dFrmd is mainly expressed in a subset of neurons, consistent with single-cell sequencing data.24,25 To reveal the projections of dFrmd-expressing neurons, we used the dFrmdCRIMIC-TG4 allele to drive UAS-mCD8::RFP (a membrane-bound red fluorescent protein). As shown in Figure 3E, RFP (dFrmd) labels neuropils of the central brain and ventral nerve cord in the larval CNS (Figure 3E). In the adult brain, RFP signals are observed in the optic lobes, antennal lobes, and mushroom bodies as well as other brain regions (Figure 3F). Altogether, these data show that dFrmd is specifically expressed in the neurons, in agreement with the prominent expression of FRMD5 in the human CNS.26
Figure 3.

dFrmd is expressed in neurons in the CNS
(A) Schematic of the whole Drosophila larva highlighting the CNS.
(B) Expression pattern of dFrmd in whole third instar (L3) larva of the indicated genotype. Note that mCherry (dFrmd) is mainly expressed in the larval CNS. Scale bar, 1 mm.
(C and D) Expression pattern of dFrmd in the L3 larval CNS (C) and adult brain (D) is visualized using dFrmdCRIMIC-TG4 allele-driven expression of UAS-mCherry.NLS co-stained with markers for neurons (Elav) or glia (Repo). Single-layer confocal images from the dashed squares indicate that mCherry is co-localized with Elav (C’, D’) but not Repo (C’’, D’’). Scale bars, 100 μm.
(E and F) The dFrmdCRIMIC-TG4 allele-driven expression of UAS-mCD8::RFP (membrane-bound RFP) confirmed the broad expression of dFrmd in L3 larval CNS (E) and the adult brain (F). Scale bars, 100 μm.
Loss of dFrmd in flies causes heat-induced seizures and is rescued by the human FRMD5 reference, but the variants rescue poorly
To explore the role of FRMD5 in the nervous system, we assessed phenotypes associated with dFrmd loss in flies. We generated dFrmd LoF mutant flies by crossing dFrmdCRIMIC-TG4 to a deficiency (Df) line lacking dFrmd. The dFrmdCRIMIC-TG4/Df mutants are viable and fertile and do not show obvious morphological abnormalities. Given that the probands exhibit seizures, we induced seizure-like behaviors in flies by mechanical stimulation (bang sensitivity assay)27 or exposure to 42°C (heat shock assay).28,29 The dFrmdCRIMIC-TG4/Df mutants do not show obvious bang sensitivity but are very sensitive to heat shock, which induces severe seizures (Figures 4A and 4B). The mutant flies cannot climb properly and display wing fluttering, leg twitching, and abdominal muscle contractions (Video S1), and loss of dFrmd causes a slow recovery after heat shock (Figures S3A and S3B and Video S2). The heat-induced seizures are rescued by a genomic rescue (GR) construct that carries a copy of the dFrmd locus (Figures 2C, 4A, and S3A), indicating that the loss of dFrmd is the cause of the heat-sensitivity phenotype.
Figure 4.

Loss of dFrmd in flies causes heat-induced seizures and specific ERG defects and is rescued by FRMD5 reference but less so by the variants
(A and B) dFrmd LoF mutants exhibit heat-induced seizures. The percentage of dFrmd LoF mutant flies with seizures is significantly higher than controls (w1118/w1118) after exposure to a 42°C water bath for 30 s. The phenotype can be fully rescued with a genomic rescue (GR). The heat-induced seizures can be significantly rescued by fly dFrmd WT or human FRMD5 Ref, but the human FRMD5 variants have significantly reduced rescue abilities when compared to the Ref at 25°C (A) and 22°C (B). Flies were raised at 25°C (A) or 22°C (B) and tested at 14–15 days. Each dot represents an independent test of 5–8 flies.
(C–E) dFrmd LoF mutants show age-dependent ERG defects. The mutants do not show any ERG defect at day 10 (C), show decreased On transients at 20–21 days (D), and show decreased On and Off transients at day 30 (E). Flies were raised at 22°C.
