

Abstract
To characterize the gut bacteriome, mycobiome and serum metabolome profiles in polycystic ovary syndrome (PCOS) patients with normal/overweight individuals and evaluate a potential microbiota-related diagnostic method development for PCOS, 16S rRNA and ITS2 gene sequencing using 88 fecal samples and 87 metabolome analysis from serum samples are conducted and PCOS classifiers based on multiomics markers are constructed. There are significant bacterial, fungal community and metabolite differences among PCOS patients and healthy volunteers with normal/overweight individuals. Healthy individuals with overweight/obesity display less abnormal metabolism than PCOS patients and uniquely higher abundance of the fungal genus Mortierella. Nine bacterial genera, 4 predicted pathways, 11 fungal genera and top 30 metabolites are screened out which distinguish PCOS from healthy controls, with AUCs of 0.84, 0.64, 0.85 and 1, respectively. The metabolite-derived model is more accurate than the microbe-based model in discriminating normal BMI PCOS (PCOS-LB) from normal BMI healthy (Healthy-LB), PCOS-HB from Healthy-HB. Featured bacteria, fungi, predicted pathways and serum metabolites display higher associations with free androgen index (FAI) in the cooccurrence network. In conclusion, our data reveal that hyperandrogenemia plays a central role in the dysbiosis of intestinal microecology and the change in metabolic status in patients with PCOS and that its effect exceeds the role of BMI. Healthy women with high BMI showed unique microbiota and metabolic features.The priority of predictive models in discriminating PCOS from healthy status in this study were serum metabolites, fungal taxa and bacterial taxa.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13048-022-01051-8.
Keywords: PCOS, Mycobiome, Diagnostic model, Metabolome, Obesity
Introduction
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder that causes infertility in reproductive-aged women [1], which is also associated with insulin resistance (IR), type 2 diabetes mellitus (T2DM), dyslipidemia, nonalcoholic fatty liver disease (NAFLD) and cardiovascular disease [2, 3]. To date, the precise underlying triggers for PCOS remain unclear, but multifactorial factors, including genetics, intrauterine environment, lifestyle and gut microbiota, are thought to be involved in its development.
Microbiota dysbiosis is associated with endocrine and metabolic diseases such as obesity [4], diabetes [5] and PCOS [6, 7]. Recent studies have shown that gut microbiota of PCOS patients differs from that of healthy controls. Qi et al. [7] found that Bacteroides vulgatus was markedly elevated in PCOS individuals. Yang et al. [8] showed that Bacteroides is a key microbial biomarker for PCOS and even has diagnostic value. The dynamic change of predominant bacteria in PCOS may be affected by race, lifestyle [9], disease severity and the sample size. Previously, we found that Prevotella_9, Dorea, Maihella and Slackia were significantly changed in PCOS patients [6]. Nevertheless, whether the gut microbiota contributes to the occurrence and development of PCOS needs further investigation.
Recent evidence suggests that fungi influence local and peripheral immune responses, enhance relevant disease status [10], trigger the occurrence and development of diseases, such as colitis [11], colorectal cancer [12], primary sclerosing cholangitis [13], alcohol-associated liver disease [14] and COVID-19 [15]. Although limited reports have indicated that fungi play a certain role in endocrine and metabolic diseases, the illumination of their role is still in its infancy. Mar Rodríguez et al. [16] firstly revealed gut fungal composition alterations in obese patients. Honkanen et al. [17] showed that bacterial and fungal dysbiosis may be associated with the development of type 1 diabetes mellitus (T1D) in children with beta-cell autoimmunity. Patients with coronary heart disease complicated with NAFLD show an increase in the abundance of Preussia, Xylodon and Cladorrhinum and a reduction of Candida glabrata and Ganoderma [18].
Metabolomics is a useful method to discover and identify metabolites involved in disease [19, 20]. Our previous study showed that Prevotella_9 correlates positively with Lysopc 18:1, Glu-Gln, lysophosphatidyl choline (LPC) 22:1, PC(14:1E/8:0), and LPC 17:2 [6] but negatively with estrone sulfate [6]. The correlation between the gut microbiota and plasma metabolites suggests that the former may participate in the metabolic pathway of PCOS. Therefore, integrated analysis of multiomics data from the gut bacteriome, mycobiome, metabolome and phenome may provide a clue to mechanistic links between PCOS and the gut microbiota. Furthermore, metabolomics provides potential metabolic markers for the prognosis and diagnosis of PCOS [19, 20]. Daan et al. reported that retinol-binding protein 4 (RBP-4), dipeptidyl peptidase IV (DPP-IV) and adiponectin are potential discriminative markers for PCOS with obvious hyperandrogenemia [21].
PCOS may have different etiological causes, and currently we can see that PCOS patients with normal/overweight individuals also differ in clinical phenotypes, such as levels of insulin and polyunsaturated fatty acids including arachidonic acid [22]. Dapas et al. [23] showed that reproductive and metabolic subtypes of PCOS appeared to have distinct genetic architecture and these two subtypes had obvious difference of BMI. Therefore, we are of great concern about the etiological differences in obese and lean PCOS patients and the differences in their gut microbes and plasma metabolites.
In this study, PCOS patients and healthy volunteers with different body mass index (BMI) levels were used to determine gut bacteria and fungi by 16S and ITS2 gene sequencing methods, and serum metabolites from widely targeted metabolomes. The microbiome and metabolome data were then integrated to distinguish PCOS-related alterations in fecal microbial and serum metabolic features, and potential links and roles of the microbiome and metabolome in disease diagnosis were assessed.
Methods
Subject recruitment
The study was a cross-sectional study and all experimental procedures were approved by the Ethics Committee of the First Affiliated Hospital Shantou University Medical College. The biological sample banks, including plasma, DNA and fecal samples were established. Written informed consent was obtained from all participants. The inclusion and exclusion criteria for the healthy volunteers and PCOS patients were the same as those described previously [6]. Patients with PCOS were diagnosed according to the 2003 Rotterdam criteria, which require the presence of at least two of the following: oligo-ovulation and/or anovulation; clinical and/or biochemical signs of hyperandrogenism; and ultrasound findings of polycystic ovaries in 1 or 2 ovaries, ≥ 12 follicles measuring 2 to 9 mm in diameter, and/or ovarian volume ≥ 10 mL. Diagnoses of PCOS were made after the exclusion of other etiologies for hyperandrogenemium or ovulatory dysfunction including Cushing syndrome, 21-hydroxylase deficiency, thyroid disease, androgen-secreting tumors, congenital adrenal hyperplasia and hyperprolactinemia. The healthy volunteers with regular menstrual cycles, normal ovarian morphology, and normal level of androgen were from the general community. A total of eighty-eight participants were recruited between June 2019 and November 2020. The cohort was divided into four groups: Healthy-LB (Healthy individuals, BMI < 24) (n = 21), PCOS-LB (PCOS patients, BMI < 24) (n = 22), Healthy-HB (Healthy individuals, BMI ≥ 24) (n = 20) and PCOS-HB (PCOS patients, BMI ≥ 24) (n = 25). All necessary clinical parameters and DNA methylation determination were determined as previously reported [6]. All the participants were asked to come to our department during days 2–4 of the menstrual period after an overnight fast. Peripheral blood samples were collected from all subjects for the parameters measurements and then oral glucose tolerance test and insulin releasing test were performed. Further blood samples were taken at 120 min for measurement of glucose and insulin.
16S rRNA gene amplicon sequencing and data processing
DNA from stool samples was extracted using HiPure Stool DNA Kits B (D3141-03B, Guangzhou Meiji Biotechnology Co., Ltd., China). 16S rRNA gene amplification [24, 25], DNA library concentration validation, multiplexing and Illumina sequencing were performed as previously described [6]. Raw sequencing data were trimmed for quality and length, and Illumina adapters were removed using Fastp (version 0.19.6) with the following criteria: (i) reads containing ≥ 10% N bases; (ii) more than 50% of the base with a quality score < 20; (iii) the adaptor sequence and its subsequent sequence; (iv) truncated reads < 200 bp. If one read in each paired-end reads reaches the filtering standard, the paired reads will be removed then.The QIIME2 pipeline [26] was applied to process and analyze 16S rRNA gene sequencing data (QIIME2, version 2019.4). The sequence file processed by Fastp was imported into QIIME2 for subsequent data filtering. The filtering steps were as follows: (i) remove primers with default parameters by cutadapt function in QIIME2; (ii) DADA2 was used to remove interference sequence, chimeric sequence, etc. The parameters –p-trunc-len-f and –p-trunc-len-r were set to 0, and default parameters were used for other analysis parameters. The representative sequence and abundance table were obtained by DADA2 [27]. Taxonomy was assigned to sequences using q2-feature-classifier classifysklearn [28] against Silva [29]. Microbial community functional composition was predicted based on 16S rRNA sequences using PICRUSt software [30].