(F and G) The decreased On transients at 20–21 days can be rescued by human FRMD5 Ref but not the variants. Representative ERG curves are shown in (F), and the quantitative data are shown in (G). Green annotations show the amplitude measurement of On/Off transients and depolarization. Flies were raised at 22°C.
For (A)–(E) and (G), total fly numbers are shown in the columns. Data are represented as mean + SEM. Unpaired t tests. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; n.s., no significance.
Flies in empty vials are submerged in a 42°C water bath for 30 s. Video recording from 12 s is shown. Flies were raised at 25°C and tested at 14–15 days. For genotypes, left vial: dFrmdCRIMIC-TG4/Df;UAS-Empty; right vial: dFrmdCRIMIC-TG4/Df;GR.
Flies in empty vials are allowed to recover after being submerged in a 42°C water bath for 30 s. Video recording from 5 s after water bath is shown. Flies were raised at 25°C and tested at 14–15 days. For genotypes, left vial: dFrmdCRIMIC-TG4/Df;UAS-Empty; right vial: dFrmdCRIMIC-TG4/Df;GR.
Next, we attempted to rescue the heat-induced seizures of dFrmd LoF mutants by expressing human FRMD5 reference (Ref) or variant cDNAs. We generated the UAS-dFrmd wild-type (WT) and UAS-FRMD5 transgenic fly lines and crossed them into the dFrmdCRIMIC-TG4/Df background. Both the UAS-dFrmd WT and UAS-FRMD5 Ref cDNA transgenes fully rescued the phenotype of dFrmd LoF mutants, at 25°C (Figure 4A) and 22°C (Figure 4B). In contrast, the three tested FRMD5 variants exhibit significantly reduced rescue abilities when compared to the Ref (Figures 4A, 4B, and S3B), indicating that the tested FRMD5 variants (c.1051A>G, c.1054T>C, and c.1637A>G) are partial LoF variants.
Loss of dFrmd in flies causes specific ERG defects
We also explored if FRMD5 affects synaptic transmission or phototransduction. We performed electroretinogram (ERG) recordings to assess the ability of the photoreceptors (PRs) to capture and transduce light signals and to assess if the PRs communicate properly with postsynaptic cells.30,31 ERG recordings of the dFrmd LoF mutants did not show obvious defects at 10 days post-eclosion (Figure 4C) but started to show reduced On transients at 20–21 days (Figure 4D), and obviously decreased On and Off transients were observed in the dFrmd LoF mutants on day 30 (Figure 4E) when compared to the GR rescued flies. These data indicate that dFrmd is required to maintain proper synaptic transmission between the presynaptic photoreceptors and the postsynaptic lamina cells in an age-dependent manner. The On transient phenotype is fully rescued by expression of the FRMD5 Ref cDNA, but the two variants that were tested (c.1051A>G and c.1637A>G) show significantly reduced rescue abilities (Figures 4F and 4G), again indicating that they are partial LoF variants.
Ectopic expression of human FRMD5 Ref is toxic, whereas the variants are less toxic
To further investigate the nature of the FRMD5 variants, we performed ectopic expression assays by expressing UAS-FRMD5 cDNAs using different GAL4 drivers at different temperatures, as the GAL4 expression increases with temperature.32,33 Interestingly, ubiquitous expression of FRMD5 Ref using daughterless-GAL4 (da-GAL4) causes semi-lethality at 18°C and full lethality at 22°C and 25°C (Figure S4A). Furthermore, the expression of FRMD5 Ref using a wing-specific nubbin-GAL4 (nub-GAL4) causes semi-lethality at 18°C and 22°C and full lethality at 25°C (Figures 5A and 5B), and the surviving flies show wing defects (Figure 5C). These data indicate that overexpression of FRMD5 Ref is toxic in a dose-dependent manner. Similarly, overexpression of dFrmd WT using da-GAL4 causes semi-lethality at 25°C (Figure S4A). Moreover, nub-GAL4-induced dFrmd WT expression causes wing defects at 25°C (Figures 5A and 5C). Six FRMD5 variants (c.340T>C, c.1045A>C, c.1051A>G, c.1053C>G, c.1054T>C, and c.1637A>G) were tested, and they all showed decreased toxicity when compared to the Ref (Figures 5A, 5B, S4A, and S4B). When the Ref causes a toxic phenotype and the variants are less toxic, the variants are classified as LoF alleles.22,34, 35, 36, 37, 38, 39, 40 In contrast, gain-of-function variants often cause more severe phenotypes in ectopic expression assays.41 Hence, all assays that we carried out argue that the FRMD5 variants are partial LoF variants. It’s worth noting that the ectopic expression assays indicate that the c.1051A>G is the most severe variant, consistent with the human phenotype since proband 2 with the variant exhibits the most severe symptoms. Also, for the c.1637A>G that could not be confirmed to be de novo, the rescue assay and ectopic expression assays consistently show that it is a partial LoF allele.
Figure 5.

FRMD5 variants are less toxic, and some have dominant-negative effects
(A) Summary of the lethality phenotype of wing-specific expression of dFrmd, FRMD5 Ref, and variants at different temperatures. Note that the FRMD5 Ref causes a more severe phenotype than the variants at 18°C. Some of the surviving flies exhibit wing defects and are noted as “1”. The variants in red could not be confirmed to be de novo.
(B) Quantitative data at 18°C are shown. The survival rate is calculated when compared to nub-GAL4>UAS-Empty. Each dot represents an independent cross.
(C) Wing-specific overexpression of dFrmd and FRMD5 Ref causes similar vein loss and blistery wing phenotypes. The defects are highlighted in red dashed circles.
(D) The heat shock assays for flies with dFrmdCRIMIC-TG4 allele-driven expression of FRMD5 cDNAs in the dFrmd LoF (dFrmdCRIMIC-TG4/Df) background. The percentage of dFrmd LoF mutant flies with seizures was ∼80% after exposure to a 42°C water bath for 30 s. The phenotype can be significantly rescued by human FRMD5 Ref, but the tested FRMD5 variants significantly reduced the rescue ability of FRMD5 Ref. Flies were raised at 25°C and tested at 14–15 days. Each dot represents an independent test of 5–8 female flies.
For (B) and (D), total fly numbers are shown in the columns. Data are represented as mean + SEM. Unpaired t tests. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; n.s., no significance.
FRMD5 variants disrupt the function of FRMD5 in a dominant-negative manner
A previous study showed that FRMD5 interacts with ROCK1 via the FA domain and inhibits the ROCK1 kinase activity.7 Hence, FRMD5 regulates actin-based cytoskeletal remodeling by modulating the kinase activity of ROCK1.7 Since the seven variants are missense, and five are clustered in the FA domain, these variants may disrupt FRMD5 function in a dominant-negative manner. To address if the variants are dominant negative, we expressed one copy of FRMD5 Ref together with one copy of the FRMD5 variants in the dFrmd LoF background (dFrmdCRIMIC-TG4/Df). If a variant is a LoF variant, the expression of the FRMD5 Ref should suppress the phenotype of the dFrmd LoF mutant, whereas the presence of a dominant-negative variant should reduce the rescue ability of the Ref.42 As shown in Figure 5D, expression of the FRMD5 Ref decreases the heat sensitivity from ∼80% to ∼20%. In contrast, co-expression of the Ref with either c.1051A>G or c.1054T>C causes an intermediate phenotype (Figure 5D). These data suggest that the tested variants impair the rescue ability of FRMD5 Ref. These data indicate that the variants act in a dominant-negative manner.