ITS2 gene amplicon sequencing and data processing
DNA was extracted from stool samples using E.Z.N.A.® Stool DNA Kit (Omega Biotek, Norcross, GA, U.S.). The ITS2 gene was amplified by PCR with barcoded forward primers (ITS1F: 5’-CTTGGTCATTTAGAGGAAGTAA-3’) and reverse primers (ITS2R: 5’-GCTGCGTTCTTCATCGATGC-3’) [31]. Pooled DNA products were used to construct an Illumina Pair-End library following Illumina’s genomic DNA library preparation procedure, and the amplicon library was paired-end sequenced (2 × 250 bp) using the Illumina platform (Shanghai BIOZERON Biotech. Co., Ltd). The bioinformatics analysis procedure for ITS2 data was similar to that for 16S rRNA data, the UNITE (ITS) reference database was used [32].
Serum wide targeted metabolomics profiling and data preprocessing
The serum sample extracts were analyzed using an LC–ESI–MS/MS system (UPLC, ExionLC AD, https://sciex.com.cn/; MS, QTRAP® System, https://sciex.com/). LIT and triple quadrupole (QQQ) scans were acquired with a triple quadrupole-linear ion trap mass spectrometer (QTRAP), QTRAP® LC–MS/MS System, equipped with an ESI Turbo Ion-Spray interface, operating in positive and negative ion mode and controlled by Analyst 1.6.3 software (Sciex). The ESI source operation parameters were as follows: source temperature 500 °C; ion spray voltage (IS) 5500 V (positive), -4500 V (negative); ion source gas I (GSI), gas II (GSII), curtain gas (CUR) set at 55, 60, and 25.0 psi, respectively; and high collision gas (CAD). Instrument tuning and mass calibration were performed with 10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. A specific set of MRM transitions was monitored for each period according to the metabolites eluted within this period.
Statistical analysis
All statistical tests were performed using R (version 4.0.2). The α diversity index was calculated using the R program package ‘vegan’ (version 2.5.7). Group comparisons were conducted by ANOVA tests, and the LDuncan method (package laercio, 1.0–1) was used to group differences. Differences in community were determined by principal co-coordinates analysis (PCoA) and RDA (package vegan, 2.5.7). PERMANOVA and ANOSIM were conducted to assess statistical significance. Mantel tests were carried out to examine Spearman’s rank correlation between bacterial, fungal, and functional pathways, metabolites and the clinical index matrix (vegan package). Linear discriminant analysis effect size (LEfSe) analysis was performed [33, 34]. Kruskal–Wallis tests among multiple groups and Wilcoxon tests between paired groups were conducted. Multiple hypothesis tests were adjusted using the Benjamini-Hochberg (B-H) false discovery rate (FDR).
Metabolome data were analyzed by PLS-DA with the package mixOmics (6.14.1). Characterized metabolites were screened out based on (1) variable importance in projection (VIP) > 1, (2) fold change of > 2 or < 0.5 and (3) FDR adjusted p < 0.05 (Wilcox test). Randomforest [35] algorithms were trained with the multiomics data using the randomForest R package [36] and graphed by pROC package. Input features were excavated on the basis of Wilcox test comparison and the mean decrease in Gini by random forest importance parameter evaluation. Data were assigned to training (80%) and test (20%) datasets after the whole dataset was shuffled. Subsequently, we further explored the complicated network interaction of discriminative features derived from multiomics data using Cytoscape (v3.5.1).
Results
Clinical characteristics of the participants
The participant design and demographics are shown in Fig. 1a and Table 1. Compared with the Healthy group, PCOS patients displayed significantly higher luteinizing hormone (LH), follicle-stimulating hormone (FSH), estrogen (E2), androstenedione (AD), total testosterone (TT), free androgen index (FAI), glucose level at fasting status (G0), insulin level at 120 min after glucose load (I120), and glucose level at 120 min after glucose load (G120) while lower progesterone (PROG) and sex hormone-binding globulin (SHBG) (FDR adjusted p < 0.05) (Table S1). PCOS-HB patients (PCOS patients, BMI ≥ 24) had significantly higher LH, LH/FSH, AD, TT, dehydroepiandrosterone (DHEA), and FAI while lower SHBG than that of Healthy-HB subjects (FDR adjusted p < 0.05) (Fig. 1b, Table 1). There were significant differences in the clinical parameter structure among the four groups based on PCoA (Fig. 1c) (PERMANOVA: p < 0.001; ANOSIM: p < 0.05).
Fig. 1.

Clinical parameter distribution. a Overview of the study design. b Differences in clinical index. Data are shown as the mean ± SD, and an error bar is shown. p values denote the significance among groups. Letters indicate ANOVA grouping. c Differences in clinical index structures as revealed by principal co-ordinates analysis (PCoA)
Table 1.
Clinical characteristics baseline in the BMI-related healthy and PCOS groups
| Parameter | Characteristic | Healthy-LB (n = 21) | PCOS-LB (n = 22) | p value | FDR | Healthy-HB (n = 20) | PCOS-HB (n = 25) | p value | FDR | Four groups p value | Four groups FDR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Demographic characteristics | Age (years) | 28.53 ± 3.19 | 28.96 ± 3.87 | 0.6 | 0.832 | 29.9 ± 2.98 | 29.04 ± 3.89 | 0.283 | 0.498 | 0.555 | 0.666 |
| BMI (kg/m2) | 20.01 ± 1.83 | 20.68 ± 2.17 | 0.23 | 0.504 | 29.03 ± 3.06 | 28.19 ± 2.82 | 0.332 | 0.541 | 0.001 | 0.001 | |
| Liver function | LDH (U/L) | 174.2 ± 29.51 | 174.96 ± 44.35 | 0.942 | 0.972 | 183.8 ± 30.79 | 198.88 ± 30.71 | 0.121 | 0.272 | 0.013 | 0.025 |
| AST (U/L) | 20.2 ± 6.09 | 20.1 ± 5.73 | 0.808 | 0.972 | 19.62 ± 4.9 | 24.88 ± 10.51 | 0.071 | 0.209 | 0.121 | 0.166 | |
| ALT (U/L) | 14.77 ± 7.95 | 18.19 ± 11.92 | 0.526 | 0.827 | 19.96 ± 10.31 | 32.35 ± 26.24 | 0.043 | 0.144 | 0.002 | 0.004 | |
| GGT (U/L) | 20 ± 9.69 | 20.91 ± 9.58 | 0.551 | 0.832 | 21.97 ± 9.72 | 28.92 ± 14.32 | 0.044 | 0.144 | 0.021 | 0.039 | |
| ALP (U/L) | 69.67 ± 16.8 | 69 ± 16.12 | 0.444 | 0.748 | 76.29 ± 16.11 | 78.04 ± 23.73 | 0.592 | 0.723 | 0.114 | 0.162 | |
| CHE (U/mL) | 7.32 ± 1.49 | 7.96 ± 1.35 | 0.294 | 0.579 | 9.02 ± 1.32 | 9.6 ± 1.54 | 0.371 | 0.583 | 0.001 | 0.001 | |
| MAO (U/L) | 3.29 ± 0.91 | 3 ± 1.12 | 0.303 | 0.579 | 5.42 ± 2.74 | 4.1 ± 1.47 | 0.128 | 0.272 | 0.001 | 0.001 | |
| AFU (U/L) | 23.34 ± 4.34 | 27.23 ± 6.57 | 0.043 | 0.142 | 26.95 ± 5.69 | 29.32 ± 6.5 | 0.326 | 0.541 | 0.031 | 0.056 | |
| TP (g/L) | 76.28 ± 4.51 | 77.01 ± 3.86 | 0.972 | 0.972 | 76.77 ± 4.77 | 76.56 ± 3.83 | 0.955 | 1 | 0.998 | 0.998 | |
| ALB (g/L) | 44.1 ± 2.12 | 44.79 ± 1.94 | 0.338 | 0.619 | 45.69 ± 6.92 | 44.74 ± 2.14 | 0.53 | 0.707 | 0.676 | 0.782 | |
| GLB (g/L) | 32.19 ± 3.03 | 32.22 ± 2.89 | 0.674 | 0.899 | 32.41 ± 3.92 | 31.82 ± 2.79 | 0.576 | 0.723 | 0.952 | 0.987 | |
| ALB/GLB | 1.38 ± 0.11 | 1.4 ± 0.12 | 0.601 | 0.832 | 1.39 ± 0.18 | 1.41 ± 0.15 | 0.479 | 0.68 | 0.822 | 0.928 | |
| TBIL (μmol/L) | 10.87 ± 4.56 | 10.94 ± 4.37 | 0.971 | 0.972 | 11.84 ± 5.65 | 10.45 ± 2.93 | 0.743 | 0.861 | 0.932 | 0.987 | |
| DBIL (μmol/L) | 2.05 ± 0.94 | 1.96 ± 0.81 | 0.933 | 0.972 | 2.51 ± 2.4 | 1.77 ± 0.57 | 0.15 | 0.3 | 0.56 | 0.666 | |
| IBIL (μmol/L) | 8.82 ± 3.7 | 8.99 ± 3.67 | 0.846 | 0.972 | 9.33 ± 3.75 | 8.67 ± 2.46 | 1 | 1 | 0.964 | 0.987 | |
| Renal function | CO2 (mmol/L) | 25.17 ± 2.48 | 26.22 ± 2.19 | 0.178 | 0.421 | 25.42 ± 7.61 | 25.62 ± 2.56 | 0.098 | 0.239 | 0.099 | 0.15 |
| BUN (mmol/L) | 4.8 ± 1.28 | 4.09 ± 0.71 | 0.032 | 0.12 | 4.74 ± 0.94 | 4.59 ± 1.14 | 0.418 | 0.634 | 0.067 | 0.114 | |
| Cr (μmol/L) | 75.62 ± 7.22 | 71.19 ± 8.67 | 0.103 | 0.282 | 72.5 ± 11.08 | 71.68 ± 9.5 | 0.561 | 0.723 | 0.105 | 0.153 | |
| Metabolic index | UA (μmol/L) | 346.39 ± 89.7 | 355.47 ± 74.5 | 0.605 | 0.832 | 404.96 ± 138.48 | 428.22 ± 79.55 | 0.234 | 0.428 | 0.005 | 0.009 |
| TC (mmol/L) | 4.74 ± 0.83 | 4.74 ± 0.61 | 0.913 | 0.972 | 4.84 ± 0.72 | 4.8 ± 0.87 | 0.508 | 0.699 | 0.884 | 0.972 | |
| TG (mmol/L) | 0.97 ± 0.39 | 1.06 ± 0.45 | 0.452 | 0.748 | 1.37 ± 0.65 | 1.43 ± 0.78 | 0.991 | 1 | 0.049 | 0.085 | |
| HDL-C (mmol/L) | 1.58 ± 0.22 | 1.46 ± 0.29 | 0.096 | 0.282 | 1.1 ± 0.18 | 1.16 ± 0.16 | 0.226 | 0.428 | 0.001 | 0.001 | |
| LDL-C (mmol/L) | 2.89 ± 0.66 | 2.94 ± 0.42 | 0.762 | 0.972 | 3.18 ± 0.58 | 3.15 ± 0.67 | 0.811 | 0.91 | 0.24 | 0.301 | |
| Sex hormone | LH (mIU/mL) | 6.12 ± 1.65 | 12.23 ± 7.73 | 0.004 | 0.027 | 3.17 ± 1.69 | 6.89 ± 3.29 | 0.001 | 0.001 | 0.001 | 0.001 |
| FSH (mIU/mL) | 4.58 ± 2.11 | 6.6 ± 2.04 | 0.001 | 0.015 | 5.64 ± 1.59 | 6.27 ± 1.68 | 0.085 | 0.225 | 0.002 | 0.003 | |
| LH/FSH | 2.05 ± 3.15 | 1.93 ± 1.16 | 0.156 | 0.402 | 0.56 ± 0.2 | 1.23 ± 0.95 | 0.001 | 0.002 | 0.001 | 0.001 | |
| PRL (ng/mL) | 26.15 ± 16.61 | 15.84 ± 7.79 | 0.015 | 0.064 | 20.5 ± 13.46 | 18.15 ± 10.28 | 0.624 | 0.742 | 0.092 | 0.146 | |
| E2 (pg/mL) | 28.58 ± 18.32 | 69.08 ± 50.55 | 0.001 | 0.001 | 32.97 ± 14.52 | 44.13 ± 15.74 | 0.033 | 0.144 | 0.001 | 0.001 | |
| PROG (nmol/L) | 0.9 ± 0.3 | 0.64 ± 0.37 | 0.008 | 0.039 | 0.64 ± 0.44 | 0.54 ± 0.48 | 0.087 | 0.225 | 0.001 | 0.003 | |
| Androgen | AD (ng/mL) | 1.2 ± 0.37 | 1.99 ± 0.9 | 0.004 | 0.027 | 0.87 ± 0.32 | 1.8 ± 0.65 | 0.001 | 0.001 | 0.001 | 0.001 |
| TT (ng/mL) | 0.31 ± 0.13 | 0.51 ± 0.28 | 0.033 | 0.12 | 0.19 ± 0.08 | 0.46 ± 0.23 | 0.001 | 0.001 | 0.001 | 0.001 | |
| DHEA (ng/mL) | 10.11 ± 5.27 | 11.02 ± 8.88 | 0.953 | 0.972 | 5.57 ± 4.41 | 9.72 ± 4.71 | 0.005 | 0.031 | 0.004 | 0.008 | |
| FAI | 1.83 ± 0.99 | 5.18 ± 3.35 | 0.001 | 0.002 | 2.32 ± 1.27 | 8.78 ± 4.93 | 0.001 | 0.001 | 0.001 | 0.001 | |
| SHBG (nmol/L) | 68.1 ± 31.72 | 40.3 ± 17.5 | 0.002 | 0.022 | 32.91 ± 13.34 | 21.75 ± 11.81 | 0.005 | 0.031 | 0.001 | 0.001 | |
| Glucose tolerance | G0 (mmol/L) | 4.77 ± 0.38 | 5.16 ± 0.42 | 0.005 | 0.027 | 5.19 ± 0.69 | 5.67 ± 1.34 | 0.479 | 0.68 | 0.001 | 0.001 |
| G120 (mmol/L) | 5.76 ± 1.33 | 7.41 ± 2.44 | 0.007 | 0.039 | 6.98 ± 1.68 | 9.18 ± 4.09 | 0.023 | 0.123 | 0.001 | 0.001 | |
| Insulin | I0 (mIU/L) | 8.33 ± 3.65 | 12.36 ± 14.26 | 0.459 | 0.748 | 18.63 ± 9.21 | 18.52 ± 10.68 | 0.848 | 0.91 | 0.001 | 0.001 |
| I120 (mIU/L) | 52.3 ± 34.95 | 92.41 ± 78.14 | 0.046 | 0.142 | 106.17 ± 85.51 | 134.32 ± 71.02 | 0.13 | 0.272 | 0.001 | 0.002 | |
| HOMA-IR | 1.8 ± 0.87 | 2.94 ± 3.73 | 0.269 | 0.564 | 4.27 ± 2.13 | 4.67 ± 2.7 | 0.847 | 0.91 | 0.001 | 0.001 | |
| Methylation | FKBP5-Met1 | 51.86 ± 3.4 | 50.61 ± 2.41 | 0.182 | 0.421 | 54.59 ± 6.13 | 51.24 ± 4.78 | 0.041 | 0.144 | 0.093 | 0.146 |
| FKBP5-Met2 | 80.41 ± 4.41 | 80.51 ± 4.07 | 0.857 | 0.972 | 84.81 ± 8.54 | 79.72 ± 7.3 | 0.046 | 0.144 | 0.161 | 0.208 | |
| FKBP5-Met | 66.14 ± 3.64 | 65.56 ± 2.67 | 0.839 | 0.972 | 69.7 ± 7.06 | 65.48 ± 5.79 | 0.045 | 0.144 | 0.153 | 0.203 |
The data are shown as the mean ± SD
BMI Body mass index, LDH Lactate dehydrogenase, AST Aspartate aminotransferase, ALT Alanine aminotransferase, GGT Glutamyltransferase, ALP Alkaline phosphatase, CHE Cholinesterase, MAO Monoamine oxidase, AFU α-L-fucosidase, TP Total protein, ALB Albumin, GLB Globulin, TBIL Total bilirubin, DBIL Direct bilirubin, IBIL Indirect bilirubin, BUN urea nitrogen, Cr Creatinine, UA Uric acid, TG Triglycerides, TC Total cholesterol, HDL-C High-density lipoprotein cholesterol, LDL-C Low-density lipoprotein cholesterol, LH Luteinizing hormone, FSH Follicle stimulating hormone, PRL Prolactin, E2 Estrogen, PROG Progesterone, AD Androstenedione, TT Total testosterone, DHEA Dehydroepiandrosterone, FAI free androgen index, SHBG Sex hormone-binding globulin, G0 glucose level at fasting status, G120 Glucose level at 120 min after glucose load, I0 insulin level at fasting status, I120 insulin level at 120 min after glucose load, HOMA-IR homeostasis model assessment for IR, FKBP5-Met1 FKBP5 DNA methylation at CpG 35,657,180/hg19, FKBP5-Met2 FKBP5 DNA methylation at CpG 35,657,202/hg19, FKBP5-Met average of FKBP5 DNA methylation at CpG 35,657,180 and 35,657,202/hg19
Altered bacterial diversity and community in PCOS patients