Since the structure of the protein is fundamental for its function, we explored if the FRMD5 variants lead to significant conformational changes in the protein when compared to the Ref. We predicted the three-dimensional structure of FRMD5 for the variants using the AlphaFold Protein Structure Database.43,44 The variants in the FA domain are clustered in a loop (Figure S5A). Modeling based on AlphaFold did not show any obvious structural differences between the Ref and the variants, not only for the variants in the FA domain, but also for the two other variants (Figures S5B and S5D). Moreover, based on the ectopic expression assays, there are no significant functional differences between the variants clustering in the FA domain and the two variants that do not map to the FA domain (Figures 5B and S4B). These data suggest that all these variants may have dominant-negative effects.
Among the ∼50 FDCPs, there are eight proteins with their names containing “FRMD” in human (FRMD1, 3, 4A, 4B, 5, 6, 7, and 8),2,45 and two genes encoding FRMD4A and FRMD7 have been associated with human diseases. A homozygous frameshift mutation of the FRMD4A (MIM: 616305) in multiple affected individuals in a family is associated with severe neurologic symptoms, which include microcephaly and intellectual disability (MIM: 616819).46 FRMD4A is a scaffolding protein that regulates epithelial cell polarity by connecting the small GTPase ADP-ribosylation factor 6 (ARF6) and the par-3 family cell polarity regulator (PARD3).47 Suppression of PARD3 (MIM: 606745) expression disrupts the polarity distribution of human neural progenitor cells.48 Interestingly, the ankyrin repeat and LEM domain containing 2 (ANKLE2)-PAR complex pathway is conserved from flies to humans, and previous work showed that bi-allelic mutations in ANKLE2 (MIM: 616062) are associated with microcephaly in humans.49 Moreover, loss of Ankle2 leads to loss of neuroblasts and disrupted asymmetric cell division of neuroblasts and causes microcephaly.50
Mutations in FRMD7 (MIM: 300628) cause X-linked idiopathic congenital nystagmus (MIM: 310700).51 FRMD7 is shown to activate GTPase RAC1 signaling in vitro52 and co-localizes with actin in the growth cones of differentiated NEURO2A cells.53 Knockdown of FRMD7 during neuronal differentiation leads to disrupted actin cytoskeleton and results in altered neurite outgrowth.53 However, little is known about the function of FRMD7 in animal models. Interestingly, the roundabout guidance receptor 1 (ROBO1), another protein localized to growth cones of neurons, controls axonal guidance in the Drosophila CNS,54 and human individuals who are homozygous for LoF variants of ROBO1 (MIM: 602430) exhibit nystagmus.55
It is striking that five of the seven FRMD5 variants are clustered within very few amino acids (aa 349–354) in the FA domain. Although our knowledge about the structure and function of the FA domain is limited, a previous study showed that the FA domain of FRMD5 is required for FRMD5-ROCK1 interaction, and FRMD5 regulates actin-based cytoskeletal rearrangements by inhibiting the ROCK1 kinase activity.7 Our data based on ERGs suggest that dFrmd is required to maintain proper synaptic transmission. Further studies examining the precise biological mechanisms will lead to a better understanding of the disease pathogenesis.
Acknowledgments
We thank the probands and families for agreeing to participate in this study. We thank Ms. Hongling Pan for transgenic fly lines. We thank the Bloomington Drosophila Stock Center (BDSC) for numerous stocks and Drosophila Genomics Resource Center (DGRC, supported by NIH Grant 2P40OD010949) for the fly cDNA clone. We thank the Chigene (Beijing) Translational Medicine Research Center Co. Ltd. for the technical support. This work was supported by the Howard Hughes Medical Institute (HHMI), the Huffington Foundation, and the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital to H.J.B. Further support was obtained from The Office of Research Infrastructure Programs of the NIH (R24 OD022005 and R24 OD031447) to H.J.B. We thank the Deciphering Developmental Disorders (DDD) study for the referral of two probands. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003). This study makes use of DECIPHER, which is funded by Wellcome (grant number 223718/Z/21/Z); see www.ddduk.org/access.html for full acknowledgement. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. We thank AlphaFold for structural predictions. The molecular graphics and analyses were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. Please see the supplemental information for full acknowledgments.