An average of 143 observed OTUs/sample were obtained from 88 samples after 16S rRNA gene sequencing (Table S2). Significantly reduced α-diversity was observed in PCOS-HB compared with Healthy-HB (p < 0.05) (Fig. 2a). There were no significant relationships between the bacterial diversity index and key clinical variables(p > 0.05) (Table S3). PCoA revealed significant differences (Fig. 2b) among the groups (PERMANOVA: p < 0.05; ANOSIM, p < 0.05). There were no significant correlations between key clinical variables and bacterial community (p > 0.05) (Fig. 2c).Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria accounted for more than 90% of the total phylum (Figure S1a). Bacteroides, Prevotella_9, Faecalibacterium, and Roseburia were the dominant bacterial genera (Figure S1b). LEfSe analysis revealed Ruminococcus torques,Escherichia/Shigella, Allisonella, Eggerthella and Hungatella as PCOS-featured genera (Fig. 2d). Lactobacillus, Coprococcus_1, Coprococcus_3 and Catenibacterium showed suggestive differences between Healthy-LB and PCOS-LB (p < 0.05) (Table S4, Figure S2a). Moreover, genus comparison between Healthy-HB and PCOS-HB by the Wilcoxon signed-rank test and LEfSe analysis revealed 25 significant taxa (Table S4, Figure S2b). Bacterial taxa enriched in Healthy-LB were Blautia (4.10%), Dorea (1.50%), whereas Agathobacter (3.28%) and Lactobacillus (0.42%) were enriched in PCOS-LB patients. In the Healthy-HB group, Alistipes (2.10%), Ruminococcus_1 (1.38%) were enriched. Lachnoclostridium (1.66%) and Erysipelotrichaceae_UCG-003 (0.67%) were overrepresented in the PCOS-HB group (Fig. 2e). Coprobacillus and Coprococcus_1 were significantly positively related to SHBG (p < 0.05), and HOMA-IR displayed a positive correlation with Lachnospiraceae_UCG-001 (Fig. 2f).
Fig. 2.

Fecal bacterial characteristic variations associated with PCOS. a α diversity. p values denote the significance among groups. Letters indicate ANOVA grouping. b Differences in bacterial structures as revealed by PCoA analysis. c RDA analyses reflecting differences in gut microbiota structures fitted with significantly correlated clinical properties. d Characteristic bacterial taxa based on LDA effect size (LEfSe) analysis between PCOS patients and healthy individuals. e The distinguished bacterial genera screened by Kruskal–Wallis tests. f The heatmap depicts the relationship between distinguished bacterial genera (screened by Kruskal–Wallis tests) and key clinical parameters
Altered fecal fungal profile in PCOS patients
Healthy-HB exhibited significantly higher Chao1 and Shannon indices than PCOS-LB (Fig. 3a). A significantly positive relationship for BMI and negative relationships for FAI, LH, AD, TT, HDL-C and DHEA with observed OTUs and Chao1 were detected (p < 0.05) (Table S3). Overall, there were significant alterations in the gut fungal community among the four groups (Fig. 3b) (PERMANOVA: p < 0.05; ANOSIM: p < 0.05). TT exerted a significantly higher influence on the fungal community structure (r2 = 0.08, p < 0.05) (Fig. 3c). Ascomycota, Basidiomycota, and Mortierellomycota were the dominant fungal phyla and Mortierellomycota was uniquely higher in Healthy-HB (Figure S3a). Fungal genera, Candida depletion while Fusarium, Mortierella and Solicoccozyma enrichment was observed in Healthy-HB (Figure S3b). Multiple group comparison revealed 16 significantly differential fungal genera in Healthy-LB versus PCOS-LB and 75 taxa in Healthy-HB versus PCOS-HB (Fig. 3d, Table S5). Candida was positively related to HDL-C, and Mortierella was negatively related to BMI but adversely to LH, AD, TT, HDL-C and DHEA (Fig. 3e). PCOS featured fungal indicators in genus level were Candida, Malassezia, Kazachstania, Microascus, Coniochaeta, Xepicula, Paraphoma, Pyrenochaetopsis, Cephaliophora, Epicoccum and Sclerophora (Fig. 3f). PCOS-HB had more distinguished fungal genera as indicators than that of PCOS-LB (Figure S4,S5).
Fig. 3.

Fecal fungal characteristic variations associated with PCOS. a α diversity measured by Chao1 and Shannon indices was highest in Healthy-HB. p values denote the significance among groups. Letters indicate ANOVA grouping. b Differences in fungal structures among Healthy-LB, Healthy-HB, PCOS-HB and PCOS-LB, as revealed by PCoA analysis. c RDA analyses reflecting differences in gut fungal structures fitted with significantly correlated clinical properties. d Distinguished fungal genera screened by Kruskal–Wallis tests. e Correlation between distinguished fungal genera and key clinical parameters. f Characteristic fungal taxa based on LDA effect size (LEfSe) analysis between PCOS patients and healthy individuals
Serum metabolomics alterations in PCOS patients
A total of 601 metabolites were identified and quantified, with significant differences among the four groups (PERMANOVA:p < 0.05; ANOSIM: p < 0.05) (Fig. 4a). BMI, FAI, LH, AD, TT, SHBG and DHEA exerted significantly higher influences on the metabolite community structure (p < 0.05) (Figure S6a). Multiple group comparisons revealed 284 significantly differential metabolites in Healthy-LB versus PCOS-LB and 358 metabolites in Healthy-HB versus PCOS-HB (p < 0.05) (Table S6). Metabolite indicators associated with PCOS, PCOS-LB and PCOS-HB were also revealed by LEfSe analysis (Fig. 4f, Figure S7, S8). The healthy and PCOS groups showed totally different metabolite patterns (Fig. 4b, Figure S6 b,c,d). VIP scores obtained by PLS-DA analysis (partial least squares discriminant analysis) (Fig. 4c, 4d), together with the threshold of FDR adjusted p < 0.05 and |log2FC|> 1, were set as thresholds to screen out featured metabolites. Ten commonly featured serum metabolites of the top VIP value were both observed between comparison pairs of Healthy-LB versus PCOS-LB and Healthy-HB versus PCOS-HB. 3-Hydroxy-2-methyl-4H-pyran-4-one, furfuryl alcohol, iminodiacetic acid, L-dihydroorotic acid, hydroxyacetone, L-ascorbate, myoinositol and pyrroloquinoline quinone were enriched in PCOS patients, and Phe-Phe and Asp-Phe were abundant in healthy individuals (Fig. 4e).
Fig. 4.

Serum metabolite variations associated with PCOS. a Differences in fungal structures as revealed by PCoA analysis. b Heatmap showing the relative abundance of the 40 metabolites screened by Kruskal–Wallis tests. c PLS-DA plot revealing the differential metabolite pattern between Healthy-LB and PCOS-LB. d PLS-DA plot revealing the differential metabolite pattern between Healthy-HB and PCOS-HB. e The distribution of the top 10 metabolites as VIP values ranked as shared by comparison between Healthy-LB and PCOS-LB and between Healthy-HB and PCOS-HB. f Characteristic metabolites based on LDA effect size (LEfSe) analysis between PCOS patients and healthy controls
Integrative multiomic signatures in the diagnosis of PCOS
We built random forest models based on multiomics characteristics individually or their combination to discriminate PCOS patients, PCOS-LB patients and PCOS-HB patients from the corresponding healthy individuals. A five-fold cross-validated random forest model was further employed to select key discriminatory characteristics after first screening by Wilcoxon signed-rank test comparison (Figure S9, Figure S10). This method identified 9 bacterial genera, 4 predicted pathways, 11 fungal genera and the top 30 metabolites that distinguished PCOS from healthy individuals with AUCs of 0.84, 0.64, 0.85 and 1, respectively, and their combination also reached an AUC of 1 in the training dataset (Fig. 5a). Robust efficacy was verified by fungal taxa and metabolites in the test dataset (Fig. 5b). Similarly, the metabolite-derived model was more accurate than the microbe-based model in discriminating PCOS-LB from Healthy-LB (metabolites, AUC = 1; 16S, AUC = 0.75; predicted pathway, AUC = 0.91; ITS2, AUC = 0.88) (Fig. 5c). Functional pathway performed weaker in the test dataset (Fig. 5d). The use of 16S and predicted pathway data showed good performance in distinguishing PCOS-HB from Healthy-HB (AUC = 0.88 and 0.92, respectively), but poor performance (AUC = 0.67 and 0.58) was observed in the test dataset (Fig. 5e, 5f). The findings indicated that metabolites alone can achieve great performance in distinguishing disease from health, much better than microbiota-driven features. Nevertheless, fungal features performed better than bacterial genera in discriminating PCOS from health.
Fig. 5.

Disease status classification using disease-associated taxa and/or metabolites. (a, c, e) Random forest classifiers were constructed to discriminate PCOS and healthy, PCOS-LB and Healthy-LB, PCOS-HB and Healthy-HB, respectively, in the training dataset. (b, d, f) Random forest classifiers composed of bacterial and fungal genera, metabolites, predicted pathways and their combinations were constructed to discriminate PCOS and healthy, PCOS-LB and Healthy-LB, PCOS-HB and Healthy-HB in the test dataset. ROC, receiver operating characteristic curve. AUC, area under the curve. The input features were excavated on the basis of Wilcox test comparison and the mean decrease in Gini by random forest importance parameters. Data were assigned to training (80%) and test (20%) datasets after the whole dataset was shuffled
Multiomic data interaction associated with PCOS patients
The Mantel test indicated that only the metabolite distance matrix, and not 16S or ITS2, predicted pathway distance, remained significantly positively correlated with phenotypic distance (r = 0.15, p = 0.001) (Table S7). Although there was a significantly positive relationship between the 16S and ITS2 data matrices (r = 0.07, p = 0.015), the bacterial diversity showed no significant linear correlation with fungal diversity (Figure S11).
We tested for the correlations of different multiomics characteristics derived from the above-mentioned diagnostic study based on Spearman’s correlation test (∣r∣ > 0.4). In healthy vs. PCOS, fecal bacteria, fungi, and predicted pathways displayed more associations with the clinical indices FAI, PROG and AD. For example, higher levels of FAI were accompanied by increased cytidine-5-monophosphate (p < 0.05, r = 0.67) and iminodiacetic acid (p < 0.05, r= 0.63) and lower allopurinol (p < 0.05, r = -0.63), lactulose (p < 0.05, r= -0.62), lactose (p < 0.05, r = -0.62) and maltose (p < 0.05, r = -0.62) levels. The fungal taxa Solicoccozyma, Tetracladium, Malassezia, Knufia and Exophiala were also center-connected (Fig. 6a). Therefore, higher levels of Solicoccozyma were negatively related to 2,4-hexadienoic acid but positively to L-homocystine. Malassezia showed negative correlations with L-homocystine (p < 0.05, r = -0.59) but positive correlations with 2-c-methyl-d-erythritol 2,4-cyclodiphosphate (p < 0.05, r = 0.54).
Fig. 6.
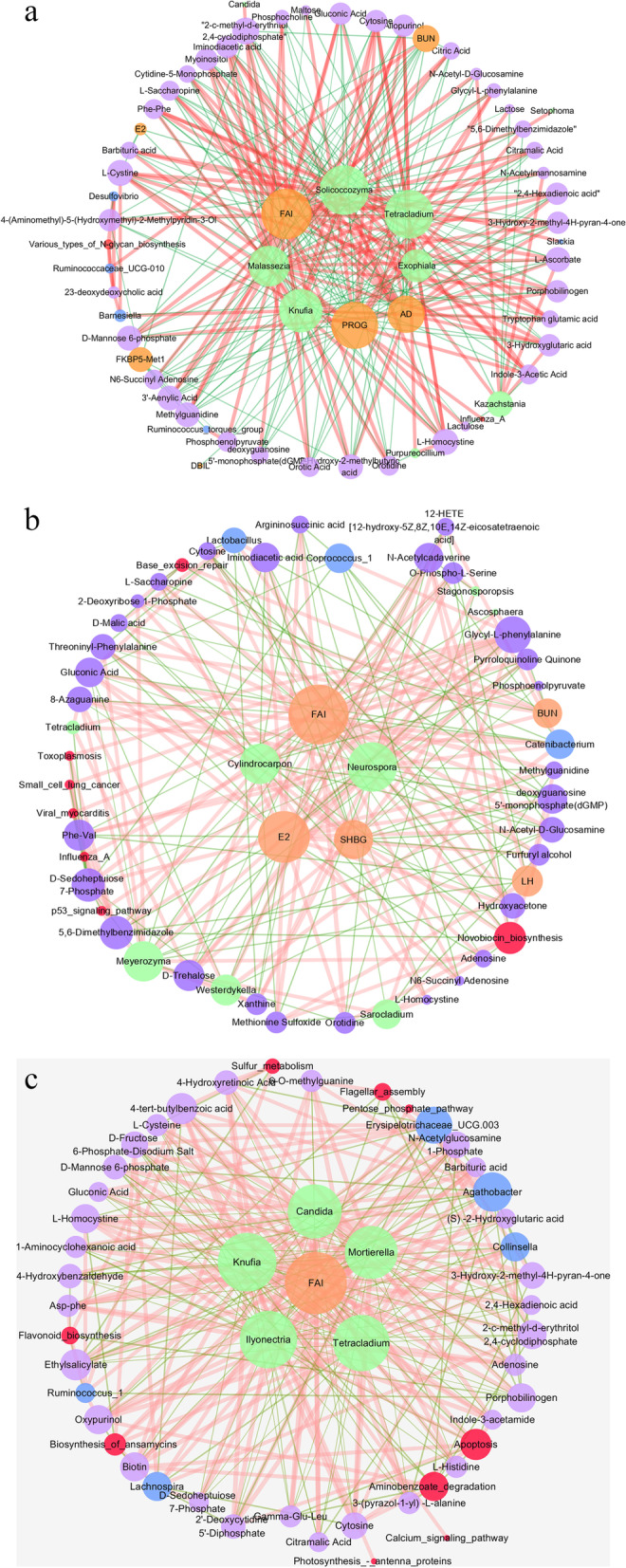
Integrative co-occurrence network reflecting multiomic-phenotype interactions. (a,b,c) Network revealed both significant (p < 0.05) and suggestive correlations (∣r∣ > 0.4, Spearman analysis) between differentially abundant bacterial, fungal, predicted pathway, metabolites and clinical indices in PCOS and healthy, PCOS-LB and Healthy-LB, PCOS-HB and Healthy-HB. Nodes represent characteristics. Purple, blue, red, green and yellow nodes denote metabolites, bacterial taxa, predicted pathways, fungal taxa and clinical parameters. Lines connecting nodes indicate positive (red) or negative (green) correlations
In Healthy-LB vs. PCOS-LB, we observed that many metabolic features were associated with FAI, E2 and SHBG. For example, FAI was positively associated with methylguanidine (p < 0.05, r = 0.76) and hydroxyacetone (p < 0.05, r = 0.70) but negatively correlated with 2-deoxyribose 1-phosphate (p < 0.05, r = -0.70). Featured bacterial taxa, Catenibacterium, Coprococcus_1 and Lactobacillus, were highly related to Westerdykella, gluconic acid and xanthine individually. The fungal taxa Cylindrocarpon and Neurospora exhibited positive and negative relationships with D-malic acid (Fig. 6b). With regard to Healthy-HB vs. PCOS-HB, increased Candida was linked to higher levels of 3-hydroxy-2-methyl-4H-pyran-4-one (p < 0.05, r = 0.87) and 2-c-methyl-d-erythritol 2,4-cyclodiphosphate (p < 0.05, r = 0.83) and lower levels of barbituric acid (p < 0.05, r = -0.82). Mortierella showed a highly negative correlation with D-fructose 6-phosphate-disodium salt (p < 0.05, r = -0.82) and D-mannose 6-phosphate (p < 0.05, r = -0.82) but a positive correlation with cytosine (p < 0.05, r = 0.83) and barbituric acid (p < 0.05, r = 0.75). The FAI was negatively related to Agathobacter (p < 0.05, r = -0.53) but positively with Erysipelotrichaceae_UCG-003 (p < 0.05, r = 0.43) (Fig. 6c).