Declaration of interests
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories. M.L. is a salaried employee and shareholder of Invitae Corp.
Published: October 6, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.09.005.
Contributor Information
Yuwei Dai, Email: daiyuwei_1996@163.com.
Hugo J. Bellen, Email: hbellen@bcm.edu.
Web resources
AlphaFold, https://alphafold.ebi.ac.uk/
DECIPHER, www.deciphergenomics.org
MARRVEL, http://www.marrvel.org/
OMIM, http://www.omim.org/
UCSF Chimera, https://www.rbvi.ucsf.edu/chimera/
Supplemental information
Data and code availability
All reagents developed in this study are available upon request. Some of the variants were submitted to ClinVar (GenBank: NM_032892.5): c.1053C>G, SCV002564145.1; c.1054T>C, SCV002564146.1; c.1045A>C, SCV002564147.1; c.1637A>G, SCV002564148.1. The exome datasets supporting this study have not been deposited in a public repository due to privacy and ethical/legal issues.
References
- 1.Chishti A.H., Kim A.C., Marfatia S.M., Lutchman M., Hanspal M., Jindal H., Liu S.C., Low P.S., Rouleau G.A., Mohandas N., et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem. Sci. 1998;23:281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- 2.Bosanquet D.C., Ye L., Harding K.G., Jiang W.G. FERM family proteins and their importance in cellular movements and wound healing (review) Int. J. Mol. Med. 2014;34:3–12. doi: 10.3892/ijmm.2014.1775. [DOI] [PubMed] [Google Scholar]
- 3.Frame M.C., Patel H., Serrels B., Lietha D., Eck M.J. The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010;11:802–814. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- 4.Patel H., König I., Tsujioka M., Frame M.C., Anderson K.I., Brunton V.G. The multi-FERM-domain-containing protein FrmA is required for turnover of paxillin-adhesion sites during cell migration of Dictyostelium. J. Cell Sci. 2008;121:1159–1164. doi: 10.1242/jcs.021725. [DOI] [PubMed] [Google Scholar]
- 5.Amberger J.S., Hamosh A. Searching Online Mendelian Inheritance in Man (OMIM): a knowledgebase of human genes and genetic phenotypes. Curr. Protoc. Bioinformatics. 2017;58:1.2.1–1.2.12. doi: 10.1002/cpbi.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T., Pei X., Zhan J., Hu J., Yu Y., Zhang H. FERM-containing protein FRMD5 is a p120-catenin interacting protein that regulates tumor progression. FEBS Lett. 2012;586:3044–3050. doi: 10.1016/j.febslet.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Hu J., Niu M., Li X., Lu D., Cui J., Xu W., Li G., Zhan J., Zhang H. FERM domain-containing protein FRMD5 regulates cell motility via binding to integrin beta5 subunit and ROCK1. FEBS Lett. 2014;588:4348–4356. doi: 10.1016/j.febslet.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Mao X., Tey S.K., Ko F.C.F., Kwong E.M.L., Gao Y., Ng I.O.L., Cheung S.T., Guan X.Y., Yam J.W.P. C-terminal truncated HBx protein activates caveolin-1/LRP6/beta-catenin/FRMD5 axis in promoting hepatocarcinogenesis. Cancer Lett. 2019;444:60–69. doi: 10.1016/j.canlet.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Zhu C., Yamaguchi K., Ohsugi T., Terakado Y., Noguchi R., Ikenoue T., Furukawa Y. Identification of FERM domain-containing protein 5 as a novel target of beta-catenin/TCF7L2 complex. Cancer Sci. 2017;108:612–619. doi: 10.1111/cas.13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amberger J.S., Bocchini C.A., Schiettecatte F., Scott A.F., Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J., Al-Ouran R., Hu Y., Kim S.Y., Wan Y.W., Wangler M.F., Yamamoto S., Chao H.T., Comjean A., Mohr S.E., et al. MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am. J. Hum. Genet. 2017;100:843–853. doi: 10.1016/j.ajhg.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDonald J.R., Ziman R., Yuen R.K.C., Feuk L., Scherer S.W. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinodoz M., Peter V.G., Cisarova K., Royer-Bertrand B., Stenson P.D., Cooper D.N., Unger S., Superti-Furga A., Rivolta C. Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am. J. Hum. Genet. 2022;109:457–470. doi: 10.1016/j.ajhg.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma M., Moulton M.J., Lu S., Bellen H.J. 'Fly-ing' from rare to common neurodegenerative disease mechanisms. Trends Genet. 2022;38:972–984. doi: 10.1016/j.tig.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N., Mohr S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinf. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee P.T., Zirin J., Kanca O., Lin W.W., Schulze K.L., Li-Kroeger D., Tao R., Devereaux C., Hu Y., Chung V., et al. A gene-specific T2A-GAL4 library for Drosophila. Elife. 2018;7:e35574. doi: 10.7554/eLife.35574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu S., Hernan R., Marcogliese P.C., Huang Y., Gertler T.S., Akcaboy M., Liu S., Chung H.L., Pan X., Sun X., et al. Loss-of-function variants in TIAM1 are associated with developmental delay, intellectual disability, and seizures. Am. J. Hum. Genet. 2022;109:571–586. doi: 10.1016/j.ajhg.2022.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcogliese P.C., Deal S.L., Andrews J., Harnish J.M., Bhavana V.H., Graves H.K., Jangam S., Luo X., Liu N., Bei D., et al. Drosophila functional screening of de novo variants in autism uncovers damaging variants and facilitates discovery of rare neurodevelopmental diseases. Cell Rep. 2022;38:110517. doi: 10.1016/j.celrep.2022.110517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davie K., Janssens J., Koldere D., De Waegeneer M., Pech U., Kreft Ł., Aibar S., Makhzami S., Christiaens V., Bravo González-Blas C., et al. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell. 2018;174:982–998.e20. doi: 10.1016/j.cell.2018.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Janssens J., De Waegeneer M., Kolluru S.S., Davie K., Gardeux V., Saelens W., David F.P.A., Brbić M., Spanier K., et al. Fly Cell Atlas: A single-nucleus transcriptomic atlas of the adult fruit fly. Science. 2022;375:eabk2432. doi: 10.1126/science.abk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganetzky B., Wu C.F. Indirect Suppression Involving Behavioral Mutants with Altered Nerve Excitability in Drosophila melanogaster. Genetics. 1982;100:597–614. doi: 10.1093/genetics/100.4.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burg M.G., Wu C.F. Mechanical and temperature stressor-induced seizure-and-paralysis behaviors in Drosophila bang-sensitive mutants. J. Neurogenet. 2012;26:189–197. doi: 10.3109/01677063.2012.690011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L., Gilligan J., Staber C., Schutte R.J., Nguyen V., O'Dowd D.K., Reenan R. A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. J. Neurosci. 2012;32:14145–14155. doi: 10.1523/JNEUROSCI.2932-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolph P., Nair A., Raghu P. Electroretinogram recordings of Drosophila. Cold Spring Harb. Protoc. 2011;2011 doi: 10.1101/pdb.prot5549. pdb.prot5549. [DOI] [PubMed] [Google Scholar]
- 31.Vilinsky I., Johnson K.G. Electroretinograms in Drosophila: a robust and genetically accessible electrophysiological system for the undergraduate laboratory. J. Undergrad. Neurosci. Educ. 2012;11:A149–A157. [PMC free article] [PubMed] [Google Scholar]