Discussion
In this study, we outlined landscapes and interaction networks of clinical indices, differential gut bacteria, fungi and serum metabolites in PCOS patients with different BMIs. According to integrated analysis of multiomics data, the most important index is FAI, which is used as a common indicator of hyperandrogenemia [37]. Overall, androgen plays an important leading role in the changes in gut bacteria, gut fungi and metabolites in patients with PCOS. Moreover, we identified and independently validated a combinatorial marker panel that was able to distinguish PCOS from non-PCOS subjects with high accuracy.
Saccharomyces cerevisiae, Malassezia restricta, Candida albicans, Candida sake, Cyberlindnera jadinii, Cladosporium spp., Penicillium spp., and Galactomyces candidum are the most prevalent fungi in the human gut [38]. Mar Rodríguez et al. [16] showed that patients with and without obesity could be distinguished by their specific fungal composition. Candida, Nakaseomyces and Penicillium were the most abundant genera detected in patients with obesity, and Mucor was the most prevalent genera in patients without obesity. Chacón et al. [39] paid attention to the compositions of Mucor spp. and found that subjects with obesity and undetectable Mucor spp. showed a significantly worse cardiovascular risk profile. Research on gut fungi in metabolic diseases is still very limited to date. Jayasudha et al. [40] showed that Mucoromycota was the only phylum showing significantly decreased abundance in T2DM compared to control mycobiomes, and the median abundance of Candida along with Cladosporium, Kodamaea, Meyerozyma and Mortierella were increased in people with T2DM. In our study, Ascomycota, Basidiomycota, and Mortierellomycota were the dominant fungal phyla, and Mortierellomycota was uniquely higher in healthy HB individuals than in the other groups. Reduced fungal family diversity has been demonstrated in individuals with obesity prepiously [16]. In our study, Non-PCOS patients with overweight/obesity seemed less metabolically abnormal than those with PCOS, and whether the composition of different gut fungi exerts metabolic protection needs to be further assessed. Overall, the human gut mycobiome has been poorly studied and characterized in patients with PCOS. Our study showed that PCOS patients are featured in fungal indicators, such as Candida, Malassezia, Kazachstania, Microascus, Coniochaeta, Xepicula, Paraphoma, Pyrenochaetopsis, Cephaliophora, Epicoccum and Sclerophora, while PCOS-HB patients having more distinguished fungal genera as indicators.
Several factors in the host will have an effect on mycobiome composition and variations, including genotype, physiology, immune system, and lifestyle [41, 42]. Among them, diet is an important factor [43]. For example, Methanobrevibacter and Candida are positively associated with diets high in carbohydrates but negatively associated with diets high in amino acids, protein, and fatty acids [44]. A plant-based diet is also associated with enrichment in Candida spp., whereas an animal-product-based diet is associated with enrichment in Debaryomyces spp. and Penicillium spp [43]. It is widely recognized that gut fungal diversity is significantly lower than bacterial diversity [45], but each fungal cell genome is approximately 100-fold larger than that of bacterial cells, which represents a significant biomass with numerous functions [46]. Bacteria are fundamental to maintain a balanced gut microbiota and to avoid fungal overgrowth [46], whereas imbalance in gut fungi leads to an abnormal composition of gut bacteria. Hoffmann et al. [44] reported that Candida and Saccharomyces are both positively associated with Methanobrevibacter and that both fungal genera are negatively associated with Nitrososphaera. Wheeler et al.[10] showed that fungi, including Penicillium brevicompactum and Candida tropicalis, are significantly decreased with antifungal treatment in mice but that this treatment leads to relative expansion of Aspergillus amstelodami, Epicoccum nigrum, and Wallemia sebi. Our study shows that in patients with PCOS, the change in gut fungi is more significant than that in gut bacteria. According to Mims et al. [47], jejunal fungal communities are indeed dynamic and more susceptible to environmental influences than bacteria in healthy mice. The significance and value of gut fungi in the occurrence and development of PCOS are still not very clear, but previous studies may give some hints. For example, Candida albicans is considered as the major inducer of human antifungal Th17 cells [48] and increases interleukin-22 (IL-22) production [49]. Qi el at. [7] reported that Bacteroides vulgatus is markedly elevated in the gut microbiota and that the level of IL-22 is reduced. Taken together, both gut bacteria and gut fungi can affect the level of IL-22 and promote the occurrence and development of PCOS.At present, many hospitals and clinical centers use the Rotterdam standard for the diagnosis of PCOS. Haoula et al. [50] tentatively identified lipid biomarkers of PCOS, which may be useful in distinguishing PCOS according to targeted lipomics analysis. To find more effective and simple indicators used for the diagnosis and screening of PCOS, we built random forest models based on multiomics characteristics individually or their combination to discriminate PCOS from healthy controls. The findings showed that metabolites alone can achieve great performance in distinguishing disease from health, much better than microbiota-driven features, and that fungal features still perform better than bacterial genera in discriminating a PCOS status from a healthy status.
Our study still had limitations. Firstly, this is a single-center study with limited sample size, future multicenter research involving multiple different geographic areas are necessary to verify the data before clinical application. Secondly, although 16S rRNA gene sequencing analysis was conducted, higher taxa resolution until species level by metagenomics sequencing would enhance the results power. Thirdly, although multiomics data were used to reveal functional links between microbiome, metabolome and phenotype, cause-and-effect evidence which would address the chicken-and-egg debate problem need to be further clarified in animal studies. Last but not the least, fecal metabolomic analysis is also valuable to deeply understand how the gut microbiome metabolize directly.
In conclusion, integrated analysis of multiomics data from the gut bacteriome, mycobiome, metabolome and phenome showed that hyperandrogenemia plays a central role in the dysbiosis of intestinal microecology and the change in metabolic state in patients with PCOS and that its effect exceeds the role of BMI. Gut bacteria, gut fungi and their interactions may be important in the occurrence and development of PCOS. The priority of predictive models in discriminating PCOS status in this study were serum metabolites, fungal taxa and bacterial taxa.
Supplementary Information
Additional file 1: Table S1. Clinical characteristics in healthy and PCOS group; Table S2. Sequence statistics and bacterial diversity indices from 16S rRNA and ITS2 gene sequencing; Table S3. The correlation between bacterial or fungal diversity and key clinical parameters; Table S4. The relative abundance comparison of bacterial genera among different groups; Table S5. The relative abundance comparison of fungal genera among different groups; Table S6. The relative abundance comparison of metabolites among different groups; Table S7. Mantel test between pairs of distance matrix
Additional file 2: Figure S1. Distribution of bacterial taxa at the phylum level and genus level; Figure S2. Reveal of characteristic bacterial taxa based on LDA Effect Size (LEfSe) analysis between Healthy-LB and PCOS-LB, between Healthy-HB and PCOS-HB; Figure S3. Distribution of fungal taxa at the phylum level and genus level; Figure S4. Reveal of characteristic fungal taxa based on LDA Effect Size (LEfSe) analysis between Healthy-LB and PCOS-LB; Figure S5. Reveal of characteristic fungal taxa based on LDA Effect Size (LEfSe) analysis between Healthy-HB and PCOS-HB; Figure S6. Serum metabolome changes in disease and healthy subjects; Figure S7. Reveal of characteristic metabolites based on LDA Effect Size (LEfSe) analysis between Healthy-LB and PCOS-LB; Figure S8. Reveal of characteristic metabolites based on LDA Effect Size (LEfSe) analysis between Healthy-HB and PCOS-HB; Figure S9. The bacterial genera and predicted pathway features were excavated on the base of Wilcox test comparison and Mean Decrease Gini by random forest importance parameter; Figure S10. The fungal genera and metabolites were excavated on the base of Wilcox test comparison and Mean Decrease Gini by random forest importance parameter; Figure S11. Correlation between bacterial diversity and fungal diversity.