- 32.Duffy J.B. GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- 33.Nagarkar-Jaiswal S., Lee P.T., Campbell M.E., Chen K., Anguiano-Zarate S., Cantu Gutierrez M., Busby T., Lin W.W., He Y., Schulze K.L., et al. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. Elife. 2015;4:e05338. doi: 10.7554/eLife.05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ansar M., Chung H.L., Al-Otaibi A., Elagabani M.N., Ravenscroft T.A., Paracha S.A., Scholz R., Abdel Magid T., Sarwar M.T., Shah S.F., et al. Bi-allelic variants in IQSEC1 cause intellectual disability, developmental delay, and short stature. Am. J. Hum. Genet. 2019;105:907–920. doi: 10.1016/j.ajhg.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanca O., Andrews J.C., Lee P.T., Patel C., Braddock S.R., Slavotinek A.M., Cohen J.S., Gubbels C.S., Aldinger K.A., Williams J., et al. De Novo variants in WDR37 are associated with epilepsy, colobomas, dysmorphism, developmental delay, intellectual disability, and cerebellar hypoplasia. Am. J. Hum. Genet. 2019;105:672–674. doi: 10.1016/j.ajhg.2019.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu N., Schoch K., Luo X., Pena L.D.M., Bhavana V.H., Kukolich M.K., Stringer S., Powis Z., Radtke K., Mroske C., et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum. Mol. Genet. 2018;27:2454–2465. doi: 10.1093/hmg/ddy146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcogliese P.C., Shashi V., Spillmann R.C., Stong N., Rosenfeld J.A., Koenig M.K., Martínez-Agosto J.A., Herzog M., Chen A.H., Dickson P.I., et al. IRF2BPL is associated with neurological phenotypes. Am. J. Hum. Genet. 2018;103:245–260. doi: 10.1016/j.ajhg.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravenscroft T.A., Phillips J.B., Fieg E., Bajikar S.S., Peirce J., Wegner J., Luna A.A., Fox E.J., Yan Y.L., Rosenfeld J.A., et al. Heterozygous loss-of-function variants significantly expand the phenotypes associated with loss of GDF11. Genet. Med. 2021;23:1889–1900. doi: 10.1038/s41436-021-01216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Splinter K., Adams D.R., Bacino C.A., Bellen H.J., Bernstein J.A., Cheatle-Jarvela A.M., Eng C.M., Esteves C., Gahl W.A., Hamid R., et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 2018;379:2131–2139. doi: 10.1056/NEJMoa1714458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uehara T., Sanuki R., Ogura Y., Yokoyama A., Yoshida T., Futagawa H., Yoshihashi H., Yamada M., Suzuki H., Takenouchi T., et al. Recurrent NFIA K125E substitution represents a loss-of-function allele: Sensitive in vitro and in vivo assays for nontruncating alleles. Am. J. Med. Genet. 2021;185:2084–2093. doi: 10.1002/ajmg.a.62226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodman L.D., Cope H., Nil Z., Ravenscroft T.A., Charng W.L., Lu S., Tien A.C., Pfundt R., Koolen D.A., Haaxma C.A., et al. TNPO2 variants associate with human developmental delays, neurologic deficits, and dysmorphic features and alter TNPO2 activity in Drosophila. Am. J. Hum. Genet. 2021;108:1669–1691. doi: 10.1016/j.ajhg.2021.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller H.J. Further studies on the nature and causes of gene mutations. Proc. 6th Int. Congr. Genet. 1932;1:213–255. [Google Scholar]
- 43.Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A., et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi: 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moleirinho S., Tilston-Lunel A., Angus L., Gunn-Moore F., Reynolds P.A. The expanding family of FERM proteins. Biochem. J. 2013;452:183–193. doi: 10.1042/BJ20121642. [DOI] [PubMed] [Google Scholar]