Acknowledgements
The author would like to thank Nature Research Editing Service for technical language editing of the manuscript.
Institutional review board statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the First Affiliated Hospital Shantou University Medical College.
Informed consent statement
Written informed consent was obtained from all participants.
Abbreviations
- AD
Androstenedione
- AFU
α-L-fucosidase
- ALB
Albumin
- ALP
Alkaline phosphatase
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- BMI
Body mass index
- BUN
Urea nitrogen
- CHE
Cholinesterase
- Cr
Creatinine
- DBIL
Direct bilirubin
- DHEA
Dehydroepiandrosterone
- DPP-IV
Dipeptidyl peptidase IV
- E2
Estrogen
- FSH
Follicle stimulating hormone
- FAI
Free androgen index
- FDR
False discovery rate
- FKBP5-Met1
FKBP5 DNA methylation at CpG 35,657,180/hg19
- FKBP5-Met2
FKBP5 DNA methylation at CpG 35,657,202/hg19
- FKBP5-Met
Average of FKBP5 DNA methylation at CpG 35,657,180 and 35,657,202/hg19
- G0
Glucose level at fasting status
- G120
Glucose level at 120 min after glucose load
- GGT
Glutamyltransferase
- GLB
Globulin
- HDL-C
High-density lipoprotein cholesterol
- HOMA-IR
Homeostasis model assessment for insulin resistance
- IBIL
Indirect bilirubin
- I0
Insulin level at fasting status
- I120
Insulin level at 120 min after glucose load
- LDH
Lactate dehydrogenase
- LDL-C
Low-density lipoprotein cholesterol
- LH
Luteinizing hormone
- LPC
Lysophosphatidyl choline
- MAO
Monoamine oxidase
- NAFLD
Nonalcoholic fatty liver disease
- PCoA
Principal co-coordinates analysis
- PCOS
Polycystic ovary syndrome
- PRL
Prolactin
- PROG
Progesterone
- RBP-4
Retinol-binding protein 4
- SHBG
Sex hormone-binding globulin
- T2DM
Type 2 diabetes mellitus
- TBIL
Total bilirubin
- TC
Total cholesterol
- TG
Triglycerides
- TP
Total protein
- TT
Total testosterone
- UA
Uric acid
- VIP
Variable importance in projection
Authors’ contributions
Conceived and designed the study: ZRC, GSY, YSC, JLR and FC; Participated in investigation: MJC, and LC; Performed formal analysis: ZRC, MC, GSY and FC; Collected the resources: GSC, XPY, QXH, MJC, WCZ, MQO, HL, and MC; Curated the data: ZRC, JLR and HZX; Wrote the manuscript: GSY and ZRC; Supervised the study: ZRC, YSC, JLR and GSY. All authors approved the final version of the manuscript and agreed for publication once accepted. All authors read and approved the final manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (81200619, 81900541), the Medical Scientific Research Foundation of Guangdong Province of China (A2020390) and Guangdong Provincial Science and Technology Special Fund (210715106900958).
Availability of data and materials
The 16S rRNA gene sequencing dataset supporting the conclusions of this article is available in the NCBI Sequence Read Archive repository under the accession number PRJNA786067 (https://submit.ncbi.nlm.nih.gov/subs/sra/SUB10762045/overview) and the ITS2 gene sequencing dataset is available in the SRA repository PRJNA786056 (https://submit.ncbi.nlm.nih.gov/subs/sra/SUB10756642/overview). The metabolome data is available in www.ebi.ac.uk/metabolights/MTBLS5982. Code and scripts used in the analyses are available upon request.
Declarations
Competing interests
The authors declare no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Guoshu Yin and Fu Chen contributed equally to this work.
Contributor Information
Guoshu Yin, Email: yinguoshu@126.com.
Jianlin Ren, Email: renjianl@xmu.edu.cn.
Yongsong Chen, Email: yongsongchen@126.com.
Zhangran Chen, Email: zhangran22105@163.com.
References
- 1.Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14(5):270–284. doi: 10.1038/nrendo.2018.24. [DOI] [PubMed] [Google Scholar]
- 2.Chen F, Liao Y, Chen M, Yin H, Chen G, Huang Q, et al. Evaluation of the Efficacy of Sex Hormone-Binding Globulin in Insulin Resistance Assessment Based on HOMA-IR in Patients with PCOS. Reprod Sci. 2021;28(9):2504–2513. doi: 10.1007/s43032-021-00535-0. [DOI] [PubMed] [Google Scholar]
- 3.Zhu T, Cui J, Goodarzi MO. Polycystic Ovary Syndrome and Risk of Type 2 Diabetes, Coronary Heart Disease, and Stroke. Diabetes. 2021;70(2):627–637. doi: 10.2337/db20-0800. [DOI] [PubMed] [Google Scholar]
- 4.Vallianou N, Stratigou T, Christodoulatos GS, Dalamaga M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Obesity and Obesity-Associated Metabolic Disorders: Current Evidence and Perspectives. Curr Obes Rep. 2019;8(3):317–332. doi: 10.1007/s13679-019-00352-2. [DOI] [PubMed] [Google Scholar]
- 5.Hartstra AV, Bouter KE, Bäckhed F, Nieuwdorp M. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care. 2015;38(1):159–165. doi: 10.2337/dc14-0769. [DOI] [PubMed] [Google Scholar]
- 6.Chen F, Chen Z, Chen M, Chen G, Huang Q, Yang X, et al. Reduced stress-associated FKBP5 DNA methylation together with gut microbiota dysbiosis is linked with the progression of obese PCOS patients. NPJ Biofilms Microbiomes. 2021;7(1):60. doi: 10.1038/s41522-021-00231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi X, Yun C, Sun L, Xia J, Wu Q, Wang Y, et al. Gut microbiota-bile acid-interleukin-22 axis orchestrates polycystic ovary syndrome. Nat Med. 2019;25(8):1225–1233. doi: 10.1038/s41591-019-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang YL, Zhou WW, Wu S, Tang WL, Wang ZW, Zhou ZY, et al. Intestinal Flora is a Key Factor in Insulin Resistance and Contributes to the Development of Polycystic Ovary Syndrome. Endocrinology. 2021;162(10):bqab118. doi: 10.1210/endocr/bqab118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Zhang L, Zhai Q, Zhao J, Zhang H, Lee YK, et al. Chinese gut microbiota and its associations with staple food type, ethnicity, and urbanization. NPJ Biofilms Microbiomes. 2021;7(1):71. doi: 10.1038/s41522-021-00245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe. 2016;19(6):865–873. doi: 10.1016/j.chom.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sovran B, Planchais J, Jegou S, Straube M, Lamas B, Natividad JM, et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome. 2018;6(1):152. doi: 10.1186/s40168-018-0538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coker OO, Nakatsu G, Dai RZ, Wu W, Wong SH, Ng SC, et al. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut. 2019;68(4):654–662. doi: 10.1136/gutjnl-2018-317178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rühlemann MC, Solovjeva M, Zenouzi R, Liwinski T, Kummen M, Lieb W, et al. Gut mycobiome of primary sclerosing cholangitis patients is characterised by an increase of Trichocladium griseum and Candida species. Gut. 2020;69(10):1890–1892. doi: 10.1136/gutjnl-2019-320008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M, et al. Intestinal Fungal Dysbiosis and Systemic Immune Response to Fungi in Patients With Alcoholic Hepatitis. Hepatology. 2020;71(2):522–538. doi: 10.1002/hep.30832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuo T, Zhan H, Zhang F, Liu Q, Tso E, Lui G, et al. Alterations in Fecal Fungal Microbiome of Patients With COVID-19 During Time of Hospitalization until Discharge. Gastroenterology. 2020;159(4):1302–10.e5. doi: 10.1053/j.gastro.2020.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mar Rodríguez M, Pérez D, Javier Chaves F, Esteve E, Marin-Garcia P, Xifra G, et al. Obesity changes the human gut mycobiome. Sci Rep. 2015;5:14600. doi: 10.1038/srep14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honkanen J, Vuorela A, Muthas D, Orivuori L, Luopajärvi K, Tejesvi M, et al. Fungal Dysbiosis and Intestinal Inflammation in Children With Beta-Cell Autoimmunity. Front Immunol. 2020;11:468. doi: 10.3389/fimmu.2020.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J, Zhang Y, Wang X, Ren X, Liu Y. Changes and roles of intestinal fungal microbiota in coronary heart disease complicated with nonalcoholic fatty liver disease. Am J Transl Res. 2020;12(7):3445–3460. [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Tan P, Zhuang Z, Wang Z, Zhu L, Qiu R, et al. Untargeted metabolomic approach to study the serum metabolites in women with polycystic ovary syndrome. BMC Med Genomics. 2021;14(1):206. doi: 10.1186/s12920-021-01058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alesi S, Ghelani D, Mousa A. Metabolomic Biomarkers in Polycystic Ovary Syndrome: A Review of the Evidence. Semin Reprod Med. 2021;39(3–04):102–110. doi: 10.1055/s-0041-1729841. [DOI] [PubMed] [Google Scholar]
- 21.Daan NM, Koster MP, de Wilde MA, Dalmeijer GW, Evelein AM, Fauser BC, et al. Biomarker Profiles in Women with PCOS and PCOS Offspring; A Pilot Study. PLoS ONE. 2016;11(11):e0165033. doi: 10.1371/journal.pone.0165033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Chu Q, Ma J, Sun Y, Tao T, Huang R, et al. Discovery of Novel Lipid Profiles in PCOS: Do Insulin and Androgen Oppositely Regulate Bioactive Lipid Production. J Clin Endocrinol Metab. 2017;102(3):810–821. doi: 10.1210/jc.2016-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dapas M, Lin F, Nadkarni GN, Sisk R, Legro RS, Urbanek M, et al. Distinct subtypes of polycystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis. PLoS Med. 2020;17(6):e1003132. doi: 10.1371/journal.pmed.1003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong Y, Sun H, Yang W, Ma S, Du B, Xu H. The Effect of Inulin on Lifespan, Related Gene Expression and Gut Microbiota in InRp5545/TM3 Mutant Drosophila melanogaster: A Preliminary Study. Nutrients. 2019;11(3):636. doi: 10.3390/nu11030636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanif MA, Guo Z, Moniruzzaman M, He D, Yu Q, Rao X, et al. Plant Taxonomic Diversity Better Explains Soil Fungal and Bacterial Diversity than Functional Diversity in Restored Forest Ecosystems. Plants (Basel) 2019;8(11):479. doi: 10.3390/plants8110479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6(1):90. [DOI] [PMC free article] [PubMed]
- 29.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mueller RC, Paula FS, Mirza BS, Rodrigues JL, Nüsslein K, Bohannan BJ. Links between plant and fungal communities across a deforestation chronosequence in the Amazon rainforest. ISME J. 2014;8(7):1548–1550. doi: 10.1038/ismej.2013.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42:D643-8. [DOI] [PMC free article] [PubMed]
- 33.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ijaz MU, Ahmed MI, Zou X, Hussain M, Zhang M, Zhao F, et al. Beef, Casein, and Soy Proteins Differentially Affect Lipid Metabolism, Triglycerides Accumulation and Gut Microbiota of High-Fat Diet-Fed C57BL/6J Mice. Front Microbiol. 2018;9:2200. doi: 10.3389/fmicb.2018.02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.A Liaw MW Classification and regression by randomForest. R news. 2002;2:18–22. [Google Scholar]
- 36.Kuhn M. Building predictive models in R using the caret package. J STAT SOFTW. 2008;5:1. [Google Scholar]
- 37.Chen F, Chen M, Zhang W, Yin H, Chen G, Huang Q, et al. Comparison of the efficacy of different androgens measured by LC-MS/MS in representing hyperandrogenemia and an evaluation of adrenal-origin androgens with a dexamethasone suppression test in patients with PCOS. J Ovarian Res. 2021;14(1):32. doi: 10.1186/s13048-021-00781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. 2017;5(1):153. doi: 10.1186/s40168-017-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chacón MR, Lozano-Bartolomé J, Portero-Otín M, Rodríguez MM, Xifra G, Puig J, et al. The gut mycobiome composition is linked to carotid atherosclerosis. Benef Microbes. 2018;9(2):185–198. doi: 10.3920/BM2017.0029. [DOI] [PubMed] [Google Scholar]
- 40.Jayasudha R, Das T, KalyanaChakravarthy S, Sai Prashanthi G, Bhargava A, Tyagi M, et al. Gut mycobiomes are altered in people with type 2 Diabetes Mellitus and Diabetic Retinopathy. PLoS ONE. 2020;15(12):e0243077. doi: 10.1371/journal.pone.0243077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui L, Morris A, Ghedin E. The human mycobiome in health and disease. Genome Med. 2013;5(7):63. doi: 10.1186/gm467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Zhan H, Xu W, Yan S, Ng SC. The role of gut mycobiome in health and diseases. Therap Adv Gastroenterol. 2021;14:17562848211047130. doi: 10.1177/17562848211047130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD, Lewis JD, Bushman FD. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE. 2013;8(6):e66019. doi: 10.1371/journal.pone.0066019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forbes JD, Bernstein CN, Tremlett H, Van Domselaar G, Knox NC. A Fungal World: Could the Gut Mycobiome Be Involved in Neurological Disease. Front Microbiol. 2018;9:3249. doi: 10.3389/fmicb.2018.03249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Musumeci S, Coen M, Leidi A, Schrenzel J. The human gut mycobiome and the specific role of Candida albicans: where do we stand, as clinicians. Clin Microbiol Infect. 2021. [DOI] [PubMed]
- 47.Mims TS, Abdallah QA, Stewart JD, Watts SP, White CT, Rousselle TV, et al. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun Biol. 2021;4(1):281. doi: 10.1038/s42003-021-01820-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell. 2019;176(6):1340–55.e15. doi: 10.1016/j.cell.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 49.Wolk K, Witte E, Hoffmann U, Doecke WD, Endesfelder S, Asadullah K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn’s disease. J Immunol. 2007;178(9):5973–81. [DOI] [PubMed]
- 50.Haoula Z, Ravipati S, Stekel DJ, Ortori CA, Hodgman C, Daykin C, et al. Lipidomic analysis of plasma samples from women with polycystic ovary syndrome. Metabolomics. 2015;11(3):657–666. doi: 10.1007/s11306-014-0726-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Clinical characteristics in healthy and PCOS group; Table S2. Sequence statistics and bacterial diversity indices from 16S rRNA and ITS2 gene sequencing; Table S3. The correlation between bacterial or fungal diversity and key clinical parameters; Table S4. The relative abundance comparison of bacterial genera among different groups; Table S5. The relative abundance comparison of fungal genera among different groups; Table S6. The relative abundance comparison of metabolites among different groups; Table S7. Mantel test between pairs of distance matrix
Additional file 2: Figure S1. Distribution of bacterial taxa at the phylum level and genus level; Figure S2. Reveal of characteristic bacterial taxa based on LDA Effect Size (LEfSe) analysis between Healthy-LB and PCOS-LB, between Healthy-HB and PCOS-HB; Figure S3. Distribution of fungal taxa at the phylum level and genus level; Figure S4. Reveal of characteristic fungal taxa based on LDA Effect Size (LEfSe) analysis between Healthy-LB and PCOS-LB; Figure S5. Reveal of characteristic fungal taxa based on LDA Effect Size (LEfSe) analysis between Healthy-HB and PCOS-HB; Figure S6. Serum metabolome changes in disease and healthy subjects; Figure S7. Reveal of characteristic metabolites based on LDA Effect Size (LEfSe) analysis between Healthy-LB and PCOS-LB; Figure S8. Reveal of characteristic metabolites based on LDA Effect Size (LEfSe) analysis between Healthy-HB and PCOS-HB; Figure S9. The bacterial genera and predicted pathway features were excavated on the base of Wilcox test comparison and Mean Decrease Gini by random forest importance parameter; Figure S10. The fungal genera and metabolites were excavated on the base of Wilcox test comparison and Mean Decrease Gini by random forest importance parameter; Figure S11. Correlation between bacterial diversity and fungal diversity.
Data Availability Statement
The 16S rRNA gene sequencing dataset supporting the conclusions of this article is available in the NCBI Sequence Read Archive repository under the accession number PRJNA786067 (https://submit.ncbi.nlm.nih.gov/subs/sra/SUB10762045/overview) and the ITS2 gene sequencing dataset is available in the SRA repository PRJNA786056 (https://submit.ncbi.nlm.nih.gov/subs/sra/SUB10756642/overview). The metabolome data is available in www.ebi.ac.uk/metabolights/MTBLS5982. Code and scripts used in the analyses are available upon request.