- 46.Fine D., Flusser H., Markus B., Shorer Z., Gradstein L., Khateeb S., Langer Y., Narkis G., Birk R., Galil A., et al. A syndrome of congenital microcephaly, intellectual disability and dysmorphism with a homozygous mutation in FRMD4A. Eur. J. Hum. Genet. 2015;23:1729–1734. doi: 10.1038/ejhg.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ikenouchi J., Umeda M. FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc. Natl. Acad. Sci. USA. 2010;107:748–753. doi: 10.1073/pnas.0908423107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X., An Y., Gao Y., Guo L., Rui L., Xie H., Sun M., Lam Hung S., Sheng X., Zou J., et al. Rare Deleterious PARD3 variants in the apkc-binding region are implicated in the pathogenesis of human cranial neural tube defects via disrupting apical tight junction formation. Hum. Mutat. 2017;38:378–389. doi: 10.1002/humu.23153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto S., Jaiswal M., Charng W.L., Gambin T., Karaca E., Mirzaa G., Wiszniewski W., Sandoval H., Haelterman N.A., Xiong B., et al. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159:200–214. doi: 10.1016/j.cell.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Link N., Chung H., Jolly A., Withers M., Tepe B., Arenkiel B.R., Shah P.S., Krogan N.J., Aydin H., Geckinli B.B., et al. Mutations in ANKLE2, a ZIKA virus target, disrupt an asymmetric cell division pathway in drosophila neuroblasts to cause microcephaly. Dev. Cell. 2019;51:713–729.e6. doi: 10.1016/j.devcel.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tarpey P., Thomas S., Sarvananthan N., Mallya U., Lisgo S., Talbot C.J., Roberts E.O., Awan M., Surendran M., McLean R.J., et al. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat. Genet. 2006;38:1242–1244. doi: 10.1038/ng1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z., Mao S., Pu J., Ding Y., Zhang B., Ding M. A novel missense mutation in the FERM domain containing 7 (FRMD7) gene causing X-linked idiopathic congenital nystagmus in a Chinese family. Mol. Vis. 2013;19:1834–1840. [PMC free article] [PubMed] [Google Scholar]
- 53.Betts-Henderson J., Bartesaghi S., Crosier M., Lindsay S., Chen H.L., Salomoni P., Gottlob I., Nicotera P. The nystagmus-associated FRMD7 gene regulates neuronal outgrowth and development. Hum. Mol. Genet. 2010;19:342–351. doi: 10.1093/hmg/ddp500. [DOI] [PubMed] [Google Scholar]
- 54.Kidd T., Brose K., Mitchell K.J., Fetter R.D., Tessier-Lavigne M., Goodman C.S., Tear G. Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell. 1998;92:205–215. doi: 10.1016/s0092-8674(00)80915-0. [DOI] [PubMed] [Google Scholar]
- 55.Huang Y., Ma M., Mao X., Pehlivan D., Kanca O., Un-Candan F., Shu L., Akay G., Mitani T., Lu S., et al. Novel dominant and recessive variants in human ROBO1 cause distinct neurodevelopmental defects through different mechanisms. Hum. Mol. Genet. 2022;31:2751–2765. doi: 10.1093/hmg/ddac070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flies in empty vials are submerged in a 42°C water bath for 30 s. Video recording from 12 s is shown. Flies were raised at 25°C and tested at 14–15 days. For genotypes, left vial: dFrmdCRIMIC-TG4/Df;UAS-Empty; right vial: dFrmdCRIMIC-TG4/Df;GR.
Flies in empty vials are allowed to recover after being submerged in a 42°C water bath for 30 s. Video recording from 5 s after water bath is shown. Flies were raised at 25°C and tested at 14–15 days. For genotypes, left vial: dFrmdCRIMIC-TG4/Df;UAS-Empty; right vial: dFrmdCRIMIC-TG4/Df;GR.
Data Availability Statement
All reagents developed in this study are available upon request. Some of the variants were submitted to ClinVar (GenBank: NM_032892.5): c.1053C>G, SCV002564145.1; c.1054T>C, SCV002564146.1; c.1045A>C, SCV002564147.1; c.1637A>G, SCV002564148.1. The exome datasets supporting this study have not been deposited in a public repository due to privacy and ethical/legal issues.